metabolic disorders knh 413. metabolic disorders inborn errors of metabolism – group of diseases...
TRANSCRIPT

Metabolic Disorders
KNH 413

Metabolic Disorders
Inborn errors of metabolism – group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty acids, sugars or metals caused by a defect in the activity of an enzyme

Metabolic Disorders
InheritanceMost inborn errors are autosomal recessive
Carrier parents have a 25% chance of an affected childMutations – permanent, transmissible changes in the
genetic material Differences in degree of stability and activity of enzyme Severity described by time of onset Classical form most severe

Metabolic Disorders
Impaired Metabolism - PathophysiologyDeficient or absent enzyme activity orChanges in binding site of cofactorPrecursors accumulated d/t block or impaired
feedback inhibitionToxic metabolites produced as a result of the build upOr deficiency of needed end productSecondary nutritional deficiencies


Metabolic Disorders
Diagnosis/ Newborn ScreeningNonselective screening – screening all newborns for a
limited number of common inborn errorsSelective – testing of an individual known to be at
increased risk (e.g. sibling)All states screen for PKU, variability in other disorders
screenedTandem mass spectroscopy – allows clinicians to
screen for > 30 disorders

Metabolic DisordersClinical manifestations
Usually appear 24 hours or more after birth, attributed to ingestion of precursor substrate of defective enzyme
CNS symptoms, poor growth, failure to thrive, developmental delays, specific neurological deficits
May have blatant signs (i.e. unusual odor)


Metabolic DisordersClinical manifestations – diagnosis
Laboratory studies Routine
Hypoglycemia, acid-base balance, hyperammonemia, ketosis
Specialized studies Require special lab Directed analysis for amino acids or organic acids

© 2007 Thomson - Wadsworth

Metabolic DisordersApproaches to Treatment
Acute therapy Correction of acid-base balance and hydration of
immediate importance Maintenance of adequate kcal to prevent tissue
catabolism Offending metabolites restricted

Metabolic DisordersApproaches to Treatment
Chronic Therapy Restriction of precursors Replacement of end products Providing alternate substrates for metabolism Use of scavenger drugs to remove toxic by-products Supplementation of vitamins or other cofactors

Amino Acid Disorders
Phenylketonuria (PKU)
Isovaleric acidemia (IVA)
Maple syrup urine disease (MSUD)
Others

© 2007 Thomson - Wadsworth

Amino Acid DisordersPhenylketonuria (PKU) – most common
Absence of phenylalanine hydroxylase enzyme Inability to convert phenylalanine to tyrosineTyrosine becomes conditionally essential


Amino Acid DisordersPhenylketonuria (PKU)
Results in metal retardation, severe behavioral problems, seizures, eczema
Musty or mousy odorToxic to brain – demyelination of white matterDecreased production of serotonin, epinephrine,
norepinephrine, dopamine, GABA

Amino Acid DisordersPKU – Nutrition Interventions
Restriction of dietary proteinSynthetic formula supplying all essential amino acids
except offending amino acidsBlood phenylalanine target levels more restrictive for
children up to age 12

Amino Acid DisordersPKU – Nutrition Interventions
Assess kcal and protein needsAmount of allowed phenylalanine determined by
enzymatic activity and blood levelsAllow as much protein as possible for adequate growth
from fruits, vegetables, limited amounts of grainsBalance provided by metabolic formulas



Amino Acid DisordersPKU – Nutritional Concerns
Risk for nutritional deficienciesGrowth retardationBone statusAmino acid deficienciesOverrestrictionMetabolic control during pregnancy

Amino Acid DisordersPKU – Adjunct Therapies
AntibioticsCarnitineSodium benzoateSodium phenylbutyrate

Urea Cycle Disorders
Impaired capacity to excrete nitrogen in the form of urea
Cascade of enzymatic reactions which converts ammonia to urea can be blocked
Or a depletion of an amino acid essential to the function of the cycle can result
Causing hyperammonemia

© 2007 Thomson - Wadsworth

© 2007 Thomson - Wadsworth

Urea Cycle Disorders
Hyperammonia may cause loss of appetite, cyclical vomiting, lethargy, learning difficulties, behavioral abnormalities, severe retardation
May require daily assistance, tube feedings, and wheelchairs

Urea Cycle DisordersAcute Treatment
Hemodialysis Sodium benzoate and sodium phenylacetate to
scavenge excess ammonia IV fluids, avoiding overhydrationCaloric supplementationGlucose, intralipidsComplete protein restriction for 24-48 hours

Urea Cycle DisordersNutrition Interventions
Protein adjustment to account for severity, age, growth rate, and individual preferences without any extra
Supplemental arginine for mostMay use essential amino acid mixture to replace
natural sources25-30% of protein intake should be essential amino
acids


Urea Cycle DisordersNutrition Concerns
Amino acid intake must be balancedRisk of micronutrient deficiency
Iron, zinc
Adequate energy intakeNutrition support may be neededContinuous monitoringSee flow sheet example


Urea Cycle DisordersAdjunct therapies
Liver transplantationAlternative pathway therapy

Mitochondrial Disorders
Results from defects either in the respiratory chain or from defects affecting overall number and function of the mitochondria
MELAS or NARP

© 2007 Thomson - Wadsworth

Mitochondrial Disorders
DiagnosisDNA mutation testingSkin and muscle tissue histological and biochemical
analysis
Disorders includeFatty acid transport disordersFatty acid oxidation defectsPyruvate complex disordersRespiratory chain defects

Mitochondrial Disorders
Respiratory Chain Five complexes that undergo changes in their
oxidative state to produce ATP
Defects lead to: Decreased energy production Hypotonia, developmental delay, failure to thrive


Mitochondrial Disorders
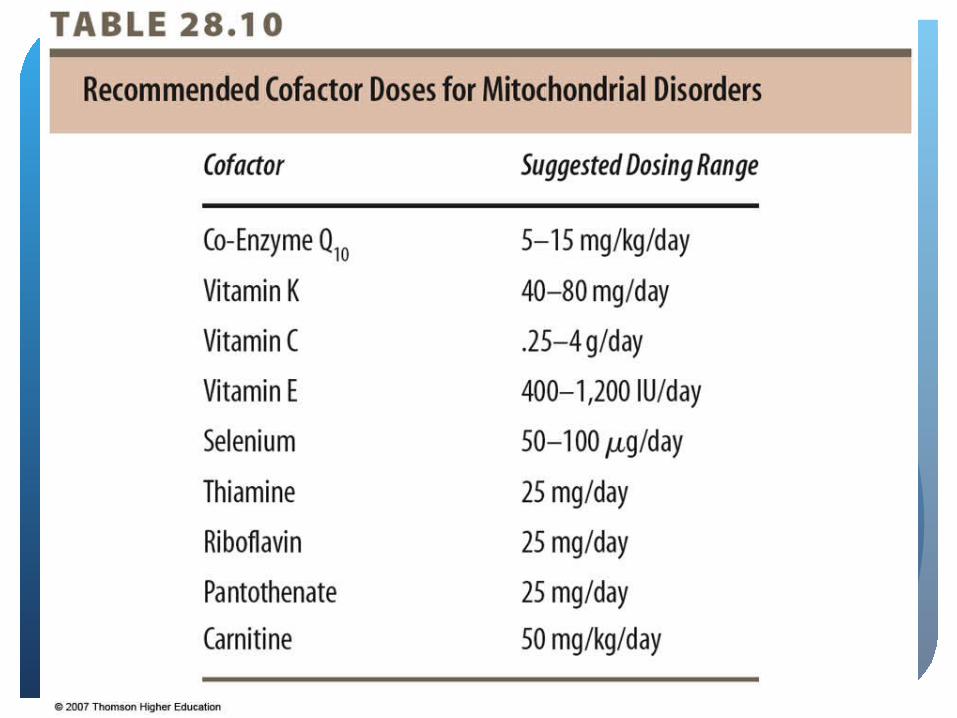
Nutrition InterventionNo definite treatmentUse of vitamin cofactors in pharmacological
amounts 100-1000 times DRI for ageRiboflavin and thiamin – cofactorsVitamin E and lipoic acid – antioxidantsVitamins C, K, CoQ10 – artificial electron receptors
and transporters
Frequent feedings recommended
© 2007 Thomson - Wadsworth

Mitochondrial Disorders
Adjunct therapiesCarnitine and glycine – conjugate with toxic
metabolites, removing them from body

Disorders of Vitamin Metabolism
Needed as cofactors for enzymatic reactions, antioxidants, or electron receptors
Pharmacologic dose may be sufficient to maintain normal enzymatic function

Disorders of Vitamin Metabolism
Nutritional InterventionsMethylmalonic acidemia – responsive to B12
Holocarboxylase synthetase deficiency and biotinidase deficiency - responsive to biotin

© 2007 Thomson - Wadsworth

Disorders of Vitamin Metabolism
Nutritional ConcernsPharmacological doses of vitamins should be treated
as “drugs”Use of “megavitamin” supplements in random
fashion discouragedToxicity a concern for fat-soluble vitaminsComplianceCost

Disorders of Carbohydrate Metabolism
Problems processing simple sugars galactose and fructose, or glycogen storage diseases
Summary of disorders and clinical symptoms


Galactosemia
Enzyme defect in galactose metabolism leading to failure to thrive, hepatomegaly, life-threatening sepsis in newborn periodVomiting, jaundice upon initiation of milk
feedingsAnorexia, failure to gain weight or growCirrhosis, ascites, edema, bleeding problems,
enlarged spleen if milk feedings continue


Galactosemia
Many states screen for it
Defect is in conversion of galactose to glucose 1 phosphate
G1P accumulates in tissue
Clinical manifestations result

Galactosemia
Nutrition InterventionsExclusion of galactose/ lactose from diet
Immediate reversal of symptoms results
Exclusion of human milk, cow’s milk …Substitution of casein hydrolysate-containing
formulaInfant soy formulasLearn other potential dietary and drug
sources of galactoseSee Table 28.12

© 2007 Thomson - Wadsworth

Galactosemia
Nutrition concernsProvision of alternative sources of missing nutrients:
vitamin D, calciumCalcium supplementsMeet kcal, protein, vitamin and mineral needs

Hereditary Fructose Intolerance
Deficiency of fructose 1 phosphate aldolase
Accumulation in tissues containing fructokinase, causing depletion of inorganic phosphate and ATP
Fructose-induced hypoglycemia
d/t ingestion of fructose, sucrose, or sorbitol in diet

Hereditary Fructose Intolerance
Clinical manifestationsVomitingPoor feeding, diarrhea, failure to thriveHepatomegaly, bleeding tendency, jaundice, edema,
ascites

Hereditary Fructose Intolerance
Nutrition InterventionWith fructose-free diet vomiting and bleeding
tendency disappear immediatelyHepatomegaly and steatosis will disappear between
5-10 years

Hereditary Fructose Intolerance
Nutrition ConcernsVitamin supplement may be indicatedRequires strict avoidance for life of all dietary
fructose and sucroseAversion to sweets may develop

Glycogen Storage DiseasesDeficiencies of enzymes that regulate the synthesis
or degradation of glycogen (8 types)
Most related to deficient activity in conversion of glycogen to glucose 6 phosphate
Results in abnormal glycogen deposition in liver and muscle




Glycogen Storage DiseasesGSD1 most commonly diagnosed
Deficiency of enzyme glucose 6 phosphatase resulting in hypoglycemia
Low blood glucose results in short periods of fasting (2-4 hours)
Elevations in lipids, lactate, uric acid
Hepatomegaly
Chronic lactic acidosis, poor growth
Osteoporotic bones, delayed bone age


Glycogen Storage DiseasesNutrition Interventions – GSD1
Frequent oral feedings, high in CHO to maintain glucose > 70 mg/dL
Daytime meals followed by continuous drip nocturnal enteral feedings
Cornstarch - 1-2 g/kg body weight every 3-6 hours

Glycogen Storage DiseasesNutrition Concerns – GSD1
Availability of high-CHO snacks at all times Illness can be life threateningAdjustment to decreased oral intakeMultivitamin/ mineral supplementCalcium and iron supplementation

Disorders of Fat Metabolism
Defect in enzymes which allows transport of fatty acids into the mitochondria; specific to short-, medium- or long-chain fatty acids
Fatty acids not utilized resulting in hypoglycemia, hyperammonemia, death
MCADD most common
Deficiencies of carnitine metabolism

© 2007 Thomson - Wadsworth

© 2007 Thomson - Wadsworth

Disorders of Fat Metabolism
Nutrition InterventionPrevention of fastingLimiting intake of fatty acidsProviding alternate substrate for metabolism (CHO,
protein) Include complex CHO vs. simple to maintain
euglycemia

Disorders of Fat Metabolism
Nutrition InterventionLCHADD – restrict long-chain fatty acids to no more
than 15% of kcalSupplement with MCTMCADD – avoidance of fasting, feed every 3 hoursMonitor blood glucose levelsDo not use MCT oil


Disorders of Fat Metabolism
Nutrition ConcernsOverrestriction of fatEssential fatty acid deficiencyExcessive weight gainMaximize fluid intakeCarnitine used to detoxify, given as supplement