matlab bioinformatics toolkit evaluation kanishka bhutani
TRANSCRIPT

Matlab Bioinformatics Toolkit Matlab Bioinformatics Toolkit EvaluationEvaluation
Kanishka BhutaniKanishka Bhutani

What I expected ??What I expected ??
Local/Global sequence alignments.Local/Global sequence alignments. Multiple sequence alignments.Multiple sequence alignments. Choice of different scoring matrices Choice of different scoring matrices
(BLOSUM, PAM) for evaluation.(BLOSUM, PAM) for evaluation. Build Hidden Markov Models. Build Hidden Markov Models. Easily import sequences from databases Easily import sequences from databases
(PFAM,PDB, Swissprot)(PFAM,PDB, Swissprot)

What I found ??What I found ??
Most of the features.Most of the features. ““Bonus” =Bonus” =
Microarray normalization tools.Microarray normalization tools.
Microarray Visualization tools including box Microarray Visualization tools including box plots, heat maps.plots, heat maps.

Any surprises ?Any surprises ?
No “Multiple sequence alignments”No “Multiple sequence alignments” Avg./Std Dev. of hydrophobicity, solvent Avg./Std Dev. of hydrophobicity, solvent
accessibility : Command ?accessibility : Command ? ““Proteinplot”- GUI for protein structure Proteinplot”- GUI for protein structure
analysis.analysis. Import your file to view, select parameters Import your file to view, select parameters
and display stats. and display stats.

What all I tried?What all I tried?
Local alignment, Global alignment.Local alignment, Global alignment. For short sequences:For short sequences: swalign(‘seq1’,’seq2’)swalign(‘seq1’,’seq2’) nwalign(‘seq1’,’seq2’)nwalign(‘seq1’,’seq2’)seq1,seq2: AA or NT sequences.seq1,seq2: AA or NT sequences. For ‘imported’ long sequences:For ‘imported’ long sequences:Convert seq into a vector of integer valuesConvert seq into a vector of integer valuesCommands: nt2int, aa2intCommands: nt2int, aa2int

Pairwise Sequence alignmentPairwise Sequence alignment
S = getgenbank(‘NM_00001’)S = getgenbank(‘NM_00001’) M= getgenbank(‘NM_00002’)M= getgenbank(‘NM_00002’) Output : Header and a sequence.Output : Header and a sequence. K=nt2int(S.Sequence)K=nt2int(S.Sequence) B=nt2int(M.Sequence)B=nt2int(M.Sequence)[sc,align] = nwalign [K,B][sc,align] = nwalign [K,B]
Alignment Score Alignment Score Aligned seq.Aligned seq.

Getting sequences : V Easy !Getting sequences : V Easy !
‘‘getgenbank’: Retrieve sequence getgenbank’: Retrieve sequence information from Genbank database.information from Genbank database.
‘‘getembl’: Retrieve seq. information from getembl’: Retrieve seq. information from EMBL database.EMBL database.
‘‘getpept’: Retrieve seq information from getpept’: Retrieve seq information from Genpept database.Genpept database.
‘‘gethmmprof’: Get HMM from the PFAM gethmmprof’: Get HMM from the PFAM database.database.

ExperimentExperiment hmmodel = gethmmprof(‘PF00001’)hmmodel = gethmmprof(‘PF00001’)

Visualization of modelVisualization of modelShowhmmprof (hmmodel,’scale’,’logodds’)

Get GPCR seq’sGet GPCR seq’s
S = getgenbank (‘NM_024531’)S = getgenbank (‘NM_024531’) disp (S.Sequence)disp (S.Sequence)

Alignment of the seq’sAlignment of the seq’s var = gethmmalignment var = gethmmalignment
(‘PF00001,’type’,’seed’)(‘PF00001,’type’,’seed’)
disp [char(var.Header) char (var.Sequence)]disp [char(var.Header) char (var.Sequence)]

For GPCR Family CFor GPCR Family C Similarly for diff families.Similarly for diff families. Multiple aligned sequences retrieved.Multiple aligned sequences retrieved.

GUI proteinplotGUI proteinplot
User friendly.User friendly. Avg./ Std. dev values for:Avg./ Std. dev values for:
Hydrophobicity.Hydrophobicity.
Secondary structure propensity (Alpha Secondary structure propensity (Alpha helices or beta strands)helices or beta strands)
Accessibility (accessible and buried Accessibility (accessible and buried residues)residues)

Mglur1 plot (Proteinplot)Mglur1 plot (Proteinplot)
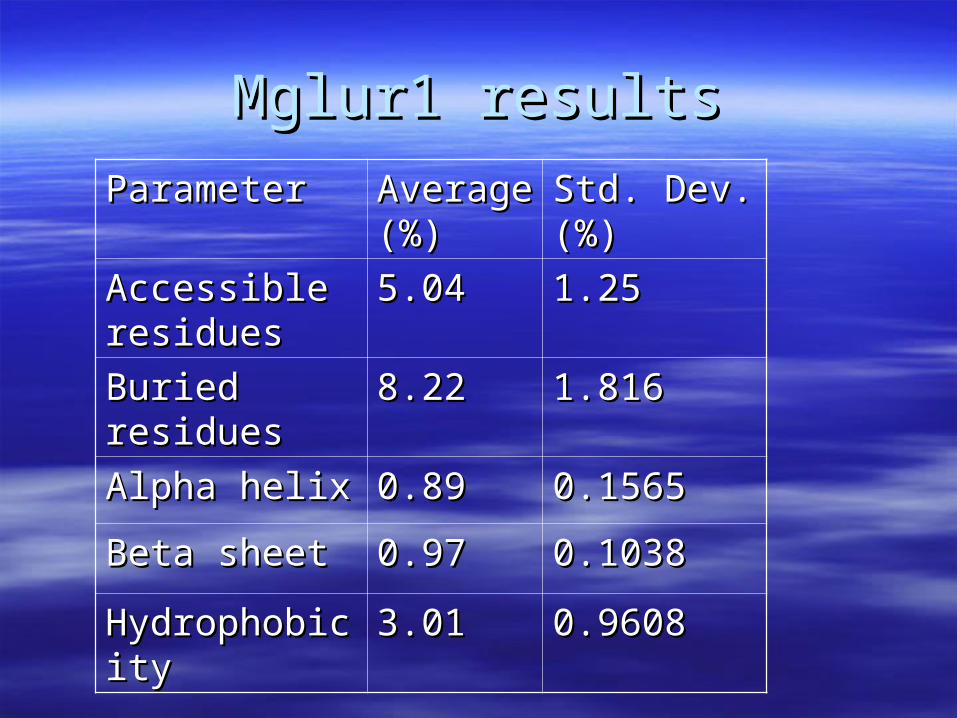
Mglur1 resultsMglur1 results
ParameterParameter Average Average (%)(%)
Std. Dev.Std. Dev.(%)(%)
Accessible Accessible residuesresidues
5.045.04 1.251.25
Buried Buried residuesresidues
8.228.22 1.8161.816
Alpha helixAlpha helix 0.890.89 0.15650.1565
Beta sheetBeta sheet 0.970.97 0.10380.1038
HydrophobicityHydrophobicity 3.013.01 0.96080.9608

Test a seq. with HMMTest a seq. with HMM
Retrieve mglur1 from GenbankRetrieve mglur1 from Genbank mgr = getgenbank (‘NM_012407’)mgr = getgenbank (‘NM_012407’) glusequence = mgr.sequenceglusequence = mgr.sequence Test it with the HMM model class ATest it with the HMM model class A [a.sglu] = hmmprofalign (model A, [a.sglu] = hmmprofalign (model A,
glusequence,’showscore’,true)glusequence,’showscore’,true) Score = -203.53Score = -203.53 Seq = Seq =

Log odd score plot for best pathLog odd score plot for best path

Difficulties & questionsDifficulties & questions
No multiple sequence alignment.No multiple sequence alignment. Demos: Not very helpful.Demos: Not very helpful. Difficult to view the sequences as no “disp” command Difficult to view the sequences as no “disp” command
found.found. Bugs:Bugs: Storing huge sequences (GPCR A) in a file, parsing error.Storing huge sequences (GPCR A) in a file, parsing error. HMMprofdemo command abruptly stops and gives errors. HMMprofdemo command abruptly stops and gives errors. Proteinplot (GUI) hangs the machine often.Proteinplot (GUI) hangs the machine often. Verify the sequences using the HMM models ??Verify the sequences using the HMM models ?? Regular expression matches and highlighting those Regular expression matches and highlighting those
positions??positions??

Suggestions of experimentSuggestions of experiment
Given an unknown sample dataset of proteins, Given an unknown sample dataset of proteins, known dataset of proteins (known structural known dataset of proteins (known structural information).information).
Utilize the BLMT to extract ‘over expressed’ 4 Utilize the BLMT to extract ‘over expressed’ 4 Grams in a protein sequence or a group of protein Grams in a protein sequence or a group of protein sequences from the known set.sequences from the known set.
Use “search for regular expression” function in the Use “search for regular expression” function in the Matlab toolkit to look for those ‘4 Grams’ in Matlab toolkit to look for those ‘4 Grams’ in unknown proteins and hence predict their unknown proteins and hence predict their structure.structure.