lysosomal storage disease module 755 the brain in health and disease sean sweeney
TRANSCRIPT

Lysosomal Storage Disease
Module 755
The Brain in Health and Disease
Sean Sweeney

Lysosomal Storage Disease (Amaurotic Idiocy)
c.a. 45 autosomal recessive diseases
Individually rare
Collectively occur c.a. 1/8000 live births
Cause death in early to late childhood (after normal infancy)
Varying involvement of the nervous system
All ‘store’ material in the lysosome due to defects insubstrate degradation or biogenesis of the lysosome

The Lysosome
subcellular electron dense organelle
filled with c.a. 70 hydrolytic enzymes: will break down all biological macromolecules
low pH (~4.0), membrane bound
Considered the ‘gut’ or garbage disposal unit of cell
Material for degradation trafficked to lysosome via endocytosisor autophagy
Lysosomal enzymes trafficked to lysosome via M6P receptorpathway

Endosome to lysosome:decreasing pH, membranelimited.
Autophagy: controls cell size,used during caloric restriction,Phagocytosis:- degrades ‘dead’ cells, pathogensAutophagy and phagocytosismeet in the PhagolysosomeProfessional Phagocytes:macrophages, neutrophils
Delivering material for degradation to the lysosome:endocytosis and autophagy

Endocytosis in the nervous system
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
The polarised and extendedstructure of the neuron creates a trafficking problem for neurons:
‘lysosomes’ (as we know them!)not present at synapse.
Late endosomal markers present:fuse with lysosomes in the soma

Delivering degradative enzymes and co-factors to the lysosome, the M6P/M6PR pathway.
Mannose-6-phosphate group added to lysosomalhydrolases via N-linked oligosaccharides ashydrolases transit through cis-golgi
M6P recognised by M6P-receptors in trans-golgi:delivers them to late endosome
Lower pH causes dissociation
M6PR then retrieved in late endosome and trafficked for re-use in trans-golgi(recognised via C-terminal tail).

General outline of LSD dysfunction:
Mutations arising in hydrolytic enzyme, co-factor or factor essential of enzyme delivery to lysosome
Also, factors essential for lysosome function and biogenesis (membrane proteins, channels and proteins of unknown function) plus factors for protein traffic to lysosome
Material (substrate) continues to be delivered to lysosome resulting in ‘stored’ material, usually ‘primary’ and ‘secondary’ leads to swollen lysosomes
Developmental dysfunction and early death: symptoms v. variable, varying involvement of different tissues

General Cellular Phenotype:
Swollen, multilammellar ‘osmiophilic endosomes/lysosomes (function? pH?)
Accumulation of lipofuscin/ceroid ‘ageing pigment’
Defects in autophagy (?)
Appearance of meganeurites (variable)
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Cellular phenotype contd.
Excessive synaptogenesis/dendritogenesis (MPS and sphingolipidoses)
Shrinkage of the CNS (variable)
Mistrafficking of cholesterol(cholesterol recycling?)

Why are symptoms and effects in different organs variable?
tissue turnover rates?
presence (or relative abundance) of substrate?
sensitivity of cell type (neurons and polarity)?
What is the ‘pathogenic cascade’?
(volume of substrate not key!!!)

Classification :
Mucopolysaccharidoses (variable nervous system involvement)Mucolipidoses (originally considered an MPS)GlycoproteinosesGlycogen storageSphingolipidosesLipid storage disordersMultiple enzyme defectsTransport defectsBatten Disease
(Red = nervous system involvement)

Mucopolysaccharides • Defective metabolism and accumulation of GAGs • Most abundant polysaccharides • Long unbranchedstructure containing disaccharide units: • High viscosity + rigidity • Excellent lubricators and shock absorbers • Important component of cell membranes

QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.

Mucopolysaccharidoses: Enzyme Defective
MPS-I: (Hurler, Sheie, Hurler/Sheie) iduronidaseMPS-II: (Hunter) iduronate-2-sulfataseMPS-III: (Sanfilippo)
IIIA heparan-N-sulfataseIIIB N-acetyl-glucosaminidaseIIIC Acetyl Co-A glucosamine
N-acetyl transferaseIIID N-acetyl-glucosamine-6-sulfatase
MPS-IV (Morquio)IVA N-acetyl-galactosamine 6-sulfataseIVB ß-galactosidase
MPS-VI (Maroteaux Lamy) N-acetyl-galatosamine 4-sulfataseMPS-VII (Sly) ß-glucuronidaseMPS-IX hyaluronidase

Sanfilippo Syndrome (MPS III)
Four types: A,B,C,D, cannot break down Heparan sulfateMost common MPS, 1/70,000 births
hepatosplenomegaly (may resume normal size with age)HyperactivitySpeech delayMental retardationJoint stiffness, bone defects (dystosis multiplex)Coarse features (dysmorphismDeath in middle teens
Screening: GAGs in urineDiagnostic: WBC enzyme assay or plasma enzyme assay
Prognosis: No effective treatment to date.

Mucolipidosis (I-Cell disease) and MPS-IV
Mucolipidosis-III-Cell (Pseudo-Hurler): first described 1967I = Inclusion, stored material mucolipid MPS and sphingolipidOccurrence: 1/640,000 live birthsSymptoms: Developmental delay, psychomotor deterioration, dysmorphia, death in
early childhood
Genetic defect: N-acetylglucosaminyl-1-phosphotransferase
Prognosis: v. poor, limited treatment (nutritional), death by 10 years of age.
Mucolipidosis-IV
Storage material: mucolipids, MPS and sphingolipidsOccurrence: carriers in Ashkenazim Jewish population, 1/90 to 1/100Symptoms: Psychomotor retardation, corneal opacity, retinal degeneration, iron deficiency, improper stomach pH (achloridia)
Genetic defect: Mucolipin-1 (MCOLN1), a TRP channel (TRPML-1)Involved in Fe2+ efflux from lysosomes? (Dong et al., (2008) Nature, 455, 992-6)
Prognosis: v. poor. Nutritional supplements, physcial and speech therapy

Sphingolipids: a major component of neural tissue
O
OH
NH
CH2O H
O
OH
NH
CH2O P O (CH2)2 N+
O
O-
CH3
CH3
CH3
O
OH
NH
CH2O Glcn
Ceramide
Sphingomyelin
Glycosphingolipids
STRUCTURE
microdomains (?)trafficking
SIGNALLING
Apoptosis proliferation stress
- Sphingomyelin - Ceramide - Sphingosine - Sphingosine-1-phosphate - Cerebrosides - Gangliosides

Sphingolipids aretightly associated withcholesterol

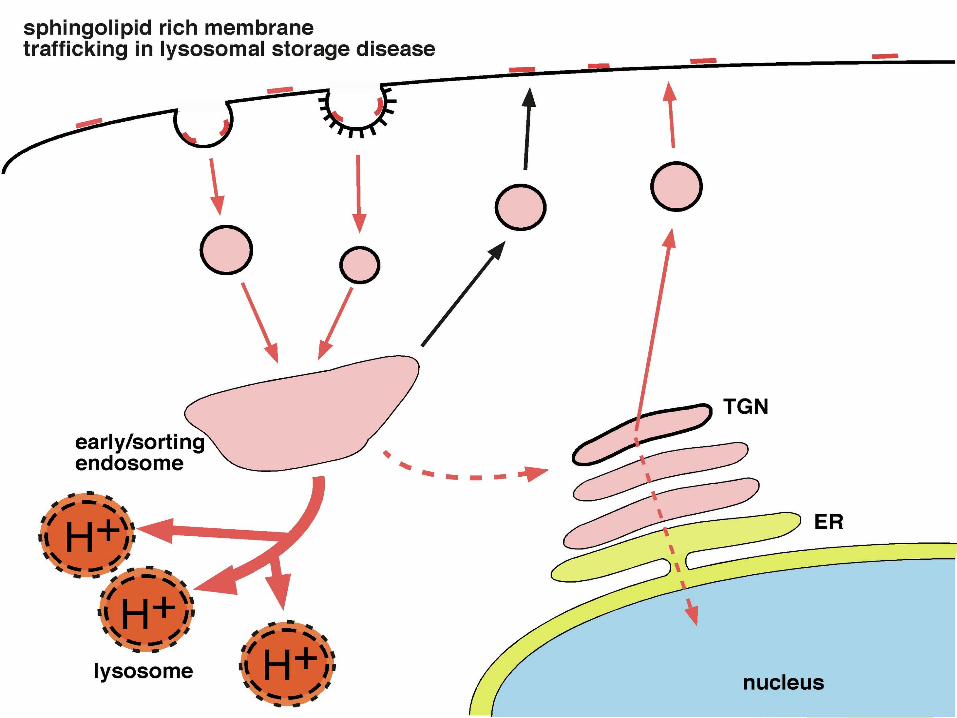

The sphingolipidoses: Tay-Sachs (GM2-gangliosidosis)
First described in 1880’s from ‘cherry-red’ spot in fundus (retina) (lipid deposition in bipolarganglion cells)
Infantile (death ~ 5yrs), Juvenile (death between 5 and 15yrs) and ‘Late-onset’ forms (v. rare)All present with increasing neurological and deterioration (ataxia, atrophy, spasticity)
Occurrence: 1/27 to 1/30 Ashkenazim Jews are carriers, also: Acadians, Cajuns
Genetic defect: Hexosaminidase A (HEXA)
storage material: GM2 ganglioside, globoside, glycolipids
cf: Sandhoff Disease: HEXB mutations and GM2 gangliosidosis(mutations in GM2 activator protein)
Glial Involvement!
Prognosis: early death, ameliorated by treatment
Enzyme Replacement TherapySubstrate Reduction Therapy
Population Screening (model of genetic screening for recessive condition)
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Other cellular defects:
Niemann-Pick disease:
occurrence: A, B collectively- 1/1000 Ashkenazim Jews are carriers, type C no ethnic distributiontype A accounts for 85% of cases
Symptoms: enlarged spleen and liver, enlarged lymph nodes, darkening of skin, neurologic impairment (not in B), cherry red spot
genetic defect: A and B, mutant for sphingomyelinaseType C mutants: two loci, two proteins, multi-transmembrane protein (related tohedgehog receptor ‘patched’ and small co-protein(cholesterol binding protein/carrier?). Homolog NPCL1 involved in cholesterol absorption in gut.
storage material: sphingomyelin, cholesterol and sphingolipids
Diagnosis: ‘filipin’ staining
cell biology (and diagnosis): mislocalised unesterified cholesterol, neurofibrillary tangles
Endosomal trafficking jam? cholesterol and sphingolipids required to organise endosomaltrafficking steps. Cholesterol recycled from lysosome.
Drosophila models reveal cholesterol is ‘limited’

Batten disease
A family of closely related disorders9 forms: congenital, infantile, late infantile, juvenileadultAKA: Neuronal Ceroid Lipofuscinosis (NCL)
Incidence: global with hotspots for some loci
Loci: ‘CLN’ genes CLN1, CLN2, CLN3, CLN5, CLN6, CLN8 CTSDcloned so far, others remain to be mapped.
occurrence: most common childhood neurodegeneration 1/8000 livebirths
Symptoms: visual defects, seizures, stumbling, echolalia, eventual loss of sight speech and motor skills, early death after blindness, dementia.
storage material: Lipofuscin/ceroid, subunit C of mitochondrial ATP synthase
Phenotype: multilamellar inclusions, selective brain cell death (glia mediated)infiltration of neuronal tissue with antibodies (defective BBB?)
Prognosis and treatment: anti-convulsives, therapy. Death in childhood
Batten (1903)
QuickTime™ and aTIFF (LZW) decompressorare needed to see this picture.

Locus Disease Protein deficiency Function
CLN1 infantile NCL palmitoyl protein thioesterase de-palmitoylationLysosome
CLN2 late infantile NCL tripeptidyl peptidase proteaselysosome
CLN3 juvenile NCL transmembrane protein ?lysosome
CLN4 adult (Kuf’s) Not identified
CLN5 late infantile NCL transmembrane protein ?(Finnish variant) LE/lysosome
CLN6 late infantile variant transmembrane protein ER protein
CLN7 late infantile variant Not Identified
CLN8 EPMR transmembrane protein ER, ER/Golgi
CTSD Ovine NCL cathepsin D proteaselysosome

Endocytosis in the nervous system
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
Lysosomes (hydrolases!)not present at synapse
Many of NCL proteins foundat synapse
NPC protein, others?
Identification of proteins involvedin neurodegeneration helpto describe functions in the neuron

Treatment:BMT (membrane proteins)
enzyme replacement (BBB?)
gene therapy
substrate reduction- Miglustat (monosaccharidemimetic-imino sugar)
Neuronal stem cells (membrane proteins?)
Chemical chaperone therapy
Neuroinflammation

Economic cost
ERT is current most effective treatment (non neurodegenerative LSDs):
Disease Treatment Annual Cost (per patient in $)
Gaucher ERT 145,000 - 290,000
Gaucher SRT 91,000
Fabry ERT 156,000
Hurler-Scheie (MPS-I) ERT 340,000
Maroteaux-Lamy (MPS-VI) ERT 377,000
Reasons:High regulatory costsCost of researchLack of competition (Orphan Drug Act 1983, US)

Studying the Lysosomal Storage Diseases:
Model Organisms
Sheep (Batten)sheepdogs (Batten)mouse (Batten, Tay-Sachs, Sandhoff, NPC)zebrafish (Batten)Drosophila (MPS, NPC, Batten, others)C. elegans (MPS, NPC)Yeast (cerviseae, pombe) Batten, NPC
Reverse Genetics (qv Tay-Sachs)
Forward Genetics

http://132.236.112.18/fruitfly/shaker/physiology/
The Drosophila neuromuscular junction: A model glutamatergic synapse

spinster synapses are overgrown
spinstersuppressessynaptic growth
spinster mutantshave a shortenedlifespan

spinster encodes a twelve transmembrane transporter
4 transcripts = 12 TM domains1 transcript = 8 TM domains

Spin localises to a low pH late-endosomal compartment

A low pH compartment is expanded in spin mutants


WT spin4/spin5
Loss of spinster induces a redistribution of cholesterol
filipin

spinster identifies a novel component of the late endosome/lysosome that when mutated givesrise to all of the hallmarks of lysosomal storage disease spinster potentially identifies a signalling pathway driving synaptic overgrowth

Summary
Lysosomal storage disease are caused by defects in lysosomal hydrolases and proteinsessential to lysosomal biogenesis/function
LSD lysosomal defects give rise to swollen lysosomes, developmental and degenerativedefects with varying involvement of the nervous system due to ‘storage’ of materialin the lysosome.
Lysosomal storage diseases identify proteins essential to lysosomal function
LSDs cause death in childhood (generally) after normal infancy
LSDs are essentially incurable, but some are treatable to varying degrees.
Model organisms are helping to define the biology of the LSDs, in particular the ‘pathogenic cascade’