lma - edisciplinas.usp.br
TRANSCRIPT
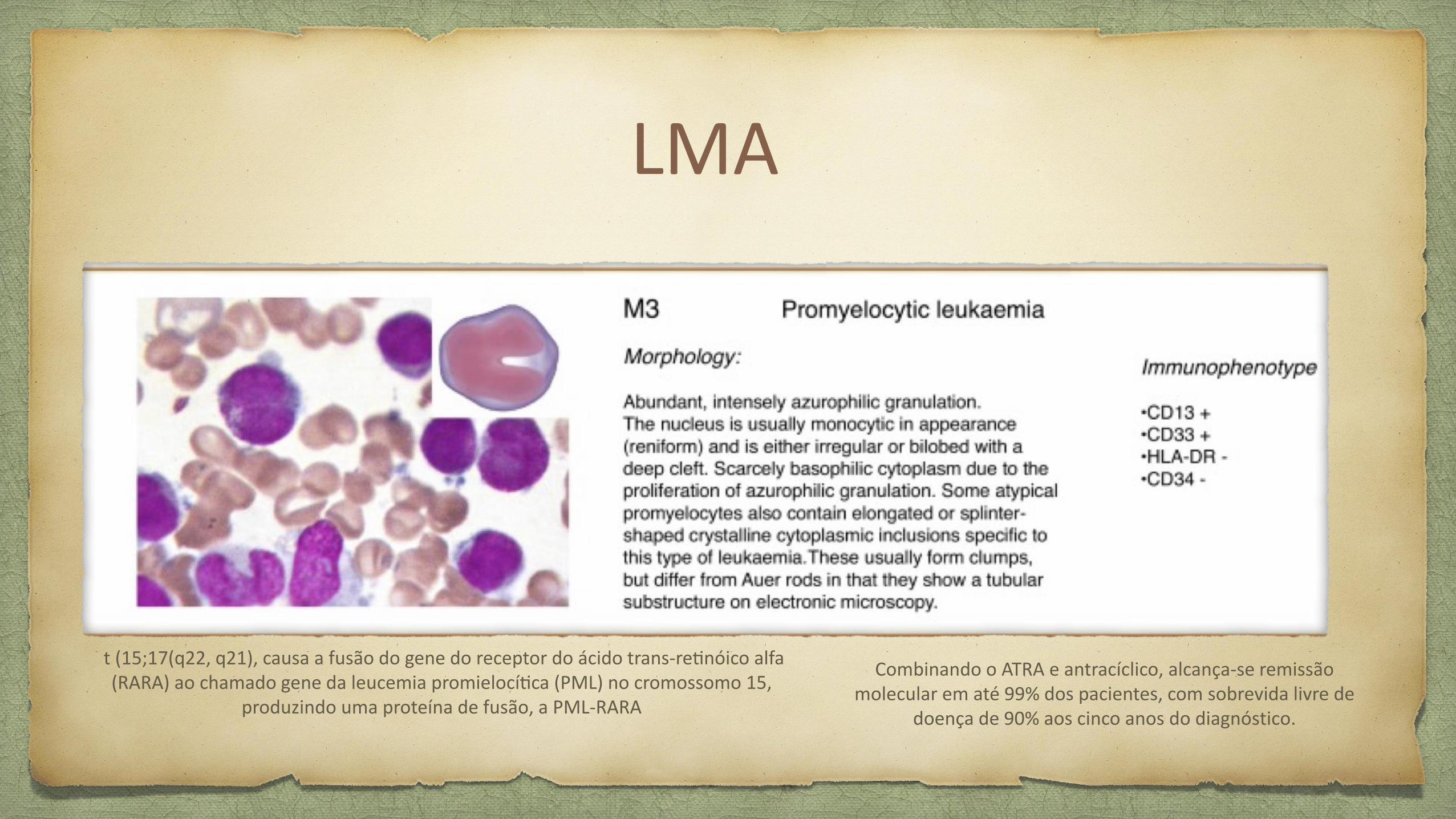
LMA
t (15;17(q22, q21), causa a fusao do gene do receptor do acido trans-retinoico alfa (RARA) ao chamado gene da leucemia promielocitica (PML) no cromossomo 15,
produzindo uma proteina de fusao, a PML-RARA
Combinando o ATRA e antraciclico, alcanca-se remissao molecular em ate 99% dos pacientes, com sobrevida livre de
doenca de 90% aos cinco anos do diagnostico.
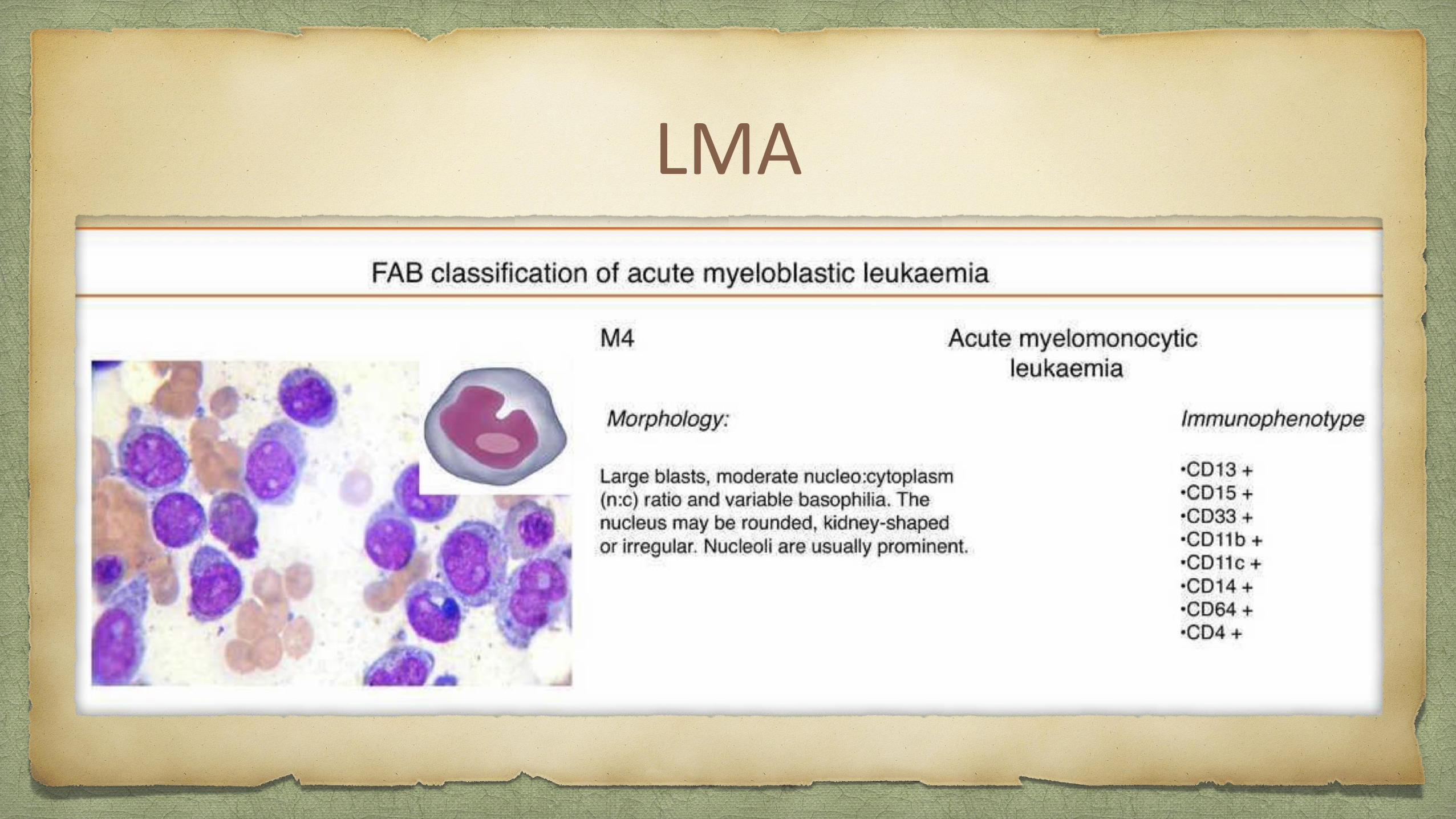
LMA
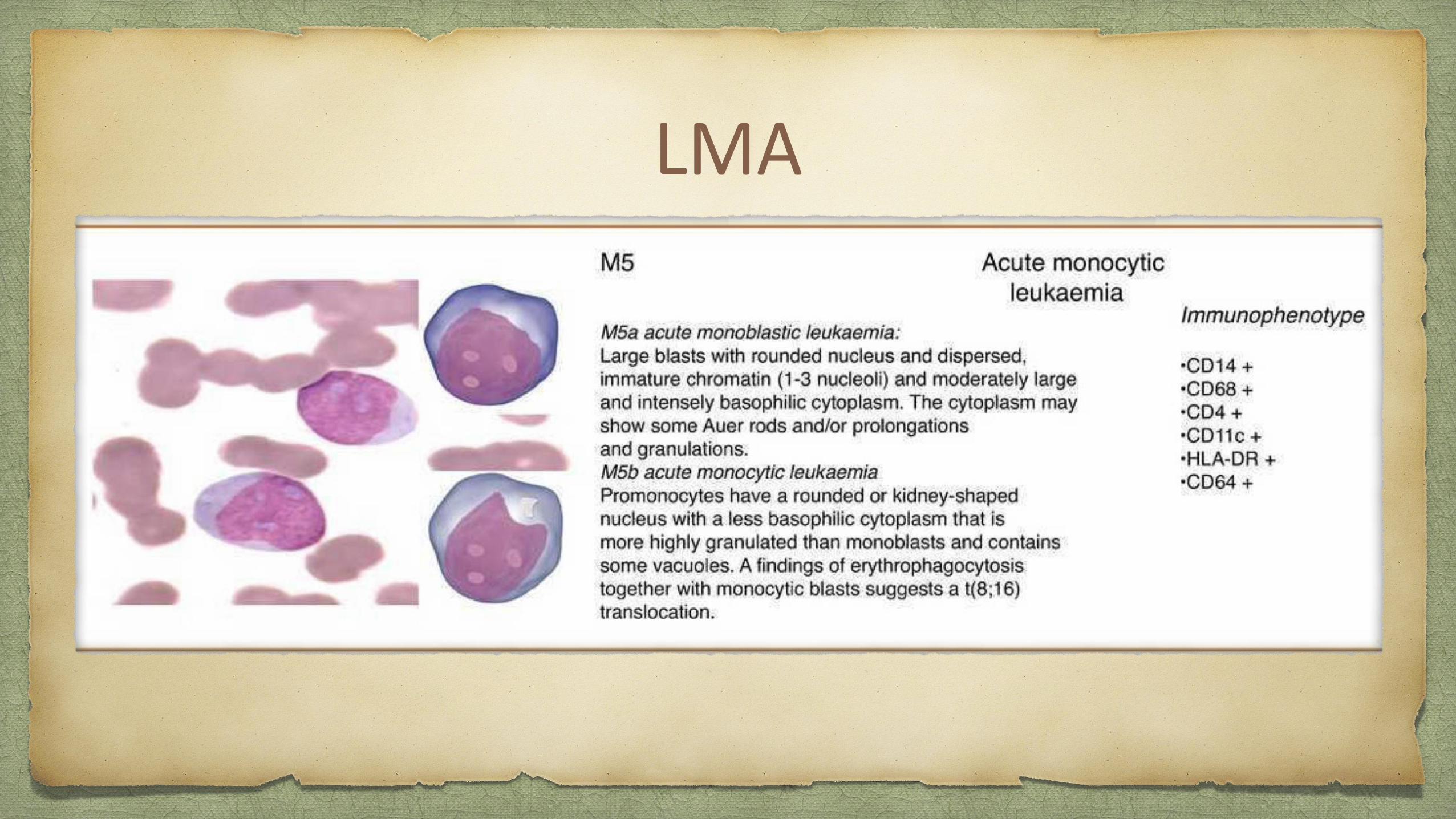
LMA
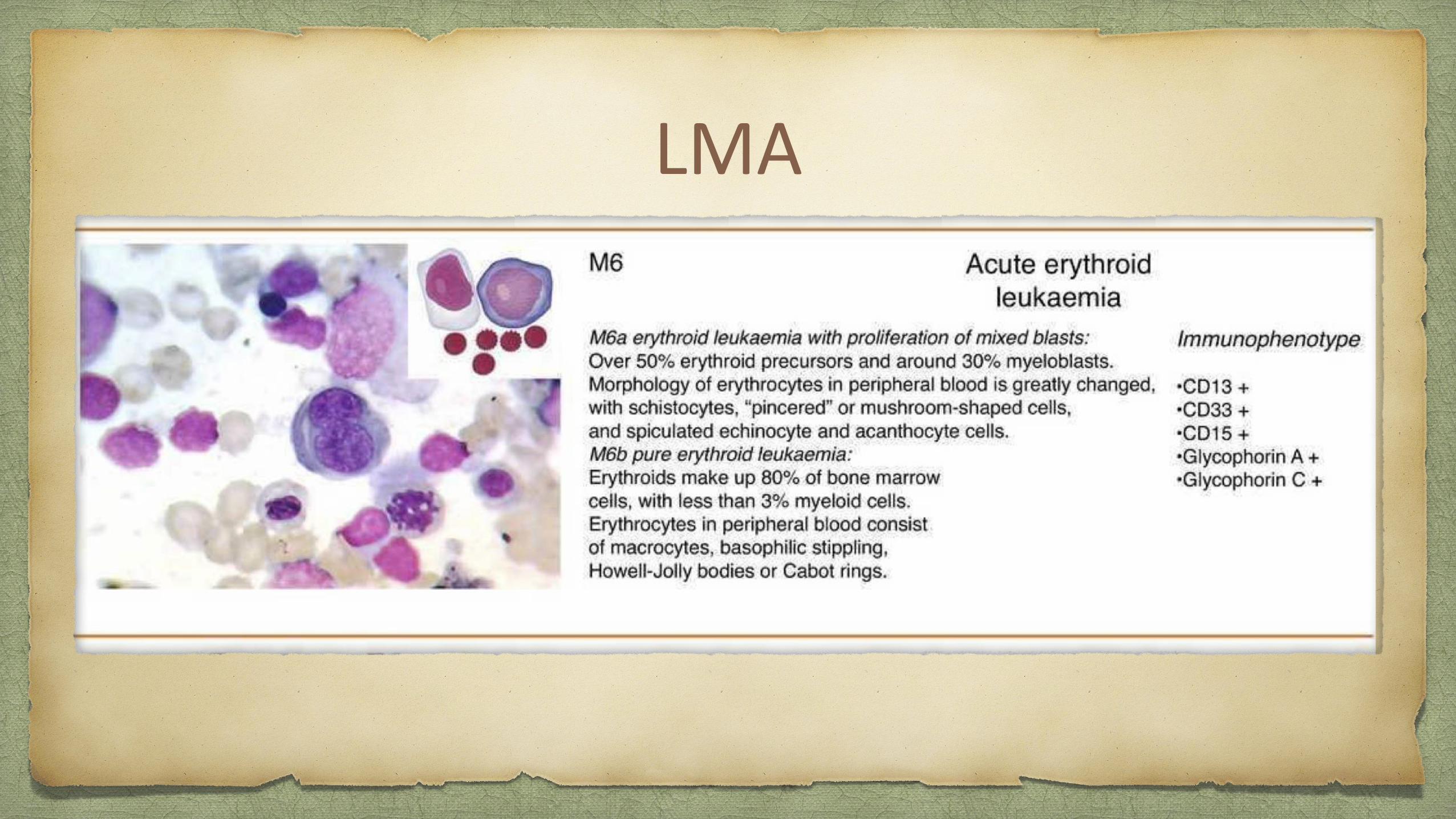
LMA

LMA
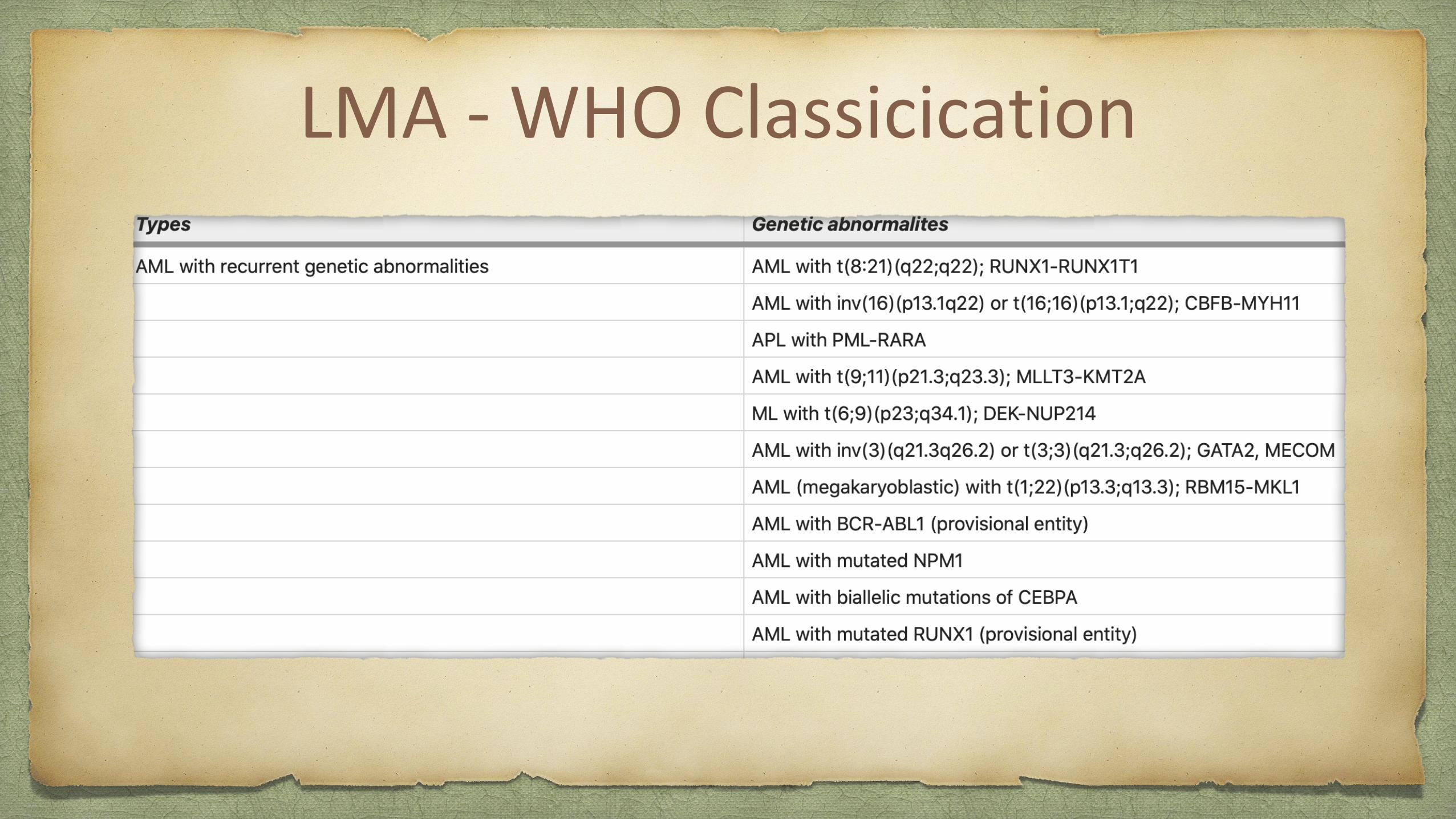
LMA - WHO Classicication
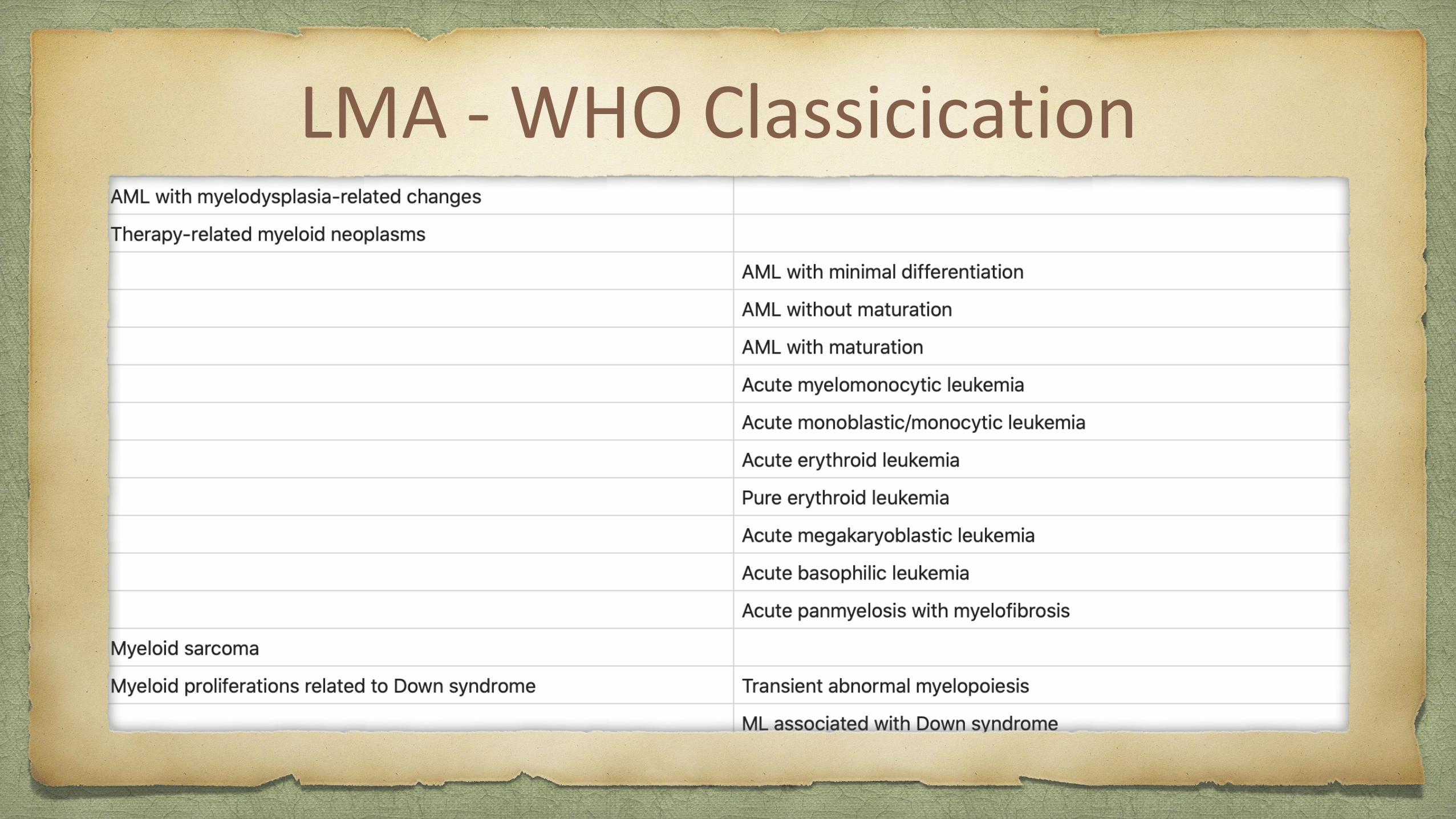
LMA - WHO Classicication

AML Prognostic factors
Treatment-related mortality
Standard x increased treatment intensity
Consolidation chemotherapy x allogenic hematopoietic stem cell
transplant
Established x investigational therapies

AML Prognostic factorsClinical factors
Increased age
Poor performance status
Lower rates of complete remission (CR) and decreased overall survival (OS)
=> risk of TRM
platelet count, serum creatinine or albumin
TR AML and AML-MDS
Cytogenetic changes:
Single strongest prognostic factor for CR and OS in AML
…

LMA - Prognostic risk group
with a 3 year OS of 66% and 33% in patients
younger and older than 60 years
c-KIT mutation significantly increases the risk
of relapse, and decreases OS to levels
comparable to those of patient with
intermediate-risk AML

LMA - Prognostic risk group

LMA - Prognostic risk group
significantly higher risk of treatment failure and death

AML Established treatmentsEligible patients first undergo induction therapy to achieve CR.
Unfortunately, minimal residual disease often persists in CR, and
relapse will inevitably occur if treatment is discontinued.
Induction therapy should be followed by consolidation therapy in
order to eradicate any residual disease and achieve lasting
remission.
‘7+3’ regimen, which combines 7 days of continuous infusion
cytarabine with 3 days of anthracycline.

AML Established treatments
Intention-to-treat analyses:
Favorable Prognosis:
No benefit to allo-HSCT when compared with chemotherapy in
patients with cytogenetically favorable AML in first CR.

AML Established treatments
Intermediary:???
allo-HSCT significantly prolongs RFS and OS in some patients with
intermediate-risk and in most with adverse-risk AML

AML Established treatments
High Risc: Allo-HSCT has been shown to prolong RFS and improve
OS in patients with CN-AML and a high FLT3-ITD allelic ratio.

AML Novel agents
FLT3-ITD inhibitors ( inhibition of tyrosine kinase-TKI)
- Sorafenib (TKI of RAF kinase, c-KIT, VGFR, PGFR and FLT3-ITD)
Treatment failure: Emergence of D835Y and D835H mutations within the
FLT3 TKD
An encouraging phase II trial of sorafenib and azacitidine in 43 patients with
relapsed/refractory AML reported a response rate of 46%, including 16% CR and
27% CR
- Midostaurin
- Quizartinib
- Crenolanib

AML Novel agentsSTAT inhibitors
- STAT3 tyrosine phosphorylation is upregulated in up to 50% of AML cases and confers a worse
prognosis
IDH1/IDH2 small molecule inhibitors
- Enzymes are found in approximately 20% of cases.
Clofarabine
- Second-generation purine nucleoside analog
Monoclonal antibodies
CART therapy
- Synthetic T-cell receptors with antibody-like specificity

AML ConclusionAML is a biologically and clinically heterogeneous disease.
Although advances overall long-term survival remains poor.
Elderly patients are more likely to present with an adverse cytogenetic profile.
Increased risk of TRM
Novel targeted therapies offer the promise of effective anti-leukemic activity with reduced toxicity
from off-target effects.
Molecular diversity of AML: single ‘magic bullet’ ?
Improved genetic profiling, risk stratification and pathology
Development of well-tolerated oral therapies, such as oral clofarabine.
We are looking to a new era in the treatment of AML to begin with novel agents so we can
achieve better responses with prolong OS particularly for patients with relapsed or refractory
diseases and poor cytogenetic features.

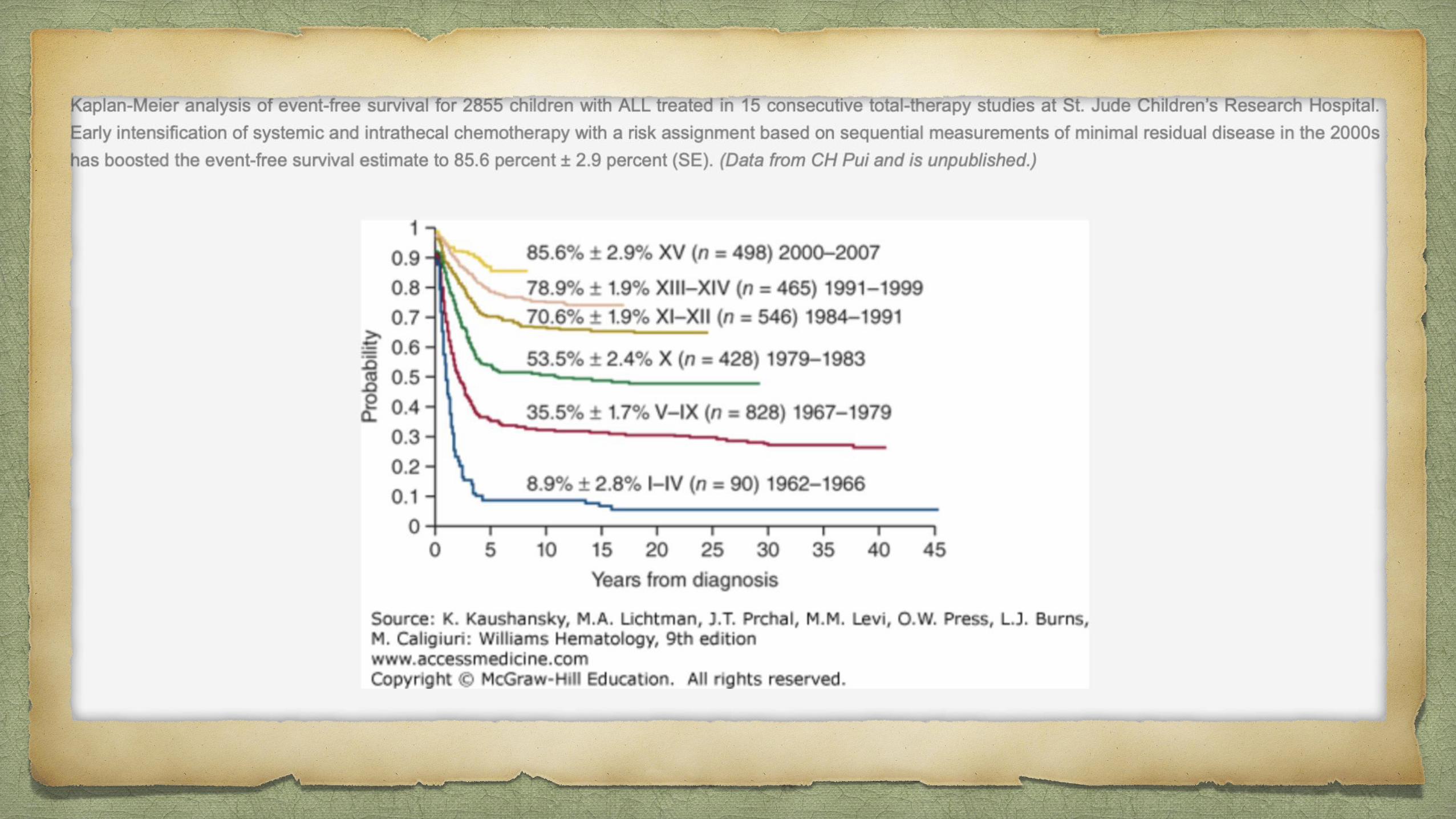

ALL
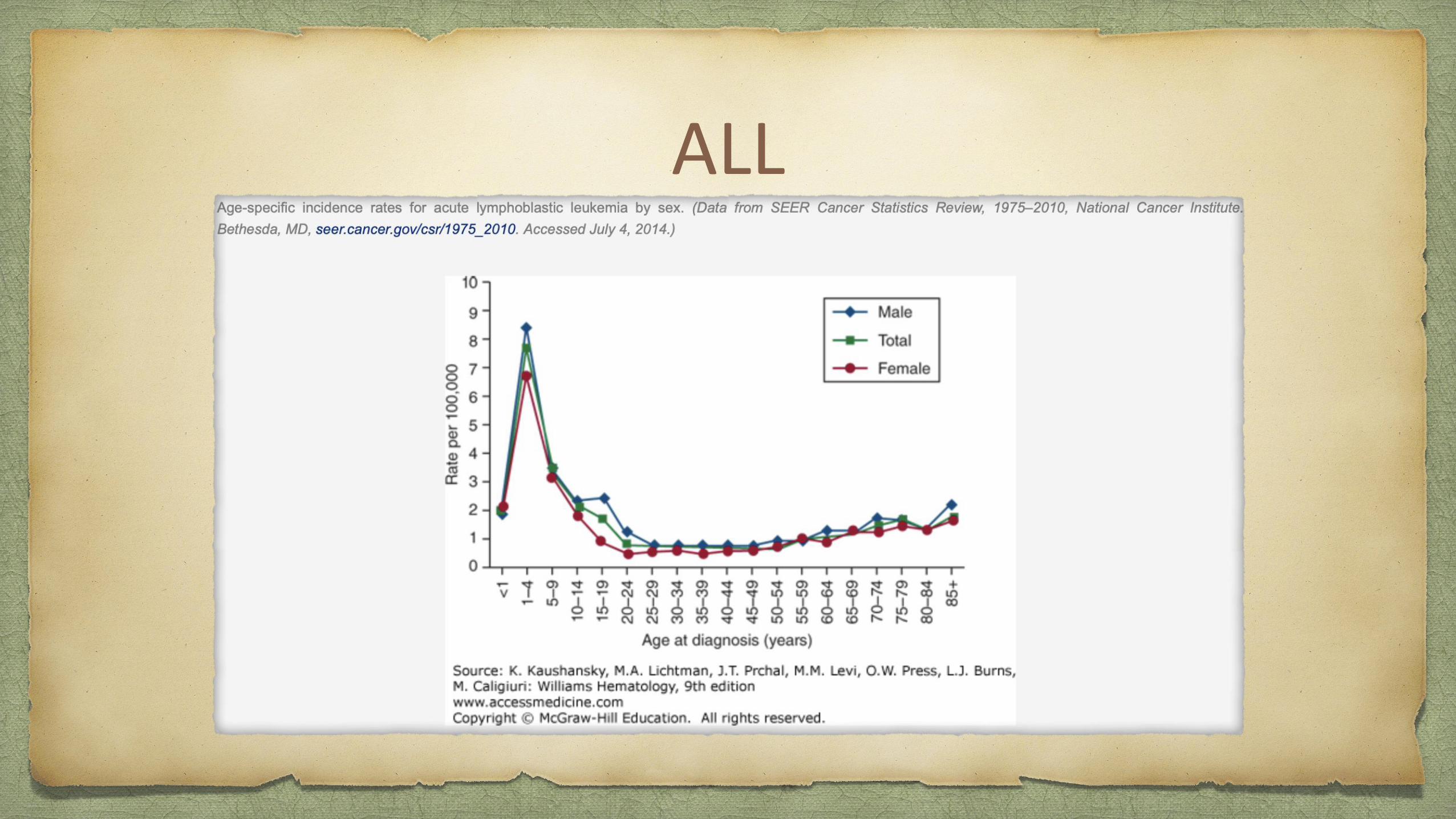
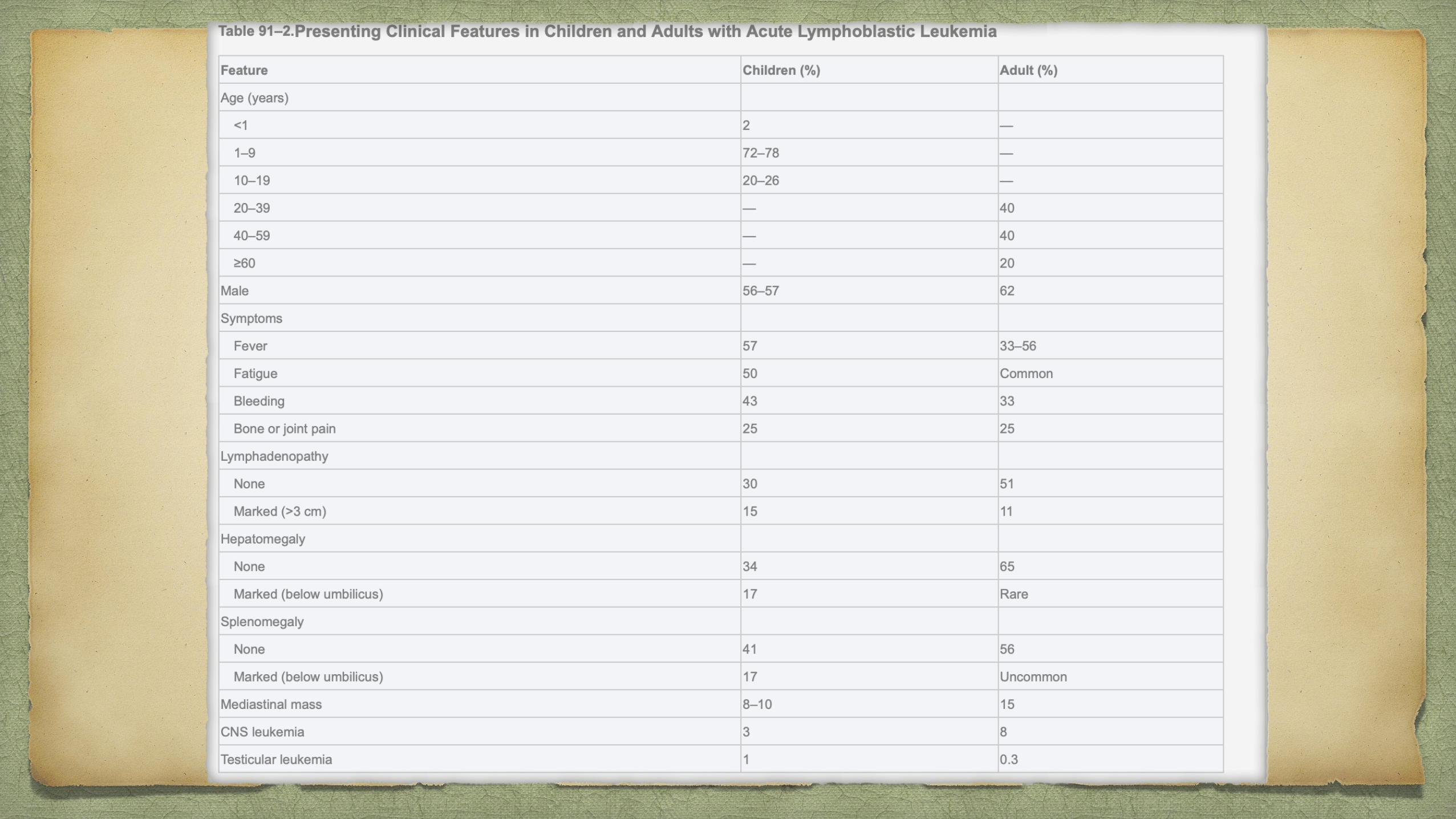
80% of ALL occurs in children, it represents a devastating disease when it occurs in adults
ALL

ALLPathophysiology
Down syndrome
Fanconi anemia
Bloom syndrome
Ataxia telangiectasia
Nijmegen breakdown syndrome.
Ionizing radiation
Pesticides
Certain solvents
Epstein-Barr Virus
Human Immunodeficiency Virus
De novo malignancy

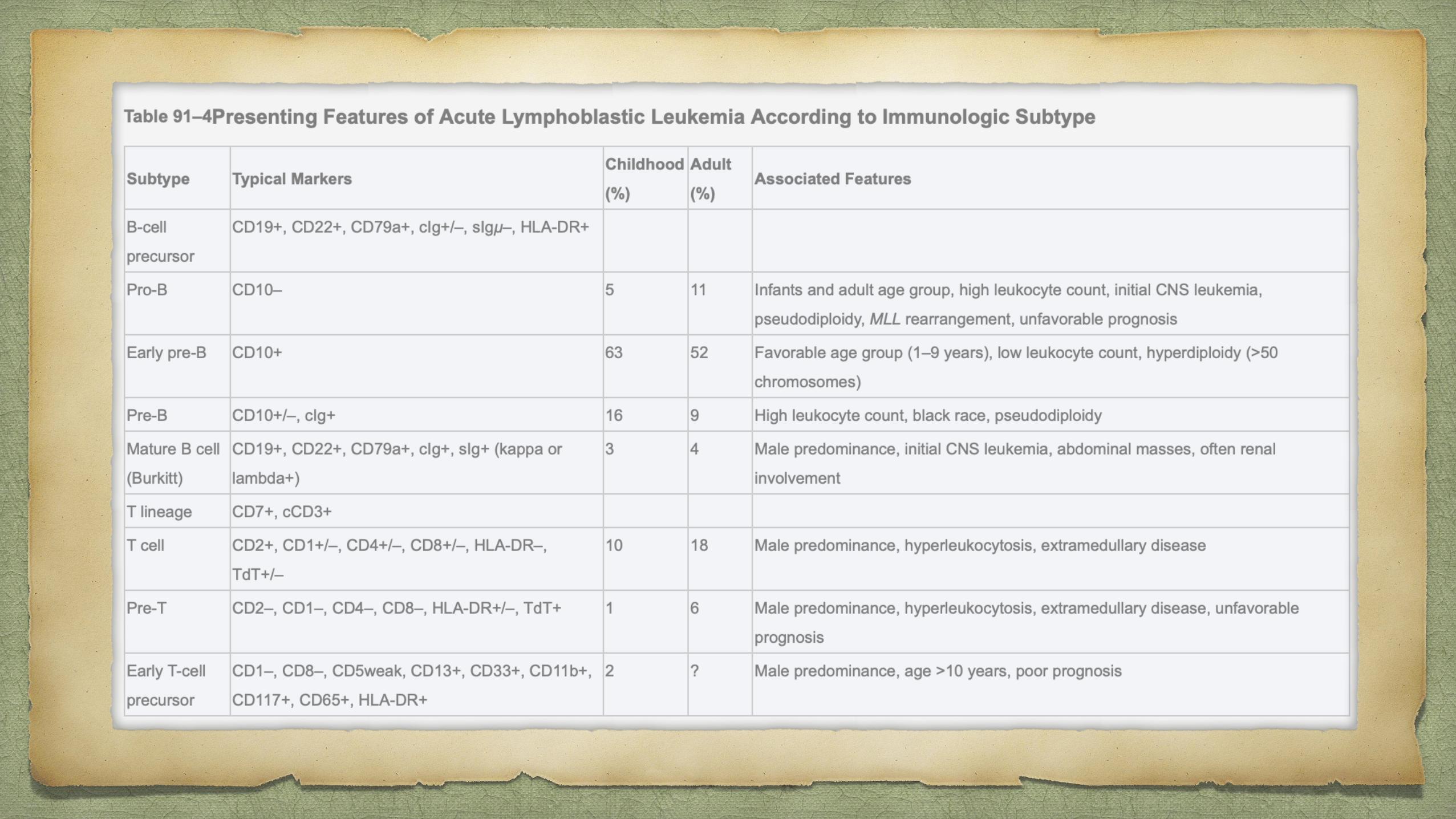
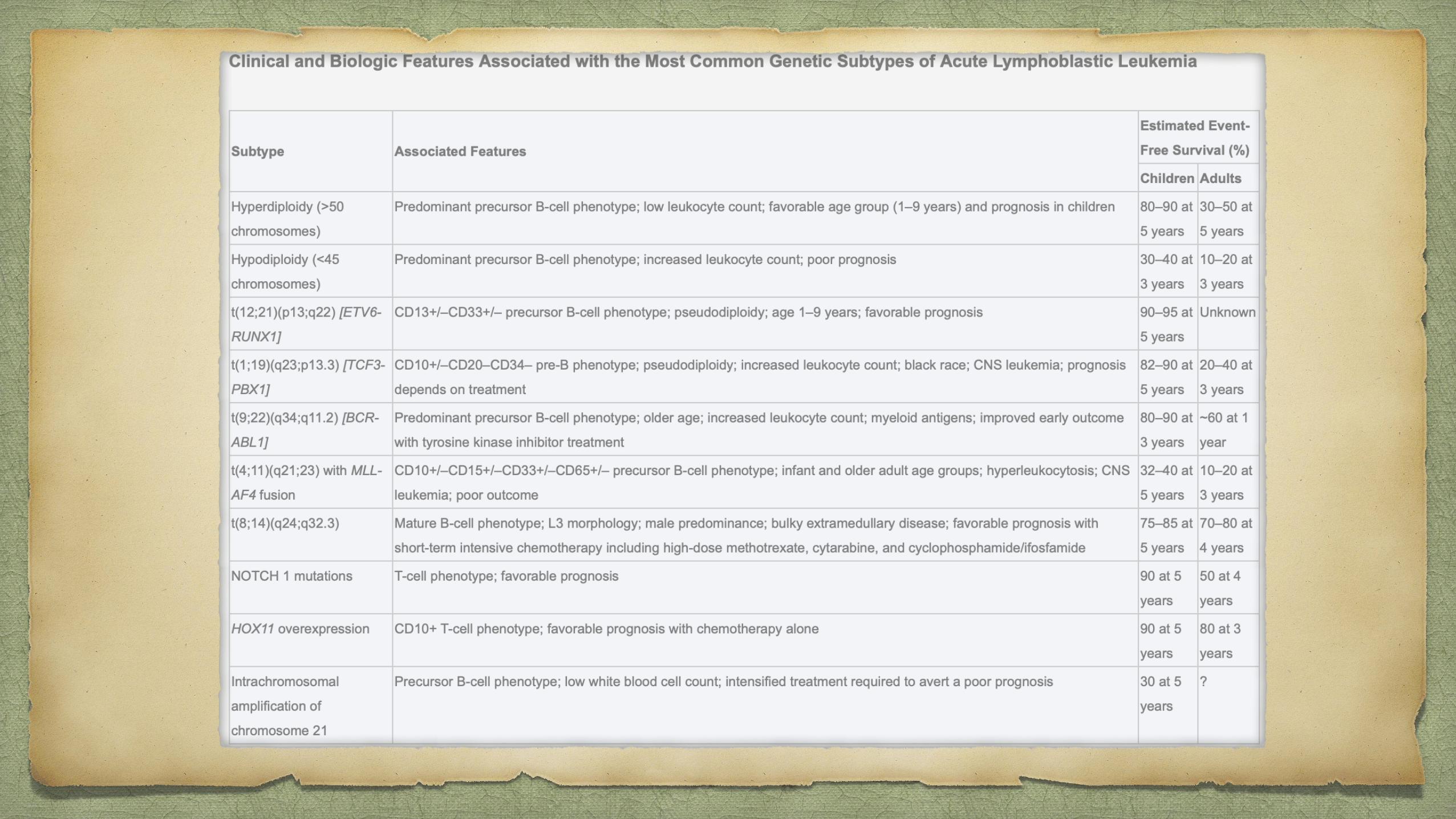
ALLPathophysiology
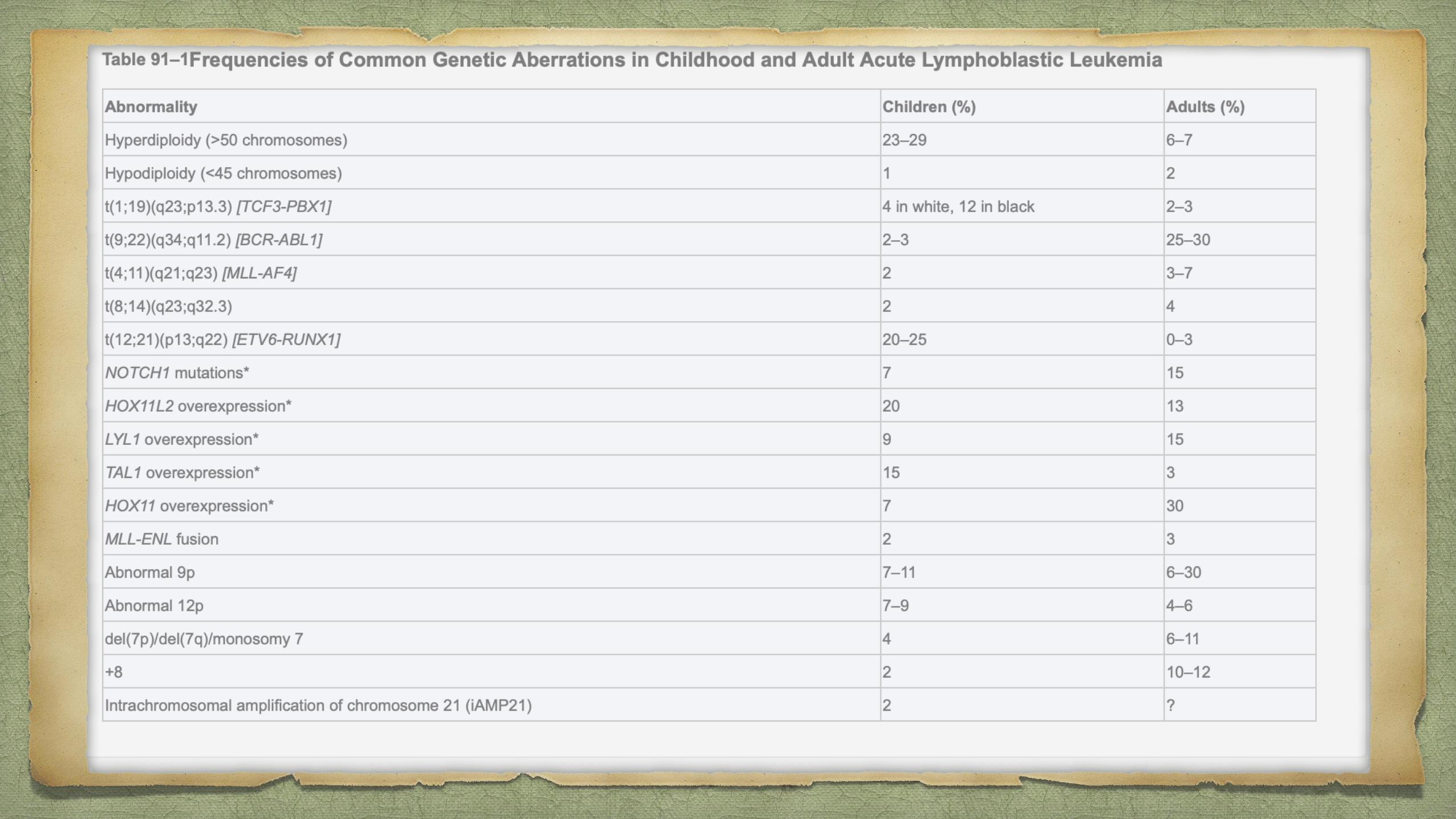
Chromosomal aberrations are the hallmark of ALL, but are not sufficient to generate
leukemia.
Characteristic translocations:
t(12;21) [ETV6-RUNX1], t(1;19) [TCF3-PBX1], t(9;22) [BCR-ABL1] and
rearrangement of MLL.
Similar gene expression profile to (Philadelphia) Ph-positive ALL but without the
BCR-ABL1 rearrangement has been identified. In more than 80% of cases of this so-
called Ph-like ALL, the variant possesses deletions in key transcription factors
involved in B-cell development including IKAROS family zinc finger 1 (IKZF1),
transcription factor 3 (E2A), early B-cell factor 1 (EBF1) and paired box 5 (PAX5).12

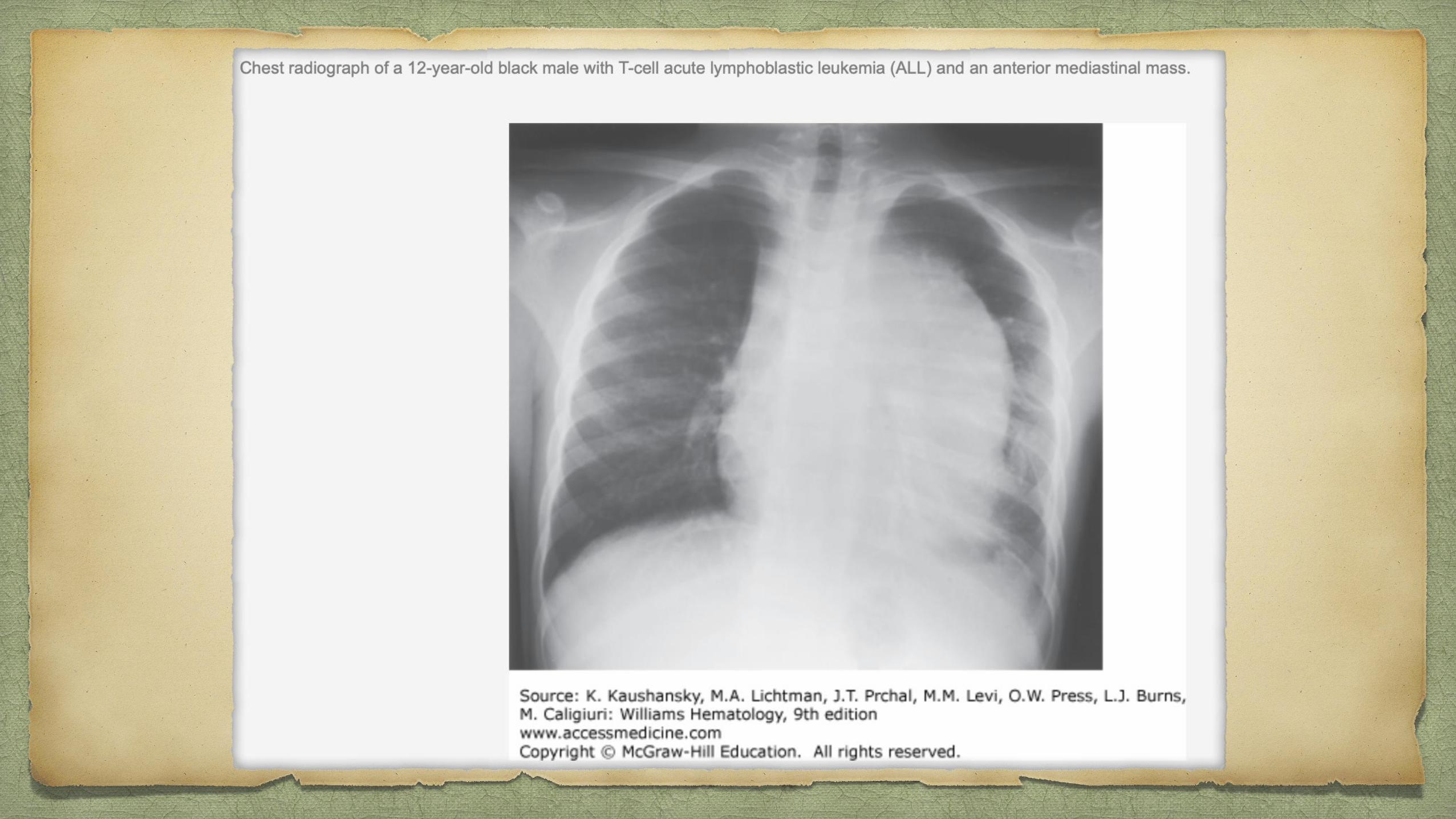
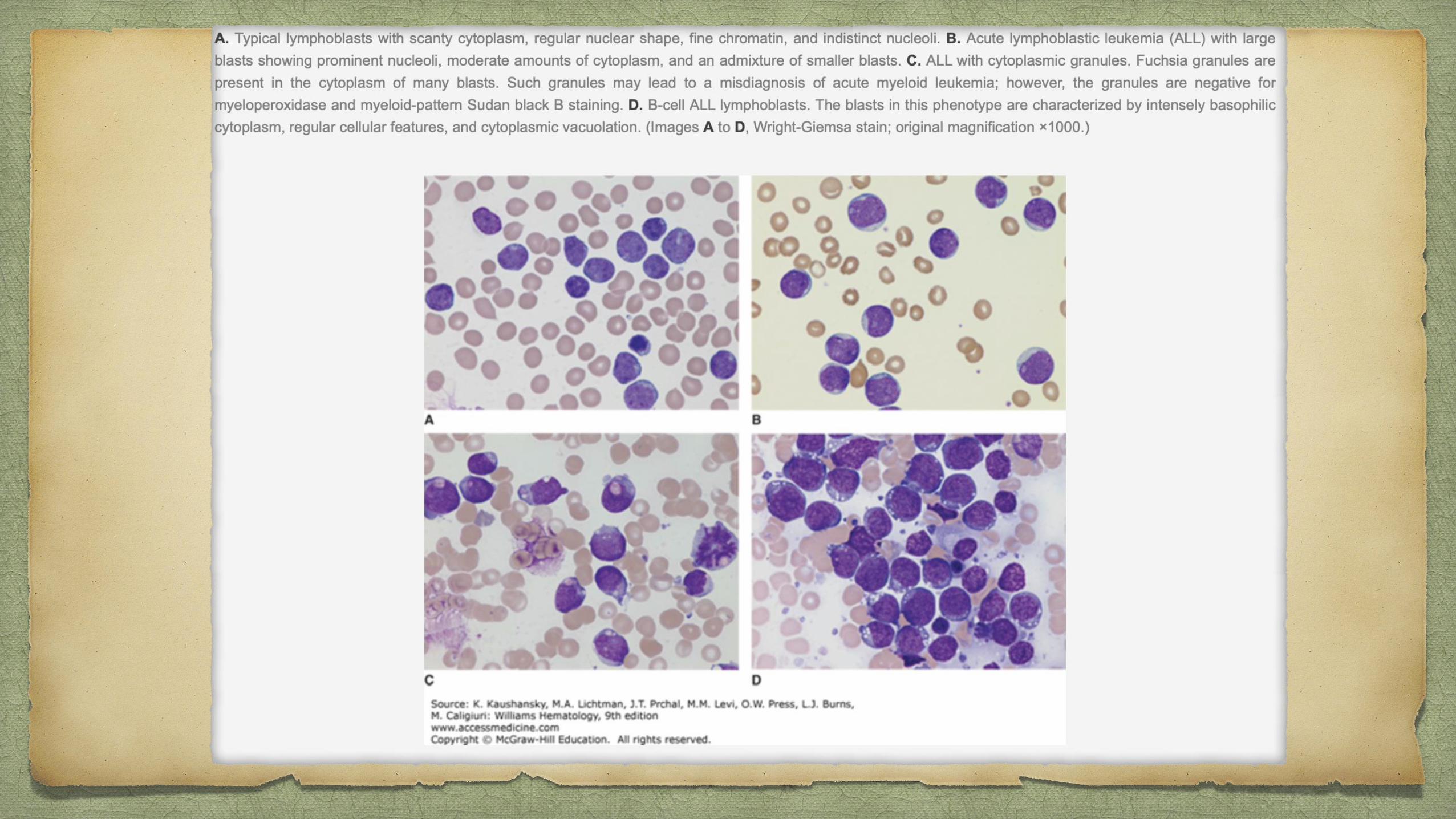

Petechiae, ecchymoses, and bleeding - PTI
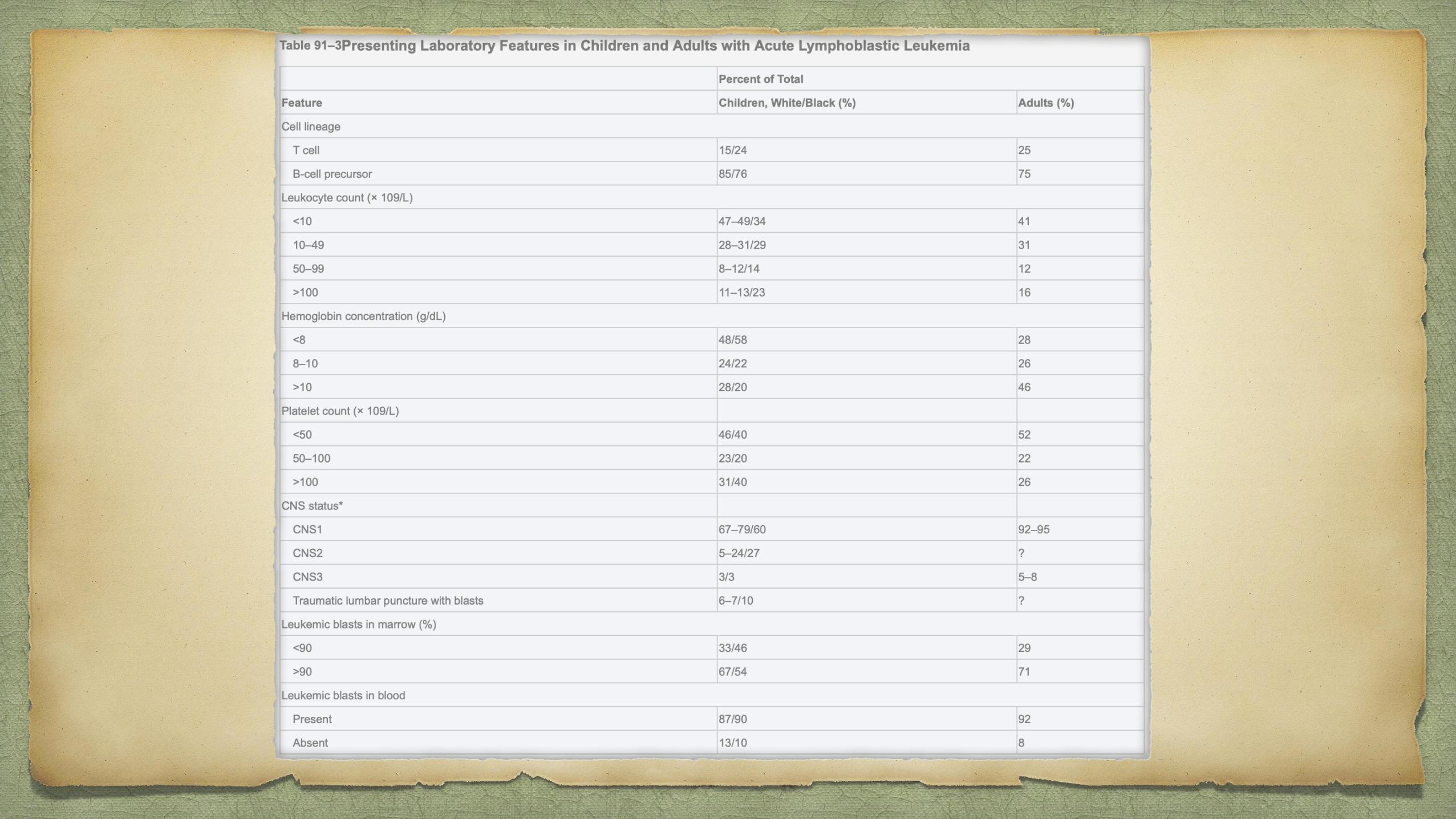
Patients with ALL, or promyelocytic leukemia, or aplastic anemia can present with pancytopenia
In aplastic anemia, hepatosplenomegaly and lymphadenopathy are rare
skeletal pain associated with ALL is absent.
BMA hypocellular marrow that is later replaced by lymphoblasts. polymerase chain reaction (PCR)
ALL should be considered in the differential diagnosis of patients with hypereosinophilia, can precede its diagnosis by
several months.
Occasionally, hematogones in a regenerative marrow may mimic leukemic blast cells; flow cytometry
Infectious mononucleosis and other viral infections, especially those associated with thrombocytopenia or hemolytic
anemia.
Acute infectious lymphocytosis, pertussis, or parapertussis can have marked lymphocytosis, as high as 50 × 109/L, BUT
WITH mature lymphocytes
Bone pain, arthralgia, and occasionally arthritis mimic juvenile rheumatoid arthritis, rheumatic fever, other collagen
diseases, or osteomyelitis. BMA-CCE?
ALL should be distinguished from small, round cell tumors involving the marrow, including neuroblastoma,
rhabdomyosarcoma, and retinoblastoma.(PRIMARY TUMOR; IFT)
ALL DIFFERENTIAL DIAGNOSIS

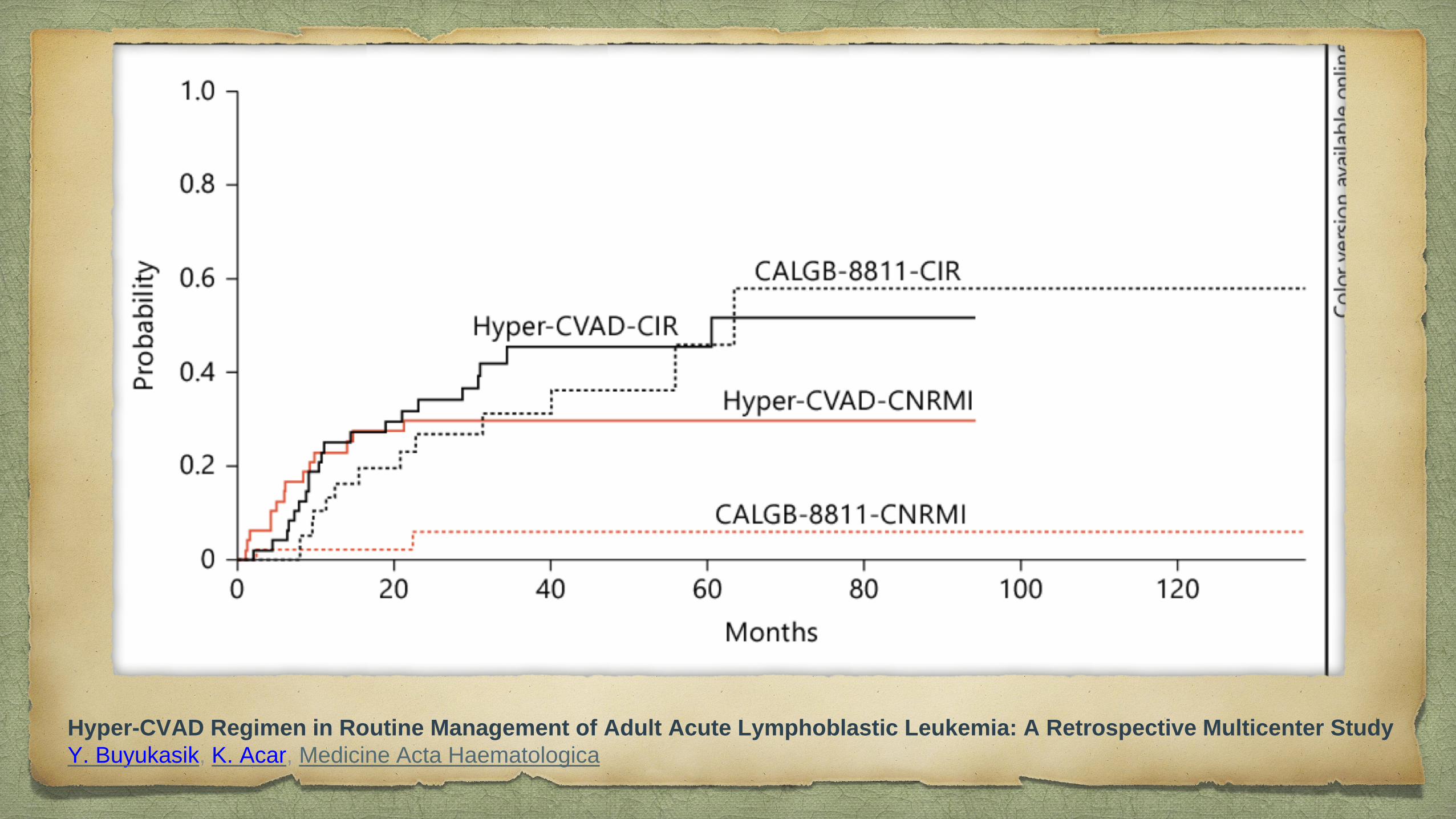
Hyper-CVAD Regimen in Routine Management of Adult Acute Lymphoblastic Leukemia: A Retrospective Multicenter Study
Y. Buyukasik, K. Acar, Medicine Acta Haematologica