layer-by-layer polypeptide macromolecular assemblies-mediated synthesis of mesoporous silica and...
TRANSCRIPT

Published: February 14, 2011
r 2011 American Chemical Society 2834 dx.doi.org/10.1021/la103923c | Langmuir 2011, 27, 2834–2843
ARTICLE
pubs.acs.org/Langmuir
Layer-by-Layer Polypeptide Macromolecular Assemblies-MediatedSynthesis of Mesoporous Silica and Gold Nanoparticle/MesoporousSilica Tubular NanostructuresJeng-Shiung Jan,* Tzu-Han Chuang, Po-Jui Chen, and Hsisheng Teng
Department of Chemical Engineering, National Cheng Kung University, No. 1, University Rd., Tainan, Taiwan 70101, Taiwan
bS Supporting Information
ABSTRACT: A simple and versatile approach is proposed touse the LbL-assembled polypeptide macromolecular assem-blies as mediating agents and templates for directed growth ofgold nanoparticles and biomimetic silica mineralization, allow-ing the synthesis of polypeptide/silica and polypeptide/goldnanoparticle/silica composite materials, as well as mesoporoussilica (meso-SiO2) and gold nanoparticle/mesoporous silica(AuNP/meso-SiO2). The formation of tubular nanostructureswas demonstrated by silicification and growth of gold nano-particles within macromolecular assemblies formed by poly(L-lysine) (PLL) and poly(L-glutamic acid) (PLGA) using polycarbo-nate membranes as templates. The experimental data revealed that the silicified macromolecular assemblies adopted mainly sheet/turn conformation. The as-prepared mesoporous silica materials possessed well-defined tubular structures with pore size andporosity depending on the size of sheet/turn aggregates, which is a function of the molecular weight of polypeptides. The directedgrowth of Au NP and subsequent silica mineralization in the macromolecular assembly resulted in Au NP/meso-SiO2 tubes withuniform nanoparticle size and the as-preparedmaterials exhibited promising catalytic activity toward the reduction of p-nitrophenol.This approach provides a facile and general method to synthesize organic-inorganic composite materials, oxide and metal-oxidenanomaterials with different compositions and structures.
’ INTRODUCTION
Nature has already developed the ability to fabricate materialssuch as diatoms and sponges with precisely controlled nano-structures and morphologies that exceed human engineeringcapabilities.1-6 Inspired by these silicification processes, thebiomimetic synthesis of silica materials has significant advancesin the recent years. Compared with conventional syntheses ofsilica, the advantage of biomimetic synthesis includes benignconditions such as neutral pH, room temperature, and aqueousenvironment. The silicification processes of the biological sys-tems leads to exquisite hierarchical structures and complexmorphologies, which are mediated by proteins or peptides viaself-assembly and template processes.5,6 It is for this reason thatnanoscale control of silicamorphology and structure via templateapproaches has received growing attention. A variety of self-assembled structures including vesicles,7-9 micelles,10,11 plate-lets,12 nanofibers,13-17 and nanotapes18,19 was utilized for silicadeposition. Micro- and nanostructured silicas were also synthe-sized using polypeptide chain conformation, polyamine-saltaggregates, and polyamine-nanoparticle aggregates as tem-plates.20-24 Micro- and nanopatterned silica at surfaces havebeen demonstrated using amine-containing templates by lithogra-phy, microcontact printing, and grafting technologies.25-30 Syn-thetic amine-containing macromolecules were commonly used tomediate the formation of nanoscale silica. The biomimetic synthesis
of silica mediated by amine-containing macromolecules is still animportant subject of study, which will continuously be pursued.
The synthesis of nanostructured materials using the synergybetween biomimetic mineralization and other techniques mayopen up new possibilities for these materials in a range of fieldsincluding catalysis, separations, gene and drug delivery, encap-sulation, and so forth. LbL assembly technique is a simple andversatile approach to deposit a variety of materials on differentsubstrates. Using this technique, free-standing structures such asfilms, tubes, and capsules can be obtained by depositing desiredmaterials and subsequent removing templates.31 One approachcombining LbL assembly technique and biomimetic mineraliza-tion has been utilized to prepare organic-inorganic compositemicrocapsules under benign conditions for cell and enzymeencapsulation.32-34 Unlike previously described composite LbLmultilayer films composed of preprepared inorganic particles,35-37
the deposited inorganic layer formed by the this approach iscontinuous and intact, which is important for many applicationssuch as encapsulation.
Recently, several groups reported that some biomolecules(e.g., L-tyrosine and tyrosine-containing peptides) can serve both
Received: March 29, 2010Revised: December 31, 2010

2835 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
as stabilizing and reducing agents to mediate the growth of metalnanoparticles in alkaline solutions.38-40 Peptide-mediated synthe-sis was also applied to obtain gold and silver nanoparticles on orwithin polyelectrolyte multilayers, organogel networks, and silkfibers.41-44 This facilitates the incorporation of functional na-noparticles into the as-prepared materials through one-stepdirected growth. Taking the advantage of LbL self-assemblytechnique and peptide-mediated synthesis of hard materials, itis a possible strategy to prepare inorganic materials with differentmorphologies and to incorporate additional functionality such asporosity and functional nanoparticle into the as-prepared mate-rials, which will be essential for many applications.
We have demonstrated using self-assembled polypeptides todirect the formation of inorganic materials,8,9,45 which haveheightened our interest in the synthesis of complex inorganicmaterials using biological macromolecules as templates. Particu-larly relevant to this work, however, only a few studies reportedthe templated synthesis of porous silica under benign con-ditions.9,24 In this paper, we report on a polypeptide-mediatedstrategy that utilizes surface macromolecular assemblies tosynthesize mesoporous silica through silicification under benignconditions, as well as the gold nanoparticle/mesoporous silicathrough directed growth of gold nanoparticles and subsequentsilicification. Layer-by-layer (LbL) self-assembly technique facil-itates the buildup of preorganized molecular templates withtailored structure and composition as well as the wall thicknessto nanometer scale. To demonstrate the feasibility of thisstrategy, the synthetic cationic and anionic polypeptides werealternatively deposited on the walls of polycarbonate (PC)membrane pores via LbL assembly for mediating the formationof gold nanoparticles and silicas. The synthetic cationic andanionic polypeptides, poly(L-lysine) (PLL) and poly(L-glutamicacid) (PLGA), serve as silica mineralizing and gold reducingagent, respectively. The LbL assembled polypeptide macromo-lecular assemblies that possess molecular organizations (i.e., β-sheet) can serve as templates to control silica nanostructures andconfine the growth of gold nanoparticles. The structure andfunctionality of as-synthesized materials are determined by themolecular organization, size, shape, and morphology of the dualtemplates (i.e., macromolecular assembly and PC membrane)used.
’EXPERIMENTAL SECTION
Materials. PC membranes (product catalogue No.: HTTP04700,47 mm diameter) with a pore diameter of 0.4 μm and a membranethickness about 10-20 μm were obtained from Millipore. THF (ACSReagent, Merck) and Diethyl ether (Anhydrous, ACS Reagent, J. T.Backer) were dried usingNametal. Hexane (ACSReagent, EM Science)was dried using calcium hydride. The amino acids used in this work Nε-Z-L-lysine (∼99%, Z: carboxybenzyl) and γ-benzyl-L-glutamic acid(>99%) were used as received from Aldrich. Bis(1,5-cyclooctadiene)-nickel(0) (98þ%), 2,20-bipyridyl (99þ%,), hydrogen tetrachloroaurate-(III) trihydrate (ACS, 99.99%), and p-nitrophenol (spectrophotometricgrade) were used as received from Sigma-Aldrich. Iodotrimethylsilane(Me3SiI, 97%, saturated with Copper) and trifluoroacetic acid (99%)were supplied by Alfa Aesar. Triphosgene (98%, Merck), tetramethylorthosilicate (99%), hydrogen bromide (33 wt % in acetic acid), andNaBH4 (96%) were used as received from Fluka.Polypeptide Synthesis. The polypeptide synthesis was per-
formed using the zerovalent nickel initiator 2,20-bipyridyl-Ni-(1,5-cyclooctadiene) (BpyNiCOD) to polymerize Nε-Z-L-lysine andγ-benzyl-L-glutamic acid NCAs by following the literature reported
procedures.46-48 Poly(Z-L-lysine) (PZLL) and poly(γ-benzyl-L-glutamic) (PBLG) acid were deprotected by using HBr and Me3SiI,respectively. The notations for poly(L-lysine) (PLL) and poly(L-gluta-mic acid) (PLGA) used throughout are Lysm and Glun, respectively,where m and n are the number of amino acids in one chain. PLL withdifferent chain lengths used in this study were Lys145 (Mn = 37800,Mw/Mn = 1.14), Lys210 (Mn = 542000, Mw/Mn = 1.02), and Lys340 (Mn =89200, Mw/Mn = 1.10), respectively. And PLGA with different chainlengths were Glu125 (Mn = 27100,Mw/Mn = 1.19), Glu190 (Mn = 42000,Mw/Mn = 1.02), and Glu370 (Mn = 81000,Mw/Mn = 1.11), respectively.The polypeptides (PZLL and PBLG) with different molecular weightswere synthesized and characterized by GPC (Figure S1 and S2,Supporting Information). NMR measurements confirmed that thepolypeptides (PZLL, PBLG, PLL, and PLGA) were prepared (FigureS3, Supporting Information).PLL/PLGA Multilayer Assembly. Method I. The PLL-coated
membranes were first prepared by immersing the PC membranes into aPLL solution (1 mg/mL in 0.5 M NaCl), followed by immediatelysonicating for 2 min and allowing 10 min for adsorption. Afterthoroughly rinsing with 0.5 M NaCl aqueous solution, negativelycharged PLGA was then adsorbed by immersing the PLL-coatingmembranes into a PLGA solution (1 mg/mL in 0.5 M NaCl) usingthe same procedure. The desired number of layers was deposited bycyclic adsorption of PLL and PLGA.
Method II. The PLL and PLGA solutions were dissolved in sodiumphosphate buffer (pH 7.4, Pierce) for depositing desired number oflayers. The samples were denoted as followed. For example, the sample,(Lys210/Glu190)9I, was obtaining from silicification of 9-layer Lys210/Glu190 coated PC membrane (that is, (Lys210/Glu190)9) using LbLassembly method I.SiO2 Tube Formation. The PLL/PLGA multilayer coated mem-
branes were then inserted in a freshly prepared 0.5 M orthosilicic acidsolution for 10-12 h to allow precipitation of silica in the PLL/PLGAmultilayer. After thoroughly rinsing with DI water and drying at roomtemperature, the film coating on the membrane surface was removedpartially using fine sandpaper. The as-prepared silica/PLL/PLGAmulti-layer membranes were slowly heat to 95 �C for 24 h, following byremoving the membrane using dichloromethane. Pure silica tubes wereobtained by calcining the silica/PLL/PLGA composite tubes in air at500 �C for 10 h. (heating rate 2 K min-1).Gold/Silica Tube Formation. The PLL/PLGA multilayer coated
membranes were immersed in a HAuCl4 solution (1.5� 10-4 M, 20 mL)at pH 7 (or 11.5) for 12 h, and the solution was adjusted to pH 7 (or 11.5)and 200 μL of Au ion solution (1.5� 10-2 M) was added to the solutionevery 12 h. After the formation of Au NPs, the membranes were taken outfrom the solution and inserted in a freshly prepared 0.35Morthosilicic acidsolution for 6-12 h to allow precipitation of silica in the Au NP/PLL/PLGA multilayer. After thoroughly rinsing with DI water and drying atroom temperature, the as-preparedAuNP/meso-SiO2/PLL/PLGAmulti-layer membranes were slowly heat to 95 �C for 24 h and the PCmembranes were removed by dissolving in dichloromethane. Pure AuNP/meso-SiO2 tubes were obtained by calcining at 500 �C for 10 h in air(heating rate 2 K min-1).Gold/Silica Catalytic Tests. First 1 mg of Au NP/meso-SiO2
catalyst was dispersed in 30 mL of DI water and the suspension wasstirred for overnight. Then, 30 mL of 2� 10-2 M NaBH4 and 30 mL of4 � 10-4 M p-nitrophenol aqueous solution were then added in thesuspension and the catalytic activity was evaluated as the percentagedisappearance of p-nitrophenol at a wavelength of 400 nm usingUV-visspectrophotometer.Instrumentation and Characterization. Gel permeation
chromatography (GPC) measurements were performed at 55 �C beforedeprotection of the polypeptide using a Viscotek system equipped withthree detectors, which are RI (VE3580, Viscotek), right angle light

2836 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
scattering, and viscometer (Dual 270, Viscotek). TwoViscoGEL I-Seriescolumns (catalog numbers I-MBHMW-J012906 and I-MBLMW-H110211, Viscotek) were used for efficient separation using 0.1 M LiBrin DMF as eluent. The eluent flow rate was 1 mL/min. 1H NMR spectrawere recorded at 300MHz on aMercury 300 Varian spectrometer usingd-TFA as solvent. Field-Emission Scanning Electron Microscopy (FE-SEM) and energy dispersive X-ray (EDX) measurements were per-formed using a JEOL JSM-6700F microscope operating at 1-10 kV.Samples were collected via centrifugation, air-dried, and mounted oncarbon tape for imaging. Transmission electron microscopy (TEM)measurements were performed on a Hitachi H7500 microscope with aTungsten lamp and an excitation voltage of 120 kV. The samples weredispersed in methanol (100%, Aldrich) and placed on a 400-meshcopper grid. Infrared spectroscopy was performed on a Thermo NicoletNexus 670 FTIR. Background spectra were collected after 10 min ofevacuation. A powder mixture of mass ratio 0.01 sample: 0.99 potassiumbromide (Aldrich) was pelletized and analyzed after 10 min of evacua-tion. Nitrogen adsorption measurements were performed using aMicromeritics 2010 ASAP instrument at 77 K. Surface areas werecalculated by the Brunauer-Emmett-Teller (BET) method. Porevolumes and pore size distributions were determined from nitrogenadsorption isotherm data using the t-plot and Barrett-Joyner-Halenda(BJH) method. Thermal gravimetric analyses (TGA) were performedusing a TGA7 Instrument from Perkin-Elmer over a temperature rangeof 25 to 800 �C using oxygen as a carrier gas and temperature rampingrate of 5 �C min-1. The UV-vis measurements were carried outon SCINCO S-3100 UV-vis spectrophotometer and the UV-ATRmeasurements were performed on JASCO V-670 UV-vis spectro-photometer.
’RESULTS AND DISCUSSION
Mesoporous tubular silicas with tunable pore size and porositywere synthesized by controlling the preorganized moleculartemplates, which are the polypeptide macromolecular assem-blies. Different molecular weights of poly(L-lysine) (PLL) andpoly(L-glutamic acid) (PLGA) were selected to deposit on thewalls of membrane pores. The PC membranes and LbL-assembled PLL/PLGA multilayer films on walls of membrane
pores were used as sacrificial templates for the synthesis of silicahollow tubes. As shown in Scheme 1, the PLL/PLGA multilayerfilms were coated on porous PC membrane via LbL method.Then the PLL/PLGA-coated PC membrane was immersed in afreshly prepared orthosilicic acid solution (0.5 M, pH 3.8) for10-12 h. The silicified PLL/PLGA-coated membranes wereheated to 95 �C for 24 h and the silica/PLL/PLGA compositetubes were obtained by dissolving PC membranes. Then thecomposite tubes were calcined at elevated temperature toremove organic materials. As a control experiment, porous PCmembranes without deposited PLL/PLGAmultilayer films wereimmersed in a freshly prepared orthosilicic acid solution for 12 hand no silica deposition on the bare PC membranes. It is knownthat PLL can induce polymerization of orthosilicic acid due toelectrostatic interactions or hydrogen bonding between theamine group on the polymer chain and orthosilicic acid. There-fore, it is expected that the deposited PLL/PLGAmultilayer filmscan induce polymerization of orthosilicic acid as well and theprecipitated silicas can translate the molecular organization.
PLL and PLGA were deposited on the walls of membranepores using two methods. For method I, PLL and PLGA wereseparately dissolved in 0.5 M NaCl aqueous solution for thepolypeptide film assembly. As a comparison to method I, phos-phate buffer solution (pH 7.4) was used as solvent in method II.After coating polypeptide on the membrane pores, the LbLassembled PLL/PLGA tubes were obtained and TEM character-ization reveals that the PLL/PLGA tubes with well-definedstructures and highly uniform coatings were observed (forexample, Figure S4, Supporting Information). FTIR spectra ofthe PLL/PLGA tubes made by using Method I and II show thatamide I and amide II characteristic peaks are at 1626-28 and1545 cm-1, respectively (Figure S5, Supporting Information). Itindicates that the PLL/PLGA macromolecular assemblies arepredominantly in β-sheet conformation. In addition, theshoulder at 1650 cm-1 suggests that random coil and/or R-helixconformations with lower relative percentage were also presentin the macromolecular assemblies. Our results are consistent
Scheme 1. Procedure Used for Preparing Mesoporous Silica and Au NP/Meso-Silica Tubes
2837 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
with previous studies reported by Boulmedais and co-work-ers.49,50 We have attempted to measure the secondary conforma-tion adopted by the polypeptide macromolecular assembliesusing circular dichroism (CD). However, the measurementswere hampered by the poor solubility and dispersion of thepolypeptide tubes in aqueous solution.
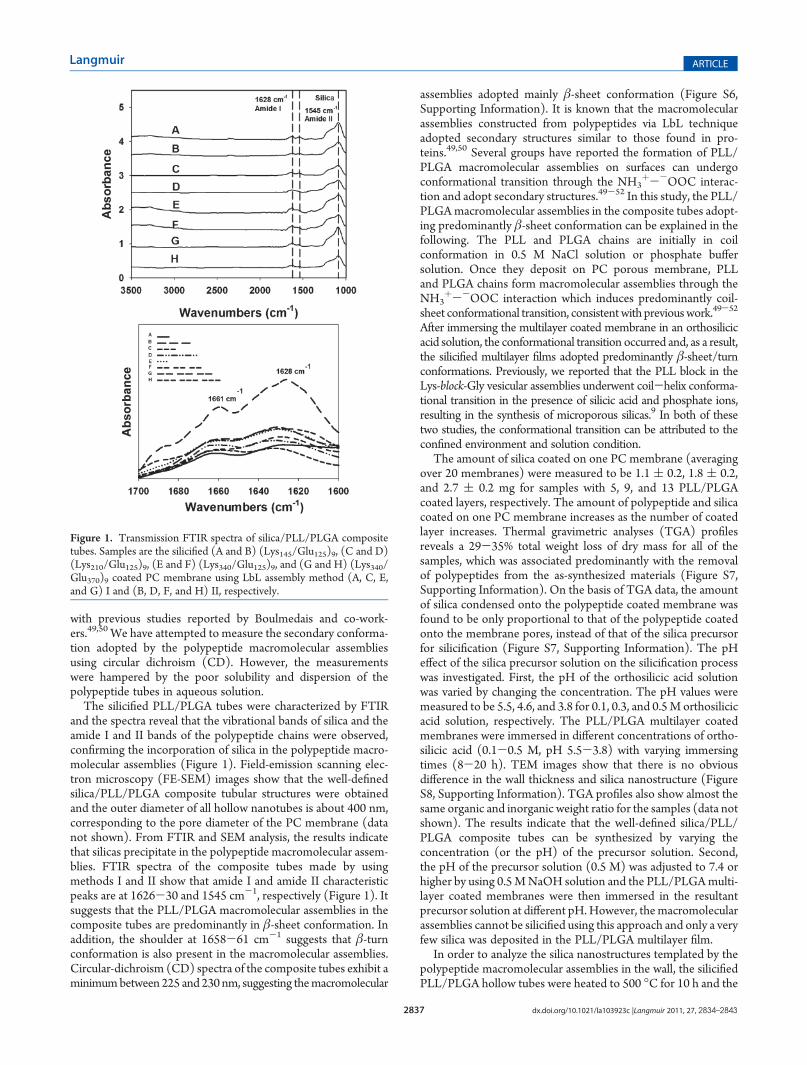
The silicified PLL/PLGA tubes were characterized by FTIRand the spectra reveal that the vibrational bands of silica and theamide I and II bands of the polypeptide chains were observed,confirming the incorporation of silica in the polypeptide macro-molecular assemblies (Figure 1). Field-emission scanning elec-tron microscopy (FE-SEM) images show that the well-definedsilica/PLL/PLGA composite tubular structures were obtainedand the outer diameter of all hollow nanotubes is about 400 nm,corresponding to the pore diameter of the PC membrane (datanot shown). From FTIR and SEM analysis, the results indicatethat silicas precipitate in the polypeptide macromolecular assem-blies. FTIR spectra of the composite tubes made by usingmethods I and II show that amide I and amide II characteristicpeaks are at 1626-30 and 1545 cm-1, respectively (Figure 1). Itsuggests that the PLL/PLGA macromolecular assemblies in thecomposite tubes are predominantly in β-sheet conformation. Inaddition, the shoulder at 1658-61 cm-1 suggests that β-turnconformation is also present in the macromolecular assemblies.Circular-dichroism (CD) spectra of the composite tubes exhibit aminimumbetween 225 and 230 nm, suggesting themacromolecular
assemblies adopted mainly β-sheet conformation (Figure S6,Supporting Information). It is known that the macromolecularassemblies constructed from polypeptides via LbL techniqueadopted secondary structures similar to those found in pro-teins.49,50 Several groups have reported the formation of PLL/PLGA macromolecular assemblies on surfaces can undergoconformational transition through the NH3
þ--OOC interac-tion and adopt secondary structures.49-52 In this study, the PLL/PLGAmacromolecular assemblies in the composite tubes adopt-ing predominantly β-sheet conformation can be explained in thefollowing. The PLL and PLGA chains are initially in coilconformation in 0.5 M NaCl solution or phosphate buffersolution. Once they deposit on PC porous membrane, PLLand PLGA chains form macromolecular assemblies through theNH3
þ--OOC interaction which induces predominantly coil-sheet conformational transition, consistentwith previouswork.49-52
After immersing the multilayer coated membrane in an orthosilicicacid solution, the conformational transition occurred and, as a result,the silicified multilayer films adopted predominantly β-sheet/turnconformations. Previously, we reported that the PLL block in theLys-block-Gly vesicular assemblies underwent coil-helix conforma-tional transition in the presence of silicic acid and phosphate ions,resulting in the synthesis of microporous silicas.9 In both of thesetwo studies, the conformational transition can be attributed to theconfined environment and solution condition.
The amount of silica coated on one PC membrane (averagingover 20 membranes) were measured to be 1.1 ( 0.2, 1.8 ( 0.2,and 2.7 ( 0.2 mg for samples with 5, 9, and 13 PLL/PLGAcoated layers, respectively. The amount of polypeptide and silicacoated on one PC membrane increases as the number of coatedlayer increases. Thermal gravimetric analyses (TGA) profilesreveals a 29-35% total weight loss of dry mass for all of thesamples, which was associated predominantly with the removalof polypeptides from the as-synthesized materials (Figure S7,Supporting Information). On the basis of TGA data, the amountof silica condensed onto the polypeptide coated membrane wasfound to be only proportional to that of the polypeptide coatedonto the membrane pores, instead of that of the silica precursorfor silicification (Figure S7, Supporting Information). The pHeffect of the silica precursor solution on the silicification processwas investigated. First, the pH of the orthosilicic acid solutionwas varied by changing the concentration. The pH values weremeasured to be 5.5, 4.6, and 3.8 for 0.1, 0.3, and 0.5M orthosilicicacid solution, respectively. The PLL/PLGA multilayer coatedmembranes were immersed in different concentrations of ortho-silicic acid (0.1-0.5 M, pH 5.5-3.8) with varying immersingtimes (8-20 h). TEM images show that there is no obviousdifference in the wall thickness and silica nanostructure (FigureS8, Supporting Information). TGA profiles also show almost thesame organic and inorganic weight ratio for the samples (data notshown). The results indicate that the well-defined silica/PLL/PLGA composite tubes can be synthesized by varying theconcentration (or the pH) of the precursor solution. Second,the pH of the precursor solution (0.5 M) was adjusted to 7.4 orhigher by using 0.5MNaOH solution and the PLL/PLGAmulti-layer coated membranes were then immersed in the resultantprecursor solution at different pH.However, themacromolecularassemblies cannot be silicified using this approach and only a veryfew silica was deposited in the PLL/PLGA multilayer film.
In order to analyze the silica nanostructures templated by thepolypeptide macromolecular assemblies in the wall, the silicifiedPLL/PLGA hollow tubes were heated to 500 �C for 10 h and the
Figure 1. Transmission FTIR spectra of silica/PLL/PLGA compositetubes. Samples are the silicified (A and B) (Lys145/Glu125)9, (C and D)(Lys210/Glu125)9, (E and F) (Lys340/Glu125)9, and (G and H) (Lys340/Glu370)9 coated PC membrane using LbL assembly method (A, C, E,and G) I and (B, D, F, and H) II, respectively.

2838 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
organic compounds (PLL and PLGA) were burned off. FE-SEMimages of all calcined silica/polypeptide tubes show that the well-defined tubular structures were obtained and the outer diametersof all silica hollow nanotubes are about 400 nm after heatingat higher temperature, similar to the silica/PLL/PLGA compo-site tubes (for example, Figure 2, parts A and B). No obviousshrinkage or collapse is observed after calcination and the tubelength corresponds to the thickness of the original PC mem-branes. TEM images further reveal that the silica hollow tubes areporous, but the porous structure do not possess long-rang order(Figure 2, parts C and D, and Figure S9, Supporting In-formation). From SEM and TEM analysis, the results indicatethat a highly uniform and conformal coating was successfullyachieved and the average wall thickness of as-synthesized silicatubes is dependent on the number of deposited layer linearly.Themean wall thickness of silica tubes is calculated to be about 9( 0.5 nm per PLL/PLGA layer. Previously, the thickness of thePLL/PLGA multilayer assembled at pH 8.4 in Mes-Tris buffersolution was measured with mean thickness about 5.2 nm perbilayer.49 In our study, it is reasonable to assume the thickness ofeach bilayer should be 5-6 nm, indicating a thickness incrementof ca. 3-4 nm for deposition of silica. It indicates the orthosilicicacid/silicate oligomers infiltrate into the coated polypeptide film,resulting in the swelling of polypeptide film and, in turn, theincrement of thickness. Silicas precipitate in the film due to thepresence of PLL, which is evident that the LbL-assembled PLL/PLGA macromolecular assembly acts as a template for theformation of silica. It is known that the presence of protonatedamines (NH3
þ) of the lysine side chain interacts strongly withthe orthosilicic acid/silicate oligomers, further enhancing silicapolymerization.8,9,53 Rather silica polymerization is relativelyslow in the presence of neutral PLL due to only hydrogenbonding interactions between negatively charged silicate and
electrically neutral Nε-amine groups (NH2).21 In this study, LbL
assembled PLL/PLGA multilayer films were fabricated viaelectrostatic association (NH3
þ--OOC salt bridging). Hencepart of protonated amines was neutralized and the diffusion oforthosilicic acid/silicate oligomers was hindered in the polypep-tide multilayers. This explains why silica polymerization in thefilm is slow (on the order of hours), which is consistent withprevious studies.29,54
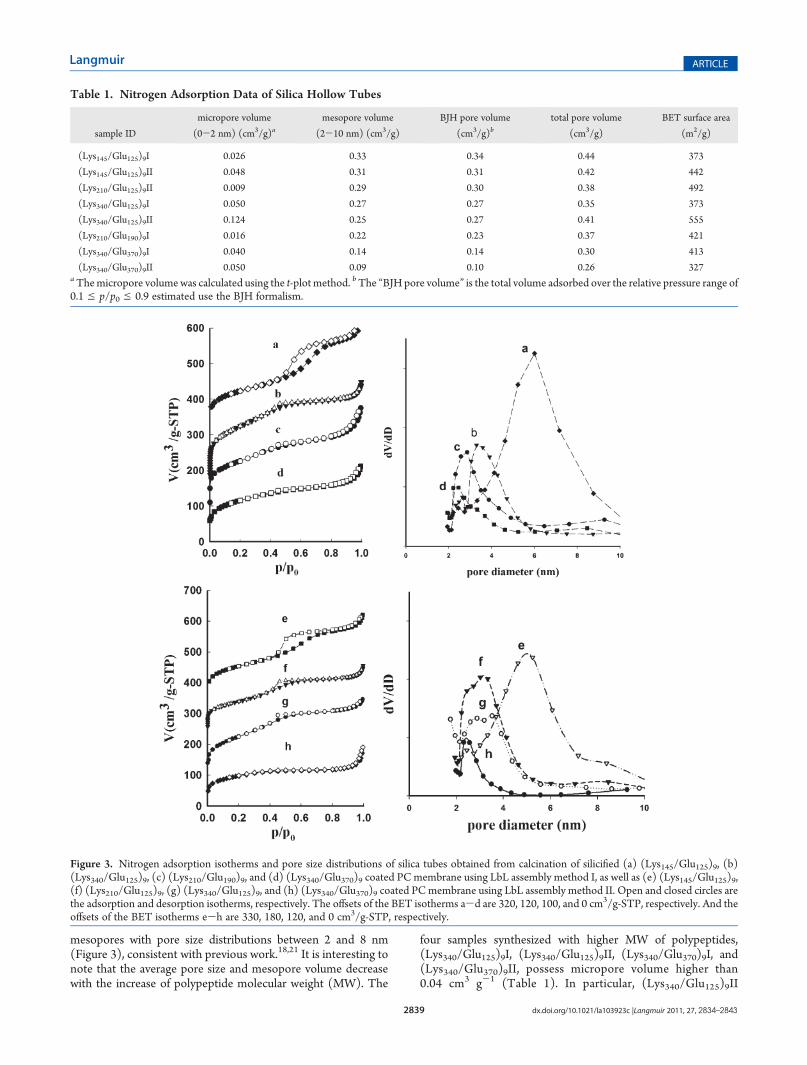
Previous studies have demonstrated that the uniform and well-defined silica hollow tubes can be synthesized by depositing silicamaterials on the walls of membrane pores using sol-gelmethod.55,56 However, the as-synthesized silica hollow tubesdid not possess porosity. On the basis of the results from TEManalysis, the as-synthesized silica hollow tubes are porous and thepore size of the calcined silica/(Lys340/Glu125)9 tubes is smallerthan 5 nm (Figure 2C and Figure S9A, Supporting Information),whereas the calcined silica/(Lys145/Glu125)9 tubes possess largerpore size mostly (Figure 2D and Figure S9C, SupportingInformation). It indicates that the size of aggregates is a functionof the PLL and PLGA molecular weight, which correlates withthe size of β-sheet/turn aggregates. Nitrogen adsorption mea-surements were further performed to determine the influence ofthe LbL assemble solution condition and polypeptide molecularweight on the pore size and porosity. From the adsorption data,the total pore volumes for all samples range between 0.26 and0.45 cm3g-1, which is the amount of nitrogen adsorbed at p/p0value of 0.98 (Table 1). the mesopore (2-10 nm) volumes formost samples, representing nitrogen adsorbed in the pore sizebetween 2 and 10 nm, range between 0.2 and 0.35 cm3 g-1,except (Lys340/Glu370)9I and (Lys340/Glu370)9II (Table 1). Theadditional nitrogen adsorbed beyond the mesopore volume isdue to nitrogen adsorbed on the surface of the tubes. On the basisof BJH analysis, all of the as-synthesized silica tubes possess
Figure 2. FE-SEM (A, B) and TEM (C, D) images of silica tubes obtained from silicification of (A) (Lys210/Glu190)5, (B) (Lys210/Glu190)9, (C)(Lys340/Glu125)9, and (D) (Lys145/Glu125)9 coated PC membrane using LbL assembly method I after calcination.
2839 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
mesopores with pore size distributions between 2 and 8 nm(Figure 3), consistent with previous work.18,21 It is interesting tonote that the average pore size and mesopore volume decreasewith the increase of polypeptide molecular weight (MW). The
four samples synthesized with higher MW of polypeptides,(Lys340/Glu125)9I, (Lys340/Glu125)9II, (Lys340/Glu370)9I, and(Lys340/Glu370)9II, possess micropore volume higher than0.04 cm3 g-1 (Table 1). In particular, (Lys340/Glu125)9II
Table 1. Nitrogen Adsorption Data of Silica Hollow Tubes
sample ID
micropore volume
(0-2 nm) (cm3/g)amesopore volume
(2-10 nm) (cm3/g)
BJH pore volume
(cm3/g)btotal pore volume
(cm3/g)
BET surface area
(m2/g)
(Lys145/Glu125)9I 0.026 0.33 0.34 0.44 373
(Lys145/Glu125)9II 0.048 0.31 0.31 0.42 442
(Lys210/Glu125)9II 0.009 0.29 0.30 0.38 492
(Lys340/Glu125)9I 0.050 0.27 0.27 0.35 373
(Lys340/Glu125)9II 0.124 0.25 0.27 0.41 555
(Lys210/Glu190)9I 0.016 0.22 0.23 0.37 421
(Lys340/Glu370)9I 0.040 0.14 0.14 0.30 413
(Lys340/Glu370)9II 0.050 0.09 0.10 0.26 327aThemicropore volumewas calculated using the t-plotmethod. bThe “BJH pore volume” is the total volume adsorbed over the relative pressure range of0.1 e p/p0 e 0.9 estimated use the BJH formalism.
Figure 3. Nitrogen adsorption isotherms and pore size distributions of silica tubes obtained from calcination of silicified (a) (Lys145/Glu125)9, (b)(Lys340/Glu125)9, (c) (Lys210/Glu190)9, and (d) (Lys340/Glu370)9 coated PCmembrane using LbL assembly method I, as well as (e) (Lys145/Glu125)9,(f) (Lys210/Glu125)9, (g) (Lys340/Glu125)9, and (h) (Lys340/Glu370)9 coated PCmembrane using LbL assembly method II. Open and closed circles arethe adsorption and desorption isotherms, respectively. The offsets of the BET isotherms a-d are 320, 120, 100, and 0 cm3/g-STP, respectively. And theoffsets of the BET isotherms e-h are 330, 180, 120, and 0 cm3/g-STP, respectively.

2840 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
possesses the highest micropore volume (0.12 cm3 g-1) amongall of these samples. The combined micropore and mesoporevolumes for those samples range between 0.23 and 0.37 cm3 g-1,except (Lys340/Glu370)9I and (Lys340/Glu370)9II. The silicatubes synthesized with lower MW of polypeptides have largerpore size and higher pore volume than those synthesized withhigher MW of polypeptides. The results show that PLL andPLGA with lower MW form larger sheet/turn aggregates on thesurface, which result in the templated silica tubes with larger poresize. PLL and PLGA with high MW tend to form smaller sheet/turn aggregates due to the chain entanglement and sterichindrance. The low MW polypeptide chains have less entangle-ment and steric hindrance as compared with the high MW onesand this can allow the peptide chains to associate with each othervia electrostatic interaction along the chains and, in turn, formlarger sheet/turn aggregates (Scheme 2). The results indicatethat the silicification process and polypeptide molecular weightinfluence the molecular organization of PLL/PLGA macromo-lecular assemblies and subsequently the resultant pore size andporosity of the as-synthesized silica tubes. In addition, the pre-sence of ions and the adsorption of polypeptides on the surfacecan regulate and confine PLL/PLGA macromolecular assem-blies, subsequently leading to the formation of much uniformsheet/turn aggregates.
We are not the first one to study the synthesis of porous silicausing polypeptide secondary structures as templates.9,18,21 Pre-viously, Jan and Shantz reported the synthesis of microporoussilica nanoparticles and platelets using both the self-assembledstructures formed by Lys-b-Gly block copolypeptides and R-helical PLL as templates under benign condition. The coil-helixtransition of PLL block and simultaneous silica precipitation inthe presence of both silicic acid and phosphate ions led toR-helixtemplated silicas.9 B€orner group utilized the self-assembled PEO-peptide nanotapes with a β-sheet core as templates for silicifica-tion in ethanol/water solution and the resulting silica possessedpore size between 2 and 8 nm.18 Shantz group reported that themicroporous silica synthesized with solvated R-helical PLLpossesses cylindrical pores of approximately 1.5 nm diameterand the mesoporous silica synthesized using PLL chains adoptedβ-sheet conformation as templates possesses larger pores withsizes between 2 and 8 nm.21 The pore size of β-sheet templatedsilica depends on the PLL concentration, or namely the size ofaggregates. However, these reported β-sheet templated silicaswere not synthesized under benign condition and the morphology
and pore size of the as-prepared silicas cannot be controlled. Inthis study, the porous silica tubes can be synthesized using thesimple synthesis process (i.e., LbL assemble and silicificationprocesses) under benign condition. In addition, the average poresize can be controlled simply by using the polypeptides with dif-ferent MWs. Some of the as-synthesized silica tubes possess highermesopore volume and narrower pore size distribution than thosereported by B€orner and Shantz group. It is also worth to note thatporous silicas with different morphologies such as films andcapsules can be easily prepared due to the versatility of the LbLtechnique and feasibility of polypeptide-mediated synthesis.
Metal nanoparticles such as gold are attractive catalysts whichhave been successfully applied to a variety of reactions such ashydrogenation, oxidation-reduction, and reforming.57,58 Usinga mesoporous silica support to immobilize gold nanoparticles havebeenwildly used for catalytic applications inmany researches.59-62
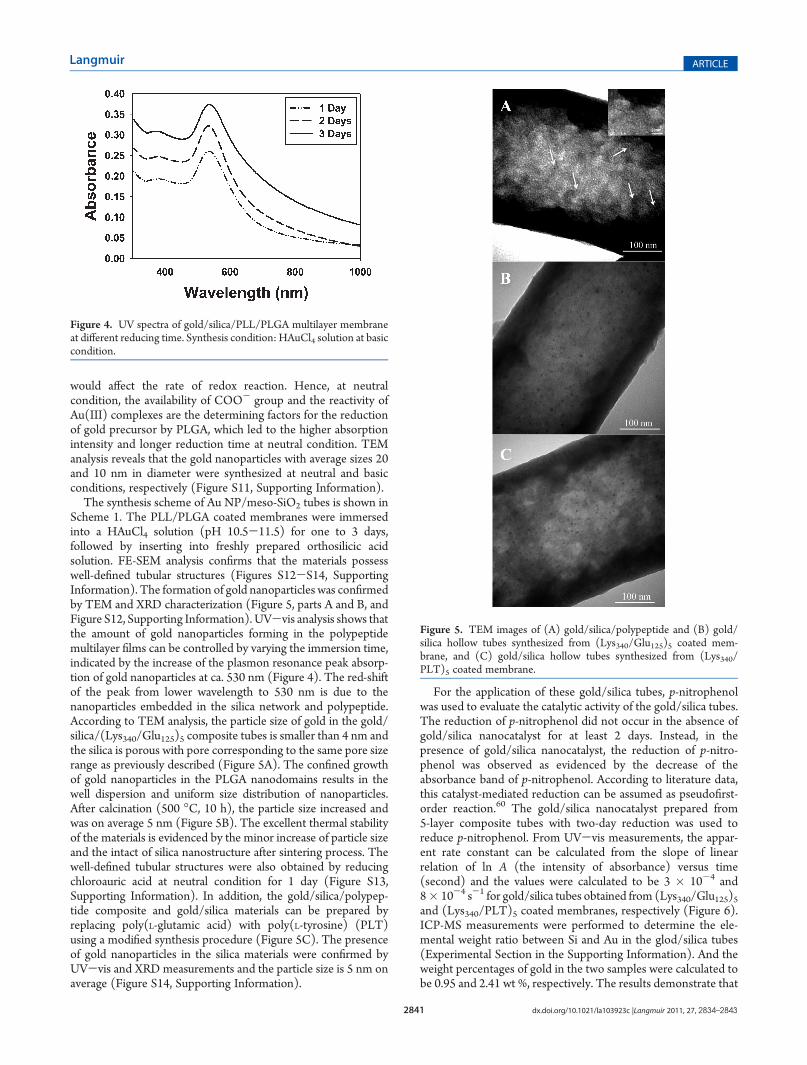
Here we first demonstrate that PLGA can serve as both reducingand stabilizing agents in aqueous solution to form monodis-persed gold nanoparticles at neutral and basic conditions asevidenced by the turning of colorless solution to pink and theappearance of plasmon resonance peak of gold nanoparticles atthe wavelength of 520 nm, indicating the formation of goldnanoparticles. Instead, PLL does not possess reducing capabilityin aqueous solution according to our study. The time-resolvedUV-vis measurements were conducted to study the kineticgrowth of gold nanoparticles in the presence of PLGA at neutraland basic conditions (Figure S10, Supporting Information). Atbasic condition, the absorption at 520 nm was recorded as afunction of time and the time course shows a rapid growth ofnanoparticles between 0-100 min, followed by a plateau in-dicating the end of reaction. Rather, at neutral condition thecontinuous increase of the absorption at 520 nm is observed evenafter 3 days. The pH-dependent reduction potential of Au(III)complexes has been reported in the literature.63-65 The Au(III)complexes, AuCl2(OH)2
- and Au(OH)4-, are the dominating
form at neutral and basic conditions, respectively. The reactivityof AuCl2(OH)2
-, which is an indication for the reduction ofAu(III) complexes, is higher than that of Au(OH)4
-.65 Therelatively low absorption intensity at basic condition suggeststhat only a portion of Au(III) complexes was reduced by PLGA,which can be probably attributed to the low reactivity of Au-(OH)4
-. In contrast, much more Au(0) atoms were formed atneutral condition as indicated by the high absorption intensity. Inaddition, the deprotonation of COOH group at different pH
Scheme 2. Illustration of the Formation of Polypeptide Macromolecular Assemblies by PLL and PLGA with Different MolecularWeights
2841 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
would affect the rate of redox reaction. Hence, at neutralcondition, the availability of COO- group and the reactivity ofAu(III) complexes are the determining factors for the reductionof gold precursor by PLGA, which led to the higher absorptionintensity and longer reduction time at neutral condition. TEManalysis reveals that the gold nanoparticles with average sizes 20and 10 nm in diameter were synthesized at neutral and basicconditions, respectively (Figure S11, Supporting Information).
The synthesis scheme of Au NP/meso-SiO2 tubes is shown inScheme 1. The PLL/PLGA coated membranes were immersedinto a HAuCl4 solution (pH 10.5-11.5) for one to 3 days,followed by inserting into freshly prepared orthosilicic acidsolution. FE-SEM analysis confirms that the materials possesswell-defined tubular structures (Figures S12-S14, SupportingInformation). The formation of gold nanoparticles was confirmedby TEM and XRD characterization (Figure 5, parts A and B, andFigure S12, Supporting Information). UV-vis analysis shows thatthe amount of gold nanoparticles forming in the polypeptidemultilayer films can be controlled by varying the immersion time,indicated by the increase of the plasmon resonance peak absorp-tion of gold nanoparticles at ca. 530 nm (Figure 4). The red-shiftof the peak from lower wavelength to 530 nm is due to thenanoparticles embedded in the silica network and polypeptide.According to TEM analysis, the particle size of gold in the gold/silica/(Lys340/Glu125)5 composite tubes is smaller than 4 nm andthe silica is porous with pore corresponding to the same pore sizerange as previously described (Figure 5A). The confined growthof gold nanoparticles in the PLGA nanodomains results in thewell dispersion and uniform size distribution of nanoparticles.After calcination (500 �C, 10 h), the particle size increased andwas on average 5 nm (Figure 5B). The excellent thermal stabilityof the materials is evidenced by the minor increase of particle sizeand the intact of silica nanostructure after sintering process. Thewell-defined tubular structures were also obtained by reducingchloroauric acid at neutral condition for 1 day (Figure S13,Supporting Information). In addition, the gold/silica/polypep-tide composite and gold/silica materials can be prepared byreplacing poly(L-glutamic acid) with poly(L-tyrosine) (PLT)using a modified synthesis procedure (Figure 5C). The presenceof gold nanoparticles in the silica materials were confirmed byUV-vis and XRD measurements and the particle size is 5 nm onaverage (Figure S14, Supporting Information).
For the application of these gold/silica tubes, p-nitrophenolwas used to evaluate the catalytic activity of the gold/silica tubes.The reduction of p-nitrophenol did not occur in the absence ofgold/silica nanocatalyst for at least 2 days. Instead, in thepresence of gold/silica nanocatalyst, the reduction of p-nitro-phenol was observed as evidenced by the decrease of theabsorbance band of p-nitrophenol. According to literature data,this catalyst-mediated reduction can be assumed as pseudofirst-order reaction.60 The gold/silica nanocatalyst prepared from5-layer composite tubes with two-day reduction was used toreduce p-nitrophenol. From UV-vis measurements, the appar-ent rate constant can be calculated from the slope of linearrelation of ln A (the intensity of absorbance) versus time(second) and the values were calculated to be 3 � 10-4 and8� 10-4 s-1 for gold/silica tubes obtained from (Lys340/Glu125)5and (Lys340/PLT)5 coated membranes, respectively (Figure 6).ICP-MS measurements were performed to determine the ele-mental weight ratio between Si and Au in the glod/silica tubes(Experimental Section in the Supporting Information). And theweight percentages of gold in the two samples were calculated tobe 0.95 and 2.41 wt %, respectively. The results demonstrate that
Figure 5. TEM images of (A) gold/silica/polypeptide and (B) gold/silica hollow tubes synthesized from (Lys340/Glu125)5 coated mem-brane, and (C) gold/silica hollow tubes synthesized from (Lys340/PLT)5 coated membrane.
Figure 4. UV spectra of gold/silica/PLL/PLGA multilayer membraneat different reducing time. Synthesis condition: HAuCl4 solution at basiccondition.

2842 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
the Au NP/meso-SiO2 tubes synthesized using the currentprocedure exhibit promising catalytic property. It is expectedthat the structure and property of gold/silica tubes can beimproved by optimizing the synthesis procedure and the sinter-ing process. Previously, the gold nanoparticle/polyelectrolytefilms were coated in the membrane pores and used as catalyticmembranes for p-nitrophenol reduction.66 Using this approach,the catalytic membranes coated with gold nanoparticle/meso-porous silica in the pores can be synthesized with the advantageof robustness and durability.
’CONCLUSION
We have demonstrated that silica/polypeptide, gold nanopar-ticle/polypeptide, and gold nanoparticle/mesoporous silica/polypeptide composite tubes can be prepared by polypeptide-mediated formation of silica andmetal nanoparticle. Mesoporoussilica and gold nanoparticle/mesoporous silica tubes can beobtained by subsequent calcination. PLL/PLGA macromolecu-lar assemblies act not only as mediating agents for formation ofmetal nanoparticles and silica, but also templates for directingmesoporous silica formation and confining the growth of metalnanoparticles. The as-prepared hollow tubes have well-defineddiameters and lengths that are determined by that of the templatepores, uniform, and conformal walls with thicknesses that arefinely controlled by the number of deposited layers, and, mostimportantly, porosities that are dependent on the aggregate sizeof the sheet/turn-like PLL/PLGA aggregates, which is a functionof polypeptide molecular weight. The successful preparation ofmetal nanoparticles with uniform size distribution in the silicaporous support is also worth noting and the as-prepared gold/silica tubes exhibit good thermal stability and catalytic activitytoward the reduction of p-nitrophenol. Because of the versatilityof the LbL technique and feasibility of polypeptide-mediatedsynthesis, it is expected the present method can be easilyextended to the synthesis of multicomponent organic-inorganiccomposite films and capsules. Also, silica and gold nanoparticle/silica micro/nanopatterns on surfaces can be obtained via thecombination of this method and soft lithography. In addition,this study will provide insight on the directed growth of metalsand mineralization of oxides in some biological systems.
’ASSOCIATED CONTENT
bS Supporting Information. Details of all materials andmeasurements. This material is available free of charge via theInternet at http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
’ACKNOWLEDGMENT
J.-S. Jan acknowledges funding support from National ScienceCouncil grant NSC98-2218-E-006-010, NSC99-2628-E-006-003, and Center for Frontier Materials and Micro/Nano Scienceand Technology (CFMMNST) at National Cheng Kung Uni-versity (D98-2700). J.-S. Jan also acknowledges T.-C. Wen foraccess to the glovebox, H. Teng for access to the nitrogen porosi-metry, and J.-J. Wu for access to the UV-vis spectrophoto-meters.
’REFERENCES
(1) Estroff, L. A.; Hamilton, A. D. Chem. Mater. 2001, 13, 3227–3235.
(2) Poulsen, N.; Sumper, M.; Kroger, N. Proc. Natl. Acad. Sci. U.S.A.2003, 100, 12075–12080.
(3) Aizenberg, J.; Weaver, J. C.; Thanawala, M. S.; Sundar, V. C.;Morse, D. E.; Fratzl, P. Science 2005, 309, 275–278.
(4) Sanchez, C.; Arribart, H.; Guille, M. M. G. Nat. Mater. 2005, 4,277–288.
(5) Jensen, M.; Keding, R.; Hoche, T.; Yue, Y. Z. J. Am. Chem. Soc.2009, 131, 2717–2721.
(6) Cha, J. N.; Shimizu, K.; Zhou, Y.; Christiansen, S. C.; Chmelka,B. F.; Stucky, G. D.; Morse, D. E. Proc. Natl. Acad. Sci. U.S.A. 1999, 96,361–365.
(7) Cha, J. N.; Stucky, G. D.; Morse, D. E.; Deming, T. J. Nature2000, 403, 289–292.
(8) Jan, J.-S.; Lee, S.; Carr, C. S.; Shantz, D. F.Chem.Mater. 2005, 17,4310–4317.
(9) Jan, J.-S.; Shantz, D. F. Adv. Mater. 2007, 19, 2951–2956.(10) Yuan, J. J.; Mykhaylyk, O. O.; Ryan, A. J.; Armes, S. P. J. Am.
Chem. Soc. 2007, 129, 1717–1723.(11) Li, Y. T.; Du, J. Z.; Armes, S. P. Macromol. Rapid Commun.
2009, 30, 464–468.(12) Tomczak, M. M.; Glawe, D. D.; Drummy, L. F.; Lawrence,
C. G.; Stone,M.O.; Perry, C. C.; Pochan, D. J.; Deming, T. J.; Naik, R. R.J. Am. Chem. Soc. 2005, 127, 12577–12582.
(13) Meegan, J. E.; Aggeli, A.; Boden, N.; Brydson, R.; Brown, A. P.;Carrick, L.; Brough, A. R.; Hussain, A.; Ansell, R. J. Adv. Funct. Mater.2004, 14, 31–37.
(14) Yuan, J. J.; Zhu, P. X.; Fukazawa, N.; Jin, R. H.Adv. Funct. Mater.2006, 16, 2205–2212.
(15) Yuwono, V. M.; Hartgerink, J. D. Langmuir 2007, 23, 5033–5038.
Figure 6. Plot of ln A versus time for the reduction of p-nitrophenol with NaBH4 by the Au NP/meso-SiO2 tubes obtained from (A) (Lys340/Glu125)5or (B) (Lys340/PLT)5 coated PC membrane. Initial condition: [p-nitrophenol] = 1.33 � 10-4 M, [NaBH4] = 6.67 � 10-3 M.

2843 dx.doi.org/10.1021/la103923c |Langmuir 2011, 27, 2834–2843
Langmuir ARTICLE
(16) Holmstrom, S. C.; King, P. J. S.; Ryadnov, M. G.; Butler, M. F.;Mann, S.; Woolfson, D. N. Langmuir 2008, 24, 11778–11783.(17) Altunbas, A.; Sharma, N.; Lamm, M. S.; Yan, C. Q.; Nagarkar,
R. P.; Schneider, J. P.; Pochan, D. J. ACS Nano 2010, 4, 181–188.(18) Kessel, S.; Thomas, A.; B€orner, H. G. Angew. Chem., Int. Ed.
2007, 46, 9023–9026.(19) Kessel, S.; B€orner, H. G. Macromol. Rapid Commun. 2008, 29,
419–424.(20) Wong,M. S.; Cha, J. N.; Choi, K. S.; Deming, T. J.; Stucky, G. D.
Nano Lett. 2002, 2, 583–587.(21) Hawkins, K.M.;Wang, S. S.-S.; Ford, D.M.; Shantz, D. F. J. Am.
Chem. Soc. 2004, 126, 9112–9119.(22) Rana, R. K.; Murthy, V. S.; Yu, J.; Wong,M. S.Adv. Mater. 2005,
17, 1145–1150.(23) Patwardhan, S. V.; Maheshwari, R.; Mukherjee, N.; Kiick, K. L.;
Clarson, S. J. Biomacromolecules 2006, 7, 491–497.(24) Begum, G.; Rana, R. K.; Singh, S.; Satyanarayana, L. Chem.
Mater. 2010, 22, 551–556.(25) Brott, L. L.; Naik, R. R.; Pikas, D. J.; Kirkpatrick, S. M.; Tomlin,
D. W.; Whitlock, P. W.; Clarson, S. J.; Stone, M. O. Nature 2001, 413,291–293.(26) Coffman, E. A.; Melechko, A. V.; Allison, D. P.; Simpson, M. L.;
Doktycz, M. J. Langmuir 2004, 20, 8431–8436.(27) Tahir, M. N.; Theato, P.; Muller, W. E. G.; Schroder, H. C.;
Borejko, A.; Faiss, S.; Janshoff, A.; Huth, J.; Tremel, W. Chem. Commun.2005, 5533–5535.(28) Kim, D. J.; Lee, K. B.; Lee, T. G.; Shon, H. K.; Kim, W. J.; Paik,
H. J.; Choi, I. S. Small 2005, 1, 992–996.(29) Wu, J. C.; Wang, Y. L.; Chen, C. C.; Chang, Y. C. Chem. Mater.
2008, 20, 6148–6156.(30) Gautier, C.; Lopez, P. J.; Hemadi, M.; Livage, J.; Coradin, T.
Langmuir 2006, 22, 9092–9095.(31) Wang, Y.; Angelatos, A. S.; Caruso, F. Chem. Mater. 2008, 20,
848–858.(32) Jiang, Y. J.; Yang, D.; Zhang, L.; Sun, Q. Y.; Sun, X. H.; Li, J.;
Jiang, Z. Y. Adv. Funct. Mater. 2009, 19, 150–156.(33) Yang, S. H.; Lee, K. B.; Kong, B.; Kim, J. H.; Kim, H. S.; Choi,
I. S. Angew. Chem., Int. Ed. 2009, 48, 9160–9163.(34) Li, J.; Jiang, Z. Y.; Wu, H.; Zhang, L.; Long, L. H.; Jiang, Y. J. Soft
Matter 2010, 6, 542–550.(35) Wang, Y. J.; Caruso, F. Chem. Mater. 2005, 17, 953–961.(36) Caruso, F.; Caruso, R. A.; Mohwald, H. Chem. Mater. 1999, 11,
3309–3314.(37) Caruso, R. A.; Susha, A.; Caruso, F. Chem. Mater. 2001, 13,
400–409.(38) Xie, J. P.; Lee, J. Y.; Wang, D. I. C.; Ting, Y. P. ACS Nano 2007,
1, 429–439.(39) Bhargava, S. K.; Booth, J. M.; Agrawal, S.; Coloe, P.; Kar, G.
Langmuir 2005, 21, 5949–5956.(40) Selvakannan, P. R.; Swami, A.; Srisathiyanarayanan, D.; Shirude,
P. S.; Pasricha, R.; Mandale, A. B.; Sastry, M. Langmuir 2004, 20, 7825–7836.(41) Ray, S.; Das, A. K.; Banerjee, A. Chem. Commun. 2006, 2816–
2818.(42) Lee, H.; Lee, Y.; Statz, A. R.; Rho, J.; Park, T. G.; Messersmith,
P. B. Adv. Mater. 2008, 20, 1619-þ.(43) Dong, Q.; Su, H. L.; Zhang, D. J. Phys. Chem. B 2005, 109,
17429–17434.(44) Kharlampieva, E.; Slocik, J. M.; Tsukruk, T.; Naik, R. R.;
Tsukruk, V. V. Chem. Mater. 2008, 20, 5822–5831.(45) Jan, J.-S.; Shantz, D. F. Chem. Commun. 2005, 2137–2139.(46) Deming, T. J. Nature 1997, 390, 386–389.(47) Deming, T. J.; Curtin, S. A. J. Am. Chem. Soc. 2000, 122, 5710–
5717.(48) Gaspard, J.; Silas, J. A.; Shantz, D. F.; Jan, J.-S. Supramol. Chem.
2010, 22, 178–185.(49) Boulmedais, F.; Schwinte, P.; Gergely, C.; Voegel, J. C.; Schaaf,
P. Langmuir 2002, 18, 4523–4525.
(50) Boulmedais, F.; Bozonnet, M.; Schwinte, P.; Voegel, J. C.;Schaaf, P. Langmuir 2003, 19, 9873–9882.
(51) Haynie, D. T.; Balkundi, S.; Palath, N.; Chakravarthula, K.;Dave, K. Langmuir 2004, 20, 4540–4547.
(52) Yang, C. T.; Wang, Y. L.; Yu, S.; Chang, Y. C. I. Biomacromo-lecules 2009, 10, 58–65.
(53) Menzel, H.; Horstmann, S.; Behrens, P.; Barnreuther, B.;Krueger, I.; Jahns, M. Chem. Commun. 2003, 2994–2995.
(54) Xu, M. J.; Gratson, G. M.; Duoss, E. B.; Shepherd, R. F.; Lewis,J. A. Soft Matter 2006, 2, 205–209.
(55) Kovtyukhova, N. I.; Mallouk, T. E.; Mayer, T. S. Adv. Mater.2003, 15, 780–785.
(56) Chen, C. C.; Liu, Y. C.; Wu, C. H.; Yeh, C. C.; Su, M. T.; Wu,Y. C. Adv. Mater. 2005, 17, 404–407.
(57) Arcadi, A. Chem. Rev. 2008, 108, 3266–3325.(58) Bond, G. C.; Thompson, D. T. Catal. Rev.—Sci. Eng. 1999, 41,
319–388.(59) Gerolamo, B.; Avelino, C. A. Angew. Chem., Int. Ed. 2006, 45,
3328–3331.(60) Lee, J.; Park, J. C.; Song, H. Adv. Mater. 2008, 20, 1523–1528.(61) Wu, Z. L.; Zhou, S. H.; Zhu, H. G.; Dai, S.; Overbury, S. H.
Chem. Commun. 2008, 3308–3310.(62) Gajan, D.; Guillois, K.; Delichere, P.; Basset, J. M.; Candy, J. P.;
Caps, V.; Coperet, C.; Lesage, A.; Emsley, L. J. Am. Chem. Soc. 2009, 131,14667–14669.
(63) Yu, Y.-Y.; Chang, S.-S.; Lee, C.-L.; Wang, C. R. C. J. Phys. Chem.B 1997, 101, 14667–14669.
(64) Pei, L.; Mori, K.; Adachi, M. Langmuir 2004, 20, 7837–7843.(65) Ji, X.; Song, X.; Li, J.; Bai, Y.; Yang, W.; Peng, X. J. Am. Chem.
Soc. 2007, 129, 13939–13948.(66) Dotzauer, D. M.; Dai, J.; Sun, L.; Bruening, M. L. Nano Lett.
2006, 6, 2268–2272.