kortikale demyelinisierung bei entzündlichen
TRANSCRIPT

Aus dem Institut für Neuropathologie
(Prof. Dr. med. W. Brück)
der Medizinischen Fakultät der Universität Göttingen
Kortikale Demyelinisierung bei entzündlichen, neoplastischen und
metabolischen ZNS-Erkrankungen
INAUGURAL-DISSERTATION
zur Erlangung des Doktorgrades
der Medizinischen Fakultät der
Georg-August-Universität zu Göttingen
vorgelegt von
Jadwiga Zyta Wozniak
geboren in
Tarnowskie Gory (Polen)
Göttingen 2018

Dekan: Prof. Dr. rer. nat. H. K. Kroemer
Referent: Prof. Dr. med. W. Brück
Ko-Referent/in: Prof. Dr. J. Gärtner
Drittreferent/in: Prof. Dr. M. Oppermann
Tag der mündlichen Prüfung: 27.11.2018

Hiermit erkläre ich, die Dissertation mit dem Titel "Kortikale Demyelinisierung bei
entzündlichen, neoplastischen und metabolischen ZNS-Erkrankungen" eigenständig
angefertigt und keine anderen als die von mir angegebenen Quellen und Hilfsmittel
verwendet zu haben.
Göttingen, den …………………………… ……………………………………………

Inhaltsverzeichnis
I
Inhaltsverzeichnis
Abkürzungsverzeichnis IV
Abbildungsverzeichnis VI
Tabellenverzeichnis VI
1 Einleitung .................................................................................................................. 1
1.1 Zielsetzung der Arbeit ................................................................................................. 1
1.2 Einführung in demyelinisierende Erkrankungen ........................................................ 2
1.3 Kategorisierung der ZNS-Erkrankungen mit kortikaler Pathogenese ........................ 3
1.3.1 Autoimmun-entzündliche Erkrankungen ........................................................... 3
1.3.1.1 MS .................................................................................................................. 3
1.3.1.1.1 Epidemiologie/Ätiologie ....................................................................... 3
1.3.1.1.2 Befallmuster/ Diagnosestellung ........................................................... 5
1.3.1.1.3 Klinik/ Verlaufsformen/Progression ..................................................... 5
1.3.1.1.4 Behandlung ........................................................................................... 7
1.3.1.1.5 Pathophysiologie der Demyelinisierung ............................................... 7
1.3.1.1.6 Histologie .............................................................................................. 9
1.3.1.2 NMO ............................................................................................................. 12
1.3.1.3 ADEM ........................................................................................................... 13
1.3.2 Infektiöse Erkrankungen ................................................................................... 14
1.3.2.1 Virale Infektionen ......................................................................................... 14
1.3.2.2 Bakterielle Infektionen ................................................................................. 17
1.3.3 Neoplastische Erkankungen ............................................................................. 20
1.3.4 Metabolische Erkrankungen ............................................................................. 20
2 Material und Methoden .......................................................................................... 22
2.1 Autopsiekollektiv ...................................................................................................... 22
2.2 Histologische und immunhistologische Methoden .................................................. 24
2.2.1 Hämatoxylin-Eosin-Färbung ............................................................................. 25
2.2.2 LFB-PAS ............................................................................................................. 25

Inhaltsverzeichnis
II
2.2.3 Silberimprägnation nach Bielschowsky ............................................................ 25
2.2.4 Immunhistochemie ........................................................................................... 26
2.3 Läsions- und Infiltratklassifikation ........................................................................... 28
3 Ergebnisse ............................................................................................................... 29
3.1 Klassisch demyelinisierende Erkrankungen .............................................................. 31
3.1.1 Multiple Sklerose .............................................................................................. 31
3.1.2 Keine subpiale Demyelinisierung bei ADEM und NMO .................................... 32
3.1.3 PML und supiale Demyelinisierung .................................................................. 34
3.1.4 Kortikale Entmarkung bei der extrapontinen Myelinolyse .............................. 35
3.2 Infektiöse Erkrankungen ........................................................................................... 37
3.2.1 Akute meningeale Entzündungen .................................................................... 37
3.2.1.1 Akute bakterielle Infektionen ...................................................................... 37
3.2.1.2 Virale Infektionen außer PML ...................................................................... 37
3.2.2 Chronische Infektionen ..................................................................................... 38
3.2.2.1 Chronisch granulomatöse bakterielle Infektionen ...................................... 38
3.2.2.2 Kortikale Pathologie bei chronisch viralen Infektionen ............................... 39
3.3 Neoplastische Erkrankungen der Hirnhäute ............................................................ 41
3.3.1 Lymphome und Plasmozytome ........................................................................ 41
3.3.2 Meningeosis carcinomatosa ............................................................................. 41
4 Diskussion ............................................................................................................... 43
4.1 Einleitung: Demyelinisierung als krankheitsspezifisches Phänomen ....................... 43
4.2 Zytokine und Chemokine prägen ein krankheitsspezifisches Milieu ........................ 43
4.4 Genetische Grundlagen bedingen das Demyelinisierungsverhalten ........................ 47
4.5 Erhebliche Unterschiede zwischen weißer und grauer Substanz ............................. 48
4.6 Myelinschädigung bei hypoxisch-ischämischen Geschehen .................................... 49
4.7 Subpiale bandförmige Läsionen sind MS-spezifisch ................................................. 49
4.8 Ausblick ..................................................................................................................... 51

Inhaltsverzeichnis
III
5 Zusammenfassung ................................................................................................... 52
6 Abstract .................................................................................................................. 54
7 Anhang ................................................................................................................... 55
8 Literaturverzeichnis ................................................................................................. 61

Abkürzungsverzeichnis
IV
Abkürzungsverzeichnis
ADEM Akut dissemenierte Enzephalomyelitis
AEC 3-Amino-9-Ethylcarbazol
AIDS acquired immune deficiency syndrome
APP amyloid precursor protein
APZ Antigen-präsentierende Zelle
AQP4 Aquaporin-4
BDNF brain-derived neurotrophic factor
CFA Freunds complete adjuvans
CMV Zytomegalievirus
CNP 2',3'-cyclic nucleotide 3'-phosphodiesterase
EPM Extrapontine Myelinolyse
CPM Zentrale Pontine Myelinolyse
CXCL chemokin C-X-C motif ligand
CCL chemokin C-C motif ligand
DAB 3, 3'-Diaminobenzidin
dest. Destilliert
DLBCL diffuse large B-cell lymphoma
DNA Desoxyribonukleinsäure
EAE Experimentelle autoimmune Enzephalomyelitis
EDTA Ethylendiamintetraacetat
FFPE formalin fixed paraffin embedded
GFAP glial fibrillary acidic protein
GM grey matter
gp120 HIV-1 envelope glycoprotein
HE Hämatoxylin-Eosin
HIV human immunodeficiency virus
HLA human leukocyte antigen
HPF high power field
HSV Herpes-simplex-Virus
iNOS NO-Synthase

Abkürzungsverzeichnis
V
IHC Immunhistochemie
IFN-γ Interferon-γ
Ig Immunglobulin
IL Interleukin
LFB-PAS luxol fast blue-periodic acid schiff
MAG myelin associated glycoprotein
MBP myelin basic protein
MHC major histocompatibility complex
MOG myelin oligodendrocyte glycoprotein
MRT Magnetresonanztomographie
MS Multiple Sklerose
NADP Nicotinsäureamid-Adenin-Dinukleotid-Phosphat
NMO Neuromyelitis Optica
NO Stickstoffmonoxid
P0 Protein Null
PBS Phosphatgepufferte Salzlösung/engl.: Phosphate Buffered Saline
PCNSL primary central nervous system lymphoma
PLP Proteolipid-Protein
PML Progressive multifokale Leukenzephalopathie
PNS Peripheres Nervensystem
RNA Ribonukleinsäure
ROS Reactive Oxygen Species
ssp. Subspecies
SSPE Subakute sklerosierende Panenzephalitis
Tat transactivator of transcription
TGF transforming growth factor
TLR Toll-like-Rezeptoren
TMEV Theiler's Murine Enzephalomyelitis Virus
TNF Tumornekrosefaktor
WM white matter
ZNS Zentrales Nervensystem

Abbildungs- und Tabellenverzeichnis
VI
Abbildungsverzeichnis
Abbildung 1: Semiquantitative Beurteilung der zellulären meningealen Infiltration in allen
untersuchten Autopsiefällen .................................................................................................... 30
Abbildung 2: Kortikal demyelinisierte MS-Läsionen ................................................................ 31
Abbildung 3: Leukokortikale Demyelinisierung bei der ADEM ............................................... 33
Abbildung 4: Kortikale Demyelinisierung in der PML .............................................................. 35
Abbildung 5: Kortiko-subkortikale Demyelinisierung in der extrapontinen Myelinolyse ....... 36
Abbildung 6: Subpiale Myelinscheiden sind bei akuten bakteriellen und viralen Infektionen
(HSV) erhalten .......................................................................................................................... 38
Abbildung 7: Intaktes subpiales Myelin bei der meningealen TBC ......................................... 39
Abbildung 8: Kein Hinweis kortikaler Demyelinisierung in der SSPE ...................................... 40
Abbildung 9: Erhaltene subpiale Myelinscheiden in einer Vielzahl von Erkrankungen mit
meningealer neoplastischer Zellinfiltration ............................................................................. 42
Tabellenverzeichnis
Tabelle 1: Untersuchte Krankheitsentitäten des ZNS .............................................................. 23
Tabelle 2: Antikörper der IHC und Färbeverfahren ................................................................. 27
Tabelle 3: Ausführliche Beschreibung der untersuchten Autopsiefälle .................................. 55

1 Einleitung
1
1 Einleitung
1.1 Zielsetzung der Arbeit
Die kortikale Demyelinisierung ist ein häufiges Phänomen bei der Multiplen Sklerose (MS),
auch Enzephalomyelitis disseminata genannt (Albert et al. 2007; Kutzelnigg und Lassmann
2005). Dabei stellen subpiale Läsionen bezogen auf die Fläche den größten Anteil der
Läsionen im Kortex dar, was die subpiale Demyelinisierung im Allgemeinen als ein
Charakteristikum der MS erscheinen lässt (Bo et al. 2003 b). Die kortikale und insbesondere
subpiale Pathologie ist dabei deutlich mit meningealer Entzündung assoziiert (Howell et al.
2011; Lucchinetti et al. 2011; Magliozzi et al. 2007). Damit liegt die Überlegung nahe, dass
möglicherweise auch andere Erkrankungen mit meningealen, zellulär infiltrativen Prozessen
des zentralen Nervensystems (ZNS), inflammatorischer oder auch neoplastischer Genese, zu
einer kortikalen Schädigung des Myelins führen können und welches Gewebemilieu
grundsätzlich eine subpiale/kortikale Myelinschädigung nach sich zieht. Beschrieben ist, dass
in seltenen Fällen das primäre ZNS-Lymphom mit fokaler Demyelinisierung assoziiert ist
(Alderson et al. 1996; Husseini et al. 2012), wobei eine kortikale Demyelinisierung in diesem
Zusammenhang bisher nicht festgestellt werden konnte (Hart und Earle 1975).
Besonders bei lang anhaltenden kortikalen Pathologien, die mit einer chronischen Infiltration
durch Entzündungszellen oder neoplastische Zellkomponente einhergehen, wie das
beispielsweise bei der Syphilis, der Tuberkulose (TBC) oder der subakuten sklerosierenden
Panenzephalitis (SSPE) der Fall ist, stellt sich die Frage, ob auch diese Erkrankungen zu einer
subpialen bzw. kortikalen Demyelinisierung führen können.
Um zu untersuchen, dass subpiale Entmarkung gegebenenfalls neben der MS auch in
anderen demyelinisierenden Erkrankungen auftritt oder einzigartig für diese Erkrankung ist,
wurden Autopsiefälle der Neuromyelitis optica (NMO/Devic), der akuten disseminierten
Enzephalomyelitis (ADEM), der progressiven multifokalen Leukenzephalopathie (PML) und
der extrapontinen Myelinolyse in diese Studie miteinbezogen. Seltene pathologische
Bedingungen, wie Vergiftungen oder genetische Erkrankungen, die eine Demyelinisierung
verursachen können, sind in diese Arbeit nicht miteingeflossen.
Ziel dieser Dissertation war es, eine große Kohorte unterschiedlicher ZNS-Erkrankungen mit
einer Vielzahl an kortikalen zellulären Infiltrations- und Zytokinmustern hinsichtlich der

1 Einleitung
2
Integrität der kortikalen Myelinscheiden zu untersuchen und diese zu vergleichen. Durch die
Erkenntnisse dieser Arbeit können entscheidende Rückschlüsse auf die Pathophysiologie des
kortikalen Myelinverlustes, insbesondere bei der MS, getroffen werden.
1.2 Einführung in demyelinisierende Erkrankungen
Demyelinisierung bezeichnet den isolierten Verlust der Myelinscheiden bei relativem
Axonerhalt als Auswirkung auf einen Schaden des Myelins oder der Myelin bildenden Zellen
(Adams und Kubik 1952). Der Prozess der Demyelinisierung wird durch verschiedene
Mechanismen und unterschiedliche Konditionen hervorgerufen, die Oligodendrozyten oder
die Myelinscheide schädigen. Dadurch kann eine Einteilung demyelinisierender
Erkrankungen nach ihrer Pathogenese erfolgen. Die gängige Klassifikation dieser
Erkrankungen umfasst ein großes Spektrum: Inflammatorische Demyelinisierung
hervorgerufen durch autoimmune oder virale Prozesse, metabolische oder toxische Formen
der Entmarkung und Myelinschädigung durch hereditäre Erkrankungen (Lassmann 2001;
Love 2006).
Im Folgenden soll ein kurzer Überblick über bekannte demyelinisierende Erkrankungen
gegeben werden, die im Rahmen dieser Arbeit untersucht wurden.
Die am weitesten verbreitete demyelinisierende Erkrankung ist die MS, bei der eine
autoimmun-inflammatorische Genese wahrscheinlich ist. Zu Sonderformen zählen die
konzentrische Sklerose (Balo) und die myelinoklastische diffuse Sklerose (Enzephalitis
periaxialis diffusa Schilder). Andere, bereits erwähnte entzündlich demyelinisierende
Erkrankungen sind die ADEM, die eine perivaskuläre Entmarkung um kleine Gefäße
verursacht und para- bzw. postinfektiös auftreten kann und die NMO, bei der AQP-4-
Antikörper eine astrozytäre Pathologie mit sekundärer Entmarkung auslösen (Love 2006).
Zu den viralen Auslösern eines selektiven fokalen Myelinverlustes im ZNS gehört beim
Menschen vor allem die PML, die insbesondere bei immunkompromittierten Patienten
durch eine Infektion von Oligodendrozyten mit dem John-Cunningham-Virus (JC-Virus) eine
Entmarkung hervorruft. Auch bei der SSPE, einer chronischen ZNS-Erkrankung, die durch
Masernviren verursacht wird, ist Demyelinisierung beschrieben (Love 2006). Des Weiteren
kann eine HIV-Infektion (human immunodeficiency virus) unter Umständen zur Enzephalitis
und Myelinschädigung der weißen Substanz führen (Langford et al. 2002; Corral et al. 2004;
Gray et al. 2003).

1 Einleitung
3
Eine metabolisch bedingte Demyelinisierung findet sich bei der zentralen pontinen oder
extrapontinen Myelinolyse (CPM/EPM), bei der intra- und extrazelluläre
Elektrolytverschiebungen im ZNS eine ursächliche Rolle spielen. Dabei führt ein häufig
iatrogener, zu schneller Ausgleich einer länger bestehenden Hyponatriämie zur osmotisch
bedingten Oligodendrozytenschädigung in der Pons, aber auch in anderen Regionen des ZNS
(Martin 2004).
Zu den demyelinisierenden Erkrankungen toxischer Genese zählen weiterhin die
Intoxikationen mit Kohlenstoffmonoxid oder mitochondrialen Toxinen, wie Cyaniden oder
Schwefelwasserstoffen (Wilson 1983; Solnyshkova und Shakhlamov 2002).
Können Myelinscheiden nicht adäquat gebildet oder erhalten werden, spricht man von
Leukodystrophien. Dies sind sehr seltene, genetisch bedingte neurometabolische
Erkrankungen, die im Kindesalter auftreten und durch die verschiedenen
Stoffwechselprozesse des ZNS eine unterschiedliche Neuropathologie aufweisen (Kuhlmann
2012).
1.3 Kategorisierung der ZNS-Erkrankungen mit kortikaler Pathogenese
1.3.1 Autoimmun-entzündliche Erkrankungen
1.3.1.1 MS
Die MS ist die am weitesten verbreitete demyelinisierende Erkrankung des ZNS und stellt die
häufigste Ursache einer neurologischen Behinderung im jungen Erwachsenenalter dar
(Noseworthy et al. 2000). In Deutschland wird die Zahl der Erkrankten auf 150.000–180.000
geschätzt. Weltweit sind es etwa 2,5 Mio. Menschen, die an MS leiden (Hein und
Hopfenmüller 2000). Am häufigsten sind junge Frauen mittleren Alters betroffen.
1.3.1.1.1 Epidemiologie/Ätiologie
Bisher konnte die Genese der Erkrankung nicht zufriedenstellend geklärt werden.
Pathogenetisch nimmt man jedoch an, die MS sei ein autoimmuner Prozess mit Reaktionen
gegen Myelinbestandteile. Dabei spielt wahrscheinlich eine Kombination aus genetischen
Faktoren und Umwelteinflüssen in der Ätiologie der MS als heterogene Erkrankung eine
wichtige Rolle.

1 Einleitung
4
Es gibt Hinweise auf eine familiäre Häufung der Erkrankung. Das Risiko, an MS zu erkranken,
steigt für Eltern oder Geschwister eines Patienten um ein 20-40-Faches. Bei einem
Gesamtbevölkerungsrisiko von 0,1 % steigt das Risiko dabei auf 2 % eine MS zu entwickeln.
Bei monozygoten Zwillingen vervielfacht sich das Risiko sogar bis zu 200-fach im Vergleich
zur Gesamtbevölkerung (Sadovnick et al. 1999; Sawcer et al. 2011). Bekannte Gene und
genetische Polymorphismen werden als MS-prädisponierend angesehen. Einen wichtigen
Suszeptibilitätslokus für die MS stellt der Haupthistokompatibilitätskomplex (human
leukocyte antigen, HLA) auf Chromosom 6p21.3 dar, der für den Major-Histocompatibility-
Complex (MHC) kodiert und dadurch eine entscheidende Rolle in der Antigenpräsentation
spielt (Oksenberg et al. 2008). Allele wie HLA-DRB2 können bei homozygoten Trägern die
Erkrankungswahrscheinlichkeit und die Anfälligkeit für einen drastischen Krankheitsverlauf
erhöhen (Barcellos et al. 2003). Weitere bekannte Risikofaktoren an MS zu erkranken sind
das Vorhandensein von Polymorphismen in den Genen, die für CD58, Interleukin-2- und
Interleukin-7-Rezeptoren auf Chromosom 5p13 kodieren (Hafler et al. 2007; International
Multiple Sclerosis Genetics Consortium (IMSGC) 2008). Zudem liefern Studien zur Migration
und Prävalenz bei MS einen deutlichen Hinweis auf die Bedeutung von Umwelteinflüssen
und genetischen Faktoren als Trigger der Erkrankung, auch wenn bisher keine
Umweltexposition als alleinige Ursache der Erkrankung ergründet werden konnte (Marrie
2004). Beispielsweise ist das Vorkommen der Erkrankung in nördlichen Regionen, wie den
USA, Kanada und dem nördlichen Europa im Vergleich zu äquatornahen Regionen gehäuft.
Eine Migration im Kindes- und Jugendalter in ein Land mit niedrigerer Prävalenz kann die
Erkrankungswahrscheinlichkeit sogar verringern (Lowis 1988; Rosati 2001). Die Länder mit
niedrigerer Prävalenz sind Länder mit stärkerer Sonneneinstrahlung, was eine vieldiskutierte
mögliche Ursache der unterschiedlichen Erkrankungshäufigkeit darstellt. Dabei wird die
inaktive Form von Vitamin D durch UV-Strahlung/Sonnenlicht in seine aktive Form überführt.
Aktives Vitamin D kann die TGF-β-1- (transforming growth factor) und Interleukin-4-
Produktion (IL-4) fördern, was die pro-entzündliche T-Zellaktivität hemmen kann (Deluca
und Cantorna 2001). Im experimentellen MS-Mausmodell, der experimentellen
autoimmunen Enzephalomyelitis (EAE), bei der durch die Immunisierung von Mäusen mit
MOG-Peptiden (Myelin-Oligodendrozyten-Glykoprotein) und der zusätzlichen Injektion von
komplettem Freundschem Adjuvans (Freunds complete adjuvans/CFA) eine
Entzündungsreaktion im Rückenmark ausgelöst werden kann, konnte die Gabe von

1 Einleitung
5
Vitamin D die Verschlechterung der Enzephalomyelitis vermindern (Deluca und Cantorna
2001). Auch beim Menschen scheint Vitamin D als Regulator der T-Zell- und
Makrophagenaktivität das Erkrankungsrisiko (Munger et al. 2006), die Schubrate und die
Behinderung bei der MS (Smolders et al. 2008) positiv zu beeinflussen.
1.3.1.1.2 Befallmuster/ Diagnosestellung
Bei der MS entstehen demyelinisierte Herde sowohl in der weißen als auch in der grauen
Substanz des ZNS. Bevorzugte Lokalisationen dieser Läsionen, die in der Bildgebung mittels
Kernspintomographie sichtbar werden, befinden sich in der periventrikulären und
subkortikalen weißen Substanz. Nervus opticus, Hirnstamm, Kleinhirn und Rückenmark sind
ebenso Prädilektionsstellen der Entmarkung. Diagnostisch wird neben der
Kernspintomographie auch der Liquor hinsichtlich entzündlicher Veränderungen untersucht.
Um eine Diagnose anhand der klinischen Symptomatik in Verbindung mit der Bildgebung zu
ermöglichen und zu erleichtern, wurden unterschiedliche Kriterien entwickelt. Die am
weitesten verbreiteten sind dabei die McDonald-Kriterien (McDonald et al. 2001) und ihre
Revisionen 2005 und 2010 .
1.3.1.1.3 Klinik/ Verlaufsformen/Progression
Der Krankheitsverlauf der MS ist individuell sehr variabel und die Symptomatik breit
gefächert. Häufige Erstsymptome sind die Retrobulbärneuritis oder Parästhesien. Die
Bandbreite der Symptome erstreckt sich von Sensibilitäts- und Koordinationsstörungen über
kognitive Symptome wie Fatigue und Depression bis hin zu Paresen und Ausfällen
vegetativer Zentren.
Man unterscheidet vier verschiedene mögliche Krankheitsverläufe (Lublin und Reingold
1996). Der mit 80% häufigste Verlauf ist der schubförmig-remittierende, bei dem die
Patienten eine akute, schubförmige Symptomatik zeigen, die sich ganz oder zumindest
teilweise zurückbildet. Dieser Verlauf kann in die sekundär-progrediente Form übergehen,
die durch eine kontinuierliche Verschlechterung der klinischen Symptomatik charakterisiert
ist. Ist eine klare Abgrenzung von Schüben bereits im frühen Stadium der Erkrankung nicht
möglich, spricht man vom primär-progredienten Verlauf. Zusammengenommen sind 40%
der Verläufe (primär) progredient oder gehen (sekundär) in einen progredienten Verlauf
über.

1 Einleitung
6
Die Mechanismen, die zu einer primären oder sekundär progressiven Form der MS führen,
sind ungeklärt. Es fällt jedoch auf, dass die Schubrate während der frühen Krankheitsphase
den Beginn der Progression beeinflusst (Confavreux et al. 2000). Das morphologische
Korrelat der progredienten klinischen Symptomatik ist im Wesentlichen ein fortschreitender
axonaler Verlust (Davie et al. 1997; Davie et al. 1995; Losseff et al. 1996; Truyen et al. 1996;
De Stefano et al. 1998). Die akute Demyelinisierung führt zu einer Beeinträchtigung der
Reizweiterleitung in den Axonen, was als Leitungsblock bezeichnet wird (McDonald und
Sears 1970). Dabei sind die Mechanismen, die zu axonaler Schädigung führen, noch nicht
ausreichend verstanden. Es wurde gezeigt, dass durch Entzündungsprozesse bedingter
oxidativer Stress die Mitochondrien, Axone und Neurone schädigt und damit letztendlich ein
Trigger der Krankheitsprogression darstellen könnte (Lassmann et al. 2012). Angriffspunkte
freier Sauerstoff- und NO-Radikale (Stickstoffmonoxid) sind dabei mitochondriale und
zelluläre DNA sowie Phospholipide. In der Folge werden Oligodendrozyten und neuronale
Strukturen beeinträchtigt (Haider et al. 2011). Dieser Effekt scheint beispielsweise durch
altersabhängige Eisenspeicherung des Gehirns noch zusätzlich verstärkt zu werden
(Lassmann et al. 2012).
Durch den Rückgang der für Axone protektiven Myelinscheiden sind die Axone schädigenden
Einflüssen vermehrt ausgesetzt, was im Verlauf der Erkrankung zu einer wesentlichen
Reduktion der axonalen Leitungsbahnen im ZNS führt. Axonale Degeneration tritt nicht nur
in den späten Phasen der Multiplen Sklerose auf, sondern ist bereits in den frühen Episoden
entzündlicher Demyelinisierung ein entscheidender Prozess (Bitsch et al. 2000b). Während
der frühen Krankheitsstadien zeigt sich dabei in den Läsionen sogar eine höhere Dichte an
axonalen Sphäroiden (als Maß für die akute axonale Schädigung) als im späteren Verlauf der
Erkrankung (Kuhlmann 2012). Neben der axonalen Schädigung kommt es zur Bildung von
Glianarben und der Erschöpfung des Oligodendrozytenvorläuferpools, was die
Regenerationsfähigkeit des ZNS im Verlauf der Erkrankung vermindert (Noseworthy et al.
2000). Das sich formende pathologische, chronisch entzündliche Milieu führt zu einer
stetigen Neurodegeneration und ist von einer weit verbreiteten Mikrogliaaktivierung in
weißer und grauer Substanz dominiert (Compston und Coles 2008).

1 Einleitung
7
1.3.1.1.4 Behandlung
In den vergangenen drei Jahrzehnten wurden wichtige Fortschritte im Verständnis des
Entzündungsprozesses und der pathophysiologischen Mechanismen bei der MS erzielt.
Damit sind wirksame entzündungshemmende und immunmodulatorische Behandlungen,
vor allem für Patienten im Stadium der schubförmig-remittierenden MS, verfügbar. Eine
kausale Therapie ist dagegen nach wie vor nicht vorhanden (Lassmann et al. 2012).
1.3.1.1.5 Pathophysiologie der Demyelinisierung
Hinweisend darauf, dass es sich bei MS um eine Autoimmunerkrankung handelt, sind das
Vorhandensein autoreaktiver T-Zellen gegen körpereigenes Myelin und pathologische
Ähnlichkeiten zwischen der MS und den experimentellen Tiermodellen. Die Vermutung liegt
nahe, dass aktivierte, autoreaktive T-Zellen in den Läsionen den chronischen Entzündungs-
prozess des ZNS in Gang setzen und Makrophagen aktivieren, die das geschädigte Myelin
abräumen (Weiner 2004). Autoreaktive T-Zellen, die auch bei Gesunden nachweisbar sind
(Babbe et al. 2000), werden bei MS-Erkrankten in der Körperperipherie aktiviert, wobei der
Trigger dieser Aktivierung immer noch nicht eindeutig aufgefunden werden konnte.
Unterschiedliche Ursachen wie chronische Entzündungsprozesse werden debattiert (Kroner-
Milsch 2012). Studien suggerieren den Einfluss von Viren, wie dem Epstein-Barr- oder
Masernvirus, auf die Pathogenese der MS (Wagner et al. 2004; Alvarez-Lafuente et al. 2004).
Aufbau der Myelinscheiden
Myelinscheiden sind lipidreiche Biomembranen, welche die Axone der Nervenzellen
mehrfach umhüllen. Durch die elektrische Isolation der Axone ermöglichen sie die schnelle,
saltatorische Erregungsleitung und sind damit eine Voraussetzung für die Funktion höher
entwickelter Nervensysteme. Im ZNS wird das Myelin von Oligodendrozyten, im peripheren
Nervensystem (PNS) von Schwann-Zellen gebildet. Die Oligodendrozyten durchlaufen eine
ausgedehnte Entwicklung und Differenzierung, um die Fähigkeit der Myelinbildung zu
erlangen. Dabei sind sie metabolisch sehr aktiv und weisen durch verschiedene
zellspezifische Eigenschaften eine hohe Vulnerabilität auf (Bradl und Lassmann 2010).
Myelin besteht vorwiegend aus Lipiden mit nur 15% Proteingehalt, was die Myelinscheiden
von anderen Membranen unterscheidet. Die Proteine sind myelinspezifisch und
unterscheiden sich in der Zusammensetzung im zentralen und peripheren Nervensystem.

1 Einleitung
8
Schwann-Zellen exprimieren vorwiegend die Myelinproteine Protein Null (P0) und das Myelin
Basic Protein (MBP), während Oligodendrozyten Proteolipid-Protein (PLP) und MBP bilden
(Lemke 1992). MBP -als Myelinprotein des ZNS und PNS- ermöglicht die Ausbildung der
zytoplasmatischen Oberfläche des kompakten Myelins und seine strukturelle
Aufrechterhaltung (Harauz et al. 2004). Zentral hat es einen Anteil von 30% und peripher von
5-15% an den Gesamtmyelinproteinen. Hauptmyelinprotein des Zentralnervensystems ist
das Proteolipid Protein (PLP), das etwa 50% der Proteinkomponente der zentralen
Myelinscheiden bildet (Lemke 1992).
Neben diesen „Hauptproteinen“ gibt es in den Myelinscheiden zahlreiche andere
Myelinproteine. Dabei wurde beobachtet, dass das Myelin-assoziierte Glykoprotein (MAG)
unter bestimmten Umständen selektiv herabreguliert (oder zerstört) werden kann. So wird
in hypoxischen Läsionen nach einem Schlaganfall vorrangig ein MAG-Verlust beobachtet,
während die übrigen Myelinproteine zunächst noch erhalten blieben (Aboul-Enein et al.
2003). Des Weiteren findet sich auch bei der MS ein selektiver Myelinproteinverlust von
MAG in früh-aktiven Subtyp-III-Läsionen (Lucchinetti et al. 2000) (siehe unten). MAG ist
sowohl zentral, als auch peripher zu finden und gilt als ein Zelladhäsionsmolekül zwischen
Oligodendrozyten und Neuronen, dessen Struktur mit dem der Immunglobuline (Ig)
verwandt ist. Die MAG-Expression kann bereits in der frühsten Phase der Myelinisierung der
Nervenfasern detektiert werden, bevor andere Myelinproteine auftreten. Somit scheint
MAG eine Rolle in der frühen Myelinisierung zu spielen (Lemke 1992).
Antikörper gegen das Myelinprotein MOG können im Serum insbesondere bei kindlichen
MS-Patienten in einem hohen Prozentsatz der Erkrankten nachgewiesen werden (Meinl et
al. 2011). MOG macht den geringsten Anteil der Proteine der Myelinscheide aus und wird
ausschließlich im ZNS exprimiert. Es befindet sich an der Oberfläche der Myelinhülle und den
Zellkörpern der Oligodendrozyten und ist damit direkten Angriffen auf das Myelin durch
Antikörper ausgesetzt. Daher ist es auch ein dominantes Zielantigen beim häufig
angewandten Tiermodell der MS, der EAE bei Nagetieren. Diese extrazelluläre Domäne ist
das einzige bekannte Antigen, das bei mit MOG immunisierten EAE-Tieren sowohl eine T-
Zell-, als auch eine demyelinisierende Antikörperantwort hervorruft (Berger und Reindl
2007).

1 Einleitung
9
1.3.1.1.6 Histologie
Die Entmarkungsherde zeigen bei der MS, insbesondere in frühen Läsionsstadien, eine große
histologische Vielfalt. Um dieser immunopathologischen Bandbreite der MS-Läsionen
gerecht zu werden, können die Regionen der Gewebeschädigung (Plaques) nach ihrer
inflammatorischen und demyelinisierenden Aktivität eingeteilt werden (Love 2006). Je nach
Läsionsstadium ist das Entzündungsinfiltrat variabel. In der Regel geht die aktive
Demyelinisierung (frühes Läsionsstadium) mit perivaskulärer Inflammation einher, wobei
sich das entzündliche Infiltrat hauptsächlich aus CD4+- und CD8+-T-Lymphozyten, einigen B-
Zellen und Plasmazellen, aktivierten Makrophagen bzw. Mikrogliazellen zusammensetzt
(Lassmann et al. 1998). Um aktive Plaques zu ermitteln, ist dabei die Suche nach
Makrophagen mit Myelinabbauprodukten im Zytoplasma die sicherste Methode
(Noseworthy et al. 2000). Diese Ablagerungen in Makrophagen werden beispielsweise durch
die LFB-PAS-Färbung (Luxol Fast Blue - Periodic Acid Schiff) oder die immunhistochemische
Färbung der Myelinproteine identifiziert (Lassmann et al. 1998). Solche Makrophagen lassen
sich häufig auch im stark infiltrierten peripheren Läsionssaum der chronisch aktiven Plaques
nachweisen, deren Zentrum jedoch hypozellulär und dicht gliotisch ist. Inaktive Plaques sind
durch ein nur noch geringes Infiltrat von Entzündungszellen und eine starke Fasergliose
gekennzeichnet und weisen eine reduzierte Axondichte auf. Eine weitere Form der inaktiven
Plaques sind so genannte Shadow-Plaques, in denen eine Remyelinisierung der
demyelinisierten Axone in einem begrenzten Umfang stattfindet. Diese Regionen sind scharf
begrenzt und zeigen eine geringere Myelinfärbung, die die dünnen Myelinscheiden an
remyelinisierten Axonen repräsentiert.
Aktive Läsionen: Immunhistologische Subtypen I-IV
Histologisch lassen sich bei früh aktiven MS-Plaques vier Subtypen (pattern) der
Demyelinisierung unterscheiden. Infolgedessen wird diskutiert, ob verschiedene Patienten
einen individuellen immunpathogenetischen Verlauf entsprechend den histopathologischen
Läsionstypen aufweisen. Eine Erklärungsmöglichkeit der vier identifizierten histo-
pathologischen Läsionstypen ist, dass die Subtypen entweder verschiedenen
Pathomechanismen entsprechen oder dass sie unterschiedliche Stadien der
Läsionsentstehung widerspiegeln. Auch könnten patientenspezifische Unterschiede der
Immunantwort zu Variationen der Histomorphologie führen (Lucchinetti et al. 2000).

1 Einleitung
10
Trotz einer gewissen Variabilität früh aktiver Läsionen, konnten einheitliche Kriterien für vier
folgende immunpathologische Schädigungsmuster erarbeitet werden. Die Expression von
Myelinproteinen, die Struktur und Ausdehnung der Läsionen, ihre Lokalisation,
Oligodendrozytendegeneration sowie Komplementaktivierung werden dabei miteinbezogen.
Entzündungszellen variablen Ausmaßes, wie Makrophagen bzw. Mikrogliazellen und CD3+-T-
Lymphozyten, sind ein Bestandteil aller Läsionen (Lucchinetti et al. 2000). Das
Hauptmerkmal, das die Subtyp-I-Läsionen von den Subtyp-II-Läsionen unterscheidet, sind die
zusätzlichen Immunglobulin- und Komplementablagerungen (C9neo-Antigen) in den
Regionen der aktiven Myelinzerstörung. Diese sind nur bei Subtyp II zu finden. Eine scharfe
Plaquegrenze mit Läsionslokalisation um kleine Venen oder Venolen zeigt sich häufig bei
Subtyp I und Subtyp II. Die unscharf begrenzten Subtyp-III-Läsionen sind durch eine
ausgeprägte Oligodendrozytenapoptose und einen überproportionalen MAG-Verlust (im
Vergleich zu Myelinproteinen wie MOG, PLP oder MBP) gekennzeichnet. Das
Charakteristikum von Läsionen des Subtyp IV ist ein Untergang der Oligodendrozyten in
einem schmalen Rand nahe der aktiven Zone der Läsion. Dieser Läsionentyp konnte bisher
nur in primär progredienten Krankheitsverläufen gefunden werden (Lucchinetti et al. 2000).
Kortikale Demyelinisierung
Kortikale Läsionen sind ein Phänomen, das wie der axonale Schaden bereits während
früher Krankheitsstadien und nicht nur in chronischen Stadien der MS anzutreffen ist
(Lucchinetti et al. 2011). Im Gegensatz zu Läsionen der weißen Substanz sind diese Läsionen
grundsätzlich durch eine geringer ausgeprägte Entzündung, weniger Gliose und effizientere
Myelinreparaturmechanismen charakterisiert (Kutzelnigg und Lassmann 2005). Ausgedehnte
kortikale Demyelinisierung geht mit neuronalem und synaptischem Verlust einher (Wegner
et al. 2006), der mit kortikaler Atrophie korreliert (Kutzelnigg und Lassmann 2005). Daher
werden kortikale MS-Läsionen für nicht-fokale Symptome, wie neuropsychologische und
kognitive Dysfunktion, verantwortlich gemacht (Kutzelnigg und Lassmann 2006; Rinaldi et al.
2010). Diese Symptomatik ist ein Merkmal der chronischen Phase der MS (Amato et al.
2004).
Der prozentuale Anteil des entmarkten Kortex liegt im Verlauf der MS im Durchschnitt bei
15% und kann sogar die Ausdehnung der Demyelinisierung der weißen Substanz

1 Einleitung
11
überschreiten. In extremen Fällen können bemerkenswerterweise sogar bis zu 70% der
grauen Substanz demyelinisiert sein. Demyelinisierte Läsionen der grauen Substanz treten
im Verlauf in etwa 90% der chronischen MS-Patienten (primär und sekundär-chronische MS)
auf (Albert et al. 2007) und sind häufig mit entzündlicher meningealer Infiltration assoziiert
(Howell et al. 2011; Magliozzi et al. 2007). Die Bedeutung meningealer Entzündung wurde
zusätzlich dadurch fundiert, dass sie auch bei der primär-progredienten MS zu finden ist
(Choi et al. 2012). Im meningealen Infiltrat sind dabei follikelähnliche Immunzellaggregate
mit T-Zellen, Makrophagen, B- und Plasmazellen zu finden (Serafini et al. 2004). Dabei hängt
das Ausmaß der meningealen Inflammation mit dem axonalen Schaden der Infiltrat-nahen
Kortexschichten (Magliozzi et al. 2010) und der kortikalen Demyelinisierung zusammen
(Magliozzi et al. 2007). Wahrscheinlich korreliert sogar die Geschwindigkeit der
Krankheitsprogression mit dem Ausmaß der meningealen Entzündung (Stadelmann 2013).
Daher scheint es möglich zu sein, dass meningeale, entzündliche Aggregate sowohl zur
kortikalen Demyelinisierung, als auch zur MS-Progression beitragen.
Arten der kortikalen Demyelinisierung bei der MS – Kortikale Läsionen Typ 1-3
Die kortikalen Läsionen bei der MS wurden in unterschiedlichen Studien anhand ihrer
Lokalisation in den kortikalen Schichten eingeteilt. Heute wird allgemein eine Klassifikation
angewendet, anhand derer die Läsionen in drei Typen gruppiert werden (Bo et al. 2003 a;
Lucchinetti et al. 2011). Typ-1-Läsionen (kortiko-subkortikale oder leukokortikale Läsionen)
liegen an der Grenze zwischen grauer und weißer Substanz. Typ-2-Läsionen finden sich
intrakortikal in der Umgebung kleiner Gefäße. Typ-3-Läsionen, in subpialer Lage, zeigen eine
umschriebene oder bandfömige Demyelinisierung der oberen kortikalen Schichten, die sich
häufig über benachbarte Gyri ausbreitet (Howell et al. 2011; Kutzelnigg und Lassmann 2005).
Bevorzugt Sulci des Gyrus cinguli, der Inselrinde, des temporalen und frontalen Kortex sind
von subpialer Demyelinisierung betroffen. Folglich entstand die Hypothese, dass Mediatoren
aus dem stagnierenden Liquor in den Sulci eine Demyelinisierung begünstigen können
(Peterson et al. 2001). Subpiale Läsionen stellen flächenmäßig den größten Anteil der
kortikalen Läsionen dar (etwa 65% aller kortikalen Läsionen) und erstrecken sich auf bis zu
67% des gesamten demyelinisierten kortikalen Bereichs (Bo et al. 2003 b). Damit steht die
subpiale Entmarkung in einem besonderen Fokus. Aufgrund des häufigen Auftretens bei
Patienten mit langjähriger MS wird die subpiale Demyelinisierung auch als pathologisches

1 Einleitung
12
Korrelat der Krankheitsprogression angesehen (Albert et al. 2007; Kutzelnigg und Lassmann
2005).
Pathophysiologie kortikaler Läsionen
Die pathophysiologischen Mechanismen, die zu kortikaler Demyelinisierung führen, sind
nach wie vor nicht zufriedenstellend geklärt. Myelinschäden können durch verschiedene
Mechanismen wie immunvermittelte Entzündung, Stoffwechselveränderungen oder
ischämische/exzitotoxische Gewebeveränderungen verursacht werden (Lassmann 2001). Im
Falle der MS werden unterschiedliche Ursachen für die autoimmune Demyelinisierung, wie
ein Antikörper- bzw. Komplementangriff, Entzündungsmediatoren (Schwab und McGeer
2002; Bo et al. 2003 a) oder zytotoxische T-Zellen (Ruijs et al. 1990) diskutiert.
1.3.1.2 NMO
Auch die NMO wird als inflammatorisch demyelinisierende Erkrankung angesehen.
Gekennzeichnet ist die NMO durch Optikusneuritiden und Myelitiden, wobei ihre Symptome
häufig schwer von einer MS abgrenzbar sind, weil es teilweise auch schubförmige NMO-
Verläufe gibt (Wingerchuk et al. 1999). Diagnostisch zeigen sich in der Magnetresonanz-
tomographie (MRT) ausgedehnte spinale Läsionen oft über drei Wirbelkörper hinaus. Häufig
beobachtet man eine örtliche, jedoch keine zeitliche Dissemination der Läsionen in der
Bildgebung. Der kranielle MRT-Befund zeigt sich dabei häufig unauffällig. In aktuellen MRT-
Studien wurde allerdings beschrieben, dass NMO-Patienten auch supraspinale
Auffälligkeiten entwickeln können, die sich sowohl in der weißen als auch grauen Substanz
detektieren lassen (Rocca et al. 2004). Bei der NMO sind neben Demyelinisierung auch
Neurodegeneration und kognitive Beeinträchtigung aufzufinden. Ein erheblicher diffuser
Neuronenverlust und umfangreiche meningeale Inflammation ohne kortikale
Demyelinisierung können beobachtet werden (Saji et al. 2013). Gleichermaßen zeigt eine
Studie, dass die zuvor berichteten kognitiven und kortikalen Anomalien in der Bildgebung
mittels MRT nicht auf kortikale Demyelinisierung zurückzuführen sind. Das Fehlen kortikaler
Demyelinisierung ist dabei ein wesentliches Merkmal, das die NMO von der MS
unterscheidet (Popescu et al. 2010).
Serologisch sind bei den betroffenen Patienten Autoantikörper (NMO-Immunoglobulin G)
gegen Aquaporin-4 (AQP4) nachweisbar. AQP4 ist ein Wasserkanal, der eine hohe Dichte an
Astrozytenendfüßchen aufweist, die die Blut-Hirn-Schranke bilden (Lennon et al. 2004).

1 Einleitung
13
Charakteristischerweise zeigt die Histopathologie in den früh-aktiven Läsionen der NMO
neben der Demyelinisierung perivaskuläre Immunglobulin- und Komplementablagerungen
(C9neo Antigen) einhergehend mit einem Astrozyten- und Aquaporin-4-Verlust. Die
umfangreiche Demyelinisierung der weißen Substanz ist mit akutem axonalem Schaden
assoziiert, der sehr ausgeprägt sein kann. Das entzündliche Infiltrat der aktiven Läsionen
besteht aus zahlreichen Makrophagen, einigen perivaskulären neutrophilen und
eosinophilen Granulozyten, sowie CD3+-T-Zellen, wobei sich ein Teil davon als zytotoxische
CD8+-T-Zellen präsentiert. Häufig ist auch eine ausgeprägte Fibrose und Hyalinisierung der
Gefäßwände zu beobachten (Mandler et al. 1993). In einigen aktiven supraspinalen Läsionen
der AQP4-IgG-seropositiven NMO Patienten fanden sich Komplementaktivierungsprodukte
in Makrophagen, apoptotische Oligodendrozyten sowie ein dominierender MAG-Verlust.
Diese Merkmale tauchen in ähnlicher Form bei der MS in den immunhistologischen
Subtypen II und III wieder auf (Brück et al. 2012). Pathologische Charakteristika chronischer
Läsionen sind extensive Gliose und Atrophie der betroffenen Regionen (Kuhlmann2012).
1.3.1.3 ADEM
Die ADEM ist eine autoimmunvermittelte, demyelinisierende ZNS-Erkrankung, die häufig
Kinder oder junge Erwachsene betrifft und parainfektiös oder seltener nach Impfungen
auftritt (Tenembaum et al. 2007). Durch den wiederholten zeitlichen Zusammenhang mit
Infektionen werden diese als Ursache einer Autoimmunreaktion vermutet. Wie bei der NMO
ist auch die ADEM-Symptomatik schwer von der MS zu unterscheiden und wird
hauptsächlich durch den meist monophasischen Verlauf von den beiden anderen
Erkrankungen abgegrenzt. Die Patienten sind oft von einer diffusen, vielfältigen
neurologischen Symptomatik betroffen. Prognostisch gesehen kommt es innerhalb kurzer
Zeit häufig zu einer Rückbildung der Beeinträchtigungen mit oft nur geringer oder keiner
Residualsymptomatik (Tenembaum et al. 2007).
Vorwiegend befällt die ADEM die weiße Substanz und das Rückenmark. Angrenzend an die
graue Substanz finden sich häufig subkortikale Läsionen (Kroner-Milsch 2012). Das Merkmal
der pathologischen Befunde ist eine auf perivenöse Areale begrenzte Demyelinisierung mit
einer wallartigen Infiltration von Lymphozyten und vorwiegend schaumzelligen
Makrophagen, die vereinzelt auch konfluiert. Bioptisch lassen sich diese Entmarkungsherde
zum Teil nur schwer von MS-Läsionen abgrenzen (Kuhlmann 2012). Bisher wurde subpiale

1 Einleitung
14
Demyelinisierung in der Literatur in drei Fällen mit perivenöser Demyelinisierung
beschrieben (Young et al. 2010). Weitere Veränderungen der weißen und grauen Substanz
sind Hyperämie, endotheliale Schwellung und Gefäßwandinvasion durch Entzündungszellen
(Garg 2003).
1.3.2 Infektiöse Erkrankungen
Zentralnervöse Infektionen können sich topographisch unterschiedlich ausbreiten. Während
eine Leptomeningitis nur die Pia und Arachnoidea mater und eine Pachymeningitis die
harten Hirnhäute betrifft, befindet sich das entzündliche Infiltrat bei einer Enzephalitis im
Gehirnparenchym. Ist die graue Substanz involviert spricht man von einer Polioenzephalitis,
eine Beeinträchtigung der weißen Substanz wird als Leukenzephalitis und der Befall der
weißen und grauen Substanz als Panenzephalitis bezeichnet. Ist das Rückenmark betroffen,
spricht man von einer Myelitis.
Vor allem die bakteriellen Meningitiden bzw. Enzephalitiden können sich mit einer
klassischen Trias von Fieber, Nackensteifigkeit und Bewusstseinsstörungen präsentieren und
gehen mit Kopfschmerzen einher (van de Beek et al. 2006).
1.3.2.1 Virale Infektionen
Die häufigsten ZNS-Infektionen viraler Genese sind durch Enteroviren bedingt und bedürfen
oftmals keiner Therapie. Enteroviren verursachen eine milde Meningitis und erreichen das
ZNS meist durch hämatogene Streuung (Deckert 2012).
Verschiedene morphologische Charakteristika sind den viralen ZNS-Infektionen gemeinsam.
Klassischerweise sind virale ZNS-Infektionen initial polymorphonukleär und im Verlauf
lymphozytär dominiert. Es gelangen T- und B-Lymphozyten, Plasmazellen, Monozyten mit
Makrophagen in die Meningen oder das Hirnparenchym. Da z.B. Herpesviren praktisch alle
neuronalen Zellen befallen können, kommt es nach der Invasion des Erregers zur
Neuronophagie. In den Zielzellen zeigen sich darüber hinaus Virusbestandteile in
zytoplasmatischen oder nukleären Einschlüssen. Üblicherweise führt eine virale ZNS-
Infektion zur ausgeprägten residualen Astrozyten- und Mikrogliaaktivierung, auch teilweise
in Form von Mikrogliaknötchen (Deckert 2012). Bei Panenzephalitiden kommt es häufig zu
ausgedehnten hämorrhagischen Nekrosen.

1 Einleitung
15
Herpes-simplex-Virus (HSV)
Das HSV verursacht häufig eine latente Infektion in den Ganglien des Menschen. Von dort
aus kann es bei Reaktivierung anterograd den Nervenbahnen folgen und unter Umständen
Symptome wie beispielsweise Effloreszenzen an den Lippen (durch Transport vom Ganglion
trigeminale in den Nervus trigeminus) auslösen. Durch retrograden Transport über Axone
können die neurotropen Viren vom Ganglion trigeminale in das das Gehirnparenchym,
bevorzugt in temporale Regionen des Gehirns, einwandern. Damit kann das
neuroanatomische Befallsmuster hinweisend auf die Art der Virusinfektion sein (Deckert
2012).
Poliomyelitis
Das Polio-Virus wird fäkal-oral übertragen, breitet sich danach hämatogen aus und führt zu
bleibenden Lähmungserscheinungen oder sogar zum Tod. Überwiegend sind dabei Kinder
betroffen. Das Virus hat eine hohe Affinität zum Rückenmark, infiltriert dort die
α-Motoneurone im Vorderhorn und befällt auch Motoneurone des Pons und der Olive.
Dadurch kommt es zur neurogenen Atrophie der Muskulatur. Histologisch findet eine
ausgeprägte lymphozytäre Entzündung der grauen Substanz und der Meningen statt, die mit
Mikrogliaknötchen einhergeht (Deckert 2012).
Subakute skleorsierende Panenzephalitis (SSPE)
Eine weitere virale Infektion des ZNS mit letalem Ausgang ist die SSPE, eine mögliche,
seltene Spätkomplikation einer Masernvireninfektion, deren Entstehung nicht vollständig
geklärt ist. Vier bis zehn Jahre nach einer Maserninfektion tritt die Erkrankung auf und
verläuft langsam progredient in drei Stadien über mehrere Monate. Das erste Stadium ist
durch psychische Störungen und Persönlichkeitsveränderungen gekennzeichnet, das zweite
durch Myoklonien, epileptische Anfälle und Demenz. Final geht das dritte Stadium mit einem
Dezerebrationssyndrom bis hin zum Koma und Mutismus einher (Garg 2008; Schonberger et
al. 2013). Serologisch ist eine starke Antikörperaktivierung nachweisbar, die auch im Liquor
vorzufinden ist. Die SSPE manifestiert sich als eine chronisch progrediente Enzephalopathie
mit Einschlusskörpern im Kern der virusinfizierten Zielzellen. Es zeigt sich eine perivaskuläre
Inflammation und Gliose sowohl in der weißen als auch in der grauen Substanz. Das

1 Einleitung
16
entzündliche Infiltrat setzt sich aus CD4+- und CD8+-T-Zellen, sowie Monozyten und
Plasmazellen zusammen (Deckert 2012). Teilweise wurde überwiegend subkortikal ein
ungleichmäßiger, fleckenförmiger oder sogar diffuser Myelinverlust bei relativ erhaltenen
Axonen beobachtet (Love 2006).
Human immunodeficiency virus (HIV)
Das HI-Virus führt nach einer mehrjährigen symptomlosen Latenzphase unbehandelt zu
einem Immundefizienzsyndrom „Acquired Immune Deficiency Syndrome“ (AIDS). Das Virus
befällt vorwiegend T-Lymphozyten, in denen es sich vermehrt und die im Verlauf der
Infektion zugrunde gehen (Fauci 1996). Die antivirale Therapie hat heutzutage das
Fortschreiten der Erkrankung stark gebremst und das Überleben der Patienten wesentlich
verlängert (Mocroft et al. 2003). Die HI-Virusinfektion kann entweder direkt zur Beteiligung
des ZNS führen oder durch die immunsupprimierenden Folgen der Erkrankungen zu
Veränderungen des ZNS beitragen. Durch die HI-Virus induzierte Immunsuppression sind die
Patienten anfällig für opportunistische Infektionen, die sich häufig auch im ZNS abspielen
(Deckert 2012). Da das HI-Virus oft das ZNS infiltriert und eine Meningitis auslöst, lassen sich
bei 20–30% der Patienten, die an AIDS versterben, neuropathologische Veränderungen
nachweisen. Es kann bereits früh zu einer aseptischen Meningitis oder im Verlauf zu einer
HIV-Enzephalopathie kommen. Ist das Rückenmark beteiligt, spricht man von einer
vakuolären Myelopathie (Epstein und Gendelman 1993). Bei über der Hälfte der
asymptomatischen HIV-Patienten ist im Liquor eine leichte, vorwiegend lymphozytäre
Pleozytose beschrieben worden, die mit einer intrathekalen Immunreaktion gegen die HIV-
Infektion zusammenzuhängen scheint. Später klingt die Pleozytose jedoch wieder ab (Feiden
2012). Wie bei allen Entzündungen des ZNS kommt es zu einer Mikroglia- und
Makrophagenaktivierung mit nachfolgender Freisetzung von proinflammatorischen
Mediatoren, die neurotoxisch wirken, sowie die Apoptose von Oligodendrozyten,
Endothelzellen und auch Gliazellen verursachen können (Epstein und Gendelman1993).
Klinisch folgen häufig neurokognitive Störungen wie eine Demenz (AIDS-Demenz-Komplex)
(Gendelman et al. 1997).
Kennzeichnend für die HIV-Enzephalopathie sind mehrkernige Riesenzellen und
Mikrogliaknötchen, die neben der diffusen lymphozytären Infiltration vorherrschen (Gray et
al. 2003). Unter Umständen kann eine HIV-Infektion sogar zur diffusen oder multifokal

1 Einleitung
17
entzündlichen Entmarkung der weißen Substanz führen (Langford et al. 2002; Corral et al.
2004). Durch die effektive retrovirale Therapie scheint es zu einem Wandel des
Infitrationsmusters bei der HIV-Leukenzephalopathie zu kommen. Die Invasion des
Hirnparenchyms durch Monozyten/Makrophagen, der Grad der Entzündung und die
Zerstörung der weißen Substanz stellt sich umfangreicher dar (Langford et al. 2003).
Progressive multifokale Leukenzephalopathie (PML)
Die PML ist eine opportunistische virale ZNS-Infektion, die vorwiegend bei HIV-Patienten
vorkommt, aber auch beispielsweise bei Patienten unter stark immunsupprimierender MS-
oder Leukämietherapie zu finden ist (Brew et al. 2010). Diese demyelinisierende Erkrankung
wird durch das JC-Virus verursacht, das auch bei Gesunden asymptomatisch in B-
Lymphozyten oder in der Niere persistiert (Dorries 1998) und bei 75% der Erwachsenen
nachweisbar ist (Love 2006). Bei immundefizienten Patienten kann das Virus reaktiviert
werden und führt so zur Erkrankung. Die neurologische Symptomatik ist vielfältig und
beeinflusst motorische und sprachliche Fähigkeiten oder auch die Kognition und
Persönlichkeit (Brew et al. 2010).
Die Läsionen enthalten zahlreiche schaumzellige Makrophagen und vereinzelt Lymphozyten
(Love 2006). Bei der PML sind in der grauen Substanz intrakortikale und leukokortikale,
jedoch keine subpialen Läsionen beobachtet worden. Die kortikalen demyelinisierenden
Herde weisen weniger entzündliches Infiltrat als Läsionen der weißen Substanz auf (Moll et
al. 2008). Es zeigt sich bei der PML bei den Myelinproteinen ein dominierender MAG-Verlust,
der histologisch dem Subtyp III der MS-Läsionen ähnelt (Lucchinetti et al. 2000; Gendelman
et al. 1985). Darüber hinaus können, assoziiert mit den demyelinisierenden Arealen, JC-Virus
infizierte Zellen gefunden werden (Moll et al. 2008). In der Nachbarschaft entzündlicher
Läsionen befinden sich oft Oligodendrozyten mit vergrößertem, stark basophilem Zellkern
und nukleären Einschlüssen mit Virusbestandteilen. Auch die Astrozyten können in Form und
Größe bizarr verändert sein und einen vergrößerten, polymorphen, hyperchromatischen
Nukleus aufweisen (Deckert 2012).
1.3.2.2 Bakterielle Infektionen
Das Erregerspektrum der bakteriellen ZNS-Infektionen ist altersabhängig. Während
Neugeborene unter einem Monat eher durch gramnegative Bakterien, Gruppe-B-

1 Einleitung
18
Streptokokken und Listerien gefährdet sind, werden Meningitiden bei Kindern und
Jugendlichen vorwiegend durch Haemophilus influenzae, Meningokokken (Neisseria
meningitidis) und Pneumokokken (Streptococcus pneumoniae) hervorgerufen. Bei
Erwachsenen nimmt die Infektionshäufigkeit mit dem Alter durch Haemophilus stark ab und
durch Listerien wiederum zu (Schuchat et al. 1997). Die genannten Bakterien sind für ca.
80% der bakteriellen Infektionen verantwortlich und führen unbehandelt zu neurologischen
Langzeitfolgen oder sogar zum Tod. Der Verlauf dieser bakteriellen Meningitiden ist meist
höchst akut und mit einem Hirnödem und eitrigem Exsudat im Subarachnoidalraum
assoziiert. Mikroskopisch lassen sich im Infiltrat vorwiegend neutrophile Granulozyten
beobachten. Die intrathekale Entzündung resultiert in manchen Fällen in thrombotischen
Verschlüssen meningealer Gefäße, die sich histopathologisch in ischämischen Arealen mit
Nekrosen und einer Aktivierung von Mikroglia und Astrozyten darstellen. Bei persistierender
Infektion ändert sich die Zellzusammensetzung des entzündlichen Infiltrats während der
ersten Woche und es treten Makrophagen, Lymphozyten, Plasmazellen und Fibroblasten in
Erscheinung (Deckert 2012).
Syphilis
Syphilis ist eine chronische entzündliche Erkrankung, die von dem Bakterium Treponema
pallidum hervorgerufen wird. Die Beteiligung des ZNS kann Jahre oder Jahrzehnte nach der
Primärinfektion auftreten. Die Syphilis manifestiert sich in drei Stadien (Miklossy 2008). Nach
dem Primäraffekt (Stadium 1), einer schmerzlosen Papel (harter Schanker), die den ersten
Kontakt des Erregers zur Haut darstellt, verbreitet sich die Infektion auf hämatogenem Weg
bis ins ZNS. In 10 % der Fälle kommt es zu einer syphilitischen Meningitis (Stadium 2), die
durch ausgedehnte zelluläre Infiltration der Meningen mit Lymphozyten und Plasmazellen
gekennzeichnet ist (Miklossy 2008). Dieses Stadium, das vorwiegend die Leptomeningen
betrifft (Kinnier Wilson 1954), klingt nach mehreren Wochen ab und kann in eine latente
Phase münden. Die latente Phase kann innerhalb von Jahren bis Jahrzenten in die tertiäre
Neurosyphilis, die sich in mehreren Formen darstellt, übergehen (Miklossy 2008). Die
unterschiedlichen Neurosyphilisformen sind häufig gemischt anzutreffen. Die
meningovaskuläre Neurosyphilis, eine chronische Meningitis mit multifokaler Arteriitis
(Heubner-Arteriitis), manifestiert sich häufig in Form von Schlaganfällen, verbunden mit
einem allmählich fortschreitenden vaskulären Syndrom. Histologisch ist neben dem

1 Einleitung
19
entzündlichen Infiltrat die Verdickung der Meningen und der Arterien aller Größen zu sehen
(Deckert 2012). Die parenchymatöse Neurosyphilis resultiert aus der Invasion des Erregers in
das Hirngewebe, die zur ausgedehnten Parenchymschädigung führt. Infolgedessen kommt
es zu unterschiedlichen neurologischen und psychiatrischen Manifestationen, wie der
progressiven Paralyse mit Demenz oder Tabes dorsalis. Diese Neurosyphilisform mit
chronischer, frontal betonter Meningoenzephalitis stellt sich durch eine spärliche
perivaskuläre und parenchymatöse Zellinvasion dar und führt zu einer generalisierten
Hirnrindenatrophie mit Verlust von Neuronen (Deckert 2012). Das entzündliche Infiltrat
enthält Lymphozyten, Plasmazellen und Makrophagen, die zu epitheloiden Zellen
transformieren können. Im Verlauf können sich Granulome in Form von syphilitischen
Gummen mit zentraler Nekrose und spärliche Infiltration mit mehrkernigen Riesenzellen
bilden (Pilleri et al. 1974; Prange 1987). Des Weiteren wurden teilweise fokale Ischämien
beobachtet (Deckert 2012).
In der Literatur werden bei der Syphilis diffuse Bereiche mit Myelinschaden und abgeblasste
Regionen der periventrikulären weißen Substanz beschrieben (Miklossy 2008). Auch in der
Bildgebung zeigen sich Areale mit geschädigtem Myelin in der weißen Substanz (Brinar und
Habek 2006).
Tuberkulose (TBC)
Die Tuberkulose manifestiert sich primär pulmonal über die Inhalation des Erregers
Mycobacterium tuberculosis (Primärtuberkulose), kann jedoch sekundär praktisch alle
Organsysteme des Körpers betreffen. Nach der Ansteckung kann das Bakterium, auch ohne
zu Symptomen zu führen, noch jahrelang im Körper eines Patienten persistieren (latente
Tuberkulose) und unter Umständen, beispielsweise durch eine Immunsuppression,
reaktiviert werden. Entwickelt ein Patient nach seiner Erstinfektion erneut Symptome,
spricht man von einer postprimären bzw. sekundären Tuberkulose (Gideon und Flynn 2011).
Durch hämatogene Dissemination im Rahmen der Primärtuberkulose gelangt das
Mycobacterium tuberculosis bei immundefizienten Patienten auch in das ZNS und löst so
zunächst eine Meningitis aus. Diese häufig basal gelegene Meningitis verläuft in der Regel
chronisch. Im Laufe der Zeit kann die ausgeprägte Entzündung auf das Hirnparenchym
übergreifen und zu einer Meningoenzephalitis führen (Deckert 2012).

1 Einleitung
20
Klassischerweise entwickeln sich im entzündlichen Infiltrat mit der Zeit verkäsende
Granulome bzw. Tuberkulome. Diese Granulome sind von Lymphozyten, Makrophagen,
Epitheloidzellen, Fibroblasten und mehrkernigen Riesenzellen (Langhans-Typ) umgeben.
Durch die Ziehl-Neelsen-Färbung lässt sich das sogenannte säurefeste Stäbchenbakterium
histologisch nachweisen. Myelinschäden der weißen Substanz und ADEM ähnliche
perivaskuläre Demyelinisierung wurde in seltenen Fällen beschrieben (Dastur et al. 1995).
1.3.3 Neoplastische Erkankungen
Die häufigsten Primärtumore, die ins ZNS und die Meningen metastasieren sind
Adenokarzinome (Mamma- oder Lungenkarzinome), aber auch maligne Melanome, sowie
Lymphome, Leukämien oder seltener das Plasmozytom. Sind die Meningen von einer
diffusen metastatischen Tumorzellaussaat betroffen, spricht man von einer Meningeosis
neoplastica. Bei Karzinomen als Primarius bezeichnet man die neoplastische Infiltration als
Meningeosis carcinomatosa oder bei Lymphomen als Meningeosis lymphomatosa (Bigner
1992).
Primäre Non-Hodgkin-Lymphome des ZNS (PCNSL) treten gehäuft bei immunsupprimierten
Patienten auf. In den allermeisten Fällen wird dabei ein diffus großzelliges B-Zell-Lymphom
(DLBCL) mittels Probebiopsie histologisch diagnostiziert (Feiden und Milutinovic 2002), das
ein angiozentrisches Tumorinfiltrationsmuster zeigt. Je nach Ausprägung ist eine
Gehinparenchymreaktion mit einer Mikrogliaaktivierung bis hin zur Makrophageninfiltration
möglich (Paulus und Hasselblatt 2012). Bekannt ist, dass seltene Fälle des PCNSL mit fokaler
Demyelinisierung assoziiert sind (Alderson et al. 1996; Husseini et al. 2012; Kuhlmann et al.
2001). Auch ischämisch bedingte Myelinschäden wurde im Rahmen von ZNS-Lymphomen
beschrieben (Sadahira et al. 2000).
1.3.4 Metabolische Erkrankungen
Ein Beispiel für metabolische Demyelinisierung ist die zentrale pontine bzw. die extrapontine
Myelinolyse (Laureno und Karp 1997). Elektrolytverschiebungen durch Alkoholmissbrauch
und ein zu rapider Hyponatriämieausgleich (z.B. iatrogen) sind bekannte Ursachen dieser
Erkrankung. Dabei kommt es zu osmotischen Vorgängen im ZNS, die über einen bisher
unbekannten Mechanismus zur Demyelinisierung führen (Martin 2004). Etwa 25-35% der
Patienten mit einer zentralen pontinen Myelinolyse zeigen auch eine extrapontine

1 Einleitung
21
Manifestation. In bis zu 25% der extrapontinen Fälle ist sogar keine pontine
Demyelinisierung zu finden. Extrapontin sind meist das Cerebellum und die weiße Substanz
im Großhirn betroffen. Klinisch zeigen sich nach wenigen Tagen Bewusstseinsstörungen bis
hin zum Koma, Schwäche der Extremitäten bis hin zur Tetraparese und Störung der
Hirnstammfunktionen. Bei einer isolierten pontinen Myelinolyse stehen pseudobulbäre
Symptome im Vordergrund. Histologisch sind die entmarkten Herde scharf begrenzt und
beinhalten Lipid beladene Makrophagen, wenige oder keine Lymphozyten und zahlreiche
reaktive Astrozyten (Love 2006; Oehmichen 2012).

2 Material und Methoden
22
2 Material und Methoden
2.1 Autopsiekollektiv
Die Studie wurde an Hirngewebe eines Autopsiekollektivs von 156 Fällen mit einer Vielfalt an
entzündlichen, neoplastischen und metabolischen Erkrankungen durchgeführt (Tabelle 1,
Tabelle 3). Die Erkrankungen der untersuchten Fälle wurden am Institut für Neuropathologie
der Universitätsmedizin Göttingen diagnostiziert. Weiterhin wurden die neuropatho-
logischen Diagnosen aller Patienten von drei Neuropathologen des Instituts (Prof. Dr. med.
Wolfgang Brück, Prof. Dr. med. Christine Stadelmann-Nessler und Dr. med. Andreas Junker)
überprüft. Die Autopsien der 33 MS-Patienten, 3 ADEM-Fälle und 6 NMO-Patienten
stammen aus dem Institut für Neurologie der McGill University in Montreal, Kanada und
dem Institut für Neuropathologie der Universitätsmedizin Göttingen.
Insgesamt wurden 457 Hirngewebeblöcke der 156 Autopsien untersucht und die
diagnostizierten Erkrankungen in 6 Kategorien mit 19 Krankheitsentitäten (Tabelle 1)
eingeteilt. Die Gewebeblöcke entstammen den frontalen, parietalen, temporalen oder
occipitalen Kortexregionen.
Darüber hinaus wurden auch die Krankengeschichte und die vorhandenen Ergebnisse der
jeweiligen gesamten Autopsie überprüft. Die kortikalen Läsionen aus der MS-Kohorte
wurden bereits in einer früheren Studie (Albert et al. 2007) detailliert analysiert. Diese MS-
Patienten hatten eine mittlere Krankheitsdauer von 17 Jahren (Mittelwert ±
Standardabweichung: 17,8 ± 6,4, Median: 20,5 Jahre).
Alle Untersuchungen wurden in Übereinstimmung mit Gesetzen und Richtlinien der Georg-
August-Universität durchgeführt und von der Ethikkommission genehmigt.

2 Material und Methoden
23
Tabelle 1: Untersuchte Krankheitsentitäten des ZNS
Kategorie Diagnose Autopsie-
fälle
Zahl der untersuchten Gewebeblöcke mit Kortex
autoimmune Demyelinisierung MS 33 180
autoimmune Demyelinisierung ADEM 3 14
autoimmune Demyelinisierung NMO 6 21
viral HIV 2 4
viral JC-Virus (PML) 11 1 39
viral SSPE 2 7
viral Poliomyelitis 5 10
viral virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
8 16
bakteriell TBC 13 22
bakteriell Syphilis 3 16
bakteriell bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
12 20
Entzündung/Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis ohne bekannten Erreger
6 12
Entzündung/Infektion ohne bekannten Erreger
akute granulozytäre Meningitis / Meningoenzephalitis ohne bekannten Erreger
5 10
Entzündung/Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der Meningen oder des Hirnparenchyms
6 13
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
18 30
neoplastisch Hodgkin-Lymphom 3 4
neoplastisch Plasmozytom 3 5
neoplastisch Meningeosis carcinomatosa 11 20
metabolische Erkrankungen Zentrale pontine Myelinolyse 6 14
Kategorienzahl:
6
Gesamtzahl der Krankheitsentitäten:
19
Gesamtfallzahl:
156
Gesamtblockzahl:
457
1 beinhaltet einen Biopsiefall mit umfangreichen kortikalen Arealen

2 Material und Methoden
24
2.2 Histologische und immunhistologische Methoden
Um Gewebe unter dem Mikroskop untersuchen zu können, muss es zunächst fixiert,
geschnitten und anschließend gefärbt werden. Direkt im Anschluss an die Autopsie wurde
das Hirngewebe mit Formalin fixiert und später in Paraffin eingebettet (FFPE/ formalin fixed
paraffin embedded). Die Gewebeschnitte (ca. 3 µm dick) der in Paraffin eingebetteten
Autopsieblöcke wurden mit einem Schlittenmikrotom (Leica) angefertigt und in einem 37 °C
warmen Wasserbad gestreckt. Danach wurden sie auf Objektträger aufgezogen und in einem
Brutschrank über Nacht bei 30 °C getrocknet. Die vor der Färbung notwendige
Entparaffinierung wurde thermisch (60 °C Brutschrank, 45 min) und chemisch (Xylol)
durchgeführt. Direkt danach erfolgte die Rehydrierung durch eine absteigende Alkoholreihe
(100%, 90%, 70%, 50%, Aqua. Bidest, je 5 min) und anschließend die jeweilige Färbung. Um
die gefärbten Schnitte permanent zu erhalten, wurden sie durch eine aufsteigende
Ethanolreihe bis 100% dehydriert, in Xylol getaucht und mit DePex mounting media (BDH
England) eingedeckt.
Alle Färbungen wurden nach Routinelaborstandards des neuropathologischen Instituts
durchgeführt und sind im folgenden Abschnitt (s. u.) näher erläutert. Mit der Hämatoxylin-
Eosin-Färbung (HE) wurden Übersichtsfärbungen angefertigt, um Pathomorphologien wie
Entzündungen beurteilen zu können. Um Demyelinisierung zu erkennen wurde mit der LFB-
PAS-Färbung das Myelin gefärbt und mit der Silberimprägnation nach Bielschowsky die
Axone dargestellt. Immunhistochemische (IHC) Färbungen wurden hinzugezogen, um das
Myelin, verschiedene neuronale Zelltypen und das zelluläre Infiltrat zu beurteilen oder
Erreger nachzuweisen. Da es schwierig sein kann, kortikale Läsionen in der LFB-PAS-Färbung
zu sehen und zu beurteilen, wurden zusätzlich die Myelinproteine PLP, MBP und MOG
immunhistochemisch gefärbt. Dadurch wurden die Myelinscheiden deutlicher visualisiert, so
dass auch Myelindichteminderungen detektiert werden konnten. Alle Blöcke wurden einer
detaillierten neuropathologischen Untersuchung unterzogen und dabei die Myelinintegrität
der grauen und weißen Substanz beurteilt. Darüber hinaus half die immunhistochemische
Darstellung des Amyloid Percursor Proteins (APP), axonalen Schaden zu detektieren. Intakte
axonale Neurofilamente konnten durch die NF200-IHC dargestellt werden.
Der Nachweis von grampositiven Bakterien wurde auf Grundlage der Gram-Färbung
vorgenommen. Weiterhin zeigte die Ziehl-Neelsen-Färbung die Anwesenheit von

2 Material und Methoden
25
säurefesten Mykobakterien und die Chloracetat-Esterase-Färbung wurde durchgeführt, um
Granulozyten zu visualisieren.
2.2.1 Hämatoxylin-Eosin-Färbung
Die Schnitte wurden mit Mayers Hämalaun (Merck), das die Zellkerne blau färbt, behandelt,
in 1% HCL-Alkohol differenziert und anschließend unter fließendem Leitungswasser gebläut.
Nach der Spülung mit Aqua dest. folgte die Gegenfärbung mit Eosin 1%, welches das
Bindegewebe und das Zytoplasma rosa erscheinen lässt.
2.2.2 LFB-PAS
Bei dieser Färbung wurden die Schnitte nicht vollständig rehydriert (absteigende
Alkoholreihe bis 90% Ethanol) und in LFB-Lösung, die das Myelin blau färbt, bei 60 °C im
Brutschrank über Nacht inkubiert. Die Differenzierung der Schnitte erfolgte in drei Schritten.
Zuerst wurden die Schnitte in 0,05% Lithiumcarbonat getaucht, danach in 70% Ethanol
getränkt und anschließend in destilliertem Wasser gespült. Anschließend folgte die PAS-
Färbung, indem die Schnitte zunächst 5 Minuten in 1% Periodsäure verblieben und danach
unter fließendem Leitungswasser und später in Aqua dest. gespült wurden. Die Schnitte
wurden 20-30 Minuten im Schiffs Reagenz belassen, mit Leitungswasser gespült, kurz in
Mayers Hämalaun getränkt und nachfolgend in Aqua dest. getaucht. Nach der
Differenzierung mit Alkohol wurde erneut mit Leitungswasser gebläut.
2.2.3 Silberimprägnation nach Bielschowsky
Die Schnitte wurden nach der Entparaffinierung in eine Küvette mit Aqua bidest. überführt.
20% Silbernitratlösung wurde tropfenweise mit Ammoniak versetzt und die Schnitte darin 15
Minuten im Dunkeln inkubiert. Danach wurden die Schnitte in Aqua bidest. mit Ammoniak
geschwenkt. Der Ammoniak-/Silberlösung wurde ein Entwickler zugefügt und die Schnitte
darin getränkt. Die rasch ablaufende Entwicklung wurde mit destilliertem Wasser gestoppt,
die Schnitte in 2% Natriumthiosulfat getaucht und vor der Alkoholreihe zu allerletzt mit
Leitungswasser gespült.
Diese Färbung versilbert Neurofilamente. Damit erscheinen Neurone und ihre Axone
schwarz.

2 Material und Methoden
26
2.2.4 Immunhistochemie
Durch Antikörper können in Gewebeschnitten spezifische Antigene markiert und
anschließend durch Färbung visualisiert werden (Immunhistochemie, IHC). Voraussetzung
für die Bindung des verwendeten Antikörpers ist das „Freiliegen“ des Gewebeantigens
(Epitops). Durch das oben beschriebene Fixieren des Gewebes werden Proteine vernetzt und
es kann zur „Maskierung“ der Epitope kommen. Je nach Antikörper (Tabelle 2) muss deshalb
eine Antigendemaskierung durch Mikrowellenbehandlung für 15 Minuten (800 Watt) in
Citrat oder EDTA (Ethylendiamintetraacetat)-Puffer durchgeführt werden. Im Anschluss an
diese Vorbehandlung wurde die endogene Peroxidaseaktivität durch die Inkubation der
Gewebeschnitte in 3% H2O2 in phosphatgepufferter Salzlösung (PBS/phosphate buffered
saline) blockiert. Um eine unspezifische Antikörperbindung zu verhindern, mussten die
Schnitte darüber hinaus mit 10% fetalem Kälberserum in PBS für 10 Minuten bei
Raumtemperatur blockiert werden. Die gewaschen Schnitte wurden mit den in der Tabelle
aufgelisteten Antikörpern (Tabelle 2) über Nacht inkubiert und danach unter Anwendung
einer Avidin-Biotin-Technik mit 3,3'-Diaminobenzidin (DAB) oder 3-Amino-9-Ethylcarbazol
(AEC) als Chromogene visualisiert. Abschließend wurden die Schnitte zusätzlich mit Mayers
Hämalaun gegengefärbt, mit Alkohol differenziert und mit Wasser gespült, um die Kerne der
Zellen sichtbar zu machen.

2 Material und Methoden
27
Tabelle 2: Antikörper der IHC und Färbeverfahren
Antigen Tierart der Antikörper
Verdünnung Vorbehandlung Firma Zielstruktur
MBP rabbit pc 1:2000 - DAKO REF A0623 Myelinprotein
MOG rat pc 1:1000 Citrat Spende von Prof. D. Merkler,Universität Geneva, Schweiz
Myelinprotein
PLP clone plpc1 mouse mc 1:500 Citrat ABD Serotec MCA 829G Myelinprotein
GFAP rabbit pc 1:1000 - DAKO REF 0334 Astrozyten
Nogo A rabbit pc 1:1000 Citrat Santa Cruz Oligodendrozyten
NF 200 clone 52 mouse mc 1:400 Citrat Sigma Produktnummer: N0142
Neurofilamente
APP mouse mc 1:2000 Citrat Chemicon MAB348 Axonschaden, Sphäroide
KiM1P mouse mc 1:5000 Citrat Spende von Prof. HJ Radzun, Universität Göttingen
Makrophagen
2B4 mouse mc 1:500 Citrat Spende von Prof. JL Bennett, Universität Colorado, Denver, USA
NK-(Natural Kil-ler) Zellen-Ligand CD244
CD 3 rabbit mc 1:50 Citrat DAKO REF A0452 T-Zellen
CD 8 clone C8/144B
mouse mc 1:50 Citrat DAKO REF M7103 Zytotoxische T-Zellen
CD 20 clone C26 mouse mc 1:100 - DAKO REF M0755 B-Lymphozyten
CD 138 clone MI15
mouse mc 1:100 EDTA DAKO REF M7228 Plasmazellen
HSV1 rabbit pc 1:500 Citrat DAKO REF 0114 Virusnachweis

2 Material und Methoden
28
2.3 Läsions- und Infiltratklassifikation
Die Anzahl und die Lage der demyelinisierten Läsionen (in Gewebeblöcken der MS, PML,
NMO, extrapontinen Myelinolyse), die Zusammensetzung des entzündlichen Infiltrats
(Granulozyten, Lymphozyten, Makrophagen) und das Vorhandensein von meningealer
Entzündung und kortikalen Beteiligung wurden dokumentiert.
Die meningeale Zellinfiltration wurde in allen Autopsiefällen semiquantitativ beurteilt, indem
die infiltrierenden Zellen pro Gesichtsfeld bei 400-facher Vergrößerung (sog. high power
field, HPF) gezählt wurden. Zuletzt wurde mithilfe von vier verschiedenen Kategorien die
Infiltratdichte eingestuft (Abbildung 1).

3 Ergebnisse
29
3 Ergebnisse
Kortikale und besonders subpiale Demyelinisierung sind ein wesentliches Kennzeichen der
MS. In dieser Studie wurde untersucht, ob innerhalb anderer entmarkender Erkrankungen
sowie bei weiteren entzündlichen, neoplastischen oder metabolischen Störungen ebenfalls
eine kortikale respektive subpiale Entmarkung zu finden ist. Des Weiteren wurde die
Integrität des kortikalen Myelins in Verbindung mit der darüber liegenden entzündlichen
oder neoplastischen Hirnhautinfiltration untersucht. Diese Untersuchung erfolgte unter der
Annahme, dass lösliche Mediatoren, die durch intrathekale Zellinfiltrate frei werden und in
den Kortex diffundieren, kortikales Myelin und Oligodendrozyten schädigen (Tabelle 2 und
Abbildung 1).
Die viralen und bakteriellen Meningitiden bzw. Enzephalitiden zeigten die am
schwerwiegendsten ausgeprägte meningeale Zellinfiltration, während kaum Infiltration in
den Fällen der PML, der extrapontinen Myelinolyse und bei HIV auftrat (Abbildung 1).
Neben der MS konnte auch bei der PML subpiale Demyelinisierung detektiert werden, die
morphologisch jedoch nicht den subpialen MS-Läsionen entspricht. In keiner anderen der
untersuchten Fallkategorien konnte subpiale Demyelinisierung nachgewiesen werden. Die
kortikale Demyelinisierung wurde hingegen bei der ADEM und der extrapontinen
Myelinolyse beobachtet.

3 Ergebnisse
30
Abbildung 1: Semiquantitative Beurteilung des zellulären meningealen Infiltrats in allen
untersuchten Autopsiefällen
Das zelluläre meningeale Infiltrat dargestellt in vier Kategorien: (0) Keine infiltrierenden Zellen, (1) spärliche
meningeale Infiltration (weniger als 50 infiltrierende Zellen pro Gesichtsfeld bei 400-facher Vergrößerung/HPF),
(2) moderate Zellinfiltration (50 bis 200 infiltrierende Zellen pro HPF), (3) dichte Infiltration (mehr als 200
infiltrierende Zellen pro HPF). Der Mittelwert der Hirnhautinfiltration aller Schnitte pro Kategorie ist in der
Abbildung dargestellt. Die dichteste meningeale Infiltration wurde in den Fällen mit viraler oder bakterieller
Meningitis/Meningoenzephalitis gesehen.

3 Ergebnisse
31
3.1 Klassisch demyelinisierende Erkrankungen
3.1.1 Multiple Sklerose
Kortikale Demyelinisierung trat wie zuvor beschrieben bei der Mehrheit der chronischen MS-
Patienten der untersuchten Kohorte auf (Albert et al. 2007). Es wurden 180 MS-
Gewebeblöcke der 33 verschiedenen MS-Fälle untersucht und darunter in 29 Fällen kortikal
entmarkte Läsionen gefunden. Dabei fanden sich leukokortikale Läsionen in der
Myelinprotein-IHC an der Grenze zwischen weißer und grauer Substanz (Typ I), perivaskuläre
Läsionen in der grauen Substanz (Typ II) und subpiale bandförmige Läsionen (Typ III). Fokale
meningeale lymphozytäre Aggregate waren in der Nähe von Typ-III-Läsionen selten zu
finden. Die subpial demyelinisierten Läsionen der untersuchten Autopsiefälle zeigten nur
spärliche Lymphozyteninfiltration und moderate Mikrogliaaktivierung. Am häufigsten traten
diese Typ-III-Läsionen im frontalen Kortex auf (64% aller frontalen kortikalen Läsionen)
(Albert et al. 2007).
Typische kortikale MS-Läsionen kommen in den Abbildungen durch die immunhistochemische Färbung des
Myelinproteins MBP zur Darstellung. A zeigt eine subpiale bandförmig demyelinisierte Läsion der oberen
kortikalen Schichten, die sich auf benachbarte Gyri erstreckt. B zeigt eine gut abgegrenzte kleine perivenöse
kortikalen Läsionen und in C ist eine leukokortikale Läsion an der Grenze der grauen und weißen Substanz
dargestellt. (WM=weiße Substanz, GM=graue Substanz)
Abbildung 2: Kortikal demyelinisierte MS-Läsionen

3 Ergebnisse
32
3.1.2 Keine subpiale Demyelinisierung bei ADEM und NMO
Kortikale Areale von 3 ADEM- und 6 NMO-Autopsiefällen, deren Diagnosen histologisch und
klinisch belegt waren, wurden untersucht.
Ein ADEM-Fall zeigte in beiden verfügbaren Gewebeblöcken leukokortikale Läsionen. Die
kleinen perivaskulären entzündlichen Läsionen wiesen erhaltene axonale Integrität und
fokalen Myelinverlust (Abbildung 3) auf. Makrophagen und aktivierte Mikroglia zeigten die
typische Anordnung um kleine Gefäße. Des Weiteren war im entzündlichen Infiltrat eine
geringe Anzahl an T-Zellen anzutreffen. In den oberen kortikalen Schichten fanden sich keine
auffälligen perivaskulären entzündlichen Infiltrate und keine subpiale oder intrakortikale
Demyelinisierung.
In den beiden anderen untersuchten ADEM-Fällen war keine Beeinträchtigung der kortikalen
oder leukokortikalen Bereiche erkennbar. Nur in der weißen Substanz wurden
demyelinisierte Areale beobachtet.
In den 6 NMO-Fällen mit 21 Gewebeblöcken waren neben aktiven auch chronisch
demyelinisierte Läsionen zu beobachten, die sich jedoch auf die weiße Substanz
beschränkten. Dieser Befund deckt sich mit der Literatur (Popescu et al. 2010). Die noch
vorhandenen Astrozyten zeigten einen deutlichen fokalen Aquaporin-4-Verlust. Die
umfangreiche Demyelinisierung der weißen Substanz war mit akutem axonalen Schaden
assoziiert. Daneben bestand das entzündliche Infiltrat der aktiven Läsionen aus zahlreichen
schaumzelligen Makrophagen und einzelnen perivaskulären Granulozyten. Spärlich
eingestreut zeigten sich CD3+- und CD8+-T-Zellen. Darüber hinaus war das Merkmal der
chronischen Läsionen eine extensive Gliose.

3 Ergebnisse
33
In LFB-PAS gefärbten Serienschnitten konnten kleine perivaskuläre demyelinisierte Herde an der
leukokortikalen Grenze (A) identifiziert werden. B zeigt eine typische perivaskuläre Zellinfiltration und
Demyelinisierung der weißen Substanz in der LFB-PAS-Färbung. Die MBP-IHC (C und D) zeigt leukokortikal
demyelinisierte Läsionen. Es wurde keine subpiale oder intrakortikale Demyelinisierung beobachtet. E zeigt das
erhaltene, aber aufgelockerte axonale Gerüst in der Silberimprägnierung nach Bielschowsky. In F und G
kommen schaumzelligeige Makrophagen und aktivierte Mikrogliazellen, immunhistochemisch mit dem
Antikörper-Klon-KiM1P gefärbt, zur Darstellung. Einige wenige perivaskuläre CD3+-T-Lymphozyten sind in H zu
sehen.
Abbildung 3: Leukokortikale Demyelinisierung bei der ADEM

3 Ergebnisse
34
3.1.3 PML und supiale Demyelinisierung
Alle 10 untersuchten PML-Fälle zeigten demyelinisierte Herde der weißen Substanz.
Insgesamt enthielten 32 Gewebeblöcke der unterschiedlichen Fälle entmarkte Herde in
dieser Region. Typische aktive PML-Läsionen enthielten schaumzellige Makrophagen, einige
Lymphozyten (vor allem CD8+) und Astrozyten mit bizarr geformten Zellkörpern und
Kernformen. Die Oligodendrozyten hatten teilweise milchglasartige Zellkerne und waren in
ihrer Anzahl reduziert. Mittels SV40-IHC, die einen positiven Virusnachweis im Zytoplasma
und den Zellkernen mehrerer Oligodendrozyten in den Läsionen zeigte, wurde die JC-Virus-
Infektion (Abbildung 4, D) verifiziert.
Intrakortikale demyelinisierte Läsionen mit unscharfen Grenzen traten bei 6 von 10
Autopsiefällen auf; ferner wurden in einer hinzukommenden Biopsie ebenfalls kortikale
Läsionen nachgewiesen. Leukokortikale Läsionen waren in 7 von 10 Fällen vorhanden.
Darüber hinaus zeigten zwei Autopsiefälle mit kortikaler Inflammation und hoher kortikaler
Viruslast subpiale demyelinisierte Areale. Diese Bereiche waren fokal, klein, rundlich und
unscharf begrenzt. Die betroffenen Läsionen zeichneten sich durch das komplette Fehlen
des subpialen Myelins und durch viele schaumzellige Makrophagen mit
Myelinabbauprodukten im Zytoplasma aus. Die axonalen Strukturen waren in den oberen
kortikalen Schichten erhalten, zeigten jedoch viele intraaxonale APP+-Sphäroide als Zeichen
eines akuten axonalen Schadens. Es imponierte eine außergewöhnliche Viruslast in den
oberen kortikalen Arealen, die bis in die subpialen Bereiche reichte. Interessanterweise
traten nur wenige, überwiegend perivaskulär lokalisierte T-Zellen in diesen Läsionen auf,
während zahlreiche schaumzellige Makrophagen die betroffenen subpialen und oberen
kortikalen Regionen besiedelten. Einige Astrozyten zeigten ein hohes Maß an
Kernpolymorphien. Auffallend waren bei einigen Oligodendrozyten die unregelmäßigen
Zellformen und die Dichteminderung in mehreren Arealen. Darüber hinaus fand sich auch in
der untersuchten Biopsie ein Areal mit subpialer Demyelinisierung.

3 Ergebnisse
35
3.1.4 Kortikale Entmarkung bei der extrapontinen Myelinolyse
Kortikale Demyelinisierung konnte auch bei der metabolischen Erkrankung, der
extrapontinen Myelinolyse, festgestellt werden. Bei einem der sechs Patienten mit zentraler
pontiner Myelinolyse (Tabelle 1), die sich mit typischen pontinen Läsionen dargestellte,
wurden extrapontine demyelinisierte Bereiche mit Myelinverlust und Axonerhalt beobachtet
(Abbildung 5). Diese Läsionen erstreckten sich nur in geringem Ausmaß auf die subkortikale
weiße Substanz, aber erfassten bandförmig umfangreiche tiefe kortikale Schichten. Gemäß
der Läsionstypisierung der kortikalen MS-Pathologie entsprächen diese Läsionen am ehesten
Typ-1-Läsionen. Sie enthielten zahlreiche schaumzellige Makrophagen mit einigen
hauptsächlich CD8+-T-Zellen (Abbildung 5).
Abbildung 4: Kortikale Demyelinisierung in der PML
Kleine, unregelmäßig demyelinisierte subpiale Herde können in den Serienschnitten der MBP-IHC (A)
identifiziert werden. B zeigt eine subpial demyelinisierte Läsion von A im Detail. C beinhaltet die geschädigten,
aber erhaltenen axonalen Strukturen (NF200). Die hohe Viruslast ist in D (SV40 IHC) sichtbar. Die geringe CD3+ T-
Lymphozyteninfiltration ist in E dargestellt (Pfeile zeigen T-Lymphozyten im Parenchym). Schaumzellige
Makrophagen sind in F mit der KiM1P-IHC hervorgehoben. Astrozyten mit bizarren Zellkernen (G – GFAP-IHC)
und geschwollene Oligodendrozyten mit milchglasartigen Kernen sind erkennbar (H – NogoA-IHC).

3 Ergebnisse
36
Bandförmige kortiko-subkortikale Demyelinisierung wurde in einem Fall der extrapontinen Myelinolyse
beobachtet (A: LFB-PAS; B: PLP-IHC). C zeigt Neurone im demyelinisierten Bereich in einer höheren
Vergrößerung (Pfeile), außerdem eine beträchtliche Anzahl schaumzelliger Makrophagen mit PAS+-Granula in
ihrem Zytoplasma (Pfeilspitzen - einige Beispiele) (LFB -PAS). Eine dichte Infiltration durch CD8+-T-Zellen (D)
und KiM1P+-Makrophagen bzw. aktivierte Mikroglia (E) ist in demyelinisierten Arealen zu finden. F stellt die
intensive reaktive Astrogliose dar (GFAP-IHC). Die demyelinisierten Bereiche sind reich an Nogo A+ -
Oligodendrozyten (G). H zeigt weitgehend erhalte axonale und neuronale Strukturen (NF200-IHC). Die akute
axonale Schädigung wird durch den Marker APP (I) dargestellt.
Abbildung 5: Kortiko-subkortikale Demyelinisierung in der extrapontinen Myelinolyse

3 Ergebnisse
37
3.2 Infektiöse Erkrankungen
3.2.1 Akute meningeale Entzündungen
3.2.1.1 Akute bakterielle Infektionen
Fälle mit akuter bakterieller Meningitis zeichneten sich durch eine ausgedehnte
Hirnhautinfiltration mit Granulozyten, Makrophagen und einigen Lymphozyten aus. In den
meisten Fällen (>90%) wurden auch kortikale Bereiche durch die Entzündungszellen fokal
infiltriert und etwa 50% der Fälle zeigten herdförmige kortikale Nekrosen. Bei Patienten, die
in einem späteren Krankheitsstadium starben, wurde die meningealen Infiltrate durch
Lymphozyten und Makrophagen mit wenigen Granulozyten dominiert. Eine Schädigung der
Myelinscheiden konnte unabhängig von nekrotischen Arealen nicht beobachtet werden.
(Abbildung 5 A und B).
3.2.1.2 Virale Infektionen außer PML
Charakteristisch für die akute virale Meningitis zeigten sich mononukleäre Infiltrate in den
Meningen. Neben lymphozytären Meningitiden, bei denen kein Erreger identifiziert werden
konnte, fanden sich ebenso mehrere Gewebeproben der Autopsiefälle mit HSV-
Meningoenzephalitiden (Abbildung 6 C und D). Dort wurden Nekrosen im Kortex und in der
weißen Substanz, sowie eine Infiltration durch Granulozyten beobachtet. Der HSV-Nachweis
gelang in Neuronen mittels IHC. Eine mäßige Anzahl an Lymphozyten und Makrophagen
erstreckte sich von der Pia durch den Kortex bis in die tiefe weiße Substanz. Eine kortikale
Demyelinisierung wurde dabei nicht beobachtet.
Eine weitere akute Virusinfektion, die Infektion durch Zytomegalieviren (CMV), zeigte
charakteristische Zellen mit Einschlusskörpern, Ependymitis und Gewebsnekrosen. Auch hier
konnte keine Schädigung des Myelins oder der Oligodendrozyten festgestellt werden.
Die Infektion mit HIV kann unter Umständen schon in frühen Stadien der Erkrankung zu
einer ZNS-Beteiligung führen. In den untersuchten HIV-Autopsiefällen waren weder
meningeale Zellinfiltrationen, noch demyelinisierte Areale erkennbar.
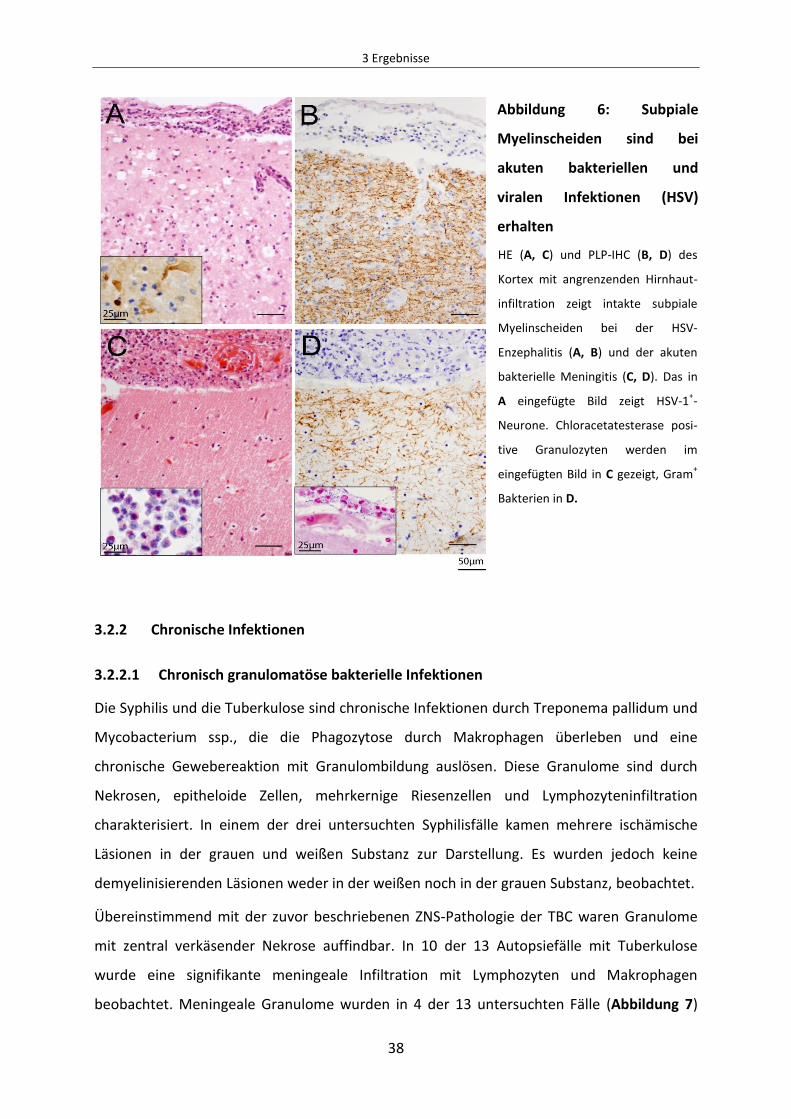
3 Ergebnisse
38
HE (A, C) und PLP-IHC (B, D) des
Kortex mit angrenzenden Hirnhaut-
infiltration zeigt intakte subpiale
Myelinscheiden bei der HSV-
Enzephalitis (A, B) und der akuten
bakterielle Meningitis (C, D). Das in
A eingefügte Bild zeigt HSV-1+-
Neurone. Chloracetatesterase posi-
tive Granulozyten werden im
eingefügten Bild in C gezeigt, Gram+
Bakterien in D.
3.2.2 Chronische Infektionen
3.2.2.1 Chronisch granulomatöse bakterielle Infektionen
Die Syphilis und die Tuberkulose sind chronische Infektionen durch Treponema pallidum und
Mycobacterium ssp., die die Phagozytose durch Makrophagen überleben und eine
chronische Gewebereaktion mit Granulombildung auslösen. Diese Granulome sind durch
Nekrosen, epitheloide Zellen, mehrkernige Riesenzellen und Lymphozyteninfiltration
charakterisiert. In einem der drei untersuchten Syphilisfälle kamen mehrere ischämische
Läsionen in der grauen und weißen Substanz zur Darstellung. Es wurden jedoch keine
demyelinisierenden Läsionen weder in der weißen noch in der grauen Substanz, beobachtet.
Übereinstimmend mit der zuvor beschriebenen ZNS-Pathologie der TBC waren Granulome
mit zentral verkäsender Nekrose auffindbar. In 10 der 13 Autopsiefälle mit Tuberkulose
wurde eine signifikante meningeale Infiltration mit Lymphozyten und Makrophagen
beobachtet. Meningeale Granulome wurden in 4 der 13 untersuchten Fälle (Abbildung 7)
Abbildung 6: Subpiale
Myelinscheiden sind bei
akuten bakteriellen und
viralen Infektionen (HSV)
erhalten

3 Ergebnisse
39
entdeckt. Das benachbarte Parenchym zeigte zahlreiche aktivierte Mikrogliazellen und
aktivierte Astrozyten. Darüber hinaus wurden zahlreiche, mit den Granulomen assoziierte
Epitheloidzellen und mehrkernige Riesenzellen beobachtet. Eine kortikale und insbesondere
eine subpiale Demyelinisierung fand sich hingegen nicht.
Meningeale Infiltration mit Lymphozyten, Epitheloid- und Riesenzellen sowie verkäsende Nekrose
charakterisiert die Tuberkulose (A, KiM1P-IHC). Mykobakterien sind durch die Ziehl-Neelsen-Färbung (kleines
Bild) visualisiert. Die PLP-IHC demonstriert das erhaltene subpiale Myelin unmittelbar neben einem
meningealen Tuberkulom (B).
3.2.2.2 Kortikale Pathologie bei chronisch viralen Infektionen
Es wurden Gewebeproben zweier Patienten untersucht, die an einer SSPE mit
nachgewiesenen Masern-Virus-Infektion (Abblildung 8) starben. Die T-Lympho-
zyteninfiltration war nur spärlich, ging jedoch mit massiver Mikrogliaaktivierung, teilweise in
Form von Mikrogliaknötchen, im gesamten Kortex und der weiße Substanz einher. Das
Masern-Virus-Antigen wurde in vielen Oligodendrozyten, in intrazytoplasmatischen oder
Abbildung 7: Intaktes subpiales Myelin bei der meningealen TBC

3 Ergebnisse
40
nukleären Einschlüssen, sowie in einigen kortikalen Neuronen nachgewiesen. Die
Myelinprotein-IHC zeigte jedoch keine Demyelinisierung im Hirnparenchym (Abbildung 8).
In A ist eine ausgeprägte Mikrogliaaktivierung in der grauen und weißen Substanz mit Mikrogliaknötchen in
einem repräsentativen Fall der Masernvirus induzierten SSPE zu sehen. Das Masern-Virus-Antigen wurde durch
die IHC in Oligodendrozyten und Neuronen (Einschub in B) nachgewiesen. Es fand sich in der IHC für MAG (B)
und PLP (C) keinen Hinweis für Demyelinisierung.
Ebenso konnte in Fällen mit unspezifischer chronischer Plasma- und B-Zell reicher
Entzündung der Meningen ohne Erregernachweis keine subpiale Demyelinisierung
nachgewiesen werden.
Abbildung 8: Kein Hinweis kortikaler Demyelinisierung in der SSPE

3 Ergebnisse
41
3.3 Neoplastische Erkrankungen der Hirnhäute
3.3.1 Lymphome und Plasmozytome
Auch bei ausgeprägter Hirnhautinfitration mit neoplastischen Plasmazellen bei
Plasmozytomen fand sich im untersuchten Kollektiv keine kortikale Demyelinisierung.
Obgleich im Rahmen des primären Non-Hodgkin-Lymphoms im ZNS Demyelinisierung
beschrieben worden ist (Alderson et al. 1996; Husseini et al. 2012; Kuhlmann et al. 2001),
war in der Studienkohorte von 18 NHL-Patienten (Tabelle 1) keine Demyelinisierung zu
detektieren, auch wenn 11 Autopsiefälle kortikale perivaskulär akzentuierte und teilweise
disseminierte oder meningeale Infiltration zeigten. Einer der 3 Patienten mit Hodgkin-
Lymphom imponierte mit einer dichten neoplastischen Infiltration (Tabelle 3). Jedoch waren
auch hier die Myelinscheiden gut erhalten (Abbildung 9 A-D).
3.3.2 Meningeosis carcinomatosa
In 11 Autosiefällen mit Meningeosis carcinomatosa infiltrierten maligne Zellen diffus oder in
drüsigen Zellverbänden den Subarachnoidalraum. Das untersuchte Autopsiekollektiv
umfasste Fälle mit Mamma-, Bronchial-, Ovarial-, und Magen-Darm-Karzinom (Tabelle 3).
Die subpialen kortikalen Schichten enthielten einige aktivierte Astrozyten, aktivierte
Mikrogliazellen und nur wenige Lymphozyten. Die subpialen Myelinschichten zeigten in
keinem der untersuchten Fälle eine Schädigung (Abbildung 9 E und F).

3 Ergebnisse
42
Abbildung 9: Erhaltene subpiale Myelinscheiden in einer Vielzahl von Erkrankungen mit
meningealer neoplastischer Zellinfiltration
HE (A, C, E) und PLP-IHC (B, D, F) des Kortex mit angrenzender Hirnhautinfiltration zeigt unbeschädigte subpiale
Myelinscheiden bei Meningeosis lymphomatosa (A, B), Plasmozytom (C, D) und Meningeosis carcinomatosa (E,
F).

4 Diskussion
43
4 Diskussion
4.1 Einleitung: Demyelinisierung als krankheitsspezifisches Phänomen
Kortikale Demyelinisierung und besonders der umfangreiche bandförmige subpiale
Myelinverlust ist ein Merkmal der MS. Bei diesem Charakteristikum bestünde die
Möglichkeit, es als spezifisches diagnostisches Merkmal dieser Erkrankung (z. B. in der
Histopathologie oder der Bildgebung) zu nutzen. Andere demyelinisierende Erkrankungen
wie die ADEM, die PML und Erkrankungen mit akuter oder chronischer, entzündlicher oder
neoplastischer kortikaler Pathologie könnten potentiell kortikale oder subpiale
Myelinveränderung aufweisen. Daher sollte anhand dieser groß angelegten Studie
verdeutlicht werden, ob die subpialen und kortikalen Läsionsformen der MS-spezifisch für
diese Erkrankung sind und ein krankheitsspezifisches Muster aufweisen. Demzufolge wurde
ein breites Erkrankungsspektrum ausgiebig untersucht, das mit einer Vielzahl an zellulären
Infiltrations- und Zytokinmustern im Kortex einhergeht. Allerdings wurde keine bandförmige
subpiale Myelinschädigung in der Autopsiekohorte, außer bei der MS, nachgewiesen. Dies
deutet darauf hin, dass krankheitsspezifische Mechanismen der Demyelinisierung bei der MS
vorherrschen.
4.2 Zytokine und Chemokine prägen ein krankheitsspezifisches Milieu
Subpiale kortikale Demyelinisierung bei der MS wurde mit lymphozytärer meningealer
Infiltration in Verbindung gebracht (Magliozzi et al. 2007; Serafini et al. 2004). Die
Hypothese, dass inflammatorische Mediatoren, die von Lymphozyten des meningealen
Kompartiments freigesetzt werden, zur Pathologie der grauen Substanz und damit zur
Zunahme klinischer Behinderung führen, wurde bereits mehrfach geäußert (Gardner et al.
2013; Magliozzi et al. 2010). Zusammen mit der Aktivierung von B- und Plasmazellen wurde
eine Hochregulation von Ig-Molekülen bei MS-Patienten im Kortex beobachtet (Torkildsen et
al. 2010). Dennoch konnte die enge örtliche Verbindung der Entzündungszellen mit
kortikaler Entmarkung nicht in allen Studien bestätigt werden (Kooi et al. 2009). Auch in
dieser Studie waren in der MS-Kohorte meningeale Infiltrate nicht konsistent mit kortikalen
Typ-III-Läsionen assoziiert. Dies weist darauf hin, dass möglicherweise eine generalisierte
Entzündungsreaktion innerhalb der Liquorräume für eine Demyelinisierung erforderlich ist

4 Diskussion
44
(Stadelmann et al. 2008). Die MS könnte als fokale Entzündung beginnen und in eine
chronifizierte diffuse Entzündungreaktion des gesamten Parenchyms münden.
Es bleibt abzuwägen, ob ein komplexes krankheitsspezifisches ZNS-Milieu demyelinisierende
Vorgänge auslösen kann. Die kortikale stereotaktische Injektion von Tumornekrosefaktor-α
(TNF-α) und Interferon-γ (IFN-γ) in MOG-immunisierten Ratten im EAE-Modell verursacht
fokale demyelinisierte Läsionen an mehreren Stellen im Kortex und verdeutlicht so die
Bedeutung der Zytokine. Bemerkenswerterweise hatten diese Läsionen histologische
Ähnlichkeiten mit kortikalen Läsionen der MS und zeigten typische Muster der
intrakortikalen und subpialen Demyelinisierung samt inflammatorischer Infiltration,
Komplementablagerungen und akuter axonaler Schädigung (Merkler et al. 2006).
MS-Läsionen beinhalten ein komplexes Zytokin-/Chemokin-Milieu. An dieser Stelle zeigt eine
aktuelle Studie die Hochregulation von inflammatorischen Genen in aktiven
demyelinisierenden MS-Läsionen des Kortex und unterstreicht damit ihre entzündliche
Komponente (Fischer et al. 2013). Dabei vermitteln immunologische Faktoren wie TNF-α,
Interleukin-1β (IL-1β) oder IFN-γ die akuten entzündlichen Prozesse der frühen MS-Läsionen
(Bitsch et al. 2000a; Argaw et al. 2006; Navikas und Link 1996). Andere Chemokine oder
Chemokinliganden wie CXCL12 (Chemokin C-X-C motif ligand), CXCL13 oder CCL19
(Chemokin C-C motif ligand) und inflammatorische Zytokine wie CXCL10, CCL2 und CCL3
begünstigen die Immunzellmigration in das erkrankte ZNS-Gewebe bei der MS (Meinl et al.
2006). Die Chemokine, die in MS-Läsionen gefunden werden konnten, sind
interessanterweise wichtige Faktoren bei der Immunzellantwort im Lymphgewebe (Drayton
et al. 2006). Darüber hinaus zeigen aktive und chronische MS-Läsionen eine erhebliche
Überexpression von TGF-β (De Groot et al. 1999).
Die Studienkohorte umfasst Patienten mit akuten sowie chronischen inflammatorischen
Krankheitsprozessen. Erstaunlicherweise weisen mehrere dieser Erkrankungen eine
Überlappung in ihrer Zytokin- und Chemokinexpression mit der MS auf. IL1-β und TNF-α sind
Zytokine, die im Liquor bei akuten entzündlichen Infektionen des ZNS mit Bakterien und
Mykobakterien (IL1-β, TNF) oder bei viralen Meningitiden/Meningoenzephalitiden (IL1-β)
hochreguliert werden (Akalin et al. 1994). TGF-β ist in reaktiven Astrozyten bei infektiösen
Geschehen wie der akuten bakteriellen Hirnhautentzündung (Ata et al. 1997), HIV-1-
Infektion oder PML (Johnson und Gold 1996) überexprimiert. Das aktuelle Konzept der
Granulom- und Riesenzellbildung favorisiert verschiedene Zytokine wie z. B. IFN-γ als

4 Diskussion
45
ursächliche Mediatoren (Sakai et al. 2012). Ebenso findet sich bei der akuten entzündlichen
Reaktion in frühen MS-Läsionen eine deutliche IFN-γ Ausschüttung.
Obwohl sich Entzündungsmediatoren, die durch verschiedene ZNS-Erkrankungen mit
kortikaler Pathologie freigesetzt werden, zumindest mit den Signalproteinen bei der MS
überschneiden, konnte in keinem der untersuchten Fälle eine kortikale Demyelinisierung
beobachtet werden. Jedes komplexe und spezifische Zytokinmilieu einer ZNS-Erkrankung ist
mit einer bestimmten glialen Genexpression verbunden (John et al. 2003), was offensichtlich
ein krankheitsspezifisches Gewebemilieu prägt.
CXCL13 in Kombination mit IL10 ist beispielsweise bei primären ZNS-Lymphomen
hochreguliert und in dieser speziellen Kombination hoch spezifisch für diese Erkrankung
(Rubenstein et al. 2013). Bei manchen ZNS-Lymphomen finden sich vor Ausbruch der
eigentlichen Erkrankung sogenannte entmarkende „Sentinel lesions“, die Ähnlichkeiten mit
der Demyelinisierung bei der MS aufzeigen (Alderson et al. 1996; Husseini et al. 2012;
Kuhlmann et al. 2001). Diese „Sentinel-lesions“ weisen ebenfalls darauf hin, dass Faktoren,
die von den Lymphomzellen abgesondert werden, zu Myelin- und Oligodendrozytenschäden
führen könnten, auch wenn kein Patient in dieser Studie eine Demyelinisierung zeigte. Einige
metastatische Hirntumoren bergen übereinstimmend mit der MS gleichermaßen eine
Hochregulation von Zytokinen wie CXCL13 (Rubenstein et al. 2013). Dennoch konnte in den
untersuchten Autopsiefällen mit metaststischem Infiltrat keine Myelinschädigung in der
Umgebung der Tumorzellen dargestellt werden.
In dieser Studie konnte dargelegt werden, dass zahlreiche chronische sowie akute
Entzündungsreaktionen und Krankheitsgeschehen per se nicht ausreichend sind, um
(kortikale) Demyelinisierung zu verursachen. Jede einzelne Erkrankung scheint typische,
krankheitsspezifische Verhältnisse zu prägen, was im Einzelfall, wie bespielsweise bei der
PML, ausreichend für Demyelinisierung ist.
4.3 Oxidativer Stress als proinflammatorischer Induktor
Bei der MS wurde die Induktion einer transkriptionsregulierten NO-Synthase (nitric oxide
synthase/iNOS) benachbart an aktive MS-Läsionen in Makrophagen bzw. Mikroglia, in
reaktiven Astrozyten (De Groot et al. 1997) und unauffälliger weißer Substanz nachgewiesen
(Liu et al. 2001; Oleszak et al. 1998). Die Expression dieses Enzyms wird durch eine
Kombination von proinflammatorischen Signalen, wie Liganden von Toll-like-Rezeptoren

4 Diskussion
46
(TLR) und Zytokinen, wie IL-1, TNF-α und IFN-γ, reguliert (Nathan und Xie 1994). Somit
setzen Makrophagen und Astrozyten Radikale frei, die möglicherweise zur Myelinschädigung
und zur Beeinträchtigung der Oligodendrozytenfunktion beitragen könnten. Bei der MS
wurde gezeigt, dass die akute Zellschädigung und der Zelltod von Oligodendrozyten mit
hochgradigem zytoplasmatischen und nukleären oxidativen Schaden verknüpft sind. Dies
untermauert die Ansicht, dass oxidativer Stress während der aktiven MS eine wichtige Rolle
bei der Destruktion des Myelins und der Oligodendrozyten spielt (Haider et al. 2011).
Oxidativer Stress mit strukturschädigenden Auswirkungen findet sich darüber hinaus auch
bei anderen Erkankungen. Beispielsweise sind reaktive Stickstoffspezies (Reactive Oxygen
Species/ROS) an pathophysiologischen Vorgängen der bakteriellen Meningitis des Menschen
beteiligt (Kastenbauer et al. 2002). Die Messung oxidierter Lipide bei einer induzierten
Meningitis im Rattenmodell durch Streptokokken der Gruppe B belegte auch dort die
Anwesenheit von ROS (Leib et al. 1996). Andere Daten zeigen, dass das Mycobacterium
tuberculosis, der Erreger der TBC, die proinflammatrorische Mikrogliareaktion durch die
NADPH (Nicotinsäureamid-Adenin-Dinukleotid-Phosphat)-Oxidase-abhängige Bildung von
ROS aktiv induziert (Yang et al. 2007).
Daneben führen auch virale Infektionen zu oxidativem Stress im Gewebe. Als Reaktion auf
eine HSV-Infektion wird die Entzündungsreaktion ebenso hier durch die NADPH-Oxidase-
abhängigen ROS moduliert (Hu et al. 2011) und in erster Linie oxidativer Schaden von
Neuronen induziert (Valyi-Nagy et al. 2000). Während einer HSV-1-Infektion spielen dabei
die TLR 2 der Mikrogliazellen bei der Vermittlung von oxidativem Stress,
proinflammatorischen Mediatoren und neuronaler Schädigung eine wichtige Rolle
(Schachtele et al. 2010).
Auch bei der HIV-Infektion überwiegt das Ungleichgewicht zugunsten von reaktiven
Sauerstoffspezies. Bei HIV-infizierten Patienten findet sich im Verlauf häufig eine
Neurodegeneration, die eine fortschreitende Demenz nach sich ziehen kann. Dabei scheinen
einige HIV-Proteine wie Tat (transactivator of transcription) und das HIV-1 envelope
glycoprotein (gp120) die Produktion von ROS zu stimulieren, die Bluthirnschranke zu
schädigen und auf diese Weise die Neurodegeneration zu fördern (Bagashev und Sawaya
2013; Banerjee et al. 2010). Eine weitere Studie zeigte weiterhin, dass HIV-1-Tat exponierte
Endothelzellen zu einer dosisabhängigen Erhöhung des oxidativen Stressspiegels und zu

4 Diskussion
47
einer Abnahme des intrazellulären Glutathions, eines Antioxidans, führen (Toborek et al.
2003).
In all diesen Studien wurde bei den beschriebenen Erkrankungen trotz des Einflusses von
oxidativem Stress keine Entmarkung beobachtet. Demzufolge kann oxidativer Stress unter
Umständen mit Entmarkung assoziiert sein, führt aber nicht zwangsläufig dazu. Es werden
offensichtlich krankheitsspezifische Muster von Faktoren benötigt, die die Demyelinisierung
hervorrufen können.
4.4 Genetische Grundlagen bedingen das Demyelinisierungsverhalten
Manche Individuen sind offensichtlich aufgrund genetischer Eigenschaften empfänglicher für
Demyelinisierung. Die genetischen Voraussetzungen scheinen beim Menschen das
demyelinisierende Verhalten bei Erkrankung wie der MS entscheidend zu beeinflussen.
Interessanterweise bestimmen Gene des MHC-Komplexes (Major Histocompatibility
Complex), ob ein Tierstamm bei der EAE für kortikale Demyelinisierung und pathologische
Veränderungen anfällig ist oder nicht. Die Kombination der MHC I- und II-Allele bei
unterschiedlichen Rattenstämmen beeinflusst die Häufigkeit und das Ausmaß kortikaler
Demyelinisierung (Storch et al. 2006). Es wurde des Weiteren gezeigt, dass bestimmte Gene
des MHC-Locus, die wahrscheinlich die Mikrogliareaktivität beeinflussen, bei bestimmten
Rattenstämmen prädisponierend für das Ausmaß axonalen Schadens sind. Darüber hinaus
wurde auch eine genetische Prädispositition für einen Makrophagen bzw. Mikroglia
dominierten Läsionsphänotyp entdeckt (Storch et al. 2002).
Der starke Einfluss der genetischen Grundlagen auf die Demyelinisierung ist ebenfalls in
einer experimentellen Infektion von Mäusen mit dem „Theiler's Murine Enzephalomyelitis
Virus“ (TMEV), die eine akute Polioenzephalomyelitis induziert, deutlich erkennbar. Tiere,
die sich erholen, sind häufig persistierend infiziert und entwickeln fokale
Demyelinisierungsherde im ZNS. Bemerkenswert ist, dass die TMEV-Infektion eine
Demyelinisierung nur in bestimmten Mausstämen herbeiführt (bei SJL/J-Mäusen, jedoch
nicht bei C57/Bl6-Mäusen). Daneben konnte festgestellt werden, dass auch bei Mäusen, die
gleiche klinische Auffälligkeiten zeigten, deutliche Unterschiede in der histopathologischen
Untersuchung der Läsionen zum Vorschein kamen (Dal Canto et al. 1995). Diese Ergebnisse
stützen die These, dass ein komplexes Netzwerk adaptiver und angeborener Immunität in
Abhängigkeit von entsprechenden genetischen Grundlagen eine Voraussetzung für die

4 Diskussion
48
Entstehung kortikaler demyelinisierender Läsionen ist und davon abhängig auch der Grad
der Behinderung und Regenerationsfähigkeit variiert. So zeigt sich, dass nicht allein das Level
von Zytokinen und Chemokinen eine kortikale bzw. subpiale Demyelinisierung auslösen
kann.
4.5 Erhebliche Unterschiede zwischen weißer und grauer Substanz
Die Pathologie und pathophysiologische Aktivität der Entzündung und Degeneration in
Läsionen der grauen Substanz unterscheiden sich bei der MS grundlegend von den Läsionen
der weißen Substanz. In einer Studie von Pham et al. wurden im Mausmodell der MS, der
EAE, Astrozyten der grauen und weißen Substanz verglichen und wiesen selbst zu einem sehr
frühen Zeitpunkt der Deymelinisierung ausgeprägte Unterschiede auf (Pham et al. 2009).
Im Cuprizon-Modell, einem häufig verwendeten Mausmodell der MS, wurden ebenfalls
erhebliche Unterschiede zwischen Kortex und Marklager entdeckt. Bei diesem Modell wird
Mäusen der Kupferchelator Cuprizone verabreicht, was eine ausgeprägte Demyelinisierung
des Balkens und in geringerer Ausprägung auch anderer Bereiche des ZNS nach sich zieht.
Wird die Verabreichung von Cuprizone gestoppt, folgt eine kurze, wenige Tage anhaltende,
intensive Phase der Remyelinisierung im ZNS. So können de- und remyelinisierende Prozesse
untersucht werden, wie sie in ähnlicher Form bei der MS auftreten (Hiremath et al. 1998;
Skripuletz et al. 2011). Verschiedene Wachstumsfaktoren, wie Brain-derived neurotrophic
factor (BDNF) sind im Corpus callosum während der Phase der Remyelinisierung
hochreguliert, aber vermindert in kortikalen Bereichen vorhanden, obwohl diese in dem
Modell auch von Demyelinisierung betroffen sind (Gudi et al. 2011). Infiltrierende
Immunzellen im ZNS und Neurone sind das Produkt von Wachstumsfaktoren wie BDNF.
BDNF ist in die gliale und neuronale Entwicklung und deren Überleben involviert. Zudem ist
es auch bei ZNS-Entzündungen in Läsionen anzutreffen und wirkt damit neuroprotektiv
(Kerschensteiner et al. 1999; Linker et al. 2010; Stadelmann et al. 2002). Neben BDNF gibt es
noch zahlreiche andere Faktoren, die ein unterschiedliches Milieu zwischen grauer und
weißer Substanz bewirken. Die Mikroglia- und Astrozytenaktivierung zeigen beispielsweise
auch deutliche Unterschiede in diesen zwei Kompartimenten (Gudi et al. 2009). Dies
bedeutet, dass nicht nur bei einzelnen demyelinisierenden Erkrankungen nach spezifischen
Pathomechanismen gesucht werden muss, sondern auch bei der Demyelinisierung selbst

4 Diskussion
49
regional unterschiedliche Milieufaktoren zwischen grauer und weißer Substanz
vorherrschen.
4.6 Myelinschädigung bei hypoxisch-ischämischen Geschehen
Hypoxisch-ischämische Vorgänge können histopathologisch bei unterschiedlichen
Erkankungen ein buntes Bild hervorrufen. Diffuse Bereiche des Myelinverlustes und
abgeblasste Regionen der periventrikulären weißen Substanz wurden bei der Syphilis
beschrieben. Kleine multifokale Läsionen mit Myelinverlust im Kortex wurden als
ischämische Schäden interpretiert (Miklossy 2008). Darüber hinaus konnten ADEM ähnliche
perivaskuläre Myelinschäden der weißen Substanz in einzelnen Fällen auch bei der
Neurotuberkulose dargestellt werden (Dastur et al. 1995). Mehrfach wurden vor allem bei
der Syphilis in der Bildgebung diffuse Schädigungen der weißen Substanz gesehen. In bis zu
25% der Patienten wurden zudem auch Infarkte diagnostiziert (Brightbill et al. 1995; Fadil et
al. 2006). Die Abgrenzung zu demyelinisierenden Läsionen ist jedoch radiologisch häufig als
schwierig einzuschätzen. Grundsätzlich imitiert die neurologische Manifestation der Syphilis
zahlreiche radiologische Befunde zerebraler Erkrankungen und zeigt vielfältige
Erscheinungsformen (Fadil et al. 2006). In den Untersuchungen dieser Studie fand sich
hingegen weder bei der Syphilis, noch bei der Tuberkulose eine Demyelinisierung. Die
Histopathologie zeigte lediglich ausgeprägte Nekrosen im Kortex und in der weißen Substanz
(TBC) oder ischämische Läsionen (Syphilis). Prinzipiell kann bei einer Nekrose bzw. einem
Schlaganfall das benachbarte Gehirnparenchym aufgelockert oder abgeblasst erscheinen.
Auch chronische Virusinfektionen wie bei der SSPE haben im Autopsiekollektiv dieser Studie
zu keiner Demyelinisierung in Hirnarealen, sondern allein zu Mikrogliaaktivierung und
Astrozytose geführt.
4.7 Subpiale bandförmige Läsionen sind MS-spezifisch
Über kortikale demyelinisierende Läsionen in einer Kohorte von biopsierten MS-Patienten in
frühen Stadien der Erkrankung wurde umfassend berichtet (Lucchinetti et al. 2011). Es
konnten leukokortikale (Typ-I-Läsionen der grauen Substanz), intrakortikale (Typ-II-Läsionen
der grauen Substanz) und subpiale Läsionen (Typ-III-Läsionen der grauen Substanz)
nachgewiesen werden. Damit wurde gezeigt, dass die kortikale Demyelinisierung ein
krankheitsspezifisches Frühphänomen der MS widerspiegelt.

4 Diskussion
50
In der Studienkohorte der hier durchgeführten Arbeit wurden neben Autopsien von MS-
Erkrankten, Autopsien von Patienten mit PML, ADEM, NMO und extrapontiner Myelinolyse
untersucht. Erkrankungen, bei denen die Demyelinisierung ein krankheitsspezifisches
Ereignis darstellt, welches auch in dieser Studie histopathologisch dargelegt wird. Somit
zeigten sich bei PML-Patienten intrakortikale, leukokortikale und sogar subpiale
Demyelinisierung. Subpiale Läsionen wurden trotz vieler unterschiedlicher Pathologien nur
bei der PML in zwei Autopsiefällen und einem Biopsiefall nachgewiesen. Doch diese Läsionen
hatten eine andere Morphologie als die der MS-Läsionen. Die Entmarkung zeigte sich als
ausnahmslos fokal, klein und wenig scharf abgegrenzt und war in kortikalen Bereichen, in
denen eine deutliche Viruslast herrschte, anzutreffen. Ferner wurde kortikale
Demyelinisierung in zwei weiteren Krankheitsentitäten nachgewiesen. Zwei ADEM-Patienten
der Studie zeigten leukokortikale Läsionen, jedoch keine supialen Herde. Bisher wurde
subpiale Demyelinisierung bei der ADEM in der Literatur in drei Fällen mit perivenöser
Demyelinisierung beschrieben (Young et al. 2010). Einer der untersuchten Fälle des
Studienkollektivs mit extrapontiner Myelinolyse wies ausgedehnte bandförmige
leukokortikale demyelinisierte Läsionen auf.
Alle diese demyelinisierenden Krankheiten beinhalten spezifische kortikale Umstände, die
durch das enge Zusammenspiel von meningealen Entzündungsmediatoren und
Parenchymzellen im Kortex, wie Mikroglia, Astrozyten und Oligodendrozyten, entstehen.
Daneben sind zudem vor allem bei MS-Patienten prädisponierende genetische Faktoren
beteiligt und bilden einen heterogenen Faktorenkomplex. Grundsätzlich liegt die Vermutung
nahe, dass zur Demyelinisierung bestimmte pathogenetische Faktoren notwendig sind, die
sich von Krankheitsursache zu Krankheitsursache unterscheiden. Diese Hypothese wird
dadurch untermauert, dass Demyelinisierung spezifisch für bestimmte Krankheiten ist und
bei anderen praktisch nicht beobachtet werden kann.
Die Studienergebnisse lassen darauf schließen, dass die bandförmige subpiale
Demyelinisierung ein MS-spezifisches Läsionsmuster und Demyelinisierung im Allgemeinen
eine krankheitsspezifische pathognomonische Erscheinung darstellt. Da diese
charakteristische Läsionsform der MS bereits in der frühen Phase der Erkrankung auftritt,
könnten diese Erkenntnisse zu neuen differentialdiagnostischen Überlegungen in der
Patientenversorgung führen.

4 Diskussion
51
4.8 Ausblick
Bisher ist nur wenig über die kortikale Demyelinisierung in den frühen Stadien der MS
bekannt, da u. a. mittels MRT viele subpiale und kortikale entmarkte Läsionen derzeit noch
nicht ausreichend darstellbar sind (Calabrese et al. 2013). Da die Kernspintomographie
jedoch ständiger Verbesserung unterliegt und immer vielfältigere hochauflösende
Sequenzen durchführbar sind (Tallantyre et al. 2010; Tardif et al. 2010), ist es
möglicherweise nur eine Frage der Zeit, dass kortikale Entmarkungsherde in der Bildgebung
besser detektiert werden und deren prognostische und diagnostische Relevanz so zur
Geltung kommen kann. Die fortschrittliche Bildgebung wird, vor allem im Hinblick auf die
Relevanz der bandförmigen bzw. kortikalen Demyelinisierung, die Basis für weitere klinisch
pathologische Studien bieten. Weiterhin können molekulare Untersuchungen zur
Pathogenese der kortikalen Demyelinisierung und zu Reparaturmechanismen der grauen
Substanz die heterogenen Pathomechanismen der MS-Läsionen zunehmend aufklären.
Insbesondere werden vergleichende Untersuchungen zum oxidativen Stress bei
unterschiedlichen kortikalen ZNS-Pathologien, basierend auf dieser Studie, durchgeführt
werden, um mögliche Milieuunterschiede und die spezifische Rolle dieser Mechanismen
aufzudecken. Vor allem sind Unterschiede der neurotoxischen Verhältnisse zwischen den
MS- und PML-Läsionen von großem Interesse

5 Zusammenfassung
52
5 Zusammenfassung
Kortikale demyelinisierte Läsionen sind bei Patienten mit chronischer Multipler Sklerose
(MS) häufig und weit verbreitet und könnten zur Krankheitsprogression beitragen. Es
wurden kortikale und subpiale Läsionen in frühen Stadien der MS beobachtet. Dies weist
darauf hin, dass diese Läsionen ein inhärentes und frühes Merkmal des Krankheitsprozesses
darstellen. Des Weiteren wurde die Hochregulation entzündungsassoziierter Gene in frühen
akuten Läsionen des Kortex nachgewiesen. Dennoch bleiben die genauen Mechanismen, die
zur Entstehung subpialer kortikaler Demyelinisierung beitragen, ungeklärt. Um zu prüfen, ob
kortikale und vor allem subpiale Demyelinisierung in anderen Erkrankungen als bei der MS
auftreten, untersuchten wir eine große Autopsiekohorte von Patienten mit Erkrankungen
autoimmuner, infektiöser, neoplastischer oder metabolischer Genese, die den Kortex
beeinträchtigen. Miteinbezogen wurden unter anderem Patienten mit MS, akuter
disseminierter Enzephalomyelitis (ADEM), Neuromyelitis optica (NMO), akuter und
chronischer, viraler und bakterieller Meningitis, progressiver multifokaler Leukoenzephalitis
(PML), subakuter sklerosierender Panenzephalitis (SSPE), Meningeosis lymphomatosa sowie
carcinomatosa und Stoffwechselerkrankungen wie der extrapontinen Myelinolyse, um ein
breites Erkrankungsspektrum mit einer Vielzahl an zellulären Infiltrations- und
Zytokinmustern im Kortex zu erfassen. Mittels Immunhistochemie für Myelin-basisches
Protein (MBP), Proteolipidprotein (PLP) und Myelin-Oligodendrozyten-Glykoprotein (MOG)
fanden wir kortikale Demyelinisierung nicht nur bei der MS, sondern auch bei der ADEM, der
PML und der extrapontinen Myelinolyse. Bedeutend ist, dass jede dieser Erkrankungen ein
krankheitsspezifisches kortikales Deymelinisierungsmuster aufweist und ausgedehnte
bandförmige subpiale Demyelinisierung nur bei der MS beobachtet wurde. Subpiale
Demyelinisierung trat insbesondere bei der PML auf, war aber im Gegensatz zu MS-Läsionen
durch ihre kleine fokale Beschaffenheit charakterisiert. Die Daten dieser Studie erweitern
und bestätigen frühere Studien und legen nahe, dass extensive bandförmige subpiale
Demyelinisierung MS-spezifisch ist und dies damit ein wichtiges diagnostisches Kriterium
darstellen könnte. Offensichtlich sind krankheitsspezifische Milieus oder heterogene
Faktoren, wie bestimmte Zytokine, Chemokine und genetischen Grundlagen, notwendig, um
Demyelinisierung zu verursachen. Damit sind weitere molekulare Untersuchungen

5 Zusammenfassung
53
erforderlich, um die spezifischen Mechanismen der Demyelinisierung bei MS und anderen
demyelinisierenden Erkrankungen zu analysieren.

6 Abstract
54
6 Abstract
Cortical demyelinated lesions are frequent and widespread in chronic multiple sclerosis (MS)
patients, and may contribute to disease progression. Cortical and subpial lesions were found
in early MS stages indicating that they may be an inherent and early feature of the disease
process. Moreover, inflammation‐associated genes were shown to be upregulated in early
cortical lesions. Yet, the precise mechanisms leading to the formation of subpial cortical
demyelination remain elusive. To test whether cortical demyelination and especially subpial
demyelination occurs in diseases other than MS, we studied a large cohort of patients
pathologically diagnosed with diseases of autoimmune, infectious, neoplastic or metabolic
origin affecting the cortex. Included were, among others, patients with MS, acute
disseminated encephalomyelitis (ADEM), neuromyelitis optica (NMO), acute and chronic
viral and bacterial meningoencephalitis, progressive multifocal leukoencephathy (PML),
subacute sclerosing panencephalitis (SSPE), meningeal carcinomatosis, lymphomatous
meningitis, and metabolic disorders such as extrapontine myelinolysis, thus encompassing a
wide range of adaptive and innate cytokine signatures. Using immunohistochemistry for
myelin basic protein (MBP), proteolipid protein (PLP) and myelin oligodendrocyte
glycoprotein (MOG) we found cortical demyelination besides MS in ADEM, PML and
extrapontine myelinolysis. Importantly, each of these diseases had a disease specific cortical
demyelination pattern and bandlike extensive subpial demyelination was only found in MS.
In particular subpial demyelination occurred also in PML but in contrast to MS lesions had a
small focal character. These study data extend and confirm previous findings and suggest
that extensive bandlike subpial demyelination is specific for MS, thus providing an important
diagnostic cue. Obviously, disease‐specific milieus or heterogeneous triggers are necessary
causing demyelination such as specific cytokines, chemokines and genetic background.
Further molecular studies are required to dissect the specific mechanisms of demyelination
in MS and other demyelinating diseases.

7 Anhang
55
7 Anhang
Tabelle 3: Ausführliche Beschreibung der untersuchten Autopsiefälle
Kategorie der ZNS-Erkankungen
Subkategorie Diagnose und Begleiterkrankungen
Ge
sch
lech
t
Alt
er in
Jah
ren
Erkr
anku
ngs
dau
er
u =
un
bek
ann
t
Tod
esu
rsac
he
ab
hän
gig
von
ZN
S-
Erkr
anku
ng
/un
abh
ängi
g u
= u
nb
ekan
nt
Ge
web
eblo
ckza
hl
(mit
gra
uer
Su
bst
anz)
1 m
enin
geal
e
Infi
ltra
tio
n
virale Infektionen
HIV
HIV-Infektion; Pneumonie; Kaposisarkom; Toxoplasmose
m 34 5 Jahre krankheitsabhängig 2(2) 0,0
virale Infektionen
HIV HIV-Infektion w 37 u u 2(2) 0,0
virale Infektionen
JC-Virus JC-Virus-Infektion nach Nierentransplantation w 59 u krankheitsabhängig 2(2) 0,0
virale Infektionen
JC-Virus PML w 78 u krankheitsabhängig 4(4) 0,0
virale Infektionen
JC-Virus PML m 27 4
Wochen krankheitsabhängig 3(3) 0,3
virale Infektionen
JC-Virus PML m 45 3.5
Monate krankheitsabhängig 4(4) 0,0
virale Infektionen
JC-Virus PML; HIV-Infektion
m 51 2
Monate krankheitsabhängig 5(5) 0,0
virale Infektionen
JC-Virus PML nach Chemotherapie; oropharyngealer Tumor
w 58 u krankheitsabhängig 4(4) 0,0
virale Infektionen
JC-Virus PML; HIV-Infektion m 36 u krankheitsabhängig 6(6) 0,0
virale Infektionen
JC-Virus PML; lange Glukokortikoidbehandlung m 55 2
Monate krankheitsabhängig 5(5) 0,2
virale Infektionen
JC-Virus PML; chronische lymphozytäre Leukämie; Aspergillus Pneumonie
m 63 u u 3(3) 0,7
virale Infektionen
JC-Virus PML; HIV-Infektion w 40 u krankheitsabhängig 2(2) 0,0
virale Infektionen
subakut sklerosierende Panenzephalitis (SSPE) subakut sklerosierende Panenzephalitis (SSPE); Zustand nach MasernInfektion
m 10 3
Monate krankheitsabhängig 2(2) 1,0
virale Infektionen
subakut sklerosierende Panenzephalitis (SSPE) subakut sklerosierende Panenzephalitis (SSPE); Zustand nach MasernInfektion; Kardiale Insuffizienz
m 8 u krankheitsabhängig 5(5) 0,8
virale Infektionen
Poliomyelitis Poliomyelitis; Polioenzephalitis m 20
Monate 7 Tage krankheitsabhängig 1(1) 1,0
virale Infektionen
Poliomyelitis Poliomyelitis; Polioenzephalitis m 28
Monate 4 Tage krankheitsabhängig 2(2) 0,0
virale Infektionen
Poliomyelitis Zustand nach Poliomyelitis; Hypoglykämie
w 41 u krankheitsunabhängig 2(2) 0,0
virale Infektionen
Poliomyelitis Zustand nach Poliomyelitis; Herzinwarkt
m 61 53 Jahre krankheitsunabhängig 2(2) 0,0
virale Infektionen
Poliomyelitis Zustand nach Poliomyelitis m 66 28 Jahre u 3(3) 0,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
virale Meningitis; Immundewizienz w 5
Monate 6 Tage krankheitsabhängig 2(2) 1,0
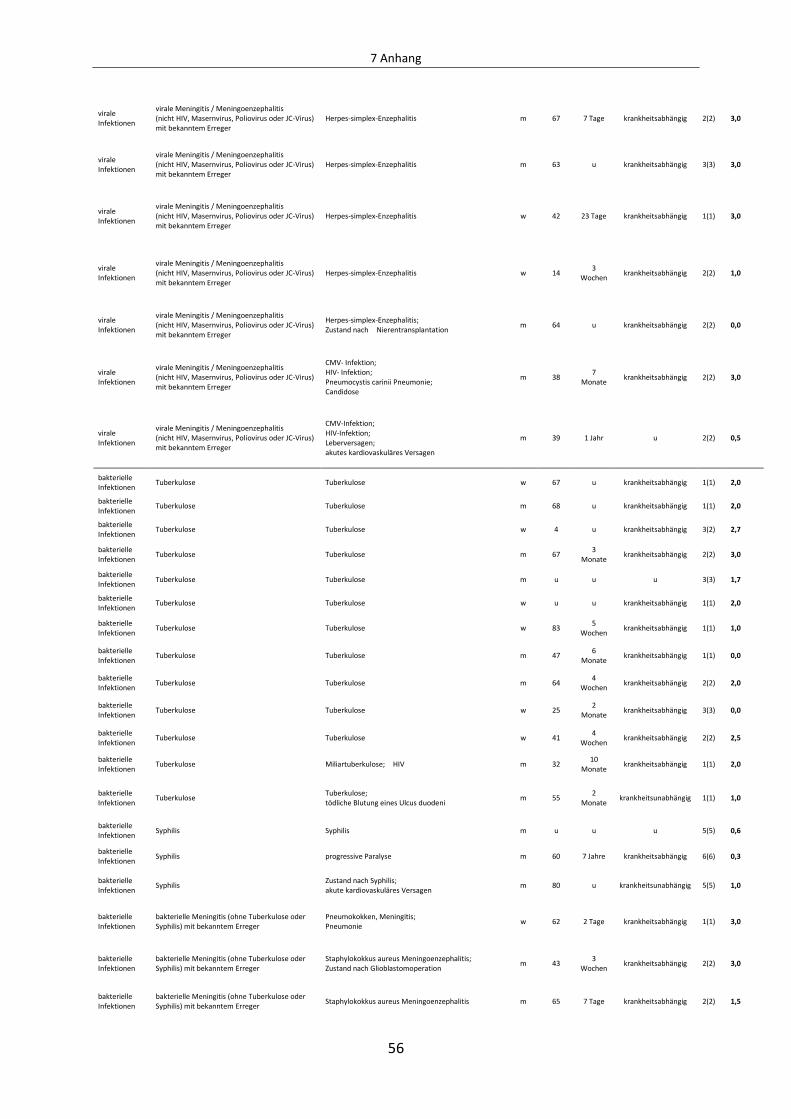
7 Anhang
56
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
Herpes-simplex-Enzephalitis m 67 7 Tage krankheitsabhängig 2(2) 3,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
Herpes-simplex-Enzephalitis m 63 u krankheitsabhängig 3(3) 3,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
Herpes-simplex-Enzephalitis w 42 23 Tage krankheitsabhängig 1(1) 3,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
Herpes-simplex-Enzephalitis w 14 3
Wochen krankheitsabhängig 2(2) 1,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
Herpes-simplex-Enzephalitis; Zustand nach Nierentransplantation
m 64 u krankheitsabhängig 2(2) 0,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
CMV- Infektion; HIV- Infektion; Pneumocystis carinii Pneumonie; Candidose
m 38 7
Monate krankheitsabhängig 2(2) 3,0
virale Infektionen
virale Meningitis / Meningoenzephalitis (nicht HIV, Masernvirus, Poliovirus oder JC-Virus) mit bekanntem Erreger
CMV-Infektion; HIV-Infektion; Leberversagen; akutes kardiovaskuläres Versagen
m 39 1 Jahr u 2(2) 0,5
bakterielle Infektionen
Tuberkulose Tuberkulose w 67 u krankheitsabhängig 1(1) 2,0
bakterielle Infektionen
Tuberkulose Tuberkulose m 68 u krankheitsabhängig 1(1) 2,0
bakterielle Infektionen
Tuberkulose Tuberkulose w 4 u krankheitsabhängig 3(2) 2,7
bakterielle Infektionen
Tuberkulose Tuberkulose m 67 3
Monate krankheitsabhängig 2(2) 3,0
bakterielle Infektionen
Tuberkulose Tuberkulose m u u u 3(3) 1,7
bakterielle Infektionen
Tuberkulose Tuberkulose w u u krankheitsabhängig 1(1) 2,0
bakterielle Infektionen
Tuberkulose Tuberkulose w 83 5
Wochen krankheitsabhängig 1(1) 1,0
bakterielle Infektionen
Tuberkulose Tuberkulose m 47 6
Monate krankheitsabhängig 1(1) 0,0
bakterielle Infektionen
Tuberkulose Tuberkulose m 64 4
Wochen krankheitsabhängig 2(2) 2,0
bakterielle Infektionen
Tuberkulose Tuberkulose w 25 2
Monate krankheitsabhängig 3(3) 0,0
bakterielle Infektionen
Tuberkulose Tuberkulose w 41 4
Wochen krankheitsabhängig 2(2) 2,5
bakterielle Infektionen
Tuberkulose Miliartuberkulose; HIV m 32 10
Monate krankheitsabhängig 1(1) 2,0
bakterielle Infektionen
Tuberkulose Tuberkulose; tödliche Blutung eines Ulcus duodeni
m 55 2
Monate krankheitsunabhängig 1(1) 1,0
bakterielle Infektionen
Syphilis Syphilis m u u u 5(5) 0,6
bakterielle Infektionen
Syphilis progressive Paralyse m 60 7 Jahre krankheitsabhängig 6(6) 0,3
bakterielle Infektionen
Syphilis Zustand nach Syphilis; akute kardiovaskuläres Versagen
m 80 u krankheitsunabhängig 5(5) 1,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Pneumokokken, Meningitis; Pneumonie
w 62 2 Tage krankheitsabhängig 1(1) 3,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Staphylokokkus aureus Meningoenzephalitis; Zustand nach Glioblastomoperation
m 43 3
Wochen krankheitsabhängig 2(2) 3,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Staphylokokkus aureus Meningoenzephalitis m 65 7 Tage krankheitsabhängig 2(2) 1,5

7 Anhang
57
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
akute Haemophilus influenzae Meningitis m 2 2 Tage krankheitsabhängig 3(3) 3,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Staphylokokkus Meningoenzephalitis w 75 8 Tage krankheitsabhängig 1(1) 1,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Klebsiellen, Pneumonie, Meningitis m 81 u krankheitsabhängig 1(1) 2,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Meningitis, Escherichia coli m 23 Tage u krankheitsabhängig 2(2) 3,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
Pseudomonas aeruginosa Meningoenzephalitis; Pneumonie nach Nierentransplantation
m 25 u krankheitsabhängig 2(2) 2,0
bakterielle Infektionen
bakterielle Meningitis (ohne Tuberkulose oder Syphilis) mit bekanntem Erreger
basale Meningitis nach Abszessdrainage des Gehirns m 44 6
Wochen krankheitsabhängig 3(3) 2,3
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
bakterielle Meningitis mit Beweis von Enterokokken und Klebsiellen, Pneumonie
m 53 u krankheitsabhängig 1(1) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis /
Meningoenzephalitis Temporale Meningoenzephalitis m 25 20 Tage krankheitsabhängig 2(2) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
akute virale Meningitis; HIV-Infektion; Heroinsucht
m 33 u u 1(1) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
lymphozytäre Meningoenzephalitis; Strangulation
w 75 u krankheitsunabhängig 1(1) 0,0
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
lymphozytäre Meningoenzephalitis w 20 3
Wochen krankheitsabhängig 2(2) 1,0
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
lymphozytäre Meningoenzephalitis; Gastroenteritis
m 3
Monate 2 Tage krankheitsabhängig 2(2) 0,5
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
lymphozytäre Meningoenzephalitis; akute respiratorischese Erkrankung
m 1 Jahr u u 1(1) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute lymphozytäre Meningitis / Meningoenzephalitis
virale Meningitis w 53 6 Tage krankheitsabhängig 4(4) 0,8
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäreMeningitis / Meningoenzephalitis
Staphylokokkus epidermidis Meningoenzephalitis w 78 1
Woche krankheitsabhängig 2(2) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäre Meningitis / Meningoenzephalitis
Haemophilus influencae Meningoenzephalitis; Otitis media
w 2
Monate 3 Tage krankheitsabhängig 2(2) 3,0
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäreMeningitis / Meningoenzephalitis
bakterielle Meningitis; Nierenversagen; Gallenblasenkarzinom
w 69 1 Tag krankheitsabhängig 1(1) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäreMeningitis / Meningoenzephalitis
akute bakterielle Meningitis w 20 2 Tage krankheitsabhängig 2(2) 2,0
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäreMeningitis / Meningoenzephalitis
bakterielle Meningitis; bronchial Karzinom; respiratorisches Versagen
w 47 u krankheitsunabhängig 2(2) 2,0

7 Anhang
58
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäreMeningitis / Meningoenzephalitis
Streptokokken Meningoenzephalitis m 5 Tage 2 Tage krankheitsabhängig 1(1) 3,0
Entzündung/ Infektion ohne bekannten Erreger
akute granulozytäre Meningitis / Meningoenzephalitis
bakterielle Meningitis; bakterielle Hypophysitis; rezidivierende Otitis media
w 37 u krankheitsabhängig 2(2) 0,5
Entzündung/ Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der
Meningen oder des Hirnparenchyms
chronische meningeale Infiltration concomitant to a brain abscess; renal Versagen; Pneumonie
m 69 u krankheitsunabhängig 2(2) 3,0
Entzündung/ Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der
Meningen oder des Hirnparenchyms
chronische meningeale Infiltration; Nierenversagen; Pancreatitis
m 41 u krankheitsunabhängig 2(2) 2,0
Entzündung/ Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der
Meningen oder des Hirnparenchyms
opportunistische Enzephalitis; abdominales Aortenaneurysma; Lungenenmbolie; akutes kardiovaskuläres Versagen
m 77 u u 4(4) 0,7
Entzündung/ Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der
Meningen oder des Hirnparenchyms
Zustand nach viraler Meningitis; Nierenversagen; Herzversagen
m 77 u u 2(2) 0,5
Entzündung/ Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der
Meningen oder des Hirnparenchyms
Hämorrhagische disseminierte chronische Meningoenzephalitis
m 47 u u 2(2) 0,0
Entzündung/ Infektion ohne bekannten Erreger
Unspezifische/nicht akute Entzündung der
Meningen oder des Hirnparenchyms
inflammatorischer ZNS-Prozess; Herzinfarkt
m 45 u krankheitsunabhängig 1(1) 2,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkankungen
Meningeosis lymphomtosa; malignes B-Zell Non-Hodgkin-Lymphom
m 67 6
Monate krankheitsabhängig 1(1) 3,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkankungen
EBV-assoziiertes B-Zell Non-Hodgkin-Lymphom w 71 u krankheitsabhängig 1(1) 0,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkankungen
Meningeosis lymphomtosa; malignes B-Zell-Non-Hodgkin-Lymphom
w 26 5
Monate krankheitsabhängig 2(2) 0,5
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkankungen
malignes B-Zell Non-Hodgkin-Lymphom; akute kardiovaskuläres Versagen
m 53 1 Jahre krankheitsabhängig 3(3) 0,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
malignes B-Zell Non-Hodgkin-Lymphom; Lungenödem; Nierenversagen
w 50 11
Monate krankheitsabhängig 3(3) 0,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
malignes B-Zell Non-Hodgkin-Lymphom w 57 u krankheitsabhängig 3(3) 0,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
malignes B-Zell Non-Hodgkin-Lymphom w 77 1 Jahre krankheitsabhängig 1(1) 1,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
malignes B-Zell Non-Hodgkin-Lymphom; Pneumonie; septischer Schock
m 39 u krankheitsabhängig 1(1) 0,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
follikuläres Lymphom; Wieber
w 57 2 Jahre krankheitsunabhängig 3(3) 0,0
neoplastisch Non-Hodgkin-Lymphom, maligne lymphoproliferative Erkrankungen
T-Zell Non-Hodgkin-Lymphom; respiratorisches Versagen
m 69 4 Jahre krankheitsabhängig 2(2) 0,0
neoplastisch Hodgkin-Lymphom Hodgkin-Lymphom w 18 1 Jahr krankheitsabhängig 1(1) 0,0
neoplastisch Hodgkin-Lymphom Hodgkin-Lymphom w 34 u krankheitsabhängig 1(1) 0,0
neoplastisch Hodgkin-Lymphom Hodgkin-Lymphom m 71 2
Monate krankheitsabhängig 2(2) 3,0

7 Anhang
59
neoplastisch Plasmozytom Plasmozytom;
Herzversagen w 66 9 Jahre krankheitsunabhängig 2(2) 2,5
neoplastisch Plasmozytom Plasmozytom; Herzversagen
w 61 6
Monate krankheitsabhängig 2(2) 1,0
neoplastisch Plasmozytom Plasmozytom m 42 6
Monate krankheitsabhängig 1(1) 0,0
neoplastisch Leukämie meningeale Infiltration einer chronischen lymphozytären Leukämie
w 66 1 Jahre krankheitsabhängig 1(1) 1,0
neoplastisch Leukämie meningeale Infiltration Infiltration einer chronischen lymphozytären Leukämie; Pneumonie
w 72 1.5
Jahre krankheitsunabhängig 1(1) 1,0
neoplastisch Leukämie akute lymphozytäre Leukämie; akutes kardiovaskuläres Versagen
w 69 20 Tage krankheitsabhängig 1(1) 0,0
neoplastisch Leukämie meningeale Infiltration bei chronischer lymphozytärer Leukämie; Pneumonie; akutes kardiovaskuläres Versagen
m 75 15
Monate krankheitsabhängig 2(2) 0,5
neoplastisch Leukämie akute myelotische Leukämie w 53 6
Monate krankheitsabhängig 2(2) 1,5
neoplastisch Leukämie akute undifferenzierte Leukämie; Meningeosis leucaemica
m 12 u krankheitsabhängig 1(1) 3,0
neoplastisch Leukämie meningeale Infiltration bei chronisch myelotischer Leukämie; Pneumonie
m 71 8
Wochen krankheitsabhängig 1(1) 1,0
neoplastisch Leukämie akute myelotische Leukämie m 4
Monate u krankheitsabhängig 1(1) 3,0
neoplastisch Meningeosis carcinomatosa Nierenzellkarzinom, Ösophaguskarzinom m 63 2 Jahre krankheitsabhängig 2(2) 2,0
neoplastisch Meningeosis carcinomatosa Pancoasttumor m 48 8
Monate krankheitsabhängig 1(1) 3,0
neoplastisch Meningeosis carcinomatosa Mammakarzinom w 42 u krankheitsabhängig 1(1) 1,0
neoplastisch Meningeosis carcinomatosa Maligner gemischter mesodermaler Tumor w 84 u krankheitsabhängig 1(1) 2,0
neoplastisch Meningeosis carcinomatosa Bronchialkarzinom m 43 3 Jahre krankheitsabhängig 1(1) 3,0
neoplastisch Meningeosis carcinomatosa Meningeosis carcinomatosa; Mammakarzinom
w 64 6
Monate krankheitsabhängig 2(2) 2,0
neoplastisch Meningeosis carcinomatosa Meningeosis carcinomatosa; Mammakarzinom
w 34 7
Monate krankheitsabhängig 3(3) 2,0
neoplastisch Meningeosis carcinomatosa Meningeosis carcinomatosa; esophageal cancer
m 60 18
Monate krankheitsabhängig 3(3) 1,7
neoplastisch Meningeosis carcinomatosa Siegelringzellkarzinom w 76 1
Monate krankheitsabhängig 2(2) 2,0
neoplastisch Meningeosis carcinomatosa Meningeosis carcinomatosa; Mammakarzinom
w 41 5 Jahre krankheitsabhängig 2(2) 1,0
neoplastisch Meningeosis carcinomatosa Meningeosis carcinomatosa; Ovarialkarzinom
w 74 9 Jahre krankheitsabhängig 2(2) 1,0
metabolische Erkankungen
zentrale pontine Myelinolyse zentrale pontine Myelinolyse; Sepsis w 65 u krankheitsunabhängig 1(1) 0,0
metabolische Erkankungen
zentrale pontine Myelinolyse zentrale pontine Myelinolyse; Sepsis; Multiorganversagen
m 55 u u 1(1) 0,0
metabolische Erkankungen
zentrale pontine Myelinolyse zentrale pontine Myelinolyse; alkoholinduzierte Zirrhose
m 31 u u 3(3) 0,0

7 Anhang
60
metabolische Erkankungen
zentrale pontine Myelinolyse zentrale pontine Myelinolyse; akutes kardiovaskuläres Versagen
m 31 u u 2(2) 0,0
metabolische Erkankungen
zentrale pontine Myelinolyse zentrale pontine Myelinolyse; bakterielle Meningitis
w 58 13 Tage krankheitsabhängig 3(3) 2,3
metabolische Erkankungen
zentrale pontine Myelinolyse Alkoholismus; Elektrolytinbalance m 39 1 Monat krankheitsabhängig 4(4) 0,3
1 meningeale Infiltration als Mittelwert aller untersuchten Gewebeblöcke des jeweiligen Autopsiefalls 0= keine Infiltration 1= wenig meningeale Infiltration (weniger als 50 infiltrierende Zellen pro Gesichtsfeld (HPW) = 400-wacher Vergrößerung) 2= moderate intrathekale Zellinfiltration (mehr als 50 und weniger als 200 infiltrierenden Zellen pro Gesichtsfeld)
3= dichte Infiltration (mehr als 200 infiltrierenden Zellen pro Gesichtsfeld)

8 Literaturverzeichnis
61
8 Literaturverzeichnis
Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Brück W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H (2003): Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62, 25-33
Adams RD, Kubik CS (1952): The morbid anatomy of the demyelinative disease. Am J Med 12, 510-46
Akalin H, Akdis AC, Mistik R, Helvaci S, Kilicturgay K (1994): Cerebrospinal fluid interleukin-1 beta/interleukin-1 receptor antagonist balance and tumor necrosis factor-alpha concentrations in tuberculous, viral and acute bacterial meningitis. Scand J Infect Dis 26, 667-74
Albert M, Antel J, Brück W, Stadelmann C (2007): Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol 17, 129-38
Alderson L, Fetell MR, Sisti M, Hochberg F, Cohen M, Louis DN (1996): Sentinel lesions of primary CNS lymphoma. J Neurol Neurosurg Psychiatry 60, 102-5
Alvarez-Lafuente R, De lH, V, Bartolome M, Picazo JJ, Arroyo R (2004): Relapsing-remitting multiple sclerosis and human herpesvirus 6 active infection. Arch Neurol 61, 1523-7
Amato MP, Bartolozzi ML, Zipoli V, Portaccio E, Mortilla M, Guidi L, Siracusa G, Sorbi S, Federico A, De Stefano N (2004): Neocortical volume decrease in relapsing-remitting MS patients with mild cognitive impairment. Neurology 63, 89-93
Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, Raine CS, Brosnan CF, John GR (2006): IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol 177, 5574-84
Ata AK, Funa K, Olsson Y (1997): Expression of various TGF-beta isoforms and type I receptor in necrotizing human brain lesions. Acta Neuropathol 93, 326-33
Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S (2000): Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 192, 393-404
Bagashev A, Sawaya BE (2013): Roles and functions of HIV-1 Tat protein in the CNS: an overview. Virol J 10, 358 doi: 10.1186/1743-422X-10-358
Banerjee A, Zhang X, Manda KR, Banks WA, Ercal N (2010): HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: potential role of the thiol antioxidant N-acetylcysteine amide. Free Radi Biol Med 48, 1388-98

8 Literaturverzeichnis
62
Barcellos LF, Oksenberg JR, Begovich AB, Martin ER, Schmidt S, Vittinghoff E, Goodin DS, Pelletier D, Lincoln RR, Bucher P (2003): HLA-DR2 dose effect on susceptibility to multiple sclerosis and influence on disease course. Am J Hum Genet 72, 710-6
Berger T, Reindl M (2007): Multiple sclerosis: disease biomarkers as indicated by pathophysiology. J Neurol Sci 259, 21-6
Bigner SH (1992): Cerebrospinal fluid (CSF) cytology: current status and diagnostic applications. J Neuropathol Exp Neurol 51, 235-45
Bitsch A, Kuhlmann T, Da CC, Bunkowski S, Polak T, Brück W (2000a): Tumour necrosis factor alpha mRNA expression in early multiple sclerosis lesions: correlation with demyelinating activity and oligodendrocyte pathology. Glia 29, 366-75
Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W (2000b): Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 123, 1174-83
Bo L, Vedeler CA, Nyland H, Trapp BD, Mork SJ (2003a): Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler 9, 323-31
Bo L, Vedeler CA, Nyland HI, Trapp BD, Mork SJ (2003b): Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol 62, 723-32
Bradl M, Lassmann H (2010): Oligodendrocytes: biology and pathology. Acta Neuropathol 119, 37-53
Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A (2010): Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 6, 667-79
Brightbill TC, Ihmeidan IH, Post MJ, Berger JR, Katz DA (1995): Neurosyphilis in HIV-positive and HIV-negative patients: neuroimaging findings. Am J Neuroradiol 16, 703-11
Brinar VV, Habek M (2006): Dementia and white-matter demyelination in young patient with neurosyphilis. Lancet 368, 2258
Brück W, Popescu B, Lucchinetti CF, Markovic-Plese S, Gold R, Thal DR, Metz I (2012): Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol 72, 385-94
Calabrese M, Favaretto A, Martini V, Gallo P (2013): Grey matter lesions in MS: from histology to clinical implications. Prion 7, 20-7
Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, Nicholas R et al (2012): Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 135, 2925-37
Compston A, Coles A (2008): Multiple sclerosis. Lancet 372, 1502-17
Confavreux C, Vukusic S, Moreau T, Adeleine P (2000): Relapses and progression of disability in multiple sclerosis. N Engl J Med 343, 1430-8

8 Literaturverzeichnis
63
Corral I, Quereda C, Garcia-Villanueva M, Casado JL, Perez-Elias MJ, Navas E, Ariza A et al (2004): Focal monophasic demyelinating leukoencephalopathy in advanced HIV infection. Eur Neurol 52, 36-41
Dal Canto MC, Melvold RW, Kim BS, Miller SD (1995): Two models of multiple sclerosis: experimental allergic encephalomyelitis (EAE) and Theiler's murine encephalomyelitis virus (TMEV) infection. A pathological and immunological comparison. Microsc Res Tech 32, 215-29
Dastur DK, Manghani DK, Udani PM (1995): Pathology and pathogenetic mechanisms in neurotuberculosis. Radiol Clin North Am 33, 733-52
Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, McDonald WI, Miller DH (1995): Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain 118, 1583-92
Davie CA, Barker GJ, Thompson AJ, Tofts PS, McDonald WI, Miller DH (1997): 1H magnetic resonance spectroscopy of chronic cerebral white matter lesions and normal appearing white matter in multiple sclerosis. J Neurol Neurosurg Psychiatry 63, 736-42
De Groot CJ, Ruuls SR, Theeuwes JW, Dijkstra CD, van der Valk P (1997): Immunocytochemical characterization of the expression of inducible and constitutive isoforms of nitric oxide synthase in demyelinating multiple sclerosis lesions. J Neuropathol Exp Neurol 56, 10-20
De Groot CJ, Montagne L, Barten AD, Sminia P, van der Valk P (1999): Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J Neuropathol Exp Neurol 58, 174-87
De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, Antel JP, Arnold DL (1998): Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain 121, 1469-77
Deckert M: Infektionen des ZNS. In: Klöppel G, Kreipe H, Remmele W, Paulus W, Schröder JM (Hrsg.): Pathologie: Neuropathologie. 3. Auflage; Springer, Berlin 2012, 303-330
Deluca HF, Cantorna MT (2001): Vitamin D: its role and uses in immunology. FASEB J 15, 2579-85
Dorries K (1998): Molecular biology and pathogenesis of human polyomavirus infections. Dev Biol Stand 94, 71-9
Drayton DL, Liao S, Mounzer RH, Ruddle NH (2006): Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol 7, 344-53
Epstein LG, Gendelman HE (1993): Human immunodeficiency virus type 1 infection of the nervous system: pathogenetic mechanisms. Ann Neurol 33, 429-36

8 Literaturverzeichnis
64
Fadil H, Gonzalez-Toledo E, Kelley BJ, Kelley RE (2006): Neuroimaging findings in neurosyphilis. J Neuroimaging 16, 286-9
Fauci AS (1996): Host factors and the pathogenesis of HIV-induced disease. Nature 384, 529-34
Feiden W: Zytologie des Liquor cerebrospinalis. In: Klöppel G, Kreipe H, Remmele W, Paulus W, Schröder JM (Hrsg.): Pathologie: Neuropathologie. 3. Auflage; Springer, Berlin 2012, 29-42
Feiden W, Milutinovic S (2002): Primary CNS lymphomas. Morphology and diagnosis. Pathologe 23, 284-91
Fischer MT, Wimmer I, Hoftberger R, Gerlach S, Haider L, Zrzavy T, Hametner S, Mahad D, Binder CJ, Krumbholz M (2013): Disease-specific molecular events in cortical multiple sclerosis lesions. Brain 136, 1799-815
Gardner C, Magliozzi R, Durrenberger PF, Howell OW, Rundle J, Reynolds R (2013): Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain 136, 3596-608
Garg RK (2003): Acute disseminated encephalomyelitis. Postgrad Med J 79, 11-7
Garg RK (2008): Subacute sclerosing panencephalitis. J Neurol 255, 1861-71
Gendelman HE, Pezeshkpour GH, Pressman NJ, Wolinsky JS, Quarles RH, Dobersen MJ, Trapp BD, Kitt CA, Aksamit A, Johnson RT (1985): A quantitation of myelin-associated glycoprotein and myelin basic protein loss in different demyelinating diseases. Ann Neurol 18, 324-8
Gendelman HE, Persidsky Y, Ghorpade A, Limoges J, Stins M, Fiala M, Morrisett R (1997): The neuropathogenesis of the AIDS dementia complex. AIDS 11, Suppl A: 35-45
Gideon HP, Flynn JL (2011): Latent tuberculosis: what the host "sees"? Immunol Res 50, 202-12
Gray F, Chretien F, Vallat-Decouvelaere AV, Scaravilli F (2003): The changing pattern of HIV neuropathology in the HAART era. J Neuropathol Exp Neurol 62, 429-40
Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M (2009): Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res 1283, 127-38
Gudi V, Skuljec J, Yildiz O, Frichert K, Skripuletz T, Moharregh-Khiabani D, Voss E, Wissel K, Wolter S, Stangel M (2011): Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS One 6, e22623
Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ (2007): Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357, 851-62

8 Literaturverzeichnis
65
Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, Esterbauer H et al (2011): Oxidative damage in multiple sclerosis lesions. Brain 134, 1914-24
Harauz G, Ishiyama N, Hill CM, Bates IR, Libich DS, Fares C (2004): Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron 35, 503-42
Hart MN, Earle KM (1975): Haemorrhagic and perivenous encephalitis: a clinical-pathological review of 38 cases. J Neurol Neurosurg Psychiatry 38, 585-91
Hein T, Hopfenmüller W (2000): Projection of the number of multiple sclerosis patients in Germany. Nervenarzt 71, 288-94
Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK (1998): Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol 92, 38-49
Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, Serafini B et al (2011): Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 134, 2755-71
Hu S, Sheng WS, Schachtele SJ, Lokensgard JR (2011): Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J Neuroinflammation 8, 123 doi: 10.1186/1742-2094-8-123
Husseini L, Saleh A, Reifenberger G, Hartung HP, Kieseier BC (2012): Inflammatory demyelinating brain lesions heralding primary CNS lymphoma. Can J Neurol Sci 39, 6-10
International Multiple Sclerosis Genetics Consortium (IMSGC) (2008): Refining genetic associations in multiple sclerosis. Lancet Neurol 7, 567-9
John GR, Lee SC, Brosnan CF (2003): Cytokines: powerful regulators of glial cell activation. Neuroscientist. 9, 10-22
Johnson MD, Gold LI (1996): Distribution of transforming growth factor-beta isoforms in human immunodeficiency virus-1 encephalitis. Hum Pathol 27, 643-9
Kastenbauer S, Koedel U, Becker BF, Pfister HW (2002): Oxidative stress in bacterial meningitis in humans. Neurology 58, 186-91
Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R et al (1999): Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 189, 865-70
Kinnier Wilson SA: Neurology. 2 Bände. Hrsg. v. Bruce AN. Butterworth, London 1954
Kooi EJ, Geurts JJ, van HJ, Bo L, van d, V (2009): Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol 68, 1021-8

8 Literaturverzeichnis
66
Kroner-Milsch A, Kleinschnitz C, Stadelmann-Nessler C, Raab P, Stangel M, Trebst C, Voß E, Cordes AL: Multiple Sklerose und andere autoimmune ZNS-Erkrankungen. In: Stangel M. Mäurer M (Hrsg.): Autoimmunerkrankungen in der Neurologie: Diagnostik und Therapie. Springer, Berlin 2012, 1-100
Kuhlmann T: Multiple Sclerose und verwandte Erkrankungen. In: Klöppel G, Kreipe H, Remmele W, Paulus W, Schröder JM (Hrsg.): Pathologie: Neuropathologie. 3. Auflage; Springer, Berlin 2012, 353-364
Kuhlmann T, Schroter A, Dechent P, Weber F, Rustenbeck HH, Fuzesi L, Brück W, Ehrenreich H, Frahm J (2001): Diagnosis of a multifocal B cell lymphoma with preceding demyelinating central nervous system lesions by single voxel proton MR spectroscopy. J Neurol Neurosurg Psychiatry 70, 259-62
Kutzelnigg A, Lassmann H (2005): Cortical lesions and brain atrophy in MS. J Neurol Sci 233, 55-9
Kutzelnigg A, Lassmann H (2006): Cortical demyelination in multiple sclerosis: a substrate for cognitive deficits? J Neurol Sci 245, 123-6
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H (2005): Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128, 2705-12
Langford TD, Letendre SL, Marcotte TD, Ellis RJ, McCutchan JA, Grant I, Mallory ME et al (2002): Severe, demyelinating leukoencephalopathy in AIDS patients on antiretroviral therapy. AIDS 16, 1019-29
Langford TD, Letendre SL, Larrea GJ, Masliah E (2003): Changing patterns in the neuropathogenesis of HIV during the HAART era. Brain Pathol 13, 195-210
Lassmann H (2001): Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr Opin Neurol 14, 253-8
Lassmann H, Raine CS, Antel J, Prineas JW (1998): Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol 86, 213-7
Lassmann H, van HJ, Mahad D (2012): Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8, 647-56
Laureno R, Karp BI (1997): Myelinolysis after correction of hyponatremia. Ann Intern Med 126, 57-62
Leib SL, Kim YS, Chow LL, Sheldon RA, Tauber MG (1996): Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest 98, 2632-9
Lemke G: Myelin and Myelination. In: Hall ZW (Hrsg.): An introduction to molecular neurobiology. Sinauer Associated Inc., Sunderland 1992, 281-312

8 Literaturverzeichnis
67
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I et al (2004): A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364, 2106-12
Linker RA, Lee DH, Demir S, Wiese S, Kruse N, Siglienti I, Gerhardt E et al (2010): Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: therapeutic implications in a model of multiple sclerosis. Brain 133, 2248-63
Liu JS, Zhao ML, Brosnan CF, Lee SC (2001): Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol 158, 2057-66
Losseff NA, Webb SL, O'Riordan JI, Page R, Wang L, Barker GJ, Tofts PS et al (1996): Spinal cord atrophy and disability in multiple sclerosis. A new reproducible and sensitive MRI method with potential to monitor disease progression. Brain 119, 701-8
Love S (2006): Demyelinating diseases. J Clin Pathol 59, 1151-9
Lowis GW (1988): Ethnic factors in multiple sclerosis: a review and critique of the epidemiological literature. Int J Epidemiol 17, 14-20
Lublin FD, Reingold SC (1996): Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46, 907-11
Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000): Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47, 707-17
Lucchinetti CF, Popescu BFG, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Brück W et al (2011): Inflammatory Cortical Demyelination in Early Multiple Sclerosis. N Engl J Med 365, 2188-97
Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, Reynolds R, Aloisi F (2007): Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 130, 1089-104
Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, Aloisi F, Reynolds R (2010): A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 68, 477-93
Mandler RN, Davis LE, Jeffery DR, Kornfeld M (1993): Devic's neuromyelitis optica: a clinicopathological study of 8 patients. Ann Neurol 34, 162-8
Marrie RA (2004): Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol 3, 709-18
Martin RJ (2004): Central pontine and extrapontine myelinolysis: the osmotic demyelination syndromes. J Neurol Neurosurg Psychiatry 75 Suppl 3 iii22-8

8 Literaturverzeichnis
68
McDonald WI, Sears TA (1970): The effects of experimental demyelination on conduction in the central nervous system. Brain 93, 583-98
McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, McFarland HF, Paty DW, Polman CH, Reingold SC (2001): Recommended diagnostic criteria for multile sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol 50, 121-7
Meinl E, Krumbholz M, Hohlfeld R (2006): B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol 59, 880
Meinl E, Derfuss T, Krumbholz M, Probstel AK, Hohlfeld R (2011): Humoral autoimmunity in multiple sclerosis. J Neurol Sci 306, 180-2
Merkler D, Ernsting T, Kerschensteiner M, Brück W, Stadelmann C (2006): A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain 129, 1972-83
Miklossy J (2008): Biology and neuropathology of dementia in syphilis and Lyme disease. Handb Clin Neurol 89, 825-44 doi: 10.1016/S0072-9752(07)01272-9
Mocroft A, Ledergerber B, Katlama C, Kirk O, Reiss P, d'Arminio MA, Knysz B, Dietrich M, Phillips AN, Lundgren JD (2003): Decline in the AIDS and death rates in the EuroSIDA study: an observational study. Lancet 362, 22-9
Moll NM, Rietsch AM, Ransohoff AJ, Cossoy MB, Huang D, Eichler FS, Trapp BD et al (2008): Cortical demyelination in PML and MS: Similarities and differences. Neurology 70, 336-43
Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A (2006): Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 296, 2832-8
Nathan C, Xie QW (1994): Regulation of biosynthesis of nitric oxide. J Biol Chem 269, 13725-8
Navikas V, Link H (1996): Review: cytokines and the pathogenesis of multiple sclerosis. J Neurosci Res 45, 322-33
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000): Multiple sclerosis. N Engl J Med 343, 938-52
Oehmichen M: Intoxikation. In: Klöppel G, Kreipe H, Remmele W, Paulus W, Schröder JM (Hrsg.): Pathologie: Neuropathologie. 3. Auflage; Springer, Berlin 2012, 365-402
Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL (2008): The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet 9, 516-26
Oleszak EL, Zaczynska E, Bhattacharjee M, Butunoi C, Legido A, Katsetos CD (1998): Inducible nitric oxide synthase and nitrotyrosine are found in monocytes/macrophages and/or astrocytes in acute, but not in chronic, multiple sclerosis. Clin Diagn Lab Immunol 5, 438-45

8 Literaturverzeichnis
69
Paulus W, Hasselblatt M: Tumoren. In: Klöppel G, Kreipe H, Remmele W, Paulus W, Schröder JM (Hrsg.): Pathologie: Neuropathologie. 3. Auflage; Springer, Berlin 2012, 481-552
Peterson JW, Bo L, Mork S, Chang A, Trapp BD (2001): Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50, 389-400
Pham H, Ramp AA, Klonis N, Ng SW, Klopstein A, Ayers MM, Orian JM (2009): The astrocytic response in early experimental autoimmune encephalomyelitis occurs across both the grey and white matter compartments. J Neuroimmunol 208, 30-9
Pilleri G, Lechi A, Carreras M (1974): Symmetric syphilitic gummas of the frontal lobes. Archiv für Psychiatrie und Nervenkrankheiten 219, 207-21
Popescu BF, Parisi JE, Cabrera-Gomez JA, Newell K, Mandler RN, Pittock SJ, Lennon VA, Weinshenker BG, Lucchinetti CF (2010): Absence of cortical demyelination in neuromyelitis optica. Neurology 75, 2103-9
Prange H : Neurosyphilis. VCH Verlagsgesellschaft, Weinheim 1987
Rinaldi F, Calabrese M, Grossi P, Puthenparampil M, Perini P, Gallo P (2010): Cortical lesions and cognitive impairment in multiple sclerosis. Neurol Sci 31, 235-7
Rocca MA, Agosta F, Mezzapesa DM, Martinelli V, Salvi F, Ghezzi A, Bergamaschi R, Comi G, Filippi M (2004): Magnetization transfer and diffusion tensor MRI show gray matter damage in neuromyelitis optica. Neurology 62, 476-8
Rosati G (2001): The prevalence of multiple sclerosis in the world: an update. Neurol Sci 22, 117-39
Rubenstein JL, Wong VS, Kadoch C, Gao HX, Barajas R, Chen L, Josephson SA et al (2013): CXCL13 plus interleukin 10 is highly specific for the diagnosis of CNS lymphoma. Blood 121, 4740-8
Ruijs TC, Freedman MS, Grenier YG, Olivier A, Antel JP (1990): Human oligodendrocytes are susceptible to cytolysis by major histocompatibility complex class I-restricted lymphocytes. J Neuroimmunol 27, 89-97
Sadahira Y, Wada H, Nakamura E, Terayama K, Sugihara T, Yamada O, Mikami Y et al (2000): Nasal NK/T cell lymphoma presenting as transverse myelopathy. Virchows Arch 436, 393-7
Sadovnick AD, Dircks A, Ebers GC (1999): Genetic counselling in multiple sclerosis: risks to sibs and children of affected individuals. Clin Genet 56, 118-22
Saji E, Arakawa M, Yanagawa K, Toyoshima Y, Yokoseki A, Okamoto K, Otsuki M et al (2013): Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol 73, 65-76

8 Literaturverzeichnis
70
Sakai H, Okafuji I, Nishikomori R, Abe J, Izawa K, Kambe N, Yasumi T et al (2012): The CD40-CD40L axis and IFN-gamma play critical roles in Langhans giant cell formation. Int Immunol 24, 5-15
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A et al (2011): Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214-9
Schachtele SJ, Hu S, Little MR, Lokensgard JR (2010): Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J Neuroinflammation 7, 35
Schonberger K, Ludwig MS, Wildner M, Weissbrich B (2013): Epidemiology of subacute sclerosing panencephalitis (SSPE) in Germany from 2003 to 2009: a risk estimation. PLoS One 8, e68909
Schuchat A, Robinson K, Wenger JD, Harrison LH, Farley M, Reingold AL, Lefkowitz L, Perkins BA (1997): Bacterial meningitis in the United States in 1995. Active Surveillance Team. N Engl J Med 337, 970-6
Schwab C, McGeer PL (2002): Complement activated C4d immunoreactive oligodendrocytes delineate small cortical plaques in multiple sclerosis. Exp Neurol 174, 81-8
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F (2004): Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14, 164-74
Skripuletz T, Gudi V, Hackstette D, Stangel M (2011): De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected. Histol Histopathol 26, 1585-97
Smolders J, Menheere P, Kessels A, Damoiseaux J, Hupperts R (2008): Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult Scler 14, 1220-4
Solnyshkova TG, Shakhlamov VA (2002): Ultrastructural and morphometric characteristics of nerve cells and myelinated fibers in the cerebral cortex after chronic exposure to natural gas containing hydrogen sulfide in low concentrations. Bull Exp Biol Med 134, 411-3
Stadelmann C: Pathologie und Immunpathogenese der Multiplen Sklerose In: Berger T, Linnebank M, Wiendl H (Hrsg.): Betaferon®: 25 Jahre Multiple Sklerose Forschung. Springer, Wien 2013, 13-26
Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H (2002): BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 125, 75-85
Stadelmann C, Albert M, Wegner C, Brück W (2008): Cortical pathology in multiple sclerosis. Curr Opin Neurol 21, 229-34

8 Literaturverzeichnis
71
Storch MK, Bauer J, Linington C, Olsson T, Weissert R, Lassmann H (2006): Cortical demyelination can be modeled in specific rat models of autoimmune encephalomyelitis and is major histocompatibility complex (MHC) haplotype-related. J Neuropathol Exp Neurol 65, 1137-42
Storch MK, Weissert R, Steffer A, Birnbacher R, Wallstrom E, Dahlman I, Ostensson CG, Linington C, Olsson T, Lassmann H (2002): MHC gene related effects on microglia and macrophages in experimental autoimmune encephalomyelitis determine the extent of axonal injury. Brain Pathol 12, 287-99
Tallantyre EC, Morgan PS, Dixon JE, Al-Radaideh A, Brookes MJ, Morris PG, Evangelou N (2010): 3 Tesla and 7 Tesla MRI of multiple sclerosis cortical lesions. J Magn Reson Imaging 32, 971-7
Tardif CL, Collins DL, Eskildsen SF, Richardson JB, Pike GB (2010): Segmentation of cortical MS lesions on MRI using automated laminar profile shape analysis. Med Image Comput Comput Assist Interv 13, 181-8
Tenembaum S, Chitnis T, Ness J, Hahn JS (2007): Acute disseminated encephalomyelitis. Neurology 68, 23-36
Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A (2003): HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem 84, 169-79
Torkildsen O, Stansberg C, Angelskar SM, Kooi EJ, Geurts JJ, van der Valk, Myhr KM, Steen VM, Bo L (2010): Upregulation of immunoglobulin-related genes in cortical sections from multiple sclerosis patients. Brain Pathol 20, 720-9
Truyen L, van Waesberghe JH, van Walderveen MA, van Oosten BW, Polman CH, Hommes OR, Ader HJ, Barkhof F (1996): Accumulation of hypointense lesions ("black holes") on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology 47, 1469-76
Valyi-Nagy T, Olson SJ, Valyi-Nagy K, Montine TJ, Dermody TS (2000): Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology 278, 309-21
van de Beek D, de GJ, Tunkel AR, Wijdicks EF (2006): Community-acquired bacterial meningitis in adults. N Engl J Med 354, 44-53
Wagner HJ, Munger KL, Ascherio A (2004): Plasma viral load of Epstein-Barr virus and risk of multiple sclerosis. Eur J Neurol 11, 833-4
Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM (2006): Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 67, 960-7
Weiner HL (2004): Multiple sclerosis is an inflammatory T-cell-mediated autoimmune disease. Arch Neurol 61, 1613-5

8 Literaturverzeichnis
72
Wilson J (1983): Cyanide in human disease: a review of clinical and laboratory evidence. Toxicol Sci 3, 397-9
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG (1999): The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 53, 1107-14
Yang CS, Lee HM, Lee JY, Kim JA, Lee SJ, Shin DM, Lee YH, Lee DS, El-Benna J, Jo EK (2007): Reactive oxygen species and p47phox activation are essential for the Mycobacterium tuberculosis-induced pro-inflammatory response in murine microglia. J Neuroinflammation 4, 27
Young NP, Weinshenker BG, Parisi JE, Scheithauer B, Giannini C, Roemer SF, Thomsen KM, Mandrekar JN, Erickson BJ, Lucchinetti CF (2010): Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain 133, 333-48

Danksagung
73
Danksagung
Ich möchte mich an dieser Stelle bei vielen Personen bedanken, die mich bei dieser
Dissertationsarbeit sehr unterstützt haben.
Im Besonderen bedanke ich mich bei Herrn Prof. Dr. med. Wolfgang Brück als meinen
Doktorvater für die Möglichkeit, meine Dissertation in der Abteilung der Neuropathologie zu
verfassen und für die Begutachtung dieser Arbeit.
Mein großer Dank gilt Dr. med. Andreas Junker als Betreuer meiner Dissertation. Er hat mich
während meiner ganzen Promotionsphase begleitet, betreut und stand mir mit Rat und Tat
zur Seite. Wir haben gemeinsam viel durchgestanden.
Weiterhin bedanke ich mich bei Frau Prof. Dr. Christine Stadelmann-Nessler für die vielen
Anregungen und die konstruktive Kritik, die zum guten Gelingen dieser Arbeit entscheidend
beigetragen haben.
Der MTA Uta Scheidt danke ich unendlich für die tatkräftige Unterstützung im Labor. Ohne
sie und ihre jahrelange Erfahrung hätte ich das nie schaffen können.
Zuletzt gilt mein besonderer Dank meiner Familie und Juuso Tiihonen für die endlose
Unterstützung und Motivation in allen Lebenslagen.

Lebenslauf
74
Lebenslauf
Ich, Jadwiga Wozniak, wurde am 30.12.1986 in Tarnowskie Gory (Polen) geboren und zog
1989 nach Deutschland.
Ich wuchs in Rottenburg am Neckar auf, wo ich das katholische freie Gymnasium St. Meinrad
besuchte, an dem ich im Jahr 2006 die allgemeine Hochschulreife erwarb.
Vor Beginn meines Studiums absolvierte ich ein Freiwilliges Soziales Jahr am
Universitätsklinikum Tübingen in der Kinderkardiologie und Kinderchirurgie.
Zum Sommersemester 2008 nahm ich das Studium der Humanmedizin an der Georg-August-
Universität zu Göttingen auf, welches ich im Herbst 2014 beendete.
Im Februar 2012 begann ich mit den histologischen Arbeiten zur vorliegenden
Dissertationsarbeit in der Arbeitsgruppe von Dr. Andreas Junker in der Abteilung
Neuropathologie der Medizinischen Fakultät der Universität Göttingen unter der Leitung von
Herrn Prof. Dr. med. Wolfgang Brück.
Seit April 2015 arbeite ich als Assistenzärztin in der Anästhesie und Intensivmedizin bei den
Vivantes Kliniken in Berlin.