klf4suppressestumorformationingeneticand pharmacological mouse models of...
TRANSCRIPT

Oncogenes and Tumor Suppressors
KLF4 Suppresses Tumor Formation inGenetic andPharmacological Mouse Models of ColonicTumorigenesisAmr M. Ghaleb1, Enas A. Elkarim1, Agnieszka B. Bialkowska1, and Vincent W.Yang1,2
Abstract
The zinc finger transcription factor Kr€uppel-like factor 4 (KLF4)is frequently downregulated in colorectal cancer. Previous studiesshowed that KLF4 is a tumor suppressor in the intestinal tract andplays an important role in DNA damage-repair mechanisms.Here, the in vivo effects of Klf4 deletion were examined from themouse intestinal epithelium (Klf4DIS) in a genetic or pharmaco-logical setting of colonic tumorigenesis: ApcMin/þ mutation orcarcinogen treatment with azoxymethane (AOM), respectively.Klf4DIS/ApcMin/þ mice developed significantly more colonic ade-nomas with 100% penetrance as compared with ApcMin/þ micewith intact Klf4 (Klf4fl/fl/ApcMin/þ). The colonic epithelium ofKlf4DIS/ApcMin/þ mice showed increased mTOR pathway activity,together with dysregulated epigenetic mechanism as indicatedby altered expression of HDAC1 and p300. Colonic adenomasfrom both genotypes stained positive for gH2AX, indicatingDNA double-strand breaks. In Klf4DIS/ApcMin/þ mice, this was
associated with reduced nonhomologous end joining (NHEJ)repair and homologous recombination repair (HRR) mechan-isms as indicated by reduced Ku70 and Rad51 staining, respec-tively. In a separatemodel, following treatmentwith AOM,Klf4DIS
mice developed significantly more colonic tumors than Klf4fl/fl
mice, with more Klf4DIS mice harboring K-Ras mutations thanKlf4fl/fl mice. Compared with AOM-treated Klf4fl/fl mice, adeno-mas of treated Klf4DIS mice had suppressed NHEJ and HRRmechanisms, as indicated by reduced Ku70 and Rad51 staining.This study highlights the important role of KLF4 in suppressingthe development of colonic neoplasia under different tumor-promoting conditions.
Implications: The study demonstrates that KLF4 plays a signifi-cant role in the pathogenesis of colorectal neoplasia.MolCancer Res;14(4); 385–96. �2016 AACR.
IntroductionColorectal cancer is a major cause of cancer mortality in the
United States. Several factors play a role in ultimately causingcolorectal cancer development. These include mutations, epi-genetic changes, and DNA damage. The majority of colorectalcancers contain mutations in the adenomatous polyposis coli(APC) tumor suppressor gene (1). APC is found in thenormal intestinal mucosa with an increasing gradient ofexpression in mature epithelial cells located in the uppercrypt region (2). In addition, APC antagonizes the pro-pro-liferative Wnt pathway by negatively regulating the steady-state level of intracellular b-catenin (3, 4). When APC isinactivated by mutation (which usually leads to a truncatedprotein), Wnt signaling is unimpeded, resulting in the nuclearaccumulation of b-catenin and subsequent activation ofdownstream target genes, such as cyclin D1 and c-Myc, thatpromote cell proliferation (5,6). In many cancer types,
including colorectal cancer, accumulation of DNA damagehas been linked to cancer, and genetic deficiencies in DNAdamage repair mechanisms are associated with susceptibilityto tumor development (7). Additionally, epigenetic changesthat lead to mutations or to silencing of DNA repair genesmay promote tumorigenesis (7).
The nuclear transcription factor Kr€uppel-like factor 4 (KLF4;also known as gut-enriched Kr€uppel-like factor or GKLF) ishighly expressed in the terminally differentiated, post-mitoticintestinal epithelial cells and is an inhibitor of cell proliferation(8,9). We have previously shown that in vitro, Klf4 is requiredfor the prevention of genomic instability (10). In the intestine,the KLF4 promoter has been shown to be regulated by APC in aCdx2-dependent manner, the latter being an intestine-specifictranscription factor that controls intestinal development (11).Conversely, KLF4 has been shown to regulate colonic cellgrowth by inhibiting b-catenin activity (12, 13). Accordingly,studies have demonstrated a potentially causal relationshipbetween KLF4 and several kinds of human cancers. For exam-ple, expression of KLF4 is often reduced in tumors of thegastrointestinal tract (14–16). In addition, loss of heterozygos-ity (LOH) and promoter hypermethylation have been identi-fied as possible reasons for the reduced expression of KLF4 in asubset of colorectal cancers (16). However, whether KLF4 playsa causal role in the in vivo development of colonic tumors hasnot been definitively established.
In the current study, we investigated the in vivo role of KLF4 incolonic tumorigenesis in two different systems: the setting ofApcMin mutation and the chemically induced DNA mutations.
1Department of Medicine, Stony Brook University, Stony Brook, NewYork. 2Department of Physiology and Biophysics, Stony Brook Uni-versity, Stony Brook, New York.
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
Corresponding Author: Vincent W. Yang, Stony Brook University School ofMedicine, HSC T-16, Room 020, Stony Brook, NY, 11794. Phone: 631-444-2066;Fax: 631-444-3144; E-mail: [email protected]
doi: 10.1158/1541-7786.MCR-15-0410
�2016 American Association for Cancer Research.
MolecularCancerResearch
www.aacrjournals.org 385
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

Materials and MethodsMice
All animal studieswere approvedby the StonyBrookUniversityInstitutional Animal Care and Use Committee (IACUC). MaleC57BL/6J ApcMin/þ founders were originally purchased from TheJackson Laboratory. Mice with floxed Klf4 gene (Klf4fl/fl) andintestine-specific villin-Cre–driven Klf4 deletion (Klf4DIS) werepreviously described (17). ApcMin/þmales were mated with eitherKlf4fl/fl or Klf4DIS females to obtain ApcMin/þ mice with intact Klf4allele (Klf4fl/fl/ApcMin/þ) or with intestine-specific Klf4 deletion(Klf4DIS/ApcMin/þ).
Azoxymethane (AOM) treatmentKlf4fl/fl and Klf4DIS mice were injected with AOM (10 mg/kg),
i.p., once a week for 4 consecutive weeks. Mice were euthanized at4 or 12 weeks after the last injection for tumor developmentassessment.
Tissue harvesting and tumor assessmentFor all groups, following euthanasia with CO2 asphyxiation
and cervical dislocation, the entire small intestine and colonwere dissected out and flushed with modified Bouin's fixative(50% ethanol/5% acetic acid) then cut open longitudinally.The intestines were examined under a dissecting microscope forthe presence of adenomas. The number and size of adenomasin both the small and large intestine were recorded as describedpreviously (18).
Tissue preparation and immunostainingFollowing macroscopic examination for tumor formation in
harvested tissue, the intestines were Swiss-rolled, fixed in bufferedformalin, embedded in paraffin, and 5-mm sections were cut forhistological hematoxylin and eosin (H&E) characterization, andfor immunostaining.
For immunostaining, sections were deparaffinized in xylene,rehydrated in ethanol, and then treatedwith 10mmol/LNa citratebuffer, pH 6.0, at 120�C for 10 minutes in a pressure cooker. Thehistological sections were incubated with blocking buffer [3%bovine serum albumin, and 0.01% Tween 20 in 1X Tris-bufferedPBS (TTBS)] for 1hour at 37�C. Primary antibodies goat anti-KLF4(1:300; R&D), rabbit anti-cyclin D1 (1:200; Biocare Medical),rabbit monoclonal anti-Ki67 (1:200; Biocare Medical), rabbitanti-gH2AX (1:400; Abcam), rabbit anti-Rad51 (1:200; Abcam),mouse anti-Ku70 (1:200; Abcam), mouse anti-b-catenin(1:1,000; BD), mouse anti-Cdx2 (1:200; LS Bio), rabbit anti-HDAC1 (1:200; Santa Cruz Biotechnology), rabbit anti-Pp44/42 MAPK (p-ERK; 1:200; Cell Signaling Technology), and mouseanti-p300 (1:200; Santa Cruz Biotechnology) were added at 4�Covernight. For IF, appropriate AlexaFluor-labeled secondary anti-bodies (Molecular Probes) were added at 1:500 dilution inblocking buffer for 30 minutes at 37�C, counterstained withHoechst 33258, mounted with Prolong gold (Molecular Probes),and cover slipped. IHC detection of cyclin D1, Ki67, and gH2AXwas done using goat anti-mouse or anti-rabbit HRP-labeled(Jackson Immuno Research) secondary antibody at 1:500 dilu-tion in blocking buffer for 30 minutes at 37�C, followed by washand then DAB color development. For Klf4 and p-ERK detectionby IHC, secondary unconjugated rabbit anti-goat antibody andgoat anti-rabbit antibody (Jackson ImmunoResearch)was added,respectively, at 1:500 dilution in blocking buffer for 30minutes at
37�C. After washing, goat anti-rabbit HRP and donkey anti-goatHRP–labeled tertiary antibodies (Jackson Immuno Research)were then added, respectively, at 1:500 dilution in blocking bufferfor 30 minutes at 37�C, followed by DAB color development.Detection of all other primary antibodies for IHC was carried outusing either Mach3 rabbit or Mach3 mouse HRP-polymer detec-tion as per manufacturer's recommendations (Biocare Medical).Color development in all IHC staining was followed by hema-toxylin counterstaining and mounting.
Anaphase bridging index (ABI) and mitotic indexThe ABI was determined as described before (19). A minimum
of 30 anaphases per mouse (4 per group) were scored from H&Esections. For Mitotic index, the number of cells undergoingmitosis per crypt (minimum of 60 per mouse) was scored fromH&E sections (3 mice per group).
Measurement of LOH of the Apcþ locusDNA was extracted from paraffin-embedded colon tissues of
Klf4fl/fl/ApcMin/þ andKlf4DIS/ApcMin/þmice, and LOHwas analyzedas described before (18,20). In brief, five 10-mm-thick sections permouse were cut and collected in a test tube. Tissue was thendeparaffinized in 1 mL xylenes for 30 minutes at room temper-ature. The xylenes was then discarded, and tissue was washedtwice in 100% ethanol followed by 2 times wash in 70% ethanoland 2 times PBS, then spun down and PBS discarded. DNA wasextracted frompelleted tissue using the REDExtract-N-Amp TissuePCR kit (Sigma) and purified using the QIAmp DNA Micro Kit(Qiagen). Equal amount of DNA was used for PCR amplificationof the Apc locus followed byHindIII digestion as described before(20). Twenty microliters of each HindIII digestion reaction waselectrophoresed through a 2.5% agarose gel. Bands were quan-tified using ImageJ software (21). Determination of LOH wascarried out as described before (18,20).
In vitro overexpression or suppression of KLF4The colon cancer cell line HCT116 was used. The cell line was
originally purchased from ATCC. Cells thawed from frozen stockwere used, and all experiments were carried out within 6 monthsof thawing. We routinely carried out morphology checks on allcell lines and we only passaged the cell lines for three months.In addition, the cell lines were tested by PCR for Mycoplasmacontamination. Furthermore, each experiment had appropriatecontrols to ensure the behavior of tested cell lines. For Klf4overexpression, plasmid containing pEGFP-Klf4 that expressesEGFP–Klf4 fusion protein was used to transfect the cells asdescribed before (10). For KLF4 suppression, KLF4-specific siRNA(Ambion) was used to transfect cells as permanufacturer's recom-mendations. For all transfection experiments, the cells wereharvested 24 hours after transfection for immunoblot analysis.
ImmunoblottingCells were lysed in complete Laemmli buffer and vortexed for 3
to 4 minutes for homogenization. Insoluble material wasremoved by centrifugation at 12,000 rpm for 5 minutes, and thesupernatant was collected and heated at 95-100�C for 10minutes,then cooled down to room temperature before loading for gelelectrophoresis. For mouse tissues (the entire length of the colon;normal epithelium and tumors), deparaffinization and hydrationwas done as described above. Reversal of crosslinking was then
Ghaleb et al.
Mol Cancer Res; 14(4) April 2016 Molecular Cancer Research386
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

carried out by adding 1mLof 10mmol/LNa citrate buffer, pH6.0,per sample, and heating at 95�C for 20minutes. Tissues were thenspun down, citrate buffer was discarded, washed twice in PBS,then spun down and PBS discarded. Pelleted tissue was then re-suspended in complete Laemmli buffer and heated at 95 to 100�Cfor 10minutes. Extracted protein from cells or tissuewere resolvedusing SDS-PAGE gel electrophoresis. For all protein samples,following protein transfer, the membranes were immunoblottedwith the following primary antibodies: rabbit anti-KLF4 (SantaCruz Biotechnology), rabbit anti-HDAC1 (Santa Cruz Biotech-nology), rabbit anti-p53 (Santa Cruz Biotechnology), rabbit anti-mTOR (Cell Signaling Technology), rabbit anti-phosphorylatedmTOR (Cell Signaling Technology), rabbit anti-phosphorylatedp70S6K1 (pS371; Cell Signaling Technology), rabbit anti-p27(Santa Cruz Biotechnology), mouse anti-p21 (BD), mouse anti-Bax (Santa Cruz Biotechnology), mouse anti-b-actin (Sigma-Aldrich), and mouse anti-GAPDH (Sigma-Aldrich) overnight at4�C. After being washed in TTBS, the blots were then incubatedwith appropriate horseradish peroxidase–conjugated secondaryantibodies for 1 hour at room temperature. The antibody–antigencomplex was visualized by ECL chemiluminescence (Millipore).
PCR and direct DNA sequencingDNA from AOM-treated Klf4fl/fl and Klf4DIS mice was extracted
from paraffin-embedded tissue sections as described above andpurified using the standard phenol/chloroform/isopropanolmethod. Thus, the DNA extracted represents the entire length ofthe colon (normal epithelium and tumors) present in the sectioncollected. PCR primers for detection of b-catenin and K-Rasmuta-tionswere designed to amplify the following: exon3of theCtnnb1gene containing the consensus sequence for GSK-3b phosphor-ylation and exon 1 of K-Ras (22). PCR amplification conditionswere used as described before (22) using RED-Taq Ready PCRMix(Sigma). Amplified fragments were then electrophoresed in 1%agarose gel, excised, and purified using theQIAEX II gel extractionkit (Qiagen). Sequencing was done at the Genomic Core Facility,Stony Brook University. Thus, the DNA sequenced represents theentire length of the colon (normal epithelium and tumors)present in the section collected.
Counting positively stained cells and statistical analysisFor p300, positively stained colonic epithelial cells were
counted in a minimum of 20 crypts per mouse (3 mice/group).For Ku70 and Rad51 in adenomas, positively stained cells werecounted permouse (3mice/group), per adenoma, per section, perfield at 400� magnification. Four fields were counted per ade-noma. Where applicable, differences in Rad51 IHC stainingintensity in adenomas (2 mice/group) were quantified usingImageJ software (22). Statistical significance between groups wasdone using paired two-tailed Student t test or one-way ANOVA.Where applicable, box plot for data was done using GraphPadPrism version 5.00 for Windows (GraphPad Software).
ResultsIncreased adenoma burden and elevated Wnt signaling level inthe setting of ApcMinmutation following intestinal epithelium–
specific deletion of Klf4We previously showed that haploinsufficiency of Klf4 in
ApcMin/þ mice leads to a significant increase in the number ofadenomas formed in the small intestine but with no significant
effect on adenoma development in the colon (18). In the currentstudy, we investigated whether complete deletion of Klf4 in theintestinal epithelium (Klf4DIS) in the setting of ApcMin mutationhas an influence on adenoma development in the colon, inaddition to the small intestine. We first compared the numberand size of the adenomas that developed in Klf4fl/fl/ApcMin/þ
(which contain intactKlf4 loci) andKlf4DIS/ApcMin/þmice between16 and 20 weeks of age. Klf4fl/fl/ApcMin/þ mice developed on theaverage 19.88� 14.75 adenomas per mouse (N¼ 8) in the smallintestine (Supplementary Fig. S1A). In comparison, the averagenumber of adenomas in the small intestine of Klf4DIS/ApcMin/þ
mice was 67 � 27.21 adenomas per mouse (N ¼ 7; Supplemen-tary Fig. S1A). Examining the size of adenomas formed, Klf4DIS/ApcMin/þmice hadhigher numbers of adenomas in each of the sizecategories than the Klf4fl/fl/ApcMin/þ mice, with significantly moreadenomas of 1 to 2 mm (Supplementary Fig. S1B). There was nosignificant difference in the distribution of tumor sizes betweenthe two groups of mice.
Because KLF4 has been shown to repressWnt/b-catenin activity(13), we examined the levels of b-catenin, Ki67 and cyclin D1 byimmunohistochemistry in the normal-appearing intestinal tis-sues and in adenomas of age-matched Klf4fl/fl/ApcMin/þ andKlf4DIS/ApcMin/þ mice. Both genotypes showed stronger b-cateninstaining in adenomas as compared with their respective normal-appearing intestinal epithelial cells (Supplementary Fig. S2A–S2D). However, Klf4DIS/ApcMin/þ showed an overall strongerb-catenin staining as compared with Klf4fl/fl/ApcMin/þmice. A sim-ilar pattern of differential staining for Ki67 (Supplementary Fig.S2E–S2H) and cyclin D1 (Supplementary Fig. S2I–S2L) wasobserved in both mouse genotypes.
Increased tumor burden and penetrance of colonic polypsfollowing deletion of Klf4 in the setting of ApcMin mutation
Analysis of adenoma number in the colon revealed thatKlf4DIS/ApcMin/þ mice developed on average 3 adenomas percolon, while the Klf4fl/fl/ApcMin/þ mice had on average less than 1adenoma (Fig. 1A). ApcMin/þ mice are known to develop ade-nomas mostly in the small intestine and very few in the colon.Importantly, 100% (7/7) of the Klf4DIS/ApcMin/þ mice developedcolonic adenomas as compared with about only 50% (4/8) ofthe Klf4fl/fl/ApcMin/þmice (Fig. 1A). Also, unlike in the smallintestine, deletion of Klf4 from the colonic epithelium (Fig.1Ba and Bb) did not have an overall effect on b-catenin expres-sion in Klf4DIS/ApcMin/þ mice as compared with Klf4fl/fl/ApcMin/þ
mice in normal tissue (Fig. 1Bc and 1Bd). Similar finding wasobserved for b-catenin and cyclin D1 staining in adenomas ofboth mouse genotypes. However, adenomas of Klf4DIS/ApcMin/þ
mice tended to have darker staining Ki67 than adenomas ofKlf4fl/fl/ApcMin/þmice (Supplementary Fig. S3).
Increased LOH of wild-type Apc locus and increased anaphasebridge index (ABI) in colons of Klf4DIS/ApcMin/þ mice
Normal-appearing cells harboring pro-cancer genetic abnor-malities are prone to develop into tumor cells if they are noteliminated, such as the casewithApcMin/þmice. Consequently, wefocused on examining normal-appearing colonic epithelial cellsto determine the occurrence of preexisting abnormalities thatmight help explain the increased penetrance of colonic adenomaformation in Klf4DIS/ApcMin/þ versus Klf4fl/fl/ApcMin/þ mice. Aspolyps are initiated by LOH at Apc in ApcMin/þ mice (20), we
KLF4 and Colonic Tumor Suppression
www.aacrjournals.org Mol Cancer Res; 14(4) April 2016 387
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

determined the Apc genotype in the colon of Klf4fl/fl/ApcMin/þandKlf4DIS/ApcMin/þ mice, using tissues representing the entire lengthof the colon. In all mice examined, the amplified band for thewild-typeApc allelewasweaker than that of themutated allele.Onthe other hand, the gain in the mutated allele was significantlyhigher in Klf4DIS/ApcMin/þ mice than in Klf4fl/fl/ApcMin/þmice (Fig.2A and B). LOH in ApcMin/þ mice is caused by homologousrecombination near the centromere (23). Additionally, a muta-tion in DNA helicase or telomerase in ApcMin/þmice increases thenumber of intestinal polyps because of high frequency of recom-bination and chromosomal instability (24). In the anaphase ofcell cycle, such chromosomal instability can be assessed by thefrequency of "anaphase bridges," extended chromosome bridgingbetween two spindle poles (24,25). We have previously shownthat KLF4 suppresses genomic instability (10). To determine theeffect of Klf4 deletion on genomic instability in mice colons, wescored ABI in the colonic epithelium (Fig. 2C).We found very fewanaphase bridges in wild-typemice, whereas Klf4fl/fl/ApcMin/þmice
had significantly more (ABI mean � SD ¼ 26.3% � 3.17%; Fig.2D). In contrast, the Klf4DIS/ApcMin/þ mice had ABI almost twicethat ofKlf4fl/fl/ApcMin/þmice (46.5%�2.4%; Fig. 2D). This result isconsistent with previous reports that chromosomal instability isenhanced bymutations in Apc (26,27) or by deletion of Klf4 (10).
Deletion of Klf4 in colonic epithelium has no effect on Cdx2expression in mouse colons
Previous studies have shown that increased tumor burden inthe colon in amousemodel withApcmutation isCdx2 dependent(19). Additionally, Cdx2was shown to regulate Klf4 expression incolon cancer lines (11). To determine whether Klf4 deletion hasany effect on Cdx2 expression that may have led to the increase incolonic adenoma incidence in Klf4DIS/ApcMin/þ mice, we stainedfor Cdx2. As shown in Fig. 3A and B,we observed no differences instaining between Klf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þ mice,where there is a decreasing gradient fromproximal to distal colon.
Figure 1.Klf4DIS/ApcMin/þ mice have increased colonic adenoma burden andpenetrance than Klf4fl/fl/ApcMin/þ. Klf4fl/fl and Klf4DIS mice were bred toApcMin/þ mice. Mice were sacrificed at 16 to 20 weeks, and the number ofadenomas was counted and compared between the two genotypes. A,comparison of the number of adenomas per mouse and of penetrance in thecolon between Klf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þmice. Each mark on thegraph represents the number of adenomas per mouse. P < 0.001 by pairedtwo-tailed t test between the two genotypes. B, IHC staining of normal-appearing colonic tissue for Klf4 (a and b) and for b-catenin (c and d). Panelsa1, b1, c1 and d1 are enlarged insets of panels a, b, c and d, respectively.
Figure 2.Normal-appearing colonic tissue of Klf4DIS/ApcMin/þ mice has increased LOHof Apc and increased ABI than Klf4fl/fl/ApcMin/þ. A, example of LOH resultfrom three Klf4fl/fl/ApcMin/þ mice (lanes 1–3) and three Klf4DIS/ApcMin/þ mice(lanes 4–6). B, densitometric analysis comparison of the relative ratio of WT:mutant band intensity shown in A. C, H&E staining showing an example of amitotic colonic epithelium cell with normal anaphase (i) and another withanaphase bridging (ii). D, graphical representation of ABI as a percentage ofanaphase cells in normal-appearing colonic tissue that harbors anaphasebridging. For BandD, analysiswasdone usingpaired two-tailed Student t testand one-way ANOVA, respectively. ��� , P < 0.001.
Ghaleb et al.
Mol Cancer Res; 14(4) April 2016 Molecular Cancer Research388
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410
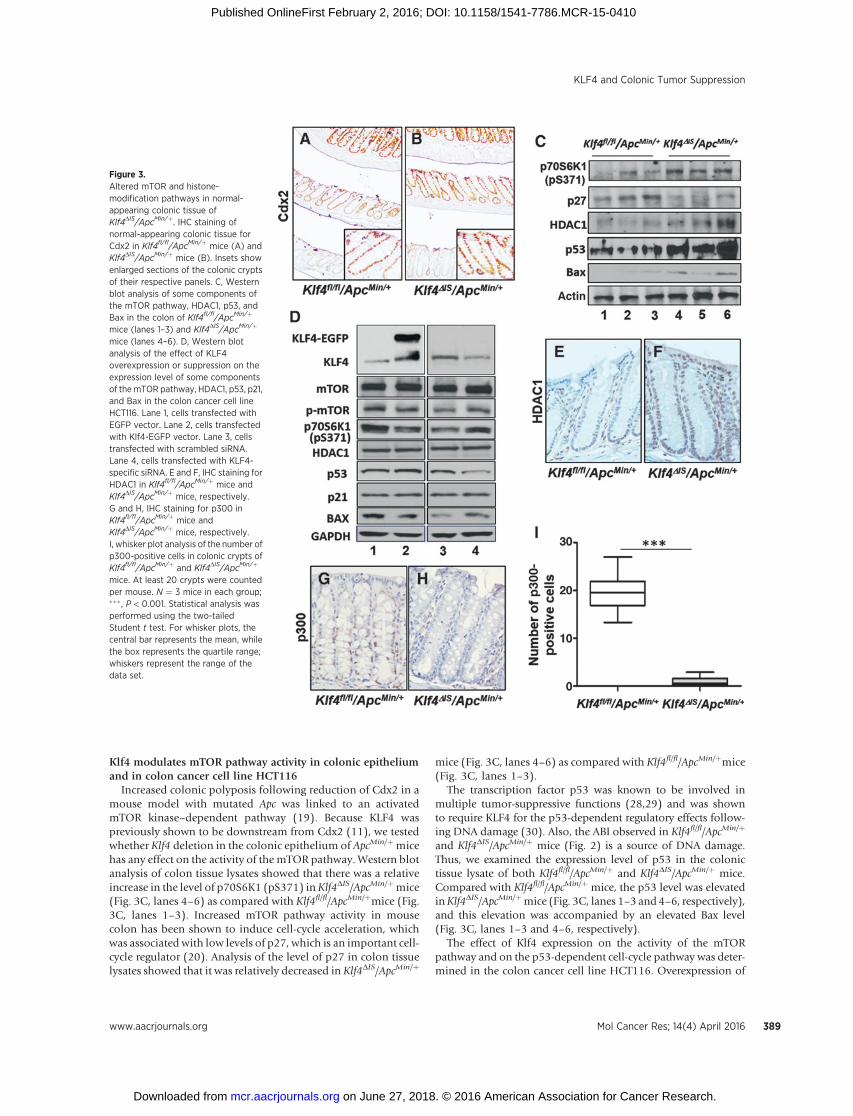
Klf4 modulates mTOR pathway activity in colonic epitheliumand in colon cancer cell line HCT116
Increased colonic polyposis following reduction of Cdx2 in amouse model with mutated Apc was linked to an activatedmTOR kinase–dependent pathway (19). Because KLF4 waspreviously shown to be downstream from Cdx2 (11), we testedwhether Klf4 deletion in the colonic epithelium of ApcMin/þmicehas any effect on the activity of the mTOR pathway. Western blotanalysis of colon tissue lysates showed that there was a relativeincrease in the level of p70S6K1 (pS371) inKlf4DIS/ApcMin/þmice(Fig. 3C, lanes 4–6) as compared with Klf4fl/fl/ApcMin/þmice (Fig.3C, lanes 1–3). Increased mTOR pathway activity in mousecolon has been shown to induce cell-cycle acceleration, whichwas associated with low levels of p27, which is an important cell-cycle regulator (20). Analysis of the level of p27 in colon tissuelysates showed that it was relatively decreased in Klf4DIS/ApcMin/þ
mice (Fig. 3C, lanes 4–6) as compared with Klf4fl/fl/ApcMin/þmice(Fig. 3C, lanes 1–3).
The transcription factor p53 was known to be involved inmultiple tumor-suppressive functions (28,29) and was shownto require KLF4 for the p53-dependent regulatory effects follow-ing DNA damage (30). Also, the ABI observed in Klf4fl/fl/ApcMin/þ
and Klf4DIS/ApcMin/þ mice (Fig. 2) is a source of DNA damage.Thus, we examined the expression level of p53 in the colonictissue lysate of both Klf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þ mice.Compared with Klf4fl/fl/ApcMin/þ mice, the p53 level was elevatedinKlf4DIS/ApcMin/þmice (Fig. 3C, lanes 1–3 and 4–6, respectively),and this elevation was accompanied by an elevated Bax level(Fig. 3C, lanes 1–3 and 4–6, respectively).
The effect of Klf4 expression on the activity of the mTORpathway and on the p53-dependent cell-cycle pathway was deter-mined in the colon cancer cell line HCT116. Overexpression of
Figure 3.Altered mTOR and histone-modification pathways in normal-appearing colonic tissue ofKlf4DIS/ApcMin/þ. IHC staining ofnormal-appearing colonic tissue forCdx2 in Klf4fl/fl/ApcMin/þ mice (A) andKlf4DIS/ApcMin/þ mice (B). Insets showenlarged sections of the colonic cryptsof their respective panels. C, Westernblot analysis of some components ofthe mTOR pathway, HDAC1, p53, andBax in the colon of Klf4fl/fl/ApcMin/þ
mice (lanes 1–3) and Klf4DIS/ApcMin/þ
mice (lanes 4–6). D, Western blotanalysis of the effect of KLF4overexpression or suppression on theexpression level of some componentsof themTOR pathway, HDAC1, p53, p21,and Bax in the colon cancer cell lineHCT116. Lane 1, cells transfected withEGFP vector. Lane 2, cells transfectedwith Klf4-EGFP vector. Lane 3, cellstransfected with scrambled siRNA.Lane 4, cells transfected with KLF4-specific siRNA. E and F, IHC staining forHDAC1 in Klf4fl/fl/ApcMin/þ mice andKlf4DIS/ApcMin/þ mice, respectively.G and H, IHC staining for p300 inKlf4fl/fl/ApcMin/þ mice andKlf4DIS/ApcMin/þ mice, respectively.I, whisker plot analysis of the number ofp300-positive cells in colonic crypts ofKlf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þ
mice. At least 20 crypts were countedper mouse. N ¼ 3 mice in each group;��� , P < 0.001. Statistical analysis wasperformed using the two-tailedStudent t test. For whisker plots, thecentral bar represents the mean, whilethe box represents the quartile range;whiskers represent the range of thedata set.
KLF4 and Colonic Tumor Suppression
www.aacrjournals.org Mol Cancer Res; 14(4) April 2016 389
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

Klf4 suppressed the activity of the mTOR pathway as indicated bylower levels of phosphorylated mTOR and p70S6K1 (pS371) ascompared with control (Fig. 3D, lanes 1–2). In contrast, suppres-sion of KLF4 leads to a relative increase in the phosphorylatedmTOR and p70S6K1 (pS371) level (Fig. 3D, lanes 3–4). The effectof the Klf4 expression level on p53 expression was previouslyshown to be cell-type dependent (31,32). Consistent with previ-ous reports (32), the p53 level was elevated and suppressedfollowing Klf4 overexpression and suppression, respectively (Fig.3D, lanes 1–4). The differences observed for the p53 expressionlevel in response to suppressed KLF4 expression in the HCT116cell line versus mouse colon lysates could be due to mutations insome regulatory genes harbored by the colon cancer cell lineHCT116 as compared with those in the mouse colon tissuelysates. While the p21 level was not affected by the KLF4 expres-sion level (Fig. 3D), there was an inverse correlation between theKLF4 and Bax levels (Fig. 3D, lanes 1–4), consistent with previousreports (33).
Altered expressionofHDAC1andp300 in colonic epitheliumofKlf4DIS/ApcMin/þ mice
Acetyl modifications at histone tails constitute a major epige-netic mechanism that regulates chromatin structure and geneexpression in cancer development (34). Klf4 was previouslyshown to suppress genomic instability in vitro (10), and to directlyinteract with modifiers of histone acetylation such as p300, amember of the histone acetyltransferase family (34). To deter-minewhether deletion ofKlf4 in the colonic epitheliumofApcMin/
þ mice has any effect on the histone acetylation mechanism, wefirst examined the level of histone deacetylase 1 (HDAC1) and ofp300, a histone acetyltransferase, byWestern blot in colonic tissuelysates. While we could not detect p300, the HDAC1 level waselevated inKlf4DIS/ApcMin/þmice (Fig. 3C, lanes 4–6) as comparedwith Klf4fl/fl/ApcMin/þmice (Fig. 3C, lanes 1–3). In HCT116 cells,theHDAC1 levelwas not affected by theKlf4 expression level (Fig.3D), and we could not detect p300. To confirm the change inHDAC1 as observed by Western blot in colonic tissue lysates, westained for HDAC1 and p300. Consistent with Western blotresults, Klf4DIS/ApcMin/þ mice showed more colonic epitheliumcells staining for HDAC1 as compared with Klf4fl/fl/ApcMin/þmice(Fig. 3E and F). On the other hand, while p300 was detected inKlf4fl/fl/ApcMin/þmice, it was significantly lower in Klf4DIS/ApcMin/þ
mice (Fig. 3G, H, and I).
Suppressed DNA damage repair mechanisms in colonicadenomas of Klf4DIS/ApcMin/þ mice
Wehave previously identified an important role of Klf4 inDNAdamage repair response and in maintaining genomic stability(10). Cells have evolved various strategies to promote genomestability through the precise repair of DNA double-strand breaks(DSB) and other lesions that are encountered during normalcellular metabolism and from exogenous insults (35,36). Giventhe role of Klf4 inmediatingDNAdamage repair, and thatKlf4DIS/ApcMin/þ mice have higher markers of genomic instability com-pared with Klf4fl/fl/ApcMin/þmice (Fig. 2), we compared the level ofgH2AX staining between Klf4fl/fl/ApcMin/þand Klf4DIS/ApcMin/þ
mice. Normal-appearing colonic crypts of both genotypes hadfew cells staining positive for gH2AX (Fig. 4A andG, respectively).However, adenomas formed in both mouse genotypes stainedstrongly for gH2AX (Fig. 4D and J, respectively). To determine
whether there is any difference between the two genotypes in theDNA damage repair mechanism, we stained for Rad51 and Ku70,representing homologous recombination repair (HRR) and non-homologous end joining (NHEJ) repairmechanisms, respectively(37). Both mouse genotypes stained negative for Ku70 in thenormal-appearing colonic epithelium (Fig. 4B and H, respective-ly). On the other hand, adenomas of Klf4fl/fl/ApcMin/þmice stainedpositive for Ku70, while adenomas of Klf4DIS/ApcMin/þ mice hadsignificantly lower staining (Fig. 4E and K, respectively, and 4M).For Rad51, bothmouse genotypes stained positive in the normal-appearing colonic epithelium (Fig. 4C and I, respectively), withno observed differences between them. However, adenomas ofKlf4fl/fl/ApcMin/þmice had significantly more positive cells stainingfor Rad51 as compared with adenomas of Klf4DIS/ApcMin/þ mice(Fig. 4F and L, respectively, and 4N). Our results suggest animportant role of Klf4 in promoting repair of DNA DSB bymodulating the HRR and NHEJ mechanisms.
Susceptibility of Klf4DIS mice to AOM-induced colonic tumorformation
AOM is a colon carcinogen that is commonly used in rodents tostudy the pathogenesis of sporadic colorectal cancer. In mice,AOM-induced tumors have been attributed to mutations inb-catenin, while rare mutations in K-Ras are also observed(22,38). To determine whether deletion of Klf4 in the colonicepithelium has any effect on the pathogenesis of AOM-inducedcolorectal cancer, both Klf4fl/fl and Klf4DIS mice were treated withAOM (4 injections, once a week) and then analyzed for tumordevelopment at 4 and 12 weeks after the last injection. At 4 weeksafter injection, no difference was found between treated Klf4fl/fl
andKlf4DISmicewhen examinedmacroscopically. However, H&Estaining of the colonic sections revealed the formation of micro-adenomas in 25% versus 75% of Klf4fl/fl in comparison to Klf4DIS
mice (Supplementary Fig. S4Aa and S4Ab). The number ofmicroadenomas per section averaged 0.5 � 0.57 and 2 � 0.82inKlf4fl/fl andKlf4DISmice, respectively (data not shown). Stainingfor b-catenin (Supplementary Fig. S4Ac and S4Ad) showed slight-ly lighter staining in the normal-appearing colonic epitheliumof Klf4fl/fl mice compared with Klf4DIS mice, and darker stainingin the microadenomas as compared with the surrounding nor-mal-appearing colonic epithelium. While there was no observeddifference in cyclin D1 staining at the basal level between Klf4fl/fl
and Klf4DIS mice, more intense staining was found in the micro-adenomas as compared with the normal-appearing colonic epi-thelium (Supplementary Fig. S4Ae and S4Af).
At 12 weeks after AOM treatment, Klf4DIS mice developedsignificantly more colonic adenomas as compared with Klf4fl/fl
mice (Fig. 5A). Adenomas of bothKlf4fl/fl andKlf4DISmice showeddarker b-catenin staining than those of the surrounding normalcolon epithelium. Although there was variation in b-cateninstaining in adenomas of Klf4DIS mice, adenomas of Klf4DIS micehad an overall more intense and nuclear staining as comparedwith adenomas of Klf4fl/fl mice (Figs. 5Ba and 5Bb). Cyclin D1showed a similar staining pattern between the two groups as inb-catenin (Figs. 5Bc and 5Bd). We also observed an increase incyclin D1 staining in the normal-appearing colonic epithelium ofboth genotypes when compared with mice at 4 weeks (data notshown). Because AOM treatment was shown to have a minimumeffect on inducing K-Ras mutations in mice, we set to determinewhether Klf4 deletion would have any effect on the K-Ras path-way. When stained for p44/42 MAPK (p-ERK), which is a
Ghaleb et al.
Mol Cancer Res; 14(4) April 2016 Molecular Cancer Research390
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

downstream effector of K-Ras, only 20% (1/5) of Klf4fl/fl miceshowed any positive staining for p-ERK in the tumors, while 75%(3/4) ofKlf4DISmice showed positive staining for p-ERK (Figs. 5Beand 5Bf, respectively).
Analysis of AOM-induced b-catenin and K-Ras mutationsshowed no mutations in b-catenin in either genotype, contraryto what was reported before (22,38). However, consistent with
previous reports (22,38), one AOM-treated Klf4fl/fl mouse (1/4)had K-Ras mutations (Fig. 5C), while 3 of 4 of AOM-treatedKlf4DIS mice had K-Ras mutations (Fig. 5C; SupplementaryFig. S4B). These mutations were detected at 12 weeks afterinjection. Codons affected were 14, 16–18, 22 and 23 in theAOM-treated Klf4fl/fl mouse, and codons 14, 17–19, 21, and 22in AOM-treated Klf4DIS mice.
Figure 4.Klf4 differentially regulates NHEJ andHRR DNA DSB repair mechanisms inthe colonic adenomas of Klf4fl/fl/ApcMin/þ mice versus Klf4DIS/ApcMin/þ
mice. IHC staining of gH2AX (A, D, G,and J), Ku70 (B, E, H, and K) and Rad51(C, F, I, and L) and in normal-appearingcolonic crypts of Klf4fl/fl/ApcMin/þ mice(A–C) and Klf4DIS/ApcMin/þ mice (G–I)and in adenomas formed in theirrespective genotypes (D–F) and (J–L).gH2AX is an indicator of DNA DSBs,while Ku70 and Rad51 are indicatorsof active NHEJ and HRR repairmechanisms, respectively. M,quantification of Ku70-positive cells inadenomas of Klf4fl/fl/ApcMin/þ andKlf4DIS/ApcMin/þ. N, quantification ofRad51 positive cells in adenomas ofKlf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þ
mice. �� , P < 0.01; ��� , P < 0.001.Statistical analysis was performedusing two-tailed Student t test. Errorbars, standard error.
KLF4 and Colonic Tumor Suppression
www.aacrjournals.org Mol Cancer Res; 14(4) April 2016 391
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

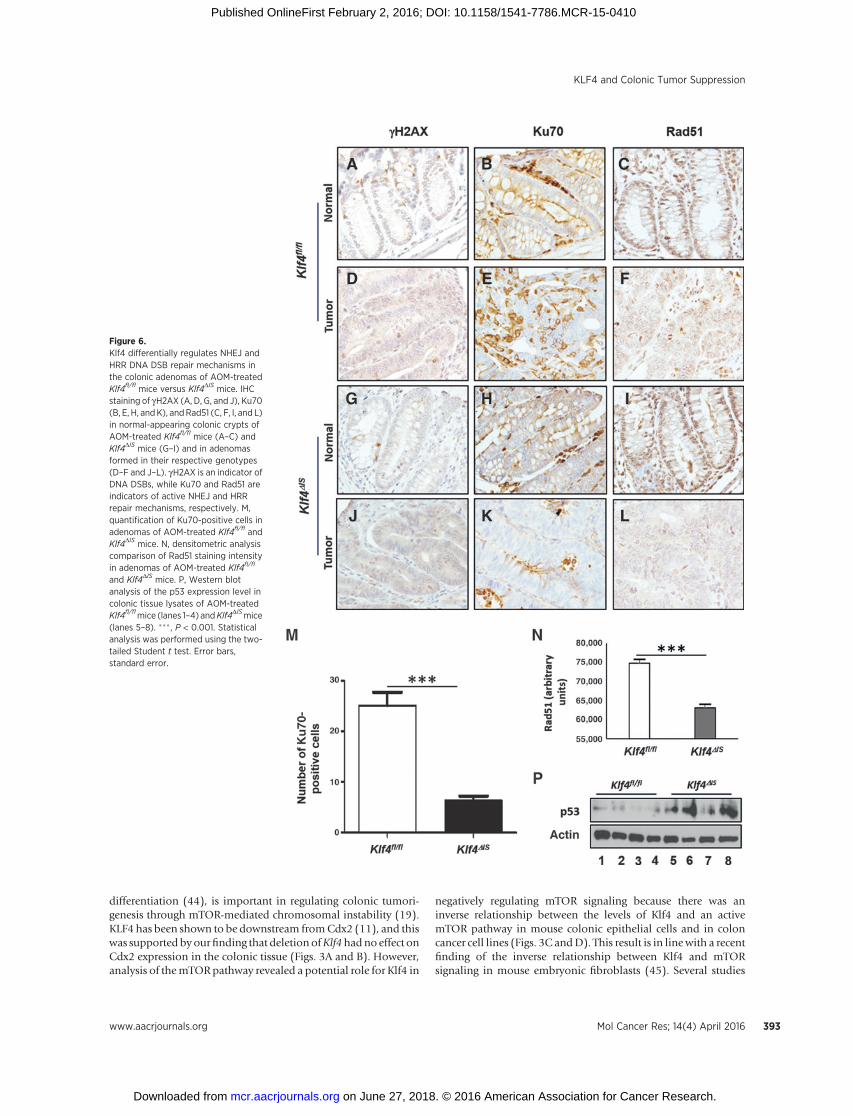
We also examined the DSB repair mechanism in the AOMmodel of colonic tumorigenesis. Normal-appearing coloniccrypts of AOM-treatedmice of both genotypes had few cells/cryptstaining positive for gH2AX (Fig. 6A and G, respectively). Ade-nomas formed in both mouse genotypes stained positive forgH2AX (Fig. 6D and J, respectively). Both mouse genotypesstained negative for Ku70 in the normal-appearing colonic epi-thelium (Fig. 6B and H, respectively). While adenomas of AOM-treated Klf4fl/flmice stained positive for Ku70, adenomas of AOM-treated Klf4DIS mice stained significantly lower (Fig. 6E and K,respectively, and 6M). For Rad51, both mouse genotypes stainedpositive in the normal-appearing colonic epithelium (Fig. 6Cand I, respectively). On the other hand, adenomas of AOM-treatedKlf4fl/flmice hadRad 51-positive staining, while adenomasof AOM-treated Klf4DISmice showed significantly weaker stainingfor Rad51 (Fig. 6F and L, respectively, and 6N). Our results areconsistent with the results shown in Fig. 4, demonstrating animportant role of Klf4 in promoting repair of DNA DSBby modulating HRR and NHEJ mechanisms. Additionally,Western blot analysis of the p53 level in colonic tissue lysates of
AOM-treated Klf4fl/fl and Klf4DIS mice indicated that, comparedwith Klf4fl/fl mice (Fig. 6P, lanes 1–4), Klf4DIS mice had a higherp53 level (Fig. 6P, lanes 5–8). This result is consistent with ourobservation on p53 in Klf4fl/fl/ApcMin/þand Klf4DIS/ApcMin/þ mice(Fig. 4).
DiscussionMany factors such as mutations, epigenetic changes, and DNA
damage contribute to the development of colorectal cancer. Inhumans, familial adenomatous polyposis is a disorder in whichgermline mutations of the APC gene lead to florid colonic poly-posis early in life (1). The ApcMin/þ mouse carries a nonsensegermline mutation in the murine Apc gene (39). A key differencebetween the human disorder and the mouse model is thatadenomas mostly occur in the small intestine compared with thecolon in ApcMin/þ mice (39), whereas it is the reverse in humans.We have previously shown that heterozygous whole-body dele-tion of Klf4 in the setting of ApcMin/þmutation leads to significantincrease in intestinal adenomas as compared with ApcMin/þ mice(18). In support of our previous finding, our current results showthat complete deletion of Klf4 in the intestinal epithelium ofApcMin/þ mice (Klf4DIS/ApcMin/þ) leads to a significant increase inthe number of intestinal adenomas but not in size as comparedwith ApcMin/þ mice (Klf4fl/fl/ApcMin/þ; Supplementary Fig. S1).These results indicate that in the small intestine Klf4 plays a rolein adenoma multiplicity (and possibly initiation) rather thantheir progression.
The Wnt signaling pathway is hyperactivated in the setting ofApc mutation, both in humans and in mice (40,41). KLF4 waspreviously shown to negatively regulate the Wnt signaling path-way (13,42). Additionally, wehave shown thatApcMin/þmicewithKlf4 haploinsufficiency have higher levels of Wnt signaling com-ponents, such as b-catenin and cyclin D1 (18). Consistent withthese findings, Klf4DIS/ApcMin/þ had higher levels of b-catenin,proliferation, and cyclin D1 in both the normal-appearing intes-tinal epithelium and adenomas, as compared with Klf4fl/fl/ApcMin/
þ (Supplementary Fig. S2). These results indicate an importantrole of Klf4 in suppressing the Wnt signaling pathway in theintestinal epithelium, and thus in adenoma formation, in thesetting of ApcMin/þ mutation.
Mice with ApcMin/þ mutation develop adenomas mainly in thesmall intestine, withmuch lower incidence andmultiplicity in thecolon (39,41). We have previously shown that there was nosignificant difference in colonic adenoma incidence and multi-plicity between ApcMin/þ mice and ApcMin/þ mice with Klf4 hap-loinsufficiency (18). However, an analysis of adenoma develop-ment in Klf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þ mice showed asignificant increase in colonic adenoma number and penetrancein Klf4DIS/ApcMin/þ as compared with Klf4fl/fl/ApcMin/þ mice (Fig.1A), suggesting that Klf4 might be a key factor in suppressingcolonic tumorigenesis. In mice, factors that determine tumordevelopment in the small intestine versus the colon are not verywell understood. Analysis of chromosomal instability in thecolonic tissues Klf4DIS/ApcMin/þ mice showed significant increaseboth in LOH of the Apc locus and in aberrant mitosis as indicatedby elevated ABI (Fig. 2). These in vivo results lend support to ourprevious finding about the role of KLF4 in the regulation ofchromosomal instability in vitro (10). It was previously reportedthat Cdx2, a mouse homolog of Drosophilamelanogaster caudal1(43) and a key transcription factor for intestinal development and
Figure 5.Increased number of colonic tumors and b-catenin and cyclin D1 expressionlevels and activated K-Ras pathway inKlf4DISmice treatedwith AOMalone. A,graphical comparison of the number of tumors per mouse in the colonbetween AOM-treated Klf4fl/fl and Klf4DIS mice. B, IHC staining for b-catenin(a and b), cyclin D1 (c and d) and p-ERK (e and f) in colonic tumors of AOM-treatedKlf4fl/fl andKlf4DISmice. C, consensus sequenceof exon 1 of themouseK-Ras andmutations detected in the colonic tissue of AOM-treated Klf4fl/fl (in1 mouse) and Klf4DIS mice (in 3 of 4 mice).
Ghaleb et al.
Mol Cancer Res; 14(4) April 2016 Molecular Cancer Research392
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410
differentiation (44), is important in regulating colonic tumori-genesis through mTOR-mediated chromosomal instability (19).KLF4 has been shown to be downstream fromCdx2 (11), and thiswas supported byourfinding that deletion ofKlf4hadno effect onCdx2 expression in the colonic tissue (Figs. 3A and B). However,analysis of themTORpathway revealed a potential role for Klf4 in
negatively regulating mTOR signaling because there was aninverse relationship between the levels of Klf4 and an activemTOR pathway in mouse colonic epithelial cells and in coloncancer cell lines (Figs. 3C andD). This result is in linewith a recentfinding of the inverse relationship between Klf4 and mTORsignaling in mouse embryonic fibroblasts (45). Several studies
Figure 6.Klf4 differentially regulates NHEJ andHRR DNA DSB repair mechanisms inthe colonic adenomas of AOM-treatedKlf4fl/fl mice versus Klf4DIS mice. IHCstaining of gH2AX (A, D, G, and J), Ku70(B, E, H, andK), andRad51 (C, F, I, and L)in normal-appearing colonic crypts ofAOM-treated Klf4fl/fl mice (A–C) andKlf4DIS mice (G–I) and in adenomasformed in their respective genotypes(D–F and J–L). gH2AX is an indicator ofDNA DSBs, while Ku70 and Rad51 areindicators of active NHEJ and HRRrepair mechanisms, respectively. M,quantification of Ku70-positive cells inadenomas of AOM-treated Klf4fl/fl andKlf4DIS mice. N, densitometric analysiscomparison of Rad51 staining intensityin adenomas of AOM-treated Klf4fl/fl
and Klf4DIS mice. P, Western blotanalysis of the p53 expression level incolonic tissue lysates of AOM-treatedKlf4fl/flmice (lanes 1–4) andKlf4DISmice(lanes 5–8). ��� , P < 0.001. Statisticalanalysis was performed using the two-tailed Student t test. Error bars,standard error.
KLF4 and Colonic Tumor Suppression
www.aacrjournals.org Mol Cancer Res; 14(4) April 2016 393
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

have implicated a positive cross-interaction between mTOR sig-naling and epigenetic events, such as histone acetylation, regu-lated by HDAC1 (46,47). Additionally, an inverse interplaybetween Klf4 and HDAC1 has been previously reported (48).Our assessment of histone acetylation regulation in normalcolonic epithelial cells of both Klf4fl/fl/ApcMin/þ and Klf4DIS/Apc-Min/þ mice indicated elevated HDAC1 and reduced p300 staininglevels in Klf4DIS/ApcMin/þ as compared with Klf4fl/fl/ApcMin/þ mice(Fig. 3). Taken together, our findings are strongly suggestive of arole for KLF4 in suppressing colon tumorigenesis, in an ApcMin/þ
setting, through the modulation of the mTOR signaling pathwayand epigenetic changes via the regulation of HDAC1 and p300and consequently chromosomal instability.
Carcinogens such as AOM are known to induce colonictumors in mice by promoting mutations in b-catenin and toa lesser extent in K-Ras (22,38). In our model, AOM-treatedC57BL/6 Klf4fl/fl and Klf4DIS mice, though both developedcolonic tumors with Klf4DIS mice having significantly morethan Klf4fl/fl mice, yet neither group had b-catenin mutations,nor did they have K-Ras mutations in the commonly affectedcodons 12 and 13 (38). This could either be due to differencesin the mouse strain used in our study versus those used in otherstudies (22,38), or due to differences in treatment regimen andtissue collection time point (22,38). Our results indicate thatthe increase in b-catenin expression in tumors of AOM-treatedC57BL/6 mice is independent of b-catenin mutations, and inKlf4DIS mice corresponds to an absence of Klf4. Additionally,our results show that following exposure to certain carcinogens
such as AOM, Klf4 is important in suppressing mutations inpro-oncogenic genes such as K-Ras.
DNA damage normally occurs in cells as a consequence of bothenvironmental and endogenous insults. During the normalcourse of DNA replication or following exposure to DNA dam-aging agents, DSBs may arise and are considered one of the mostcytotoxic forms of DNA damage (49). Deficiencies in DSB repaircan lead to mutations and chromosomal aberrations that ulti-mately may result in genomic instability and tumorigenesis (49).Consequently, cells have effective mechanisms for the accurateand timely repair ofDSBs inDNA (49). Inmice, the importance ofKlf4 is demonstrated by its ability to modulate DSB events andregulate DSB repair mechanisms (Figs. 4 and 6). Our resultsindicate that in the mouse intestinal epithelium, under basalconditions, the NHEJ repair mechanism is inactive, while theHRR mechanism is active and is independent of Klf4 expression(Figs. 4 and 6). However, in colonic adenomas both NHEJ andHRR mechanisms are active, and are dependent on Klf4 expres-sion as both mechanisms were suppressed in the absence of Klf4.This differential activation of HRR versus NHEJ in normal epi-thelium versus adenomas points to a crucial role for Klf4 inregulating DNA damage repair pathways under pathologic con-ditions. Several factors play important roles in regulating DNAdamage repair mechanisms, for example, the tumor suppressorp53. p53 acts as an important link between upstream signalingand activation of downstream signaling cascades depending onthe extent of DNA damage and can activate cell-cycle arrest andallow the damage to be repaired, or it could transactivate genes
Figure 7.Schematic diagram of Klf4 and itsproposed targets in three differentmodels that lead to tumor formation. Inthe setting of ApcMin/þ mutation, Klf4might be playing a role in preventingmTOR pathway activation andresultant increase in LOH. Additionally,Klf4 may be important in preventingepigenetic alterations via theregulation of histone acetylation. Thebroken-line box outlines the generalarea of the pathway that is possiblytargeted by Klf4. Under conditionswhere DNA mutations are induced byexogenous factors, such as AOM, therole of Klf4 is proposed to be insuppressing gene mutations, such as inK-Ras, that may result in tumorformation. Klf4 is also proposed to playa role in DNA DSB repair through thedifferential regulation of NHEJ and HRRDNA repair mechanisms.
Ghaleb et al.
Mol Cancer Res; 14(4) April 2016 Molecular Cancer Research394
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

involved in the apoptotic machinery (50). Surprisingly, the pri-mary effect of p53 on DNA repair mechanisms was shown be theinhibition of the HRR and of NHEJ repair mechanisms (50). Ourprevious work has shown Klf4 to be a crucial mediator of p53-dependent cell-cycle arrest and DNA damage repair machineryand to suppress p53-dependent apoptosis (30,33). Our data fromKlf4DIS/ApcMin/þ mice, where there was an elevated p53 level andsuppressedHRRandNHEJmechanisms in the absence of Klf4, arein line with these reports. Elevated p53 level and suppressed HRRand NHEJ repair mechanisms in the absence of Klf4 were alsoobserved in AOM-treated mice, adding validation to our obser-vation on the p53 level in Klf4fl/fl/ApcMin/þ and Klf4DIS/ApcMin/þ
mice. Together, our data hint to a potential mechanism by whichKlf4 promotes damaged DNA repair. It is possible that one wayKlf4 promotes DNA damage repair, at least in the colonic epi-thelium, is by counteracting the suppressive effects of p53 onHRRandNHEJ repairmechanisms, thus allowing for theDNA repair toproceed. The exact mechanism by which Klf4 differentially reg-ulates HRR and/orNHEJ DNADSB repair pathways remains to beinvestigated.
Our findings are summarized in Fig. 7, where it is shown thatKlf4 suppresses colonic tumor formation in association withApcMin/þ mutation by both suppressing the mTOR pathway andreducing rates of precancerous epigenetic alterations. In responseto the colon carcinogen AOM, Klf4 suppresses carcinogen-inducedK-Rasmutations. In bothmousemodels, Klf4 is involvedin DNADSB repair by differential regulation of HRR versus NHEJin normal versus tumor tissue. The exact mechanism by whichKlf4 suppresses tumorigenesis under these seemingly differentconditions requires further investigation. In conclusion, in themouse intestinal mucosa, KLF4 plays an integral role in suppres-sing tumor formation under conditions of genetic mutations,
epigenetic alterations, aberrant DNA damage repair, and carcin-ogen-inducedmutations. Thus,modulation ofKLF4 expression inthe intestinal epithelium represents a potential therapeuticapproach to prevent colon cancer.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: A.M. Ghaleb, V.W. YangDevelopment of methodology: A.M. Ghaleb, V.W. YangAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): A.M. Ghaleb, E.A. Elkarim, V.W. YangAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): A.M. Ghaleb, V.W. YangWriting, review, and/or revision of the manuscript: A.M. Ghaleb, A.B. Bialk-owska, V.W. YangAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): A.M. Ghaleb, V.W. YangStudy supervision: A.M. Ghaleb, V.W. Yang
AcknowledgmentsThe authors thank pathologist Dr. Kenneth R. Shroyer, Department of
Pathology, Stony Brook University, for examining stage of adenomas formed.
Grant SupportThis work was supported by grants from the NCI (CA084197 and
DK052230) awarded to V.W. Yang.The costs of publication of this articlewere defrayed inpart by the payment of
page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received October 1, 2015; revised January 27, 2016; accepted January 27,2016; published OnlineFirst February 2, 2016.
References1. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell
1996;87:159–702. Smith KJ, Johnson KA, Bryan TM, Hill DE, Markowitz S, Willson JK, et al.
The APC gene product in normal and tumor cells. Proc Natl Acad Sci U S A1993;90:2846–50.
3. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al.Activation of beta-catenin-Tcf signaling in colon cancer by mutations inbeta-catenin or APC. Science 1997;275:1787–90.
4. Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabi-lization of beta-catenin by genetic defects in melanoma cell lines. Science1997;275:1790–2.
5. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 incolon carcinoma cells. Nature 1999;398:422–6.
6. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al.Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509–12.
7. Lahtz C, PfeiferGP. Epigenetic changes ofDNA repair genes in cancer. JMolCell Biol 2011;3:51–8.
8. Shields JM, Christy RJ, Yang VW. Identification and characterization of agene encoding a gut-enriched Kruppel-like factor expressed during growtharrest. J Biol Chem 1996;271:20009–17.
9. Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM,Yang VW. Kruppel-like factors 4 and 5: the yin and yang regulators ofcellular proliferation. Cell Res 2005;15:92–6.
10. El-Karim EA, Hagos EG, Ghaleb AM, Yu B, Yang VW. Kruppel-like factor 4regulates genetic stability in mouse embryonic fibroblasts. Mol Cancer2013;12:89.
11. Dang DT, Mahatan CS, Dang LH, Agboola IA, Yang VW. Expression of thegut-enriched Kruppel-like factor (Kruppel-like factor 4) gene in the human
colon cancer cell line RKO is dependent on CDX2. Oncogene 2001;20:4884–90.
12. Stone CD, Chen ZY, Tseng CC. Gut-enriched Kruppel-like factor regulatescolonic cell growth through APC/beta-catenin pathway. FEBS Lett2002;530:147–52.
13. ZhangW,ChenX,Kato Y, Evans PM,Yuan S, Yang J, et al.Novel cross talk ofKruppel-like factor 4 and beta-catenin regulates normal intestinal homeo-stasis and tumor repression. Mol Cell Biol 2006;26:2055–64.
14. Wang N, Liu ZH, Ding F, Wang XQ, Zhou CN, Wu M. Downregulation ofgut-enriched Kruppel-like factor expression in esophageal cancer. World JGastroenterol 2002;8:966–70.
15. Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, et al. Drasticdownregulation of Kruppel-like factor 4 expression is critical in humangastric cancer development and progression. Cancer Res 2005;65:2746–54.
16. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW.Identification of Kruppel-like factor 4 as a potential tumor suppressor genein colorectal cancer. Oncogene 2004;23:395–402.
17. Ghaleb AM, McConnell BB, Kaestner KH, Yang VW. Altered intestinalepithelial homeostasis in mice with intestine-specific deletion of theKruppel-like factor 4 gene. Dev Biol 2011; 349:310–20.
18. Ghaleb AM, McConnell BB, Nandan MO, Katz JP, Kaestner KH, Yang VW.Haploinsufficiency of Kruppel-like factor 4 promotes adenomatous poly-posis coli dependent intestinal tumorigenesis. Cancer Res 2007;67:7147–54.
19. Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. Colonic polyposiscaused by mTOR-mediated chromosomal instability in Apcþ/Delta716Cdx2þ/- compound mutant mice. Nat Genet 2003;35:323–30.
20. Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apcþ in intestinaladenomas from Min mice. Cancer Res 1994;54:5947–52.
www.aacrjournals.org Mol Cancer Res; 14(4) April 2016 395
KLF4 and Colonic Tumor Suppression
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

21. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years ofimage analysis. Nat Methods 2012;9:671–5.
22. Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent muta-tions of the beta-catenin gene in mouse colon tumors induced by azox-ymethane. Carcinogenesis 2000;21:1117–20.
23. Haigis KM, Dove WF. A Robertsonian translocation suppresses a somaticrecombination pathway to loss of heterozygosity. Nat Genet 2003;33:33–9.
24. Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunc-tion and evolution of intestinal carcinoma inmice and humans. Nat Genet2001;28:155–9.
25. Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L,Wiegant J, et al. Chromosomal breakage-fusion-bridge events causegenetic intratumor heterogeneity. Proc Natl Acad Sci U S A 2000;97:5357–62.
26. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al.Mutations in the APC tumour suppressor gene cause chromosomal insta-bility. Nat Cell Biol 2001;3:433–8.
27. Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS. A role forthe adenomatous polyposis coli protein in chromosome segregation. NatCell Biol 2001;3:429–32.
28. Levine AJ.p53, the cellular gatekeeper for growth and division. Cell1997;88:323–31.
29. Levine AJ, Finlay CA, Hinds PW. P53 is a tumor suppressor gene. Cell2004;116:S67–9, 1 p following S9.
30. Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH,et al. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediatesthe transactivating effect of p53 on the p21WAF1/Cip1 promoter. J BiolChem 2000;275:18391–8.
31. Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is atranscriptional repressor of p53 that acts as a context-dependent oncogene.Nat Cell Biol 2005;7:1074–82.
32. Yoon HS, Ghaleb AM, Nandan MO, Hisamuddin IM, Dalton WB, YangVW. Kruppel-like factor 4 prevents centrosome amplification followinggamma-irradiation-induced DNA damage. Oncogene 2005;24:4017–25.
33. Ghaleb AM, Katz JP, Kaestner KH, Du JX, Yang VW. Kruppel-like factor 4exhibits antiapoptotic activity following gamma-radiation-induced DNAdamage. Oncogene 2007;26:2365–73.
34. Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers2013;5:676–713.
35. Moynahan ME, Jasin M. Mitotic homologous recombination maintainsgenomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol2010;11:196–207.
36. Murphy CG, Moynahan ME. BRCA gene structure and function in tumorsuppression: a repair-centric perspective. Cancer J 2010;16:39–47.
37. Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechan-ismsofmammalianDNA repair and theDNAdamage checkpoints. AnnualRev Biochem 2004;73:39–85.
38. TakahashiM,Wakabayashi K. Genemutations and altered gene expressionin azoxymethane-induced colon carcinogenesis in rodents. Cancer science2004;95:475–80.
39. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes tomultiple intestinal neoplasia in the mouse. Science 1990;247:322–4.
40. Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressorprotein with catenins. Science 1993;262:1734–7.
41. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al.Multiple intestinal neoplasia caused by amutation in themurine homologof the APC gene. Science 1992;256:668–70.
42. Sellak H, Wu S, Lincoln TM. KLF4 and SOX9 transcription factors antag-onize beta-catenin and inhibit TCF-activity in cancer cells. Biochim Bio-phys Acta 2012;1823:1666–75.
43. Mlodzik M, Gehring WJ. Expression of the caudal gene in the germ line ofDrosophila: formation of an RNA and protein gradient during earlyembryogenesis. Cell 1987;48:465–78.
44. Lorentz O, Duluc I, Arcangelis AD, Simon-Assmann P, Kedinger M, FreundJN. Key role of the Cdx2 homeobox gene in extracellular matrix-mediatedintestinal cell differentiation. J Cell Biol 1997;139:1553–65.
45. Liu C, DeRoo EP, Stecyk C, Wolsey M, Szuchnicki M, Hagos EG. Impairedautophagy in mouse embryonic fibroblasts null for Kruppel-like Factor 4promotes DNA damage and increases apoptosis upon serum starvation.Mol Cancer 2015;14:101.
46. Makarevic J, Tawanaie N, Juengel E, Reiter M, Mani J, Tsaur I, et al. Cross-communication between histone H3 and H4 acetylation and Akt-mTORsignalling in prostate cancer cells. J Cell Mol Med 2014;18:1460–6.
47. Citro S, Miccolo C, Meloni L, Chiocca S. PI3K/mTOR mediate mitogen-dependent HDAC1 phosphorylation in breast cancer: a novel regulation ofestrogen receptor expression. J Mol Cell Biol 2015;7:132–42.
48. Huang Y, Chen J, Lu C, Han J, Wang G, Song C, et al. HDAC1 and Klf4interplay critically regulates human myeloid leukemia cell proliferation.Cell Death Disease 2014;5:e1491.
49. Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett 2010;584:3703–8.
50. Menon V, Povirk L. Involvement of p53 in the repair of DNA double strandbreaks: multifaceted Roles of p53 in homologous recombination repair(HRR) and nonhomologous end joining (NHEJ). Subcell Biochem 2014;85:321–36.
Mol Cancer Res; 14(4) April 2016 Molecular Cancer Research396
Ghaleb et al.
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410

2016;14:385-396. Published OnlineFirst February 2, 2016.Mol Cancer Res Amr M. Ghaleb, Enas A. Elkarim, Agnieszka B. Bialkowska, et al. Pharmacological Mouse Models of Colonic TumorigenesisKLF4 Suppresses Tumor Formation in Genetic and
Updated version
10.1158/1541-7786.MCR-15-0410doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2016/03/10/1541-7786.MCR-15-0410.DC1
Access the most recent supplemental material at:
Cited articles
http://mcr.aacrjournals.org/content/14/4/385.full#ref-list-1
This article cites 50 articles, 15 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/14/4/385To request permission to re-use all or part of this article, use this link
on June 27, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst February 2, 2016; DOI: 10.1158/1541-7786.MCR-15-0410