kaposi sarcoma-associated herpesvirus latency-associated
TRANSCRIPT

Kaposi Sarcoma-associated Herpesvirus Latency-associatedNuclear Antigen Inhibits Interferon (IFN) � Expression byCompeting with IFN Regulatory Factor-3 for Binding toIFNB Promoter*□S
Received for publication, May 8, 2009, and in revised form, December 22, 2009 Published, JBC Papers in Press, January 4, 2010, DOI 10.1074/jbc.M109.018838
Nathalie Cloutier1 and Louis Flamand2
From the Laboratory of Virology, Rheumatology and Immunology Research Center, Centre Hospitalier Universitaire de QuebecResearch Center and Faculty of Medicine, Laval University, Quebec City, Quebec G1V 4G2, Canada
Host cells respond to viral infections by synthesizing and pro-ducing antiviral molecules such as type I interferons (IFN). TheKaposi sarcoma-associated herpesvirus (KSHV) encodes multi-ple proteins expressed during the lytic replication cycle thatalter the antiviral response of the host. Considering that inKaposi sarcoma lesions and primary effusion lymphoma cellsKSHV is latent in the vastmajority of cells, wewere interested indetermining whether latently expressed viral proteins have theability to modulate IFN synthesis. The latency-associatednuclear antigen (LANA-1) is a large nuclear protein that plays arole in the establishment and maintenance of latent KSHV epi-some in the nucleus of infected cells. LANA-1 is also describedto modulate the cellular transcription. Here, we report thatLANA-1 inhibits IFN-� transcription and synthesis by compet-ing with the binding of interferon regulatory factor-3 (IRF3) tothe IFNB promoter. Using mutants of LANA-1, we have identi-fied the central acidic repeated region as the domain essentialfor interfering with the binding of IRF3 to the positive regula-tory domains I–III of the IFNB promoter. In addition, thenuclear localization of LANA-1 proved essential for IFN-� inhi-bition. Thus, LANA-1 interferes with the formation of IFN-�enhanceosome by competing with the fixation of IRF3 and byinhibiting the expression of the CREB-binding protein. Theability of LANA-1 to inhibit IFNB gene expression highlights anew role for this protein in cellular gene modulation andimmune evasion strategies.
Kaposi sarcoma-associated herpesvirus (KSHV),3 also calledhuman herpesvirus 8, is an oncogenic virus associated with the
development of Kaposi sarcoma and two lymphoproliferativediseases, primary effusion lymphoma and multicentric Castle-man disease (1). Most (�90%) cells recovered from thesetumors are latently infected with KSHV and express only asmall subset of viral genes (2). One such gene product, encodedby ORF73, is the latency-associated nuclear antigen (LANA-1).LANA-1 is a large (1162 amino acids) multifunctional and con-stitutively expressed protein that plays critical roles during theKSHV life cycle. LANA-1 is required for viral episome mainte-nance in proliferating cells (3). LANA-1 modulates the cellulartranscription program by altering the functions of various tran-scription factors, likemSin3A (4), CREB-binding protein (CBP)(5), RING3 (6), GSK-3b (7), andATF4/CREB2 (8). LANA-1 alsofunctions as a potent transcriptional repressor and can sup-press activation of heterologous promoters, suggesting thatLANA-1 can act on chromatin structure and/or general tran-scriptional machinery (9). LANA-1 inhibits p53-mediated apo-ptosis (10) and stabilizes c-Myc, which is frequently deregu-lated in cancer (11). Within the KSHV genome, LANA-1up-regulates its own promoter (12) and represses the transcrip-tion of ORF50, a transcriptional activator regulating the switchfrom latency to lytic replication cycle (13).The early innate host response to viral infections is charac-
terized by production of interferon (IFN) as well as pro-inflam-matory chemokines and cytokines that exert direct antiviralactivity and regulate cellular processes important for antiviralhost defense (14, 15). The antiviral response is initiated by cel-lular sensors that recognize viral molecules (RNA or DNA) andactivate an intracellular signal transduction cascade leading toactivation of genes having antiviral functions (16). Such cellularsensors include the cytoplasmic RNA helicases (retinoic acid-inducible gene I and melanoma differentiation antigen 5), thecytoplasmic dsDNA-dependent activator of IFN regulatory fac-tors, and the Toll-like receptors. Although several pathways areactivated following recognition of viral nucleic acids, threeprincipal cellular pathways are known to coordinate the activityof type I IFNs as follows: the nuclear factor (NF)-�B, the mito-gen-activated protein kinase, and the IFN regulatory factor(IRF) pathways (16).
* This work was supported in part by a grant from the National Cancer Insti-tute of Canada (to L. F.).
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Table 1.
1 Recipient of a Canadian Institutes of Health Research doctoral award.2 Supported as a senior scholar from the Fonds de la Recherche en Sante du
Quebec. To whom correspondence should be addressed: Rheumatologyand Immunology Research Center, Centre Hospitalier Universitaire deQuebec Research Center, 2705 Laurier Blvd., Rm. T1-49, Quebec City, Que-bec G1V 4G2, Canada. Tel.: 418-656-4141 (ext. 46164); Fax: 418-654-2765;E-mail: [email protected].
3 The abbreviations used are: KSHV, Kaposi sarcoma-associated herpesvirus;CBP, CREB-binding protein; CREB, cAMP-response element-binding pro-tein; IFN, interferon; LANA-1, latency-associated nuclear antigen; NF-�B,nuclear factor-�B; NLS, nuclear localization signal; PRD, positive-regulatorydomain; SeV, Sendai virus; TBK1, TANK-binding kinase 1; HAU, hemagglu-
tination unit; shRNA, short hairpin RNA; GAPDH, glyceraldehyde-3-phos-phate dehydrogenase; dsDNA, double strand DNA; HA, hemagglutinin; aa,amino acid; EMSA, electromobility shift assay; ELISA, enzyme-linked immu-nosorbent assay; DTT, dithiothreitol; QPCR, quantitative PCR; RT, reversetranscription; WT, wild type; PARP, poly(ADP-ribose) polymerase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 285, NO. 10, pp. 7208 –7221, March 5, 2010© 2010 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
7208 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Members of Herpesviridae virus are sensitive to the antiviraleffects of IFN-�/�. To minimize the antiviral responses trig-gered following viral entry, these viruses have evolved variousmechanisms to counteract the cellular reaction. Microarrayanalysis performed during early KSHV infection revealed thatseveral IFN-responsive genes were rapidly up-regulated (17).Interestingly, innate immune response gene activation isinduced following contact between recombinant KSHV gpK8.1and cellular receptors, but such an antiviral response was notobserved when cells are challenged with infectious KSHV viri-ons (18). This suggests that KSHV possesses mechanisms todifferently modulate the expression of IFN. Because KSHVLANA-1 is constantly expressed in infected cells and known tomodulate the cellular transcription program, we have examinedthe effects of LANA-1 expression on the synthesis of type IIFN. We report that by itself LANA-1 has no effect on IFNBgene activation. However, we observed that LANA-1 could effi-ciently block IFNB gene inductionwhen known inducers of IFNsynthesis were used. LANA-1 does not affect the phosphoryla-tion status of IRF3 but prevents the binding of this transcriptionfactor to the IFNB promoter. The central acidic region ofLANA-1 is required for the inhibition of IFNB synthesis.
EXPERIMENTAL PROCEDURES
Cells and Virus—HEK-293T cells (ATCC, Manassas, VA)and HEC-IB cells (ATCC), which lack a functional �/� inter-feron receptor (19), were cultured in Dulbecco’s modifiedEagle’s medium (Sigma) containing 10% fetal bovine serum.HEK-Blue IFN-�/� cells (InvivoGen, San Diego) were culturedin Dulbecco’s modified Eagle’s medium supplemented with 30�g/ml blasticidin and 100 �g/ml Zeocin. HEK-293T-E1 werecultured in Dulbecco’s modified Eagle’s medium containing10% fetal bovine serum supplemented with 150 �g/ml hygro-mycin (20). A549 cells (ATCC) were cultured in F-12K/Ham’smedium (Hyclone, Quebec, Canada) containing 10% fetalbovine serum. Sendai virus (SeV) (Cantell strain) was obtainedfrom Charles River Laboratories (St-Constant, Quebec,Canada).Plasmids and Constructs—The primers used to generate
LANA-1 WT and mutant vectors are listed in supplementalTable 1. Full-length LANA-1 sequence corresponding to nucle-otides 123793 to 127300 (GenBankTM U75698) was amplifiedfrom genomic DNA of BC3 cells by PCR and digested by EcoRIand XbaI. This PCR product was cloned in-frame with threeN-terminal hemagglutinin (HA) tags into the pCMV3T vectordigested by EcoRI and XbaI. The pCMV3T vector represents amodified pCMV2N3T (a kind gift from Didier Trouche, Uni-versity Paul Sabatier, Toulouse, France) in which the twonuclear localization signals (NLS) were removed. Three C-ter-minal deletion mutants were generated by introducing a stopcodon by site-directed mutagenesis according to the manufac-turer’s instructions (QuikChange site-directedmutagenesis kit,Stratagene, La Jolla, CA) as follows: G996X (LANA-1 1–996),E875X (LANA-1 1–875), and C300X (LANA-1 1–300). Threeothers mutants were made by PCR amplification of specificLANA-1 domains as follows: LANA-1 319–1162, LANA-1854–1162, and LANA-1 888–1162. In brief, 50 nM of eachprimer, 20 �M dNTPs, 1� Expand buffer, and 1 unit of Expand
DNA polymerase (Roche Applied Science) were mixed with 50ng of pCMV3T-LANA-1, followed by PCR amplification.These PCR products were digested by EcoRI and XbaI andcloned into the same restriction enzyme sites in-frame with thethree HA tags into pCMV2N3T vector containing two NLSsignals. Another mutant was generated by site-directedmutagenesis from LANA-1 319–1162 to create LANA-1 319–892 and was cloned in-frame with the three HA tags intopCMV2N3T vector. pIFN-�-Luc and positive regulatorydomain (PRD)-I–III-Luc were obtained from Tom Maniatis(Harvard University) (21). Expression vectors for TBK1(TANK-binding kinase-1), Myc-IRF3, FLAG-IRF3, andIRF3–5D were obtained from J. Hiscott and Rongtuan Lin(Lady Davis Institute, Canada) (22, 23). Expression vector forp50 was obtained through the National Institutes of Health,AIDS Research and Reference Reagent Program, Division ofAIDS, NIAID; pRSV-NF-�B1 (p50) was from Dr. Gary Nabeland Dr. Neil Perkins (24). CBP expression vector was obtainedfrom Didier Trouche (25). The sequence targeting LANA-1(small interfering RNA-LANA-1) was described by Godfrey etal. (26). One hundred pmol of each LANA-1-specific oligonu-cleotide (5�-GTC CCA CAG TGT TCA CAT CCG GGC-3�)was phosphorylated using T4 polynucleotide kinase in a totalvolume of 50 �l for 30 min. To anneal the oligonucleotides, themixture was incubated at 95 °C for 5min and allowed to cool toroom temperature. One �l of this mixture was ligated intopTER vector (27) that had been digested with BglII and HindIIIand treated with shrimp alkaline phosphatase. In the sameway,an irrelevant shRNA control (shRNA-NS) was designed fromthe HHV-6 IE1 mRNA (28). Two others vectors encodingshRNA against LANA-1 (pSUPER-shRNA LANA-1-N andpSUPER-shRNA LANA-1-C) were obtained from Dr. DianeHayward (29). An unrelated control specific to Renilla lucifer-ase, pSUPER-NS, was obtained from Dr. Shoji Yamaoka (30).Transfections and Infections—A549 and HEC-1B cells were
plated 1 day prior to transfection at 150,000 cells/well of a6-well plate. Cells were transfected using the TransiT-LT rea-gent (Mirus Corp.) according to the manufacturer’s instruc-tions. 24 h post-transfection, cells were infected with 20HAU/ml SeV for 8 h. HEK-293T cells were seeded at 75,000cells/well of a 24-well plate 1 day prior to transfection using thecalcium phosphate precipitation procedure. For some experi-ments, HEK-293T cells were co-transfected with IFNB geneactivators, such as TBK1 or IRF3–5D plasmids, together withincreasing quantities of LANA-1 expression vector. The level ofDNAwas kept constant at 1.25 �g by the addition of the emptypCMV3T vector. In some experiments, cells were infected with10 HAU/ml of SeV or transfected 10 �g of polydeoxyade-nosine-deoxythymidine (poly(dA-dT)) DNA (Amersham Bio-sciences) by using Lipofectamine (Invitrogen) 24 h after trans-fection with pCMV3T-LANA-1. HEK-293T-E1 were seededand transfected as HEK-293T cells.RT-QPCR—Total RNA was extracted from HEK-293T,
HEK-293T-E1, or A549 cells using TRIzol reagent (Invitrogen).All RNA samples were treated with DNase I to eliminate resid-ual genomic DNA prior to amplification. cDNA was synthe-sized, and real time quantitative PCR analysis was performedusing a Rotorgene apparatus (Montreal Biotech Inc., Canada)
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7209
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

and QuantiTect Multiplex PCR kit (Qiagen, Ontario, Canada)for GAPDH and IFNB gene expressions. The following primerpairs have been used: GAPDH forward (5�-CGA GAT CCCTCC AAA ATC AA-3�), GAPDH reverse (5�-TTC ACA CCCATG ACG AAC AT-3�), and GAPDH gene probe (5�-hexa-chloro-6-carboxyfluorescein-TGGAGAAGGCTGGGGCTCAT-black hole quencher 1–3�); IFNB forward (5�-AAA CTCATG AGC AGT CTG CA-3�), IFNB reverse (5�-AGG AGATCT TCA GTT TCG GAG G-3�), IFNB gene probe (5�-hexa-chloro-6-carboxyfluorescein-ATGGTCCAGGCACAGTGACTG TCC TC-black hole quencher 1–3�). Levels of ISG15mRNA were determined by using SYBR Green dye and thefollowing primer pair: ISG15 forward (5�-CAT GGG CTGGGA CCT GAC G-3�) and ISG15 reverse (5�-CGC CAA TCTTCT GGG TGA TCT G-3�).Interferon � Detection—HEK-293T cells were co-transfected
with HA-LANA-1 and TBK1 expression vectors with totalDNA levels kept constant at 1.25 �g per well by the addition ofpCMV3T control plasmid. Forty eight hours after transfection,supernatant was collected and used for IFN-� detection.According to the manufacturer’s instructions, 20 �l of eachsample was added to a flat bottom 96-well plate containing50,000 HEK-Blue IFN-�/� cells per well and incubated at 37 °Cfor 24 h. Forty �l of induced HEK-Blue IFN-�/� supernatantwere then collected and incubated with 160 �l of QUANTI-Blue (InvivoGen) per well for 1 h at 37 °C. The activation of theIFN pathway is made through the inclusion of a reporter geneexpressing a secreted form of embryonic alkaline phosphataseunder the control of the IFN-responsive ISG54 promoter.Secreted embryonic alkaline phosphatase levels were deter-mined using a spectrophotometer at 650 nm and comparedwith a standard curve made by serial dilutions of the IFN-�protein.Nuclear Extracts—HEK-293T were transfected with Myc-
IRF3, p50, CBP, and HA-LANA-1 expression vectors in 10-cmplates. One day after transfection, cells were infected with 10HAU of SeV. After 18 h, cells were collected, and the pelletswere resuspended in 400 �l of buffer A (10 mMHEPES, pH 7.9,10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, and 0.5mM phenylmethylsulfonyl fluoride) and incubated for 10 minon ice. Twenty five �l of 10% Nonidet P-40 were added, andnucleiwere sedimented by centrifuging the samples at 12,000�g for 30 s. The nuclear pellets were resuspended in 50 �l ofbuffer B (20mMHEPES, pH 7.9, 0.4 MNaCl, 1 mM EDTA, 1mM
EGTA, 1 mM DTT, and 1 mM phenylmethylsulfonyl fluoride)and incubated on a rocker for 15 min at 4 °C. The suspensionswere clarified by centrifugation at 12,000 � g for 5 min. Thesupernatants containing nuclear extracts were collected, andthe protein content was determined using the Pierce BCA pro-tein assay kit.Cell Extracts—A549 cells were transfected with the expres-
sion vector of LANA-1 for 36 h, infected with 20 HAU of SeVfor 8 h, lysed in 150�l of lysis buffer (50mMHEPES, pH 7.4, 150mM NaCl, 5 mM EDTA, 10% glycerol, 1% Nonidet P-40, Com-plete-Lysis tablets (Roche Applied Science), 0.1 �M phenyl-methylsulfonyl fluoride, and 1 mM DTT) and 50 �l of 3�Laemmli buffer, and resolved by SDS-PAGE followed by immu-noblotting. HEC-1B cells were transfected with LANA-1 and
IRF3 expression vectors for 36 h and infectedwith 200HAU/mlSeV for 6 h. Cells were collected and frozen at �80 °C. Frozencell pelletswere resuspended in 4 volumes of lysis buffer (20mM
HEPES, pH 7.9, 0.2 mM EDTA, 0.2 mM EGTA, 10% glycerol, 10mM sodium molybdate, 2 mM sodium pyrophosphate, 2 mM
sodium orthovanadate, 0.5 mM spermidine, 0.15 mM spermine,50 �M L-1-tosylamido-2-phenylethyl chloromethyl ketone, 25�M 1-chloro-3-tosylamido-7-amino-2-heptanone, 1 �g/mleach of aprotinin, pepstatin A, and leupeptin, 0.5 mM benzami-dine, 1 mM DTT, and 0.5 mM phenylmethylsulfonyl fluoride).KCl was added to 400mM final, and the extracts were rotated at4 °C for 30 min and centrifuged at 10,000 � g for 5 min. Theprotein content was determined using the Pierce BCA proteinassay kit.Western Blot Analysis—Samples were lysed in Laemmli
buffer, boiled, and electrophoresed on SDS-polyacrylamide gel,and separated proteins were transferred onto polyvinylidenedifluoride membranes. Membranes were incubated in 5% drymilk in TBS-T saline (0.25 M Tris-HCl, pH 7.6, 0.19 M NaCl,0.1% Tween 20) for 1 h to block nonspecific sites. Blots werethen incubated in 0.2% milk/TBST solution containing eithermouse anti-HA (12CA5), mouse anti-Myc (9E10), mouse anti-p50, rabbit anti-ATF2, rabbit anti-c-Jun/AP1, rabbit anti-CBP,mouse anti-actin antibodies (all from Santa Cruz Biotechnol-ogy), rat anti-LANA-1 (Advanced Biotechnology), and mouseanti-PARP-1 (31). Some blots were incubated in 5% bovineserum albumin/TBST solution containing anti-rabbit IRF3 ser-ine 396 (a kind gift of Nathalie Grandvaux, University of Mon-treal, Montreal, Canada). Membranes were washed twice withTBS-T and treated with either a horseradish peroxidase-linkedgoat anti-mouse or anti-rabbit or anti-rat antibody. Reactiveproteins were visualized by enhanced chemiluminescence(PerkinElmer Life Sciences).DNA Affinity Binding Assay—The DNA affinity binding
assay method was described previously by Severa et al. (32) andadapted by Lefort et al. (33). Briefly, biotinylated oligonucleo-tides containing the IFNB promoter sequence (5�-Bio-CCCCCC AAA TGA CAT AGG AAA ACT GAA AGG GAG AAGTGA AAG TGG GAA ATT CC-3�) or the PRD of the IFNBpromoter (PRD-I–III, 5�-Bio-CCC CCCGAAAAC TGAAAGGGA GAA GTG AAA GTG-3�; PRD-II, 5�-Bio-CCC CCCAGTGGGAAATTCCTCTGA-3�; and PRD-IV, 5�-Bio-CCCCCC AAA TGT AAA TGA CAT AGG-3�) were annealed withthe corresponding antisense oligonucleotides in 1� SSC buffer(0.15 M NaCl and 15 mM sodium citrate). Biotinylated DNAoligonucleotides were mixed with 150 �g of nuclear extracts in500�l of binding buffer (20mMTris-HCl, pH 7.5, 75mMKCl, 1mM DTT, 5 mg/ml bovine serum albumin, and 13% glycerol).Twenty �g of poly(dI-dC) were added, and samples were incu-bated for 25 min at room temperature. Streptavidin magneticbeads (Roche Applied Science), washed three times with bind-ing buffer, were added to the reaction mixtures and incubatedfor 30 min at 4 °C and for 10 min at room temperature withmixing by rotation. The beads were collected using a magnetand washed three times. The bound proteins were eluted fromthe beads by boiling in Laemmli sample buffer and wereresolved by SDS-PAGE followed by immunoblotting.
KSHV LANA-1 Inhibits Interferon � Expression
7210 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Electromobility Shift Assays (EMSA)—The methodologyused was as described by Wathelet et al. (34). EMSA were per-formed using 10 �g of cell extracts from HEC-1B cells trans-fected with pCMV3T-LANA-1 and FLAG-IRF3 vectors andinfected or not with SeV. Extracts were incubated with 40 fmolof 32P-labeled double-stranded oligonucleotides correspondingto the ISG15 promoter (5�-CCT AGG GCC TCG GGA AAGGGA AAC CGA AAC TGA A-3�). For the competition exper-iments, excess unlabeled ISG15 promoter double-stranded oli-gonucleotides were used. For supershift experiments, 2 �g ofanti-FLAG (IRF3)were added to the binding reactions. Protein-DNAcomplexeswere resolved on 5%polyacrylamide gel (acryl-amide/bisacrylamide, 75:1) at 4 °C in 1% glycerol, 0.5� TBE.Detection of Active IRF3—The amount of IRF3 capable of
binding to the consensus IRF3-binding site in the absence orpresence of LANA-1 was measured by TransAMTM IRF3 tran-scription factor ELISA (Active Motif). The amount of LANA-1capable of binding to consensus IRF3-binding site was mea-sured by substituting the anti-IRF3 primary antibody for a ratanti-LANA-1 (Advanced Biotechnology) diluted 1:1000.Extracts from LANA-1-transfected and/or SeV-infected 293Tcells were prepared using the protocol provided (ActiveMotif).Reporter Assay—HEK-293T cells were co-transfected with
50 ng of reporter plasmids and expression vectors with totalDNA levels kept constant at 1.25 �g per well by the addition ofpCMV3T control plasmid. Cells were lysed 48 h after transfec-tion, and the luciferase activity was measured using an MLXMicrotiter plate luminometer (Dynex Technologies, Chantilly,VA). Values were normalized using Pierce BCA protein assay(Thermo Fisher Scientific Inc., Rockford, IL).Immunofluorescence Analysis—HEK-293T cells were cul-
tured on coverslips in 6-well plates for 24 h (600,000 cells/well)and transfected using TransiT-LT reagent (Mirus Corp.)according to the manufacturer’s instructions. Twenty fourhours post-transfection, coverslips were washed twice in phos-phate-buffered saline and fixed with cold acetone for 5 min.The slides were then incubated with mouse anti-HA (12CA5)for 1 h at room temperature, washed twice in phosphate-buff-ered saline, and incubated with Alexa 488-labeled goat anti-mouse antibody (Invitrogen) in phosphate-buffered saline for1 h at room temperature. Slides were washed three times inphosphate-buffered saline and mounted with SlowFade anti-fade with 4�,6-diamidino-2-phenylindole (Invitrogen) beforeexamination. Images were captured by a CoolSNAP HQ cam-era mounted on an Olympus BX-51 upright microscope(Olympus America, Melville, NY) using a �60 objective andprocessed with ImagePro 4.5.1 software (Media Cybernetics,Silver Spring, MD).Statistical Analysis—Statistical analysis was performed with
the aid of GraphPad InStat software using unpaired Student’s ttest. All experiments were done in triplicate and repeated atleast three times.
RESULTS
KSHV LANA-1 Inhibits IFNB Gene Induction—KSHVLANA-1 is described as a nuclear protein able to modulate thetranscriptional activity of the human immunodeficiency virus-long terminal repeat promoter and to inhibit KSHV viral lytic
replication by inhibiting the expression and the functions of Rta(13). Considering that LANA-1 is constitutively expressed ininfected cells during latent as well as lytic infection (36), and thefact that neither IFN-� expression nor a functional antiviralresponse is observed in cells infected with KSHV virions (18),we hypothesized that LANA-1 proteinmay interfere with IFNBgene induction. To determine whether LANA-1 influencesIFNB gene expression, we transfected a LANA-1 expressionvector into HEK-293T cells and studied IFNB gene expressionin response to a variety of stimuli, including expression ofTBK1or IRF3–5D, transfection of poly(dA-dT), or infection withSendai virus. Total RNA was extracted, and endogenous IFNBgene expression was monitored by RT-QPCR. As shown in Fig.1A, in the absence of LANA-1, IFNB gene induction was effi-ciently triggered by all of the inducers tested. By itself, LANA-1did not modulate the IFNB synthesis. In contrast, in LANA-1-expressing cells, IFNB gene activation in response to all IFN-�inducers tested was strongly reduced. These results indicatedthat LANA-1 could efficiently block IFNB gene inductionwhenthe IFN pathway is triggered through different mechanisms.We next confirmed that the reduced IFNB mRNA levelsobserved correlated with a reduction in biologically activeIFN-� protein being secreted. HEK-293T cells were co-trans-fected with LANA-1, and TBK1 expression vectors and thesupernatant were collected and assayed for IFN secretion usingHEK-Blue IFN-�/� cells (Fig. 1B). These cells were engineeredto monitor the activation of the JAK-STAT pathway upon type IIFN stimulation. In the absence of LANA-1, the JAK-STATpathwaywas strongly activated by TBK1 indicating that biolog-ically active IFN was present in the supernatant. In contrast, inLANA-1-expressing cells, the signaling pathwaywas reduced tobasal levels indicating that only a small amount of IFN waspresent (Fig. 1B). To confirm that physiological levels ofLANA-1 were capable of reducing IFNBmRNA expression, wetransfected varying quantities of pCMV3T-LANA-1 in HEK-293T cells (0.5, 5, and 20 ng) and studied IFNB gene expressionin response to TBK1 (Fig. 1C). With as little as 5 ng of LANA-1vector, we were able to reduce IFNB gene expression, and sta-tistically significant inhibitionwas observed following transfec-tion of 20 ng of LANA-1 vector. Levels of LANA-1 expressionfollowing transfection with 5 or 20 ng of LANA-1 vector werecomparable with LANA-1 expression levels in HEK-293T-E1cells carrying a recombinant KSHV (Fig. 1D) (20). Expression ofLANA-1 inHEK-293T-E1 cells is less abundant than inBCBL-1cells (Fig. 1E), suggesting that the LANA-1 doses used are phys-iological and suggest that at physiological levels LANA-1 iscapable of inhibiting the expression of IFNB mRNA. Overall,these results indicate that LANA-1 inhibits the expression ofIFNBmRNA resulting in reduced synthesis and release of bio-logically active IFN-� protein.Under resting conditions, IRF3 is located in the cell cyto-
plasm. Once the cell detects an invading pathogen, IRF3 getsphosphorylated by the I�K-like kinases TBK1 and IKK� (21) onserine and threonine residues located within the C terminus(37, 38), and it homodimerizes and enters the nucleus to acti-vate the IFNB gene in cooperation with NF-�B. To determinewhether LANA-1 inhibits IFNB gene expression by interferingwith the phosphorylation of IRF3, we transfected A549 cells
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7211
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
with the LANA-1 expression vector and infected them withSendai virus. We chose to use A549 cells for two reasons. First,to demonstrate reproducibility in another cell type, and secondbecause the detection of phospho-IRF3 is facilitated in this cellline. A549 cells were transfected with LANA-1 expression vec-tor followed by SeV infection. Total RNA was isolated, andendogenous IFNB gene expression was monitored byRT-QPCR. As shown in Fig. 2A, LANA-1 did not modulateIFNB gene expression by itself, but it effectively inhibited theactivation of this gene in response to SeV infection. Lowertransfection efficiencies of A549 cells compared withHEK-293T cells explain the slightly diminished potential ofLANA-1 at inhibiting IFNB gene expression in response to SeVinfection. The phosphorylation status of IRF3 was monitoredbyWestern blot analysis. As shown in Fig. 2B, SeV infection ledto the phosphorylation of IRF3, and the presence of LANA-1did not modify this cellular state. This confirms the results
obtained using the phosphomimetic and constitutively activeIRF3 (IRF3–5D), which does not rely on cellular kinases for itsactivation (Fig. 1A). To confirm that the absence of effect ofLANA-1 on the phosphorylation of IRF3 was not caused by thelower transfection efficiency of A549 cells, HEK-293T cells wereco-transfected with LANA-1 and Myc-IRF3 expression vectors,and phospho-IRF3 levels uponSeV infectionwere determined.Asshown in Fig. 2C, LANA-1 did not interfere with the phosphory-lationof IRF3 inducedby SeV infection.UsingHEK-293T-E1 cellsthat are infectedwith a recombinantKSHV, similar levels of phos-pho-IRF3 were also detected following SeV infection (data notshown). These results suggest that LANA-1 inhibits the synthesisof IFNB, and this effect is not mediated through impaired phos-phorylation of IRF3.LANA-1 Interferes with the Fixation of IRF3 on the IFNB
Promoter—The activation of the IFNB promoter requires theformation of an enhanceosome, a multicomponent complex of
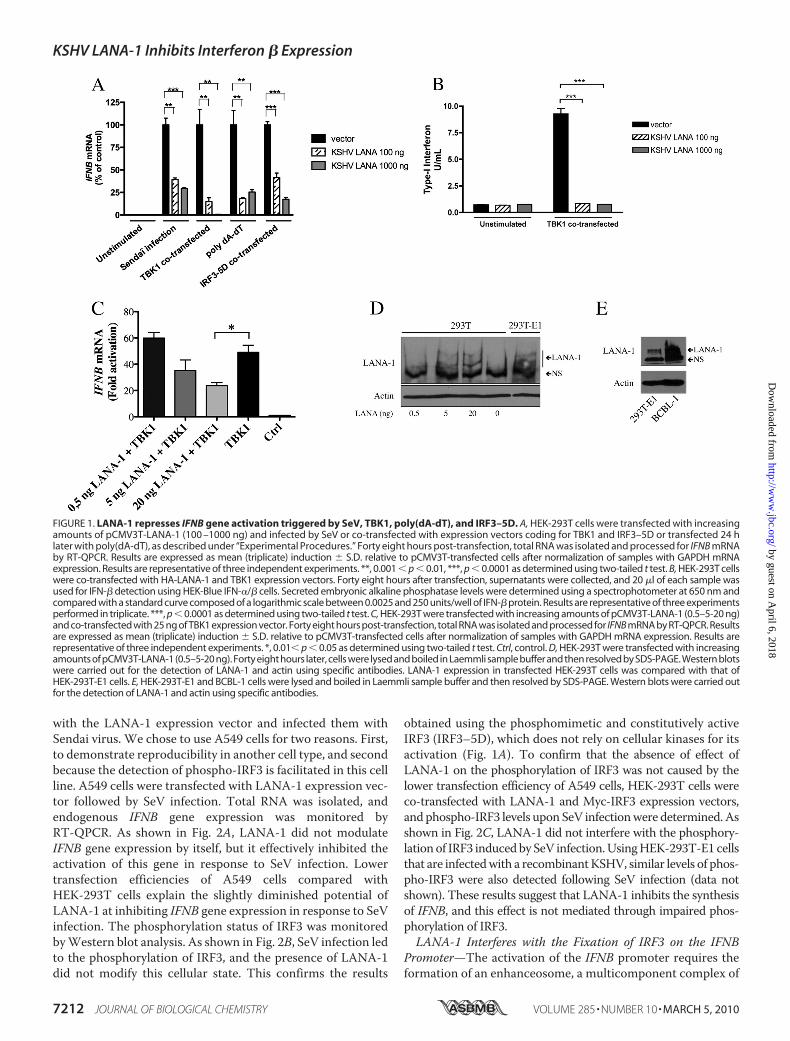
FIGURE 1. LANA-1 represses IFNB gene activation triggered by SeV, TBK1, poly(dA-dT), and IRF3–5D. A, HEK-293T cells were transfected with increasingamounts of pCMV3T-LANA-1 (100 –1000 ng) and infected by SeV or co-transfected with expression vectors coding for TBK1 and IRF3–5D or transfected 24 hlater with poly(dA-dT), as described under “Experimental Procedures.” Forty eight hours post-transfection, total RNA was isolated and processed for IFNB mRNAby RT-QPCR. Results are expressed as mean (triplicate) induction � S.D. relative to pCMV3T-transfected cells after normalization of samples with GAPDH mRNAexpression. Results are representative of three independent experiments. **, 0.001 � p � 0.01, ***, p � 0.0001 as determined using two-tailed t test. B, HEK-293T cellswere co-transfected with HA-LANA-1 and TBK1 expression vectors. Forty eight hours after transfection, supernatants were collected, and 20 �l of each sample wasused for IFN-� detection using HEK-Blue IFN-�/� cells. Secreted embryonic alkaline phosphatase levels were determined using a spectrophotometer at 650 nm andcompared with a standard curve composed of a logarithmic scale between 0.0025 and 250 units/well of IFN-� protein. Results are representative of three experimentsperformed in triplicate. ***, p � 0.0001 as determined using two-tailed t test. C, HEK-293T were transfected with increasing amounts of pCMV3T-LANA-1 (0.5–5-20 ng)and co-transfected with 25 ng of TBK1 expression vector. Forty eight hours post-transfection, total RNA was isolated and processed for IFNB mRNA by RT-QPCR. Resultsare expressed as mean (triplicate) induction � S.D. relative to pCMV3T-transfected cells after normalization of samples with GAPDH mRNA expression. Results arerepresentative of three independent experiments. *, 0.01� p � 0.05 as determined using two-tailed t test. Ctrl, control. D, HEK-293T were transfected with increasingamounts of pCMV3T-LANA-1 (0.5–5-20 ng). Forty eight hours later, cells were lysed and boiled in Laemmli sample buffer and then resolved by SDS-PAGE. Western blotswere carried out for the detection of LANA-1 and actin using specific antibodies. LANA-1 expression in transfected HEK-293T cells was compared with that ofHEK-293T-E1 cells. E, HEK-293T-E1 and BCBL-1 cells were lysed and boiled in Laemmli sample buffer and then resolved by SDS-PAGE. Western blots were carried outfor the detection of LANA-1 and actin using specific antibodies.
KSHV LANA-1 Inhibits Interferon � Expression
7212 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
transcription factors and enhancers that operate as a functionalunit. This promoter is activated by the fixation of p50/p65 onthe NF-�B-binding site (PRD-II), the fixation of IRF3 on thePRD-I/III, and the fixation of ATF-2/c-Jun on the PRD-IVdomains (39). The local protein-protein contacts suggest thatcooperative occupancy of the enhanceosome comes frombind-ing-induced changes in DNA conformation and interactionswith additional co-activators such as CBP/p300 (40) andenables the RNA polymerase II machinery access to the pro-moter. To determine how LANA-1 inhibits IFNB gene activa-tion, we tested whether LANA-1 could affect the binding ofthese transcription factors to the IFNB promoter. Nuclearextracts of resting and SeV-infected cells co-transfected withexpression vectors for IRF3, LANA-1, p50, and CBP were ana-lyzed by Western blot for IRF3 (Myc), LANA-1 (HA), ATF-2,c-Jun, p50, CBP, and PARP-1 to compare the input of the dif-ferent nuclear extracts (Fig. 3A). The nuclear input is similarbetween conditions except for HA-LANA-1 and CBP. Theexpression of LANA-1 was restricted to the conditions wherethe protein is expressed upon pCMV3T-LANA-1 transfection.
The expression of CBP in nuclear extracts is reduced in thepresence of LANA-1 confirming the results of Lim et al. (5)describing transcriptional inhibition of CBP by LANA-1. Dou-ble-stranded oligonucleotides containing the entire IFNB pro-moter were incubated with the nuclear extracts, and the pro-teins bound to the promoter sequence were analyzed byWestern blot (Fig. 3B). LANA-1 was able to bind to the pro-moter in a dose-dependent manner, and SeV infection did notaffect LANA-1 attachment. The binding of ATF-2 and c-Junwas neither affected by the presence of LANA-1 nor by SeVinfection. The fixation of IRF3 to the IFNB promoter wasinduced following SeV infection. IRF3 efficiently bound theIFNB promoter in the absence of LANA-1 but much less sowhen LANA-1 was present. In fact, transfection of cells with 5�g of LANA-1 expression vector strongly reduced the fixationof IRF3, whereas transfection with 20 �g of LANA-1 abolishedthe binding of IRF3 to the IFNB promoter. LANA-1 did notaffect the fixation of p50 (Fig. 3B) or the binding of p65 (data notshown) to the IFNB promoter. The apparent reduction in CBPbinding to the IFNB promoter is a consequence of the reducedlevels of CBP in the nuclear extracts (Fig. 3A). These resultsindicate that LANA-1 negatively affects the binding of IRF3 tothe IFNB promoter. Furthermore, the results indicate thatLANA-1 can bind (directly or indirectly) to the IFNB promoterand impair the recruitment of IRF3 following activation by SeVinfection. These results could explain, at least in part, the inhi-bition of IFNB gene expression caused by the presence ofLANA-1 in stimulated cells.To determine on which part of the IFNB promoter LANA-1
was able to bind, we incubated the same nuclear extracts ana-lyzed in Fig. 3A with three different double-stranded oligonu-cleotides containing each of the three PRDs of the IFNB pro-moter (PRD-I–III, PRD-II, and PRD-IV) and identified thebound proteins by Western blot (Fig. 3, C–E). As expected,IRF3 efficiently bound to the PRD-I–III domain, p50 bound tothe PRD-II domain, and c-Jun bound to the PRD-IV domain.LANA-1was found capable of binding to the PRD-I–III domain(Fig. 3C) and did not bind to other IFNB promoter domains.The fixation of LANA-1 to the PRD-I–III affected the bindingof IRF3. At the highest dose tested, the co-expression ofLANA-1 eliminated IRF3 binding to the PRD-I–III promoter.The fixation of p50 to the PRD-II domain seemed to be slightlyaffected in the presence of the highest dose of LANA-1 (Fig.3D). However, this diminution was not significant consideringthe fact that there was also a diminution of p50 in nuclearextracts in the same samples (Fig. 3A). The fixation of c-Jun onPRD-IV was similar under all conditions tested indicating thatLANA-1 did not affect the fixation c-Jun to the IFNB promoter(Fig. 3E). These results confirm the results obtained in Fig. 3B.They suggest that LANA-1 binds to the PRD-I–III domain ofIFNB promoter and impairs the recruitment of IRF3 when acti-vated by SeV infection.Next, we confirmed that LANA-1 reduced IRF3 fixation to
the promoter by performing EMSA. It is a known fact that theaffinity of IRF3 for the IFNB promoter is somewhat low and�200-fold lower than that of the ISG15 promoter (34). Ourfailed attempts to obtain reliable gel shifts using the IFNB pro-moter sequences support this observation. To alleviate this
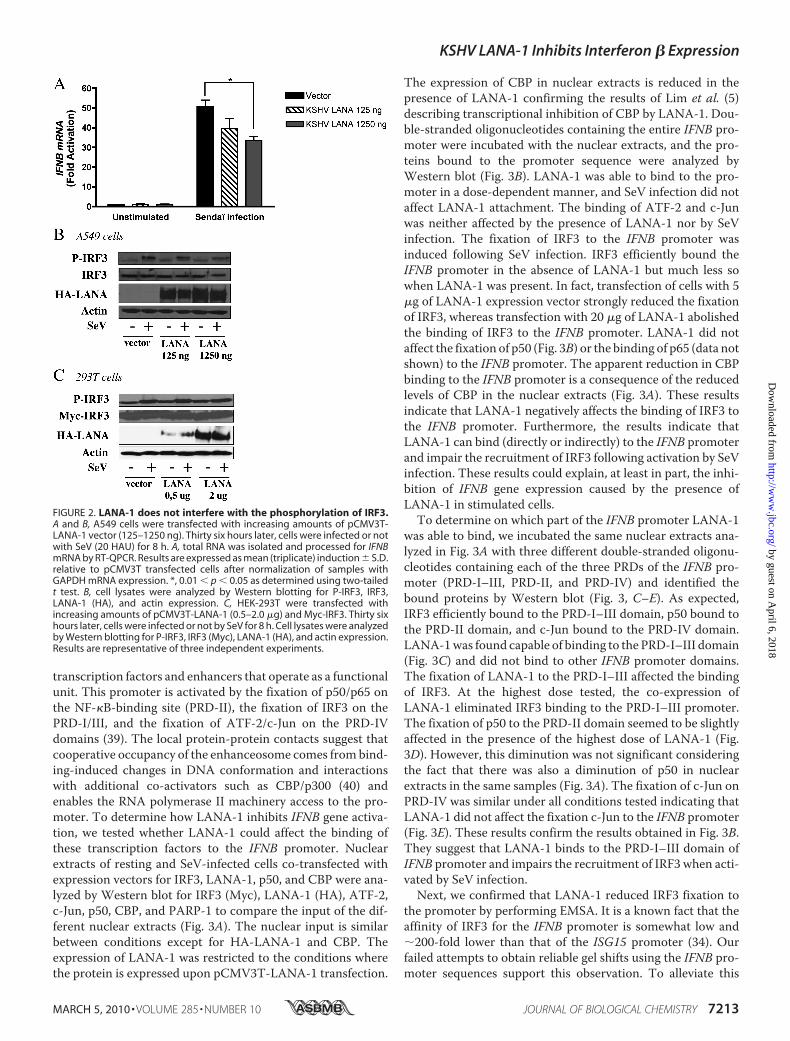
FIGURE 2. LANA-1 does not interfere with the phosphorylation of IRF3.A and B, A549 cells were transfected with increasing amounts of pCMV3T-LANA-1 vector (125–1250 ng). Thirty six hours later, cells were infected or notwith SeV (20 HAU) for 8 h. A, total RNA was isolated and processed for IFNBmRNA by RT-QPCR. Results are expressed as mean (triplicate) induction � S.D.relative to pCMV3T transfected cells after normalization of samples withGAPDH mRNA expression. *, 0.01 � p � 0.05 as determined using two-tailedt test. B, cell lysates were analyzed by Western blotting for P-IRF3, IRF3,LANA-1 (HA), and actin expression. C, HEK-293T were transfected withincreasing amounts of pCMV3T-LANA-1 (0.5–2.0 �g) and Myc-IRF3. Thirty sixhours later, cells were infected or not by SeV for 8 h. Cell lysates were analyzedby Western blotting for P-IRF3, IRF3 (Myc), LANA-1 (HA), and actin expression.Results are representative of three independent experiments.
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7213
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
KSHV LANA-1 Inhibits Interferon � Expression
7214 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

problem, we used the ISG15 promoter sequence as a dsDNAprobe for our EMSA experiments. ISG15 is the first reportedtarget of IRF3 (41). However, knowing that the ISG15 gene canalso respond to secreted IFNs, EMSAs were performed usingnuclear extracts from HEC-1B, which do not respond to IFNstimulation (42). Upon infection of these cells, phosphorylatedIRF3 translocates into the nucleus and activates the expressionof IFN-stimulated genes by binding to the ISRE/IRFE sequence(43). HEC-1B cells were transfected with FLAG-IRF3,pCMV3T, or pCMV3T-LANA-1 vectors that were infected ornot with SeV for 8 h. As shown in Fig. 3F, a binding complex onISG15 promoter (*) was observed in SeV-infected pCMV3T cellextracts (lane 2). In the presence of LANA-1, the complex inSeV-infected cells (Fig. 3F, lane 4) was strongly reduced (35% oflane 2). The specificity of the bound complex was ascertainedby homologous competition using excess unlabeled (25�) wildtype ISG15 (Fig. 3F, lanes 5–8). Then we tested whether thiscomplex (*) could be supershifted by the addition of anti-FLAG(IRF3) antibodies (Fig. 3F, lanes 9 and 10 and lanes 12 and 13reduced exposition). A new complex (**) with reducedmobilitywas detected in pCMV3T-SeV-infected cells (Fig. 3F, lanes 9and 12), and the presence of LANA-1 (lanes 10 and 13) reducedthe formation of this complex (100 to 83%). Using the sameextracts, we next tested the capacity of LANA-1 to inhibit IFNBand ISG15 expression in HEC-1B cells (Fig. 3, G and H). Theinhibitions of IFNB and ISG15 expression in these cells wereweak compared with HEK-293T, a likely consequence of lowertransfection efficiencies of HEC-1B cells. Overall, these resultsare in agreement with those obtained using the DNA-affinitybeads assay (Fig. 3B) and suggest that LANA-1 interferes withthe fixation of IRF3 to consensus DNA sequences.Binding of LANA-1 to the IFNB promoter and inhibition of
IRF3 binding to the IFNB promoter was further confirmedusing a commercially available modified ELISA kit allowing thedetection and binding of activated IRF3 to consensus bindingsites (Fig. 3I). The primary antibody used to detect interferonregulatory factor recognizes an epitope on IRF3 protein uponDNA binding. Incubations of nuclear extracts from resting andSeV-infected HEK-293T cells transfected with HA-LANA-1expression vector were carried out, and the results obtainedindicate that in the presence of LANA-1 endogenous IRF3binding is severely diminished (Fig. 3I). As control, transfectionof an expression vector (20 �g) encoding for an irrelevant HA-tagged protein had no effect on IRF3 binding (data not shown).
Furthermore, by substituting the anti-IRF3 primary antibodyfor anti-LANA-1, we could demonstrate binding of LANA-1 tothe IRF3 consensus binding site (Fig. 3J). LANA-1 binding wasnot modulated by SeV infection (data not shown). Binding ofnuclear extracts from control transfected cells yielded back-ground levels of reactivity (data not shown). Taken together,these results are in agreement with our DNA beads bindingassay and indicate that LANA-1 binds and affects the binding ofIRF3 to consensus sites and subsequent activation of IRF3-re-sponsive genes, such as IFNB.The specificity of LANA-1 for the PRD-I–III element was fur-
ther confirmed by transfecting HEK-293T cells with expressionvector for LANA-1 together with luciferase reporters whoseexpressions are driven by the IFNB promoter (IFN-�-Luc) orPRD-I–III domain (PRD-I–III-Luc). The IFN-� induction path-way was triggered by transfecting dsDNA (poly(dA-dT)), an IFNinducer relevant to dsDNA viruses such as herpesviruses. Indeed,recent reports indicate that cells possess a DNA-dependent acti-vator of IFN regulatory factors capable of activating innateimmune system genes (44, 45). DNA-dependent activator of IFN-regulatory factors recruits TBK1 and IRF3 transcription factor,bothofwhichplaycritical roles in the inductionof type I IFNgenes(44).Aspresented inFig.4A, poly(dA-dT)activatedboth IFNBandPRD-I–III promoters efficiently, whereas LANA-1, by itself, dem-onstrated no effect on these promoters. A dose-dependent inhibi-tion of both promoters was observed in the presence of increasingamounts of LANA-1. Compared with the PRD-I–III reporter, theIFNB promoter was more severely affected by the presence ofLANA-1. Similar results were obtained using SeV infectioninstead of poly(dA-dT) stimulation (data not shown). PRD-II andPRD-IV promoters were not affected by the presence of LANA-1(data not shown). Expressionof LANA-1 and actin in cell lysates isshown in Fig. 4B confirming that similar quantities of extractswere analyzed.Repeated Region of LANA-1 Interferes with the Binding of
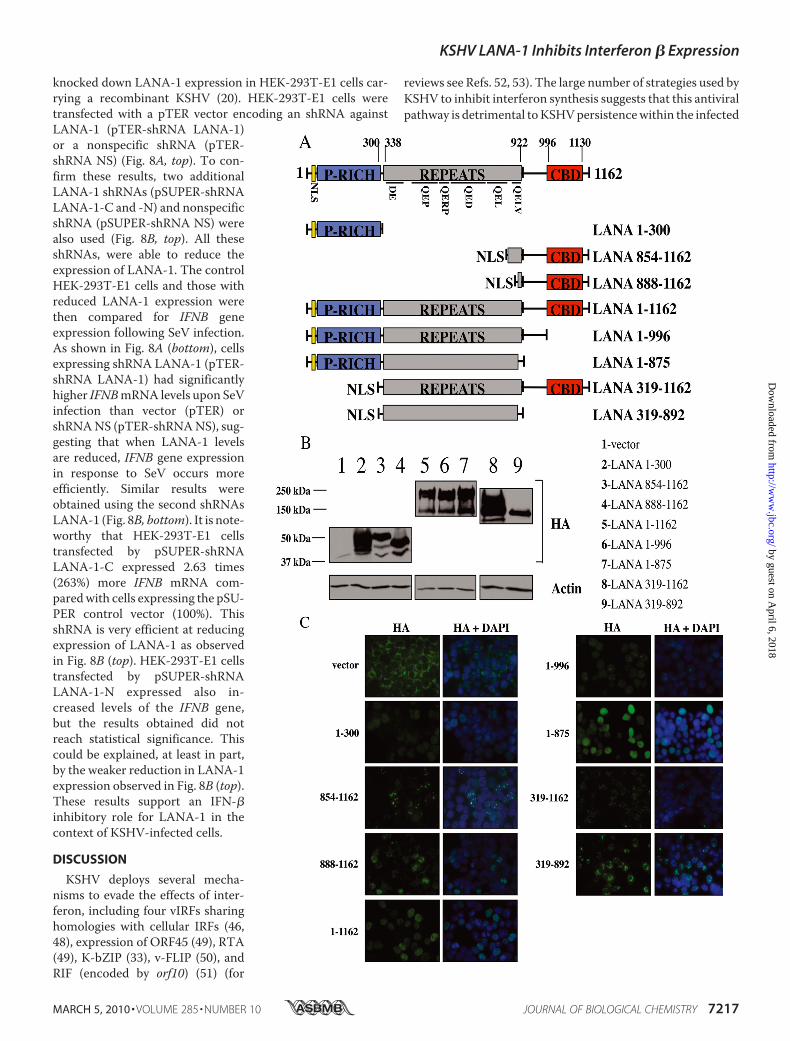
IRF3 on the IFNB Promoter—The LANA-1 protein containsthreemain regions based on amino acid composition as follows:a central region composed of a variable number of highly acidicamino acid repeats, a C-terminal more basic region involved inDNA binding and oligomerization, and an N-terminal regionimplicated in chromatin attachment and the recruitment ofco-repressors (Fig. 5A) (9, 46, 47). To determine whichregion(s) of LANA-1 is(are) responsible for inhibiting the bind-ing of IRF3 on the IFNB promoter, we generated various dele-
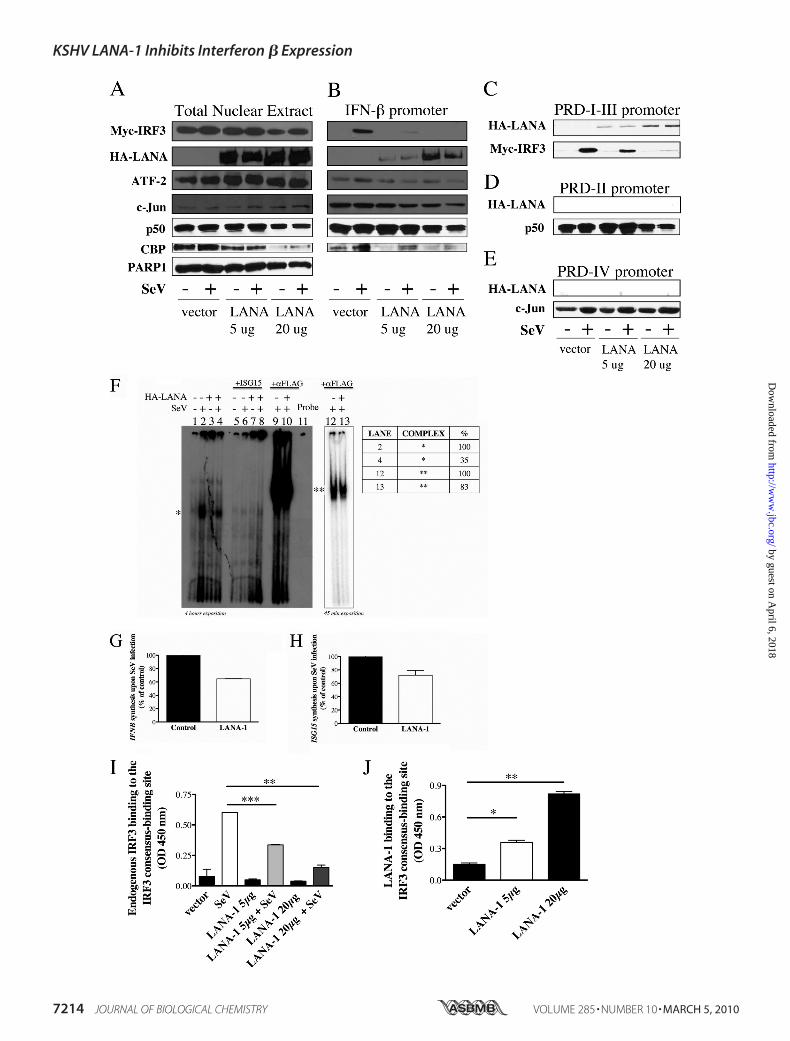
FIGURE 3. LANA-1 binds to the PRD-I–III region and interferes with the fixation of IRF3 to the IFNB promoter. A–E, HEK-293T cells were transfected withpCMV3T-LANA-1 (5–20 �g), Myc-IRF3, p50, and CBP vectors. Twenty four hours later, half of the cells were infected with SeV for 18 h. A, nuclear extracts weremade and analyzed by Western blot (5% of input used in the binding reaction). The nuclear extracts were incubated with biotinylated oligonucleotidescontaining either the IFN-� (B), the PRD-I–III (C), PRD-II (D), or the PRD-IV (E) promoter sequences. Streptavidin beads were added for 40 min, washed threetimes, boiled in Laemmli sample buffer, and resolved by SDS-PAGE. Western blots were carried out for the detection of IRF3 (Myc), LANA-1 (HA), ATF-2, c-Jun,p50, and CBP using specific antibodies. PARP-1 was used as a control to demonstrate the equal amounts of nuclear proteins between the different samples.F, binding of IRF3 on the ISG15 promoter in cell extracts of HEC-1B cells transfected with 500 ng of FLAG-IRF3 and 2 �g of pCMV3T or pCMV3T-LANA-1 andinfected or not by 200 HAU of SeV for 6 h was analyzed by EMSA. Specificity of binding was confirmed by homologous competition using excess (25 times)unlabeled ISG15 oligonucleotides. Supershift experiments were done using 2 �g of anti-FLAG (IRF3) antibody. * and ** denote different protein complexes onthe ISG15 promoter. G and H, fraction of the cells used for EMSA (F) were kept to analyze the IFNB (G) and ISG15 (H) mRNA content by RT-QPCR. Results areexpressed as mean (triplicate) induction � S.D. relative to pCMV3T- transfected cells after normalization of samples with GAPDH mRNA expression. I and J,HEK-293T cells were transfected with 2 �g of pCMV3T-LANA-1 in a 10-cm dish. Twenty four hours later, half of the cells were infected with 20 HAU of SeV for18 h. Nuclear extracts were isolated and measured using BCA assay, and 5 �g were used to determine the binding activity. I, endogenous IRF3 binding activitydetermined using the TransAMTM IRF3 ELISA (Active Motif). Results are expressed mean � S.D. of IRF3 binding (OD 450 nm). J, LANA-1 binding on the IRF3consensus binding site was determined using a modified TransAMTM ELISA by substituting the anti-IRF3 primary antibody for anti-LANA-1 diluted 1:1000. *,0.01 � p � 0.05; **, 0.001 � p � 0.01; ***, p � 0.0001 as determined using two-tailed t test.
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7215
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
tion mutants of LANA-1 (Fig. 5A). All of these mutants werecloned in-framewith three HA tags at the N terminus. Both theN- and C-terminal regions of the protein contain nuclear local-ization sequences, but, in accordancewith the study of Schwamet al. (9), onlymutants containing theN-terminal regions local-ized in the nucleus suggesting that the C-terminal NLS is non-functional in our hands. Consequently, we decided to addNLS atthe N-terminal region of these deletion mutants: LANA-1 854–1162, LANA-1 888–1162, LANA-1 319–1162, and LANA-1319–892. All mutants were sequenced and analyzed for expres-sion byWestern blot using anti-HA antibodies (Fig. 5B).We nextverified the cellular localization of these mutants, and all wereexpressed in the nucleus (Fig. 5C). All mutants were expresseduniformly in the nucleus except LANA-1 854–1162 , LANA-1888–1162 ,andLANA-1319–1162thatwereexpressedasnuclearspeckles. Schwam et al. (9) described that the deletion of residues1020–1089prevents formationof nuclear speckles.Our immuno-fluorescence results confirm this fact, with the exception thatLANA-1 1–1162 (WT)was expressed uniformly in the nucleus inHEK-293T cells. Cell transfected with the pCMV3T (HA) controlvector displayed cytoplasmic staining.To determine whether these LANA-1 mutants influenced
IFNB gene expression, we transfected them into HEK-293Tcells alone or in combination with TBK1. Total RNA wasextracted, and endogenous IFNB gene expression was moni-tored by RT-QPCR. As shown in Fig. 6, TBK1 expressioninduced IFNB gene expression efficiently in the absence ofLANA-1. In contrast, in cells expressing LANA-1 1–1162, theactivation of the IFNB gene upon TBK1 co-transfection wasstrongly reduced, confirming the results shown in Fig. 1A.LANA-1mutants 1–996, 1–875, 319–1162, and 319–892were
equally effective at suppressing the activation of IFNB induced byTBK1. As a control, we performed an experiment using LANA-1mutant 319–892 cloned into pCMV3T vector. This mutant ofLANA-1, which lacks an NLS and does not enter the nucleus,could no longer inhibit IFNB gene induction upon TBK1 expres-sion suggesting an essential role for nuclear localization ofLANA-1 (data not shown). LANA-1 mutants 1–300, 854–1162,and 888–1162 displayed weak inhibitory activities toward TBK1-activated IFNB gene expression. These three mutants lack thehighly repetitive central domain of LANA-1, suggesting a role forthis domain in the inhibition of IFN-� synthesis.We next tested whether inhibition of IFNB gene expression
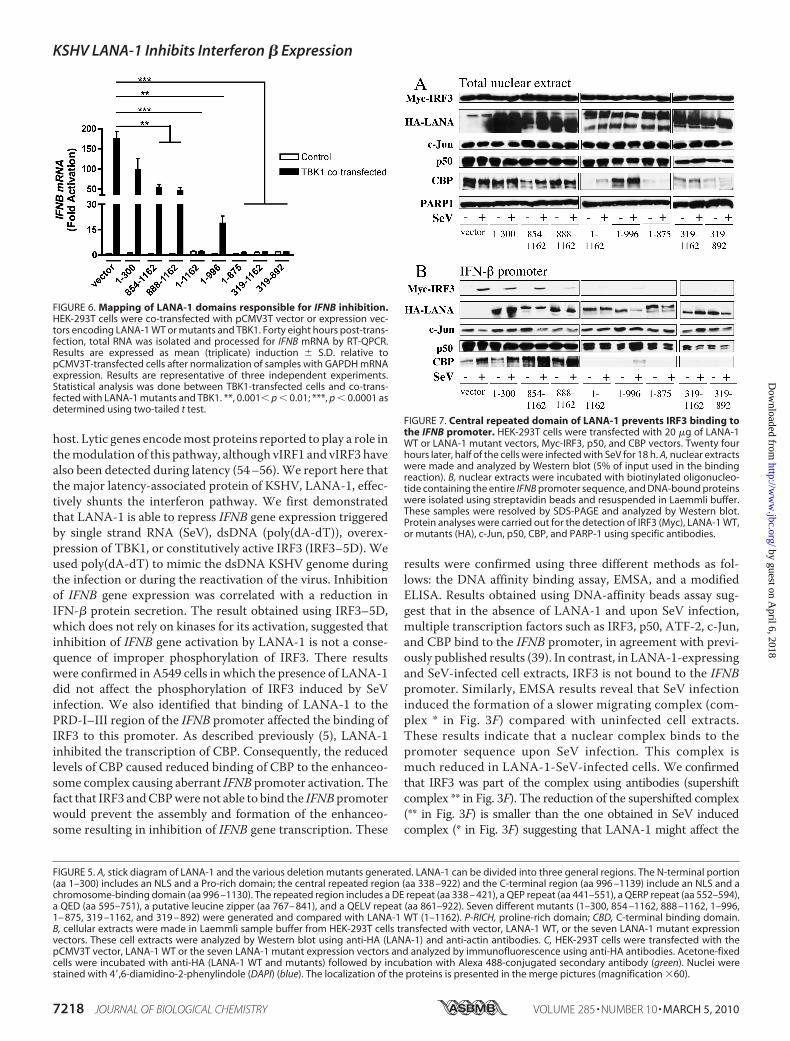
by the LANA-1mutants could be correlatedwith their ability tobind to the PRD-I–III domain. As described above (Fig. 3),nuclear extracts from resting and SeV-infected cells co-trans-fected with expression vectors of Myc-IRF3, HA-LANA-1mutants, p50, and CBP were analyzed by Western blot usinganti-Myc, anti-HA, anti-c-Jun, anti-p50, anti-CBP, and anti-PARP-1 antibodies to compare nuclear inputs (Fig. 7A). In gen-eral, with the exception of LANA-1 expression and CBP, thenuclear inputs were similar between all conditions. CBP wasstrongly reduced in nuclear extracts in the presence of LANA-11–1162 and LANA-1 mutants 1–875, 319–1162, and 319–892. Interestingly, LANA-1 mutant 1–996 did not reduce theexpression of CBP. Double-stranded oligonucleotides contain-ing the entire IFNB promoter were incubated with the nuclearextracts, and the proteins bound to this promoter were ana-lyzed byWestern blot (Fig. 7B). As shown, all LANA-1mutantswere capable of binding the promoter to some degree, suggest-ing that multiple domains of LANA-1 can bind the IFNB pro-moter, and SeV infection did not affect this binding. In responseto SeV infection, IRF3 could efficiently attach to the IFNB pro-moter in pCMV3T nuclear extracts, but this binding varied inthe presence of various LANA-1 mutants. LANA-1 1–1162abolished the fixation of IRF3 to this promoter. LANA-1mutants 1–996, 1–875, 319–1162, and 319–892 acted likeLANA-1 1–1162 and strongly reduced the binding of IRF3 tothe promoter. All of thesemutants possess the LANA-1 centralrepeated domain (Fig. 5A) and strongly reduced the IFNB geneinduction caused by TBK1 (Fig. 6). The LANA-1 mutants1–300, 854–1162, and 888–1162 reduced slightly the fixationof IRF3 to the IFNB promoter, but these mutants but were notas effective as LANA-1 1–1162. The binding of c-Jun was notsignificantly affected by the presence of LANA-1 mutants. Thefixation of p50 to the IFNB promoter wasminimally affected bythe presence of LANA-1 1–1162 and mutants. The binding ofCBP to IFNB promoter was affected with LANA-1 1–1162 andLANA-1 mutants 1–996, 1–875, 319–1162, and 319–892.However, considering the varying levels of nuclear CBP, a firmconclusion is difficult to make. These results suggest that thecentral repeated domain of LANA-1 is responsible for inhibit-ing the binding of IRF3 to the IFNB promoter. Compared withIRF3, the binding of p50 was only minimally affected. By occu-pying the IRF3-binding site, LANA-1 prevents the properassembly of the enhanceosome causing inefficient IFNB geneactivation.We next attempted to demonstrate the IFN inhibitory role of
endogenous LANA-1 in KSHV-infected cells. To do so, we
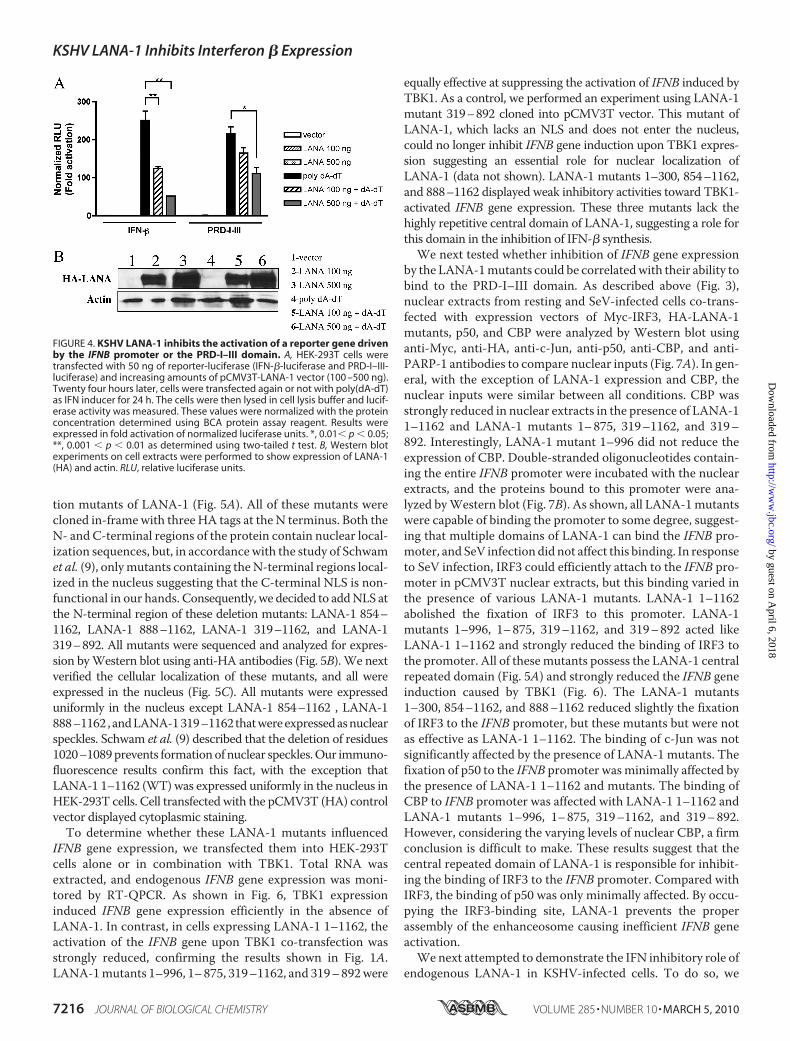
FIGURE 4. KSHV LANA-1 inhibits the activation of a reporter gene drivenby the IFNB promoter or the PRD-I–III domain. A, HEK-293T cells weretransfected with 50 ng of reporter-luciferase (IFN-�-luciferase and PRD-I–III-luciferase) and increasing amounts of pCMV3T-LANA-1 vector (100 –500 ng).Twenty four hours later, cells were transfected again or not with poly(dA-dT)as IFN inducer for 24 h. The cells were then lysed in cell lysis buffer and lucif-erase activity was measured. These values were normalized with the proteinconcentration determined using BCA protein assay reagent. Results wereexpressed in fold activation of normalized luciferase units. *, 0.01� p � 0.05;**, 0.001 � p � 0.01 as determined using two-tailed t test. B, Western blotexperiments on cell extracts were performed to show expression of LANA-1(HA) and actin. RLU, relative luciferase units.
KSHV LANA-1 Inhibits Interferon � Expression
7216 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
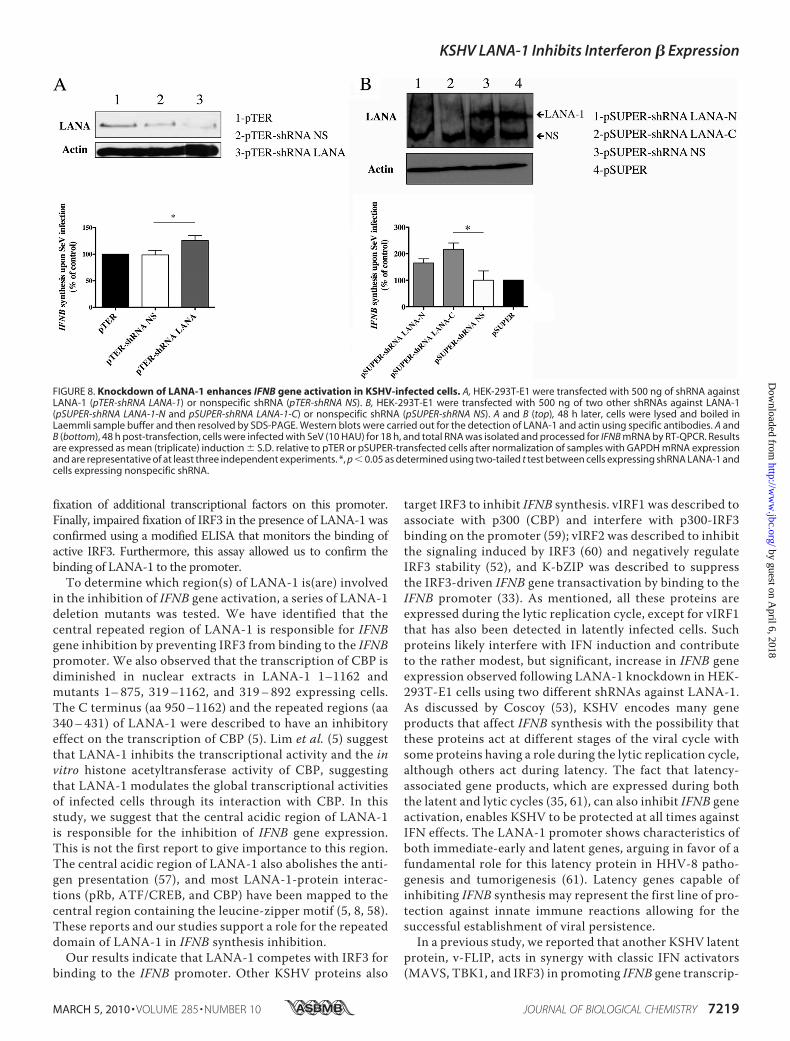
knocked down LANA-1 expression in HEK-293T-E1 cells car-rying a recombinant KSHV (20). HEK-293T-E1 cells weretransfected with a pTER vector encoding an shRNA againstLANA-1 (pTER-shRNA LANA-1)or a nonspecific shRNA (pTER-shRNA NS) (Fig. 8A, top). To con-firm these results, two additionalLANA-1 shRNAs (pSUPER-shRNALANA-1-C and -N) and nonspecificshRNA (pSUPER-shRNA NS) werealso used (Fig. 8B, top). All theseshRNAs, were able to reduce theexpression of LANA-1. The controlHEK-293T-E1 cells and those withreduced LANA-1 expression werethen compared for IFNB geneexpression following SeV infection.As shown in Fig. 8A (bottom), cellsexpressing shRNA LANA-1 (pTER-shRNA LANA-1) had significantlyhigher IFNBmRNA levels upon SeVinfection than vector (pTER) orshRNANS (pTER-shRNANS), sug-gesting that when LANA-1 levelsare reduced, IFNB gene expressionin response to SeV occurs moreefficiently. Similar results wereobtained using the second shRNAsLANA-1 (Fig. 8B, bottom). It is note-worthy that HEK-293T-E1 cellstransfected by pSUPER-shRNALANA-1-C expressed 2.63 times(263%) more IFNB mRNA com-paredwith cells expressing the pSU-PER control vector (100%). ThisshRNA is very efficient at reducingexpression of LANA-1 as observedin Fig. 8B (top). HEK-293T-E1 cellstransfected by pSUPER-shRNALANA-1-N expressed also in-creased levels of the IFNB gene,but the results obtained did notreach statistical significance. Thiscould be explained, at least in part,by the weaker reduction in LANA-1expression observed in Fig. 8B (top).These results support an IFN-�inhibitory role for LANA-1 in thecontext of KSHV-infected cells.
DISCUSSION
KSHV deploys several mecha-nisms to evade the effects of inter-feron, including four vIRFs sharinghomologies with cellular IRFs (46,48), expression of ORF45 (49), RTA(49), K-bZIP (33), v-FLIP (50), andRIF (encoded by orf10) (51) (for
reviews see Refs. 52, 53). The large number of strategies used byKSHV to inhibit interferon synthesis suggests that this antiviralpathway is detrimental toKSHVpersistencewithin the infected
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7217
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
host. Lytic genes encodemost proteins reported to play a role inthemodulation of this pathway, although vIRF1 and vIRF3 havealso been detected during latency (54–56).We report here thatthe major latency-associated protein of KSHV, LANA-1, effec-tively shunts the interferon pathway. We first demonstratedthat LANA-1 is able to repress IFNB gene expression triggeredby single strand RNA (SeV), dsDNA (poly(dA-dT)), overex-pression of TBK1, or constitutively active IRF3 (IRF3–5D). Weused poly(dA-dT) to mimic the dsDNA KSHV genome duringthe infection or during the reactivation of the virus. Inhibitionof IFNB gene expression was correlated with a reduction inIFN-� protein secretion. The result obtained using IRF3–5D,which does not rely on kinases for its activation, suggested thatinhibition of IFNB gene activation by LANA-1 is not a conse-quence of improper phosphorylation of IRF3. There resultswere confirmed in A549 cells in which the presence of LANA-1did not affect the phosphorylation of IRF3 induced by SeVinfection. We also identified that binding of LANA-1 to thePRD-I–III region of the IFNB promoter affected the binding ofIRF3 to this promoter. As described previously (5), LANA-1inhibited the transcription of CBP. Consequently, the reducedlevels of CBP caused reduced binding of CBP to the enhanceo-some complex causing aberrant IFNB promoter activation. Thefact that IRF3 andCBPwere not able to bind the IFNBpromoterwould prevent the assembly and formation of the enhanceo-some resulting in inhibition of IFNB gene transcription. These
results were confirmed using three different methods as fol-lows: the DNA affinity binding assay, EMSA, and a modifiedELISA. Results obtained using DNA-affinity beads assay sug-gest that in the absence of LANA-1 and upon SeV infection,multiple transcription factors such as IRF3, p50, ATF-2, c-Jun,and CBP bind to the IFNB promoter, in agreement with previ-ously published results (39). In contrast, in LANA-1-expressingand SeV-infected cell extracts, IRF3 is not bound to the IFNBpromoter. Similarly, EMSA results reveal that SeV infectioninduced the formation of a slower migrating complex (com-plex * in Fig. 3F) compared with uninfected cell extracts.These results indicate that a nuclear complex binds to thepromoter sequence upon SeV infection. This complex ismuch reduced in LANA-1-SeV-infected cells. We confirmedthat IRF3 was part of the complex using antibodies (supershiftcomplex ** in Fig. 3F). The reduction of the supershifted complex(** in Fig. 3F) is smaller than the one obtained in SeV inducedcomplex (* in Fig. 3F) suggesting that LANA-1 might affect the
FIGURE 5. A, stick diagram of LANA-1 and the various deletion mutants generated. LANA-1 can be divided into three general regions. The N-terminal portion(aa 1–300) includes an NLS and a Pro-rich domain; the central repeated region (aa 338 –922) and the C-terminal region (aa 996 –1139) include an NLS and achromosome-binding domain (aa 996 –1130). The repeated region includes a DE repeat (aa 338 – 421), a QEP repeat (aa 441–551), a QERP repeat (aa 552–594),a QED (aa 595–751), a putative leucine zipper (aa 767– 841), and a QELV repeat (aa 861–922). Seven different mutants (1–300, 854 –1162, 888 –1162, 1–996,1– 875, 319 –1162, and 319 – 892) were generated and compared with LANA-1 WT (1–1162). P-RICH, proline-rich domain; CBD, C-terminal binding domain.B, cellular extracts were made in Laemmli sample buffer from HEK-293T cells transfected with vector, LANA-1 WT, or the seven LANA-1 mutant expressionvectors. These cell extracts were analyzed by Western blot using anti-HA (LANA-1) and anti-actin antibodies. C, HEK-293T cells were transfected with thepCMV3T vector, LANA-1 WT or the seven LANA-1 mutant expression vectors and analyzed by immunofluorescence using anti-HA antibodies. Acetone-fixedcells were incubated with anti-HA (LANA-1 WT and mutants) followed by incubation with Alexa 488-conjugated secondary antibody (green). Nuclei werestained with 4�,6-diamidino-2-phenylindole (DAPI) (blue). The localization of the proteins is presented in the merge pictures (magnification �60).
FIGURE 6. Mapping of LANA-1 domains responsible for IFNB inhibition.HEK-293T cells were co-transfected with pCMV3T vector or expression vec-tors encoding LANA-1 WT or mutants and TBK1. Forty eight hours post-trans-fection, total RNA was isolated and processed for IFNB mRNA by RT-QPCR.Results are expressed as mean (triplicate) induction � S.D. relative topCMV3T-transfected cells after normalization of samples with GAPDH mRNAexpression. Results are representative of three independent experiments.Statistical analysis was done between TBK1-transfected cells and co-trans-fected with LANA-1 mutants and TBK1. **, 0.001� p � 0.01; ***, p � 0.0001 asdetermined using two-tailed t test.
FIGURE 7. Central repeated domain of LANA-1 prevents IRF3 binding tothe IFNB promoter. HEK-293T cells were transfected with 20 �g of LANA-1WT or LANA-1 mutant vectors, Myc-IRF3, p50, and CBP vectors. Twenty fourhours later, half of the cells were infected with SeV for 18 h. A, nuclear extractswere made and analyzed by Western blot (5% of input used in the bindingreaction). B, nuclear extracts were incubated with biotinylated oligonucleo-tide containing the entire IFNB promoter sequence, and DNA-bound proteinswere isolated using streptavidin beads and resuspended in Laemmli buffer.These samples were resolved by SDS-PAGE and analyzed by Western blot.Protein analyses were carried out for the detection of IRF3 (Myc), LANA-1 WT,or mutants (HA), c-Jun, p50, CBP, and PARP-1 using specific antibodies.
KSHV LANA-1 Inhibits Interferon � Expression
7218 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
fixation of additional transcriptional factors on this promoter.Finally, impaired fixation of IRF3 in the presence of LANA-1 wasconfirmed using a modified ELISA that monitors the binding ofactive IRF3. Furthermore, this assay allowed us to confirm thebinding of LANA-1 to the promoter.To determine which region(s) of LANA-1 is(are) involved
in the inhibition of IFNB gene activation, a series of LANA-1deletion mutants was tested. We have identified that thecentral repeated region of LANA-1 is responsible for IFNBgene inhibition by preventing IRF3 from binding to the IFNBpromoter. We also observed that the transcription of CBP isdiminished in nuclear extracts in LANA-1 1–1162 andmutants 1–875, 319–1162, and 319–892 expressing cells.The C terminus (aa 950–1162) and the repeated regions (aa340–431) of LANA-1 were described to have an inhibitoryeffect on the transcription of CBP (5). Lim et al. (5) suggestthat LANA-1 inhibits the transcriptional activity and the invitro histone acetyltransferase activity of CBP, suggestingthat LANA-1 modulates the global transcriptional activitiesof infected cells through its interaction with CBP. In thisstudy, we suggest that the central acidic region of LANA-1is responsible for the inhibition of IFNB gene expression.This is not the first report to give importance to this region.The central acidic region of LANA-1 also abolishes the anti-gen presentation (57), and most LANA-1-protein interac-tions (pRb, ATF/CREB, and CBP) have been mapped to thecentral region containing the leucine-zipper motif (5, 8, 58).These reports and our studies support a role for the repeateddomain of LANA-1 in IFNB synthesis inhibition.
Our results indicate that LANA-1 competes with IRF3 forbinding to the IFNB promoter. Other KSHV proteins also
target IRF3 to inhibit IFNB synthesis. vIRF1 was described toassociate with p300 (CBP) and interfere with p300-IRF3binding on the promoter (59); vIRF2 was described to inhibitthe signaling induced by IRF3 (60) and negatively regulateIRF3 stability (52), and K-bZIP was described to suppressthe IRF3-driven IFNB gene transactivation by binding to theIFNB promoter (33). As mentioned, all these proteins areexpressed during the lytic replication cycle, except for vIRF1that has also been detected in latently infected cells. Suchproteins likely interfere with IFN induction and contributeto the rather modest, but significant, increase in IFNB geneexpression observed following LANA-1 knockdown in HEK-293T-E1 cells using two different shRNAs against LANA-1.As discussed by Coscoy (53), KSHV encodes many geneproducts that affect IFNB synthesis with the possibility thatthese proteins act at different stages of the viral cycle withsome proteins having a role during the lytic replication cycle,although others act during latency. The fact that latency-associated gene products, which are expressed during boththe latent and lytic cycles (35, 61), can also inhibit IFNB geneactivation, enables KSHV to be protected at all times againstIFN effects. The LANA-1 promoter shows characteristics ofboth immediate-early and latent genes, arguing in favor of afundamental role for this latency protein in HHV-8 patho-genesis and tumorigenesis (61). Latency genes capable ofinhibiting IFNB synthesis may represent the first line of pro-tection against innate immune reactions allowing for thesuccessful establishment of viral persistence.In a previous study, we reported that another KSHV latent
protein, v-FLIP, acts in synergy with classic IFN activators(MAVS, TBK1, and IRF3) in promoting IFNB gene transcrip-
FIGURE 8. Knockdown of LANA-1 enhances IFNB gene activation in KSHV-infected cells. A, HEK-293T-E1 were transfected with 500 ng of shRNA againstLANA-1 (pTER-shRNA LANA-1) or nonspecific shRNA (pTER-shRNA NS). B, HEK-293T-E1 were transfected with 500 ng of two other shRNAs against LANA-1(pSUPER-shRNA LANA-1-N and pSUPER-shRNA LANA-1-C) or nonspecific shRNA (pSUPER-shRNA NS). A and B (top), 48 h later, cells were lysed and boiled inLaemmli sample buffer and then resolved by SDS-PAGE. Western blots were carried out for the detection of LANA-1 and actin using specific antibodies. A andB (bottom), 48 h post-transfection, cells were infected with SeV (10 HAU) for 18 h, and total RNA was isolated and processed for IFNB mRNA by RT-QPCR. Resultsare expressed as mean (triplicate) induction � S.D. relative to pTER or pSUPER-transfected cells after normalization of samples with GAPDH mRNA expressionand are representative of at least three independent experiments. *, p � 0.05 as determined using two-tailed t test between cells expressing shRNA LANA-1 andcells expressing nonspecific shRNA.
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7219
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

tion (50). This effect was strictly dependent on NF-�B and ismediated through the PRD-II of the IFNB promoter. Duringinfection, LANA-1, v-cyclin, and v-FLIP are simultaneouslytranscribed from the same promoter on polycistronic orbicistronic (v-cyclin and v-FLIP) transcripts (61). Our resultssuggest that LANA-1 and v-FLIP may have opposing effectsin IFNB gene modulation. Preliminary results suggest thatwhen LANA-1 and v-FLIP are co-expressed, the synthesis ofbiologically active IFN-� protein is reduced suggesting thatthe LANA-1 effects are dominant. The LANA-1 promotercould also achieve a balancing effect between these two pro-teins through the regulation of the polycistronic/bicistronictranscripts. A cellular model of latency-infected cells usingknock-out virus or small interfering RNA will help us tounderstand more the role of LANA-1 in the synthesis ofIFNB. When available, the use of an animal model will beessential to further our understanding of the interplaybetween KSHV and the type I IFN signaling pathway.
Acknowledgments—We thank Didier Trouche, John Hiscott, Rong-tuan Lin, TomManiatis, Shou-Jiang Gao, Nathalie Grandvaux, andDiane Hayward for generously providing plasmids, antibodies, or cellline.
REFERENCES1. Cesarman, E., Chang, Y., Moore, P. S., Said, J. W., and Knowles, D. M.
(1995) N. Engl. J. Med. 332, 1186–11912. Zhong, W., Wang, H., Herndier, B., and Ganem, D. (1996) Proc. Natl.
Acad. Sci. U.S.A. 93, 6641–66463. Ballestas,M. E., Chatis, P. A., andKaye, K.M. (1999) Science 284, 641–6444. Krithivas, A., Young, D. B., Liao, G., Greene, D., andHayward, S. D. (2000)
J. Virol. 74, 9637–96455. Lim, C., Gwack, Y., Hwang, S., Kim, S., and Choe, J. (2001) J. Biol. Chem.
276, 31016–310226. Platt, G.M., Simpson, G. R., Mittnacht, S., and Schulz, T. F. (1999) J. Virol.
73, 9789–97957. Liu, J., Martin, H., Shamay, M., Woodard, C., Tang, Q. Q., and Hayward,
S. D. (2007) J. Virol. 81, 4722–47318. Lim, C., Sohn, H., Gwack, Y., and Choe, J. (2000) J. Gen. Virol. 81,
2645–26529. Schwam, D. R., Luciano, R. L., Mahajan, S. S., Wong, L., andWilson, A. C.
(2000) J. Virol. 74, 8532–854010. Friborg, J., Jr., Kong, W., Hottiger, M. O., and Nabel, G. J. (1999) Nature
402, 889–89411. Bubman, D., Guasparri, I., and Cesarman, E. (2007) Oncogene 26,
4979–498612. Jeong, J. H., Orvis, J., Kim, J.W.,McMurtrey, C. P., Renne, R., andDittmer,
D. P. (2004) J. Biol. Chem. 279, 16822–1683113. Lan, K., Kuppers, D. A., Verma, S. C., and Robertson, E. S. (2004) J. Virol.
78, 6585–659414. Malmgaard, L. (2004) J. Interferon Cytokine Res. 24, 439–45415. Rasmussen, S. B., Jensen, S. B., Nielsen, C., Quartin, E., Kato, H., Chen,
Z. J., Silverman, R. H., Akira, S., and Paludan, S. R. (2009) J. Gen. Virol. 90,74–78
16. Kawai, T., and Akira, S. (2006) Nat. Immunol. 7, 131–13717. Naranatt, P. P., Krishnan, H. H., Svojanovsky, S. R., Bloomer, C., Mathur,
S., and Chandran, B. (2004) Cancer Res. 64, 72–8418. Perry, S. T., and Compton, T. (2006) J. Virol. 80, 11105–1111419. Fuse, A., Ashino-Fuse, H., and Kuwata, T. (1984) Gann 75, 379–38420. Zhou, F. C., Zhang, Y. J., Deng, J. H.,Wang, X. P., Pan,H. Y., Hettler, E., and
Gao, S. J. (2002) J. Virol. 76, 6185–619621. Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E.,
Golenbock, D. T., Coyle, A. J., Liao, S. M., and Maniatis, T. (2003) Nat.
Immunol. 4, 491–49622. Lin, R., Yang, L., Nakhaei, P., Sun, Q., Sharif-Askari, E., Julkunen, I., and
Hiscott, J. (2006) J. Biol. Chem. 281, 2095–210323. Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R., and His-
cott, J. (2003) Science 300, 1148–115124. Duckett, C. S., Perkins, N. D., Kowalik, T. F., Schmid, R. M., Huang,
E. S., Baldwin, A. S., Jr., and Nabel, G. J. (1993) Mol. Cell. Biol. 13,1315–1322
25. Ramirez, S., Ait-Si-Ali, S., Robin, P., Trouche, D., Harel-Bellan, A., and AitSi Ali, S. (1997) J. Biol. Chem. 272, 31016–31021
26. Godfrey, A., Anderson, J., Papanastasiou, A., Takeuchi, Y., and Boshoff, C.(2005) Blood 105, 2510–2518
27. van deWetering, M., Oving, I., Muncan, V., Pon Fong, M. T., Brantjes, H.,van Leenen, D., Holstege, F. C., Brummelkamp, T. R., Agami, R., andClevers, H. (2003) EMBO Rep. 4, 609–615
28. Lefort, S., and Flamand, L. (2009) J. Virol. 83, 5869–588029. Fujimuro, M., Wu, F. Y., ApRhys, C., Kajumbula, H., Young, D. B., Hay-
ward, G. S., and Hayward, S. D. (2003) Nat. Med. 9, 300–30630. Saitoh, T., Tun-Kyi, A., Ryo, A., Yamamoto, M., Finn, G., Fujita, T., Akira,
S., Yamamoto, N., Lu, K. P., and Yamaoka, S. (2006) Nat. Immunol. 7,598–605
31. Lamarre, D., Talbot, B., de Murcia, G., Laplante, C., Leduc, Y., Mazen, A.,and Poirier, G. G. (1988) Biochim. Biophys. Acta 950, 147–160
32. Severa, M., Coccia, E. M., and Fitzgerald, K. A. (2006) J. Biol. Chem. 281,26188–26195
33. Lefort, S., Soucy-Faulkner, A., Grandvaux, N., and Flamand, L. (2007)J. Virol. 81, 10950–10960
34. Wathelet, M. G., Lin, C. H., Parekh, B. S., Ronco, L. V., Howley, P. M., andManiatis, T. (1998)Mol. Cell 1, 507–518
35. Cloutier, N., Gravel, A., and Flamand, L. (2004) J. Virol. Methods 122,1–7
36. Dittmer, D., Lagunoff,M., Renne, R., Staskus, K., Haase, A., andGanem,D.(1998) J. Virol. 72, 8309–8315
37. Hiscott, J., Grandvaux, N., Sharma, S., Tenoever, B. R., Servant, M. J., andLin, R. (2003) Ann. N.Y. Acad. Sci. 1010, 237–248
38. Yoneyama,M., Suhara,W., and Fujita, T. (2002) J. Interferon Cytokine Res.22, 73–76
39. Maniatis, T., Falvo, J. V., Kim, T. H., Kim, T. K., Lin, C. H., Parekh, B. S.,and Wathelet, M. G. (1998) Cold Spring Harbor Symp. Quant. Biol. 63,609–620
40. Panne, D., Maniatis, T., and Harrison, S. C. (2007) Cell 129, 1111–112341. Au,W. C., Moore, P. A., Lowther,W., Juang, Y. T., and Pitha, P. M. (1995)
Proc. Natl. Acad. Sci. U.S.A. 92, 11657–1166142. Wathelet, M. G., Clauss, I. M., Content, J., and Huez, G. A. (1988) Eur.
J. Biochem. 174, 323–32943. Hiscott, J., Pitha, P., Genin, P., Nguyen, H., Heylbroeck, C., Mamane, Y.,
Algarte, M., and Lin, R. (1999) J. Interferon Cytokine Res. 19, 1–1344. Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., Lu, Y.,
Miyagishi, M., Kodama, T., Honda, K., Ohba, Y., and Taniguchi, T. (2007)Nature 448, 501–505
45. Stetson, D. B., and Medzhitov, R. (2006) Immunity 24, 93–10346. Russo, J. J., Bohenzky, R. A., Chien,M.C., Chen, J., Yan,M.,Maddalena,D.,
Parry, J. P., Peruzzi, D., Edelman, I. S., Chang, Y., and Moore, P. S. (1996)Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867
47. Ganem, D. (2006) Annu. Rev. Pathol. 1, 273–29648. Rezaee, S. A., Cunningham, C., Davison, A. J., and Blackbourn, D. J. (2006)
J. Gen. Virol. 87, 1781–180449. Zhu, F. X., King, S. M., Smith, E. J., Levy, D. E., and Yuan, Y. (2002) Proc.
Natl. Acad. Sci. U.S.A. 99, 5573–557850. Cloutier, N., Grandvaux, N., and Flamand, L. (2007) Eur. J. Immunol. 37,
2772–277851. Bisson, S. A., Page, A. L., and Ganem, D. (2009) J. Virol. 83, 5056–506652. Areste, C., and Blackbourn, D. J. (2009) Trends Microbiol. 17, 119–12953. Coscoy, L. (2007) Nat. Rev. Immunol. 7, 391–40154. Parravicini, C., Chandran, B., Corbellino, M., Berti, E., Paulli, M., Moore,
P. S., and Chang, Y. (2000) Am. J. Pathol. 156, 743–74955. Fakhari, F. D., and Dittmer, D. P. (2002) J. Virol. 76, 6213–6223
KSHV LANA-1 Inhibits Interferon � Expression
7220 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 10 • MARCH 5, 2010
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

56. Rivas, C., Thlick, A. E., Parravicini, C., Moore, P. S., and Chang, Y. (2001)J. Virol. 75, 429–438
57. Zaldumbide, A., Ossevoort, M., Wiertz, E. J., and Hoeben, R. C. (2007)Mol. Immunol. 44, 1352–1360
58. Radkov, S. A., Kellam, P., and Boshoff, C. (2000) Nat. Med. 6,1121–1127
59. Lin, R., Genin, P., Mamane, Y., Sgarbanti, M., Battistini, A., Harrington,W. J., Jr., Barber, G. N., and Hiscott, J. (2001) Oncogene 20, 800–811
60. Fuld, S., Cunningham, C., Klucher, K., Davison, A. J., and Blackbourn, D. J.(2006) J. Virol. 80, 3092–3097
61. Talbot, S. J., Weiss, R. A., Kellam, P., and Boshoff, C. (1999) Virology 257,84–94
KSHV LANA-1 Inhibits Interferon � Expression
MARCH 5, 2010 • VOLUME 285 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 7221
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Nathalie Cloutier and Louis Flamand PromoterIFNBFactor-3 for Binding to
Expression by Competing with IFN RegulatoryβInhibits Interferon (IFN) Kaposi Sarcoma-associated Herpesvirus Latency-associated Nuclear Antigen
doi: 10.1074/jbc.M109.018838 originally published online January 4, 20102010, 285:7208-7221.J. Biol. Chem.
10.1074/jbc.M109.018838Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2010/01/20/M109.018838.DC1
http://www.jbc.org/content/285/10/7208.full.html#ref-list-1
This article cites 61 references, 30 of which can be accessed free at
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from