investigating the role of a natural reca mutation on bcg ... · investigating the role of a natural...
TRANSCRIPT

Investigating the Role of a Natural RecA Mutation on BCG-Russia Vaccine Properties
by
Mark Ng
A thesis submitted in conformity with the requirements for the degree of Master’s of Science
Department of Molecular Genetics University of Toronto
© Copyright by Mark Ng 2016

ii
Investigating the Role of a Natural RecA Mutation on BCG-Russia
Vaccine Properties
Mark Ng
Master’s of Science
Graduate Department of Molecular Genetics
University of Toronto
2016
Abstract
The Bacille Calmette-Guérin (BCG) vaccine is the only vaccine for tuberculosis (TB) control.
Although effective in preventing disseminated forms of TB in children, its efficacy in preventing
pulmonary TB in adults is highly variable. BCG-Russia, one of the most widely used substrains
to produce the vaccine, is a natural RecA mutant. Whether the presence of functional RecA
affects the virulence and efficacy of BCG-Russia remain unknown. Here I show that a
recombinant BCG-Russia expressing functional RecA, rBCG-RecA, is more attenuated than its
parental counterpart in SCID mice and this difference is due to slower growth within the animals.
I also demonstrate that rBCG-RecA confers equivalent protection against M. tb challenge as the
parental strain in guinea pigs with similar bacterial burden in the organs. Taken together, the
presence of RecA in BCG-Russia allows for a safer yet equally protective vaccine. This work
provides new insight to future TB vaccine design.

iii
Acknowledgments
First and foremost, I would like to thank my supervisor, Dr. Jun Liu, for the opportunity to work
on this project and for his ongoing support over the last three years. Without your expertise and
guidance, this work would not be possible. I would also like to thank my committee members,
Dr. Alex Ensminger and Dr. John Brumell, for taking the time to provide constructive comments
and critiques which helped to shape my project. My gratitude goes to all the DCM/BSL3 staff for
their contribution to this work.
Thank you to all the past and present members of the Liu lab, especially Steven Ahn and Ming
Li for their assistance in the guinea pig experiments. Also I want to thank Dr. Howard Song, Dr.
Vanessa Tran, and Jackie Liu for teaching me and for providing me with their insightful advice
which was instrumental in bringing this work into success.
I would also like to thank my family and friends for their constant support and encouragement
throughout all stages of my life as I wouldn’t be where I am today without you.

iv
Table of Contents
Acknowledgments.......................................................................................................................... iii
Table of Contents ........................................................................................................................... iv
List of Figures ................................................................................................................................ vi
List of Abbreviations .................................................................................................................... vii
Chapter 1 General Introduction .......................................................................................................1
1.1 Tuberculosis .........................................................................................................................1
1.1.1 Mycobacterium tuberculosis ....................................................................................1
1.1.2 Pathogenesis of Mycobacterium tuberculosis ..........................................................3
1.1.3 Global burden of TB and increase in anti-mycobacterial resistance .......................6
1.2 The Bacille Calmette-Guérin vaccine ..................................................................................7
1.2.1 History of the BCG vaccine .....................................................................................8
1.2.2 The mechanism and attenuation of BCG .................................................................9
1.2.3 Variation in the clinical properties of BCG ...........................................................11
1.3 BCG-Russia, a natural RecA mutant .................................................................................12
1.3.1 Properties of BCG-Russia ......................................................................................12
1.3.2 Characteristics of Mycobacterium RecA ...............................................................14
1.4 Current vaccine development ............................................................................................16
1.4.1 Recombinant and attenuated M.tb vaccines ...........................................................16
1.4.2 Subunit vaccines ....................................................................................................18
1.4.3 DNA vaccines ........................................................................................................19
1.5 Rationale and hypothesis ...................................................................................................21
Chapter 2 Materials and Methods ..................................................................................................23
2.1 Bacterial strains and culture conditions .............................................................................23
2.2 Cloning of recombinant BCG-Russia RecA ......................................................................23

v
2.3 Mitomycin C tolerance assay .............................................................................................24
2.4 In vitro growth assay ..........................................................................................................24
2.5 Virulence in SCID mice .....................................................................................................25
2.6 Protection against M. tb challenge in guinea pigs ..............................................................25
2.7 Histological analysis on animal tissue ...............................................................................26
Chapter 3 Results ...........................................................................................................................28
3.1 Confirmation of functional RecA in rBCG-RecA .............................................................28
3.2 rBCG-RecA does not exhibit an inherent growth defect ...................................................28
3.3 RecA attenuates BCG-Russia in SCID mice .....................................................................31
3.4 Organ weights of SCID mice suggest lower virulence of rBCG-RecA .............................33
3.5 The attenuation is due to slower replication of rBCG-RecA in the host ...........................35
3.6 rBCG-RecA does not have an intracellular growth defect in macrophages ......................37
3.7 rBCG-RecA confers protection similar to that of the parental strain ................................39
3.8 rBCG-RecA-vaccinated guinea pigs exhibit disease phenotypes similar to the
parental-vaccinated animals ...............................................................................................41
3.9 Comparable lung pathology observed in rBCG-RecA-vaccinated and parental-
vaccinated animals .............................................................................................................43
Chapter 4 Discussion .....................................................................................................................46
4.1 General Discussion ............................................................................................................46
4.2 Summary of key findings and conclusion ..........................................................................50
4.3 Future directions ................................................................................................................50
References ......................................................................................................................................52

vi
List of Figures
Figure 1. Schematic diagram of the mycobacterial cell wall. ......................................................... 2
Figure 2. Overview of TB Infection. .............................................................................................. 4
Figure 3. Genealogy of current BCG substrains. .......................................................................... 10
Figure 4. The recA mutation in BCG-Russia. ............................................................................... 13
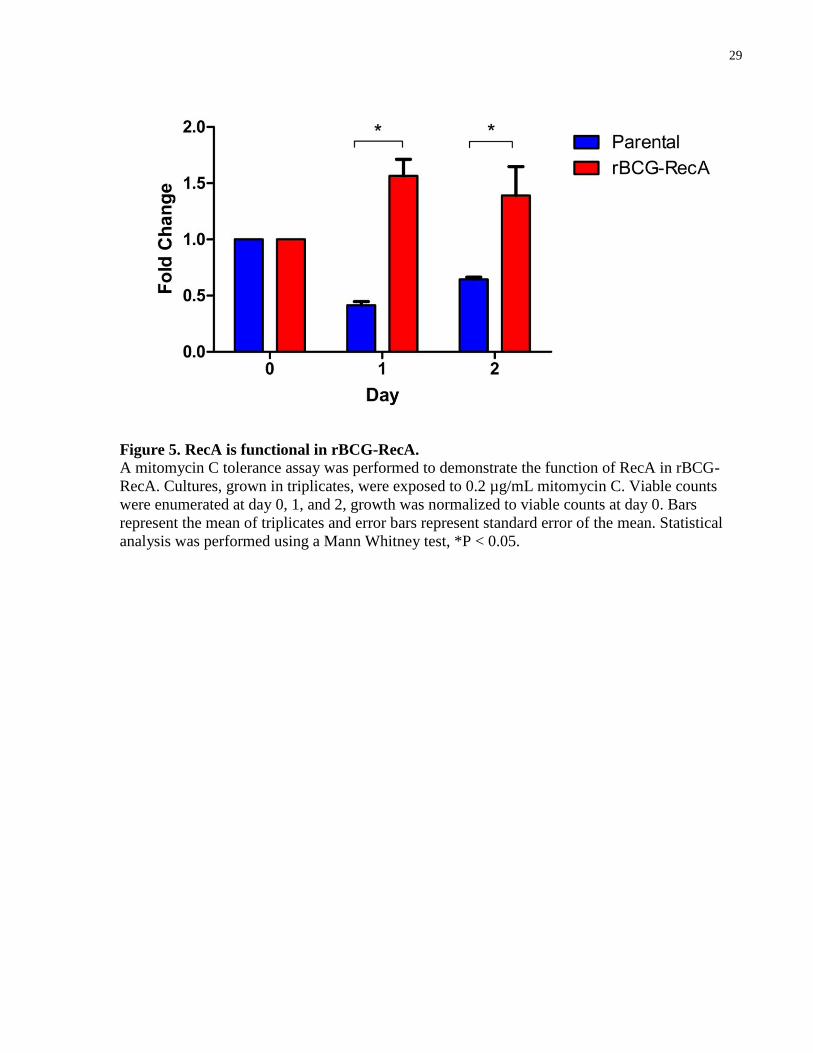
Figure 5. RecA is functional in rBCG-RecA. ............................................................................... 29
Figure 6. rBCG-RecA does not exhibit a growth defect. .............................................................. 30
Figure 7. rBCG-RecA is more attenuated than the parental strain in SCID mice. ....................... 32
Figure 8. SCID mice endpoint lungs and spleens. ........................................................................ 34
Figure 9. Organ weight and bacterial burden in SCID mice......................................................... 36
Figure 10. rBCG-RecA does not have a defect in intracellular survival. ..................................... 38
Figure 11. Bacterial burden in the lungs and spleens of guinea pigs. ........................................... 40
Figure 12. Guinea pig body weight change and organ weight. .................................................... 42
Figure 13. Histological analysis on guinea pig lungs and spleen. ................................................ 44

vii
List of Abbreviations
ADC albumin-dextrose-catalase
ATP adenosine triphosphate
BCG Bacille Calmette-Guérin
bp base pair
CFP-10 culture filtrate protein 10
CFU colony forming unit
dsDNA double stranded deoxyribonucleic acid
DU duplication
ESAT-6 early secretory antigenic target 6
ESX early secretory antigenic target 6 system
H&E hematoxylin and eosin
HIV Human Immunodeficiency Virus
IFNγ interferon-gamma
kb kilobase
LAM lipoarabinomannan
LTBI latent tuberculosis infection
MDR multidrug-resistant
MOI multiplicity of infection

viii
M. tb Mycobacterium tuberculosis
MTBC Mycobacterium tuberculosis complex
OADC oleic acid-albumin-dextrose-catalase
PBS phosphate buffered saline
PCR polymerase chain reaction
PDIM phthiocerol dimycocerosate
PGL phenolic glycolipid
RD1 Region of Difference 1
RD2 Region of Difference 2
RNI reactive nitrogen intermediates
ROI reactive oxygen intermediates
ssDNA single stranded deoxyribonucleic acid
TB tuberculosis
TNF tumour necrosis factor
WHO World Health Organization
XDR extensively drug-resistant

1
Chapter 1
General Introduction
1.1 Tuberculosis
1.1.1 Mycobacterium tuberculosis
Mycobacterium tuberculosis (M. tb) is the causative agent of tuberculosis (TB) and is part of the
Mycobacterium tuberculosis complex (MTBC) along with M. africanum, M. bovis, M. microti,
M. canetti, M. caprae, and M. pinnipedii. Members of the MTBC share over 99.9% sequence
identity and are all intracellular pathogens that cause tuberculosis disease (Brosch, Gordon et al.
2002). Although M. tb is a gram-positive bacillus, it contains a complex cell wall exterior to its
peptidoglycan layer. Notably, the cell wall is rich in mycolic acids containing 70-90 carbons, a
characteristic of the mycobacterium genus (Liu, Rosenberg et al. 1995). Mycolic acids are
complex branch-chained hydroxyl lipids, covalently linked to arabinogalactan that contribute to
the impermeability of the cell wall (Liu, Rosenberg et al. 1995, Liu, Barry et al. 1996) and
consequently, to the decreased susceptibility of M. tb to antibiotics (Gebhardt, Meniche et al.
2007), rendering M. tb one of the most difficult pathogens to treat. The high amount of mycolic
acid in the cell wall is also the basis of the Ziehl-Neelsen stain commonly used to identify
mycobacteria due to retention of the carbol-fuchsin stain by the thick lipid-rich layer. Other
components present in the cell wall of mycobacteria include lipoarabinomannan (LAM),
phthiocerol dimycoserate (PDIM), and phenolic glycolipids (PGL), all of which have been
implicated in the pathogenesis and virulence of mycobacterium (Yu, Tran et al. 2012, Fukuda,
Matsumura et al. 2013) (Figure 1).

2
Figure 1. Schematic diagram of the mycobacterial cell wall.
In contrast to most gram-positive bacteria, Mycobacterium tuberculosis has a thick, waxy cell
wall exterior to its peptidoglycan layer and serves as the basis for the Ziehl-Neelsen staining. The
cell wall contains large amounts of mycolic acids, lipoarabinomannan (LAM), arabinogalactan,
acyl glycolipids, and complex free lipids (PDIM and PGL). Figure from (Riley 2006).

3
The 4 million base pair, GC-rich genome of Mycobacterium tuberculosis contains approximately
4000 genes (Cole, Brosch et al. 1998). Approximately 40% of the genes have been assigned
precise functions and another 44% of these genes have similarities to other known proteins in
other species. Encoded in these genes are 11 two-component systems, thirteen sigma factors,
approximately 250 unique genes implicated in fatty acid metabolism, and 5 ESX systems (Gey
Van Pittius, Gamieldien et al. 2001) which are type-VII secretion systems, two of which (ESX-1
and ESX-5) have been shown to be involved in virulence (Abdallah, Bestebroer et al. 2011)..
About 10% of the M. tb genome encodes two families of proteins, the PE and PPE family
proteins, which have highly conserved N-termini and variable C-termini. The functions of these
proteins are poorly understood but evidence suggests they may be implicated in various cell
processes including persistence within the host (Nair, Ramaswamy et al. 2009) and virulence
(Goldstone, Goonesekera et al. 2009).
1.1.2 Pathogenesis of Mycobacterium tuberculosis
The primary route of transmission of Mycobacterium tuberculosis is through respiration. Thus,
M. tb infection often begins with the inhalation of aerosols containing the bacilli generated by a
person with active disease (Figure 2A). Once bacilli reach the lungs, it is quickly taken up by
alveolar macrophages and very few bacilli (<10 bacteria) are sufficient to successfully establish
infection (Figure 2B). Individuals infected with M. tb are often able to control the initial
infection and appear clinically asymptomatic for decades in which the bacteria remain in a
dormant, inactive state. These individuals are considered to have latent TB infection (LTBI) and

4
Figure 2. Overview of TB Infection.
Typically, (A) Naïve individuals are infected with M. tb via inhalation of bacilli containing
aerosols generated from an individual with active disease. (B) Once the bacilli reach the lungs, it
is quickly taken up by alveolar macrophages. Antigen-presenting dendritic cells eventually
migrate to the draining lymph nodes and (C) recruit antigen-specific T lymphocytes to the site of
infection. The recruited T lymphocytes activate the macrophages via IFNγ and TNF to enable
effective killing of the pathogen. Cooperatively, the innate immune cells and recruited adaptive
immune cells contain M. tb in a structure called granuloma. (D) Weakening of the host’s
immune system allows for the reactivation of the bacterium, causing active TB disease, and
rendering the individual infectious. Figure from (Cambier, Falkow et al. 2014).

5
are non-contagious. In approximately 5-10% of these individuals, the dormant M. tb will
reactivate during their lifetime causing active TB disease. It is important to mention that the risk
of reactivation increases substantially to 5-10% annually for immunocompromised individuals
such as those co-infected with HIV (Selwyn, Alcabes et al. 1992).
M. tb has a multitude of strategies to overcome host defenses and persist within the host,
enabling it to become a successful intracellular pathogen. One such strategy involves the
inhibition of phagosome-lysosome fusion. Lipoarabinomannan (LAM), a major component of
the M. tb cell wall has been reported to inhibit phagosomal maturation by blocking the increase
in cytosolic Ca2+
, effectively disrupting the regulation of PI3P and the downstream recruitment
of EEA1 which is responsible for the trafficking of lysosomal components to the phagosome
(Vergne, Chua et al. 2003), allowing the bacilli to survive within the non-acidified compartment.
Also implicated in the inhibition of phagosomal maturation are effector proteins ESAT6 and
CFP10 secreted by the ESX-1 secretion system (Tan, Lee et al. 2006). In addition, ESAT-6 has
been suggested to exhibit membrane-lysing activity to facilitate phagosomal escape into the
cytosol (de Jonge, Pehau-Arnaudet et al. 2007) and induce apoptosis (Derrick and Morris 2007).
Eventually, dendritic cells migrate to the draining lymph nodes and recruit antigen-specific T
lymphocytes to the site of infection (Figure 2C). The host’s cell-mediated immunity by Th1-type
CD4+ and CD8+ cells are crucial for controlling the infection. Activation of the host
macrophages by IFNγ and TNF are necessary to overcome the M. tb inhibition of phagosomal
maturation and for effective killing of the bacteria via reactive oxygen intermediates (ROI) or
reactive nitrogen intermediates (RNI) (Flesch and Kaufmann 1990, Flynn, Chan et al. 1993,
Herbst, Schaible et al. 2011), highlighting the important role of the two cytokines in the control
of M. tb within the host and providing an explanation as to why the rate of reactivation is

6
significantly increased in immunocompromised individuals. Animal models and humans with
deficiencies in these two cytokines have been shown to be more susceptible to TB infection
(Flynn, Chan et al. 1993, Ottenhoff, Kumararatne et al. 1998, Keane, Gershon et al. 2001).
Granulomas, although not exclusive to TB, is a hallmark of TB disease and is a result of the
interplay between the M. tb and the host’s immune system. It is generally described as a caseous,
necrotic centre containing the bacteria, with low pH and oxygen, surrounded by multi-nucleated
giant cells, epitheliod cells, B cells, and a T-cell cuff (Via, Lin et al. 2008, Ramakrishnan 2012).
Whether granuloma formation is beneficial for M. tb or the host is still a matter of debate.
Although the granuloma sequesters the bacteria and allow for control of the infection, it also
provides a niche microenvironment in which the bacteria can persist without the perturbation of
the host’s immune system. Weakening of the host’s immune system, and consequently its ability
to contain the bacteria, is a likely cause of reactivation (Figure 2D).
1.1.3 Global burden of TB and increase in anti-mycobacterial resistance
Tuberculosis remains a major global health threat and one of the world's most deadly infectious
diseases along with malaria and HIV. In 2014, WHO reported 1.5 million deaths, 0.4 million of
these in HIV-positive individuals, and 9.6 million new cases caused by the bacilli (WHO 2015).
It is estimated that one third of the world's population are latently infected with tuberculosis and
these individuals act as reservoirs for the bacteria. Potential reactivation from these individuals
may contribute to the further spread of the disease. Furthermore, we have seen an unprecedented
increase in anti-mycobacterial resistance over the last decade. The current treatment for active
TB disease is a six month regiment of four first-line antibiotics: rifampicin, isoniazid,

7
pyrazinamide, and ethambutol. Poor adherence to the lengthy treatment and inappropriate use of
antibiotics are suggested to be partially responsible for the emergence in multidrug-resistant,
defined as resistant to isoniazid and rifampicin, and extensively drug-resistant strains, defined as
resistant to isoniazid, rifampicin, and a second line drug. In 2014, an estimated 480,000
multidrug-resistant cases have been reported globally and 9.7% of these cases were extensively
drug-resistant (WHO 2015). This clearly indicates the pressing need for an alternative strategy,
in addition to chemotherapy, for tuberculosis control.
1.2 The Bacille Calmette-Guérin vaccine
The Bacille Calmette-Guérin (BCG) vaccine is the only licensed vaccine for tuberculosis and is
the most widely used vaccine with more than 100 million doses administered annually. It is a
live, attenuated vaccine obtained from in vitro passaging of pathogenic Mycobacterium bovis, a
close relative of M. tuberculosis isolated from cattle, which shares >99% genetic identity with M.
tb (Brosch, Gordon et al. 2002). It has demonstrated superior efficacy in preventing disseminated
forms of TB in children (Colditz, Berkey et al. 1995), including miliary and tubercular
meningitis, but offers highly variable protection against pulmonary TB in adults (Colditz, Brewer
et al. 1994) which is the most prevalent form of the disease. Furthermore, the protection offered
by BCG wanes over time (Sterne, Rodrigues et al. 1998). Although BCG is attenuated, it is still a
live vaccine nonetheless and is able to cause severe infection in infants, especially in those that
are immunocompromised (Hesseling, Marais et al. 2007). Recognizing that the risks may
potentially outweigh the benefits of vaccination for the immunocompromised, WHO revised its
policy in 2007 and recommends that the BCG vaccine is only administered to HIV-negative

8
infants in regions with high TB incidence or HIV-negative infants at high risk of exposure to TB
(WHO 2007). Interestingly, BCG has found use in areas besides TB control. Currently, it is used
in immunotherapy for early, non-invasive bladder cancer which has been demonstrated to
improve the prognosis of the disease and reduce the rate of relapse although the mechanisms
behind this are poorly understood (Herr, Schwalb et al. 1995).
1.2.1 History of the BCG vaccine
The BCG vaccine was developed by the two scientists, Albert Calmette and Camille Guérin, who
recognized that the continual in vitro passaging of M. bovis resulted in decreased virulence in a
guinea pig model. From 1908 to 1921, M. bovis was passaged 230 times on bile-potato-glycerol
medium to generate a strain attenuated enough to be used in humans, the M. bovis BCG. This
attenuated strain was first used as a vaccine in humans, given orally, in an infant born to a
mother who succumbed to TB shortly after his birth. The infant did not exhibit any adverse
reactions to the vaccine nor did he eventually develop TB demonstrating the safety and efficacy
of BCG in humans. By 1924, 664 oral vaccinations in infants had been reported, and from 1924-
1928, 114,000 infants were vaccinated with no serious complications observed. Importantly, the
protection efficacy of this 1921 M. bovis BCG was shown to be >80% (Calmette, Guerin et al.
1924). Starting from 1924, the BCG was distributed to various laboratories around the world but
due to different culturing conditions, the 1921 M. bovis BCG had undergone further in vitro
evolution and diversified into genetically distinct substrains (Brosch, Gordon et al. 2007) (Figure
3). This in vitro evolution continued in their respective laboratories until 1966 when the seed-lot
system was established to prevent further deviation from the original BCG. Consequently, it is
important to recognize that the current BCG is not isogenic but rather refers to the collection of

9
BCG strains, each named after the country from where it was cultured. In 1974, WHO
incorporated the BCG into their Expanded Program on Immunization to vaccinate infants on a
global scale.
1.2.2 The mechanism and attenuation of BCG
Although the benefits of BCG vaccination are undeniable, the underlying mechanisms of these
benefits remain poorly understood. The primary attenuation of the BCG occurred from the loss
of the RD1 region during the in vitro passaging between 1908 and 1921 (Figure 3). This region is
present in virulent M. tb and M. bovis but is lost in all known strains of BCG. This 9.5kb RD1
region is comprised of 9 genes, including the two effector proteins ESAT-6 and CFP-10, and
components of the ESX-1 secretion system (Cole, Brosch et al. 1998). Interestingly, deletion of
this region in M. tb leads to attenuation but not to the same level as BCG (Lewis, Liao et al.
2003). Consistently, complementation of the RD1 region in BCG does not restore virulence to
the same level as M. tuberculosis suggesting other genetic alterations may contribute to the
attenuation of BCG (Pym, Brodin et al. 2002). Comparative genome analysis has revealed
numerous genetic differences, including deletions, duplications, and single nucleotide
polymorphisms, between substrains (Brosch, Gordon et al. 2007) (Figure 3). These genetic
differences acquired in their respective laboratories after being distributed from the Institute
Pasteur may have contributed to further attenuation.

10
Figure 3. Genealogy of current BCG substrains.
The current BCG consists of over a dozen genetically distinct substrains that diversified from the
original 1921 M. bovis BCG. A number of genetic alterations including duplications and
deletions have been outlined here. Figure from (Brosch, Gordon et al. 2007).

11
1.2.3 Variation in the clinical properties of BCG
The differential virulence of the current BCG strains in humans are well documented with less
risk of adverse reactions associated with vaccinations observed in BCG-Japan, -Moreau, -Glaxo,
and Prague compared to BCG-Danish and BCG-Pasteur (Lotte, Wasz-Hockert et al. 1984).
Evidence from animal studies have also supported this notion. A study directly comparing the
virulence of 5 BCG strains in golden hamsters had demonstrated that the ability of the strains to
overwhelm the animal were markedly different (Bunch-Christensen, Ladefoged et al. 1970). One
possible explanation for this is the loss of known virulence factors such as PDIM and PGL in
BCG-Japan, -Moreau, and -Glaxo (Chen, Islam et al. 2007) as well as a phoP mutation in BCG-
Prague (Leung, Tran et al. 2008), a response regulator that regulates a number of genes, some of
which are known virulence factors.
Clinical trials of BCG vaccination have demonstrated highly variable efficacies (0-80%) against
TB in humans (Brewer 2000). Besides the setting and populations of the clinical studies, the
heterogeneity of BCG strains used in these trials may have also contributed to the large variation.
Animal studies have also suggested that there may be a difference in protection efficacies
between substrains. A study in a mice model comparing 10 substrains of BCG found that the
substrains conferred various levels of protection and immunogenicity (Castillo-Rodal, Castanon-
Arreola et al. 2006). It is important to note that studies directly comparing the efficacy of BCG
substrains in humans are unavailable. Despite the large variation in protection efficacies, there is
no better alternative than the BCG vaccine for tuberculosis control.

12
1.3 BCG-Russia, a natural RecA mutant
1.3.1 Properties of BCG-Russia
BCG-Russia is the earliest strain to have diverged from the parental 1921 M. bovis BCG (Figure
3). The vaccine was distributed from the Institute Pasteur to Russia in 1924 where it was further
cultured and administered. BCG-Russia, along with BCG-Japan and BCG-Danish, is one of the
three strains used to produce the vaccine today (Luca and Mihaescu 2013). Based on
comparative genomic analysis, BCG-Russia has acquired the least genetic alterations relative to
the 1921 M. bovis BCG (Keller, Bottger et al. 2008). BCG-Russia has lost the RD1 region, a
common characteristic of all current BCG substrains, but contains a large duplication (DU2).
From examining the current BCG strains, the DU2 region is present in one of four forms,
nominally DU2-I to IV, and BCG-Russia contains a DU2-I (Brosch, Gordon et al. 2007). In
addition, BCG-Russia contains two IS6110 elements while substrains from after 1925 only have
one copy. One of the copies of IS6110 lies inversely in the promoter of the phoP gene though its
effect on BCG-Russia remains unknown (Leung, Tran et al. 2008). In 2008, Keller and
colleagues had attempted to make recA mutants from the current BCG substrains in hopes of
generating genetically-stable strains, a property favourable for live vaccines. They were
successful in making knockouts in all the substrains attempted except for BCG-Russia which
prompted further investigation. This led to the discovery that BCG-Russia is a natural RecA
mutant due to a single nucleotide insertion, a cytosine, at position 414 of the open reading frame
of the recA gene (Keller, Bottger et al. 2008) (Figure 4). This frameshift mutation results in a
premature stop codon and a truncated RecA protein.

13
Figure 4. The recA mutation in BCG-Russia.
BCG-Russia is a natural recA mutant due to a cytosine insertion at position 414 of the recA open
reading frame. This frameshift mutation leads to a premature stop codon at residue 140 and
results in a truncated protein. Figure from (Keller, Bottger et al. 2008).

14
1.3.2 Characteristics of Mycobacterium RecA
The mycobacterium RecA protein is 790 amino acids in length and is highly conserved among
species. There exists a functional homolog in almost every species (Roca and Cox 1990) and the
homolog in eukaryotes is the Rad51 protein (Kawabata, Kawabata et al. 2005). RecA has been
implicated in a variety of cell processes including homologous recombination (Chen, Yang et al.
2008) and in the induction of the SOS response (Nautiyal, Patil et al. 2014). Homologous
recombination is the major driving force behind genetic exchange, and consequently bacterial
evolution, which could explain why BCG-Russia, a natural RecA mutant, has such a high degree
of genomic stability. To facilitate homologous recombination, RecA forms nucleoprotein
filaments, binding ssDNA, dsDNA, and ATP simultaneously (Bell 2005). The RecA protein
searches along the dsDNA to find a complementary region to the ssDNA although the
mechanism behind this is poorly understood. Once the complementary sequence has been found,
RecA facilitates the exchange of strands in an ATP-dependent manner. After strand exchange, a
heteroduplex region is formed and subsequently resolved by other Rec proteins involved in
homologous recombination. This pathway is important not only for the exchange of genetic
elements or bacterial evolution but for DNA repair as well. The critical role of RecA in the
induction of the SOS response to repair double stranded breaks has been demonstrated in other
organisms with decreased survival in RecA mutants exposed to UV irradiation or other DNA
damaging agents (Huang and Chen 2006). Under normal conditions, LexA, a transcriptional
repressor, binds the promoter of SOS response genes and represses their expression to varying
degrees. In response to DNA damage, RecA aids in the auto-catalytic cleavage of LexA allowing
for expression of the regulated SOS response genes including those responsible for nucleotide

15
excision repair, homologous recombination, and the error-prone DNA repair polymerase Pol V
(Michel 2005).
The mycobacterial recA gene resides in an operon upstream of recX and BCG_2748c. RecX is a
negative regulator of homologous recombination (Cardenas, Carrasco et al. 2012) while
BCG_2748c is a putative protein. recA is under control of two separate promoters, both induced
by DNA damage; one promoter is regulated classically by LexA/RecA whereas the other
promoter is LexA/RecA independent (Davis, Springer et al. 2002). It has also been demonstrated
in M. smegmatis that overexpression of RecA in the absence of RecX is toxic for cells
(Papavinasasundaram, Colston et al. 1998).
The crystal structure of the M. tb RecA has been solved previously. The RecA mature protein
consists of a 30 residue α-helical N-terminus that contacts neighbouring RecA proteins in the
filament (Bell 2005), followed by a 239 residue central region with two disordered loops
implicated in ATP-binding and ssDNA binding (Roca and Cox 1990, Malkov and Camerini-
Otero 1995), and a 59 residue C-terminus implicated in dsDNA binding (Aihara, Ito et al. 1997).
Interestingly, there is a large intervening sequence (PI-MtuI) found only in pathogenic
mycobacterium (Davis, Sedgwick et al. 1991), termed intein, within the RecA precursor. This
precursor undergoes a self protein-splicing reaction in which the intein is excised to produce the
mature 38 kDa RecA. It has been shown that the Mn2+
and ATP-dependent excision of the intein
is necessary for the function of the RecA protein (Kumar, Vaze et al. 1996, Guhan and
Muniyappa 2002).

16
1.4 Current vaccine development
With the cases of multi-drug resistant and extensively-drug resistant TB on the rise,
chemotherapy is quickly becoming obsolete and focus is being turned to preventative measures.
The highly variable protection efficacy and safety of the current BCG demonstrates that it is far
from an ideal vaccine. Scientists in the field have responded to the need for a better vaccine and
here I describe several of the recent vaccine candidates currently in clinical trials.
1.4.1 Recombinant and attenuated M.tb vaccines
Recombinant vaccines are live vaccines that are modified through recombinant technology to
either modify the expression of an already existing protein within the bacterium or express a
heterologous protein from another species. Genes selected for this purpose often include known
antigens, virulence factors, and regulatory proteins. The logic behind this is that the recombinant
protein, through some mechanism, will stimulate a strong immune response which yields greater
protection. Here I discuss three recombinant vaccines used for TB control, rBCG30, VPM1002,
and MTBVAC, two of which are currently in the pipeline for clinical trials (WHO 2015).
Interestingly, one of these vaccines, MTBVAC, is a first of its kind, live attenuated M. tb
vaccine.
rBCG30 is the first recombinant vaccine to be significantly attenuated and demonstrates superior
protection to BCG in animal models. This recombinant strain overexpresses Ag85B in a BCG-
Tice background. Further genetic modifications were made to increase its safety for use in
humans. The resulting strain, rBCG(mbtB)30, also contains a deletion in the mbtB gene which

17
disrupts the mycobactin siderophore and consequently, the strain’s ability to acquire iron. As a
result, the strain is replication-limited in the host for increased safety in humans, especially
immunocompromised individuals. In a guinea pig model, this strain showed 0.33log10 CFU
reduction of virulent M. tb in the lungs compared to the parental strain and almost 0.5log10 CFU
reduction in the spleen (Tullius, Harth et al. 2008). rBCG(mbt)30 is also highly attenuated in a
SCID mice model compared to the parental strain as demonstrated by increased survival of SCID
mice infected with the strains. The bacterial load in the lungs and spleens of SCID mice were
significantly reduced compared to that of the parental strain as well. A variation,
rBCG(panCD)30, that is also replication limited and must be supplemented with pantothenate
also demonstrated attenuation in SCID mice but equivalent levels of protection as that offered by
the parental BCG. This vaccine did not enter clinical trials due to the presence of an antibiotic
resistance marker carried in the vaccine.
VPM1002 is a recombinant vaccine generated in a BCG-Prague background. The strain
expresses the hly gene from Listeria monocytogenes and inactivates ureC. The idea behind this
vaccine is that the expression of Listeriolysin O, encoded by the hly gene, could facilitate
phagosomal escape into the cytosol which would enhance MHC-I and cross presentation as well
as induce apoptosis. The purpose of inactivating ureC is to provide the optimal pH for the
function of Listeriolysin O within the phagosome. The recombinant strain was attenuated in a
SCID mice model of infection and showed increased survival compared to parental strain
(Kaufmann, Cotton et al. 2014). In BALB/c mice, the recombinant strain was shown to have
greater protection with 1log10 and 0.5log10 reduced bacterial load in the lungs and spleen,
respectively, compared to the parental strain. This vaccine is currently in phase II clinical trials.

18
MTBVAC is the first live-attenuated M. tb vaccine to be used in clinical trials. It is based on
SO2, a previously established live-attenuated M. tb vaccine generated from an insertion of a
kanamycin resistant marker within the phoP gene. The SO2 vaccine was abandoned due to the
establishment of the Geneva consensus which stated that live mycobacterial vaccines must
contain two independent mutations with no antibiotic resistance markers present. MTBVAC
contains two deletions in the phoP and fadD26 genes with no antibiotic resistance markers. In a
guinea pig model, MTBVAC demonstrated a 2log10 and a 4log10 reduction in bacterial load in
the lungs and spleens, respectively, compared to unvaccinated controls (Arbues, Aguilo et al.
2013). In the C57BL/6 mice model, MTBVAC offered 0.5log10 reduction in both the lungs and
spleens compared to BCG-Danish. Attenuation of MTBVAC was demonstrated in a SCID mice
model with bacterial loads in the lungs and spleens comparable to BCG-Danish and BCG-
Pasteur. This vaccine is currently in phase II clinical trials.
1.4.2 Subunit vaccines
Subunit vaccines are vaccines consisting of purified proteins that stimulate the immune response.
Proteins that are often used include antigens and surface markers of pathogens as these are
molecular factors that the immune cells will recognize during infection. Typically, only the
epitope of the antigen that the receptor of immune cells will bind to is required. Subunit vaccines
are often given with an adjuvant to boost the immune response to the antigens. Here I discuss
two subunit vaccines, M72-AS01E and H1/IC31, which are currently in the pipeline for clinical
trials (WHO 2015).

19
The M72-AS01E is a vaccine consisting of the two antigens, Mtb39a and Mtb32a, and
formulated in an AS01E adjuvant. A point mutation in Mtb32a allows improved stability of the
fusion protein. This subunit vaccine was developed to boost BCG or M. tb primed immune
responses. When primed with BCG, the M72 fusion protein formulated in AS02A adjuvant
elicits a strong Th1 response and IFNγ response. The M72-AS02A, or when delivered as a DNA
vaccine, shows improved protection efficacy against M. tb challenge in guinea pig models as
measured by increased survival (Brandt, Skeiky et al. 2004). M72-AS01E induced higher CD4+
T cell responses than the M72-AS02A vaccine but did not induce a strong CD8+ T cell response
(Leroux-Roels, Forgus et al. 2013). The safety and immunogenicity of M72-AS01E has been
demonstrated in humans in a high TB burden area (Penn-Nicholson, Geldenhuys et al. 2015).
This vaccine is now in phase IIb clinical trials.
The H1/IC31 vaccine is a recombinant subunit vaccine consisting of a fusion protein of ESAT6
and Ag85B formulated in an IC31 adjuvant system. This vaccine has demonstrated greater
protection efficacy than when using Ag85B alone in a guinea pig model, but is inferior to the
protection conferred by BCG. Similarly, H1/IC31 induces a stronger immune response than
when using Ag85B alone in both naïve and BCG-vaccinated/tuberculosis infected individuals
(van Dissel, Arend et al. 2010, van Dissel, Soonawala et al. 2011). This vaccine is now in Phase
II clinical trials.
1.4.3 DNA vaccines
DNA vaccines are vaccines administered in the form of DNA and often encode antigens of the
pathogen in question. One common way to deliver this is by use of viral vectors as it greatly

20
enhances the uptake of the vaccine into the host cells. The concept behind these vaccines is that
immune cells in the host will be able to acquire pieces of the DNA vaccine and express the
antigen in the host cells. These cells then present the antigen on its surface in order to develop an
antigen-specific immune response against this pathogen. There are several DNA vaccines for TB
control reported to be undergoing phase I trials in the current pipeline of clinical trials including
Ad5 MVA85A, Crucell Ad35/MVA85A, and ChAdOx1.85A/MVA85A (WHO 2015). Here I
briefly discuss the MVA85A as it is a common component to the DNA vaccines mentioned. The
MVA85A is a recombinant strain of Modified Vaccinia Ankara that expresses Ag85A from M.
tb. MVA85A was found to be well tolerated and immunogenic in adolescents. In addition, the
vaccine induced a potent CD4+ T cell response in the study population (Scriba, Tameris et al.
2011). These CD4+ T cell responses to the MVA85A are persistent in both healthy,
immunocompetent individuals and HIV-postive individuals who were undergoing anti-retroviral
therapy at the time of vaccination (Tameris, Geldenhuys et al. 2014). These findings showed
great promise for MVA85A to be a potential TB vaccine. Subsequently, a phase IIb prospective
clinical trial was conducted to evaluate MVA85A as a booster in addition to BCG priming for
enhanced protective efficacy. In the study population of 2797 infants, boosting with MVA85A
demonstrated no additional benefit against tuberculosis or against M. tb infection than when
BCG was used alone, as measured by tuberculosis incidence and seroconversion, respectively
(Tameris, Hatherill et al. 2013). Furthermore, animal studies evaluating the efficacy of MVA85A
as a booster also found no evidence to support MVA85A as an effective booster for BCG
(Kashangura, Sena et al. 2015).

21
1.5 Rationale and hypothesis
Tuberculosis remains one of the deadliest infectious diseases globally and represents an immense
burden on global health. Chemotherapy as a treatment of active TB disease is quickly becoming
ineffective due to the emergence of multi-drug and extensively-drug resistant strains of M. tb. A
preventative measure is the only way to halt the spread and to effectively eradicate TB. BCG is
the only vaccine available for TB control but its protection efficacy against pulmonary TB varies
greatly, from 0-80%. Furthermore, development of disseminated disease due to BCG vaccination
in immunocompromised individuals poses another challenge. This demonstrates the pressing
need for a better vaccine. Examining and interrogating the role of molecular factors will
contribute to our understanding of whether and how these factors affect the virulence and
protection efficacy of the BCG vaccine.
Based on the literature, live vaccines confer protection superior to that of any subunit or DNA
vaccines when used alone. BCG-Russia is a relevant background to construct further genetic
modifications as BCG-Russia is one of the three most widely used strains to produce the vaccine
today. It is a natural RecA mutant due to a frameshift mutation resulting in a truncated RecA
protein. The RecA protein has been implicated in homologous recombination as well as in the
induction of the SOS response to DNA damage. In addition, BCG-Russia is also genetically the
most similar to the 1921 M. bovis BCG which reported >80% protection efficacy (Calmette,
Guerin et al. 1924), likely due to the defect in homologous recombination which prevented
further in vitro evolution, and this would be desirable as some have raised concerns regarding the
overattenuation of the current BCG resulting in decreased efficacy (Behr and Small 1997).
Whether the presence or absence of RecA affects the two main properties, virulence and
protection efficacy, of BCG-Russia remain unknown.

22
I propose to generate a recombinant BCG-Russia expressing functional RecA under its natural
promoter, rBCG-RecA. Subsequently, I will evaluate the recombinant strain’s virulence and
protection efficacy using a SCID mice and guinea pig model, respectively. I hypothesize that
rBCG-RecA will exhibit increased virulence and increased protection against M. tb challenge.
The rationale behind this hypothesis is that by restoring functional RecA to BCG-Russia, it will
have an intact SOS response to better tolerate the oxidative burst and persist within the host. The
prolonged survival of the bacterium would stimulate a greater immune response and as a result,
there will be greater protection efficacy when challenged with virulent M. tb.

23
Chapter 2
Materials and Methods
2.1 Bacterial strains and culture conditions
Mycobacterium bovis BCG strain, BCG-Russia, was grown at 37°C in DifcoTM
Middlebrook
7H9 broth (BD Biosciences) supplemented with 0.2% glycerol, 10% albumin-dextrose-catalase
(ADC; BD Biosciences), and 0.05% Tween-80 (Bioshop) or on DifcoTM
7H11 agar (BD
Biosciences) supplemented with 0.5% glycerol and 10% oleic acid-albumin-dextrose-catalase
(OADC; BD Biosciences). Escherichia coli strain DH5α was used for routine manipulation and
propagation of plasmid DNA. E. coli DH5α was grown at 37°C in LB broth (BioBasic) or on LB
agar (BioBasic). Antibiotics were added as required: kanamycin, 50 µg/mL for E. coli and 25
µg/mL for BCG; polymyxin B 5.5 µg/mL carbenicillin 50 µg/mL, amphotericin 4 µg/mL, and
trimethoprim 2 µg/mL for M. tuberculosis H37Rv.
2.2 Cloning of recombinant BCG-Russia RecA
A 2838-bp fragment containing the recA (BCG_2750c) gene and its upstream sequence,
presumably containing its natural promoter, was amplified by PCR using Mycobacterium bovis
BCG-Pasteur 1173P genomic DNA as the template and the forward primer 5’-
GGCCAGGCTAGCGGTGTTGAGCAGATCGTC-3’ and reverse primer 5’-
CGCCCGTCTAGACCTGGACTGAACCCATTGTT-3’, which contains a NheI and XbaI site
(underlined), respectively. The PCR product was cloned into the NheI and XbaI sites of the

24
kanamycin-selectable Mycobacterium/E. Coli shuttle vector pME, generating pME-RecA. pME-
RecA or the empty vector pME was electroporated into BCG-Russia and transformants were
selected on 7H11 agar plates containing 25 µg/mL kanamycin to generate rBCG-RecA or the
parental strain, respectively. Plasmids were extracted from transformants and the presence of the
intact construct was confirmed by DNA sequencing (TCAG, Toronto).
2.3 Mitomycin C tolerance assay
Cultures were grown to mid-log phase and subcultured into 10 mL cultures of OD600 = 0.4.
Subcultures were allowed to grow overnight at 37°C. Mitomycin C was made fresh and added to
each culture to a final concentration of 0.2 µg/mL. 100 µl of each culture was serially diluted in
PBS and plated on 7H11 agar to quantify the amount of bacteria at day 0, 1, and 2. Viable counts
were obtained after incubation at 37°C for 3 weeks. Bacterial numbers were normalized to day 0
counts and reported as fold change.
2.4 In vitro growth assay
Cultures were grown to mid-log phase and subcultured into 10 mL cultures of OD600 = 0.1. 100
µl of each culture were serially diluted in PBS and plated on 7H11 agar at day 0, 1, 2, 4, 6, 8, and
12. Viable counts were obtained after incubation at 37°C for 3 weeks.

25
2.5 Virulence in SCID mice
All of the animal procedures were approved by the University of Toronto Animal Care. Female
Fox Chase CB17 SCID mice were purchased from Charles River Laboratories and age-matched
(7 weeks) within each experiment. Groups of twenty-four mice were infected intravenously via a
lateral tail vein with either 107 CFU of the parental strain or rBCG-RecA in 0.2 mL of PBS, or
with PBS alone as a control. Ten mice from each group were used to determine bacterial load
within the lungs and spleen of the mice at predetermined time points; day 1 (2 mice per group),
week 4 (4 mice per group), and week 10 (4 mice per group). The lower right lobe of each lung
and a portion of each spleen were immediately fixed in 10% PBS-buffered formalin after
euthanasia for histological analysis. Weights of organs were measured using a Mettler- Toledo
Ab104-S analytical balance. The remaining tissue were homogenized in PBS, serially diluted,
and plated on 7H11 agar plates. Viable counts were performed after incubation at 37°C for 3
weeks. Fourteen mice from each group were used to determine survival of the SCID mice using
20% body weight loss from maximal body weight as a humane endpoint. At endpoint or at the
end of the experiment, lungs and spleens were subjected to bacterial load quantification, weight
measurement, and histological analysis as described above.
2.6 Protection against M. tb challenge in guinea pigs
Female Hartley guinea pigs, weighing between 200 and 250g, were ordered from Charles River
Laboratories. Groups of 6 guinea pigs were vaccinated subcutaneously with either 5x104 CFU of
the parental strain or rBCG-RecA in 0.2 mL PBS, or with PBS. Ten weeks post-vaccination,
guinea pigs were infected with a high aerosol dose (~1000 CFU retained dose in the lung) of M.

26
tb H37Rv. Aerosol challenge was conducted using a Glass-Col inhalation exposure system.
Animals were weighed on a weekly basis and monitored for health, using 20% body weight loss
from maximal body weight as a humane endpoint. 12 weeks post-infection, all animals were
euthanized via cardiac puncture. The bottom right lobe of the lung and a portion of the spleen
were immediately fixed in 10% PBS-buffered formalin for histological analysis. The remaining
tissue were weighed, homogenized in PBS, and plated on 7H11 agar plates for bacterial load
quantification. Viable counts were performed after incubation at 37°C for 3 weeks.
2.7 Histological analysis on animal tissue
Formalin-fixed tissue were processed and embedded into paraffin by the Toronto Centre for
Phenogenomics (Ontario, Canada). 5 µm thick sections were cut with a Leica RM2235
microtome and mounted on glass microscope slides for staining. Slides were deparaffinized in
three changes of xylene for 3 min each and rehydrated through a series of graded alcohols (100%
for 5 min twice, 95% for 2 min, and 70% for 2 min) into distilled water. For hematoxylin and
eosin staining, rehydrated slides were stained in Harris aluminum hematoxylin solution (Harleco)
for 6 min, differentiated in 1% acid alcohol for 30 sec, and counterstained in eosin Y solution
(Harleco) for 2 min. Slides were then dehydrated in graded alcohols (95% for 3 min, 100% for 3
min twice), cleared in three changes of xylene for 3 min each, and mounted with ShuriMount
(TBS Inc.), a xylene based mounting medium. For Ziehl-Neelsen staining, the Acid Fast Stain
Kit (Leica) was used following the recommended protocol. Briefly, rehydrated slides were
stained in carbol-fuchsin for 20 min then subsequently differentiated and counterstained in
malachite green solution for 3 min. After allowing to air dry, slides were cleared in three changes

27
of xylene for 5 min each and mounted with ShuriMount (TBS Inc.). Slides were examined using
a Leica microscope and imaged using the Openlab software. Areas of perceived infiltration and
granulomatous lesions were quantified using ImageJ.

28
Chapter 3
Results
3.1 Confirmation of functional RecA in rBCG-RecA
To confirm that the RecA cloned into rBCG-RecA is functional, I performed a mitomycin C
tolerance assay. Mitomycin C is a known DNA damaging agent and an inducer of the SOS
response. As the parental strain does not have a functional RecA, it is expected to grow less than
the recombinant strain expressing functional RecA. rBCG-RecA and the parental strain, both
grown in triplicates, were exposed to mitomycin C at a concentration of 0.2 µg/mL over a period
of 48 hours. After 24 hours of exposure to mitomycin C, viable bacterial counts for the two
strains were significantly different with the recombinant strain increased by 1.6 fold in
comparison to the parental strain which decreased 2.4 fold (Figure 5). After 48 hours, the viable
bacterial counts remained significantly different with the recombinant strain increased by 1.4
fold and the parental strain decreased 1.6 fold. These results demonstrate that rBCG-RecA is able
to grow when exposed to mitomycin C while the parental strain cannot, suggesting that the RecA
in the recombinant strain is indeed functional.
3.2 rBCG-RecA does not exhibit an inherent growth defect
To determine whether the presence of RecA impacted the growth of BCG-Russia, I performed an
in vitro growth assay. Cultures of the parental strain and rBCG-RecA, both grown in triplicates,
were calibrated to an initial OD600 of 0.1 then allowed to grow at 37°C. Bacterial counts were
29
Figure 5. RecA is functional in rBCG-RecA.
A mitomycin C tolerance assay was performed to demonstrate the function of RecA in rBCG-
RecA. Cultures, grown in triplicates, were exposed to 0.2 µg/mL mitomycin C. Viable counts
were enumerated at day 0, 1, and 2, growth was normalized to viable counts at day 0. Bars
represent the mean of triplicates and error bars represent standard error of the mean. Statistical
analysis was performed using a Mann Whitney test, *P < 0.05.
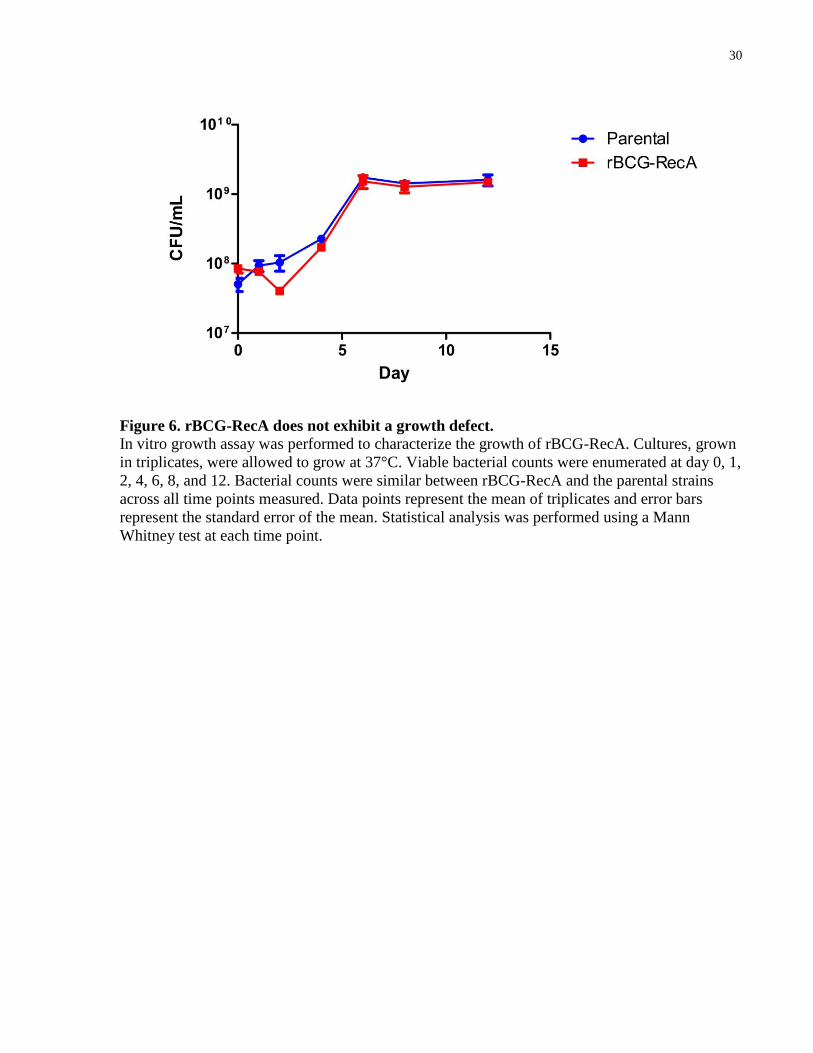
30
Figure 6. rBCG-RecA does not exhibit a growth defect.
In vitro growth assay was performed to characterize the growth of rBCG-RecA. Cultures, grown
in triplicates, were allowed to grow at 37°C. Viable bacterial counts were enumerated at day 0, 1,
2, 4, 6, 8, and 12. Bacterial counts were similar between rBCG-RecA and the parental strains
across all time points measured. Data points represent the mean of triplicates and error bars
represent the standard error of the mean. Statistical analysis was performed using a Mann
Whitney test at each time point.

31
obtained at various timepoints and plated to quantify viable counts. No significant differences
were observed between the two strains at any of the time points measured (Figure 6). These
results suggest that the presence of RecA in BCG-Russia does not cause an inherent growth
defect in the strain.
3.3 RecA attenuates BCG-Russia in SCID mice
To determine if the presence of RecA impacted the virulence of BCG-Russia, I used a SCID
mice model of infection. SCID mice are a well-established animal model and widely used in the
field of BCG vaccine development for evaluating the virulence of live vaccines. They are inbred
animals that are deficient in functional B and T lymphocytes due to a homozygous scid mutation
(Bosma and Carroll 1991). Briefly, mice were infected intravenously via the lateral tail vein with
107 CFU of the parental strain, rBCG-RecA, or PBS as a control. The primary outcome measured
was survival of the animals with 20% body weight loss from maximal body weight as a humane
endpoint for the study. The median survival of the rBCG-RecA group was 16 weeks compared to
the parental group of 11 weeks (Figure 7A). This difference was highly significant (P < 0.0001)
according to the Mantel-Cox test. A replicate of the experiment was conducted to verify these
findings. In the replicate, the median survival of the rBCG-RecA group was 13 weeks compared
to the parental group of 11 weeks (Figure 7B). This difference was also highly significant (P <
0.0001) by the Mantel-Cox test. Taken together, these results suggest that rBCG-RecA is more
attenuated than its parental strain in the SCID mice model of infection. In order to verify that the
death in these animals were actually due to BCG infection, the bacterial load in the lungs and
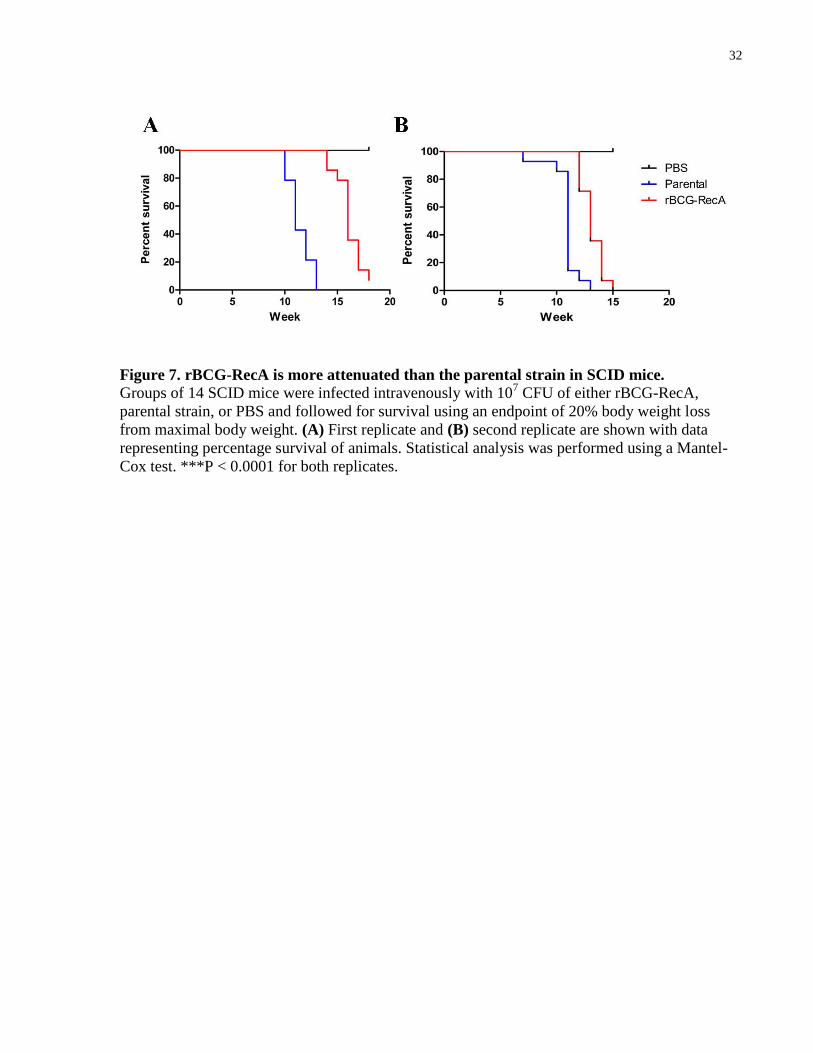
32
Figure 7. rBCG-RecA is more attenuated than the parental strain in SCID mice.
Groups of 14 SCID mice were infected intravenously with 107 CFU of either rBCG-RecA,
parental strain, or PBS and followed for survival using an endpoint of 20% body weight loss
from maximal body weight. (A) First replicate and (B) second replicate are shown with data
representing percentage survival of animals. Statistical analysis was performed using a Mantel-
Cox test. ***P < 0.0001 for both replicates.

33
spleens were evaluated at endpoint. The bacterial load was comparable in the lungs of the
parental group (3.81 x 108 ± 1.74 x 10
8 CFU) and rBCG-RecA group (2.48 x 10
8 ± 5.16 x 10
7
CFU) (Figure 8A). Comparable bacterial loads were also seen in the spleens of the parental (1.68
x 107 ± 5.05 x 10
6) and rBCG-RecA group (2.43 x 10
7 ± 4.45 x 10
6) (Figure 8B). In contrast, no
bacteria were found in the lungs and spleens of PBS control animals (Figure 8A,B). This
suggests that the death of the mice were indeed due to the BCG infection. The lungs in both
groups of infected animals were also found to be significantly heavier than the PBS control,
although no difference is seen between the two infected groups (Figure 8C). Similarly,
splenomegaly, an indicator of systemic infection and BCG virulence (Bourassa, Forget et al.
1985), is observed in the two groups of infected mice and not in the control animals (Figure 8D).
The weight of the spleens from the parental (310.1 ± 15.47 mg) and rBCG-RecA group (320.9 ±
7.491 mg) remained significantly different (P = 0.0035 for both) than the PBS control (39.33 ±
1.417 mg) but was comparable between the two strains (P = 0.3701) (Figure 8D). This is further
supported by histological data in which lungs and spleens from the mice infected with the
parental strain or rBCG-RecA both showed a high number of acid-fast positive bacilli in the
lungs (Figure 8F,G) and spleens (Figure 8I,J). As a control, no acid-fast positive bacilli were
observed in the lungs (Figure 8E) or spleen (Figure 8H) of the PBS mice.
3.4 Organ weights of SCID mice suggest lower virulence of
rBCG-RecA
The attenuation of rBCG-RecA is further supported by organ weight data obtained from the
SCID mice. As mentioned previously, splenomegaly is often used as an indicator of systemic

34
Figure 8. SCID mice endpoint lungs and spleens.
Bacterial burden within the (A) lungs and (B) spleens of SCID mice were enumerated at
endpoint. The weight of the (C) lungs and (D) spleens of animals were also evaluated at this
time. Bars represent the mean (N=14 for parental and rBCG-RecA group, N=2 for PBS) and
error bars represent the standard of the mean. Statistical analysis was performed using a Mann
Whitney test. *P < 0.05, **P < 0.01. Acid fast positive bacteria were observed in the lungs
(arrows), 50x magnification, of (F) parental-infected and (G) rBCG-RecA-infected animals but
not in the (E) PBS control. Consistently, acid-fast positive bacteria were observed in the spleens
(arrows), 50x magnification, of (I) parental-infected and (J) rBCG-RecA-infected mice but not in
(H) PBS animals at endpoint.

35
infection and the health of the animals. At week 4 post-infection, highly significant differences
(P < 0.001) in the weights of the spleen were observed in the parental group (155.1 ± 20.96 mg)
and rBCG-RecA group (138.0 ± 25.28 mg) when compared to the PBS control (25.55 ± 4.798
mg), although no significant difference was observed between the two infected groups (Figure
9B). By week 10, splenomegaly in the two infected groups became more severe and a larger
difference was seen between the parental group (388.1 ± 24.27 mg) or rBCG-RecA group (278.4
± 10.84 mg) compared to the PBS control (34.95 ± 5.519 mg). Furthermore, a significant
difference (P = 0.0062) was observed between the two infected groups. Interestingly, the weight
of the lungs in the parental (273.7 ± 31.44 mg) and rBCG-RecA groups (247.1 ± 11.83 mg) were
also significantly increased (P = 0.0210 and 0.0034, respectively) compared to the PBS group
(169.6 ± 11.61 mg) at week 10 (Figure 9A). These findings provide further evidence that the
recombinant strain may be less virulent than the parental strain.
3.5 The attenuation is due to slower replication of rBCG-RecA in
the host
The attenuation of the rBCG-RecA in SCID mice prompted further investigation into possible
reasons for the attenuation. The bacterial load in the lungs and spleens were evaluated at day 1,
week 4, and week 10 for the growth of the bacteria in vivo. Bacterial load at day 1 was used to
determine the actual inoculation dose received by the animal. The mice from the parental group
received a similar dose as the rBCG-RecA group, 1.09 x 105 ± 4.1 x 10
4 CFU and

36
Figure 9. Organ weight and bacterial burden in SCID mice.
(A) Lung and (B) spleen weights of SCID mice infected with either the parental strain (blue),
rBCG-RecA (red), or PBS (white) at day 1, week 4, and week 10 post-infection were evaluated
(N=2 for day 1, N=4 for week 4 and 10). The bacterial burden in the (C) lungs and (D) spleens
were also quantified at these timepoints. Bars represent the mean and errors bars represent the
standard error of the mean. Statistical analysis was performed using a Mann-Whitney test. *P <
0.05, **P < 0.01, ***P < 0.001.

37
7.2 x 104 ± 8.1 x 10
3 CFU, respectively as determined by the bacterial load in the spleen (Figure
9D). Comparable levels of bacteria were also seen in the lungs of these mice at day 1, 1.94 x 104
± 2.33 x 103 CFU and 2.27 x 10
4 ± 927 CFU, respectively (Figure 9C). By week 4 post-infection,
there are significantly (P = 0.0286) less bacteria in the lungs of the rBCG-RecA group compared
to the parental group, 1.75 x 106 ± 3.37 x 10
5 CFU and 6.34 x 10
6 ± 3.31 x 10
5 CFU, respectively
(Figure 9C). In contrast, no significant difference in the bacterial load of the spleens was
observed between the two groups at week 4 (Figure 9D). By week 10 post-infection, the bacterial
load in the lungs and spleens of the parental group are 2.3 fold and 3.5 fold higher, respectively,
than the rBCG-RecA group (Figure 9C,D). It is important to note that the bacterial load in the
organs at week 10 are similar to the bacterial loads at endpoint (Figure 8A,B) and may suggest
that the bacteria have reached the maximum capacity of the host. Although the differences are
not statistically significant, they may be biologically important. Taken together, these findings
suggest that the rBCG-RecA may grow slower within the host and may be the reason for the
partial attenuation of the strain in mice.
3.6 rBCG-RecA does not have an intracellular growth defect in
macrophages
To investigate whether the slower growth of rBCG-RecA in mice was due to an inability to
survive in the macrophage, the primary host of the bacilli, I performed a macrophage
intracellular survival assay. Murine RAW macrophages were infected at an MOI of 10 with the
parental strain, rBCG-RecA, or PBS as a control. At day 0, 2, 4, and 6 post-infection,
macrophages were lysed and viable bacterial counts were obtained. Viable bacterial counts for

38
Figure 10. rBCG-RecA does not have a defect in intracellular survival.
Murine RAW macrophages were infected at an MOI of 10 with the parental strain (blue), rBCG-
RecA (red), or PBS (not shown) as a control. At day 0, 2, 4, and 6 post-infection, macrophages
were lysed and intracellular bacteria were enumerated. Data points represent the mean of three
technical replicates and error bars represent the standard error of the mean.

39
rBCG-RecA remained similar throughout the course of the experiment, similar to the parental
strain (Figure 10). At day 2 post-infection, the bacterial counts of the parental strain and rBCG-
RecA were 3 x 105 ± 1.2 x 10
4 CFU and 3.53 x 10
5 ± 2.6 x 10
4 CFU, respectively. At day 4 post-
infection, the bacterial count for rBCG-RecA and the parental strain were similar to those seen at
day 2 (3.97 x 105 ± 3.8 x 10
4 CFU and 5.93 x 10
5 ± 4.3 x 10
4 CFU, respectively). At day 6 post-
infection, the viable counts in the parental strain and rBCG-RecA were 1.41 x 106 ± 1.1 x 10
5
CFU and 4.8 x 105 ± 3.1 x 10
4 CFU, respectively. A replicate of this experiment was conducted
and similar results were observed. These findings taken together suggest that the slower growth
of rBCG-RecA in mice is not due to a defect in intracellular survival within macrophages.
3.7 rBCG-RecA confers protection similar to that of the parental
strain
To evaluate if the presence of functional RecA in BCG-Russia would impact the protection
efficacy against M. tb challenge, I used a well-established and widely used aerosol challenge
model in guinea pigs. The aerosol route of challenge was preferred as it mimics the natural route
of M. tb infection. Guinea pigs were selected as the model of choice due to the high similarity of
pathological and clinical features to human infection as well as the increased sensitivity to M. tb
infection. Briefly, groups of six guinea pigs were vaccinated subcutaneously with 5 x 104 CFU of
either the parental strain or rBCG-RecA, or PBS/0.01% Tween 80 as a control. Additionally,
four guinea pigs were also vaccinated with PBS/0.01% Tween 80 which was to be used for
determining the retention dose of the M. tb H37Rv challenge. 10 weeks post-vaccination, the
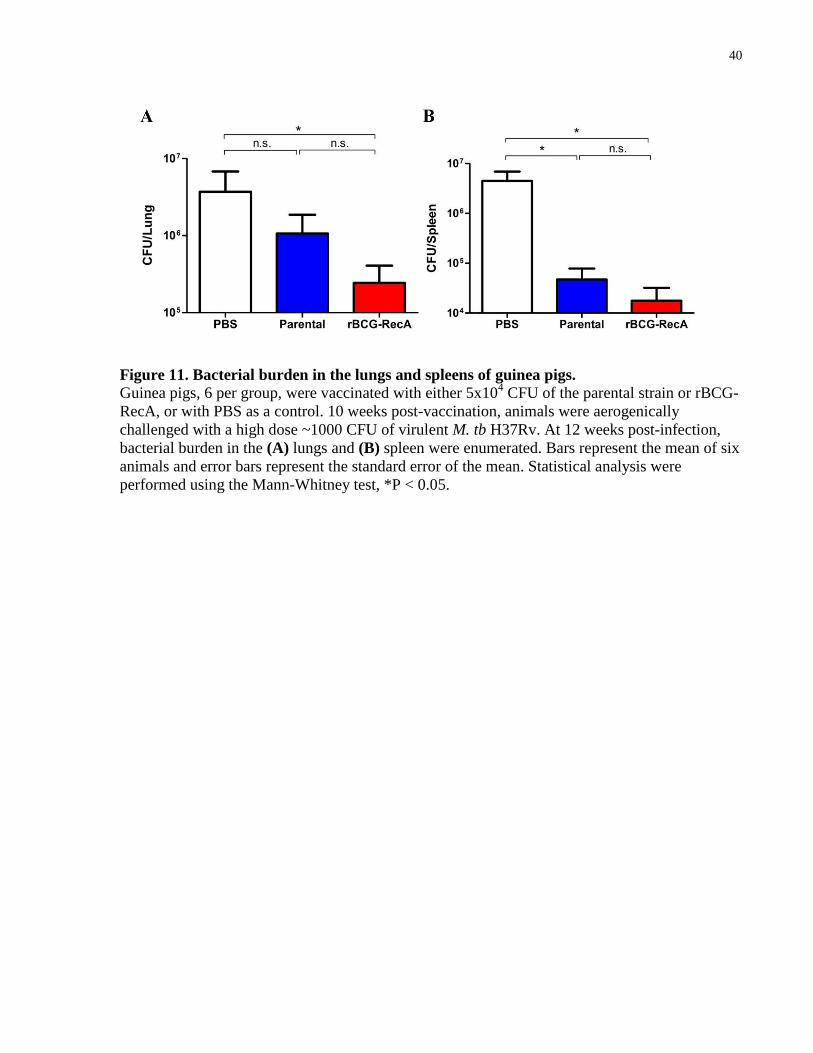
40
Figure 11. Bacterial burden in the lungs and spleens of guinea pigs.
Guinea pigs, 6 per group, were vaccinated with either 5x104 CFU of the parental strain or rBCG-
RecA, or with PBS as a control. 10 weeks post-vaccination, animals were aerogenically
challenged with a high dose ~1000 CFU of virulent M. tb H37Rv. At 12 weeks post-infection,
bacterial burden in the (A) lungs and (B) spleen were enumerated. Bars represent the mean of six
animals and error bars represent the standard error of the mean. Statistical analysis were
performed using the Mann-Whitney test, *P < 0.05.

41
animals were aerogenically challenged with M. tb H37Rv to obtain a retention dose of ~1000
CFU per animal. Four guinea pigs from the PBS group were euthanized day 1 post-infection and
the retention dose in the lungs was determined to be 1281 CFU/animal (data not shown). The
remainder of the animals, six per group, were used to determine bacterial load in the lungs and
spleen at week 12 post-infection. It was previously reported that the short term reduction in
bacterial burden in guinea pigs correlates with the long term survival of the animals
(Wiegeshaus, McMurray et al. 1970). It is important to note that one guinea pig from the
unvaccinated group had reached a pre-determined endpoint prior to the end of the experiment
and had to be euthanized at week 9 of the experiment. At 12 weeks post-infection, the parental-
vaccinated group yielded 3.5 fold less bacteria in the lungs (P = 0.18) and 94 fold less bacteria in
the spleen (P = 0.03) when compared to the unvaccinated control (Figure 11A). The rBCG-RecA
group yielded significantly less bacteria in both the lungs and spleen compared to the
unvaccinated control, 15 fold (P = 0.026) and 25 fold (P = 0.0152), respectively (Figure 11A,B).
When comparing the two vaccinated groups, the rBCG-RecA group yielded 4.3 fold less bacteria
in the lungs (P = 0.63)(Figure 11A) and 2.7 fold less bacteria in the spleen (P = 0.94)(Figure
11B) than the parental group. These findings suggest that the recombinant strain may offer
similar levels of protection against M.tb as the parental strain in guinea pigs.
3.8 rBCG-RecA-vaccinated guinea pigs exhibit disease
phenotypes similar to the parental-vaccinated animals
Observable disease phenotypes in the guinea pigs further support that rBCG-RecA offer similar
protection as the parental strain. A well documented phenotype of M. tb infection is the body
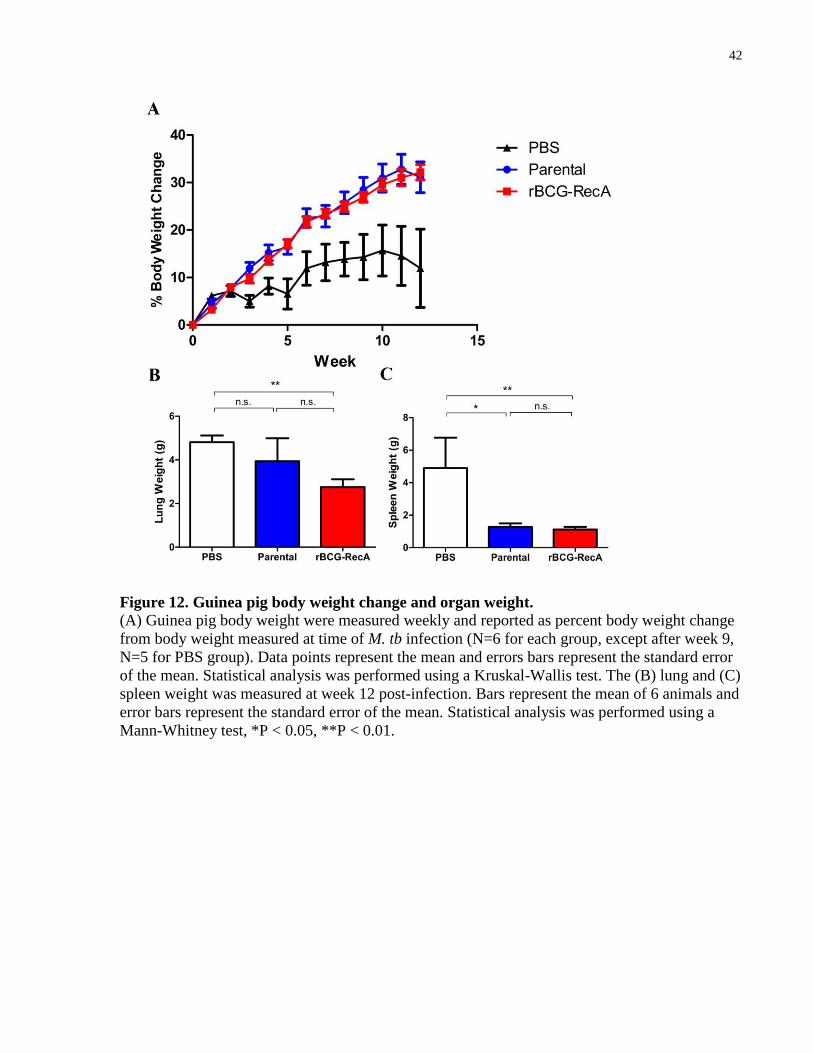
42
Figure 12. Guinea pig body weight change and organ weight.
(A) Guinea pig body weight were measured weekly and reported as percent body weight change
from body weight measured at time of M. tb infection (N=6 for each group, except after week 9,
N=5 for PBS group). Data points represent the mean and errors bars represent the standard error
of the mean. Statistical analysis was performed using a Kruskal-Wallis test. The (B) lung and (C)
spleen weight was measured at week 12 post-infection. Bars represent the mean of 6 animals and
error bars represent the standard error of the mean. Statistical analysis was performed using a
Mann-Whitney test, *P < 0.05, **P < 0.01.

43
weight loss and consumption of the host (Padilla-Carlin, McMurray et al. 2008). A clear and
significant difference (P = 0.0369) in body weight change of vaccinated and unvaccinated
animals emerged over the course of the experiment (Figure 12A). The unvaccinated group
showed weight gain to a much lesser extent when compared to the vaccinated groups. No
difference in body weight changes was observed between the two vaccinated groups and weight
gain was observed until the end of the experiment. Furthermore, a significant decrease (P =
0.0152 and P = 0.0043) was observed when comparing the spleen weights of parental-vaccinated
animals (1.278 ± 0.2111 g) or rBCG-RecA-vaccinated animals (1.122 ± 0.1651 g) to the
unvaccinated control (4.891 ± 1.882 g) (Figure 12C). As mentioned previously, splenomegaly is
a disease phenotype that correlates with systemic infection. No difference in spleen weight was
observed between the two vaccinated groups. Interestingly, the rBCG-RecA-vaccinated group
(2.753 ± 0.3569 g) exhibited lower lung weight than the unvaccinated animals (4.811 ± 0.3078
g) while the parental-vaccinated group (3.936 ± 1.059 g) did not (Figure 12B). Taken together,
these findings also suggest that the recombinant strain offers similar levels of protection as the
parental strain.
3.9 Comparable lung pathology observed in rBCG-RecA-
vaccinated and parental-vaccinated animals
Histological analysis on the lungs and spleens of the guinea pigs were performed to obtain
further evidence on the protective efficacy of the rBCG-RecA. Tissue samples, obtained 12
weeks post-infection, were processed and subjected to H&E staining for quantitative analysis.

44
Figure 13. Histological analysis on guinea pig lungs and spleen.
Representative images, 50x magnification, of H&E staining in the lungs (A, B, C) and spleens
(D, E, F) of unvaccinated (A,D), parental-vaccinated (B, E), and rBCG-RecA-vaccinated (C, F)
guinea pigs show visible granulomatous lesions (arrows) in the organs. Severe infiltration is seen
in all lung samples examined. Quantitative analysis was performed to evaluate the percentage
area of (G) granulomatous lesions and (H) infiltration in the lungs. (I) Number of granulomas
seen in the spleen of the animals were evaluated as well. Bars represent the mean of 6 slides and
error bars represent the standard of the mean. Statistical analysis was performed using a Mann-
Whitney test, *P < 0.05.

45
Severe lung pathology was observed in all animals including extensive infiltration of the alveolar
space and visible granulomatous lesions (Figure 13A-F). The perceived area of granulomatous
lesions and infiltration in the lungs were quantified separately to evaluate the lung pathology. A
significant difference (P = 0.0152) was seen in the percent of granulomatous lesions of total lung
tissue between the rBCG-RecA-vaccinated animals (5.688 ± 1.668 %) and the unvaccinated
control (15.40 ± 2.568 %) (Figure 13G). This difference was not observed between the parental-
vaccinated animals (6.859 ± 2.585 %) and the unvaccinated control or the rBCG-RecA-
vaccinated animals. No significant difference was seen between any of the groups with regards
to the percentage of infiltration observed in the lungs (Figure 13H). I also performed quantitative
analysis on the number of granulomatous lesions observed in the spleen as it considers
dissemination of M. tb as a factor. Visible granulomatous lesions were observed in the spleens of
all groups but both parental- and rBCG-RecA-vaccinated animals have significantly less
granulomatous lesions (P = 0.0435 and P = 0.0128, respectively) observed in the spleen than the
unvaccinated control, though no difference is seen between the two vaccinated groups (Figure
13I). Taken together, these findings further support that rBCG-RecA may offer equivalent BCG-
mediated protection in guinea pigs compared to the parental strain.

46
Chapter 4
Discussion
4.1 General Discussion
Tuberculosis is a major global health threat that claims over a million lives every year and causes
illness in many more. The large population of latently infected individuals along with the highly
contagious nature of the pathogen proves to be a large obstacle for the eradication of TB. This is
further complicated since regions with high risk of TB also coincide with regions with high risk
of HIV. Immunocompromised individuals that are co-infected with M. tb have been shown to
have significantly increased risks of reactivation (Selwyn, Hartel et al. 1989). Traditional
chemotherapy as treatment for TB infection is quickly becoming ineffective due to the
emergence of multidrug and extensively-drug resistant strains demonstrating the pressing need
for better preventative measures for TB control. Aside from improving diagnosis of TB
infection, a potent vaccine is needed to prevent further spread of the disease. The BCG is the
only available vaccine for TB control. Although it demonstrates superior efficacy in preventing
disseminated forms of the disease in children, it shows highly variable efficacies in preventing
pulmonary TB in adults. Several hypotheses have been posed to explain this, one of which
concerns the heterogeneity of BCG. It has been described that the 1921 M. bovis BCG has
diversified into over a dozen of genetically distinct substrains and that these genetic differences
between substrains have led to phenotypical and immunological variations (Castillo-Rodal,
Castanon-Arreola et al. 2006, Rodriguez-Alvarez, Mendoza-Hernandez et al. 2009). Evidence in
animal studies has suggested that the substrains may exhibit different clinical properties
including immunogenicity, vaccine virulence and protection efficacy (Bunch-Christensen,

47
Ladefoged et al. 1970, Junior and Gontijo Filho 1979, Castillo-Rodal, Castanon-Arreola et al.
2006), though head-to-head comparison of substrains in humans are not available. Understanding
these molecular factors and their impact on BCG vaccine properties will allow the development
of better TB vaccines.
In this work, I have generated a recombinant vaccine since no other types of vaccine, neither
protein subunit or DNA vaccines, show greater protection efficacy than live vaccines when used
alone. A genetic background of BCG-Russia was chosen as it is the earliest strain to have
diverged from the original 1921 M. bovis BCG, which was shown to have >80% efficacy
(Calmette, Guerin et al. 1924), and is the most genetically conserved amongst all the current
BCG substrains. It was previously demonstrated that BCG-Russia is a RecA mutant and is likely
the reason for the high genomic stability. RecA plays a critical role in homologous
recombination and in the induction of the SOS response. BCG-Russia is also one of the three
strains used to produce the vaccine today (Luca and Mihaescu 2013), making it a very relevant
background for further investigation. The recombinant strain used in this study was named
rBCG-RecA which expresses functional RecA in a BCG-Russia background.
In contrast to what I had originally hypothesized, rBCG-RecA was more attenuated than the
parental strain. In a SCID mice model, animals infected with rBCG-RecA demonstrated
increased survival when compared to the parental strain. Consistently, pathological indicators
such as spleen weight also suggested that mice infected with the recombinant strain were
healthier than their parental counterparts. Bacterial burden in the organs suggest that rBCG-
RecA may grow slower within the host compared to the parental strain. This defect in in vivo
growth is not due to an inherent growth defect or the ability to survive intracellularly within the
macrophage, its primary host. Although the mechanism for the growth defect within the animal

48
host remains unknown, I propose a possible explanation for this observation. RecA has been
known to be an important cell cycle checkpoint in DNA damage and repair. DNA damage in
bacteria results in RecA-dependent cleavage of LexA repressor to allow expression of SOS
response genes. The SOS response leads to cell cycle arrest via induction of SfiA (Autret, Levine
et al. 1997) so DNA repair may be performed before cell replication. The parental strain that has
a non-functional RecA protein may suffer sublethal DNA damage but the defect in the SOS
response allows bacterial replication to continue despite oxidative damage. In contrast, the
recombinant strain with functional RecA confers an intact SOS response, even to sublethal
oxidative damage, resulting in the halt of bacterial replication until the DNA is repaired. This
sublethal damage is likely due to SCID mice lacking functional lymphocytes resulting in
impaired activation of macrophages and other innate immune cells which are necessary for
effective killing of the pathogen via reactive oxygen species and reactive nitrogen species. This
growth defect was not observed in the intracellular survival assay in resting RAW murine
macrophages possibly because other factors present in vivo that partially contribute to pathogen
elimination were absent in the in vitro assay. As such, activated macrophages may be a better
suited model to study this and further investigation is warranted.
I have also demonstrated that rBCG-RecA confers protection efficacy equivalent, if not superior,
to that of the parental strain in a guinea pig model. rBCG-Russia induced greater BCG-induced
protection in the lungs with reduced bacterial loads compared to both the parental-vaccinated and
unvaccinated groups. This reduction rivals that seen from animal trials involving vaccines
currently undergoing clinical trials. Surprisingly, the parental-vaccinated group did not show
reduced bacterial burden in the lungs when compared to the unvaccinated animals. Consistently,
the amount of granulomatous lesions observed was significantly reduced in the recombinant-
vaccinated strain compared to the two other groups, which had no difference between them. In

49
contrast, the extent of infiltration observed in the lungs was comparable between all three groups.
Similar bacterial burdens were observed in the spleens of guinea pigs between the two
vaccinated strains and were markedly reduced compared to the unvaccinated strain suggesting
the ability of the vaccine to reduce dissemination of the virulent M. tb from the primary site of
infection to distant organs. Consistently, the number of granulomas observed in the spleen was
similar between the two vaccinated strains but significantly reduced compared to the
unvaccinated strain. The ability to prevent disseminated infection is an important quality of a
vaccine and should not be overlooked. Taken together, this demonstrates that rBCG-RecA, at
least, provides protection equivalent to that conferred by the parental strain.
Previously, it has been shown that the deletion of RecA in BCG-Pasteur increased its genetic
stability but did not impact the virulence or protection efficacy against virulent M. tb challenge in
mice models (Sander, Papavinasasundaram et al. 2001, Sander, Bottger et al. 2003). This is
somewhat in contrast to the findings I have presented here in that RecA in BCG-Russia leads to
attenuation of the strain while maintaining its protective efficacy. This is likely due to the genetic
differences between BCG-Russia and BCG-Pasteur, further supporting that the genetic
background is an important factor to consider when choosing which substrains to use for
developing vaccines. Therefore, the findings in this work can only be interpreted in the context
of BCG-Russia. Further investigation is needed to elucidate the impact of RecA in other BCG
substrains.
Due to the nature of rBCG-RecA containing a functional RecA, it is more genetically unstable
than its parental counterpart, which is an unfavourable property for a vaccine. Furthermore, it
contains an antibiotic resistance marker, thus, careful consideration must be taken with regards to
the potential use of rBCG-RecA as a human vaccine.

50
4.2 Summary of key findings and conclusion
In this work, I report the successful generation of a recombinant BCG-Russia expressing
functional RecA, rBCG-RecA. This strain does not exhibit any inherent growth defects or a
defect in intracellular survival within resting murine macrophages. This recombinant strain is
attenuated in a SCID mice model of infection when compared to the parental strain. Consistently,
splenomegaly was less severe in rBCG-RecA infected mice compared to the parental-infected
animals. This attenuation is likely a result of slower growth of the bacteria in vivo although the
mechanism is not fully understood. The protection against aerosol M. tb challenge conferred by
rBCG-RecA in guinea pigs is equivalent to that offered by the parental strain demonstrated by
similar levels of bacterial burden were observed in the lungs and spleens of the animals. In
addition, pathological indicators such as weight loss, splenomegaly, extent of infiltration, and
amount of granulomatous lesions are similar for both vaccinated groups which further support
this notion. The findings presented here will provide a better understanding of molecular factors
and their impact on the clinical properties of the BCG vaccine, providing new insight to the
development of future TB vaccines.
4.3 Future directions
Based on my findings here, investigation into the molecular differences of rBCG-RecA and
further characterization of the strain will be important to fully understand the impact of
functional RecA on BCG-Russia’s vaccine properties. Immunogenicity studies, which are

51
lacking in this present work, are worth pursuing in in vitro and in the animal models since
reagents that enable immunological studies in guinea pigs have recently become available. These
studies will allow the elucidation of the mechanism behind the attenuation and protection
conferred by rBCG-RecA. Transcriptome analysis on rBCG-RecA may identify differentially
regulated genes that may also contribute to understanding the molecular differences of rBCG-
RecA.
rBCG-RecA has been shown to be attenuated without compromising BCG-mediated protection
in this work. Though further studies are needed to evaluate the safety and protection efficacy in
humans, this is very promising news for the use of rBCG-RecA as a potential vaccine in
immunocompromised individuals. Editing of the genome to restore functional RecA in BCG-
Russia will circumvent the issue of the vaccine containing an antibiotic resistance marker.
Moreover, development into the further attenuation or improved protection efficacy will greatly
increase the potency of rBCG-RecA as a TB vaccine.

52
References
Abdallah, A. M., J. Bestebroer, N. D. Savage, K. de Punder, M. van Zon, L. Wilson, C. J.
Korbee, A. M. van der Sar, T. H. Ottenhoff, N. N. van der Wel, W. Bitter and P. J. Peters (2011).
"Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and
inflammasome activation." J Immunol 187(9): 4744-4753.
Aihara, H., Y. Ito, H. Kurumizaka, T. Terada, S. Yokoyama and T. Shibata (1997). "An
interaction between a specified surface of the C-terminal domain of RecA protein and double-
stranded DNA for homologous pairing." J Mol Biol 274(2): 213-221.
Arbues, A., J. I. Aguilo, J. Gonzalo-Asensio, D. Marinova, S. Uranga, E. Puentes, C. Fernandez,
A. Parra, P. J. Cardona, C. Vilaplana, V. Ausina, A. Williams, S. Clark, W. Malaga, C. Guilhot,
B. Gicquel and C. Martin (2013). "Construction, characterization and preclinical evaluation of
MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials."
Vaccine 31(42): 4867-4873.
Autret, S., A. Levine, I. B. Holland and S. J. Seror (1997). "Cell cycle checkpoints in bacteria."
Biochimie 79(9-10): 549-554.
Behr, M. A. and P. M. Small (1997). "Has BCG attenuated to impotence?" Nature 389(6647):
133-134.
Bell, C. E. (2005). "Structure and mechanism of Escherichia coli RecA ATPase." Mol Microbiol
58(2): 358-366.
Bosma, M. J. and A. M. Carroll (1991). "The SCID mouse mutant: definition, characterization,
and potential uses." Annu Rev Immunol 9: 323-350.
Bourassa, D., A. Forget, M. Pelletier, E. Skamene and R. Turcotte (1985). "Cellular immune
response to Mycobacterium bovis (BCG) in genetically-susceptible and resistant congenic mouse
strains." Clin Exp Immunol 62(1): 31-38.
Brandt, L., Y. A. Skeiky, M. R. Alderson, Y. Lobet, W. Dalemans, O. C. Turner, R. J. Basaraba,
A. A. Izzo, T. M. Lasco, P. L. Chapman, S. G. Reed and I. M. Orme (2004). "The protective
effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the
Mycobacterium tuberculosis 72-kilodalton fusion polyprotein Mtb72F in M. tuberculosis-
infected guinea pigs." Infect Immun 72(11): 6622-6632.
Brewer, T. F. (2000). "Preventing tuberculosis with bacillus Calmette-Guerin vaccine: a meta-
analysis of the literature." Clin Infect Dis 31 Suppl 3: S64-67.
Brosch, R., S. V. Gordon, T. Garnier, K. Eiglmeier, W. Frigui, P. Valenti, S. Dos Santos, S.
Duthoy, C. Lacroix, C. Garcia-Pelayo, J. K. Inwald, P. Golby, J. N. Garcia, R. G. Hewinson, M.
A. Behr, M. A. Quail, C. Churcher, B. G. Barrell, J. Parkhill and S. T. Cole (2007). "Genome

53
plasticity of BCG and impact on vaccine efficacy." Proc Natl Acad Sci U S A 104(13): 5596-
5601.
Brosch, R., S. V. Gordon, M. Marmiesse, P. Brodin, C. Buchrieser, K. Eiglmeier, T. Garnier, C.
Gutierrez, G. Hewinson, K. Kremer, L. M. Parsons, A. S. Pym, S. Samper, D. van Soolingen and
S. T. Cole (2002). "A new evolutionary scenario for the Mycobacterium tuberculosis complex."
Proc Natl Acad Sci U S A 99(6): 3684-3689.
Bunch-Christensen, K., A. Ladefoged and J. Guld (1970). "The virulence of some strains of
BCG for golden hamsters. Further studies." Bull World Health Organ 43(1): 65-70.
Calmette, A., C. Guerin and B. Weill-Halle (1924). "Essai d'immunisation contre l'infection
tuberculeuse." Bull Acad Med. 91: 787-796.
Cambier, C. J., S. Falkow and L. Ramakrishnan (2014). "Host evasion and exploitation schemes
of Mycobacterium tuberculosis." Cell 159(7): 1497-1509.
Cardenas, P. P., B. Carrasco, C. Defeu Soufo, C. E. Cesar, K. Herr, M. Kaufenstein, P. L.
Graumann and J. C. Alonso (2012). "RecX facilitates homologous recombination by modulating
RecA activities." PLoS Genet 8(12): e1003126.
Castillo-Rodal, A. I., M. Castanon-Arreola, R. Hernandez-Pando, J. J. Calva, E. Sada-Diaz and
Y. Lopez-Vidal (2006). "Mycobacterium bovis BCG substrains confer different levels of
protection against Mycobacterium tuberculosis infection in a BALB/c model of progressive
pulmonary tuberculosis." Infect Immun 74(3): 1718-1724.
Chen, J. M., S. T. Islam, H. Ren and J. Liu (2007). "Differential productions of lipid virulence
factors among BCG vaccine strains and implications on BCG safety." Vaccine 25(48): 8114-
8122.
Chen, Z., H. Yang and N. P. Pavletich (2008). "Mechanism of homologous recombination from
the RecA-ssDNA/dsDNA structures." Nature 453(7194): 489-484.
Colditz, G. A., C. S. Berkey, F. Mosteller, T. F. Brewer, M. E. Wilson, E. Burdick and H. V.
Fineberg (1995). "The efficacy of bacillus Calmette-Guerin vaccination of newborns and infants
in the prevention of tuberculosis: meta-analyses of the published literature." Pediatrics 96(1 Pt
1): 29-35.
Colditz, G. A., T. F. Brewer, C. S. Berkey, M. E. Wilson, E. Burdick, H. V. Fineberg and F.
Mosteller (1994). "Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of
the published literature." JAMA 271(9): 698-702.
Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K.
Eiglmeier, S. Gas, C. E. Barry, 3rd, F. Tekaia, K. Badcock, D. Basham, D. Brown, T.
Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd,
T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A.
Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J.
E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell (1998). "Deciphering the biology of
Mycobacterium tuberculosis from the complete genome sequence." Nature 393(6685): 537-544.

54
Davis, E. O., S. G. Sedgwick and M. J. Colston (1991). "Novel structure of the recA locus of
Mycobacterium tuberculosis implies processing of the gene product." J Bacteriol 173(18): 5653-
5662.
Davis, E. O., B. Springer, K. K. Gopaul, K. G. Papavinasasundaram, P. Sander and E. C. Bottger
(2002). "DNA damage induction of recA in Mycobacterium tuberculosis independently of RecA
and LexA." Mol Microbiol 46(3): 791-800.
de Jonge, M. I., G. Pehau-Arnaudet, M. M. Fretz, F. Romain, D. Bottai, P. Brodin, N. Honore, G.
Marchal, W. Jiskoot, P. England, S. T. Cole and R. Brosch (2007). "ESAT-6 from
Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic
conditions and exhibits membrane-lysing activity." J Bacteriol 189(16): 6028-6034.
Derrick, S. C. and S. L. Morris (2007). "The ESAT6 protein of Mycobacterium tuberculosis
induces apoptosis of macrophages by activating caspase expression." Cell Microbiol 9(6): 1547-
1555.
Flesch, I. E. and S. H. Kaufmann (1990). "Activation of tuberculostatic macrophage functions by
gamma interferon, interleukin-4, and tumor necrosis factor." Infect Immun 58(8): 2675-2677.
Flynn, J. L., J. Chan, K. J. Triebold, D. K. Dalton, T. A. Stewart and B. R. Bloom (1993). "An
essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection." J Exp
Med 178(6): 2249-2254.
Fukuda, T., T. Matsumura, M. Ato, M. Hamasaki, Y. Nishiuchi, Y. Murakami, Y. Maeda, T.
Yoshimori, S. Matsumoto, K. Kobayashi, T. Kinoshita and Y. S. Morita (2013). "Critical roles
for lipomannan and lipoarabinomannan in cell wall integrity of mycobacteria and pathogenesis
of tuberculosis." MBio 4(1): e00472-00412.
Gebhardt, H., X. Meniche, M. Tropis, R. Kramer, M. Daffe and S. Morbach (2007). "The key
role of the mycolic acid content in the functionality of the cell wall permeability barrier in
Corynebacterineae." Microbiology 153(Pt 5): 1424-1434.
Gey Van Pittius, N. C., J. Gamieldien, W. Hide, G. D. Brown, R. J. Siezen and A. D. Beyers
(2001). "The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-
positive bacteria." Genome Biol 2(10): RESEARCH0044.
Goldstone, R. M., S. D. Goonesekera, B. R. Bloom and S. L. Sampson (2009). "The
transcriptional regulator Rv0485 modulates the expression of a pe and ppe gene pair and is
required for Mycobacterium tuberculosis virulence." Infect Immun 77(10): 4654-4667.
Guhan, N. and K. Muniyappa (2002). "Mycobacterium tuberculosis RecA intein possesses a
novel ATP-dependent site-specific double-stranded DNA endonuclease activity." J Biol Chem
277(18): 16257-16264.
Herbst, S., U. E. Schaible and B. E. Schneider (2011). "Interferon gamma activated macrophages
kill mycobacteria by nitric oxide induced apoptosis." PLoS One 6(5): e19105.

55
Herr, H. W., D. M. Schwalb, Z. F. Zhang, P. C. Sogani, W. R. Fair, W. F. Whitmore, Jr. and H.
F. Oettgen (1995). "Intravesical bacillus Calmette-Guerin therapy prevents tumor progression
and death from superficial bladder cancer: ten-year follow-up of a prospective randomized trial."
J Clin Oncol 13(6): 1404-1408.
Hesseling, A. C., B. J. Marais, R. P. Gie, H. S. Schaaf, P. E. Fine, P. Godfrey-Faussett and N.
Beyers (2007). "The risk of disseminated Bacille Calmette-Guerin (BCG) disease in HIV-
infected children." Vaccine 25(1): 14-18.
Huang, T. W. and C. W. Chen (2006). "A recA null mutation may be generated in Streptomyces
coelicolor." J Bacteriol 188(19): 6771-6779.
Junior, L. P. and P. P. Gontijo Filho (1979). "Comparative immunogenicity of BCG substrains
for mice." Rev Bras Pesqui Med Biol 12(4-5): 311-316.
Kashangura, R., E. S. Sena, T. Young and P. Garner (2015). "Effects of MVA85A vaccine on
tuberculosis challenge in animals: systematic review." Int J Epidemiol.
Kaufmann, S. H., M. F. Cotton, B. Eisele, M. Gengenbacher, L. Grode, A. C. Hesseling and G.
Walzl (2014). "The BCG replacement vaccine VPM1002: from drawing board to clinical trial."
Expert Rev Vaccines 13(5): 619-630.
Kawabata, M., T. Kawabata and M. Nishibori (2005). "Role of recA/RAD51 family proteins in
mammals." Acta Med Okayama 59(1): 1-9.
Keane, J., S. Gershon, R. P. Wise, E. Mirabile-Levens, J. Kasznica, W. D. Schwieterman, J. N.
Siegel and M. M. Braun (2001). "Tuberculosis associated with infliximab, a tumor necrosis
factor alpha-neutralizing agent." N Engl J Med 345(15): 1098-1104.
Keller, P. M., E. C. Bottger and P. Sander (2008). "Tuberculosis vaccine strain Mycobacterium
bovis BCG Russia is a natural recA mutant." BMC Microbiol 8: 120.
Kumar, R. A., M. B. Vaze, N. R. Chandra, M. Vijayan and K. Muniyappa (1996). "Functional
characterization of the precursor and spliced forms of RecA protein of Mycobacterium
tuberculosis." Biochemistry 35(6): 1793-1802.
Leroux-Roels, I., S. Forgus, F. De Boever, F. Clement, M. A. Demoitie, P. Mettens, P. Moris, E.
Ledent, G. Leroux-Roels, O. Ofori-Anyinam and M. S. Group (2013). "Improved CD4(+) T cell
responses to Mycobacterium tuberculosis in PPD-negative adults by M72/AS01 as compared to
the M72/AS02 and Mtb72F/AS02 tuberculosis candidate vaccine formulations: a randomized
trial." Vaccine 31(17): 2196-2206.
Leung, A. S., V. Tran, Z. Wu, X. Yu, D. C. Alexander, G. F. Gao, B. Zhu and J. Liu (2008).
"Novel genome polymorphisms in BCG vaccine strains and impact on efficacy." BMC
Genomics 9: 413.
Lewis, K. N., R. Liao, K. M. Guinn, M. J. Hickey, S. Smith, M. A. Behr and D. R. Sherman
(2003). "Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin
attenuation." J Infect Dis 187(1): 117-123.

56
Liu, J., C. E. Barry, 3rd, G. S. Besra and H. Nikaido (1996). "Mycolic acid structure determines
the fluidity of the mycobacterial cell wall." J Biol Chem 271(47): 29545-29551.
Liu, J., E. Y. Rosenberg and H. Nikaido (1995). "Fluidity of the lipid domain of cell wall from
Mycobacterium chelonae." Proc Natl Acad Sci U S A 92(24): 11254-11258.
Lotte, A., O. Wasz-Hockert, N. Poisson, N. Dumitrescu, M. Verron and E. Couvet (1984). "BCG
complications. Estimates of the risks among vaccinated subjects and statistical analysis of their
main characteristics." Adv Tuberc Res 21: 107-193.
Luca, S. and T. Mihaescu (2013). "History of BCG Vaccine." Maedica (Buchar) 8(1): 53-58.
Malkov, V. A. and R. D. Camerini-Otero (1995). "Photocross-links between single-stranded
DNA and Escherichia coli RecA protein map to loops L1 (amino acid residues 157-164) and L2
(amino acid residues 195-209)." J Biol Chem 270(50): 30230-30233.
Michel, B. (2005). "After 30 years of study, the bacterial SOS response still surprises us." PLoS
Biol 3(7): e255.
Nair, S., P. A. Ramaswamy, S. Ghosh, D. C. Joshi, N. Pathak, I. Siddiqui, P. Sharma, S. E.
Hasnain, S. C. Mande and S. Mukhopadhyay (2009). "The PPE18 of Mycobacterium
tuberculosis interacts with TLR2 and activates IL-10 induction in macrophage." J Immunol
183(10): 6269-6281.
Nautiyal, A., K. N. Patil and K. Muniyappa (2014). "Suramin is a potent and selective inhibitor
of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target
for antibacterial drug discovery." J Antimicrob Chemother 69(7): 1834-1843.
Ottenhoff, T. H., D. Kumararatne and J. L. Casanova (1998). "Novel human immunodeficiencies
reveal the essential role of type-I cytokines in immunity to intracellular bacteria." Immunol
Today 19(11): 491-494.
Padilla-Carlin, D. J., D. N. McMurray and A. J. Hickey (2008). "The guinea pig as a model of
infectious diseases." Comp Med 58(4): 324-340.
Papavinasasundaram, K. G., M. J. Colston and E. O. Davis (1998). "Construction and
complementation of a recA deletion mutant of Mycobacterium smegmatis reveals that the intein
in Mycobacterium tuberculosis recA does not affect RecA function." Mol Microbiol 30(3): 525-
534.
Penn-Nicholson, A., H. Geldenhuys, W. Burny, R. van der Most, C. L. Day, E. Jongert, P. Moris,
M. Hatherill, O. Ofori-Anyinam, W. Hanekom, T. Vaccine Study, A. Bollaerts, M. A. Demoitie,
A. K. Kany Luabeya, E. De Ruymaeker, M. Tameris, D. Lapierre and T. J. Scriba (2015).
"Safety and immunogenicity of candidate vaccine M72/AS01E in adolescents in a TB endemic
setting." Vaccine 33(32): 4025-4034.
Pym, A. S., P. Brodin, R. Brosch, M. Huerre and S. T. Cole (2002). "Loss of RD1 contributed to
the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium
microti." Mol Microbiol 46(3): 709-717.

57
Ramakrishnan, L. (2012). "Revisiting the role of the granuloma in tuberculosis." Nat Rev
Immunol 12(5): 352-366.
Riley, L. W. (2006). "Of mice, men, and elephants: Mycobacterium tuberculosis cell envelope
lipids and pathogenesis." J Clin Invest 116(6): 1475-1478.
Roca, A. I. and M. M. Cox (1990). "The RecA protein: structure and function." Crit Rev
Biochem Mol Biol 25(6): 415-456.
Rodriguez-Alvarez, M., G. Mendoza-Hernandez, S. Encarnacion, J. J. Calva and Y. Lopez-Vidal
(2009). "Phenotypic differences between BCG vaccines at the proteome level." Tuberculosis
(Edinb) 89(2): 126-135.
Sander, P., E. C. Bottger, B. Springer, B. Steinmann, M. Rezwan, E. Stavropoulos and M. Joseph
Colston (2003). "A recA deletion mutant of Mycobacterium bovis BCG confers protection
equivalent to that of wild-type BCG but shows increased genetic stability." Vaccine 21(27-30):
4124-4127.
Sander, P., K. G. Papavinasasundaram, T. Dick, E. Stavropoulos, K. Ellrott, B. Springer, M. J.
Colston and E. C. Bottger (2001). "Mycobacterium bovis BCG recA deletion mutant shows
increased susceptibility to DNA-damaging agents but wild-type survival in a mouse infection
model." Infect Immun 69(6): 3562-3568.
Scriba, T. J., M. Tameris, N. Mansoor, E. Smit, L. van der Merwe, K. Mauff, E. J. Hughes, S.
Moyo, N. Brittain, A. Lawrie, H. Mulenga, M. de Kock, S. Gelderbloem, A. Veldsman, M.
Hatherill, H. Geldenhuys, A. V. Hill, G. D. Hussey, H. Mahomed, W. A. Hanekom and H.
McShane (2011). "Dose-finding study of the novel tuberculosis vaccine, MVA85A, in healthy
BCG-vaccinated infants." J Infect Dis 203(12): 1832-1843.
Selwyn, P. A., P. Alcabes, D. Hartel, D. Buono, E. E. Schoenbaum, R. S. Klein, K. Davenny and
G. H. Friedland (1992). "Clinical manifestations and predictors of disease progression in drug
users with human immunodeficiency virus infection." N Engl J Med 327(24): 1697-1703.
Selwyn, P. A., D. Hartel, V. A. Lewis, E. E. Schoenbaum, S. H. Vermund, R. S. Klein, A. T.
Walker and G. H. Friedland (1989). "A prospective study of the risk of tuberculosis among
intravenous drug users with human immunodeficiency virus infection." N Engl J Med 320(9):
545-550.
Sterne, J. A. C., L. C. Rodrigues and I. N. Guedes (1998). "Does the efficacy of BCG decline
with time since vaccination?" International Journal of Tuberculosis and Lung Disease 2(3): 200-
207.
Tameris, M., H. Geldenhuys, A. K. Luabeya, E. Smit, J. E. Hughes, S. Vermaak, W. A.
Hanekom, M. Hatherill, H. Mahomed, H. McShane and T. J. Scriba (2014). "The candidate TB
vaccine, MVA85A, induces highly durable Th1 responses." PLoS One 9(2): e87340.
Tameris, M. D., M. Hatherill, B. S. Landry, T. J. Scriba, M. A. Snowden, S. Lockhart, J. E. Shea,
J. B. McClain, G. D. Hussey, W. A. Hanekom, H. Mahomed, H. McShane and M. A. T. S. Team
(2013). "Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously

58
vaccinated with BCG: a randomised, placebo-controlled phase 2b trial." Lancet 381(9871):
1021-1028.
Tan, T., W. L. Lee, D. C. Alexander, S. Grinstein and J. Liu (2006). "The ESAT-6/CFP-10
secretion system of Mycobacterium marinum modulates phagosome maturation." Cell Microbiol
8(9): 1417-1429.
Tullius, M. V., G. Harth, S. Maslesa-Galic, B. J. Dillon and M. A. Horwitz (2008). "A
Replication-Limited Recombinant Mycobacterium bovis BCG vaccine against tuberculosis
designed for human immunodeficiency virus-positive persons is safer and more efficacious than
BCG." Infect Immun 76(11): 5200-5214.
van Dissel, J. T., S. M. Arend, C. Prins, P. Bang, P. N. Tingskov, K. Lingnau, J. Nouta, M. R.
Klein, I. Rosenkrands, T. H. Ottenhoff, I. Kromann, T. M. Doherty and P. Andersen (2010).
"Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium
tuberculosis specific T cell responses in naive human volunteers." Vaccine 28(20): 3571-3581.
van Dissel, J. T., D. Soonawala, S. A. Joosten, C. Prins, S. M. Arend, P. Bang, P. N. Tingskov,
K. Lingnau, J. Nouta, S. T. Hoff, I. Rosenkrands, I. Kromann, T. H. Ottenhoff, T. M. Doherty
and P. Andersen (2011). "Ag85B-ESAT-6 adjuvanted with IC31(R) promotes strong and long-
lived Mycobacterium tuberculosis specific T cell responses in volunteers with previous BCG
vaccination or tuberculosis infection." Vaccine 29(11): 2100-2109.
Vergne, I., J. Chua and V. Deretic (2003). "Tuberculosis toxin blocking phagosome maturation
inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade." J Exp Med 198(4): 653-659.
Via, L. E., P. L. Lin, S. M. Ray, J. Carrillo, S. S. Allen, S. Y. Eum, K. Taylor, E. Klein, U.
Manjunatha, J. Gonzales, E. G. Lee, S. K. Park, J. A. Raleigh, S. N. Cho, D. N. McMurray, J. L.
Flynn and C. E. Barry, 3rd (2008). "Tuberculous granulomas are hypoxic in guinea pigs, rabbits,
and nonhuman primates." Infect Immun 76(6): 2333-2340.
WHO (2007). "Revised BCG vaccination guidelines for infants at risk for HIV infection."
Weekly Epidemiological Record 82(21): 193-196.
WHO (2015). Global tuberculosis report 2015. WHO Report 2015.
Wiegeshaus, E. H., D. N. McMurray, A. A. Grover, G. E. Harding and D. W. Smith (1970).
"Host-parasite relationships in experimental airborne tuberculosis. 3. Relevance of microbial
enumeration to acquired resistance in guinea pigs." Am Rev Respir Dis 102(3): 422-429.
Yu, J., V. Tran, M. Li, X. Huang, C. Niu, D. Wang, J. Zhu, J. Wang, Q. Gao and J. Liu (2012).
"Both phthiocerol dimycocerosates and phenolic glycolipids are required for virulence of
Mycobacterium marinum." Infect Immun 80(4): 1381-1389.