introduction à la microanalyse x quantitativemicro.icaunais.free.fr/ea01_quanti.pdf · de...
TRANSCRIPT

1
J. Ruste
CACEMI – Stage EA01 – Microscopie Électronique à Balay age et Microanalyse
Introduction à la microanalyse Xquantitative
http://micro.icaunais.free.fr
( ) ∫∞
θµρ−
αα ρρφπ
Ω++ω=0
sin
z
FR
CK
jj0AjAe
Aj zde)z()E(D
4I1)C1(Z)E(Q
ACnI
0N

Mais, il faut avoir conscience que quelque soit votr e échantillon, son état de surface, vos conditions opératoires…que vous ayez travaillé avec le plus grand soin ou
de façon extrêmement brouillonne…votre instrument vous donnera des informations qualitat ives et
des résultats quantitatifs sans le moindre état d’âm e!
Mais, que valent ces résultats ? Quelle en sera la précision ?
Comment faire pour que votre analyse soit correcte ? …
Les spectromètres de rayons X (EDS et WDS) sont des in struments de qualité, aptes à fournir des informations précises
sur votre échantillon.
Il en est de même des logiciels de quantification
2
! La microanalyse X quantitative peut sembler très simple à réaliser en EDS :on « clique » sur « quantification » et on obtient un tab leau de valeurs…

3
Microanalyse X quantitative
Analyse qualitative : déterminer quels sont les élé ments présents
Analyse quantitative : en préciser la quantité (tene ur chimique)
En microanalyse X, la teneur est exprimée en titre massique(nombre sans dimension)
et non en « concentration » !
∑=
== n
1ii
A
total
AA
m
mmm
C

La microanalyse X quantitative en EDS, comment ça ma rche ?
+1 – On sélectionne la zone d’analyse
2 – On choisit les paramètres d’analyse
3 – On fait l’acquisition d’un spectre
4 – On identifie les raies du spectre
5 – On soustrait le fond continu
6 – On recherche d’éventuelles interférences et on « déconvolue »
7 – On lance la quantificationen choisissant le cas échéant les options :- la méthode de correction (ZAF, PhiRoz…)- le type d’analyse :
- avec ou sans témoin- élémentaire, oxydes, élément non dosé … 4

Et en final on obtient généralement un tableau de ce genre :
Comment le lire et l’interpréter ?
k-ratio :rapport de l’intensité mesuréesur l’échantillon à l’intensité
« mesurée » sur un témoin pur« teneur » massique de l’élémentanalysé : rapport de la masse de
l’élément analysé à la masse totale analysée
Wt% : titre massique
∑=
=
.
.
.
C,B,Aii
A
mm1
00
%Wt
Ici la somme destitres massiquesest normaliséece qui n’est pas
toujours le cas…A éviter si possible
facteurs correctifs
k=(Z.A.F.) Wt
[ ]∑=
=
.
.
.
C,B,Aiii A/ )A%
(
Wt
A/%Wt)A%
(
At 5
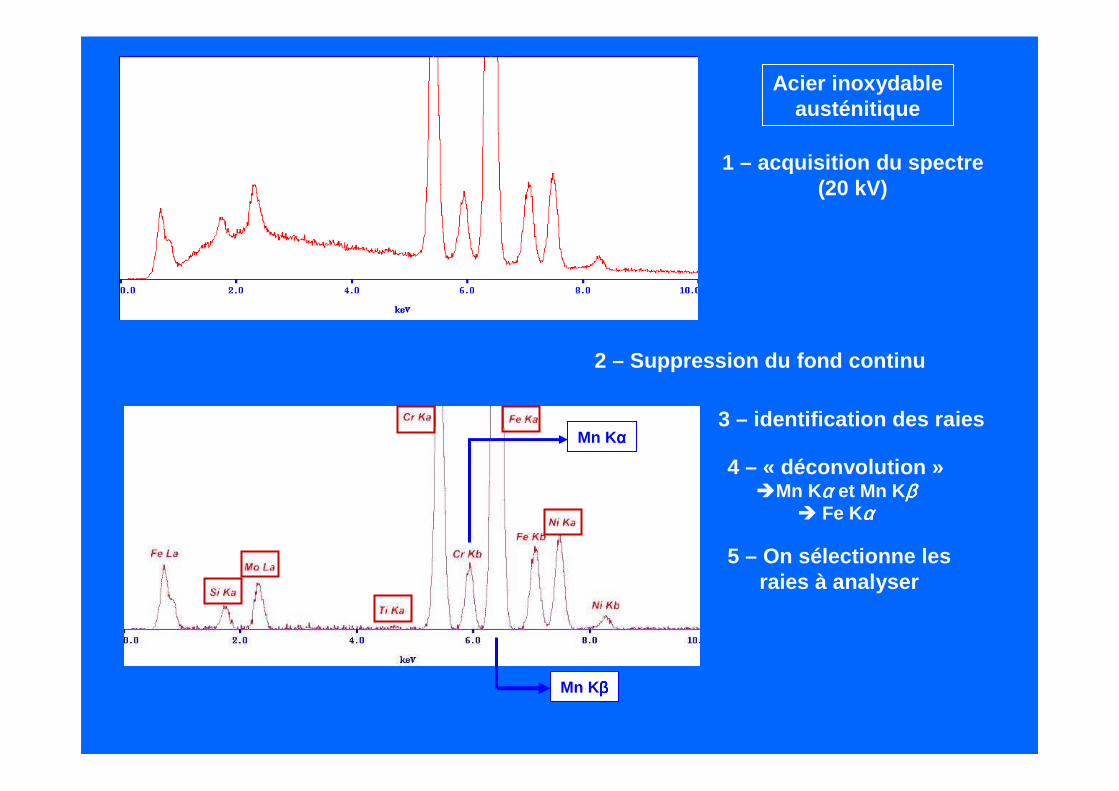
Acier inoxydableausténitique
1 – acquisition du spectre(20 kV)
2 – Suppression du fond continu
3 – identification des raiesMn K αααα
Mn Kββββ
4 – « déconvolution »Mn Kαααα et Mn K ββββ
Fe Kαααα
5 – On sélectionne lesraies à analyser

Identificationdes raies
Déterminationdu fond continu
Recherche desinterférences
« déconvolution »Quantification
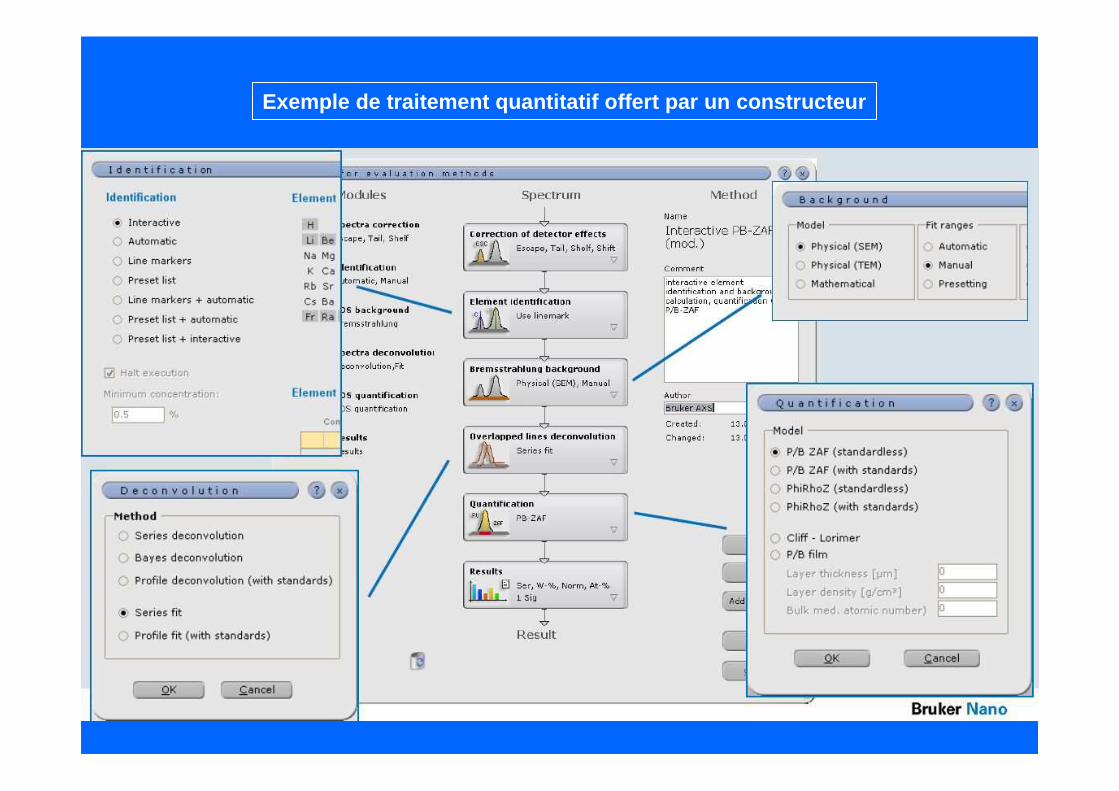
Exemple de traitement quantitatif offert par un con structeur

9
Principes de l’analyse quantitative
CA, CB, CC… : composition de l’échantillon titres massiques réels (inconnus)
[ ]COR
AtemACBAAA FC)C,
.
.
.,
C,C,C(fCk ==
f(CA…) : « calculs de correction » (ZAF, PhiRoz…)
[ ] 1COR
AA FkC −=
« terme correctif »
t : temps de comptage
)N(N
)N(Nktem
echB
FAtemA
BFA
echA
A −−=
kA : « k-ratio » ou titre apparent(grandeur mesurée)
tIN echA
echA =Spectre échantillon :
Spectre témoin : tIN temA
temA =

10
Question : comment calculer le terme correctif [F COR]-1 ?
« Calculs de corrections »- méthode ZAF- procédures PhiRoz
En toute rigueur, tous les éléments doivent être an alysés !
)C,...,
C,C,C(fCk temACBAAA =
Exceptions :
- Un élément peut être calculé par différence : ∑−
=
−=1n
1iix C1C
- Eléments traces (%<1)
On peut se contenter d’analyser uniquement ces élém ents si on peut introduire la composition des éléments majeurs comm e donnée.
- Un ou plusieurs éléments peuvent être calculé par s tœchiométrie
(oxygène, carbone…)
Il faut connaitre les formules chimiques (A n0m…)
Contrainte :

Analyse quantitative : quels sont les paramètres infl uents ?
réglage et position du détecteur X (EDS)
• distance de travail• distance du détecteur• résolution• efficacité de détection• calibration en énergie• gamme d’analyse (0-10, 0-20 keV…)• énergie par canal (10 eV, 20 eV…)
Paramètres d’analyse- choix des raies d’analyse - tension d’accélération- intensité du faisceau électronique- temps d’acquisition des spectres
Mode d’analyse
- avec ou sans témoin- élément(s) dosé(s) par différence
ou par stœchiométrie ?
Procédure de quantification- suppression du fond continu- méthode de « déconvolution »-choix de la méthode : ZAF, Phiroz, autre…
Question : avez-vous le choix ?
Optimiser ces paramètres, ce n’est pas toujours cho se facile !
Paramètresinfluents
échantillon
- homogène dans la zone analysée- plan- poli- conducteur- dense …
témoins
origine et qualité des témoinsréels ? bibliothèques ?
« constructeur » ou « personnelle » ?
Analyse en pression contrôléepression et nature du gaz
11

Dans le cas d’échantillons fortement rugueux, le re lief peut entraîner des effets d’absorption rendant même l’analyse qual itative fausse…
Quant à l’analyse quantitative !!!Faut même pas y penser !!!
2 – Influence du relief
12
1 – Homogène dans la zone d’analyse
I - L’échantillon
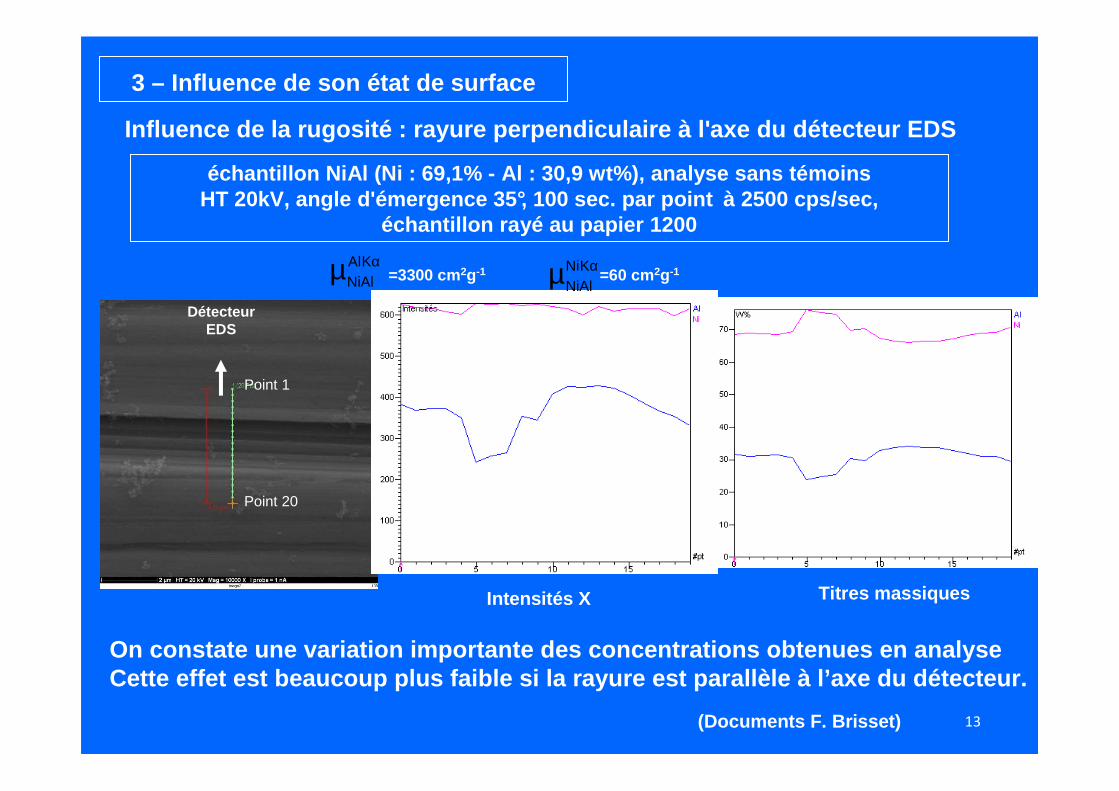
3 – Influence de son état de surface
échantillon NiAl (Ni : 69,1% - Al : 30,9 wt%), analys e sans témoinsHT 20kV, angle d'émergence 35°, 100 sec. par point à 2500 cps/sec,
échantillon rayé au papier 1200
Influence de la rugosité : rayure perpendiculaire à l 'axe du détecteur EDS
Détecteur EDS
Point 1
Point 20
On constate une variation importante des concentrat ions obtenues en analyseCette effet est beaucoup plus faible si la rayure e st parallèle à l’axe du détecteur.
Intensités X
Ni
Al
Titres massiques
Ni
Al
(Documents F. Brisset)
αµ KAlNiAl =3300 cm 2g-1 αµ KNi
NiAl=60 cm 2g-1
13

limite deDuane-Hunt
effets decharge
4 – Influence de la conductivité
Un manque de conductivité (matériaux peu conducteur, mauvaise mise à lamasse…) peut entraîner une diminutionde la tension d’accélération au niveau de l’échantillon.
On peut vérifier ce risque en observant(en échelle logarithmique) la limite de Duane-Hunt (limite supérieurede l’énergie du spectre continu).
O
Y Y
Y Y
Zr
Zr
ZrZr
keV0
10
210
310
0 5 10 15
échantillon métallisé
échantillon non-métallisé
(document F. Brisset)14

D E Newbury, Microsc. & Microanal.10 (2004) 739
E0= 3keV; SiCO(isolant) non métallisé
EK (Si) = 1,8 keV à l’état stationnaire
Evolution du spectre EDSau cours du temps
(de 10 sec en 10 sec)
Spectres cumulés
85 sec 500 sec
15

O
Na
MgAl
Si
K K Ca
Ca keV0
500
1000
1500
2000
2500
1 2 3 4
Acquisition sur champ50 µm x 40 µmbalayage rapide, 60 sec.
Les verres au sodium, potassium
Acquisition en mode ponctuel
sonde fixe, 60 sec.
O
Na
MgAl
Si
K K Ca
CakeV0
500
1000
1500
2000
2500
1 2 3 4
50 x 40
balayage
sonde
fixe
"ex. : Na ~11% at. ~ 4 % at."
Superposition des 2 spectres pour Na
O
Na
MgAl
Si
keV0
500
1000
1500
2000
0.5 1.0 1.5 2.0
5 – Modification de la composition sous le faisceau
Exemple : la migration des ions alcalins sous le fa isceau électronique…
(document F. Brisset)16

Nimonic 90
58,6% Ni19,4% Cr17,0% Co2,4% Ti1,1% Al0,5% Si0,3% Mn0,2% Fe
30 kV A une tension donnée, la section efficace d’ionisation est très différente selon les éléments analysés...
II - Influence de la tension d’accélération
15 kV
5 kV
La forme des spectres est directement liée
à la tension d’accélération…

( )68,168,10 EE
1064,0z −
ρ=
(Andersen)
Plus la tension sera élevée, et plus la zone analysée sera grande...
Selon la tension et la nature de la cible, les dimensions de la zone
analysés (latéralement et en profondeur)peuvent varier de quelques dixième à
quelques dizaines de µm…
variation du volume d’analyseen fonction de la tension
d’accélération pour différentes valeurs de la masse volumique
0,5 µm
5 µm
20 keV
acier 1,2 µm
tension d’accélération et résolution spatiale
z en µmE en keVρρρρ en g.cm -2
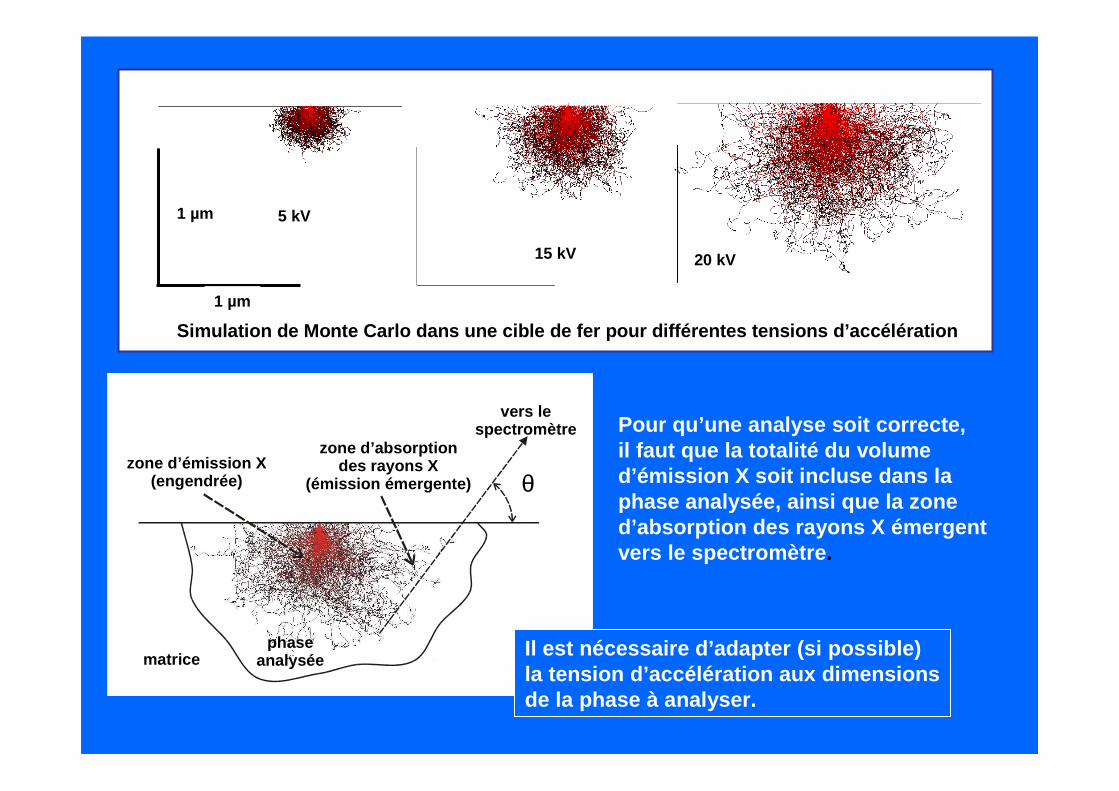
1µm
1µm
5 kV
15 kV 20 kV
Simulation de Monte Carlo dans une cible de fer pou r différentes tensions d’accélération
1 µm
1 µm
θ
vers le spectromètre
zone d’émission X(engendrée)
zone d’absorptiondes rayons X
(émission émergente)
phaseanalyséematrice
Pour qu’une analyse soit correcte,il faut que la totalité du volume d’émission X soit incluse dans la phase analysée, ainsi que la zoned’absorption des rayons X émergentvers le spectromètre .
Il est nécessaire d’adapter (si possible)la tension d’accélération aux dimensionsde la phase à analyser.

θ
vers le spectromètre
zone d’émission X(engendrée)
zone d’absorptiondes rayons X
(émission émergente)
phaseanalysée
matrice
θ
vers le spectromètre
zone d’émission X(engendrée)
zone d’absorptiondes rayons X
(émission émergente)
phaseanalysée
matrice
La phase est nettement plus petiteque le volume d’émission, la matriceparticipera à l’émission totale.
La phase semble plus grande que le volume d’émission, mais son épaisseur n’est pas suffisante : là aussi, la matrice participeraà l’émission totale.
Ne pas oublier également que dans le volume d’émiss ion X la phase doit êtrehomogène.

III – Choix de l’intensité électronique (et du temps de comptage)
Compromis entre :- obtenir une précision statistique suffisante- garder la meilleure résolution du détecteur
a) Précision statistique (sur l’émission X) : loi d e Poisson
écart-type : t.I2=σI : intensité Xt : temps de comptage
rapport « signal-sur-bruit » : t.I5,0t.I2
t.IRSB
==
En réalité le calcul précis de cette précision est u n peu plus complexe !
b) Résolution du détecteur
- choisir la plus grande constante de mise en forme d e l’amplificateur- limiter l’intensité électronique pour que le coeffic ient de temps mort
reste inférieur à 20%
En résumé, on règle l’intensité électronique (temps mort ≤ 20%)puis le temps de comptage pour avoir la précision r echerchée(pour la plus faible teneur…)

Traitement des spectres EDS
Pour quantifier un spectre il faut en extraire l’information utile
(émission caractéristique)
- identification des raies- suppression de l’émission de fond continu- « déconvoluer » (extraire l’information cachée)- mesurer l’émission caractéristique (ROI ou aire de pic)
Moyens

5 10 15
Sr Kα
Sr Kα
Sr Kβ
Sr Kβ
pics d’échapement
9,874 keV
énergie (keV)
Identification des raies
But : 1) identifier toutes les raies caractéristique s présentes, visibles ET cachées…2) rejeter toutes les raies parasites (pics d’empil ement, pics d’échappement…)
Attention aux méthodes de recherche et d’identifica tion automatiques…
L’identification doit se faire sur l’ensemble du sp ectre émis (même s’il faut élargir lagamme d’énergie à 0-20 keV)
La présence de pics d’échappement peutinduire des erreurs d’identification, en particulier si les pics principaux ne sontpas visualisés sur l’écran.
0 – 10 keV
Sr Lαααα(1,806 keV)
2Sr Lαααα
23

Traitement des spectres EDS
Pour quantifier un spectre il faut en extraire l’information utile
(émission caractéristique)
- identification des raies- suppression de l’émission de fond continu- « déconvoluer » (extraire l’information cachée)- mesurer l’émission caractéristique (ROI ou aire de pic)
Moyens
- Modélisation- Filtrage en fréquence (Transformée de Fourrier)- Application d’un filtre numérique
Trois méthodes :

a) modélisation du fond continu
Cette technique consiste à calculer la variation réelle de l’intensité de fond continu par des modèles théoriques et des ajustements expérimentaux.
(Ware, Reed, Lifshin)
paramètres ajustables expérimentalement
)E(D4E
)EE(b
EEE
a)(fZk)E(I2
00
πΩ
−+−χ=
nombre atomiquemoyen de la cible
correctiond’absorption
rendement de détectionde l’EDS
Suppression du fond continu

b) - filtrage par transformée de Fourrier
Si on fait l’analyse en « fréquences* » d’un spectre E DS, on peut distinguer 3 domaines :- un domaine de faibles fréquences qui correspond à l ’émission de fond continu- un domaine de fréquences moyennes et qui correspon d aux raies caractéristiques- un domaine de hautes fréquences qui correspond au bruit, électronique et physique.
* il ne s’agit pas des fréquences liées aux rayonne ments X mais les fréquences de distributions desinformations dans le spectre
On peut obtenir cette distribution par une transformée de Fourier du spectre.
On passe d’un espace « énergie » à un espace « fréquence ».
On élimine les fréquences liées au fond continu et au bruit, puis on applique une transforméeinverse qui restitue le spectre en énergie mais sans le fond continu et sans le bruit.
L’inconvénient de cette technique est quele filtre peut éliminer également, en raison du chevauchement des 3 composantes une partiedes informations utiles et entraîner des distorsion s.
N( )ν
fréquence ( )νfond continu
pics caractéristiques
bruit
filtre
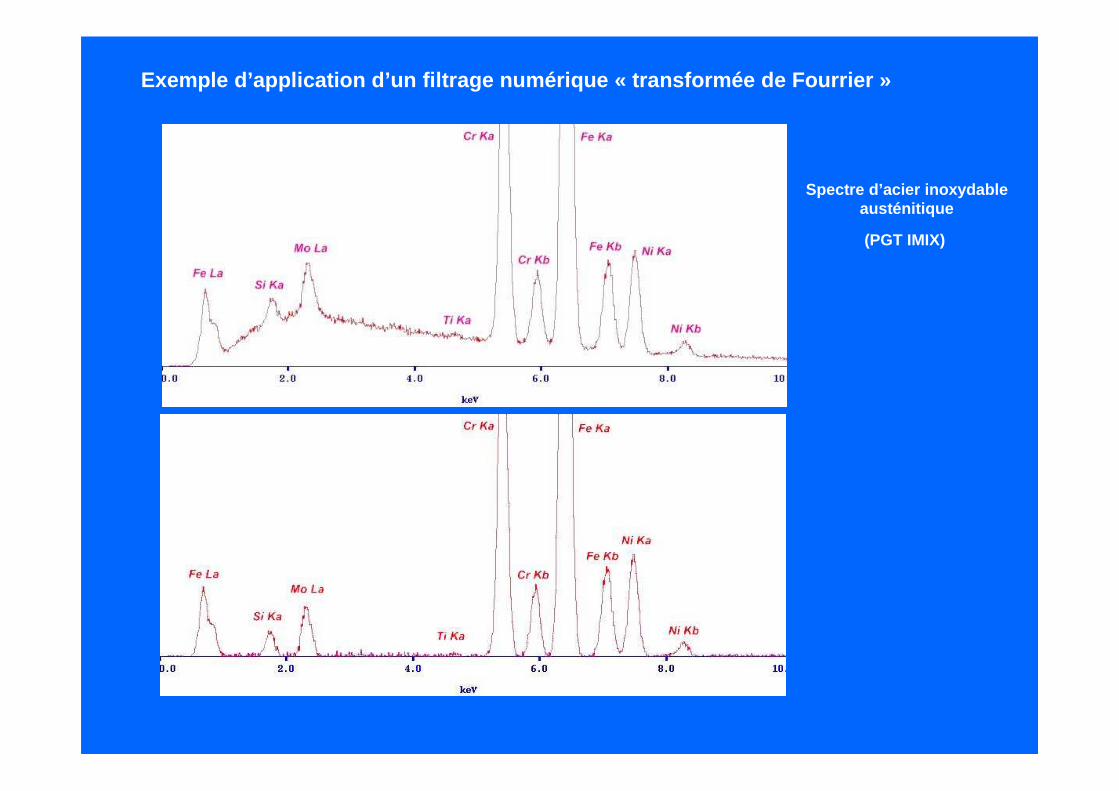
Spectre d’acier inoxydableausténitique
Exemple d’application d’un filtrage numérique « tran sformée de Fourrier »
(PGT IMIX)

c) - filtre numérique passe-bande (« top-hat »)
L
canal
(canaux utilisés par le filtre)
- -
+
A chaque canal on applique un filtre numériquedit « top-hat » (ou « chapeau haut-de-forme »)
Ce filtre consiste à remplacer pour chaque canal sa valeur par la somme moyennée d’un certainnombre de canaux situés de part et d’autre ducanal considéré, diminuée par la somme moyennéede deux zones situées de part et d’autre de la zonecentrale.
La longueur totale du filtre est égale au doublede la largeur à mi-hauteur de la raie du Mn K ααααLe nombre des canaux de la zone centrale est égaleou légèrement supérieure à la somme des deux zonesadjacentes.
Ce filtre est équivalent à une dérivée seconde. Il n’apporte pas de distorsions importantes sur le spectre et préserve l’information.
Il a l’avantage d’être simple mais manque d’efficac itédans le cas de pics de faibles amplitudes situés au pied de pics intenses.
Application du filtre « top-hat »sur une raie gaussienne

Filtrage numérique sur spectre témoin
Filtrage sur spectre échantillon
Le pic est reconstruit en ajoutant à la partie posit ive les 2 parties négatives.
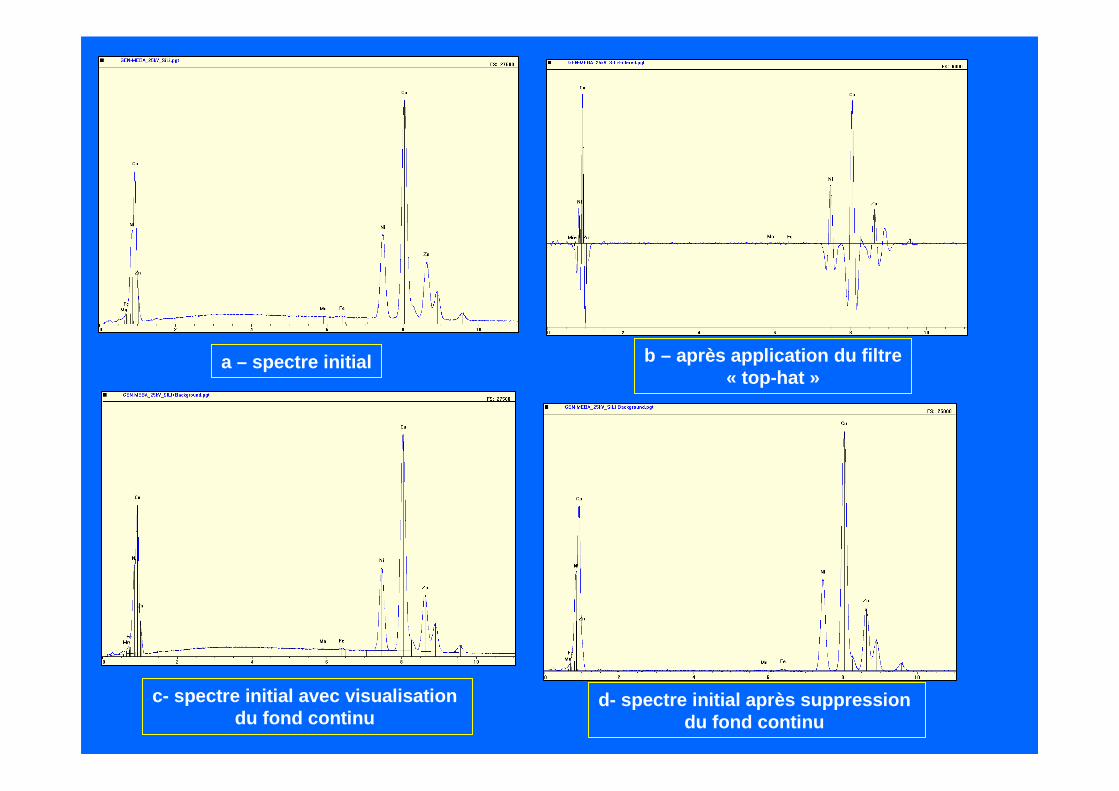
a – spectre initial b – après application du filtre« top-hat »
c- spectre initial avec visualisation du fond continu
d- spectre initial après suppression du fond continu

Traitement des spectres EDS
Pour quantifier un spectre il faut en extraire l’information utile
(émission caractéristique)
- identification des raies- suppression de l’émission de fond continu- « déconvoluer » (extraire l’information cachée)- mesurer l’émission caractéristique (ROI ou aire de pic)
Moyens
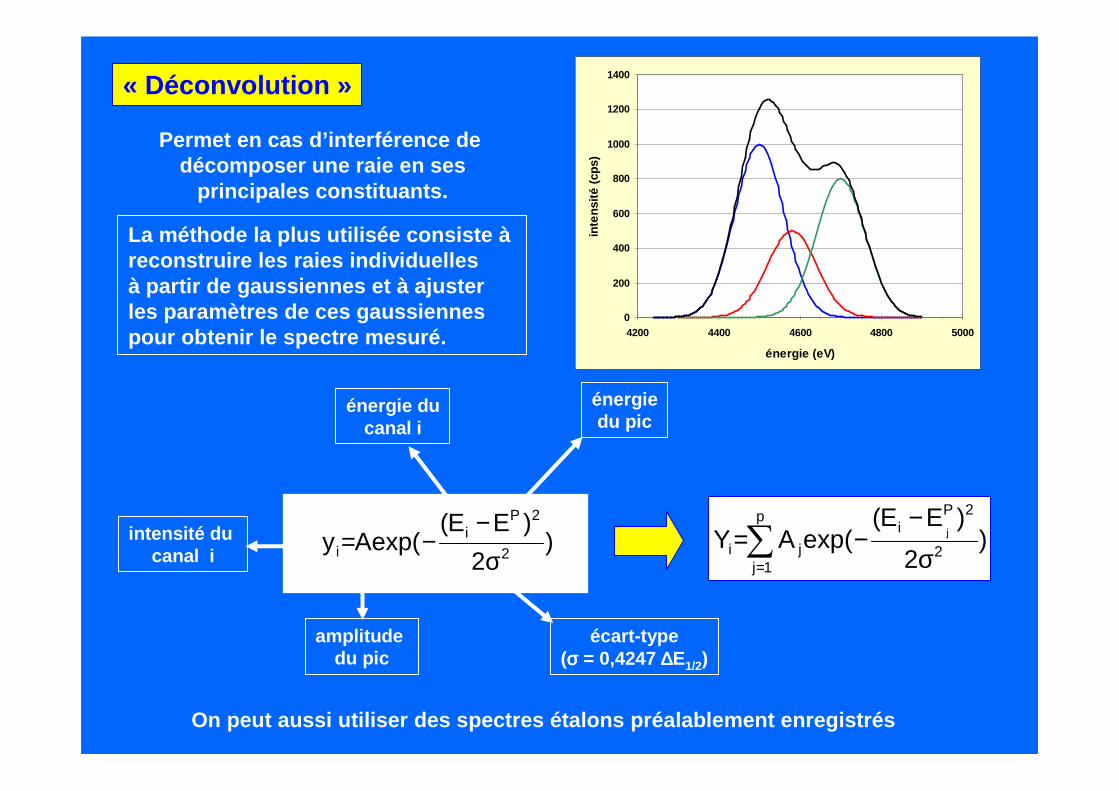
Permet en cas d’interférence de décomposer une raie en ses
principales constituants.
0
200
400
600
800
1000
1200
1400
4200 4400 4600 4800 5000
énergie (eV)
inte
nsité
(cp
s)
« Déconvolution »
La méthode la plus utilisée consiste àreconstruire les raies individuellesà partir de gaussiennes et à ajuster les paramètres de ces gaussiennespour obtenir le spectre mesuré.
intensité du canal i
amplitude du pic
énergie ducanal i
énergiedu pic
écart-type(σσσσ = 0,4247 ∆∆∆∆E1/2)
)2
)EE(exp(Ay 2
2Pi
i σ−
−=
On peut aussi utiliser des spectres étalons préalab lement enregistrés
∑= σ
−−=
p
1j2
2Pi
ji )2
)EE(exp(AY j

Si χχχχ=1 à 5 ajustement parfait, de 5 à 20, ajustement acceptable,au delà de 50, il y a un problème...
Importante : s’assurer de ne pas avoir oublié un élé ment en comparant le spectremesuré avec le spectre reconstitué après « déconvoluti on »
La qualité de l’ajustement est donnée par un Khi2 normalisé
Acier inoxydable
Mn K ααααMn Kββββ
Exemple : l’analyse d’un acierinoxydable où il y a interférence entreles raies Cr K ββββ et Mn K αααα et les raiesMn Kββββ et Fe K αααα
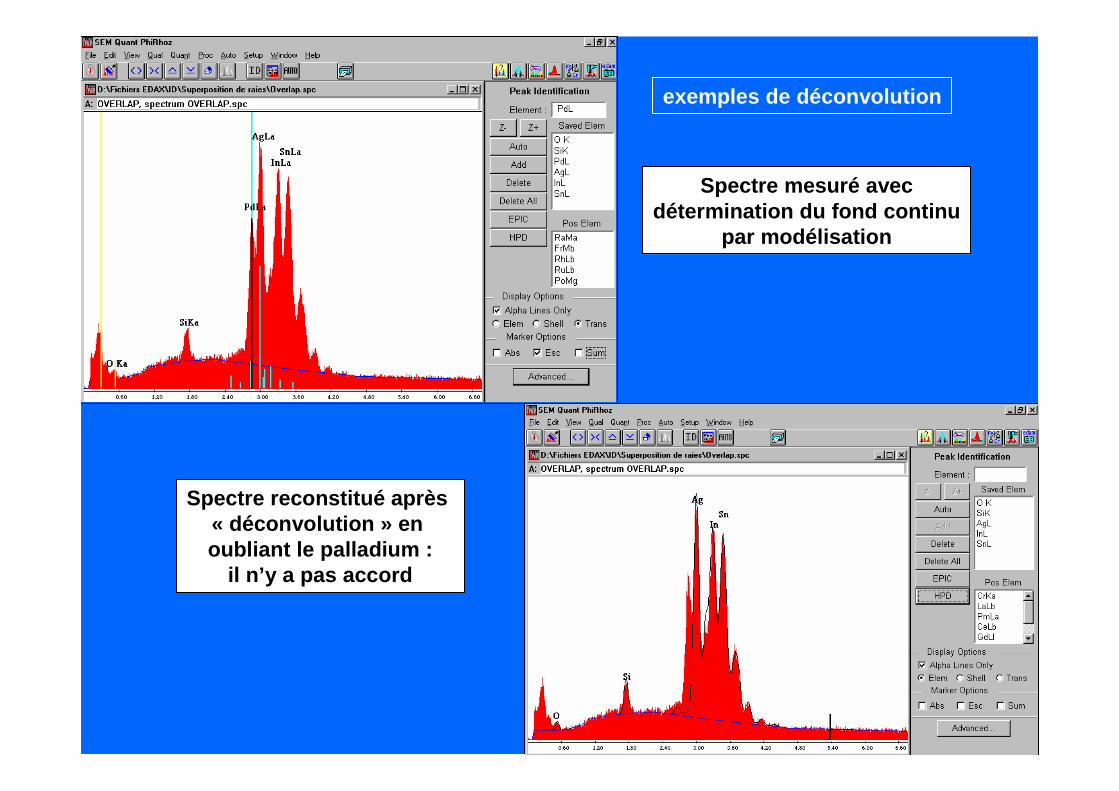
Spectre mesuré avecdétermination du fond continu
par modélisation
Spectre reconstitué après « déconvolution » en oubliant le palladium :
il n’y a pas accord
exemples de déconvolution

Spectre brut (acier inoxydable)
Spectre reconstitué(Cr pur)
Spectre reconstitué(Cr + Mn)

Autre exemple : échantillon contenant du Mo et du S

Reconstructions (après soustraction du fond continu ) :
1 – Soufre pur (0,13%) :mauvais !
2 – molybdène pur (0,29%) :meilleur mais imparfait
spectre réel
spectre reconstitué
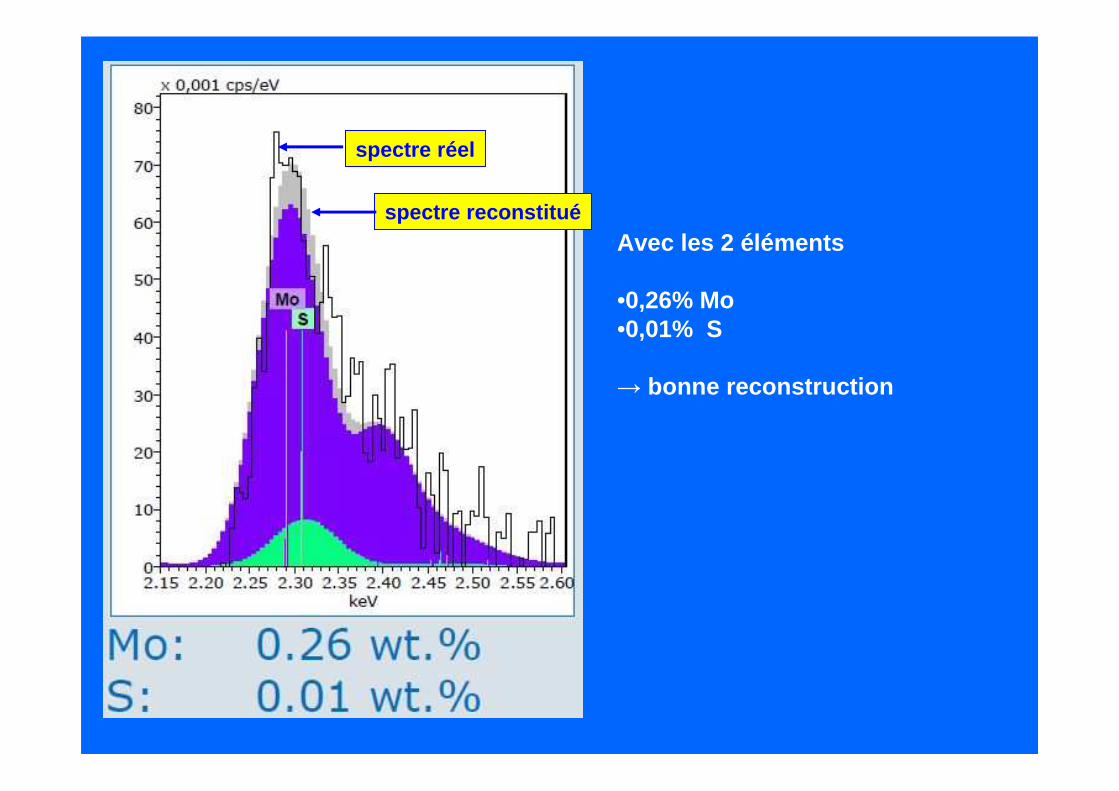
Avec les 2 éléments
•0,26% Mo •0,01% S
→ bonne reconstruction
spectre réel
spectre reconstitué

Dans un acier austénitique, Les raies des éléments principaux (Fe, Cr, Ni) cachent les raies d’émissions d’autres éléments, moins abon dants :
V, Mn, Co, Cuqu’il est nécessaires d’extraire pour l’analyse qua ntitative.

En résumé :- acquisition du spectre- identification des raies et des éléments constituan ts l’échantillon inconnu- suppression du fond continu- recherche éventuelle d’interférences et « déconvolut ion »
On dispose alors pour chaque élément de son intensité X caractéristique
SL
BK
AK II,I ααα L
Pour obtenir C A, CB,…CS à partir de ces intensités, il faut faireappliquer un programme dit « de corrections » qui va teni rcompte de l’ensemble des phénomènes physiques qui re lie l’intensité X détectée au titre massique…L’ensemble des traitements est automatique et transp arentpour l’utilisateur ! c’est souvent une « boîte noire »…

Choix d’un programme de correction…
Échantillon massif
Méthode Z.A.F. Procédures Phi(Roz).
PAP (1984)
XPP (1988)
XPHI (1995)
PROZA96 (1996)
(P/B ZAF)
Pouchou_Pichoir
Pouchou
Merlet
Bastin
41
Échantillon mincemétallique, minéral… Cliff-Lorimer
biologique… Hall

42
ionisation Q (E)AK
perte d’énergieA dE/dρρρρz
absorptionphotoélectrique
µKαααα
émissions X ωωωωK, ZKαααα
A Kαααα
détecteur
ΩΩΩΩD(E)
θθθθ
A Kαααα
émission Xpar fluorescence
Aionisation parfluorescence
BB Kαααα
si EB K αααα > seuil d’ionisation de K A
émissionsrétrodiffusée
Rpertes d’ionisation
les interactions rayonnement-matière et les princip aux paramètres pertinents...
électron primaire incident
trajectoiresélectroniques
CA
IA
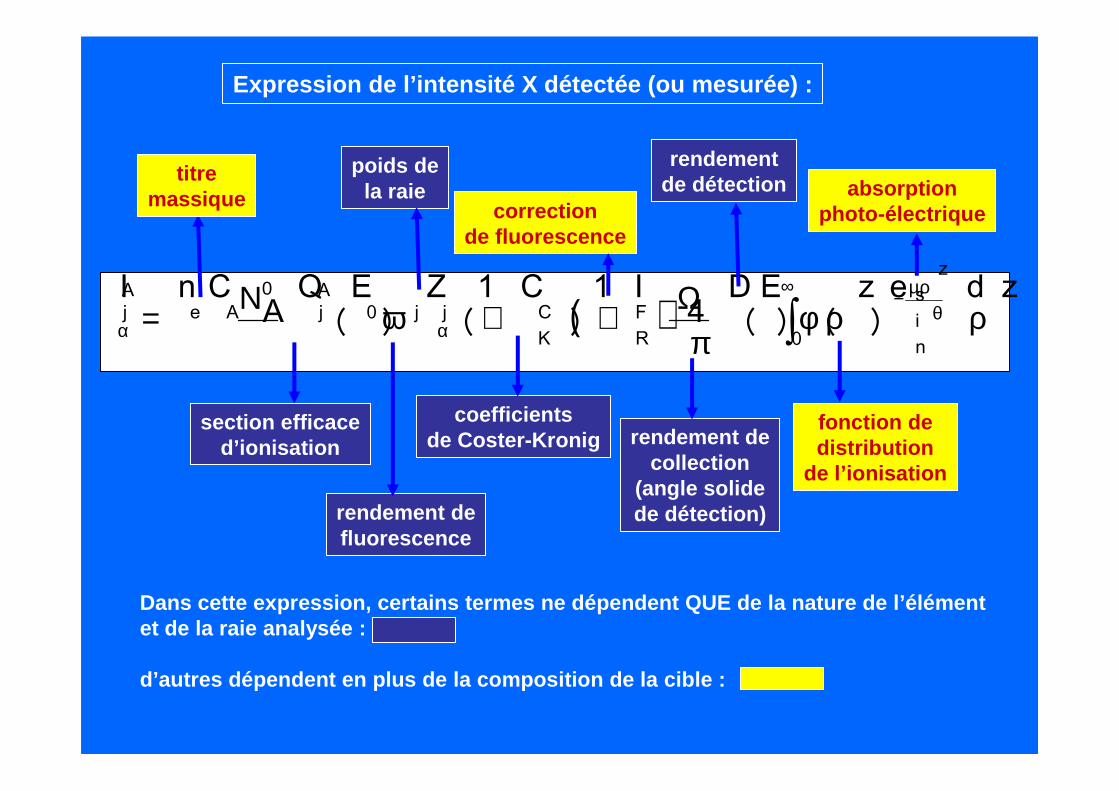
( ) ∫∞
θµρ−
αα ρρφπ
Ω++ω=0
sin
z
FR
CK
jj0AjAe
Aj zde)z()E(D
4I1)C1(Z)E(Q
ACnI
0N
section efficaced’ionisation
rendement defluorescence
poids dela raie
coefficientsde Coster-Kronig
correctionde fluorescence
rendement decollection
(angle solidede détection)
rendementde détection
fonction dedistribution
de l’ionisation
absorptionphoto-électrique
Expression de l’intensité X détectée (ou mesurée) :
Dans cette expression, certains termes ne dépendent QUE de la nature de l’élémentet de la raie analysée :
d’autres dépendent en plus de la composition de la cible :
titremassique

( )
( )tem
0FR
Atem
ech
0FR
Atem
A
ech
A
zd )zexp
()z(I1C
zd )zexp
()z(I1
C)I(
)I(
ρχρ−ρφ+
ρχρ−ρφ+
=
∫
∫∞
∞
Compte tenu des imprécisions avec lesquelles certai ns termes sont connus…
On calcule le rapport de l’intensité émise par l’éc hantillon inconnu avec celle d’un étalon de référence de composition connu, ce q ui simplifie l’écriture :
Cette expression (appelée « concentration apparente » ou « k-ratio ») peut être simplifiée en prenant un étalon pur :
1CAtem
=
0)I1( tem
FR
≈+ en négligeant la fluorescence de fond continu
Méthodes correctives : - la plus ancienne : méthode ZAF- plus récentes : les procédures PhiRoz

Méthode ZAF :
Développée à partir de la thèse de R. Castaing, en p articulier par R. Tixier et J. Philibert (1960)Elle consiste à décomposer le calculs des correction s en 3 étapes indépendantes etsuccessives :
-le calcul de l’émission X engendrée (correction « de Z »)-le calcul des effets d’absorption (correction « A »)-le calcul des effets de fluorescence X (correction « F »)
Procédures « PhiRoz »
Plus récentes (1985 …)
Elles utilisent une expression correcte de la loi d e distribution de l’ionisationen profondeur φφφφ(ρρρρz)
Ces expressions sont un ajustement expérimentales d ’expressionspurement mathématiques, représentant la forme obser vée de cettefonction.
Il existe un certain nombre de ces fonctions qui ne diffèrent que par la courbe mathématique (gaussienne, parabole, exponentielle, mixte…)
PAP, XPP, XPHI, PROZA96…

Question : quel témoin utiliser ?
En microanalyse EDS on a le choix entre :
1 – Méthode rigoureuse : on utilise de vrais témoins …
Pas toujours facile… le MEB ne dispose pas comme la microsonde de blocsd’étalons de référence in-situ… il faut les préparer et les introduire…
méthode la plus précise mais peu utilisée…
2 – On dispose dans l’ordinateur d’une bibliothèque de spectres pré-enregistrés(analyse « à témoins cachés » )
1 – A l’aide d’un échantillon de référence (Cu par e x.) on ré-étalonne les spectrespar rapport à l’intensité électronique,2 – Les spectres doivent avoir été enregistrés dans l es mêmes conditions…
Attention aux « spectres constructeurs »…si on ignore comment ils ont été obtenus …
3 – Analyse « sans témoins »
Le logiciel calcule à l’aide de la formule précédent e un témoin virtuel…
Attention : compte tenu des nombreuses incertitudes , cette méthode ne donne souventqu’un ordre de grandeur, le risque d’erreur peut êt re important…
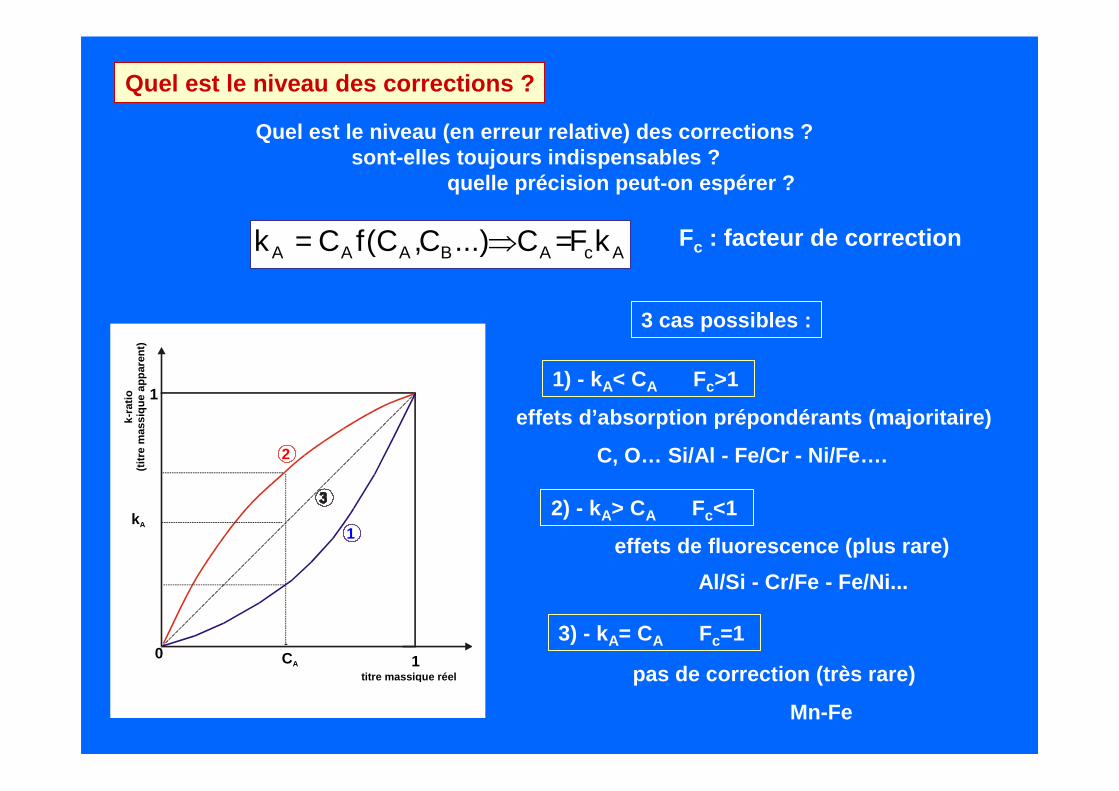
Quel est le niveau des corrections ?
Quel est le niveau (en erreur relative) des correct ions ?sont-elles toujours indispensables ?
quelle précision peut-on espérer ?
k-ra
tio
(tit
re m
assi
qu
e ap
par
ent)
titre massique réel
0
1
1
kA
CA
1
2
3 cas possibles :
AcABAAA kFC...)C,C(fCk =⇒= Fc : facteur de correction
1) - kA< CA Fc>1
effets d’absorption prépondérants (majoritaire)
C, O… Si/Al - Fe/Cr - Ni/Fe….
2) - kA> CA Fc<1
effets de fluorescence (plus rare)
Al/Si - Cr/Fe - Fe/Ni...
3) - kA= CA Fc=1
pas de correction (très rare)
Mn-Fe

Exemple :analyse d’un acier inoxydable
(Fe-16%Cr-14%Ni) (20kV - θθθθ=40°)
0.5
1
1.5
2
2.5
3
0 5 10 15 20 25 30
tension d'accélération (kV)
fact
eur
corr
ectif
Mo La
Si Ka
Cr Ka
Fe KaNi Ka
Mn Ka
Variation du facteur correctifavec la tension d’accélération
Acier inoxydable0,4% Si - 16%Cr - 1,7% Mn - 14% Ni - 1,5% Mo
A de rares exceptions, les facteurs correctifs sont loin d’être
négligeables...
Élément F c taux (%)
Mn 1 -Fe 1,02 +2%Cr 0,88 -12%Mo 1,32 +32%Si 1,8 +80%Al 2 +100%C 2,5 +150%

3 exemples de niveau de correction
1 – Ni-Cr (90-10%)
très peu de correction :- Ni : absorption- Cr : fluorescence
2 – Ni-Al (70-30%)
Ni : peu de correctionAl : forte absorption
3 – Si-C (70 – 30%)
Si : légère absorptionC : très forte absorption

CONCLUSIONS
Si la microanalyse X permet une analyse quantitativ e précise, ce n’est en touterigueur qu’avec une microsonde de Castaing…
Le MEB n’est pas a priori un instrument conçu pour ce genre d’analyse :- pas de régulation d’intensité de faisceau- pas de blocs d’étalons- utilisation de spectromètres EDS
Cependant, on peut en prenant beaucoup de précautio ns et en travaillanttrès soigneusement obtenir en MEB-EDS de bonnes ana lyses quantitatives.
Les analyses sans témoins offrent une solution rapi de et pratiquepour obtenir une information sur la composition rée lle de son échantillon.
C’est de toutes façons mieux que de l’estimer « au p if » à partir du spectre !
Mais, ce n’est quelquefois qu’un ordre de grandeur…
Il faut alors être très prudent dans l’interprétati on des résultats .