interaction of rhodanese with mitochondrial nadh dehydrogenase
TRANSCRIPT

278 Biochimica et Biophysics Acta, 742 (1983) 278-284
Elsevier Biomedical Press
BBA 31449
INTERACTION OF RHODANESE WITH MITOCHONDRIAL NADH DEHYDROGENASE
SILVIA PAGAN1 * and Y.M. GALANTE **
Department of Biochemistry Scripps Clinic and Research Fondation, La Jolla, CA 92037 (U.S.A.)
(Received August 2nd, 1982)
Key words: NADH dehydrogenase; Rhodanew; Sulfur transfer; Reactivation; Fe-S protein; (Bovine heart and liver mitochondria)
NADH dehydrogenase is an iron-sulfur flavoprotein which is isolated and purified from Complex I (mitochondrial NADH: ubiquinone oxidoreductase) by resolution with NaClO,. The activity of the enzyme (followed as NADH : 2-methylnaphthoquinone oxidoreductase) increases linearly with protein concentration (in the range between 0.2 and 1.0 mg/ml) and decreases with aging upon incubation on ice. In the present work a good correlation was found between enzymic activity and labile sulfide content, at least within the limits of sensitivity of the assays employed. Rhodanese (thiosulfate:cyanide sulfurtransferase (EC 2.8.1.1) purified from bovine liver mittihondria was shown to restore, in the presence of thiosulfate, the activity of the partly inactivated NADH dehydrogenase. Concomitantly, sulfur was transferred from thiosulfate to the flavoprotein and incorporated as acid-labile sulfide. Rhodanese-mediated sulfide transfer was directly demonstrated when the reactivation of NADH dehydrogenase was performed in the presence of radioactive thiosulfate (labeled in the outer sulfur) and the 35S-loaded flavoprotein was re-isolated by gel filtration chromatography. The results indicated that the [“S]sulfide was inserted in NADH dehydrogenase and appeared to constitute the structural basis for the increase in enzymic activity.
Introduction
Little is known about the biological mechanism of assembling of the iron-sulfur centers of iron- sulfur proteins. Previous work of others has indi- cated that sulfur transferases may be involved in the formation of the iron-sulfur clusters [ 1,2]. More recent studies on the interaction of rhodanese with mitochondrial succinate dehydrogenase, an iron- sulfur flavoprotein, showed that sulfur was trans- ferred to the dehydrogenase and inserted as acid- labile sulfide [3]. It was also proposed that all three centers of succinate dehydrogenase were in-
* Permanent address: University of Milan, Department of General Biochemistry, Faculty of Agriculture, Milan, Italy.
** To whom inquiries should be addressed at (current address): Centre de Recherche, Departement de Pharmacologic, Merck Sharp & Dohme-Chibret, 63203 Riom CCdex, France.
0167~4838/83/0000-0000/$03.00 0 1983 Elsevier Biomedical Press
volved [4]. A rhodanese-mediated sulfide insertion
into the iron-sulfur cluster of ferredoxin from
spinach and Clostridium pasteurianum was also demonstrated [5,6]. Furthermore, it was shown that no significant sulfide transfer occurred with proteins which, in their native state, contain no
iron-sulfur centers, such as bovine serum albumin and yeast alcohol dehydrogenase [3,6].
We have investigated the effect of rhodanese on NADH dehydrogenase from bovine heart mito- chondria. NADH dehydrogenase is an iron-sulfur flavoprotein which is extracted from Complex I (NADH : ubiquinone oxidoreductase) by resolu- tion with chaotropic agents and purified by am- monium sulfate fractionation. The enzyme was shown to consist of three polypeptide subunits of molecular weight 5 1000, 24 000 and 9 000, present in equimolar amount; and to contain FMN, non- heme iron and labile sulfide in a molar ratio of

1 : 5-6 : 5-6 [7,8]. A preliminary characterization of the flavoprotein by EPR has been reported [9,10]. In this communication, we wish to report: (a) on the relation that we found between enzymic activity of NADH dehydrogenase and its labile sulfide content; (b) on the rhodanese-mediated reactivation, in the presence of thiosulfate, of the partly inactivated enzyme, and (c) on the insertion of sulfur (as sulfide) in the flavoprotein by the action of rhodanese. The results show a direct relation between sulfide insertion and enzyme re- activation, and point to a possible role of rhodanese in the formation of the clusters of mitochondrial iron-sulfur proteins.
Methods and Materials
Complex I and soluble NADH dehydrogenase were prepared as described previously [8,11]. The enzyme was stored as an ammonium sulfate pellet at -70°C. Before each set of experiments one pellet (about 4-5 mg of enzyme) was suspended at a protein concentration of 5-6 mg /ml in 50 mM Tris-HC1 (pH 7.8), subdivided into small aliquots and kept in liquid nitrogen. Each aliquot was thawed only once, immediately before use. Activ- ity of NADH dehydrogenase was measured by following the oxidation of NADH by 2-methyl- naphthoquinone (menadione) at 38°C in 50 mM potassium phosphate (pH 8.0)/1 mM EDTA/0.15 mM NADH/0 .2 mM menadione as before [8]. Rhodanese (EC 2.8.1.1) was purified from beef liver and was crystallized in the presence of 1 mM thiosulfate as described by Blumenthal and Hein- rikson [12]. Sulfur transferase activity of rhodanese was determined according to Srrbo [13] by follow- ing the rate of thiocyanate formation from thio- sulfate and cyanide. Rhodanese concentration was estimated as in Ref 3. The molecular weight used in all calculations was 32900 [14]. Treatment of NADH dehydrogenase with rhodanese in the pres- ence of radioactive thiosulfate was carried out as follows. NADH dehydrogenase, at a protein con- centration of 0.75 mg/ml, was incubated at 0°C with rhodanese in the ratio of 4.4 mol dehydro- genase to 1 mol rhodanese in 50 mM Tris-HC1 (pH 7.8)/1 mM [35S]thiosulfate. After 20 min incubation, the mixture was filtered through a Sephadex G-75 fine column (55 × 2 cm), equi-
279
librated and eluted with 50 mM Tris-HC1 (pH 7.8)/1 mM dithiothreitol. NADH dehydrogenase appeared in the void volume, while rhodanese was included.
Acid-labile sulfide was determined by the method of Fogo and Popowski [15]. In the pres- ence of interfering thiosulfate, the procedure of Gilboa-Garber [16] was used. The portion of the total sulfide incorporated as radioactive sulfide was estimated as described by Bonomi et al. [3]. Protein concentration was determined by the method of Lowry et al. [17], or by the method of Bensadoun and Wenstein [18] to avoid inter- ference by dithiothreitol and /o r detergents.
Radioactivity was measured using a Beta Blend cocktail (West Chem, San Diego) in a Beckman LS 250 liquid scintillation counter. Protein-containing samples were previously digested with Protosol (New England Nuclear).
Radioactive sodium thiosulfate, labeled in the outer sulfur, was purchased from Amersham Inter- national and had a specific radioactivity of 42 000 dpm/nmol .
Results
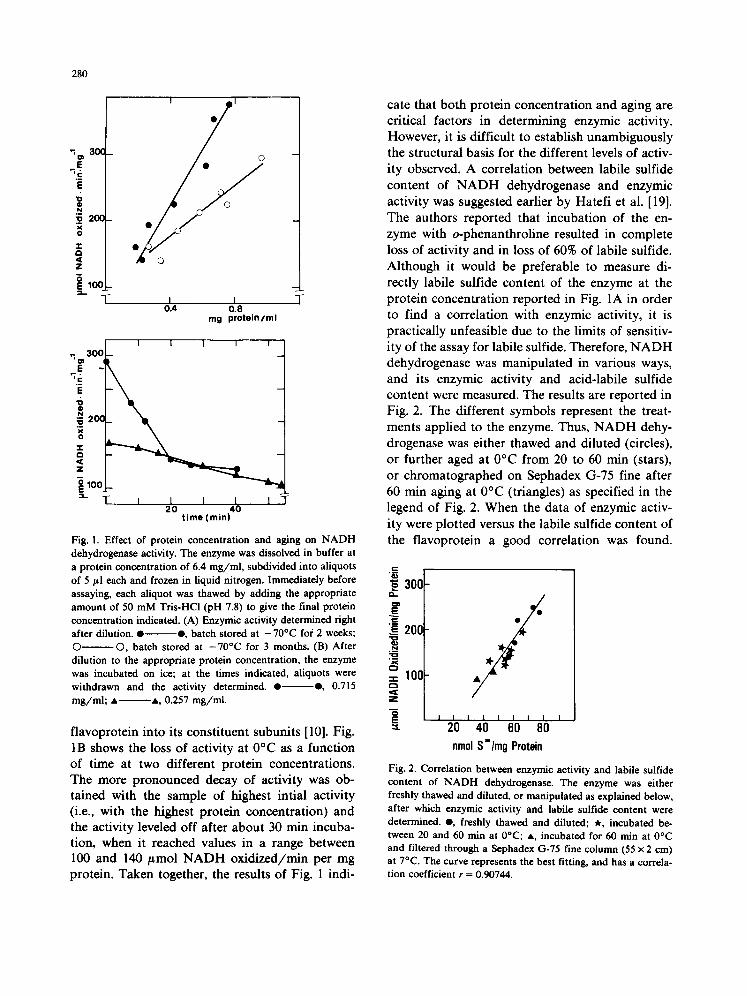
The enzymic activity of NADH dehydrogenase is very unstable, varies somewhat from prepara- tion to preparation, decreases upon prolonged storage even at -70°C, and is extremely sensitive to repeated freezing and thawing [8,10]. The ex- periments reported below were carried out with different ammonium sulfate pellets of a prepara- tion of freshly made enzyme stored for a period of up to 3 months. The most suitable conditions for measurement of enzymic activity were explored. As shown in Fig. 1A, with two batches of enzyme stored at - 7 0 ° C for different lengths of time, it was found that the activity exhibited a linear dependence on enzyme concentration when the dehydrogenase, immediately prior to assaying, was diluted at 4°C to the concentrations indicated. The range explored was between 0.17 and 0.96 mg/ml. Below 0.17 mg /ml the relation between enzymic activity and enzyme concentration was not linear. If the enzyme was subsequently frozen in liquid nitrogen and thawed at room temperature, the activity decreased significantly. This effect could also be due to a partial resolution of the
280
• ~ 3OG 01 E ";.E E
~ 2OO
Z
Z
0
I I 0.4 0.8
mg p r o t e i n / m l
300
%: T=
200
==
~=_100.
I I I I I
I I I I I 2 0 4 0
t i m e (rain)
Fig. 1. Effect of protein concentration and aging on N A D H dehydrogenase activity. The enzyme was dissolved in buffer at a protein concentration of 6.4 mg / ml , subdivided into aliquots of 5 #l each and frozen in liquid nitrogen. Immediately before assaying, each aliquot was thawed by adding the appropriate amount of 50 m M Tris-HCl (pH 7.8) to give the final protein concentration indicated. (A) Enzymic activity determined fight after dilution. • • , batch stored at - 70°C for 2 weeks; © ©, batch stored at - 7 0 ° C for 3 months. (B) After dilution to the appropriate protein concentration, the enzyme was incubated on ice; at the times indicated, aliquots were withdrawn and the activity determined. • • , 0.715 m g / m l ; • • , 0.257 mg / ml .
flavoprotein into its constituent subunits [10]. Fig. 1B shows the loss of activity at 0°C as a function of time at two different protein concentrations. The more pronounced decay of activity was ob- tained with the sample of highest intial activity (i.e., with the highest protein concentration) and the activity leveled off after about 30 rain incuba- tion, when it reached values in a range between 100 and 140 #mol NADH oxidized/min per mg protein. Taken together, the results of Fig. 1 indi-
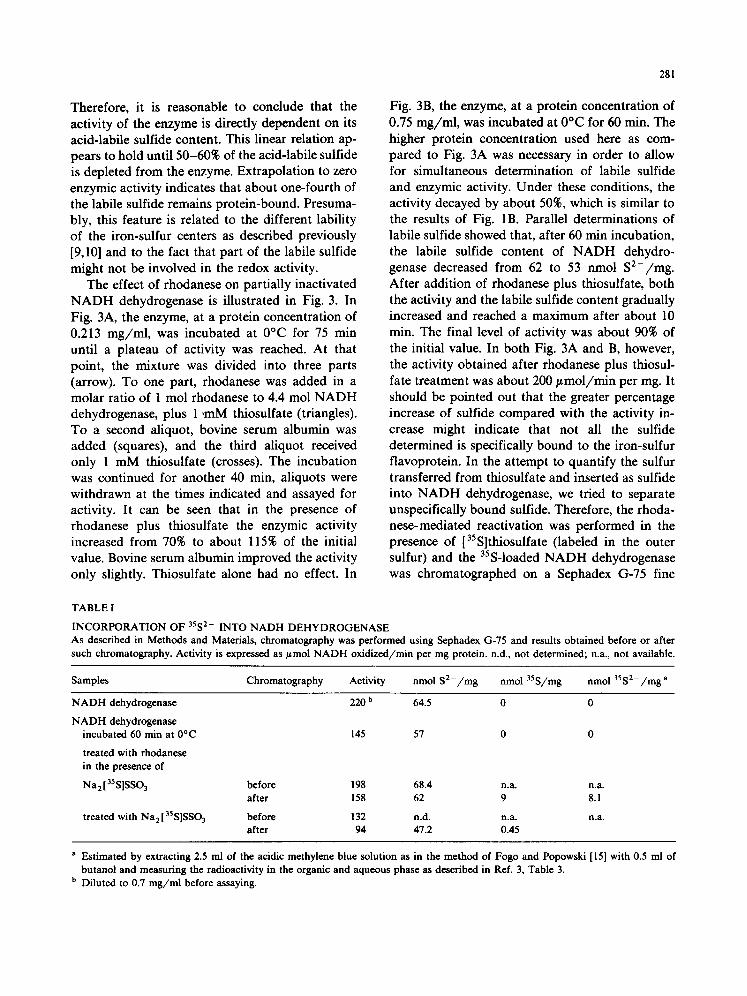
cate that both protein concentration and aging are critical factors in determining enzymic activity. However, it is difficult to establish unambiguously the structural basis for the different levels of activ- ity observed. A correlation between labile sulfide content of NADH dehydrogenase and enzymic activity was suggested earlier by Hatefi et al. [19]. The authors reported that incubation of the en- zyme with o-phenanthroline resulted in complete loss of activity and in loss of 60% of labile sulfide. Although it would be preferable to measure di- rectly labile sulfide content of the enzyme at the protein concentration reported in Fig. 1A in order to find a correlation with enzymic activity, it is practically unfeasible due to the limits of sensitiv- ity of the assay for labile sulfide. Therefore, NADH dehydrogenase was manipulated in various ways, and its enzymic activity and acid-labile sulfide content were measured. The results are reported in Fig. 2. The different symbols represent the treat- ments applied to the enzyme. Thus, NADH dehy- drogenase was either thawed and diluted (circles), or further aged at 0°C from 20 to 60 rain (stars), or chromatographed on Sephadex G-75 fine after 60 rain aging at 0°C (triangles) as specified in the legend of Fig. 2. When the data of enzymic activ- ity were plotted versus the labile sulfide content of the flavoprotein a good correlation was found.
.=
E"
-.i-
, ~ Z
E =1.
30C
20E
10C
' 2'o ' 4b ' do ' 8'o nmol S'/mg Protein
Fig. 2. Correlation between enzymic activity and labile sulfide content of N A D H dehydrogenase. The enzyme was either freshly thawed and diluted, or manipulated as explained below, after which enzymic activity and labile sulfide content were determined, e , freshly thawed and diluted; * , incubated be- tween 20 and 60 rain at 0°C; • , incubated for 60 rain at 0°C and filtered through a Sephadex G-75 fine column (55 × 2 cm) at 7°C. The curve represents the best fitting, and has a correla- tion coefficient r = 0.90744.
281
Therefore, it is reasonable to conclude that the activity of the enzyme is directly dependent on its acid-labile sulfide content. This linear relation ap- pears to hold until 50-60% of the acid-labile sulfide is depleted from the enzyme. Extrapolation to zero enzymic activity indicates that about one-fourth of the labile sulfide remains protein-bound. Presuma- bly, this feature is related to the different lability of the iron-sulfur centers as described previously [9,10] and to the fact that part of the labile sulfide might not be involved in the redox activity.
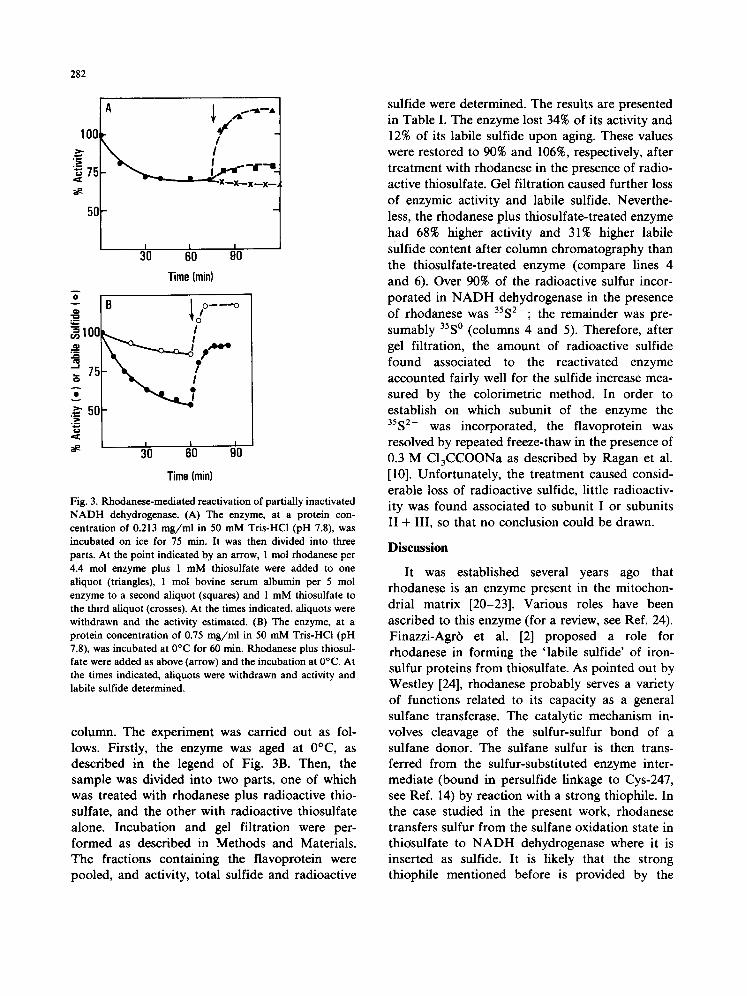
The effect of rhodanese on partially inactivated NADH dehydrogenase is illustrated in Fig. 3. In Fig. 3A, the enzyme, at a protein concentration of 0.213 mg/ml, was incubated at 0°C for 75 min until a plateau of activity was reached. At that point, the mixture was divided into three parts (arrow). To one part, rhodanese was added in a molar ratio of 1 mol rhodanese to 4.4 mol NADH dehydrogenase, plus 1 ,mM thiosulfate (triangles). To a second aliquot, bovine serum albumin was added (squares), and the third aliquot received only 1 mM thiosulfate (crosses). The incubation was continued for another 40 min, aliquots were withdrawn at the times indicated and assayed for activity. It can be seen that in the presence of rhodanese plus thiosulfate the enzymic activity increased from 70% to about 115% of the initial value. Bovine serum albumin improved the activity only slightly. Thiosulfate alone had no effect. In
Fig. 3B, the enzyme, at a protein concentration of 0.75 mg/ml, was incubated at 0°C for 60 min. The higher protein concentration used here as com- pared to Fig. 3A was necessary in order to allow for simultaneous determination of labile sulfide and enzymic activity. Under these conditions, the activity decayed by about 50%, which is similar to the results of Fig. lB. Parallel determinations of labile sulfide showed that, after 60 min incubation, the labile sulfide content of NADH dehydro- genase decreased from 62 to 53 nmol S2- /mg. After addition of rhodanese plus thiosulfate, both the activity and the labile sulfide content gradually increased and reached a maximum after about 10 min. The final level of activity was about 90% of the initial value. In both Fig. 3A and B, however, the activity obtained after rhodanese plus thiosul- fate treatment was about 200/~mol/min per mg. It should be pointed out that the greater percentage increase of sulfide compared with the activity in- crease might indicate that not all the sulfide determined is specifically bound to the iron-sulfur flavoprotein. In the attempt to quantify the sulfur transferred from thiosulfate and inserted as sulfide into NADH dehydrogenase, we tried to separate unspecifically bound sulfide. Therefore, the rhoda- nese-mediated reactivation was performed in the presence of [35S]thiosulfate (labeled in the outer sulfur) and the 35S-loaded NADH dehydrogenase was chromatographed on a Sephadex G-75 fine
TABLE I
INCORPORATION OF 3552- INTO NADH DEHYDROGENASE As described in Methods and Materials, chromatography was performed using Sephadex G-75 and results obtained before or after such chromatography. Activity is expressed as ttmol NADH oxidized/min per mg protein, n.d., not determined; n.a., not available.
Samples Chromatography Activity nmol S 2 - / m g nmol 35S/mg nmol 35S2-/mg a
NADH dehydrogenase
NADH dehydrogenase incubated 60 rain at 0°C
treated with rbodanese in the presence of
Na 2 [ 3s S]SSO3
treated with Na2135S]SSO3
220 b 64.5 0 0
145 57 0 0
before 198 68.4 n.a. n.a. after 158 62 9 8.1
before ! 32 n.d. n.a. n.a. after 94 47.2 0.45
a Estimated by extracting 2.5 ml of the acidic methylene blue solution as in the method of Fogo and Popowski [15] with 0.5 ml of butanol and measuring the radioactivity in the organic and aqueous phase as described in Ref. 3, Table 3.
b Diluted to 0.7 mg /ml before assaying.
282
100~
~ 75
5O
/
I I I 30 60 90
Time (rain)
B |4/- ---o I00 ~ Y'*¢~
~ 511 "N
I I t 30 60 90
Time (rain)
Fig. 3. Ru~_odanese-mediated reactivation of partially inactivated N A D H dehydrogenase. (A) The enzyme, at a protein con- centration of 0.213 m g / m l in 50 mM Tris-HCl (pH 7.8), was incubated on ice for 75 rain. It was then divided into three parts. At the point indicated by an arrow, 1 tool rhodanese per 4.4 mol enzyme plus 1 mM thiosulfate were added to one aliquot (triangles), 1 mol bovine serum albumin per 5 mol enzyme to a second aliquot (squares) and 1 mM thiosulfate to the third aliquot (crosses). At the times indicated, aliquots were withdrawn and the activity estimated. (B) The enzyme, at a protein concentration of 0.75 m g / m l in 50 mM Tris-HCl (pH 7.8), was incubated at 0°C for 60 min. Rhodanese plus thiosul- fate were added as above (arrow) and the incubation at 0°C. At the times indicated, aliquots were withdrawn and activity and labile sulfide determined.
column. The experiment was carried out as fol- lows. Firstly, the enzyme was aged at 0°C, as described in the legend of Fig. 3B. Then, the sample was divided into two parts, one of which was treated with rhodanese plus radioactive thio- sulfate, and the other with radioactive thiosulfate alone. Incubation and gel filtration were per- formed as described in Methods and Materials. The fractions containing the flavoprotein were pooled, and activity, total sulfide and radioactive
sulfide were determined. The results are presented in Table I. The enzyme lost 34% of its activity and 12% of its labile sulfide upon aging. These values were restored to 90% and 106%, respectively, after treatment with rhodanese in the presence of radio- active thiosulfate. Gel filtration caused further loss of enzymic activity and labile sulfide. Neverthe- less, the rhodanese plus thiosulfate-treated enzyme had 68% higher activity and 31% higher labile sulfide content after column chromatography than the thiosulfate-treated enzyme (compare lines 4 and 6). Over 90% of the radioactive sulfur incor- porated in N A D H dehydrogenase in the presence of rhodanese was 35S2-; the remainder was pre- sumably 35S° (columns 4 and 5). Therefore, after gel filtration, the amount of radioactive sulfide found associated to the reactivated enzyme accounted fairly well for the sulfide increase mea- sured by the colorimetric method. In order to establish on which subunit of the enzyme the 35S2- was incorporated, the flavoprotein was resolved by repeated freeze-thaw in the presence of 0.3 M CIaCCOONa as described by Ragan et al. [10]. Unfortunately, the treatment caused consid- erable loss of radioactive sulfide, little radioactiv- ity was found associated to subunit I or subunits II + III, so that no conclusion could be drawn.
Discussion
It was established several years ago that rhodanese is an enzyme present in the mitochon- drial matrix [20-23]. Various roles have been ascribed to this enzyme (for a review, see Ref. 24). Finazzi-Agr6 et al. [2] proposed a role for rhodanese in forming the 'labile sulfide' of iron- sulfur proteins from thiosulfate. As pointed out by Westley [24], rhodanese probably serves a variety of functions related to its capacity as a general sulfane transferase. The catalytic mechanism in- volves cleavage of the sulfur-sulfur bond of a sulfane donor. The sulfane sulfur is then trans- ferred from the sulfur-substituted enzyme inter- mediate (bound in persulfide linkage to Cys-247, see Ref. 14) by reaction with a strong thiophile. In the case studied in the present work, rhodanese transfers sulfur from the sulfane oxidation state in thiosulfate to N A D H dehydrogenase where it is inserted as sulfide. It is likely that the strong thiophile mentioned before is provided by the

283
iron-sulfur protein itself. However, the redox mechanism which allows for the reduction of sulfur from the sulfane to the sulfide oxidation state is not clear.
Soluble N A D H dehydrogenase catalyzes the oxidation of NADH by artificial electron accep- tors. The iron-sulfur clusters of the flavoprotein were recently studied by electron paramagnetic resonance and chaotropic resolution of the holoen- zyme [9,10]. It was concluded that the enzyme contains at least two different types of iron-sulfur cluster. One, a slowly relaxing species, has features of center N- lb found in Complex I [9] and was tentatively identified as a tetranuclear (or even trinuclear) cluster present on subunit I [10]. The other, a rapidly relaxing species, should probably be a binuclear species and be present on subunit II. Because NADH dehydrogenase is an extremely labile enzyme, application of harsh treatments to deplete it substantially of its labile sulfide content would also cause an irreversible loss of flavin and of enzymic activity. The experiments of Fig. 1, however, indicate that by mild manipulations (such as dilution and brief incubation on ice) it is possi- ble to bring about only partial inactivation of the enzyme without apparent loss of flavin. While the effect of protein concentration on activity might be related to the state of aggregation of the en- zyme (a decrease in resolution of the enzyme into its subunits in C13CCOONa with increasing pro- tein concentration was observed previously, Ref. 10) it seems that aging, freeze-thaw and column chromatography all cause a significant loss of labile sulfide. Fig. 2 shows a good correlation between sulfide content and enzymic activity. Reactivation of the partially inactivated NADH dehydrogenase was accomplished in the presence of rhodanese plus thiosulfate (Fig. 3A). Dithiothreitol (not shown) or thlosulfate alone had no effect. From Fig. 3B it can be seen that about twice the amount of sulfide lost during the inactivation phase was incorporated in the enzyme by the action of rhodanese plus thiosulfate. It cannot be excluded, however, that a portion of the incorporated sulfide does not involve an iron-sulfur center. The experi- ment reported in Table I (i.e., rhodanese-mediated reactivation in the presence of radioactive thiosul- fate) was designed with two aims in mind: firstly, to eliminate non-protein bound sulfur and corre-
late radioactive determination of sulfide to the colorimetric measurement; secondly, to identify the subunit(s) involved in the reactivation after resolution of the holoenzyme with chaotropic agent. Although gel filtration caused, as also observed before with freshly prepared enzyme [8], loss of labile sulfide even in the presence of di- thiothreitol, most of the sulfide loaded on the enzyme before chromatography (about 11 n m o l / mg protein) appeared as radioactive sulfide after G-75 gel filtration (about 8 nmol /mg protein). This is reflected in an activity more than 60% higher than the control. The results would indicate that rhodanese-mediated transfer of sulfide to the flavoprotein indeed takes place at the level of an iron-sulfur cluster. Resolution of 35S-loaded N A D H dehydrogenase with C13CCOONa and separation of subunit I and subunit II + III caused loss of about 80% of the radioactive sulfide. Thus, identification of the subunit(s) bearing the cluster(s) involved in the inactivation/activation cycle could not be made by this approach. How- ever, we suggest that the center N-1 type, shown before to be slowly relaxing and extremely sensi- tive to handling [9,10], is the cluster primarily involved in the phenomenon described in this paper.
Acknowledgments
We are indebted to Dr. Y. Hatefi, in whose laboratory the experiments were carried out, for suggestions and encouragement during the course of this work. We also wish to thank Dr. P. Cerletti for reviewing the manuscript. The preparation of mitochondria and mitochondrial extracts by Mr. C. Munoz is gratefully acknowledged. The work was supported by N IH Grant GM 27812 to Y.M.G. and by a short-term grant from the Na- tional Research Council of Italy (C.N.R.) to S.P.
References
1 Taniguchi, T. and Kimura, T. (1974) Biochim. Biophys. Acta 364, 284-295
2 Finazzi-Agr6, A., CanneUa, C., Graziani, M.T. and Caval- lini, D. (1979) FEBS Lett. 16, 172-174
3 Bonomi, F., Pagani, S., Cerletti, P. and Cannella, C. (1977) Eur. J. Biochem. 72, 17-24

284
4 Bonomi, F., Pagani, S. and Cerletti, P. (1980) in Flavins and Flavoproteins (Yagi, K. and Yamano, T., eds.), pp. 227-235, Scientific Societies Press, Tokyo
5 Bonomi, F., Pagani, S. and Cerletti, P. (1977) FEBS Lett. 84, 149-152
6 Pagani, S., Bonomi, F. and Cerletti, P. (1982) Biochim. Biophys. Acta 700, 154-164
7 Hatefi, Y. and Stempel, K.E. (1969) J. Biol. Chem. 244, 2350-2357
8 Galante, Y.M. and Hatefi, Y. (1979) Arch. Biochem. Bio- phys. 192, 559-568
90hnishi, T., Blum, H., Galante, Y.M. and Hatefi, Y. (1981) J. Biol. Chem. 256, 9216-9220
l0 Ragan, C.I., Galante, Y.M., Hatefi, Y. and Ohnishi, T. (1982) Biochemistry 21,590-594
I I Hatefi, Y, Haavik, A.G. and Griffiths, D.E. (1962) J. Biol. Chem. 237, 1676-1680
12 Blumenthal, K.H. and Heinrikson, R. (1971) J. Biol. Chem. 246, 2430-2437
13 Sorbo, B.H. (1953) Acta Chem. Scand 7, 1129-136
14 Ploegman, J.H., Drent, G., Kalk, K.H., Hoi, W.G.J., Hein- rikson, R.L., Keim, P., Weng, L. and Russell, J. (1978) Nature 273, 124-129
15 Fogo, J.K. and Popowski, M. (1949) Anal. Chem. 21, 732-734
16 Gilboa-Garber, N. (1971) Anal. Biochem. 43, 129-133 17 Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall,
R.J. (1951) J. Biol. Chem. 193, 265-275 18 Bensadoun, A. and Weinstein, D. (1976) Anal. Biochem. 70,
241-250 19 Hatefi, Y., Stempel, K.E. and Hanstein, W.G. (1969) J.
Biol. Chem. 244, 2358-2365 20 Sorbo, B.H. (1951) Acta Chem. Scand. 5, 724-734 21 De Duve, C., Pressman, B.C., Gianetto, R., Wattiaux, R.
and Appelmans, F. (1955) Biochem. J. 60, 604-617 22 Koj, A., Frendo, J. and Wojtczac, L. (1975) FEBS Left. 57,
42-46 23 Dudek, M., Frendo, J. and Koj, A. (1980) Comp. Biochem.
Physiol. 65B, 383-386 24 Westley, J. (1973) Adv. Enzymol. 39, 237-368