intein-mediated biotinylation of proteins and its ... · really appreciate a/p yao shao qin for his...
TRANSCRIPT

Intein-Mediated Biotinylation of Proteins and its Application
in Protein Microarray
Lue Yee Peng Rina (B.Sc. (Hons), NUS)
A THESIS SUBMITTED
FOR THE DEGREE OF MASTER OF SCIENCES
DEPARTMENT OF BIOLOGICAL SCIENCES
NATIONAL UNIVERSITY OF SINGAPORE
2004

Acknowledgements
First, I would like to thank the National University of Singapore (NUS) and the Agency
for Science, Technology and Research (A*STAR) of Singapore for the funding support. I
also thank the Department of Biological Sciences (NUS) for granting me the Research
Scholarship that financially supported me through my post-graduate days. I would also
like to thank Joan and Reena for their advice on most of the administrative matters. I
really appreciate A/P Yao Shao Qin for his guidance and moral support. As the supervisor
for both my honors and master’s projects, he has always provided me with insightful
discussion. His continuing vision is the main key to the success of this project. Special
thanks to Grace for organizing the enjoyable lab outings. Besides that, she has also given
me lots of technical advice and assistance on the project. Thanks also to Dr Zhu Qing for
synthesizing the cysteine-biotin probe. Last but not the least, I would like to thank all the
people working in the Functional Genomic Laboratory (FGL) and my fellow lab mates in
the Department of Chemistry for their valuable friendship.
i

Table of Contents
Acknowledgements i
Table of Contents ii
Summary iv
List of Tables vi
List of Figures vii Abbreviations ix
1. Introduction 1
2. Materials & Methods 11
2.1. Chemical synthesis of the cysteine-biotin 11
2.1.1. Using Boc-protected cysteine 11
2.1.2. Using Fmoc-protected cysteine 11
2.1.3. Purification and identification of cysteine-biotin 12
2.2. Cloning of target genes into pTYB1 & pTWIN expression vector 12
2.3. Site-directed mutagenesis of pTYB1-wtEGFP (Lys239)-intein 14
2.4. Expression of intein-fused proteins 14
2.5. Affinity purification & C-terminal biotinylation of 15 recombinant proteins
2.6. SPR analysis 16
2.7. In vivo protein biotinylation in E. coli. 16
2.8. In vivo protein biotinylation of in mammalian cells 18
2.9. Generation of protein microarray 19
2.10. Cell free synthesis and biotinylation of MBP 20
3. Results & Discussion 21
ii

3.1. General features of pTYB expression vectors 21
3.2. Intein-Mediated Biotinylation of three model proteins 24
3.2.1. Cloning of target genes into pTYB1 expression vector 24
3.2.2. Expression and extraction of fusion proteins 25
3.2.3. Affinity purification and on-column biotinylation 26
3.3. Protein microarray application 29
3.4. Immobilization of biotinylated proteins onto 34 self-assembled monolayers (SAM) in SPR analysis
3.5. Influence of C-terminal residues on biotinylation 37
3.6. High-throughput expression and biotinylation of yeast proteins 43
3.6.1. Cloning of yeast gene into pTYB1 expression vector 43
3.6.2. Expression, purification & biotinylation of yeast proteins 45
3.7. In vivo biotinylation of proteins 48
3.7.1. In bacterial cells 48
3.7.2. In mammalian cells 51
3.7.2.1. Construction of mammalian expression plasmid, 51 pT-Rex-DEST30-EGFP-Sce VMA intein-CBD
3.7.2.2. Expression and in vivo biotinylation of EGFP 52
in HEK 293 cells
3.7.3. Protein microarray generation using crude bacterial cell lysate 57
3.8. Protein biotinlyation using different inteins 59
3.9. Protein biotinylation in a cell-free system 63
4. Conclusion 66
5. References 68
iii

Summary
The post-genome era has led us to a new frontier of proteomics that requires us to
gain information on the millions of proteins encoded by these identified genes. The
challenge ahead therefore lies in the development of protein microarray that would enable
us to unravel the biological function of proteins in a massively parallel fashion. This
high-throughput screening technique would allow thousands of functional molecules to
be analyzed simultaneously, possibly leading to a better understanding of how these
molecules affect cellular functions. It can be used for discovery of novel protein
functions, screening of protein-protein interactions, detecting enzyme-substrate
interactions and identifying protein targets of biologically active small molecules. Beside
basic protein expression studies, application of the protein microarray technology has
also evolved to diagnostics, mutation analysis, and toxicology in recent years. The idea of
a protein microchip is to immobilize tens of thousands of protein molecules (e.g.
antibodies, receptors, enzymes) onto a solid surface such as glass slides. Each of these
proteins is geared towards identifying and binding of specific targets, thus it is necessary
to immobilize them in its native conformation and correct orientation to preserves their
functional sites. There are several reported strategies of immobilizing proteins onto solid
surfaces but many of these mode of attachments are unspecific, causing the molecules to
be immobilized in the ‘wrong’ orientation. In this report, we present an intein-mediated
approach for efficient and site-specific immobilization of proteins. The reactive C-
terminal thioester generated from intein-assisted protein splicing, either in vitro or in live
cells, served as an attractive, as well as exclusive site for attaching cysteine-containing
biotin. Using this novel biotinylation strategy, we were able to biotinylate many proteins
from different biological sources in a potentially high-throughput fashion. These proteins
iv

were subsequently immobilized onto different avidin-functionalized solid surfaces for
applications such as protein microarray and surface plasmon resonance (SPR)
spectroscopy. We highlighted the numerous advantages of using biotin over other tags
(e.g. GST, His tag etc) as the method of choice in protein purification/immobilization. In
addition, our intein-mediated strategies also provided critical advantages over other
protein biotinylation strategies in a number of different ways. We successfully
demonstrated that, for the first time, intein-mediated protein biotinylation proceeded
inside both bacterial and mammalian living cells, as well as in a cell-free protein
synthesis system. Taken together, our results indicate the versatility of these intein-
mediated strategies, which should provide invaluable tools for potential high-throughput
proteomics applications. They may also serve as useful tools for various biochemical and
biophysical studies of proteins both in vitro and in vivo.
v

List of Tables
Table Page
1. The influence of C-terminal residues on the in vivo cleavage of 39 EGFP-intein and on-column cleavage/biotinylation of EGFP
vi

List of Figures
Figure Page
1. Mechanism of protein splicing 6
2. Biotin-tagging of protein via IPL reaction 8
3. Chemical structure of cysteine-biotin derivative 9
4. Three intein-mediated protein biotinylation strategies 10
5. Map and multiple cloning sites (MCS) of 22 pTYB1 & pTYB2 expression vector
6. Cloning of gene fragment into pTYB1 & pTYB2 23
7. Affinity purification of MBP 28
8. On-column biotinylation of MBP 28
9. Site-specific immobilization of biotinylated, functionally 29 active proteins onto avidin slides
10. Integrity of biotinylated proteins immobilized on 30 avidin-functionalized glass surface 11. Chemical structure of glutathione, natural ligand of GST 30
12. Biotinylated GST on an avidin slide treated with 32 different washing conditions 13. Overview of the on-column biotinylation strategy and 33 site-specific immobilization procedure 14. SPR data showing immobilization of biotinylated MBP on 35 avidin-functionalized sensor chip
15. SPR response of anti-MBP through the MBP-coated sensor chip 36
16. Influence of the C-terminal amino acid residue 40
17. Effect of an extra glycine residue on intein-mediated biotinylation 41
vii

18. DNA fragments obtained from PCR amplification of the 44 yeast cDNA 19. DNA fragments obtained from NdeI and SapI 44 digestion of the TA plasmid 20. Cloning of yeast gene fragment into pTYB1 45
21. High-throughput expression and biotinylation of yeast proteins 47
22. Purification and biotinylation of a yeast protein (YAL012W) 47
23. Optimizing in vivo biotinylation conditions in bacterial cells 50
24. In vivo biotinylation efficiency in bacterial cells 50
25. In vivo biotinylation of proteins in bacterial cells shown 51 by anti-biotin blot 26. Construction of mammalian expression plasmid 54 using GatewayTM Technology 27. Expression of EGFP-Sce VMA intein-CBD in 55 different mammalian cell line 28. In vivo biotinylation of EGFP in mammalian cells 55 shown by anti-biotin blot 29. In vivo biotinylation efficiency in mammalian cells 56
30. Site-specific immobilization of biotinylated proteins 57 onto avidin slides using bacterial cell lysate 31. Schematic representation of pTWIN vectors 60
32. Recovery of the intein fusion proteins from the cell extract 61 and its cleavage efficiency with MESNA and cysteine-biotin 33. Yield of EGFP from the different intein fusion 62
34. In vivo biotinylation of EGFP with different intein fusion 62
35. Protein biotinylation in a cell-free system 65
viii

Abbreviations AA Amino acid
Ala Alanine
Ampr Ampicillin resistant
Arg Arginine
Asn Asparagine
Asp Aspartic acid
Boc t-Butoxycarbonyl
CBD Chitin binding domain
Cys Cysteine
DCM Dichloromethane
DMEM Dulbecco’s modified Eagle’s medium
DMF Dimethylformamide
DTT 1,4-dithiothreitol
E.coli. Escherichia coli
ECL Enhanced ChemiLuminescent
EDTA Ethylenediaminetetraacetic acid
EGFP Enhanced green fluorescent protein
FITC Fluorescein Isothiocyanate
Fomc 9-Fluorenylmethoxycarbonyl
GFP Green fluorescent protein
Gln Glutamine
Glu Glutamic acid
Gly Glycine
ix

GST Glutathione-S-transferase
His Histidine
HOBt N-Hydroxybenzotriazole
HPLC High pressure liquid chromatography
HRP Horse-radish peroxidase
Ile Isoleucine
IPL Intein-mediated protein ligation
IPTG Isopropyl thiogalactosidase
Kanr Kanamycin resistant
LB Luria Bertani
Leu Leucine
Lys Lysine
MBP Maltose binding protein
MeOH Methanol
MESNA 2-mercaptoenthanesulfonic acid
Met Methioine
mutEGFP Mutant EGFP
Ni-NTA Nickel nitrilotriacetic
NMR Nuclear magnetic resonance
ORF Open reading frame
PBS Phosphate buffer saline
PCR Polymerase chain reaction
Phe Phenylalanine
PMSF Phenylmethylsulfonyl fluoride
x

Pro Proline
RTS Rapid Translation System
SAM Self-assembled monolayers
SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis
Ser Serine
SPR Surface plasmon resonance
TBTU O-(Benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
tetrafluoroborate
TCEP Tris-(2-carboxyethyl)phosphine
TFA Tifluoroacetic acid
Thr Threonine
Tris (hydroxymethyl)-aminomethane
Trp Tryptophan
Tyr Tyrosine
Val Valine
wtEGFP Wild type EGFP
xi

1. Introduction With the completion of the Human Genome Project, one may estimate that the number
of proteins in human could vary from approximately 40,000 to as many as 1,000,000.1
This poses an even greater challenge ahead - to identify the structures and functions of all
proteins encoded by the human genome. Traditionally, the function of a protein is
elucidated through its structure using NMR, X-ray crystallography and other related
techniques. Although structural features in a protein can help in determining it
biochemical functions, they are not as useful in defining its biological functions (e.g. its
interacting partners and/or biological pathways). This calls for new methods that allow
high-throughput determination of protein functions and/or interactions. Protein microarray
technologies satisfy many of these criteria, and in the past few years, have emerged as the
uprising technology in the field of proteomics.1-5 Success stories from the DNA
microarray in the last decade have propelled the rapid development of the protein
microarray, providing a potential means for high-throughput identification and
quantification of proteins from biological samples.6-13 Due to fundamental differences
between proteins and DNA, however, the protein array technology is currently in its
infancy. Unlike DNA, which is highly stable and robust, proteins are known to lose its
functional integrity upon immobilization onto a solid surface. Furthermore, there is
presently no known technique, which can effectively amplify proteins as in DNA. Existing
methods for protein expression have many limitations. The inevitable chemical, physical
and structural variation among different proteins results in their non-specific absorption to
solid surfaces, thus creating further problems for their immobilization in a microarray.
Despite these technical hurdles, several research groups have successfully demonstrated
1

the functional use of protein microarrays using a wide variety of surface substrates and
attachment chemistries.6,7,14-25
The simplest way to immobilize proteins on a solid support relies on non-covalent
interactions such hydrophobic or van der Waals interactions, hydrogen bonding or
electrostatic forces. Examples of electrostatic immobilization include the use of materials
such as nitrocellulose and poly-lysine- or aminopropyl silane-coated surfaces.14-17 Protein
microarrays were also fabricated by means of physical adsorption onto porous gel pads.18-
22 A major advantage of these non-covalent immobilization concepts is their ease of use.
Usually no protein modification is needed prior to imprinting onto the surface. The
disadvantage is that proteins often get denatured on these fairly undefined surfaces due
non-specific interactions between the protein and the surface material. On top of that,
physical adsorption of proteins onto surfaces may also lead to de-adsorption of proteins
during biochemical assays, which can lead to signal loss. Covalent attachment of proteins
onto NHS-activated glass surface, via nucleophiliic groups (-NH2, -SH, -OH) located on
protein surface, has been described by MacBeath and Schreiber.23 Other surfaces such as
epoxide surfaces have also been used to capture proteins covalently.24 However, in these
cases, the immobilization is random which may lead to deactivation of the protein
molecules on the array. Ideally, the proteins should be site-specifically immobilized on the
surface to obtain a homogeneous orientation. Oriented immobilization was first reported
by Zhu et al who expressed more than 90 % of proteins encoded by the Saccharomyces
cerevisiae ORFs using a double-tagging system. The yeast proteins were expressed
laboriously in the form of fusion proteins containing both histidine and GST tags before
affinity purification through the glutathione (GSH) column. These proteins were
2

subsequently immobilized on a single 25 x 75 mm Ni-NTA coated glass slide to generate
a ‘yeast proteome chip’.25 Unfortunately, this strategy of immobilization is extremely
tedious and time consuming, requiring multiple steps of sample processing. Moreover,
protein immobilization via His-tag/Ni-NTA interaction was shown to be neither strong nor
robust enough to withstand harsh wash condition, thereby limiting the downstream
application of the protein microarray. Avidin-biotin technology has gained much
prominence in research due to the remarkable affinity between avidin (or streptavidin-its
bacteria relative from streptomyces avidinii) and biotin (vitamin H, 0.24kDa).26 With a Kd
of 10-15 M, avidin/biotin binding is the strongest non-covalent interaction known in nature.
Consequently, avidin/biotin systems have been exploited for a variety of diverse
applications in modern biology including peptide and protein microarray technology.26-32
In a recent example, Peluso et al. were able to site-specifically immobilized biotinylated
antibodies and antibody fragments onto streptavidin surfaces leading to an increase in
sensitivity over random attachment in a microarray assay.32
An essential prerequisite for the success of avidin-biotin technology is the
incorporation of biotin moiety into experimental system. Historically, biotinylation of
proteins has been carried out by standard bioconjugate techniques using biotin-containing
chemicals. This leads to random biotinylation of proteins and in many cases, the
subsequent inactivation of some protein biological activities.33 Alternative techniques
have been developed which allows for site-specific labeling of proteins with biotin. 34,35 A
stretch of amino acids sequences has been identified by Cronan for site-specific tagging of
biotin to proteins.35 The covalent attachment of biotin to specific lysine residue on the tag
3

sequence was catalyzed by biotin ligase (EC 6.3.4.10), a 35.5 kDa monomeric enzyme
encoded by the birA gene36, in a 2 step reaction as follows;
Step 1: Biotin + ATP ↔ Biotinoyl-AMP + PPi
Step 2: Biotinoyl-AMP + apoprotein → Biotinoyl-protein + AMP
These sequences, however, are typically quite large (> 63 AAs) and thus may interfere
with the biological activity of the proteins they are fused to. Further optimization of these
sequence tags revealed that smaller tags (15-30 AAs) may be used.37 In general, proteins
fused with these tag sequences could be biotinylated either in vitro or in vivo by biotin
ligase.38,39 Unfortunately, in vivo biotinylation of proteins catalyzed by biotin ligase is
often inefficient and cell toxicity is likely to occur due to decrease biotinylation of
important endogenous proteins within the host cells.39 Recent advances in the field
however, have partially rectified this problem, and at the same time unequivocally
demonstrated numerous advantages associated with protein biotinylation in live cells.40 In
vitro biotinylation is used when in vivo expression of the soluble fusion protein is
insufficient. This however, also faces with problems such as proteolytic degradation of tag
sequences and inhibitory effects of commonly used reagents towards biotin ligase.38 To
overcome these drawbacks, we have developed an intein-based system to incorporate
biotin moiety exclusively at the C-terminus of protein.
According to the central dogma of gene expression, genetic information flows from
DNA to RNA through transcription, and is then translated and expressed as protein.
4

However, more genetic information appears to be present within the chromosomal DNA
other then the gene that actually encodes for the protein product. These excess genetic
information are known to be introns, which get excised post-transcriptionally during RNA
splicing. In 1990, two groups independently reported the existence of protein splicing
elements that is capable of excising themselves post-translationally through a process
analogous to RNA splicing.41,42 These protein “introns”, known as intein, are found within
genes of other proteins and translated as a single polypeptide chain. After translation, the
intein initiates an autocatalytic event to excise itself and join the flanking host segments
with a new peptide bond to form the final protein product (Figure 1).43 To date, over 100
inteins have been discovered in unicellular organisms from all three domains of life.44,45
They can be divided into four basic classes: (1) the bifunctional/maxi-inteins, which
contain an endonuclease domain inserted into the splicing domain46,47; (2) the mini-
inteins, which lack the endonuclease domain48-52; (3) the trans-splicing inteins, which is
splitted in the splicing domain and each precursor fragment is present as a different
primary translation product53-57; and (4) the newly discovered alanine inteins, which
contain a naturally occurring N-terminal alanine residue58.
5
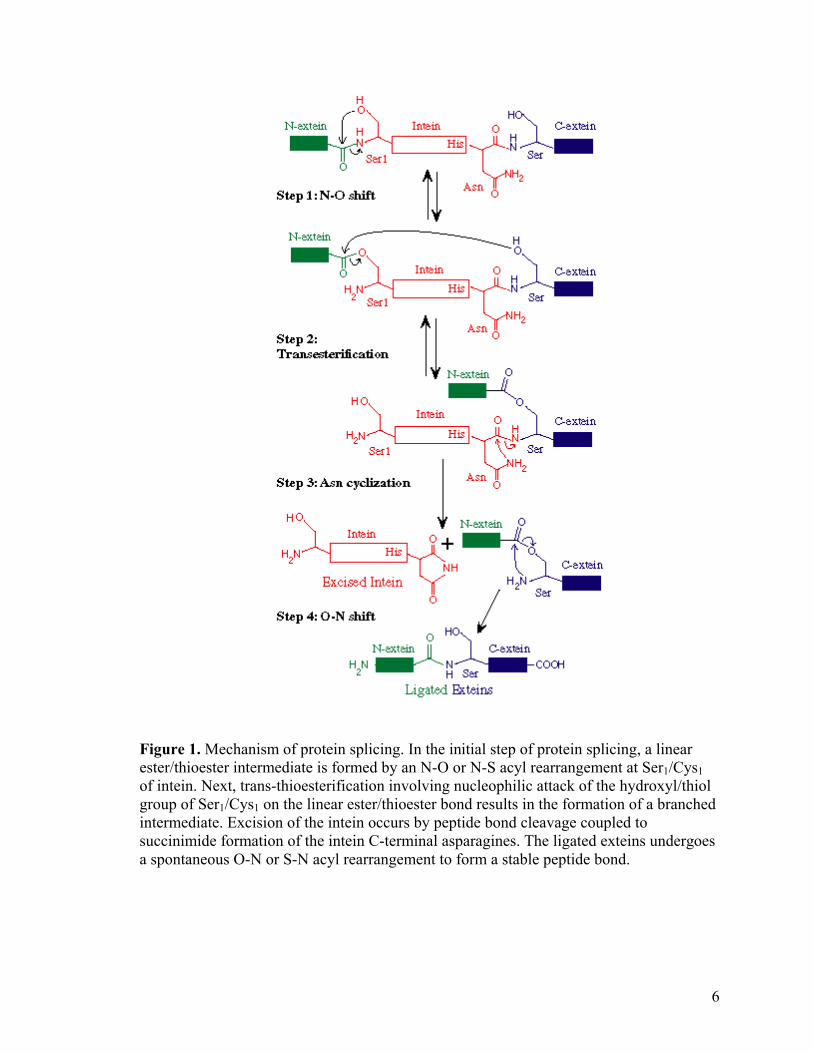
Figure 1. Mechanism of protein splicing. In the initial step of protein splicing, a linear ester/thioester intermediate is formed by an N-O or N-S acyl rearrangement at Ser1/Cys1 of intein. Next, trans-thioesterification involving nucleophilic attack of the hydroxyl/thiol group of Ser1/Cys1 on the linear ester/thioester bond results in the formation of a branched intermediate. Excision of the intein occurs by peptide bond cleavage coupled to succinimide formation of the intein C-terminal asparagines. The ligated exteins undergoes a spontaneous O-N or S-N acyl rearrangement to form a stable peptide bond.
6

The discovery of inteins and elucidation of its self-splicing mechanism has triggered
the research of new applications and techniques for protein chemistry and engineering. For
example, through identification of the residues directly participating in the breakage and
peptide bond formation, Chong et al were able to engineer inteins with controllable
cleavage at single splice junctions.59 By fusing the chitin binding domain (CBD, 5kD) of
Bacillus circulans60 to one terminus of the intein, they developed an intein-mediated
affinity purification system that eliminates the use of protease, which may further
complicate the downstream purification process. Protein purification occurs within a
single chitin beads packed column due the self-catalytic activity of the fused intein.61,62
The ability of intein to generate C-terminal thioester and N-terminal cysteine protein
during bond breakage also greatly expands the utility of native chemical ligation
chemistry in protein engineering. Intein-mediated protein ligation (IPL) is an extremely
useful method for protein synthesis with a variety of peripheral applications.63-67 It has
been used to incorporate noncoded amino acids into a protein sequences64, purify
cytotoxic proteins65, study protein structure/function relationship by segmental isotopic
labeling of proteins for NMR analysis66, and introduce fluorescent probes into a protein
sequence67. Intein fusion system has also been employed to generate both complementary
reactive groups on the same protein resulting in either inter- or intramolecular ligation,
leading to multimeric or cyclic protein species, respectively.68,69 Trans-splicing between
two foreign protein sequences enable in vitro fusion of two protein sequences via a simple
peptide bond thus creating a protein chimera with new added properties.70 The utility of
IPL can be expanded to a wide range of proteomic application by a variety of
functionalities, depending on the experimental requirements.71,72 Herein, we described a
7

novel and highly efficient approach for site-specific biotin-tagging of proteins using IPL
(Figure 2).
NH
OHS
InteinS
O
H2N
HS SO3-
NH2
HS
S
O
SO3-
S
O H2N
NH
O
Intein
N-S acyl shiftThiol induced cleavage
Chemoselective reaction
S-N acyl shift
Figure 2. Biotin-tagging of protein via IPL reaction. Engineered intein, fused with the protein of interest, catalyzed the formation of thioester. The intein gets cleaved off in a thioester exchange reaction with a thiol compounds, such as 2-mercaptoethanesulfonic (MESNA), generating proteins with an active C-terminal thioester. IPL occurs between the thioester protein and the cysteine-containing biotin tag resulting in a native peptide bond formation.
In our work with 3 model proteins, namely MBP (Maltose Binding Protein), EGFP
(Enhanced Green Fluorescent Protein) and GST, we demonstrated that site-specific
biotinylation of proteins could be efficiently carried out by applying a cysteine-containing
biotin tag (Figure 3) to the intein-fused protein purified and bound onto a chitin column
(Method B in Figure 4).29 Since both cleavage and biotin tagging of the proteins were
carried out in a single column, the eluted proteins can therefore be immobilized directly
onto an avidin-coated glass slide to generate a protein microarray, without further
purification step. To further validate the feasibility of this biotinylation strategy for protein
array and other high-throughput proteomic applications, we went on to examine the
versatility of this biotin-tagging approach for many other different classes of yeast
8

proteins. In addition, we also demonstrate for the first time, that this intein-mediated
biotinylation strategy could be successfully implemented in both bacterial and mammalian
cells to generate in vivo biotinylated proteins. The cell lysate containing the biotin-tagged
proteins were subsequently used to generate corresponding protein array in a single step
without further downstream processing (Method A in Figure 4). Beside cell expressed
recombinant proteins, intein-mediated biotinylation strategy may also be extended to
biotinylate proteins synthesized in a cell-free synthesis system (Method C in Figure 4).31
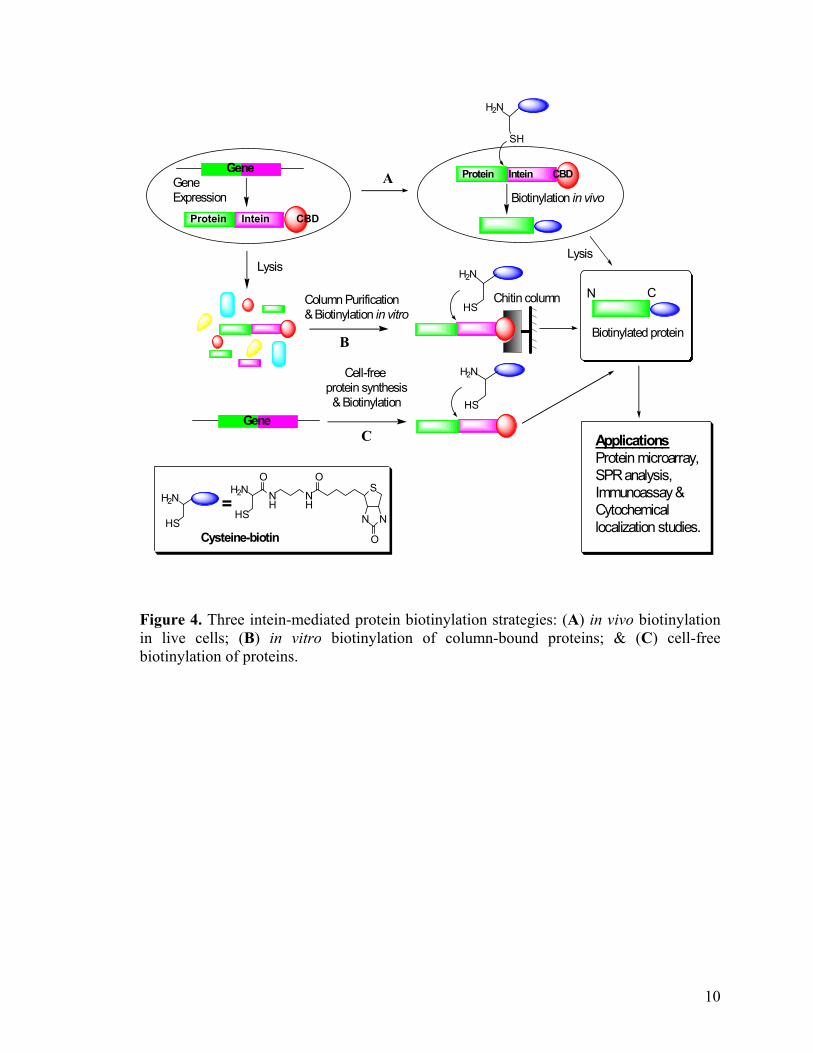
Figure 4 summarizes the 3 intein-mediated protein biotinylation strategies described.
H2N NH
HN
SH
O
OS
HNNH
O
Figure 3. Chemical structure of cysteine-biotin derivative
9
S
H2N
SH
H2N
HS
NH
O
N N
S
O
NH
H2NO
HS
H2N
HS
=
Cell-free protein synthesis & Biotinylation
Protein Intein CBD
Gene
Gene
Biotinylated protein
A
B
C
Cysteine-biotin
Column Purification& Biotinylation in vitro
Lysis
Biotinylation in vivo
Protein Intein CBD
N C
Lysis
H2N
HS
ApplicationsProtein microarray,SPR analysis, Immunoassay &Cytochemical localization studies.
Chitin column
GeneExpression
Figure 4. Three intein-mediated protein biotinylation strategies: (A) in vivo biotinylation in live cells; (B) in vitro biotinylation of column-bound proteins; & (C) cell-free biotinylation of proteins.
10

2. Materials and Methods 2.1. Chemical synthesis of the cysteine-biotin. Cysteine-biotin derivatives (Figure 3) can be synthesized with either with (1) Boc-
protected, or (2) Fmoc-protected cysteine:
2.1.1. Using Boc-protected cysteine
N- -t-Boc-S-trityl-L-cysteine (1.2 g, 2.6 mmol), TBTU (1.0 g, 3.10 mmol), and HOBt
(0.60 g, 3.9 mmol) were dissolved in 50 ml of dry DMF. This mixture was stirred under
argon for 20 min at room temperature before addition of 4-methyl morpholine (0.75 g, 7.8
mmol) and biotinylethylenediamine (0.75 g, 2.6 mmol). The reaction was stirred further
for 3 h, followed by evaporation in vacuo. The crude product was dissolved in 200 ml of
CH2Cl2, extracted with 3 x 200 ml of H2O, dried over MgSO4, and concentrated in vacuo.
Further purification was done by flash chromatography (4-8% MeOH in CH2Cl2) to give
the protected form of 1, which was deprotected by first stirring in a solution containing
trifluoroacetic acid (50 ml), H2O (1.6 ml), and triisopropylsilane (1.2 g, 7.8 mmol) for 30
min, and then evaporation in vacuo. The resulting residue was taken in a mixture of 1:1
H2O/CH2Cl2 (200 ml), and the aqueous layer was extracted with 3 x 100 ml of CH2Cl2
before evaporation to dryness.
2.1.2. Using Fmoc-protected cysteine
N-Fmoc-S-Trityl-L-cysteine (0.996 g, 1.7 mmol), TBTU (0.674 g, 2.1 mmol) and
HOBt (0.3989 g, 2.6 mmol) were dissolved in 17 ml of DMF. After stirring for 30 minutes
at room temperature, biotinylethylenediamine (0.5 g, 1.7 mmol) and triethylamine (0.515
11

g, 5.1 mmol) were added. The reaction was carried out under nitrogen for 3 hours at room
temperature, followed by concentration in vacuo. The resulting residue was dissolved in
ethyl acetate (50 ml), and extracted with 1.0 M HCl (50 ml), 10% Na2CO3 (50 ml),
saturated NaCl (50 ml), dried over MgSO4, and then evaporated to dryness. A solution of
20% piperdine in DMF (15 ml) was added to the resulting residue and stirred for 30
minutes at room temperature. Following evaporation, the residue was dissolved in ethyl
acetate and washed with 2 x 10% Na2CO3 (50 ml), saturated NaCl (50ml), dried over
MgSO4, and then evaporated to dryness. The residue was taken in 15 ml of
TFA/EDT/H2O (9/0.5/0.5), stirred for 1 hour, and then evaporated to dryness. The residue
was taken in 100 ml of 1:1 DCM/H2O and insoluble solid was removed by filtration.
2.1.3. Purification and identification of cysteine-biotin
Final purification of the product from both syntheses was done using HPLC with a
C18 reverse-phase column to give the final product as a white solid (39% overall yield).
1H NMR (400 MHz, D2O) 4.57 (dd, 1H, J = 7.8, 5.0), 4.39 (dd, 1H, J = 7.8, 5.0), 4.12 (t,
1H, J = 5.4), 3.45 (m, 1H), 3.33-3.24 (m, 4H), 3.03 (dd, 1H, J = 14.9, 5.4), 3.00-2.93 (m,
2H), 2.74 (d, 1H, J = 13.2), 2.22 (t, 2H, J = 7.3), 1.72-1.50 (m, 4H), 1.48-1.31 (m, 2H);
13C NMR 179.62, 170.46, 64.53, 62.70, 57.79, 56.01, 42.16, 42.12, 41.45, 37.96, 30.39,
30.12, 27.50, 27.30; ESI 390.2 (MH+).
2.2. Cloning of target genes into pTYB1 & pTWIN expression vector
To construct intein fusion proteins, target gene fragments were first PCR amplified
from pEGFP (CLONTECH), pGEX-4T1 (Pharmacia Biotech), and yeast ex-clones
(Invitrogen) respectively. PCR amplification for both EGFP and GST gene fragments
12

utilized upstream primers (5’-GGC GGC CAT ATG GTG AGC AAG GGC GAG-3’) &
(5’-GGC GGC CAT ATG TCC CCT ATA CTA GGT-3’) containing an NdeI site with a
translation initiation codon (ATG), and downstream primers (5’-GGC GGC TGC TCT
TCC GCA CTT GTA CAG CTC-3’) & (5’-GGC GGC TGC TCT TCC GCA GTC ACG
ATG CGG-3’) containing a SapI site, respectively. A common upstream primer (5’-GGC
GGC CAT ATG GAA TTC CAG CTG ACC ACC-3’) and individual downstream primers
(5’-GGC GGC TGC TCT TCC GCA ACC ACC N15-18-3’) were used to amplify the yeast
gene fragments from the Yeast ExClonesTM, and at the same time introduce 2 extra Gly
residues to the C-terminus of the yeast gene. A standard PCR mixture contained 1x
HotStarTaq DNA polymerase buffer (Qiagen), 0.2 mM of each dNTPs (NEB), 0.5 µM of
each primer, 100 ng of plasmid DNA template and 2 units of HotStarTaq DNA
polymerase (Qiagen). Amplification was carried out with a DNA Engine™ thermal cycler
(MJ Research) at 94 °C for 45 sec, 65 °C for 45 sec and 72 °C for 1 min, for 25 cycles for
the EGFP and GST gene fragments, and at 94°C for 45 sec, 55°C for 45 sec and 72°C for
2 min, for 25 cycles for the yeast gene fragments. The PCR products were then cloned into
pCR2.1-TOPO using TOPO TA cloning kit (Invitrogen) prior to double digestion with
NdeI and SapI (NEB). Digested EGFP, GST and yeast gene fragments of correct sizes
were gel-purified and cloned into either pTYB1 or pTWIN expression vector (NEB,
USA), via NdeI and SapI sites to yield the intein-fused constructs. The C-terminal residue
of GST in pTYB1-GST-intein was site-mutagenized from Cys to Gly using using
QuickChange™ XL Site–Directed Mutagenesis Kit (Stratagene) with upstream primer
(5'-CGG CCG CAT CGT GGG TGC TTT GCC AA-3’) and downstream primer (5'-TT
GGC AAA GCA CCC ACG ATG CGG CCG-3'); Gly is underlined in the primers. The
13

pTYB1 construct containing the MBP gene, pMYB5, is commercially available (NEB).
The resulting T7-driven expression plasmids, shown to be free of mutation by automated
DNA sequencing (Applied Biosystems), were then transformed into E.coli ER2566 host
(NEB) for protein expression.
2.3. Site-directed mutagenesis of pTYB1-wtEGFP (Lys239)-intein
The C-terminal residue of wtEGFP in pTYB1-wtEGFP (Lys239)-intein was site-
mutagenized from the original Lys239 to the other 19 amino acids using QuickChange™
XL Site–Directed Mutagenesis Kit. 19 sets of primers, each containing a primer (5'-GAC
GAG CTG TAC NNN TGC TTT GCC AA-3’) and a complementary primer (5'-TT GGC
AAA GCA N’N’N’ GTA CAG CTC GTC-3'), were used, in which NNN (and N’N’N’) in
each set of primers represents an amino acid to which Lys239 in pTYB1-wtEGFP (Lys239)-
intein was replaced. Upon confirmation by DNA sequencing, the mutated plasmids (e.g.
pTYB1-mutEGFP (AA239)-intein, where AA represents a corresponding mutated amino
acid) were transformed into ER2566 E. coli.. EGFP (Asp239)-intein and EGFP (Cys239)-
intein were also cloned into pTYB-2 vector via NdeI and SmaI site based on ImpactTM-CN
protocols (NEB).
2.4. Expression of intein-fused proteins
The transformed E. coli host was grown in Luria Bertani (LB) medium supplemented
with 100 µg/ml ampicillin at 37 °C in a 250 rpm shaker to an OD600 of about ~0.5. Protein
expression was induced overnight at room temperature using 0.3 mM isopropyl
thiogalactosidase (IPTG). Upon harvest (4000 rpm, 15 min, 4°C), cells were resuspended
14

in lysis buffer (20 mM Tris-HCl pH 8.0, 0.5 M NaCl, 1 mM EDTA, 1 % CHAPS, 1 mM
TCEP and 1 mM PMSF) and lysed by glass beads (Sigma). The cell debris was pelleted
down by centrifugation (20,000 × g, 30 min, 4 °C) to give a clear lysate ready for loading
onto a column packed with chitin affinity resin (NEB, USA) for purification and
biotinylation.
2.5. Affinity purification & C-terminal biotinylation of recombinant proteins
Microspin columns were pre-packed with 100 µl of chitin resin and pre-equilibrated
with 1 ml of column buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl and 1 mM EDTA).
To purify the fusion protein, the clarified cell lysate was incubated on the column for 30
min at 4 ˚C with gentle agitation to ensure maximum protein binding. Unbound impurities
were then washed away with 2 ml of column buffer. To biotinylate recombinant proteins,
200 µl of the column buffer containing 50 mM MESNA (Sigma) and 5 mM cysteine-
biotin was passed through the column to distribute it evenly throughout the resin before
the flow was stopped and the column was incubated at 4°C overnight. The resulting
biotinylated protein was eluted with 100 µl of column buffer, and analyzed by 12 - 15%
SDS-PAGE gel. Resin-bound proteins were analyzed by first boiling the resin with DTT-
free SDS-PAGE loading buffer, then separated by SDS-PAGE. Silver or coomassie
staining of the gel was done to visualize the separate proteins bands. Premature in vivo
cleavage and on-column cleavage of the intein-fusion was determined from the stained
SDS-PAGE gel. To determine the ratio between the biotinylated and the non-biotinylated
protein in the eluted fraction, an absorption experiment with streptavidin beads was
performed. The eluted fraction was first incubated with excessive Streptavidin
15

MagneSphere® Paramagnetic Particles (Promega) for 1 h at 4 oC to ensure all biotinylated
proteins were absorbed onto the beads. Both eluents, before and after streptavidin
adsorption, were then analyzed by SDS-PAGE. Western blots with horseradish
peroxidase (HRP)-conjugated anti-biotin antibody (NEB) and the Enhanced
ChemiLuminescent (ECL) Plus kit (Amersham) were performed to confirm the presence
of biotin-tagged proteins.
2.6. SPR analysis.
All SPR experiments were performed with a BIAcore X instrument (Biacore).
Biotinylated MBP was prepared as described above. Surface activation of the CM5 sensor
chip (Biacore) was done using standard amino-coupling procedures according to
manufacture’s instructions. 1.75 µg of avidin in 10 nM acetate (pH 4.5) and 0.125 M
NaCl was passed over the activated chip surface. Excess reactive groups were then
deactivated with 1 M ethanolamine hydrochloride (pH 8.5) before injection of 35 µl
biotinylated MBP (10 µg/ml) to the avidin-functionalized surface. Subsequently, 10 µl of
anti-MBP antibody (0.1 mg/ml) was injected at a flow rate of 1 µl/min to confirm the
immobilization of MBP onto the chip surface. 10 mM HCl was used to regenerate the chip
surface before subsequent rounds of antibody injections. The Kd of the anti-MBP/MBP
binding was determined by BioEvaluation software installed on the BIAcore X.
2.7. In vivo protein biotinylation in E. coli.
For in vivo biotinylation of proteins in E. coli., pMYB5 and pTYB-1 constructs
containing two yeast proteins (YAL012W & YGR152C) were used. Liquid cultures of
16

ER2566 carrying the intein-fusion construct were grown to OD600 of ~0.6 in LB medium
supplemented with 100 µg/ml of ampicillin. Expression of MBP and yeast protein fusions
was induced with 0.3 mM IPTG at room temperature overnight. MESNA and cysteine-
biotin were subsequently added to final concentrations of 10 mM and 5 mM, respectively.
Other concentrations of MESNA/cysteine-biotin were also tested but these conditions
gave the best in vivo bintinylation efficiency while maintaining viability of the cells. In
vivo biotinylation was allowed to proceed overnight at 4˚C with gentle agitation. Cells
were harvested and washed thoroughly with PBS to remove excess MESNA/cysteine-
biotin before lysed with glass beads. Clear lysates containing the desired biotinylated
proteins were collected by centrifugation, and used without further purifications. The
entire process was monitored by SDS-PAGE and western blots with anti-MBP. In vivo
protein biotinylation was unambiguously confirmed with HRP-conjugated anti-biotin
antibody. Additionally, to confirm the affinity of the in vivo biotinylated protein towards
avidin/streptavidin and to determine the ratio of the biotinylated/non-biotinylated proteins
generated in vivo, an absorption experiment with streptavidin beads was performed. Clear
cell lysates were incubated with Streptavidin MagneSphere® Paramagnetic Particles
(Promega) at 4°C for 30 min. The beads were then thoroughly washed with PBS to
remove unbound proteins, and subsequently analyzed by boiling in SDS-PAGE loading
buffer, then resolved on a 12% SDS-PAGE gel, followed by immunoblotting with HRP-
conjugated anti-biotin antibody. Cell lysates before and after streptavidin absorption were
also separated on a 12% SDS-PAGE gel followed by western blots with anti-MBP and
anti-biotin antibodies.
17

2.8. In vivo protein biotinylation of in mammalian cells.
EGFP-intein was cloned into pT-Rex-DEST30 (Invitrogen) mammalian expression
vector by Gateway™ cloning technology. EGFP-Sce VMA intein-CBD was amplified
from pTYB1-wtEGFP-intein using upstream primer (5’-GGGG ACA AGT TTG TAC
AAA AAA GCA GGC TTC GAA GGA GAT AGA ACC ATG GTG AGC AAG GGC
GAG GAG-3’) and downstream primer (5’-GGG GAC CAC TTT GTA CAA GAA AGC
TGG GTC TCA TTG AAG CTG CCA CAA GGC -3’), where the underline nucleotides
represent the attB recombination sites. The amplified EGFP-Sce VMA intein-CBD gene
was first cloned into the pDONRTM 201 donor vector then to the final pT-Rex-DEST30
destination vector using BP and LR ClonasesTM Mix (Invitrogen). The final expression
plasmid was transfected into HEK 293 cells, grown in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 units/ml)
and streptomycin (100 µg/ml), using PolyFect Transfection Reagent (Qiagen). The
mammalian cells were seeded at 2.4 x 106 cells per 100 mm tissue culture plate the day
before transfection. After 48 h of transient expression, the culture medium was changed
to DMEM containing 10 mM MESNA and 1 mM cysteine-biotin, and further incubated at
37 °C overnight. These biotinylation conditions were optimized to ensure cell viability
and maximum biotinylation efficiency. Mammalian cells were then harvested, washed
thoroughly with PBS to remove excess biotin, and lysed by glass beads. The entire
biotinylation process was monitored by SDS-PAGE and western blots with anti-EGFP.
The biotinylated protein in the mammalian cell lysates was purified using Streptavidin
MagneSphere® Paramagnetic Particles before unambiguously confirmed by
immunoblotting using HRP-conjugated anti-biotin antibody as described earlier.
18

2.9. Generation of protein microarray.
Glass slides were cleaned in a piranha solution and derivatized with a 1% solution of
3-glyicidoxypropyltrimethoxisilane (95 % ethanol, 16 mM acetic acid) for 1 hr and cured
at 150 °C for 2 hours. The epoxy slides were reacted with a solution of 1 mg/ml avidin in
10 mM NaHCO3 for 30 minutes, washed with water, air dried, and the remaining epoxides
were quenched with a solution of 2 mM aspartic acid in a 0.5 M NaHCO3 buffer, pH 9.
Trace amount of cysteine-biotin in the eluted protein sample, from on-column
biotinylation, and the clarified cell lysate, from in vivo biotinylation, did not seem to affect
the spotting quality, as NAP-5 treated protein samples did not seem to improve the array
quality. Therefore, protein samples from both sources can be directly spotted onto the
avidin-functionalized slides using an ESI SMA arrayer (Toronto), without any
additional purification step. No incubation was necessary before the slide were further
processed by washing with PBS and drying in air. Sequence specific monoclonal
antibodies, anti-EGFP (Clontech) and anti-MBP (Santa Cruz Biotechnology), were
labeled with Cy3-NHS (λEx = 548 nm; λEm = 562 nm) and Cy5-NHS (λEx = 646 nm; λEm =
664 nm)(Amersham Biosciences) respectively. The antibody was reacted with the dye for
one hour in 0.1 M NaHCO3, pH 9, according to manufacturer’s protocols and purified
with a NAP5 column (Amersham Pharmacia). The anti-GST was purchased as a FITC-
conjugate (λEx = 490 nm; λEm = 528 nm)(Molecular Probes). The spotted slides were
incubated with the labeled antibody (or mixture of antibodies) for 1 hour, washed 4 times,
each time for 15 min with PBST (PBS + 0.1 % Tween 20), dried and scanned with an
ArrayWoRx microarray scanner (Applied Precision). To show selective binding of
glutathione to GST on the protein array, the N-terminal amine group of glutathione was
19

first labeled with Cy3-NHS by reacting the molecule overnight with the dye in sodium
phosphate buffer at pH 7. The reaction was subsequently quenched with ethanolamine for
12 hours to degrade the remaining Cy3-NHS, and any glutathione labeled at its cysteinyl
thiol. Avidin slides, immobilized with biotinylated GST as described earlier, were
incubated with the Cy3-labeled glutathione for 1 hour, and washed with PBST. Finally,
the slides were dried and specific binding between GST and glutathione was visualized
with the microarray scanner.
2.10. Cell free synthesis and biotinylation of MBP.
The pMYB5 plasmid was used as the DNA template in the Rapid Translation System
(RTS) 100 E. coli. HY kit (Roche) for cell-free protein synthesis. Based on the
manufacturer’s protocol, the reaction was performed at 30˚C for 4 h, based on the
manufacturer’s protocol in a 25 µl reaction with 500 ng DNA as the template. At the end
of protein synthesis, MESNA and cysteine-biotin were added to the lysate to final
concentrations of 50 mM and 5 mM, respectively, to induce cleavage/biotinylation of
MBP at 4°C overnight. Cell lysates were precipitated with acetone and analyzed by SDS-
PAGE. Biotinylation of MBP was unambiguously confirmed by western blots with HRP-
conjugated anti-biotin antibody.
20

3. Results and Discussion
3.1. General features of pTYB expression vectors
pTYB vectors are commercially available from New England Biolabs for expression
and isolation of proteins processing a C-terminal thioester. The target gene is inserted into
the polylinker region of each vector such that the C terminus of the target protein is fused
in-frame to the N terminus of the Sce VMA intein (from Saccharomyces cerevisiae
VMA1 gene). Transcription of the fusion gene is initiated from the pTYB T7 promoter73
under the tight control of a lac operon. Binding of the lac repressor (encoded by the lac I
gene in the same vector) to the lac operator sequences immediately downstream of the T7
promoter, suppresses basal expression of the fusion gene in the absence of IPTG
induction. A T7 transcription terminator is located downstream of the CBD to prevent
continued transcription. pTYB vectors also carries an ampicillin resistance gene (Ampr)
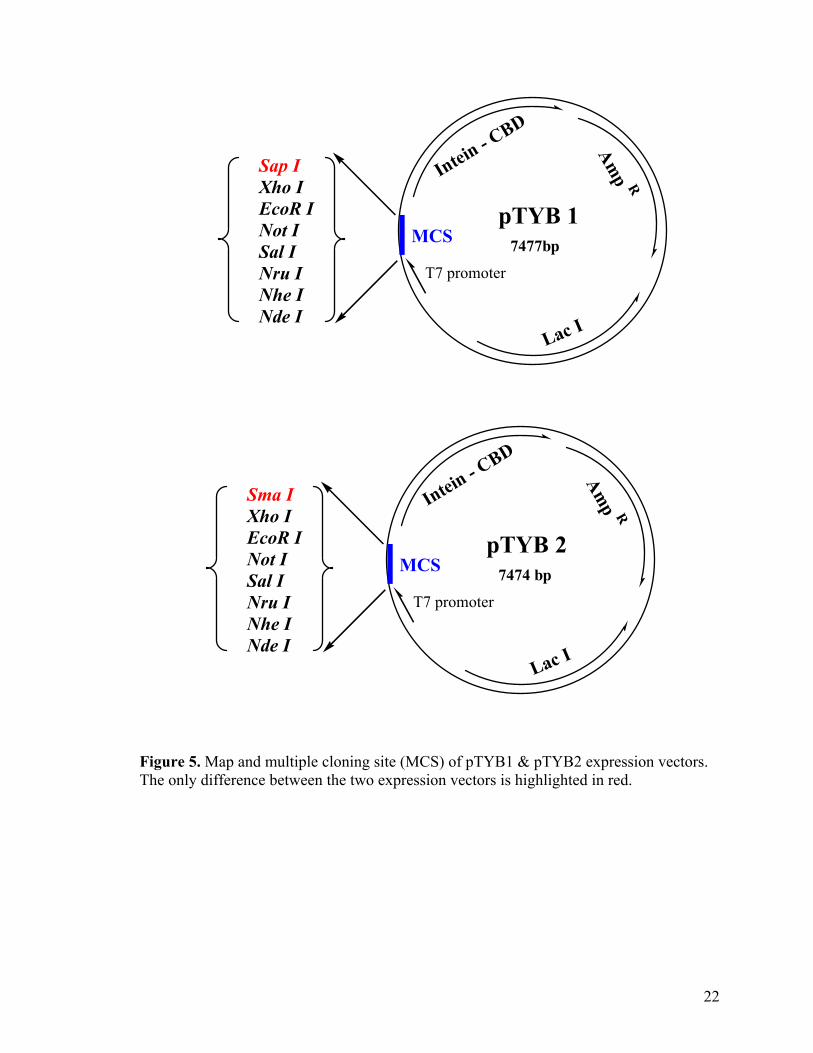
for selection of transformed host strain. Both pTYB1 and pTYB2 contain an NdeI site for
cloning the 5’ end of a target gene. The ATG codon of the NdeI site is used to initiate
translation of the fusion protein The only difference between the two vector lies within the
3’ end restriction site, just before the start of the intein gene. pTYB1 and pTYB2 contains
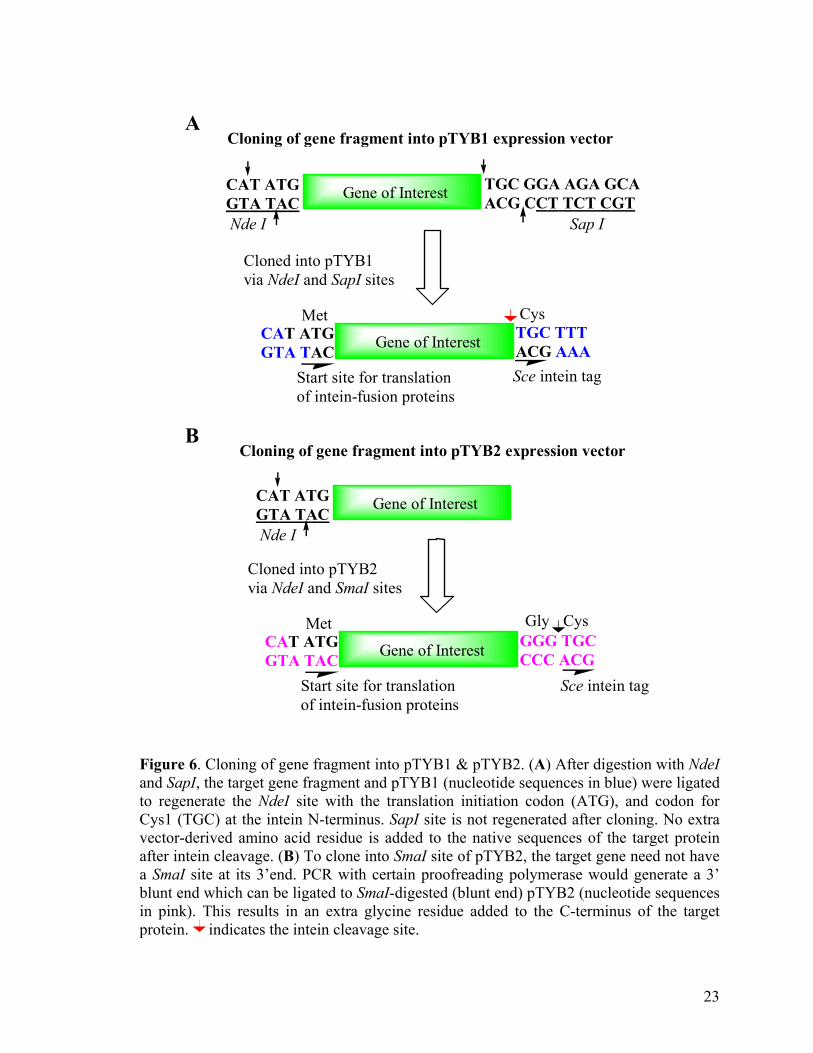
SapI and SmaI sites at their 3’ ends, respectively (Figure 5). The use of SapI site in
pTYB1 allows the C-terminus of the target protein to be fused directly next to the intein
cleavage site, whilst the use of SmaI site in pTYB2 adds an extra glycine residue to the C-
terminus of the target proteins (Figure 6A & B).
21
MCS
T7 promoter
Intein - CBD
Amp R
Lac I
Xho IEcoR INot ISal INru INhe INde I
pTYB 1 7477bp
Sap I
Sma I
MCS
T7 promoter
Intein - CBD
Amp R
Lac I
Xho IEcoR INot ISal INru INhe INde I
pTYB 2 7474 bp
Figure 5. Map and multiple cloning site (MCS) of pTYB1 & pTYB2 expression vectors. The only difference between the two expression vectors is highlighted in red.
22
A
B Figure and Sapto regenCys1 (Tvector-dafter inta SmaI blunt enin pink)protein.
CAT ATGGTA TAC
Sap INde I
TGC GGA AGA GCAACG CCT TCT CGTGene of Interest
Cloned into pTYB1via NdeI and SapI sites
Gene of Interest TGC TTTACG AAA
CAT ATGGTA TAC
CysMet
Start site for translationof intein-fusion proteins
Sce intein tag
Cloning of gene fragment into pTYB1 expression vector
CAT ATGGTA TACNde I
Gene of Interest
Cloned into pTYB2via NdeI and SmaI sites
Gene of Interest GGG TGCCCC ACG
CAT ATGGTA TAC
CysMet
Start site for translationof intein-fusion proteins
Sce intein tag
Cloning of gene fragment into pTYB2 expression vector
Gly
6. Cloning of gene fragment into pTYB1 & pTYB2. (A) After digestion with NdeI I, the target gene fragment and pTYB1 (nucleotide sequences in blue) were ligated erate the NdeI site with the translation initiation codon (ATG), and codon for GC) at the intein N-terminus. SapI site is not regenerated after cloning. No extra erived amino acid residue is added to the native sequences of the target protein ein cleavage. (B) To clone into SmaI site of pTYB2, the target gene need not have site at its 3’end. PCR with certain proofreading polymerase would generate a 3’ d which can be ligated to SmaI-digested (blunt end) pTYB2 (nucleotide sequences . This results in an extra glycine residue added to the C-terminus of the target indicates the intein cleavage site.
23

3.2. Intein-Mediated Biotinylation of three model proteins
3.2.1. Cloning of target genes into pTYB1 expression vector
In the proof-of-concept experiment, the gene fragments of three model proteins,
namely MBP, EGFP and GST were cloned into pTYB1 to generate thioester-tagged
proteins for biotinylation. Restriction enzyme digestion of PCR product is often less
efficient than releasing a fragment from a vector and may result in lower cloning
efficiency. Consequently, the PCR fragment of target gene was first cloned into a T-vector
to facilitate the cloning process.74 To add a single deoxyadenosine to the 3’ end of PCR
product, the target gene sequence had to be PCR-amplified using HotStar Taq polymerase
with proofreading ability. The PCR product was then ligated to the corresponding T-
vector containing 3’ deoxythymidine overhangs. The choice of restriction sites in the
primers determines the extra amino acids residues that may be attached to the target
protein after intein cleavage. Therefore to obtain target protein with no extra vector-
dervied residues, we decided to clone the target gene between the NdeI and SapI sites in
pTYB1 (Figure 6A). NdeI and SapI restriction sites, absent in the target gene, are
incorporated into the forward and reverse primers, respectively. The TA clone containing
the PCR fragment was double digested with NdeI and SapI and the target gene fragment,
isolated by agarose gel electrophoresis, was then ligated to the NdeI/SapI digested pTYB1.
Clones containing the target gene insert were identified by restriction digestion and colony
PCR. The final expression plasmid, verified by DNA sequencing, was transferred to
ER2566 for protein expression. This E.coli stain carries a chromosomal copy of the T7
RNA polymerase gene, under the control of the lac promoter. In the presence of IPTG,
expression of T7 RNA polymerase is activated which in turn initiate the transcription of
the fusion gene.
24

3.2.2. Expression and extraction of fusion proteins
Expression level of fusion protein from the pTYB vector is greatly influence by: 1)
bacterial cell line, 2) nature of the fusion protein and 3) induction condition (temperature,
duration and IPTG concentration). ER2566 is the E.coli strain supplied by NEB for
expression of fusion protein from a pTYB vector but other commercially available strain
(e.g. BL21) may be tested for optimal expression level of the fusion protein. Different
induction conditions (e.g. 30°C for 3hrs, 20-25°C for 6-16hrs or 12-16°C for overnight)
were tested out for the 3 fusion proteins (MBP-intein-CBD, EGFP-intein-CBD, GST-
intein-CBD) to optimize expression of soluble fusion protein and minimize proteolysis.
After expression, the bacterial cells were harvested and lysed in simple lysis buffer,
containing Tris-HCL & NaCl & EDTA, using glass beads. Beside glass beads, the E.coli
cells can also be broken either by sonication or french press or freeze-thawing method.
The type of mechanical lysis method used greatly depends on the amount of lysis buffer
resuspending the induced bacterial cells. Sonication and french press method is more
efficient but requires large volume of suspension for lysis. Lysozyme is not the preferred
cell lysis method for extracting protein fused to an intein tag, since it is known to bind and
digest chitin beads. However, if no alternative method is available, low level of lysozyme
can still be used (incubate at 4 ºC for 1hr) for cell lysis.
25

3.2.3. Affinity purification and on-column biotinylation
The intein-fused proteins were purified and biotinylated, in a single step, by first
loading the clarified cell lysate onto a column pe-packed with chitin beads, then flushing
the column with MESNA and cysteine-biotin, to obtain the C-terminally biotinylated
proteins. The affinity purification process was monitored by SDS-PAGE with coomassie
and silver stain. Figure 7 shows the SDS-PAGE result of MBP purification through the
chitin column. The full-length fusion precursor (97 kDa) and a small amount of the
cleaved intein tag (55 kDa) were found to bind to the chitin resin after the lysate was
passed through the column (Figure 7, lane 6). MBP was co-eluted with minute amount of
contaminating proteins after the thiol-induced cleavage and its purity was estimated to be
about 95 % (Figure 7, lane 8 - 9). The high affinity of CBD for the chitin beads has
allowed a better recovery of the fusion protein from the crude extract and the use of
stringent wash conditions (e.g. high salt concentration and detergent) to reduce non-
specific binding while increasing purity of the eluted MBP. About 2 mg of MBP was
yielded from a 200 ml of bacteria cell culture. Less than 5 % of the intact fusion protein
was found to remain bound on the chitin beads after the cleavage indicating the
effectiveness of on-column cleavage with MESNA (Figure 7, lane 10). Biotinylation of
the eluted MBP was unambiguously confirmed by western blotting as shown in Figure 8A.
Immunoblot result indicates specific biotin-tagging of the affinity purified MBP in the
presence of cysteine-biotin. No biotinylation was observed for MBP eluted in the absence
cysteine-biotin derivatives. Streptavidin adsorption experiment was used to determine the
on-column biotinylation efficiency with respect to the total amount of MBP eluted (Figure
8B). More than 95 % of the eluted MBP were adsorbed to the streptavidin matrix,
suggesting most eluted proteins were biotinylated following cysteine-biotin/MESNA
26

treatment. The on-column biotin-tagging process is highly efficiency (> 95% efficiency),
hence, we equate cysteine-biotin/MESNA-induced cleavage efficiency of a target protein
to its biotinylation efficiency for subsequent experiment. Among the 3 fusion proteins,
only a small amount of the intact GST(Asp719) -intein-CBD fusion protein was detected on
the chitin beads after cell extraction. Most of the fusion proteins were cleaved prematurely
within the bacterial cells leaving mostly the intein-CBD tag to bind onto the affinity beads
during the purification process. We eventually found out that the high in vivo cleavage of
GST(Asp719) -intein-CBD was mainly due to the C-terminal aspartic acid (Asp) of GST,
which will be further explained in the later section of this report. To minimize pre-mature
cleavage of GST-intein-CBD inside the bacterial cells, we mutate the C-terminal Asp
residue of GST to glycine (Gly) by PCR-based site-directed mutagenesis. The mutant
pTYB1-GST(Gly719)-intein construct was then transformed back to ER2566 for protein
expression. SDS-PAGE gel of the purification process showed higher amount of
GST(Gly716)-intein-CBD binding to the chitin beads, resulting in higher yield of the
biotinylated GST (data not shown). Lastly, to generate corresponding protein array,
eluted protein fractions were spotted directly onto an avidin-functionalized slide, without
further downstream processing.
27
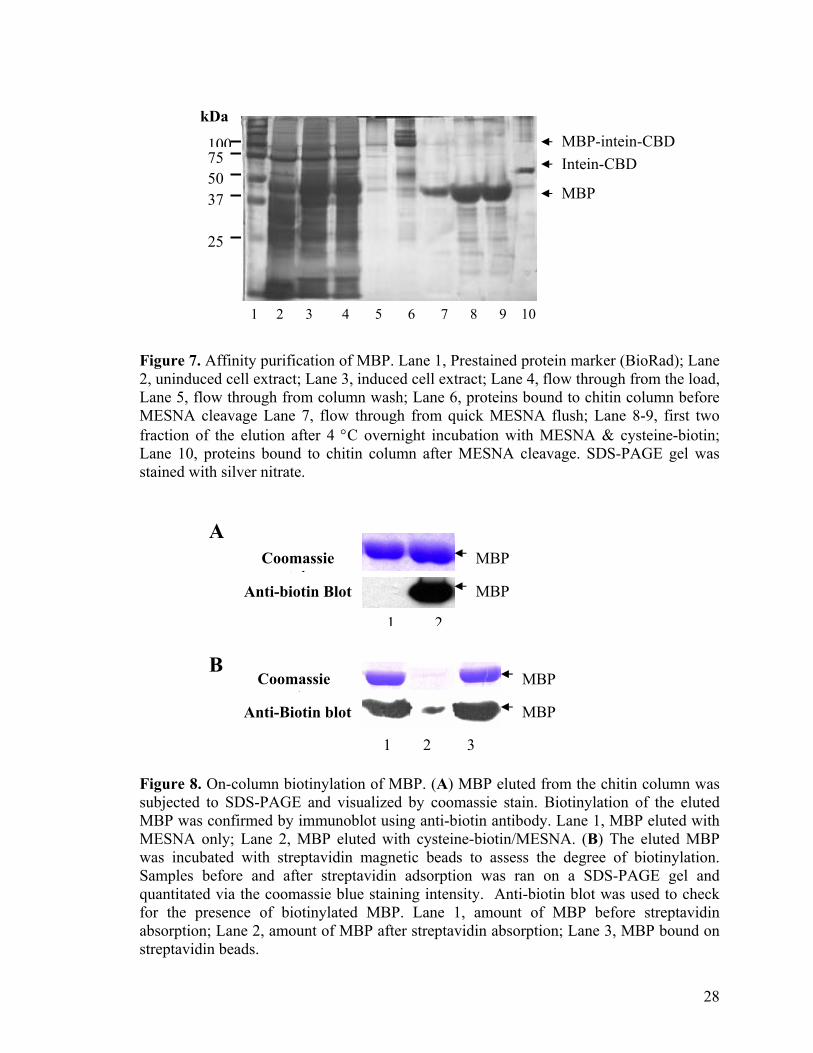
kDa
MBP
Intein-CBD MBP-intein-CBD
25
37 50 75 100
1 2 3 4 5 6 7 8 9 10
Figure 7. Affinity purification of MBP. Lane 1, Prestained protein marker (BioRad); Lane 2, uninduced cell extract; Lane 3, induced cell extract; Lane 4, flow through from the load, Lane 5, flow through from column wash; Lane 6, proteins bound to chitin column before MESNA cleavage Lane 7, flow through from quick MESNA flush; Lane 8-9, first two fraction of the elution after 4 °C overnight incubation with MESNA & cysteine-biotin; Lane 10, proteins bound to chitin column after MESNA cleavage. SDS-PAGE gel was stained with silver nitrate. A
MBP
MBP
1 2
Coomassie t i
Anti-biotin Blot
B Coomassie
t iAnti-Biotin blot
MBP
MBP
1 2 3 Figure 8. On-column biotinylation of MBP. (A) MBP eluted from the chitin column was subjected to SDS-PAGE and visualized by coomassie stain. Biotinylation of the eluted MBP was confirmed by immunoblot using anti-biotin antibody. Lane 1, MBP eluted with MESNA only; Lane 2, MBP eluted with cysteine-biotin/MESNA. (B) The eluted MBP was incubated with streptavidin magnetic beads to assess the degree of biotinylation. Samples before and after streptavidin adsorption was ran on a SDS-PAGE gel and quantitated via the coomassie blue staining intensity. Anti-biotin blot was used to check for the presence of biotinylated MBP. Lane 1, amount of MBP before streptavidin absorption; Lane 2, amount of MBP after streptavidin absorption; Lane 3, MBP bound on streptavidin beads.
28

3.3. Protein microarray generation
A protein array was generated with the biotinylated EGFP, MBP and GST, and probed
with Cy3-anti-EGFP, Cy5-anti-MBP and FITC-anti-GST, respectively. Three
corresponding non-biotinylated proteins were also spotted onto the same slide, as controls.
The array, incubated with either individual antibodies, or a mixture of all the three
antibodies, showed specific binding of the fluorescence-labeled monoclonal antibodies to
their corresponding biotinylated proteins (Figure 9A & B), indicating the specificity of the
immobilization. No fluorescence signal was observed with the non-biotinylated control
proteins (data not shown) and this proves that the biotin tag is essential for the
immobilization of proteins onto avidin-functionalized slides.
Figure 9. Site-specific immobilization of biotinylated, functionally active proteins onto avidin slides. (A) EGFP, MBP and GST were individually detected with Cy3-anti-EGFP (green), Cy5-anti-MBP (red) and FITC-anti-GST (blue), respectively; (B) specific detection of all three proteins with a mixture containing all three antibodies.
29

The most critical issue in generating a protein array is to ensure that proteins
maintain their native activity, as it is previously known that proteins tend to denature on
glass surfaces. Native fluorescence of EGFP observed on the slide indicates the proper
folding of the EGFP protein on the glass surface (Figure 10A). No loss of EGFP
fluorescence intensity was observed after prolonged incubation at 4 0C, suggesting that
folding of the protein was properly maintained on the slide. To further confirm the
integrity of the biotinylated proteins immobilized on the glass surface, a slide immobilized
with EGFP, MBP and GST was incubated with Cy3-labeled glutathione, a known natural
ligand of GST (Figure 11). Array scans result showed exclusive binding between GST and
glutathione, further indicating the retention of GST native conformation on the slide
(Figure 10B).
Figure 10. Integrity of biotinylated proteins immobilized on avidin-functionalized glass surface. (A) Fluorescence from the native EGFP; and (B) specific binding between GST and its Cy3-labeled natural ligand, glutathione.
HN
NH
SH
COOH
O
O
COOHH2N
Figure 11. Chemical structure of glutathione, natural ligand of GST
30

Site-specific immobilization of His-tag proteins on slides functionalized with Ni-NTA
has been used by Zhu et al to generate yeast protein array.25 However, the binding
between his-tag proteins and Ni-NTA complex is not very strong, and incompatible with
many commonly used chemical reagents such as DTT, SDS, EDTA and etc.75 The binding
of His-tag proteins are also depleted outside the pH range of 4 to 10, or when the buffer
contains high concentrations of common salts. On the other hand, the binding between
biotin and avidin is one of the strongest known in nature, and is stable under most
stringent conditions.76 Avidin is also extremely stable77, making it an ideal agent for slide
functionalization. To confirm the stability of avidin-biotin linkage for protein microarray
application, avidin slides immobilized with biotinylated GST were first subjected to a
number of harsh washing conditions, and then incubated with FITC-labeled anti-GST to
detect for any loss of GST on the glass surface. No significant loss of fluorescence signal
was observed even after the slide has been treated with 1M acetic acid at pH 3.3, 60 0C
water and 4 M GuHCl for prolonged time (Figure 12), suggesting the robustness of the
protein array generated via biotin-avidin interaction. For comparison, we expressed a GFP
protein fused with a His-tag, and spotted it onto Ni-NTA slides as described.25
Immobilization of the Hs-tag protein on the Ni-NTA was completely removed when this
GFP-containing slide was treated with any of the above harsh conditions. More recent
experiments have indicated that the His-tag/Ni-NTA immobilization does not even sustain
simple aqueous washings.
31

1 M acetic acid solution
pH 3.3
60 ºC water
4 M GuHCl pH 3.3
No treatment
Figure 12. Biotinylated GST on an avidin slide treated with different washing conditions: 1 M acetic acid solution pH 3.3, 60 ºC water, 4 M GuHCl, all for 30 min and control slide with no treatment. Slides were probed with FITC-anti-GST.
Findings described here present a novel strategy for site-specific immobilization of
proteins on glass surfaces. The protein array generated has optimally oriented proteins
with retain native conformation for subsequent biological screenings. The advantage of
avidin/biotin linkage over His-tag/Ni-NTA strategies for protein immobilization is
highlighted by its ability to withstand a variety of chemical conditions, thus making it
compatible with most biological assays. Further improvement may be readily made by
using streptavidin as the immobilization agent on the slide in place of avidin, which is a
glycoprotein and known to have higher nonspecific binding characteristics.78 Figure 13
shown below outline the overall on-column biotinylation strategy and site-specific
immobilization procedures on the protein array.
32
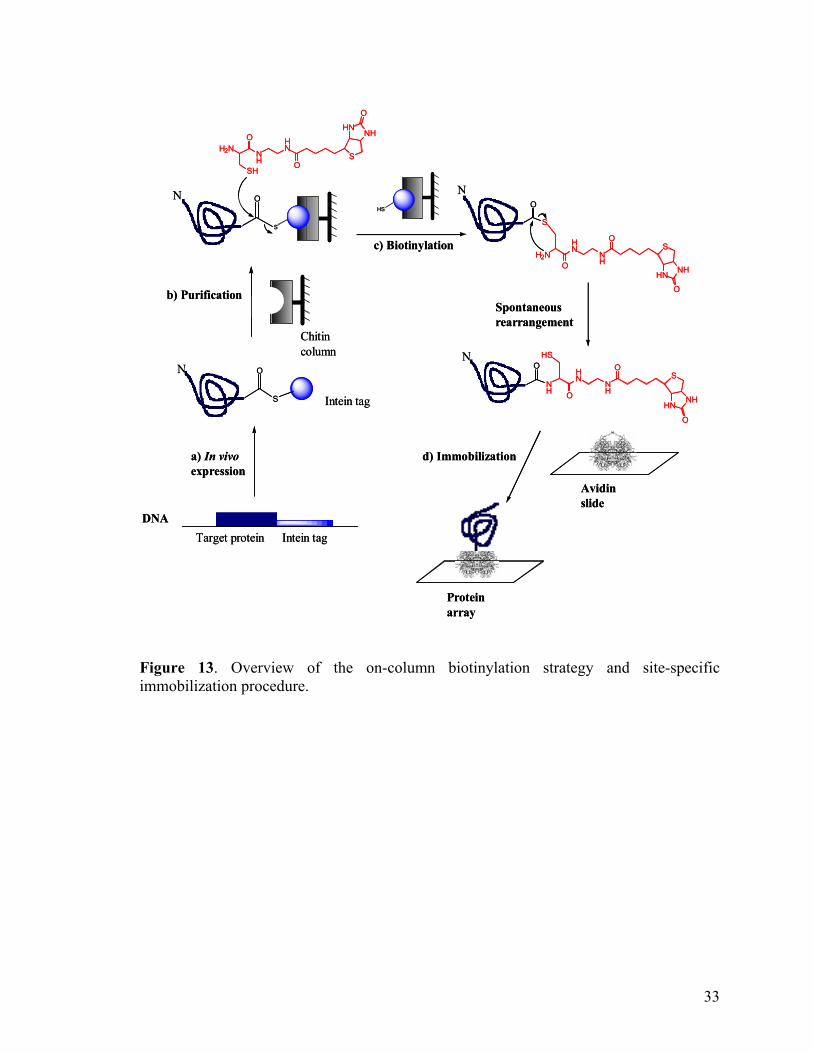
DNADNA
S
HS
S
OO
O
S
O
Target protein Intein tag
N
Chitin column
N
N
a) In vivoexpression
c) Biotinylation
d) Immobilization
Protein array
Avidin slide
Spontaneous rearrangement
b) Purification
N
Intein tag
S
HS
S
OO
O
S
O
Target protein Intein tag
N
Chitin column
N
N
a) In vivoexpression
c) Biotinylation
d) Immobilization
Protein array
Avidin slide
Spontaneous rearrangement
b) Purification
N
Intein tag
H2N NH
HN
SH
O
OS
HNNH
O
H2NHN
NHO
OS
HN NH
O
S
HN
NHO
OS
HNNH
O
NH
HS
H2N NH
HN
SH
O
OS
HNNH
O
H2NHN
NHO
OS
HN NH
O
S
HN
NHO
OS
HNNH
O
NH
HS
Figure 13. Overview of the on-column biotinylation strategy and site-specific immobilization procedure.
33

3.4. Immobilization of biotinylated proteins onto self-assembled monolayers
(SAM) in SPR analysis.
Having demonstrated the feasibility of our intein-mediated strategy for generation of
functional protein array, we would also like to examine its biochemical applications.
Herein, we show that biotin-tagged proteins may also be immobilized onto other surfaces,
such as that of self-assembled monolayers (SAM). The advantage of using SAM on gold-
coated surface is that SPR and mass spectrometry can potentially be integrated as
detection methods to monitor the dynamics of the reactions, or to identify the captured
molecules, respectively. This approach provides the opportunity to study dynamics of
biochemical reactions in a high-throughput fashion, and has great potential in drug and
drug-target discovery and biomedical research.79-81 We used Surface Plasmon Resonance
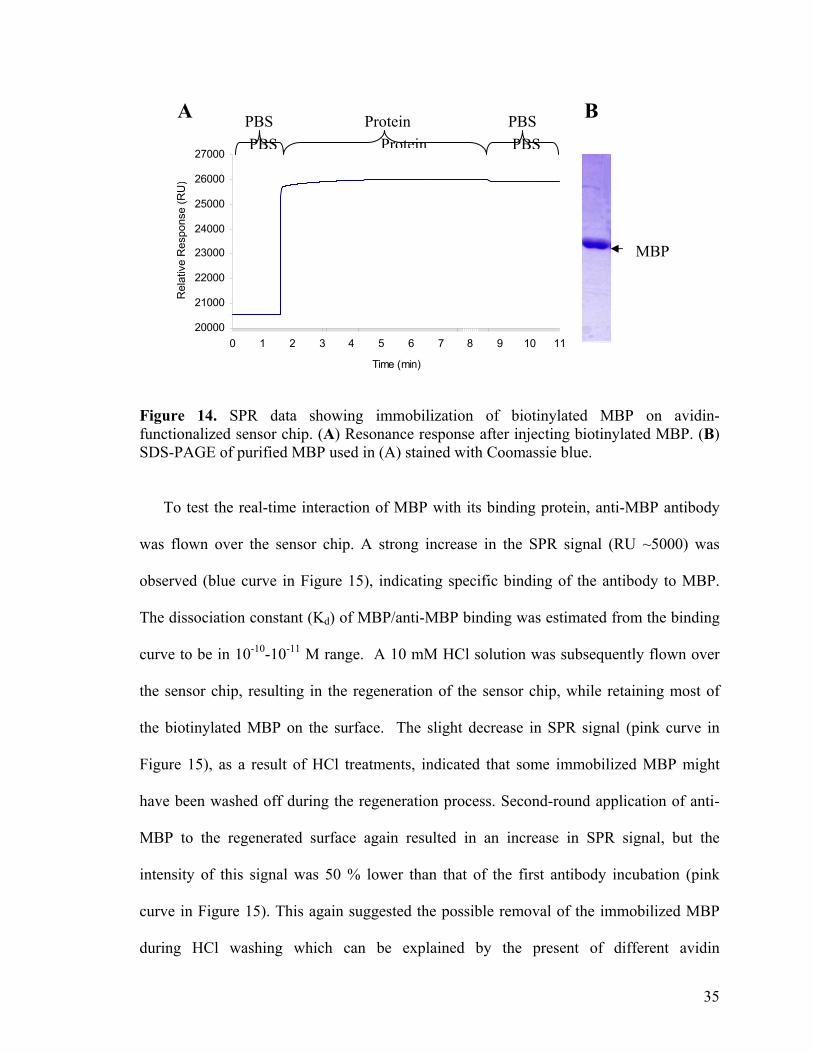
(SPR) spectroscopy to follow the immobilization of the biotinylated protein to an avidin-
functionalized SAM surface. SPR allows direct visualization of protein immobilization,
and its subsequent interaction with other proteins in real time.82 MBP, expressed and
biotinylated as described earlier (Figure 14B), was passed over an avidin-functionalized
sensor chip. Its instantaneous interaction with the sensor chip was evident, as shown by a
rapid increase in the SPR signal (Figure 14A). Subsequent washes with PBS did not
reduce the SPR signal significantly, indicating a stable immobilization of the biotinylated
protein to the avidin surface.
34
20000
21000
22000
23000
24000
25000
26000
27000
0 1 2 3 4 5 6 7 8 9 10 11
Time (min)
Rel
ativ
e R
espo
nse
(RU
)
A PBS Protein PBSPBSProteinPBS
B
MBP
Figure 14. SPR data showing immobilization of biotinylated MBP on avidin-functionalized sensor chip. (A) Resonance response after injecting biotinylated MBP. (B) SDS-PAGE of purified MBP used in (A) stained with Coomassie blue.
To test the real-time interaction of MBP with its binding protein, anti-MBP antibody
was flown over the sensor chip. A strong increase in the SPR signal (RU ~5000) was
observed (blue curve in Figure 15), indicating specific binding of the antibody to MBP.
The dissociation constant (Kd) of MBP/anti-MBP binding was estimated from the binding
curve to be in 10-10-10-11 M range. A 10 mM HCl solution was subsequently flown over
the sensor chip, resulting in the regeneration of the sensor chip, while retaining most of
the biotinylated MBP on the surface. The slight decrease in SPR signal (pink curve in
Figure 15), as a result of HCl treatments, indicated that some immobilized MBP might
have been washed off during the regeneration process. Second-round application of anti-
MBP to the regenerated surface again resulted in an increase in SPR signal, but the
intensity of this signal was 50 % lower than that of the first antibody incubation (pink
curve in Figure 15). This again suggested the possible removal of the immobilized MBP
during HCl washing which can be explained by the present of different avidin
35
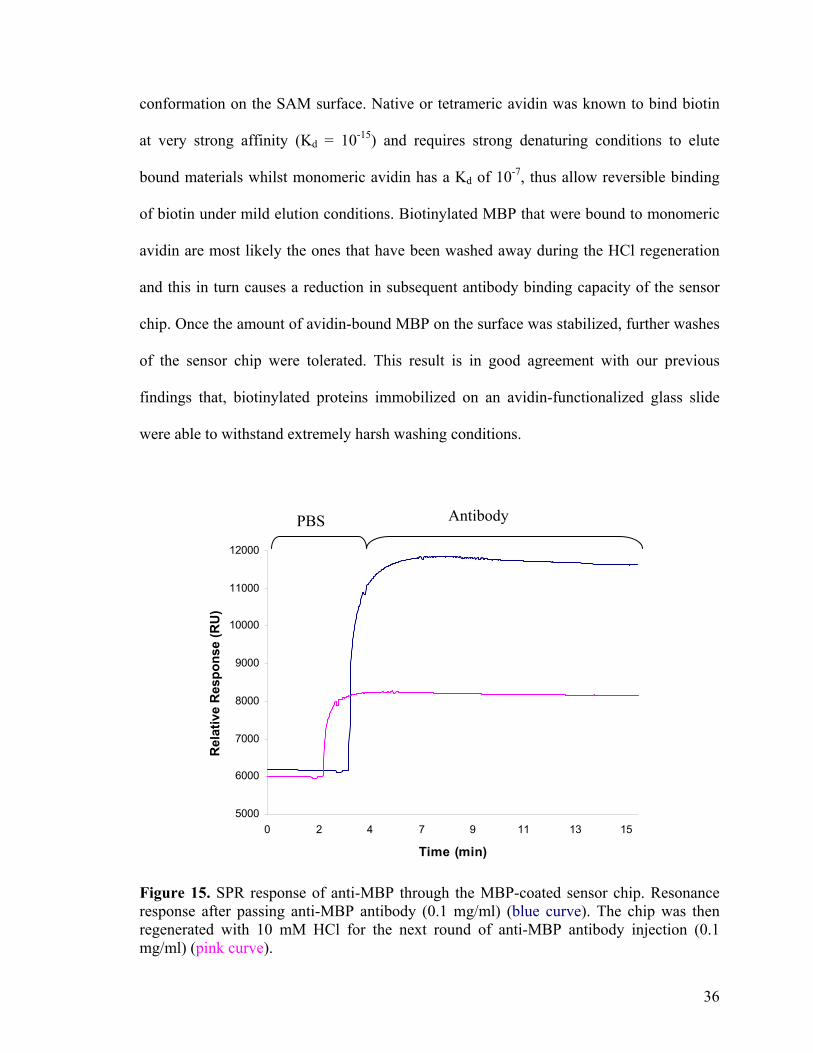
conformation on the SAM surface. Native or tetrameric avidin was known to bind biotin
at very strong affinity (Kd = 10-15) and requires strong denaturing conditions to elute
bound materials whilst monomeric avidin has a Kd of 10-7, thus allow reversible binding
of biotin under mild elution conditions. Biotinylated MBP that were bound to monomeric
avidin are most likely the ones that have been washed away during the HCl regeneration
and this in turn causes a reduction in subsequent antibody binding capacity of the sensor
chip. Once the amount of avidin-bound MBP on the surface was stabilized, further washes
of the sensor chip were tolerated. This result is in good agreement with our previous
findings that, biotinylated proteins immobilized on an avidin-functionalized glass slide
were able to withstand extremely harsh washing conditions.
5000
6000
7000
8000
9000
10000
11000
12000
0 2 4 7 9 11 13 15
Time (min)
Rel
ativ
e R
espo
nse
(RU
)
Antibody PBS
Figure 15. SPR response of anti-MBP through the MBP-coated sensor chip. Resonance response after passing anti-MBP antibody (0.1 mg/ml) (blue curve). The chip was then regenerated with 10 mM HCl for the next round of anti-MBP antibody injection (0.1 mg/ml) (pink curve).
36

3.5. Influence of C-terminal residues on biotinylation.
The final yield of an in vitro biotinylated protein is primarily dependent upon the
amount of the intein fusion recovered from cell extract and its subsequent on-column
cleavage/biotinylation efficiency. From previous reports and our experiences with
GST(Asp719)-intein-CBD, it was known that the C-terminal amino acid residue of the
fused protein at the intein cleavage site has great effect on the cleavage efficiency of
intein.72 To examine how this differential cleavage effect would influence the on-column
biotinylation efficiency of the intein-fused protein, pTYB1-wtEGFP (Lys239)-intein, which
contains EGFP fused to the Sce VMA intein tag via the original C-terminal residue of
EGFP, Lys239, was site-directed mutagenized. Lys239 was mutated to the other 19 amino
acids and the mutant constructs, confirmed by DNA sequencing, were transformed back to
ER2566 for protein expression. The intein-fused proteins were overexpressed in E. coli.
before harvesting. After cell lysis, the fusion proteins were bound to chitin column and
their in vivo cleavage and on-column cleavage efficiency was assessed by SDS-PAGE gel
stained with coomassie blue. The overall results are tabulated in Table 1. Figure 16A
shows the amount of fusion protein cleaved in vivo prior to on-column cleavage for the
different amino acids adjacent to the intein cleavage site. Nearly all the fusion proteins got
cleaved off within the cells when aspartic acid (highlighted red in Figure 16A) is present
next to the intein cleavage site, leaving only the intein tag to bind on the chitin beads.
SDS-PAGE results showed that acidic amino acids (e.g. Asp and Glu) at the C-terminus of
EGFP caused almost complete pre-mature cleavage (~100%) of the fusion protein inside
the bacterial cells, whilst some other residues (e.g. Arg, His and Tyr) caused substantial
amount of in vivo cleavage (> 50%)(column 2 in Table 1). Majority of the C-terminal
residues, however, caused less in vivo cleavage (< 50%), thus allowing sufficient amount
37

of fusion proteins to be obtained prior to subsequent on-column cleavage. By streptavidin
adsorption experiments, it was determined that more than 95% of biotinylated proteins
were consistently obtained in the eluted fractions following cysteine-biotin/MESNA
treatments. Consequently, the amount of on-column protein cleavage was taken to
quantitate the relative biotinylation efficiency for the respective EGFP mutants (column 3
in Table 1). Figure 16B shows the amount of fusion protein cleaved by cysteine-
biotin/MESNA, thus indicating the in vitro cleavage/biotinylation efficiency for the
respective amino acid residues (assumed 100% biotinylation of all cleaved product). Most
of the column-bound fusion proteins were not cleaved when cysteine (highlighted blue in
Figure 16B) was place adjacent to the intein cleavage site, suggesting the ineffectiveness
of cysteine-biotin/MESNA-induced cleavage on the mutant. Most amino acids substituted
at the cleavage site retained relatively high degrees of protein biotinylation (> 50%),
except for some residues (e.g. Asn, Cys, Ile & Val) that generate less amounts of the
biotinylated protein (< 25%) as shown in Figure 16C.
38
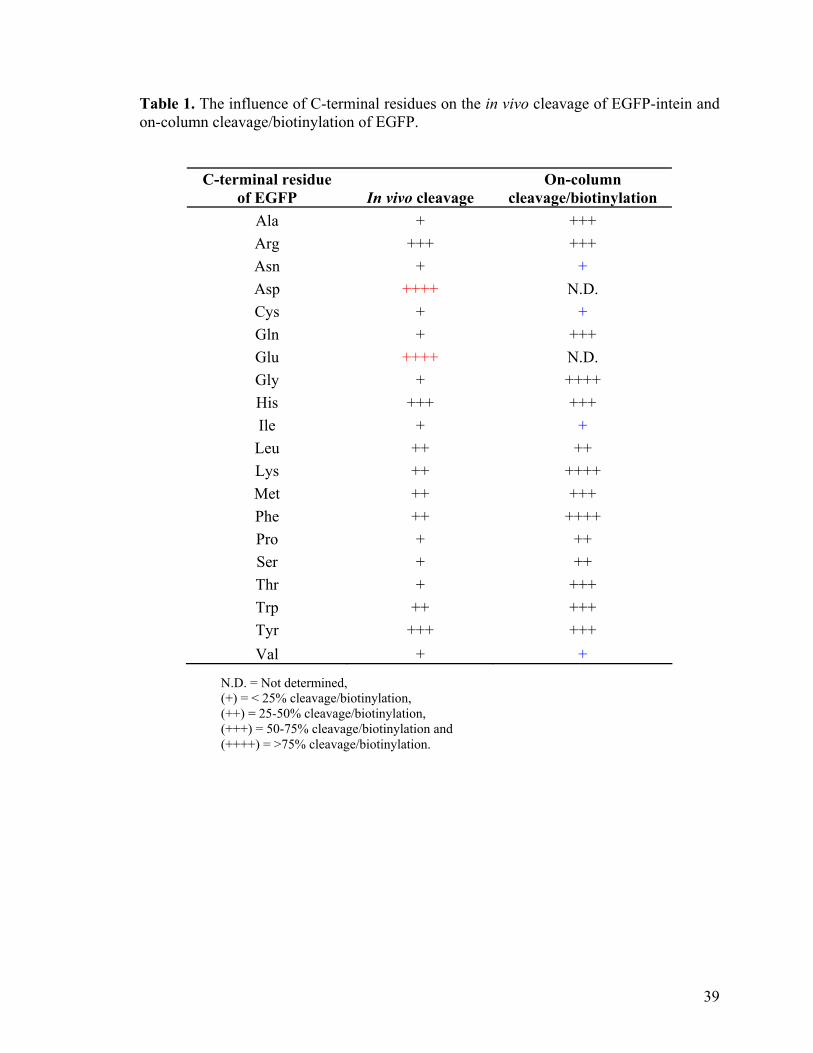
Table 1. The influence of C-terminal residues on the in vivo cleavage of EGFP-intein and on-column cleavage/biotinylation of EGFP.
C-terminal residue of EGFP In vivo cleavage
On-column cleavage/biotinylation
Ala + +++ Arg +++ +++ Asn + + Asp ++++ N.D. Cys + + Gln + +++ Glu ++++ N.D. Gly + ++++ His +++ +++ Ile + + Leu ++ ++ Lys ++ ++++ Met ++ +++ Phe ++ ++++ Pro + ++ Ser + ++ Thr + +++ Trp ++ +++ Tyr +++ +++ Val + +
N.D. = Not determined, (+) = < 25% cleavage/biotinylation, (++) = 25-50% cleavage/biotinylation, (+++) = 50-75% cleavage/biotinylation and (++++) = >75% cleavage/biotinylation.
39

A
55 81
81 55
Thr Lys Ile Gly Gln Cys Asp Ala MW
kDa
EGFP 27
kDa
kDa
Intein-CBD
EGFP-intein-CBD
Intein-CBD
EGFP-intein-CBD
B
C
Figure 16. Influence of the C-terminal amino acid residue. (A) Proteins bound on chitin beads before cysteine-biotin/MESNA cleavage. Data are summarized in Column 2 of Table 1. (B) Proteins remaining on the chitin beads after on-column cysteine-biotin/MESNA cleavage. Data are summarized in Column 3 of Table 1. (C) Eluted EGFP. The yield of EGFP from the on-column cleavage is shown here. All SDS-PAGE gels shown here are stained with coomassie blue.
In order for our strategy to be generally applicable for biotinylation of most
proteins in high-throughput proteomics applications, the “C-terminal effect” on the
cleavage of the fusion protein needs to be minimized. Based on above mutagenesis
experiments with the EGFP-intein fusion (Table 1), having a Gly residue at the cleavage
site seem to minimize the pre-mature cleavage of the fusion in the bacterial cells, and at
the same time maximizing the subsequent on-column cleavage/biotinylation efficiency.
We therefore reasoned that, introduction of one or two extra Gly residues at the C
terminus of a protein having undesired residues (e.g. Asp & Glu) may optimize the on-
column biotin-tagging process, and at the same time have negligible effect on the protein
function. To prove this hypothesis, we cloned two EGFP mutants (i.e. EGFP(Asp239) and
EGFP(Cys239)), containing C-terminal Asp and Cys, respectively, into the pTYB2 vector.
40

The resulting constructs, i.e. pTYB2-EGFP(Asp239)-intein and pTYB2-EGFP(Cys239)-
intein, were the same as their pTYB1 counterparts except the addition of an extra Gly at
the C-terminus of the EGFP mutants. Although, protein expression from the two pTYB2
constructs (Figure 17) revealed that the addition of an extra Gly only substantially lowered
the in vivo cleavage of the fusion proteins (70% for pTYB-2 construct vs ~100% for
pTYB-1 construct of EGFP(Asp239)-intein mutant), but significant improvement on the
biotinylation efficiency of the proteins (i.e. up ~ 80% for pTYB-2 construct vs 0% pTYB-
1 construct of EGFP(Cys239)-intein mutant) was observed, thereby validating our
hypothesis. Consequently, extra Gly residues were introduced in all of our subsequent
experiments (vide infra).
Asp Cys
kDa
27
5581
EGFP-intein Intein-CBD EGFP
B A E B A E Figure 17. Effect of an extra glycine residue on intein-mediated biotinylation. Fusion proteins, EGFP(Asp239)-intein and EGFP(Cys239)-intein, were expressed, extracted and incubated with chitin beads. After washing, bound proteins were incubated with MESNA and cysteine-biotin. B: Proteins bound on chitin beads before cysteine-biotin/MESNA elution, A: proteins remaining on chitin beads after cysteine-biotin/MESNA elution, E: and eluted EGFP. Coomassie blue staining of the SDS gel are presented.
41

In summary, our cleavage studies with EGFP mutants showed that amino acid residues
adjacent to the intein cleavage site have adverse effects on the cleavage of the fusion
proteins. Some amino acids (e.g. Asp) cause pre-mature cleavage of the fusion protein,
whilst others (e.g. Cys) reduce cleavage efficiency of fusion protein by the thiol
compound. Biotinylation efficiency of the target protein is greatly reduced in both cases,
thus it is crucial to ensure appropriate amino acids residues are present at the C-terminus
of the target proteins for intein-mediated biotinylation. Cloning of a target gene into
pTYB2 results in the addition of an extra Gly residue next to the intein cleavage site,
which may be necessary to either prevent in vivo cleavage or improve cleavage efficiency
of target proteins for cases whereby unfavorable amino acid residues are present adjacent
to the cleavage site. Alternatively, one can also ensure the presence of a suitable C-
terminal amino acid residue next to the intein cleavage site with appropriate design of the
3’ end primers and the choice of restriction sites, used for cloning target genes into the
pTYB expression vectors.
42

3.6. High-throughput expression and biotinylation of yeast proteins.
3.6.1. Cloning of yeast gene into pTYB1 expression vector.
To validate our in vitro biotinylation strategy for potential high-throughput protein
expression, we cloned ~100 different yeast proteins in the form of intein fusions. Yeast
proteins were chosen in our studies as their DNA sources are readily available from the
Yeast ExClonesTM.83 cDNA plasmids, containing the yeast gene, were extracted from
yeast exclones before transforming them into TOP10 E.coli. host. Plasmid DNA, extracted
from the transformed E.coli. cells in 96-well format, was used as the template for
subsequent PCR amplification reaction. All the yeast gene fragments within the cDNA
plasmid were flanked by two consensus DNA sequences at their 5’ and 3’ end. For ease
of PCR amplification, we designed a common upstream primer, which recognize the 3’
end consensus sequences on the yeast cDNA, and 100 gene specific downstream primers
to amplify the respective yeast genes from the cDNA plasmids. Individual downstream
primers were used to avoid the stop codon present on the yeast cDNA plasmid. To ensure
maximal yield of the biotinylated yeast proteins, two extra Gly residues were conveniently
introduced at the C-terminus of each yeast protein with the addition of 6 nucleotides
sequences (ACC ACC) into the coding region of the downstream primers. The amplified
PCR products (Figure 18) were then cloned into T-vectors to obtain TA clones of the
yeast gene fragment. The TA plamsids were double digested with NdeI and SapI
restriction enzymes and separated on DNA gels (Figure 19). Gene fragments of the correct
size were isolated and ligated to NdeI/SapI digested pTYB1 to obtain the final intein-fused
construct. Figure 20 illustrate the overall cloning of yeast gene fragment into pTYB1
expression vector.
43
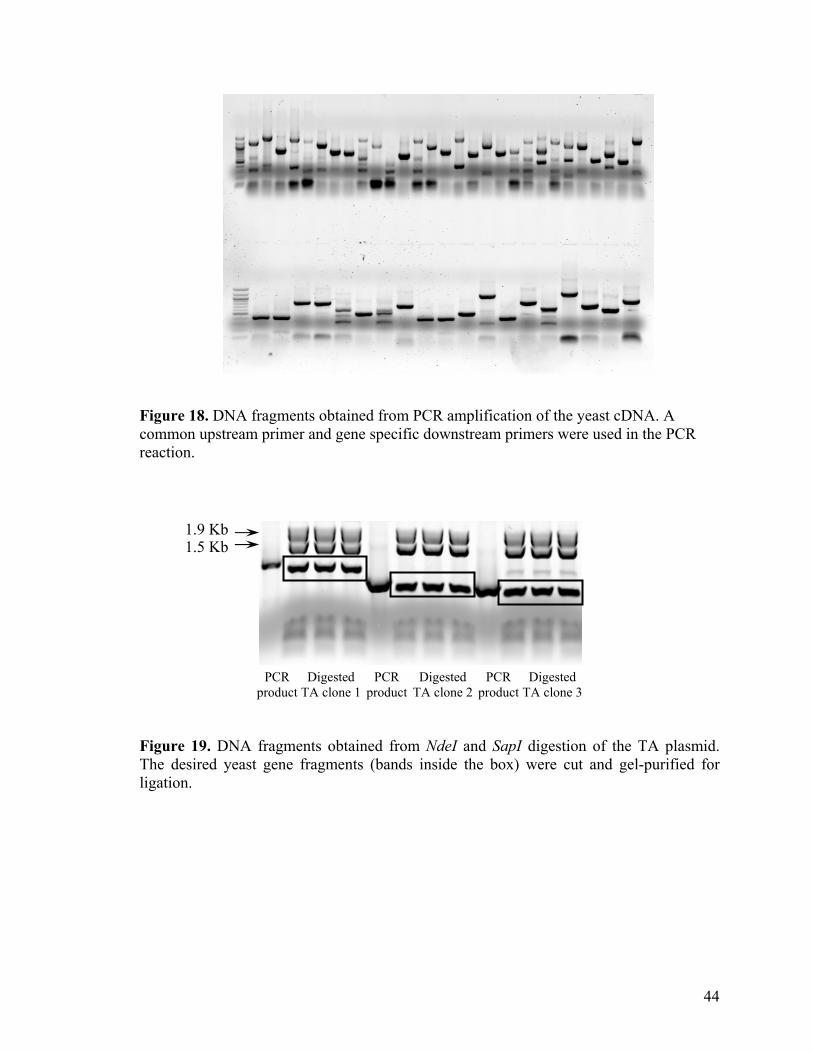
Figure 18. DNA fragments obtained from PCR amplification of the yeast cDNA. A common upstream primer and gene specific downstream primers were used in the PCR reaction. 1.9 Kb
1.5 Kb PCR
product Digested
TA clone 1 PCR
productDigested
TA clone 2 PCR
product Digested
TA clone 3 Figure 19. DNA fragments obtained from NdeI and SapI digestion of the TA plasmid. The desired yeast gene fragments (bands inside the box) were cut and gel-purified for ligation.
44

GGC GGC CAT ATGCCG CCG GTA TAC
Sap INde I
GGT GGT TGC GGA AGA GCA GCC GCCCCA CCA ACG CCT T CT CGT CGG CGG
Gene of Interest
Gene of Interest
CCA CCA ACG CCT TCT CGT CGG CGG
GGC GGC CAT ATGNde I
Sap I
Gly Gly
3'
3'5'
5'3'
3'
5'
5'
PCR amplification from yeast cDNA
Gene of Interest GGT GGT TGC TTTCCA CCA ACG AAA
CAT ATGGTA TAC
CysMet
Sce intein tag
Gene fragment cloned into pTYB1
Consensus SequenceStop codon
Figure 20. Cloning of yeast gene fragment into pTYB1 (nucleotide sequences in blue). indicates the intein cleavage site. Gly-Gly respresents the extra glycine residues added at the C-terminus of each yeast proteins.
3.6.2. Expression, purification & biotinylation of yeast proteins.
All expression clones of intein-fused yeast proteins were induced at room temperature
overnight. Overexpression of eukaryotic fusion protein in E.coli. often results in the
formation of inclusion bodies therefore 1 % CHAP were added in the lysis buffer to
improve the folding and overall solubility of the yeast fusion proteins. Protease inhibitor
(1 mM PMSF) and reducing agent (1 mM TCEP) were also added to ensure stability of
the fusion protein during extraction. TCEP [tris-(2-carboxyethyl)phosphine) substitutes
the use of thiol reagents such as β-mercaptoethanol and 1,4-dithiothreitol (DTT), which
would cause premature cleavage of the fusion protein resulting in the lost of target protein
45

prior to affinity purification. Clarified cell lysates were loaded onto chitin columns and
after several washes with column buffer, the yeast proteins were simply eluted and
biotinylated with MESNA/cysteine-biotin.
We found that, the cloning/protein expression/biotinylation could be readily adopted
in 96-well formats, enabling high-throughput generation of potentially large numbers of
different proteins. Roughly half of the clones (~50) were further expressed, and 31 of
which were successfully biotinylated (Figure 21). The remaining ones (~20) failed to
express as soluble proteins in E. coli., thus were not pursued further. Despite the
introduction of 2 extra Gly residues at the C-termini of some yeast proteins, a substantial
amount (70%) of in vivo cleavage was still observed in the cell lysate (Figure 22). This
indicates that the addition of Gly-Gly does not satisfactorily solve the in vivo cleavage
problem, thus other approaches must be sought to allow true high-throughput biotinylation
via this strategy. Fortunately, we were able to isolate sufficient amounts of the intact
fusion for most of our yeast proteins. In most cases, subsequent on-column
cleavage/biotinylation steps typically eluted the desired biotinylated proteins as the
predominant products with acceptable yields (Figures 21 & 22). Variable degrees of
protein biotinylation were observed for the different yeast proteins (Figure 21), which
might have been caused by a number of different factors, including differences in the
expression level of the fusion protein, the extent of in vivo self-cleavage and different
degrees of on-column cleavage/biotinylation, etc. Of the 31 biotinylated yeast proteins,
many are yeast enzymes, covering a wide rage of biological activities (i.e. 4 kinases, 4
dehydrogenases, 4 phosphatases, 2 transferases, 2 lyases, 1 protease, 14 others) and
46
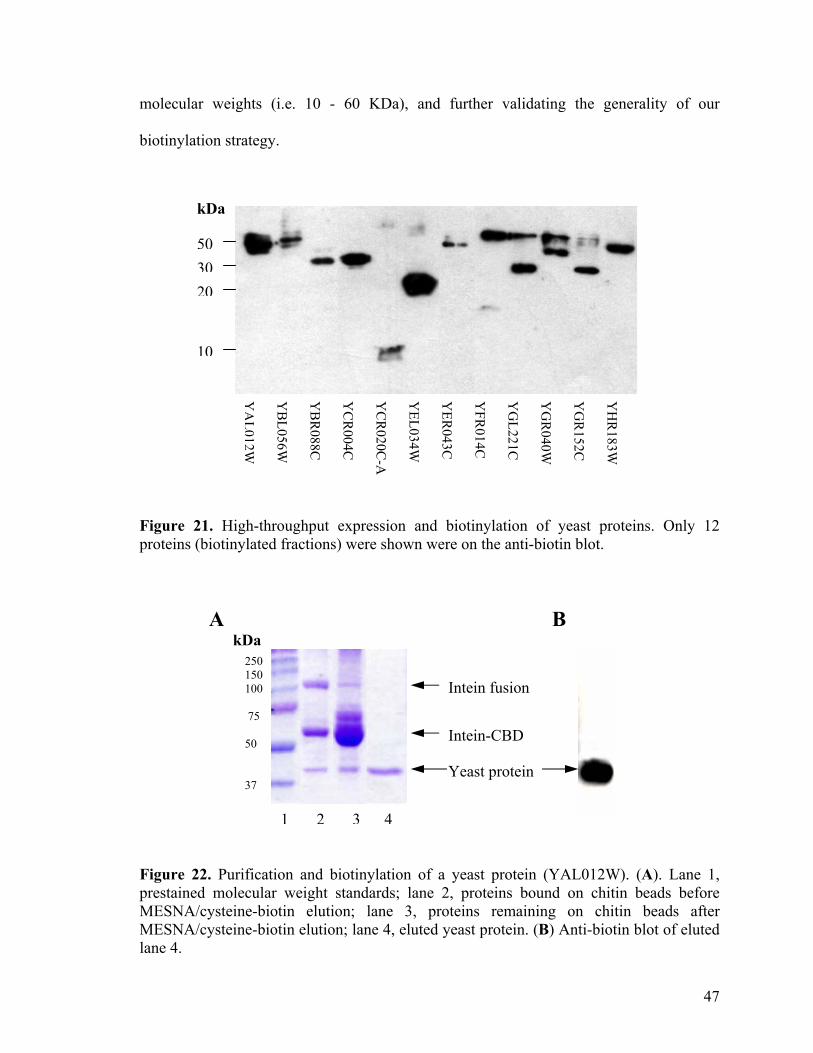
molecular weights (i.e. 10 - 60 KDa), and further validating the generality of our
biotinylation strategy.
kDa
50 30
20
10
YH
R183W
Y
GR
152C
YG
R040W
Y
GL221C
Y
FR014C
Y
ER043C
Y
EL034W
YC
R020C
-A Y
CR
004C
YB
R088C
Y
BL056W
Y
AL012W
Figure 21. High-throughput expression and biotinylation of yeast proteins. Only 12 proteins (biotinylated fractions) were shown were on the anti-biotin blot.
A B
kDa
Yeast protein
Intein-CBD
Intein fusion
1 2 3 4
250 150 100 75 50 37
Figure 22. Purification and biotinylation of a yeast protein (YAL012W). (A). Lane 1, prestained molecular weight standards; lane 2, proteins bound on chitin beads before MESNA/cysteine-biotin elution; lane 3, proteins remaining on chitin beads after MESNA/cysteine-biotin elution; lane 4, eluted yeast protein. (B) Anti-biotin blot of eluted lane 4.
47

3.7. In vivo biotinylation of proteins.
We next extended our intein-mediated biotinylation strategy to living cells. Although
intein-mediated protein splicing is part of the naturally occurring processes in cells, its
utilities in protein engineering have thus far been limited to in vitro applications.72 The
only exceptions are in the engineering of circulated proteins, where head-to-tail native
chemical ligation occurred intramolecularly within live cells.68-69 A recent report by Giriat
et al. indicated that intein-mediated protein semi-synthesis was also possible in live cells
between two designer protein fragments.84 We reasoned that, if our cysteine-biotin tag is
sufficiently cell-permeable, it may be able to cross the membrane of cells overexpressing
the desired protein-intein fusion, cleave the fusion and at the same time biotinylate the
target protein (method A in Figure 4).
3.7.1. In bacterial cells
To validate this, we first tested the in vivo biotinylation of MBP in bacterial cells
(ER2566) carrying the pMYB5 construct. It was found that, following IPTG induction to
overexpress MBP-intein-CBD in the growing bacterial cells, the addition of cysteine-
biotin/MESNA to the growth media followed by further incubation of the cells resulted in
substantial biotinylation of the MBP protein. Modifications of the cell growth, as well as
the in vivo biotinylation conditions, further increased the level of protein biotinylation in
the bacterial cells (Figure 23A & 23B). The optimal concentration of MESNA and
cysteine-biotin required for the in vivo tagging process was determined to be about 5 – 30
mM and 5 – 6 mM, respectively. Biotinylation was observed in the target protein ONLY if
both cysteine-biotin and MESNA were concomitantly added to the cell media (lane 1 & 2
in Figure 23A & 23B). Upon treatments with cysteine-biotin/MESNA, more than 40% of
48

the fusion protein was shown to be cleaved (lane 4 in Figure 24A). No significant increase
in the cleavage of MBP-intein-CBD was observed for the bacterial culture incubated with
MESNA or cysteine-biotin alone (lane 2 & 3 in Figure 24A). There is an estimated 20 -
40% decrease in the amount of MBP after streptavidin adsorption (Figure 24B), indicating
that the majority of MBP present within the cells were non-biotinylated. Taken together,
it was estimated that the overall biotinylation efficiency in E. coli. was between 20 - 40%
of all overexpressed MBP. We also demonstrated that proteins from different biological
sources (i.e. MBP and the two yeast proteins shown in Figure 25A & 25B, respectively)
could be efficiently biotinylated in live bacterial cells. The purity of the in vivo
biotinylated proteins was confirmed by first incubating crude cell lysates with
paramagnetic streptavidin beads, then analyzing the bead-bound proteins by SDS-PAGE
and western blots. In all cases, the desired biotinylated protein could be isolated with high
purity. The only impurity detected was acetyl-CoA carboxylase - an endogenous
biotinylated protein known in E. coli.85 (Figure 25A and 25B).
49

50
+
1030 10510.5-- MESNA (mM)
++ +++++- 3 mM Cys-biotin
B 5 mM MESNA - + + + + + + +
Cys-biotin (mM) - - 0.5 1 2 3 4 6
+
5
A
MBP
MBP
Figure 23. Optimizing in vivo biotinylation conditions in bacterial cells. Induced bacterial cells incubated with different concentration of MESNA and cysteine-biotin were lysed and subjected to SDS-PAGE. Biotinylation efficiency was analysis from the anti-biotin blot of the cell lysate. (A) Increasing MESNA concentration. (B) Increasing cysteine-biotin concentration.
A B
Anti-biotin blotAnti-biotin blot
42 MBP 42
97kDa
42
97 kDa
42 MBP
MBP-intein
Anti-MBP blot Anti-MBP blot
MBP-intein
MBP
MBP
1 2 3 4 1 2 Figure 24. In vivo biotinylation efficiency in bacterial cells. Bacterial cells expressing MBP-intein were incubated with cysteine-biotin/MESNA. (A) The clarified cells lysate was then analyzed by SDS-PAGE followed by anti-MBP and anti-biotin blot. Lane 1: Lysate of IPTG induced bacterial culture; lane 2: lyaste of bacterial culture incubated with cysteine-biotin only; lane 3: lysate of bacterial culture incubated with MESNA only; lane 4: lysate of bacterial culture incubated with cysteine-biotin/MESNA. (B) The cell lysate was also incubated with streptavidin magnetic beads to assess the degree of biotinylation inside the bacterial cells. Cell lyaste before and after streptavidin adsorption was ran on a SDS-PAGE gel and blotted with anti-MBP and anti-biotin. Lane 1, Lysate before streptavidin adsorption; Lane 2, Lysate after streptavidin adsorption.
50

kDa
MBP
A
*
3043
20
kDa B
42
20
YAL012W YGR152C
*
1 2 3 4 1 2
Figure 25. In vivo biotinylation of proteins in bacterial cells shown by anti-biotin blot. (A) MBP was used as the model protein to demonstrate in vivo biotinylation. Lane 1, lysate of uninduced bacterial culture; lane 2, lysate of IPTG induced bacterial culture; lane 3, lysate of bacterial culture incubated with MESNA only; lane 4, lysate of bacterial culture incubated with MESNA & cysteine-biotin. (B) In vivo biotinylation of yeast proteins (lane 1: YAL012W; lane 2: YGR152C). The 20 kDa protein bands (*) in (A) and (B) correspond to acetyl-CoA carboxylase - the only biotinylated protein present in E. coli..
3.7.2. In Mammalian cells
3.7.2.1. Construction of mammalian expression plasmid, pT-Rex-DEST30- EGFP-Sce VMA intein-CBD.
We next tested the biotinylation strategy in mammalian cells. The mammalian
expression vector, pT-Rex-DEST30-EGFP-Sce VMA intein-CBD, was constructed by
homologous recombination using Gateway™ cloning technology. GatewayTM Technology
is a universal cloning technology that provides a high-throughput route to
cloning/subcloning of DNA segments. Based on the well-characterized site-specific
recombination system of phage l, GatewayTM Technology allows the transfer of DNA
segments between different cloning vectors while maintaining orientation and reading
frame, effectively replacing the use of restriction endonucleases and ligase. It is a
51

powerful method for highly efficient, directional cloning of PCR products.86-88 The
Gateway™-adapted T-REx™ destination vectors pT-REx™-DEST30 is designed for
regulated expression from the complete cytomegalovirus (CMV) promoter enhancer. The
CMV enhancer-promoter sequence containing two copies of the tetracycline operator
TetO2 sequence for high-level regulated expression of the target protein. Neomycin
resistance gene is incorporated for easy selection of stable cells. Figure 26 illustrates the
cloning steps taken to obtain our final expression clone, pT-Rex-DEST30-EGFP-Sce
VMA intein-CBD.
3.7.2.2. Expression and in vivo biotinylation of EGFP in HEK 293 cells.
Transient expression of the expression construct in mammalian cells resulted in an
overexpression of green fluorescent proteins, which could be readily followed under an
UV lamp. HEK (Human Embryonic Kidney) 293 cells were used in all our biotinylation
experiment as it was found to be the most suitable cell line for expression of EGFP-Sce
VMA intein-CBD (Figure 27). Addition of cysteine-biotin/MESNA in basal media
containing the transfected cells resulted in appearance of a new biotinylated protein band
(lane 4 in Figure 28, Mw ~ 27 KDa), corresponding to the apparent molecular weight of
biotinylated EGFP. In addition, only three other biotinylated proteins were detected,
which were also present in untreated cells, and they were identified to be the known
naturally biotinylated proteins: pyruvate carboxylase, methylcrotonyl CoA carboxylase
and propionyl CoA carboxylase. As shown in Figure 28 (lane 4), the expression of
naturally biotinylated proteins appeared to be enhanced with the addition cysteine-biotin.
This is probably caused by artifacts in our western blots due to extremely low protein
expression level inherent to transient transfection experiments, although we could not
52

completely rule out the possibility that the expression level of endogenous biotinylated
proteins was enhanced with the addition of cysteine-biotin/MESNA. Further experiments
need to be conducted to verify this observation.
Attempts were also made to quantitate the amounts of uncleaved EGFP-intein fusion,
the self-cleaved, as well as the biotinylated EGFP by western blots using anti-EGFP and
anti-biotin antibodies. There was a significant self-cleavage of EGFP-fusion even before
the cysteine-biotin/MESNA treatments as shown in lane 2 of figure 29. Upon incubation
of the cells with cysteine-biotin/MESNA, however, biotinylated EGFP was
unambiguously detected by anti-biotin blot (Figure 29). There is no significant difference
in the amount of EGFP-intein and EFGP with the addition of cysteine-biotin/MESNA,
indicating that the biotinylation process was not very efficient. Less than 10% of the
expressed EGFP were biotinylated as estimated from the immunoblot results (Figure 29).
For this strategy to be useful in mammalian cells, further optimization needs to be done to
improve the in vivo biotinylation efficiency.
53
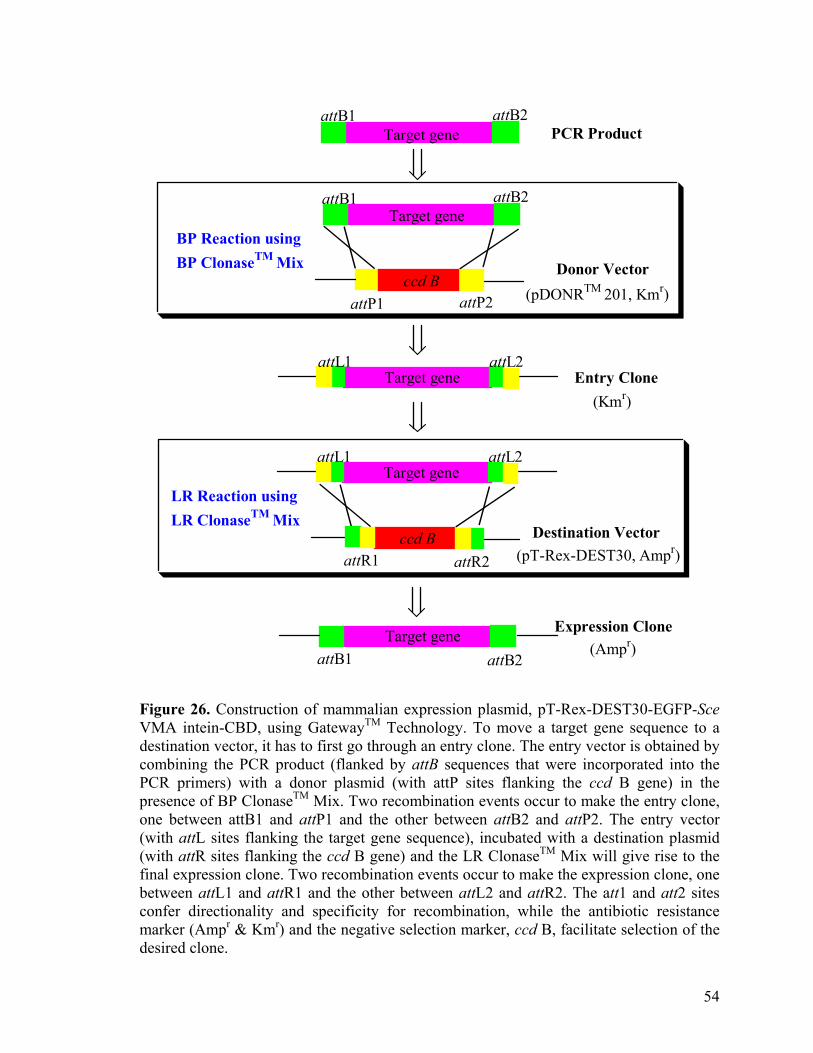
ccd B
ccd B
Target gene
Target gene
Target gene
Target gene
BP Reaction using BP ClonaseTM Mix
LR Reaction using LR ClonaseTM Mix
attB2attB1PCR Product
Expression Clone(Ampr)
Destination Vector(pT-Rex-DEST30, Ampr)
attB2attB1
attL2attL1
attP2attP1
attR2attR1
attB2attB1
(pDONRTM 201, Kmr)
Target gene Entry Clone(Kmr)
attL2attL1
Donor Vector
Figure 26. Construction of mammalian expression plasmid, pT-Rex-DEST30-EGFP-Sce VMA intein-CBD, using GatewayTM Technology. To move a target gene sequence to a destination vector, it has to first go through an entry clone. The entry vector is obtained by combining the PCR product (flanked by attB sequences that were incorporated into the PCR primers) with a donor plasmid (with attP sites flanking the ccd B gene) in the presence of BP ClonaseTM Mix. Two recombination events occur to make the entry clone, one between attB1 and attP1 and the other between attB2 and attP2. The entry vector (with attL sites flanking the target gene sequence), incubated with a destination plasmid (with attR sites flanking the ccd B gene) and the LR ClonaseTM Mix will give rise to the final expression clone. Two recombination events occur to make the expression clone, one between attL1 and attR1 and the other between attL2 and attR2. The att1 and att2 sites confer directionality and specificity for recombination, while the antibiotic resistance marker (Ampr & Kmr) and the negative selection marker, ccd B, facilitate selection of the desired clone.
54
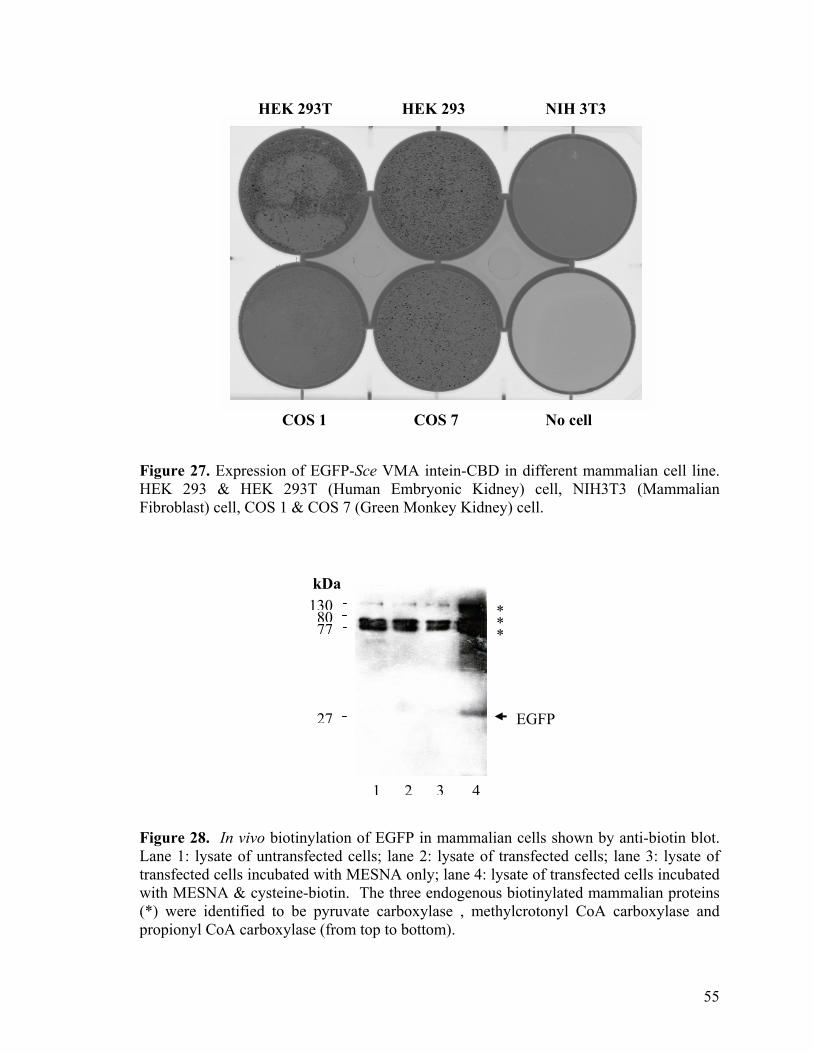
HEK 293T HEK 293 NIH 3T3
COS 1 COS 7 No cell
Figure 27. Expression of EGFP-Sce VMA intein-CBD in different mammalian cell line. HEK 293 & HEK 293T (Human Embryonic Kidney) cell, NIH3T3 (Mammalian Fibroblast) cell, COS 1 & COS 7 (Green Monkey Kidney) cell.
kDa
***
27
7713080
EGFP
1 2 3 4
Figure 28. In vivo biotinylation of EGFP in mammalian cells shown by anti-biotin blot. Lane 1: lysate of untransfected cells; lane 2: lysate of transfected cells; lane 3: lysate of transfected cells incubated with MESNA only; lane 4: lysate of transfected cells incubated with MESNA & cysteine-biotin. The three endogenous biotinylated mammalian proteins (*) were identified to be pyruvate carboxylase , methylcrotonyl CoA carboxylase and propionyl CoA carboxylase (from top to bottom).
55

Anti-biotin blot
kDa
27
97
27
EGFP-intein
Anti-EGFP blot
EGFP
EGFP
1 2 3 Figure 29. In vivo biotinylation efficiency in mammalian cells. Mammalian cells expressing EGFP-intein were incubated with cysteine-biotin/MESNA, lysed and analyzed by SDS-PAGE and western blots with anti-EGFP and anti-biotin antibodies. Lane 1: lysate of untransfected cells; lane 2: lysate of transfected cells; lane 3: lysate of transfected cells incubated with cysteine-biotin/MESNA.
56

3.7.3. Protein microarray generation using crude bacterial cell lysate
Next, we examined whether in vivo biotinylated proteins in the crude cell lysate could
be used directly for protein microarray applications. We first in vivo biotinylate, as
described above, three model proteins (EGFP, GST & MBP). Following cell harvest and
lysis, the crude lysates were spotted directly onto avidin-functionalized glass slides,
washed and detected either by their native fluorescence (for EGFP) (Figure 30A) or with
FITC-anti-GST and Cy5-anti-MBP, respectively (Figure 30B). Native fluorescence of
EGFP and specific bindings between the biotinylated proteins and their corresponding
antibodies were observed. This further confirms the binding specificity of the biotinylated
proteins to the avidin-fucntionalized slides thus suggesting the possible elimination of
extra purification step prior to spotting on a protein microarray. It should be pointed out
that, one of the major challenges in protein array technologies is the ability of retaining the
functional activity of proteins immobilized on the glass surface. In our experiments, the
native fluorescence of the immobilized EGFP could be retained on the glass slide for
weeks if stored properly at 4 oC, thereby highlighting the potential of our biotinylation
strategies in protein microarray generation.
EGFP GST MBP
Figure 30. Site-specific immobilization of biotinylated proteins onto avidin slides using bacterial cell lysate. Native fluorescence signal of EGFP (green) was observed while GST and MBP were individually detected with FITC-anti-GST (red) and Cy5-anti-MBP (blue) respectively.
57

Taken together, these data demonstrated that, for the first time, the intein-mediated
protein biotinylation could proceed within both mammalian and bacterial cells. Compared
to the in vitro method (method B in Figure 1), the intein-mediated, in vivo protein
biotinylation strategy presented herein is less efficient, requiring considerable refinements
before it becomes useful for high-throughput proteomic applications. Nevertheless, it
provides several obvious advantages: (1) pre-mature cleavage of the fusion protein in vivo
may be alleviated by addition of cysteine-biotin/MESNA to growing cells, thus potentially
maximizing the yield of the biotinlyated protein obtained; (2) excess biotin tag may be
introduced during in vivo biotinylation, and readily removed at the end by simple washes
of the cells; (3) following simple harvest and lysis of the cells, crude lysates containing the
desired biotinylated proteins (together with other endogenous cellular proteins) may be
used, without further purifications, for subsequent immobilization and downstream
applications, i.e. one could simply spot crude lysates onto an avidin-coated glass slide to
generate a protein array. Non-biotinylated proteins in the cell lysate could be washed
away on-chip in an efficient (e.g. protein immobilization and purification are done in a
single step) and highly-parallel (e.g. thousands of different protein spots could be
processed simultaneously on a single glass slide) fashion, resulting in purified proteins
immobilized on the microarray (vide infra). This is true because of the rare occurrence of
naturally biotinylated proteins in the cell, and the highly specific and strong nature of
biotin/avidin interaction, which can withstand extremely stringent washing/purification
conditions otherwise impossible with other affinity tags. Beside biotinylation, this tagging
strategy is also useful for in vivo labeling of mammalian cells using cysteine-containing
fluorescent probes.
58

3.8. Protein biotinlyation using different inteins.
It is believed that each intein has evolved in the context of its own host protein
sequence. Therefore, different inteins may proceed at different reaction rates in response
to the alternative amino acids residues adjacent to its cleavage site as well as to various
expression and reaction conditions, such as cell types, reagents, pH and the temperature.
The availability of a large number of inteins (> 100) offers high possibility of selecting an
optimal intein fusion partner suitable for our biotin-tagging strategy. Of particular interest
are the mini-inteins, which range in size from 134 to 198 amino acid residues. These
naturally occurring mini-inteins, lacking the homing endonuclease domain but process the
two important terminal regions required for splicing, have shown to cleave more
proficiently with MENSA than Sce VMA intein (454 amino acids), resulting in higher
yield of the thioester proteins for IPL. Currently, two modified mini-inteins are present in
commercially available E.coli. expression vectors, namely; Mxe GyrA intein (found in the
Mycobacterium xenopi gyrA gene)49 and Mth RIR1 intein (found Methanobacterium
thermoautotrophicum ribonucleoside diphosphate reductase gene).50 To select the most
suitable intein for our biotinylation strategy, we cloned the EGFP gene into two
expression vector containing the modified mini-inteins, pTWIN1 and pTWIN2. pTWIN1
contains a CBD-Ssp DnaB mini-intein fusion gene upstream of the Mxe GyrA intein-CBD
(198 amino acids) coding region. The use of NdeI and SapI sites for cloning results in
deletion of the CBD-Ssp DnaB mini-intein sequence and fusion of the target gene to the N
terminus of the Mxe GyrA intein-CBD coding region (Figure 31). pTWIN2 carries the 134
amino acids Mth RIR1 intein in place of Mxe GyrA intein in pTWIN1.
59

CBD CBDIntein 1 Intein 2MCS
T7 promoter
NdeI SapI
Target gene
CBDTarget gene Intein 2
T7 promoter
Cloning into pTWIN vectors via NdeI and Sap I sites
Figure 31. Schematic representation of pTWIN vectors. The pTWIN vectors are designed for the generation of an N-terminal cysteine residue and/or a C-terminal thioester on the target protein. Both pTWIN1 and pTWIN2 carry the 154 amino acid residues Ssp DnaB mini-intein (Intein 1) for the production of an N-terminal cysteine. pTWIN1 vector differs from pTWIN2 vectors at the intein 2 coding region, with Mth RIR1 intein (134 amino acid residues) in place of Mxe GyrA intein (198 amino acid residues). Cloning a target gene into pTWIN vectors via NdeI and SapI restriction sites, result in the fusion of the target protein to the N terminus of intein 2. indicates the intein cleavage site.
After cloning, the two pTWIN constructs were transformed into ER2566 for protein
expression. For comparison, EGFP-Sce intein-CBD was expressed from pTYB1 construct.
Fusion proteins were extracted and purified within the chitin column and biotin-tagged
EGFP was eluted following the on-column cleavage/biotinylation reaction. Higher
recovery of the intact EGFP-Mxe intein-CBD fusion in the cell lysate was observed
(Figure 32), resulting in an increase yield of the biotinylated EGFP as shown in Figure 33.
In contrast, the high in vivo cleavage activity of EGFP-Mth-intein-CBD greatly reduces
the final yield of the biotin-tagged EGFP from the column purification step (Figure 32 &
33). Beside on-column biotin-tagging, we also tried in vivo biotinylation of EGFP using
the two pTWIN construct. MESNA and cysteine-biotin were added to bacterial cells
60
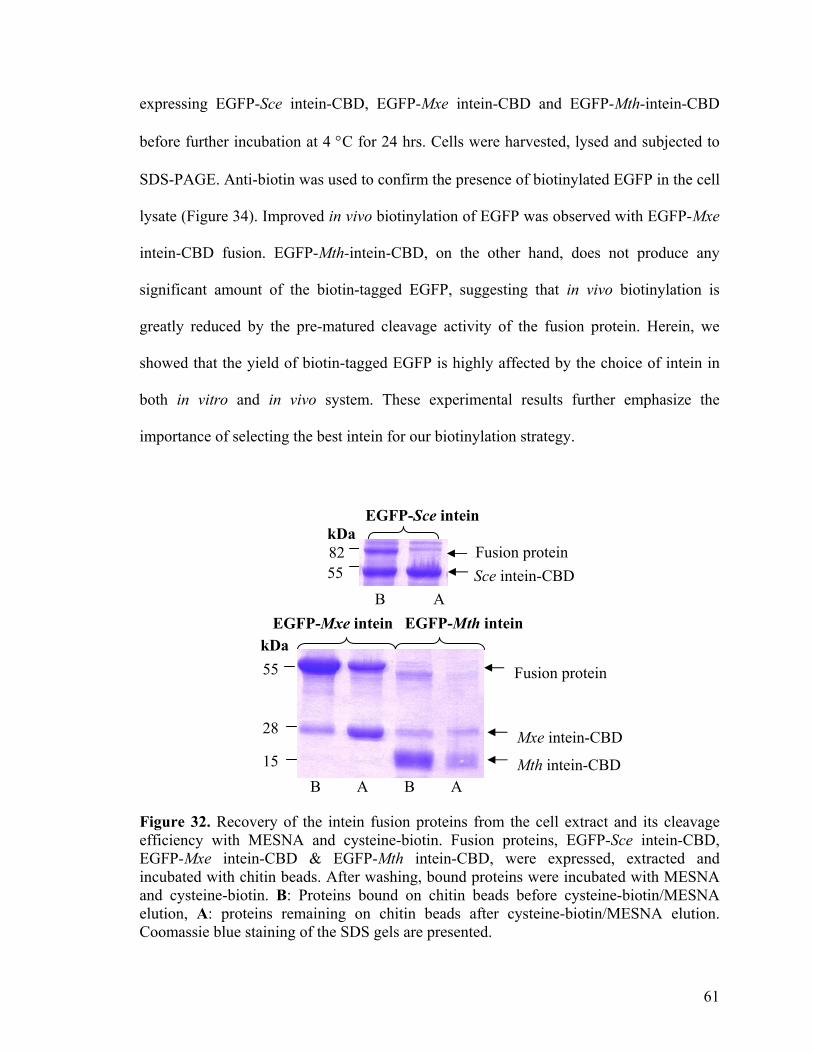
expressing EGFP-Sce intein-CBD, EGFP-Mxe intein-CBD and EGFP-Mth-intein-CBD
before further incubation at 4 °C for 24 hrs. Cells were harvested, lysed and subjected to
SDS-PAGE. Anti-biotin was used to confirm the presence of biotinylated EGFP in the cell
lysate (Figure 34). Improved in vivo biotinylation of EGFP was observed with EGFP-Mxe
intein-CBD fusion. EGFP-Mth-intein-CBD, on the other hand, does not produce any
significant amount of the biotin-tagged EGFP, suggesting that in vivo biotinylation is
greatly reduced by the pre-matured cleavage activity of the fusion protein. Herein, we
showed that the yield of biotin-tagged EGFP is highly affected by the choice of intein in
both in vitro and in vivo system. These experimental results further emphasize the
importance of selecting the best intein for our biotinylation strategy.
Fusion protein
Fusion protein Sce intein-CBD
kDa 55
B A
B A B A 15
28
kDa8255
EGFP-Mth intein EGFP-Mxe intein
EGFP-Sce intein
Mxe intein-CBD
Mth intein-CBD Figure 32. Recovery of the intein fusion proteins from the cell extract and its cleavage efficiency with MESNA and cysteine-biotin. Fusion proteins, EGFP-Sce intein-CBD, EGFP-Mxe intein-CBD & EGFP-Mth intein-CBD, were expressed, extracted and incubated with chitin beads. After washing, bound proteins were incubated with MESNA and cysteine-biotin. B: Proteins bound on chitin beads before cysteine-biotin/MESNA elution, A: proteins remaining on chitin beads after cysteine-biotin/MESNA elution. Coomassie blue staining of the SDS gels are presented.
61
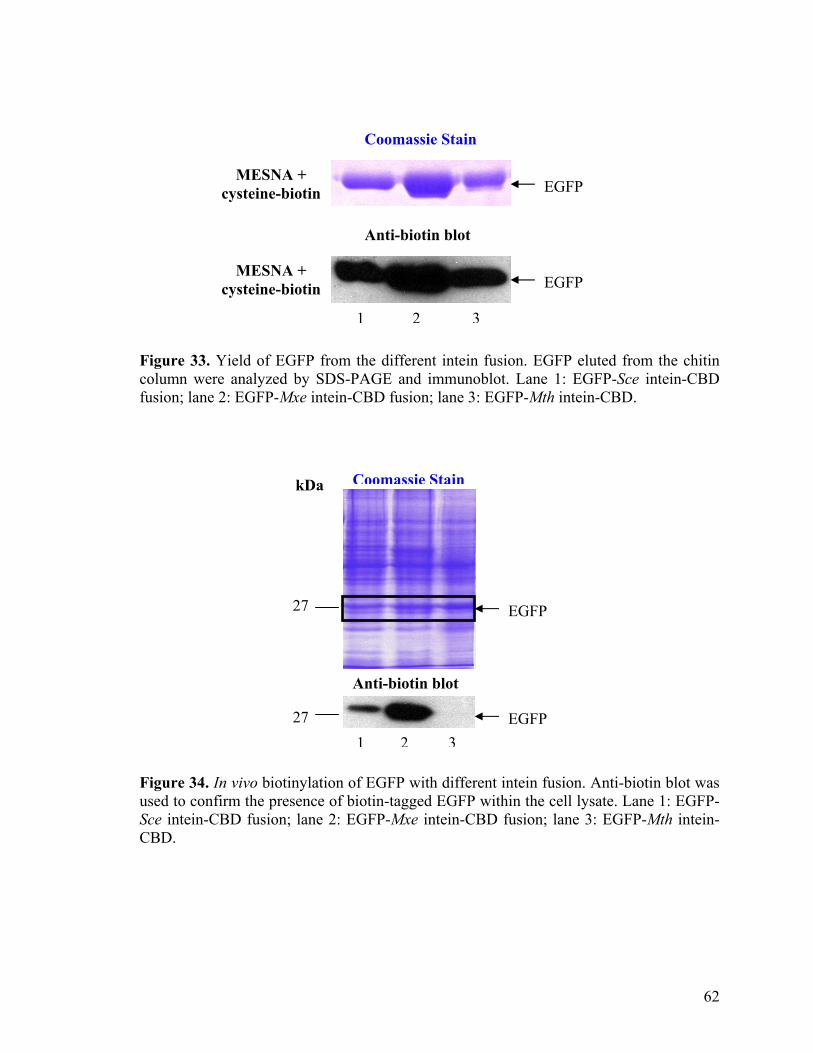
Coomassie Stain
Anti-biotin blot
MESNA + cysteine-biotin EGFP
MESNA + cysteine-biotin EGFP
1 2 3
Figure 33. Yield of EGFP from the different intein fusion. EGFP eluted from the chitin column were analyzed by SDS-PAGE and immunoblot. Lane 1: EGFP-Sce intein-CBD fusion; lane 2: EGFP-Mxe intein-CBD fusion; lane 3: EGFP-Mth intein-CBD.
kDa
27
27
Anti-biotin blot
Coomassie Stain
EGFP
EGFP 1 2 3
Figure 34. In vivo biotinylation of EGFP with different intein fusion. Anti-biotin blot was used to confirm the presence of biotin-tagged EGFP within the cell lysate. Lane 1: EGFP-Sce intein-CBD fusion; lane 2: EGFP-Mxe intein-CBD fusion; lane 3: EGFP-Mth intein-CBD.
62

3.9.Protein biotinylation in a cell-free system.
We have thus far successfully demonstrated the utilities of intein-mediated biotinylation
strategies in both in vitro and in vivo settings. In both cases, intein-fused proteins need to
be successfully expressed in soluble forms in the host cell before biotinylation (either in
vitro or in vivo) could take place. However, numerous problems may arise during protein
expression in a host cell. The formation of inclusion bodies is one. This is especially true
when one attempts to express eukaryotic proteins in prokaryotic hosts. Other problems
include potential proteolytic degradation of the protein by endogenous proteases, as well
as expression of proteins toxic to the host cell. Cell-free protein synthesis provides an
attractive alternative for protein expression which may potentially overcome many of
these problems (method C, Figure 4), and is well-suited for protein microarray
applications because (1) minute quantities of proteins generated in cell-free system are
sufficient for spotting in a protein array, and (2) the method could be easily adopted in 96-
and 384-well formats with a conventional PCR machine for potential high-throughput
protein synthesis.89-91
To assess whether our intein-mediated strategy is suitable for biotinylation of proteins
synthesized in a cell-free system, pMYB5, the plasmid expressing MBP-intein-CBD
fusion under the transcription control of T7 promoter, was used as the DNA template in a
Rapid Translation System (RTS) 100 E. coli. HY kit. Optimal temperature for most
protein synthesis is at 30 °C, however, lower temperatures may be used for synthesis of
proteins that tend to aggregate at that temperature. Protein synthesis can proceed for up to
6 h but the synthesis reaction is usually 90% complete after 4 h. After cell-free protein
63

synthesis, the reaction was incubated with cysteine-biotin/MESNA, followed by analysis
with SDS-PAGE and western blots (Figure 35A). The presence of a 42 kDa band on the
anti-biotin immunoblot, and not any other band (Figure 35A, lane 2), indicated successful
and exclusive biotinylation of the MBP protein synthesized in the cell-free system. It
should be noted that, among three protein biotinylation strategies presented herein (e.g.
Figure 4), the cell-free method seems to be the simplest of all. In our hands, however, it is
also the least reliable: the efficiency of protein expression as well as the subsequent
protein biotinylation depends greatly on a number of different factors, including the
nature of the protein itself, the amount and quality of the DNA template used (Figure 35
B) and the kind of cell lysates used for protein expression, etc. For optimized performance
of this biotin-tagging strategy in cell-free system, more experiments have to be done to
further assess some of these issues.
64
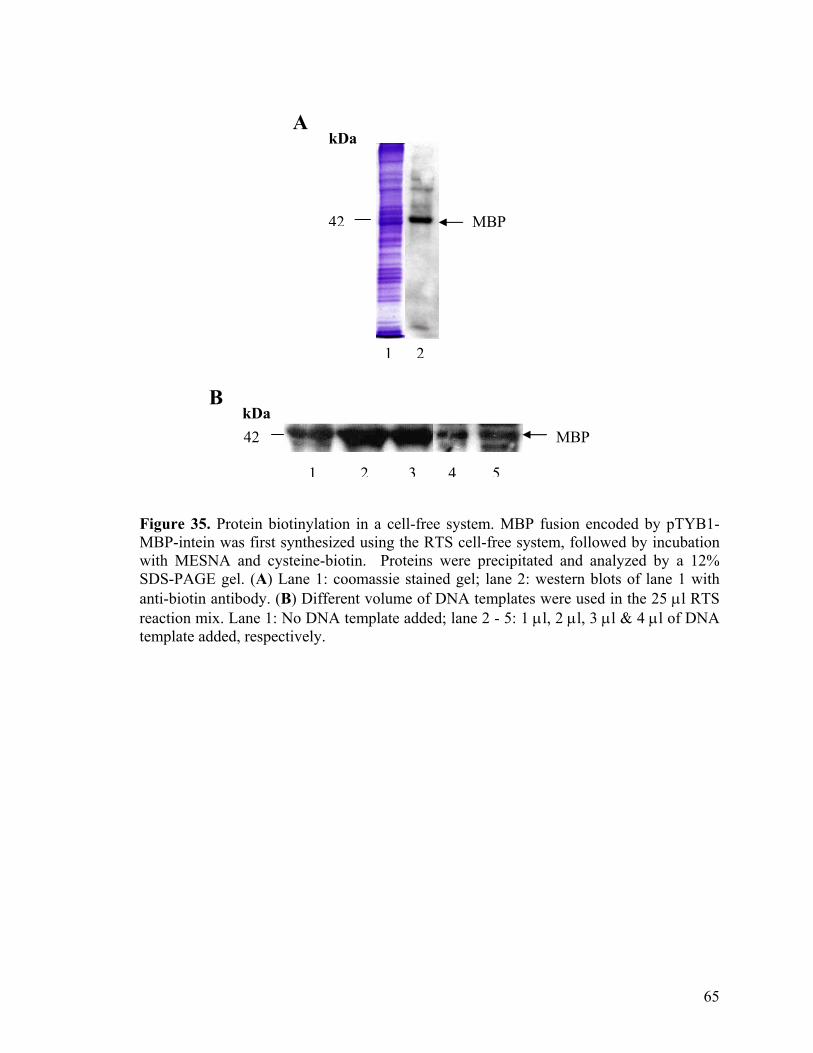
kDa 42
1 2
MBP 42
kDa A
B
MBP
1 2 3 4 5
Figure 35. Protein biotinylation in a cell-free system. MBP fusion encoded by pTYB1-MBP-intein was first synthesized using the RTS cell-free system, followed by incubation with MESNA and cysteine-biotin. Proteins were precipitated and analyzed by a 12% SDS-PAGE gel. (A) Lane 1: coomassie stained gel; lane 2: western blots of lane 1 with anti-biotin antibody. (B) Different volume of DNA templates were used in the 25 µl RTS reaction mix. Lane 1: No DNA template added; lane 2 - 5: 1 µl, 2 µl, 3 µl & 4 µl of DNA template added, respectively.
65

4. Conclusions
Our intein-mediated biotinylation strategies have several advantages over other
traditional methods in which biotin ligase is used. First, the precise splicing mechanism of
intein allows coupling of biotin moiety to the C-terminus of proteins without introduction
of additional amino acids sequences that otherwise may compromise the native protein
activity. Second, most commonly used biochemical reagents do not inhibit the intein-
mediated ligation reaction, thus enabling purification/biotinylation of the desired protein
to be done efficiently in a single step. Third, cell toxicity due to over-expression of fusion
proteins (unless the target protein is toxic to the host strain itself) is unlikely, since there is
no competition of endogenous biotin consumption. Finally, since protein biotinylation is
solely dependent on the cleavage of the fusion protein from the intein tag, use of
expensive enzyme is not required and co-expression of biotin ligase is not necessary for in
vivo biotinylation of proteins.
Our findings herein indicate that the intein-mediated biotinylation approaches are
sufficiently general and versatile, enabling proteins from different biological sources to be
site-specifically biotinylated under different conditions, and subsequently used in a wide
range of avidin/biotin technologies. Expressed proteins fused to an intein tag could be
efficiently purified and biotinlyated, in vitro, in a single purification step. We also
showed that the strategy proceed in both live bacterial and mammalian cells. We further
showed intein-fused proteins synthesized from a cell-free system could undergo intein-
mediated biotinylation reaction as well. We emphasized the unique utilities of our
strategies in the area of protein microarray, as this technology may be one of the most
66

powerful tools for high-throughput analysis of protein functions. Several essential aspects
of a protein microarray were addressed using our intein-mediated biotinylation strategies.
Firstly, protein biotinylation/immobilization was site-specific, leading to uniform
orientation, and more importantly retention of the functional integrity of proteins
immobilized on the array. Secondly, no extra macromolecular tag was introduced in the
immobilized protein, further ensuring the biological activity of proteins was minimally
perturbed. Thirdly, avidin/biotin interaction was extremely stable, enabling immobilized
proteins to be thoroughly washed to remove cellular contaminants, and subsequently
screened under even the most stringent conditions. Finally, the strategy upon
modifications, does not involve tedious protein purification/elution steps and allows the
facile generation of biotinylated proteins, making it possible for proteins in crude lysates
to be spotted directly onto a protein array (e.g. in live cells or in a cell-free system). This
enables expression, without further processing (e.g. purification and elution, etc), of a
large array of biotinylated, ready-to-spot proteins in a truly high-throughput, high-content
fashion.
Key challenges remain with the strategies presented herein, and none is more pressing
than to further improve the efficiency of protein biotinylation in live cells, especially
mammalian cells. In addition to biotin tagging, we are also exploring the feasibility of
incorporating a variety of cysteine-containing molecular probes at the C-terminus of
proteins (e.g. fluorescent and photo-crossing probes) for various biochemical and
biophysical studies of proteins, both in vitro and in vivo. 92-94
67

5. References
1. Chen, G.Y.J., Uttamchandani, M., Lesaicherre, M.L., Lue, Y.P.R., and Yao, S.Q. (2003). Array-based technologies and their applications in proteomics. Curr. Top. Med. Chem. 3, 705-724.
2. Zhu, H., Bilgin, M., and Snyder, M. (2003). Proteomics. Annu. Rev. Biochem. 72, 783-812.
3. Zhu, H., and Snyder, M. (2003). Protein chip technology. Curr. Opin. Chem. Biol. 7 (1), 55-63.
4. Seong, S.Y., and Choi, C.Y. (2003) Current status of protein chip development in terms of fabrication and application. Proteomics 3 (11), 2176-2189.
5. Schweitzer, B., Predki, P., and Snyder, M.(2003). Microarrays to characterize protein interactions on a whole-proteome scale. Proteomics 3 (11): 2190-2199.
6. Haab, B.B., Dunham, M.J., and Brown, P.O. (2001). Protein microarray for highly parallel detection and quantitation of specific proteins and antibiodies in complex solution. Genome Biol. 2, 1-13.
7. Schweitzer, B., and Kingsmore, S. (2002). Measuring proteins on microarray. Curr. Opin. Biotechnol. 13, 14-19.
8. Sreekumar, A., Chinnaiyan, A.M. (2002) Protein microarrays: A powerful tool to study cancer. Curr Opin. Mol. Ther. 4 (6), 587-593.
9. Espina, V., Mehta, A.I., and Winters, M.E. (2003). Protein microarrays: Molecular profiling technologies for clinical specimens. Proteomics 3 (11), 2091-2100.
10. Coleman, M.A., Miller, K.A., and Beernink, P.T. (2003). Identification of chromatin-related protein interactions using protein microarrays. Proteomics 3 (11), 2101-2107.
11. Nam, M.J., Madoz-Gurpide, J., and Wang, H. (2003). Molecular profiling of the immune response in colon cancer using protein microarrays: Occurrence of autoantibodies to ubiquitin C-terminal hydrolase L3. Proteomics 3 (11), 2108-2115.
12. Tannapfel, A., Anhalt, K., Hausermann, P. (2003). Identification of novel proteins associated with hepatocellular carcinomas using protein microarrays. J. Pathol. 201 (2), 238-249.
13. Nishizuka, S., Chen, S.T., and Gwadry, F.G. (2003). Diagnostic markers that distinguish colon and ovarian adenocarcinomas: identification by genomic, proteomic, and tissue array profiling. Cancer Res. 63 (17), 5243-5250.
68

14. De Wildt, R. M., Mundy, C.R., Gorick, B.D., and Tomlinson, I.M. (2000). Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat. Biotechnol. 18, 989-994.
15. Xu, Y.Q., Bao, G. (2003). A filtration-based protein microarray technique. Anal. Chem. 75 (20): 5345-5351.
16. Huang, R.P., Huang, R., Fan, Y., and Lin, Y. (2001). Simultaneous detection of multiple cytokines from conditioned media and patient's sera by an antibody-based protein array system. Anal. Biochem. 294, 55-62.
17. Martin, B. D., Gaber, B. P., Patterson, C. H., and Turner, D. C. (1998) Direct Protein Microarray Fabrication Using a Hydrogel "Stamper". Langmuir 14, 3971-3975.
18. Guschin, D., Yershov, G., Zaslavsky, A., Gemmell, A., Shick, V., Proudnikov, D., Arenkov, P., and Mirzabekov , A. (1997). Manual manufacturing of oligonucleotide, DNA, and protein microchips. Anal. Biochem. 250, 203–211.
19. Afanassiev, V., Hanemann, V., and Wolfl, A. (2000). Preparation of DNA and protein micro arrays on glass slides coated with an agarose film. Nucleic Acids Res. 28, e66.
20. Arenkov P., Kukhtin A., Gemmell A., Voloshchuk S., Chupeeva V., and Mirzabekov A. (2000). Protein Microchips: Use for Immunoassay and Enzymatic Reactions. Anal. Biochem. 278 (2), 123-131.
21. Cho, E.J., Tao, Z., Tehan, E.C., and Bright, F.V. (2002) Multianalyte pin-printed biosensor arrays based on protein-doped xerogels. Anal. Chem. 74, 6177-6184.
22. Rupcich, N., and Brennan, J.D. (2003). Coupled enzyme reaction microarrays based on pin-printing of sol-gel derived biomaterials. Anal. Chim. Acta. 500, 3-12.
23. MacBeath, G., and Schreiber, S.L. (2000). Printing proteins as microarrays for high-throughput function determination. Science 289, 1760-1763.
24. Zhu, H., Klemic, J.F., Chang, S., Bertone, P., Casamayor, A., Klemic, K.G., Smith, D., Gerstein, M., Reed, M.A., and Snyder, M. (2000). Analysis of yeast protein kinases using protein chips. Nat. Genetics 26, 283-289.
25. Zhu, H., Bilgin, M., Bangham, R., Hall, D., Casamayor, A., Bertone, P., Lan, N., Jansen, R., Bidlingmaier, S., Houfek, T., Mitchell, T., Miller, P., Dean, R.A,, Gerstein, M., and Snyder, M. (2001). Global analysis of protein activities using proteome chips. Science 293, 2101-2105.
69

26. Wilchek, M., and Bayer, E.A. (1990). Introduction to Avidin-Biotin Technology. Methods Enzymol. 184, 5-13.
27. Wilchek, M., and Bayer, E.A. (1990). Applications of Avidin-Biotin Technology: Literature Survey. Methods Enzymol. 184, 14-15.
28. Lesaicherre, M.L., Uttamchandani, M., Chen, G.Y.J., and Yao, S.Q. (2002) Developing site-specific immobilization strategies of peptides in a microarray. Bioorg. Med. Chem Lett. 12, 2079-2083.
29. Lesaicherre, M.L., Lue, R.Y.P., Chen, G.Y.J., Zhu, Q. and Yao, S.Q. (2002) Intein-mediated biotinylation of proteins and its application in a protein microarray. J. Am. Chem. Soc. 124, 8768-8769.
30. Uttamchandani, M., Chan, E.W.S., and Chen, G.Y.J. (2003). Combinatorial peptide microarrays for the rapid determination of kinase specificity. Bioorg. Med. Chem Lett. 13 (18), 2997-3000.
31. Lue, R.Y.P., Chen, G.Y.J., Hu, Y., Zhu, Q., and Yao, S.Q. (2004). Versatile Protein Biotinylation Strategies for Potential High-Throughput Proteomics. J. Am. Chem. Soc. In press.
32. Peluso, P., Wilson, D.S., Do, D., Tran, H., Venkatasubbaiah, M., and Quincy, D.
(2003). Optimizing antibody immobilization strategies for the construction of protein microarrays. Anal. Biochem. 312, 113-24.
33. Bayer, E.A., and Wilchek, M. (1990). Protein Biotinylation. Methods Enzymol. 184, 139-160.
34. Schwarz, A., Wandrey, C., Bayer, E.A. and Wilchek, M. (1990). Enzymatic C-Terminal Biotinylation of Proteins. Methods Enzymol. 184, 160-162.
35. Cronan, J.E. (1990) Biotinylation of proteins in vivo. A post-translational modification to label, purify, and study proteins. J. Biol. Chem. 265, 10327-10333.
36. Samols, D., Thornton, C.G., Murtif, V.L., Kumar, G.K., Haase, F.C. & Wood, H.G. (1988) Evolutionary conservation among biotin enzymes. J. Biol. Chem. 263, 6461-6464.
37. Schatz, P.J. (1993). Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology 11, 1138-1143.
38. Cull, M.G. & Schatz, P.J. (2000) Biotinylation of proteins in vitro and in vivo using small peptide tags. Methods Enzymol. 326, 430-440.
70

39. Cronan, J.E. & Reed, K.E. (2000) Biotinylation of proteins in vivo: A useful posttranslational modification for protein analysis. Methods Enzymol. 326, 440-458.
40. Boer, E., Rodriguez, P., Bonte, E., Krijgsveld, J., Katsantoni, E., Heck, A., Grosveld, F. & Strouboulis, J. (2003). Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 7480-7485.
41. Cooper, A.A., Chen, Y., Lindorfer, M.A. and Stevens, T.H. (1993) Protein splicing of the yeast TFP1 intervening protein sequence: a model for self-excision. EMBO J. 12, 2575-2583.
42. Perler, F. B., Comb, D. G., Jack, W. E., Moran, L. S., Qiang, B., Kucera, R. B., Benner, J., Slatko, B. E., Nwankwo, D. O., Hempstead, S. K., Carlow, C. K. S. and Jannasch, H. (1992) Intervening sequences in an Archaea DNA polymerase gene. Proc. Natl. Acad. Sci. USA. 89, 5577-5581.
43. Evan, T.C., and Xu, M.Q. (2002). Mechanistic and Kinetic Considerations of Protein Splicing. Chem. Rev. 51, 4869-4883.
44. Perler, F.B. (1999). InBase, the New England Biolabs Intein Database. Nucleic Acids Res. 27, 346-347.
45. Perler, F.B., Davis, E.O., Dean, G.E., Gimble, F.S., Jack, W.E., Neff, N., Noren, C.J., Thorner, J., and Belfort, M. (1994). Protein splicing elements; intein and exteins a definition of terms and recommended nomenclature. Nucleic Acids Res. 22, 1125-1127.
46. Gimble, F.S., and Thorner, J. (1992). Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature 357, 301-306.
47. Belfort, M., and Roberts, R.J. (1997). Homing endonucleases: keeping the house in order. Nucleic Acids Res. 25, 3379-3388.
48. Shingledecker, K., Jiang, S., and Paulus, H. (1998). Molecular dissection of the Mycobacterium tuberculosis RecA intein: design of a minimal intein and of a trans-splicing system involving two intein fragments. Gene 207, 187-195.
49. Derbyshire, V., Wood, D.W., Wu, W., Dansereau, J.T., Dalgaard, J.Z., Belfort, M., (1997). Genetic definition of a protein-splicing domain: Functional mini-inteins support structure predictions and a model for intein evolution. Proc. Natl. Acad. Sci. U.S.A. 94, 11466-11471.
50. Chong, S., and Xu, M.Q. (1997). Protein Splicing of the Saccharomyces cerevisiae VMA Intein without the Endonuclease Motifs. J. Biol. Chem. 272, 15587-15590.
71

51. Telenti, A., Southworth, M., Alcaide, F., Daugelat, S., Jacobs, J., William, R., Perler, F.B. (1997). The Mycobacterium xenopi GyrA protein splicing element: characterization of a minimal intein. J. Bacteriol. 179, 6378-6382.
52. Evan, T.C., Benner, J., and Xu, M.Q. (1999) The in vitro ligation of bacterially expressed proteins using an Intein form Methanobacterium thermoautotrophicum. J. Bio. Chem. 274, 3923-3926.
53. Wu, H., Hu, Z., and Liu, X.Q. (1998). Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U.S.A. 95, 9226-9231.
54. Wu, H., Xu, M.Q., Liu, X.Q. (1998). Protein trans-splicing and functional mini-inteins of a cyanobacterial dnaB intein.. Biochim. Biophys. Acta 35732, 1-11.
55. Mills, K.V., Lew, B.M., Jiang, S., Paulus, H. (1998). Protein splicing in trans by purified N- and C-terminal fragments of the Mycobacterium tuberculosis RecA intein. Proc. Natl. Acad. Sci. U.S.A. 95, 3543.
56. Southworth, M.W., Adam, E., Panne, D., Byer, R., Kautz, R., Perler, F.B. (1998). Control of Protein Splicing by Intein Fragment Reassembly. EMBO J. 17, 918-926.
57. Evans, J.T.C., Martin, D., Kolly, R., Panne, D., Sun, L., Ghosh, I., Chen, L.,Benner, J., Liu, X.Q., Xu, M.Q. (2000). Protein Trans-splicing and Cyclization by a Naturally Split Intein from the dnaE Gene of Synechocystis Species PCC6803. J. Biol. Chem. 275, 9091-9094.
58. Gorbalenya, A.E., (1998). Non-canonical inteins. Nucleic Acids Res. 26, 1741-1748.
59. Chong, S.R., Shao, Y., Paulus, H., Benner, J., Perler, J.B. and Xu, M.Q. (1996). Protein splicing involving the Saccharomyces cerevisiae VMA intein. The steps in the splicing pathway, side reactions leading to protein cleavage, and establishment of an in vitro splicing reaction. J. Biol. Chem. 271, 22159-22168.
60. Watanabe, T., Ito, Y., Yamada, T., Hashimoto, M., Sekine, S., and Tanaka, H. (1994). The roles of the C-terminal domain and type III domains of chitinase A1 from Bacillus circulans WL-12 in chitin degradation. J. Bacteriol. 176, 4465-4472.
61. Chong, S.R., Mersha, F.B., Comb, D.G., Scott, M.E., Landry D., Vence, L.M., Perler, F.B., Benner, J., Kucera, R.B., Hirvonen, C.A., Pelletier, J.J., Paulus, H., and Xu, M.Q. (1997) Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene 192, 271-281.
72

62. Chong, S.R., Montello, G.E., Zhang, A., Cantor, E.J., Liao, W., Xu, M.Q., and Benner, J. (1998). Utilizing the C-terminal cleavage activity of a protein splicing element to purify recombinant proteins in a single chromatographic step. Nucleic Acids Res. 26, 5109-5115.
63. Muir ,T.W. (2003) Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 72, 249-289 2003.
64. Ayers, B., Blaschke, U.K, Camarero, J.A., Cotton, G.J., Holford, M., and Muir, T.W. (1999). Introduction of unnatural amino acids into proteins using expressed protein ligation. Biopolymers 51, 343-354.
65. Evan, T.C., Benner, J., and Xu, M.Q. (1998). Semisynthesis of cytotoxic proteins using modified protein splicing element. Protein Science 7, 2256-2264.
66. Blaschke, U.K., Cotton, G.J., and Muir, T.W. (2000). Synthesis of Multi-Domain proteins using expressed protein liagtion: Strategies for segmental isotopic labeling of internal regions. Tetrahedron 56, 9461-9470.
67. Cotton, G.J., and Muir, T.W. (2000). Generation of a dual-labeled fluorescence biosensor for Csk-II phosphorylation using solid-phase expressed protein ligation. Chemistry & Biology 7, 253-261.
68. Scott, C.P., Abel-Santos, E., Wall, M., Wahnon, D.C., and Benkovic, S.J. (1999) Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. USA. 96, 13638-13643.
69. Siebold, C., and Erni, B. (2002). Intein-mediated cyclization of a soluble and a membrane protein in vivo: function and stability. Biophys. Chem. 96, 163-171.
70. Muir, T.W., Sondhi, D., and Cole, P.A. (1998). Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. U.S.A. 98, 6705-6710.
71. Muir, T.W. (2001). Development and application of expressed protein ligation. Synlett 6, 733-740.
72. Xu, M.Q., and Evan, T.C. (2001). Intein-mediated ligation and cyclization of expressed proteins. METHODS 24, 257-277.
73. Studier, F. W., and Moffatt, B. A. (1986) Use of Bacteriophage T7 RNA Polymerase to direct selective high-level expression of cloned genes. J.Mol. Biol. 189, 113-130.
74. Marchuk, D., Drumm, M., Saulino, A., and Collins F.S. (1991). Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res. 19, 1154.
73

75. Paborsky, L. R., Dunn, K. E., Gibbs, C. S., and Dougherty, J. P. (1996). A Nickel Chelate Microtiter Plate Assay for Six Histidine-Containing Proteins. Anal. Biochem. 234, 60-65.
76. Green, N. M., and Toms, E. J. (1973). The properties of subunits of avidin coupled to sepharose. Biochem. J. 133, 687-700.
77. Savage, D., Mattson, G., Nielander, G., Morgensen, S., and Conklin, E. Avidin-Biotin Chemistry: A Handbook, 2nd Ed.; Pierce Chemical Co.:Illinois, USA, 1994.
78. Reznik, G. O., Vajda, S., Cantor, C. R., and Sano, T. (2001). A streptavidin mutant useful for directed immobilization on solid surfaces. Bioconjugate Chem. 12, 1000-1004.
79. Houseman, B.T., Huh, J.H., Kron, S.J., and Mrksich, M. (2002) Peptide chips for the quantitative evaluation of protein kinase activity. Nat. Biotechnol. 20, 270–274.
80. Rich, R.L., Day, Y.S., Morton, T.A., and Myszka, D.G. (2001). High-resolution and high-throughput protocols for measuring drug/human serum albumin interactions using BIACORE. Anal. Biochem. 296, 197–207.
81. Schaeferling, M., Schiller, S., Paul, H., (2002). Application of self-assembly techniques in the design of biocompatible protein microarray surfaces. Electrophoresis 23, 3097-3105.
82. Hodneland, C.D., Lee, Y.S., Min, D.H., and Mrksich, M. (2002). Selective Immobilization of Protein to Self-Assembled Monolayers Presenting Active Site Directed Capture Ligands. Proc. Natl. Acad. Sci. U.S.A. 99, 5048-5052.
83. James, R., H., Elliott, P., D., Kimberly L.R., Cynthia H.J., Daniel L., Diana C., Christian L., James R. H., Steven J. S., Rodney R., and Stanley, F. (1997) The Complete Set of Predicted Genes from Saccharomyces cerevisiae in a Readily Usable Form. Genome Res. 7, 1169-1173.
84. Giriat, I., and Muir, T.W. (2003). Protein semi-synthesis in living cells. J. Am. Chem. Soc. 125, 7180-7181.
85. Choi-Rhee, E., and Cronan, J.E. (2003). The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase. J. Biol. Chem. 278, 30806-30812.
86. Walhout et al. (2000). Gateway™ Recombinational Cloning: Application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 328, 575-592.
74

75
87. Ohara, O., and Temple, G. (2001). Directional cDNA library construction assisted by the in vitro recombination reaction. Nucleic Acids Research. 29(4), e22.
88. Hartley, J., Temple, G., and Brasch, M.A. (2000). DNA cloning using in vitro site-specific recombination. Genome Research 10, 1788-1795.
89. He, M.Y., and Taussig, M.J. (2001). Single step generation of protein arrays from DNA by cell-free expression and in situ immobilisation. Nucleic Acids Res. 29, e73.
90. Kawahashi, Y., Doi, N., and Takashima, H, (2003). In vitro protein microarrays for detecting protein-protein interactions: Application of a new method for fluorescence labeling of proteins. Proteomics 3, 1236-1243.
91. Oleinikov, A.V., Gray, M.D., and Zhao, J. (2003). Self-assembling protein arrays using electronic semiconductor microchips and in vitro translation. J. Proteome Res. 2 , 313-319.
92. Tolbert, T., and Wong, C.-H. J. (2000) Intein-mediated synthesis of proteins containing carbohydrates and other molecular probes. J. Am. Chem. Soc. 122, 5421-5428.
93. Kapanidis, A.N., Weiss, S. (2002). Fluorescent probes and bioconjugation chemistries for single-molecule fluorescence analysis of biomolecules. J. Chem. Phys. 117, 10953-10964.
94. Yeo, S.Y.D., Srinivasan, R., Uttamchandani, M., Chen, G.Y.J., Zhu, Q., Yao, S.Q. (2003) Cell-permeable small molecule probes for site-specific labeling of proteins. Chem. Commun., 2870-2871.