inhibition of mtor suppresses uvb-induced keratinocyte...
TRANSCRIPT

Research Article
Inhibition of mTOR Suppresses UVB-Induced KeratinocyteProliferation and Survival
Theresa D. Carr1, John DiGiovanni2, Christopher J. Lynch1, and Lisa M. Shantz1
AbstractUV radiation is the major risk factor for developing skin cancer, the most prevalent cancer worldwide.
Several studies indicate that mTOR signaling is activated by UVB and may play an important role in skin
tumorigenesis. mTOR exists in two functionally and compositionally distinct protein complexes: the
rapamycin-sensitive mTOR complex 1 (mTORC1) and the rapamycin-resistant mTOR complex 2
(mTORC2). The purpose of these studies was to investigate the roles of the two mTOR complexes in
UVB-mediated proliferation and apoptosis in the skin. We used rapamycin, a pharmacologic inhibitor
of mTORC1, and an inducible mTOR-deficient (K5-CreERT2;mTORfl/fl) mouse model that allows
epidermal-specific disruption of mTOR following topical treatment with 4-hydroxytamoxifen (4OHT).
Rapamycin blocked UVB-induced phosphorylation of S6K, the downstream target of mTORC1, and
significantly reduced UVB-stimulated epidermal proliferation and cell-cycle progression, but had no
effect on cell death. In contrast, mTOR deletion, which attenuated UVB-induced phosphorylation of
both S6K and the mTORC2 target AKTSer473, significantly increased apoptosis both in vivo and in
keratinocyte cultures, in addition to reducing hyperproliferation following UVB irradiation. The role of
mTORC2 in UVB-induced prosurvival signaling was verified in Rictor�/� mouse embryo fibroblasts,
which lack functional mTORC2 and were more sensitive to UVB-induced apoptosis than controls. These
studies show that mTORC1 and mTORC2 play unique but complementary roles in controlling
proliferation and apoptosis in the skin. Our findings underscore the importance of both mTOR
complexes in mediating UVB-induced signaling in keratinocytes and provide new insight into the
pathogenesis of skin cancer. Cancer Prev Res; 5(12); 1394–404. �2012 AACR.
IntroductionNonmelanoma skin cancer (NMSC) is the most frequent
malignancy worldwide, with more than 1 million casesdiagnosed each year in the United States alone (1). Squa-mous cell carcinoma (SCC) and basal cell carcinoma are themost common forms of NMSC and account for more than40% of newly diagnosed cancers (1). The most importantrisk factor for NMSC is UV radiation exposure, which is theleading cause of skin cell damage, photoaging, and malig-nant transformation (2). UV radiation from sunlight iscomposed of UVB (280–315 nm) and UVA (315–400 nm).UVBwavelengths are themost energetic and account for the
majority of the biologically damaging effects from sunexposure. UVB is a complete carcinogen and has beenshown to act as a tumor initiator by inducing mutationsin critical target genes and a tumor promoter by activatingsignal transduction pathways mediated by kinases andtranscription factors that induce cell-cycle progression andproliferation (3).
mTOR, an essential serine/theronine kinase conserved inall eukaryotes, is at the center of an increasingly intricatesignaling network that responds to nutrients, growth factors,and cellular stress (4). Hyperactivation of mTOR signalingthrough mutational activation of upstream signals, such asRas or phosphoinositide 3-kinase (PI3K), occurs in manycancers and contributes to increased proliferation andreduced sensitivity to apoptotic stimuli (4, 5). mTOR existsin at least two functionally and compositionally distinctprotein signaling complexes: the rapamycin-sensitivemTORcomplex 1 (mTORC1) and the rapamycin-resistant mTORcomplex 2 (mTORC2). The mTORC1 complex, consistingof mTOR, Raptor, and mLST8, is downstream of both theRaf/MEK/ERK and PI3K/Akt pathways and controls proteinsynthesis and ribosome biogenesis by direct phosphoryla-tion of eukaryotic initiation factor 4E binding proteins(4EBPs) and p70 S6 kinase 1 (S6K), respectively (6). ThemTORC2 complex also contains mTOR and mLST8, but
Authors' Affiliations: 1Department of Cellular and Molecular Physiology,The Pennsylvania State University College of Medicine, Hershey, Penn-sylvania; and 2DivisionofPharmacology andToxicology andDepartment ofNutritional Science, The University of Texas at Austin, Austin, Texas
Note:Supplementary data for this article are available atCancer PreventionResearch Online (http://cancerprevres.aacrjournals.org/).
Corresponding Author: Lisa M. Shantz, Department of Cellular andMolecular Physiology H166, The Pennsylvania State University College ofMedicine, 500University Drive, Hershey, PA 17033. Phone: 717-531-1562;Fax: 717-531-7667; E-mail: [email protected].
doi: 10.1158/1940-6207.CAPR-12-0272-T
�2012 American Association for Cancer Research.
CancerPreventionResearch
Cancer Prev Res; 5(12) December 20121394
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

Raptor is replaced by Rictor and mSin1. While the functionof mTORC2 is less well understood, it has been shown tophosphorylate AKT at Ser473, thus promoting cell survivaland proliferation (7). Phosphorylation of AKT by mTORC2can also serve to activate mTORC1, emphasizing the inter-dependence of the two mTORCs (8).Several studies indicate that mTOR signaling may play a
critical role in NMSC development. Immunohistochemicalanalysis of human epidermal tumors showed that mTORitself as well as its downstream effectors 4EBP1, S6K, andAKTSer473 are phosphorylated at much higher levels in SCCand precancerous actinic keratosis than normal skin (9).More recently, reverse phase protein microarray analysis ofSCC, actinic keratosis, and normal skin revealed aberrantlyactivated mTOR pathways in the precancerous and trans-formed tissues (10). Data compiled frommore than 30,000kidney transplant recipients found the use of mTOR inhibi-tors asmaintenance immunosuppressive therapyproducedaremarkable reduction inNMSC incidence comparedwith thecalcineurin inhibitor cyclosporine A (CsA; ref. 11). A dra-matic decrease in the incidence of skinmalignancieswas alsoobserved in transplantpatientswhowere converted tomTORinhibitors after 3 months of treatment with CsA (the Siroli-mus Renal Conversion Trial study; ref. 12). These strikingresults have led to several ongoing prospective randomizedtrials evaluating the use of mTOR inhibitors in renal trans-plant patients, and could significantly impact prevention ofNMSC in the general population as well.Photocarcinogensis is characterized by the inhibition of
apoptosis and the enhancement of cell proliferation.Although studies have shown that UVB induces phosphor-ylation of 4EBP1, S6K, and AKT (13–15), the specific rolesof mTORC1- and mTORC2-dependent pathways in UVB-induced proliferation and apoptosis have not been defined.In the present study, we examined the effect of UVB radi-ation on mTOR signaling in cell culture and in vivo usingboth the specific mTORC1 inhibitor rapamycin and agenetic model of conditional mTOR deletion in the epider-mis, which results in inhibition of both mTORC1 andmTORC2. Our findings indicate that UVB stimulates bothmTORC1 and mTORC2 activities. While rapamycin treat-ment effectively blocked the hyperproliferation responsethat occurs with UVB exposure, keratinocytes were sensi-tized to UVB-induced apoptosis only whenmTORC2 activ-ity was downregulated. Results obtained using rictor-nullcells confirmed that intact mTORC2 is necessary to activateprosurvival signaling. These studies thus provide newinsights into the molecular mechanisms of UVB-induceddamage leading to skin aging and skin cancer.
Materials and MethodsCell culture and drug treatmentHaCaT keratinocytes (obtained fromTheGermanCancer
Research Center), were maintained in Dulbecco’s modifiedEagle’s medium (DMEM, Invitrogen) supplemented with10% heat-inactivated FBS (Atlanta Biologicals) and peni-cillin/streptomycin (100 mg/mL, Invitrogen), and culturedfor less than 20 passages. Control (RictorEx3cond/w) and
rictor-null (RictorEx3del/Ex3del) mouse embryo fibroblasts(MEF), abbreviated Rictorþ/þ and Rictor�/�, were a gener-ous gift from Dr. Mark Magnuson, Vanderbilt University(Nashville, TN; ref. 16) and maintained in DMEM supple-mented with 10% FBS and penicillin/streptomycin. Nofurther characterization of cell lineswas conducted. Primarymouse keratinocytes were isolated from 1- to 3-day-oldpups as described previously (17). Briefly, full thicknessskin was floated overnight at 4�C in Caþ2-free 0.25%Trypsin without EDTA (Cellgro) to separate the epidermisfrom the dermis. Epidermal sheets wereminced and the cellsuspension was strained (100-mm nylon filter, Fisher) andplated in keratinocyte growth medium [calcium-free mini-mum essential medium (MEM) Eagle with Earle’s balancedsalt solution (BSS), Gln and nonessential amino acids(Lonza), 8% chelexed FBS (Gibco), and penicillin/streptomycin]. Cells were maintained in 7% CO2 at 37�C,and the medium was changed every other day. Rapamycin(Developmental Therapeutics Program, National CancerInstitute, Bethesda, MD) or vehicle dimethyl sulfoxide(DMSO) was added to medium 1 hour before UVB treat-ment. Primary keratinocyte cultures from transgenic animals(K5-CreERT2;mTORfl/fl and mTORfl/fl) were supplementedwith 5 nmol/L of 4-hydroxytamoxifen (4OHT, Sigma) for 4days to induce recombination.
Animals and drug treatmentAll experiments involving mice were carried out in com-
pliance with the Guide for the Care and Use of LaboratoryAnimals and protocols were approved by the Animal Careand Use Committee of the Pennsylvania State UniversityCollege of Medicine (Hershey, PA).
Formouse experiments, the dorsal surface of FVB/Nmice(6–8 weeks) was shaved with electrical clippers and micewere allowed to rest for 24 to 48 hours before all experi-ments. For rapamycin in vivo studies, mice were treatedtopically with 100 nmol rapamycin [in 100 mL DMSO:Acetone (1:9), D:A] or vehicle 1 hour before UVB exposure.We used an inducible Cre-LoxP mouse model to ablatemTOR in the epidermis. Floxed mTOR mice (mTORfl/fl)contain LoxP sites flanking exons 49 and 50 of the mTORgene (18); recombination results in a frameshift mutationand loss of the essential kinase domain. K5-CreERT2 mice(19) express a tamoxifen-activated Cre recombinase fusedto a modified estrogen receptor in the basal layer of theepidermis under the control of the keratin 5 promoter. K5-CreERT2 mice were bred with mTORfl/fl mice to ultimatelygenerate mice hemizygous for the K5-Cre-ERT2 transgeneand homozygous for the mTOR floxed allele (K5-CreERT2;mTORfl/fl). K5-CreERT2;mTORfl/fl, and mTORfl/fl controlswere treated topically with 1 mg 4OHT (in 100 mL D:A) orvehicle daily for 5 consecutive days. All animals were back-crossed for at least 9 generations onto the FVB/N back-ground. Primer sequences used for PCR are as follows:mTOR 1 (wild-type, LoxP, DLoxP): GTC CAC CAA CTCGGG CCT CAT T, mTOR 2 (wild-type, LoxP): GCA TGGCGA GGA CAT GTC A, and mTOR 3 (DLoxP): CCA CGCATG GCC CAC TGT CTT T.
mTOR Affects the Keratinocyte Response to UVB
www.aacrjournals.org Cancer Prev Res; 5(12) December 2012 1395
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

UVB treatmentCells were cultured for 48 to 72 hours until 70% conflu-
ence on 60-mmplates under normal culture conditions. Forcell-cycle experiments (low-dose UVB), medium wasreplaced with 0.1% FBS for 24 hours before UVB treatment.Cells were washed twice with PBS, then in a minimalvolume of PBS exposed to UVB (FS20 UVB bulbs, NationalBiological) emitting UV light between 290 to 320 nm. Theirradiation intensity was monitored using a UVB 500Cmeter (National Biological). After irradiation, PBS wasremoved and saved medium with drug treatments wasadded back. For in vivo studies, 3 to 4 mice were used foreach treatment group and housed together. Mice wereexposed to UVB irradiation from UVB lamps (FS20 UVBbulbs, National Biological) at a dose of 120 mJ/cm2. Bulbintensity was measured at the beginning of eachexperiment.
Western blottingFor in vitro studies, cells were washed twice with cold PBS
and harvested by scraping into the radioimmunoprecipita-tion assay (RIPA) buffer (Santa Cruz Biotechnology), cen-trifuged, and supernatants collected. In selected experi-ments examining apoptosis, detached cells were collectedvia centrifugation of medium and combined with theattached cells in RIPA buffer. For in vivo studies, dorsal skinwas treated with a depilatory agent for 3 minutes, washed,excised, and underlying fat and connective tissue wereremoved. Whole skin samples were flash-frozen and laterprocessed in RIPA buffer by homogenizing for 30 secondson ice using a Polytron homogenizer, centrifuged at 30,000� g for 30 minutes at 4�C, and supernatants collected.Epidermal samples were collected by scraping the surfaceof excised skin with a razor blade and placed into ice-coldRIPA buffer. Protein concentrations were determined usingBio-Rad assay. Equal amounts of protein were subjected toelectrophoresis. Western blotting was conducted asdescribed previously (20). Antibodies used include AKT,p-AKTSer473, S6K, p-S6KThr389, caspase-3, cleaved caspase-3,mTOR, actin (all from Cell Signaling, 1:1,000) and glycer-aldehyde-3-phosphate dehydrogenase (GAPDH; Protein-tech, 1:2,000).
Flow cytometryFor cell-cycle analysis, cells were harvested 18 hours after
UVB and fixed with ice-cold ethanol overnight. Cells werewashed twice with PBS and then incubated with propidiumiodide (20 mg/mL) containing RNase A (Sigma). The DNAcontent was determined using a FACSCalibur cytometer(Beckman Coulter) and analyzed with Modfit LT software(Verify Software). Apoptosis was assessed with flow cyto-metry using the PEAnnexin VApoptosis Detection Kit I (BDBiosciences) according to the manufacturer’s instructions.
Cell viabilityThe cell viability at 24 hours after UVB exposure was
determined colorimetrically by MTS assay (CellTiter 96Aqueous Proliferation Assay, Promega) according to the
manufacturer’s instructions andmonitored at 490nmusinga model 3550-UV plate reader (Bio-Rad). Each experimentwas carried out in triplicate and the experiments wererepeated at least 3 times.
Histologic analysisTissue sections were collected from 3 to 4 mice for each
condition at each timepoint (mock animals were not irra-diated and collected at 24 hours). Dorsal skin was treatedwith a depilatory agent for 3 minutes, washed, excised, andfixed overnight in 10% neutral buffered formalin. Skin wasembedded in paraffin and 5 mm sections were cut forimmunohistochemistry. The effect of rapamycin andmTORdeletion onUVB-mediated epidermal hyperplasia was stud-ied by histopathologic examination of hematoxylin andeosin (H&E)-stained tissue sections. Epidermal thicknesswas measured at 5 locations in 4 different sections for eachmouse. Epidermal proliferationwas assessed in vivo using 5-bromo-2-deoxyuridine (BrdUrd) incorporation. Micereceived intraperitoneal injections of BrdUrd (Sigma) at100 mg/g body weight in 0.9% NaCl 1 hour before killing.Sections were deparaffinized, rehydrated, and stained withan anti-BrdUrd antibody as described previously (21).Epidermal proliferation index (PI) was determined by cal-culating the percentage of basal cells positive for BrdUrd.A minimum of 1,000 basal cells were counted. Apoptosiswas assessed using immunohistochemical staining withcleaved caspase-3 antibody. Interfollicular apoptotic kera-tinocytes were counted microscopically in at least 5 non-overlapping low-power fields.
Statistical analysisData are expressed as the mean of at least 3 independent
experiments analyzedby2-sidedStudent t test. AP valueof<0.05 was considered significant.
ResultsRapamycin inhibits UVB-induced cell-cycleprogression and proliferation
In photocarcinogenesis, UVB-stimulated activation ofserine/theronine and tyrosine kinase signaling pathwaysand transcription factors mediate a number of pathologicchanges in the skin. The resulting keratinocyte prolifer-ation is responsible for epidermal hyperplasia and tumorpromotion (3). We first sought to investigate the role ofmTORC1 in UVB-induced cell-cycle progression and pro-liferation using rapamycin. It was previously shown thatsubapoptotic doses of UVB (2.5–10 mJ/cm2) activate EGFreceptor and AKT and induce G1–S cell-cycle progressionin serum-deprived HaCaT cells (22). To determine wheth-er mTORC1 contributes to UVB-induced cell-cycle pro-gression, HaCaT cells were serum-starved to synchronizethem in G0 and then subjected to low-dose UVB. Asshown in Fig. 1A, low-dose UVB activates both mTORC1and mTORC2 signaling pathways as measured by phos-phorylation of S6K and AKTSer473, respectively. To deter-mine whether mTORC1 activation is involved in UVB-mediated cell-cycle progression, cells were incubated with
Carr et al.
Cancer Prev Res; 5(12) December 2012 Cancer Prevention Research1396
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

50 nmol/L rapamycin to block mTORC1 activation.Rapamycin suppressed UVB-induced S6K phosphoryla-tion, but did not alter phosphorylation of AKTSer473 (Fig.1B). Low-dose UVB stimulated cell-cycle progression invehicle-treated cells, as measured by the percentage ofcells in S-phase at 18 hours after irradiation (Fig. 1C). Theproportion of cells in the S-phase decreased significantlyin cells treated with rapamycin (Fig. 1C).Because cellular proliferation in response to UVB is a
key feature of photocarcinogenesis, we next sought toexplore the role of mTORC1 in UVB-mediated responsesin vivo. Wild-type FVB/N adult mice were treated topicallywith rapamycin (100 nmol) 1 hour before irradiationwith a single doses of UVB (120 mJ/cm2), and whole-skinsamples were subjected to immunoblot analysis at 6hours after UVB exposure to assess mTOR activation.Concentrations of p-S6K and p-AKTS473 were too low inmock-irradiated samples to discern any noticeable inhib-itory effect of rapamycin (Fig. 2A). However, there was adramatic increase in p-S6K and p-AKTSer473 levels invehicle-treated skin after UVB irradiation. Rapamycinpretreatment prevented UVB-stimulated phosphorylationof S6K, but had no inhibitory effect on p-AKTSer473.Epidermal hyperplasia (as measured by epidermal thick-ness) and proliferation (as measured by BrdUrd incor-poration) were examined in skin sections at 24 and 48hours after UVB exposure. Rapamycin had no effect onepidermal thickness or proliferation in mock-irradiatedanimals. There was noticeable epidermal thickening invehicle-treated animals at 24 and 48 hours following UVBexposure, but this effect was significantly reduced inanimals treated with rapamycin (Fig. 2B and C). Similar
blocking effects were seen with rapamycin when epider-mal thickening was stimulated by treatment with thechemical tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA, see Supplementary Fig. S1). BrdUrdlabeling of actively proliferating cells at 48 hours showeda similar pattern. Vehicle-treated mice showed a signifi-cant increase in BrdUrd-positive cells at 48 hours afterUVB exposure, and this effect was significantly reducedby rapamycin treatment (Fig. 2D, E). Collectively, theseresults and those in Fig. 1 suggest that mTORC1 plays animportant role in keratinocyte cell-cycle progression andepidermal hyperproliferation following UVB irradiation.
Inducible mTOR deficiency inhibits UVB-stimulatedepidermal proliferation
We next explored the consequences of deletion of mTORin the basal layer of the skin, targeting both mTORC1 andmTORC2 signaling, to analyze the contribution of bothmTOR-signaling complexes in UVB-mediated responses.Because homozygous deletion of mTOR is lethal in utero(23), we used a transgenic system with 4OHT-inducibledeletion ofmTOR in the epidermis (K5-CreERT2;mTORfl/fl).We verified that the topical treatment with 4OHT, but notvehicle, leads to CreERT2-dependent recombination of themTOR allele (DLoxP) by PCR analysis using DNA from thedorsal epidermis harvested frommice 2weeks after the final4OHT treatment (Fig. 3A). No recombinationwas observedin the absence of either CreERT2 expression or 4OHT induc-tion (data not shown). Western blot analysis confirmedreduction of mTOR protein levels in 4OHT-treated K5-CreERT2;mTORfl/fl animals (Fig. 3B). Downregulation ofboth mTORC1 and mTORC2 signaling pathways was
Figure 1. Rapamycin attenuates UVB-induced cell-cycle progression in HaCaT cells. HaCaT cells at 70% confluence in 60-mm dishes were starved in 0.1%FBS DMEM for 24 hours and then exposed to 2.5, 5, or 10 mJ/cm2 or mock UVB irradiation. A, immunoblot analysis of mTORC1 and mTORC2activation markers in HaCaT cells exposed to sublethal doses of UVB (2.5, 5, 10 mJ/cm2). B, immunoblot analysis of mTOR activation markers at 2 hourspostirradiation of HaCaT cells pretreated with rapamycin (Rapa, 50 nmol/L) or vehicle (DMSO). C, cell cycle analysis of S-phase analyzed by flow cytometry18 hours following UVB radiation exposure (mean � SEM). �, P < 0.05; ��, P < 0.01. All data are representative from 2 to 5 independent experiments.
mTOR Affects the Keratinocyte Response to UVB
www.aacrjournals.org Cancer Prev Res; 5(12) December 2012 1397
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

verified by examining phosphorylation of S6K andAKTSer473 in whole skin protein extracts. In the absenceof UVB stimulation of mTOR pathways, there was noapparent difference between the vehicle-treated and4OHT-treated animals (Fig. 3C). However, p-S6K andp-AKTser473 levels were dramatically increased at 6 hoursafter UVB (120 mJ/cm2) in vehicle-treated mice. This effectwas significantly attenuated upon treatment of K5-CreERT2;mTORfl/fl animals with 4OHT (Fig. 3C), confirming that4OHT treatment in K5-CreERT2;mTORfl/fl mice was suffi-cient to block UVB-induced activation of both mTORC1andmTORC2.Histologic evaluation of epidermal thicknessandproliferation index inmock-irradiated animals revealedno differences between vehicle-treated and 4OHT-treatedanimals (Fig. 3D–G). UVB irradiation caused a significantincrease in epidermal thickness in vehicle-treated K5-CreERT2;mTORfl/fl mice at 24 and 48 hours, whereas micetreated with 4OHT (Fig. 3D and E) showed much lessepidermal thickening in response to UVB. K5-CreERT2;mTORfl/flmice treated with 4OHT also showed a significant
reduction in proliferation index at 48 hours after UVBtreatment compared with vehicle-treated controls (Fig. 3Fand G). All mice used as controls in these experimentsresponded similarly to each other and to wild-type miceat all timepoints, including mTORfl/fl mice treated with4OHT, mTORfl/fl mice treated with vehicle, and K5-CreERT2;mTORfl/fl mice treated with vehicle (see Supple-mentary Fig. S2).
Rapamycin does not alter UVB-mediated cell deathBecause one of the major activities of AKT is to promote
cell survival (24), we sought to investigate whether UVB-induced mTOR activation of AKT-dependent pathwaysplays a role in preventing cell death following high-doseUVB. We investigated mTOR signaling pathways after 50mJ/cm2 UVB exposure by examining the expression of totalandphosphorylated S6KandAKT innormalmouseprimarykeratinocytes by Western blot analysis. Enhanced signalingthrough both mTORC1 and mTORC2 was indicated byincreased levels of p-S6K and p-AKTSer473 at 30 minutes
Figure 2. Rapamycin inhibits UVB-induced epidermal hyperproliferation in vivo. FVB/N mice at 7 weeks of age were treated topically with rapamycin(Rapa; 100 nmol) or vehicle 1 hour before being exposed to 120 mJ/cm2 or mock UVB irradation. A, representative immunoblot analysis of mTORC1and mTORC2 activation markers in whole-skin extract harvested 6 hours following UVB radiation exposure from 3 independent experiments. B,representative H&E images of skin sections (scale bar ¼ 50mm). C, quantification of epidermal thickness (mean � SEM) for 3 to 4 mice/group; �, P < 0.05.D, representative BrdUrd staining images (scale bar ¼ 50mm). E, quantification of BrdUrd PI (mean � SEM) for 3 to 4 mice/group; �, P < 0.05.
Carr et al.
Cancer Prev Res; 5(12) December 2012 Cancer Prevention Research1398
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

after 50 mJ/cm2 UVB and remained upregulated at 2hours (Fig. 4A). Pretreatment of cells with rapamycin for1 hour completely blocked UVB-induced activation ofmTORC1 signaling (p-S6K), but had little, if any, effecton mTORC2 activity (p-AKTSer473; Fig. 4B). To determinewhether inhibition of mTORC1 could sensitize keratino-cytes to apoptosis, cells exposed to various rapamycinconcentrations were harvested 24 hours after UVB expo-sure and assayed for viability. Treatment with a widerange of rapamycin concentrations did not alter cellviability in mock-irradiated cells (Fig. 4C). Exposure to50 mJ/cm2 UVB resulted in an obvious decrease in cellviability (54% � 3.6%). However, rapamycin did notenhance UVB-mediated cell death in wild-type primarykeratinocytes (Fig. 4C), and this same result was seen inHaCaT cells (Supplementary Fig. S3A). In addition,increasing the pretreatment time with rapamycin to 24hours did not enhance UVB-mediated cell death (Sup-plementary Fig. S3B and S3C). Western blot analysisof cleaved caspase-3 verified that there was no increasein UVB-mediated apoptosis with rapamycin treatment(Fig. 4D).
Inducible mTOR deficiency sensitizes keratinocytes toUVB-induced apoptosis
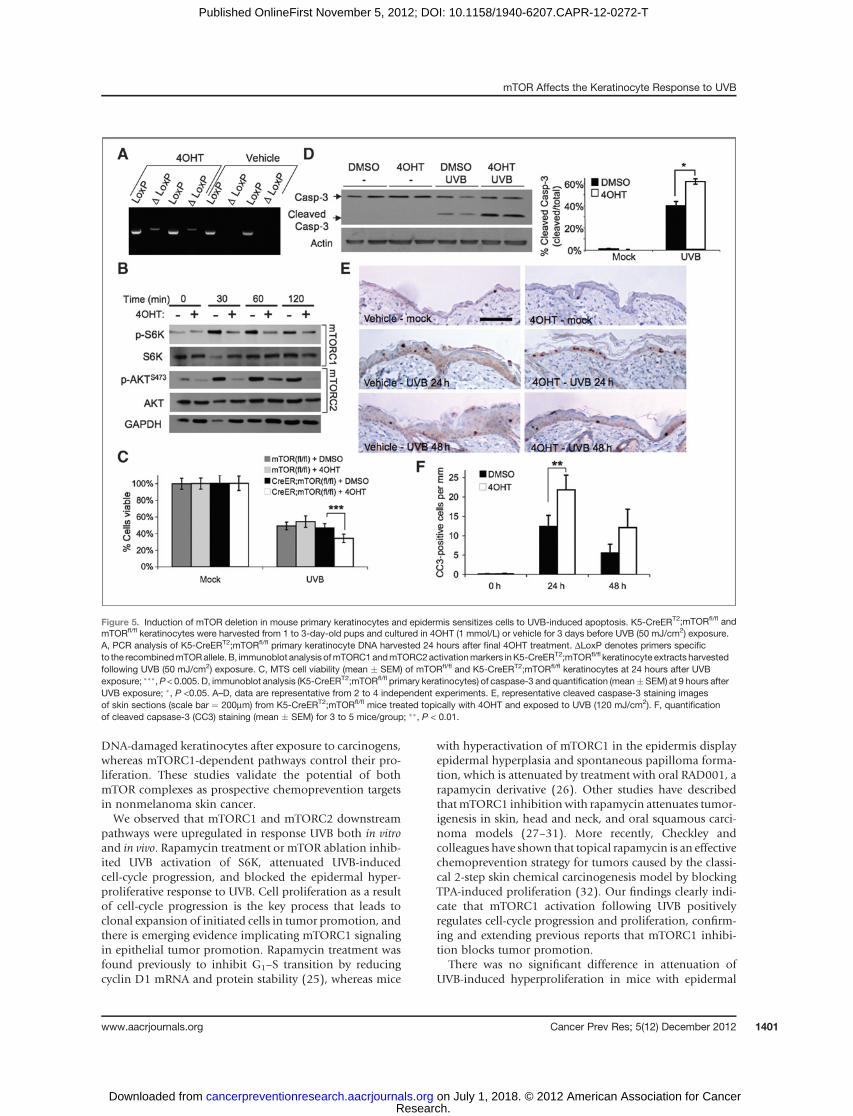
To investigate the possible role of mTORC2 in suppres-sing apoptosis after UVB exposure, we used primary kera-tinocytes isolated from K5-CreERT2;mTORfl/fl mice. PCRanalysis verified recombination of themTOR allele (DLoxP)in cells culturedwith 4OHT, but not vehicle (Fig. 5A). Therewas no obvious difference in mTORC1 and mTORC2activities in 4OHT-treated keratinocytes not exposed toUVB(0 minutes), as measured by p-S6K and p-AKTSer473 (Fig.5B).However, when keratinocyteswere exposed toUVB (50mJ/cm2) to activate both mTOR complexes, a dramaticreduction in phosphorylation of both S6K and AKTser473
was observed in 4OHT-treated cells compared with vehicle,confirming downregulation of both mTORC1 andmTORC2 signaling (Fig. 5B).Unlike inhibition ofmTORC1with rapamycin (Fig. 4C), deletion of mTOR enhancedUVB-induced cell death (Fig. 5C). Though there was nodifference in cell viability in mock-irradiated cells, K5-CreERT2;mTORfl/fl keratinocyte cultures treated with 4OHTcontained significantly fewer viable cells 24 hours after UVBexposure compared with both vehicle-treated K5-CreERT2;
Figure 3. Induction of mTOR deletion in mouse epidermis suppresses UVB-stimulated epidermal proliferation. K5-CreERT2;mTORfl/fl mice at 7 weeks of agewere treated topically with 4OHT (1 mg) or vehicle (D:A) daily for 5 days. A, PCR analysis of epidermal DNA harvested 14 days after final 4OHT treatment.DLoxP denotes primers specific to the recombined mTOR allele. B, immunoblot analysis of mTOR in epidermal extracts harvested at 7 or 14 daysafter final 4OHT treatment. C, immunoblot analysis of mTORC1 and mTORC2 activation markers in whole-skin extracts harvested 6 hours following UVB(120 mJ/cm2) radiation exposure. A–C, data are representative of 2 to 3 independent experiments. D, representative H&E images of skin sections(scale bar ¼ 50mm). E, quantification of epidermal thickness (mean � SEM) for 3–4 mice/group; �, P < 0.05; ���, P < 0.005. F, representative BrdUrd stainingimages (scale bar ¼ 50mm). G, quantification of BrdUrd PI (mean � SEM) for 3 to 4 mice/group; ��, P < 0.01.
mTOR Affects the Keratinocyte Response to UVB
www.aacrjournals.org Cancer Prev Res; 5(12) December 2012 1399
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

mTORfl/fl and 4OHT-treated mTORfl/fl cells. To determinewhether the significant decrease in cell viability was due toapoptosis, immunoblot analysis of cleaved caspase-3 wasexamined (Fig. 5D). Cell extracts harvested at 9 hours afterUVB showed significantly higher levels of cleaved caspase inmTOR-ablated keratinocytes than in vehicle-treated K5-CreERT2;mTORfl/fl cells.
We further investigated the effects ofmTORdeficiency onUVB-meditated apoptosis in vivo using our K5-CreERT2;mTORfl/fl mice. UVB irradiation increased the number ofepidermal cleaved caspase-3–positive cells 24 and 48 hoursafter irradiation in K5-CreERT2;mTORfl/fl mice treated withvehicle. The number of apoptotic cells was significantlyincreased in the epidermis of mTOR-deficient mice (4OHTtreated) comparedwith vehicle controls at 24 hours (Fig. 5Eand F).
mTORC2 disruption sensitizes cells to UVB-inducedapoptosis
Our results show that K5-CreERT2;mTORfl/fl primary ker-atinocytes treated with 4OHT to induce mTOR deletionhave enhanced sensitivity to UVB-induced apoptosis (Fig.5), but wild-type primary keratinocytes treated with themTORC1 inhibitor do not (Fig. 4). These data suggest thatmTORC2, but not mTORC1, influences cell survival signal-ing following UVB exposure. To further elucidate the spe-cific role of mTORC2 in UVB-induced apoptosis, we usedrictor-null MEFs (Rictor�/�). UVB exposure (50 mJ/cm2)increased phosphorylation of S6K and AKTSer473 in wild-type MEFs (Rictorþ/þ; Fig. 6A). UVB induced p-S6K in a
similar manner in Rictor�/� cells, but p-AKTSer473 wascompletely absent, illustrating the loss of mTORC2 signal-ing (Fig. 6A). Significantly, fewer rictor-null cells were viablecompared with wild-type cells at 24 hours after UVB expo-sure (Fig. 6B). To determine whether this represents anincreased sensitivity to apoptosis in the absence ofmTORC2 signaling, cells were analyzed using Annexin Vby flow cytometry. There was a significant increase in thepercentage of apoptotic Rictor�/� cells following UVB com-pared with wild-type cells (Fig. 6C). Themarked increase inapoptosis was verified byWestern blot analysis of caspase-3.The results show that cleaved caspase-3 begins to accumu-late in Rictor�/� cells by 6 hours and continues to increaseup to 12 hours after UVB exposure (Fig. 6D). In contrast,wild-type MEFs show considerably less caspase-3 cleavageover the same time course. Taken together with the resultspresented in Figs. 4 and 5, these data are consistent with theidea that downregulation of mTORC2 signaling sensitizescells to UVB-induced apoptosis.
DiscussionBetter understanding of the signal transduction path-
ways activated by UVB in keratinocytes is essential foreffective prevention of skin cancer. Using rapamycin toinhibit mTORC1, and a Cre/LoxP approach to block bothmTORC1 and mTORC2 signaling, the present study showsthat the two mTOR complexes play distinct roles in medi-ating UVB-induced proliferation and prosurvival sig-naling in the epidermis. Our results fit a model in whichmTORC2-dependent pathways maintain the survival of
Figure 4. Rapamycin does notsensitize keratinocytes to UVB-induced cell death. Wild-typeprimary keratinocytes wereharvested from 1- to 3-day-oldpups and plated in low-calciummedium. When confluent, cellswere treated with rapamycin(Rapa) for 1 hour and exposed to50 mJ/cm2 UVB irradiation. A,immunoblot analysis of mTORC1andmTORC2activationmarkers inprimary keratinocytes exposed toUVB. B, immunoblot analysis ofmTOR activation markers at 2hours postirradiation of cellspretreated with various doses ofrapamycin. C, MTS cell viability at24 hours after UVB exposure. D,immunoblot analysis of cleavedcaspase-3, a marker of apoptosis,at 9 hours after UVB exposure. Alldata are representative from 3 to 5independent experiments.
Carr et al.
Cancer Prev Res; 5(12) December 2012 Cancer Prevention Research1400
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T
DNA-damaged keratinocytes after exposure to carcinogens,whereas mTORC1-dependent pathways control their pro-liferation. These studies validate the potential of bothmTOR complexes as prospective chemoprevention targetsin nonmelanoma skin cancer.We observed that mTORC1 and mTORC2 downstream
pathways were upregulated in response UVB both in vitroand in vivo. Rapamycin treatment or mTOR ablation inhib-ited UVB activation of S6K, attenuated UVB-inducedcell-cycle progression, and blocked the epidermal hyper-proliferative response to UVB. Cell proliferation as a resultof cell-cycle progression is the key process that leads toclonal expansion of initiated cells in tumor promotion, andthere is emerging evidence implicating mTORC1 signalingin epithelial tumor promotion. Rapamycin treatment wasfound previously to inhibit G1–S transition by reducingcyclin D1 mRNA and protein stability (25), whereas mice
with hyperactivation of mTORC1 in the epidermis displayepidermal hyperplasia and spontaneous papilloma forma-tion, which is attenuated by treatment with oral RAD001, arapamycin derivative (26). Other studies have describedthatmTORC1 inhibitionwith rapamycin attenuates tumor-igenesis in skin, head and neck, and oral squamous carci-noma models (27–31). More recently, Checkley andcolleagues have shown that topical rapamycin is an effectivechemoprevention strategy for tumors caused by the classi-cal 2-step skin chemical carcinogenesis model by blockingTPA-induced proliferation (32). Our findings clearly indi-cate that mTORC1 activation following UVB positivelyregulates cell-cycle progression and proliferation, confirm-ing and extending previous reports that mTORC1 inhibi-tion blocks tumor promotion.
There was no significant difference in attenuation ofUVB-induced hyperproliferation in mice with epidermal
Figure 5. Induction of mTOR deletion in mouse primary keratinocytes and epidermis sensitizes cells to UVB-induced apoptosis. K5-CreERT2;mTORfl/fl andmTORfl/fl keratinocytes were harvested from 1 to 3-day-old pups and cultured in 4OHT (1 mmol/L) or vehicle for 3 days before UVB (50 mJ/cm2) exposure.A, PCR analysis of K5-CreERT2;mTORfl/fl primary keratinocyte DNA harvested 24 hours after final 4OHT treatment. DLoxP denotes primers specificto the recombinedmTORallele. B, immunoblot analysis ofmTORC1andmTORC2activationmarkers inK5-CreERT2;mTORfl/fl keratinocyte extracts harvestedfollowing UVB (50 mJ/cm2) exposure. C, MTS cell viability (mean � SEM) of mTORfl/fl and K5-CreERT2;mTORfl/fl keratinocytes at 24 hours after UVBexposure; ���,P < 0.005. D, immunoblot analysis (K5-CreERT2;mTORfl/fl primary keratinocytes) of caspase-3 and quantification (mean�SEM) at 9 hours afterUVB exposure; �, P <0.05. A–D, data are representative from 2 to 4 independent experiments. E, representative cleaved caspase-3 staining imagesof skin sections (scale bar ¼ 200mm) from K5-CreERT2;mTORfl/fl mice treated topically with 4OHT and exposed to UVB (120 mJ/cm2). F, quantificationof cleaved capsase-3 (CC3) staining (mean � SEM) for 3 to 5 mice/group; ��, P < 0.01.
mTOR Affects the Keratinocyte Response to UVB
www.aacrjournals.org Cancer Prev Res; 5(12) December 2012 1401
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

mTOR deletion compared with wild-type FVB/N micereceiving topical rapamycin treatment (P¼0.11), suggestingUVB-induced cell-cycle progression and proliferation arenot mediated by mTORC2 signaling. This does not, how-ever, preclude a role for mTORC2 in this process. Althoughwe see no evidence of mTORC2 inhibition in the presenceof rapamycin in this model, it is difficult to compare phar-macologic inhibition to genetic ablation. The effects ofmTOR deletion may be dampened because it is unlikelythat recombination and deletion occurred in all keratino-cytes within the epidermis. In addition, topical rapamycinhas been shown to decrease infiltration of dermal inflam-matory cells (32),whichmay contribute to inhibitory effectson keratinocyte proliferation and confound comparisonsto our genetic model of mTOR deletion in the skin.
NMSC pathogenesis is characterized by both enhance-ment of cell proliferation and inhibition of apoptosis.Induction of apoptosis following DNA damage is an essen-tial protective mechanism, ensuring the removal of dam-aged cells that may harbor oncogenic mutations. However,UVB also activates signaling cascades that promote thesurvival of these potentially cancerous cells. Previous workin othermousemodels has shown that targeting prosurvivalpathways induced by UVB increases the sensitivity of DNA-damaged keratinocytes to apoptotic signaling, and is suffi-cient to inhibit skin carcinogenesis (33). The studiesreported here investigate whether activation of mTORC1andmTORC2downstream effectors byUVB lead to changesin keratinocyte prosurvival signaling. The effects ofmTORC1 inhibition on apoptosis vary greatly dependingon the system used. Enhancement of AKT signaling canoccur in the presence of rapamycin due to relief of an S6K-dependent negative feedback loop targeting PI3K (34, 35).Loss of this feedback inhibition is thought to be responsiblefor increased mTORC2/AKT activation and decreased sen-sitivity to apoptotic stimuli in certain malignancies treated
with rapamycin (35).On theotherhand, there are anumberof studies that report enhancement of apoptosis by rapa-mycin (36–41). It has been shown that prolonged rapamy-cin treatment reducesmTORC2 complex assembly and AKTactivation in approximately 20% of cancer cell lines (42),which could have a direct affect on apoptosis pathways. It isthus possible that the proapoptotic effects of rapamycinseen in some previous studies are the result of mTORC2inhibition rather than a direct affect on mTORC1. Thisrationale is supported by our results. We see no affect ofrapamycin on mTORC2-dependent pathways in our sys-tem, and rapamycin treatment does not result in enhancedactivation of apoptosis in UVB-treated cells. In contrast,4OHT-induced mTOR deletion resulted in a significantincrease in apoptosis following UVB exposure in bothkeratinocyte culture and mouse epidermis. Furthermore,rictor-null cells were more sensitive to UVB-induced apo-ptosis than their wild-type counterparts. These results indi-cate that mTORC2 activation by UVB plays a critical role inmediating pathways that control keratinocyte survival, andreinforce previous observations of aberrant AKT activationin mouse models of NMSC (43, 44). mTORC2 was alsoidentified as a therapeutic target in prostate cancer inducedby loss of the tumor suppressor PTEN, using mice withconditional deletion of either mTOR or rictor (45, 46). Therole of mTORC2 in these tumor types may be to maintainhigh levels of p-AKTSer473, which results in decreased tran-scription of a number of FOXO1/3-dependent cell-cyclearrest and apoptotic genes (47, 48).
In summary, this is, to our knowledge, the first study toreport that mTORC1- and mTORC2-dependent pathwaysare both activated by UVB, and play unique roles in con-trolling proliferation and apoptosis in the skin. Theseresults emphasize the need to further elucidate the rolesof mTORC1 and mTORC2 in photocarcinogensis andtheir links to cell proliferation, apoptosis, and tumor
Figure 6. Loss of Rictor increasessensitivity of MEFs to UVB-induced apoptosis. Rictor wild-type (þ/þ) and knockout (-/-) MEFswere exposed to UVB (50 mJ/cm2)at 70% confluence. A, immunoblotanalysis of mTORC1 and mTORC2activation markers in cells exposedto UVB. B, MTS cell viability(mean � SEM) at 24 hours afterUVB exposure; ��, p < 0.01.C, Annexin-V flow cytometry(mean � SEM) at 24 hours afterUVB exposure. D, immunoblotanalysis of caspase-3 andquantification (mean � SEM);�, P < 0.05. All data arerepresentative from 2 to4 independent experiments.
Carr et al.
Cancer Prev Res; 5(12) December 2012 Cancer Prevention Research1402
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

development. Our data provide compelling evidence tosupport the novel hypothesis that both mTORC1 andmTORC2 act as critical mediators of UVB-activated signaltransduction in keratinocytes and suggest that the com-bined targeting of both mTOR complexes, or alternativelymTORC1 and AKT, may be an effective chemopreventionstrategy against photocarcinogensis.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: T.D. Carr, L.M. ShantzDevelopment of methodology: T.D. Carr, L.M. ShantzAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): T.D. Carr, J. DiGiovanni, C.J. Lynch, L.M. ShantzAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis): T.D. Carr, C.J. Lynch, L.M. Shantz
Writing, review, and/or revision of the manuscript: T.D. Carr, J. DiGio-vanni, C.J. Lynch, L.M. ShantzAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): T.D. CarrStudy supervision: L.M. Shantz
AcknowledgmentsThe authors thank Patricia Welsh for excellent technical support and Dr.
David J. Feith for his helpful suggestions and critical reading of themanuscript.
Grant SupportThis work was supported by NIH grants CA133945 (to L.M. Shantz),
ES19242 (to L.M. Shantz), DK62880 (to C.J. Lynch), CA37111 (to J.DiGiovanni), and the Pennsylvania Department of Health Tobacco CUREfunds (to L.M. Shantz). T.D. Carr is the recipient of an MD/PhD pre-doctoral fellowship (F30 ES19809).
Received June 22, 2012; revised September 25, 2012; acceptedOctober 10,2012; published OnlineFirst November 5, 2012.
References1. Bowden GT. Prevention of non-melanoma skin cancer by targeting
ultraviolet-B-light signaling. Nat Rev Cancer 2004;4:23–35.2. ErbP, Ji J, KumpE,MielgoA,WernliM.Apoptosis andpathogenesis of
melanoma and nonmelanoma skin cancer. Adv Exp Med Biol2008;624:283–95.
3. AnanthaswamyHN,PierceallWE.Molecularmechanismsof ultravioletradiation carcinogenesis. Photochem Photobiol 1990;52:1119–36.
4. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. CancerCell 2007;12:9–22.
5. Albanell J, Dalmases A, Rovira A, Rojo F. mTOR signalling in humancancer. Clin Transl Oncol 2007;9:484–93.
6. FingarDC,Blenis J. Target of rapamycin (TOR): an integrator of nutrientand growth factor signals and coordinator of cell growth and cell cycleprogression. Oncogene 2004;23:3151–71.
7. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation andregulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307:1098–101.
8. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response tocontrol cell growth and survival. Cell 2003;115:577–90.
9. Chen SJ, Nakahara T, Takahara M, Kido M, Dugu L, Uchi H, et al.Activation of themammalian target of rapamycin signalling pathway inepidermal tumours and its correlation with cyclin-dependent kinase 2.Br J Dermatol 2009;160:442–5.
10. Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, WarnekeJ,KrouseR, et al. Functional protein pathwayactivationmapping of theprogression of normal skin to squamous cell carcinoma. Cancer PrevRes 2012;5:403–13.
11. Kauffman HM, Cherikh WS, Cheng Y, Hanto DW, Kahan BD. Mainte-nance immunosuppression with target-of-rapamycin inhibitors isassociated with a reduced incidence of de novomalignancies. Trans-plantation 2005;80:883–9.
12. Alberu J, PascoeMD, Campistol JM, Schena FP, Rial Mdel C, PolinskyM, et al. Lower malignancy rates in renal allograft recipients convertedto sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation 2011;92:303–10.
13. LiuG, Zhang Y, Bode AM,MaWY,Dong Z. Phosphorylation of 4E-BP1is mediated by the p38/MSK1 pathway in response to UVB irradiation.J Biol Chem 2002;277:8810–6.
14. HuangC, Li J, KeQ, LeonardSS, JiangBH, ZhongXS, et al. Ultraviolet-induced phosphorylation of p70(S6K) at Thr(389) and Thr(421)/Ser(424) involves hydrogen peroxide and mammalian target of rapamycinbut not Akt and atypical protein kinase C. Cancer Res 2002;62:5689–97.
15. Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultravioletirradiation activates PI 3-kinase/AKT survival pathway via EGF recep-tors in human skin in vivo. Int J Oncol 2001;18:461–6.
16. Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelicdisruption of the rictor gene in mice reveals that mTOR complex 2 isessential for fetal growth and viability. Dev Cell 2006;11:583–9.
17. Lichti U, Anders J, Yuspa SH. Isolation and short-term culture ofprimary keratinocytes, hair follicle populations and dermal cellsfrom newborn mice and keratinocytes from adult mice for in vitroanalysis and for grafting to immunodeficient mice. Nat Protoc2008;3:799–810.
18. Lang CH, Frost RA, Bronson SK, Lynch CJ, Vary TC. Skeletalmuscle protein balance in mTOR heterozygous mice in responseto inflammation and leucine. Am J Physiol Endocrinol Metab 2010;298:E1283–E94.
19. Kataoka K, Kim DJ, Carbajal S, Clifford JL, DiGiovanni J. Stage-specific disruption of Stat3 demonstrates a direct requirement duringboth the initiation and promotion stages of mouse skin tumorigenesis.Carcinogenesis 2008;29:1108–14.
20. Origanti S, Shantz LM. Ras transformation of RIE-1 cells activates cap-independent translation of ornithine decarboxylase: regulation by theRaf/MEK/ERK and phosphatidylinositol 3-kinase pathways. CancerRes 2007;67:4834–42.
21. FeithDJ,BolDK,Carboni JM, LynchMJ,Sass-KuhnS,ShoopPL, et al.Induction of ornithine decarboxylase activity is a necessary step formitogen-activated protein kinase kinase-induced skin tumorigenesis.Cancer Res 2005;65:572–8.
22. Han W, He YY. Requirement for metalloproteinase-dependent ERKand AKT activation in UVB-induced G1-S cell cycle progression ofhuman keratinocytes. Photochem Photobiol 2009;85:997–1003.
23. Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF,et al. Disruption of the mouse mTOR gene leads to early postimplan-tation lethality and prohibits embryonic stem cell development. MolCell Biol 2004;24:9508–16.
24. Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: ahard Akt to follow. Trends Biochem Sci 2001;26:657–64.
25. Hashemolhosseini S, NagamineY,MorleySJ, DesrivieresS,Mercep L,Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated byeffects on cyclin D1 mRNA and protein stability. J Biol Chem 1998;273:14424–9.
26. Lu ZH, Shvartsman MB, Lee AY, Shao JM, Murray MM, Kladney RD,et al. Mammalian target of rapamycin activator RHEB is frequentlyoverexpressed in human carcinomas and is critical and sufficient forskin epithelial carcinogenesis. Cancer Res 2010;70:3287–98.
27. AmornphimolthamP, Leelahavanichkul K,Molinolo A, Patel V, GutkindJS. Inhibition of Mammalian target of rapamycin by rapamycin causesthe regression of carcinogen-induced skin tumor lesions. Clin CancerRes 2008;14:8094–101.
28. Wulff BC, Kusewitt DF, VanBuskirk AM, Thomas-Ahner JM, DuncanFJ,Oberyszyn TM. Sirolimus reduces the incidence andprogression of
mTOR Affects the Keratinocyte Response to UVB
www.aacrjournals.org Cancer Prev Res; 5(12) December 2012 1403
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

UVB-induced skin cancer in SKH mice even with co-administration ofcyclosporine A. J Invest Dermatol 2008;128:2467–73.
29. de Gruijl FR, Koehl GE, Voskamp P, Strik A, Rebel HG, Gaumann A,et al. Early and late effects of the immunosuppressants rapamycin andmycophenolate mofetil on UV carcinogenesis. Int J Cancer 2010;127:796–804.
30. AmornphimolthamP, Patel V, Sodhi A,NikitakisNG, Sauk JJ, SausvilleEA, et al. Mammalian target of rapamycin, a molecular target insquamous cell carcinomas of the head and neck. Cancer Res 2005;65:9953–61.
31. Raimondi AR,Molinolo A,Gutkind JS. Rapamycin prevents early onsetof tumorigenesis in an oral-specific K-ras and p53 two-hit carcino-genesis model. Cancer Res 2009;69:4159–66.
32. Checkley LA, Rho O, Moore T, Hursting S, DiGiovanni J. Rapamycin isa potent inhibitor of skin tumor promotion by 12-O-tetradecanoyl-phorbol-13-acetate. Cancer Prev Res 2011;4:1011–20.
33. Kim DJ, Kataoka K, Sano S, Connolly K, Kiguchi K, Digiovanni J.Targeted disruption of Bcl-x(L) in mouse keratinocytes inhibits bothUVB- and chemically induced skin carcinogenesis. Mol Carcinog2009;48:873–85.
34. Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al.Activation of Akt and eIF4E survival pathways by rapamycin-medi-ated mammalian target of rapamycin inhibition. Cancer Res 2005;65:7052–8.
35. O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTORinhibition induces upstream receptor tyrosine kinase signaling andactivates Akt. Cancer Res 2006;66:1500–8.
36. AvellinoR,RomanoS,ParasoleR,Bisogni R, Lamberti A, Poggi V, et al.Rapamycin stimulates apoptosis of childhood acute lymphoblasticleukemia cells. Blood 2005;106:1400–6.
37. Treeck O, Wackwitz B, Haus U, Ortmann O. Effects of a combinedtreatment with mTOR inhibitor RAD001 and tamoxifen in vitro ongrowth and apoptosis of human cancer cells. Gynecol Oncol 2006;102:292–9.
38. Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S. Rapamycin andUCN-01 synergistically induce apoptosis in human leukemia cellsthrough a process that is regulated by the Raf-1/MEK/ERK, Akt,
and JNK signal transduction pathways. Mol Cancer Ther 2005;4:457–70.
39. Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O'Reilly T,et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell2005;120:747–59.
40. ThimmaiahKN,Easton J,HuangS, VeverkaKA,GermainGS,HarwoodFC, et al. Insulin-like growth factor I-mediated protection from rapa-mycin-induced apoptosis is independent of Ras-Erk1-Erk2 and phos-phatidylinositol 30-kinase-Akt signaling pathways. Cancer Res2003;63:364–74.
41. Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S,et al. Survival signalling by Akt and eIF4E in oncogenesis and cancertherapy. Nature. 2004;428:332–7.
42. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF,et al. Prolonged rapamycin treatment inhibits mTORC2 assembly andAkt/PKB. Mol Cell 2006;22:159–68.
43. Suzuki A, Itami S, Ohishi M, Hamada K, Inoue T, Komazawa N, et al.Keratinocyte-specificPten deficiency results in epidermal hyperplasia,accelerated hair follicle morphogenesis and tumor formation. CancerRes 2003;63:674–81.
44. Segrelles C, Lu J, Hammann B, Santos M, Moral M, Cascallana JL,et al. Deregulated activity of Akt in epithelial basal cells inducesspontaneous tumors and heightened sensitivity to skin carcinogene-sis. Cancer Res 2007;67:10879–88.
45. Guertin DA, StevensDM,SaitohM,Kinkel S, Crosby K, Sheen JH, et al.mTOR complex 2 is required for the development of prostate cancerinduced by Pten loss in mice. Cancer Cell 2009;15:148–59.
46. Nardella C, Carracedo A, Alimonti A, Hobbs RM,Clohessy JG, Chen Z,et al. Differential requirement of mTOR in postmitotic tissues andtumorigenesis. Sci Signal 2009;55:1–10.
47. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Aktphosphorylation and substrate specificity. Cell 2006;127:125–37.
48. Greer EL, Brunet A. FOXO transcription factors at the interfacebetween longevity and tumor suppression. Oncogene 2005;24:7410–25.
Carr et al.
Cancer Prev Res; 5(12) December 2012 Cancer Prevention Research1404
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T

2012;5:1394-1404. Published OnlineFirst November 5, 2012.Cancer Prev Res Theresa D. Carr, John DiGiovanni, Christopher J. Lynch, et al. Proliferation and SurvivalInhibition of mTOR Suppresses UVB-Induced Keratinocyte
Updated version
10.1158/1940-6207.CAPR-12-0272-Tdoi:
Access the most recent version of this article at:
Material
Supplementary
DC1
http://cancerpreventionresearch.aacrjournals.org/content/suppl/2012/11/05/1940-6207.CAPR-12-0272-T.Access the most recent supplemental material at:
Cited articles
http://cancerpreventionresearch.aacrjournals.org/content/5/12/1394.full#ref-list-1
This article cites 48 articles, 20 of which you can access for free at:
Citing articles
http://cancerpreventionresearch.aacrjournals.org/content/5/12/1394.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerpreventionresearch.aacrjournals.org/content/5/12/1394To request permission to re-use all or part of this article, use this link
Research. on July 1, 2018. © 2012 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst November 5, 2012; DOI: 10.1158/1940-6207.CAPR-12-0272-T