inherited kidney diseases zehra eren m.d. nephrology department
TRANSCRIPT

Inherited Kidney
Diseases
Zehra Eren M.D.Nephrology Department

LEARNING OBJECTIVES
Recognize
•Renal cystic disorders
-Autosomal dominant polycystic kidney disease
-Autosomal recessive polycystic kidney disease
-Medullary sponge kidney
-Medullary cystic kidney disease
-Tuberous sclerosis cpmplex
-von Hippel-Lindau disease

LEARNING OBJECTIVES
•Alport’s Syndrome
•Thin basement membrane disease
•Anderson-Fabry disease
•Nail-Patella Syndrome
•Sicle Cell Nephropathy



•EpidemiologyThe most common renal hereditary disease, affects 1 in 400 to 1,000 live births
Affects all races with equal frequence
AUTOSOMAL DOMINANT POLYCYSTIC
KIDNEY DISEASE (ADPKD)

Autosomal dominant polycystic kidney disease
•Prevalence: 1:300 to 1:1000
•90% of cases are inherited, 10% are
sporadic
•Only1 to 5% nephrons developed cysts
•Cysts are in medulla and cortex
•ADPKD causes symptoms in third or fourth
decade
•50% of patients developing ESRD by age 60
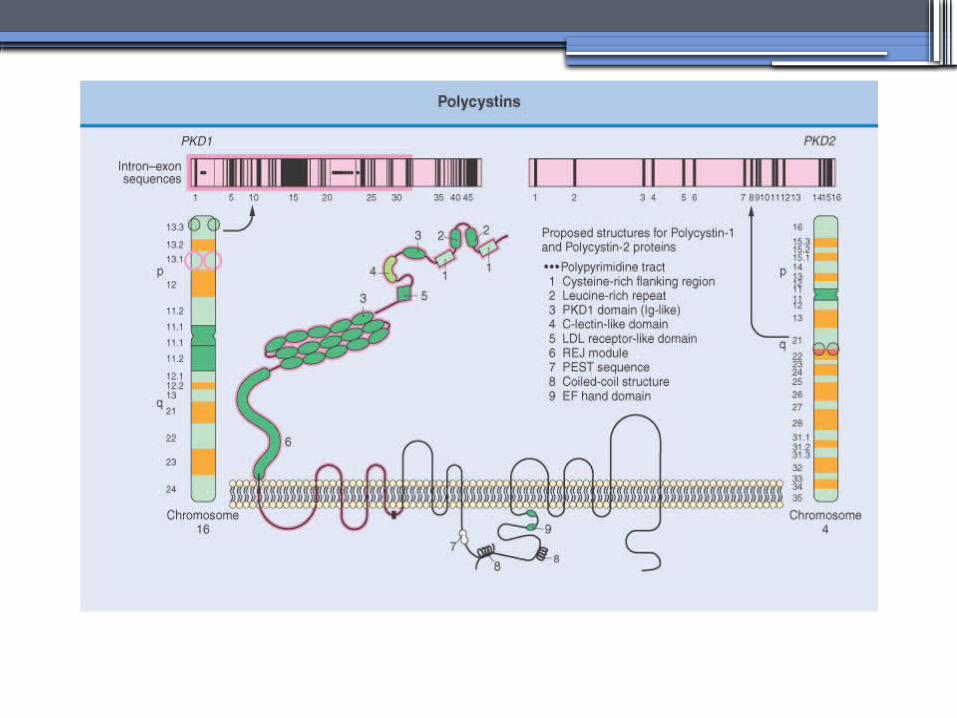

AUTOSOMAL DOMINANT POLYCYSTICKIDNEY DISEASE (ADPKD)
• Inheritance- Mutations in the polycystic kidney disease (PKD)1 gene:85%, located on the short arm of chromosome 16 (16p.3.3), codes for a 4,304-amino-acid protein (polycystin 1)
- Mutations in the PKD2 gene:15%, located on chromosome 4 (4q.21.2), codes for a 968-amino-acid protein (polycystin 2)

ADPKD
•Pathology- massive enlargement of the kidneys secondary to cyst growth and development
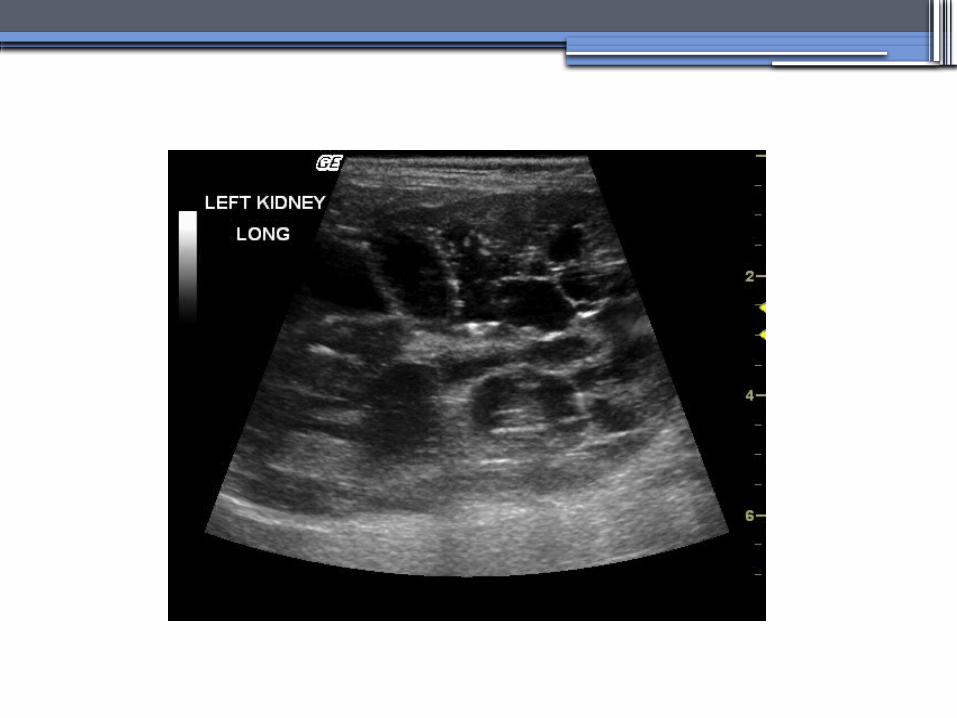
•Diagnosis-RadiologicRadiological imaging is required (ultrasoundis the current imaging modality of choice in at-risk individuals (positive family history in a parent).
-Genetic
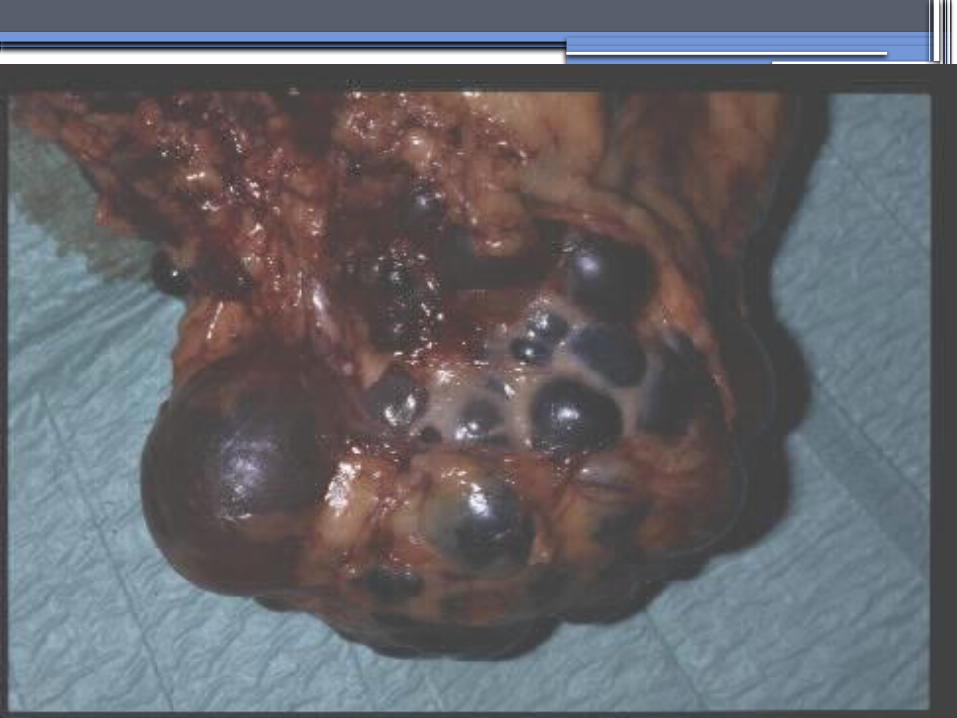


ADPKD
•Pathology- massive enlargement of the kidneys secondary to cyst growth and development
•Diagnosis-RadiologicRadiological imaging is required (ultrasoundis the current imaging modality of choice in at-risk individuals (positive family history in a parent).
-Genetic

Ravine Criteria for Diagnosing ADPKD

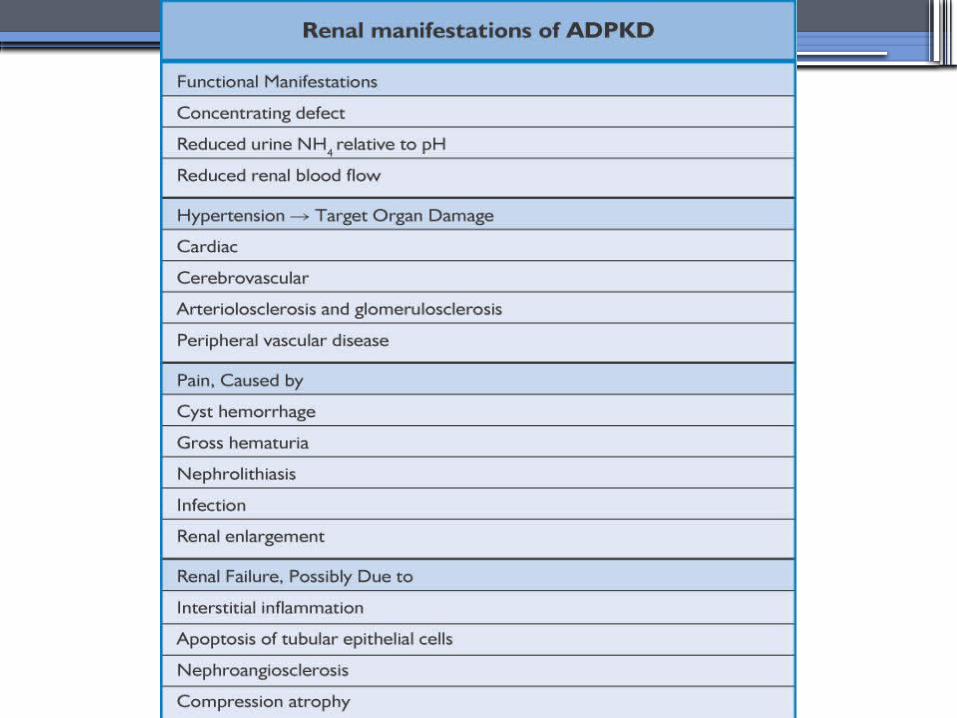

Clinical Manifestations• Renal
Chronic flank pain
Acute pain indicates:
infection (pyelonephritis- pyocyst)
urinary tract obstruction
sudden hemorrhage into cysts
Hematuria
Impaired renal concentrating ability
Nephrolitiasis in 15%to 20%
Hypertension in 75% adults

Clinical Manifestations
• Extrarenal
Hepatic cysts: Liver cysts present approximately 10 years after
renal cysts
Intracranial aneurysms:Occur in 4% to 8% of symptomatic
ADPKD patients
Cardiac disease:Mitral and aortic prolapse and regurgitation
Diverticular disease:
Hernias:abdominal and inguinal hernias (up to 45% of ADPKD
patients)
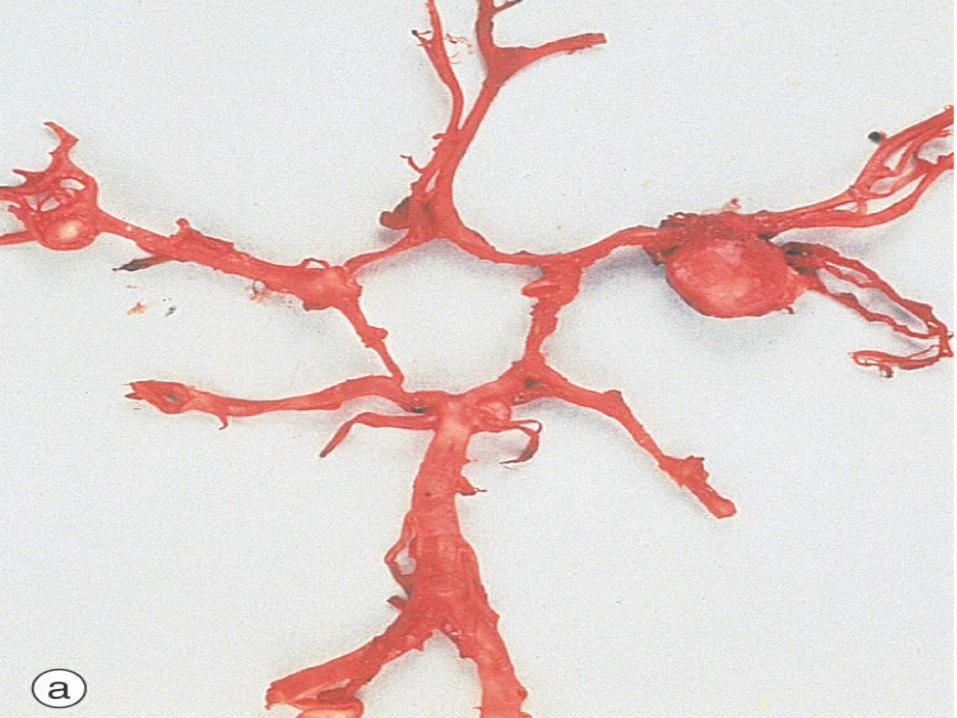
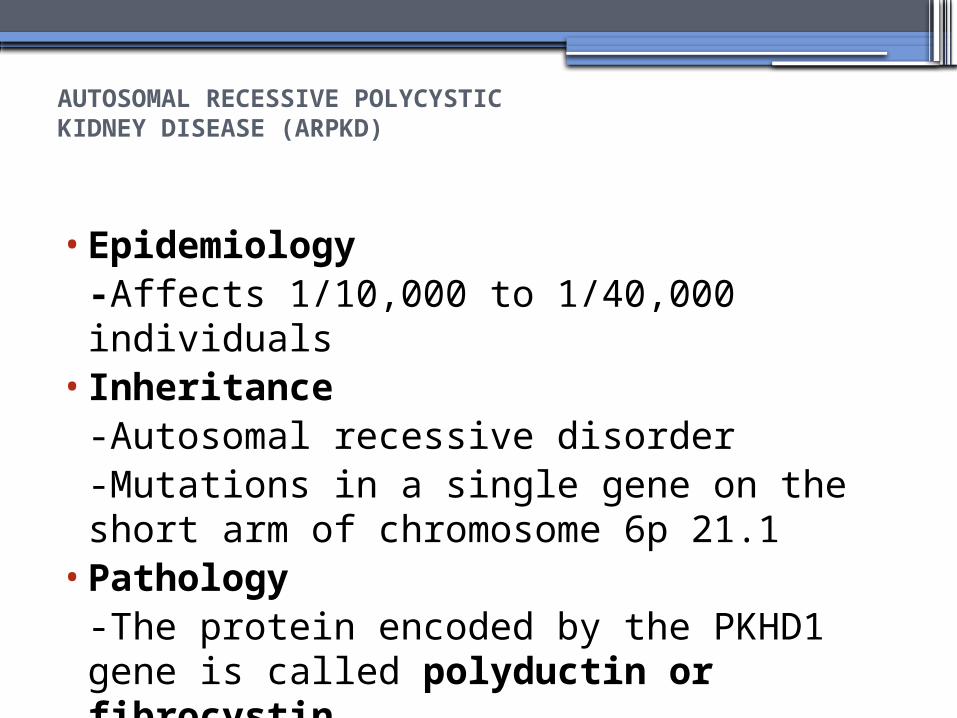
AUTOSOMAL RECESSIVE POLYCYSTICKIDNEY DISEASE (ARPKD)
•Epidemiology-Affects 1/10,000 to 1/40,000 individuals
• Inheritance-Autosomal recessive disorder-Mutations in a single gene on the short arm of chromosome 6p 21.1
•Pathology-The protein encoded by the PKHD1 gene is called polyductin or fibrocystin.
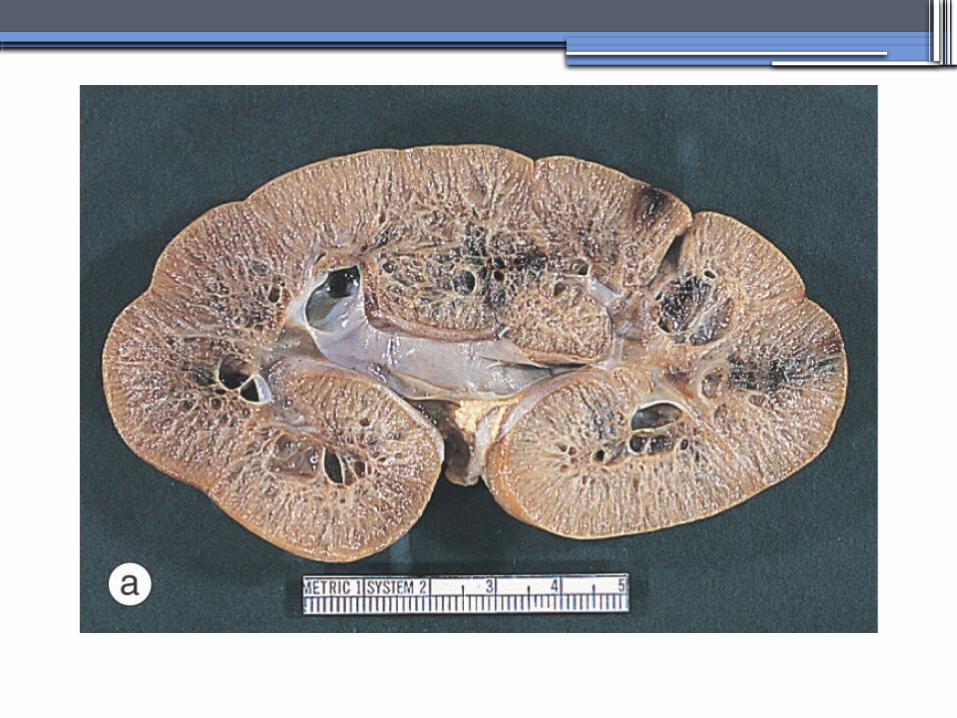

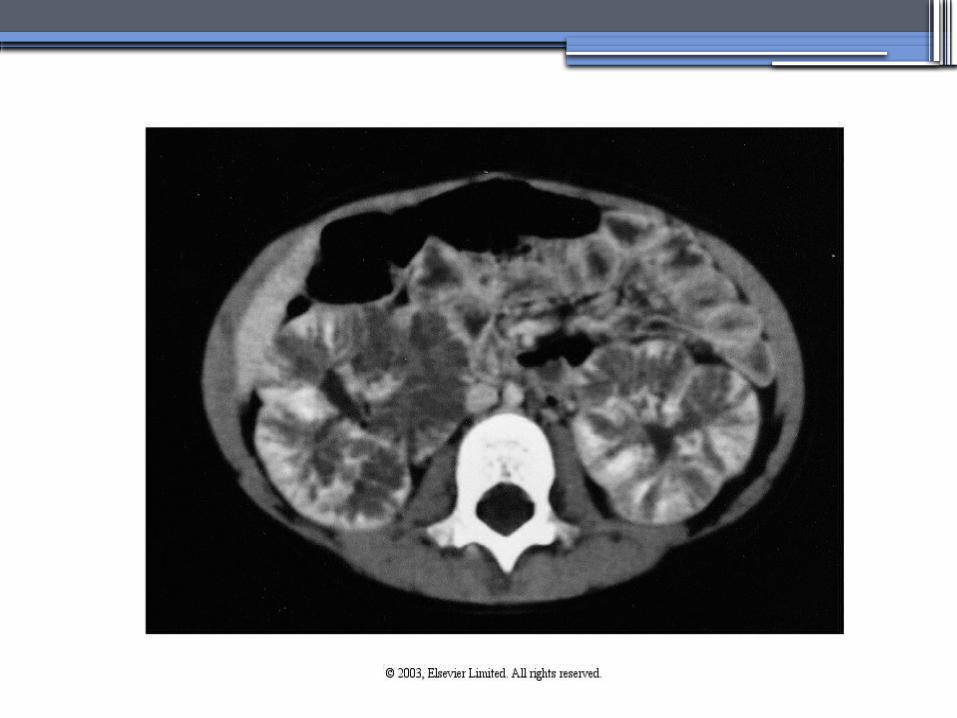

Diagnosis and Clinical Manifestations
● Prenatally: Oligohydramnios, enlarged kidneys, and lung hypoplasia with resultant Potter facies
● Infancy: Pneumomediastinum, pneumothorax, HTN, cardiac hypertrophy,endomyocardiofibrotic congestive heart failure, and renal failure
● Older children: Hepatic fibrosis, portal HTN, and complications of variceal bleeding, thrombocytopenia, and anemia predominate
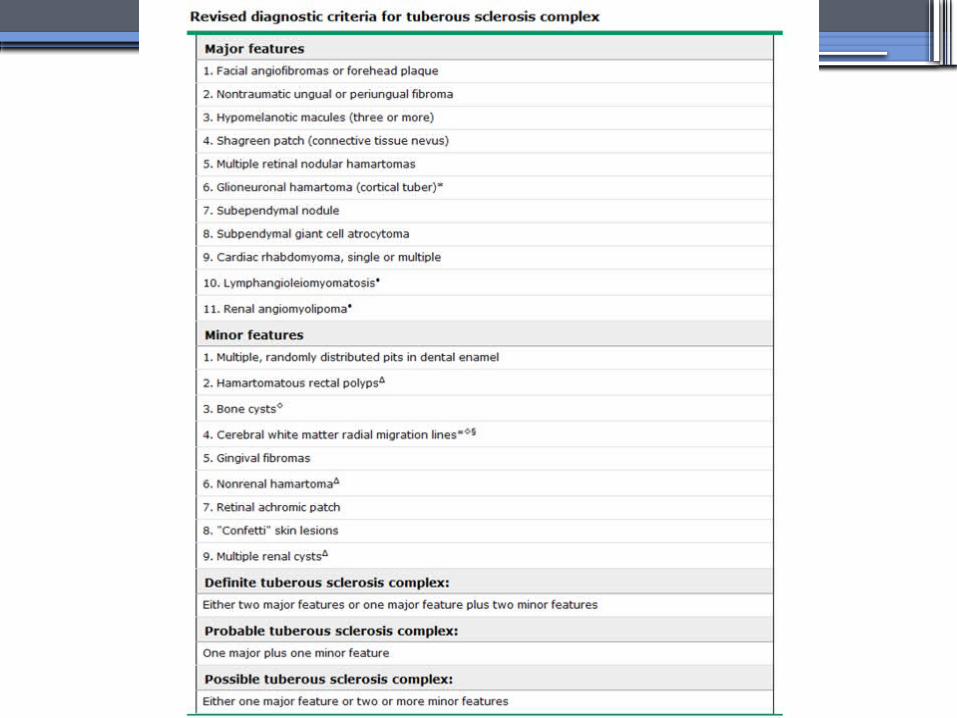
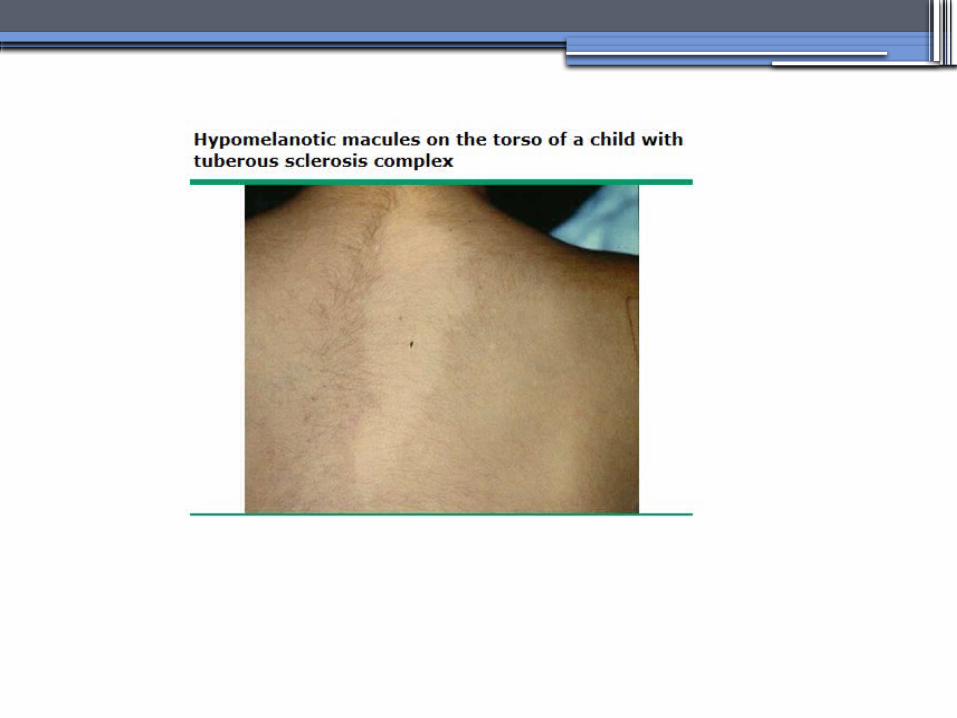
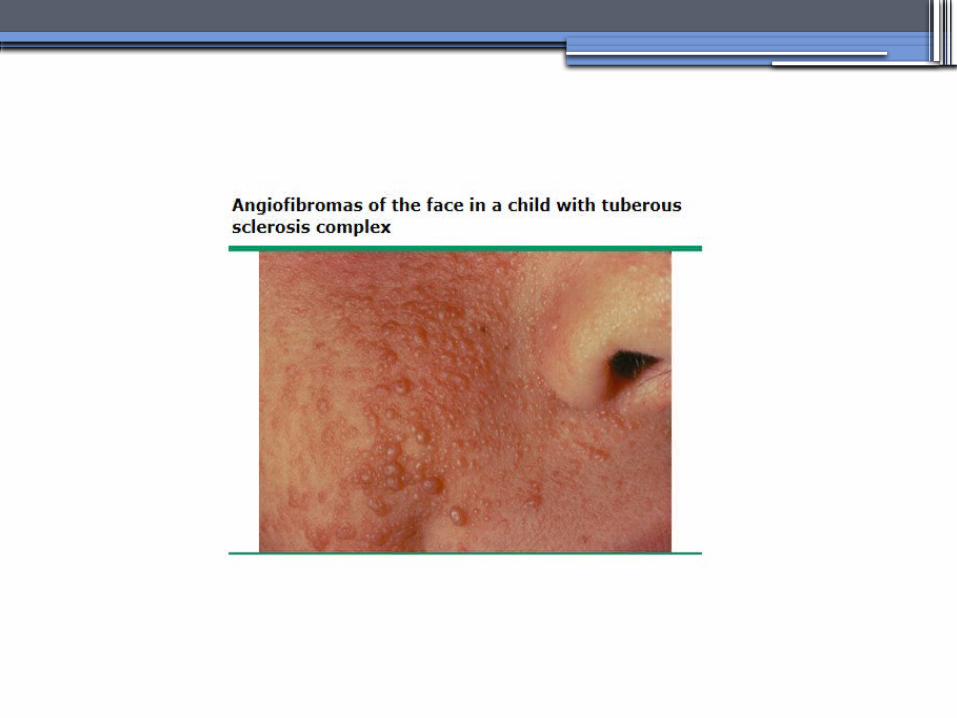
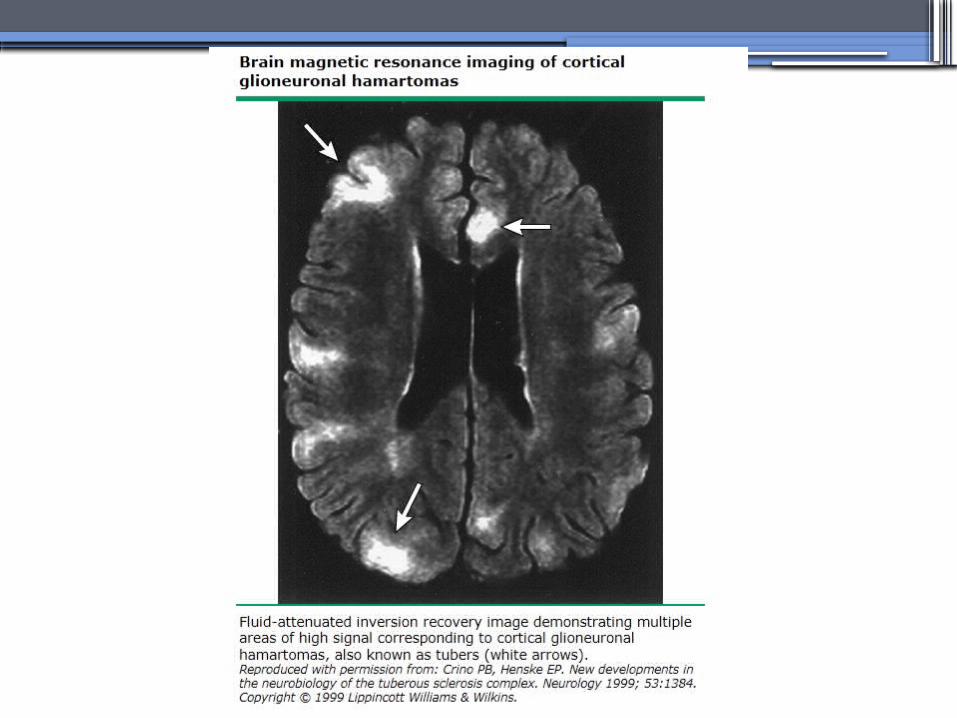
TUBEROUS SCLEROSIS COMPLEX (TSC)
•Genetics-autosomal dominant pattern -high rate of spontaneous mutations (65% to 75% of patients)
•TSC1 →9 (9q34), its protein product is called Hamartin
•TSC2→16 (16p13.3),its protein product is named Tuberin

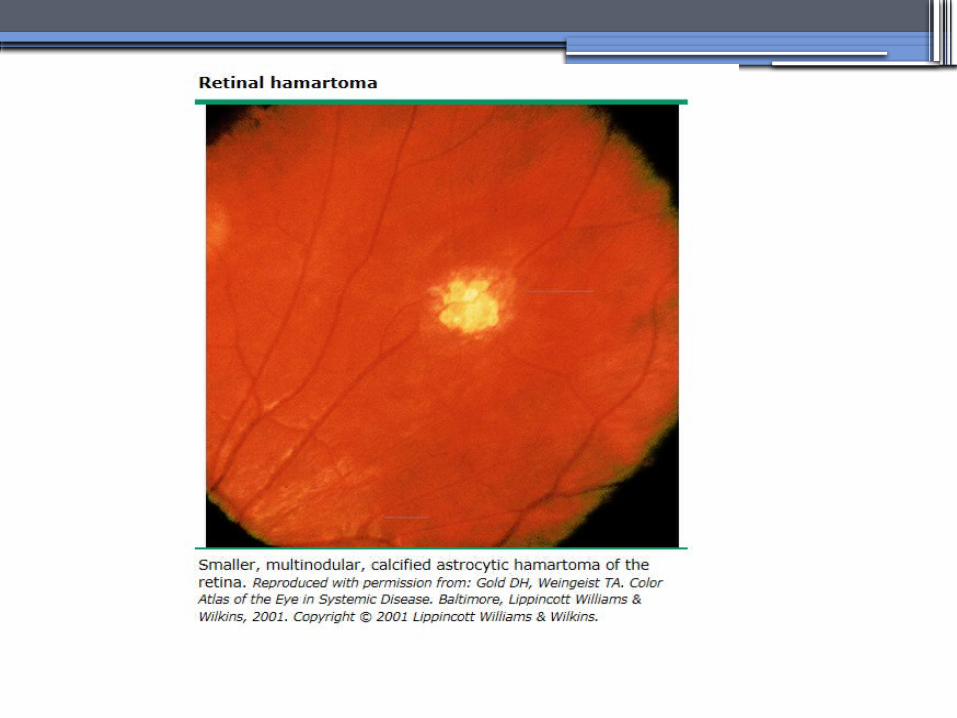
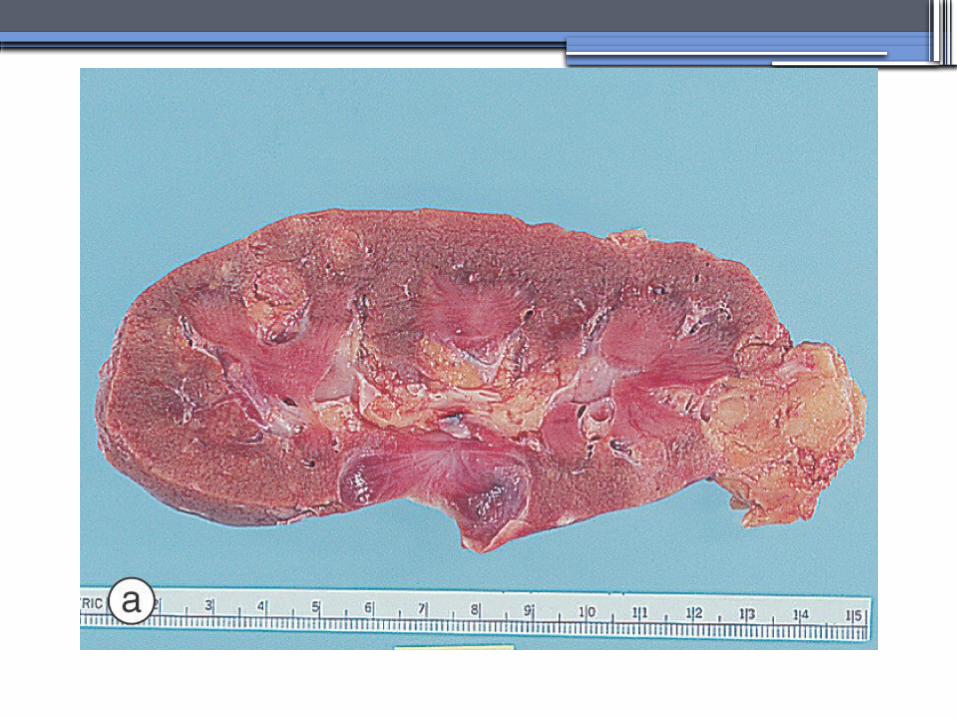
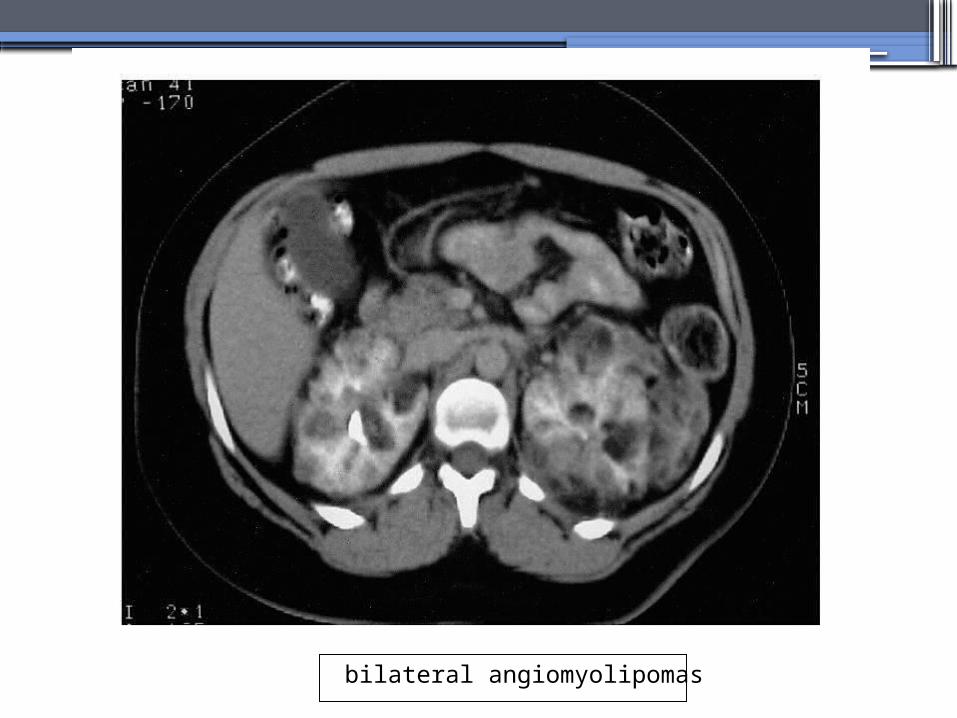
bilateral angiomyolipomas

MEDULLARY CYSTIC KIDNEY DISEASE(MCKD)
•Rare inherited cystic disease, autosomal dominant inheritance:- MCKD1 was mapped to chromosome 1 (1q21) and accounts for the minority of cases
- MCKD2 was mapped more recently to chromosome 16 (16p12) and accounts for mutations in most cases

MEDULLARY CYSTIC KIDNEY DISEASE(MCKD)
•Pathology-normal- to small sized kidneys
-cysts located at the corticomedullary junction and in the medulla. However, the presence of cysts is not universal
-Microscopically: diffuse tubulointerstitial inflammation, hypertrophied and dilated tubules. Glomeruli are usually normal

Diagnosis and Clinical Course
•Clinical features with a family history•The presence of cysts supports the
diagnosis but is not essential•Computed tomography scan is the most
sensitive technique for cyst detection•Polyuria and polydipsia •Progressive renal failure ultimately leads
to ESRD

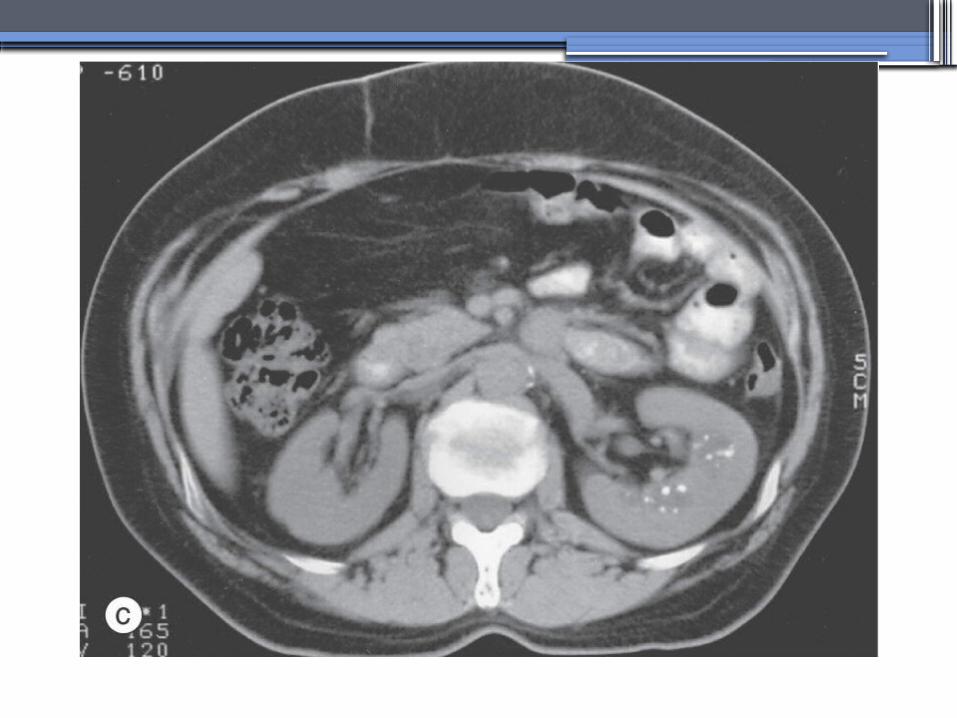

von Hippel-Lindau disease
•autosomal dominant syndrome manifested by a variety of benign and malignant tumors
•VHL gene abnormality is present in about
1 in 36,000 newborns


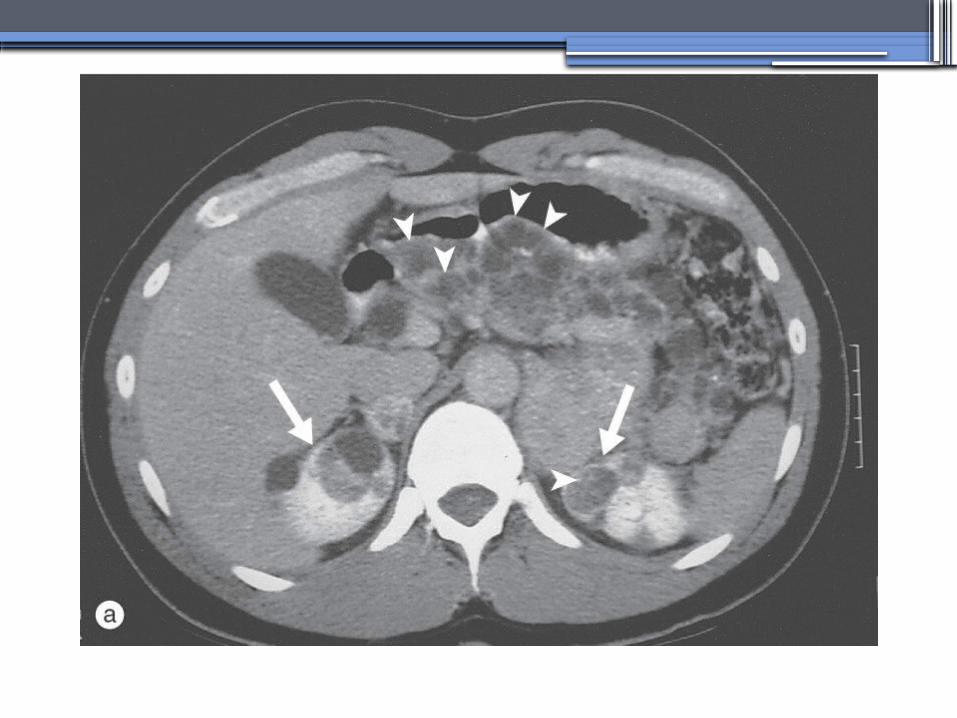
von Hippel-Lindau disease
•Clear cell RCCs occur in approximately 70 percent of VHL patients who survive to 60 years of age.
•Annual imaging of the kidneys with MRI or CT is indicated to establish the diagnosis
•For patients in whom an RCC is diagnosed, we recommend a nephron-sparing approach to remove lesions that are 3 cm or larger whenever possible
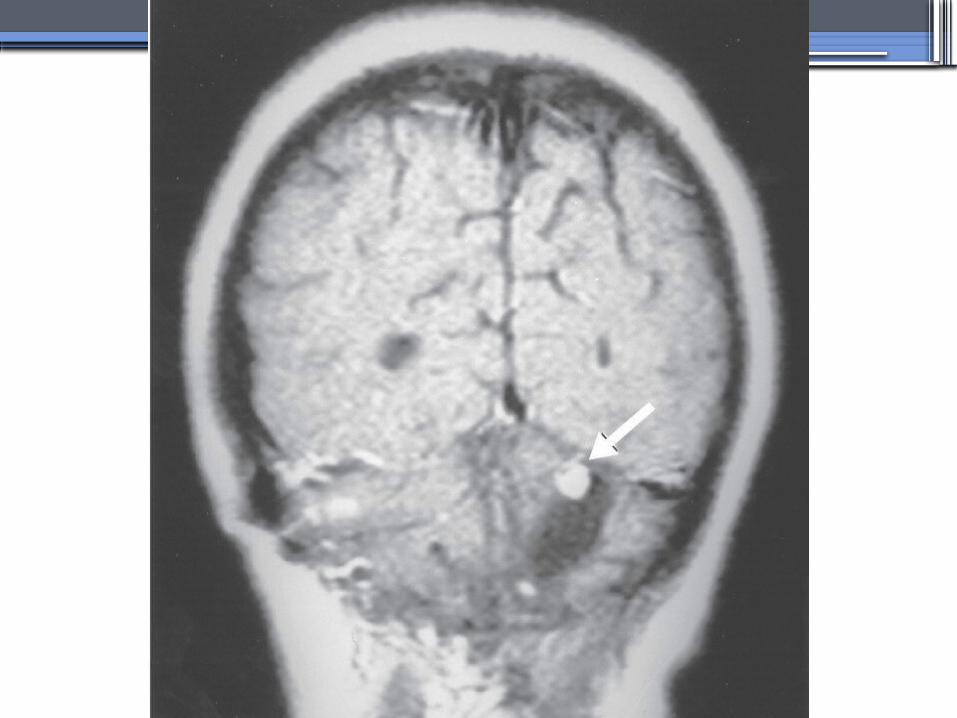
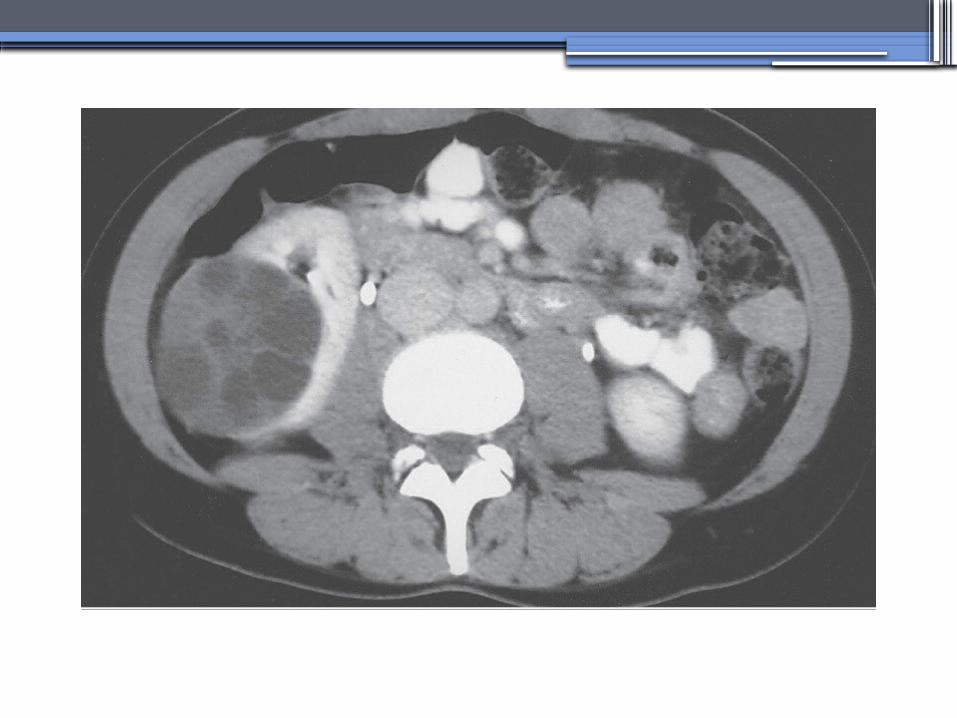

ALPORT’S SYNDROME OR HEREDITARY NEPHRITIS
•Genetics-Prevalence of genetic mutation estimated at 1 in 5,000 to 1 in 10,000.-Accounts for 1% to 2% of ESRD cases.-X-linked inheritance in almost all cases (85%)-Of the non–X-linked cases, most are autosomal recessive
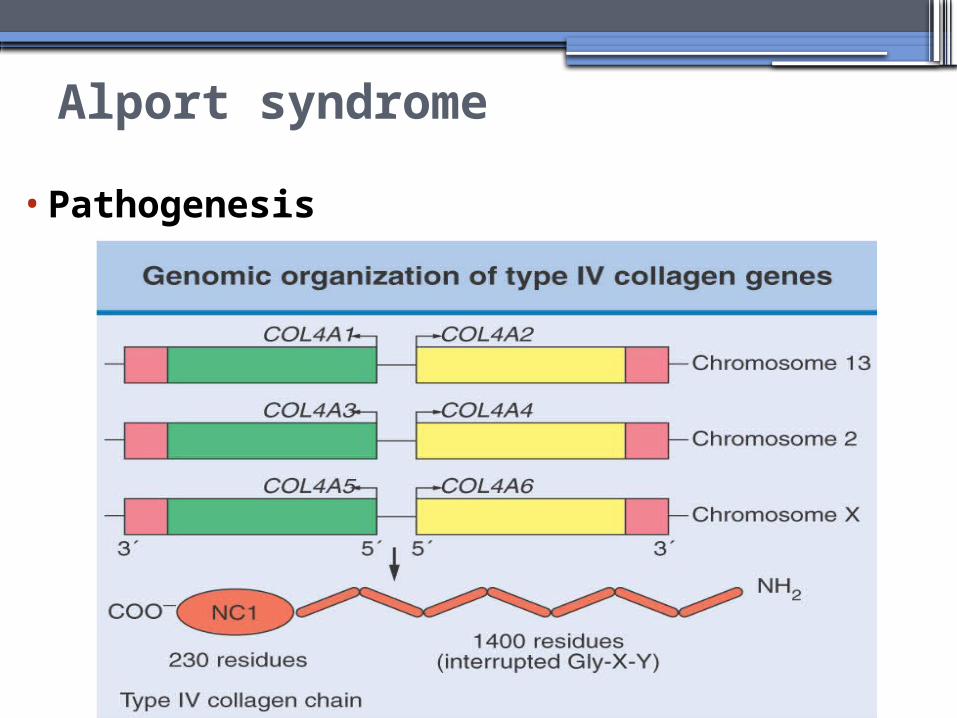
Alport syndrome
•Pathogenesis

ALPORT SYNDROME
•Renal Manifestations-Hematuria: the characteristic clinical feature of Alport’s syndrome-Proteinuria
•Extrarenal Manifestations-Sensorineural hearing loss-Ocular defects: anterior lenticonus-Leiomyomatosis of the esophagus and genitalia

THIN BASEMENT MEMBRANE DISEASE(TBMD)
• Inherited renal disease of the GBM clinically characterized by persistent microscopic hematuria
• inherited in an autosomal dominant fashion, is not accompanied by extrarenal manifestations, and has a benign course
•The diagnosis is established by renal biopsy and electron microscopy

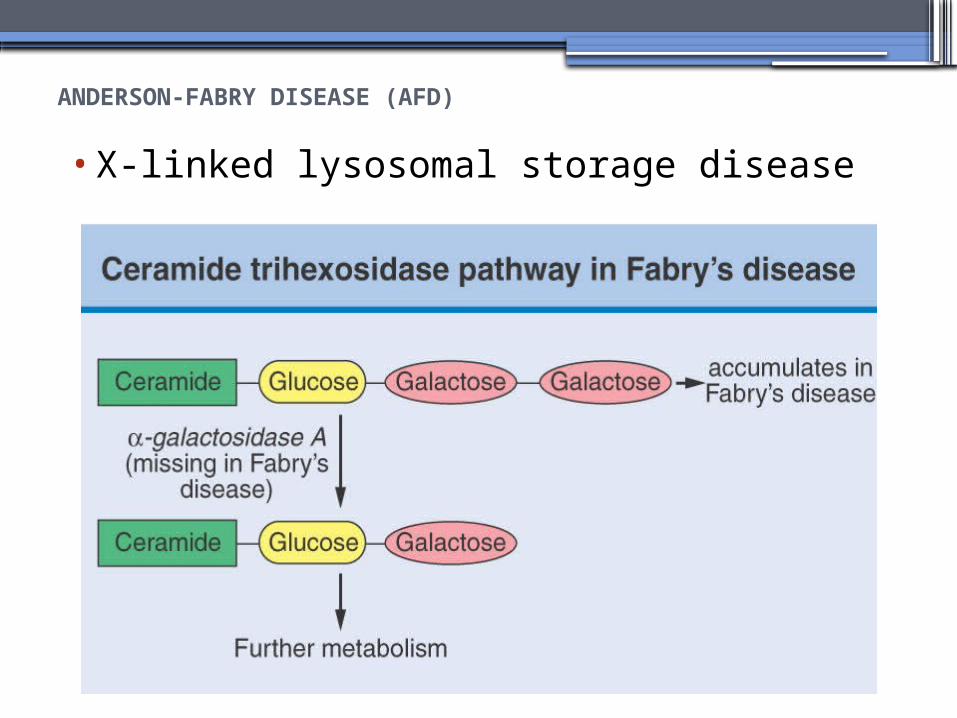
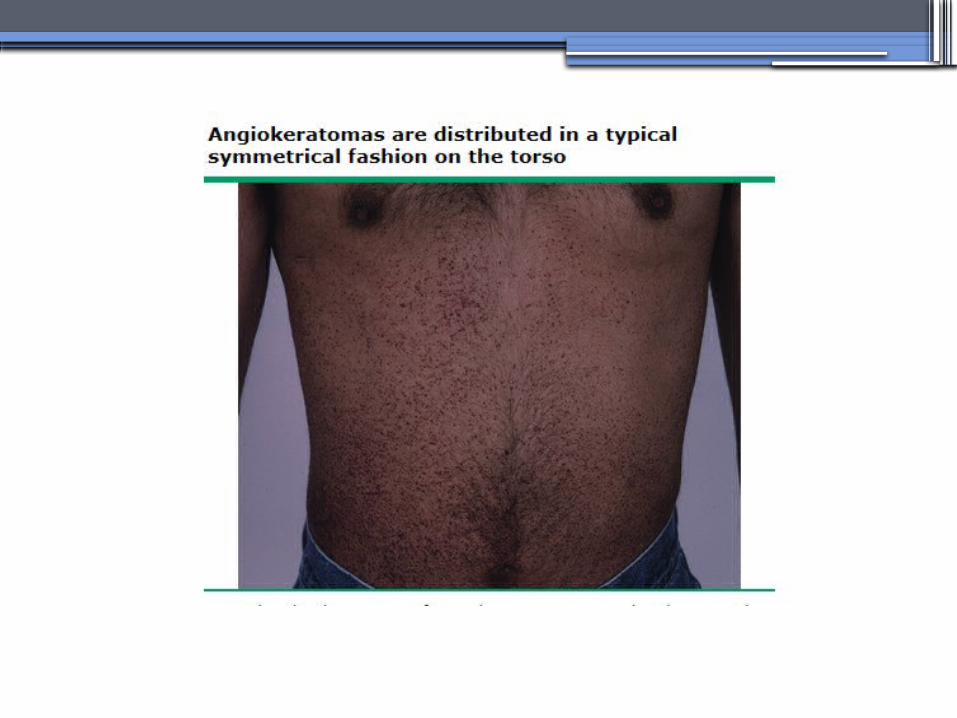
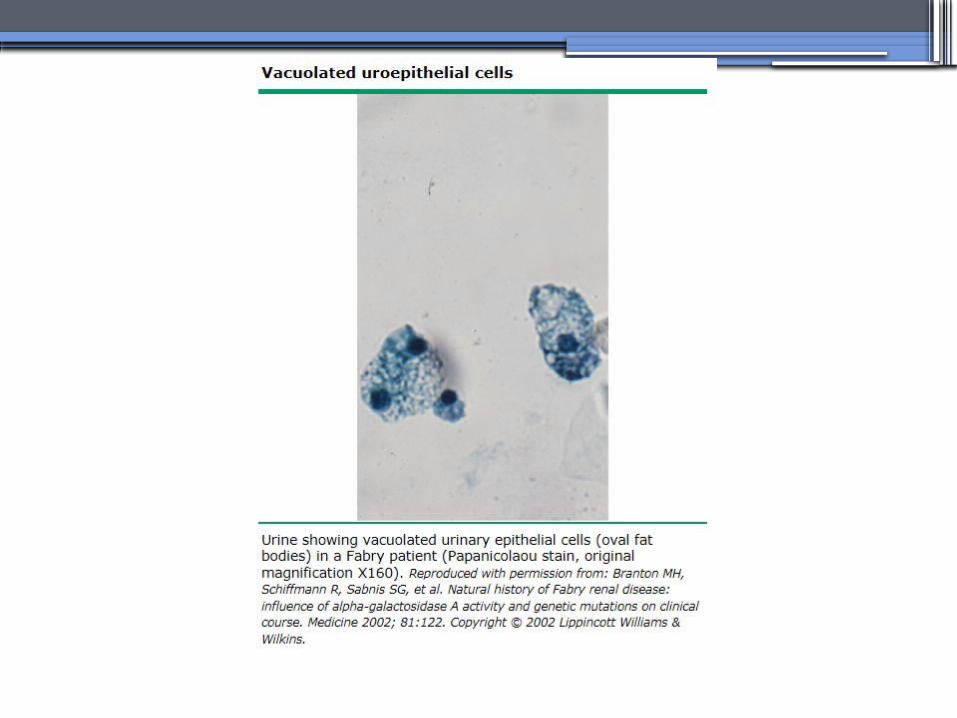
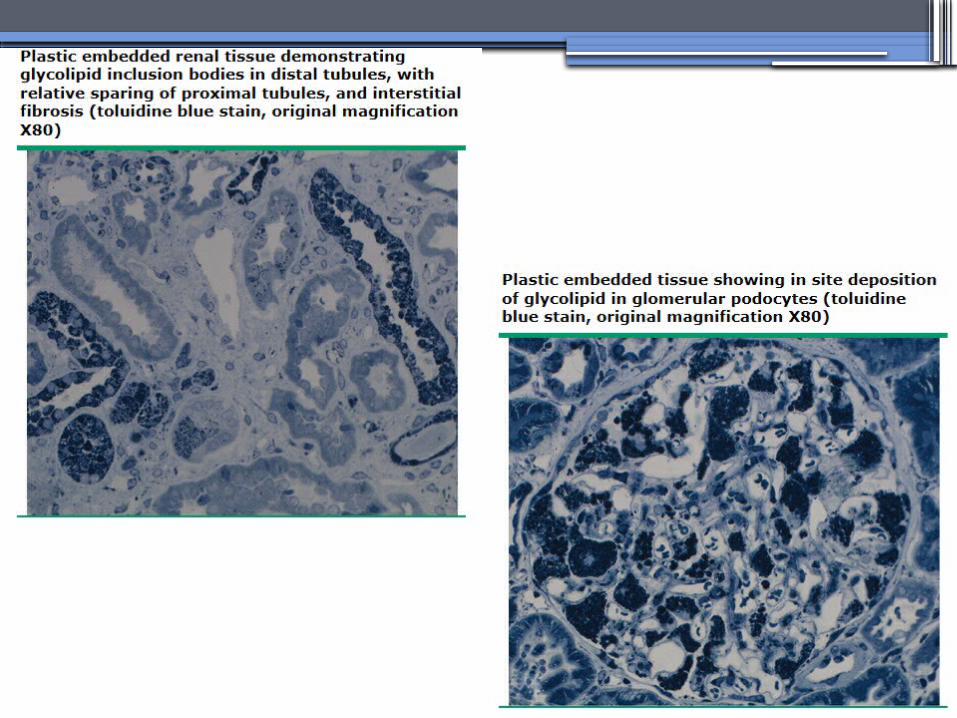
ANDERSON-FABRY DISEASE (AFD)
•X-linked lysosomal storage disease

ANDERSON-FABRY DISEASE (AFD)
•Diagnosis: measurement of plasma or leukocyte α-GALAactivity, skin biopsy, examination of urine sediment, or sequencing of the defective gene
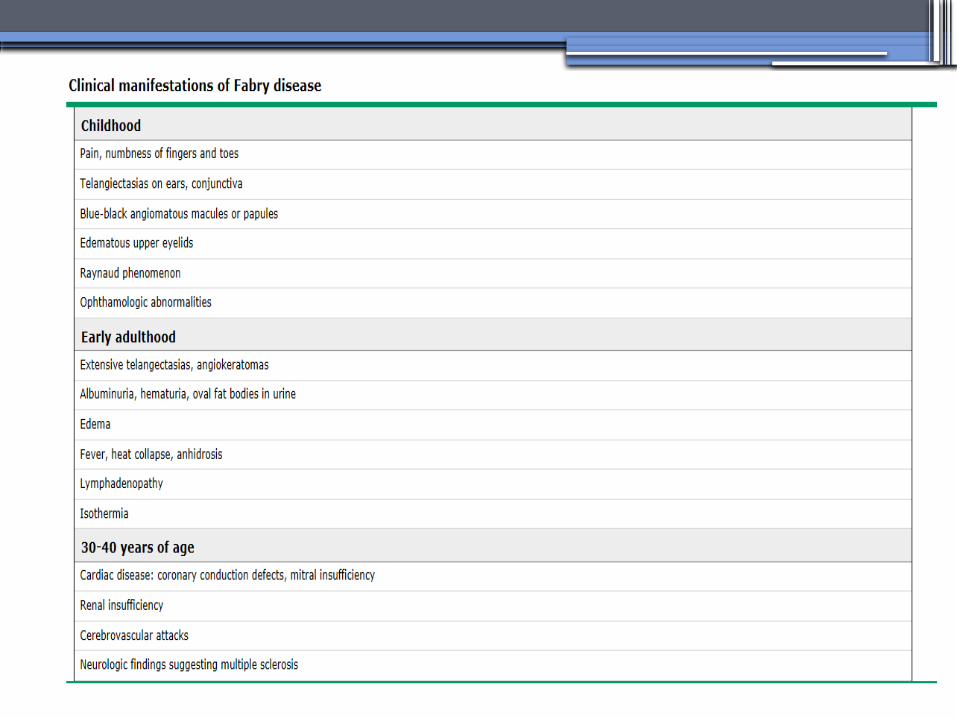
•Clinical Manifestations▫Renal▫Cardiac:
cardiomyopathy Valvular disease Coronary artery disease



Nail-Patella syndrom (NPS)osteo-onychodysplasia
•Genetics-mutations of the LMX1B gene located at the distal end of the long arm of chromosome 9 -LMX1B is a transcription factor of the LIM-homeodomain type that plays an important role for limb and renal development-incidence:22 per million

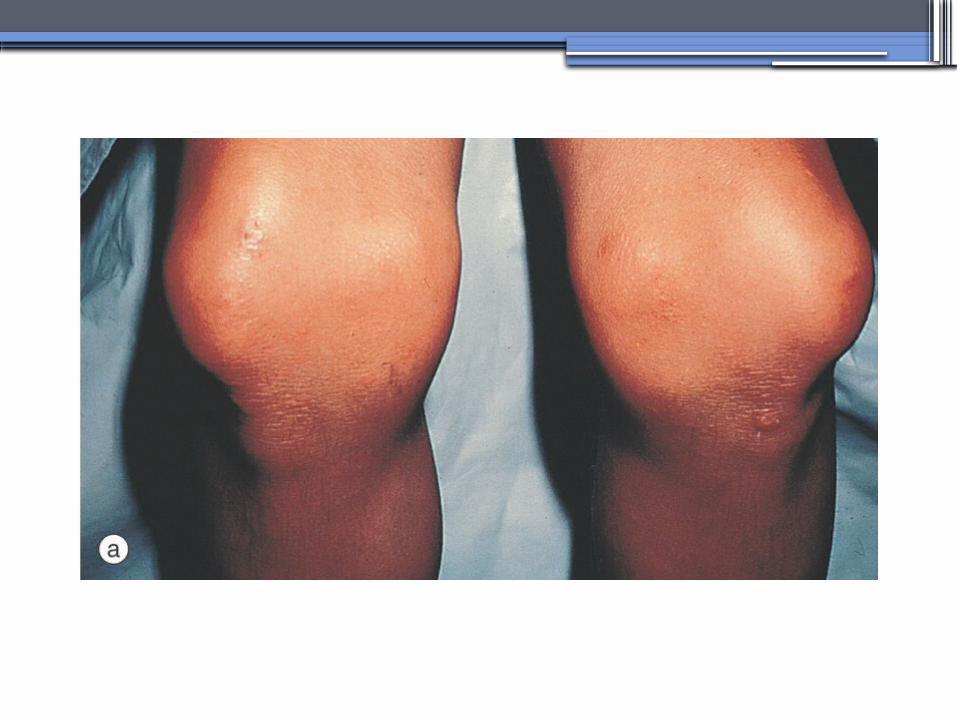
Nail-Patella syndrom (NPS)
•Renal manifestations: •present in one-half of patients- proteinuria (sometimes nephrotic range proteinuria)
- Hematuria- Hypertension- Impaired urinary concentration• About 30 percent of patients with renal
manifestations will develop end-stage renal disease (ESRD)

Sickle Cell Nephropathy
• Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of sickle cell disease
• The primary event leading to renal involvement appears to be sickling of erythrocytes in the vasa recta capillaries leading to microthrombotic infarction and extravasation of blood in the medulla
• Renal failure may ensue in over 10 percent of affected patients, but other clinically significant renal manifestations occur much more frequently

Renal Findings in SCN•Hematuria-medullary congestion-renal papillary necrosis-medullary calcification-medullary carcinoma
•Tubular Dysfunction-concentration defect-increased sodiım and phosphate reabsorption-decreased proton and potassium secretion-increased urate secretion
•Glomerular sclerosis -hyperfiltration-glomerular hypertrophy-proteinuria/nephrotic syndrome-Focal segmental glomerulosclerosis


SUGGESTED READING
•Goldman's Cecile Medicine, Goldman L, Schafer AI
•Case files Internal Medicine, Toy Patlan
•Current Medical Diagnosis and Treatment, Maxine A. Papadakis, Stephen J. McPhee, Eds. Michael W. Rabow, Associate
Ed.
•Current Diagnosis & Treatment:
Nephrology & Hypertension Edgar V. Lerma, Jeffrey S.
Berns, Allen R. Nissenson