influence of zeolite structure on the activity and ... · 2 ecn-rx--02-062 abstract selective...
TRANSCRIPT

December 2002 ECN-RX--02-062
INFLUENCE OF ZEOLITE STRUCTURE ON THEACTIVITY AND DURABILITY OF CO-PD-ZEOLITECATALYSTS IN THE REDUCTION OF NOX WITH
METHANE
J.A.Z. PieterseR.W. van den Brink
S. BooneveldF.A. de Bruijn
Accepted for publication in ‘Applied Catalysis, 2003’, Place, date
Revisions
A final version 17December 2002
B
Made by:
J.A.Z. Pieterse
Approved by:
J.W. Erisman
Checked by:
F. A. de Bruijn
Issued by:
C. A. M. van der Klein
ECN Clean Fossil Fuels

2 ECN-RX--02-062
AbstractSelective catalytic reduction of NO with CH4 was studied over ZSM-5, MOR, FER and BEAzeolite-based cobalt (Co) and palladium (Pd) catalysts in the presence of oxygen and water. Ascompared to other catalytic systems reported in literature for CH4-SCR in the presence of water,zeolite supported Co-Pd combination catalysts are very active and selective. The most activecatalysts, based on MOR and ZSM-5, are characterised by well-dispersed Pd ions in the zeolitethat activate methane. Wet ion-exchange is a good method to achieve high dispersion of Pdprovided that it is carried out in a competitive manner. The presence of cobalt (Co3O4, Co-oxoions) boosts SCR activity by oxidising NO to NO2. The activity of the zeolite-based Co-Pdcombination catalysts decreases with prolonged times on stream. The severity of thedeactivation was found to be different for different zeolite topologies. The characterisation andevaluation of freshly calcined catalysts and spent catalysts show two things that occur duringreaction: 1) zeolite solvated metal cations disappear in favour of (inactive) metal oxides andpresumably larger metal entities, i.e. loss of dispersion, 2) loss of crystallinity affiliated withsteam-dealumination and the concomitant formation of extra-framework aluminium in thepresence of water. Both phenomena strongly depend on the (reaction) temperature. Thedeactivation of Co-Pd-zeolite resembles the deactivation of Pd-zeolite. Hence, future researchcould encompass the stabilisation of Pd (cations) in the zeolite pores by exploring additivesother than cobalt. For this, detailed understanding on the siting of Pd in zeolites is important.
KeywordsNOx reduction, Methane, Selective reduction, Palladium, Cobalt, Durability, ZSM-5, MOR,FER, BEA.

ECN-RX--02-062 3
CONTENTS
LIST OF TABLES 4
LIST OF FIGURES 4
1. INTRODUCTION 5
2. EXPERIMENTAL 72.1 Materials 72.2 Characterisation 72.3 Activity measurements 8
3. RESULTS AND DISCUSSION 93.1 Influence of zeolite structure on the activity 93.2 Influence of zeolite structure on the durability 123.3 Characterisation of fresh and spent samples 14
4. CONCLUSIONS 19
REFERENCES 21

4 ECN-RX--02-062
LIST OF TABLES
Table 2.1 Materials.......................................................................................................................7Table 2.2 Catalyst Codes ..............................................................................................................7Table 3.1 Dispersion of Pd in Pd-zeolite ....................................................................................17
LIST OF FIGURES
Figure 3.1 NOx and methane conversion over Pd(WIE) - zeolites, 5% H2O, 5% O2, 2500ppm CH4, 500 ppm NO, 10.000 h-1 .............................................................................9
Figure 3.2 NOx and methane conversion over Co(Imp)-Pd(WIE)-zeolites, 5% H2O, 5% O2,2500 ppm CH4, 500 ppm NO, 20.000 h-1...................................................................10
Figure 3.3 NOx conversion over CoMOR (10.000h-1), PdMOR (10.000h-1) and CoPdMOR(20.000h-1) at 450 °C, 5% H2O, 5% O2, 2500 ppm CH4, 500 ppm NO.....................13
Figure 3.4 Stability of Co(Imp)-Pd(WIE) zeolites, NOx conversion at 450 °C, 20.000h-1, 5%H2O, 5% O2, 2500 ppm CH4, 500 ppm NO ...............................................................14
Figure 3.5 [CH4/NOx]consumed of Co(Imp)-Pd(WIE) zeolites as a function of the time onstream, 450 °C and 20.000h-1.5% H2O, 5% O2, 2500 ppm CH4, 500 ppm NO..........16
Figure 3.6 a) DRIFT analysis of FER samples. The lines denote the band at ca. 3630 cm-1
and the Bm band at ca. 960 - 900 cm-1
b) DRIFT analysis of ZSM-5 samples. The lines denote the band at ca. 3630 cm-1
and the Bm band at ca. 960 - 900 cm-1 .......................................................................17Figure 3.7 a) H2-TPR spectra of Co(Imp) (top), Pd(WIE) and Co(Imp)-Pd(WIE)FER, grey
colour relates to spent sample for at least 50 hour, 5% H2O, 5% O2, 2500 ppmCH4, 500 ppm NO. The second Y-axis represents the temperature programb) H2-TPR spectra of Co(Imp) (top), Pd(WIE) en Co(Imp)-Pd(WIE)MOR, greycolour relates to spent sample for at least 50 hour, 5% H2O, 5% O2, 2500 ppmCH4, 500 ppm NO. The second Y-axis represents the temperature program ............18

ECN-RX--02-062 5
1. INTRODUCTION
Catalytic methods to reduce NOx, a major contributor to the acidification of the atmosphere andsoil [1,2], have received much attention. In this respect, the Selective Catalytic Reduction withhydrocarbons (SCR-HC) is of special interest. Zeolite supported metal-cation catalysts [3] werefound to have superior activity and selectivity as compared to alternative support materials inSCR-HC. Nevertheless, the limited hydrothermal stability of zeolites [4,5,6] may limit the useof zeolites to applications without large temperature excursions, e.g. in the field of the stationarypollution control. The high activity that can be accomplished with zeolites lowers the (required)minimum temperature of operation and therefore poses fewer demands with regards to(hydro)thermal stability of the material.
The use of methane – in the form of readily available natural gas – is very attractive. A fewexamples of potential markets for zeolite based CH4-SCR are treatment of flue gases stemmingfrom nitric acid factories, small sized boiler installations, combined-cycle devices, caprolactamproduction and (lean-burn) gas engines and transformers.
The use of methane is, however, restricted: the activation of the strong C-H bond oftennecessitates high reaction temperature. Zeolite-supported cobalt and palladium catalysts areamong the most capable ones [3,4]. Moreover, several studies have indicated the stronginfluence of zeolite structure on the activity of palladium in zeolites [4, 7, 8] and cobalt inzeolites [4, 9-11]. In-depth analysis of the zeolite structure in relation to cobalt-cation siting hasincreased the understanding of the activity differences observed among the various zeolites [12-15]. Despite the possibility of tuning the activity (and selectivity) by using various zeolitestructures, the activity of monometallic cobalt-zeolites and palladium-zeolites remainedinsufficient under practical conditions [4].
Ogura et al. [16] studied the activity of a catalyst comprising both cobalt and palladium plantedin ZSM-5 and quoted a remarkable increase of activity. In a recent paper we continued toexplore the performance of ZSM-5 supported cobalt-palladium combination catalysts in CH4-SCR [17]. Emphasis was put on durability in the presence of high concentration water andoxygen. As compared to alternative catalytic systems reported in literature for CH4-SCR in thepresence of water, ZSM-5 supported Co-Pd combination catalysts were found very active andselective. The activity of the ZSM-5-based Co-Pd combination catalysts, however, decreasedstrongly with time on stream. The deactivation is more pronounced at higher reactiontemperatures. The observations are consistent with a temperature-induced mechanism of ionmigration and sintering. No evidence was found for a significant role in the deactivation bysteam-dealumination of the zeolite.
The present study aims to study how zeolite topology affects both the activity and durability ofthe bimetallic cobalt-palladium zeolite catalyst for CH4-SCR in the presence of water andoxygen. This study is part of a larger effort to develop a catalyst for a practical application inexhaust gases. The catalytic performance of zeolites MOR, ZSM-5, FER and BEA will beevaluated and discussed in the light of characterisation data.

6 ECN-RX--02-062

ECN-RX--02-062 7
2. EXPERIMENTAL
2.1 MaterialsThe materials used for the preparation of catalysts are listed in Table 2.1. Pd-zeolite wasprepared by competitive wet ion exchange in air (24 h at 80°C) from NH4-zeolite powder and anacidified solution of palladium nitrate. Additional NH4NO3 was added and the NH4
+/Pd2+ ratioinitially in the liquid amounted to ca. 20. The pH was 2.2 and was allowed to decrease to 2during exchange. Following the ion exchange, the catalyst was filtered, washed thoroughly withdemineralised water and dried for 16 h at 80°C. For the Co-Pd combination catalysts, cobalt wasadded to Pd-H-zeolite by pore-volume impregnation (added volume of cobalt precursor solutionequals the pore volume) (coded as ‘Co(Imp)’). Finally, the catalysts were dried for 16 h at 80°Cand calcined at 450°C in situ. This procedure ensured the transition of NH4-zeolite into H-zeolite with the release of NH3. Combinations of the different metals and preparation methodshave led to the catalysts described in Table 2.2. Mordenite CBV21a was found to contain anadditional 0.05 Wt% iron as an impurity. Sodium levels were in all cases below <0.03 Wt%.
Table 2.1 MaterialsZeolites Si/Al Code Supplier Metal salts SupplierMORBEAZSM5FER
101112
9
CBV21aCP814ESM27
HSZ-700
ZeolystZeolyst
Alsi PentaTosoh
Pd(NO3)2.3H2OCo(NO3) 2.6H2O
AldrichMerck
2.2 CharacterisationThe metal loading of the catalyst was examined using ICP elemental analysis. The results arelisted in Table 2.2.
Table 2.2 Catalyst CodesZeolites Wt% Pd Wt% CoPd(WIE)-HZSM5 0.4
Co(Imp)-Pd(WIE)-HZSM5 0.4 2.3
Pd(WIE)-HMOR 0.4
Co(Imp)-HMOR 2.2
Co(Imp)-Pd(WIE)- HMOR 0.4 2.2
Pd(WIE)-HBEA 0.4
Co(Imp)-Pd(WIE)-HBEA 0.4 2.2
Pd(WIE)-HFER 0.35
Co(Imp)-Pd(WIE)-HFER 0.35 2.2

8 ECN-RX--02-062
H2-TPR spectra were recorded with an Altamira AMI-1 apparatus equipped with a TCDdetector and a Balzers MS-detector, applying 30 ml/min flow of 10% H2 in Argon at a heatingrate of 20°C min-1. Possible deposits on the catalyst surface were traced for by simultaneousanalysis of C, H, N, and S using a LECO CHNS-932 analyser.
The same AMI-1 apparatus was also utilised for pulse-chemisorption of CO to determine metaldispersion. The Pd-zeolites were pre-reduced applying 30 ml/min flow of H2 at 200°C for 40minutes. The samples were cooled down to 35°C under hydrogen and CO was pulsed untilsaturation was reached.
Infrared spectroscopic (IR) measurements were performed with diffuse reflectance spectroscopy(DRIFT) on a BIORAD FTS-175 spectrometer equipped with a Harrick HV-DR2 flow cell.Spectra were recorded in the Kubelka-Munk mode at 350°C. The spectra were recorded with aspectral resolution of 4 cm -1.
2.3 Activity measurementsCatalytic tests were carried out in a computer-controlled flow set-up. The quartz reactor with aninternal diameter of 0.6, 0.8 or 1 cm is placed in an oven. The catalyst sieve fraction (0.25–0.5mm) is placed on a quartz grid. The catalyst bed height to reactor diameter ratio was keptconstant to exclude axial dispersion effects. Space velocities (GHSV) are reported at roomtemperature and atmospheric pressure.
Unless stated otherwise, the feed consists of 500 ppm NO, 2500 ppm CH4, 5% O2 and 5% H2Oin nitrogen. The quantitative analysis of the gas-phase components is performed using a BomenMB100 Fourier transform infrared (FTIR) spectrometer equipped with a model 9100 gasanalyser. At the start of each activity experiment, the reactor temperature is increased to 175°Cat 3°C. min -1 under N2 flow and flushed for 2 h. Subsequently, a background IR scan is madeand the reaction gas mixture is then applied and fed to the catalyst. Preconditioning was set for20 min at each temperature. Unless stated differently in the text, data are collected at ascendingtemperature using a ramp of 5°C/min to maximal 500°C. FTIR analysis averages 150 scans(resolution 1 cm -1) and is performed twice at each temperature.
Conversion of NOx is defined (1 – (NO2 + NO)t)/(NO2 + NO)o * 100 %, conversion methane isdefined (1 – (CH4)t/(CH4)o) * 100 %, based on dry flow.
The methane-based selectivity, in order to distinguish reaction with NOx via SCR and methaneoxidation by oxygen, is defined by means of consumed methane and NO molecules, i.e.,[CH4/NOx]consumed.

ECN-RX--02-062 9
3. RESULTS AND DISCUSSION
3.1 Influence of zeolite structure on the activityFigure 3.1 shows the NOx and CH4 conversions obtained with the Pd loaded zeolites at a GHSVof 10,000 h-1. The NOx removal efficiency at T ≥ 350°C decreases in the order Pd-HZSM-5 >Pd-HMOR > Pd-HBEA > Pd-FER. The conversion of methane follows an identical order.Methane combustion activity is rather low: only Pd-HMOR appears to have a SCR-selectivitylimiting methane combustion activity at temperatures over 450°C.
10
20
3040
50
60
70
8090
100
300 350 400 450 500
Temperature [°C]
NO
x co
nver
sion
[%]
NOx MOR
NOx ZSM5
NOx BEA
NOx FER
0
a
102030405060708090
100
300 350 400 450 500
Temperature [°C]
CH
4 co
nver
sion
[%]
CH4 MOR
CH4 ZSM5
CH4 BEA
CH4 FER
0
bFigure 3.1 NOx and methane conversion over Pd(WIE) - zeolites, 5% H2O, 5% O2, 2500 ppm
CH4, 500 ppm NO, 10.000 h-1

10 ECN-RX--02-062
The addition of cobalt to the Pd-zeolites results in a significant increase of the activity of allcatalysts. Figure 3.2 evaluates the conversion levels of the various Co-Pd-zeolites. Co-Pd-zeolite catalysts were prepared by pore-volume impregnation of Pd-zeolite. As long as thecobalt loading does not exceed Co/Al=0.35 this preparation method does not result in cloggingof the micropores. The thus-created catalysts are characterised by cobalt species of the form ofCo3O4, Co-oxo ions [34,35] and bare Co2+ as shown from H2-TPR and DRIFT analysis. In theCH4-SCR catalysis with the onset at around 300°C, Pd plays the key role in the rate determiningstep, i.e. the activation of methane [3]. Co-oxo ions and Co2+ can co-activate methane,presumably at (slightly) higher temperature. Co3O4 (and Co-oxo ions) oxidises NO to NO2 [17].The oxidation of NO to NO2 as part of the mechanism is important to achieve high activity[4,16,17] by surface recombination with activated methane. This results in the formation ofhighly active CN species intermediate in the formation of N2 [18].
10
20
3040
50
6070
80
90100
300 350 400 450 500
Temperature [°C]
NO
x co
nver
sion
[%]
NOx MOR
NOx ZSM5
NOx BEA
NOx FER
0
a
2030405060708090
100
300 350 400 450 500
Temperature [°C]
CH
4 co
nver
sion
[%]
CH4 MOR
CH4 ZSM5
CH4 BEA
CH4 FER
010
bFigure 3.2 NOx and methane conversion over Co(Imp)-Pd(WIE)-zeolites, 5% H2O, 5% O2, 2500
ppm CH4, 500 ppm NO, 20.000 h-1

ECN-RX--02-062 11
The catalysts based on MOR and ZSM-5 are most active. NOx conversion passes through amaximum for MOR as a result of the decreasing availability of the reductant with increasingtemperature, i.e. CH4 is consumed by direct oxidation. A patent by Gaz de France [19] andOgura et al. [16,20] quoted the enhanced catalytic performance compared to the single metalsystems of zeolite supported Co-Pd combination catalysts. Synergistic effects between cobaltand palladium were proposed to explain the enhanced performance compared to the single metalsystems.
The positive effect of adding cobalt is largest in case of Pd-FER. Also, the effect of addingcobalt was found larger for Pd-MOR than for Pd-ZSM-5. Obviously, in case of MOR and FERthe bifunctional path we reported for Co-Pd-ZSM-5 [17], with distinct functions of thecomponents, may not be adequate. Here we need to acknowledge the ability of cobalt-FER andcobalt-MOR to catalyse the NOx reduction reaction by themselves. We have shown before [17]that pore-volume impregnated ZSM-5 with cobalt results in a significant amount of Co2+, whichare able to activate methane, situated at the exchange site positions of the zeolite. In comparisonto Co-ZSM-5, Co-BEA, Co-MOR and Co-FAU, Co-FER revealed the highest activity [4,10].Moreover, Garcia et al. [9] studied CH4-SCR over Co-zeolites and found the activity to followthe order of Co-MOR > Co-FER > Co-ZSM-5. The activities were related to the total NOxconversion rather than to TOF based activities. TOF based activities determined at low Co/Alratio by Kauckcy et al. [13] led to the reverse order. The reason for the discrepancy is that athigh Co/Al ratio, the activity depends on Co2+ and the synergy between Co2+, Co-oxo species[34,35] and Co3O4. Also, the presence of cobalt and palladium in the vicinity of each other, as isthe case in our work, induces additional complexity in activity assignments. Notwithstanding,these studies indicate the strong influence of zeolite structure on the activity of cobalt sites.
Moreover, Li et al. reported that the position, environment and co-ordination of Co2+ ions have asignificant influence [11], Wichterlova and co-workers accumulated vast evidence for thepresence of topological different cobalt siting in zeolites and their occupation and accessibilityfor reaction varied among zeolites [12-15]. While these studies have delivered importantknowledge on cobalt siting in zeolites and their catalytic consequences, similar studies on Pd(ion) siting are lacking. The size of the Pd2+ ion exceeds the size of Co2+ and this is likely toresult in different spatial limitations and interaction with the walls of the pores. As aconsequence, the siting, i.e. the occupation of α, β and γ sites (following the classification ofKauckcy et al.[12]) may be different and depends on the zeolite topology.
There is no consensus in literature on the exact nature of the active Pd site for CH4-SCR. Lobreeet al. [21], based on density functional theory studies, propose H+[Pd(OH)]+ adducts thatcompensate the negative charge affiliated with two exchange sites. This view is supported byPommier et al. [22] who suggest the formation of isolated oxo and/or hydroxo Pd(II) complexes.Recently, Sachtler and co-workers [23] supported this view, albeit they observe under steady-state conditions the co-existence of clusters of metallic palladium. The latter observationemphasises the operating of a redox mechanism over Pd as opposed to cobalt-zeolites for whichthe reduction of Co2+ under the reaction conditions is unlikely.
Several authors have quoted the difference in performance between Pd loaded onto zeolites ofdifferent topology [7,8]. Ohtsuka et al. reported the superior performance of Pd-MOR over Pd-ZSM-5 both in the presence and absence of water.
Montes de Correa et al. [8] studied the influence of zeolite topology on the reduction of NOxwith methane. It was found that (under dry conditions) the NOx conversion activity at 450°Cfollowed the order of Pd-HZSM-5 > Pd-HMOR > Pd-HFER. The methane combustion activityof these catalysts is very high and much higher than the SCR activity.

12 ECN-RX--02-062
The high methane combustion activity observed in [8] could be affiliated with the impregnationmethod used to prepare Pd-zeolites. The method of loading palladium is important. The (pore-volume) impregnation method is believed to result in PdO particles [17, 23], presumably on theexternal surface of the zeolite. PdO is known for the high methane combustion activity [24,25]and is believed to be responsible for the high methane oxidation activity at the expense of SCRactivity observed with the impregnated Pd(IMP)-zeolite [17].
The ion-exchange procedure guarantees a high level of dispersion by co-ordination of Pd ionswith charge compensating oxygen sites in the zeolite. In a recent paper [17] we compared theactivity of 0.4 wt% Pd loaded ZSM-5 using pore-volume impregnation and wet ion-exchangemethod. Clearly, the NOx conversion was much higher when Pd is loaded using the ion-exchange method. It is anticipated that the relative concentrations of protons, Pd2+ and Pd-oxoions will depend on the catalyst preparation technique. DRIFT analysis of the ion-exchangedPd-zeolite shows the presence of a band around 930 cm-1 (see also Figure 7a and b). This is theso-called Bm band that originates from the shift of the unperturbed antisymmetric vibration ofthe framework T-O bonds at ca. 1100 cm-1 due to perturbation by cations [15]. This bandindicates the co-ordination of palladium cations to framework oxygens [22]. As with the H-zeolite reference samples, this band is not observed on the impregnated sample.
Obviously, it is essential to disperse Pd cations into the zeolite pores for high NOx conversion[22,26]. For this to achieve, it is also important to carry out the ion-exchange in a competitivemanner [27]. Non-competitive ion-exchange was seen to result in poorly dispersed Pd withconcomitant low SCR-activity [23].
3.2 Influence of zeolite structure on the durabilityRecently, in a comprehensive overview of SCR of NOx with hydrocarbons, Traa et al. [4]stresses the need for tests on the time-on-stream behaviour of metal-loaded zeolites in realexhaust gas for a good evaluation of their potential for practical application. In this respect, thepresence of water in the test conditions is a prerequisite.
In a previous paper we have studied the durability of Co-Pd-ZSM-5 catalysts in the presence ofoxygen and water. The activity of the ZSM-5-based Co-Pd combination catalysts decreasedwith prolonged times on stream. It was shown that the deactivation of Co-Pd-ZSM-5 is mainlythe deactivation of Pd-ZSM-5: the deactivation was found to be almost independent of thepresence of cobalt [17]. This observation holds true also for the other zeolite topologies. As anexample, Figure 3.3 shows the NOx conversion over Co-MOR, Pd-MOR and Co-Pd-MOR.From Figure 3.3 one can see that 2.3 Wt% Co-MOR is much less active in CH4-SCR than 0.4Wt% Pd-MOR. Strikingly, the NOx conversion of Co-Pd-MOR and Pd-MOR both increaseduring the first hours on stream. Similar behaviour was previously reported by Descorme [28]who attributed this phenomenon to the migration of Pd ions to the more accessible positions.

ECN-RX--02-062 13
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50
Time [hr]
NO
x co
nver
sion
[%]
CoMOR 10.000 h-1PdMOR 10.000 h-1
CoPdMOR 20.000h-1
Figure 3.3 NOx conversion over CoMOR (10.000h-1), PdMOR (10.000h-1) and CoPdMOR(20.000h-1) at 450 °C, 5% H2O, 5% O2, 2500 ppm CH4, 500 ppm NO
Unlike one could conclude from Figure 3.3, MOR-based Co and Pd catalysts are not stablecatalysts for CH4-SCR. The total methane conversion decreases from time zero. It should beremarked that also, after about 35 – 40 hours time on stream plotted in the Figure 3.3, theconversion of NOx over Co-Pd-MOR and Pd-MOR decreases.
Stability tests carried out with the different zeolites loaded with cobalt and palladium in thepresence of 5 % water are presented in Figure 3.4. During the first 50 hours BEA and ZSM-5are the most severe deactivating catalysts. FER deactivates much less, albeit that it operates atlow conversion level. Co-Pd-HZSM-5 appears to be less stable than Co-Pd-HMOR. This is inagreement with earlier observations made by Ohtsuka et al. [29] with the Pd-zeolites. As theselectivity for SCR over methane combustion increases with time (see Figure 3.5, decreasing[CH4/NOx]consumed), this means that the total methane conversion deactivates stronger than theNOx conversion. Note that the conversion of NOx over Co-Pd-MOR eventually decreases againafter about 40 hours time on stream. Notwithstanding, compared to the other zeolites, MORbased catalysts maintain the highest SCR activity over the longest period of time. ComparingFigures 4 and 5, it also follows that MOR has the highest overall (SCR + combustion) activity.In this respect, the complete conversion of methane is of course a matter of much concern too.Residual methane should be avoided and stoichiometry of the reaction is desired. The NOxconversion was, however, found to increase with the methane concentration over Pd-ZSM-5 andPd-MOR [30,31].

14 ECN-RX--02-062
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50
Time [hr]
NO
x co
nver
sion
[%]
CoPd FER
CoPd BEA
CoPd MOR
CoPd ZSM5
Figure 3.4 Stability of Co(Imp)-Pd(WIE) zeolites, NOx conversion at 450 °C, 20.000h-1, 5%H2O, 5% O2, 2500 ppm CH4, 500 ppm NO
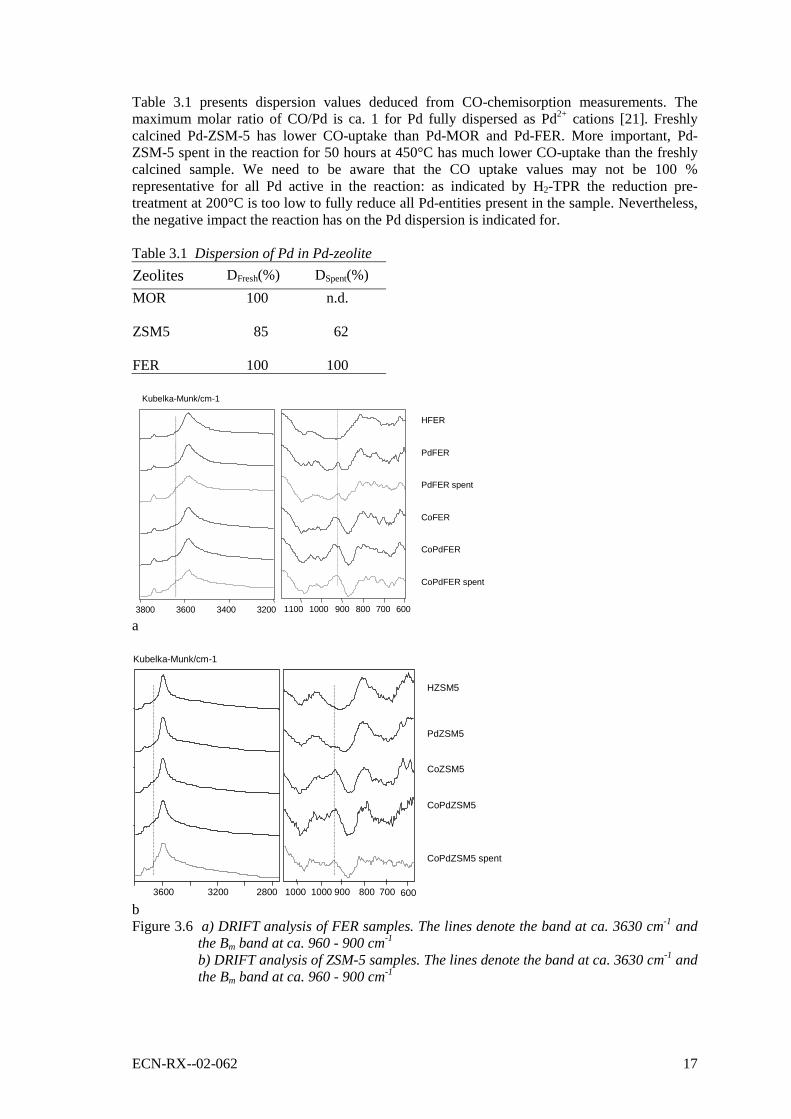
3.3 Characterisation of fresh and spent samplesDRIFT analysis, hydrogen-TPR and CO-chemisorption is applied to shed some light on thedeactivation mechanism(s). Figure 6a and 6b show the DRIFT spectra of FER and ZSM-5,respectively. Important features in the spectra of calcined metal loaded FER include thebridging OH situated at around 3590 cm-1 and the peak that centres at around 930 cm-1. The ca.930 cm-1 peak stems from the shift of the unperturbed antisymmetric vibration of the frameworkT-O bonds at ca. 1100 cm-1 due to perturbation by cations forming the so-called Bm band [15].This peak is characteristic for the cation co-ordination at the exchange (proton) site of thezeolite. In case of Co-FER the peak is nearly symmetrical, centres on 930 cm-1 and spans theregion between 970 and 890 cm-1. It indicates that also the pore volume impregnation methodbrings about co-ordination of (part of) the cobalt cations at the exchange site positions. In caseof Pd-FER the peak is nearly symmetrical and centres on 915 cm-1. The presence of the peakindicates that a substantial amount of the palladium replaced the protons and is of ionic nature.The peak changes its shape and increases in intensity upon loading cobalt. Close analysis of thethus-created asymmetry of the peak using the curve-fit tool of GRAMS/32 software suggeststhat the additional cobalt cations induce a second, much larger peak at ca. 930 cm-1. Besides, theBm region is broadened to ca. 900 cm-1 and is slightly increased in its intensity. Wichterlova andco-workers [12-15] have defined three typical cationic sites for divalent transition metal ionsreferred to as of α, β and γ sites. Recently, Drozdova et al. [15] has classified the occupation ofα, β and γ sites by cobalt in Co-HFER with a Co/Al loading of ca. 0.05. They quote that mainlyβ sites at ca. 940 cm-1 and γ sites at ca. 905 cm-1 are occupied [15]. The α sites in FER are foundat ca. 920 cm-1 and are least favourable for occupation by cobalt. Combining our observationswith the classifications of Drozdova et al. it seems likely to us that Pd favourably situates nearthe α positions in FER. The loading of Pd by means of wet ion-exchange in ZSM-5 gives rise toa small Bm band that centres at ca. 930 cm-1 (Figure 6b). The loading of cobalt by means ofpore-volume impregnation in ZSM-5 and PdZSM-5 gives rise to a broad and intense Bm bandthat covers the region between 980 and 900 cm-1 and is centred between 945 and 930 cm-1
(Figure 7b). At a Co/Al of 0.4 Drozdova et al. concluded that it are the α sites at ca. 935 cm-1
and the β sites at ca. 970 cm-1 that are occupied. Like in the case of FER, we infer that Pd ionsare most abundant at the α sites. Note that the accessibility and activity of cobalt at the α sites inZSM-5 and FER were found to be different [13]. The α sites in FER are situated in the main

ECN-RX--02-062 15
channel (10-membered ring channel) and are the most open and weakly bound [13]. The α sitesin ZSM-5 are situated in the straight, 10-membered ring channels but were concluded less activethan the β sites of ZSM-5, located at the intersection of the straight and sinusoidal channels.
Comparing the fresh ZSM-5 samples with spent Co-Pd-ZSM-5 (50 hours exposure to thestandard gas at 450°C) it is clear that the Bm band as well as the fine structural features below900 cm-1 (the framework asymmetrical and symmetrical stretch vibrations [35]) became muchless intense. Also, the bridging OH at 3590 cm-1 became less intense and instead a new bandevolved at slightly higher wavenumber, 3630-3660 cm-1 (marked by the line in Figure 6). Thisnew band is attributed to extra-framework aluminium (EFAL) and indicates the removal offramework aluminium toward the exchange site position [32]. The phenomenon is usuallyobserved after exposure to water at high temperatures. Altogether, it is clear that the crystallinityof the zeolite has been affected negatively by reaction. In case of FER it is difficult to judge onany changes in the cation co-ordinations in the Bm band region. This observation relates wellwith the fact that less deactivation was observed with FER as compared to ZSM-5 (see Figure3.4). Nevertheless, the Al-OH vibrator affiliated with the extra-framework aluminium is clearlypresent in spent FER samples.
With these observations, the generation of defect sites in the zeolite during reaction is apparent.Recently, we reported on a FTIR measurement of spent Co-Pd-ZSM-5 that did show the loss ofbridging OH that retained after the loading with Co and Pd but the concomitant evolution of the3630-3660 cm-1 band, affiliated with the formation of EFAL, was not observed [17]. In contrast,in the present study we do observe the band related to EFAL on the Co-Pd-zeolite used in thereaction at 450°C. The explanation of this discrepancy could be the temperature of reaction: theCo-Pd-ZSM-5 sample studied by IR in [17] was spent at 500°C instead of 450°C here. Wespeculate that the higher temperature caused some of the metal to co-ordinate/replace thehydroxy of EFAL. As a result, this vibrator disappeared in the IR spectrum. Relocation andconcomitant loss of active metal species as a function of the temperature was indicated by theworsened deactivation at 500°C as compared to 450°C [17]. The co-ordination of the metal-cations as a function of the temperature will be subjected to further investigation.
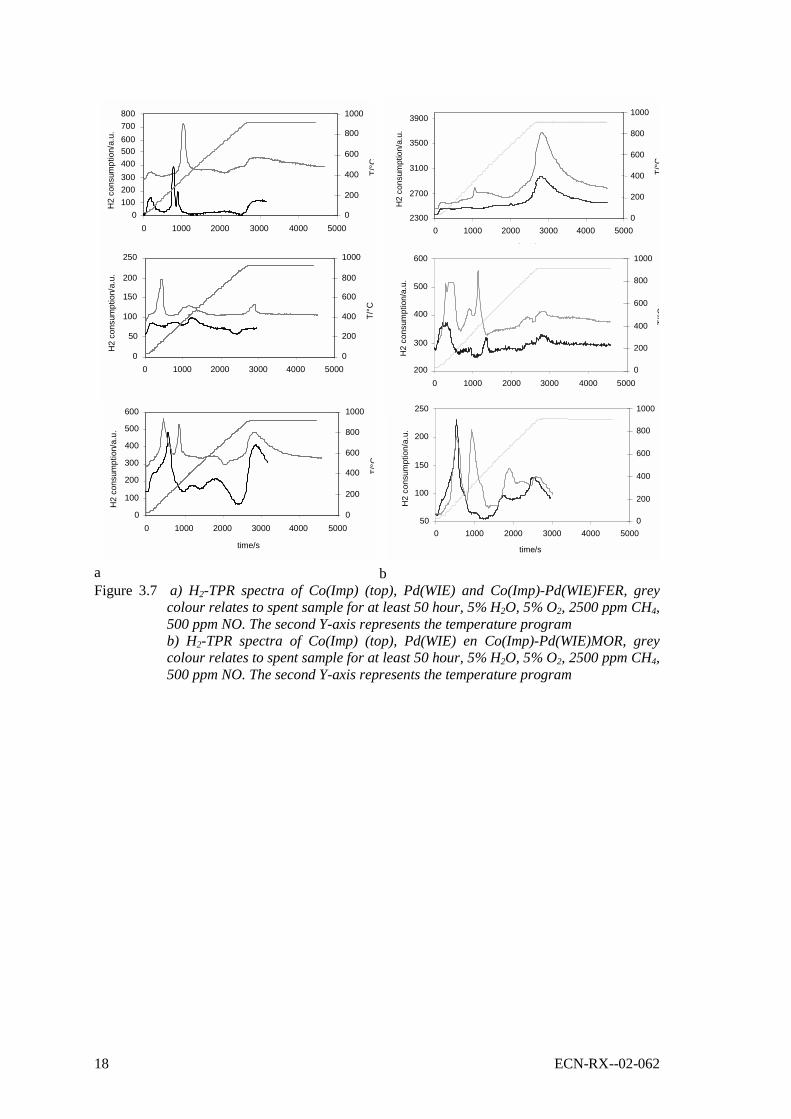
H2-TPR contributes to the analysis of the various palladium and cobalt phases present in thezeolite [34,35] and is appropriate to detect variations in the metal species brought about by(reaction) temperature. Figures 7a and 7b show H2-TPR spectra of palladium and cobalt loadedFER and MOR zeolites, respectively.
A detailed hydrogen and CO (in order to distinguish between ions and oxides) TPR analysis ofCo/zeolite is given by Sachtler et al. [34,35] and the peaks will be assigned accordingly. Co3O4can be found at around 380°C. Peaks at around 600 to 640°C were assigned to (CoO)x in thepores of the zeolite, peaks around 700 and higher were assigned to Co2+ ions associated tocharge compensating (exchange) sites. In the presence of Pd, the high temperature peaksassigned to ionic cobalt (Co2+) are shifted to about a 100°C lower temperature. Presumably,most of the Pd is reduced at temperatures below 450°C and the presence of metallic Pd eases thereduction of cobalt species. Pd contributions can be found at 110-150°C (hydrated, i.e.Pd(H2O)2+) and around 300-400°C (ions).

16 ECN-RX--02-062
H2-TPR analysis confirms the transformation of metal phases. The shift of the reduction peakstoward lower temperatures on spent samples suggest that cations transformed to oxides or thatmetal migrated toward more easy reducible places in the zeolite microstructure. This is alsoindicated for by the higher consumption of moles of hydrogen on spent samples as compared tofresh samples [17,34]. Reduction of Pd-zeolite reveals a peak at around 900°C, which indicatesa very difficult reducible Pd species. The importance of this species for CH4-SCR remainsunclear. The relation of the deactivation of zeolyst-based Pd catalysts with the formation of PdOaggregates is reported in literature [7,16,20,29]. In case of Pd containing catalyst, catalystdegradation corresponds mainly to the evolution of a peak around 200-250°C, presumablycharacteristic for the formation of some Pd of oxidic nature. The formation of PdO is reported tooccur irrespective to the presence of water [7].
Simultaneous on-line MS analysis of the gas phase arisen during reduction of spent Co-Pd-zeolite showed the presence of hydrocarbons. As a consequence, the consumption of hydrogenbetween 250 and 350°C cannot be fully attributed to reduction and is, for a small part, caused byhydrogenation of the carbon. In case of ZSM-5, FER and MOR the amount of carbon detectedby a CHNS analyser after 50 hours on stream is by far too little (100-200 ppm) to significantlycontribute to the deactivation. However, in case of BEA the sample turned black during reactionand the MS analysis now indicated large amounts of carbon. Therefore, carbon deposition islikely to contribute to the deactivation mechanism over Co-Pd-BEA. We speculate that theunique (lewis) acidic nature of BEA [32] (characterised by the band around 3780 cm-1 as seenwith IR) is somehow involved in the carbon deposition. The deviating mechanism ofdeactivation for BEA as compared to ZSM-5, MOR and FER is reflected by the selectivitydecrease seen in Figure 3.5.
0
1
2
3
4
5
6
7
8
9
10
0 10 20 30 40 50
Time (hr)
[CH
4/NO
x]con
sum
e
CoPdFER
CoPdBEA
CoPdMOR
CoPdZSM5
Figure 3.5 [CH4/NOx]consumed of Co(Imp)-Pd(WIE) zeolites as a function of the time on stream,450 °C and 20.000h-1.5% H2O, 5% O2, 2500 ppm CH4, 500 ppm NO
ECN-RX--02-062 17
Table 3.1 presents dispersion values deduced from CO-chemisorption measurements. Themaximum molar ratio of CO/Pd is ca. 1 for Pd fully dispersed as Pd2+ cations [21]. Freshlycalcined Pd-ZSM-5 has lower CO-uptake than Pd-MOR and Pd-FER. More important, Pd-ZSM-5 spent in the reaction for 50 hours at 450°C has much lower CO-uptake than the freshlycalcined sample. We need to be aware that the CO uptake values may not be 100 %representative for all Pd active in the reaction: as indicated by H2-TPR the reduction pre-treatment at 200°C is too low to fully reduce all Pd-entities present in the sample. Nevertheless,the negative impact the reaction has on the Pd dispersion is indicated for.
Table 3.1 Dispersion of Pd in Pd-zeoliteZeolites DFresh(%) DSpent(%)MOR
ZSM5
FER
100
85
100
n.d.
62
100
HFER
PdFER
PdFER spent
CoFER
CoPdFER
CoPdFER spent
Kubelka-Munk/cm-1
3800 3600 3400 3200 1100 1000 900 800 700 600
a
HZSM5
PdZSM5
CoZSM5
CoPdZSM5
CoPdZSM5 spent
3600 3200 2800 1000 800 600
Kubelka-Munk/cm-1
1000 900 700
bFigure 3.6 a) DRIFT analysis of FER samples. The lines denote the band at ca. 3630 cm-1 and
the Bm band at ca. 960 - 900 cm-1
b) DRIFT analysis of ZSM-5 samples. The lines denote the band at ca. 3630 cm-1 andthe Bm band at ca. 960 - 900 cm-1
18 ECN-RX--02-062
0100200300400500600700800
0 1000 2000 3000 4000 5000
H2
cons
umpt
ion/
a.u.
0
200
400
600
800
1000
T/°C
0
100
200
300
400
500
600
0 1000 2000 3000 4000 5000
time/s
H2
cons
umpt
ion/
a.u.
0
200
400
600
800
1000
T/°C
0
50
100
150
200
250
0 1000 2000 3000 4000 5000
H2
cons
umpt
ion/
a.u.
0
200
400
600
800
1000
T/°C
a
2300
2700
3100
3500
3900
0 1000 2000 3000 4000 5000
time /s
H2
cons
umpt
ion/
a.u.
0
200
400
600
800
1000
T/°C
200
300
400
500
600
0 1000 2000 3000 4000 5000
time/sH
2 co
nsum
ptio
n/a.
u.
0
200
400
600
800
1000
T/°C
50
100
150
200
250
0 1000 2000 3000 4000 5000
time/s
H2
cons
umpt
ion/
a.u.
0
200
400
600
800
1000
bFigure 3.7 a) H2-TPR spectra of Co(Imp) (top), Pd(WIE) and Co(Imp)-Pd(WIE)FER, grey
colour relates to spent sample for at least 50 hour, 5% H2O, 5% O2, 2500 ppm CH4,500 ppm NO. The second Y-axis represents the temperature programb) H2-TPR spectra of Co(Imp) (top), Pd(WIE) en Co(Imp)-Pd(WIE)MOR, greycolour relates to spent sample for at least 50 hour, 5% H2O, 5% O2, 2500 ppm CH4,500 ppm NO. The second Y-axis represents the temperature program

ECN-RX--02-062 19
4. CONCLUSIONS
Zeolite supported Co-Pd combination catalysts are very active and selective catalysts for theSelective Catalytic Reduction of NO with CH4 in the presence of oxygen and water. The mostactive catalysts are based on MOR and ZSM-5 zeolites. The activity is characterised by well-dispersed Pd (ions) in the zeolite that activates methane. Wet ion-exchange is a good method toachieve high dispersion of Pd provided that it is carried out in a competitive manner. Thepresence of cobalt (Co3O4, Co-oxo ions) boosts SCR activity by oxidising NO to NO2.Unfortunately, the activity of the zeolite-based Co-Pd combination catalyst decreases withprolonged times on stream. The severity of the deactivation was found different for differentzeolite topologies. The characterisation and evaluation of freshly calcined catalysts and spentcatalysts point at several causes of deactivation: 1) formation of (SCR-inactive) metal oxidesfrom zeolite solvated metal cations, 2) sintering of metal (cations) i.e. loss of dispersion, 3) lossof crystallinity affiliated with dealumination and the formation of extra-framework aluminiumand 4) additional poisoning by carbon species is important for BEA. The relation betweendealumination and metal migration and agglomeration is not yet clear. MOR proves to be themost appropriate structure to accommodate palladium and cobalt giving high activity forprolonged periods of time on stream in wet exhaust gas. The deactivation of Co-Pd-zeoliteresembles the deactivation of Pd-zeolite. Hence, future research should encompass thestabilisation of Pd cations in the zeolite pores by exploring additives other than cobalt. For this,the detailed understanding of Pd siting in zeolite frameworks is also important.

20 ECN-RX--02-062

ECN-RX--02-062 21
REFERENCES
[1] Lox, E.S.J., B.H. Engler: Environmental catalysis. Ed. G.Ertl, H.Knozinger,J.Weitkamp, Wiley 1999.
[2] Armor, J.N.: Catal. Today 38 (1997) 163.
[3] Misono, M.: CatTech 6 (1998) 53.
[4] Traa, Y., B. Burger, J. Weitkamp: Micr. Mes. Mater. 30 (1999) 3 and references therein.
[5] Chen, L., T. Horiuchi, T. Mori: Catal. Letters 72. (2001) 1.
[6] Horiuchi, T., L. Chen, T. Osaki, T. Mori: Catal. Letters 72. (2001) 77.
[7] Ohtsuka, H., T. Tabata: Appl. Catal. B 21. (1999) 133.
[8] Montes de Correa, C., F. Cordoba, F. Bustamante: Micr. Mes. Mat. 40. (2000) 149.
[9] Garcia M., E.A., L.F. Bustamante, C. Montes de Correa: Revista Colombiana deQuimica, 28 1. (1999) 38.
[10] Hansel, J.G., S.V. Raman, J.L. Stolz, J.N. Armor, Y. Li: US Patent 5 451 385. (1995),to Air Products and Chemicals Inc.
[11] Li, Y., J.N. Armor: J. Catal. 150. (1994) 388.
[12] Kaucky, D., J. Dedecek, A. Vondrova, Z. Sobalik: Collect. Czech. Chem. Commun. 63.(1998) 1781.
[13] Kaucky, D., A.Vondrova, J. Dedecek, B. Wichterlova: J. Catal. 194. (2000) 318.
[14] Sobalik, Z., J. Dedecek, L. Ikonnikov, B. Wichterlova: Micr. Mes. Mater. 21. (1998)525.
[15] Drozdova, L., R. Prins, J. Dedecek, Z. Sobalik, B. Wichterlova: J. Phys. Chem. B 106.(2002) 2240.
[16] Ogura, M., Y. Sugiura, M. Hayashi, E. Kikuchi: Catal. Letters 42. (1996) 185.
[17] Pieterse, J.A.Z., R.W. van den Brink, S. Booneveld, F.A. de Bruijn: Appl. Catal. B 39.(2002) 167.
[18] Lobree, L.J., A.W. Aylor, J.A. Reimer, A.T. Bell: J. Catal. 169. (1997) 188.
[19] Hamon, C., O. Le Lamer, N. Morio, J. Saint-Just: US Patent 6 063 0351. (1998), to Gazde France.
[20] Ogura, M., S. Kage, M. Hayashi, M. Matsukata, E. Kikuchi E.: Appl. Catal. B 27.(2000) L213.
[21] Lobree, L.J., A.W. Aylor, J.A. Reimer, A.T. Bell: J. Catal. 181. (1999)189.
[22] Pommier, B., P. Gélin: Phys. Chem. Chem. Phys. 1. (1999) 1665.
[23] Wen, B., Q. Sun, W.M.H. Sachtler: J. Catal. 204. (2001) 314.
[24] Au-Yeung, J., K. Chen, A.T. Bell, E.J. Iglesia: J. Catal. 188. (1999) 132.
[25] Gélin, P., M. Primet: Appl. Catal. B 39. (2002) 1.
[26] Loughran, C.J., D.E. Resasco: Appl. Catal. B 7. (1995) 113.

22 ECN-RX--02-062
[27] Che, M., O. Clause and Ch. Marcilly: Preparation of solid catalysts. Ed. G.Ertl, H.Knozinger, J. Weitkamp, Wiley 1999.
[28] Descorme, C., A. Fakche, E. Garbowski, M. Primet, C. Lecuyer: D.A. Dolenc (Ed.),Proc. Int Gas Res. Conf., vol. 2. GRI Chicago, (1995) 1862.
[29] Ohtsuka, H., T. Tabata: Appl. Catal. B 26. (2000) 275.
[30] Nishizaka, Y., M. Misono: Chem. Letters. (1993) 1295.
[31] Hansel, J.G.: US Patent 5 524 432. (1996), to Air Products.
[32] Pieterse, J.A.Z., K. Seshan and J.A. Lercher: Stud. Surf. Sci. Catal. 130. (2000) 2567.
[33] Flanigen, E.M., H. Khatami, H.A. Seymenski: Adv. Chemistry Series 101. E.M.Flanigen, L.B. Sand Ed., American Chemical Society, Washington D.C. 1971.
[34] Wang, X., H. Chen, W.M.H. Sachtler: Appl. Catal. B 26. (2000) L227.
[35] Wang, X., H. Chen, W.M.H. Sachtler: Appl. Catal. B 29. (2000) 47.