in vitro cell migration and invasion assays
TRANSCRIPT

Mutation Research 752 (2013) 10–24
Review
In vitro cell migration and invasion assays
Nina Kramer a, Angelika Walzl a, Christine Unger a, Margit Rosner a, Georg Krupitza b,Markus Hengstschlager a, Helmut Dolznig a,*a Institute of Medical Genetics, Medical University of Vienna, A-1090 Vienna, Wahringer Strasse 10, Austriab Institute of Pathology, Medical University of Vienna, A-1090 Vienna, Wahringer Gurtel, Austria
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.1. Definition of migration and invasion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.2. Different modes of cell motility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.3. Importance of migration/invasion in cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2. Migration assays . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2.1. Transwell migration assay (Boyden chamber assay) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2.2. In vitro wound-healing assay (scratch assay) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.3. Cell exclusion zone assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.4. Fence assay (ring assay) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.5. Microcarrier bead assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.6. Spheroid migration assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.7. Capillary chamber migration assays (microfluidic chamber assays) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3. Capillary tube migration assays . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3.1. Leukocyte migration agarose technique assay (LMAT assay) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
3.2. Single cell motility assay (colloidal particle assay, colloidal gold single cell migration assay) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
3.3. Time-lapse/cell tracking . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
A R T I C L E I N F O
Article history:
Received 29 November 2011
Received in revised form 14 August 2012
Accepted 16 August 2012
Available online 23 August 2012
Keywords:
Migration
Invasion
Spheroid
Migration assay
Invasion assay
Extracellular matrix
A B S T R A C T
Determining the migratory and invasive capacity of tumor and stromal cells and clarifying the
underlying mechanisms is most relevant for novel strategies in cancer diagnosis, prognosis, drug
development and treatment. Here we shortly summarize the different modes of cell travelling and
review in vitro methods, which can be used to evaluate migration and invasion. We provide a concise
summary of established migration/invasion assays described in the literature, list advantages,
limitations and drawbacks, give a tabular overview for convenience and depict the basic principles
of the assays graphically. In many cases particular research problems and specific cell types do not leave
a choice for a broad variety of usable assays. However, for most standard applications using adherent
cells, based on our experience we suggest to use exclusion zone assays to evaluate migration/invasion.
We substantiate our choice by demonstrating that the advantages outbalance the drawbacks e.g. the
simple setup, the easy readout, the kinetic analysis, the evaluation of cell morphology and the feasibility
to perform the assay with standard laboratory equipment. Finally, innovative 3D migration and invasion
models including heterotypic cell interactions are discussed. These methods recapitulate the in vivo
situation most closely. Results obtained with these assays have already shed new light on cancer cell
spreading and potentially will uncover unknown mechanisms.
� 2012 Elsevier B.V. All rights reserved.
Contents lists available at SciVerse ScienceDirect
Mutation Research/Reviews in Mutation Research
jo u rn al h om epag e: ww w.els evier .c o m/lo cat e/ rev iew sm rCo mm un i ty ad dr es s : w ww.els evier . co m/lo c ate /mu t r es
Abbreviations: ECM, extracellular matrix; BME, basal membrane extract; EMT, epithelial mesenchymal transition; PET, polyethylenterephthalat; PC, polycarbonat; ECIS,
electric cell–substrate impedance sensing; EHS, Engelbreth–Holm–Swarm tumor; HTS, high throughput screening; MMP, matrix metalloprotease; CAF, cancer associated
fibroblast; LMAT, leukocyte migration agarose technique; FFPE, formalin fixed paraffin embedded; EC, endothelial cell; SEM, scanning electron microscopy.
* Corresponding author.
E-mail address: [email protected] (H. Dolznig).
1383-5742/$ – see front matter � 2012 Elsevier B.V. All rights reserved.
http://dx.doi.org/10.1016/j.mrrev.2012.08.001

N. Kramer et al. / Mutation Research 752 (2013) 10–24 11
4. Invasion assays . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1. Transwell invasion assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.2. Platypus invasion assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.3. 3D cell tracking. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.4. Gelatin degradation assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
4.5. Vertical gel 3D migration/invasion assays . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
4.6. Spheroid/monodispersed cell invasion assay. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
4.7. Spheroid confrontation assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
4.8. Spheroid gel invasion assays . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
4.9. Conclusions and perspectives. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
1. Introduction
Normal processes of cell migration/invasion include gastrula-tion [1] embryonic morphogenesis, branching morphogenesis (e.g.
breast ducts), development of the nervous system [2], vascularsprouting [3], placental development, wound healing or immune-cell trafficking [4]. There are also pathological situations in whichderegulated cell movement is highly significative. Tumor cellmigration and invasion is for example clinically most relevant.Cancer is the second leading cause of death and accounted for 7.6million deaths worldwide by 2008 [5]. Metastasis represents themajor problem in the treatment of cancer, is indicative for poorprognosis and has dramatic effects on the survival of patients.Several models of tumor invasion and metastasis have beenproposed. These are for example based on multi-step progression[6], ‘‘intrinsic’’ metastasis [7] or metastatic dissemination [8]although no clear mechanisms were defined. Metastasis involvesmultiple processes such as infiltrative growth through theextracellular matrix (ECM), cell migration through blood or lymphvessels and rise of distant colonies [9]. It is strictly associated withtissue invasion of the primary tumor. Matrix metalloproteases(MMPs) have been identified to break down the ECM. Conse-quently, small molecular weight compounds, which block MMPfunction, have been developed. Unfortunately, these therapyapproaches failed in clinical trials. Since then novel insight intothe mechanisms of cancer cell spreading has been gained, showingthat tumor cells could escape the MMP inhibition by using proteaseindependent mechanisms to travel through the ECM [10,11].Hence, therapeutically targeting proteases in combination withadditional features of tumor cell invasion and migration remainpotent concepts for anti-cancer strategies.
Therefore, it is of great interest and of potential therapeuticimportance to understand the basic principles and molecularpathways, which are altered in cancer cells to drive the first step ofmetastatic dissemination.
1.1. Definition of migration and invasion
Migration is often used as umbrella term in biology to describeany directed cell movement within the body. The ability to migrateallows cells to change their position within tissues or betweendifferent organs.
In pathology, invasion of carcinomas is defined as the penetrationof tissue barriers, such as passing the basement membrane andinfiltration (intrusion) into the underlying interstitial tissues bymalignant tumor cells. Intestinal cancers for example are classifiedas invasive, when the tumor mass has crossed the basal membraneand entered the submucosal muscle layer. Defining invasiveness innon-epithelial cancers is more difficult, since there is no clear-cutseparation of for example mesenchymal cells by a basementmembrane. An explicit discrimination of migration from invasionis not made in many fields of biology dealing with the in vivo
description of directed cell movement.
Migration and invasion are clearly separated terms inexperimental cell biology. Migration is defined as the directedmovement of cells on a substrate such as basal membranes, ECMfibers or plastic plates. Therefore, migration is occurring on 2Dsurfaces without any obstructive fiber network (an example for acollagen I network is shown in Fig. 1A). Invasion is defined as cellmovement through a 3D matrix, which is accompanied by arestructuring of the 3D environment. In order to travel through thematrix, a cell must modify its shape and interact with the ECM,which on the one hand provides the cell attachment substrate, onthe other hand represents a barrier toward the moving cell body(Fig. 1B). Invasion requires adhesion, proteolysis of extracellular-matrix components and migration [12], therefore invading cellsremodel the ECM (Fig. 1C). However, passenger leukocytes canmigrate through 3D tissues and do not require proteolytic actionand tissue remodeling [13]. Thus, the term ‘‘invasion’’ solelydescribes the restructuring or destructive movement of cellsthrough a 3D barrier, whereas ‘‘3D migration’’ is used herein todescribe non-destructive, non-proteolytic movement in 3D tissuesor matrices.
Despite the difficulties in defining different modes of migrationit is a fact that the ability to migrate is a prerequisite to invade; acell cannot invade without migration but can move withoutinvasion. In analogy, migration can be seen as walking on a pave,while invasion would correspond to moving through a thorn hedge(requiring all the tools needed to bushwhack including the abilityto walk).
1.2. Different modes of cell motility
The basic features occurring in a migrating cell have alreadybeen clarified. These cells display directional polarity, with aleading edge at the front and a lagging edge at the back of the cellbody [14]. Common to all migration modes is the actomyosincytoskeleton-mediated change of the cell body shape [15].However, external clues such as the physical and molecularcharacteristics of the environment and intrinsic cellular determi-nants influence the mode of migration. The two main types arecollective migration of multicellular compartments and single cellmovement [12].
The migration of single cells can be subdivided into amoeboid-and mesenchymal types of movement.
Amoeboid movement is characterized by cells moving asrounded, ellipsoid bodies without the involvement of focaladhesions and cell attachment but with the aid of propulsingblebs. This is called blebby amoeboid migration [13,16,17] and isused for example by embryonic cells, leukocytes migratingthrough the ECM (3D migration) or by certain cancer cells. Thesecond form of amoeboid migration is characterized by outgrowthof actin-rich filopodia and weak substrate interaction [18,19]for example in moving neutrophils and dendritic cells or tumorcells [20].

Fig. 1. Invasion. (A) Scanning electron microscopy (SEM) illustrates the structure of collagen I fibers assembled in vitro. This picture exemplifies that the dense ECM structure
represents an appreciable barrier for a moving cell. Scale bar 5 mm. (B) SEM of a mesenchymal cell (pseudocolored in blue) invading through a collagen I (white fibers) matrix
by fibroblast like movement. The cell modifies its shape and attaches to the ECM in order to squeeze through pores in between the fibers. Scale bar 5 mm. (C) An embryonic
intestinal fibroblast embedded in collagen I gel attaches to, restructures and remodels the ECM fibers on its movement through as indicated by a Cason’s trichrome staining of
a section of a formalin-fixed paraffin-embedded collagen I gel. Collagen fibers are stained in blue whereas the cell body is reddish-brown. Scale bar 5 mm.
N. Kramer et al. / Mutation Research 752 (2013) 10–2412
Mesenchymal migration involves strong focal attachment to theextracellular matrix, cytoskeletal contractility and elongatedspindle-like cell bodies [21]. Fibroblasts, sarcoma cells and highlydedifferentiated tumor cells, which have undergone epithelial tomesenchymal transition (EMT) [22], use this mode of motility. EMThas not only been proposed to play a key role in the acquisition of amigratory or invasive phenotype of many carcinoma cells, but isalso discussed to increase the proportion of cancer stem cells[23,24]. Epithelial cells could therefore dedifferentiate via EMT tocancer stem cells. This might have important clinical consequencesfor the curative treatment of cancers as the increased plasticitywithin tumors will require a combination of cancer stem cell andnon-cancer stem cell targeting [25].
Collective migration is characterized by the movement of acellular cohort through the ECM with the preservation offunctional cell–cell junctions. This type of group movement ofcells, which are physically attached to each other, occurs either on2D surfaces, when epithelial sheets travel collectively across basalmembranes (wound healing [26,27]). Alternatively, collectivemigration happens in 3D as branches (mammary gland [28]),tubes (vascular sprouting [29]), strands or clusters (lateral linemigration in fish [30]) or cancer cell invasion [31,32]). Here,leading cells predominantly travel invasively and the followingcells move on pre-paved routes through the ECM.
1.3. Importance of migration/invasion in cancer
Metastasis is by far the main cause of cancer lethality; 90% ofdeaths from solid tumors can be ascribed to metastatic dissemi-nation. It is obvious that there is need for novel therapeuticstrategies in the clinic to avoid metastatic spreading. New anti-metastatic drugs can only be developed upon knowledge of thebasic principles of metastasis. To date the major challenge toclinically monitor inhibition of migration, invasion and metastasisis far from being solved. As metastasis is a multistage process andtakes place over months if not years, a clear-cut clinical readout isextremely difficult to obtain and will represent a major challengefor clinically testing anti-metastatic drugs.
As already described above, metastasis involves multipleprocesses; however, it is strictly connected to initial tissueinvasion at the primary tumor site. Therefore, it is essential toknow, how cancer cells acquire an invasive phenotype and tounderstand the respective molecular mechanisms. Functionalinterferences with different modes of invasion are necessary tocounteract the different strategies cancer cells have evolved tomove through tissues and organs. These interfering treatmentstrategies need to be established, which can either be achieved bythe use of in vivo and/or in vitro models. Here we will focus on in
vitro assays that measure the migratory potential and/or theinvasive property of cells and give an overview of commonly usedexperimental setups. The main advantages of in vitro assays aretheir relatively easy handling and high reproducibility. Moreover,they often allow the examination and phenotypic analysis duringthe assay. Furthermore, in vitro assays are suitable for highthroughput drug testing (high throughput screening, HTS) and areless expensive than in vivo assays. In addition, animal experimentsraise ethical concerns, which can be reduced by the use of in vitro
models. A summary of the features of these assays is given inTables 1 and 2. In vivo migration/invasion assays will not besummarized at this point as these assays were excellentlyreviewed recently [33]. However, it should be noted that so farnone of the described assays could fairly recapitulate all essentialsteps in metastasis, but only address parts of it. It seems that acomprehensive in vitro metastasis assay is far from beingtechnically achievable in the near future.
2. Migration assays
2.1. Transwell migration assay (Boyden chamber assay)
The transwell assay was originally introduced by Boyden (and istherefore often called Boyden chamber assay) to analyze thechemotactic responses of leukocytes [34]. Improved, simplifiedand disposable versions of the original chambers were developed.The principle of this assay is based on two medium containingchambers separated by a porous membrane through which cellstransmigrate (Fig. 2A). The size of the cells to be analyzed,determines the required pore size of the membranes. It is essentialto choose a pore diameter, which allows an active transmigrationi.e. being smaller than the cell diameter to avoid unspecificdropping of the cells. Membranes are available with porediameters between 3 and 12 mm. Generally, cells are seeded inmedium in the upper part and can migrate in vertical directionthrough the pores of the membrane into the lower compartment,in which medium containing an attractant or simply higher serumcontent is present. Of note, a major determinant is the phase ofhorizontal migration of the cells until a pore is being actuallyreached. There are two possibilities to detect and quantify themigrated cells: first, the cells that passed the membrane can befixed on the membrane, stained and quantified. It is important toindividually determine the proper incubation time, until the motilecells appear at the other side of the filter, since it significantlyvaries between different cell types. Thereafter, the membrane isfixed and non-migrated cells remaining on the topside of the filterare removed with a cotton swab. The migrated cells are stainedwith cytological dyes (such as hematoxylin, toluidin blue or crystal

Fig. 2. Schemes of commonly used migration assays. An overview of the technical setup is schematically drawn for each assay and a close-up view is given right to it (inside
the big circles). Arrows indicate the direction of cell movement. Hatched areas symbolize ECM. (A) Transwell migration assay (Boyden chamber assay). Migration of a cell
through a pore in the membrane is depicted. (B) Wound healing assay. Scratching off cells from a dense monolayer produces a cell free area. This is often performed with a
vertically held pipette tip. The close up view depicts that cells can be seeded on plastic or glass surfaces or on ECM coated surfaces (hatched areas). (C) Cell exclusion zone
assay (Platypus migration assay). The cell free area is produced by cell exclusion upon seeding by using silicone stoppers, which are removed before starting the experiment.
(D) Fence assay. Cells are seeded inside a ring-shaped plastic device, which is placed on a cell culture dish. After cell attachment the ring is removed and the cells migrate from
the circular area to the non-covered surrounding. Migration is measured as the increase of the area covered by moving cells. (E) Microcarrier bead assay. Microcarrier beads
are coated with cells to confluence and subsequently the cell-coated beads are placed onto cell culture dishes and incubated. Cells from the microbead attach to the plate
surface and perform radial movement, the area of which can be measured. (F) Spheroid migration assay. Multicellular spheroids of a certain cell type are produced and put
onto conventional tissue culture dishes. The spheroids attach to the surface and cells start moving concentrically outward. The increase in the spreading area can be measured
over time. (G) Horizontal capillary assay (Dunn chamber, Zigmond chamber). The cells migrate along a stable gradient of a chemoattractant within a thin bridging capillary.
None or special surface coating can be used (hatched areas, as in B and C). (H) Capillary tube migration assay. In small capillaries, leukocytes migrate out of the ‘‘buffy coat
area’’ into serum. The migration capacity of the cells can be directly measured by determining the position of the migration front in correlation to the starting line. (I)
Leukocyte migration agarose technique assay. Holes of a defined diameter and distance are punched out of agarose gels; in one of these holes leukocytes are seeded, which
migrate underneath the agarose layer toward a medium reservoir containing a chemoattractant (or repellent), whereas migration toward medium alone serves as control. The
area of migration (dotted line) is visible and the difference in migration distance is determined. (J) Single cell motility assay. Normal tissue culture plates are preincubated
with colloidal gold particles and thereafter cells are seeded at low density above. On their way across the plate, the cells clear the gold particles on their routes leaving bright
tracks behind, which can be measured.
N. Kramer et al. / Mutation Research 752 (2013) 10–24 13
violet), and the number of cells that have migrated is determined.For the second method, the migrated cells are stained fluorescent-ly, removed from the membrane by dissociation (using celldissociation agents such as trypsin) and quantified using afluorescent reader. Alternatively, dark colored porous membranesare available, which block light transmission (FluoroBlok, BectonDickinson) from unmigrated cells. The detection of the migratedlabeled cells is therefore simplified and there is no need to removethe remaining cells from the top-side of the membrane. Theadvantages are the availability of different cell culture inserts andsizes, the relative easiness of the experimental setup and a shortlasting medium/cytokine/chemokine gradient between the upperand lower culture medium reservoir. This assay is most frequentlyused to assess cell migration. Except the cell culture inserts nospecial equipment is needed. The disadvantages are that it is an
endpoint assay; the optimal time of analysis has to be determinedindividually for each cell type tested. If histological cell stainingprocedures are used there are several precautions to make: non-invaded cells, which stayed on the upper side of the transwellinsert, have to be removed prior to staining of the invasive cells atthe bottom of the membrane. This is commonly done by getting ridof the cells with a cotton swab, which often turns out difficult, non-quantitative and of variable success. We strongly recommendusing fluorescent dyes, lysing the cells and quantifying them in aplate reader or to rely on the light blocking membranes andquantify migrated cells without lysis. The transwell migrationassay is suitable for many different cell types including epithelial[35], mesenchymal [36] and brain [37] cancer cell lines as well asmany primary cells from all three germ layers. Conventional cellculture inserts or whole transwell migration kits are commercially

Table 1Comparison of common in vitro migration assays.
Trans-well
migration assay
Wound healing
assay
Cell exclusion
zone assay
Fence assay Micro-carrier
bead assay
Spheroid
migration
assay
Capillary
chamber
assays
Capillary
tube
assays
Leukocyte
migration
agarose
technique assay
Colloidal
particle
assay
Time-lapse
cell tracking
Dimensionality 2D 2D 2D 2D 2D/(3D) 2D/3D 2D 2D 2D 2D 2D
Chemotaxis + � � � � � + + + � Assay dependent
Stable chemokine
gradient
� � � � � � + + + � Assay dependent
Measurement Cell count,
fluorescence
Migration area Migration area Migration
area
Migration area Migration area Cell count,
migration area
Migration
distance
Migration
distance, area
Cell migration
path
Cell migration
path
Live imaging � + + + + + + + + + +
IF, IHC � + + + + + + � n.d. � +
Substrate PC, PET membrane
or coated
Plastic, glass, or
coated
Plastic, glass,
or coated
Plastic, glass, or
coated
Plastic or
coated
Plastic or
coated
Glass or
coated
Glass Plastic, glass,
or coated
Plastic, glass
or coated
Plastic, glass,
or coated
Direction of movement Horizontal then
vertical
Horizontal Horizontal Horizontal Vertical then
horizontal
Mixed, then
horizontal
Horizontal Horizontal Horizontal Horizontal Horizontal
HTS + + + +/� +/� +/� � � � + +
Type of analysis Endpoint Kinetic Kinetic Kinetic Kinetic Kinetic Kinetic Endpoint
(kinetic)
Endpoint
(kinetic)
Kinetic Kinetic
Recapitulated ‘‘in vivo’’
migration mode
Single cell migration,
chemotaxis
Collective
migration of
epithelial sheets,
EMT
Collective
migration of
epithelial sheets,
EMT
Collective
migration of
epithelial sheets,
EMT
Attachment
and migration
Migration from
cell cluster,
established
cell–cell
interaction
Single cell
migration,
chemotaxis
Leukocyte
migration,
single cell
migration
Leukocyte
migration
Single cell
migration
Assay dependent
Notes a b c d e f g h i j k
References [36,37,52,103] [38–43,45–52,97] [34,52,54–56] [57–61] [62,63] [64,65] [67–71] [72–74] [75–77] [78–86] [87–91]
Abbreviations: 3D, three-dimensional; �, not suitable; +, suitable; PC, polycarbonate; PET; polyethylenterephthalat; BME, basal membrane extract; IF, immunofluorescence; IHC, immunohistochemistry; HTS, high throughput
screening; n.d., not done so far.a For adherent and suspension cells, no special equipment needed, technically non-demanding.b For adherent cells only, any plate can be used, movement in defined direction, no special equipment needed, variation in wound area, damaged cells, damaged ECM.c For adherent cells only, pre-defined assay formats, simple technical setup, no expensive equipment needed, exactly defined cell free area, no damaged cells.d For adherent cells only, custom-made rings needed, technically non-demanding, directed movement.e For adherent cells only, microbeads needed, technically medium-demanding.f For adherent cells only, spheroid formation capacity required, technically medium-demanding.g For adherent and suspension cells, technically non-demanding, special chambers needed, directed movement.h For suspension cells only, technically non-demanding.i Only for suspension cells tested, any plate can be used, movement in defined direction, no special equipment needed, directed movement.j Simple, technically non-demanding, individual cell paths, simple readout, time consuming if non-automated analysis.k For adherent and suspension cells, individual cell movement measurable, path detection, expensive equipment, advanced technical setup, non-directed movement.
N.
Kra
mer
et a
l. /
Mu
tatio
n R
esearch
75
2 (2
01
3)
10
–2
41
4

N. Kramer et al. / Mutation Research 752 (2013) 10–24 15
available from many providers (e.g. Life Technologies, BectonDickinson, Merck Millipore and many others). Multiwell transwellassays for high throughput screening of migration were developedin a 96-well format (e.g. the disposable ChemoTx system fromNeuro Probe Inc., Gaithersburg, USA; www.neuroprobe.com).
2.2. In vitro wound-healing assay (scratch assay)
This popular, technically non-demanding and cheap assay canbe used to study migration of cells on 2D surfaces. A confluentplate of any type of attached cells is ‘‘wounded’’ by scraping off anarea of cells, which is most easily done using a plastic pipette tip(Fig. 2B) [38,39]. Cell migration can subsequently be monitoredmicroscopically, as cells travel from the intact zones into thescratched region. Cell movement can be calculated by measuringthe decrease of the uncovered region at different time points untilthe ‘‘wound’’ is closed. Coating of the plates with e.g. collagen I,collagen IV, fibronectin, or basal membrane extract (BME, oftenalso called Matrigel) prior to cell seeding offers the possibility tostudy migration on different substrates. A long term wound-healing assay (>24 h) cannot distinguish cell proliferation andchanges in cell survival from cell motility [40]. Cells can eithermigrate as single cells, as loosely connected population (mesen-chymal cells) or collectively as sheets of cells (epithelial cells). Awide variety of cells have been analyzed for migration with thisassay e.g. epithelial and mesenchymal cancer cells [41], kerati-nocytes [42], normal epithelial cells [43], endothelial cells [44]and fibroblasts [45,46]. The advantages of the assay are its simpleand rapid setup, easy readout and analysis and of course itscheapness. One drawback of the assay is that the scratch is oftenunevenly thick. The migration speed of cells just prior to woundclosure is often increased; therefore variations in gap width priorto the migration start of the cells are critical. In addition, somecells keep attached to the border of the scratch after wounding.These cells often reattach to the plate and move into the woundedarea, which leads to adulterated results. Third, the plastic surfaceor the ECM substrate is scraped off in an uncontrolled manner [47]and artifacts might be induced by mechanical cell damage. Tocircumvent these problems new techniques have been developedto ensure invariable wound sizes with defined edges. One way togenerate wound areas of defined size without significant variationis electric cell–substrate impedance sensing (ECIS) [48,49] as forexample provided by Applied BioPhysics (New York, USA;www.biophysics.com). Instead of mechanically disrupting thecell layer and following the migration microscopically, electricsignals are employed to both, wound the cell monolayer and tomonitor the re-population of the cell-free area. Electricalwounding is applied to cells in contact with a small ECIS electrodeby a pulse of high current, which leads to cell death. Thus a well-defined 250 mm wound is created. The insulating property ofliving cells is lost in the treated region and the impedance drops.Thereafter, the migration of cells into the cleared area is measuredby increased impedance in real time as the wound proceeds toclose [50]. The advantage of the system is that it can be automatedand is feasible for medium to high throughput assays. Alterna-tively, wounding can be achieved by laser ablation of the cells in adefined area [51]. One drawback of both systems might be cellremnants, which are left after the electric ablation in thewounding area [52] and possibly influence wound closure. Toour knowledge simple scratch assays, which are based onmechanical wounding by hand, are not commercially offered.Electric cell ablation assays can be bought from AppliedBiophysics as described in more detail above. Of note, a freesoftware tool called ‘‘TScratch’’ (www.chaton.ethz.ch/software) isavailable, which allows simple automated image analysis of anywound healing and cell exclusion zone assay [53].
2.3. Cell exclusion zone assay
A possibility to circumvent the above described cell remnants isto create cell exclusion zones at the time of cell seeding with e.g.
microstencils [52] or by applying an electrical fence (AppliedBioPhysics, New York, USA; www.biophysics.com). PlatypusTechnologies Inc. (Madison, USA; www.platypustech.com)designed small silicone stoppers that fit into each well of a 96-well plate. These stoppers are positioned prior to seeding of thecells and create an exclusion zone with the tip of the stopper(OrisTM cell migration assay). The cell density is adjusted that cellsare fully confluent. After cell adhesion, the stoppers are removedcreating a 2 mm diameter circular cell-free area, wherein the cellsthen will migrate (Fig. 2C). The advantages of this setup are woundzones of reproducibly similar sizes and sharp borders. In addition,there is no damage to the cells from mechanical scraping or electricablation (see previous section). Moreover, the assay is well-standardized, easy to set up and does not need special equipmentfor analysis. This assay is only suited for adherent cells. Care mustbe taken that the stoppers are tightly attached; otherwise cellsenter the cell exclusion zone beneath the silicone device. Afterremoval of the stoppers, the wells have to be washed carefully withmedium in order to prevent attachment of floating cells in the cellexclusion area, which would compromise the results. A derivativeof the assay was recently developed and employs precastdissolvable biocompatible gels instead of the silicone stoppers(OrisTM Pro cell migration assay). Of note, this assay is available in a96-well and 384-well setting. The standard silicone stopper assayhas been widely used e.g. for assessing epithelial [54,55] or smoothmuscle cell migration [56]. Results generated with the OrisTM Promigration assay have not been published so far. Similar replace-ment devices are also available from other companies (e.g. CellBiolabs, Inc., San Diego, USA; www.cellbiolabs.com). One of thesedevices (Ibidi GmbH, Martinsried, Germany; www.ibidi.com)provides two cell culture reservoirs when placed on a cell culturedish surface. The reservoirs are separated by a 500 mm thick wall.Seeding cells in both reservoirs and removing the silicon insertfrom the surface results in two defined cell patches separated by acell free gap of 500 mm width. This setup also allows investigatingthe migratory behavior and interaction of two different cellpopulations seeded separately into the two different wells. Asalready described above, migration can be measured by photomi-crography or quantified using microplate readers if fluorescentlylabeled cells are used. For this a mask is used to cover the areas,which are populated by the cells before starting the experiment. Ascells move, they leave the masked area and can be detected andquantified in fluorescence microplate readers. The detectedfluorescence signal is proportional to the amount of migrated cells.
2.4. Fence assay (ring assay)
The fence assay is in principle a reversion of the cell exclusionzone assay described above. Here, cells are seeded into the innerarea of a Teflon, glass or metal fence (ring) placed on a standard cellculture dish (Fig. 2D). The cell attachment area is restricted to theinner part encircled by the ring device: After the ring is detached,non-attached cells are removed by gently washing and the cells areincubated and allowed to migrate from the circular area in a radialway outward. The rate of movement is measured as the increase ofthe newly area covered by migrating cells, which is often donedigitally by automated image analysis [57,58]. Using this assay themigratory potential of human endothelial cells was quantified[59,60]. In a slightly different assay cell aggregates of defined sizeformed by centrifugation can be placed onto microscopic slidesand the subsequent migration evading out of the structure can bemeasured [61]. Due to the same principles the advantages and

N. Kramer et al. / Mutation Research 752 (2013) 10–2416
drawbacks are similar to the cell exclusion zone assays. Metal/silicone fences are commercially available from e.g. Aix ScientificsCRO (Aachen, Germany, www.aix-scientifics.co.uk).
2.5. Microcarrier bead assay
This assay measures cell motility based on migration of the cellsfrom microcarrier beads onto 2D cell culture vessel surfaces [62](Fig. 2E). Therefore microcarrier beads (e.g. DEAE Dextran beads;e.g. Cytodex beads from GE Healthcare, Chalfont St. Giles, UK) arecoated with cells. These cells can be grown to confluence on thesurface of the beads. Thereafter the cell-coated beads are placedonto cell culture dishes and incubated for a defined period.Subsequently the beads are removed by suction. The cells, whichmoved to the cell culture vessel surface area, are fixed, stained andmicroscopically evaluated or densitometrically measured. Herethe advantage is that cells established close cell–cell interactionson the beads, which is more closely mimicking the tight contact ofcells in vivo. In addition, due to the limited space and little cell sizevariation a fairly constant amount of cells are present on the beadsurface (e.g. approximately 400 HUVECs on collagen coatedCytodex beads), when confluence is reached. Cell coating can beeasily monitored with conventional light microscopy. There arealways some beads, which are not or insufficiently covered by cells.These beads should be identified and not used for analysis. Anotherdrawback of the assay is that the Cytodex beads are ratherexpensive. The initially described assay is an endpoint assay;however, we have used live cell microscopy to monitor cellmigration off the beads in kinetic experiments without fixationand staining of the cells. The model was rarely used as a simplemigration assay in the literature. For example it was deployed todemonstrate the negative effects of PAI-1 on cell migration [63].The cell coated beads are more often embedded into ECM andinvasion is monitored (see below). There are no commerciallyavailable kits on the market.
2.6. Spheroid migration assay
This assay combines 3D with 2D technologies by placingmulticellular tumor cell spheroids of a certain cell type on top of aconventional cell culture dish. The assay principle closelyresembles that of the above mentioned microcarrier bead assay.After attachment of the spheroid to the plastic surface, the cellsstart to migrate and the area of attachment is increasedconcentrically as the cells move out. Cell movement can bemeasured microscopically (Fig. 2F) over time. One major advan-tage compared to the above described assays is that the 3Dstructure of the spheroid represents a more physiologic tissue-likemorphology with close cell–cell contacts and different cellularstatuses as nutrient and oxygen supply is concerned. In respect totumor biology for example, the migration of cells out of smallcancer clusters can be closely mimicked. Of note, this assay is onlypossible if the cells investigated are able to form spheroids. Somepre-experience with spheroid formation is necessary, but this canbe easily achieved. The method was used to demonstrate thattissue factor pathway inhibitor-2 (TFPI-2) expression inhibited cellmigration and invasion in prostate cancer cells [64]. In a differentstudy spheroid migration was used to prove that valproic acid haseither pro- or anti-migratory effects on malignant gliomas,dependent on different cell lines analyzed [65].
We currently use a modified spheroid migration assay, whichreflects tumor–stroma interaction. Spheroids from fluorescentlylabeled tumor cells (genetically modified to express fluorescentproteins e.g. GFP or labeled with fluorescent cell tracker dyes like 5-chloromethylfluorescein diacetate, CMFDA) are formed andapplied to confluent fibroblasts (e.g. cancer associated fibroblasts,
CAFs), which are grown on a conventional plastic dish. This setupcan be used to analyze the migratory changes upon interactionbetween the two cell types. Upon attachment, the tumor cells startto interact with the monolayer cells. The close interaction ensuresparacrine crosstalk and direct cell–cell interaction at the migrationfront of the tumor cells. Migration can again be measured asincrease of the attached tumor cell area over time. Fluorescentlabeling helps to distinguish tumor cells from the CAFs (Dolzniget al., unpublished results). The general feasibility of the assay toanalyze the interaction of cancer cell spheroids with differentstromal cell types was demonstrated using colon carcinoma cellspheroids co-cultured with monocytes and fibroblasts [66].However the authors did not specifically address migration. Toour knowledge there is no commercial assay available on themarket.
2.7. Capillary chamber migration assays (microfluidic chamber
assays)
In a horizontal setting two chambers are linked side-by-side bya narrow connecting bridge (e.g. Zigmond chamber [67], Dunnchamber [68,69]). One of the chambers is loaded with cellsresuspended in medium, whereas the other is filled with mediumcontaining a chemoattractant. The system is covered by a glassslide. A stable concentration gradient develops between the tworeservoir chambers in the connecting capillary area and thenumber of migrating cells is counted on the surface of theconnection by light microscopy (Fig. 2G). These assays are oftenused for leukocyte migration studies. The advantage lies in thesmall assay volumes, making these assays well suitable for studieswith rare cell types and to test expensive compounds. The use ofsmall media volumes demands daily changes and carefulhumidification of the incubator. In addition, automation processesare difficult to set up with the available systems. The horizontalcapillary migration chamber was further advanced to the mSlideChemotaxis assay (Ibidi, Martinsried, Germany, www.ibidi.com). Itconsists of two medium reservoirs and a perpendicular channel,where cells can be seeded. A gradient can be applied via the tworeservoir chambers and the movement of cells can be monitoredmicroscopically. The main advantage to the above-mentionedmethods is the easy handling and the assessment ofdirected chemotaxis along a gradient. Recently, the capillarychamber assay was used to prove that the Src family kinase Fyn isinvolved in HGF-mediated chemotaxis of prostate cancer cells [70].For HTS, 96-well and 192-well formats have been developed [71]and are commercially available (BellBrook Labs, WI, USA,www.bellbrooklabs.com).
3. Capillary tube migration assays
The capillary tube migration assay was developed manydecades ago to address leukocyte migration [72]. For this assay,glass capillaries are filled with blood, sealed and centrifuged. Fromthe leukocyte ‘‘buffy coat’’, which is located above the red bloodcell pellet, leukocytes start to move out to the plasma layer(Fig. 2H). The glass capillaries are placed onto a microscopic slideand the distance of migration can be measured by microscopy withocular micrometer devices. This assay only needs small samplevolumes and the use of multiple capillaries leads to properstatistical analyses. In an advancement of the assay, the capillariesare cut at the buffycoat/erythrocyte interface and the leukocytecontaining part is placed onto plastic culture dishes. Subsequentlyit is covered with test medium and cells start to migrate. The size ofthe area of cells that migrated out of the capillary onto the plasticsurface is a measure of cell migration [73]. Neutrophil migration inresponse to an anti-inflammatory response was reported using this

N. Kramer et al. / Mutation Research 752 (2013) 10–24 17
assay [74]. This specialized migration test for white blood cellsseems not to be used any more, maybe because it vanished intooblivion. It is described here for reasons of completeness and todemonstrate its potential. Companies do not offer this assay type.
3.1. Leukocyte migration agarose technique assay (LMAT assay)
Another assay to analyze white blood cell migratory potentialwas introduced in 1968 [75]. This method involves a pre-cast thinagarose layer on a tissue culture dish into which circular wellswere cut out in defined distances. Leukocytes are loaded into one ofthese wells, whereas the adjacent holes are filled with differentmedia containing either chemotactic factors, or inhibitors orcontrols [76]. The leukocytes migrate below the agarose layertoward the different media reservoirs (Fig. 2I) and the distance ofmovement toward the test substance (divided by the migrationdistance toward the control medium) defines the migratory index.The advantages for this assay are the easy and cheap setup as wellas the simple detection method. The assay seems to be outdated;only a very limited number of citations still using this assay can befound. In addition, this method is restricted to analyze themigration of white blood cells. The LMAT assay was employed toshow that monocytes and macrophages increase the migration ofneutrophils in the presence of glucocorticoid [74] and CXCL8 mightbe involved in reduced neutrophil migration [77]. We could notfind any commercially available LMAT assay.
3.2. Single cell motility assay (colloidal particle assay, colloidal gold
single cell migration assay)
A simple way to measure migration on the single cell level is touse colloidal gold particle coated surfaces [78–80]. Tissue cultureplates are coated with colloidal gold particles and cells are seededonto these plates at low density (1 � 103 cells/ml). The goldcolloidal particles are seen as a homogenous layer of small darkdots under the microscope. The migrating single cells phagocytosethe gold particles and thereby remove them from the plasticsurface, resulting in white tracks, which can be photographed andthe cleared areas can be evaluated quantitatively (Fig. 2J). Thissingle cell motility assay was used to track the migratory paths ofe.g. keratinocytes [81], fibroblasts [82], epithelial cancer cells[83,84]. This assay has also been used to demonstrate thatrapamycin reduced motility of tumor cells at the single cell level[85]. The method is suitable for automation and high throughputassays and the gold particles can be replaced by quantum dots [86].The advantages of this assay are: single cell tracking; undirectedmovement (chemokinesis) can be monitored; real-time pathdetection is possible and therefore the absolute speed of migrationcan be determined. Disadvantages are: intense analysis labor orautomated system needed; rather small sample sizes (one cell–onetrack). We are not aware of a currently available assay sold by lifescience companies.
3.3. Time-lapse/cell tracking
Another possibility to analyze single cell migration is thetracking of individual cells with videomicroscopy in time-lapseexperiments. The selection and tracking of the cells can either bedone manually, semi-automatic or fully automatic. For automatedcell tracking many algorithms have been developed which cancope with the recording of the migration path of several dozens ofcells simultaneously. The automated analysis can also cope withcell division of the moving cells occurring within the recordingtime interval [87,88]. A full description of the novel automatedtracking systems goes far beyond the scope of this review and isgiven elsewhere [89]. Essentially, the individual movement of
many cells can be analyzed at once and the recorded migrationpaths provides information about the total movement length aswell as the direction and velocity at a given time point. Time lapsecell tracking has been used for example to analyze myogenic cellmotility [90] or to show that collective migration is responsible forpattern formation in co-cultures of different keratocytes [91]. Aswe are no specialists in this field we do not comment on pros andcons of these systems and again refer to specialized review articles(for example [89]).
4. Invasion assays
4.1. Transwell invasion assay
The principal technical setup of the transwell invasion assayequals the transwell migration assay (see above). In addition, theporous filter is overlaid by a thin layer of ECM, before seeding thecells into the top chamber [92–94]. The ECM occludesthe membrane pores, blocking non-invasive cells from migratingthrough. By contrast, invasive cells can degrade the matrix andmove through the ECM layer and adhere to the bottom of the filter(Fig. 3A). Depending on the detection method, the invasive cells,which crossed the membrane pores, are either stained and countedwith a light microscope, or detached, stained and lysed usingfluorimetric detection. The ECM can be variable and often BMEobtained from Engelbreth–Holm–Swarm (EHS) mouse sarcomas orcollagen I is used. It is recommended to calculate the ratio ofinvaded (passed through the ECM coated filters) against themigrated cells (non-coated filters, see migration assay), the so-called ‘‘invasive index’’. This determines the relative contributionof invasion to the overall motility speed [93]. The advantages of themethod are the broad availability of different cell culture insertsand sizes; the relative ease of the experimental setup and the –albeit only short – medium or cytokine/chemokine gradientbetween the upper (cell culture insert) and lower (culture vessel)growth medium. This method is an endpoint assay and there aresome further drawbacks, if simple cell staining proceduresare used. Non-invaded cells, which stayed on the upper side ofthe transwell insert, have to be eliminated prior to staining of theinvasive cells at the bottom of the membrane. This is generallydone by removing the cells with a cotton swab, which often turnsout difficult, non-quantitative and of variable success. We stronglyrecommend to use fluorescent dyes, lyse the cells and quantifythem in a plate reader, for reliable results. The transwell invasionassay is the most frequently used invasion assay and has forexample been employed to analyze human trophoblast [95],melanoma [96], or colon cancer invasion [97]. Transwell invasionassay kits provided by many cell biology companies (e.g. BectonDickinson, Merck-Millipore, Corning) are widely used, due to theirwell-standardized assay conditions and protocols.
4.2. Platypus invasion assay
The Platypus invasion assay (OrisTM cell invasion assay,Platypus Technologies, Madison, USA; www.platypustech.com)uses the same equipment as the migration assay describedpreviously in the cell exclusion zone assay section: small siliconestoppers fitting into 96-well plates. However, the setup of themethod is quite different. First, the bottom of individual wells iscovered by a thin layer of BME. Subsequently the silicone stoppersare positioned and create an exclusion zone when cells are seeded.After cell adhesion on top of the first layer of BME, the stoppers areremoved and the cells as well as the cell-free circular center regionare overlaid by a thicker second layer of BME. Thereby a layer ofcells embedded between two sheets of ECM is made and a centralcell-free area filled with ECM is generated (Fig. 3B). Invasive cells
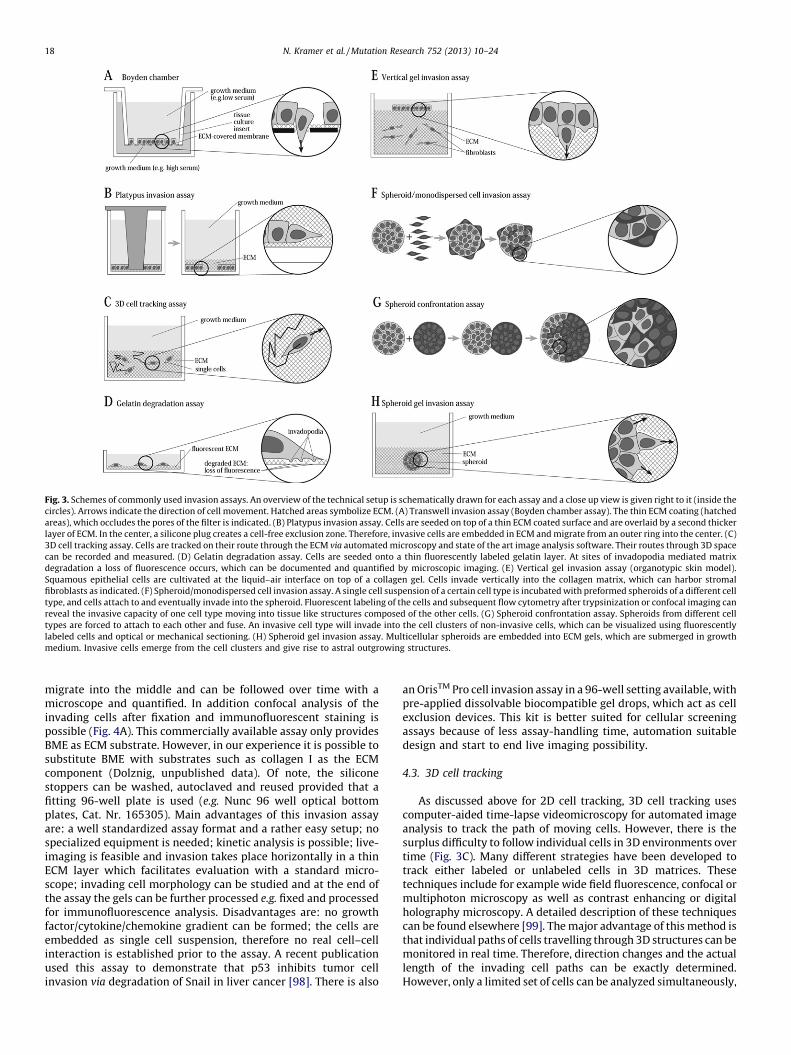
Fig. 3. Schemes of commonly used invasion assays. An overview of the technical setup is schematically drawn for each assay and a close up view is given right to it (inside the
circles). Arrows indicate the direction of cell movement. Hatched areas symbolize ECM. (A) Transwell invasion assay (Boyden chamber assay). The thin ECM coating (hatched
areas), which occludes the pores of the filter is indicated. (B) Platypus invasion assay. Cells are seeded on top of a thin ECM coated surface and are overlaid by a second thicker
layer of ECM. In the center, a silicone plug creates a cell-free exclusion zone. Therefore, invasive cells are embedded in ECM and migrate from an outer ring into the center. (C)
3D cell tracking assay. Cells are tracked on their route through the ECM via automated microscopy and state of the art image analysis software. Their routes through 3D space
can be recorded and measured. (D) Gelatin degradation assay. Cells are seeded onto a thin fluorescently labeled gelatin layer. At sites of invadopodia mediated matrix
degradation a loss of fluorescence occurs, which can be documented and quantified by microscopic imaging. (E) Vertical gel invasion assay (organotypic skin model).
Squamous epithelial cells are cultivated at the liquid–air interface on top of a collagen gel. Cells invade vertically into the collagen matrix, which can harbor stromal
fibroblasts as indicated. (F) Spheroid/monodispersed cell invasion assay. A single cell suspension of a certain cell type is incubated with preformed spheroids of a different cell
type, and cells attach to and eventually invade into the spheroid. Fluorescent labeling of the cells and subsequent flow cytometry after trypsinization or confocal imaging can
reveal the invasive capacity of one cell type moving into tissue like structures composed of the other cells. (G) Spheroid confrontation assay. Spheroids from different cell
types are forced to attach to each other and fuse. An invasive cell type will invade into the cell clusters of non-invasive cells, which can be visualized using fluorescently
labeled cells and optical or mechanical sectioning. (H) Spheroid gel invasion assay. Multicellular spheroids are embedded into ECM gels, which are submerged in growth
medium. Invasive cells emerge from the cell clusters and give rise to astral outgrowing structures.
N. Kramer et al. / Mutation Research 752 (2013) 10–2418
migrate into the middle and can be followed over time with amicroscope and quantified. In addition confocal analysis of theinvading cells after fixation and immunofluorescent staining ispossible (Fig. 4A). This commercially available assay only providesBME as ECM substrate. However, in our experience it is possible tosubstitute BME with substrates such as collagen I as the ECMcomponent (Dolznig, unpublished data). Of note, the siliconestoppers can be washed, autoclaved and reused provided that afitting 96-well plate is used (e.g. Nunc 96 well optical bottomplates, Cat. Nr. 165305). Main advantages of this invasion assayare: a well standardized assay format and a rather easy setup; nospecialized equipment is needed; kinetic analysis is possible; live-imaging is feasible and invasion takes place horizontally in a thinECM layer which facilitates evaluation with a standard micro-scope; invading cell morphology can be studied and at the end ofthe assay the gels can be further processed e.g. fixed and processedfor immunofluorescence analysis. Disadvantages are: no growthfactor/cytokine/chemokine gradient can be formed; the cells areembedded as single cell suspension, therefore no real cell–cellinteraction is established prior to the assay. A recent publicationused this assay to demonstrate that p53 inhibits tumor cellinvasion via degradation of Snail in liver cancer [98]. There is also
an OrisTM Pro cell invasion assay in a 96-well setting available, withpre-applied dissolvable biocompatible gel drops, which act as cellexclusion devices. This kit is better suited for cellular screeningassays because of less assay-handling time, automation suitabledesign and start to end live imaging possibility.
4.3. 3D cell tracking
As discussed above for 2D cell tracking, 3D cell tracking usescomputer-aided time-lapse videomicroscopy for automated imageanalysis to track the path of moving cells. However, there is thesurplus difficulty to follow individual cells in 3D environments overtime (Fig. 3C). Many different strategies have been developed totrack either labeled or unlabeled cells in 3D matrices. Thesetechniques include for example wide field fluorescence, confocal ormultiphoton microscopy as well as contrast enhancing or digitalholography microscopy. A detailed description of these techniquescan be found elsewhere [99]. The major advantage of this method isthat individual paths of cells travelling through 3D structures can bemonitored in real time. Therefore, direction changes and the actuallength of the invading cell paths can be exactly determined.However, only a limited set of cells can be analyzed simultaneously,

Table 2Comparison of common in vitro invasion assays.
Transwell invasion
assay
Platypus invasion
assay
3D cell tracking Gelatin degradation
assay
Vertical gel invasion
assay
Spheroid/monodispersed
cell invasion assay
Spheroid
confrontation assay
Spheroid invasion
assays
Chemotaxis + � Assay dependent � � n.d. n.d. �Stable chemokine
gradient
� � � � � � � �
Measurement Number of cells Invasion area Invasion distance Area of matrix
degradation
Invasion deepness Number of cells n.a. Invasion area
Live imaging � + + + � � � +
IF, IHC � + +/� + + + + +
Substrate BME, collagen I BME any ECM gelatin BME, collagen I, fibrin. . . n.a. n.a. BME, collagen I, fibrin. . .
Direction of movement Vertical Horizontal Any Vertical Vertical Any Any Any
HTS + + + � � � � �Type of analysis Endpoint Kinetic Kinetic (Kinetic) endpoint Endpoint Endpoint Endpoint Kinetic
Recapitulated ‘‘in vivo’’
migration mode
Invasion through
thin matrix
Single cell or
collective cell
invasion
Single cell migration,
chemotaxis
Proteolytic ECM
degradation
Squamous carcinoma
invasion, leukocyte
infiltration
(White blood) cell
invasion into tissues
Cancer cell invasion
into tissues
Single cell or collective
cell invasion from cell
clusters
Notes a b c d e f g h
References [92–97] [98] [99–101] [102–106] [109,110,112,113] [116,119–122,143] [123–128] [118,129–137]
Abbreviations: 3D, three-dimensional; �, not suitable; +, suitable; PC, polycarbonate; PET, polyethylenterephthalat; BME, basal membrane extract; IF, immunofluorescence; IHC, immunohistochemistry; HTS, high throughput
screening; n.d., not done so far; n.a., not applicable.a For adherent and suspension cells, no special equipment needed, technically non-demanding, commercially available.b For adherent cells, no special equipment needed, technically non-demanding, commercially available.c Special equipment needed, technically highly demanding, individual cell paths detectable, non-directed cell movement.d Subcellular invasion areas detectable, no long-term movement detection, commercially available, fluorescence microscope needed.e Used for leukocyte and epithelial cell invasion, quantification using radioactive labeled cells or microscopy, heterotypic cell interaction possible, quantification needs confocal microscope or fixation and sectioning.f Heterotypic cell interaction, in vivo like condition, invasion through tissue-like structures with cell–cell contacts, difficult to distinguish attachment and invasion, quantification by FACS.g Heterotypic cell interaction, in vivo like condition, invasion through tissue-like structures with cell–cell contacts, confocal microscopy or fixation and sectioning needed, time consuming.h Heterotypic cell interaction possible but not required, in vivo like condition, invasion through ECM, confocal microscopy or fixation and sectioning needed, time consuming, technically demanding.
N.
Kra
mer
et a
l. /
Mu
tatio
n R
esearch
75
2 (2
01
3)
10
–2
4
19

Fig. 4. In vitro invasion assays. (A) Platypus invasion assay. Mesenchymal cells invade from an outer ring into the cell-free inner zone. The dashed white line indicates the rim
of the cells prior to invasion. Cells are stained with F-actin in red. (B) Spheroid gel invasion assay. Invasive mesenchymal cells disseminate from spheroids into the
surrounding collagen I gel. The dashed white line indicates the outer rim of the spheroid. Cells are stained with F-actin in red. (C) Scanning electron microscopy (SEM) of a
similar spheroid as shown in B. The initial size of the spheroid is indicated in red. Incubation time 24 h. Astral outgrowth of invasive cells can be detected.
N. Kramer et al. / Mutation Research 752 (2013) 10–2420
a specialized microscope for live imaging is necessary as well asthere is need for advanced knowledge in data processing. The 3D celltracking was essential to demonstrate that migration of tumor cellsis directed by the stiffness of the 3D matrix in addition to celladhesion properties to and proteolysis of the matrix [100]. Extensive3D cell tracking analysis was also used to prove that rapid amoeboidmigration of leukocytes is integrin independent and relies on F-actinmediated protrusions and a Myosin II reliant squeezing of thenucleus in narrow pores [101]. This study used ImarisTrack (BitplaneAG, Zurich, Switzerland; www.bitplane.com), a commerciallyavailable software for 3D cell tracking.
4.4. Gelatin degradation assay
This assay allows visualizing and quantifying invasion at the sub-cellular level rather than analyzing the invasive behavior of wholecells. The higher resolution of this method led to the discovery ofcellular protrusions called invadopodia and podosomes [102], whichdegrade the ECM. The principle of the assay is to seed cells on top of athin layer of fluorescently labeled matrix and to record and measureregions where the cells degraded the matrix leaving behind areasthat lack fluorescence (Fig. 3D) [103,104]. The plus of this method isthat sub-cellular high resolution data of invading structures can bemade visible. On the other hand, cells cannot be followed as a wholeduring their movement. In addition the assay is not a real 3Dinvasion assay, since cells are attached to a thin layer of ECM andtherefore adapt a 2D cell shape. Therefore invadopodia structuresmight be not exactly reflecting the 3D reality. The gelatindegradation assay was used in a study, which demonstrated thatthe Src substrate Tks5 is implicated in podosome mediatedmacrophage invasion [105]. In another report the method wasemployed to demonstrate that bioactive laminin-derived peptidesincreased invadopodia activity in adenoid cystic carcinoma [106].This assay is also commercially available, providing either red orgreen fluorescent labeled gelatin as extracellular matrix (QCMTM
Gelatin Invadopodia Assay, Millipore).
4.5. Vertical gel 3D migration/invasion assays
3D migration into collagen gels were first described usingleukocytes, which were placed on top of few millimeter thickcollagen gels [107]. The vertical 3D migration was monitored byoptical sectioning and counting the cells or by radioactive labeling ofthe cells and scintillation counting [108]. Vertical invasion fromepithelial cell layers can be followed by plating carcinoma cells ontop of a collagen gel layer (Fig. 3E). Therefore, thick collagen plugs areprepared and cells are seeded on top of the gel surface (Fig. 3E). Avariant of the vertical invasion assay is the upward movement ofcells from a monolayer, onto which a layer of ECM has been poured.
The first mentioned assay is often used as organotypic skin model(skin equivalents) to study skin cancer cell invasion [109–111]. Here,most studies are performed in the presence of stromal subcutaneousfibroblasts embedded in the collagen gel (Fig. 3E). Invasion can bequantified by (immuno)histochemical staining [112] and quantifi-cation of the invasive areas using image analysis software. Theadvantage is that the organotypic skin cancer model most faithfullymimics the in vivo situation for invasion. It combines 3D invasioninto ECM with heterotypic cell–cell interaction of epithelial cancercells and stromal fibroblasts. Best analysis of invasion is possiblewith formalin fixed paraffin embedded (FFPE) samples, which arecut perpendicular to the surface. This sample processing alsoprovides information about histological properties and cellularmorphology. On the other hand, there is the drawback that samplepreparation is quite labor intense and needs special equipmentincluding embedding stations and microtomes as well as FFPEcutting experience. Using the organotypic skin cancer model it wasshown that stromal fibroblasts lead to collective invasion ofepithelial cells [113] in squamous cell carcinomas. Recently, basedon the vertical invasion assay it was shown that inhibition ofautophagy resulted in impaired invasion in a glioma cell line,whereas cell viability, proliferation and migration were unaffected[114]. One impressive organotypic 3D model investigates invasionof squamous cell carcinoma cell lines seeded on top of primaryhuman myoma tissue instead of the collagen gel [115]. Here the in
vivo situation is recapitulated even more closely. So far there is novertical invasion assay available on the market.
4.6. Spheroid/monodispersed cell invasion assay
In vivo tumor cells invade the surrounding tissue from cancercell clusters. These tumor clusters are 3D structures, which can bemimicked by small aggregates of cells formed in vitro, calledmulticellular spheroids [116]. Cells grown as multicellularspheroids closely recapitulate the in vivo situation of solid cancers[117–119]. The single cell/spheroid invasion model is a model tostudy the invasive property of a certain cell type (cell type A, whichin most cases is a non-malignant cell) into a tissue like structurecomposed of a different cell type (cell type B). However, it isimportant to note that this assay might also be used to monitor theinvasion of malignant cells into spheroid structures made ofnormal or non-malignant cells. In principle, spheroids of cell type Bare co-cultivated with a single cell suspension of cell type A, whichattach to the spheroid surface and eventually start to invade intothe spheroid (Fig. 3F). The use of fluorescently labeled cellsprovides a tool to analyze 3D migration or invasion by (confocal)fluorescence microscopy. The quantification of cells attached to orinvading into the spheroids can be done by flow cytometry aftertrypsinization. Another possibility is to fix the cells, make slices

N. Kramer et al. / Mutation Research 752 (2013) 10–24 21
and perform immunofluorescence or immunohistochemical anal-ysis. The main advantage of this method is that the barrier to beinvaded is composed of tightly arranged multicellular 3Dstructures with established cell–cell interactions as it is the casein vivo. It is ideally suited to study immune cell infiltration in vitro.However, a prerequisite must be fulfilled: the cell type B has to becapable of forming spheroids, which some cell lines do not. Thequantification of invading cells is not straight forward and requireseither deep confocal imaging or sample preparation for immuno-histochemistry including paraffin embedding and tissue sectioningequipment as well as technical expertise. If using flow cytometryfor quantitative detection a prior trypsinization step is required toremove the outmost cells prior to single cell dissemination. This isimportant to distinguish cells attached to the spheroid surfacefrom truly invading cells. A standard protocol for this has not beendeveloped yet. For example, the spheroid/monodispersed cellinvasion assay was used to study T-cell infiltration in pancreaticcarcinoma [120]; to demonstrate that tumor cell spheroids caninfluence monocyte differentiation to tumor associated macro-pghages [121]; or to prove that T-cadherin increased the invasionof endothelial cells into melanoma spheroids [122]. To the best ofour knowledge this assay is not offered commercially.
4.7. Spheroid confrontation assay
The interaction and invasion behavior of two different 3D cellaggregates can be addressed with the spheroid confrontation assay[123]. In this approach, two preformed spheroids derived fromdifferent cell types (one being invasive the other one non-invasive)are cultivated side by side and eventually start to fuse (Fig. 3G).Thereafter, cells either infiltrate the opposing spheroid as singlecells, or collectively, or display a non-invasive phenotype. Inthe non-invasion case a well-defined border is formed at theinterface between the two cell types. One important advantage ofthis method is that the invasive properties of cells grown in 3Dwith well-established cell–cell interactions into another tissue-like structure composed of a different cell type can be studied. Thisis closely reflecting the in vivo situation in case of a carcinoma isinvading a certain tissue or organ. The quantification of invadingcells requires either confocal imaging or preparation for immuno-histochemistry including paraffin embedding and tissue section-ing. If the cells express distinct markers, one can differentiate themimmunohistochemically. Alternatively, the invading cell type canbe fluorescently labeled prior to the confrontation and analyzedthereafter either by live imaging or after fixation and furtherprocessing. Therefore the main drawback of the assay is that itrequires extensive post-experimental processing and specialequipment. The prerequisite to form multicellular spheroids couldrepresent another disadvantage. There are primary cells and celllines, which do not form spheroids and therefore are not suitablefor this kind of invasion assay. The spheroid confrontation methodwas first described to study the invasive properties of cells derivedfrom human brain tumor explants into dermal cell spheroidsrepresenting the non-invasive, normal counterpart [124]. Anotherstudy analyzed the invasive capacity of colon carcinoma cells incontact with normal skin fibroblasts [125]. Recently, the spheroidconfrontation assay was employed to demonstrate that NM23-H1regulates contact inhibition of locomotion in invading glioblasto-ma cells [126]); that CXCL16 induces invasion of glial progenitors[127]; and that Endoglin enhances breast cancer cell invasion[128]. This assay format is not commercially available.
4.8. Spheroid gel invasion assays
When multicellular spheroids are embedded into 3D ECM such ascollagen I or BME gels (Fig. 3H), non-invasive cancer cell lines stay as
compact spheroids with a distinct border to the surrounding ECMand do not show any signs of invasion even after 2 weeks ofcultivation [129]. Invasive cell lines (such as the HT-1080 sarcomacell line, the ovarian carcinoma cell line SK-OV-3 or the breast cancerline MDA-MB-231) or endothelial cells start to invade into thesurrounding matrix and display astral outgrowth from the spheroid[130] (Figs. 3H and 4B and C). Invasion can be followed by liveimaging and quantified by measuring the invasive area over time inphotomicrographs. The gels with the invading structures can befixed and processed for immunofluorescence staining and confocalmicroscopy [118]. Alternatively, the gels can be enzymaticallydegraded and the cells isolated for flow cytometry analysis. A thirdpossibility is to make protein lysates and perform Western blotanalysis (Kramer, Walzl unpublished). The main advantage of thisassay is that cell movement through a 3D matrix closely mimicsinvasion in vivo. Importantly, invasion occurs from cell clusters withwell-established cell–cell interactions rather than from single cells,as it is the situation in human cancers. A wide range of different ECMgels (e.g. collagen I, BME, fibrin) can be commercially obtained intheir liquid forms and quickly solidified chemically or physically.Therefore different substrates are readily available depending on theresearch question. The outer border of spheroids placed in the gelcan be easily detected in a standard inverted light microscope. In liveimaging experiments kinetic measurements of cell invasion can bemade. Finally, it is compatible with molecular biology analysismethods. Care must be taken to distinguish real invasion from cellmovement on the surface of the gel. Some spheroids are occasionallyembedded at the gel-medium interface or at the bottom in contactwith the tissue culture plate and migrating cells always take theroute of least resistance. If cells are situated at the gel-mediuminterface they migrate along the surface instead of taking the routethrough the gel. This falsely gives the impression of rapid invasion ofthat particular spheroid. These events have to be excluded fromthe analysis. Some experience is needed to distinguish the surfacemigration, which is characterized by movement of cells in a singleplane and 2D cell morphology, from 3D invasion. Additionallythe experimental effort is rather high compared to other assays andsome pre-experience with 3D gel systems is required. The spheroidinvasion system has already been used to define essential molecularpathways during 3D invasive growth [118,131] or to measureTGFbeta induced invasion [132]. In a very similar assay cell-coatedmicrocarrier beads (see above in the migration section) can beembedded into ECM gels and invading cells can be detected. Thisassay format was used to investigate the destructive 3D motility introphoblast-like cells [133]. It is mostly used to measure theformation of endothelial cell (EC) networks in 3D gels e.g. todemonstrate the secretion of pro-angiogenic factors by mesenchy-mal stem cells [134] or to show that ECM density influences ECcapillary formation [135]. We employed this model to analyze EMTin hepatocellular carcinoma [136] and to clarify the function ofhVps37A in ovarian carcinoma [137]. Using an advancement of themethod we determined invasive properties and the molecularpathways activated by the interaction of colon carcinoma cells withstromal fibroblasts [129]. For that we dispersed fibroblasts in thecollagen gel in addition to colon cancer spheroids. This setup allowsthe interaction of tumor cells with stromal fibroblasts in 3D, whichinduced cancer cell invasion in otherwise non-invasive lines.Currently we are using the spheroid invasion model to addressthe function of the mTOR signaling pathway [138–140] duringinvasion. So far there is no spheroid invasion assay commerciallyavailable.
4.9. Conclusions and perspectives
Since the acquisition of a migratory phenotype is theprerequisite for metastatic spreading, the determination of the

N. Kramer et al. / Mutation Research 752 (2013) 10–2422
migratory and invasive potential of tumor cells and the molecularmechanisms behind this process is fundamental for proposingnovel clinical strategies in cancer diagnosis, prognosis and drugdevelopment.
A wide variety of assays assess the migratory or invasivepotential and activity of cells in vitro. These assays range from verysimple and inexpensive ones to technical demanding or expensivesolutions. However, the suitability of a specific method may belimited when considering a certain cell type(s) or specific researchquestion. In this review, we offer researchers a concise andinformative description of all available methods to measureinvasion and migration potentials of cancer cells to employ intheir investigations. We have in particular provided pragmaticdescription of all the available techniques and tools to choose from,including a depiction of the assay principle, advantages, drawbacksand limitations that will aid relative ‘‘newcomers’’ to the field inchoosing the most appropriate methods for their work. To evaluatecell migration, with reference to our own experience, werecommend using the exclusion zone assay for several reasons.(1) There is no need for advanced cell biology technical knowledge.Cells are mostly seeded in 96 well plates as monolayers; the cellexclusion zone is defined by a physical barrier, avoiding damage tothe cells. (2) The cell free zone is reproducibly of the same averagesize. (3) No special technical equipment is required; an invertedcell culture microscope equipped with a digital camera issufficient. During migration of cells into the cell exclusion zonethereby decreasing it, the wells can be photographed usingconventional phase contrast microscopy; the cell free area can bedetermined by image analysis tools (e.g. ImageJ, which candownloaded free of charge from http://www.rsbweb.nih.gov/ij/).(4) Optionally cells can be fluorescently labeled before seeding orat the end of the experiment; a mask can be applied and afluorescence signal, which is proportional to the amount ofmigrated cells, can be detected in a microplate reader; this allowshigh throughput screening. (5) The assay allows kinetic assess-ment of migration and time lapse videomicroscopy is possible ifdesired. Therefore, actual migration velocity can be determinedand the phenotype of the moving cells is recorded during theexperiment. From our point of view these facts make the cellexclusion zone assay superior to the also widely used transwellmigration assay, which reliably detects differences in cellmigration, however offers less readout options for roughly thesame cost input and work load. For a simple technique to measureinvasion the same features pointed out above we support the use ofthe Platypus invasion assay instead of the widespread transwellinvasion assay, despite being more laborious. If a close mimicry ofthe in vivo situation in tumors is desired, the following consider-ations should be taken into account. First, tissues and tumors arestructures with established cell–cell contacts in the threedimensional space. Before being able to move out of thesestructures cells have to first bear down the close physical contactto their neighbors or move collectively and attach to a differentsubstrate. This is best mimicked by the multicellular spheroidmigration assay, which is from our experience a good compromisebetween a fairly close reflection of the in vivo situation andmanageable experimental effort. Second, it is now obvious that thecellular components present in the interstitial space, whichtogether comprise the tumor stroma (i.e. fibroblasts, myofibro-blasts, pericytes, endothelial cells, immune cells) influence theinvasive potential of cancer cells [113,141,142]. These interactionsshould be considered in in vitro assays to recapitulate the in vivo
situation as close as possible. Some of the assays described here –i.e. the spheroid migration assay, the vertical gel invasion assay,spheroid gel invasion assay – can be used with heterotypiccell interaction and have already been demonstrated towork. Undoubtedly, these assays are labor intense and therefore
error-prone and cannot be used in cellular high throughputscreening. However we are convinced that they are the mostsophisticated models to address cell migration and invasion in ahighly relevant physiological setting and are therefore worth theadditional effort.
Conflict of interest
There are no conflicts of interest.
Acknowledgments
Research in our laboratories was supported by the EuropeanCommunity, FP6 036097-2 (MH), Osterreichische NationalbankP13423 (MH), Herzfelder’sche Familienstiftung (HD) and NOForschungs-und Bildungsges.m.b.H. (NFB) (HD).
References
[1] R. Keller, Cell migration during gastrulation, Curr. Opin. Cell Biol. 17 (2005) 533–541.[2] M.E. Hatten, Central nervous system neuronal migration, Annu. Rev. Neurosci. 22
(1999) 511–539.[3] J. Rossant, L. Howard, Signaling pathways in vascular development, Annu. Rev. Cell
Dev. Biol. 18 (2002) 541–573.[4] P. Friedl, B. Weigelin, Interstitial leukocyte migration and immune function, Nat.
Immunol. 9 (2008) 960–969.[5] W. International Agency for Research on Cancer (IARC), Globocan 2008, 2008.[6] D. Hanahan, R.A. Weinberg, The hallmarks of cancer, Cell 100 (2000) 57–70.[7] D.X. Nguyen, P.D. Bos, J. Massague, Metastasis: from dissemination to organ-
specific colonization, Nat. Rev. Cancer 9 (2009) 274–284.[8] I.J. Fidler, G. Poste, The seed and soil hypothesis revisited, Lancet Oncol. 9 (2008) 808.[9] G.P. Gupta, J. Massague, Cancer metastasis: building a framework, Cell 127 (2006)
679–695.[10] K. Wolf, I. Mazo, H. Leung, K. Engelke, U.H. von Andrian, E.I. Deryugina, A.Y.
Strongin, E.B. Brocker, P. Friedl, Compensation mechanism in tumor cell migra-tion: mesenchymal–amoeboid transition after blocking of pericellular proteoly-sis, J. Cell Biol. 160 (2003) 267–277.
[11] J.B. Wyckoff, S.E. Pinner, S. Gschmeissner, J.S. Condeelis, E. Sahai, ROCK- andmyosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo, Curr. Biol. 16 (2006) 1515–1523.
[12] P. Friedl, K. Wolf, Plasticity of cell migration: a multiscale tuning model, J. CellBiol. 188 (2010) 11–19.
[13] O.T. Fackler, R. Grosse, Cell motility through plasma membrane blebbing, J. CellBiol. 181 (2008) 879–884.
[14] A.J. Ridley, M.A. Schwartz, K. Burridge, R.A. Firtel, M.H. Ginsberg, G. Borisy, J.T.Parsons, A.R. Horwitz, Cell migration: integrating signals from front to back,Science 302 (2003) 1704–1709.
[15] K. Keren, Z. Pincus, G.M. Allen, E.L. Barnhart, G. Marriott, A. Mogilner, J.A. Theriot,Mechanism of shape determination in motile cells, Nature 453 (2008) 475–480.
[16] J.P. Trinkaus, Surface activity and locomotion of Fundulus deep cells duringblastula and gastrula stages, Dev. Biol. 30 (1973) 69–103.
[17] G. Charras, E. Paluch, Blebs lead the way: how to migrate without lamellipodia,Nat. Rev. Mol. Cell Biol. 9 (2008) 730–736.
[18] L.A. Smith, H. Aranda-Espinoza, J.B. Haun, M. Dembo, D.A. Hammer, Neutrophiltraction stresses are concentrated in the uropod during migration, Biophys. J. 92(2007) L58–L60.
[19] K. Yoshida, T. Soldati, Dissection of amoeboid movement into two mechanicallydistinct modes, J. Cell Sci. 119 (2006) 3833–3844.
[20] G. Gadea, V. Sanz-Moreno, A. Self, A. Godi, C.J. Marshall, DOCK10-mediated Cdc42activation is necessary for amoeboid invasion of melanoma cells, Curr. Biol. 18(2008) 1456–1465.
[21] F. Grinnell, Fibroblast biology in three-dimensional collagen matrices, Trends CellBiol. 13 (2003) 264–269.
[22] J.P. Thiery, Epithelial–mesenchymal transitions in tumour progression, Nat. Rev.Cancer 2 (2002) 442–454.
[23] A.P. Morel, M. Lievre, C. Thomas, G. Hinkal, S. Ansieau, A. Puisieux, Generation ofbreast cancer stem cells through epithelial–mesenchymal transition, PLoS One 3(2008) e2888.
[24] S.A. Mani, W. Guo, M.J. Liao, E.N. Eaton, A. Ayyanan, A.Y. Zhou, M. Brooks, F.Reinhard, C.C. Zhang, M. Shipitsin, L.L. Campbell, K. Polyak, C. Brisken, J. Yang, R.A.Weinberg, The epithelial–mesenchymal transition generates cells with propertiesof stem cells, Cell 133 (2008) 704–715.
[25] P.B. Gupta, C.L. Chaffer, R.A. Weinberg, Cancer stem cells: mirage or reality? Nat.Med. 15 (2009) 1010–1012.
[26] R.B. Vaughan, J.P. Trinkaus, Movements of epithelial cell sheets in vitro, J. Cell Sci.1 (1966) 407–413.
[27] M. Gerharz, A. Baranowsky, U. Siebolts, S. Eming, R. Nischt, T. Krieg, C. Wick-enhauser, Morphometric analysis of murine skin wound healing: standardizationof experimental procedures and impact of an advanced multitissue array tech-nique, Wound Repair Regen. 15 (2007) 105–112.

N. Kramer et al. / Mutation Research 752 (2013) 10–24 23
[28] A.J. Ewald, A. Brenot, M. Duong, B.S. Chan, Z. Werb, Collective epithelial migrationand cell rearrangements drive mammary branching morphogenesis, Dev. Cell 14(2008) 570–581.
[29] H. Gerhardt, VEGF and endothelial guidance in angiogenic sprouting, Organogen-esis 4 (2008) 241–246.
[30] N.B. David, D. Sapede, L. Saint-Etienne, C. Thisse, B. Thisse, C. Dambly-Chaudiere,F.M. Rosa, A. Ghysen, Molecular basis of cell migration in the fish lateral line: roleof the chemokine receptor CXCR4 and of its ligand, SDF1, Proc. Natl. Acad. Sci.U.S.A. 99 (2002) 16297–16302.
[31] S. Alexander, G.E. Koehl, M. Hirschberg, E.K. Geissler, P. Friedl, Dynamic imaging ofcancer growth and invasion: a modified skin-fold chamber model, Histochem.Cell Biol. 130 (2008) 1147–1154.
[32] Y. Hegerfeldt, M. Tusch, E.B. Brocker, P. Friedl, Collective cell movement inprimary melanoma explants: plasticity of cell–cell interaction, beta1-integrinfunction, and migration strategies, Cancer Res. 62 (2002) 2125–2130.
[33] K. Wolf, S. Alexander, V. Schacht, L.M. Coussens, U.H. von Andrian, J. van Rheenen,E. Deryugina, P. Friedl, Collagen-based cell migration models in vitro and in vivo,Semin. Cell Dev. Biol. 20 (2009) 931–941.
[34] S. Boyden, The chemotactic effect of mixtures of antibody and antigen onpolymorphonuclear leucocytes, J. Exp. Med. 115 (1962) 453–466.
[35] W.L. Chen, K.T. Kuo, T.Y. Chou, C.L. Chen, C.H. Wang, Y.H. Wei, L.S. Wang, The roleof cytochrome c oxidase subunit Va in nonsmall cell lung carcinoma cells:association with migration, invasion and prediction of distant metastasis, BMCCancer 12 (2012) 273.
[36] R. Harisi, I. Kenessey, J.N. Olah, F. Timar, I. Babo, G. Pogany, S. Paku, A. Jeney,Differential inhibition of single and cluster type tumor cell migration, AnticancerRes. 29 (2009) 2981–2985.
[37] S. Qi, Y. Song, Y. Peng, H. Wang, H. Long, X. Yu, Z. Li, L. Fang, A. Wu, W. Luo, Y. Zhen,Y. Zhou, Y. Chen, C. Mai, Z. Liu, W. Fang, ZEB2 mediates multiple pathwaysregulating cell proliferation, migration, invasion, and apoptosis in glioma, PLoSOne 7 (2012) e38842.
[38] G.J. Todaro, G.K. Lazar, H. Green, The initiation of cell division in a contact-inhibited mammalian cell line, J. Cell. Physiol. 66 (1965) 325–333.
[39] C.C. Liang, A.Y. Park, J.L. Guan, In vitro scratch assay: a convenient and inexpensivemethod for analysis of cell migration in vitro, Nat. Protoc. 2 (2007) 329–333.
[40] L.G. Rodriguez, X. Wu, J.L. Guan, Wound-healing assay, Methods Mol. Biol. 294(2005) 23–29.
[41] A. Inoue, S.Y. Sawata, K. Taira, R. Wadhwa, Loss-of-function screening by ran-domized intracellular antibodies: identification of hnRNP-K as a potential targetfor metastasis, Proc. Natl. Acad. Sci. U.S.A. 104 (2007) 8983–8988.
[42] S. Chigurupati, T.V. Arumugam, T.G. Son, J.D. Lathia, S. Jameel, M.R. Mughal, S.C.Tang, D.G. Jo, S. Camandola, M. Giunta, I. Rakova, N. McDonnell, L. Miele, M.P.Mattson, S. Poosala, Involvement of notch signaling in wound healing, PLoS One 2(2007) e1167.
[43] U. Doehn, C. Hauge, S.R. Frank, C.J. Jensen, K. Duda, J.V. Nielsen, M.S. Cohen, J.V.Johansen, B.R. Winther, L.R. Lund, O. Winther, J. Taunton, S.H. Hansen, M. Frodin,RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinatepromotile/invasive gene program and phenotype in epithelial cells, Mol. Cell 35(2009) 511–522.
[44] S. Zhang, Z. Cao, H. Tian, G. Shen, Y. Ma, H. Xie, Y. Liu, C. Zhao, S. Deng, Y. Yang, R.Zheng, W. Li, N. Zhang, S. Liu, W. Wang, L. Dai, S. Shi, L. Cheng, Y. Pan, S. Feng, X.Zhao, H. Deng, S. Yang, Y. Wei, SKLB1002, a novel potent inhibitor of VEGFreceptor 2 signaling, inhibits angiogenesis and tumor growth in vivo, Clin. CancerRes. 17 (2011) 4439–4450.
[45] C.S. Wu, P.H. Wu, A.H. Fang, C.C. Lan, FK506 inhibits the enhancing effects oftransforming growth factor (TGF)-beta1 on collagen expression and TGF-beta/Smad signalling in keloid fibroblasts: implication for new therapeutic approach,Br. J. Dermatol. 167 (3) (2012) 532–541.
[46] A. Mendoza-Naranjo, P. Cormie, A.E. Serrano, R. Hu, S. O’Neill, C.M. Wang, C.Thrasivoulou, K.T. Power, A. White, T. Serena, A.R. Phillips, D.L. Becker, TargetingCx43 and N-cadherin, which are abnormally upregulated in venous leg ulcers,influences migration, adhesion and activation of Rho GTPases, PLoS One 7 (2012)e37374.
[47] Y. Kam, C. Guess, L. Estrada, B. Weidow, V. Quaranta, A novel circular invasionassay mimics in vivo invasive behavior of cancer cell lines and distinguishessingle-cell motility in vitro, BMC Cancer 8 (2008) 198.
[48] C.M. Lo, C.R. Keese, I. Giaever, Monitoring motion of confluent cells in tissueculture, Exp. Cell Res. 204 (1993) 102–109.
[49] C.M. Lo, C.R. Keese, I. Giaever, Impedance analysis of MDCK cells measured byelectric cell–substrate impedance sensing, Biophys. J. 69 (1995) 2800–2807.
[50] I. Gorshkova, D. He, E. Berdyshev, P. Usatuyk, M. Burns, S. Kalari, Y. Zhao, S.Pendyala, J.G. Garcia, N.J. Pyne, D.N. Brindley, V. Natarajan, Protein kinase C-epsilon regulates sphingosine 1-phosphate-mediated migration of human lungendothelial cells through activation of phospholipase D2, protein kinase C-zeta,and Rac1, J. Biol. Chem. 283 (2008) 11794–11806.
[51] M. Tamada, T.D. Perez, W.J. Nelson, M.P. Sheetz, Two distinct modes of myosinassembly and dynamics during epithelial wound closure, J. Cell Biol. 176 (2007)27–33.
[52] M. Poujade, E. Grasland-Mongrain, A. Hertzog, J. Jouanneau, P. Chavrier, B. Ladoux, A.Buguin, P. Silberzan, Collective migration of an epithelial monolayer in response to amodel wound, Proc. Natl. Acad. Sci. U.S.A. 104 (2007) 15988–15993.
[53] T. Geback, M.M. Schulz, P. Koumoutsakos, M. Detmar, TScratch: a novel andsimple software tool for automated analysis of monolayer wound healing assays,Biotechniques 46 (2009) 265–274.
[54] T. Omelchenko, A. Hall, Myosin-IXA regulates collective epithelial cell migration bytargeting RhoGAP activity to cell–cell junctions, Curr. Biol. 22 (2012) 278–288.
[55] P.H. Su, Y.W. Lin, R.L. Huang, Y.P. Liao, H.Y. Lee, H.C. Wang, T.K. Chao, C.K. Chen,M.W. Chan, T.Y. Chu, M.H. Yu, H.C. Lai, Epigenetic silencing of PTPRR activatesMAPK signaling, promotes metastasis and serves as a biomarker of invasivecervical cancer, Oncogene (2012), http://dx.doi.org/10.1038/onc.2012.29. [Epubahead of print].
[56] A. Fougerat, N. Smirnova, S. Gayral, N. Malet, E. Hirsch, M. Wymann, B. Perret, L.Martinez, M. Douillon, M. Laffargue, Key role of PI3Kgamma in monocyte che-motactic protein-1-mediated amplification of PDGF-induced aortic smooth mus-cle cell migration, Br. J. Pharmacol. 166 (2012) 1643–1653.
[57] E.G. Fischer, A. Stingl, C.J. Kirkpatrick, Migration assay for endothelial cells inmultiwells. Application to studies on the effect of opioids, J. Immunol. Methods128 (1990) 235–239.
[58] G. Cai, J. Lian, S.S. Shapiro, D.A. Beacham, Evaluation of endothelial cell migrationwith a novel in vitro assay system, Methods Cell Sci. 22 (2000) 107–114.
[59] S.M. Sagnella, F. Kligman, E.H. Anderson, J.E. King, G. Murugesan, R.E. Marchant, K.Kottke-Marchant, Human microvascular endothelial cell growth and migrationon biomimetic surfactant polymers, Biomaterials 25 (2004) 1249–1259.
[60] W.M. Elbjeirami, J.L. West, Angiogenesis-like activity of endothelial cells co-cultured with VEGF-producing smooth muscle cells, Tissue Eng. 12 (2006)381–390.
[61] D.R. Friedlander, D. Zagzag, B. Shiff, H. Cohen, J.C. Allen, P.J. Kelly, M. Grumet,Migration of brain tumor cells on extracellular matrix proteins in vitro correlateswith tumor type and grade and involves alphaV and beta1 integrins, Cancer Res.56 (1996) 1939–1947.
[62] E.M. Rosen, L. Meromsky, E. Setter, D.W. Vinter, I.D. Goldberg, Quantitation ofcytokine-stimulated migration of endothelium and epithelium by a new assayusing microcarrier beads, Exp. Cell Res. 186 (1990) 22–31.
[63] L. Kjoller, S.M. Kanse, T. Kirkegaard, K.W. Rodenburg, E. Ronne, S.L. Goodman, K.T.Preissner, L. Ossowski, P.A. Andreasen, Plasminogen activator inhibitor-1represses integrin- and vitronectin-mediated cell migration independently ofits function as an inhibitor of plasminogen activation, Exp. Cell Res. 232 (1997)420–429.
[64] S.D. Konduri, A. Tasiou, N. Chandrasekar, J.S. Rao, Overexpression of tissue factorpathway inhibitor-2 (TFPI-2), decreases the invasiveness of prostate cancer cellsin vitro, Int. J. Oncol. 18 (2001) 127–131.
[65] M.M. Knupfer, F. Pulzer, I. Schindler, P. Hernaiz Driever, H. Knupfer, E. Keller,Different effects of valproic acid on proliferation and migration of malignantglioma cells in vitro, Anticancer Res. 21 (2001) 347–351.
[66] S. Krueger, T. Kalinski, H. Wolf, U. Kellner, A. Roessner, Interactions betweenhuman colon carcinoma cells, fibroblasts and monocytic cells in coculture—regulation of cathepsin B expression and invasiveness, Cancer Lett. 223 (2005)313–322.
[67] S.H. Zigmond, Orientation chamber in chemotaxis, Methods Enzymol. 162 (1988)65–72.
[68] D. Zicha, G.A. Dunn, A.F. Brown, A new direct-viewing chemotaxis chamber, J. CellSci. 99 (Pt 4) (1991) 769–775.
[69] S. Chaubey, A.J. Ridley, C.M. Wells, Using the Dunn chemotaxis chamber toanalyze primary cell migration in real time, Methods Mol. Biol. 769 (2011)41–51.
[70] A.R. Jensen, S.Y. David, C. Liao, J. Dai, E.T. Keller, H. Al-Ahmadie, K. Dakin-Hache, P.Usatyuk, M.F. Sievert, G.P. Paner, S. Yala, G.M. Cervantes, V. Natarajan, R. Salgia,E.M. Posadas, Fyn is downstream of the HGF/MET signaling axis and affectscellular shape and tropism in PC3 cells, Clin. Cancer Res. 17 (2011) 3112–3122.
[71] V. Echeverria, I. Meyvantsson, A. Skoien, T. Worzella, C. Lamers, S. Hayes, Anautomated high-content assay for tumor cell migration through 3-dimensionalmatrices, J. Biomol. Screen. 15 (2010) 1144–1151.
[72] M.M. Ketchel, C.B. Favour, The acceleration and inhibition of migration of humanleucocytes in vitro by plasma protein fractions, J. Exp. Med. 101 (1955) 647–663.
[73] M. George, J.H. Vaughan, In vitro cell migration as a model for delayed hyper-sensitivity, Proc. Soc. Exp. Biol. Med. 111 (1962) 514–521.
[74] J.D. Young, A.J. Lawrence, A.G. MacLean, B.P. Leung, I.B. McInnes, B. Canas, D.J.Pappin, R.D. Stevenson, Thymosin beta 4 sulfoxide is an anti-inflammatory agentgenerated by monocytes in the presence of glucocorticoids, Nat. Med. 5 (1999)1424–1427.
[75] R.R. Carpenter, P.B. Barsales, R.P. Ganchan, Antigen-induced inhibition of cellmigration in agar gel, plasma clot, and liquid media, J. Reticuloendothel. Soc. 5(1968) 472–483.
[76] J.E. Clausen, Leukocyte migration agarose technique: some technical details, ActaAllergol. 28 (1973) 351–364.
[77] M. Abe, M. Yagi, Y. Wakasugi, H. Hattori, K. Uno, A study of elevated interleukin-8(CXCL8) and detection of leukocyte migration inhibitory activity in patientsallergic to beta-lactam antibiotics, Allergol. Int. 60 (2011) 497–504.
[78] G. Albrecht-Buehler, The phagokinetic tracks of 3T3 cells, Cell 11 (1977) 395–404.[79] K. Takaishi, T. Sasaki, Y. Takai, Cell motility assay and inhibition by Rho-GDP
dissociation inhibitor, Methods Enzymol. 256 (1995) 336–347.[80] Y. Niinaka, A. Haga, A. Raz, Quantification of cell motility: gold colloidal phago-
kinetic track assay and wound healing assay, Methods Mol. Med. 58 (2001) 55–60.[81] D.T. Woodley, P.M. Bachmann, E.J. O’Keefe, Laminin inhibits human keratinocyte
migration, J. Cell. Physiol. 136 (1988) 140–146.[82] W. Li, J. Fan, M. Chen, S. Guan, D. Sawcer, G.M. Bokoch, D.T. Woodley, Mechanism
of human dermal fibroblast migration driven by type I collagen and platelet-derived growth factor-BB, Mol. Biol. Cell 15 (2004) 294–309.
[83] M. Lin, M.M. DiVito, S.D. Merajver, M. Boyanapalli, K.L. van Golen, Regulation ofpancreatic cancer cell migration and invasion by RhoC GTPase and caveolin-1,Mol. Cancer 4 (2005) 21.

N. Kramer et al. / Mutation Research 752 (2013) 10–2424
[84] G. Sommer, C. Rossa, A.C. Chi, B.W. Neville, T. Heise, Implication of RNA-bindingprotein La in proliferation, migration and invasion of lymph node-metastasizedhypopharyngeal SCC cells, PLoS One 6 (2011) e25402.
[85] L. Liu, F. Li, J.A. Cardelli, K.A. Martin, J. Blenis, S. Huang, Rapamycin inhibits cellmotility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways, Onco-gene 25 (2006) 7029–7040.
[86] W. Gu, T. Pellegrino, W.J. Parak, R. Boudreau, M.A. Le Gros, A.P. Alivisatos, C.A.Larabell, Measuring cell motility using quantum dot probes, Methods Mol. Biol.374 (2007) 125–131.
[87] K. Jaqaman, D. Loerke, M. Mettlen, H. Kuwata, S. Grinstein, S.L. Schmid, G.Danuser, Robust single-particle tracking in live-cell time-lapse sequences,Nat. Methods 5 (2008) 695–702.
[88] R. Zantl, E. Horn, Chemotaxis of slow migrating mammalian cells analysed byvideo microscopy, Methods Mol. Biol. 769 (2011) 191–203.
[89] K. Miura, Tracking movement in cell biology, Adv. Biochem. Eng. Biotechnol. 95(2005) 267–295.
[90] A.L. Campbell, H.P. Shih, J. Xu, M.K. Gross, C. Kioussi, Regulation of motility ofmyogenic cells in filling limb muscle anlagen by Pitx2, PLoS One 7 (2012)e35822.
[91] E. Mehes, E. Mones, V. Nemeth, T. Vicsek, Collective motion of cells mediatessegregation and pattern formation in co-cultures, PLoS One 7 (2012) e31711.
[92] A. Albini, Y. Iwamoto, H.K. Kleinman, G.R. Martin, S.A. Aaronson, J.M. Kozlowski,R.N. McEwan, A rapid in vitro assay for quantitating the invasive potential oftumor cells, Cancer Res. 47 (1987) 3239–3245.
[93] A. Albini, R. Benelli, The chemoinvasion assay: a method to assess tumor andendothelial cell invasion and its modulation, Nat. Protoc. 2 (2007) 504–511.
[94] J. Marshall, Transwell((R)) invasion assays, Methods Mol. Biol. 769 (2011) 97–110.[95] C.L. Tang, H.B. Zhao, M.Q. Li, M.R. Du, Y.H. Meng, D.J. Li, Focal adhesion kinase
signaling is necessary for the cyclosporin A-enhanced migration and invasion ofhuman trophoblast cells, Placenta 33 (9) (2012) 704–711.
[96] B. Kreiseder, L. Orel, C. Bujnow, S. Buschek, M. Pflueger, W. Schuett, H. Hunds-berger, R. de Martin, C. Wiesner, Alpha-catulin downregulates E-cadherin andpromotes melanoma progression and invasion, Int. J. Cancer (2012), Jun 26.http://dx.doi.org/10.1002/ijc.27698. [Epub ahead of print].
[97] F. Zhang, Q. Yang, F. Meng, H. Shi, H. Li, Y. Liang, A. Han, Astrocyte elevated gene-1 interacts with beta-catenin and increases migration and invasion of colorectalcarcinoma, Mol. Carcinog. (2012), Mar 16. http://dx.doi.org/10.1002/mc.21894.[Epub ahead of print].
[98] S.O. Lim, H. Kim, G. Jung, p53 inhibits tumor cell invasion via the degradation ofsnail protein in hepatocellular carcinoma, FEBS Lett. 584 (2010) 2231–2236.
[99] N. Hamilton, Quantification and its applications in fluorescent microscopyimaging, Traffic 10 (2009) 951–961.
[100] M.H. Zaman, L.M. Trapani, A.L. Sieminski, D. Mackellar, H. Gong, R.D. Kamm, A.Wells, D.A. Lauffenburger, P. Matsudaira, Migration of tumor cells in 3D matricesis governed by matrix stiffness along with cell-matrix adhesion and proteolysis,Proc. Natl. Acad. Sci. U.S.A. 103 (2006) 10889–10894.
[101] T. Lammermann, B.L. Bader, S.J. Monkley, T. Worbs, R. Wedlich-Soldner, K. Hirsch,M. Keller, R. Forster, D.R. Critchley, R. Fassler, M. Sixt, Rapid leukocyte migration byintegrin-independent flowing and squeezing, Nature 453 (2008) 51–55.
[102] I. Ayala, M. Baldassarre, G. Caldieri, R. Buccione, Invadopodia: a guided tour, Eur.J. Cell Biol. 85 (2006) 159–164.
[103] W.T. Chen, K. Olden, B.A. Bernard, F.F. Chu, Expression of transformation-associated protease(s) that degrade fibronectin at cell contact sites, J. Cell Biol.98 (1984) 1546–1555.
[104] V.V. Artym, K.M. Yamada, S.C. Mueller, ECM degradation assays for analyzinglocal cell invasion, Methods Mol. Biol. 522 (2009) 211–219.
[105] K.L. Burger, A.L. Davis, S. Isom, N. Mishra, D.F. Seals, The podosome marker proteinTks5 regulates macrophage invasive behavior, Cytoskeleton 68 (2011) 694–711.
[106] C.F. Nascimento, A.S. de Siqueira, J.J. Pinheiro, V.M. Freitas, R.G. Jaeger, Laminin-111 derived peptides AG73 and C16 regulate invadopodia activity of a humanadenoid cystic carcinoma cell line, Exp. Cell Res. 317 (2011) 2562–2572.
[107] S.L. Schor, T.D. Allen, B. Winn, Lymphocyte migration into three-dimensionalcollagen matrices: a quantitative study, J. Cell Biol. 96 (1983) 1089–1096.
[108] B. Rocha, W.S. Haston, A.A. Freitas, Lymphocyte migration into collagen gels: roleof lymph, Scand. J. Immunol. 19 (1984) 297–305.
[109] E. Yamamoto, G. Kohama, H. Sunakawa, M. Iwai, H. Hiratsuka, Mode of invasion,bleomycin sensitivity, and clinical course in squamous cell carcinoma of the oralcavity, Cancer 51 (1983) 2175–2180.
[110] N.E. Fusenig, D. Breitkreutz, R.T. Dzarlieva, P. Boukamp, A. Bohnert, W. Tilgen,Growth and differentiation characteristics of transformed keratinocytes frommouse and humanskinin vitro and invivo, J. Invest. Dermatol. 81 (1983) 168s–175s.
[111] P. Timpson, E.J. McGhee, Z. Erami, M. Nobis, J.A. Quinn, M. Edward, K.I. Anderson,Organotypic collagen I assay: a malleable platform to assess cell behaviour in a3-dimensional context, J. Vis. Exp. (2011) e3089.
[112] M.L. Nystrom, G.J. Thomas, M. Stone, I.C. Mackenzie, I.R. Hart, J.F. Marshall,Development of a quantitative method to analyse tumour cell invasion inorganotypic culture, J. Pathol. 205 (2005) 468–475.
[113] C. Gaggioli, S. Hooper, C. Hidalgo-Carcedo, R. Grosse, J.F. Marshall, K. Harrington, E.Sahai, Fibroblast-led collective invasion of carcinoma cells with differing roles forRhoGTPases in leading and following cells, Nat. Cell Biol. 9 (2007) 1392–1400.
[114] R.L. Macintosh, P. Timpson, J. Thorburn, K.I. Anderson, A. Thorburn, K.M. Ryan,Inhibition of autophagy impairs tumor cell invasion in an organotypic model,Cell Cycle 11 (2012) 2022–2029.
[115] S. Nurmenniemi, T. Sinikumpu, I. Alahuhta, S. Salo, M. Sutinen, M. Santala, J.Risteli, P. Nyberg, T. Salo, A novel organotypic model mimics the tumor micro-environment, Am. J. Pathol. 175 (2009) 1281–1291.
[116] R.M. Sutherland, J.A. McCredie, W.R. Inch, Growth of multicell spheroids in tissueculture as a model of nodular carcinomas, J. Natl. Cancer Inst. 46 (1971) 113–120.
[117] L.A. Kunz-Schughart, P. Heyder, J. Schroeder, R. Knuechel, A heterologous 3-Dcoculture model of breast tumor cells and fibroblasts to study tumor-associatedfibroblast differentiation, Exp. Cell Res. 266 (2001) 74–86.
[118] K. Wolf, Y.I. Wu, Y. Liu, J. Geiger, E. Tam, C. Overall, M.S. Stack, P. Friedl, Multi-step pericellular proteolysis controls the transition from individual to collectivecancer cell invasion, Nat. Cell Biol. 9 (2007) 893–904.
[119] M. Pickl, C.H. Ries, Comparison of 3D and 2D tumor models reveals enhancedHER2 activation in 3D associated with an increased response to trastuzumab,Oncogene 28 (2009) 461–468.
[120] D. Nummer, E. Suri-Payer, H. Schmitz-Winnenthal, A. Bonertz, L. Galindo, D.Antolovich, M. Koch, M. Buchler, J. Weitz, V. Schirrmacher, P. Beckhove, Role oftumor endothelium in CD4+ CD25+ regulatory T cell infiltration of humanpancreatic carcinoma, J. Natl. Cancer Inst. 99 (2007) 1188–1199.
[121] A. Konur, M. Kreutz, R. Knuchel, S.W. Krause, R. Andreesen, Cytokine repertoireduring maturation of monocytes to macrophages within spheroids of malignantand non-malignant urothelial cell lines, Int. J. Cancer 78 (1998) 648–653.
[122] S. Ghosh, M.B. Joshi, D. Ivanov, C. Feder-Mengus, G.C. Spagnoli, I. Martin, P. Erne,T.J. Resink, Use of multicellular tumor spheroids to dissect endothelial cell–tumor cell interactions: a role for T-cadherin in tumor angiogenesis, FEBS Lett.581 (2007) 4523–4528.
[123] K. Hattermann, J. Held-Feindt, R. Mentlein, Spheroid confrontation assay: asimple method to monitor the three-dimensional migration of different celltypes in vitro, Ann. Anat. 193 (2011) 181–184.
[124] L. de Ridder, M. Cornelissen, D. de Ridder, Autologous spheroid culture: ascreening tool for human brain tumour invasion, Crit. Rev. Oncol. Hematol.36 (2000) 107–122.
[125] T. Brabletz, A. Jung, S. Reu, M. Porzner, F. Hlubek, L.A. Kunz-Schughart, R.Knuechel, T. Kirchner, Variable beta-catenin expression in colorectal cancersindicates tumor progression driven by the tumor environment, Proc. Natl. Acad.Sci. U.S.A. 98 (2001) 10356–10361.
[126] M. Tanaka, S. Kuriyama, N. Aiba, Nm23-H1 regulates contact inhibition oflocomotion which is affected by ephrin-B1, J. Cell Sci. (2012), Jun 20. [Epubahead of print].
[127] K. Hattermann, A. Ludwig, V. Gieselmann, J. Held-Feindt, R. Mentlein, Thechemokine CXCL16 induces migration and invasion of glial precursor cells viaits receptor CXCR6, Mol. Cell. Neurosci. 39 (2008) 133–141.
[128] D. Oxmann, J. Held-Feindt, A.M. Stark, K. Hattermann, T. Yoneda, R. Mentlein,Endoglin expression in metastatic breast cancer cells enhances their invasivephenotype, Oncogene 27 (2008) 3567–3575.
[129] H. Dolznig, C. Rupp, C. Puri, C. Haslinger, N. Schweifer, E. Wieser, D. Kerjaschki, P.Garin-Chesa, Modeling colon adenocarcinomas in vitro a 3D co-culture systeminduces cancer-relevant pathways upon tumor cell and stromal fibroblastinteraction, Am. J. Pathol. 179 (2011) 487–501.
[130] T. Korff, H.G. Augustin, Tensional forces in fibrillar extracellular matrices controldirectional capillary sprouting, J. Cell Sci. 112 (Pt 19) (1999) 3249–3258.
[131] F. Sabeh, R. Shimizu-Hirota, S.J. Weiss, Protease-dependent versus-independentcancer cell invasion programs: three-dimensional amoeboid movement revis-ited, J. Cell Biol. 185 (2009) 11–19.
[132] H.P. Naber, E. Wiercinska, P. Ten Dijke, T. van Laar, Spheroid assay to measureTGF-beta-induced invasion, J. Vis. Exp. (2011), Nov 16;(57). pii: 3337. http://dx.doi.org/10.3791/3337.
[133] J. McCormick, G.S. Whitley, P. Le Bouteiller, J.E. Cartwright, Soluble HLA-Gregulates motility and invasion of the trophoblast-derived cell line SGHPL-4,Hum. Reprod. 24 (2009) 1339–1345.
[134] A.I. Hoch, B.Y. Binder, D.C. Genetos, J.K. Leach, Differentiation-dependent secretionof proangiogenic factors by mesenchymal stem cells, PLoS One 7 (2012) e35579.
[135] E. Kniazeva, A.J. Putnam, Endothelial cell traction and ECM density influenceboth capillary morphogenesis and maintenance in 3-D, Am. J. Physiol. CellPhysiol. 297 (2009) C179–C187.
[136] F. van Zijl, M. Mair, A. Csiszar, D. Schneller, G. Zulehner, H. Huber, R. Eferl, H. Beug,H. Dolznig, W. Mikulits, Hepatic tumor–stroma crosstalk guides epithelial tomesenchymal transition at the tumor edge, Oncogene 28 (45) (2009) 4022–4033.
[137] M. Wittinger, P. Vanhara, A. El-Gazzar, B. Brenner, D. Pils, M. Anees, T.W. Grunt,M. Sibilia, M. Holcmann, R. Horvat, S. Michael, R. Zeillinger, C. Schofer, H. Dolznig,P. Horak, M. Krainer, hVps37A status affects prognosis and cetuximab sensitivityin ovarian cancer, Clin. Cancer Res. 17 (24) (2011) 7816–7827.
[138] M. Rosner, H. Dolznig, C. Fuchs, N. Siegel, A. Valli, M. Hengstschlager, CDKs astherapeutic targets for the human genetic disease tuberous sclerosis? Eur. J. Clin.Invest. 39 (12) (2009) 1033–1035.
[139] A. Valli, M. Rosner, C. Fuchs, N. Siegel, C.E. Bishop, H. Dolznig, U. Madel, W.Feichtinger, A. Atala, M. Hengstschlager, Embryoid body formation of humanamniotic fluid stem cells depends on mTOR, Oncogene 29 (2010) 966–977.
[140] C. Fuchs, M. Rosner, H. Dolznig, M. Mikula, N. Kramer, M. Hengstschlager,Tuberin and PRAS40 are anti-apoptotic gatekeepers during early human amni-otic fluid stem cell differentiation, Hum. Mol. Genet. 21 (5) (2011) 1049–1061.
[141] A.F. Olumi, G.D. Grossfeld, S.W. Hayward, P.R. Carroll, T.D. Tlsty, G.R. Cunha,Carcinoma-associated fibroblasts direct tumor progression of initiated humanprostatic epithelium, Cancer Res. 59 (1999) 5002–5011.
[142] A. Orimo, P.B. Gupta, D.C. Sgroi, F. Arenzana-Seisdedos, T. Delaunay, R. Naeem,V.J. Carey, A.L. Richardson, R.A. Weinberg, Stromal fibroblasts present in invasivehuman breast carcinomas promote tumor growth and angiogenesis throughelevated SDF-1/CXCL12 secretion, Cell 121 (2005) 335–348.
[143] L.A. Kunz-Schughart, Multicellular tumor spheroids: intermediates betweenmonolayer culture and in vivo tumor, Cell Biol. Int. 23 (1999) 157–161.