hydrolysis and transfer reactions catalyzed by w … · hydrolysis and transfer reactions catalyzed...
TRANSCRIPT

HYDROLYSIS AND TRANSFER REACTIONS CATALYZED BY w-AMIDASE PREPARATIONS *
BY ALTON MEISTER, LEON LEVINTOW, ROBERT E. GREENFIELD, AND PATRICIA A. ABENDSCHEIN
(From the Laboratory of Biochemistry, National Cancer Institute, National Institutes of Health, Bethesda, Maryland)
(Received for publication, December 22, 1954)
Recent studies have shown that deamidation of glutamine and aspara- gine may take place by at least several types of enzymatic reactions. These include (a) hydrolysis of glutamine or asparagine to the respective or-aminodicarboxylic acids and ammonia (1, 2), (b) hydrolysis as in (a) but dependent upon the presence of phosphate or certain other anions for activity (3-5), (c) deamidation of glutamine and asparagine by mecha- nisms involving participation of an a-keto acid (6-9), (d) deamidation of glutamine to glutamate associated with the synthesis of adenosine tri- phosphate from adenosine diphosphate and phosphate (i.e., reversal of gluta.mine synthesis) (lo), and (e) deamidation of glutamine and aspara- gine associated with w replacement reactions (11-16). Studies in this lab- oratory have shown that the deamidation reactions described under (c) are associated with transaminat.ion leading to formation of the a-amino acid analogous to the cy-keto acid (8,9). The formation of ammonia in the ar-keto acid-dependent deamidation reactions may be ascribed to enzymatic hydrolysis of the oc-keto acid w-amides, a-ketoglutaramic and cr-ketosuccin- amic acids, which appear to be intermediates in these reactions. Extracts of a number of tissues catalyze the hydrolysis of the.&-keto acid w-amides, and a purified enzyme preparation capable of catalyzing these reactions has been obtained from rat, liver (17, 18).
The present communication describes studies on certain properties of this liver enzyme as well as those of preparations of a bacterial glutaminase and guinea pig serum asparaginase. The liver enzyme catalyzes the hy- drolysis of or-ketosuccinamic, cu-ketoglutaramic, succinamic, and glutaramic acids, but does not deamidate glutamine or asparagine. It also catalyzes the formation of succinylmonohydroxamic and glutarylmonohydroxamic acids from the corresponding amides and hydroxylamine much more rapidly than the hydrolysis of these amides. On the other hand, the glutaminase and asparaginase preparations studied here catalyze hydroxamic acid for- mation from glutamine and asparagine, respectively, at rates relatively
*Presented in part before the Division of Biological Chemistry at the 127th meeting of the American Chemical Society at Cincinnati, April 1, 1955.
441
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

442 w-AMIDASE PREPARATIONS
low compared to those of hydrolysis. It is of interest that, in the presence of hydroxylamine, all of these preparations also catalyze monohydroxamic acid synthesis from the dicarboxylic acids corresponding to the susceptible amides. The findings are discussed in terms of the concept that the trans- fer and hydrolysis reactions are catalyzed by the same enzyme.
EXPERIMENTAL
Substrates-The following compounds were prepared as described : bar- ium a-ketoglutaramate (17)) sodium cY-ketosuccinamate (17)) sodium (Y- ketoadipamate (18)) barium cr-keto-dl-y-methylglutaramate (18)) D- and L-glutamine (lo), D- and L-homoglutamine (18)) cY-methyl-nn-glutamine (18)) dl-y-methyl-n-glutamine (18)) adipamide (19)) adipamic acid (20)) glutaramic acid (20), glutaramide (19), oxamic acid (21), L-y-glutamyl- hydroxamic acid (lo), succinamic acid,l sodium malonamate,l potassium succinylhydroxamate (22).
Acetamide, propionamide, valeramide, isovaleramide, caproamide, and malonamide were obtained from the Eastman Kodak Company. Bu- tyramide, isobutyramide, oxamide, and succinamide were Kahlbaum prod- ucts. n-Asparagine monohydrate ( [OI]~~ - 29.2°)2 and L-asparagine mono- hydrate ([O(]:~ +29.2”)z were obtained from the Nutritional Biochemicals Corporation. Several of the commercial preparations required recrystal- lization; all of the compounds gave theoretical values for nitrogen3
The authors are indebted to Dr. Jesse P. Greenstein, Dr. Sanford M. Birnbaum, and Dr. Milton Winitz of this Laboratory for the amino acid a-amides used in these studies, to Dr. Karl Pfister of Merck and Company for the ar-methyl-nn-asparagine, and to Dr. Simon Black of the National Institute of Arthritis and Metabolic Diseases for a sample of L-P-aspartyl- hydroxamic acid.
dl-fi-Methyl-DL-glutamic Acid-This compound was prepared from di- ethylacetamidomalonate and ethyl crotonate by a procedure analogous to those of Snyder et al. (23) and Done and Fowden (24) for glutamic and y-methylglutamic acids, respectively. 3.6 gm. of sodium were added to. 600 ml. of absolute ethanol, and, after the reaction was complete, 326 gm. of diethylacetamidomalonate were added and the mixture was boiled under a reflux. 208 gm. of ethyl crotonate were added dropwise over a 5 hour period, and boiling was continued for an additional 12 hours. Evapora- tion of the solvent yielded a white crystalline compound, which was boiled
1 Otani, T. T., and Meister, A., Abstracts, Division of Biological Chemistry, American Chemical Society, 127th meeting, Cincinnati, April I (1955).
2 9 per cent, in 3 N hydrochloric acid. 3 The microanalpses were carried out by Dr. William C. Alford and Mr. Robert J.
Koegel
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 443
under a reflux with 10 times its weight of 6 N hydrochloric acid for 18 hours. The product was isolated and crystallized as described for y-methylglu- tamic acid (18). Calculated for C6H1104N: C 44.7, H 6.9, N 8.7; found, C 44.3, H 6.8, N 8.8.
dl-~-Methyl-~~-glutamine-Carbobenzoxy-dl-~-methyl-n~-glutamic acid diamide was prepared and found to be resistant to deamidation by papain; the w-amide was therefore prepared via the carbobenzoxy w ester as de- scribed previously for a-methyl-nn-glutamine (18). Calculated for d&/3- methyl-nn-glutamic acid y-ethyl ester, CsH1604N: C 50.7, H 7.9, N 7.4; found, C 50.4, H 8.2, N 7.6. Amidation of carbobenzoxy-dl-fi-methyl-nn- glutamic acid y-ethyl ester proceeded more slowly than did amidation of the corresponding cr-methylglutamic and a-aminoadipic acid derivatives; amidation of the ,&methyl compound was complete after 4 days at room temperature. Calculated for d@-methyl-nn-glutamine, C6H1203N2: C 45.0, H 7.6, N 17.5; found, C 44.9, H 7.7, N 17.3.
dl-a-Aminomalonamic Acid-80 gm. of bromine were added dropwise over a 3 hour period to 66 gm. of malonic acid-monoethyl ester dissolved in 250 ml. of dry ether. The mixture was kept at about 25” by occasional use of an ice bath. After all the bromine was added, the mixture was stirred for an additional 30 minutes. The ether and hydrogen bromide were removed in vacua, and the crude sirupy bromo compound was cooled to -10” and added to 1 liter of cold 28 per cent aqueous ammonia. The mixture was allowed to stand for 1 day in a stoppered bottle at room tem- perature, following which the volume was reduced to about 200 ml. by evaporation in vacua. 1 liter of a 20 per cent solution of lead acetate was added, and, after standing for several hours, the precipitated lead salt was filtered and washed with cold water until the wash water was free of halogen and ammonia. The lead salt was decomposed with hydrogen sulfide, and about 30 gm. of crude ol-aminomalonamic acid were obtained by evap- oration of the solution to dryness in vacua. Paper chromatography re- vealed that the product was contaminated with glycine, glycine amide, and aminomalonic acid. Attempts to recrystallize the product from warm (50”) water resulted in conversion of the desired amide to glycine and glycine amide, and some breakdown was observed, even at room tempera- ture. Purification was carried out by chromatography on an Amberlite XE-64 column in the acid form (170 X 2.5 cm.) as follows. 2 gm. of crude material dissolved in 10 ml. of water (pH 5.2) were added to the top of the column, and elution was carried out with water, a flow rate of 0.67 ml. per minute being used. Sixty fractions of 10 ml. each were collected. Tubes 26 to 30 contained ar-aminomalonic acid and a brown material which did not give a ninhydrin reaction. Tubes 31 to 34 apparently contained only a-aminomalonamic acid. Tubes 35 to 37 did not give a ninhydrin
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

444 w-AMIDASE PREPARATIONS
reaction, Tubes 38 to 45 contained glycine, and glycine amide appeared in later fractions. The fractions containing the product were combined and lyophilized. 850 mg. of a light white powder were obtained, which gave the following analyses. Calculated for C3H603N2: C 30.5, H 5.1, N 23.7; found, C 30.1, H 5.3, N 23.7. The product was stable in aqueous solution at room temperature for only a few hours, after which time glycine amide formation was detected. When such solutions were heated at 100’ for 2 hours, complete destruction of the product with conversion to glycine amide, glycine, and ammonia was observed. Treatment with 2 N hydro- chloric acid at 100” for 2 hours yielded glycine and stoichiometric quantities of ammonia.
Procedures-In the course of purification of the Escherichia coli glu- taminase, assays were performed as follows: Enzyme solution (0.25 ml.) was mixed with 0.25 ml. of freshly prepared 0.08 M L-glutamine (in 0.1 M
sodium acetate buffer of pH 4.9) and incubated for 5 to 60 minutes at 37”. The reaction was stopped by addition of 0.5 ml. of 15 per cent trichloro- acetic acid, and ammonia was determined after aeration in the presence of potassium carbonate into sulfuric acid traps (25). The assay system used for following the preparation of asparaginase consisted of 0.5 ml. of 0.04 M
L-asparagine, 0.5 ml. of enzyme solution, and 0.5 ml. of 0.1 M sodium borate buffer of pH 8.5. With both systems, controls were employed in which enzyme and substrate were separately omitted.
The composition of the reaction mixtures used in the other studies is given in the text. Hydroxylamine hydrochloride solutions were adjusted to the desired pH with sodium hydroxide just prior to use. Hydroxamic acid formation was determined by the method of Lipmann and Tuttle (26). 1 ml. of the reaction mixture was treated with 1.5 ml. of a solution containing 0.67 N hydrochloric acid, 0.37 M ferric chloride, and 0.2 M tri- chloroacetic acid. After centrifugation, the colors were compared with those of standard solutions in a Klett-Summerson calorimeter equipped with a No. 540 filter. Freshly prepared aqueous solutions of r-glutamyl- hydroxamic, fl-aspartylhydroxamic, and succinylmonohydroxamic acids were employed as standards. Glutarylmonohydroxamic acid solut.ions were prepared by dissolving glutaric anhydride in a large excess of hy- droxylamine at pH 7 (26). The relative color values for the hydroxamic acids corresponding to glutamine, asparagine, glutaramic acid, and suc- cinamic acid were 100, 67.0, 115, and 115, respectively. In all experi- ments, controls in which enzyme was omitted were carried out.
Paper chromatography was carried out on Whatman No. 4 paper by an ascending technique and the following solvents: (a) formic acid-water- tertiary butanol (15 : 15 : 70) ; (b) phenol saturated with 10 per cent aqueous sodium carbonate; and (c) n-butanol-ethanol-water (2: 1: 1). Amino
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 445
acids were identified by dipping the dried chromatograms in acetone con- taining 0.25 per cent ninhydrin. For identification of hydroxamic acids the dried chromatograms were dipped in a solution of 95 per cent ethanol containing 1.0 N hydrochloric acid and 0.185 M ferric chloride. The RF values for p-aspartylhydroxamic, y-glutamylhydroxamic, succinylmono- hydroxamic, and glutarylmonohydroxamic acids were, respectively, as fol- lows: solvent (a) 0.15 to 0.24, 0.23 to 0.28, 0.52 to 0.57, 0.58 to 0.66; sol- vent (b) 0.15 to 0.22, 0.28 to 0.35, 0.45 to 0.50, 0.53 to 0.63; solvent (c), 0.25 to 0.32, 0.32 to 0.37, 0.54 to 0.59, 0.60 to 0.68.
Succinic, succinamic, glutaric, glutaramic, and pyrrolidonecarboxylic acids were identified on the chromatograms by application of the method of Rydon and Smith (27). In our hands, the procedure gave satisfactory re- sults when the following conditions were employed. After chromatog- raphy, the papers were dried for 10 to 20 minutes in a stream of warm air and then placed in an autoclave at 20 pounds pressure for 15 minutes. The autoclave was exhausted rapidly and steam was allowed to flow through the outer jacket for 10 minutes. The papers were exposed to an atmosphere of chlorine for 10 minutes, after which they were placed in a stream of air at room temperature for 30 minutes. The spots were visualized by spraying lightly and evenly with a fine mist of 1 per cent soluble starch in 1 per cent potassium iodide solution. The Rp values for succinamic, succinic, glutaramic, glutaric, and pyrrolidonecarboxylic acids were, respectively, as follows: solvent (a), 0.53 to 0.60, 0.70 to 0.74, 0.68 to 0.72, 0.80 to 0.85, 0.62 to 0.70; solvent (c), 0.52 to 0.58, 0.67 to 0.71, 0.62 to 0.66, 0.72 to 0.78, 0.40 to 0.45.
Preparation of PuriJied Glutaminase from E. co&-Lyophilized E. coli cells (strain W) were prepared, ground, and extracted as previously de- scribed (28). The cell-free extract contained 60 to 80 per cent of the origi- nal activity of the cells, and the specific activity was twice that of the cells. The extract obtained from 5 gm. of cells was mixed with the residue ob- tained by centrifuging 100 ml. of a solution of calcium phosphate gel (dry weight, 15 mg. per ml.). The mixture was mechanically shaken at room temperature for 30 minutes and centrifuged. The enzyme was eluted by shaking the gel residue with 40 ml. of 0.1 M potassium dihydrogen phos- phate (pH 4.5) for 20 minutes at 5“, followed by centrifugation at this temperature. The elution procedure was repeated twice, and the eluates were combined and lyophilized. The lyophilized material represented between 20 and 40 per cent of the original activity a.nd exhibited a 15- to 20-fold increase in specific activity over the cell-free extract. The prep- aration at this stage was free of glutamic decarboxylase and asparaginase activities. It was found possible to double the specific activity at the ex- pense of about half of the total activity by fractionation with sodium sul-
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

446 w-AMIDASE PREPARATIONS
fate as follows. 50 mg. of the lyophilized powder were suspended in 5 ml. of saturated sodium sulfate solution, shaken for 20 minutes at room temperature, and centrifuged. The pellet was dissolved in 1 ml. of water and the solution was stored at 5”. In contrast to the lyophilized powder obtained in the previous step, the enzyme in this form was unstable and lost considerable activity after several days. Such preparations catalyzed the hydrolysis of 10 to 15 mmoles of L-glutamine per mg. of enzyme nitro- gen per hour, representing a 50- to 75-fold increase in specific activity over the original cell suspension.
Preparation of Guinea Pig Serum Asparaginase-100 ml. of fresh guinea pig serum were mixed with an equal volume of 30 per cent sodium sulfate. After standing at room temperature for 30 minutes, the mixture was cen- trifuged at 20”. The pellet was dissolved in 50 ml. of cold water and mixed with the residue obtained by centrifuging an equal volume of calcium phosphate gel. After shaking for 30 minutes, the gel was removed by cen- trifugation, and the supernatant fluid, containing the enzyme, was stored in the frozen state. Such preparations hydrolyzed 120 to 160 pmoles of L-asparagine per mg. of protein nitrogen per hour, compared to values of 15 to 22 obtained with the original serum.
Preparation of Rat Liver Amidase (Fraction K)-This was prepared as described previously (17). In most preparations the most active fraction was obtained by elution from calcium phosphate gel at pH 6.5. However, with some batches of gel the most active fraction was obtained by elution at pH 5.5 or at pH 6.0. Because of the variability in the behavior of the gel, all of the eluates were assayed and the most active fraction was chosen for study.
Results
Non-Enzymatic Hydroxamic Acid Formation-Non-enzymatic hydrox- amic acid formation from amides was not appreciable except with relatively high (0.5 to 1.0 M) concentrations of hydroxylamine. Even with high hydroxylamine concentrations, the rates of non-enzymatic hydroxamic acid formation were considerably lower than those observed with enzyme. The values reported here for enzymatic hydroxamic acid formation were corrected by subtraction of the values observed without enzyme. The effect of pH on non-enzymatic hydroxamic acid formation with 1 M hy- droxylamine is described in Fig. 1. All of the amides exhibited maximal non-enzymatic hydroxamic acid formation at pH 6. The curve for aspara- gine is unique in that it shows a second rise in the region of pH 8.5 to 10. It is also interesting to not,e that succinamic, glutaramic, and adipamic acids reacted more rapidly t.han did the respective amino acids.
Although the corresponding dicarboxylic acids did not react non-enzy-
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 447
matically with hydroxylamine under the conditions described in Fig. 1, hydroxamic acid formation was observed at a higher temperature. Thus, when a solution of 1 ml. containing 1 mmole of hydroxylamine and 200 pmoles of succinic acid at pH 6 was placed in an autoclave at 15 pounds pressure and 120” for 30 minutes, 40 pmoles of succinylmonohydroxamic acid were formed. Under these conditions, glutaric, glutamic, and aspartic
I I I 3 4 5 6 7 8 9 IO
PH
FIG. 1. pa-dependence of the non-enzymatic formation of hydroxamic acids from glutamine (Curve I), homoglutamine (Curve 2), asparagine (Curve 3), succinamic
acid (Curve 4), glutaramic acid (Curve 5), and adipamic acid (Curve 6). The reac- tion mixtures contained 20 pmoles of amide and 1 mmole of hydroxylamine, adjusted to the appropriate value of pH with sodium hydroxide, in a final volume of 1 ml. The values are expressed as micromoles of hydroxamic acid formed per hour at 37”.
The values for adipamic acid and homoglutamine are calculated as glutarylmono- hydroxamic and r-glutamylhydroxamic acids, respectively.
acids yielded 31.4, 4.2, and 3.7 pmoles of hydroxamic acid, respectively. The paper chromatographic behavior of the respective hydroxamic acids formed non-enzymatically from the amides and from the dicarboxylic acids was identical with that of the authentic compounds and with that of the enzymatically formed hydroxamic acids.
Substrate XpeciJicity-Table I describes the susceptibility of a number of amides to hydrolysis by the glutaminase, asparaginase, and rat liver amidase preparations. The glutaminase and asparaginase preparations exhibited a relatively high degree of specificity. Of the amides studied,
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from
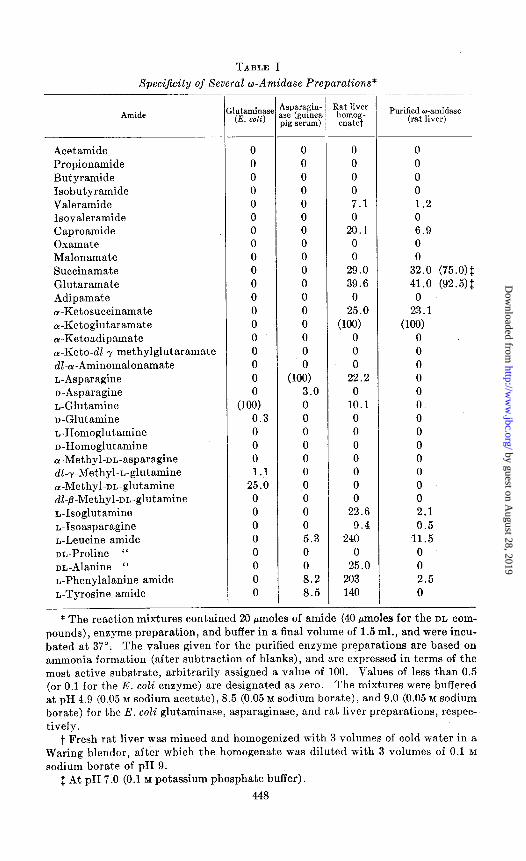
TABLE I
Specificily of Several w-Amidase Preparations*
Amide
Acetamide Propionamide
Butyramide Isobutyramide Valeramide Isovaleramide
Caproamide Oxamate Malonamate Succinamate
Glutaramate Adipamate a-Ketosuccinamate
a-Ketoglutaramate a-Ketoadipamate ru-Keto-dl-~-methylglutaramate
&a-Aminomalonamate L-Asparagine n-Asparagine
n-Glutamine n-Glutamine L-Homoglutamine n-Homoglutamine
a-Methyl-m-asparagine dl-r-Methyl-n-glutamine a-Methyl-on-glutamine
dl-B-Methyl-or,-glutamine n-Isoglutamine
n-Isoasparagine n-Leucine amide nn-Proline “
nn-Alanine “ n-Phenylalanine amide n-Tyrosine amide
lutaminasl (E. coli)
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
(lOi)
0.3 0
0 0 1.1
25.0 0 0
0 0 0
0 0 0
sparagin- se (guinea 8ig serum)
1
0 0
0 0 0
0 0 0 0
0 0 0
0 0 0
0
(10:) 3.0
0 0 0
0 0 0
0 0 0
0 5.3 0
0 8.2 8.5
<at liver homog- enatet
l-
0 0
0 0
0 0
0 0
7.1 1.2 0 0
20.1 6.9 0 0 0 0
29.0 32.0 (75.0)$ 39.6 41.0 (92.5)$
0 0 25.0 23.1
wm WJO) 0 0 0 0 0 0
22.2 0 0 0
10.1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
22.6 2.1 9.4 0.5
240 11.5 0 0
25.0 0 203 2.5 140 0
Purified w-amidase (rat liver)
-
* The reaction mixtures contained 20 pmoles of amide (40 pmoles for the DL com-
pounds), enzyme preparation, and buffer in a final volume of 1.5 ml., and were incu-
bated at 37”. The values given for the purified enzyme preparations are based on ammonia formation (after subtraction of blanks), and are expressed in terms of the
most active substrate, arbitrarily assigned a value of 100. Values of less than 0.5 (or 0.1 for the E. coli enzyme) are designated as zero. The mixtures were buffered at pH 4.9 (0.05 M sodium acetate), 8.5 (0.05 M sodium borate), and 9.0 (0.05 M sodium borate) for the E. coli glutaminase, asparaginase, and rat liver preparations, respec-
tively. t Fresh rat liver was minced and homogenized with 3 volumes of cold water in a
Waring blendor, after which the homogenate was diluted with 3 volumes of 0.1 M
sodium borate of pH 9. $ At pH 7.0 (0.1 M potassium phosphate buffer).
448
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 449
only glutamine and a-methylglutamine were attacked at an appreciable rate by the E. coli glutaminase. The asparaginase preparation hydrolyzed several amino acid a-amides and n-asparagine at low rates. Neither the asparaginase nor the glutaminase preparation hydrolyzed the next lower homologue of asparagine, ac-aminomalonamic acid. The low but definite susceptibility of the D isomers of asparagine and glutamine recorded in Table I cannot be ascribed to L isomer impurities in the substrate prepara- tion, since these products were found to contain less than 0.2 per cent of their enantiomorphs by enzymatic tests (29). Furthermore, when large amounts of enzyme were used, the D-amides were hydrolyzed to more than 50 per cent of completion. It is of interest to note that the presence of a n-specific asparaginase has been reported in certain microorganisms (30).
The rat liver homogenate hydrolyzed CY-ketoglutaramate, a-ketosuc- cinamate, succinamate, glutaramate, valeramide, caproamide, glutamine, asparagine, and several amino acid a-amides. The purified liver w-amidase fraction hydrolyzed the first four of these at approximately the same rela- tive rates, while activity toward the other amides was considerably lower or absent. For example, phenylalanine amide, which was hydrolyzed more than twice as rapidly by the homogenate as was a-ketoglutaramate, was hydrolyzed at less than 12 per cent of the rate observed with a-ketoglu- taramate by the purified w-amidase. The values for the other a-amides were much lower with the purified liver w-amidase, and there was no activ- ity toward glutamine or asparagine. These data suggest the existence of a hepatic enzyme (or enzymes) different from the amino acid ar-amidases and from the amino acid w-amidases. It is of interest that neither the purified w-amidase nor the liver homogenate hydrolyzed malonamate, oxamate, or adipamate. Although introduction of an a-keto group in the substrates of susceptible carbon chain length did not lead to inactivity, the presence of a r-methyl group apparently prevented hydrolysis. A series of homologous diamides from oxamide to adipamide was examined and found to be resistant to deamidation by all of the enzyme preparations.
The initial studies with the liver preparations were carried out at pH 9, since this value of pH was previously found to be optimal for the hydrolysis of or-ketoglutaramate and cr-ketosuccinamate (17). The pH optimum for the hydrolysis of L-leucine amide by rat liver extracts has also been re- ported to be in the neighborhood of 9 (31). However, experiments with glutaramate and succinamate revealed a relatively broad optimal pH range from 6.5 to 7.5; the rates of hydrolysis of these substrates at pH 7.0 are therefore included in Table I. The formation of succinic and glutaric acids as products of enzymatic hydrolysis of the respective amides was demonstrated by paper chromatography. Oxalacetic and cu-ketoglutaric acids were found previously to be products of hydrolysis of Lu-ketosuc- cinamic and a-ketoglutaramic acids (17).
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

450 w-AMIDASE PREPARATIONS
Synthesis of Hydroxamic Acids by PuriJied Liver w-Amidase Preparation- Enzyme systems which catalyze the exchange of the amide group of glu- tamine and asparagine with hydroxylamine and certain other amines have been found in a variety of species (11-15, 32-34), and hydroxamic acid formation has also been observed with a number of peptidase preparations (35-39). In the present investigation it was found that the liver w-amidase preparation catalyzed the formation of the corresponding hydroxamic acids
2.0
1.6
1.2
0.8
0.4
C 5 6 7 a 9
PH
4 5 6 7 a 9
PH
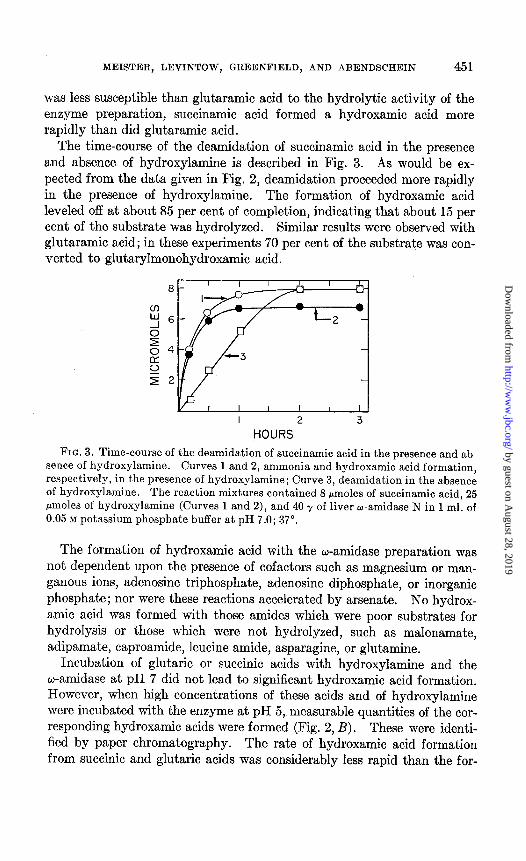
FIG. 2. A, pH-dependence of the enzymatic hydrolysis of glutaramic and succin-
amic acids and of the analogous transfer reactions. Curves 1 and 2, formation of hydroxamic acid and ammonia, respectively, from succinamic acid; Curves 3 and 4, formation of hydroxamic acid and ammonia, respectively, from glutaramic acid.
The reaction mixtures contained 10 rmoles of amide, 40 y of liver w-amidase N, 25 Mmoles of hydroxylamine (Curves 1 and 3), and buffer in a final volume of 1 ml. 0.05 M sodium acetate (pH 4.9 to 6.5), 0.05 M potassium phosphate (pH 6.8 to 7.6), and 0.05
M sodium borate (pH 7.7 to 9.1) buffers were used. The mixtures were incubated at 37” for 10 minutes (Curves 1 and 3) or 60 minutes (Curves 2 and 4), and the values are expressed as micromoles of ammonia or hydroxamic acid formed per hour. B,
pH-dependence of the formation of hydroxamic acid from glutaric and succinic acids. Curve 1, glutaric acid; Curve 2, succinic acid. The reaction mixtures contained 200 pmoles of dicarboxylic acid, 1 mmole of hydroxylamine (adjusted to the appropriate
value of pH), and 200 7 of liver w-amidase N in a final volume of 1 ml. The incuba- tion time was 30 to 60 minutes at 37”; the values are expressed as in A.
from succinamic and glutaramic acids and hydroxylamine. These were identified by paper chromatography as described above. The effect of pH on the ability of the liver o-amidase to catalyze the hydrolysis of glutaramic and succinamic acids, and to catalyze the formation of hydrox- amic acids from these amides in the presence of hydroxylamine, is described in Fig. 2, A. All of these reactions proceeded most rapidly at about pH 6.5 to 7.5. Under these conditions, the formation of succinylmonohy- droxamic acid occurred about 6 times more rapidly than did the hydrolysis of succinamic acid. With glutaramic acid, the rate of hydroxamic acid formation was about twice that of hydrolysis. Although succinamic acid
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from
MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 451
was less susceptible than glutaramic acid to the hydrolytic activity of the enzyme preparation, succinamic acid formed a hydroxamic acid more rapidly than did glutaramic acid.
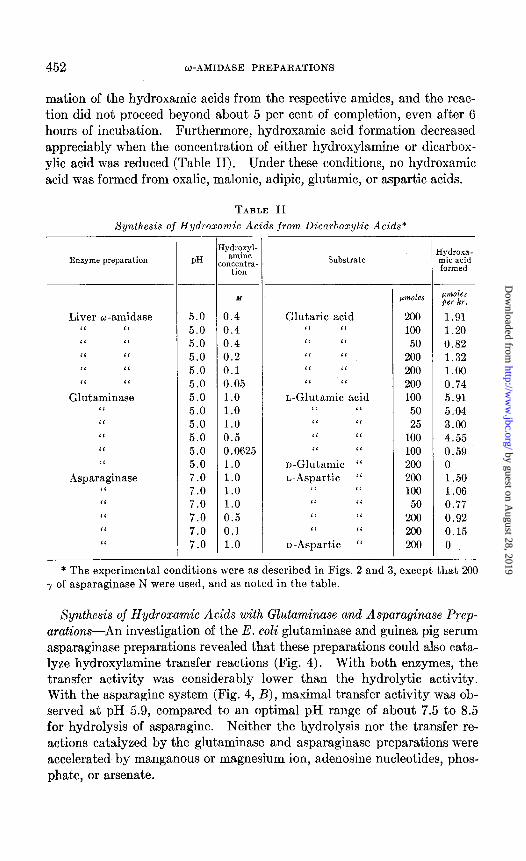
The time-course of the deamidation of succinamic acid in the presence and absence of hydroxylamine is described in Fig. 3. As would be ex- pected from the data given in Fig. 2, deamidation proceeded more rapidly in the presence of hydroxylamine. The formation of hydroxamic acid leveled off at about 85 per cent of completion, indicating that about 15 per cent of the substrate was hydrolyzed. Similar results were observed with glutaramic acid; in these experiments 70 per cent of the substrate was con- verted to glutarylmonohydroxamiic acid.
I 2 3
HOURS
FIG. 3. Time-course of the deamidation of succinamic acid in the presence and ab
sence of hydroxylamine. Curves 1 and 2, ammonia and hydroxamic acid formation, respectively, in the presence of hydroxylamine; Curve 3, deamidation in the absence
of hydroxylamine. The reaction mixtures contained 8 pmoles of succinamic acid, 25
pmoles of hydroxylamine (Curves 1 and 2), and 40 y of liver w-amidase N in 1 ml. of
0.05 M potassium phosphate buffer at pH 7.0; 37”.
The formation of hydroxamic acid with the w-amidase preparation was not dependent upon the presence of cofactors such as magnesium or man- ganous ions, adenosine triphosphate, adenosine diphosphate, or inorganic phosphate; nor were these reactions accelerated by arsenate. No hydrox- amic acid was formed with those amides which were poor substrates for hydrolysis or those which were not hydrolyzed, such as malonamate, adipamate, caproamide, leucine amide, asparagine, or glutamine.
Incubation of glutaric or succinic acids with hydroxylamine and the o-amidase at pH 7 did not lead to significant hydroxamic acid formation. However, when high concentrations of these acids and of hydroxylamine were incubated with the enzyme at pH 5, measurable quantities of the cor- responding hydroxamic acids were formed (Fig. 2, B). These were identi- fied by paper chromatography. The rate of hydroxamic acid formation from succinic and glutaric acids was considerably less rapid than the for-
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from
452 W-AMIDASE PREPARATIONS
mation of the hydroxamic acids from the respective amides, and the reac- tion did not proceed beyond about 5 per cent of completion, even after 6 hours of incubation. Furthermore, hydroxamic acid formation decreased appreciably when the concentration of either hydroxylamine or dicarbox- ylic acid was reduced (Table II). Under these conditions, no hydroxamic acid was formed from oxalic, malonic, adipic, glutamic, or aspartic acids.
TABLE II
Synthesis of Hydroxamic Acids from Dicarboxylic Acids*
Enzyme preparation PH
Liver o-amidase “ “ ‘6 ‘I “ “
“ “
I‘ “
Glutaminase “
“ “ “
“
Asparaginase “
“ I‘ “
‘I
[ydroxyl- amine
mcentra- tion
x
5.0 0.4
5.0 0.4
5.0 0.4
5.0 0.2
5.0 0.1
5.0 0.05
5.0 1.0 5.0 1.0 5.0 1.0
5.0 0.5
5.0 0.0625 5.0 1.0
7.0 1.0
7.0 1.0
7.0 1.0
7.0 0.5 7.0 0.1
7.0 1.0
Substrate
Glutaric acid “ “ L‘ ‘I “ “ “ <‘ “ “
n-Glutamic acid “ “ ‘C “ “ “
‘< “
n-Glutamic “ n-Aspartic “
<‘ “ “ “ “ “
“ ‘I
n-Aspartic “
Lmwles
200 100
50
200 200 200
100 50 25
100
100 200 200
100 50
200 200 200
I 1
pmoles per hr.
1.91 1.20
0.82 1.32 1.00 0.74
5.91 5.04 3.00
4.55 0.59 0
1.50 1.06 0.77
0.92 0.15 0
* The experimental conditions were as described in Figs. 2 and 3, except that 200 -r of asparaginase N were used, and as noted in the table.
Synthesis of Hydroxamic Acids with Glutaminase and Asparaginase Prep- arations-An investigation of the E. coli glutaminase and guinea pig serum asparaginase preparations revealed that these preparations could also cata- lyze hydroxylamine transfer reactions (Fig. 4). With both enzymes, the transfer activity was considerably lower than the hydrolytic activity. With the asparagine system (Fig. 4, B), maximal transfer activity was ob- served at pH 5.9, compared to an optimal pH range of about 7.5 to 8.5 for hydrolysis of asparagine. Neither the hydrolysis nor the transfer re- actions catalyzed by the glutaminase and asparaginase preparations were accelerated by manganous or magnesium ion, adenosine nucleotides, phos- phate, or arsenate.
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from
MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 453
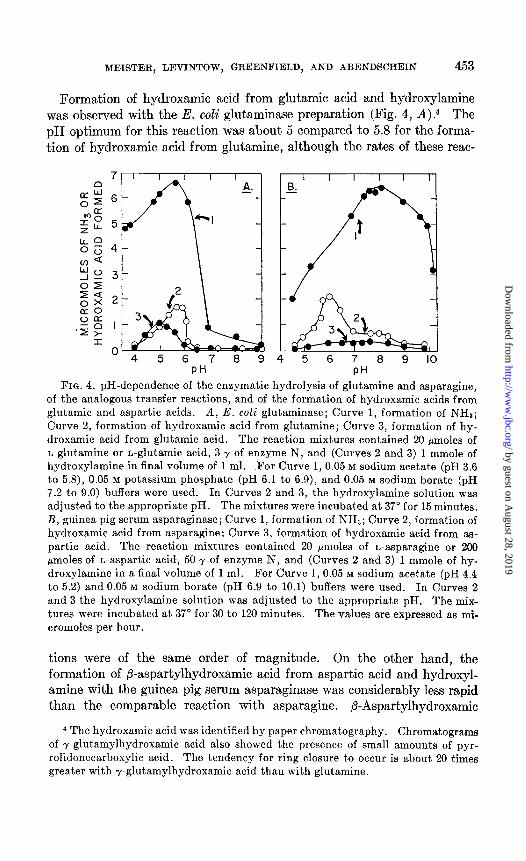
Formation of hydroxamic acid from glutamic acid and hydroxylamine was observed with the E. coli glutaminase preparation (Fig. 4, A).4 The pH optimum for this reaction was about 5 compared to 5.8 for the forma- tion of hydroxamic acid from glutamine, although the rates of these reac-
7 8 9 4 5 6 7 8 9 IO PH PH
FIQ. 4. pH-dependence of the enzymatic hydrolysis of glutamine and asparagine, of the analogous transfer reactions, and of the formation of hydroxamic acids from glutamic and aspartic acids. A, E. coli glutaminase; Curve 1, formation of NHa; Curve 2, formation of hydroxamic acid from glutamine; Curve 3, formation of hy-
droxamic acid from glutamic acid. The reaction mixtures contained 20 pmoles of
n-glutamine or n-glutamic acid, 3 y of enzyme N, and (Curves 2 and 3) 1 mmole of hydroxylamine in final volume of 1 ml. For Curve 1, 0.05 M sodium acetate (pH 3.6
to 5.8), 0.05 M potassium phosphate (pH 6.1 to 6.9), and 0.05 M sodium borate (pH 7.2 to 9.0) buffers were used. In Curves 2 and 3, the hydroxylamine solution was
adjusted to the appropriate pH. The mixtures were incubated at 37” for 15 minutes.
B, guinea pig serum asparaginase; Curve 1, formation of NHZ; Curve 2, formation of hydroxamic acid from asparagine; Curve 3, formation of hydroxamic acid from as- partic acid. The reaction mixtures contained 20 pmoles of L-asparagine or 200
pmoles of n-aspartic acid, 50 y of enzyme N, and (Curves 2 and 3) 1 mmole of hy- droxylamine in a final volume of 1 ml. For Curve 1, 0.05 M sodium acetate (pH 4.4 to 5.2) and 0.05 M sodium borate (pH 6.9 to 10.1) buffers were used. In Curves 2
and 3 the hydroxylamine solution was adjusted to the appropriate pH. The mix- tures were incubated at 37’ for 30 to 120 minutes. The values are expressed as ml-
cromoles per hour.
tions were of the same order of magnitude. On the other hand, the formation of P-aspartylhydroxamic acid from aspartic acid and hydroxyl- amine with the guinea pig serum asparaginase was considerably less rapid than the comparable reaction with asparagine. P-Aspartylhydroxamic
4 The hydroxamic acid was identified by paper chromatography. Chromatograms
of 7.glutamylhydroxamic acid also showed the presence of small amounts of pyr-
rolidonecarboxylic acid. The tendency for ring closure to occur is about 20 times greater with r-glutamylhydroxamic acid than with glutamine.
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

454 o-AMIDASE PREPARATIONS
acid formation from aspartic acid exhibited a broad optimal pH range and was dependent upon a relatively high aspartic acid concentration. Miller and Waelsch have observed a similar reaction with extracts of Proteus vulgcris (40). The dependence of these reactions on high substrate and hydroxylsmine concentrations is similar to that observed in experiments with succinic and glutaric acids and the liver w-amidase preparation. After prolonged incubation, these reactions proceeded to about 5 per cent of completion.
Enzymatic Hydrolysis qf Hydroxamic Acids-Experiments with the hy- droxamic acids corresponding to glutamine, asparagine, and succinamic acid revealed that the hydroxamic acids were hydrolyzed much more slowly by the respective enzyme preparations than were the amides (cf. (11)). In these studies the disappearance of hydroxamic acid was determined quantitatively by the ferric chloride procedure, and the formation of the corresponding dicarboxylic acids was observed by paper chromatography. nr-Glutamylhydroxamic acid was hydrolyzed by the glutaminase prep- aration at about 10 per cent of the rate observed with L-glutamine. The pH optimum was 5.0, and the reaction appeared to proceed to completion. However, there was about a 10 per cent non-enzymatic disappearance of hydroxamic acid under these conditions. Hydrolysis of L-p - aspartyl- hydroxamic acid by the asparaginase preparation occurred most rapidly at pH 8.0. At this pH value the reaction proceeded virtually to completion at about 15 per cent of the rate observed with L-asparagine. Succinyl- monohydroxamic acid was hydrolyzed by the liver w-amidase preparation at about 1.5 per cent of the rate found with succinamic acid; the pH- activity curves for hydrolysis of these compounds were similar and, in the presence of sufficient enzyme, the reaction went to completion.
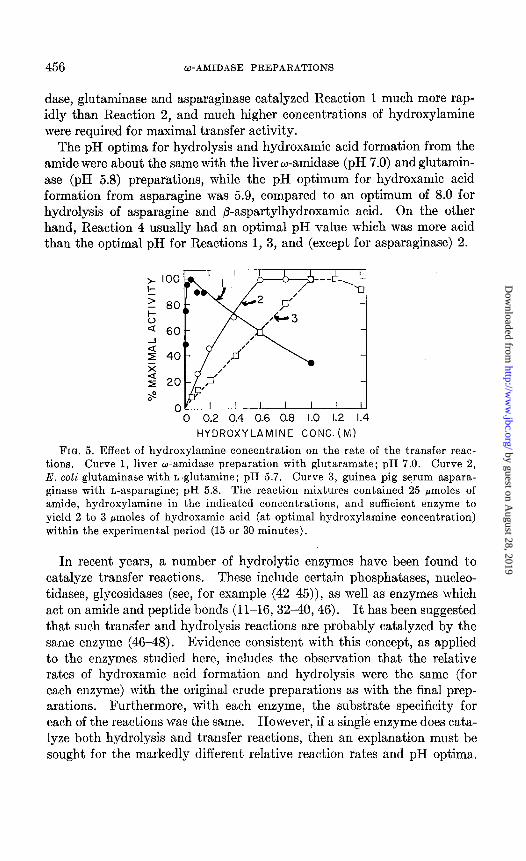
E$ect of Hyclroxy1amin.e Concentration on Hydroxamic Acid Formation from Amides-In addition to the marked difference in the relative rates of hydrolysis and transfer between the glutaminase and asparaginase prepara- tions, on the one hand, and the liver w-amidase on the other, it was ob- served that the former systems required considerably higher concentrations of hydroxylamine for optimal activity than the latter. As described in Fig. 5, maximal activity of the w-amidase preparation was achieved with 0.05 M hydroxylamine, while 12 and 20 times as much amine was required for the glutaminase and asparaginase systems, respectively. The relatively high affinity of the liver w-amidase for hydroxylamine is also indicated by the fact that the reaction proceeded rapidly with stoichiometric quantities of hydroxylamine and amide.
Studies with Other Substrates-Attempts to evaluate the formation of hydroxamic acid from a-ketosuccinamic acid with the liver w-amidase were unsuccessful, due to the fact that the keto acid itself gave a color with ferric
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 455
chloride: On the other hand, a-ketoglutaramate appeared to be active with hydroxylamine, as judged by the increase in color on treatment with ferric chloride after incubation of the keto acid with hydroxylamine and liver w-amidase. In a typical experiment with 20 pmoles of a-ketoglutaramate, 20 pmoles of hydroxylamine, and 350 y of enzyme N at pH 9.0 in a final volume of 1.0 ml., 0.85 pmole of hydroxamic acid (calculated as glutaryl- monohydroxamic acid) was formed per hour. The pH optimum for this reaction was 8.5 to 9.0, or close to that for hydrolysis of ar-ketoglutaramate (17). With higher concentrations of hydroxylamine or cr-ketoglutaramate, an increase in hydroxamic acid formation was observed. Thus, under the above conditions, the hydroxamate formation was increased by S-fold when 400 pmoles of hydroxylamine were used, and by about 4-fold when the concentrations of both amide and amine were doubled.
No detectable hydrolysis of L-aspartic acid P-ethyl ester by the aspara- ginase preparation was observed, and enzymatic hydroxamic acid forma- tion from this compound could not be detected. On the other hand, the glutaminase preparation hydrolyzed L-glutamic acid y-ethyl ester at about 0.1 per cent of the rate with L-glutamine, and catalyzed y-glutamyl- hydroxamic acid formation at approximately 4 times the non-enzymatic rate at pH 5. The susceptibility of esters to attack by peptidases has been extensively considered by Neurath and Schwert (41).
DISCUSSION
Each of the enzyme preparations studied here has been found to catalyze four reactions :
RCONHz + Hz0 + RCOOH + NH, (1)
RCONH? + NHzOH -+ RCONHOH + NH3 (2) RCONHOH + Hz0 -+ RCOOH + NH,OH (3)
RCOOH + NHzOH + RCONHOH + HqO (4)
With all of the enzyme preparations, Reaction 3 proceeded more slowly than did Reaction 1, and Reaction 4 always took place at a very low rate compared to that of Reaction 1.
A finding of considerable interest was that, with succinamic and glu- taramic acids, the liver w-amidase catalyzed Reaction 2 at a more rapid rate than Reaction 1. Furthermore, a relatively low hydroxylamine con- centration was required (Fig. 5). Although hydrolysis of glutaramic acid was faster than succinamic acid, the latter substrate reacted more rapidly with hydroxylamine than did the former (Fig. 2). The non-enzymatic formation of succinylmonohydroxamic acid was also more rapid than that of glutarylmonohydroxamic acid (Fig. 1). In contrast to the liver w-ami-
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from
456 w-AMIDASE PREPARATIONS
dase, glutaminase and asparaginase catalyzed Reaction 1 much more rap- idly than Reaction 2, and much higher concentrations of hydroxylamine were required for maximal transfer activity.
The pH optima for hydrolysis and hydroxamic acid formation from the amide were about the same with the liver w-amidase (pH 7.0) and glutamin- ase (pH 5.8) preparations, while the pH optimum for hydroxamic acid formation from asparagine was 5.9, compared to an optimum of 8.0 for hydrolysis of asparagine and @-aspartylhydroxamic acid. On the other hand, Reaction 4 usually had an optimal pH value which was more acid than the optimal pH for Reactions 1, 3, and (except for asparaginase) 2.
'6 0.2 0.4 0.6 0.8 1.0 1.2 1.4 HYDROXYLAMINE CONC. (MI
FIQ. 5. Effect of hydroxylamine concentration on the rate of the transfer reac- tions. Curve 1, liver w-amidase preparation with glutaramate; pH 7.0. Curve 2,
E. coli glutaminase with L-glutamine; pH 5.7. Curve 3, guinea pig serum aspara- ginase with L-asparagine; pH 5.8. The reaction mixtures contained 25 pmoles of amide, hydroxylamine in the indicated concentrations, and sufficient enzyme to
yield 2 to 3 pmoles of hydroxamic acid (at optimal hydroxylamine concentration) within the experimental period (15 or 30 minutes).
In recent years, a number of hydrolytic enzymes have been found to catalyze transfer reactions. These include certain phosphatases, nucleo- tidases, glycosidases (see, for example (42-45)), as well as enzymes which act on amide and peptide bonds (ll-16,3240,46). It has been suggested that such transfer and hydrolysis reactions are probably catalyzed by the same enzyme (4648). Evidence consistent with this concept, as applied to the enzymes studied here, includes the observation that the relative rates of hydroxamic acid formation and hydrolysis were the same (for each enzyme) with the original crude preparations as with the final prep- arations. Furthermore, with each enzyme, the substrate specificity for each of the reactions was the same. However, if a single enzyme does cata- lyze both hydrolysis and transfer reactions, then an explanation must be sought for the markedly different relative reaction rates and pH optima.
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, AND ABENDSCHEIN 457
Fruton and collaborators (35-39,47), in an extensive study of transpeptida- tion replacement reactions catalyzed by hydrolytic enzymes (e.g., papain, ficin, cathepsin C), observed that, in general, optimal hydrolysis occurred at about pH 5, while transpeptidation was favored by more alkaline values of pH. In our experiments, the optimal pH for hydroxamic acid formation from the amide was the same or lower than the pH optimum for hydrolysis. It is probable that the pH optimum for a transfer reaction may be in- fluenced by at least several factors, including the pK values of the base and the cation, other properties of the substrate, and the characteristics of the enzyme itself. A comparison of the pH-activity curves for the non- enzymatic formation of hydroxamic acids from glutamine and asparagine (Fig. 1) is of interest in this connection.
Assuming that the hydrolysis and transfer reactions are catalyzed by the same enzyme, the results may be explained, in analogy with Fruton’s formulation, in terms of an enzyme-amide complex which may react either with water or with hydroxylamine. That this cannot be a simple com- petitive phenomenon is evident from the fact that appreciable hydrox- amic acid is formed with a concentration of hydroxylamine which is very low compared to that of water. Furthermore, the preferential re- action of hydroxylamine can probably not be attributed entirely to par- ticipation of an enzyme, since similar reactions occur non-enzymatically. The enzymatic synthesis of a hydroxamic acid from hydroxylamine and a dicarboxylic acid (e.g., succinic, glutamic), as in Reaction 4, may be con- sidered formally as the reversal of the hydrolysis reaction (Reaction 3). The very low rate and extent of this reaction are compatible with such an interpretation. One may also consider this phenomenon in terms of an enzyme-dicarboxylic acid complex which may react either with water or with hydroxylamine. The small extent of such reactions may be ascribed to a low affinity of the enzyme for the free acid as compared to the amide; this concept is consistent with the requirement for a relatively high sub- strate concentration. It is of interest that these reactions proceeded at higher temperature without enzyme.
The transfer reactions described here differ from that reported to occur with glutamine in a number of plant, animal, and bacterial systems, in which adenosine nucleotides, phosphate, and manganese or magnesium ions are required. The latter reaction appears to be associated with the gluta- mine synthesis system, and attempts in several laboratories to separate the glutamine synthesis activity from this t’ransfer activity have not been suc- cessful (49, 50, 10). The glutamine transfer activity studied here also dif- fers from that of certain transfer systems studied by Waelsch (12) in which the glutaminase activity was found to be relatively low. We have also found that a fraction obtained from a glutamine-requiring mutant of
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

458 w-AMIDASE PREPARATIONS
Aerobacter aerogenes (51) catalyzes y-glutamylhydroxamic acid formation from glutamine much more rapidly than it hydrolyzes glutamine. No cofactors are needed, and optimal activity is achieved with a low (0.01 M)
hydroxylamine concentration. On the other hand, in studies with a puri- fied preparation of the phosphate-activated glutaminase of rat liver (5),6 we have been unable to detect hydroxamic acid formation. There are evidently a number of enzymes which catalyze hydroxamic acid formation from glutamine. At this time it would appear that enzyme preparations which exhibit such activity also catalyze (a) hydrolysis of glutamine or y-glutamyl peptides (12, 16) or (b) synthesis of glutamine. It may be postulated that an activated enzyme-substrate complex is formed in both types of reactions, and that hydroxylamine can readily react with such an. intermediate. The relative rates of hydrolysis and transfer may reflect differences in the nature of the intermediate complex.
The r81e of the liver w-amidase system in the transamination-deamida- tion reactions of glutamine and asparagine has been considered previously (52). The present observation that this enzyme can catalyze exchange re- actions leading to hydroxamic acid formation suggests a possible mechanism for a-ketosuccinamate formation; i.e., by reaction of oxalacetate or a derivative of this keto acid with ammonia or with an amino donor. The question as to whether hydroxylamine serves as a model for a natural sub- strate must await further study.
SUMMARY
1. A purified rat liver w-amidase fraction hydrolyzed a-ketoglutaramic, ac-ketosuccinamic, glutaramic, and succinamic acids, but did not deamidate glutamine, asparagine, and a number of other amides. The enzyme also catalyzed the formation of succinylmonohydroxamic and glutarylmono- hydroxamic acids from the corresponding amides and relatively low con- centrations of hydroxylamine; these reactions proceeded much more rap- idly than did the analogous hydrolysis reactions. Both hydrolysis and transfer reactions proceeded most rapidly at about pH 7.
2. Preparations of guinea pig serum asparaginase and Escherichia coli glutaminase catalyzed hydroxamic acid formation from asparagine and glutamine, respectively, at rates which were low in comparison to those of hydrolysis. In contrast to the liver w-amidase system, high concentra- tions of hydroxylamine were required for hydroxamic acid formation. The pH-dependence of these reactions was determined.
3. Succinylmonohydroxamic, p-aspartylhydroxamic, and y-glutamyl- hydroxamic acids were hydrolyzed by the respective enzyme preparations at much slower rates than observed with the corresponding amides.
6 Levintow, L., and Meister, A., unpublished. 6 Generously provided by Mr. M. C. Otey and Dr. J. P. Greenstein.
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

MEISTER, LEVINTOW, GREENFIELD, .4ND ABENDSCHEIN 459
4. In the presence of hydroxylamine, all of the enzyme preparations catalyzed hydroxamic acid synthesis from the dicarboxylic acids corre- sponding to the susceptible amides. This and the other findings are con- sidered in terms of the concept that the transfer, hydrolysis, and synthesis reactions are catalyzed by the same enzyme.
5. The pa-dependence of the non-enzymatic formation of hydroxamic acids from glutamine, homoglutamine, asparagine, succinamic acid, and glutaramic acid, and hydroxylamine at 37”, was studied. Non-enzymatic formation of monohydroxamic acids from aspartic, glutamic, succinic, and glutaric acids was observed at 120’.
6. The preparation of dl-ar-aminomalonamic acid, d&/3-methyl-m-glu- tamic acid, dl-P-methyl-nn-glutamic acid y-ethyl ester, and dl-&methyl- rm-glutamine is described.
BIBLIOGRAPHY
1. Krebs, H. A., Rio&em. J., 29, 1951 (1935). 2. Grassmann, W., and Mayr, O., Z. physiol. Chem., 214, 185 (1933). 3. Carter, C. E., and Greenstein, J. P., J. Nut. Cuncer Inst., 7,433 (1947). 4. Errera, M., and Greenstein, J. P., J. Biol. Chem., 178,495 (1949).
5. Otey, M. C., Birnbaum, S. hiI., and Greenstein, J. P., Arch. Bioehem. and Bio- phys., 49, 245 (1954).
6. Greenstein, J. P., and Carter, C. E., J. Biol. Chem., 186,741 (1946).
7. Greenst,ein, J. P., and Price, V. E., J. Biol. Chem., 178,695 (1949). 8. Meister, A., and Tice, S. V., J. Biol. Chem., 187, 173 (1950). 9. Meister, A., Sober, H. A., Tice, S. V., and Fraser, P. E., J. Biol. Chem., 197, 319
(1952). 10. Levintow, L., and Meister, A., J. Biol. Chem., 209, 265 (1954). 11. Grossowicz, N., Wainfan, E., Borek, E., and Waelsch, H., J. Biol. Chem., 187,
111 (1950). 12. Waelsch, H., Advances in Enzymol., 13, 237 (1952). 13. Stumpf, P. K., and Loomis, W. D., Arch. Biochem., 26,451 (1950).
14. Stumpf, P. K., Loomis, W. D., and Michelson, C., Arch. Biochem. and Biophys., 30, 126 (1951).
15. Delwiche, C. C., Loomis, W. D., and Stumpf, P. K., Arch. Rio&em. and Biophys., 33, 333 (1951).
16. Williams, W. J., and Thorne, C. B., J. BioZ. Chem., 210, 203 (1954).
17. Meister, A., J. Biol. Chem., 200,571 (1953). 18. Meister, A., J. BioZ. Chem., 210, 17 (1954). 19. Fischer, E., and Dilthey, A., Ber. them. Ges., 36, 844 (1902).
20. Jeffery, G. H., and Vogel, A. I., J. Chem. Sot., 1101 (1934). 21. Oelkers, L., Ber. them. Ges., 22, 1566 (1889). 22. Renfrow, W. B., Jr., and Hauser, C. R., J. Am. Chem. Sot., 69, 2308 (1937). 23. Snyder, H. R., Shekleton, J. F., and Lewis, C. D., J. Am. Chem. Sot., 67, 310
(1945). 24. Done, J., and Fowden, L., Biochem. J., 51, 451 (1952). 25. Greenstein, J. P., and Leuthardt, F., J. Nut. Cancer Inst., 5,209 (1944). 26. Lipmann, F., and Tuttle, I,. C., J. Biol. Chem., 169, 21 (1945).
27. Rydon, H. N., and Smith, P. W. G., Nature, 169,922 (1952).
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

460 w-AMIDASE PREPARATIONS
28. Rudman, D., and Meister, A., J. Biol. Chem., 200, 591 (1953). 29. Meister, A., Levintow, L., Kingsley, R. B., and Greenstein, J. P., J. Biol. Chem.,
192, 535 (1951). 30. Altenbern, R. A., and Housewright, R. D., Arch. Biochem. and Biophys., 49, 130
(1954).
31. Errera, M., and Greenstein, J. P., J. Nut. Cancer Inst., 8, 71 (1947). 32. Waelsch, H., Owades, P., Borek, E., Grossowicz, N., and Schou, M., Arch. Bio-
them., 27, 237 (1950).
33. Schou, M., Grossowica, N., Lajtha, A., and Waelsch, H., Nature, 167,818 (1951). 34. Lajtha, A., Mela, P., and Waelsch, H., J. Biol. Chem., 205,553 (1953). 35. Johnston, R. B., Mycek, M. J., and Fruton, J. S., J. Biol. &em., 185,629 (1950). 36. Johnston, R. B., Mycek, M. J., and Fruton, J. S., J. Biol. Chem., 187, 205 (1950).
37. Fruton, J. S., Johnston, R. B., and Fried, M., J. Biol. Chem., 190,39 (1951). 38. Durell, J., and Fruton, J. S., J. Biol. Chem., 207,487 (1954). 39. Jones, M. E., Hearn, W. R., Fried, M., and Fruton, J. S., J. Biol. Chem., 195,645
(1952).
40. Miller, A., and Waelsch, H., Federation Proc., 12,246 (1953). 41. Neurath, H., and Schwert, G. W., Chem. Rev., 46,69 (1950). 42. Axelrod, B., J. BioZ. Chem., 172, 1 (1948).
43. Hehre, E. J., J. Biol. Chem., 177,267 (1949). 44. Meyerhof, O., and Green, H., J. BioZ. Chem., 183, 377 (1950). 45. Heppel, L. A., Whitfeld, P. R., and Markham, R., Biochem. J., 66, p. iii (1954). 46. Hanes, C. S., Hird, F. J. R., and Isherwood, F. A., Nature, 167,818 (1951); Bio-
them. J., 61, 25 (1952). 47. Fruton, J. S., Yale J. BioZ. and Med., 22,263 (1950). 48. Hird, F. J. R., and Springell, P. H., Biochem. J., 66, 417 (1954).
49. Waelsch, H., in McElroy, W. D., and Glass, B., Phosphorus metabolism, Balti- more, 2, 109 (1952).
50. Elliott, W. H., J. BioZ. Chem., 201,661 (1953).
51. Davis, B. D., VIth International Congress of Microbiology, Symposia, Rome 23 (1953).
25. Meister, A., and Fraser, P. E., J. BioZ. Chem., 210,37 (1954).
by guest on August 28, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Greenfield and Patricia A. AbendscheinAlton Meister, Leon Levintow, Robert E.
-AMIDASE PREPARATIONSωREACTIONS CATALYZED BY
HYDROLYSIS AND TRANSFER
1955, 215:441-460.J. Biol. Chem.
http://www.jbc.org/content/215/1/441.citation
Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
alerts to choose from all of JBC's e-mailClick here
tml#ref-list-1
http://www.jbc.org/content/215/1/441.citation.full.haccessed free atThis article cites 0 references, 0 of which can be by guest on A
ugust 28, 2019http://w
ww
.jbc.org/D
ownloaded from