hemophagocytic syndrome in acute myeloid leukemia patients
TRANSCRIPT

Hemophagocytic syndrome in acute myeloid leukemia patients undergoing intensive chemotherapy
by Karen Delavigne, Emilie Bérard, Sarah Bertoli, Jill Corre, Eliane Duchayne, Cécile Demur,Véronique Mansat-De Mas, Cécile Borel, Muriel Picard, Muriel Alvarez, Audrey Sarry, Françoise Huguet, and Christian Récher
Haematologica 2013 [Epub ahead of print]
Citation: Delavigne K, Bérard E, Bertoli S, Corre J, Duchayne E, Demur C, Mansat-De Mas V, Borel C, Picard M, Alvarez M, Sarry A, Huguet F, and Récher C. Hemophagocytic syndrome in acute myeloid leukemia patients undergoing intensive chemotherapy. Haematologica. 2013; 98:xxxdoi:10.3324/haematol.2013.097394
Publisher's Disclaimer.E-publishing ahead of print is increasingly important for the rapid dissemination of science.Haematologica is, therefore, E-publishing PDF files of an early version of manuscripts thathave completed a regular peer review and have been accepted for publication. E-publishingof this PDF file has been approved by the authors. After having E-published Ahead of Print,manuscripts will then undergo technical and English editing, typesetting, proof correction andbe presented for the authors' final approval; the final version of the manuscript will thenappear in print on a regular issue of the journal. All legal disclaimers that apply to thejournal also pertain to this production process.
Copyright 2013 Ferrata Storti Foundation.Published Ahead of Print on October 18, 2013, as doi:10.3324/haematol.2013.097394.

1
Hemophagocytic syndrome in acute myeloid leukemia patients undergoing
intensive chemotherapy
Running title: Hyperinflammation in AML
Karen Delavigne,1,6 Emilie Bérard,2,3 Sarah Bertoli,1 Jill Corre,4 Eliane Duchayne,4
Cécile Demur,4 Véronique Mansat-De Mas,4,6 Cécile Borel,1,6 Muriel Picard,1
Muriel Alvarez,5 Audrey Sarry,1 Françoise Huguet,1 and Christian Récher1,6
1Service d’Hématologie, Centre Hospitalier Universitaire de Toulouse, Hôpital Purpan, 31059
Toulouse, France; 2Service d'Epidémiologie, Centre Hospitalier Universitaire de Toulouse,
Toulouse, F-31073, France; 3UMR 1027, INSERM-Université de Toulouse III, Toulouse, F-
31062, France; 4Laboratoire d’Hématologie, Centre Hospitalier Universitaire de Toulouse,
Hôpital Purpan, 31059 Toulouse, France; 5Service des Maladies Infectieuses et Tropicales,
Centre Hospitalier Universitaire de Toulouse, Hôpital Purpan, 31059 Toulouse, France;
and 6Université Toulouse III Paul Sabatier, Toulouse, France.

2
Correspondence
Christian Récher, Service d’Hématologie, CHU de Toulouse, Hôpital Purpan, place du Dr
Baylac, 31059 Toulouse cedex 9, France. Email : [email protected]
Key words: hemophagocytic lymphohistiocytosis, infection-associated hemophagocytic
syndrome, malignancy-associated hemophagocytic syndrome, macrophage activation
syndrome, ferritin, acute myeloid leukemia.
Acknowledgments
The authors would like to thank the GAEL Association (Gael Adolescent Espoir Leucémie)
for their support to patients. We also thank Sarah Scotland for the corrections of the article.

3
Abstract
Hemophagocytic lymphohistiocytosis is an immune dysregulation characterized by severe
organ damages induced by a hyperinflammatory response and uncontrolled T-cell and
macrophage activation. Secondary hemophagocytic lymphohistiocytosis typically occurs in
association with severe infections or malignancies. Acute myeloid leukemia patients may be
prone to develop hemophagocytic lymphohistiocytosis because of an impaired immune
response and a high susceptibility to severe infections. In a series of 343 patients treated by
intensive chemotherapy over a 5-year period in our centre, we identified 32 patients (9.3%)
with fevers, very high ferritin levels, and marrow hemophagocytosis (hemophagocytic
lymphohistiocytosis patients). Patients with hemophagocytic lymphohistiocytosis exhibited
hepatomegaly, pulmonary or neurological symptoms, liver abnormalities, lower platelet count
and higher levels of C-reactive protein as well as prolonged pancytopenia as compared to
patients without hemophagocytic lymphohistiocytosis. A microbial etiology was documented
in 24 patients: 14 bacterial infections, 9 Herpesviridae infections and 11 fungal infections.
Hemophagocytic lymphohistiocytosis treatment consisted of corticosteroids and/or
intravenous immunoglobulin along with adapted antimicrobial therapy. Patients with
hemophagocytic lymphohistiocytosis had a median overall survival of 14.9 months,
significantly lower than patients without hemophagocytic lymphohistiocytosis (22.1 months)
(p=0.0016). Hemophagocytic lymphohistiocytosis was significantly associated with a higher
rate of induction failures mainly due to deaths in aplasia. Hemophagocytic
lymphohistiocytosis can be diagnosed in up to 10% of acute myeloid leukemia patients
undergoing intensive chemotherapy and is associated with early mortality. Fever, very high
ferritin levels and marrow hemophagocytosis represent the cornerstone of diagnosis. Further
biological studies are needed to better characterize and recognize this syndrome in acute
myeloid leukemia patients.

4
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a dysregulation of the immune system
characterized by both severe inflammation and uncontrolled activation of T-cells and
macrophages inducing severe, sometimes fatal, organ damage observed in the bone marrow,
liver, and central nervous system.(1) Patients with HLH have rapid deterioration of their
general condition, fevers, hepatosplenomegaly, profound pancytopenia, disseminated
intravascular coagulation, very high ferritin levels and hemophagocytosis in the bone marrow,
spleen or lymph nodes. Polyadenopathy, jaundice, rash, and neurological symptoms are less
frequent. This clinical syndrome can be encountered in association with various underlying
diseases, including inherited genetic defects (primary or familial HLH), malignancies, severe
infections and autoimmune disorders (secondary HLH). Primary HLH often occurs in young
children with inherited genetic defects in genes of the perforin cytotoxic pathway such as
PRF1, UNC13D, Munc 18-2, Rab27a, STX11, SH2D1A or BIRC4. Secondary HLH typically
occurs in association with severe infections (i.e, infection-associated hemophagocytic
syndrome), malignancies (i.e, malignancy-associated hemophagocytic syndrome) or
autoimmune disorders (i.e, macrophage activation syndrome). Diagnosis criteria have evolved
over time and now combine both clinical and biological features including impaired NK-cell
activity.(2-4) However, while primary HLH has been well characterized in light of the recent
discovery of mutations in several genes involved in T-cell cytotoxic activity, it is often
challenging to distinguish between true secondary HLH in adults and severe inflammatory
conditions, in which features of hemophagocytosis are encountered (i.e, critically ill
patients).(5-7) Moreover, hypomorphic mutations in PRF1, UNC13D and STBXBP2 have
recently been discovered in sporadic cases of HLH in adults.(8) Thus, there may be an overlap
between HLH and severe inflammation in some clinical contexts. An emerging concept
suggests that HLH could be a unique syndrome associated with a continuum of underlying

5
genetic risk factors and triggered by an immune challenge of varying intensity determined by
the genetic background of patients.(6)
In hematological malignancies, HLH is classically associated with specific entities, such as T-
cell or NK/T-cell lymphoma and intravascular large B-cell lymphoma, or induced by
treatment-related bacterial, viral or fungal infections and thus, frequently described as
malignancy- or infection-associated hemophagocytic syndrome.(9-11) In patients with acute
myeloid leukemia (AML), HLH has been occasionally described in case-reports. AML
patients may be prone to develop HLH due to their disease- and/or treatment-related impaired
immune response and their high susceptibility to severe infections, which act as triggering
factors.(12) Warned by several cases of HLH in our department, we sought to determine the
frequency and patterns of HLH presentation as well as its impact on prognosis in a series of
consecutive AML patients treated by intensive chemotherapy.

6
Methods
Patients
Between Jan 1, 2006, and Dec 31, 2010, all consecutive patients with a new diagnosis of
AML (except acute promyelocytic leukemia) admitted in our centre and eligible for intensive
chemotherapy were registered for this study. Diagnosis workup and treatment modalities have
been described elsewhere.(13-14) All patients had central venous catheter, bacterial digestive
tract decontamination, antibiotherapy for febrile neutropenia (piperacilline-
tazobactam/amikacine and imipenem/vancomycin/ciprofloxacin as first and second line,
respectively) and antifungal prophylaxis with posaconazole. Clinical and biological data were
recorded by K.D, A.S, S.B and C.R. Biological data, including fibrinogen, C-reactive protein,
ferritin and triglyceride levels were assessed at diagnosis and every week thereafter. Bone
marrow aspirations were routinely performed at diagnosis, day 15 of induction chemotherapy
(for patients under 60 years of age), assessment of response (~d35), in cases of unexpected
prolonged cytopenias (> d35) or in cases with suspicion of HLH (i.e, patients with
antimicrobial treatments for febrile neutropenia, and/or sudden increases in ferritinemia
and/or unexpected cytopenias). The presence of hemophagocytosis features was recorded
regardless of clinical presentation. Routine cytological analysis of May–Grünwald–Giemsa
(MGG) stained bone marrow smears was performed. Hemophagocytosis was defined by the
presence of images exhibiting macrophage-dependant phagocytosis of erythrocytes,
leukocytes, platelets and/or their precursors, regardless of the percentage of macrophages
(supplementary figure). Results of bone marrow aspirations were discussed for each patient
between cytologists and physicians during weekly meetings. This study was approved by the
Institutional Ethics Committee.

7
Study population
Three hundred and forty three patients were included. Their characteristics at diagnosis of
AML are shown in table 1. The criteria used to assign patients in the HLH group were:
hemophagocytosis in the bone marrow with unexplained fever under broad antimicrobial
treatments, and/or sudden increases in ferritin and/or unexpected non-blastic pancytopenia.
Twenty-nine patients fulfilled these criteria. Three other patients without bone marrow
hemophagocytosis exhibited strong bio-clinical evidence for the diagnosis of HLH: (i) one
patient with fever (40°C) for 12 days, hepatomegaly, cutaneous rash, rapid rise in ferritinemia
(from 1200 to 21000 µg/L) and pancytopenia with “dry tap” on bone marrow aspiration; (ii)
one patient with fever, pancytopenia, neurological symptoms, hyperferritinemia (106000
µg/L), macrophages in the cerebrospinal fluid without features of hemophagocytosis; (iii) one
patient with fever, multi-organ failure, pancytopenia and hyperferritinemia (30000 µg/L). The
total number of patients in the HLH group was 32 (HLH+ group). A group of 22 patients with
hemophagocytosis features in the bone marrow but without any clinical or biological
symptoms of HLH was also identified (HLH-/HemoPh+ group). The remaining 289 patients
had neither criterion for HLH nor bone marrow hemophagocytosis (HLH-/HemoPh- group).
Bio-clinical data were collected at diagnosis of AML for all patients and at time of HLH or
hemophagocytosis onset for HLH+ and HLH-/HemoPh+ groups. Overall, bone marrow
aspiration in HLH+ and HLH-/HemoPh+ groups were respectively performed at: (i) clinical
suspicion of HLH (n=14, 44% vs. n=0); (ii) prolonged pancytopenia (n=13, 41% vs. n=3,
14%); (iii) systematic bone marrow aspiration at diagnosis (n=1, 3% vs. n=2, 9%) or for
response evaluation (n=4, 13% vs. n=17, 77%) (p<0.0001).
Endpoints and Statistical analysis (supplementary file)

8
Results
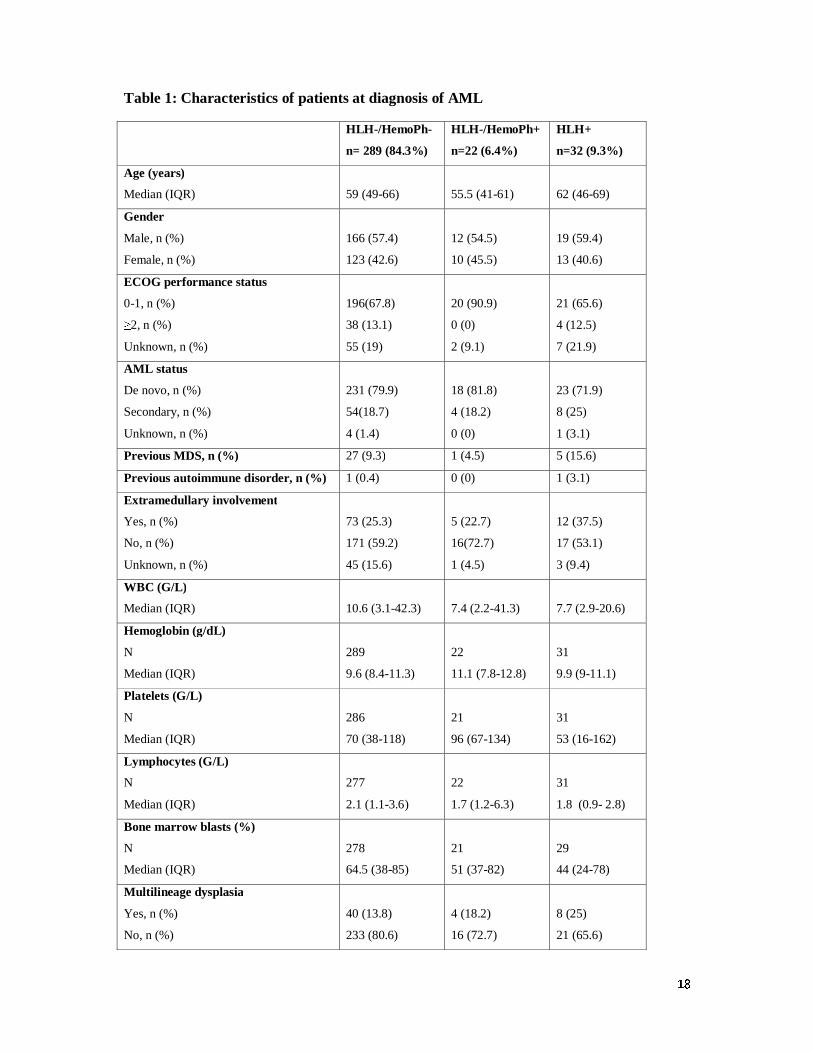
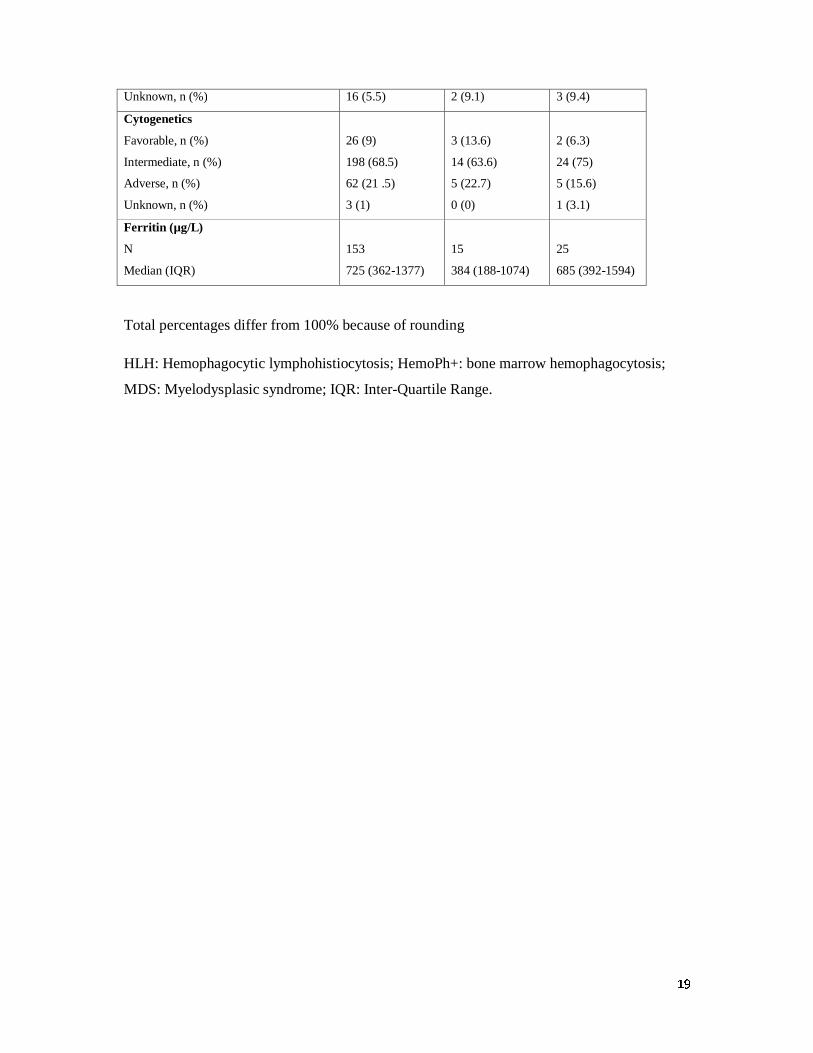
Characteristics of patients at AML diagnosis
As shown in table 1, there were no differences in the main clinical and biological parameters
between the three groups at time of AML diagnosis. Of note, the level of ferritin was above
the upper normal limit in most patients with median ferritin levels of 725 (IQR, 362-1377),
384 (IQR, 188-1074) and 685 (IQR, 392-1594) µg/L in the HLH-/HemoPh-, HLH-/HemoPh+
and HLH+ groups, respectively. All patients received intensive chemotherapy for remission
induction, including daunorubicin + cytarabine (n=146), idarubicin + cytarabine (n=50),
idarubicin + cytarabine + lomustine (n=101), time sequential induction with daunorubicin and
cytarabine (n=13) or idarubicin + high-dose cytarabine (n=11). Other drugs, such as
gemtuzumab ozogamycin (n=19), fludarabin (n=1) or imatinib (n=2) were occasionally
added.
Characteristics of patients at HLH+ or HLH-/HemoPh+ diagnosis
Twenty-two patients developed HLH during the first induction chemotherapy, 7 during
consolidation and 3 during salvage therapy after relapse. The median time from AML
diagnosis to HLH onset was 40 days (IQR, 27-71) in the 22 patients who developed HLH
during the first induction chemotherapy. We describe the characteristics of HLH+ patients
comparatively to HLH-/HemoPh+ at HLH+ or HLH-/HemoPh+ diagnosis (table 2).
Clinically, patients in the HLH+ group had significantly more often occurrences of fever
(81%), hepatomegaly (21.9%) and respiratory symptoms (59.4%) than HLH-/HemoPh+
patients (table 2). Splenomegaly (18.8%), jaundice (25%), rash (18.8%) and neurological
symptoms (12.5%) were also frequently encountered in the HLH+ group albeit not significant
compared to the HLH-/HemoPh+ group. Biologically, HLH+ patients had significantly lower
platelets count, prothrombin time and serum albumin level as well as higher levels of C-

9
reactive protein, whereas there were no differences in serum sodium level, creatinine,
triglycerides and fibrinogen levels between the two groups. Liver abnormalities were
significantly associated with HLH as indicated by higher bilirubin levels and cholestasis
markers, including γGT and alkaline phosphatase.
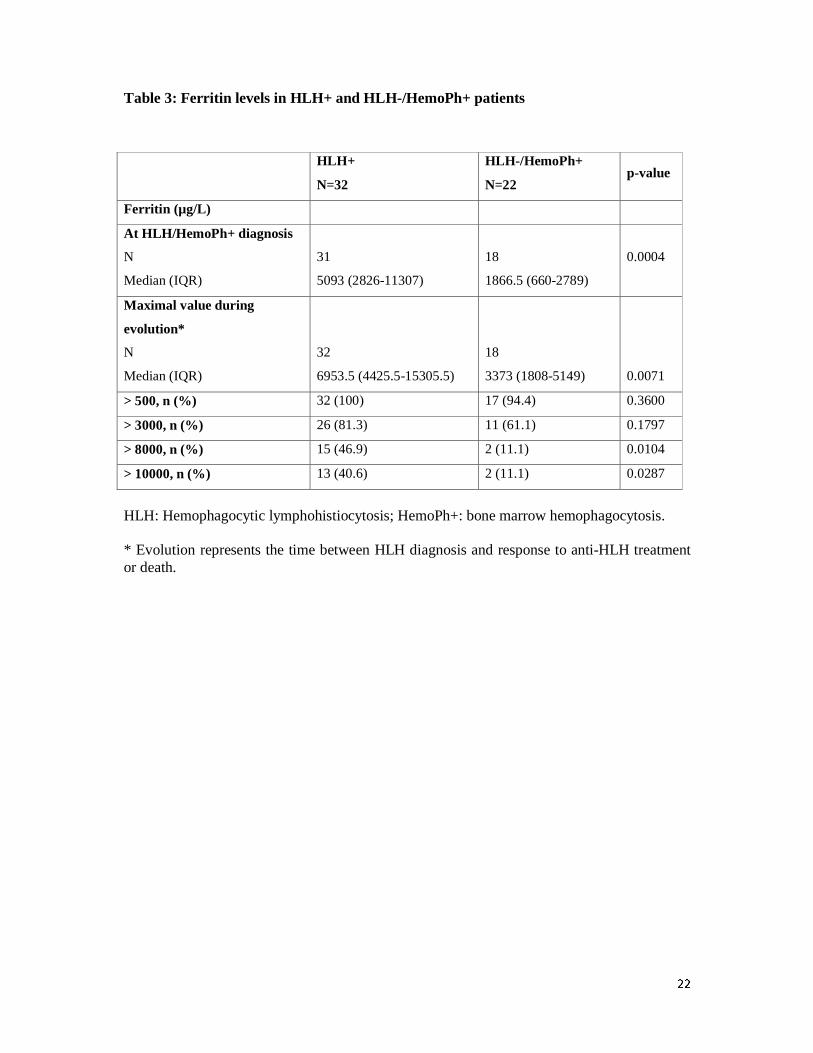
Variations of ferritin levels
At time of bone marrow aspiration performed for HLH diagnosis or response assessment, the
median serum ferritin level was 5093 µg/L (IQR, 2826-11307) in HLH+ patients compared to
1866.5 µg/L (IQR, 660-2789) in HLH-/HemoPh+ patients (p=0.0004) (table 3). During the
period following the bone marrow aspiration (until response to HLH treatment or death), the
median maximal value of ferritin was 6953.5 µg/L (IQR, 4425.5-15305.5) in the HLH+ group
compared to 3373 µg/L (IQR, 1808-5149) in the HLH-/HemoPh+ patients (p=0.0071).
Moreover, 15 patients (47%) in the HLH+ group and only 2 (11%) in the HLH-/HemoPh+
group, had a ferritin level higher than 8000 µg/L (p=0.0104). We also studied the variation in
serum ferritin either in the week prior to diagnosis of HLH in 22 patients or prior to
hemophagocytosis on bone marrow aspiration in 12 HLH-/HemoPh+ patients. The median
increase in ferritin was 1421 µg/L (IQR, -287-8746) and 73.5 µg/L (IQR, -836-802.5) in
HLH+ and HLH-/HemoPh+ patients, respectively (p=0.1395). The median number of red cell
packs received by HLH+ patients in the three-month period before the diagnosis of HLH was
14 (IQR, 8-20) compared to 7 (IQR, 2-10) in HLH-/HemoPh+ patients (p=0.0048). There was
no significant correlation between the maximal level of serum ferritin and the number of red
cell packs in HLH+ patients (Spearman correlation coefficient, Rho=-0.14; p=0.46).
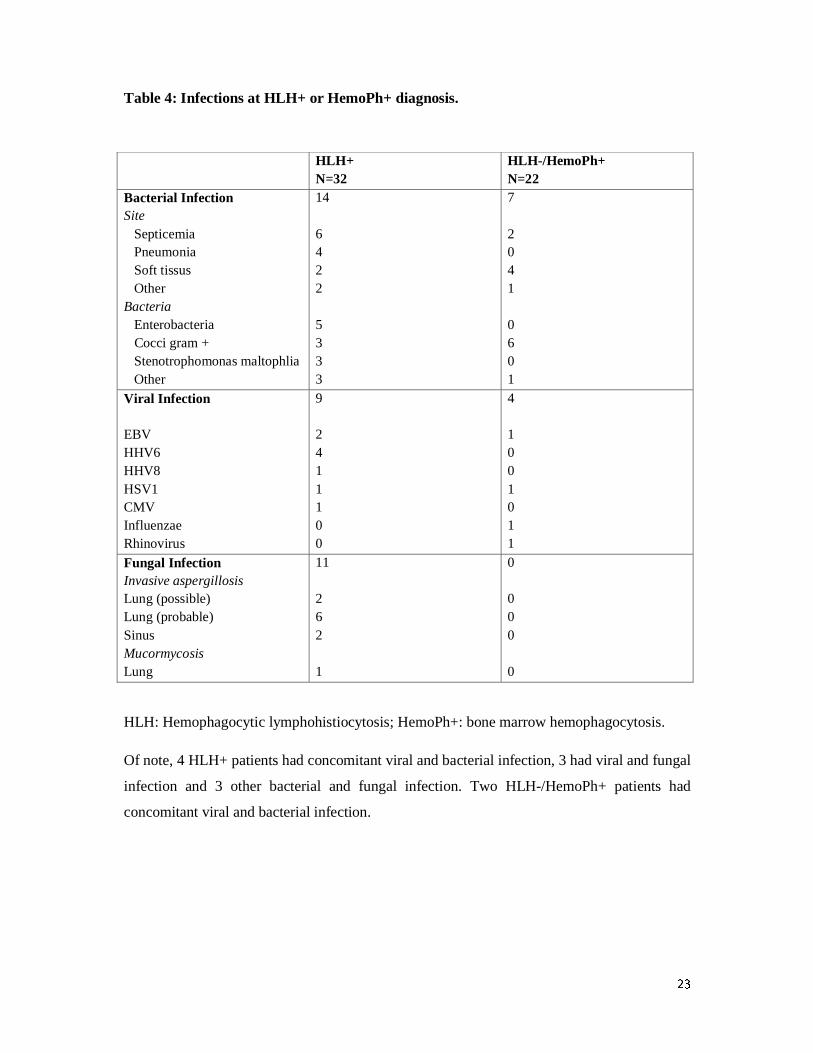
Etiology
A potential infectious etiology functioning as trigger for HLH has been found in 24 patients
(75%): 14 patients with bacterial infections, which were mainly septicemia and pneumonia, 9

10
patients with Herpesviridae infections and 11 patients with fungal infections, which were
mainly invasive aspergillosis (table 4). No mycobacterial infections have been documented.
During the induction phase, bacterial or fungal infections were documented in 15 HLH+
patients (46.9%) and in 91 HLH-/HemoPh- patients (31.5%) (p=0.079). Parenteral nutrition
and growth factors have been occasionally associated with hemophagocytosis.(15-16)
Parenteral nutrition was delivered in 10 HLH+ (31%) and 6 HLH-/HemoPh+ (27%) patients,
respectively (p=0.75). Before HLH and hemophagocytosis diagnosis, 20 HLH+ (62.5%) and
6 HLH-/HemoPh+ (27%) patients received G-CSF (p=0.01).
Treatment of HLH
Twenty nine patients (90.6%) received a specific treatment for HLH in a median time of 24
hours after diagnosis. First line treatment was corticosteroids (CS) in 5 patients, intravenous
immunoglobulins (IVIg) in 16 patients, CS + IVIg in 5 patients, CS + etoposide in 2 patients
and CS + IVIg + ciclosporine in one patient. Overall, CS and IgIV were used as the first line
treatment in 13 (44%) and 22 (76%) patients, respectively. The distribution of treatment
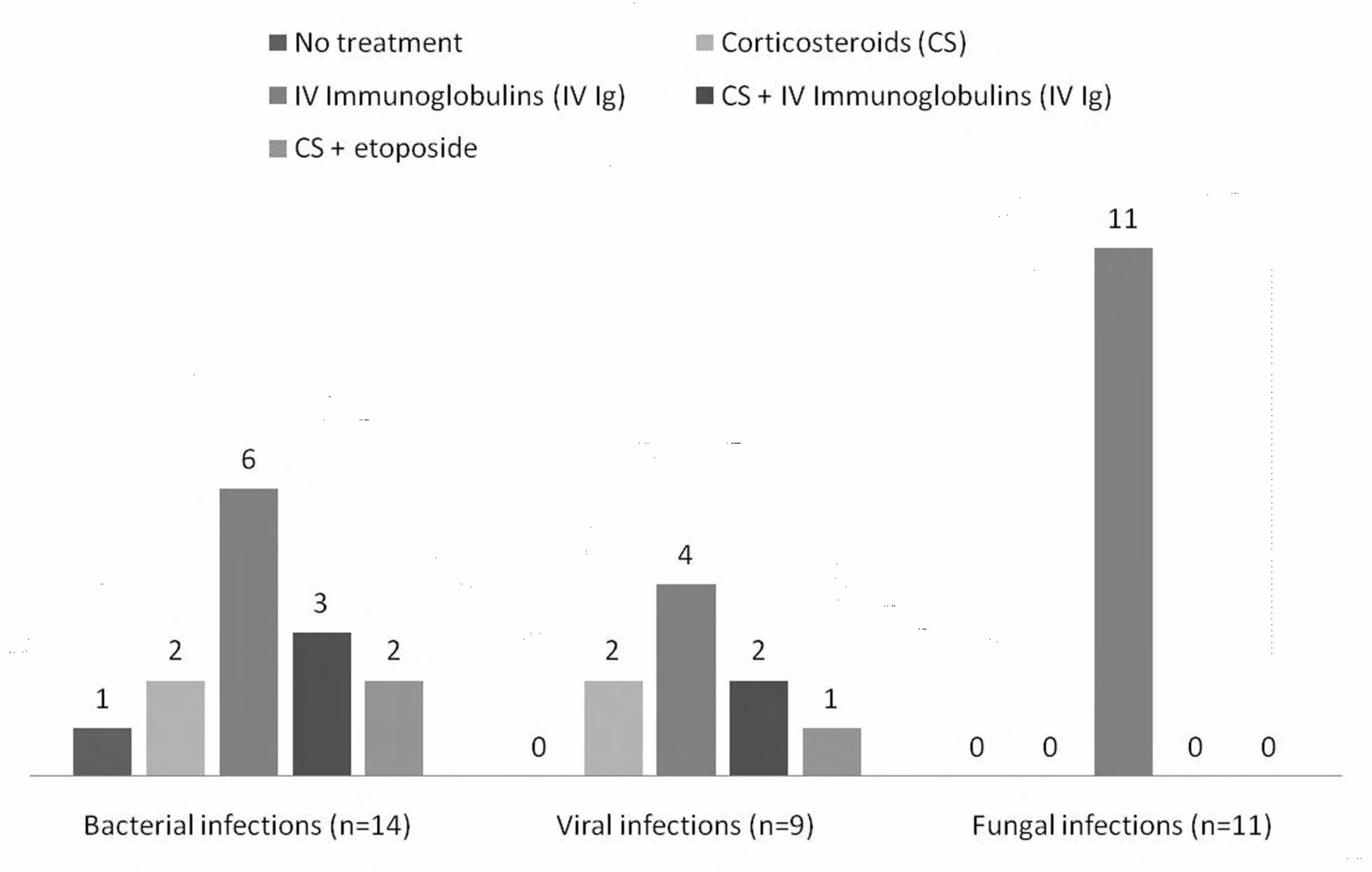
modalities according to the type of infection associated with HLH is shown in figure 1. No
patient with invasive fungal infection received CS as part of the first-line treatment. Patients
could receive second-line treatment with CS, IVIg or ciclosporine according initial response.
Although no response criteria are specifically defined for HLH, we judged that rapid fever
disappearance (within 48 hours) and resolution of cytopenias were the most relevant markers
of response. In our data, the ferritin decrease in the few days (d0 to d7 or d15) following the
treatment of HLH+ patients (n=29), did not permit to differentiate between responders and
non-responders. Of the 13 patients treated by CS, 9 (69%) had complete resolution of fever
whereas only 6 out of 16 patients (37.5%) treated by IVIg alone became apyretic.
Improvement of cytopenias was observed in 1/5 (20%) patients treated by CS alone and in
7/16 (44%) patients treated by IVIg alone. Of the 5 patients treated by CS + IVIg as the first

11
line, 4 had fever resolution and 3 had improvement of cytopenias. Of note, the response rate
was not associated with the type of underlying infection: 43% (n=6; p=0.688) in bacterial
infections, 33% (n=3; p=0.397) in viral infections and 36% (n=4; p=0.388) in fungal
infections. Overall, 20 patients (62.5%) had a resolution of symptoms after adapted antibiotic
or antifungal therapy and specific treatment of the HLH.
Outcome of HLH patients
The median overall survival was 14.9 months (IQR, 2.9-41.7) in the HLH+ group and 22.1
months (IQR, 9.2-85.6) in the HLH- group (HLH-/HemoPh- and HLH-/HemoPh+),
respectively (log-rank test, p=0.0016) (figure 2). In Cox proportional hazards models adjusted
for prognostic factors of AML, including age, secondary AML, white blood cell count,
cytogenetics, performance status and consolidation treatment by autologous or allogeneic
stem cell transplantation, HLH+ patients had a significantly increased risk of death compared
to HLH- patients (Hazard Ratio, 2.05, 95% CI, 1.31 to 3.22, p=0.002) (table 5). The adjusted
risk of HLH-/HemoPh+ patients did not significantly differ compared with HLH-/HemoPh-
patients. To describe in more detail the impact of HLH onset in the context of AML patients,
we focused on the 22 patients (and 16 patients) in whom HLH (or HLH-/HemoPh+) was
diagnosed in the course of intensive chemotherapy for first remission induction. In these
patients, the duration of neutropenia (37 days, IQR 29-59) tended to be longer than in HLH-
/HemoPh+ patients (28.5 days, IQR 24-38; p=0.158). In the HLH+ group, 9/22 patients
(40.9%) reached CR or CRi compared to 214/289 (74.1%) and 15/16 (93.8%) in the HLH-
/HemoPh- and HLH-/HemoPh+ groups, respectively (p=0.001) (table 6). The lower response
rate in the HLH+ group was associated with higher rates of hypoplastic death and resistant
disease, resulting in a significantly higher mortality rate at 3 months (36.4% in the HLH+
group compared to 12.5% in the HLH-/HemoPh- and HLH-/HemoPh+ groups, p=0.011).

12
Discussion
It has been recently suggested that hyperinflammatory disorders, such as HLH, macrophage
activating syndrome, malignancy-associated hemophagocytic syndrome or systemic
inflammatory systemic response, could be linked by a common pathophysiology involving
aberrant cytokine release, the so-called cytokine storm.(6, 17) We show here that up to 10%
of AML patients undergoing intensive chemotherapy displayed a clinical presentation
consistent with HLH although the strict criteria defined for pediatric patients were not met in
our series.(4) However, it should be emphasized that there is no current consensus on the
HLH criteria for adult patients.(18) The comparison of patients without symptoms of HLH
but hemophagocytosis in the bone marrow and HLH+ patients, showed that
hemophagocytosis itself is not a specific feature of HLH. We also acknowledge that the
distinction between HLH and other febrile conditions (i.e. pure infectious complications)
could be challenging in the setting of intensive chemotherapy course. Thus, a comparison
with other febrile, neutropenic AML patients would be necessary to better assess the specific
markers of HLH in those patients. Unfortunately, we did not collect data of all AML patients
at the time of first febrile episode during the period of this study to perform such a
comparison. However, virtually all AML patients undergoing intensive chemotherapy are
neutropenic for a long period of time (usually 21 days) and it is quite exceptional that these
patients remain afebrile. In our data base including 643 patients undergoing intensive
chemotherapy between 2000 and 2010, 136 patients had bacterial and 60 fungal documented
infections (~30%).(13) In this series, bacterial or fungal infections were documented in 31.5% of
HLH-/HemoPh- patients during the induction phase, indicating that a substantial proportion of
neutropenic patients with documented infections did not develop the particular clinical picture of
HLH+ patients. Thus, we feel confident that the clinical picture we describe here in roughly
10% of patients encompass bona fide criteria to distinguish HLH patients from other

13
neutropenic AML patients. Furthermore, most of the classical criteria of HLH seem
unsuitable when applied to leukemia due to the nature of the disease. This is especially the
case for pancytopenia related to bone marrow involvement and intensive chemotherapy and
the ferritin levels that are nearly always greater than 500 µg/L at time of AML diagnosis and
further increased by repeated red blood cell transfusions. Overall, we have identified criteria
that could help to identify HLH in AML patients (table 7). These criteria could be further
improved by specific prospective studies assessing the T-cell and NK cell activity, sCD25,
genetic screen and cytokine profiling in the setting of AML patients treated by intensive
chemotherapy. Indeed, it will be interesting to determine whether those patients have a
genetic background (mild hypomorphic mutations, polymorphism or complex polygenic
traits) predisposing them to HLH during intensive chemotherapy.(17) Additionally, a large
microbiological screen including viral testing should be part of the initial workup in HLH+
patients. In this study, the onset of HLH in the course of AML therapy had a strong impact on
prognosis as shown by the multivariate analysis. We show that HLH+ patients had a higher
rate of death in aplasia, resistant disease and a shorter overall survival. As various infectious
causes have been identified, with an overrepresentation of herpesviridae and invasive
aspergillosis, these early deaths are probably due to severe infections. However, HLH induced
liver and/or pulmonary involvement and prolonged pancytopenia that undoubtedly worsened
these infections. Although not significant in our study, it is also noteworthy that HLH+
patients had a higher rate of induction chemotherapy failure suggesting that uncontrolled
disease could have also impacted survival. The fact that less patients with HLH failed to
achieve complete response could underline the role of the malignant disease as a trigger for
HLH. One could therefore argue that HLH is a surrogate marker for failure to chemotherapy.
Alternatively and somewhat provocative, inflammatory response could also improve leukemia
cell survival and subsequent resistance to chemotherapy.

14
It is therefore critical to detect early onset of HLH in AML patients. A typical presentation is
a patient with a high fever even under broad antibiotic treatments, a rapid rise in the ferritin
level (sometimes to extraordinarily high levels) and liver abnormalities. This clinical picture
should suggest bone marrow aspiration for determination of hemophagocytosis, although this
latter criterion is not an absolute prerequisite for diagnosis. A large infectious workup is also
required to maximize antimicrobial treatment since we documented an infectious agent as a
potential trigger of HLH in 75% of cases. HLH treatments, which should be started promptly,
remain challenging, particularly in aplastic patients having received intensive chemotherapy.
Corticosteroids, etoposide-based regimens, ciclosporin and antithymocyte globulin are
currently administered in pediatric patients.(18) Immunomodulation with IVIg has also been
used in virus-associated HLH.(19) Because of the profound pancytopenia induced by
chemotherapy, etoposide has been exceptionally used in our patients. In fact, most were
treated by corticosteroids (either prednisolone or dexamethasone) or IVIg as determined by
the underlying infection. Although our study does not establish a standard of care in this
situation, we believe that the combination of IVIg and corticosteroids (short pulse then taper)
could be the treatment of choice. Indeed, corticosteroids induced a rapid improvement in
symptoms and overall condition, while IVIg were associated with a better survival in HLH+
patients (data not shown). Other more rational therapies such as anti-cytokines (e.g.,
tocilizumab) should be assessed in this setting.(20)
Our study also revealed quite unexpectedly that patients with bone marrow hemophagocytosis
but without HLH did particularly well in terms of complete response and overall survival with
a median of 85.6 months (IQR, 27.9-85.6). In most of these HLH-/HemoPh+ patients, bone
marrow aspiration was performed to assess the response to chemotherapy. Although
speculative, hemophagocytosis in this setting could be a marker of innate immunity against
leukemic cells. Indeed, macrophages express signal regulatory protein alpha (SIRPα) that

15
when activated by its ligand, CD47, induces an intracellular signalization resulting in
inhibition of phagocytosis.(21) It has been demonstrated that increased CD47 expression by
leukemic stem cells inhibits macrophage activity and is associated with poor prognosis in
AML.(22-23) Conversely, patients with low CD47 expression are more chemosensitive,
suggesting that there may be a correlation between marrow hemophagocytosis and CD47
expression in AML patients.
To conclude, HLH onset can be encountered in up to 10% of AML patients undergoing
intensive chemotherapy and is associated with induction failure and early mortality. Fever, a
very high ferritin level and marrow hemophagocytosis represent the cornerstone of diagnosis,
which must be rapidly established to adapt antimicrobial treatments and to promptly introduce
immunomodulation. Further studies are needed to better characterize this syndrome in AML
patients using genetic screening, cytokine monitoring or T-cell and NK-cell functional
studies.
Authorship and disclosures
Author’s Contributions
K.D. collected and analyzed data; E.B. performed statistical analysis and wrote the paper;
S.B. collected and analyzed data; J.C.; E.D.; C.D. and V.d.M. performed cytological analysis;
C.B; M.P.; M.A and F.H.; treated patients; A.S. collected data; C.R. treated patients, collected
and analyzed data, and wrote the paper. All the authors checked the final version of the
manuscript.
Disclosure of Conflicts of Interest: the authors declare no competing financial interests.

16
References
1. Janka GE. Hemophagocytic syndromes. Blood Rev. 2007;21(5):245-53. 2. Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10(2):197-203. 3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33. 4. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-31. 5. Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/ systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10(3):387-92. 6. Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24(1):9-15. 7. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients--a postmortem clinicopathologic analysis. Crit Care Med. 2004;32(6):1316-21. 8. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794-8. 9. Falini B, Pileri S, De Solas I, Martelli MF, Mason DY, Delsol G, et al. Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood. 1990;75(2):434-44. 10. Ferreri AJ, Dognini GP, Campo E, Willemze R, Seymour JF, Bairey O, et al. Variations in clinical presentation, frequency of hemophagocytosis and clinical behavior of intravascular lymphoma diagnosed in different geographical regions. Haematologica. 2007;92(4):486-92. 11. Han AR, Lee HR, Park BB, Hwang IG, Park S, Lee SC, et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86(7):493-8. 12. Bertozzi AI, Suc A, Rubie H, Duchayne E, Demur C, Robert A. [Hemophagocytic syndrome associated with neutropenia after chemotherapy]. Arch Pediatr. 2002;9(2):125-9. 13. Bertoli S, Berard E, Huguet F, Huynh A, Tavitian S, Vergez F, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013;121(14):2618-26. 14. LaRochelle O, Bertoli S, Vergez F, Sarry JE, Mansat-De Mas V, Dobbelstein S, et al. Do AML patients with DNMT3A exon 23 mutations benefit from idarubicin as compared to daunorubicin? A single center experience. Oncotarget. 2011;2(11):850-61. 15. Wang S, Degar BA, Zieske A, Shafi NQ, Rose MG. Hemophagocytosis exacerbated by G-CSF/GM-CSF treatment in a patient with myelodysplasia. Am J Hematol. 2004;77(4):391-6. 16. Roth B, Grande PO, Nilsson-Ehle P, Eliasson I. Possible role of short-term parenteral nutrition with fat emulsions for development of haemophagocytosis with multiple organ failure in a patient with traumatic brain injury. Intensive Care Med. 1993;19(2):111-4. 17. Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161(5):609-22. 18. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-52. 19. Chen RL, Lin KH, Lin DT, Su IJ, Huang LM, Lee PI, et al. Immunomodulation treatment for childhood virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol. 1995;89(2):282-90. 20. Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154-7. 21. Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. 2006;6(6):457-64.

17
22. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, Jr., et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286-99. 23. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271-85.
18
Table 1: Characteristics of patients at diagnosis of AML
HLH-/HemoPh-
n= 289 (84.3%)
HLH-/HemoPh+
n=22 (6.4%)
HLH+
n=32 (9.3%)
Age (years)
Median (IQR)
59 (49-66)
55.5 (41-61)
62 (46-69)
Gender
Male, n (%)
Female, n (%)
166 (57.4)
123 (42.6)
12 (54.5)
10 (45.5)
19 (59.4)
13 (40.6)
ECOG performance status
0-1, n (%)
≥2, n (%)
Unknown, n (%)
196(67.8)
38 (13.1)
55 (19)
20 (90.9)
0 (0)
2 (9.1)
21 (65.6)
4 (12.5)
7 (21.9)
AML status
De novo, n (%)
Secondary, n (%)
Unknown, n (%)
231 (79.9)
54(18.7)
4 (1.4)
18 (81.8)
4 (18.2)
0 (0)
23 (71.9)
8 (25)
1 (3.1)
Previous MDS, n (%) 27 (9.3) 1 (4.5) 5 (15.6)
Previous autoimmune disorder, n (%) 1 (0.4) 0 (0) 1 (3.1)
Extramedullary involvement
Yes, n (%)
No, n (%)
Unknown, n (%)
73 (25.3)
171 (59.2)
45 (15.6)
5 (22.7)
16(72.7)
1 (4.5)
12 (37.5)
17 (53.1)
3 (9.4)
WBC (G/L)
Median (IQR)
10.6 (3.1-42.3)
7.4 (2.2-41.3)
7.7 (2.9-20.6)
Hemoglobin (g/dL)
N
Median (IQR)
289
9.6 (8.4-11.3)
22
11.1 (7.8-12.8)
31
9.9 (9-11.1)
Platelets (G/L)
N
Median (IQR)
286
70 (38-118)
21
96 (67-134)
31
53 (16-162)
Lymphocytes (G/L)
N
277
22
31
Median (IQR) 2.1 (1.1-3.6) 1.7 (1.2-6.3) 1.8 (0.9- 2.8)
Bone marrow blasts (%)
N
Median (IQR)
278
64.5 (38-85)
21
51 (37-82)
29
44 (24-78)
Multilineage dysplasia
Yes, n (%)
No, n (%)
40 (13.8)
233 (80.6)
4 (18.2)
16 (72.7)
8 (25)
21 (65.6)
19
Unknown, n (%) 16 (5.5) 2 (9.1) 3 (9.4)
Cytogenetics
Favorable, n (%)
Intermediate, n (%)
Adverse, n (%)
Unknown, n (%)
26 (9)
198 (68.5)
62 (21 .5)
3 (1)
3 (13.6)
14 (63.6)
5 (22.7)
0 (0)
2 (6.3)
24 (75)
5 (15.6)
1 (3.1)
Ferritin (µg/L)
N
Median (IQR)
153
725 (362-1377)
15
384 (188-1074)
25
685 (392-1594)
Total percentages differ from 100% because of rounding
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+: bone marrow hemophagocytosis;
MDS: Myelodysplasic syndrome; IQR: Inter-Quartile Range.

20
Table 2: Comparisons of HLH+ and HLH-/HemoPh+ patients at HLH+ or HemoPh+ diagnosis
HLH+ n=32
HLH-/HemoPh+ n=22
p-value
Fever-n (%) 26 (81.3) 6 (27.3) .0001
Splenomegaly-n (%) 6 (18.8) 0 (0) .0706
Hepatomegaly-n (%) 7 (21.9) 0 (0) .0335
Icterus-n (%) 8 (25) 1 (4.5) .0666
Rash-n (%) 6 (18.8) 1 (4.5) .2197
Neurological symptoms-n (%) 4 (12.5) 0 (0) .1368
Respiratory symptoms-n (%) 19 (59.4) 2 (9.1) .0002 WBC (G/L)-Median (IQR) 0.4 (0.2- 1.6) 0.9 (0.5-4.1) .0723 Hemoglobin (g/dL)-Median (IQR) 9 (8.3-9.7) 9.7 (9- 10.8) .0628 Platelets (G/L)-Median (IQR) 18.5 (11- 28) 47.5 (12-155) .0494
PTT (%) N Median (IQR)
30
66.5 (57- 78)
20
84 (69.5-98)
.0013
Fibrinogen N Median (IQR)
26
5.1 (4.2-5.9)
18
4.4 (3.1-5.3)
.1042
Natremia (mmol/L) -Median (IQR) 135.5(133-138) 138 (134-139) .2351
Triglycerides (mmol/L) N Median (IQR)
30
1.3 (0.9- 2.5)
22
1.2 (0.8-1.8)
.3788
Creatinine (>1.5 x ULN)-n (%) 5 (15.6) 1 (4.5) .3826
Albumin (g/L) N Median (IQR)
29
27 (25- 30)
22
33 (29-37)
.0005
AST (UI/L)-Median (IQR) 31.5 (18- 103.5) 26 (19-38) .1835 ALT (UI/L)-Median (IQR) 31 (15.5-107.5) 29 (16-37) .2241 AST or ALT (>5 x ULN)-n (%) 7 (21.9) 0 (0) .0335
Alkaline phosphatase (UI/L) Median (IQR) >2 x ULN-n (%)
427 (251.5-635.5)
10 (31.3)
214 (175-276)
1 (4.5)
.0005 .0189
γGT (UI/L) Median (IQR) >5 x ULN-n (%)
213 (136-333.5)
19 (59.4)
59.5(34-119)
3 (13.6)
.0001 .0008
Bilirubin (µmol/L) N Median (IQR) >ULN-n (%)
30
13.5 (10-49) 13(40.6)
21
10 (8-11) 3 (13.6)
.0066 .0328
C-reactive protein (mg/L) N Median (IQR)
30
115.5 (57- 178)
19
16 (5- 92)
.0005

21
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+: bone marrow hemophagocytosis.
WBC: White Blood Cells, ULN: Upper Limit of Normal, PTT: Prothrombin time; AST:
aspartate transaminases; ALT: alanine transaminases; γGT: gamma-glutamyltranspeptidase; N
is only mentioned when there is unknown data. Bio-clinical data were collected at time of HLH or
hemophagocytosis onset for HLH+ and HLH-/HemoPh+ groups.
22
Table 3: Ferritin levels in HLH+ and HLH-/HemoPh+ patients
HLH+
N=32
HLH-/HemoPh+
N=22 p-value
Ferritin (µg/L)
At HLH/HemoPh+ diagnosis
N
Median (IQR)
31
5093 (2826-11307)
18
1866.5 (660-2789)
0.0004
Maximal value during
evolution*
N
Median (IQR)
32
6953.5 (4425.5-15305.5)
18
3373 (1808-5149)
0.0071
> 500, n (%) 32 (100) 17 (94.4) 0.3600
> 3000, n (%) 26 (81.3) 11 (61.1) 0.1797
> 8000, n (%) 15 (46.9) 2 (11.1) 0.0104
> 10000, n (%) 13 (40.6) 2 (11.1) 0.0287
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+: bone marrow hemophagocytosis. * Evolution represents the time between HLH diagnosis and response to anti-HLH treatment or death.
23
Table 4: Infections at HLH+ or HemoPh+ diagnosis.
HLH+ N=32
HLH-/HemoPh+ N=22
Bacterial Infection Site Septicemia Pneumonia Soft tissus Other Bacteria Enterobacteria Cocci gram + Stenotrophomonas maltophlia Other
14 6 4 2 2 5 3 3 3
7 2 0 4 1 0 6 0 1
Viral Infection EBV HHV6 HHV8 HSV1 CMV Influenzae Rhinovirus
9 2 4 1 1 1 0 0
4 1 0 0 1 0 1 1
Fungal Infection Invasive aspergillosis Lung (possible) Lung (probable) Sinus Mucormycosis Lung
11 2 6 2 1
0 0 0 0 0
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+: bone marrow hemophagocytosis.
Of note, 4 HLH+ patients had concomitant viral and bacterial infection, 3 had viral and fungal
infection and 3 other bacterial and fungal infection. Two HLH-/HemoPh+ patients had
concomitant viral and bacterial infection.

24
Table 5: Multivariate Cox proportional-hazards model for overall survival in HLH+
patients versus HLH- patients (including HLH-/HemoPh+ and HLH-/HemoPh-)
Hazard Ratio 95% Confidence Interval P value
HLH- 1.00
HLH+ 2.05 1.31-3.22 0.002
Age 1.01 1.00-1.02 0.028
De novo AML 1.00
Secondary AML 1.63 1.18-2.26 0.003
WBC ≤ 50 G/liter 1.00
WBC > 50 G/liter 1.64 1.16-2.30 0.005
Favorable cytogenetic risk 1.00
Intermediate cytogenetic risk 1.96 1.01-3.83 0.047
Adverse cytogenetic risk 3.52 1.72-7.19 0.001
WHO performance status=0 1.00
WHO performance status=1 1.32 0.93-1.87 0.116
WHO performance status=2 1.84 1.10-3.11 0.022
WHO performance status=3 1.92 0.89-4.15 0.096
No-SCT 1.00
Autologous-SCT 0.32 0.14-0.73 0.007
Allogeneic-SCT 0.63 0.42-0.95 0.027
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+: bone marrow hemophagocytosis.
WBC: White Blood Cell. SCT: Stem Cell Transplantation.

25
Table 6: Outcome of patients after induction remission chemotherapy (in patients with HLH+ or HLH-/HemoPh+ during induction remission chemotherapy)
HLH-/HemoPh- n= 289
HLH-/HemoPh+ n=16
HLH+ n=22
P-value
Response-n (%) CRi 33 (11.4) 0 (0) 5 (22.7) 0.093 CR 181 (62.6) 15 (93.8) 4 (18.2) <0.001* CR or CRi 214 (74.1) 15 (93.8) 9 (40.9) 0.001**
Induction failure-n (%) Early deaths 11 (3.8) 0 (0) 0 (0) 1 Deaths in aplasia 15 (5.2) 0 (0) 7 (31.8) <0.001*** Resistant disease 40 (13.9) 1 (6.3) 6 (27.3) 0.167
Relapse £ -n (%) 107 (48.2) 5 (33.3) 5 (62.5) 0.387
3-month mortality -n (%) 36 (12.5) 2 (12.5) 8 (36.4) 0.011****
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+ : bone marrow hemophagocytosis; CRi : complete response with incomplete blood count recovery ; CR : complete response.
£ In patients with CR or CRi or complete response with two cycles of induction.
*According to Bonferroni correction p-value=0.033 for HLH-/HemoPh- vs HLH-/HemoPh+, p-value<0.001 for HLH-/HemoPh- vs HLH+ and p-value<0.001 for HLH-/HemoPh+ vs HLH+.
**According to Bonferroni correction p-value=0.396 for HLH-/HemoPh- vs HLH-/HemoPh+, p-value=0.003 for HLH-/HemoPh- vs HLH+ and p-value=0.003 for HLH-/HemoPh+ vs HLH+.
***According to Bonferroni correction p-value=1.000 for HLH-/HemoPh- vs HLH-/HemoPh+, p-value<0.001 for HLH-/HemoPh- vs HLH+ and p-value=0.042 for HLH-/HemoPh+ vs HLH+.
****According to Bonferroni correction p-value=1.000 for HLH-/HemoPh- vs HLH/HemoPh+, p-value=0.018 for HLH-/HemoPh- vs HLH+ and p-value=0.429 for HLH-/HemoPh+ vs HLH+.

26
Table 7: Proposed criteria for HLH diagnosis in AML patients
AML patient undergoing intensive chemotherapy
and
Fever
Hepatosplenomegaly
Ferritin > 5000 µg/L
Prolonged neutropenia and/or thrombopenia (outside the expected range for chemotherapy-
induced myelosuppression, e.g, day 35)
Liver abnormalities
These criteria should lead to perform a bone marrow aspiration to reveal hemophagocytosis.
These proposed criteria should be improved by classical criteria for HLH such as genetic
defect of in genes of the cytotoxic pathway (PRF1, UNC13D, STXBP2, RAB27A, STX11,
SH2D1A, or XIAP), natural killer cell activity and soluble CD25.

27
Figure Legends
Figure 1: Distribution of anti-HLH treatments according to underlying infections
CS: corticosteroids (either prednisolone 1 mg/kg/d or dexamethasone 10 mg twice a day three
days then taper); IVIg: intravenous immunoglobulins (0.4 g/kg/d for 5 days or 1 g/kg/d for 2
days). Of note, 4 patients had concomitant viral and bacterial infection (1 was treated with
IVIg, 2 with CS + IVIg and 1 with CS + etoposide), 3 had viral and fungal infection treated
with IVIgand 3 other bacterial and fungal infection treated with IVIg.
Figure 2: Overall survival of HLH + and HLH- patients
HLH: Hemophagocytic lymphohistiocytosis; HemoPh+: bone marrow hemophagocytosis.


Supplementary data
Methods
Endpoints
The primary endpoint of the study was overall survival. For each participant, the length of
follow-up corresponds to the period between the date of diagnosis and July 1, 2012 or the date
of death if the patient died during the study period. The response to treatment was evaluated
after full hematological recovery (e.g, when neutrophils and platelet counts were > 1 G/L and
> 100 G/L to document complete responses), or at day 35 in cases of prolonged aplasia, and
defined according to the international consensus criteria as complete response (CR) or
complete response with incomplete blood count recovery (CRi).15 Early death was defined as
death from any cause occurring between the start of chemotherapy and the response
assessment. Resistant disease, death in aplasia and relapses were defined according to Cheson
criteria.15 The duration of neutropenia was defined as the number of days between d1 of
chemotherapy and the first day with neutrophils higher than 0.5 G/L or death for those who
died with less than 0.5 G/L.
Statistical analysis
Statistical analysis was performed on STATA statistical software, release 11.2 (STATA
Corporation, College station, TX, USA). We described patients’ characteristics using number
and frequency for qualitative data and number, median and Inter-Quartile Range (IQR) for
quantitative data. Qualitative variables were compared between groups (HLH+, HLH-
/HemoPh+ and HLH-/HemoPh- patients) using the χ2-test (or Fisher’s exact test in the case of
small expected numbers). Student’s t-test was used to compare the distribution of quantitative
data (or Mann-Whitney’s test when distribution departed from normality or when

homoscedasticity was rejected). Differences in survival functions were tested using the Log-
Rank test. The independent impact of HLH on overall survival was assessed using a Cox
model adjusted for age, secondary AML, white blood cell count, cytogenetics, performance
status and consolidation treatment by autologous or allogeneic stem cell transplantation
(SCT). HLH+ patients first become at risk of HLH+ mortality at the date of HLH+ diagnosis
and patients treated with autologous or allogeneic SCT first become at risk of SCT mortality
at the date of SCT. Since the linearity hypothesis was not fully respected for WBC, this
continuous co-factor was transformed into ordered data: ≤ 50 G/L and > 50 G/L. The
proportional-hazard assumption was tested for each covariate by the “log-log” plot method
curves ((-ln{-ln(survival)}), for each category of nominal covariate, versus ln(analysis time)).
None of the assumptions could be rejected. Interactions between HLH and the independent
covariates were tested in the survival model (none were significant). All reported p-values
were two-sided and the significance threshold was < 0.05.

Supplementary figure.
Light microscopic representative images of May–Grünwald–Giemsa (MGG) stained bone marrow smear showing macrophagescontaining hematopoietic cells in their cytoplasm (x100). A, B, C. Erythroblast. D. Platelets.
A B
C D