gains of chromosome 8 are confined to mesenchymal components in pleuropulmonary blastoma
TRANSCRIPT

ORIGINAL ARTICLES
Gains of Chromosome 8 Are Confined toMesenchymal Components inPleuropulmonary Blastoma
SARA O. VARGAS,1* VANIA NOSE,1,2 JONATHAN A. FLETCHER,1,2 ANDANTONIO R. PEREZ-ATAYDE1
1Department of Pathology, Harvard Medical School, 300 Longwood Avenue, Boston, MA 02115, USA2Department of Pathology, Brigham and Women’s Hospital, 75 Francis Street, Boston, MA 02115, USA
Received December 5, 2000; accepted April 25, 2001.
ABSTRACTPleuropulmonary blastoma, an aggressive tumor that isemerging as a distinct entity of childhood, is character-ized by mesenchymal elements (including undifferenti-ated blastema and often cartilaginous, rhabdomyoblas-tic, or fibroblastic differentiation) and epithelium-linedspaces. We investigated two patients with pleuropulmo-nary blastoma, a 3-year-old boy and an 11-year-old girl,both with large cystic masses replacing one lung. In bothchildren, the post-chemotherapy resection specimensshowed more maturation of rhabdomyoblasts and morenuclear pleomorphism in all mesenchymal cell lines,compared with biopsies sampled before treatment.Karyotypic analysis demonstrated gains in chromosome8 in both cases and 17p deletion in one case. Fluorescentin situ hybridization analysis demonstrated that thechromosome 8 gains were present in all mesenchymalelements, including undifferentiated blastematous, rh-abdomyoblastic, fibroblastic, and chondroblastic areas.Epithelial cells showed no chromosome 8 gains. Thechromosome 8 aberrations were not appreciably differ-ent in pre- versus post-chemotherapy tissue. Our find-ings substantiate previous reports that polysomy ofchromosome 8 is a consistent feature of pleuropulmo-nary blastoma. Further, they indicate that clonal prolif-eration in pleuropulmonary blastoma is restricted to the
malignant mesenchymal elements, supporting the no-tion that the epithelial components of this tumor arenon-neoplastic.
Key words: chromosomes, human, pair 8, pair 17, cy-togenetics, fluorescence in situ hybridization, lung neo-plasms, pulmonary blastoma, pleuropulmonary blas-toma, rhabdomyosarcoma
INTRODUCTIONPediatric tumors are often characterized by theproliferation of both epithelial and mesenchymalelements. Many of these lesions, such as Wilms’tumor, hepatoblastoma, and pancreatoblastoma,have been described as “dysontogenetic,” that is,resembling the developmental anatomy of the or-gan of origin [1]. General characteristics of such“embryonic” lesions are that they may be associ-ated with developmental anomalies, may appearthemselves to represent abnormally developing tis-sue, or both; they have been described as “simul-taneously both neoplasms and malformations” [2].To what extent the anomalies predispose tissues tobecome malignant or to what extent the malignantgrowth governs the anomalous development ispoorly understood.*Corresponding author
Pediatric and Developmental Pathology 4, 434–445, 2001
DOI: 10.1007/s10024-001-0080-8
Pediatric and Developmental Pathology
© 2001 Society for Pediatric Pathology

Pleuropulmonary blastoma (PPB) is a pri-mary intrathoracic malignancy that occurs mainlyin early childhood and has been termed dysonto-genetic. It is composed of immature mesenchyme,often differentiating toward skeletal muscle, carti-lage, fibrous tissue, and sometimes fat, and it mostoften includes epithelium. The mesenchymal ele-ments are regarded as malignant. Since PPB wasrecognized as a clinicopathologic entity distinctfrom adult pulmonary blastoma [3], which is char-acterized by malignant glands and malignantstroma, the epithelial elements in PPB have beendescribed as benign. This has been due to theirbland histologic appearance and their absencefrom metastases [3,4]. Various authors have im-plied that the epithelium represents a benign-ap-pearing component of the tumor, entrapped un-derlying lung, or both [5,6]. Its arrangement incystic structures has been likened to the architec-ture of congenital cystic adenomatoid malforma-tion (CCAM) [6,7].
To define the neoplastic populations in PPB,we analyzed two cases biopsied and resected at thisinstitution. Finding them to be characterizedkaryotypically by excess material from chromo-some 8, we performed fluorescent in situ hybrid-ization (FISH) for chromosome 8 to characterizethis cytogenetic aberration in each of the tumors’various components before and after treatment.
METHODSTwo instances of PPB were recognized at this in-stitution. Clinical features were reviewed. In bothcases, specimens included biopsies sampled beforetreatment as well as tissue from resections per-formed after chemotherapy. Portions were imme-diately placed in 2% glutaraldehyde for ultrastruc-tural analysis, in Hank’s solution for cytogeneticanalysis, and in 10% buffered formalin for histo-logic evaluation. Paraffin-embedded sections werestained with hematoxylin and eosin (H&E).
Immunohistochemical stains were performedon formalin-fixed, paraffin-embedded material us-ing a streptavidin-biotin-based alkaline phospha-tase detection kit with Fast Red (BioGenex Labo-ratories, San Ramon, CA) as the chromogen. Thefollowing antibodies were used: monoclonaldesmin, myoglobin, HMB-45, alpha-1-antichymot-rypsin (BioGenex Laboratories), epithelial mem-
brane antigen (EMA), AE1-AE3, muscle-specificactin (HHF-35) (Signet Laboratories, Dedham,MA), vimentin clone V-9 (Dako Corporation,Carpinteria, CA), and low-molecular-weight cyto-keratin (CAM 5.2) (Becton Dickinson, San Jose,CA). Positive and negative controls were stained inparallel.
Cytogenetic analysis was performed by minc-ing representative portions of each tumor and dis-aggregating them with collagenase. Cell cultureand metaphase harvest were performed as previ-ously described [8].
For the in situ hybridization analysis, 4-mm-thick tissue sections were cut from paraffin blocksand mounted on poly-L-resin glass slides. Theslides were baked overnight, then deparaffinized inxylene. They were dehydrated in 100% ethanol andair dried. The tissue was microwaved in 100 mMTris-base/50 mM EDTA, then washed in 23 SSCsolution. Protein digestion was conducted with Di-gest-all 3 (Zymed, San Francisco, CA). The slideswere washed again in 23 SSC, and the tissue wasfixed in 10% buffered formalin, followed again bywashes in 23 SSC. The tissue was then placed in adenaturing solution (4 ml 203 SSC, 8 ml distilledwater, 28 ml formamide). The slides were immedi-ately dehydrated in cold ethanol series, then airdried.
A chromosome 8 alpha satellite probe (D8Z1biotin-labeled probe, 10 ng/ml) (Oncor, Gaithers-burg, MD) was used. One microliter of probe wassuspended in Hybrisol VII (Oncor) and denaturedin a 70°C water bath, then transferred to ice. Then5 ml of probe was applied to the section, coveredwith a glass coverslip, sealed with rubber cement,and incubated overnight.
The detection was done after washing in 0.53
SSC at 72°C, then in PBS/T. Staining was with afluorescein-labeled avidin detection kit (Oncor),and the tissue sections were counterstained withpropidium iodide/antifade (Vector Labs, Burlin-game, CA). The FISH signals were evaluated with aZeiss Axioscope epifluorescence microscope; theentire tissue section was analyzed. Cell lineageswere classified as trisomic or polysomic if morethan 5% of nuclei showed three or more pericen-tromeric alpha-satellite signals. Representative im-ages were captured using a charge-coupled devicecamera (CCD-CH250, Photometrics, Tucson, AZ).
CHROMOSOME 8 GAINS IN PPB 435

RESULTSClinical findingsThe first patient (case 1) was a 31⁄2-year-old Cauca-sian boy with a 2-week history of cough. He was anonly child, the product of a 36-week gestation de-livered by Cesarean section for pregnancy-inducedhypertension. He had bilateral herniorrhaphies at8 weeks and an episode of croup at 9 months. A
computed tomography (CT) scan of the chest
showed a large heterogeneous mass with solid and
cystic elements occupying nearly the entire right
hemithorax with mediastinal deviation to the left.
Surgical exploration revealed a large unresectable
tumor involving the right upper lobe, displacing
the rest of the lung downward and extending su-
periorly into the mediastinum. A biopsy from a
Figure 1. A, B: Mesenchy-mal cells ranging from cellu-lar blastema to paucicellularmyxoid stroma. Interspersedepithelium-lined cysts ofvarying size are present.Some cysts (A) are sur-rounded by densely cellulartumor, case 1. C: Cellularmesenchymal tissue sur-rounding epithelium-linedspaces, with polypoid pro-jections reminiscent of bot-ryoid rhabdomyosarcoma,case 2. D: Spindle cells infascicles resembling infan-tile fibrosarcoma, case 1.
436 S.O. VARGAS ET AL.

solid portion of the tumor was performed. Threedays after surgery a year-long chemotherapycourse was begun; it consisted of three doses ofvincristine and then alternating cycles every 3weeks of vincristine/actinomycin D/cyclophospha-mide with adriamycin/cisplatinum. Three monthsinto the chemotherapy regime, a complete surgicalresection was performed via right upper and mid-dle lobectomy. At the time it was noted that thetumor had shrunk markedly, no longer extending
into the mediastinum. The patient is well and freeof disease 8 years after initial diagnosis.
The second patient (case 2) was an 11-year-old previously healthy girl who presented with a2-week history of cough, periodic vomiting, occa-sional dyspnea, and fever to 104°F. A chest radio-graph showed opacification of the right hemitho-rax. A CT scan revealed a right thoracic massextending from the anterior and middle mediasti-num to the chest wall, infiltrating and compressing
Figure 2. A: Anaplasia withinareas of immature embryonal“blastema,” case 1. B: Areas ofimmature cartilage, case 1. C:Immunoperoxidase stain fordesmin showing cytoplasmicstaining, case 1. D: Post-treat-ment specimen showing areasof bronchiole-like cystic struc-tures with intervening blandmesenchyme, with an architec-ture similar to that of type 2congenital cystic adenomatoidmalformation, case 2.
CHROMOSOME 8 GAINS IN PPB 437

the lung. An open lung biopsy was performed. Thepatient was treated with chemotherapy (adriamy-cin, actinomycin, vincristine, cyclophosphamide,and cisplatinum) followed after 4 months by ex-trapleural pneumonectomy. After histologic exam-ination showed focal extension to a diaphragmaticmargin, local radiation was administered. Threeyears after surgery, the patient is well except fordecreased exercise tolerance and point tendernessover the rib at the surgical site; there is no evidenceof recurrent tumor.
Gross findingsIn both cases, the open lung biopsy specimensconsisted of small fragments of nodular tan tissue.For case 1, the right upper and middle lobes, re-sected after treatment, together weighed 193.5 g.The tumor was 9 cm, poorly defined, and occupiedmainly the upper lobe with partial involvement ofthe middle lobe. It was soft and hemorrhagic withcysts and areas of apparent cartilage, myxoidchange, and yellow necrosis. The uninvolved lungappeared grossly normal. In case 2, the right ex-trapleural pneumonectomy specimen, excised af-ter chemotherapy, weighed 335 g. Much of thelung showed involvement by tumor and diffusemicrocystic change. Approximately 2%–5% of theparietal pleura and 15% of the visceral pleura wasstudded with tumor nodules ranging from 0.3 to 6cm in greatest dimension. There was also a singlewhite–yellow focally necrotic rubbery nodule mea-suring 1.4 cm in the lower lobe.
Histologic evaluationH&E-stained sections in pretreatment biopsy speci-mens from both cases revealed mesenchymal ele-ments surrounding epithelium-lined cystic struc-tures (Figs. 1 and 2A, B). The mesenchyme consistedof clusters of undifferentiated stromal cells resem-bling blastema. This tissue was densely cellular witha high mitotic rate, yet blended with areas that werepaucicellular, verging on myxoid. For the most part,these poorly differentiated cells were bland and uni-form, but in both cases they showed focal anaplasia,most notable in case 1, where in areas the nucleiwere very enlarged, pleomorphic, and markedly hy-perchromatic (Fig. 2A). The blastematous tissueshowed increased cellularity immediately surround-ing epithelial-lined cystic structures. These cysts
were often compressed and slit-like and showed fre-quent polypoid projections of epithelium-lined mes-enchyme within their lumens (Fig 1C). In both cases,subepithelial areas of condensed cellularity showedrhabdomyoblastic differentiation, made evident byeccentrically located pink cytoplasm, with an overallpattern histologically and antigenically (Fig. 2C; seebelow) characteristic of the cambium layer in botry-oid rhabdomyosarcoma. In more solid areas, cellsshowed an embryonal organoid appearance, and instill other areas, cells took on a spindled shape in apattern closely resembling fibrosarcoma. The epithe-lium varied from simple flat-to-cuboidal topseudostratified ciliated respiratory type.
In case 1 there were variably cellular islandsof immature cartilage (Fig. 2B) that blended withthe more myxoid areas of mesenchyme. Case 1 alsoshowed large areas of confluent necrosis in thepretreatment biopsy specimen. Case 2 had morepronounced and widespread rhabdomyoblasticdifferentiation.
The tumor resections, performed after treat-ment in both cases, showed areas of confluent ne-crosis, most extensive in case 1. Anaplasia wasmore pronounced in both cases and involved prim-itive mesenchyme, rhabdomyosarcomatous areas,and, in case 1, cartilage. Both tumors showedmarkedly increased differentiation of rhabdomyo-blasts, with abundant pink cytoplasm and occa-sional cross-striations. Large areas of both treatedtumors were devoid of histologically malignant-appearing tissue and were comprised of bronchi-ole-like cystic structures separated by bland mes-enchyme, focally containing well-differentiatedrhabdomyoblasts. These areas were reminiscent oftype 2 CCAM (Fig. 2D).
Immunohistochemical evaluationTumor cells in case 1 showed focal cytoplasmicstaining with desmin, myoglobin, and vimentin.This staining was most pronounced in the areas ofcondensed cellularity underlying epithelium-linedspaces. Cytokeratin was positive only in the liningepithelium.
Tumor cells in case 2 were positive for mus-cle-specific actin, desmin, alpha-chymotrypsin,and vimentin, and negative for CAM 5.2, AE1-AE3,EMA, and HMB-45.
438 S.O. VARGAS ET AL.

Ultrastructural examinationTissue from case 1 available for electron micro-scopic examination included only areas with rh-abdomyoblastic differentiation. No other areassuch as primitive blastema, cartilage, or epithe-lium were available for study. The tissue exam-ined showed a population of rhabdomyoblasts indifferent stages of development. Some rhab-domyoblasts were characterized by a well-devel-oped continuous basal lamina and by abundantcytoplasm containing large amounts of glycogenand numerous bundles of skeletal muscle fila-ments with Z bands and poorly formed sarco-meres. Other less differentiated rhabdomyo-blasts had high nuclear-to-cytoplasmic ratios,pleomorphic nuclei, large nucleoli, and scant cy-toplasm. These primitive cells occasionallyshowed paranuclear clusters of myosin and actinfilaments and no Z bands.
Tissue taken for electron microscopic examina-tion from a core needle biopsy in case 2 was devoid oftumor, showing only normal skeletal muscle.
Cytogenetic analysisKARYOTYPE. Twenty metaphase cells were evalu-
ated in biopsy tissue from case 1, yielding the fol-lowing karyotype: 47, Y, inv(X)(p11q28),add(4)(q31), add(6)(q22), t(7;9), (p11;q22), 18,del(17)(p11) (Fig. 3).
Seventeen metaphase cells were evaluatedin biopsy tissue from case 2, yielding the follow-ing karyotype: 62– 64, XX, 12, add (6)(q27), 17,18, 18, 18, 111, 111, 113, 113, 113, 119, 120,120, 121, 1123mar.
FLUORESCENT IN SITU HYBRIDIZATION. Morphologicdetail was easily appreciated. Mesenchymalcomponents of both cases, including blastema-tous, anaplastic, rhabdomyosarcomatous, fibro-blastic, and chondroid areas, showed a majorityof nuclei with three to nine fluorescent signalsfor chromosome 8 (Fig. 4). In case 1, the mostcommon number of signals seen in all mesenchy-mal cells was three. In case 2, polysomy withfour to six signals was most common. Chondroidareas showed lesser degrees of polysomy, with
Figure 3. Giemsa-band-ed karyotype, case 1. Ar-rows indicate clonal aber-rations.
CHROMOSOME 8 GAINS IN PPB 439
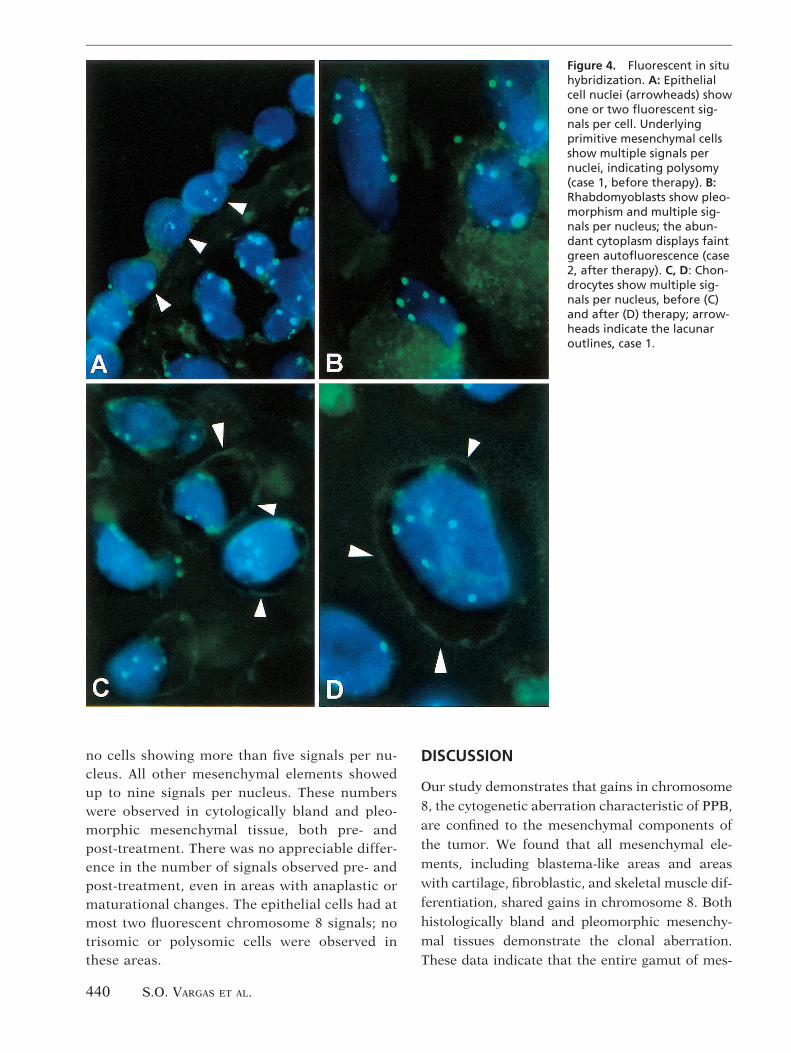
no cells showing more than five signals per nu-cleus. All other mesenchymal elements showedup to nine signals per nucleus. These numberswere observed in cytologically bland and pleo-morphic mesenchymal tissue, both pre- andpost-treatment. There was no appreciable differ-ence in the number of signals observed pre- andpost-treatment, even in areas with anaplastic ormaturational changes. The epithelial cells had atmost two fluorescent chromosome 8 signals; notrisomic or polysomic cells were observed inthese areas.
DISCUSSION
Our study demonstrates that gains in chromosome
8, the cytogenetic aberration characteristic of PPB,
are confined to the mesenchymal components of
the tumor. We found that all mesenchymal ele-
ments, including blastema-like areas and areas
with cartilage, fibroblastic, and skeletal muscle dif-
ferentiation, shared gains in chromosome 8. Both
histologically bland and pleomorphic mesenchy-
mal tissues demonstrate the clonal aberration.
These data indicate that the entire gamut of mes-
Figure 4. Fluorescent in situhybridization. A: Epithelialcell nuclei (arrowheads) showone or two fluorescent sig-nals per cell. Underlyingprimitive mesenchymal cellsshow multiple signals pernuclei, indicating polysomy(case 1, before therapy). B:Rhabdomyoblasts show pleo-morphism and multiple sig-nals per nucleus; the abun-dant cytoplasm displays faintgreen autofluorescence (case2, after therapy). C, D: Chon-drocytes show multiple sig-nals per nucleus, before (C)and after (D) therapy; arrow-heads indicate the lacunaroutlines, case 1.
440 S.O. VARGAS ET AL.

enchymal cells represents phenotypically divergentcell lines of a malignant clone. The epithelial ele-ments, lacking the clonal aberration, are mostlikely non-neoplastic. The PPB cytogenetic find-ings in this regard parallel those in other mixedlineage neoplasms; cytogenetic aberrations are re-stricted to the mesenchymal elements in pulmo-nary chondroid hamartoma, endometrial polyps,and breast adenofibromas, whereas the epithelialelements in these tumors are cytogenetically nor-mal [9–11].
PPB is an intrathoracic tumor of childhood[3,6]. Recent schemas define it as a subset of[12,13] or as an entity distinct from [3,6,14–16]pulmonary blastoma. Defining pulmonary blas-toma as a lung tumor that mimics developing lung[5], the term “pulmonary blastoma” may encom-pass the following: (1) classic biphasic blastoma,composed of malignant glands and malignantstroma [5,17]; (2) well-differentiated fetal adeno-carcinoma, comprised only [18,19] or predomi-nantly [17] of malignant epithelium; and (3) PPB,consisting of malignant stroma with or withoutaccompanying benign epithelium [3,5].
Despite past variation in interpreting the def-inition of pulmonary blastoma [5,12,13,15–17, 20],PPB appears to be emerging as a distinct entity ofchildhood with characteristic clinicopathologicfeatures that distinguish it from pulmonary blas-tomas of adulthood [3,6,21]. Children with PPB,usually less than 5 years of age, most often presentwith persistent cough and respiratory difficulty ac-companied occasionally by fever, pneumothorax,or empyema [3,4,15]. Radiographically, the tumoris characteristically large and partially cystic. PPBhas grossly and microscopically been classifiedinto three types [4,6]. In type 1, the tumor is en-tirely cystic, resembling type 1 congenital cysticadenomatoid malformation. The cysts are lined byrespiratory epithelium and the septa contain fi-brous tissue, primitive cells, and a subepithelialcambium layer with features of botryoid-type em-bryonal rhabdomyosarcoma. Type 2 PPB are bothcystic and solid. Type 3 tumors are entirely solidwith occasional secondary cystic degeneration. Intypes 1 and 2, the cystic areas are similar. The solidareas of types 2 and 3 are identical and usuallycontain primitive blastema, cartilage, and aggre-gates of pleomorphic undifferentiated sarcoma.
Chondroblastic, rhabdomyoblastic, fibroblastic,and lipoblastic differentiation are not uncommon[4,12]. The treatment of PPB is complete surgicalexcision when possible. The tumor may relapsewith local recurrence or bone, contralateral pul-monary, and central nervous system metastasesand has a mortality of approximately 40% [3,4,6].The two tumors presented here were clinically andpathologically characteristic of type 2 PPB, bothcontaining cystic and solid areas.
Adult pulmonary blastoma is characterizedby malignant glands and malignant mesenchymemimicking developing lung. It tends to occur inolder adults with smoking histories [17] and it israrely cystic [22]. Unlike PPB, characterized bykeratin immunostaining confined to the epithe-lium, adult pulmonary blastoma shows expressionof keratin in all components, including spindlecells [23]. Our finding that the epithelial compo-nents of PPB are most likely non-neoplastic furtherdistinguishes this tumor from adult pulmonaryblastoma. This pathologic distinction is diagnosti-cally important, especially in light of a recent re-port of PPB in adulthood [24] and the observationthat adult pulmonary blastoma may occur in child-hood [17].
In our view, the epithelium in PPB probablyrepresents entrapped and malformed non-neoplas-tic lung. This apparent malformative growth ofcystic structures seems to indicate a superimposeddevelopmental anomaly. In this light, PPB typifiesan important aspect of many embryonic tumors,representing both a neoplasm and a maldevelop-ment at once. It is known that mesenchymal–epi-thelial interactions are vital to branching morpho-genesis and development of the lung [25,26]. It ispossible that in PPB, maldeveloped benign epithe-lium predisposes the surrounding stroma to unreg-ulated clonal proliferation. Conversely, perhapsthe presence of neoplastic mesenchymal elementsinduces the malformation by interfering with nor-mal development.
We and others [6,7] have noted a resemblancebetween PPB and CCAM, a cystic mass of epithe-lium and stroma that resembles immature lungtissue and shows striated muscle in up to 10% ofcases [27]. CCAM is thought of as a non-neoplasticmalformation. After treatment with chemotherapyand radiation, our two cases showed areas archi-
CHROMOSOME 8 GAINS IN PPB 441

tecturally similar to CCAM, suggesting that cysticabnormalities may provide a structural frameworkfrom which PPB arises. Another possibility is thatareas resembling CCAM represent a maldevelop-ment of lung caused by the presence of the tumor.Malignant “transformation” of both epithelial andmesenchymal elements has been described previ-ously in CCAM [28–31]; malignant transformationof mesenchymal elements reported in CCAM mayrepresent part of the spectrum of PPB.
Both case 1 and case 2 showed skeletal mus-cle differentiation. Case 2 was notable in that itssolid component consisted entirely of cellularblastematous elements and rhabdomyosarcoma.This illustrates the overlap between PPB and theentity reported previously as primary pulmonaryrhabdomyosarcoma. A subset of rhabdomyosarco-mas of the lung appear to arise in childhood inassociation with congenital cystic lung disease,particularly CCAM [31–35], indicating a dysonto-genetic nature. It has previously been suggested
that primary pulmonary rhabdomyosarcoma orig-inating in a site of abnormally formed lung ininfants and children represents a distinct type oftumor [36] or a prognostically different subtype[37]. Since the recognition of PPB of childhood, weand others [6] believe that these apparent primaryrhabdomyosarcomas actually represent PPB.
The genetics of PPB are not well defined. Thetumor is associated with a high rate of familialtumors and other diseases, including pulmonarycysts, cystic nephroma, medulloblastoma, testicu-lar germ cell tumors, and embryonal rhabdomyo-sarcoma [21,38,39]. Characteristic chromosomalabnormalities are not well established. Trisomy 2[40,41] and trisomy 8 [42] both have been pro-posed as characteristic aberrations. A loss of het-erozygosity on chromosome 11p15.5 has also beensuggested [4].
Our results, together with findings from areview of previously reported karyotypes[21,40,42–44], indicate that gains in chromosome
Table 1. Cytogenetic findings in pleuropulmonary blastoma
Patient Age/sex Reference Karyotype
1 41 months/M Case 1 47, Y, inv(X)(p11q28), add(4)(q31), add(6)(q22), t(7;9)(p11;q22), 18, del(17)(p11)
2 11 years/F Case 2 62-64, XX, 12, add(6)(q27), 17, 18, 18, 18, 111,111, 113, 113, 113, 119, 120, 120, 121, 1123mar
3 45 months/M Sciot et al. [40] 48, XY, 1del(2)(q31q33), 18, del(9)(q22) (initialbiopsy)
47, XY, 1del(2)(q31q33), del(9)(q22) (treatedresection)
4 22 months/F Novak et al. [42] 47, XX, 18
5 24 months/F Novak et al. [42] 47, XX, 18, 211, 1der(11)t(8;11)(q21.2;q23)
6 42 months/F Novak et al. [42]and Priest et al. [21]
47, XY, 113, [14]/76, XY, 1X, 1X, 11, 11, 1t(1;19)(p13;p13.3) x2, 12, 12, 12, 13, 15, 15,1der(6)t(6:?)(?q11;?), 18, 110, 110, 111, 112,112, 113, 113, 114, 114, 115, 115, 116, 116,1del(17)(?p11.2) x2, 118, 219, 122, 1mar[4]/46,XY[24]
7 4 years/M Priest et al. [21] 47, XY, inv(7)(q22q36), 18, der(11)t(7;11)(q11.2;p11.2), inv(7)(q22q36), del(17)(p11.2)[18]/46,XY[2]
8 4 years/M Priest et al. [21] 46, XY, t(3;10)(q23;p15), del(6)(q16), 18[16]/48, XY,del(6)(q16), der(10)t(3;10)(q23;p15), 18, 114[3]/52, XY, del(6)(q16), t(3;10)(q23;p15), 15, 17, 18,111, 114, 121, 18[1]
9 2 years/F Kelsey et al. [44] 46, XX, der(17)t(8;17)(q11.2;p13.1)
10 3 years/F Barnard et al. [43] 48, X, 2X, 18, 1mar x2
442 S.O. VARGAS ET AL.

8 are a consistent feature of PPB. All cases show apolysomy or an excess of material from chromo-some 8 (Table 1). (However, in one case showingtrisomy 8 [case 6, Table 1] [42], the tetraploidnature of the tumor precludes interpreting the ad-ditional chromosome 8 as a relative gain.) Anotherreport, in abstract form, describes trisomy 8 in acase of PPB, although a karyotype is not provided[45]. Adding our findings to those of previous au-thors firmly implicates gains in chromosome 8 as aconsistent abnormality in PPB.
Two previous cases of PPB (case 5 and case 9,Table 1) [42,44] showed additional material de-rived from an unbalanced translocation involvingthe terminal region of 8q, the site of the c-myc
gene. The dysregulation of myc, a proto-oncogene,has a well-established role in oncogenesis and maybe a factor in the development of PPB. It is of notethat embryonal rhabdomyosarcoma, previouslydescribed in the uncle of a patient with familialPPB [21], has also been associated with trisomy 8[46,47]. This is especially interesting given the rh-abdomyosarcomatous differentiation observed inPPB and suggests a relationship between the twotumors. Other tumors characterized by gains inchromosome 8 include mesoblastic nephroma[48], infantile fibrosarcoma [49], Ewing’s sarcoma[50], myxoid liposarcoma [51], and desmoid tumor[52]. Most of these tumors share with PPB a pro-pensity to occur in childhood.
Full somatic trisomy 8 is an apparently earlylethal disorder; most individuals with trisomy 8 aremosaics [53]. The most commonly reported so-matic trisomy involving chromosome 8 is of 8p,due either to an inv dup(8p) or an unbalancedtranslocation. The most consistent features of par-tial trisomy 8p are postnatal growth delay, mentalretardation, hypotonia, spastic paraplegia, agene-sis of the corpus callosum, dysmorphic facies(prominent forehead, hypertelorism, saggingcheeks, everted lower lip, and large mouth), highor cleft palate, large ears, and cardiac defects [54].Lung abnormalities (other than one case exhibit-ing anomalous lobation) [55], cystic disease, andtumors (other than one case of congenital neuro-blastoma in situ) [56] have not been described inthis syndrome. This lends support to the observa-tion that the development of PPB may be related
specifically to gains in material from 8q, the longarm of chromosome 8.
Four cases of PPB, including our case 2, haveshown 17p deletion. This suggests a possible roleof p53, a tumor suppressor gene located at 17p13.However, immunostaining for p53, thought to re-flect mutation, was negative in three cases [13].Also of note is that medulloblastoma was describedin one patient 2 years after the diagnosis of PPBand in the uncle of another patient with PPB [21].Medulloblastoma is characterized by isochromo-some 17q [57], occurs in childhood, and may havea genetic relationship to PPB.
In our cases, the FISH observations reflectedthe karyotypic findings. Case 1, which showed onlyone extra copy of chromosome 8 by karyotype,showed fewer nuclear signals than case 2, which hadthree extra copies of chromosome 8 by karyotype. ByFISH, there was no change in signal numbers ob-served before and after chemotherapy, despite theincreased degree of anaplasia and maturation.
Our study underscores the increasing impor-tance of DNA in situ hybridization. Not only can thisnew technique be used as an adjunct in pathologicdiagnosis [58], but because it identifies cytogeneticabnormalities in specific populations of cells, it canfurther our understanding of tumors comprised oftwo or more different cell types [9–11]. In this man-ner, in situ techniques may play a role in the nextgeneration of tumor classification.
In summary, we conclude that gains in chro-mosome 8 are a consistent finding in PPB. Further,our data indicate that clonal proliferation is re-stricted to the mesenchymal elements.
R E F E R E N C E S1. Dehner LP. The evolution of the diagnosis and understand-
ing of primitive and embryonic neoplasms in children:living through an epoch. Mod Pathol 1998;11:669–685.
2. Willis RA. The Borderland of Embryology and Pathology.2nd ed. Washington; DC: Butterworths, 1962; p.422.
3. Manivel JC, Priest JR, Watterson J, et al. Pleuropulmonaryblastoma: the so-called pulmonary blastoma of childhood.Cancer 1988;62:1516–1526.
4. Priest JR, McDermott MB, Bhatia S, Watterson J, ManivelJC, Dehner LP. Pleuropulmonary blastoma: a clinicopath-ologic study of 50 cases. Cancer 1997;80:147–161.
5. Colby TV, Koss MN, Travis WD. Tumors of the LowerRespiratory Tract. Washington, DC: Armed Forces Insti-tute of Pathology, 1995.
6. Dehner LP, Watteron J, Priest J. Pleuropulmonary blasto-ma—a unique intrathoracic-pulmonary neoplasm of child-hood. Perspect Pediatr Pathol 1995;18:214–226.
7. Stocker JT. The respiratory tract. In: Stocker JT, Dehner
CHROMOSOME 8 GAINS IN PPB 443

LP, eds. Pediatric Pathology, Vol. 1. Philadelphia; Lippin-cott, 1992;505–573.
8. Fletcher JA, Kozakewich HP, Hoffer FA, et al. Diagnosticrelevance of clonal cytogenetic aberrations in malignantsoft-tissue tumors. N Engl J Med 1991;324:436–442.
9. Fletcher JA, Pinkus GS, Donovan K, et al. Clonal rear-rangement of chromosome band 6p21 in the mesenchymalcomponent of pulmonary chondroid hamartoma. CancerRes 1992;52:6224–6228.
10. Fletcher JA, Pinkus GS, Weidner N, Morton CC. Lineage-restricted clonality in biphasic solid tumors. Am J Pathol1991;138:1199–1207.
11. Fletcher JA, Pinkus JL, Lage JM, Morton CC, Pinkus GS.Clonal 6p21 rearrangement is restricted to the mesenchy-mal component of an endometrial polyp. Genes Chromo-somes Cancer. 1992;5:260–263.
12. Cohen M, Emms M, Kaschula RO. Childhood pulmonaryblastoma: a pleuropulmonary variant of the adult-typepulmonary blastoma. Pediatr Pathol 1991;11:737–749.
13. Pacinda SJ, Ledet SC, Gondo MM, et al. p53 and MDM2immunostaining in pulmonary blastomas and broncho-genic carcinomas. Hum Pathol 1996;27:542–546.
14. Dehner LP. Pleuropulmonary blastoma is the pulmonaryblastoma of childhood. Semin Diagn Pathol 1994;11:144–151.
15. Hachitanda Y, Aoyama C, Sato J, Shimada H. Pleuropul-monary blastoma in childhood—a tumor of divergent dif-ferentiation. Am J Surg Pathol 1993;17:382–391.
16. Wick MR, Ritter JH, Humphrey PA. Sarcomatoid carcino-mas of the lung: a clinicopathologic review. Am J ClinPathol 1997;108:40–53.
17. Koss MN, Hochholzer L, O’Leary T. Pulmonary blastomas.Cancer 1991;67:2368–2381.
18. Kodama T, Shimosato Y, Watanabe S, Koide T, Naruke T,Shimase J. Six cases of well differentiated adenocarci-noma simulationg fetal lung tubules in pseudoglandularstage: comparison with pulmonary blastoma. Am J SurgPathol 1984;8:735–744.
19. Kradin RL, Young RH, Dickersin GR, Kirkham SE, MarkEJ. Pulmonary blastoma with argyrophil cells and lackingsarcomatous features (pulmonary endodermal tumor re-sembling fetal lung). Am J Surg Pathol 1982;6:165–172.
20. Singh SP, Besner GE, Schauer GM. Pulmonary endoder-mal tumor resembling fetal lung: report of a case in a14-year-old girl. Pediatr Pathol Lab Med 1997;17:951–958.
21. Priest JR, Watteron J, Strong L, et al. Pleuropulmonaryblastoma: a marker for familial disease. J Pediatr 1996;128:220–224.
22. Francis D, Jacobsen M. Pulmonary blastoma. Curr TopPathol 1983;73:265–294.
23. Yousem SA, Wick MR, Randhawa P, Manivel JC. Pulmo-nary blastoma. An immunohistochemical analysis withcomparison with fetal lung in its pseudoglandular stage.Am J Clin Pathol 1990;93:167–175.
24. Hill DA, Sadeghi S, Schultz MZ, Burr JS, Dehner LP.Pleuropulmonary blastoma in an adult: an initial casereport. Cancer 1999;85:2368–2374.
25. Hogan BL, Yingling JM. Epithelial/mesenchymal interac-tions and branching morphogenesis of the lung. Curr OpinGenet Dev 1998;8:481–486.
26. Warburton D, Lee MK. Current concepts on lung develop-ment. Curr Opin Pediatr 1999;11:188–192.
27. Stocker JT, Madewell JE, Drake RM. Congenital cysticadenomatoid malformation of the lung: classification andmorphologic spectrum. Hum Pathol 1977;8:155–171.
28. Benjamin DR, Cahill JL. Bronchioloalveolar carcinoma ofthe lung and congenital cystic adenomatoid malformation.Am J Clin Pathol 1991;95:889–892.
29. Donnelly LF, Frush DP. Localized radiolucent chest le-sions in neonates: causes and differentiation. AJR Am JRoentgenol 1999;172:1651–1658.
30. Granata C, Gambini C, Balducci T, et al. Bronchioloalveo-lar carcinoma arising in a congenital cystic adenomatoidmalformation in a child: a case report and review onmalignancies originating in congenital cystic adenomatoidmalformation. Pediatr Pulmonol 1998;25:62–66.
31. Murphy JJ, Blair GK, Fraser GC, et al. Rhabdomyosar-coma arising within congenital pulmonary cysts: report ofthree cases. J Pediatr Surg 1992;27:1364–1367.
32. Allan BT, Day DL, Dehner LP. Primary pulmonary rhab-domyosarcoma of the lung in children: report of two casespresenting with spontaneous pneumothorax. Cancer 1987;59:1005–1011.
33. Krous HF, Sexauer CL. Embryonal rhabdomyosarcomaarising within a congenital bronchogenic cyst in a child.J Pediatr Surg 1981;16:506–508.
34. Shariff S, Thomas JA, Shetty N, D’Cunha S. Primary pul-monary rhabdomyosarcoma in a child, with a review ofliterature. J Surg Oncol 1988;38:261–264.
35. Ueda K, Gruppo R, Unger F, Martin L, Bove K. Rhabdo-myosarcoma of the lung arising in a congenital cysticadenomatoid malformation. Cancer 1977;40:383–388.
36. Lee SH, Rengachary SS, Paramesh J. Primary pulmonaryrhabdomyosarcoma: a case report and review of the liter-ature. Hum Pathol 1981;12:92–96.
37. Sciavetti A, Dominici C, Matrunola M, Capocaccia P, Cec-camea A, Castello MA. Primary pulmonary rhabdomyosar-coma in childhood: clinico-biologic features in two caseswith review of the literature. Med Pediatr Oncol 1996;26:201–207.
38. Delahunt B, Thomson KJ, Ferguson AF, Neale TJ, MeffanPJ, Nacey JN. Familial cystic nephroma and pleuropulmo-nary blastoma. Cancer 1993;71:1338–1342.
39. Ishida Y, Kato K, Kigasawa H, Ohama Y, Ijiri R, Tanaka Y.Synchronous occurrence of pleuropulmonary blastomaand cystic nephroma: possible genetic link in cystic lesionsof the lung and the kidney. Med Pediatr Oncol 2000;35:85–87.
40. Sciot R, Dal Cin P, Brock P, et al. Pleuropulmonary blas-toma (pulmonary blastoma of childhood): genetic linkwith other embryonal malignancies? Histopathology 1994;24:559–563.
41. Yang P, Hasegawa T, Hirose T, et al. Pleuropulmonaryblastoma: fluorescence in situ hybridization analysis indi-cating trisomy 2. Am J Surg Pathol 1997;21:854–859.
42. Novak R, Dasu S, Agamanolis D, Herold W, Malone J,Waterson J. Trisomy 8 is a characteristic finding in pleu-ropulmonary blastoma. Pediatr Pathol Lab Med 1997;17:99–103.
43. Barnard M, Bayani J, Grant R, Teshima I, Thorner P,Squire J. Use of multicolor spectral karyotyping in geneticanalysis of pleuropulmonary blastoma. Pediatr Dev Pathol2000;3:479–486.
44. Kelsey AM, McNally K, Birch J, Mitchell EL. Case of extrapulmonary, pleuro-pulmonary blastoma in a child: patho-logical and cytogenetic findings. Med Pediatr Oncol 1997;29:61–64.
45. Richardson MS, Hazen-Martin D, Cantu RE, Garvin AJ.Pleuropulmonary blastoma: characterization of a pediat-ric tumor and its resulting tumor cell line [abstract]. ModPathol 1995;8:144A.
46. Afify A, Mark HF. Trisomy 8 in embryonal rhabdomyosar-coma detected by fluorescence in situ hybridization. Can-cer Genet Cytogenet 1999;108:127–132.
47. Dietrich CU, Jacobsen BB, Starklint H, Heim S. Clonalkaryotypic evolution in an embryonal rhabdomyosarcoma
444 S.O. VARGAS ET AL.

with trisomy 8 as the primary chromosomal abnormality.Genes Chromosomes Cancer 1993;7:240–244.
48. Schofield DE, Yunis EJ, Fletcher JA. Chromosome aber-rations in mesoblastic nephroma. Am J Pathol 1993;143:714–724.
49. Schofield DE, Fletcher JA, Grier HE, Yunis EJ. Fibrosar-coma in infants and children: application of new tech-niques. Am J Surg Pathol 1994;18:14–24.
50. Maurici D, Perez-Atayde A, Grier HE, Baldini N, Serra M,Fletcher JA. Frequency and implications of chromosome 8and 12 gains in Ewing sarcoma. Cancer Genet Cytogenet1998;100:106–110.
51. Sreekantaiah C, Karakousis CP, Leong SP, Sandberg AA.Trisomy 8 as a nonrandom secondary change in myxoidliposarcoma. Cancer Genet Cytogenet 1991;51:195–205.
52. Fletcher JA, Naeem R, Xiao S, Corson JM. Chromosomeaberrations in desmoid tumors: trisomy 8 may be a predictorof recurrence. Cancer Genet Cytogenet 1995;79:139–143.
53. Jones KL. Smith’s Recognizable Patterns of Human Malfor-mation, 5th ed. Philadelphia: W. B. Saunders, 1997;24–27.
54. Plomp AS, Engelen JJ, Albrechts JC, de Die-SmuldersCE, Hamers AJ. Two cases of partial trisomy 8p andpartial monosomy 21q in a family with a reciprocaltranslocation (8;21)(p21.1;q22.3). J Med Genet 1998;35:604 – 608.
55. Pezzolo A, Biococchi MP, Zampatti C, Cuoco C, Gimelli G.Prenatal diagnosis of a partial 8p trisomy. Prenat Diagn1990;10:533–538.
56. Fryns JP, Petit P, Moerman F, Cassiman JJ, van den BergeH. 8p trisomy in a malformed fetus. Ann Genet 1982;25:162–163.
57. Giordana MT, Mighelli A, Pavanelli E. Isochromosome17q is a constant finding in medulloblastoma. An inter-phase cytogenetic study on tissue sections. NeuropatholAppl Neurobiol 1998;24:233–238.
58. Fletcher JA. DNA in situ hybridization as an adjunct intumor diagnosis. Am J Clin Pathol 1999;112:S11–S18.
CHROMOSOME 8 GAINS IN PPB 445