functional analyses of human apolipoprotein cii by site ... · abbreviations used: apocii,...
TRANSCRIPT

Running title: Apolipoprotein CII mutants
1
Functional analyses of human apolipoprotein CII by site-directed
mutagenesis. Identification of residues important for activation of
lipoprotein lipase*
Yan Shen§, #, Aivar Lookene§, ¶, Solveig Nilsson§ and Gunilla Olivecrona§, **
From the §Department of Medical Biosciences, Umeå University, SE-90 187 Umeå,
Sweden and ¶ National Institute of Chemical Physics and Biophysics, Tallinn, Estonia
*This work was supported by the Swedish Medical Research Council (727 and 12203),
by the Swedish Royal Academy of Sciences, by the Estonian Science Foundation (4925)
and by Donation Funds at the Medical Faculty, Umeå University
#Present address: Cardiovascular Research Center, Cardiology Division, Massachusetts
General Hospital, Harvard Medical School, Boston, Massachusetts, USA
Additional key words: protein interaction, lipid interaction, kinetics, liposomes, mutants,
lipase activation
Abbreviations used: ApoCII, apolipoprotein CII; ApoAI, apolipoprotein AI; CD, circular
dichroism; GdmCl, guanidinium chloride; IPTG, isopropyl-β-D-thiogalactoside; LCAT,
lecithin:cholesterol acyl transferase; LPL, lipoprotein lipase; PCR, polymerase chain
reaction
**To whom correspondence should be addressed: Prof. Gunilla Olivecrona
Tel. +46-90-7867762, Fax. +46-90-7867840, E-mail.
Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on November 21, 2001 as Manuscript M105421200 by guest on June 8, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Running title: Apolipoprotein CII mutants
2
SUMMARY
Apolipoprotein CII (apoCII) activates lipoprotein lipase (LPL). Seven residues, located on
one face of a model α-helix spanning residues 59-75, are fully conserved in apoCII from
ten different animal species. We have mutated these residues one by one. Substitution of
Ala59 by glycine, or Thr62 and Gly65 by alanine did not change the activation, indicating
that these residues are outside the LPL-binding site. Replacement of Tyr63, Ile66, Asp69
or Gln70 by alanine lowered the affinity for LPL and the catalytic activity of the
LPL/apoCII complex. For each residue several additional replacements were made. Most
mutants retained some activating ability, but replacement of Tyr63 by phenylalanine or
tryptophan and Gln70 by glutamate caused almost complete loss of activity. All mutants
bound to liposomes with similar affinity as wild-type apoCII, and they also bound with
similar affinity to LPL in the absence of hydrolyzable lipids. However, the inactive
mutants did not compete with wild-type apoCII in the activation assay. Therefore we
conclude that the productive apoCII/LPL interaction may be dependent on substrate
molecules. In summary, our data demonstrate that residues 63, 66, 69 and 70 are of
special importance for the function of apoCII, but no single amino acid residue is
absolutely crucial.
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
3
INTRODUCTION
Human apolipoprotein CII (apoCII) is a 79-residue protein synthesized in the intestine
and liver and secreted as a surface component of chylomicrons, very low density
lipoproteins and high-density lipoproteins (1-3). ApoCII plays an important role in plasma
lipid metabolism as an activator of lipoprotein lipase (LPL) (1,4), clearly illustrated by the
severe hypertriglyceridemia that is associated with genetic deficiency of apoCII (5). A
number of natural mutations affecting the apoCII gene have been identified and
characterized (6). Most mutations reported so far result in either very low levels of
plasma apoCII or major structural changes in the protein, e.g. prematurely truncated
forms. A Trp26Arg mutation was recently reported to lead to apoCII deficiency, probably
because of a disturbed lipid interaction of the activator (7). This is the only example of a
single point mutation in apoCII leading to loss of function, but the interpretation is not
straightforward since the protein was not detectable in plasma.
The structure of full-length human apoCII in complex with SDS-micelles was recently
reported based on studies by NMR (8). The protein contains three regions with helical
conformation spanning residues 16-36, 50-56 and 63-77. The lipase-activating region of
apoCII has previously been localized to the C-terminal one third of the sequence, from
about residue 56, while the N-terminal two thirds of the sequence is involved in lipid
binding (9-11). In most studies it has been found that apoCII causes an increase in the
catalytic efficiency of LPL, and not an enhanced lipid affinity of the enzyme (10, 12). In
spite of a long history of studies on the effects of apoCII, the detailed mechanism for the
activation remains unknown. It has been proposed that apoCII binds to LPL and induces
conformational changes in the lipase, or changes its orientation at the lipid-water
interface. As a result of these changes, LPL may become more efficient in binding of
individual substrate molecules or in release of lipolysis products (fatty acids) (13). It has
also been proposed that apoCII, as an amphipathic molecule, may influence the structural
organization of lipids such that substrate molecules become more available to the active
site of LPL (8, 12). However, synthetic peptides corresponding to the C-terminal part,
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
4
which does not bind to lipid, are able to activate LPL in vitro (14). The strongest evidence
for activation of LPL via formation a complex with apoCII come from data showing that
LPL and apoCII bind to each other even in the absence of a lipid/water interface (15).
In the present study we have focused on the region comprising residues 56-79 in human
apoCII. Compared to the corresponding region in apoCII from 9 other animal species,
there are only 7 fully conserved residues: Ala59, Thr62, Tyr63, Gly65, Ile66, Asp69 and
Gln70 (16). In the rest of the apoCII molecule there are only two other fully conserved
residues, Leu15 and Ala33, but both are outside the activating region. We have
systematically evaluated the functional role of each of the seven conserved residues by
using site-directed mutagenesis, followed by analyses of the secondary structure by
circular dichroism (CD) measurements and analyses of activating ability. We have also
directly studied the interaction of the mutants with LPL using energy transfer
measurements. The results suggest that the LPL-binding site in apoCII consists of mainly
4 residues located on one face of the third α-helix located closest to the C-terminus.
EXPERIMENTAL PROCEDURES
Materials
Restriction endonucleases were from Amersham Phamacia Biotech. T4 ligase was from
Boehringer Mannheim. Kanamycin was from Duchefa, Haarle, The Netherlands.
Isopropyl-β-D-thiogalactoside (IPTG), dansyl chloride and egg yolk phosphatidylcholine
were from Sigma, St. Louis, MO. Low-melting agarose and polyvinylidene difluoride
filters PVDF (0.2 µm) were from BioRad, Hercules, CA. JET sorb Gel extraction kit was
from Genomed. The expression vector pET-29a, S-protein agarose and BL21(DE3)
competent cells were from Novagen, Inc., Madison, WI. Epicurian Coli BL21-
codonPlus(DE3)-RIL competent Cells and QuikChangeTM Site-Directed Mutagenesis Kit
were from Stratagene, La Jolla, CA. Penta His-tag antibody and Ni2+-NTA agarose were
purchased from QIAGEN GmbH, Hilden, Germany. Factor Xa from bovine plasma was
from Biolads, Beverly, MA. Serva Blue G was from Serva, GmbH, Heidelberg, Germany.
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
5
Transfer Membrane was from Pall Corporation, Ann Arbor, MI. Dialysis tubes were from
spectrum Medical Industries, Houston, TX. Low molecular weight marker was from
Amersham, Buckinghamshire, UK. For the analyses of LPL activity an emulsion with a
composition corresponding to Intralipid 10 % (soy bean triacylglycerols emulsified in
egg yolk phospholipids), but containing in addition a trace amount of 3H-oleic acid-
labeled trioleoylglycerol (kindly prepared by KABI-Fresenius, Uppsala, Sweden) was
used. BCA protein Assay was from Pierce, Rockford, IL. LPL was purified from bovine
milk as described (17). The stock solutions contained about 0.5 mg LPL/ml 10 mM
Bistris, pH 6.5, 1M NaCl.1,2-Di-O-hexadecyl-sn-Glycero-3-phosphatidylcholine was
from Larodan Fine Chemicals AB, Malmö, Sweden. Oligonucleotides were synthesized
by Pharmacia Biotech, Uppsala, Sweden. An antiserum against human apoCII, purified
from plasma and coupled as hapten to keyhole limpet hemocyanin, was produced in a
rabbit by a standard immunization procedure (18). Computer-assisted analyses were
accomplished with the programs Lasergene DNA Star Inc., Madison, WI and Wisconsin
sequence analysis package, Genetics Computer Group (GCG). For secondary structure
prediction of the mutants the programs at the NPS@(dNetwork Protein Sequence
analysis) web sites were used (http://pbil.icbp.fr/cgibin/npsa_automat.
pl?page=/NPSA7npsa_seccons.html). Binding and activation curves were fitted using the
program Fig. P (BIOSOFT, Cambridge, UK).
Plasmid constructions
Two plasmids were used for expression of recombinant human apoCII: pET29a-hapoCII
and pET-histag-hapoCII.
cDNA for human apolipoprotein CII in the pUC18 vector (hapoCII, a kind gift from Dr
Steven Humphries, London) was connected to an EcoRI linker coding for the cleavage
site for Factor Xa (coding sequence for factor Xa cleavage: Ile-Glu-Gly-Arg-) attached to
the first codon of the mature apoCII cDNA sequence. The whole DNA fragment was
cloned into the EcoRI/XhoI site of the vector pET29a(+) to form the expression plasmid
pET29a-hapoCII. The recombinant was verified by EcoRI/XhoI cleavage and DNA
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
6
sequencing. The plasmid was transformed into E. coli BL21(DE3) to express the fusion
protein S-tag-“factor Xa cleavage site”-hapoCII (117 amino acid residues in total). For
production of pET-histag-hapoCII, the pET29a-hapoCII plasmid was cleaved with NdeI
and EcoRI to delete the sequence encoding the S-tag and the thrombin cleavage site. The
complementary oligonucleotides 5’ TATGCACCATCATCATCATCATG 3’ and 5’
AATTCATGATGATGATGATGGTGCA 3’, coding for an N-terminal 6xHis-tag, were
ligated to the cleaved pET29a vector. The new construct resulted in a vector with the T7
RNA polymerase promoter and the lac operator followed by sequences coding for 6xHis-
tag, Factor Xa cleavage site and human apoCII.
Site-directed mutagenesis of human apoCII
Mutant 63A was generated by overlap extension by using the polymerase chain reaction
(PCR) and four different primers (Table 1). All other mutants were generated using
QuikchangeTM Site- Directed Mutagenesis Kit. The basic procedure for generation of
each mutant utilizes a template vector with apoCII cDNA (pET29a-hapoCII or pET-
histag-hapoCII) and two synthetic oligonucleotide primers containing the desired
mutation (Table 1). The oligonucleotide primers, each complementary to opposite strands
of the vector, were extended by Pfu Turbo DNA polymerase. All mutants were sequenced
using ABI 337 DNA sequencer, Dye Terminator Cycle Sequencing Ready Reaction Kit,
Perkin Elmer.
Expression and purification of wild-type and mutated human apoCII proteins
pET29-hapoCII and its mutants (A59G, T62A, Y63A, G65A, I66A, D69A and Q70A)
were introduced into BL21(DE3) competent cell. Expression, purification and cleavage of
the fusion proteins by factor Xa were made as described (Martinez et al., manuscript in
preparation), but in LB medium.
pET-histag-hapoCII ant its mutants (Y63A, Y63T, Y63L, Y63F, Y63W, I66A, I66L,
I66V, I66F, D69A, D69S, D69N, D69E, Q70A, Q70A, Q70N, Q70E, L72A, S73A and
V74A) were transformed into Epicuran Coli BL21-CodonPlus(DE3)-RIL competent
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
7
cell. The bacteria were grown at 37°C in Terrific Broth containing kanamycin (50 µg/ml)
to an OD600nm of 1.0. At this point the temperature was lowered to 16°C and one hour
later expression of human apoCII and its variants was induced with 0.5 mM IPTG. On the
next day, the bacteria were harvested by centrifugation and purified by Ni2+-NTA agarose
as described (19). Briefly, buffers containing 8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl
were used to produce a stepwise gradient from pH 8.0 to pH 4.5. The fusion proteins that
eluted at pH 7.2 were dialyzed for 24 hours at 4°C against ammonium bicarbonate and
were then lyophilized.
High performance liquid chromatography of wild-type and mutated His-tagged
human apoCII proteins
High performance ion exchange was performed on an ÄKTA purifier 900 (Amersham-
Pharmacia) using a Mini Q PE 4.6/50 column. The fusion proteins eluted between pH 6.8
and pH 4.5 from Ni2+-NTA agarose were dialyzed against 6 M urea, 0.01 M Tris pH 8.2
and were filtered through a 0.2 µm filter (Gelman Sciences, 600 S, Wagner Rd, Ann
Arbor, MI 48103) before they were applied to the column at a pressure of 5 MPa. Proteins
were then eluted by a linear gradient of Tris-Cl (pH 8.2) from 0.01 to 0.15 M. Remaining
proteins were washed out by 1M NaCl and/or 6 M guanidinium chloride (GdmCl).
Protein concentrations in all the fractions were measured by the BCA assay. Fractions
were combined and the proteins were analyzed by electrophoresis on SDS-Tricine gels
and by N-terminal amino acid sequence analyses as previously described (16). Two forms
of apoCII were detected by matrix-assisted laser desorption/ionization time-of-flight mass
spectrometry (MALDI-TOF MS) on a homemade instrument at National Institute of
Chemical Physics and Biophysics, Tallinn, Estonia.
Proteins analyzed by Tricine SDS-PAGE and Western blotting
The expressed proteins were separated on Tricine -SDS-PAGE and were stained by
0.025% Serva-blue G in 10% acetic acid (19). The separated proteins were transferred
onto a nitrocellulose filter. The filter was put in TBST (10 mM Tris-Cl, pH 8.0, 0.5 M
NaCl and 0.05% Tween-20) containing 5% bovine serum albumin and 3% gelatin for 30
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
8
minutes at 42°C. Human apoCII and mutants were detected by incubation with the rabbit
anti-human apoCII serum (dilution 1:100). After incubation overnight the filter was
washed 3 x 10 min in TBST. Then the filter was incubated with goat anti-rabbit IgG
(conjugated to alkaline phosphatase) diluted 1:3000 in TBST. Finally the filter was
washed 3 x 10 min as before. Bound antibodies were detected by addition of substrate to
alkaline phosphatase and the reaction was stopped by rinsing the filter in H2O. For
blotting with the penta His-tag antibody, the nitrocellulose filter was handled as
recommended by the manufacturer.
Circular dichroism
Possible global changes of the secondary structure of some of the mutated apoCIIs were
investigated by measurements of far UV spectra at 25 °C on a CD6 spectrodichrograph
(Jobin-Yvon Instruments SA, Longjumeau, France) in a 0.5 mm path-length cell. For this
the protein concentrations were determined by absorbancy at 280 nm.
Lipase assays
For measurement of LPL activity we used an assay mixture of 200 µl containing 2 mg of
triacylglycerols from the lipid emulsion. The medium contained 0.1 M NaCl, 0.1 M Tris-
Cl, 20 µg heparin, and 12 mg of bovine serum albumin. The pH was 8.5. To study the
activation, wild-type apoCII and its mutants were dissolved in 5 M urea, 10 mM Tris-Cl
(pH 8.2). The protein concentration was in each case determined by the BCA protein
assay. Five µl of the stock solution of apoCII, or the same volume of dilutions in 5 M
urea, 10 mM Tris (pH 8.2), was added to the incubations. The reactions were started by
addition of LPL and were stopped after incubation for 15 min at 25 º C by addition of
organic solvents for extraction of the labeled free fatty acids (19). The lipase activity is
expressed in U/mg of LPL, where 1 U corresponds to release of 1 µmol fatty acid per
min. For studies of competition between the wild-type apoCII and mutants for activation
of LPL the experiments were carried out as described, except for the presence of 2.5 µM
Q70E, Y63F or Y63W in the incubation mixture together with wild-type apoCII.
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
9
Determination of kinetic parameters from activation curves
Quinn et al. have shown that the activation of LPL by apoCII can be described by a
random mechanism (20). Based on this mechanism, the velocity of the reaction (v) at
substrate saturation is expressed as
++⋅⋅= ⋅
d
dcat
KC
KC�Lk v
Where L corresponds to LPL and C corresponds to apo CII.
This equation was used for determination of Kd and β. Kd characterizes the apparent
affinity of apoCII for LPL, and β indicates how much more active the LPL/apoCII
complex was compared to LPL alone (fold activation). When β is equal to 1, apoCII does
not activate LPL. The kinetic constants were determined by using the program Fig. P.
Preparation of liposomes
Two different phospholipids were used in this study. One was 1,2-di-O-hexadecyl-sn-
glycero-3-phosphatidylcholine, which cannot be hydrolyzed by LPL. This ether lipid was
used for studies of the apoCII-LPL interaction. The other was egg yolk
phosphatidylcholine, used for studies of the interaction between apoCII and lipids. For
preparation of liposomes the lipids were first dissolved in chloroform and dried under
nitrogen. The dry lipid was then dispersed in 5 mM Tris-Cl, 0.15 M NaCl, 0.02% NaN3,
pH 7.4 and sonicated without break in ice-cold buffer by an MSE Soniprep 150 equipped
with a 5 mm probe. Egg yolk phosphatidylcholine was sonicated for 30 minutes and 1,2-
di-O-hexadecyl-sn-glycero-3-phosphatidylcholine was sonicated for 1 hr. The liposomes
were kept for 2-3 hours at 37 °C before they were centrifuged for 10 min at 3000 rpm at 4
°C. The supernatant was collected and dialyzed against 0.1 M Tris-Cl (pH 8.1). Any
possible large aggregates were removed by filtration through 0.2 µM filter. The
concentrations of phospholipids in the final preparations were measured using an
enzymatic kit from Wako Chemical GmbH, Germany.
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
10
Determination of equilibrium dissociation constants by dansylation of apoCII
The wild-type apoCII and the mutants (His-tagged, not cleaved) were dansylated
according to the procedure described in a previous study (19). The relative increase of
fluorescence at 490 nm on excitation at 280 nm was measured after each addition of
bovine LPL to the solution of dansylated apoCII in 0.1 M Tris-Cl (pH 8.1) with 10 µl 1,2-
di-O-hexadecyl-sn-glycero-3-phosphatidylcholine liposomes (final concentration 10 µg
/ml). For the measurements a Spex FluoroMax-2 fluorometer or a Shimadzu
spectofluorophotometer model RF500 were used. The fluorescence background of apoCII
was subtracted from the experimental values. Binding of wild-type apoCII to LPL was
also studied in the absence of liposomes.
Binding of apoCII to liposomes
Binding of apoCII to liposomes was measured by using the change of fluorescence of the
single tryptophan at residue 26 as described in our previous study (19). The increase of
fluorescence was attributed to binding of apoCII to the liposomes.
RESULTS
Expression and purification of apoCII and mutants
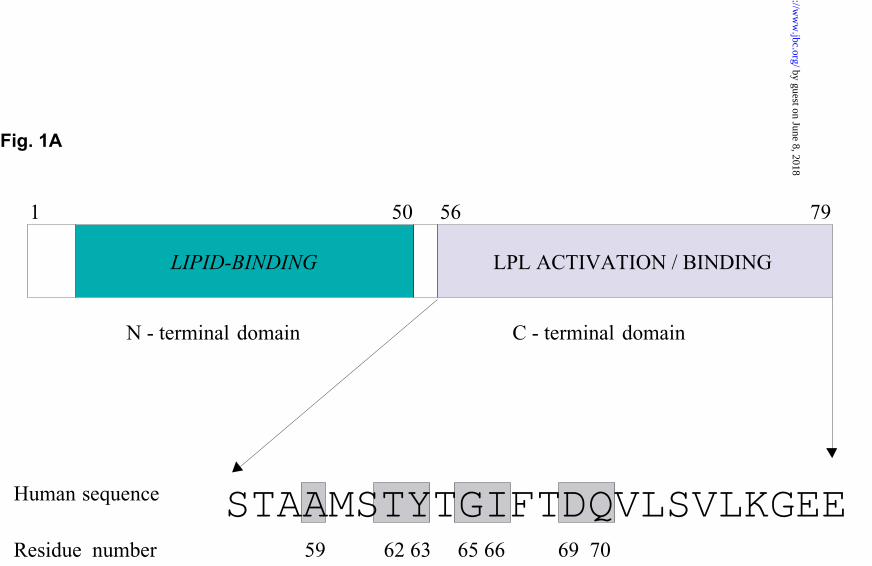
Comparison of the amino acid sequences of apoCII from the 10 different animal species
shows that there are seven fully conserved residues in the C-terminal region (Fig. 1A). All
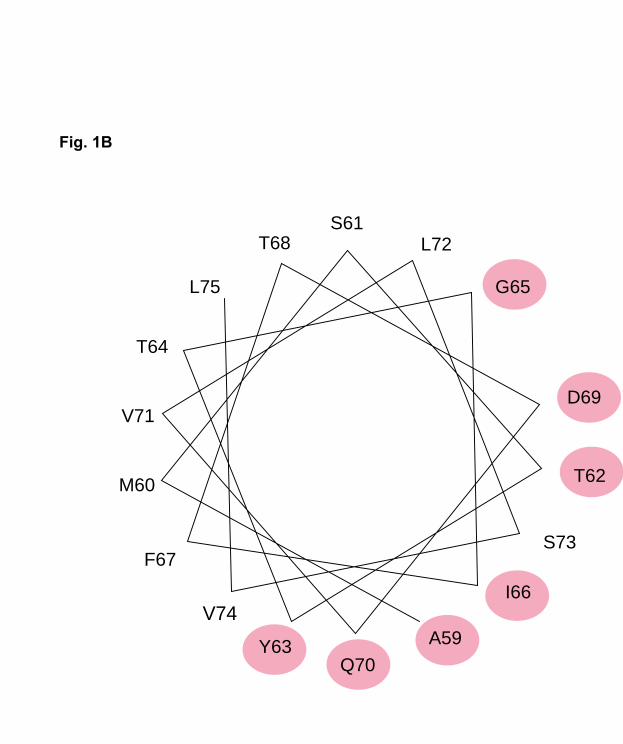
were located on the same side of a model α-helix spanning residue 59 to 75 (Fig. 1B). To
study the role of the conserved residues for the function of apoCII we first made
substitutions with alanine using the codons GCC, GCA and GCT. Ala59 was instead
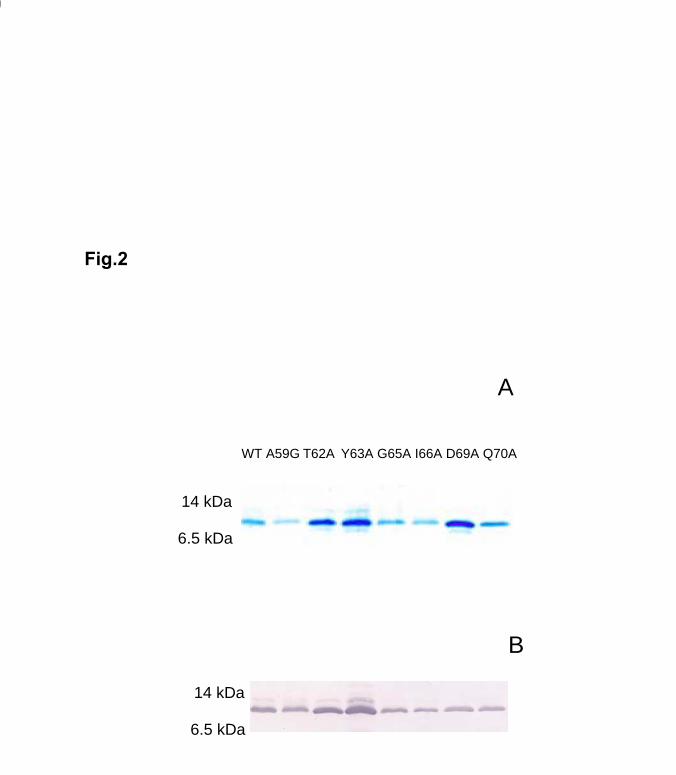
substituted with glycine. The seven mutants and the wild-type apoCII were expressed
using the pET29a-hapoCII vector, generating S-tagged apoCII proteins that were purified
on S-protein agarose and cleaved by factor Xa (Fig. 2).
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
11
In order to identify critical chemical groups or side chains, more mutants were generated
at residues 63, 66, 69 and 70. These mutants were expressed using the pET-histag-
hapoCII vector and purified on Ni2+-NTA-agarose. When the pH of the elution buffer was
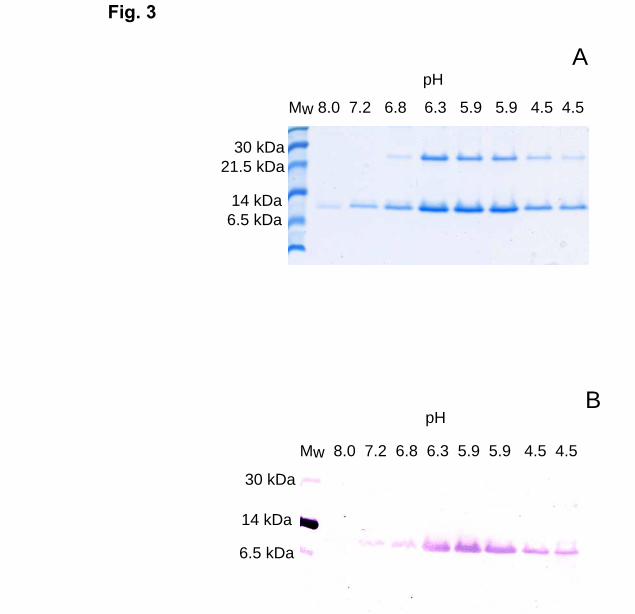
decreased to 6.8 or lower, a protein with an apparent molecular weight of 26 kDa was
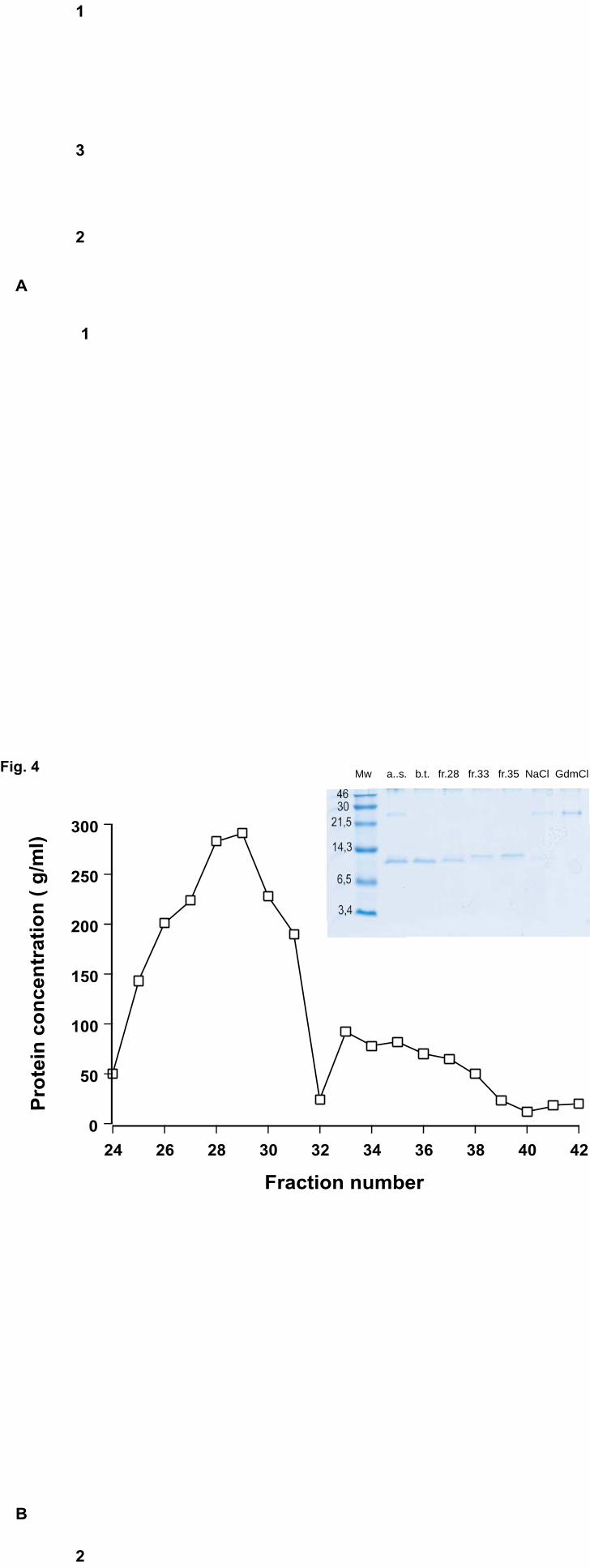
eluted in addition to apoCII (Fig. 3). For separation of this protein from apoCII, ion-
exchange chromatography on a Mini Q column was used (Fig. 4). The contaminating
protein bound stronger than apoCII to the column and was eluted by 1 M NaCl and/or 6M
GdmCl (Fig. 4, inset). N-terminal sequence analyses of the contaminant gave the
sequence KVAKDLVVS, which was found to correspond to an E. Coli protein consisting
of a domain homologous to FK506-binding proteins (21). This protein is rich in
potentially metal-binding amino acids, such as histidine, cysteine, and acidic amino acids,
explaining why it bound very strongly to the Ni2+-NTA agarose. ApoCII was separated
into two forms by the ion-exchange column (Fig. 4, inset). N-terminal amino acid
sequence analysis showed that they both had the same sequence, MHHHHH. Mass
spectrometry revealed that there was a 260 dalton difference between the two forms. This
corresponded to a lack of the last two glutamate residues at the C-terminal end (the
theoretical difference is 258.2 dalton). The expressed wild-type apoCII contained more
than 75% of the short form. Since the two forms of apoCII showed similar ability to
activate LPL (data not shown), we used a mixture of the two forms of each expressed
mutant for the following experiments. In another set of experiments we compared in
detail the ability of His-tagged apoCII with apoCII purified from human plasma with
regard to actvation of LPL. From these data we know that the His-tag at the N-terminus
does not interfere with the activation (Shen Y, Lookene A, Vija H, Olivecrona G, data
unpublished). Therefore we decided to use the His-tagged recombinant proteins without
further cleavage for the present functional studies.
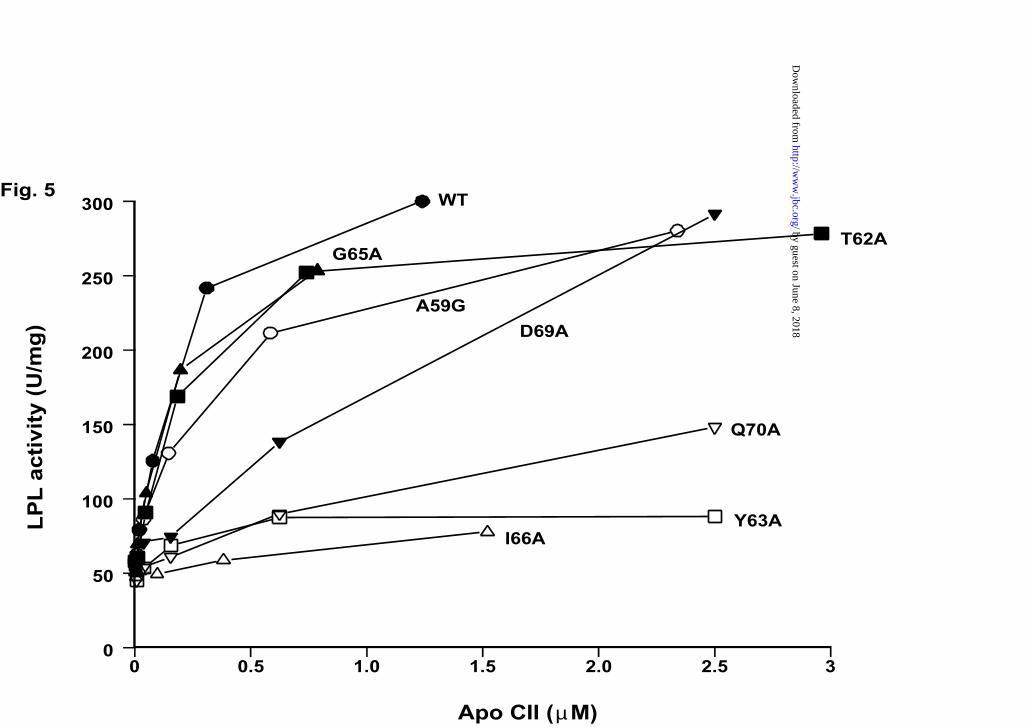
Activation of LPL by apoCII mutants
Substitution of Ala59 by glycine and residues Thr62, Gly65 by alanine did not have much
effect on the activating ability of apoCII (Fig. 5). In contrast, the mutants with alanine
substitutions at position 63, 66, 69 and 70 showed a defective function (Fig. 5). For
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
12
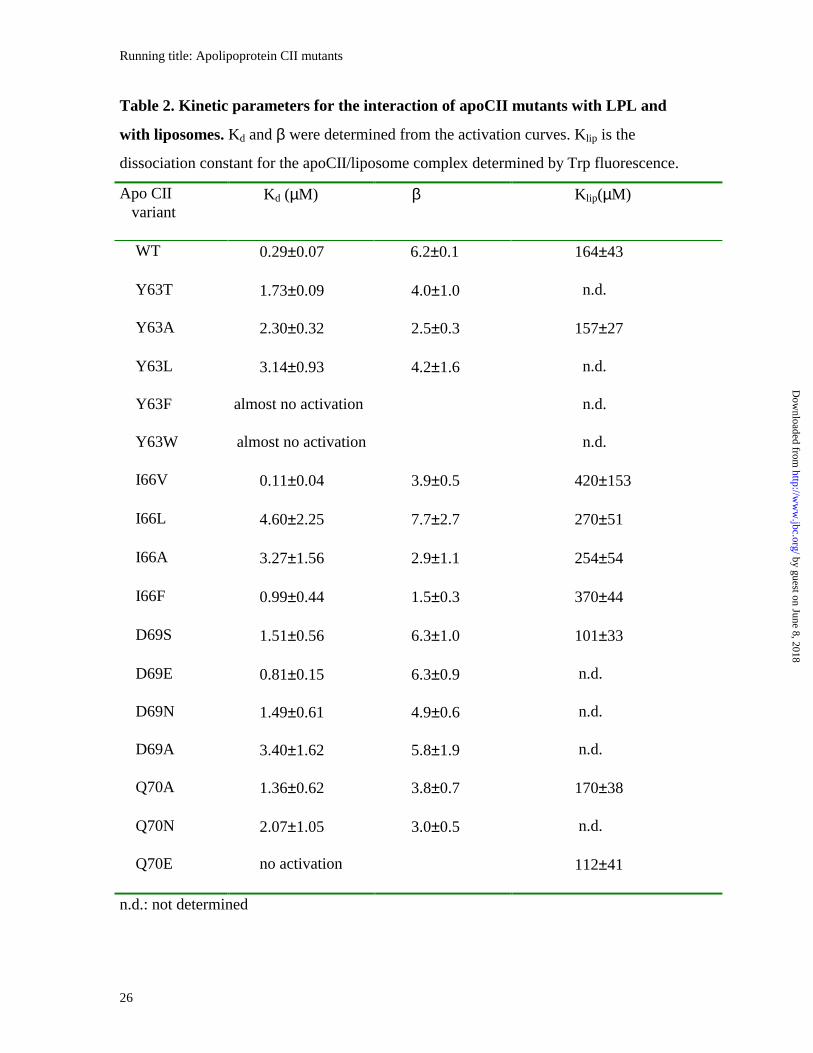
quantitative comparisons, the kinetic parameters Kd and β were determined from the
activation curves. Under our experimental conditions the parameters for wild-type apoCII
were Kd = 0.29 ± 0.07 µM and β = 6.2 ± 0.1. Corresponding data for all mutants are
shown in Table 2.
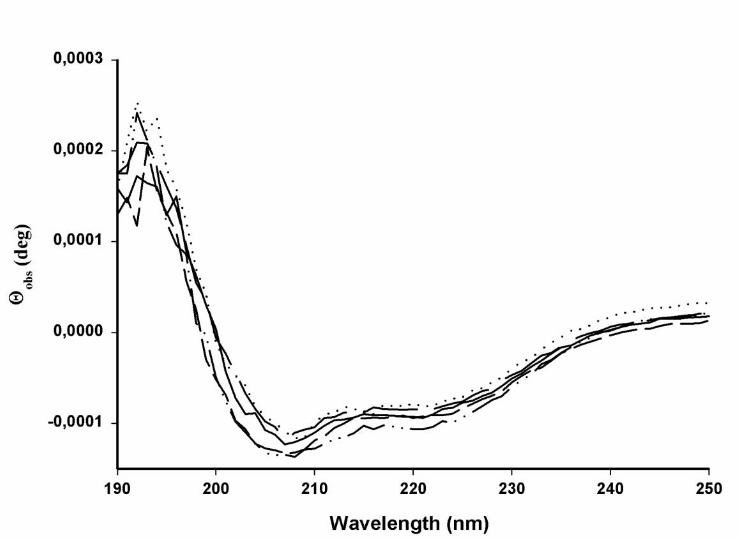
To investigate whether the markedly reduced activation ability of some of the mutants
was due to a major change of secondary structure, mutants Y63A, I66A, D69A and Q70A
were analyzed by CD measurements in the presence of liposomes of phosphatidylcholine
(Fig. 6). No major differences were found compared to the spectrum for wild-type apoCII.
All mutants retained the typical, largely helical, structure represented by double minima at
208 and 222 nm.
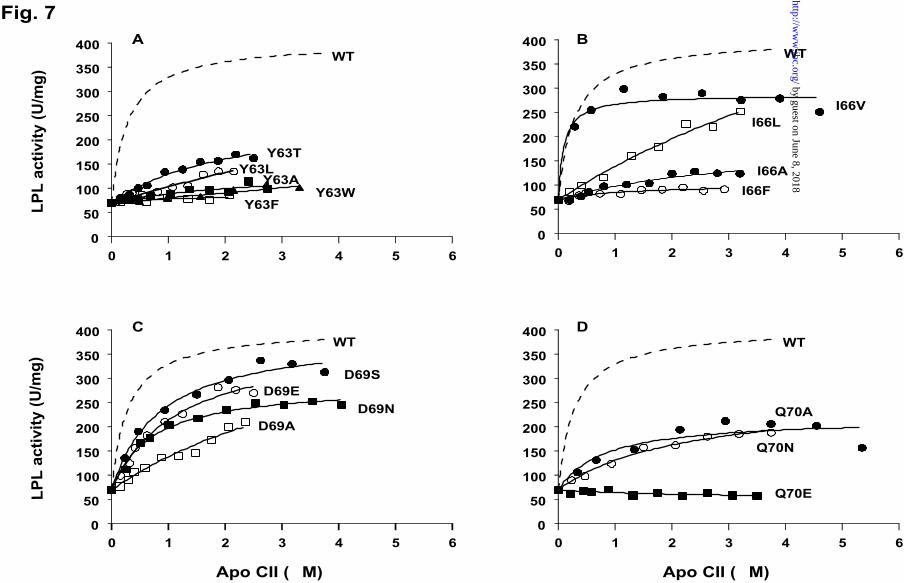
In addition to the alanine substitutions mentioned above, Tyr63 was replaced by threonine
(Y63T), leucine (Y63L), phenylalanine (Y63F) and tryptophan (Y63W) to investigate the
importance of the hydroxyl group and/or the aromatic ring (Fig. 7A). The Y63F and
Y63W mutants showed very low, though detectable, activation. Since the activation
curves remained linear even at the maximally achievable apoCII concentrations, it was
not possible to calculate Kd and β for these mutants. To elucidate whether the poor
activation was caused by low affinity of apoCII for LPL, or by low activity of the mutant
apoCII/LPL complexes, the mutants Y63W and Y63F were mixed with wild-type apoCII
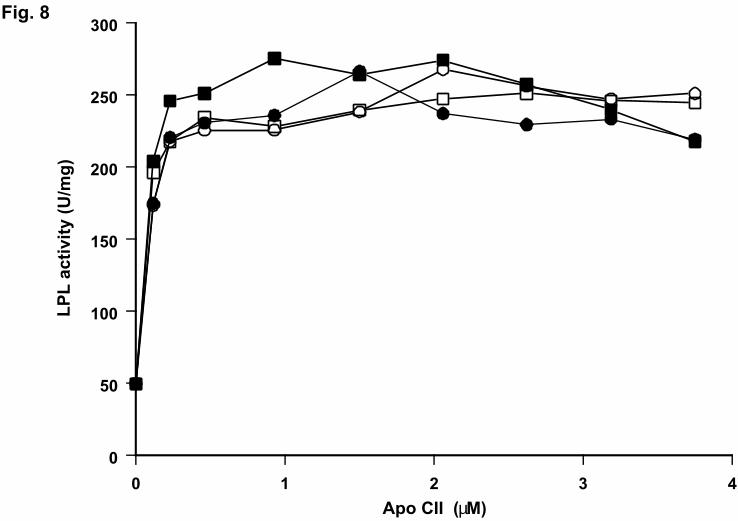
in the same assay (Fig. 8). The activity of wild-type apoCII was not affected even at a 20-
fold excess of the inactive mutants. Therefore we concluded that the low activation by
mutants Y63F and Y63W was mainly due to a reduced affinity for LPL. In contrast, the
Y63T and the Y63L mutants were more efficient that Y63A (Fig. 7A). The Y63T bound
to LPL with Kd = 1.73 µM and enhanced the catalytic activity four-fold (β= 4.0, Table 2).
Corresponding values for the Y63L mutant were 3.14 µM and 4.2.
Substitution of Ile66 by leucine (I66L), alanine (I66A) or phenylalanine (I66F) caused
significant changes in the activation properties of apoCII (Fig. 7B). The activation curve
for I66L remained close to linear up to 3 µM. Therefore the Kd and β values for this
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
13
mutant were determined with large deviation (Table 2). Also I66A exhibited low affinity
Kd=3.27 µM, and the maximal activation was about 3-fold (β= 2.9). I66V was the most
active among the mutants at position 66. It appeared to bind to LPL even more tightly
than wild-type apoCII (Kd = 0.11 µM). However, I66V could not activate LPL to the level
reached with wild-type apoCII (β = 3.9). At high concentrations (>3 µM) there was even
a tendency for inhibition of LPL by this mutant (Fig. 7B).
Substitution of Asp69 by hydrophilic residues such as serine (D69S), glutamate (D69E)
and asparagine (D69N) did not lead to prominent changes in the activating properties
(Fig. 7C). Effects were mainly seen on the Kd values, which were increased
approximately 6-fold compared to wild-type apoCII. Mutants D69S and D69E had similar
β values as wild-type apoCII, but for D69N it was somewhat lower (β = 4.9). The
weakest binding affinity was seen with the D69A mutant (Kd=3.40 µM).
Three mutants were generated at position Gln70: Q70A, Q70N and Q70E (Fig, 7D). The
most active one was Q70N with Kd = 2.07 µM and β=3.0. Both Q70A and Q70N showed
lowered binding affinity to LPL and decreased maximal activation (Table 2). In contrast
to these mutants, the Q70E mutant was completely inactive and could not compete with
the wild-type apoCII in the assay (Fig. 8). Thus, a negative charge at position 70 appeared
to block binding of apoCII to LPL.
In addition to substitutions at the fully conserved residues, three mutants were generated
at residue Leu72, Ser73 and Val74 by alanine replacement. These mutants bound to LPL
with an apparent affinity comparable to that of wild-type apoCII and they activated the
enzyme to the same extent as the wild-type apoCII did (data not shown) Thus, the
structures of these three residues did not appear to be essential for the function of apoCII.
Interaction between LPL and apoCII in the absence of hydrolyzable lipids
The interaction of apoCII mutants with LPL was studied in the presence of liposomes of a
non-hydrolysable lipid (1, 2-di-O-hexadecyl-sn-glycero-3-phosphatidylcholine). In these
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
14
experiments the apoCII mutants were dansylated, and their association to LPL was
measured by monitoring fluorescence energy transfer from LPL tryptophans to the dansyl
groups of the modified apoCII molecules. Dissociation constants (Kd’) were determined
for the apoCII/LPL complexes. All studied apoCII mutants appeared to bind to LPL with
an affinity comparable to that of wild-type apoCII. The values for the dissociation
constants were 0.3-0.8 µM (Fig. 9). For wild-type apoCII this value was similar in the
absence of liposomes (data not shown). Thus, in the absence of hydrolysable lipids the
dissociation constants (Kd’) did not vary as much as they did in the presence of the
radiolabeled emulsion of triacylglycerols, where the Kd varied������������������� ������
2). Figure 9 demonstrates that there was no correlation between the Kd and Kd’ values.
Interaction of apoCII with lipids
To elucidate whether the poor activation of LPL by some mutants was caused by a
reduced interaction with lipids, the mutants were titrated with liposomes of
phosphatidylcholine. Formation of lipsome/apoCII complexes was measured by
monitoring fluorescence of the single tryptophan (Trp26) of apoCII on excitation at 280
nm. We had previously shown that on association of wild-type apoCII with liposomes, the
tryptophan fluorescence increased and shifted the emission maximum from 345nm to 320
nm (19). In the present study, we detected similar changes in the fluorescence for all of
the studied apoCII mutants. Consequently, even the inactive mutants were able to interact
with lipids. Apparent dissociation constants (Klip) calculated from the titration curves,
ranged between 101-420 µM (Table 2).
DISCUSSION
The starting point for the present study was our previous observation that there are only
seven fully conserved residues in the C-terminal one third of the apoCII molecule, the
part to which the ability to activate LPL had previously been localized (16). This implied
that the detailed structure of this region, limited to residues 59-70, was important and had
therefore not changed during evolution. We generated a set of single-residue substitution
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
15
mutants of human apoCII, mainly at the seven fully conserved sites. ApoCII was
expressed either as a fusion protein with the S-tag or the 6xHis-tag at the N-terminal end.
The expressed apoCII appeared in two forms, one of which was due to truncation of the
last two amino acid residues at the C-terminus (Glu78-Glu79). Although the short form in
some cases constituted about 75% of the total apoCII protein, we did not further separate
the individual forms since both were shown to activate LPL. Based on sequence
comparisons of apoCII from different animal species we had previously concluded that
the negatively charged residues (Glu78-Glu79) in the C-terminal end of human apoCII are
not important for the activation (16). This was further supported by the present findings
with the recombinant, truncated forms.
Effects of mutations
There was little or no effect of some of the presently investigated mutations on the
secondary structure of apoCII. We can, however, not absolutely exclude conformational
effects for some of the other mutants, which were not produced in sufficient amounts for
CD measurements. However, theoretical structure prediction using several available
computer programs did not indicate major changes for any of the mutants studied here.
To investigate if the mutations affected formation of the apoCII/ LPL complex, the
catalytic power of the complex, or both, we derived two kinetic parameters; the apparent
dissociation constant Kd and the activation factor β. The aim was to test the hypothesis
that the LPL-interaction domain of apoCII contains two regions: one contributing mainly
to activation, while the other is mainly responsible for binding of apoCII to LPL (22). For
most of our mutants, however, both parameters were affected, indicating that
discrimination between these functional parts of apoCII was not possible.
Tyr63 appeared to be the most sensitive residue. Both Kd and β were reduced for all
studied mutants. Thus, Tyr63 is probably involved both in the binding to LPL and in the
activation. Substitution by Phe, Trp, Ala or Leu reduced the activation factor more than
substitution by Thr. This indicated that the hydroxyl group could be involved in formation
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
16
of a hydrogen bond between residue 63 and LPL during activation, but the difference
between Y63T and the wild-type was still very large. The importance of Tyr63 was
previously demonstrated by substitution with Trp and Gly in a synthetic apoCII peptide
spanning residues 44-79. This caused reduced but not lost ability to activate LPL (23).
The mutations of Ile66 suggested that at this position both shape and hydrophobicity of
the side chain are essential properties. I66A and I66F were much less efficient than I66L
and I66V, indicating that a hydrophobic aliphatic side-chain is preferred. I66V
demonstrated even stronger apparent binding affinity to LPL than wild-type apoCII. The
maximal activation level for I66V was, however, somewhat lower than that of the wild-
type apoCII. Thus, even minor structural changes at position 66 led to marked changes in
the activation. It appears as if the sterical fit of Ile66 into a narrow hydrophobic pocket in
LPL is important for full activation.
For residue Asp69, mutants D69S, D69E and D69N did not show very dramatic changes
in the activating ability, suggesting that apoCII may tolerate some variation in hydrophilic
residues at this position. The main effect was an increase in the Kd values. The D69S
mutant was practically as efficient as the D69E mutant. Thus, a negative charge at this
position did not seem to play a crucial role. In contrast to hydrophilic residues,
substitution with alanine caused a more pronounced decrease in apparent binding affinity.
One of the most dramatic effects on activation was obtained by substitution of Gln70 by
glutamate, indicating that the corresponding binding region in LPL is probably negatively
charged. Also the Q70N mutant was rather inefficient, demonstrating that the additional
carbon in the side chain of the glutamine residue is probably important for the interaction
with LPL. Substitution with alanine decreased both the binding affinity (Kd) and
activation ability (β). Thus, not only the amide group in the side chain of residue 70, but
also the length of the carbon side chain was important for the function.
LPL interactions
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
17
To further characterize the properties of our mutants, we studied the apoCII-LPL
interaction in the absence of hydrolyzable lipids. In the system with ether phospholipids
the mutants did not show significant differences with regard to LPL affinity. These results
are in conflict with the apparent LPL affinities from the activation experiments (Kd),
where several of the mutants showed little or no activation. The competition experiments
showed that the low-activity mutants could not compete with wild-type apoCII for
binding to LPL, even at a 20-fold higher concentration. Thus, the apoCII/LPL complex
observed in the absence of hydrolyzable lipid may not be the true functional complex that
can hydrolyze lipids at optimal rate. One explanation could be that the lipid substrate is
needed for induction of the right conformation of either LPL or apoCII, and that some of
the mutations affected this interaction. It is, unfortunately, not possible to study the
interaction by fluorescence with a hydrolysable lipid. The substrate would almost
immediately be consumed by the high amounts of lipase needed for the experiments.
Recently it was reported that the synthetic apoCII fragment 39-62 activates LPL to the
same extent as full-length apoCII (24). These data cannot be understood from our present
and previous experiences. In our study, this region is intact in all affected mutants. Still
their ability to activate LPL was severely reduced or absent. Therefore, the proposed
region cannot be crucial for activation, at least not as part of native apoCII. It is possible
that the peptide, by non-specific effects, causes an increase in LPL activity. From
previous experiments the activation seems to be specific and similar effects have not been
found by other peptides. We cannot exclude that the LPL-binding site is extended towards
the N-terminal end to also involve residues 59 and 62. These residues were only
substituted by glycine and alanine, respectively, with little change in the activation.
Considering the evolutionary conservation of these residues, and their location preceding
the third α-helix (8), it is possible that other substitutions would have had effects.
Lipid interactions
The investigation of the apoCII-liposome interaction showed that all mutants had lipid-
binding affinities comparable to that of wild-type apoCII. Thus, the loss or reduction in
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
18
the activation ability of some of the mutants was not due to reduced ability to interact
with lipid/water interfaces. This is consistent with the previous observations that the N-
terminal domain of apoCII is the part mainly responsible for binding to lipids (9-11). The
mutations could, however, still affect details in the steric arrangement of apoCII at the
lipid/water interface ways that were not detectable by the methods used here to measure
lipid interaction.
ApoCII-binding site
It is still not clear where the apoCII-binding site is located on LPL. Studies with lipase
chimeras of LPL and hepatic lipase suggest that apoCII binds to the N-terminal folding
region of LPL (25, 26). It was also suggested that apoCII interacts simultaneously with
regions located in the N- and C-terminal domains of opposing subunits of the LPL dimer
(27). The crystal structure of the related pancreatic lipase shows that its activator,
colipase, binds to the loop covering active site (28). It is, however, well known that
apoCII and colipase differ both in structure and functions. Recently the binding site of
apolipoprotein AI (apoAI) for lecithin:cholesterol acyl transferases (LCAT) was reported
by Roosbeek et al. (29). In apoAI three strictly conserved arginine residues point out from
the same side of an amphipathic α-helix, which on the other side interacts with lipids.
The corresponding binding site in LCAT for its activator has not yet been defined. It is
clear, however, that this interaction has a different nature than that between apoCII and
LPL, which does not appear to involve charged residues. Thus additional studies are
required to identify the apoCII-binding site on LPL.
Conclusions
Our results suggest that matching between Ile66 and LPL is important for full activation.
Introduction of other hydrophobic side chains like those of Phe, Ala, Leu and Val led to
decreased activation. The other clear result is that a negative charge cannot be tolerated in
position 70. From our data we conclude that residues 66, 69 and 70 contribute, together
with Tyr63, to the LPL-binding site of apoCII. The lack of change of the activation ability
by substitution of residues 72, 73 and 74 by alanine indicated that these residues are
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
19
outside the LPL-interaction site, even though residue 73 is situated on the same side of
the α-helix as residues 69 and 70 (8). Taken together, multiple residues (Tyr63, Ile66,
Asp69, Gln70) are involved in formation of the LPL-binding site of apoCII, but no single
amino acid was absolutely crucial for the activation.
Acknowledgements
We thank Dr. Per-Ingvar Ohlsson for the N-terminal amino acid sequence analyses and
Dr. Juhan Subbi for the analyses by mass spectrometry.
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
20
REFERENCES 1. Bier, D. M., and Havel, R. J. (1970) J. Lipid Res. 11, 565-570 2. Havel, R. J., Kane, J. P., and Kashyap, M. L. (1973) J. Clin. Invest. 52, 32-38 3. Applebaum-Bowden, D., Goldberg, A. P., Hazzard, W. R., Sherrard, D. J., Brunzell, J. D., Huttunen, J. K., Nikkilä, E. A., and Ehnholm, C. (1979) Metabolism 28, 917-924 4. LaRosa, J. C., Levy, R. I., Herbert, P., Lux, S. E., and Fredrickson, D. S. (1970) Biochem. Biophys. Res. Commun. 41, 57-62 5. Breckenridge, W. C., Little, J. A., Steiner, G., Chow, A., and Poapst, M. (1978) N. Engl. J. Med. 298, 1265-1273 6. Santamarina-Fojo, S. (1992) Curr. Opin. Lipidol. 3, 186-195 7. Kuniyoshi, A., Okamoto, Y., Tamagawa, T., Matsuyama, Y., and Fuku, H. (1999) Intern. Med. 38, 140-144 8. MacRaild, C. A., Hatters, D. M., Howelett, G. J., and Gooley, P. R. (2001) Biochemistry 40, 5414-5421 9. Sparrow, J. T., and Gotto, A. M. Jr. (1980) Ann. N. Y. Acad. Sci. 348, 187-208 10. Vainio, P., Virtanen, J. A., Kinnunen, P. K., Gotto, A. M. J., Sparrow, J. T., Pattus, F., Bougis, P., and Verger, R. (1983) J. Biol Chem. 258, 5477-5482 11. MacPhee, C.E., Howlett, G. J., and Sawyer W. H. (1999) Anal. Biochem. 275, 22-29. 12. Olivecrona, G. and Beisiegel, U. (1997) Arterioscler. Thromb. Vasc. Biol. 17, 1545-1549 13. Bengtsson, G. and Olivecrona, T. (1982) FEBS Lett. 147, 183-187 14. Voyta, J. C., Vainio, P., Kinnunen, P. K. J., Gotto.A.M.Jr., Sparrow, J. T., and Smith, L. C. (1983) J. Biol. Chem. 258, 2934-2939 15. Clarke, A. R. and Holbrook, J. J. (1985) Biochim. Biophys. Acta 827, 358-368 16. Shen, Y., Lindberg, A., and Olivecrona, G. (2000) Gene 254, 189-198 17. Bengtsson-Olivecrona, G. and Olivecrona, T. (1991) Methods Enzymol. 197, 345-356 18. Harlow, E. and Lane, D. (1988) Antibodies, A Laboratory Manual, Cold Spring
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
21
Harbor Laboratory, Cold Spring Harbor, NY 19. Andersson, Y., Lookene, A., Shen, Y., Nilsson, S., Thelander, L., and Olivecrona, G. (1997) J. Lipid Res. 38, 2111-2124 20. Quinn, D., Shirai, K., and Jackson, R. L. (1983) Prog. Lipid Res. 22, 35-78 21. Wülfing, C., Lombardero, J., and Plückthun, A. (1994) J. Biol Chem 269, 2895-2901 22. Cheng, Q., Blackett, P. R., Jackson, K. W., McConathy, W. J., and Wang, C.-S. (1990) Biochem. J. 269, 403-407 23. Smith, L. C., Voyta, J. C., Catapano, A. L., Kinnunen, P. K. J., Gotto, A. M. Jr. and Sparrow, J. T. (1980) Ann. N. Y. Acad. Sci. 348, 213-221 24. MacPhee, C. E., Hatters, D. M., Sawyer, W. H., and Howelett, G. J. (2000) Biochemistry 39, 3433-3440 25. Wong, H., Davis, R. C., Nikazy, J., Seebart, K. E., and Schotz, M. C. (1991) Proc. Natl. Acad. Sci. U. S. A. 88, 11290-11294 26. Dichek, H. L., Parrott, C., Ronan, R., Brunzell, J. D., Brewer, H. B.,Jr., and Santamarina-Fojo, S. (1993) J. Lipid Res. 34, 1393-1401 27. Hill, J. S., Yang, D., Nikazy, J., Curtiss, L. K., Sparrow, J. T., and Wong, H. (1998) J. Biol. Chem. 273, 30979-30984 28. van Tilbeurgh, H., Sarda, L., Verger, R., and Cambillau, C. (1992) Nature 359, 159-162 29. Roosbeek, S., Vanloo, B., Duverger, N., Caster, H., Breyne, J., De Beun, I., Vandekerckhove, J., Shoulders, C., and Rosseneu, M. (2001) J. Lipid Res. 42, 31-40
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
22
FIGURE LEGENDS
Fig. 1. Panel A. Schematic view of functional domains of human apoCII. The seven
fully conserved residues in the activating region (16) are shaded and numbered.
Panel B. Helical wheel model of residues 59-75 in human apoCII. The seven fully
conserved residues are shaded.
Fig. 2. Analysis of S-tagged apoCII on Tricine-SDS-PAGE
The wild-type (WT) and mutant apoCII proteins were purified on S-protein agarose and
repurified by passage over the same column after cleavage by factor Xa. Panel A shows a
gel stained by Serva Blue G. Panel B shows a Western blot of the same fractions as in
panel A using rabbit anti-human apoCII serum.
Fig. 3. Analysis of His-tagged wild-type apoCII on Tricine-SDS-PAGE
His-tagged, wild-type apoCII was eluted from the Ni2+-NTA-agarose column by a pH
gradient from pH 8.0 to pH 4.5 in 8M urea, 0.1 M Tris and 0.01 M NaH2PO4. Panel A
shows a gel stained with Serva blue G. Panel B shows Western blot of the same fractions
as in panel A using the penta-His tag antibody.
Fig. 4. Purification of His-tagged wild-type apoCII by ion-exchange
chromatography and analysis on Tricine-SDS-PAGE
The fraction eluted from Ni2+-NTA-agarose column at pH 6.8 (see Fig. 3) was applied on
a Mini-Q column. The chromatogram shows the separation of the two forms of apoCII.
The inset shows the analysis of the fractions on Tricine-SDS-PAGE. Lanes: Mw - low
molecular weight markers; a.s.- applied sample; b.t.- break through (material that was not
bound to Mini-Q); fractions that were run on gel are shown by numbers; concentrations of
NaCl and GdmCl used for elution were 1 M and 6 M, respectively.
Fig. 5. Activation of lipoprotein lipase by recombinant wild-type apoCII and
mutants produced as fusion proteins with the S-tag. Purified and cleaved wild-type
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
23
apoCII and mutants (shown in Figure 2) were incubated with LPL (7.85 ng) in a total
volume of 200 µl assay mixture. Each data point is the mean of triple determinations.
Fig. 6. Circular dichroism spectra of wild-type apoCII and alanine replacement
mutants of apoCII in the presence of liposomes of egg yolk phosphatidyl choline
The concentration of phospholipids was 240 µM and the concentration of apoCII was in
all cases 4.7 µM. The experiments were performed in 0.1 M phosphate buffer, pH 7.4, at
25oC All spectra are means of three scans. WT, ⋅ ⋅ D69A, ⋅⋅⋅⋅⋅⋅⋅⋅⋅⋅⋅⋅
Y63A, I66A, ⋅ ⋅ Q70A.
Fig. 7. Stimulation of lipoprotein lipase by His-tagged apoCII mutants at residues
63, 66, 69 and 70. Conditions for the incubations were the same as in Figure 5. Each data
point is the mean of triple determinations. Panel A shows stimulation of LPL by apoCII
and mutants at residue 63; panel B shows stimulation of LPL by mutants at residue 66;
panel C shows stimulation of LPL by mutants at residue 69; panel D shows stimulation of
LPL by mutants at residue 70. In all panels the fitted curve for wild-type apoCII is shown
by a dotted line.
Fig. 8. Competitive assay using wild-type apoCII and mutants with low activity
The incubations were carried out as described in figure 6 except for the presence of 2.5
µM of the mutants Q70E, Y63F or Y63W in the incubation mixture together with the
indicated amounts of wild-type apoCII. Filled circle, wild-type alone; open circle, in the
presence of Y63W; filled square, in the presence of Y63F; open square, in the presence of
Q70E.
Fig. 9. Lack of correlation between Kd and Kd’ values. The determination of the Kd and
Kd’ is described under Experimental procedures. Kd originates from activation curves,
while Kd’ was determined from studies of the direct interaction between apoCII and LPL
using dansylated apoCII.
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
24
Table 1. Primers used for mutagenesis. The first four primers were used for generation
of mutant Y63A by overlap extension by PCR. The other primers were used together with
QuickchangeTMSite-Directed Mutagenesis Kit.
Y63A1a 5’ GGCTGATATCGGATCCGA 3’
Y63A1b 5’ GCTCATGGCTGCTGTGCTTTTGCT 3’
Y63A2
5’GCACAGCAGCCATGAGCACTGCTACAGGCATTTTTACTGACCAAGTT 3’
T7 terminator primer 5’ CGATCAATAACGAGTCGCC 3’
A59G1 5’ GCAAAAGCACAGCAGGCATGAGCACTTACACAGG 3’
A59G2 5’ CCTGTGTAAGTGCTCATGCCTGCTGTGCTTTTGC 3’
T62A1 5’ GCAGCCATGAGCGCTTACACAGGCATTTTTACTGACC 3’
T62A2 5’ GGTCAGTAAAAATGCCTGTGTAAGCGCTCATGGCTGC 3’
Y63F1 5’ CACAGCAGCCATGAGCACTTTTACAGGCATTTTTACTGACC 3’
Y63F2 5’ GGTCAGTAAAAATGCCTGTAAAAGTGCTCATGGCTGCTGTG 3’
Y63L1 5’ CACAGCAGCCATGCGCACTCTCACAGGCATTTTTACTGACC 3’
Y63L2 5’ GGTCAGTAAAAATGCCTGTGAGAGTGCTCATGGCTGCTGTG 3’
Y63T1 5’ CACAGCAGCCATGAGCACTACCACAGGCATTTTTACTGACC 3’
Y63T2 5’ GGTCAGTAAAAATGCCTGTGGTAGTGCTCATGGCTGCTGTG 3’
Y63W1 5’ CACAGCAGCCATGAGCACTTGGACAGGCATTTTTACTGACC 3’
Y63W2 5’ GGTCAGTAAAAATGCCTGTCCAAGTGCTCATGGCTGCTGTG 3’
G65A1 5’ GCAGCCATGAGCACTTACACAGCGATTTTTACTGACCAAG 3’
G65A2 5’ CTTGGTCAGTAAAAATCGCTGTGTAAGTGCTCATGGCTGC 3’
I66A1 5’ GCCATGAGCACTTACTCAGGCGCTTTTACTGACCAAGTTC 3’
I66A2 5’ GAACTTGGTCAGTAAAAGCGCCTGTGTAAGTGCTCATGGC 3’
I66V1 5’ GCCATGAGCACTTACTCAGGCGTGTTTACTGACCAAGTTC 3’
I66V2 5’ GAACTTGGTCAGTAAACACGCCTGTGTAAGTGCTCATGGC 3’
I66L1 5’ GCCATGAGCACTTACACAGGCCTCTTTACTGACCAAGTTC 3’
I66L2 5’ GAACTTGGTCAGTAAAGAGGCCTGTGTAAGTGCTCATGGC 3’
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Running title: Apolipoprotein CII mutants
25
I66F1 5’ GCCATGAGCACTTACACAGGCTTTTTTACTGACCAAGTTC 3’
I66F2 5’ GAACTTGGTCAGTAAAAAAGCCTGTGTAAGTGCTCATGGC 3’
D69A1 5’ CACAGGCATTTTTACTGCCCAAGTTCTTTCTGTGC 3’
D69A2 5’ GCACAGAAAGAACTTGGGCAGTAAAAATGCCTGTG 3’
D69E1 5’ CTTACACAGGCATTTTTACTGAGCAAGTTCTTTCTGTGCTG 3’
D69E2 5’ CAGCACAGAAAGAACTTGCTCAGTAAAAATGCCTGTGTAAG 3’
D69S1 5’ CTTACACAGGCATTTTTACTAGCCAAGTTCTTTCTGTGCTGAAG 3’
D69S2 5’ CTTCAGCACAGAAAGAACTTGGCTAGTAAAAATGCCTGTGTAAG 3’
D69N1 5’ CTTACACAGGCATTTTTACTAACCAAGTTCTTTCTGTGCTG 3’
D69N2 5’ CAGCACAGAAAGAACTTGGTTAGTAAAAATGCCTGTGTAAG 3’
Y70A1 5’ CACAGGCATTTTTACTGACGCAGTTCTTTCTGTGCTGAAGG 3’
Y70A2 5’ CCTTCAGCACAGAAAGAACTGCGTCAGTAAAAATGCCTGTG 3’
Q70N1 5’ CACAGGCATTTTTACTGACAACGTTCTTTCTGTGCTGAAGG 3’
Q70N2 5’ CCTTCAGCACAGAAAGAACGTTGTCAGTAAAAATGCCTGTG 3’
Q70E1 5’ CACAGGCATTTTTACTGACGAGGTTCTTTCTGTGCTGAAGG 3’
Q70E2 5’ CCTTCAGCACAGAAAGAACCTCGTCAGTAAAAATGCCTGTG 3’
L72A1 5’ GCATTTTTACTGACCAAGTTGCTTCTGTGCTGAAGGGAGAG 3’
L72A2 5’ CTCTCCCTTCAGCACAGAAGCAACTTGGTCAGTAAAAATGC 3’
S73A1 5’ CTGACCAAGTTCTTGCTGTGCTGAAGGGAGAG 3’
S73A2 5’ CTCTCCCTTCAGCACAGCAAGAACTTGGTCAG 3’
V74A1 5’ GACCAAGTTCTTTCTGCGCTGAAGGGAGAGGAG 3’
V74A2 5’ CTCCTCTCCCTTCAGCGCAGAAAGAACTTGGTC 3’
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
Running title: Apolipoprotein CII mutants
26
Table 2. Kinetic parameters for the interaction of apoCII mutants with LPL and
with liposomes. Kd and β were determined from the activation curves. Klip is the
dissociation constant for the apoCII/liposome complex determined by Trp fluorescence.
Apo CII variant
Kd (µM) β Klip(µM)
WT 0.29±0.07 6.2±0.1 164±43
Y63T 1.73±0.09 4.0±1.0 n.d.
Y63A 2.30±0.32 2.5±0.3 157±27
Y63L 3.14±0.93 4.2±1.6 n.d.
Y63F almost no activation n.d.
Y63W almost no activation n.d.
I66V 0.11±0.04 3.9±0.5 420±153
I66L 4.60±2.25 7.7±2.7 270±51
I66A 3.27±1.56 2.9±1.1 254±54
I66F 0.99±0.44 1.5±0.3 370±44
D69S 1.51±0.56 6.3±1.0 101±33
D69E 0.81±0.15 6.3±0.9 n.d.
D69N 1.49±0.61 4.9±0.6 n.d.
D69A 3.40±1.62 5.8±1.9 n.d.
Q70A 1.36±0.62 3.8±0.7 170±38
Q70N 2.07±1.05 3.0±0.5 n.d.
Q70E no activation 112±41
n.d.: not determined
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
LIPID-BINDING LPL ACTIVATION / BINDING
1 50 56 79
Human sequence
Residue number 59 62 63 65 66 69 70
STAAMSTYTGIFTDQVLSVLKGEE
N - terminal domain C - terminal domain
Fig. 1A
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
A59
I66V74
F67
M60
V71
T64
L75
T68S61
L72
G65
D69
T62
S73
Y63Q70
Fig. 1B
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
A
WT A59G T62A Y63A G65A I66A D69A Q70A
6.5 kDa
14 kDa
B
6.5 kDa
14 kDa
Fig.2
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
Mw 8.0 7.2 6.8 6.3 5.9 5.9 4.5 4.5
30 kDa21.5 kDa
14 kDa 6.5 kDa
A
B
Mw 8.0 7.2 6.8 6.3 5.9 5.9 4.5 4.5
14 kDa
6.5 kDa
30 kDa
pH
pH
Fig. 3
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
24 26 28 30 32 34 36 38 40 42
0
50
100
150
200
250
300
1
2
1
2
3
A
B
Fraction number
Pro
tein
co
nce
ntr
atio
n (
g/m
l)
Mw a..s. b.t. fr.28 fr.33 fr.35 NaCl GdmClFig. 4
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
0 0.5 1.0 1.5 2.0 2.5 3
Apo CII (µ M)
0
50
100
150
200
250
300
LP
L a
cti
vit
y (
U/m
g)
WT
A59G
T62A
Y63A
G65A
I66A
D69A
Q70A
Fig. 5
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
0 1 2 3 4 5 60
50
100
150
200
250
300
350
400L
PL
act
ivit
y (U
/mg
)
0 1 2 3 4 5 60
50
100
150
200
250
300
350
400
0 1 2 3 4 5 6
Apo CII ( M)
0
50
100
150
200
250
300
350
400
LP
L a
ctiv
ity
(U/m
g)
0 1 2 3 4 5 6
Apo CII ( M)
0
50
100
150
200
250
300
350
400
WT
WTWT
WT
Q70A
Q70N
Q70E
D69SD69E
D69ND69A
I66VI66L
I66A
I66F
Y63T
Y63AY63L
Y63WY63F
A B
C D
Fig. 7
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from
0 1 2 3 4
Apo CII ( µM)
0
50
100
150
200
250
300
LP
L a
ctiv
ity
(U/m
g)
Fig. 8 by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from

Yan Shen, Aivar Lookene, Solveig Nilsson and Gunilla Olivecronamutagenesis.Identification of residues important for activation of lipoprotein lipase
Functional analyses of human apolipoprotein CII by site-directed
published online November 21, 2001J. Biol. Chem.
10.1074/jbc.M105421200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on June 8, 2018http://w
ww
.jbc.org/D
ownloaded from