fetal development of mouse oocytes and zygotes cryopreserved in a nonconventional freezing medium
TRANSCRIPT

Fetal development of mouse oocytes and zygotescryopreserved in a nonconventional freezing mediumq
James J. Stachecki,* Jacques Cohen, Tim Schimmel,and Steen M. Willadsen
Institute for Reproductive Medicine and Science of Saint Barnabas Medical Center, 101 Old Short Hills Road, Suite 501,
West Orange, NJ 07052, USA
Received 31 July 2001; accepted 2 January 2002
Abstract
This study (1) analyzed fetal development of mouse embryos after oocyte cryopreservation in CJ2, a
choline-based medium, (2) examined the effect of culture duration in vitro on subsequent fetal develop-
ment, and (3) compared survival and fetal development of zygotes frozen in embryo transfer freeze medium
(ETFM; sodium-based medium) or CJ2. Unfertilized oocytes and zygotes were cryopreserved using a slow-
cooling protocol. After thawing, oocytes were inseminated after drilling a hole in their zona, cultured in
vitro either to the two-cell or blastocyst stage, and transferred to the oviducts or uterine horns of recipient
mice. In parallel experiments, frozen–thawed zygotes were similarly cultured and transferred. Implantation
rates for transferred embryos were high (range 66–88%), regardless of whether they had been frozen as
oocytes or zygotes and whether they had been transferred to the oviduct or uterus. However, fetal de-
velopment was significantly higher when two-cell embryos were transferred. With blastocyst transfer,
control embryos implanted and produced a greater proportion of fetuses than did oocytes frozen in CJ2,
whereas transfer at the two-cell stage resulted in similar proportions of implantation sites and fetuses.
Blastocyst transfer of zygotes cryopreserved in ETFM or CJ2 produced similar fetal development rates
(23.6% vs 20.0%), but when frozen–thawed zygotes were transferred at the two-cell stage the fetal devel-
opment rates were higher in the ETFM group (53.3%) than in the CJ2 group (32.0%). A high proportion
(46.7%) of oocytes frozen in CJ2 in a nonprogrammable freezer and plunged at )20 �C developed into liveoffspring. This study shows that in the mouse (1) oocytes frozen in CJ2 can develop into viable fetuses, (2)
prolonging culture in vitro has a detrimental effect on embryo transfer outcome, and (3) CJ2 offers no
advantage for zygote cryopreservation. � 2002 Elsevier Science (USA). All rights reserved.
Keywords: Cryopreservation; Oocytes; Zygotes; Choline chloride; Mouse; Uterus; Oviduct; Embryo transfer
In previous reports we have described how the
results of cryopreserving unfertilized mouse eggs
according to a conventional slow-freezing proto-
col are substantially improved by the use of CJ2, a
choline-based medium, instead of a conventional
sodium-based medium, ETFM (Embryo Transfer
Freeze Medium; Gibco BRL, Gaithersburg, MD;
[24–26]). In the present study we examined (1)
fetal development of embryos produced from
oocytes cryopreserved in CJ2, (2) the effect of
Cryobiology 44 (2002) 5–13
www.academicpress.com
qThis work was funded by institutional sources.* Corresponding author.
0011-2240/02/$ - see front matter � 2002 Elsevier Science (USA). All rights reserved.PII: S0011 -2240 (02)00007 -X

culture duration in vitro on subsequent develop-
ment in vivo, and (3) the usefulness of CJ2 for
zygote freezing.
Our previously published results were ex-
pressed in terms of survival after thawing, fer-
tilization, and development to the blastocyst
stage in vitro. Ultimately, however, it is the
ability of embryos produced from cryopreserved
oocytes to develop into normal offspring that is
the best measure of success. Because we are
working toward developing a medium for storing
oocytes from a range of species, including hu-
mans, and because CJ2 is very different from any
other medium previously used, in being virtually
sodium-free, it is imperative that fetal develop-
ment be assessed and documented. Furthermore,
there have been less than a dozen studies that
have reported fetal development from cryopre-
served mouse oocytes [1,2,5,23]. The highest fetal
development rates obtained with slowly cooled
oocytes were 31% for two-cell oviductal transfers
and 23% for blastocyst uterine transfers [5] and a
rate of 42% for two-cell oviductal transfers [2],
although Bos-Mikich et al. [1] obtained 41% with
oviductal transfers of two-cell embryos produced
from vitrified oocytes. Of particular note is the
study by Schroeder et al. [23], in which a live pup
rate of 26% was obtained after oviductal trans-
fer. More recent studies describe oocyte cryo-
preservation success only in terms of the
blastulation rate after fertilization and culture in
vitro (for review see [10]). Relatively early in the
course of our experiments, a small trial estab-
lished that viable pups could develop from
blastocysts produced from oocytes cryopreserved
in CJ2. However, the success rate was lower than
expected from the high rates of development in
vitro (data not shown), similar to the findings of
Lane and Gardner [15] when they transferred
slowly cooled oocytes to recipients at the
blastocyst stage.
Choline and other organic osmolytes including
betaines are known to have osmoprotective and
cryoprotective effects on liposomes, erythrocytes,
plants, and mouse embryos [16,17,27]. The osmo-
lyte glycinebetaine has been found in animals,
bacteria, fungi, algae, and many drought- and
salt-tolerant plants [22]. Some of these com-
pounds protect enzymes and membranes from
cold [14,18,28], salt [7,9], and freezing damage
[8,32]. It is possible that choline has similar effects
on the membrane integrity of oocytes and em-
bryos. In support of this hypothesis, Toner et al.
[27] has shown that during cooling, zygote cell
membranes are more tolerant to hyperosmotic
stress from a mixture of sodium and choline ions
than from sodium ions alone.
In the present study we examined fetal devel-
opment to days 10–13 of gestation and briefly
examined development to term to obtain a more
clear indication of oocyte viability after cryopre-
servation in CJ2. Finally, we investigated whether
mouse embryo cryopreservation could be im-
proved by the use of CJ2, rather than ETFM, a
common sodium-based medium for freezing
mouse embryos.
Materials and methods
Collection and cryopreservation of oocytes and
zygotes
For all experiments, C57BL/6�BALB/c F1mice (The Jackson Laboratory, Bar Harbor, ME,
USA) were used [24]. Freshly ovulated metaphase
II mouse oocytes (collected 13 h after HCG) and
zygotes (collected 17 h after HCG) were treated
according to the methods described by Stachecki
et al. [24]. Oocytes and zygotes were frozen in ei-
ther CJ2 supplemented with 10% fetal bovine se-
rum or ETFM with 1.5 M 1,2-propanediol
(PrOH) and 0.1 M sucrose as cryoprotectants and
frozen using a slow-cooling protocol [24]. After
storage for at least 3 days, the straws were thawed
by exposing them to air at room temperature for
30 s followed by immersion in a 30 �C water bathfor an additional 10 s (30/10; [26]). After thawing,
the cryoprotectants were removed in five steps at
5-min intervals at 23 �C as described by Stacheckiet al. [24]: freezing medium supplemented with (I)
0.2 M sucrose and 1.0 M PrOH, (II) 0.2 M sucrose
and 0.5 M PrOH, (III) 0.2 M sucrose, (IV) 0.1 M
sucrose, and (V) freezing medium alone. Oocytes
and zygotes frozen in CJ2 were held for an addi-
tional 5 min in mCZB [3,11] for reequilibration
with a sodium-based medium and then incubated
on a slide warmer at 37 �C for 5 min. Embryosfrozen in ETFM were not moved to mCZB me-
dium, but incubated in ETFM on a slide warmer
at 37 �C for 5 min prior to being placed intoculture. The zonae of frozen–thawed oocytes was
opened during step IV (freezing medium with
0.1 M sucrose) of the thawing procedure by using
a Fertilase laser [24]. Spermatozoa were aspirated
from the epididymides of 12- to 16-week-old
C57BL/6�BALB/c F1 mice into Ham’s F10 me-dium (Sigma Chemical, St. Louis, MO, USA),
6 J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13

supplemented with 6 mg/ml bovine serum albumin
(Fraction V; Sigma) and allowed to capacitate for
1.5–2 h before insemination. Directly after cryo-
protectant removal, the oocytes were transferred
to Ham’s F10 containing approximately 5� 105motile sperm for 5–9 h. Fertilized oocytes were
cultured in S1 (a low-glucose, 0.50 mM, preim-
plantation embryo culture medium containing
nonessential amino acids; Scandinavian IVF Sci-
ence, Gothenburg, Sweden) for 2 days before be-
ing transferred to S2 (a high-glucose, 3.15 mM,
preimplantation embryo culture medium con-
taining essential and nonessential amino acids;
Scandinavian IVF Science). Zygotes were cultured
in KSOM (a conventional mouse embryo culture
medium without amino acids and with 0.20 mM
glucose; Specialty Media, Lavallette, NJ, USA),
for 2 days before being transferred to a fresh drop
of KSOM. In earlier experiments we determined
that the use of sequential media (S1/S2) was nec-
essary to maximize the production of blastocysts
from frozen–thawed oocytes [26], whereas culture
in KSOM (our standard mouse embryo culture
medium) was adequate for obtaining a high rate
of blastocyst formation from frozen–thawed zy-
gotes. Nonfrozen zygotes served as controls and
were cultured in KSOM either to the two-cell or
to the early blastocyst stage.
Embryo transfers
Two-cell embryos that developed from frozen–
thawed oocytes or zygotes were transferred to the
oviducts of Day 1 (day of copulation plug)
pseudopregnant CD1 mice (The Jackson Labo-
ratory) along with nonfrozen control two-cell
embryos collected at the zygote stage [2,5]. Re-
cipients were anesthetized with tribromoethanol
(Avertin; 2.5% solution; 0.02 ml/g body weight).
Frozen oocyte- and frozen zygote-derived ex-
panding blastocysts that appeared to be mor-
phologically normal and had a prominent
blastocoel, along with in vitro cultured, nonfrozen
control blastocysts, were transferred to the uterine
horns of Day 3 recipient mice [5]. Six to 10 em-
bryos were transplanted per recipient. Control
embryos were transferred to one oviduct or uter-
ine horn, while an equivalent number of embryos
from cryopreserved oocytes or zygotes were
transferred to the con-tralateral oviduct or uterine
horn of the same recipient. Only the number of
embryos transferred to recipients that became
pregnant are shown in the tables. The recipient
mice were sacrificed 10–13 days after embryo
transfer, and the numbers of implantation sites
and fetuses were recorded.
Cryopreservation in a nonprogrammable freezer
To test the ability of oocytes cryopreserved in
CJ2 to develop into live-born fetuses and to fur-
ther demonstrate that oocytes can withstand the
trauma of cryopreservation, mouse eggs were
frozen in a nonprogrammable freezer using a liq-
uid nitrogen plunge temperature of )20 �C. Theoocytes were exposed to cryoprotectants as de-
scribed above, sealed into straws, and transferred
into a 500-ml beaker filled with methanol at room
temperature (23 �C). The beaker was subsequentlyplaced in the freezer compartment of a conven-
tional refrigerator/freezer (approximately )24 �C).To monitor temperature changes, a thermocouple
was placed inside a control straw containing the
same freezing solution as the straws that con-
tained the oocytes. All straws were seeded at )7 �Cand plunged into LN2 after cooling to )20 �C.The average cooling rate down to )7.1 �C (theseeding temperature) was )1.76 �C/min. The av-erage rate of cooling following seeding down to
the plunge temperature of )20.3 �C was )0.36 �C/min. Based upon the results of pilot experiments
involving plunging oocytes at )20 �C (data notshown), the straws were thawed by holding them
in room temperature air for 10 s and then sub-
merging them into 30 �C water for 10 s. Aftercryoprotectant removal, insemination, and culture
overnight, the resulting two-cell embryos were
transferred to the oviducts (five embryos per tube)
of pseudopregnant recipients as described above.
Live-born young were counted and examined vi-
sually for the presence of gross morphological
abnormalities.
Statistical analysis
Within experiments, implantation and fetal
development rates in control and cryopreserved
oocytes/embryo groups were analyzed using a v2
test with Yates’ correction for small sample sizes.
Results
The main result of the present study was the
demonstration that fully viable embryos can be
produced from unfertilized oocytes frozen in CJ2.
The experiments also show that extended culture
J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13 7

in vitro reduced the viability of embryos produced
from frozen oocytes and zygotes.
Following oviductal transfers, implantation
rates were marginally different for control em-
bryos and embryos from frozen oocytes
(P ¼ 0:040; 62.3 and 73.0%, respectively; Table 1).After uterine transfer, embryos from the same two
groups had similar implantation rates (P ¼ 0:085;75.5 and 62.0%, respectively; Table 2). There was
a significant reduction (P < 0:001; 35.3% lower) infetal development when embryos from cryopre-
served oocytes were cultured in vitro to the
blastocyst stage before being transferred rather
than being transferred at the two-cell stage to re-
cipient mice (12.3% vs 47.6%, respectively). By
contrast, fetal development rates were similar for
the control and the cryopreserved groups when
oviductal transfers were used (P ¼ 0:180; 40.0 and47.6%, respectively; Table 1). The overall fetal
development rate for all oocytes frozen in CJ2 and
transferred at the two-cell stage was 40.2%
(0:844� 0:476). These data show that oocytesfrozen in CJ2 can develop into viable fetuses at
rates similar to those of control embryos.
The overall survival postcryoprotectant re-
moval (93.7 and 93.5%, respectively) and the de-
velopment rate to the two-cell stage (80.8 and
86.0%, respectively) were similar whether zygotes
were frozen in ETFM or in CJ2 (Tables 3 and 4).
Implantation rates were also similar (P > 0:05Þfor zygotes frozen in either ETFM or CJ2 and
control embryos, regardless of whether the em-
bryos were transferred to the oviduct (66.7, 66.0,
and 67.4%) or uterus (80.0, 88.3, and 87.0%, re-
spectively; Tables 3 and 4). By contrast, fetal de-
velopment rates after uterine transfer were
significantly lower for embryos cryopreserved in
ETFM (P < 0:001; 23.6%) or in CJ2 (P < 0:001;20%) than for nonfrozen control embryos (59.1%;
Table 4). Fetal development rates for embryos
transferred to the oviduct were similar for those
zygotes cryopreserved with ETFM (53.3%) and
the control embryos (54.7%) and slightly lower for
embryos frozen with CJ2 (P ¼ 0:043; 32.0%).Oocytes frozen in the nonprogrammable free-
zer and plunged at )20 �C did not survive as wellor develop as well as oocytes frozen in a BioCool
programmable freezer and plunged at )33 �C,
Table 1
Oviductal transfer results of two-cell embryos cryopreserved in CJ2 as unfertilized oocytes
Nonfrozen control Cryopreserved
Number of oocytes intact ND 833 (97.5)
Postcryoprotectant removal (%)
Two-cells (%) ND 721 (84.4)
Transferred (to mice that became pregnant) 175 185
Implanted (%) 109 (62.3) 135 (73.0)
Fetuses (%) 70 (40.0) 88 (47.6)
Note. ND, not determined. Data represent 22 straws frozen. There were 39 mice that received embryos. Of these, 33
mice were pregnant. Five control embryos were transferred to one oviduct and five embryos from cryopreserved oocytes
were transferred to the contralateral oviduct of the same recipient. There was 1 mouse that received only cryopreserved
oocytes.
Table 2
Uterine transfer results of blastocysts cryopreserved in CJ2 as unfertilized oocytes
Nonfrozen control Cryopreserved
Number of oocytes intact ND 1212 (89.1)
Postcryoprotectant removal (%)
Two-cells (%) ND 943 (69.3)
Blastocysts (%) ND 594 (43.7)
Transferred (to mice that became pregnant) 163 163
Implanted (%) 123 (75.5) 101 (62.0)
Fetuses (%) 77 (47.2) 20 (12.3)
Note. ND, not determined. Data represent 40 straws frozen. There were 46 mice that received embryos. Of these, 33
mice were pregnant. Two to five control embryos were transferred to one uterine horn, while an equivalent number of
embryos from cryopreserved oocytes were transferred to the contralateral horn of the same recipient.
8 J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13
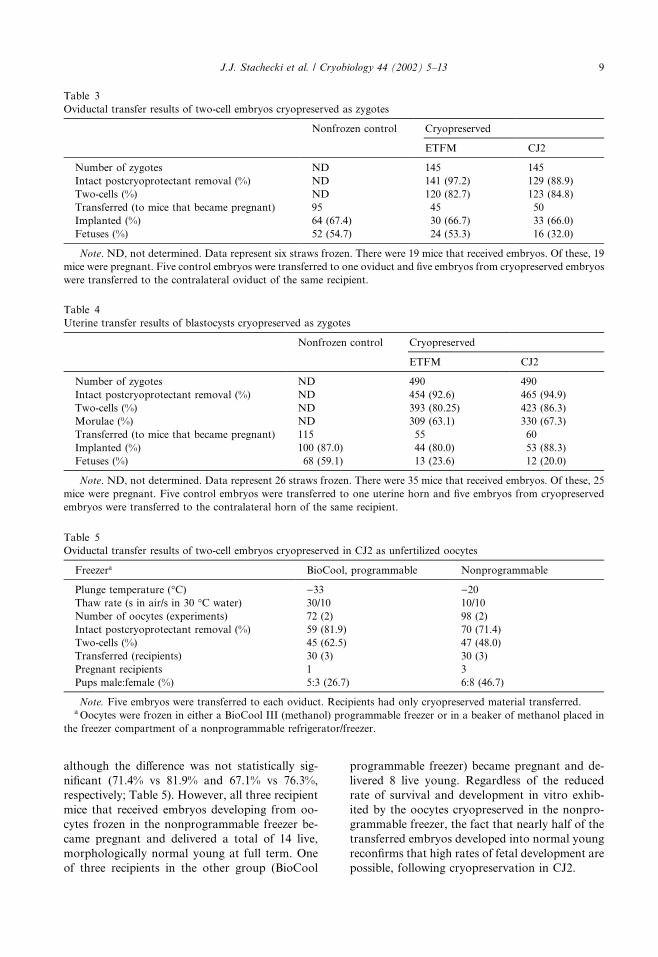
although the difference was not statistically sig-
nificant (71.4% vs 81.9% and 67.1% vs 76.3%,
respectively; Table 5). However, all three recipient
mice that received embryos developing from oo-
cytes frozen in the nonprogrammable freezer be-
came pregnant and delivered a total of 14 live,
morphologically normal young at full term. One
of three recipients in the other group (BioCool
programmable freezer) became pregnant and de-
livered 8 live young. Regardless of the reduced
rate of survival and development in vitro exhib-
ited by the oocytes cryopreserved in the nonpro-
grammable freezer, the fact that nearly half of the
transferred embryos developed into normal young
reconfirms that high rates of fetal development are
possible, following cryopreservation in CJ2.
Table 4
Uterine transfer results of blastocysts cryopreserved as zygotes
Nonfrozen control Cryopreserved
ETFM CJ2
Number of zygotes ND 490 490
Intact postcryoprotectant removal (%) ND 454 (92.6) 465 (94.9)
Two-cells (%) ND 393 (80.25) 423 (86.3)
Morulae (%) ND 309 (63.1) 330 (67.3)
Transferred (to mice that became pregnant) 115 55 60
Implanted (%) 100 (87.0) 44 (80.0) 53 (88.3)
Fetuses (%) 68 (59.1) 13 (23.6) 12 (20.0)
Note. ND, not determined. Data represent 26 straws frozen. There were 35 mice that received embryos. Of these, 25
mice were pregnant. Five control embryos were transferred to one uterine horn and five embryos from cryopreserved
embryos were transferred to the contralateral horn of the same recipient.
Table 3
Oviductal transfer results of two-cell embryos cryopreserved as zygotes
Nonfrozen control Cryopreserved
ETFM CJ2
Number of zygotes ND 145 145
Intact postcryoprotectant removal (%) ND 141 (97.2) 129 (88.9)
Two-cells (%) ND 120 (82.7) 123 (84.8)
Transferred (to mice that became pregnant) 95 45 50
Implanted (%) 64 (67.4) 30 (66.7) 33 (66.0)
Fetuses (%) 52 (54.7) 24 (53.3) 16 (32.0)
Note. ND, not determined. Data represent six straws frozen. There were 19 mice that received embryos. Of these, 19
mice were pregnant. Five control embryos were transferred to one oviduct and five embryos from cryopreserved embryos
were transferred to the contralateral oviduct of the same recipient.
Table 5
Oviductal transfer results of two-cell embryos cryopreserved in CJ2 as unfertilized oocytes
Freezera BioCool, programmable Nonprogrammable
Plunge temperature (�C) )33 )20Thaw rate (s in air/s in 30 �C water) 30/10 10/10
Number of oocytes (experiments) 72 (2) 98 (2)
Intact postcryoprotectant removal (%) 59 (81.9) 70 (71.4)
Two-cells (%) 45 (62.5) 47 (48.0)
Transferred (recipients) 30 (3) 30 (3)
Pregnant recipients 1 3
Pups male:female (%) 5:3 (26.7) 6:8 (46.7)
Note. Five embryos were transferred to each oviduct. Recipients had only cryopreserved material transferred.aOocytes were frozen in either a BioCool III (methanol) programmable freezer or in a beaker of methanol placed in
the freezer compartment of a nonprogrammable refrigerator/freezer.
J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13 9

Discussion
In previous reports we have examined the main
factors governing survival of mouse oocytes dur-
ing cryopreservation with CJ2 as the freezing me-
dium [24–26]. The freezing method used in the
present experiments is relatively simple compared
to other published methods [2,5,10] and in our
hands consistently yields oocyte survival rates in
the 90% range, i.e., equal to or higher than any so
far reported in the literature [2,23, for reviews see
5,10]. By ‘‘survival’’ it is meant that the oocyte has
an intact cell membrane and looks normal when
viewed under a microscope at powers up to 400�.However, survival thus defined does not imply full
viability. Therefore this study was undertaken to
examine the ability of these eggs to develop in vivo.
Experience leads to the expectation that in the
course of cryopreservation the oocyte’s viability
will be reduced, although not necessarily to an
extent that makes it incapable of becoming fer-
tilized and developing into a viable conceptus. It is
worth bearing in mind that at ovulation the
mammalian egg possesses developmental poten-
tial in excess of what is strictly necessary for
launching a single viable embryo. Although the
precise nature of the damage caused by cryopre-
servation remains to be determined, cryopreser-
vation severely taxes the reserves of the oocyte. In
this perspective, all the procedural steps from
thawing onward are best viewed as one long res-
cue mission, the success of which depends on the
integrated optimal use of a number of techniques
and procedures that form no part of the actual
freezing and thawing of the oocytes. These sup-
portive techniques include in vitro fertilization, in
vitro culture, and embryo transplantation, all of
which, being themselves imperfect, are sources of
variation. Others have also reported within-labo-
ratory variations in fetal development rates [1].
Expertise and organization with regard to the
supportive techniques are therefore essential to
maximize the outcome of oocyte cryopreserva-
tion. In the present study, ensuring that suitable
recipient mice would be available presented par-
ticular logistical problems, since the mice could
not be kept at the laboratory under close super-
vision, but had to be obtained on a daily basis
from a facility about 25 miles away. This ar-
rangement was by no means ideal.
The overall rate of normal fetal development
achieved after transfer of two-cell embryos from
cryopreserved oocytes in this study is at the same
level as the best reported in the literature [1,2,5,6].
This was achieved despite the fact that we cannot
be considered particularly experienced in oviduc-
tal embryo transfer in the mouse. More experi-
enced operators, with more conveniently located
mouse colonies, may achieve at least marginally
better results with the same freezing method.
However, our results are somewhat better than
those of Schroeder et al. [23]. In their study, over
1000 mouse oocytes were frozen using a standard
slow-cooling protocol and live pups were pro-
duced at a rate of 26% following two-cell transfer
to the oviduct.
Significant progress has been made in oocyte
vitrification and live offspring have been produced
from vitrifiedmouse oocytes. Early studies byKola
et al. [12], Wood et al. [30], and Nakagata [19–21]
report fetal development rates of between 5 and
36%. In addition, Kono et al. [13] demonstrated
oocyte viability after vitrification by producing 36
live pups (51% of those transferred; 25% per frozen
oocyte). In a more recent study Lane and Gardner
[15] compared the use of nylon loop vitrification to
slow-cooling using CJ1 medium (our original
choline-based freezing medium). They report some
of the highest survival, fertilization, in vitro de-
velopment, and fetal development rates (52% of the
embryos transferred; 37.9% of oocytes frozen) for
frozen mouse oocytes in the literature, using the
vitrification process. Their initial survival results
with CJ1 were similar to those in our original
publication describing the use of this medium [25],
but it is unclear why their fertilization and devel-
opment rates were significantly lower. It is also not
clear why they chose to use CJ1 instead of CJ2
(a modified choline-based medium) which we have
shown to work substantially better [24,26]. How-
ever, their fetal development rate of 11% for slowly
cooled oocytes in CJ1, 41% for their nonfrozen
controls, and 52% for their best group, the nylon
loop-vitrified oocytes, are similar to the results
presented here for slow-cooling oocytes in CJ2 and
transfer to the uterus (12.3%), nonfrozen controls
(47.2%), and our best group, slowly cooled oocytes
transferred to the oviduct (47.6%). Our data sug-
gest that their fetal rates could have been even
higher, especially for the CJ1 frozen oocytes, if the
resulting embryos were transferred to the oviduct
at the two-cell stage.
In the present experiments, delaying the
transfer of embryos from cryopreserved oocytes
(and zygotes) until they had reached the early
blastocyst stage severely reduced the rate of nor-
mal fetal development, while such an effect was
not evident in the control groups. A similar effect
10 J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13

of prolonged culture in vitro was previously ex-
perienced and discussed by George et al. [5]. Since
these are the only two reports involving uterine
transfers of embryos from slowly cooled mouse
oocytes, there seems to be a consensus that it is
preferable to transfer embryos from cryopreserved
oocytes at the two- to four-cell stage (see also [1]
and for a review [5]). In previous studies, we found
that in vitro culture after thawing and insemina-
tion of frozen oocytes is detrimental to blastocyst
development and that this can be significantly
improved by using a sequential (S1/S2) culture
media system [24–26]. Even though blastocyst
development is improved using sequential media,
the significant postimplantation loss (87.7%
[100) 12.3%]) observed when such blastocystswere transferred shows that the embryos, al-
though judged to be morphologically normal by
microscopic inspection, were in fact compromised
and that in vitro culture conditions were poor
compared to an in vivo environment [15]. Al-
though there is an apparent discrepancy between
the use of S1/S2 for the culture of cryopreserved
oocytes and the use of KSOM for control em-
bryos, this study was not aimed at optimizing
culture medium, but rather concerned (1) the
ability and rate of oocytes cryopreserved in CJ2 to
develop into viable fetuses in vivo and (2) the
differences, if any, in fetal development rates in
zygotes cryopreserved in CJ2 and ETFM. The
control embryos simply served as a reference
point by which comparisons could be made.
The ability of mouse oocytes to survive cryo-
preservation was far greater than expected, espe-
cially in the light of the literature on survival and
development with plunge temperatures higher
than )30 �C [4,29,31]. Although more oocytesdied during thawing and failed to become fertil-
ized after being cooled in a nonprogrammable
freezer and plunged at )20 �C (a breakpoint 13 �Cwarmer than those eggs cooled in the BioCool
freezer), as opposed to being cooled in a pro-
grammable freezer and plunged at )33 �C, themajority survived and became fertilized, and
nearly half of all two-cell embryos transferred
developed to term. Potentially detrimental factors
such as intracellular ice formation would have had
a much greater chance of occurring with plunging
into LN2 from )20 �C than from )33 �C, due tothe level of cellular dehydration achieved at the
respective temperatures. However, this was not
the case, indicating that lethal intracellular ice
formation was negligible even after cooling to
only )20 �C followed by submersion into LN2.
Although there have been numerous reports of
live births from embryos and oocytes plunged
from )30 �C after slow cooling, this is the firststudy to report live births from oocytes plunged
into LN2 from )20 �C. Indeed, it is still commonpractice to use plunge temperatures below )33 �C.Karlsson et al. [10] examined oocyte survival and
development with plunge temperatures ranging
from )30 to )150 �C, with )80 �C yielding max-imal recovery of oocytes. The present experiments
suggest that there are factors that lead to oocyte
demise during cryopreservation other than the
stresses induced by plunging from the relatively
high temperature of )20 �C. One such factorwould appear to be the dominance of sodium ions
in conventional cryopreservation media, but there
are most likely others.
The overall rate of fetal development obtained
when two-cell embryos from zygotes frozen in
ETFM were transferred to the oviduct was only
slightly higher than that obtained with embryos
from cryopreserved oocytes, and when comparing
the two results, it should be kept in mind that the
fertilization rate in vivo is not uniformly 100%.
However, while the oocyte group had a higher
implantation rate, fewer of the embryos that im-
planted gave rise to a fetus in this group than in
the zygote (ETFM) group (65.2% vs 80.0%); the
fetal development rate of implanted embryos from
zygotes frozen in CJ2 was lower than both
(48.5%). Thus, cryopreservation of oocytes ap-
pears to be more damaging to the cells’ ability to
form a fetus than cryopreservation of zygotes. We
are not able to adequately explain why CJ2 is
more effective as a freezing medium for oocytes
than ETFM, but it is interesting to note that no
significant advantage was observed regarding zy-
gotes with the protocol used in this study.
Acknowledgments
We thank Giles Tomkin for his critical review
of the manuscript, Anna Blasczyk for help in
preparing the mice used for these experiments,
and Dr. Fred Zander for providing the use of the
Fertilase system.
References
[1] A. Bos-Mikich, M.J. Wood, C.J. Candy, D.G.
Whittingham, Cytogenetical analysis and develop-
mental potential of vitrified mouse oocytes, Biol.
Reprod. 53 (1995) 780–785.
J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13 11

[2] J. Carroll, M.J. Wood, D.G. Whittingham, Normal
fertilization and development of frozen–thawed
mouse oocytes: Protective action of certain macro-
molecules, Biol. Reprod. 48 (1993) 606–612.
[3] C.L.Chatot,C.A.Ziomek,B.D.Bavister, J.L. Lewis,
I. Torres, An improved culture medium supports
development of random-bred 1-cell mouse embryos
in vitro, J. Reprod. Fertil. 86 (1989) 679–688.
[4] S. Friedler, L.C. Giudice, E.J. Lamb, Cryopreser-
vation of embryos and ova, Fertil. Steril. 49 (1988)
743–764.
[5] M.A. George, M.H. Johnson, S.K. Howlett, As-
sessment of the developmental potential of frozen–
thawed mouse oocytes, Hum. Reprod. 9 (1994) 130–
136.
[6] P.H. Glenister, M.J. Wood, C. Kirby, D.G. Whit-
tingham, Incidence of chromosome anomalies in
first-cleavage mouse embryos obtained from fro-
zen–thawed oocytes fertilized in vitro, Gamete Res.
16 (1987) 205–216.
[7] A.D. Hanson, B. Rathinasabapthi, J. Rivoal, M.
Burnet, M.O. Dillon, D.A. Gage, Osmoprotective
compounds in the Plumbaginaceae: A natural
experiment in metabolic engineering of stress toler-
ance, Proc. Natl. Acad. Sci. USA 91 (1994) 306–
310.
[8] J. Huang, R. Hirji, L. Adam, K.L. Rozwadowski,
J.K. Hammerlindl, W.A. Keller, G. Selvaraj, Ge-
netic engineering of glycinebetaine production to-
ward enhancing stress tolerance in plants:
Metabolic limitations, Plant Physiol. 122 (2000)
747–756.
[9] Y. Jolivet, J. Hamelin, F. Larher, Osmoregulation
in halophytic higher plants: The protective effects of
glycinebetaine and other related solutes against the
oxalate destabilization of membranes in beet root
cells, Z. Pflanzenphysiol. 109S (1983) 171–180.
[10] J.O. Karlsson, A. Eroglu, T.L. Toth, E.G. Crav-
alho, M. Toner, Fertilization and development of
mouse oocytes cryopreserved using a theoretically
optimized protocol, Hum. Reprod. 11 (1996) 1296–
1305.
[11] Y. Kimura, R. Yanagimachi, Intracytoplasmic
sperm injection in the mouse, Biol. Reprod. 52
(1995) 709–720.
[12] I. Kola, C. Kirby, J. Shaw, A. Davey, A. Trounson,
Vitrification of mouse oocytes results in aneuploid
zygotes and malformed fetuses, Teratology 38
(1988) 467–474.
[13] T. Kono, O.Y. Kwon, T. Nakahara, Development
of vitrified mouse oocytes after in vitro fertilization,
Cryobiology 28 (1991) 50–54.
[14] J.P. Krall, G.E. Edwards, C.S. Andreo, Protection
of pyruvate, Pi dikinase from maize against cold
lability by compatible solutes, Plant Physiol. 89
(1989) 280–285.
[15] M. Lane, D.K. Gardner, Vitrification of mouse
oocytes using a nylon loop, Mol. Reprod. Dev. 58
(2001) 342–347.
[16] W.A. Lloyd, J.A. Baker, C.J. Olliff, K.J. Rutt,
The evaluation of N-modified betaines as erythro-
cyte cryoprotectants, Cryo-Letters 13 (1992) 337–
348.
[17] A.W. Lloyd, C.J. Oliff, K.J. Rutt, A study of
modified betaines as cryoprotective additives, J.
Pharm. Pharmacol. 46 (1994) 704–707.
[18] R.D. Lynch, E.E. Schneeberger, R.P. Geyer, Alter-
ations in L fibroblasts lipid metabolism and mor-
phology during choline deprivation, Exp. Cell Res.
122 (1979) 103–113.
[19] N. Nakagata, High survival rate of unfertilized
mouse oocytes after vitrification, J. Reprod. Fertil.
87 (1989) 479–483.
[20] N. Nakagata, Production of normal young follow-
ing transfer of mouse embryos obtained by in vitro
fertilization between cryopreserved gametes, J. Re-
prod. Fertil. 99 (1993) 77–80.
[21] N. Nakagata, Studies on cryopreservation of em-
bryos and gametes in mice, Exp. Animals 44 (1995)
1–8.
[22] D. Rhodes, A.D. Hanson, Quartenary ammonium
and tertiary sulfonium compounds in higher plants,
Annu. Rev. Plant Physiol. Plant Mol. Biol. 44
(1993) 357–384.
[23] A.C. Schroeder, A.K. Champlin, L.E. Mobraaten,
J.J. Eppig, Developmental capacity of mouse oo-
cytes cryopreserved before and after maturation in
vitro, J. Reprod. Fertil. 89 (1990) 43–50.
[24] J.J. Stachecki, J. Cohen, S.M. Willadsen, Cryo-
preservation of unfertilized mouse oocytes: The
effect of replacing sodium with choline in the
freezing medium, Cryobiology 37 (1998) 346–
354.
[25] J.J. Stachecki, J. Cohen, S.M. Willadsen, Detri-
mental effects of sodium during oocyte cryopreser-
vation, Biol. Reprod. 59 (1998) 395–400.
[26] J.J. Stachecki, S.M. Willadsen, Cryopreservation of
mouse oocytes using a medium with low sodium
content: Effect of plunge temperature, Cryobiology
40 (2000) 4–12.
[27] M. Toner, E.G. Cravalho, J. Stachecki, T. Fitz-
gerald, R.G. Tompkins, M.L. Yarmush, D.R.
Armant, Nonequilibrium freezing of one-cell
mouse embryos: Membrane integrity and devel-
opmental potential, Biophys. J. 64 (1993) 1908–
1921.
[28] L.J. Van Winkle, N. Haghighat, A.L. Campoine,
Glycine protects preimplantation mouse concep-
tuses from a detrimental effect on development of
the inorganic ions in oviductal fluid, J. Exp. Zool.
253 (1990) 215–219.
[29] D.G. Whittingham, M. Wood, J. Farrant, H. Lee,
J.A. Halsey, Survival of frozen mouse embryos after
rapid thawing from )196 �C, J. Reprod. Fertil. 56(1979) 11–21.
[30] M.J. Wood, C. Barros, C.J. Candy, J. Carroll, J.
Melendez, D.G. Whittingham, High rates of sur-
vival and fertilization of mouse and hamster oocytes
12 J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13

after vitrification in dimethylsulphoxide, Biol. Re-
prod. 49 (1993) 489–495.
[31] M.J. Wood, J. Farrant, Preservation of mouse
embryos by two-step freezing, Cryobiology 17
(1980) 178–180.
[32] Y. Zhao, D. Aspinall, L. Paleg, Protection of
membrane integrity in Medicago sativa L. by
glycinebetaine against the effects of freezing, J.
Plant Physiol. 140 (1992) 541–543.
J.J. Stachecki et al. / Cryobiology 44 (2002) 5–13 13