factores clÍnicos, analÍticos, anatomopatolÓgicos y
TRANSCRIPT

DEPARTAMENTO DE CIRUGÍA, CIENCIAS MÉDICAS Y SOCIALES
PROGRAMA DE DOCTORADO DE CIRUGÍA
FACTORES CLÍNICOS, ANALÍTICOS, ANATOMOPATOLÓGICOS Y
MOLECULARES PREDICTIVOS DE RECIDIVA EN EL CÁNCER
DE COLON SIN AFECTACIÓN GANGLIONAR
TESIS DOCTORAL
RAQUEL GRAJAL MARINO
Madrid, 2014
DIRECTORES:
D. JOSÉ MANUEL DEVESA MÚGICA
(Jefe de Sección de Coloproctología del Servicio de Cirugía General y Digestiva del Hospital Universitario
Ramón y Cajal)
D. ADOLFO LÓPEZ BUENADICHA
(Profesor Asociado en Ciencias de la Salud del Área de Cirugía del Departamento de Cirugía de la
Universidad de Alcalá y Facultativo Especialista de Área del Servicio de Cirugía General y Digestiva del
Hospital Universitario Ramón y Cajal)


A Luis, Juan y Elia
A mis padres y mi hermana


AGRADECIMIENTOS
Al Dr. Manuel Devesa por el apoyo y la confianza depositada en mí, así como por la
paciencia a lo largo de este trabajo, y por darme las herramientas para que esta tesis
sea una realidad.
Al Dr. Javier Die, por toda labor inicial en este trabajo, y por ser un pilar profesional y
personal durante el tiempo que he compartido con él en la residencia y
posteriormente en el equipo de Coloproctología.
Al Dr. Antonio Rey, por animarme a luchar por mi carrera profesional, y estar siempre
que he necesitado un consejo.
Al Dr. Adolfo López Buenadicha por toda su labor docente a lo largo de la residencia y
por acompañarme en este trabajo.
A todas las personas que forman el Servicio de Cirugía General del Hospital
Universitario Ramón y Cajal, imprescindibles en mi aprendizaje y sin cuyo acogimiento
a lo largo de seis años, no sería la profesional que soy actualmente.
Al Dr. Manuel Morente y la Dra. María Jesus Artiga, del CNIO, sin cuya labor
desinteresada y ayuda, este trabajo no hubiera sido posible.
A Servicio de Estadística del Hospital Ramón y Cajal, en especial a Alfonso Muriel, por
su inestimable ayuda en el análisis de los datos.
Al Dr. Fernando Piniella, por darme la oportunidad de continuar desarrollándome
como cirujana en esta nueva etapa en el Hospital Santos Reyes de Aranda de Duero.
A Luis, por ser el pilar de mi vida.
A Clara, por estar siempre ahí.
A mis padres, porque sin ellos no sería quien soy, y sin su apoyo, no habría llegado
hasta aquí.


ABSTRACT
INTRODUCCIÓN. El cáncer de colon y recto es una enfermedad neoplásica que ocupa el
segundo lugar en frecuencia en mujeres y el tercero en hombres a nivel mundial. El
pronóstico del cáncer de colon sin afectación ganglionar es relativamente bueno, con
una supervivencia a los 5 años aproximada de entre el 70 y el 87 %. Las guías
oncológicas actuales no recomiendan la administración de quimioterapia adyuvante de
forma rutinaria en pacientes con cáncer de colon sin afectación ganglionar, ya que no
hay evidencia de beneficio en la supervivencia libre de enfermedad ni global.
En la actualidad se están desarrollando múltiples trabajos dirigidos a la detección de
aquellos factores de riesgo, tanto del paciente, como anatomopatológicos y
moleculares que nos permitan seleccionar los pacientes con cáncer de colon sin
afectación ganglionar que se benefician de la quimioterapia adyuvante por presentar
mayor riesgo de recidiva.
MATERIAL Y METODOS. Realizamos un estudio de cohortes retrospectivo de 141
pacientes intervenidos de cáncer de colon con el resultado anatomopatológico T2-T4
N0 en la pieza quirúrgica en el Hospital Universitario Ramón y Cajal entre los años 1975
y 2007, analizando durante el seguimiento la aparición de recidiva local o a distancia.
Se revisan factores clínicos, bioquímicos de los pacientes y factores
anatomopatológicos y moleculares de las piezas quirúrgicas.
RESULTADOS. De la cohorte de 141 pacientes, 25 (18%) presentaron recidiva durante
el seguimiento. La mediana de supervivencia global fue de 45 meses en el grupo
recidiva y de 78 meses en el grupo sin recidiva. En el análisis univariante, resultaron de
pronóstico adverso para la supervivencia libre de enfermedad con carácter
significativo: la profundidad de invasión (estadio T), la presencia de infiltración vascular
y la inestabilidad de microsatélites. La inmunohistoquímica positiva para c-Kit, c-myc y
la presencia de menos de 12 ganglios linfáticos en la pieza quirúrgica resultaron de
pronóstico adverso y muy próximos a la significación estadística.
CONCLUSIONES. La presencia de factores de riesgo como T4, infiltración vascular,
inestabilidad de microsatélites, la presencia de menos de 12 ganglios linfáticos en la
pieza o la inmunohistoquímica positiva para c-kit o c-myc en pacientes con cáncer de
colon resecado T2-T4 sin afectación ganglionar, confiere un peor pronóstico, y por lo
tanto debe plantearse tratamiento adyuvante. Se deben seguir estudiando qué
factores pueden ser pronósticos de recidiva en el estadio II (T3-T4 N0) del cáncer de
colon intervenido quirúrgicamente con intención curativa, para determinar un perfil de
riesgo individual.

ABSTRACT (EN INGLÉS)
INTRODUCTION. Cancer of the colon and rectum is the second most frecuent neoplasic
disease in women and the third in men in the world. Colon cancer prognosis when
lymph nodes are not involved is quite good, with a five-years-survival estimated
between 70-87 %. Current oncologic guides do not recommend rutinary adjuvant
chemotherapy in colon cancer patients without involvement of lymph nodes because
there is not enough evidence of the benefit in either disease-free survival or overall
survival.
Actually, there are several studies ongoing trying to detect which factors, either from
the patient, the anatomopathologic analysis of the specimen and the molecular
analysis of the tumor could give the tools to select those patients whom would benefit
from adjuvant chemotherapy when the colon cancer has not spread to the lymph
nodes.
MATERIALS AND METHODS. This is a retrospective cohorts study involving 141 patients
with the diagnosis of T2-T4 N0 colon cancer in the specimen, operated in Hospital
Universitario Ramón y Cajal (Madrid, Spain) between 1975 and 2007. We analyzed
during the surveillance the occurrence of local or distant recurrence. We study
retrospectively clinical, biochemical factors of the patients and anatomopathologic and
molecular factors of the specimens
RESULTS. From the group of 141 patients, 25 (18%) of them developed recurrence
during surveillance. The overall survival median in the recurrence group was 45
months and 78 months in the free-disease group. The univariant analysis showed
worse prognosis with significant differences in disease-free-survival in the deepness of
the tumor invasion (T status), vascular infiltration and microsatellites instability. The
immunochemistry for C-kit or c-myc and absence of 12 or more lymph nodes in the
specimen were adverse prognosis factors nearby the statistic significance.
CONCLUSIONS. The presence of several risk factors as T4, vascular infiltration,
microsatellite instability, less than 12 lymph nodes in the specimen or positive
immunochemistry to c-kit or c-myc in patients with resected T2-T4 N0 colon cancer will
give those patients a higher risk of recurrence, and so adjuvant chemotherapy should
be proposed to them. There is still a long way to go in the study of which factors could
be prognostic of recurrence in colon cancer stage II (T3-T4 N0) after oncologic surgery,
and they will determinate the individual risk of each patient.

ABREVIATURAS
AJCC American Joint Committee on Cancer
AP Anatomía Patológica
ASCO American Society of Clinical Oncology
CAP Colegio Americano de Patólogos
CC Cáncer de colon
CCR Cáncer colorrectal
CDK Quinasa dependiente de ciclina
CKI Inhibidor de CDK
CIMP CpG Islands Metilation Phenotipe
CNIO Centro Nacional de Investigaciones Oncológicas
EGFR Epithelial Growth Factor Receptor
FAP Poliposis adenomatosa familiar
HNPCC Cáncer colorrectal hereditario no polipósico
HR Hazard ratio
IAP Proteína inhibidora de la apoptosis
IC Intervalo de confianza
IMS Inestabilidad de microsatélites
Me mediana
MMR Mismatch Repair
NCCN National Comprehensive Cancer Network

Pág. Página
pRB Proteína del Retinoblastoma
SEER Surveillance, epidemiology and end results Program
SG Supervivencia Global
SLE Supervivencia libre de enfermedad
UICC Unión internacional contra el cáncer
5-FU 5 Fluorouracilo

ÍNDICE


INTRODUCCIÓN 19
1. El cáncer de colon 21
2. Factores clínico, analíticos y anatomopatológicos pronósticos en el
cáncer de colon 27
2.1. Factores clínicos 28
2.2. Factores analíticos 29
2.3. Factores anatomopatológicos 30
2.3.1. Extensión local del tumor (estadiaje T)
2.3.2. Número de ganglios linfáticos
2.3.3. Grado histológico
2.3.4. Tipo histológico
2.3.5. Invasión linfovascular
2.3.6. Invasión perineural
2.3.7. Inestabilidad de microsatélites
3. Factores genético-moleculares pronósticos en el cáncer de colon 35
3.1. El ciclo celular 35
3.1.1. Control del ciclo celular
3.1.2. Estímulos intrínsecos del ciclo celular
3.1.3. Estímulos extrínsecos del ciclo celular
3.1.4. Apoptosis
3.2. Génesis del cáncer 44
3.2.1. Los fundamentos del proceso de carcinogénesis.
3.2.2. Desregulación del ciclo celular en el cáncer.
3.2.3. Las metástasis en el cáncer
3.2.4. Vías moleculares en la transformación neoplásica del cáncer de colon
3.2.4.1. La inestabilidad cromosómica
3.2.4.2. La inestabilidad de microsatélites
3.2.4.3. La vía de la isla CG de metilación (CIMP)
3.2.5. Secuencia adenoma carcinoma en el cáncer de colon

3.3. Biomarcadores 58
3.3.1. Definición
3.3.2. Biomarcadores moleculares en el cáncer de colon
3.3.2.1. Oncogenes
3.3.2.2. Genes supresores tumorales
3.3.2.3. Genes reparadores de ADN
3.3.2.4. Proteínas de la vía de la apoptosis
3.3.2.5. Proliferación celular
3.3.2.6. Angiogénesis, metástasis e invasión tisular
3.3.2.7. Estructura celular
3.3.2.8. Diferenciación celular
4. Seguimiento, pronóstico y supervivencia del cáncer de colon 83
4.1. Seguimiento 83
4.2. Tratamiento adyuvante de cáncer de colon 85
4.3. Pronóstico del cáncer de colon. 86
HIPOTESIS Y OBJETIVOS 87
Justificación 89
Hipótesis 91
Objetivos 91
MATERIAL Y METODOS 93
1. Diseño del estudio 95
2. Esquema de diseño 96
2.1. Factores clínicos 95
2.2. Factores analíticos 96
2.3. Factores anatomopatológicos 96
2.4. Factores moleculares y biológicos 97
2.5. Momento de la determinación de las variables 99
3. Población de estudio 100
3.1. Población diana 100

3.2. Tamaño de la muestra 100
3.3. Selección de la muestra 101
3.4. Selección de los grupos de trabajo 103
4. Procedimiento 104
4.1. Recogida de datos 104
4.2. Parámetros anatomopatológicos 106
4.3. Parámetros moleculares 106
5. Descripción operativa 109
6. Limitaciones del estudio 111
7. Metodología estadística 112
7.1. Estadística descriptiva 112
7.2. Estadística analítica 112
7.3. Procesamiento de datos 113
RESULTADOS 115
1. Estadística descriptiva 117
1.1. Población estudiada 117
1.2. Variables demográficas 117
1.3. Características de los pacientes que presentaron recidiva 118
1.4. Características de los pacientes con ausencia de enfermedad 118
1.5. Variables de presentación clínica 120
1.6. CEA sérico prequirúrgico 121
1.7. Estadiaje tumoral 122
1.7.1. TNM
1.7.2. Modificado de Astler-Coller
1.7.3. Dukes
1.8. Variables anatomopatológicas 125
1.8.1. Diferenciación histológica
1.8.2. Células en anillo de sello
1.8.3. Componente mucinoso
1.8.4. Infiltración vascular
1.8.5. Infiltración linfática

1.8.6. Infiltración perineural
1.8.7. Número de ganglios linfáticos
1.8.8. Características de riesgo agrupadas
1.9. Variables moleculares 130
2. Estadística analítica 135
2.1. Seguimiento y supervivencia 137
2.2. Análisis por regresión de Cox 140
2.2.1. Variables clínicas y analíticas
2.2.2. Variables anatomopatológicas
2.2.3. Variables moleculares
DISCUSION 153
1. La situación actual del cáncer de colon en estadio II 155
2. Factores clínicos: obstrucción/perforación 158
3. Factores analíticos: CEA sérico 159
4. Factores anatomopatológicos 160
4.1. Profundidad de la invasión (Estadio T) 160
4.2. Grado histológico 162
4.3. Invasión vascular 164
4.4. Invasión linfática 165
4.5. Invasión perineural 166
4.6. Número de ganglios 168
5. Marcadores moleculares 170
5.1. C-Kit 171
5.2. C-myc 172
5.3. Inestabilidad de microsatélites 174
CONCLUSIONES 177
BIBLIOGRAFÍA 181
ANEXOS 191
I. Procesamiento inmunohistoquímico de las muestras de cáncer de colon

II. Tablas de variables demográficas.
III. Tablas de variables del estadiaje tumoral.
IV. Tablas de variables histológicas.
V. Tablas de resultados de variables moleculares.
VI. Agrupación de variables moleculares de forma dicotómica.


INTRODUCCIÓN


INTRODUCCIÓN 21
1.- El cáncer de colon
El cáncer de colon y recto (CCR) supone el 10% de los casos de cáncer diagnosticados
cada año tanto en hombres como en mujeres y constituye en la actualidad una de las
causas más importantes de morbimortalidad en Europa, EEUU y otros países
desarrollados con similares estilos de vida y hábitos dietéticos. 1
El CCR causó globalmente el 11% de las defunciones por cáncer en hombres y el 15%
en mujeres, según datos del año 2000 1. Aunque no se conocen las cifras exactas en
España se estima que el número de casos nuevos por año se sitúa en torno a los
21.000 en ambos sexos frente a 11.900 defunciones. La mortalidad es mayor en el caso
de los varones. El cáncer colorrectal constituye la segunda localización tumoral más
frecuente en mujeres y la tercera en hombres1,2.
La incidencia en España de CCR según la tasa ajustada por edad es de 35,54/100.000
en hombres y 16,46/100.000 en mujeres. A nivel europeo, nos encontramos por
debajo de las tasas promedio de Europa. Salvo Finlandia, Suecia y Grecia, el resto de
países presentan mayor incidencia de CCR que España.
El tratamiento quirúrgico es el de elección en la mayoría de los pacientes. El objetivo
de la cirugía es la resección amplia del segmento de colon afecto, junto con todo su
drenaje linfático. El número de ganglios extirpados durante la cirugía ha sido
identificado como una medida importante de la calidad de la cirugía. Una revisión de la
literatura, que incluye el análisis de 17 estudios, sugirió que el número de ganglios
evaluados tras la cirugía está asociado positivamente con la supervivencia 3.
En la clasificación TNM, el estadio I (T1-2/N0) y el estadio II del cáncer de colon (T3-
4/N0) representan aproximadamente el 39% de todos los tumores de colon
diagnosticados. Su tratamiento principal es la extirpación del tumor y de los ganglios
regionales. El estadio I tiene una supervivencia a los cinco años del 93.2%, y en el
estadio II la tasa de supervivencia a los cinco años se sitúa entre el 72 y el 85%. La
disminución en la supervivencia se produce por la aparición de recidiva local o a
distancia tras la cirugía.4,5

22 INTRODUCCIÓN
El pronóstico del CCR está condicionado fundamentalmente por el estadio al
diagnóstico, variando la supervivencia a cinco años entre un 90% en estadios
localizados a un 10% en caso de metástasis a distancia. En conjunto, se estima que la
supervivencia actual en el cáncer de colon se sitúa en torno al 65% a los cinco años en
EEUU.6
El estadiaje clásico se basa en el sistema desarrollado en 1932 por el anatomopatólogo
del hospital St. Mark´s de Londres, el Dr. Dukes. La clasificación de Dukes es bastante
simple: 7
o Estadio A: el tumor está limitado a la pared intestinal
o Estadio B: el tumor penetra la pared intestinal.
o Estadio C: el tumor invade los ganglios linfáticos.
Posteriormente se modificó la clasificación para añadir el estadio D para los tumores
con metástasis a distancia. Más adelante, apareció la clasificación de Astler-Coller que
modificó a la clasificación previa de Dukes 7.
Sin embargo, actualmente la clasificación que más se utiliza es la desarrollada por el
American Joint Committee on Cancer (AJCC) y aprobada por la Unión Internacional
contra el cáncer (UICC) 6–8. Esta clasificación, conocida como TNM (del inglés: tumor,
node, metastasis), combina la información clínica obtenida preoperatoriamente con
los datos obtenidos durante la cirugía y el análisis histológico de la pieza quirúrgica, y
aporta la ventaja de que se actualiza continuamente además de permitir su aplicación
de forma estandarizada. La clasificación actual se basa en la 7ª Edición publicada en
2010, según se describe a continuación: 9,10

INTRODUCCIÓN 23
CLASIFICACIÓN TNM
TUMOR
PRIMARIO
TX No determinado
T0 No evidencia de tumor primario
Tis Carcinoma in situ: intraepitelial o con invasión de la lámina
propia
T1 Invasión de la submucosa
T2 Invasión de la muscular propia
T3 Invasión de tejido pericólico a través de la muscular propia
T4a Invasión del peritoneo visceral
T4b Invasión directa o está adherido a otros órganos o estructuras
TABLA 1. Clasificación del estadio T del cáncer de colon según la7ª edición de la AJCC.
GANGLIOS LINFÁTICOS
REGIONALES
NX No puede ser evaluado
N0 Ausencia de metástasis en ganglios linfáticos regionales
N1 Metástasis en1-3 ganglios
N1a Metástasis en un ganglio
N1b Metástasis en2-3 ganglios
N1c
Depósitos tumorales en la subserosa, mesenterio o tejido
pericólico no peritonizado sin metástasis a ganglios linfáticos
regionales
N2 Metástasis en 4 o más ganglios
N2a Metástasis en 4-6 ganglios
N2b Metástasis en 7 o más ganglios linfáticos regionales
TABLA 2. Clasificación del estadio N del cáncer de colon según la7ª edición de la AJCC.
METÁSTASIS A
DISTANCIA
M0 Ausencia de metástasis a distancia
M1 Metástasis a distancia
M1a Metástasis confinado a un órgano o sitio
M1b Metástasis en más de un órgano o sitio o en el peritoneo
TABLA 3. Clasificación del estadio M del cáncer de colon según la7ª edición de la AJCC.

24 INTRODUCCIÓN
Las equivalencias entre la clasificación de Dukes, la de Astler-Coller Modificada y la
TNM se refleja en la siguiente tabla:
TABLA 4. ESTADIAJE ANATÓMICO/GRUPOS PRONÓSTICOS. Adaptada de la 7ª Edición de la
AJCC. 9
Estadio T N M Dukes Modificada de
Astler-Coller
0 Tis N0 M0
I T1 N0 M0 A A
T2 N0 M0 A B1
II A T3 N0 M0 B B2
II B T4a N0 M0 B B2
II C T4b N0 M0 B B3
III A T1-T2 N1/N1c M0 C C1
T1 N2a M0 C C1
III B T3-T4a N1/N1c M0 C C2
T2-T3 N2a M0 C C1/C2
T1-T2 N2b M0 C C1
III C T4a N2a M0 C C2
T3-T4a N2b M0 C C2
T4b N1-N2 M0 C C3
IV A Cualquier T Cualquier N M1a D
IV B Cualquier T Cualquier N M1b D
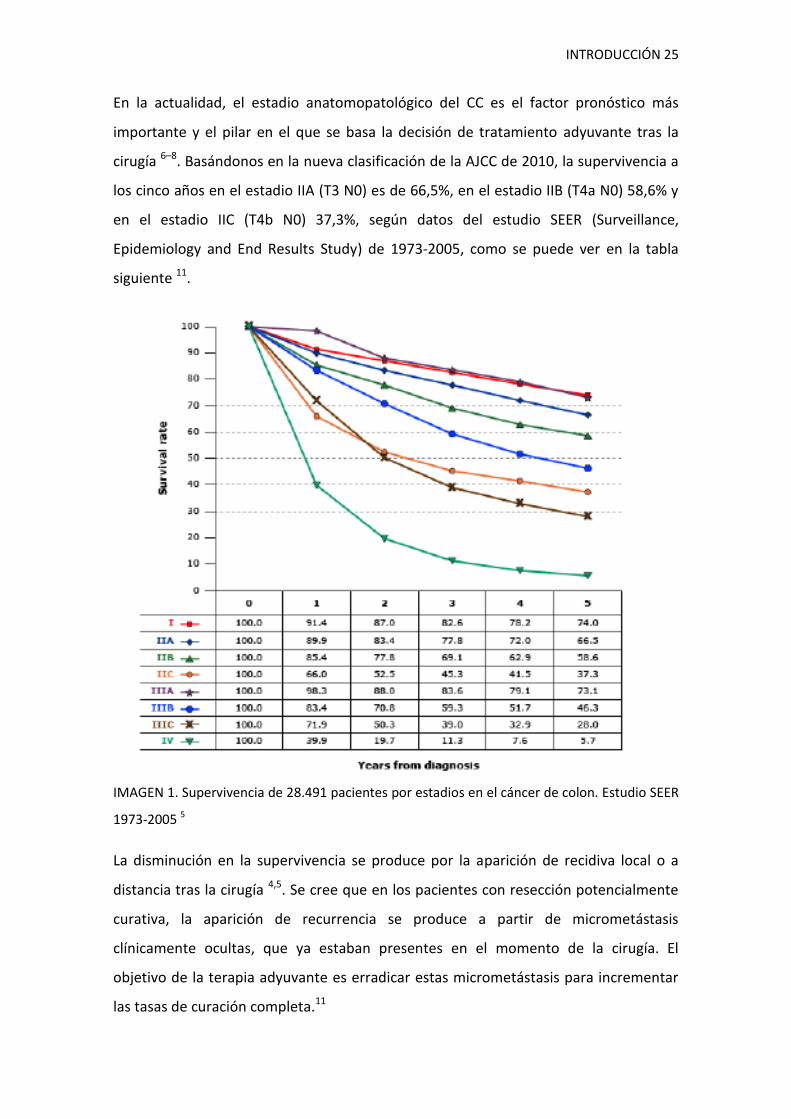
INTRODUCCIÓN 25
En la actualidad, el estadio anatomopatológico del CC es el factor pronóstico más
importante y el pilar en el que se basa la decisión de tratamiento adyuvante tras la
cirugía 6–8. Basándonos en la nueva clasificación de la AJCC de 2010, la supervivencia a
los cinco años en el estadio IIA (T3 N0) es de 66,5%, en el estadio IIB (T4a N0) 58,6% y
en el estadio IIC (T4b N0) 37,3%, según datos del estudio SEER (Surveillance,
Epidemiology and End Results Study) de 1973-2005, como se puede ver en la tabla
siguiente 11.
IMAGEN 1. Supervivencia de 28.491 pacientes por estadios en el cáncer de colon. Estudio SEER
1973-2005 5
La disminución en la supervivencia se produce por la aparición de recidiva local o a
distancia tras la cirugía 4,5. Se cree que en los pacientes con resección potencialmente
curativa, la aparición de recurrencia se produce a partir de micrometástasis
clínicamente ocultas, que ya estaban presentes en el momento de la cirugía. El
objetivo de la terapia adyuvante es erradicar estas micrometástasis para incrementar
las tasas de curación completa.11

26 INTRODUCCIÓN
Los beneficios de la quimioterapia adyuvante se han demostrado claramente en el
estadio III (es decir, N positivo), con una reducción relativa del 30% del riesgo de
recurrencia y de un 22% a un 32% de reducción relativa en la mortalidad, por lo que el
abordaje estándar en el estadio III de CC incluye la quimioterapia adyuvante tras la
cirugía con intención curativa.
Sin embargo, el beneficio de la quimioterapia adyuvante en el estadio II no está tan
claro y su uso en este grupo es variable11. El tratamiento con terapia adyuvante en
pacientes en estadio II del cáncer de colon no se recomienda de forma generalizada,
sólo en el contexto de ensayos clínicos. La ASCO (American Society of Clinical
Oncology) sugiere la utilización de terapia adyuvante basada en 5-FU en pacientes en
estadio II con al menos un factor de riesgo de mal pronóstico 7,8. Actualmente se están
estudiando diversos factores clínicos, analíticos, anatomopatológicos y moleculares
que nos permitan seleccionar mejor a los pacientes con mayor riesgo de desarrollar
una recidiva durante el seguimiento.
Este trabajo analiza factores clínicos, analíticos, anatomopatológicos y moleculares en
un grupo de pacientes intervenidos de cáncer de colon con estadio B de Astler-Coller
(correspondiente al estadio I y II de la AJCC, T2-4 N0) sin adyuvancia posterior, en el
Hospital Universitario Ramón y Cajal, y su relación con la aparición de recidivas
durante el seguimiento.

INTRODUCCIÓN 27
2.- Factores clínicos, analíticos y anatomopatológicos pronósticos en el
cáncer de colon
La actualización de la 7ª Edición de la AJCC sobre el cáncer de colon, establece cambios
en la clasificación tumoral basados en el pronóstico, tras el análisis de diversos
estudios epidemiológicos sobre el cáncer de colon.
En esta nueva actualización de la clasificación TNM el estadio T4 se divide en T4a y T4b
en función de si invade el peritoneo visceral o un órgano sólido, respectivamente. De
esta forma subdivide a los pacientes en estadio II en: IIa (T3 N0), IIb (T4a N0) y IIc (T4b
N0). Además, aunque no lo recoge en el estadiaje, recomienda la recogida de factores
pronósticos específicos como los niveles de CEA preoperatorio, la presencia o ausencia
de invasión perineural, la inestabilidad de microsatélites y el análisis del gen K-ras.11
La NCCN (National Comprehensive Cancer Network) considera los siguientes factores
como indicativos de alto riesgo de recurrencia 12:
o Obstrucción.
o Perforación.
o Márgenes positivos, próximos o indeterminados.
o Diferenciación histológica grado 3 o 4.
o Invasión perineural o linfovascular.
o Menos de 12 ganglios en la pieza quirúrgica
Los factores reflejados a continuación son de categoría I, es decir, comprobados en
ensayos clínicos amplios y que se utilizan actualmente en la toma de decisiones clínicas
en los pacientes con CC; o IIA, lo que significa que hay evidencia suficiente para ser
considerados factores pronósticos, aunque su relevancia a nivel clínico todavía no está
suficientemente probada. Ambos tipos de factores, debería venir reflejados en la
historia clínica del paciente y en el análisis anatomopatológico de la pieza quirúrgica
para la toma de decisiones más adecuada en cada paciente.
Otros factores de menor impacto, como los de categoría IIB, son aquellos que aunque
parecen estar relacionados con el pronóstico, no tienen suficiente evidencia para ser
incluidos en las categorías I ni IIA.

28 INTRODUCCIÓN
2.1. CLÍNICOS
Obstrucción/perforación colónica
La presencia de obstrucción o perforación intestinal se ha relacionado con un peor
pronóstico, independientemente del estadio. Los pacientes con lesiones obstructivas
no siempre son candidatos a una cirugía curativa y tienen mayores tasas de
morbimortalidad quirúrgica. La recidiva tras cirugía curativa también es mayor en
aquellos pacientes con obstrucción o perforación colónica. 5,8
Estos factores tienen evidencia de categoría I. Se ha visto que independientemente del
estadio y del resto de características anatomopatológicas del tumor, la presencia de
perforación u obstrucción en el debut del tumor empeora el pronóstico. 13

INTRODUCCIÓN 29
2.2. ANALÍTICOS
CEA sérico
Los niveles de CEA deberían medirse de forma rutinaria antes de la cirugía en
pacientes sometidos a resecciones curativas de cáncer de colon. Esto es por dos
motivos:10
1. Los niveles preoperatorios elevados de CEA que no se normalizan tras la
resección quirúrgica implican la presencia de enfermedad persistente y
precisan de una evaluación adicional.
2. Los niveles preoperatorios de CEA tienen significado pronóstico. Niveles
superiores a 5ng/ml tienen un efecto adverso en la supervivencia que es
independiente del estadio tumoral. En el estudio SEER con casi 18.000
pacientes y una mediana de seguimiento de 27 meses, se encontró un aumento
en el riesgo de mortalidad global (HR 1.6, 95% IC 1.46-1.76),
independientemente del estadio tumoral. Un hallazgo intrigante fue que en los
pacientes con N0 y elevación preoperatoria de los niveles de CEA tenían peor
pronóstico (HR para muerte de 1.75, 95% IC 1.48-2.09) que los que tenían N
positivo y CEA preoperatorio normal, aunque en este estudio faltaba
información sobre la quimioterapia adyuvante.
Toda esta información sugiere que la elevación de CEA preoperatorio debería
incorporarse al estadiaje convencional TNM en el cáncer de colon. La clasificación TNM
de 2010 de la AJCC no incluye el CEA en el estadiaje, pero recomienda que la
información sea recogida por su valor pronóstico.10
Esta información también apoya la idea de que el CEA preoperatorio debe tenerse en
consideración en relación con la decisión de quimioterapia adyuvante tras la cirugía,
especialmente en pacientes con ganglios negativos y CEA preoperatorio elevado, por
su aumento de riesgo. En el consenso de 2006 de la ASCO concluyeron, sin embargo,
que no había suficiente evidencia para recomendar el uso de CEA como determinante
si se debía administrar adyuvancia a los pacientes con cáncer de colon.10
El nivel de evidencia es de categoría I.

30 INTRODUCCIÓN
2.3. ANATOMOPATOLÓGICOS
Son los factores más ampliamente estudiados como pronósticos de recidiva en el
cáncer de colon estadio II. Están disponibles en la mayoría de los pacientes
intervenidos de CC, lo que facilita el análisis de los mismos. La mayor parte de ellos se
han demostrado pronósticos de recidiva, y se utilizan en la práctica habitual,
clasificando a los pacientes que presentan dichos factores como “alto riesgo” para la
toma de decisiones en relación con la administración de terapia adyuvante.13
2.3.1 Extensión local del tumor (estadio T)
La profundidad con la que penetra el tumor en las capas de la pared colónica está
relacionada con la supervivencia, de forma que a mayor profundidad de invasión, peor
es el pronóstico del paciente. Sin embargo, el estudio de los factores que determinan
el estadio T son variables, especialmente en lo que se refiere a la infiltración de la
serosa. Se ha visto que la infiltración de la serosa por el tumor es un factor patológico
adverso.
No hay guías anatomopatológicas claras que estandaricen la interpretación de la
invasión de la serosa (estadio T4), y en los casos en los que no quede clara si existe
infiltración (porque se asocie a reacción inflamatoria, erosión o ulceración, por
ejemplo), se debe categorizar como si no la hubiera, es decir estadio T3.
El nivel de evidencia es de categoría I. 10
2.3.2 Grado histológico
El grado histológico refleja el grado de diferenciación tumoral, y ha mostrado ser factor
pronóstico independiente del estadio tumoral. Sin embargo, el estadio histológico es
un parámetro subjetivo, con una variabilidad interobservador y sin un sistema
uniforme aceptado de forma general. La interpretación del grado se basa en la
valoración del tumor por completo o de la peor área, por la valoración de la cantidad

INTRODUCCIÓN 31
de formación glandular únicamente o la valoración combinada de las glándulas y de
otras características citológicas o estructurales. 6,8,10
En la mayor parte de los estudios el significado pronóstico del grado se divide en dos:
bajo grado (tumores bien y moderadamente diferenciados) y alto grado (tumores
pobremente diferenciados e indiferenciados). La formación glandular está presente de
mayor o menor manera en tumores de bajo grado. Sin embargo, en los tumores de
alto grado, no se forman estructuras glandulares bien definidas, sino más bien
cordones y hojas sólidas de células infiltrantes, a veces con marcada atipia,
pleomorfismo y una alta tasa mitótica.
Tanto la AJCC como el CAP (Colegio Americano de Patólogos) recomiendan la adopción
de un sistema dicotómico y utilizar la formación glandular como única característica
analizada (por ejemplo, mayor o menor del 50% de formación glandular). Un sistema
dicotómico con un punto de corte bien definido debería reducir las diferencias
interobservador y preservar o mejorar el poder pronóstico del grado histológico.
El nivel de evidencia es de categoría IIA. 10
2.3.3 Tipo histológico
La mayor parte de los cánceres de colon son adenocarcinomas, que posteriormente
son clasificados según el grado histológico. 8,10
Muchos tumores producen mucina, y esta sustancia puede quedarse dentro de las
células o ser secretada. La mucina extracelular se extiende a través de la pared
tumoral, ayudando a su extensión local. Los tumores productores de mucina
extracelular se clasifican como “mucinosos” cuando la proporción de mucina es más
del 50% de la masa tumoral. Este subtipo histológico ocurre en el 11-17% de los
tumores de colon y recto, y tiene predilección por el recto y el colon sigmoide.
Además, parecen diagnosticarse en un estadio más avanzado, y tener baja respuesta a
quimioradioterapia adyuvante y a tratamiento con 5-FU. 8,10

32 INTRODUCCIÓN
Como regla general, no se ha mostrado como factor pronóstico en el cáncer de colon,
a excepción de algunos subtipos de alto grado (anillo de sello, poco diferenciados o
indiferenciados). Además pocos estudios han evaluado la influencia del subtipo
histológico tras la estratificación en la presencia o ausencia de inestabilidad de
microsatélites.
El nivel de evidencia es de categoría IIB. 10
2.3.4 Invasión linfovascular
La invasión tumoral a venas o pequeños vasos sin pared muscular que pueden
representar linfáticos post-capilares o vénulas, son un factor pronóstico importante en
el CC al representar un paso fundamental en el proceso metastásico. La invasión
venosa, especialmente la de venas extramurales, es un factor pronóstico adverso
independiente.14
Este tipo de información debería ser recogida en todos los análisis anatomopatológicos
de especímenes de cáncer de colon, incluyendo pólipos malignos, y especificando la
localización intra o extramural. El diagnóstico de invasión linfovascular requiere la
identificación de células tumorales rodeadas de una lámina elástica o con un túnel de
células endoteliales. El colegio americano de patólogos (CAP) recomienda que sea
examinada con hematoxilina-eosina una sección de cada bloque tumoral,
específicamente para buscar invasión linfovascular. No hay suficiente evidencia para
recomendar el estudio a otros niveles de la pieza ni mediante otras técnicas más
específicas como la inmunohistoquímica.
El nivel de evidencia es de categoría I.8,10
2.3.5 Invasión perineural
La invasión perineural se caracteriza por la invasión de las estructuras nerviosas y
diseminación a lo largo de las vainas nerviosas. Aunque no se conoce el mecanismo

INTRODUCCIÓN 33
por el que se produce, refleja un fenotipo tumoral más agresivo, con peor pronóstico
en múltiples neoplasias, especialmente en cánceres de cabeza, cuello y próstata.15
En varios estudios, la invasión perineural por si misma estaba asociada con un peor
pronóstico en análisis multivariante. Sin embargo, actualmente no hay suficientes
estudios que avalen esta evidencia por lo que hacen que la recomendación sea de
categoría IIB.8,10
2.3.6 Número de ganglios en la pieza quirúrgica
El número total de ganglios extraídos en la pieza quirúrgica tiene una influencia directa
en el pronóstico tanto en el cáncer de colon estadio II como en el estadio III. En un
metaanálisis de 17 estudios, el número de ganglios encontrados por paciente (con o
sin invasión tumoral) estaba relacionado significativamente con la supervivencia libre
de enfermedad a los cinco años como con la supervivencia global a los cinco años, en
estadio II y III.3,10
El motivo por el que esto es así parece incierto, aunque lo más razonable sería que se
explicara como que un mayor número de ganglios obtenidos nos da una información
más veraz sobre el estadiaje real del paciente. Por otro lado, un mayor número de
ganglios puede reflejar la calidad en la cirugía y una resección más completa del
pedículo mesentérico. Alternativamente, las diferencias entre unos individuos y otros
en número de ganglios también puede reflejar un mejor sistema inmunológico con
capacidad para reconocer a las células tumorales y una supervivencia mayor en los
pacientes con un mayor número de ganglios, que es independiente del número de
ganglios positivos.10
Las guías de los grupos de expertos como el Colegio Americano de Cirujanos, el NCCN y
el ASCO recomiendan al menos 12 ganglios examinados histológicamente para
determinar con exactitud el estadio tumoral N. Este número deriva de los datos
observados empíricamente, sin estar ajustado por variables como estadio-T o grado
tumoral. 4,9,12
El nivel de evidencia es de categoría I. 10

34 INTRODUCCIÓN
2.4 MOLECULARES
2.4.1 Inestabilidad de microsatélites.
La inestabilidad de microsatélites es la expresión fenotípica de mutaciones esporádicas
o heredadas que hacen que las células pierdan la capacidad de reparar los defectos de
emparejamiento del ADN. Esta alteración se detecta en el 15% de las piezas
anatomopatológicas de los pacientes con cáncer colorrectal, correspondiendo un 3% a
la asociación con síndromes hereditarios y un 12% a la aparición de forma
esporádica16.
La inestabilidad de microsatélites, definido como más de un 30% de microsatélites
inestables, se produce por un gran número de errores de replicación en el ADN.
Aunque este tipo de tumores tienden a ser pobremente diferenciados, están asociados
a una mejor supervivencia que los tumores con microsatélites estables o con baja
inestabilidad, tanto en los casos hereditarios como el síndrome de Lynch, como en los
casos esporádicos.16
La inestabilidad de microsatélites se recoge en las recomendaciones del estadiaje TNM
de 2010, por su relevancia en la toma de decisiones terapéuticas (mejor pronóstico y
menor beneficio de la quimioterapia con 5-FU).
El nivel de evidencia es de categoría IIB. 10

INTRODUCCIÓN 35
3.- Factores genético-moleculares pronósticos en el cáncer de colon
3.1 EL CICLO CELULAR
El ciclo celular es el proceso por el cual las células se dividen para formar nuevas
células dentro del organismo. Este proceso está controlado por diversos estímulos y
mecanismos que modulan el fin último de cada célula. La mayor parte de las células
del cuerpo humano son quiescentes. Una excepción a la situación de quiescencia son
el sistema hematopoyético, la mucosa intestinal y la piel, donde las células mueren y
son reemplazadas de forma continua cada pocos días para renovar el tejido que
forman.17
El ciclo celular tiene como objetivo el crecimiento de la célula y su división en dos
células hijas idénticas, mediante una serie de pasos ordenados y controlados. Tiene
cuatro fases bien estudiadas, que hacen que una célula pueda llegar a dividirse:
G0: es el estado en el que están la mayoría de las células en el organismo. Es un
estado quiescente, desde el que la célula puede entrar a la fase G1 del ciclo
celular para comenzar el proceso de división celular.
G1: en esta fase se produce un crecimiento de la célula duplicándose los
productos celulares (proteínas y ARN).
S: durante esta fase se produce una replicación o síntesis de ADN.
G2: se continúan con la síntesis de proteínas y ARN previo a la división celular.
M: es la mitosis, en la que el material genético y los productos de la célula
progenitora, se dividen en dos partes iguales para dar lugar a dos células hijas
idénticas.
3.1.1 Control del ciclo celular
Ante la aparición de estímulos extrínsecos y intrínsecos sobre la célula, esta puede
pasar desde la quiescencia a la fase G1. Los estímulos extrínsecos pueden ser el
contacto célula-célula, la unión a la membrana basal, la exposición a factores de

36 INTRODUCCIÓN
crecimiento y/o citoquinas, etc. Los estímulos intrínsecos consisten en niveles
oscilantes de ciclinas y de la actividad de las quinasas dependientes de ciclinas (CDK).
A lo largo de todo este proceso, el ciclo celular es controlado por un sistema que vigila
que cada paso realizado se haga de manera correcta. En regiones concretas del ciclo, la
célula comprueba que se cumplan las condiciones para pasar a la etapa siguiente: de
este modo, si no se cumplen estas condiciones, el ciclo se detiene. Existen cuatro
transiciones principales:
Paso de G0 a G1: comienzo de la proliferación.
Transición de G1 a S: iniciación de la replicación.
Paso de G2 a M: iniciación de la mitosis.
Avance de metafase a anafase, dentro de la mitosis.
Los puntos de control sirven para detener temporalmente el ciclo celular y permitir de
esa manera que la célula pueda repararse, que dé tiempo a que desaparezcan
estímulos de estrés o a que aumenten los factores de crecimiento, hormonas o
nutrientes.
Los genes que regulan el ciclo celular se dividen en tres grandes grupos:
I. Genes que codifican proteínas para el ciclo: enzimas y precursores de la
síntesis de ADN, fundamentalmente.
II. Genes que codifican proteínas que regulan positivamente el ciclo:
protooncogenes. Las proteínas que codifican estos genes, activan la
proliferación celular, para que células quiescentes pasen a la fase S y entren en
división. Algunos de estos genes codifican las proteínas del sistema de ciclinas y
CDK. Las ciclinas y las CDK, son sintetizadas a partir de protooncogenes y
trabajan en cooperación para regular el ciclo positivamente.
III. Genes que codifican proteínas que regulan negativamente el ciclo: genes
supresores tumorales. Se encargan de que la mitosis no continúe si se ha

INTRODUCCIÓN 37
producido una alteración del proceso normal. Entre estos genes, también
llamados de verificación, se encuentran los que codifican:
Productos que evitan mutaciones de genes reguladores del ciclo.
Proteínas que inactivan las CDK.
Proteínas inhibidoras del ciclo de las CDK (por ejemplo, p53, p21,
p16).
Proteína del retinoblastoma.
Proteínas que inducen la salida del ciclo hacia un estado celular
diferenciado o hacia la apoptosis.
3.1.2 Estímulos intrínsecos del ciclo celular: la regulación de los complejos
ciclina/CDK 18
La progresión del ciclo celular, y en consecuencia, la de la proliferación está
controlada por la activación y/o inhibición secuencial de las CDK. La activación de CDK
precisa de la unión a la proteína reguladora (ciclina). La actividad de CDK es inhibida
por las proteínas inhibidoras de Ciclinas (CKI). El complejo CDK-ciclina es el encargado
de la activación (mediante fosforilación) de otras proteínas reguladoras del ciclo
celular.
Cada tipo de ciclina presenta un patrón de expresión específico para la fase del ciclo
celular. Por el contrario, las CDK se expresan a lo largo de todo el ciclo celular. Ciclinas,
CDK y CKI forman la unidad fundamental para la regulación de la maquinaria del ciclo
celular.
El ciclo celular tiene mecanismos de control del mismo, de forma que cuando se
produce un error durante el proceso, la célula produce una parada celular para
intentar reparar el error o comienza el proceso de apoptosis. La verificación se lleva a
cabo en los puntos de control y asegura la fidelidad de la replicación y segregación del
genoma. Algunos componentes, además de detectar fallos, pueden poner en marcha
la reparación.
El punto de restricción más importante en el ciclo celular ocurre al final de G1,
controlada por la proteína del retinoblastoma (pRb). La proteína supresora de tumores

38 INTRODUCCIÓN
p53 tiene como función principal producir la detención del ciclo celular, la senescencia
o la muerte celular, ante un estímulo de estrés celular.
El inicio de la fase G1 se produce por la activación de CDK4 y CDK6, cuyo ligando son
las ciclinas D. Al final de la fase G1, es necesaria la activación de CDK2 a través de su
unión a la ciclina E y posteriormente a la ciclina A. Posteriormente es imprescindible la
activación de CDK1 (cdc2) por ciclina B para la transición de fase G2 a M.
A su vez, hay descritos complejos inhibidores CKI como la p27 y p21 que se unen a la
ciclina y a la CDK al mismo tiempo bloqueando su función promotora del ciclo celular.
Dentro de los inhibidores de CDK/ciclina hay dos familias: la familia CIP-KIP y la familia
INK. Las proteínas de la familia INK inhiben selectivamente CDK-4 y CDK-6. La familia
CIP-KIP está formada por tres proteínas: p21, p27 y p57. Todas ellas son capaces de
inhibir el complejo ciclina-CDK.18
Durante la fase G1, la proteína del retinoblastoma, que bloquea estando activa el
promotor de genes necesarios para la entrada en fase S, se inactiva permitiendo la
activación de los genes de la transcripción.
IMAGEN 2. Mecanismos de estimulación/inhibición del ciclo celular. Abelford´s Clinical
Oncology. Control del ciclo celular, pág. 53.19

INTRODUCCIÓN 39
3.1.3 Estímulos extrínsecos del ciclo celular
Como previamente hemos dicho, estos estímulos pueden ser el contacto célula-célula,
la unión la membrana basal, la exposición a factores de crecimiento y/o citoquinas,
etc. Las moléculas de señalización extracelular regulan el tamaño y el número de
células. Los factores que promueven el crecimiento celular se dividen en:
Mitógenos, que estimulan la división celular mediante la liberación de la célula
de los controles negativos que impiden su progreso en el ciclo celular.
Factores de crecimiento, que estimulan el crecimiento celular al promover la
síntesis de proteínas y macromoléculas.
Proteínas de señalización extracelular que inhiben el crecimiento, la división y
la supervivencia celular.
Factores de supervivencia que suprimen la apoptosis.
Apoptosis.
Algunas moléculas de señalización extracelular promueven todos estos procesos,
mientras que otras sólo promueven algunos de ellos.
Mitógenos que estimulan la división celular
Su mecanismo de acción se basa en desbloquear el freno que impide el progreso en
el ciclo celular. Hay otros mitógenos que actúan estimulando la proliferación celular
como la familia del factor transformante de crecimiento (TGF-beta), que actúa como
estimulante o inhibidor e función de su concentración.
Un paso precoz en la señalización mediada por mitógenos es la activación de Ras,
que hace que aumenten los niveles de Myc. La proteína Myc promueve la entrada en
el ciclo celular por varios mecanismos superpuestos y tiene un papel principal en la
estimulación de la transcripción de genes que aumentan el crecimiento celular.

40 INTRODUCCIÓN
IMAGEN 3. Mecanismo de acción de Myc en el ciclo celular. Molecular Biology of the Cell. 4th
edition.20
Factores de crecimiento que estimulan el crecimiento celular
El crecimiento de un órgano o un organismo depende del crecimiento celular: la
división celular por sí sola no puede aumentar la masa celular sin crecimiento celular.
Los factores de crecimiento extracelular, se unen a la superficie celular activando
mecanismos de señalización intracelular que producen un acúmulo de proteínas y
macromoléculas. 20
Una de las vías de señalización intracelular más importantes activada por factores de
crecimiento es la vía de Fosfato-inositol-3 quinasa (PI3K). Esta vía regula procesos
como la proliferación, supervivencia, movilidad y cambios morfológicos en las
células. La activación de PI3K por factores de crecimiento produce una cascada de
señales que disparan la progresión del ciclo, la supervivencia celular, activan el
metabolismo, la biogénesis de ribosomas, la traducción y transcripción, y favorecen
la motilidad celular. La fosfatasa PTEN actúa inhibiendo al producto de la PI3K, la
fosfato-inositol-3 fosfato, y de esta manera frenando la vía. La activación de esta vía
se asocia a quimioresistencia. La activación de PTEN actúa favoreciendo la acción de
los agentes quimioterápicos. 21

INTRODUCCIÓN 41
La estimulación por factores de crecimiento también aumenta la producción de Myc,
estimulando a su vez el metabolismo celular, la progresión del ciclo celular y el
crecimiento celular.
Proteínas de señalización extracelular que inhiben el crecimiento, la división y la
supervivencia celular
De igual manera que hay factores extrínsecos que promueven el ciclo celular, existen
algunas moléculas extracelulares que regulan negativamente el ciclo. La proteína
más estudiada a este nivel es la familia de TGF-beta. Esta familia de proteínas inhibe
la proliferación mediante el bloqueo del ciclo celular en G1 o por estímulo del
proceso de apoptosis. La activación de TGF-beta regula la actividad de un grupo de
proteínas llamadas Smad.
La ruta de TGF-beta tiene en los tumores un papel dual: la vía, relacionada
fisiológicamente con el inicio de la apoptosis, se inhibe en fases iniciales de la
tumorogénesis, provocando el freno en la apoptosis y el inicio de la proliferación. En
estadios tardíos de la formación de tumor, elevados niveles de TGF beta promueven
el crecimiento tumoral por facilitar la migración, invasividad, angiogénesis y evasión
del sistema inmune. Parece además que los efectos de esta vía dependen mucho del
estado y del entorno de la célula.
Factores de supervivencia que suprimen la apoptosis
Las células necesitan señales de las células que les rodean, no solo para proliferar y
crecer, sino también para sobrevivir. Cuando estas señales están ausentes, se activa
la muerte celular programada (apoptosis). Esto asegura que las células sobrevivan
sólo cuando y donde sean necesarias.
Estos factores de supervivencia generalmente se unen a la superficie celular, y
activan señales para inhibir la apoptosis. En muchos casos, esta señal está mediada
por miembros de la familia Bcl-2, como ya se tratará más adelante (ver apoptosis).

42 INTRODUCCIÓN
Apoptosis
También llamado “muerte celular programada” es el proceso por el cual la célula, ante
la aparición de estímulos intracelulares o extracelulares, inicia un proceso ordenado
para su propia destrucción. Este proceso es fundamental en el ciclo celular, ya que
evita que las células dañadas genéticamente puedan continuar el proceso de división
celular. Cuando se produce un crecimiento celular descontrolado, las propias células
activan el mecanismo de apoptosis, para que se mantenga el
equilibrio en el organismo.
Para un organismo pluricelular, la apoptosis es un proceso
habitual, normal y generalmente benigno. Sin embargo,
cuando una célula adquiere por mutaciones la capacidad de
evitar los estímulos de la apoptosis, puede dividirse de forma
ilimitada y da lugar a la formación de un tumor.17,22
La idea de que los genes podían regular la apoptosis celular
surgió a mediados de los años 80 ante el descubrimiento de la
sobreexpresión del gen Bcl-2 en el linfoma folicular. La
proteína Bcl-2 inhibía la muerte programada de las células,
demostrándose así, que la inhibición de las vías de la apoptosis podía generar
neoplasias.23
El proceso de apoptosis depende de una familia de proteasas llamadas caspasas, que
se encuentran en forma de precursor inactivo en el interior de las células. Una vez
activadas, se inicia una cascada que hace que se rompa la lámina nuclear y se degrade
el ADN nuclear. De esta forma, la célula se destruye a sí misma de forma rápida y
limpia, sin afectar a las células circundantes. 22
El inicio del proceso de apoptosis se puede activar a través de receptores en la
superficie celular llamados “receptores de muerte celular”, como la proteína de unión
a Fas, producida por linfocitos. 22
La propia célula puede iniciar la apoptosis por la vía mitocondrial, en la que se activa a
la familia de proteínas Bcl-2. Esta familia de proteínas intracelulares regula la

INTRODUCCIÓN 43
activación de procaspasas, algunas estimulando la apoptosis y otras inhibiendo el
proceso. 22
Otra familia importante de reguladores intracelulares de la apoptosis es la familia IAP
(Inhibidor de la apoptosis). Estas proteínas funcionan inhibiendo la apoptosis por dos
vías: uniéndose a procaspasas para prevenir su activación y uniéndose a caspasas e
inhibiendo su actividad. 22

44 INTRODUCCIÓN
3.2 LOS ORIGENES DEL CÁNCER.
Los tumores son la consecuencia del crecimiento de las células de un tejido de forma
anormal, rápida y desorganizada, fuera del control de los múltiples mecanismos de
división y muerte celular de la célula normal. Además, cuando dichas células adquieren
la capacidad de invadir órganos adyacentes, vasos sanguíneos y el sistema linfático,
hablamos de cáncer. 24
IMAGEN 4. Transformación histológica adenoma a carcinoma.
El 90% de los tumores del cuerpo humano son carcinomas, es decir, se desarrollan a
partir de células epiteliales. Esto se debe probablemente a que los epitelios son los
tejidos con mayor recambio celular y a que están más expuestos al daño físico y
químico que favorece el cáncer.24
El cáncer se produce por la acumulación de alteraciones genéticas somáticas en las
células de un organismo. En algunos casos, estas alteraciones no son todas somáticas,
si no que parten de una mutación en la línea germinal, como es el caso de los
síndromes de cáncer hereditario. De cualquier manera, el cáncer es consecuencia de
múltiples alteraciones genéticas que contribuyen a la pérdida de control del ciclo

INTRODUCCIÓN 45
celular y a la adquisición por parte de la célula de nuevas funciones como la invasión
tisular y la angiogénesis.
En el cuerpo humano se producen más de 1014 mutaciones en las células cada día. La
posibilidad de que alguna escape a los controles celulares es muy alta, prácticamente
una realidad. Una mutación puede conferir a una célula una ventaja selectiva,
permitiendo que esta célula se divida por delante de sus vecinas y cree una colonia de
clones mutantes. El acúmulo de mutaciones y la selección de las células con mayores
ventajas para la supervivencia dentro de la población de células somáticas puede ser el
inicio del cáncer. Estos son los ingredientes básicos para la carcinogénesis, una
enfermedad en la cual las células mutantes comienzan a prosperar sobre sus
compañeras, y que termina destruyendo a las células somáticas no mutadas.24
Tanto los oncogenes como los genes supresores tumorales son diferentes caras del
mismo proceso de proliferación celular. Desde el punto de vista de la célula
cancerígena, ambos genes actúan sobre el mismo objetivo, que es la liberación de la
célula de los controles habituales y su proliferación. Esto se produce porque los
mecanismos de control celular vienen regulados por componentes de estímulo (los
proto-oncogenes) y de inhibición (los genes supresores tumorales). En lo que se refiere
al funcionamiento celular, no es tan importante la distinción entre oncogenes y genes
supresores tumorales, si no las vías de señalización bioquímica en las que actúan. 20
Algunas de las vías importantes en la señalización del cáncer se producen por señales
del entorno de la célula, otras por programas internos celulares (como el ciclo celular y
el proceso de muerte celular programada). Otras vías controlan los movimientos de la
célula y las interacciones mecánicas de la misma, lo cual es de suma importancia en la
adquisición de capacidad invasiva por parte de la célula tumoral. Muchas de estas vías
están unidas y relacionadas entre sí por un complejo entramado de interacciones. 20
3.2.1 Los fundamentos del proceso de carcinogénesis.
Para que un cáncer sea capaz de reproducirse y avanzar, es necesario que adquiera
una serie de características aberrantes en su evolución. Dependiendo del tipo de

46 INTRODUCCIÓN
cáncer, este requerirá una combinación de propiedades diferentes. De forma general,
es necesario que la célula tumoral adquiera las siguientes capacidades de forma
consecutiva:
I. Capacidad para ignorar las señales internas y externas que regulan la
proliferación celular.
II. Evitar la muerte celular por apoptosis.
III. Eludir las limitaciones de la proliferación celular, escapando de la senescencia
replicativa y evitando la diferenciación celular.
IV. Ser genéticamente inestable.
V. Escapar de sus tejidos de origen (es decir, volverse invasivos).
VI. Sobrevivir y proliferar en tejidos diferentes a los de origen (es decir,
metastatizar).
3.2.2 Desregulación del ciclo celular en el cáncer
La alteración en los mecanismos de control del ciclo celular es un distintivo de las
células tumorales. Estas alteraciones se pueden manifestar como alteración de las vías
de señalización de factores de crecimiento, desregulación de la maquinaria que
controla el núcleo celular y/o alteración de los puntos de control del ciclo celular.
Como los mecanismos de control del ciclo celular están alterados en todos los
tumores, las mutaciones de los genes relacionados con el ciclo celular son una diana
terapéutica en las células cancerígenas, frente al tejido normal.
Gran variedad de genes implicados en la carcinogénesis
La mayor parte de los genes clave para el desarrollo del cáncer están implicados de
una u otra manera en la regulación del comportamiento de las células en el organismo.
Especialmente, en aquellos mecanismos en los que las señales de las células vecinas
pueden estimular a sus compañeras a dividirse, diferenciarse o morir. De hecho,
muchos de los componentes de las vías de señalización fueron identificados en el

INTRODUCCIÓN 47
estudio de los genes causantes del cáncer, incluyendo un grupo importante de
productos derivados de proto-oncogenes y de genes supresores tumorales (proteínas
secretadas, receptores transmembrana, proteínas de unión a GTPasa, proteín-kinasas,
proteínas reguladoras de genes, etc.). Muchas mutaciones en el cáncer tienen
alterados algún componente de la vía de señalización de tal manera que mandan
señales de proliferación incluso cuando las células no las necesitan más, activando
inapropiadamente la maquinaria de crecimiento celular, replicación del ADN y división
celular
En última instancia, los genes clave del cáncer que regulan la división celular median su
efecto actuando en la maquinaria de de control del ciclo celular. Es por ello que en la
mayor parte de los cánceres se encuentran mutaciones en las características de esta
maquinaria. Un punto clave para comenzar la replicación está controlado por la
proteína del retinoblastoma, el producto del gen supresor de tumores Rb. Muchos
tumores proliferan inapropiadamente por la eliminación de Rb, o adquiriendo
mutaciones en otros componentes de la vía reguladora de Rb.
La variedad de forma en las que la maquinaria de control del ciclo celular puede
alterarse en el cáncer demuestra dos ideas importantes:
Los casos individuales de cáncer pueden tener los mismos síntomas aunque se
hayan formado a través de diferentes mutaciones: en muchos casos, diferentes
mutaciones tienen el mismo efecto en la proliferación celular.
No hay diferencias fundamentales en el proceso que afecta a los oncogenes
(que se activan al mutarse) y a los genes supresores tumorales (que se
inactivan al mutarse). Estos dos tipos de genes clave en la carcinogénesis, solo
difieren en su papel estimulador o inhibidor en una vía de señalización.
La carcinogénesis y la apoptosis
Para que se produzca crecimiento celular, no solo es necesario que las células se
dividan, sino que no entren en apoptosis, de forma que el balance entre división y
muerte celular haga que el tumor se desarrolle. El mecanismo de apoptosis se activa

48 INTRODUCCIÓN
en las células normales ante señales de daño celular y ausencia de señales de
supervivencia.
La resistencia a la apoptosis es una característica clave de las células malignas, y es
necesaria para aumentar el número de células y sobrevivir cuando no deberían.
En los tumores se han encontrado un numerosas mutaciones que inhiben la apoptosis.
De todos los genes involucrados en el control de la apoptosis, hay uno que está
alterado en la gran mayoría de los tejidos tumorales: p53.
El papel de p53 en la carcinogénesis se basa en tres pilares: el control del ciclo celular,
la apoptosis y el mantenimiento de la estabilidad genética.
3.2.3 Las metástasis en el cáncer
La diseminación de un tumor por metástasis es un proceso complejo y entendido sólo
parcialmente. Requiere múltiples pasos:
I. Las células deben separarse del tumor primario,
II. Invadir los tejidos circundantes y los vasos sanguíneos o linfáticos, y por último,
III. Ser capaces de establecer nuevas colonias celulares en otros órganos.
Cada uno de estos pasos requiere de mecanismos moleculares complejos que no están
todavía claros. El primer paso, la separación del tejido tumoral primario, es el punto
clave para la invasión de otros tejidos. Esta capacidad para invadir otros tejidos es la
característica principal de los tumores malignos, que muestran unos bordes
desorganizados y desiguales. Uno de los puntos clave en este proceso es la
interrupción de los mecanismos de adhesión celular, que normalmente mantiene a las
células unidas a las células vecinas.
El siguiente paso en el proceso de metástasis se produce de forma lenta. La célula
tumoral debe penetrar en los vasos sanguíneos o linfáticos, atravesar la lámina basal y
el revestimiento endotelial para entrar a la circulación y salir de la circulación en otra
parte del cuerpo.

INTRODUCCIÓN 49
Por último, para que se produzca una metástasis, es necesario que las células
tumorales que han llegado a la circulación sanguínea o linfática sean capaces de salir
de la misma y puedan proliferar en un nuevo tejido.
Además de que la célula sea capaz de adquirir propiedades para metastatizar, es
necesario que la célula cancerígena reciba un aporte adecuado de sangre. La
angiogénesis, es decir, la formación de nuevos vasos sanguíneos, es necesaria para el
crecimiento de tumores primarios y metástasis. Aunque los tejidos normales tienen la
capacidad para aumentar el suministro sanguíneo cuando es necesario, los tejidos
tumorales son capaces de crecer rápidamente mediante el aumento de las señales
angiogénicas. Los neovasos facilitan al tumor nutrientes y oxígeno y facilitan una ruta
de escape para las células metastásicas.24
3.2.4 Vías moleculares en la transformación neoplásica del cáncer de colon
El proceso de transformación de las células epiteliales intestinales normales en células
cancerígenas, se produce por la acumulación de múltiples mutaciones genéticas en la
célula, hasta que se crea un clon celular con capacidad maligna. Se acepta de forma
general que la mayor parte de los cánceres colónicos se desarrolla a partir de
adenomas preexistentes (ver “secuencia adenoma-carcinoma”, pág. 56). Parece claro,
sin embargo, que el cáncer colorrectal es el resultado final de varias enfermedades con
vías patológicas diferentes.25
La identificación de diferentes vías moleculares en la carcinogénesis colorrectal ha
demostrado la heterogeneidad de la naturaleza del cáncer colorrectal. Las
alteraciones genéticas y epigenéticas actúan desregulando vías de señalización
involucradas en el metabolismo celular, la proliferación, la diferenciación, la
supervivencia y la apoptosis.
Entendiendo el proceso de carcinogénesis colorrectal podremos actuar a nivel
pronóstico y terapéutico. Optimizando los protocolos de cribado y seguimiento,
mejorando la precisión del estadio e individualizando el tratamiento de los pacientes

50 INTRODUCCIÓN
en base a las características patológicas y moleculares del tumor, podremos mejorar
los resultados en cuanto a curación y a supervivencia se refiere, sin perjudicar a los
pacientes con menor riesgo de recidiva de su enfermedad.
Se han identificado múltiples mutaciones genéticas en la carcinogénesis colorrectal,
pero el papel que desempeñan cada una de ellas en la iniciación y progresión de la
enfermedad todavía no se ha confirmado. Solo un pequeño número de estos genes,
los más conocidos: APC, K-ras y p53, se han encontrado alterados en una proporción
suficiente de CCR.
La caracterización de los síndromes hereditarios de cáncer colorrectal ha permitido la
mejor compresión del proceso de carcinogénesis.
Según el modelo de Fearon y Vogelstein21,26 los tumores tienes 3 características
importantes en su formación: en primer lugar, la activación de oncogenes y
desactivación de genes supresores tumorales; segundo, mutación de al menos 4-5
genes diferentes; y tercero, la acumulación de mutaciones. Mas que el orden de las
mismas, el acúmulo de las mutaciones, es el responsable del comportamiento
biológico del tumor. En las últimas dos décadas, sin embargo, se han hecho
descubrimientos importantes como:
La inestabilidad de microsatélites: una característica del cáncer
colorrectal hereditario no polipósico o síndrome de Lynch, y presente en
el 15% de los CCR esporádicos.
El papel de la epigenética, o la silenciación de la función de algunos
genes, en particular mediante la hipermetilación de dinucleótidos en la
región promotora de múltiples genes en el que ha sido llamado CpG
Island Methylator Phenotype (CIMP). 27
Existen tres vías descritas en la transformación neoplásica de las células colónicas.
Estas son:
La vía cromosómica
La vía de los microsatélites

INTRODUCCIÓN 51
La vía de la isla CG de metilación (CIMP)
Inestabilidad cromosómica
Es la causa más común de inestabilidad genómica en el CCR, encontrándose en el 65-
70% de los CCR esporádica. Se caracteriza por la pérdida o ganancia de cromosomas o
de regiones cromosómicas comprendiendo genes implicados en el proceso de
carcinogénesis colorrectal. Además, se han encontrado ganancias genéticas focales en
regiones que contienen genes importantes para el cáncer como VEGF, c-myc, PTEN y
otros. La pérdida global de un cromosoma se produce con más frecuencia en el
cromosoma 18, mientras que otros cromosomas se ven afectados predominantemente
por pérdidas parciales. 27
Algunas alteraciones cromosómicas en el CCR son:
Oncogen K-ras: está mutado en el 30-60% de los CCR y de adenomas grandes.
La activación continua de ras afecta a múltiples vías celulares que controlan el
crecimiento celular, la diferenciación, la supervivencia, la apoptosis, la
organización del citoesqueleto, la motilidad celular, la proliferación y la
inflamación. 27
Perdida del alelo 5q: se encuentra en el 20-50% de los CCR esporádicos. Hay
dos genes importantes localizados en este alelo: el APC (Adenomatous Poliposis
Coli) y el MCC (Mutated in Colorectal Cancer). APC se considera el ”guardián”
de la proliferación celular, y es un elemento clave de la vía Wnt.27
o La vía Wnt es fundamental en la renovación celular del epitelio
intestinal. La molécula de beta-catenina se une a su ligando APC,
activándose así su degradación. La mutación de APC provoca que la
beta-catenina citoplasmática migre al núcleo, donde se une a T-cell
factor (TCF), induciendo la proliferación celular. 27
La vía Wnt tiene como diana genes que afectan a múltiples funciones
celulares, entre las que están: la proliferación celular, la regulación el

52 INTRODUCCIÓN
ciclo celular, angiogénesis y apoptosis. La alteración de esta vía no se
produce únicamente por mutación de APC, sino que también se puede
encontrar tras mutaciones de beta-catenina en las que se hace
resistente a la degradación (menos del 5% de los cánceres de colon), o
de TCF. 27
Pérdida del alelo 8p
Pérdida del alelo 17p: presente en el 75% de los CCR, pero no en adenomas.
Este segmento contiene el gen supresor tumoral p53.
Pérdida del alelo 18q: se detecta en el 50-70% de los CCR y es un marcador de
mal pronóstico en estadio II y III de CCR. Contiene varios genes supresores
tumorales de importancia como DCC, Smad2 y Smad4.
Inestabilidad de microsatélites
Los microsatélites son repeticiones cortas de secuencias de nucleótidos que se
encuentran dispersas por todo el genoma, y que están expuestos a errores durante la
replicación debido a su carácter repetitivo. El sistema de reparación de errores de
emparejamiento, conocido como mismatch repair system (MMR) reconoce y repara
errores de emparejamiento durante la replicación de ADN. La inestabilidad de
microsatélites es un reflejo de la incapacidad de este sistema para corregir estos
errores. El descubrimiento de la inestabilidad de microsatélites en 1993 y su relación
con el síndrome de Lynch, ha permitido el estudio de la vía de los microsatélites como
una vía alternativa en la carcinogénesis de colon. Los miembros de la familia MMR
identificados son: MSH2, MLH1, MSH6, PMS2, MLH3, MSH3, PMS1 y Exo1. En el caso
de CCR esporádico con inestabilidad de microsatélites, esta se suele deber a
silenciación por hipermetilación de MLH1. 27
Esta vía de carcinogénesis, hace que las características clínicopatológicas de los
pacientes tengan un fenotipo característico que incluye: mujeres mayores, con
predominancia por el colon derecho (proximal al ángulo esplénico), aumento de la
infiltración linfocítica, histología mucinosa y pobre diferenciación. 27

INTRODUCCIÓN 53
La vía de la isla CG de metilación (CIMP)
Los cambios epigenéticos son aquellos cambios en la expresión genética o en la
función del producto genético que se producen sin cambios en la secuencia del ADN.
En humanos, estos cambios se suelen relacionar con la metilación o la modificación de
histonas. La metilación y consecuente silenciación se suele producirse en el promotor
del gen, sobre el dinucleótido 5´-CG-3´ (CpG). El hábito tabáquico y la edad avanzada
se han relacionado con un aumento de la metilación. 27
La vía CIMP se encuentra alterada en el 15-20% de los CCR. Estos tumores tienen un
fenotipo característico que consiste en que es más frecuente en mujeres, pacientes
ancianos y de localización proximal. Patológicamente se caracterizan porque
frecuentemente son poco diferenciados y con histología en anillo de sello o
mucinoso.27
TIPO DE MUTACIÓN GENES IMPLICADOS TIPO DE ENFERMEDAD
Línea germinal APC Poliposis adenomatosa familiar
MMR S. Lynch (cáncer colorrectal familiar no
polipósico)
Somática Oncogenes:
myc
ras
src
erbB
Enfermedad esporádica
Genes supresores tumorales:
p53
DCC
APC
Genes MMR:
bMSH2
bMLH1
bPMS1
bPMS2
bMSH6
bMSH3
Polimorfismo genético APC Cáncer de colon familiar en judíos Askenazis
TABLA 5. Mutaciones genéticas que causan CC. Adaptado de Sabinston Textbook of Surgery (2012) pág. 1338.

54 INTRODUCCIÓN
3.2.5 Secuencia adenoma carcinoma en el cáncer de colon
Se cree que el cáncer de colon y recto tiene su origen en la mayor parte de los casos en
pólipos adenomatosos. Los pólipos adenomatosos se producen por el fallo en alguno
de los pasos de proliferación celular y apoptosis en las células epiteliales de las criptas
intestinales. Se cree que el primer error se produce en una sola cripta, en la que el
componente proliferativo, en vez de quedarse confinado a la base de la cripta, se
extiende por toda la cripta, dando lugar a un adenoma. Las células de la superficie no
se desprenden a la luz, sino que se acumulan en las capas profundas del epitelio
intestinal, interponiéndose entre las criptas normales existentes. Se piensa que,
inicialmente, el adenoma se crea por la expansión monoclonal de una única célula
alterada, y según aumenta de tamaño el adenoma, la población celular se vuelve
policlonal.
Está aceptado de forma general que la “hipótesis adenoma-carcinoma” explica el
origen del CCR. La evidencia que apoya la teoría adenoma-carcinoma se basa en
estudios epidemiológicos, clínicos, patológicos y moleculares. Por una parte, los
estudios epidemiológicos correlacionan la incidencia de adenomas colorrectales con la
incidencia de CCR. Por otra parte, desde el punto de vista clinicopatológico se apoya en
el hecho de que en la poliposis adenomatosa familiar, en la que los pacientes
desarrollan de cientos a miles de adenomas, la probabilidad de desarrollar CCR es del
100%. A nivel molecular se han observado alteraciones similares en adenomas y
células cancerígenas en el colon, aunque quizá la mejor evidencia es que el patrón de
alteraciones moleculares en células cancerígenas, y en células adenomatosas próximas
al tumor, son idénticas, excepto que las células cancerígenas poseen otras mutaciones
adicionales que probablemente sean las responsables de su comportamiento
maligno.7,28

INTRODUCCIÓN 55
IMAGEN 5. Vía de la carcinogénesis colónica. Extraído y adaptado de Sleisenger and Fordtran's Gastrointestinal and Liver Disease (2010) pág. 2161.
De esta manera, el CCR se explica por una secuencia de malignización bien establecida,
comenzando por el desarrollo de pólipos adenomatosos, que progresan a adenomas
con displasia (de bajo y alto grado). Dejado a su evolución, se transformaran en
carcinoma in situ, que con el tiempo degenerará en carcinoma invasivo.29
IMAGEN 6. Secuencia adenoma-carcinoma a nivel molecular. Adaptado de Sabiston Textbook of Surgery. Capítulo Colon and rectum, pág. 1338.
El potencial de transformación maligna de un pólipo adenomatoso de colon se
relaciona con tres características del pólipo: el tamaño, el tipo histológico y el grado de
displasia. De esta manera podemos encontrarnos con un probabilidad de malignización

56 INTRODUCCIÓN
de 1,3% en lesiones menores de 1 cm sin displasia y con tipo histológico favorable
(tubular), hasta un 35-40% en caso de lesiones de predominio velloso o con displasia
de alto grado.28
La formación de pólipos adenomatosos se produce al fallar uno a uno, los mecanismos
de control de división celular y muerte celular. El proceso de carcinogénesis se divide
en dos fases: una primera fase de iniciación en la que la mucosa normal se transforma
en un adenoma; y una fase de progresión, en la que el adenoma se transforma en
carcinoma. Se cree que el primer paso dentro de este proceso es la pérdida de función
del gen APC. Otra de las vías implicadas en las fases iniciales de transformación
neoplasia son los genes reparadores del ADN, conocidos bajo las siglas DNA MMR
(DNA mismatch repair). Las mutaciones de estas enzimas dan lugar a un fenotipo
específico denominado inestabilidad de microsatélites, que corresponde en el 85% de
los casos a pacientes con cáncer colorrectal hereditario no polipósico (HNPCC) y en el
15% restante a pacientes con tumores esporádicos.28

INTRODUCCIÓN 57
IMAGEN 7. Secuencia adenoma-carcinoma a nivel patológico. Adaptado de Sabiston Textbook
of Surgery. Capítulo Colon and rectum, pág. 1342.

58 INTRODUCCIÓN
3.3 BIOMARCADORES EN EL CÁNCER
Un biomarcador se define como una variable que es medible objetivamente y evaluada
como indicador de procesos biológicos normales, procesos patológicos o respuestas
farmacológicas a intervenciones terapéuticas. Algunos biomarcadores se han utilizado
para predecir la mejor o peor respuesta a un tratamiento; es el caso de HER2 en el
cáncer de mama, la mutación de EGFR en el cáncer de pulmón y la mutación de K-RAS
en el cáncer de colon. 30
En el cáncer de colon, los biomarcadores constituyen un área en desarrollo, tanto en la
identificación de factores pronóstico como de factores predictivos de respuesta a
terapia adyuvante. Entre estos factores se ha estudiado la delección del cromosoma
18q, la sobreexpresión y/o genotipo de timidilato sintetasa, la inestabilidad de
microsatélites/ déficit en la reparación de mismatch y la hipermetilación (inactivación
epigenética) de genes que afectan a la matriz extracelular.11
Actualmente existen baterías de análisis genéticos que identifican pacientes de riesgo,
como son la expresión de genes del “12-gene recurrence score assay” (Oncotype DX
Colon Cancer Assay), o la clasificación de 18 genes (ColoPrint), así como la
identificación de células tumorales circulantes por métodos moleculares. Todas estas
baterías de pruebas, que pueden mejorar la precisión en el pronóstico individual de los
pacientes con CCR, necesitan ser suficientemente validadas en estudios prospectivos
antes de ser usadas en la práctica clínica habitual. 11
A pesar de los esfuerzos y los resultados iniciales alentadores, no se ha encontrado un
marcador molecular único ni un grupo de marcadores o panel de expresión genética
que tenga utilidad predictiva (es decir, que identifique con precisión los grupos de
pacientes que tendrían mayor beneficio con el tratamiento adyuvante). Sin embargo,
hay una excepción a esto: la presencia de inestabilidad microsatélites parece estar
asociada a una resistencia relativa a la terapia basada en fluoropirimidinas. 11

INTRODUCCIÓN 59
3.3.1 Variables moleculares
Se han estudiado múltiples variables moleculares expresadas en tumores, y se están
buscando variables que puedan servir tanto como factores pronósticos como para
seleccionar el mejor tratamiento para cada individuo. Sin embargo, en la actualidad no
se ha conseguido demostrar el valor pronóstico en la mayor parte de ellos.
Los biomarcadores moleculares los dividimos en función de su actividad e implicación
en el proceso de carcinogénesis.
3.3.1.1 Oncogenes
Los protooncogenes son genes cuya presencia o activación a oncogenes pueden
estimular el desarrollo de cáncer. Cuando se activan exageradamente en las células
normales, provocan que ellas pierdan el control de la división y se mantengan
proliferando sin control. En este trabajo se han analizado los siguientes:
cERB-B2
Es una glicoproteína con actividad tirosin-kinasa, también conocida como Her-2
neu 31. Forma parte de la familia del factor de crecimiento epitelial (EGFR) que
puede interaccionar con otros miembros de la familia para formar la forma activa
del receptor, desencadenando de esta manera la señalización intracelular en
ausencia de ligando mitogénico.
En la forma mutada, este receptor del factor de crecimiento se encuentra
amplificado, y es habitual encontrarlo en tumores de mama, ovario, estómago y
otros carcinomas.21
Esta proteína ha sido muy estudiada en el cáncer de mama. Se ha desarrollado un
agente molecular que tiene como objetivo cERB-B2, el transtuzumab (herceptin),
que se usa actualmente en el tratamiento del cáncer de mama. 32

60 INTRODUCCIÓN
Se ha encontrado expresión de c-erbB2 en el 20-40% de los cánceres de colon,
aunque su importancia pronóstica en este tipo de tumores sigue siendo
desconocida 32. El hecho de que no se exprese con tanta frecuencia en el cáncer de
colon hace que no se hayan desarrollado ensayos clínicos con transtuzumab. 6
EGFR 32
Es el receptor del factor de crecimiento epitelial, una proteína con actividad tirosín-
kinasa. Al interaccionar con su ligando, EGFR produce proliferación y activa las vías
ERK (supervivencia) y AKT, activando la síntesis proteica y pérdida de apoptosis.
Es un factor pronóstico en un número importante de tumores sólidos, incluyendo
el carcinoma de mama y el de ovario. La frecuencia de sobreexpresión de EGFR en
carcinoma colorrectal parece ser superior a la de la glicoproteína HER-2. En el
cáncer de colon ya hay estudios que evalúan la importancia de la expresión de
EGFR, aunque son necesarios que definan la utilidad pronóstica de la detección
inmunohistoquímica de EGFR.
Los inhibidores de EGFR (cetuximab y panitumumab) se están utilizando
actualmente en ensayos clínicos para el tratamiento del cáncer de colon, aunque el
papel de EGFR como factor pronóstico independiente todavía no ha sido definido
claramente.
C-myc
Es una proteína que promueve el inicio del ciclo celular mediante varios
mecanismos superpuestos. Aumenta la transcripción de ciclinas y la actividad de
los complejos CDK-ciclina. Además promueve indirectamente la degradación del
inhibidor de CDK p27 y la inactivación de la proteína Rb. C-myc también tiene un
efecto sobre genes que aumentan el crecimiento celular.33

INTRODUCCIÓN 61
Se relaciona con el aumento de la proliferación por activación del ciclo celular (al
ser un inhibidor de p21), que se relaciona con el control del ciclo), pérdida de
control de la muerte celular en respuesta a daño genotóxico por interferir en la vía
activada por p53.
Parece tener un papel en pacientes con cáncer de colon Dukes B o C, ayudando a la
selección de aquellos candidatos a quimioterapia adyuvante. En otros estudios, se
ha encontrado que su sobreexpresión se relaciona con un peor pronóstico.
C-kit 34
Codifica un receptor de tirosin quinasa. Este receptor se activa por la interacción
de su ligando SCF (stem-cell factor), activando entonces la supervivencia,
proliferación, migración y homing. Se han descrito dos mecanismos de activación
en células malignas: la adquisición de mutaciones activadoras o la estimulación
autocrina o paracrina del receptor por su ligando SCF.
En los pacientes con cáncer colorrectal en los que se expresa KIT/SCF, se está
utilizando Imatinib mesilato, un inhibidor selectivo de tirosin quinasa (incluyendo
KIT) en combinación con la terapia estándar.
En el cáncer colorrectal, se ha encontrado que la expresión de KIT se asocia a un
peor pronóstico, aunque no está claro el significado biológico de su expresión en el
proceso de carcinogénesis de colon.
NTSR-1
Sus siglas corresponden al receptor de neurotensina 1, y es un receptor acoplado a
proteínas G que media los efectos biológicos de neurotensina (hipotensión,
hiperglucemia, hipotermia, regulación de la movilidad intestinal y secreción). Es un
péptido que estimula el crecimiento de algunos tipos celulares.

62 INTRODUCCIÓN
La neurotensina puede activar EGFR, aunque no en todos los modelos
experimentales. La activación de NTSR lleva a la activación de ERK1/2 y a la
proliferación, por mecanismos independientes o no, de la activación de EGFR.
También está implicada en la inflamación que se ve asociada a los tumores de
colon.
NTSR1 ha sido identificado recientemente como marcador pronóstico en el cáncer
de mama, de pulmón y de tumores escamosos de cabeza y cuello. La neurotensina
ha mostrado ejercer numerosos efectos oncogénicos en el crecimiento tumoral y la
diseminación metastásica. Estos efectos parecen estar mediados
fundamentalmente por NTSR1, haciendo del complejo NT-NTSR1 un personaje
principal en la progresión tumoral. 35
La neurotensina se expresa en tejido colónico fetal, pero no en el epitelio colónico
diferenciado. Su receptor, NTRS-1, es expresa escasamente o no se expresa en el
epitelio colónico del adulto, pero se expresa en la mayoría de las líneas celulares
cancerígenas en el colon. Estas observaciones sugieren que una alteración de la
señal NT-NTSR1 puede estar asociada con la transformación neoplásica en el
colon.36
En un trabajo se ha analizado la expresión de NTSR1 en colon (mucosa normal,
adenomas y adenocarcinomas), y se ha encontrado mayor expresión de NTSR-1 en
adenomas y adenocarcinomas, siendo mayor la expresión cuando se trataba de un
adenocarcinoma y mayor aún en caso de infiltración linfovascular. 36

INTRODUCCIÓN 63
3.3.1.2 Genes supresores tumorales
Los genes supresores de tumores o genes “de verificación” regulan negativamente el
ciclo celular. Se encargan de que la mitosis no continúe si se ha producido una
alteración del proceso normal. Para que ejerzan su efecto cancerígeno, es necesario
que estén mutados los dos alelos del gen. En este trabajo se han analizado los
siguientes:
p53
Es un gen supresor que codifica una proteína involucrada en la regulación del ciclo
celular, replicación del ADN y la apoptosis. El gen está localizado en el cromosoma
17p, una región de delección alélica en muchos tumores. 31
Se le conoce como el “guardián del genoma” por su papel esencial en la regulación
de la apoptosis. Es uno de los genes supresores tumorales más estudiados, y se
encuentra mutado en la mitad de las neoplasias solidas humanas. La actividad de la
proteína se induce ante la estabilización de la misma en respuesta a diferentes
estímulos, incluido el daño al ADN, desencadenando la apoptosis o la detención del
ciclo celular. El efecto antitumoral de p53 está mediado por diversos efectos a nivel
de la transcripción. Los genes dependientes de p53 tienen múltiples efectos en la
apoptosis. 7,23
p53 se encuentra mutado en el 75% de los tumores de colon, y esta mutación se
produce habitualmente tarde en la secuencia adenoma-carcinoma7. El papel de
p53 en el cáncer colorrectal como factor pronóstico no queda claro, ya que hay
trabajos que lo implican como factor pronóstico adverso para la supervivencia, y
otros que no encuentran asociación.31

64 INTRODUCCIÓN
P21
Actúa como regulador del ciclo celular en G1. Responde a varios estímulos (TGF
beta, inhibición del crecimiento por contacto, senescencia replicativa y daño al
ADN (responde a p53)). Al activarse, bloquea el complejo CDK2-Ciclina E,
inhibiendo la transición a fase S. En la transición G2/M inhibe la progresión del ciclo
en respuesta a la estimulación de p53.
Tiene un papel múltiple, tanto como factor pro-apoptosis, y también anti-
apoptosis, a través de la activación de la quinasa activada por p21 (PAK), facilitando
de esta manera la invasión tumoral y la metástasis.
Dado que p21 se relaciona con la senescencia celular y el envejecimiento de las
células madre, es posible que la influencia de la pérdida de p21 en el
comportamiento tumoral pueda ser modificada por el balance energético celular y
la edad del paciente.
La mayor parte de los estudios no han demostrado ningún papel pronóstico
independiente de p21, aunque se ha visto que la pérdida de expresión de p21 en el
cáncer de colon está asociada de forma inversa con la inestabilidad de
microsatélites. 32,37
P27
Es un inhibidor del ciclo celular, también conocido como Kip 1. Forma parte de la
familia de inhibidores de las CDK, regulando negativamente el paso de G1 a S.
Durante el ciclo celular inhibe la activación de los complejos ciclina-CDK,
controlando la transición G1/S. Es necesaria la inactivación de p27 para que el ciclo
celular pueda progresar y la célula pase de estado de quiescencia al de
proliferación. Responde a TGF beta y a la inhibición de crecimiento por contacto. 32

INTRODUCCIÓN 65
No se han descrito mutaciones inactivadoras en tumores humanos. Solo un estudio
ha encontrado relación significativa independiente en el pronóstico del cáncer
colorrectal. 38
Nm23
Nm23-H1 y Nm23-H2 son dos genes diferentes que codifican proteínas que están
involucradas en la proliferación celular y en la diferenciación, así como en la
formación de membrana basal y cese del crecimiento. 31
Se describió inicialmente por sus bajos niveles en células metastásicas. Se
considera un gen supresor de metástasis: niveles altos de expresión se suelen
asociar a un mejor pronóstico. 39
SMAD 4
Las proteínas Smad se activan en respuesta a la señalización TGF beta. La
activación de TGF-beta produce, entre otras cosas, control sobre el ciclo celular:
activa Smad3/4, y eso activa p16, p15, p27 y p21, frenando el ciclo celular.
Se han encontrado mutaciones del receptor de TGF-beta en algunos cáncer de
colon y de Smad4, el transductor de la señal intracelular de TGF-beta en el cáncer
de páncreas y otros tejidos.20
Las mutaciones o delecciones de este gen se han asociado a distintos tumores. La
presencia de gran cantidad de estroma y la disminución de la actividad de SMAD 4
predice una peor supervivencia en los pacientes con cáncer de colon estadio I-
II.39,40

66 INTRODUCCIÓN
Caveolina
Las caveolinas 1, 2 y 3 son las proteínas principales de la caveola, las invaginaciones
de la membrana plasmática.
Se consideran un supresor tumoral, ya que se encuentra en una región
frecuentemente deleccionada en el cáncer. En el colon, como en otros tejidos, se
ha visto que su expresión disminuye con la transformación neoplásica. Sin
embargo, tiene un papel dual al estar implicado en la transducción de señales vía
receptores de membrana. Según su estado de fosforilación y su entorno, su
incremento actuará como supresor o activador tumoral: la forma no activa limita la
transducción de señales mediada por receptores de membrana, pero la forma
activa promueve la migración celular, confiriendo agresividad a los tumores. 41
La caveolina 1 se ha visto asociada como factor pronóstico favorable en el cáncer
de recto. 42
p16
Llamado también CDKN2A, es un inhibidor de CDK4 y CDK6 en el ciclo celular.
Funciona como supresor tumoral al ser capaz de inducir la paralización del ciclo
celular. En G1 bloquea el complejo ciclina D-CDK4-CDK6 evitando la inactivación de
Rb y la transición a fase S. Es sensible a la senescencia replicativa.
Este gen se encuentra deleccionado en la línea germinal de los melanomas
familiares. 19 A nivel de cáncer de colon, la mutación de p16 se puede producir por
pérdida o por modificación epigenética (metilación del promotor), y aunque parece
estar relacionada con un peor pronóstico, esto no ha sido confirmado, ya que en la
mayor parte de los casos se encuentra asociado al status CIMP que de por sí
confiere mal pronóstico.43

INTRODUCCIÓN 67
Proteína del retinoblastoma (pRB)
Es una proteína que fue identificada inicialmente por su asociación al
retinoblastoma hereditario. Se comporta como un supresor tumoral con funciones
relacionadas con la proliferación, diferenciación, muerte celular y estabilidad
genómica. La forma activa, hipofosforilada, se une al factor de transcripción E2F1 y
lo reprime, llevando a la paralización del ciclo. Actúa en la transición G1/S. se
inactiva por hiperfosforilación a cargo de CDK2 y CDK4, no pudiendo unirse a E2F1,
y permitiendo la progresión del ciclo. 19
La cooperación de pRB y p27 es crucial para la regulación del ciclo celular, la
diferenciación celular y supresión tumoral. pRB puede unirse a Cdh1 y promover la
degradación de SKp2. 19
Se encuentra inactivado en un tercio de los tumores humanos esporádicos. La
inactivación de pRB en tumores suele ser concomitante a la inactivación de p53. La
hiperfosforilación es el modo fisiológico de inactivación de pRB, aunque a veces los
tumores también muestran inactivación de pRB por mutaciones de pérdida de
función de p16 (que ya no inhibe CDK4 y CDK6) o por mutaciones de ganancia de
función de ciclina D. 19
Las dos vías de inactivación de pRB (delección e hipermutación por pérdida de p16)
no son equivalentes: p53 coopera con la primera y se asocia a roturas de DNA por
desregulación de E2F1. 19
pRB controla la apoptosis: E2F1 la induce en ausencia de Rb vía múltiples
mecanismos y promueve la respuesta al daño del DNA bien por activación
transcripcional de p14 que inhibe la degradación de p53 por Mdm2 o bien vía
fosforilación de p53 independiente de p14. Se ha visto en distintos modelos que la
inducción de la apoptosis por pRB se produce en células proliferantes (por ejemplo,
tratadas con agentes que dañan el ADN) pero no en otros contextos celulares. 19

68 INTRODUCCIÓN
N-Cadherina
Es una glicoproteína transmembrana que se encuentra anclada al citoesqueleto de
actina por interacciones con los complejos de catenina. Está implicada en la
formación de uniones célula-célula. Las cadherinas se consideran supresoras
tumorales y su pérdida favorece la aparición de metástasis por conferir capacidad
migratoria, asociándose a mal pronóstico. Esto es porque la expresión de N-
cadherina hace que las células interaccionen con tejidos positivos para N-cadherina
como el estroma y el endotelio potenciando su acceso al sistema vascular y su
penetración y supervivencia en órganos secundarios.44
Molecularmente, N-cadherina interacciona con el receptor de FGF disminuyendo la
señalización y promueve la supervivencia, migración e invasividad.
Se ha descrito la implicación de N-cadherina en el cáncer de mama, páncreas,
próstata y melanoma, donde su expresión crece por la disminución de E-cadherina
(intercambio entre cadherinas), normalmente debido a modificación epigenética
(hipermetilación del promotor). Este intercambio de cadherinas es parte del
programa de reprogramación transcripcional que las células epiteliales sufren con
la desdiferenciación.
PTEN (phosphatase and tensin homolog in cromosome ten)
Es una fosfatasa que antagoniza la vía de supervivencia mediada por Akt y PI3K. Se
descubrió en tumores de próstata y glioblastoma donde se observaba con
frecuencia la pérdida de un supresor tumoral en el cromosoma 10. Actúa como
regulador negativo de Akt y frenando toda la vía. La tumorogénesis en respuesta a
la perdida de PTEN actúa a través de la desregulación de la actividad de Akt.
A menudo PTEN está mutada, deleccionada o funcionalmente inhibida en tumores,
llevando a una activación constitutiva de la vía. La pérdida de un solo alelo de PTEN
parece ser suficiente para promover la tumorogénesis. Se encuentra mutado en
multitud de tumores humanos, solo superado en frecuencia por p53.23

INTRODUCCIÓN 69
3.3.1.3 Genes reparadores del ADN
Los genes reparadores de ADN codifican proteínas implicadas en la reparación del
ADN, en concreto, la reparación de los errores de emparejamiento, llamados
mismatch. Son necesarias para la activación de los puntos de control celulares en G1/S
y G2, así como para la respuesta apoptótica en respuesta al daño del ADN. Por tanto, la
pérdida de función de estas proteínas favorece la supervivencia.
La presencia de un ADN defectuoso al no haber sido reparado, produce como
resultado alteraciones somáticas en el tamaño de las secuencias de nucleótidos
repetidos (microsatélites). Este fenómeno es conocido como inestabilidad de
microsatélites (IMS), debido a la silenciación de genes de reparación de mismatch,
principalmente los genes MLH1 y MSH2.
La inactivación de los genes reparadores del ADN puede ser heredada, como en el caso
del cáncer de colon hereditario no polipósico (también conocido como síndrome de
Lynch) o adquirida, como en los tumores en los que la silenciación del gen que codifica
la proteína reparadora de “mismatch” del ADN se produce por modificación
epigenética por metilación.29
La IMS es conocida por producir la inactivación de un número de supresores tumorales
que incluyen TGFBR2, BAX y muchos otros; sin embargo, parece estar asociada con un
mejor pronóstico en los pacientes con cáncer colorrectal. 45
La pérdida de la función de reparación de mismatch es fácil de reconocer porque se
encuentra asociada al epifenómeno de inestabilidad de microsatélites, en la que
debido a la incapacidad para reparar la hebra retrasada de la secuencia repetitiva de
ADN, cambia el tamaño de las repeticiones de mononucleótidos o dinucleótidos
(microsatélites). También puede ser reconocido mediante inmunohistoquímica, al
perderse la expresión de alguna de las proteínas de reparación de mismatch. 29
La IMS se detecta en aproximadamente el 90% de las muestras tumorales de pacientes
con cáncer colorrectal hereditario no polipósico (HNPCC), con un riesgo de cáncer de
colon y recto del 80% a lo largo de la vía en individuos no diagnosticados previamente

70 INTRODUCCIÓN
de HNPCC. Este grupo de pacientes parece tener un mejor pronóstico comparado con
los que padecen un cáncer colorrectal esporádico. Los tumores colorrectales con alta
inestabilidad parecen tener un curso menos agresivo que los tumores con baja
inestabilidad o con microsatélites estables. Parece sin embargo, que este mejor
pronóstico está atenuado por la presencia de la mutación de BRAF.7,22,37
Los tumores pueden ser caracterizados en función de la frecuencia de aparición de los
marcadores de inestabilidad en:
Alta inestabilidad: si dos o más de los cinco marcadores muestran
inestabilidad
Baja inestabilidad: si solo uno de los cinco marcadores muestra
inestabilidad.
La deficiencia de MMR es más prevalente en el estadio II del cáncer de colon
comparado con el estadio III. Los tumores con IMS se localizan característicamente en
el colon proximal y tienen una histología mucinosa con infiltración por linfocitos.16
Inicialmente, el estudio de la inestabilidad de microsatélites se utilizaba en aquellos
pacientes con sospecha de HNPCC que cumplían criterios de Bestheda o de
Amsterdam. Sin embargo, aparte de utilizarlo para esta enfermedad hereditaria, cada
vez hay más evidencia de que el análisis de la posible inestabilidad de microsatélites es
un marcador pronóstico asociado a bajo riesgo de recurrencia en el cáncer de colon
estadio II. También un marcador predictivo de ausencia de beneficio significativo de
quimioterapia adyuvante con fluoropirimidinas en régimen único en este tipo de
pacientes. La mayoría de los estudios a este respecto, aunque no todos, apoyan la idea
de que la terapia adyuvante únicamente con fluoropirimidinas es menos beneficiosa, o
incluso potencialmente dañina en pacientes con IMS o deficiencia de MMR. 11

INTRODUCCIÓN 71
Las proteínas reparadoras de los defectos de emparejamiento del ADN que se han
estudiado en este trabajo son las siguientes:
MLH1 (mutL homologue 1)
Es la proteína reparadora de los defectos de emparejamiento del ADN más
estudiados. Las consecuencias patogenéticas de la silenciación de MLH1 están bien
establecidas, pero la contribución de otros eventos epigenéticos al proceso de
carcinogénesis permanece en estudio. 29
MSH2 (mutS homologue 2)
Es una proteína reparadora de los defectos de ADN. Su silenciación está
relacionada con la aparición de cáncer de colorrectal y de otros tumores
extracolónicos.
MSH6 (mutS homologue 6)
Es una proteína reparadora de los defectos de emparejamiento del ADN. Su
defecto tiene relación con el desarrollo de cáncer colorrectal a edades más
avanzadas que con MLH1 y MSH2, y tienen más riesgo de desarrollo de cáncer de
endometrio.
H-TERT
Es una telomerasa que puede tener implicación en la progresión tumoral.23 Es una
subunidad catalítica con actividad transcriptasa reversa, estructura que protege los
telómeros. La telomerasa se expresa en células madre, y su sobreexpresión
predispone al cáncer. Además de su función en los telómeros, TERT puede también
actuar como activador transcripcional en la vía Wnt-beta-catenina, así como actuar

72 INTRODUCCIÓN
como RNA-polimerasa dependiente de RNA cuando se asocia al RNA que forma
parte de la endoribonucleasa mitocondrial RMRP. La disfunción de los telómeros
causa transformación celular por provocar inestabilidad genómica.
3.3.1.4 Proteínas de la vía de la apoptosis
BCL-2
Es un proto-oncogen que codifica una familia de proteínas que están involucradas
en la regulación de la muerte celular mediante la inhibición de la apoptosis a
través de mecanismos no definidos 32, aunque parece que desempeña un papel
clave en la integridad mitocondrial y en la muerte celular programada.23
El complejo BCL-2/BAX juega un papel clave en la regulación de la apoptosis. La
proteína BAX es un promotor de la apoptosis, mientras que el protooncogen BCL-2
codifica una proteína con propiedades antiapoptosis. De acuerdo con este
escenario biológico, la sobreexpresión de BCL-2 o la disminución de la expresión de
BAX puede tener un papel pronóstico negativo en el cáncer colorrectal. 32
La sobreexpresión de BCL-2 en carcinomas de pulmón, tiroides y mama se ha
identificado como un factor pronóstico favorable en aquellos pacientes con
sobreexpresión de bcl-2 en el tumor. En el caso de los tumores colorrectales,
parece que los tumores que expresan BCL-2 por inmunohistoquímica puedan tener
un mejor pronóstico de supervivencia. 32
3.3.1.5 Proliferación celular
Ciclina D1
Codifica una proteína reguladora celular, que se expresa en niveles altos durante la
fase G1 del ciclo celular. La ciclina D1 se une a CDK y a los antígenos nucleares de
proliferación celular. La formación de estos complejos se ha implicado en el control

INTRODUCCIÓN 73
de la proliferación celular. La ciclina D1 puede ser inhibida por los CKI como p27 y
p21 en el ciclo celular.
La ciclina D1 es una proteína esencial para la progresión de G1 a S en el ciclo celular
y ha sido estudiada en muchas neoplasias, incluyendo el cáncer de colon.
Se ha demostrado que la amplificación del gen de ciclina D1 se relaciona con un
mayor grado histológico, así como con metástasis linfáticas o hematógenas y peor
pronóstico. La expresión de ciclina D1 en el cáncer de colon está relacionada con la
inestabilidad de microsatélites, el fenotipo metilador de la isla CpG (CIMP) y
mutación de BRAF.46,47
Aunque hay dos estudios que han encontrado que la expresión de ciclina D1 se ha
asociado a peor pronóstico, la mayor parte de los estudios, no han encontrado un
valor pronóstico de ciclina D1.47
Beta-catenina
Es un componente crítico de la vía de señalización de Wnt, que regula la
proliferación y diferenciación celular.
Beta-catenina existe en tres formas diferentes dentro de la célula: anclada a la
membrana, en el citoplasma y en el núcleo. Cuando beta-catenina se fosforila vía
activación de EFGR o Ras/ERK o aumenta transcripcionalmente en la célula, entra
en el núcleo, donde se une a Tcf-4 para iniciar la transcripción de de c-myc y
ciclina, entre otros, contribuyendo a la proliferación celular.
Cuando beta-catenina está libre en el citoplasma se degrada por unión al complejo
APC. Además de degradar a beta-catenina, el complejo APC actúa también
inhibiendo su localización nuclear. Cuando APC está mutado, la beta-catenina
plasmática se acumula, se transloca al núcleo y se une al factor de transcripción
Tcf-4 (T-cell factor). La activación de esta vía previene que las células entren en
fase G1 o terminen de diferenciarse e inducen resistencia a la apoptosis. El

74 INTRODUCCIÓN
complejo beta-catenina/Tcf-4 parece tener un papel fundamental en la
diferenciación de las células del epitelio de las criptas intestinales y en la
proliferación celular. Por lo tanto, la pérdida o mutación de APC contribuye a la
localización nuclear de beta-catenina, y a la activación de la vía Wnt. 26,27,47 La
activación de c-KIT activa beta-catenina llevándola al núcleo.
IMAGEN 8. Extraído de Molecular pathways in colorectal cancer.27
La pérdida de beta-catenina de membrana y el aumento en la expresión de beta-
catenina en el núcleo en tumores colorrectales se ha asociado con el estadio
tumoral y una supervivencia más corta en varios estudios. 47
Todavía no se ha encontrado utilidad clínica a la detección de la mutación de APC o
beta-catenina en lo que a selección de tratamiento, pronóstico o detección
temprana de tumores de colon se refiere. Se están intentando desarrollar
moléculas que actúen en esta vía de la tumorogénesis del cáncer de colon; sin

INTRODUCCIÓN 75
embargo, dichos estudios están todavía en fase preclínica. Si consiguiera
desarrollarse una molécula que actuara a este nivel, la detección de la beta-
catenina nuclear mediante inmunohistoquímica sería suficiente para seleccionar
los pacientes candidatos a este tratamiento.48
CDK-2 49
Sus siglas corresponden a “cyclin dependent kinase 2”. Es la subunidad catalítica de
la quinasa dependiente de ciclina que actúa en la transición G1/S del ciclo celular.
Su inactivación lleva a la célula a la parada del ciclo celular.
Se une a la ciclina E para formar un complejo esencial para la entrada en fase S. Su
actividad es máxima antes de de entrar en fase S.
La expresión de ciclina E y CDK en focos metastásicos, la correlación de su
expresión con parámetros clinico-patológicos en cáncer primario y la relación de su
expresión con el pronóstico, es desconocido.
Se ha desarrollado ya el primer inhibidor de CDK, flavopiridol, que puede utilizarse
en pacientes con micrometástasis residual en ganglios linfáticos tras cirugía
curativa.
3.3.1.6 Angiogénesis/metástasis/invasión
A lo largo de las últimas décadas se ha estudiado la importancia de los factores
relacionados con la angiogénesis y la capacidad de invasión, sabiendo actualmente que
son de gran importancia en el desarrollo y progresión de los tumores.6
VEGF
Es el factor de crecimiento vascular endotelial, y funciona como un regulador de la
angiogénesis en células sanas y en células neoplásicas.

76 INTRODUCCIÓN
El aumento en la expresión de VEGF en el cáncer de colon es un factor de mal
pronóstico.6 La tasa de expresión de VEGF en el cáncer colorrectal y su valor
pronóstico continúa siendo desconocida, como demuestran los resultados
discordantes encontrados en la literatura. 31
Se ha desarrollado un anticuerpo monoclonal humanizado que tiene por objetivo
bloquear la actividad de VEGF, el bevacizumab. Sin embargo, el uso actual de este
tratamiento para el cáncer de colon se limita a los pacientes con enfermedad
avanzada.12
CD44
Es una glicoproteína de la superficie celular que funciona como molécula de
adhesión. Hay diversas isoformas de la parte extracelular de esta proteína, en
función del “splicing” de los diez exones que componen la proteína. Se han
identificado diferentes variantes de CD44 en las células tumorales. 31
Induce reacciones antiapoptóticas y de supervivencia tumoral cuando se activa. La
sobreexpresión de CD44 activa la vía de beta-catenina por regular la fosforilación
de AKT.
Los tumores de colon con expresión intensa de CD44 tienen peor pronóstico que
aquellos con poca expresión, independientemente del estadio de Dukes.31,50
E-Cadherina
Es una glicoproteína de adhesión implicada en la formación de uniones célula-
célula. Es una proteína transmembrana que interacciona con proteínas del
citoplasma, especialmente con beta-catenina. 31. La E-cadherina forma uniones con
su dominio extracelular, teniendo un papel fundamental en la adhesión célula-
célula. El dominio intracelular, a su vez, produce interacciones de actinas del

INTRODUCCIÓN 77
citoesqueleto, mediante la interacción con cateninas. Se postula, que la perdida de
función de las cadherinas es un factor asociado a la invasividad tumoral y a un
estadio celular tumoral avanzado. 21
Las cadherinas se consideran supresoras tumorales y su pérdida favorece la
aparición de metástasis por conferir capacidad migratoria, asociándose a mal
pronóstico. En el proceso de metástasis, las células tumorales deben adquirir la
capacidad para la angiogénesis y para la migración. Esta migración precisa de una
“transición epitelial-mesenquimal”, por la que las células adquieren características
que les facilitan la invasión tisular. Este proceso se produce por la activación de
múltiples vías que incluye la activación de TGF-beta, que a su vez activa la vía Wnt y
produce la alteración de la expresión de factores de transcripción y produce una
represión en la adhesión intracelular de E-cadherina.27
La alteración del gen APC también puede dar lugar a cambios en la adhesión célula-
célula, debido a una modificación en su unión con E-cadherina. 8
Osteonectina 31,46
También llamado SPARC (Secreted Protein Acidic and Rich in Cysteine) o BM-40, es
una glicoproteína asociada a la matriz celular que influye en la remodelación
tisular, la reparación de heridas, la migración celular y la angiogénesis. La
señalización de osteonectina en relación a su papel en la proliferación se produce
por la inhibición de la vía STAT3, que induce la parada del ciclo mediado por p16,
p21 y p27. Por tanto, al menos en determinados tipos celulares, la sobreexpresión
de osteonectina lleva a parada del ciclo y parada de la proliferación de tumores, y
también de células endoteliales.
Por su papel sobre las integrinas, favorece la migración al provocar la pérdida de
anclaje celular. Podría en conjunto provocar un estado en que la célula tumoral
deja de crecer y se prepare para migrar.

78 INTRODUCCIÓN
La expresión aumentada de osteonectina se ha asociado a un aumento del
crecimiento del tumor, las metástasis y a un peor pronóstico en varias neoplasias
que incluyen el melanoma, el cáncer de mama y el de esófago.
La osteonectina también se ha implicado en el desarrollo de pólipos colónicos y
progresión del cáncer colorrectal a través de la regulación de la cicloxigenasa-2 y la
transformación de TGF-beta.
Osteopontina
Es una proteína que fue identificada asociada a tumores en células transformadas
en cultivo. Las funciones biológicas de osteopontina todavía no se entienden de
manera completa, aunque se relaciona con diversas neoplasias, funciones inmunes
y remodelación vascular, así como en la remodelación de hueso. 51
También llamada SPP1, esta glicoproteína forma parte de la matriz extracelular e
induce la motilidad celular e inflamación persistente. Puede unirse a varios
receptores integrinas y a CD44 y está implicada en el desarrollo de enfermedades
inmunes crónicas. Según el tipo celular, se ha descrito que induce la motilidad y la
invasividad, promueve la supervivencia y protege las células contra la apoptosis.
Se ha asociado a progresión tumoral en muchos tumores sólidos y parece que tiene
relación con partes críticas de la tumorogénesis, como quimiotaxis, angiogénesis
dependiente de estrés, prevención de la apoptosis y crecimiento independiente de
sustrato. Hay una correlación entre la sobreexpresión de osteopontina y la
expresión aberrante de beta-catenina (señal de la activación de la vía Wnt), y
parece que ostepontina es una diana transcripcional de la señalización Wnt
aberrante. 52,53

INTRODUCCIÓN 79
3.3.1.7 Estructura celular
CEA
Sus siglas corresponden al antígeno carcinoembrionario. Es una glicoproteína de
membrana que une las selectinas E y L, contribuyendo al anclaje de las células a la
microvascularización. Se expresa de manera normal en el epitelio intestinal y se
sobreexpresa en procesos tumorales intestinales. Otorga así un mayor potencial
metastásico a los tumores por favorecer el “rolling” dependiente de selectina L a
través del flujo sanguíneo.
Es un marcador serológico utilizado habitualmente en el seguimiento del cáncer
colorrectal para detectar recurrencias o metástasis. No se ha encontrado utilidad
pronóstica en la detección de la tinción cuantitativa de CEA en muestras tumorales
de cáncer colorrectal y en mucosa normal.31
Algunos autores han encontrado en sus estudios que las concentraciones de CEA
en tejido tumoral no predecían el pronóstico tras la cirugía, aunque se
correlacionaban significativamente con el estadiaje de Dukes. No se encontró
relación entre el patrón tisular de la distribución de CEA y los parámetros
morfológicos de la neoplasia, excepto en los carcinomas con compromiso linfático,
que presentaban mayor distribución tisular de CEA en la parte apical que en la
citoplasmática, al compararlo con los que no tenía compromiso linfático. 54
CK19
Sus siglas corresponden a Citokeratina 19. Pertenece a una familia de queratinas
que normalmente se expresan en la superficie del tracto gastroenteropancreáticos
y hepatobiliar. Es una queratina con filamentos intermedios estructurales,
responsable de la integridad de la célula epitelial.
CK19 es positivo en la mayoría de los tumores neuroendocrinos del tracto
gastrointestinal, excepto a nivel rectal, donde es negativo.

80 INTRODUCCIÓN
CK19 se ha mostrado como factor pronóstico independiente en tumores
pancreáticos neuroendocrinos, especialmente en los tumores insulina negativos.
De hecho, se recomienda el análisis inmunohistoquímico de los tumores
pancreáticos endocrinos como parte del estudio anatomopatológico. 55
CK20
Es una queratina de filamentos intermedios estructurales, responsable de la
integridad estructural celular. CK20 es una proteína celular de enterocitos maduros
y “globet cells” del tracto digestivo, el urotelio y las células de Merckel en la piel.
El inmunofenotipo CK20+/CK7- se ha considerado característico del
adenocarcinoma colónico. 56
EMA (Epithelial Membrane Antigen)
Es una glicoproteína de membrana que está implicada en la polarización celular y la
estructura de membrana, que se proteoliza en dos fragmentos: alfa implicada en la
adhesión, y beta implicado en la señalización. Regula la progresión tumoral y
determina el destino celular en la respuesta al estrés genotóxico (reprime la
actividad de p53), a través de la regulación de las rutas de ERK, SRC y NF-kappa-B.
Compite con E-cadherina por la unión a beta-catenina. Al unirse a esta, pueden
entrar en el núcleo activando LEF1, que a su vez activa la transcripción.
EMA se expresa en los tumores colónicos al igual que CEA. En un estudio se
encontró que la expresión de EMA es más específica de neoplasia de colon, aunque
menos sensible que CEA.57

INTRODUCCIÓN 81
3.3.1.8 Diferenciación celular:
CD133
Es un marcador de células madre de cáncer (CSC). Se expresa en tejidos
indiferenciados y neoplásicos. Se asocia a un peor pronóstico y a la resistencia a
tratamientos citotóxicos. Suprime la diferenciación, pero no se conoce bien su
función. Puede que sea estructural de membrana, similar a la caveolina, porque es
capaz de unirse al colesterol y esfingolípidos y también a receptores tirosin-kinasa.
Se cree que las CSC están dotadas con una capacidad única de renovarse. Por
definición, una sola CSC puede alojarse en un órgano permisivo como el hígado, y
producir metástasis. Actualmente no es posible aislar células neoplásicas
colorrectales de forma individual, aunque algunas proteínas de superficie (como
por ejemplo CD133, CD44, CD166, y la aldehído deshidrogenasa 1) son
prometedoras.29
La positividad para CD133 tiene una relación lineal con la positividad para invasión
linfática tumoral, metástasis a distancias, clasificación de Dukes y estadiaje TNM. 50
HES-1 (Hairy and Enhancer of Split homologue 1)
Es un represor transcripcional de la familia helix-loop-helix, que reprime genes que
necesiten factores de este tipo para su transcripción. Se expresa en tumores y
células quiescentes, frenando la diferenciación. Vías como Notch, Hedgehog o JNK
activan a HES-1 para bloquear la diferenciación.
NANOG
Es un factor trascripcional fundamental en el mantenimiento de la integridad de las
células madre embrionarias e implicado en el control de la autorenovación y en el

82 INTRODUCCIÓN
bloqueo de la diferenciación propio de las células madre. Puede ser activado por
estímulos intrínsecos y extrínsecos. Entre los estímulos extrínsecos se encuentran
TGF-beta, Hedgehog y HIF; y entre los factores intrínsecos se encuentra beta-
catenina y SMAD, produciendo un aumento de Nanog. El estímulo de p53 o la
modificación epigenética del promotor de Nanog producen una inhibición del
mismo. Su activación (por ejemplo, por la activación de CD44) disminuye la
expresión de supresores de tumores, favoreciendo la supervivencia y
quimiorresistencia y disminuyendo la apoptosis.58
Se suele expresar en altos niveles en tumores, incluso en estadios muy tempranos,
pero no en células normales maduras. La expresión de la proteína Nanog en células
cancerígenas de colon se suele producir en estadios avanzados y es un signo de mal
pronóstico. 58

INTRODUCCIÓN 83
4.- Seguimiento, tratamiento y pronóstico del cáncer de colon
4.1 SEGUIMIENTO
Las pautas óptimas de seguimiento tras cirugía con intención curativa en el cáncer de
colon no quedan claras. No está bien definido cada cuanto debería ser evaluado un
paciente trasuna resección curativa por cáncer de colon.
La historia clínica y el examen físico, combinado con la determinación de CEA sérico, a
intervalos regulares, puede darnos un beneficio coste-efectivo para detectar recidivas.
La sensibilidad para detectar recidivas con TC o CEA sérico se sitúa en torno al 61%.8
Por supuesto, dada la aparición de un cáncer en el colon, todo paciente con CC
resecado se considera de alto riesgo, y debe realizarse colonoscopias de forma regular,
según las pautas de las guías clínicas.12
Aproximadamente el 85% de las recurrencias se detectan en los dos años posteriores a
la cirugía, por lo que el seguimiento debe ser especialmente intenso durante este
periodo. 7
Las recomendaciones de seguimiento del cáncer de colon resecado de la NCCN, según
la medicina basada en la evidencia, son: 12
Revisión médica cada 3-6 meses durante los primeros 2 años y posteriormente
cada 6 meses hasta los 5 años.
CEA sérico cada 3-6 meses durante 2 años y posteriormente cada 6 meses
hasta los 5 años, para tumores T2 o superior.
Considerar la realización de TC abdominal/torácica/pélvico durante 3 años en
pacientes en estadio III y en estadio II con factores de mal pronóstico (con
invasión linfática o venosa o histología pobremente diferenciado).
TC cada 3-6 meses durante 2 años, y después cada 6-12 meses hasta los 5 años
en pacientes con enfermedad metastásica (incluye N positivo).
Colonoscopia al año de la cirugía, y otra a los 3 años, posteriormente cada 5
años.

84 INTRODUCCIÓN
Aquellos pacientes con síntomas sospechosos como síndrome constitucional (fatiga,
disminución del apetito, sudoración nocturna, fiebre,…), deberán ser valorados en
busca de posible recurrencia de la enfermedad.

INTRODUCCIÓN 85
4.2 TRATAMIENTO ADYUVANTE EN EL CÁNCER DE COLON
La quimioterapia es el tratamiento inicial en pacientes con enfermedad a distancia al
diagnóstico (estadio IV), y el tratamiento estándar indicado tras la cirugía con intención
curativa en pacientes con estadio III. Las guías clínicas de la ASCO y la NCCN no
recomiendan la administración rutinaria de adyuvancia en estadio II, aunque en
determinados pacientes con factores de riesgo, si que puede valorarse esta posibilidad
tras exponer al paciente los riesgos y beneficios potenciales, y generalmente, dentro
del escenario de un ensayo clínico.11
El principio en el que se basa la terapia adyuvante es que el tratamiento es más
efectivo cuando la carga tumoral es mínima y la cinética celular es óptima. Numerosos
estudios han demostrado un retraso en la recurrencia y un aumento de supervivencia
en grupos de pacientes con cáncer de colon que han recibido quimioterapia dentro de
las 8 semanas posteriores a la cirugía. 8
El tratamiento adyuvante está indicado de rutina en pacientes con CC en estadio III y
IV, es decir con extensión tumoral a nivel ganglionar o a distancia, respectivamente.
Sin embargo, no queda claro si los pacientes con CC en estadio II deben recibir
quimioterapia adyuvante, porque no ha sido establecido el balance riesgo/beneficio.
De forma global, la administración de terapia adyuvante en el CC en estadio II produce
un beneficio menor al 5% en la supervivencia en el mejor de los casos. La ASCO en
2004 publicó que no había evidencia directa que recomendara el uso de terapia
adyuvante fuera de ensayos clínicos aleatorizados, incluso en los casos de pacientes
con CC en estadio II y factores de alto riesgo; sin embargo, también sugería que de
forma indirecta, estaba justificada el planteamiento de quimioterapia adyuvante en
relación con los beneficios encontrados en los pacientes con CC estadio III, tras
entender el paciente que el beneficio absoluto en la supervivencia es pequeño59.
En el estudio Quasar encontraron que la terapia con 5FU y leucovorin podía mejorar la
supervivencia de pacientes estadio II, aunque este beneficio era pequeño.8

86 INTRODUCCIÓN
4.3 PRONÓSTICO DEL CÁNCER DE COLON
El factor pronóstico más importante tras una resección curativa por CC es el estadiaje
del tumor primario tras la cirugía. A pesar de realizar una resección completa, los
pacientes en los que el tumor había penetrado la serosa o que tienen metástasis en los
ganglios linfáticos regionales en el momento de la cirugía tienen altas tasas de
recurrencia. El riesgo de recurrencia tras la cirugía varía de un 20-30% en el estadio II a
un 50-80% en el estadio III. 8
La supervivencia en pacientes resecados con intención curativa depende del estadiaje:
en el estadio I la supervivencia a 5 años se sitúa en torno al 90%,mientras que en el
estadio II la supervivencia a 5 años está entre el 72 al 85% a los cinco años
dependiendo de otros factores. La supervivencia en pacientes en estadio III, con
ganglios linfáticos regionales afectados, se sitúa entre el 44 y el 83% a los cinco años
tras la quimioterapia adyuvante, y en los pacientes con metástasis a distancia (estadio
IV) es menor del 5%.11
Dentro del estadio II (T3-T4 N0), la supervivencia a los cinco años en pacientes en
estadio IIA, es decir T3 N0, es del 85%, comparada con el 72% de supervivencia a 5
años de los pacientes con estadio IIB (T4a N0), que es de hecho peor que la de los
pacientes en estadio IIIA (con metástasis en ganglios regionales) cuya supervivencia se
sitúa sobre el 83% a los cinco años (teniendo en cuenta que estos pacientes sí que
reciben terapia adyuvante). La ASCO sugiere la utilización de terapia adyuvante basada
en 5-FU en pacientes en estadio II con al menos un factor de riesgo de mal pronóstico
(menos de 12 ganglios en la pieza quirúrgica, lesiones T4, histología pobremente
diferenciada o perforación intestinal).7,8

HIPÓTESIS Y
OBJETIVOS


HIPÓTESIS Y OBJETIVOS 89
Justificación
Actualmente no está indicado el tratamiento sistemático con quimioterapia tras la
cirugía de los pacientes que presentan CC estadio II de la clasificación TNM, excepto en
el contexto de ensayos clínicos. La selección de pacientes en estadio II del CC con
riesgo de recidiva supone, actualmente, un reto importante para el oncólogo y el
cirujano. Sin embargo, el beneficio de la adyuvancia no mejora la supervivencia más
allá de un 5%, por lo que es fundamental encontrar variables que nos hagan
seleccionar a los pacientes que más se pueden beneficiar de la quimioterapia
adyuvante. 11 Se han encontrado variables clínicas y anatomopatológicas que nos
pueden ayudar en la toma de decisiones 10–12
Las guías de la ASCO están en contra de la administración rutinaria de quimioterapia
sistémica adyuvante en los pacientes con cáncer de colon estadio II, excepto en el
contexto de los ensayos clínicos. A pesar de la ausencia de evidencia de beneficio a
este respecto, las guías ASCO también sugieren que los riesgos y beneficios de la
quimioterapia adyuvante sean discutidos con los pacientes con factores de alto riesgo,
como son la perforación, estadio T4, invasión linfovascular o perineural, histología
pobremente diferenciada incluyendo morfología en anillo de sello y morfología
mucinosa y la presencia de menos de 13 ganglios en la pieza quirúrgica11.
En la última década se ha avanzado de forma importante en el conocimiento de las
características moleculares y biológicas de la patogénesis del CCR. La aparición de
nuevas herramientas nos permiten en la actualidad determinar grupos de alto riesgo
de CCR y realizar un seguimiento adecuado, así como definir que pacientes se van a
beneficiar más de terapias adyuvantes, de un cribado por colonoscopia más frecuente
o incluso la utilización de una cirugía profiláctica en los casos con alto riesgo de
desarrollar cáncer a lo largo de su vida. 8
El objetivo de este estudio es determinar que variables clínicas, anatomopatológicas y
moleculares están relacionadas con las máximas posibilidades de recidiva en el cáncer
de colon en estadio T2-T4/N0 (estadio B de la clasificación modificada de Astler-Coller).

90 HIPÓTESIS Y OBJETIVOS
Si dispusiéramos de esta información con mayor precisión que la que tenemos
actualmente, nos permitiría establecer el tratamiento adyuvante y el seguimiento de
paciente de forma selectiva e individualizada.

HIPÓTESIS Y OBJETIVOS 91
Hipótesis
Existen factores clínicos, analíticos, anatomopatológicos y moleculares que nos pueden
ayudar a detectar aquellos pacientes con mayor riesgo de recidiva tras ser intervenidos
de CC en estadio anatomopatológico B de Astler-Coller (T2-T4 N0).
Objetivos
Los objetivos primarios de este estudio son:
1. Identificar factores clínicos pronósticos de recidiva en el CC T2-T4 N0.
2. Identificar factores analíticos pronósticos de recidiva en el CC T2-T4 N0.
3. Identificar factores anatomopatológicos pronósticos de recidiva en el CC T2-T4
N0.
4. Identificar marcadores moleculares pronósticos de recidiva en el CC T2-T4 N0.
Y de esta manera poder:
1. Establecer de forma individual el riesgo de recidiva en CC estadio T2-T4 N0.
2. Seleccionar aquellos pacientes candidatos al tratamiento quimioterápico tras la
cirugía, evitando el tratamiento innecesario a la mayoría de los pacientes que
en este estadio estarán curados sólo con el tratamiento quirúrgico adecuado.
3. Establecer un seguimiento intensivo según el riesgo individual de recidiva,
evitando los costes y perjuicios de las pruebas diagnósticas en los pacientes
potencialmente curados.


MATERIAL Y
MÉTODOS


MATERIAL Y MÉTODOS 95
1.- Diseño del estudio
Se trata de un estudio epidemiológico, observacional y analítico según el esquema de
cohortes retrospectivo, en el que una parte del grupo durante el seguimiento presentó
un evento, la aparición de recidiva de cáncer de colon previamente intervenido con
intención curativa.
La muestra estaba compuesta por un grupo de pacientes con diagnóstico inicial
adenocarcinoma en estadio B de la clasificación modificada de Astler-Coller (T2-T4N0)
según la anatomía patológica y que fueron intervenidos quirúrgicamente con intención
curativa.
Durante el seguimiento de los pacientes, se desarrolló recidiva del adenocarcinoma de
colon en algunos pacientes, bien de forma local o a distancia, de forma que se dividió a
los pacientes en función de si presentaron recidiva de la enfermedad (Grupo recidiva)
o no lo presentaron (Grupo ausencia de enfermedad).
Las variables analizadas han sido las características analíticas, clínicas y
anatomopatológicas de los pacientes y del tumor al diagnóstico. Así mismo se han
revisado las piezas a nivel anatomopatológico y se han estudiado una batería factores
moleculares y biológicos relacionados con la oncogénesis tumoral, el crecimiento
celular, la apoptosis, la estructura celular, la división celular, la proliferación celular y la
invasión celular.
Se recogieron los datos demográficos de todos los pacientes estudiados.

96 MATERIAL Y MÉTODOS
2.- Esquema de diseño
Mediante búsqueda por informe anatomopatológico, se seleccionaron todos los
pacientes con estadio B de la clasificación modificada de Astler-Coller intervenidos en
un periodo de tiempo de 22 años, y se analizaron los siguientes factores:
2.1. FACTORES ANALÍTICOS
CEA preoperatorio
2.2 FACTORES CLÍNICOS
Presencia de perforación tumoral
Presencia de obstrucción tumoral
2.3 FACTORES ANATOMOPATOLÓGICOS
Tamaño tumoral
Morfología tumoral.
Invasión vascular del tumor (de vaso fino o grueso)
Invasión linfática
Invasión perineural
Porcentaje de mucina en el tumor
Presencia de células en anillo de sello
Numero de ganglios extirpados

MATERIAL Y MÉTODOS 97
2.4 FACTORES MOLECULARES Y BIOLÓGICOS
Los factores moleculares que han sido estudiados en el tumor según su función
biológica son:
Oncogenes
cERB2
EGFR
C-myc
C-kit
NTSR-1
Genes supresores tumorales
P53
P21
P27
Nm23
SMAD4
Caveolina
P16
Proteína del retinoblastoma
N-Cadherina
PTEN
Genes reparadores del ADN
MLH1
MSH2
MSH6
H-TERT

98 MATERIAL Y MÉTODOS
Proteínas de la vía de la apoptosis
BCL-2
Proliferación celular
Cyclin D1
B-catenina
CDK-2
Angiogénesis/metástasis/invasión
VEGF
CD44
E-Cadherina
Osteonectina
Osteopontina
Estructura celular
CEA
CK19
CK20
EMA
Diferenciación celular
CD133
HES-1
NANOG

MATERIAL Y MÉTODOS 99
2.5 MOMENTO DE LA DETERMINACIÓN DE LAS VARIABLES
Se analizaron las muestras de la pieza quirúrgica de los pacientes intervenidos de
cáncer de colon con AP de adenocarcinoma estadio II, entre los años 1975 y 2007 por
un grupo reducido de cirujanos que utilizaron los mismos criterios oncológicos y
realizaron una técnica quirúrgica estandarizada.
Estas muestras fueron analizadas por un solo anatomopatólogo del Servicio de
Anatomía Patológica del Hospital Universitario Ramón y Cajal que determinó las
variables anatomopatológicas y posteriormente fueron remitidas al Centro Nacional de
Investigación Oncológica (CNIO) para el análisis de variables moleculares y biológicas.
En ambos casos, el análisis fue ciego a si el paciente presentó durante el seguimiento
recidiva o no.
Paralelamente se recogió y continuó el seguimiento de todos los pacientes hasta 2011
o hasta la muerte o pérdida de seguimiento del paciente, con un seguimiento mínimo
de dos años tras la cirugía para poder detectar la presencia de recidiva tumoral.

100 MATERIAL Y MÉTODOS
3. Población de estudio
3.1. POBLACIÓN DIANA
Esta tesis se ha realizado en el Servicio de Cirugía General y Digestivo del Hospital
Universitario Ramón y Cajal de Madrid. Este Hospital depende de la Comunidad de
Madrid y es hospital de referencia estatal y del Área Sanitaria 4 de la Comunidad de
Madrid. Además es Hospital Universitario vinculado a la Universidad de Alcalá de
Henares.
La población total del Área Sanitaria es de 536.071 según datos de la Memoria Anual
del Hospital de 2008. No se conocen las cifras exactas de incidencia del CCR en Madrid,
pero en el año 1997 en España se registraron, 19.166 casos nuevos con una tasa bruta
de 58,9 por 100.000 en hombres y 46,59 en mujeres.
La población diana con la que se trabaja en esta tesis son todos los pacientes
intervenidos de CCR entre los años 1975 y 2007 por un equipo de tres cirujanos
especializados en cirugía de colon dentro del Servicio de Cirugía General y del Aparato
Digestivo del Hospital Universitario Ramón y Cajal de Madrid, en los que se realizó una
cirugía oncológica radical y con intención curativa para el tratamiento de su
enfermedad y con posterior diagnóstico anatomopatológico de adenocarcinoma de
colon estadio B de la clasificación de Astler-Coller. Este grupo de pacientes son
extrapolables a la población española.

MATERIAL Y MÉTODOS 101
3.2 TAMAÑO DE LA MUESTRA
En este caso fueron incluidos todos los pacientes que cumplían los requisitos del
estudio, es decir, todos los pacientes con adenocarcinoma de colon estadio B de
Astler-Coller intervenidos en el periodo 1975 a 2007 por un equipo de tres cirujanos
colorrectales en el Hospital Universitario Ramón y Cajal.
Se trata por tanto de 141 pacientes intervenidos de adenocarcinoma de colon estadio
B de Astler-Coller en el Servicio de Cirugía General y Digestivo en el periodo de tiempo
comprendido entre Noviembre de 1975 y Diciembre de 2007. Durante ese periodo se
valora en el seguimiento la presencia o no de recidiva o metástasis tumoral y se divide
la muestra en dos grupos: 116 pacientes en el grupo de ausencia de enfermedad y 25
pacientes en el grupo de presencia de enfermedad recidivada.

102 MATERIAL Y MÉTODOS
3.3 SELECCIÓN DE LA MUESTRA
Se trata de una muestra no probabilística de la población diana. Los criterios que se
han utilizado para la selección son los siguientes:
Criterios de inclusión:
- Pacientes con diagnóstico anatomopatológico de cáncer de colon entre los
años 1975 y 2007.
- Estadiaje anatomopatológico B de Astler-Coller (T2-T4 N0 de la clasificación
TNM).
- Pacientes que no hayan recibido quimioterapia adyuvante tras la cirugía.
- Intervenidos quirúrgicamente de su proceso oncológico por un equipo de
cirujanos especializados en tratamiento del cáncer de colon durante ese
periodo.
- Información clínica completa.
-Seguimiento de al menos dos años tras la cirugía o hasta el fallecimiento o
pérdida de seguimiento.
Criterios de exclusión:
- Tumor a nivel del recto
- Haber recibido terapia adyuvante o neoadyuvante.
- Estudio anatomopatológico incompleto
- No cumplir los criterios de inclusión.

MATERIAL Y MÉTODOS 103
3.4 SELECCIÓN DE LOS GRUPOS DE TRABAJO
Se incorporaron al estudio todos los pacientes que habían sido sometidos a
intervención quirúrgica por adenocarcinoma de colon estadio B de Astler-Coller según
el estudio anatomopatológico de la pieza quirúrgica y los hallazgos intraoperatorios.
Todas las muestras anatomopatológicas fueron estudiadas en el Servicio de Anatomía
Patológica del Hospital Universitario Ramón y Cajal de Madrid.
Posteriormente se dividieron a los pacientes en dos grupos:
1. Grupo recidiva: aquellos que durante el seguimiento presentaron recaída
tumoral en forma de metástasis local o a distancia.
Criterios de inclusión:
-Demostración clínica y/o radiológica de recidiva tumoral durante el
seguimiento.
2. Grupo ausencia de enfermedad: aquellos que durante el seguimiento y hasta el
final del estudio o la pérdida de seguimiento, no presentaron recaída de su
enfermedad tumoral.
Criterios de inclusión:
-Ausencia de recidiva tumoral demostrada en el seguimiento mediante
examen físico, analítico y radiológico.

104 MATERIAL Y MÉTODOS
4. Procedimiento
4.1. RECOGIDA DE DATOS
De cada uno de los pacientes se recogió el número de historia, número de
identificación de pieza quirúrgica, edad, así como fecha y tipo de cirugía. Las variables
fueron incorporadas a una base de datos de Excel diseñada previamente.
Las variables demográficas recogidas fueron:
- Fecha de nacimiento
- Edad cuando se realizó la cirugía
- Sexo
Las variables prequirúrgicas recogidas fueron:
- CEA sérico
Las variables anatomopatológicas recogidas del estudio de la pieza quirúrgica fueron:
- Localización del tumor
o Ciego/colon derecho
o Colon transverso
o Colon izquierdo/sigma
- Obstrucción del tumor
- Perforación del tumor
- Tamaño del tumor (en centímetros)
- Grado de diferenciación tumoral:
o bien diferenciado
o moderadamente diferenciado
o indiferenciado
- Presencia o ausencia de células en anillo de sello
- Nivel de infiltración
o T2
o T3
o T4

MATERIAL Y MÉTODOS 105
- Presencia o ausencia de invasión vascular de vaso fino o grueso
- Presencia o ausencia de invasión linfática
- Presencia o ausencia de invasión perineural
- Presencia o ausencia de componente mucinoso en el tumor
- Estadificación de Dukes
- Estadificación de Astler-Coller
- Estadificación T de la clasificación TNM:
o T2 N0
o T3 N0
o T4 N0
- Número total de ganglios extraídos
En el seguimiento fueron recogidos los siguientes datos:
- Recidiva tumoral y fecha de la misma
- Lugar de la recidiva tumoral y tratamiento realizado en relación con la misma
- Fecha fin de seguimiento
- Éxitus de pacientes, causa del éxitus y fecha de la misma.
- Estado del paciente al final del seguimiento.
A partir de los datos obtenidos, se calculó en meses:
- Supervivencia libre de enfermedad (SLE)
- Supervivencia global (SG)

106 MATERIAL Y MÉTODOS
4.2 PARÁMETROS ANATOMOPATOLÓGICOS
Fueron estudiadas por el servicio de Anatomía Patológica conforme al protocolo de
estudio estándar del hospital, que incluye el tamaño tumoral, la distancia a los bordes
de resección, la estadificación TNM, Dukes y Astler-Coller, así como el número de
ganglios, la presencia de células en anillo de sello, la presencia de componente
mucinoso y la presencia de perforación u obstrucción de la pieza quirúrgica.
Posteriormente y de forma retrospectiva, un solo anatomopatólogo ciego a la
presencia o ausencia de recidiva, estudió todas las muestras para la determinación de
la infiltración vascular de vaso fino y grueso, la invasión perineural y la invasión
linfática.
4.3 PARÁMETROS MOLECULARES
Tras la selección de todas las muestras de anatomía patológica según los criterios de
inclusión y exclusión, fueron enviadas muestras de cada uno de los pacientes (de tejido
tumoral y tejido sano de la pieza quirúrgica) al CNIO donde fueron procesadas para la
determinación de la expresión de una batería de variables moleculares mediante
técnicas de inmunohistoquímica y clasificadas de manera semicuantitativa según los
propios criterios establecidos por el CNIO para la incorporación a la base de datos. En
el anexo I se encuentra la descripción técnica del análisis inmunohistoquímico.
La determinación de las variables moleculares a estudiar, fueron seleccionadas por el
CNIO en base a la expresión en estudios previos sobre tejido colónico o tumoral de
dichas variables. De forma exploratoria se realizó el estudio de las 39 variables
seleccionadas por el CNIO sobre las primeras 50 muestras. Algunas variables
moleculares no se expresaron de forma diferencial en las muestras, por lo que se
excluyeron del resto del análisis inmunohistoquímico del resto de la cohorte.
Las variables son las que siguen y se clasificaron de la siguiente manera:
I. BCL2: negativo, positivo débil o positivo intenso.

MATERIAL Y MÉTODOS 107
II. B-CATENINA: negativo, positivo débil o positivo intenso.
III. CAVEOLINA: pobreza de estroma entre glándulas, moderada presencia de
estroma entre glándulas o riqueza de estroma entre glándulas.
IV. CD133 (VALORADO EN TOTAL, EN ESTROMA Y EN TUMOR): negativo, positivo
débil, positivo intenso o positivo muy intenso.
V. CD44: negativo, positivo débil o positivo intenso.
VI. CDK2 (EN NUCLEO Y CITOPLASMA): negativo, menor del 25%, entre el 25-75%
o mayor del 75%.
VII. CEA: negativo, positivo débil o positivo intenso.
VIII. C-ERB: negativo, positivo débil o positivo intenso.
IX. CK19: negativo, positivo débil o positivo intenso.
X. CK20: negativo, positivo débil, positivo intenso o positivo muy intenso
XI. C-KIT: negativo, positivo débil o positivo intenso.
XII. C-MYC: negativo, positivo débil o positivo intenso.
XIII. COX2: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XIV. CYCLIN D1: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XV. E-CADHERINA: negativo, positivo débil o positivo intenso.
XVI. EGFR: negativo, positivo débil o positivo intenso.
XVII. EMA: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XVIII. HES1: negativo, positivo débil o positivo intenso.
XIX. HTERT: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XX. KI67: negativo, positivo débil o positivo intenso.
XXI. MLH1: negativo, positivo débil o positivo intenso.
XXII. MSH2: negativo, positivo débil o positivo intenso.
XXIII. MSH6: negativo, positivo débil, positivo intenso o positivo muy intenso.

108 MATERIAL Y MÉTODOS
XXIV. MSI (MLH1 o MSH2 o MSH6 desaparecen): inestable o estable.
XXV. NANOG: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXVI. N-CADHERINA: negativo, positivo débil o positivo intenso.
XXVII. NEUROTENSINA: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXVIII. NM23: negativo, positivo débil o positivo intenso.
XXIX. NTSR1: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXX. OSTEONECTINA: negativo, positivo débil o positivo intenso.
XXXI. OSTEOPONTINA: negativo, positivo débil o positivo intenso.
XXXII. P16: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXXIII. P21: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXXIV. P27: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXXV. P53: negativo, positivo débil o positivo intenso.
XXXVI. PTEN (en núcleo y en citoplasma): negativo, menor del 25%, entre el 25-75% o
mayor del 75%.
XXXVII. RBP: negativo, positivo débil, positivo intenso o positivo muy intenso.
XXXVIII. SMADY: negativo, menor del 25%, entre el 25-75% o mayor del 75%.
XXXIX. VEGRF: negativo, positivo débil o positivo intenso.

MATERIAL Y MÉTODOS 109
5. Descripción operativa
1. Se formuló la hipótesis principal y las secundarias.
2. A partir de ello se recogieron los datos de todos los pacientes intervenidos con
diagnóstico de adenocarcinoma de colon estadio B de Astler-Coller, según los
criterios de inclusión y exclusión.
3. Se realizó un análisis anatomopatológico ciego de cada uno de los tumores.
4. Se enviaron las muestras tumorales al CNIO para el análisis de las variables
moleculares. El análisis se realizó mediante tissue micro-arrays. En la unidad de
inmunohistoquímica del CNIO se realizó el procesamiento de todas las
muestras.
5. Se realizó el análisis de 39 marcadores moleculares, que posteriormente fueron
valoradas de forma semicuantitativa por un único investigador del CNIO.
6. Las variables que no se expresaban de forma diferencial en el análisis
preliminar, se excluyeron del análisis del resto de las muestras.
7. Se realizó el análisis estadístico preliminar y posteriormente se estudiaron las
variables moleculares agrupadas de forma dicotómica (ver anexo VI).
8. Se creó una nueva variable llamada “Riesgo” con los factores
anatomopatológicos de mal pronóstico. La variable numérica ganglios se
clasificó de forma dicotómica en menor de 12 ganglios y mayor o igual a 12
ganglios. Y la variable numérica CEA sérico se clasificó en mayor o menor de
5ng/mL.
9. Se creó una variable molecular llamada inestabilidad de microsatélites,
agrupando los resultados de las variables MLH1, MSH2 y MSH6, y clasificando
las muestras como estable o inestable (si se encontraba ausente la expresión
de alguna de las proteínas reparadoras).

110 MATERIAL Y MÉTODOS
10. Se realizó análisis univariante de los factores analíticos, clínicos,
anatomopatológicos y moleculares para analizar la supervivencia global y la
supervivencia libre de enfermedad.
11. Se realizó análisis multivariante de los factores analíticos, clínicos,
anatomopatológicos y moleculares significativos o cercanos a la significación
estadística en el análisis univariante para analizar la supervivencia global y la
supervivencia libre de enfermedad.

MATERIAL Y MÉTODOS 111
6.- Limitaciones del estudio
Este estudio por su diseño de cohortes retrospectivo, está sujeto a sesgos de selección
e información que pueden condicionar los resultados. Las asociaciones observadas
siempre deben corroborarse con estudios prospectivos, y es la observación repetida de
resultados consistentes la que sustenta la evidencia.
Además, este estudio por su planteamiento tiene otras limitaciones. La primera de las
cuales es que los pacientes del estudio no son todos los pacientes con diagnóstico de
adenocarcinoma estadio B de un área geográfica ni siquiera del centro hospitalario,
sino solo los intervenidos por un grupo de cirujanos. Esto se realizó así para disminuir
el sesgo del cirujano en el análisis por ser un grupo de cirujanos con amplia experiencia
en el tratamiento quirúrgico del cáncer de colon y con los mismos criterios y técnica
estandarizada. Sin embargo los pacientes que fueron remitidos a estos cirujanos
pueden suponer un sesgo en sí (mayor cantidad de pacientes con predisposición
genética, variabilidad en la demografía, tumores más avanzados, etc.)
Otra de las limitaciones del estudio es el número de casos, que es bajo, por lo que la
posibilidad de detectar factores pronósticos de recidiva es más difícil, al no tener
suficiente potencia estadística para poder detectarlos.
El hecho de que el análisis de las variables moleculares se realizara mediante
inmunohistoquímica y se valorara de forma semi-cuantitativa, hace que pueda
aparecer otro sesgo, el del factor observador.

112 MATERIAL Y MÉTODOS
7.- Metodología estadística
El objetivo principal del estudio fue establecer si la expresión o presencia de algún
marcador clínico, analítico, anatomopatológico o molecular nos puede hacer predecir
la posibilidad de recidiva en el adenocarcinoma de colon estadio B de la clasificación
de Astler-Coller. En caso de ser así, sería de gran interés analizar la correlación e
importancia de la expresión o presencia de estos factores con el riesgo de recidiva.
Como primer paso, se realizó un estudio descriptivo de las variables clínicas de los
pacientes estudiados:
7.1 ESTADÍSTICA DESCRIPTIVA
Las variables cuantitativas que seguían una distribución normal fueron definidas por la
media, la desviación típica y el intervalo de confianza. En aquellas variables que no
seguían una distribución normal, se utilizó la mediana como medida de centralización,
y el rango como medida de dispersión. Las variables discretas fueron definidas por el
número de casos y el porcentaje.
Para establecer si una variable era de distribución normal, se comprobó si presentaba
una curva equivalente a la campana de Gauss y se confirmó mediante el test de
Kolmogorov-Smirnoff (p>0,05).
7.2 ESTADÍSTICA ANALÍTICA
Se hizo análisis univariante y multivariante con regresión logística tipo Cox,
comparando la aparición de recidiva frente a la expresión de diferentes variables.
Posteriormente se agruparon las variables moleculares de forma dicotómica, y se
realizó regresión logística tipo Cox.

MATERIAL Y MÉTODOS 113
El análisis de supervivencia se realizó mediante el método de Kaplan–Meier, y se
compararon mediante las pruebas del long-rank, de Breslow y de Tarone-Ware.
Se consideró de significación estadísticas los resultados con p< 0.05. Todos los datos se
expresaron con el intervalo de confianza del 95%.
7.3 PROCESAMIENTO DE DATOS
El procesamiento y análisis de datos se realizó con SPSS 15.0 para Windows. Se contó
con la colaboración del Departamento de Estadística del Hospital Universitario Ramón
y Cajal.


RESULTADOS


RESULTADOS 117
1.- Estadística descriptiva
1.1 POBLACIÓN ESTUDIADA
La recogida de datos del estudio se realizó de forma retrospectiva, incluyendo 141
pacientes que fueron intervenidos de cáncer de colon con AP de adenocarcinoma T2-
T4 N0, entre enero de 1975 y enero de 2007 por un equipo de cirujanos dedicados a la
Coloproctología y bajo los mismos criterios quirúrgicos oncológicos y técnica quirúrgica
estandarizada en el Hospital Universitario Ramón y Cajal de Madrid.
Se realizó un seguimiento mínimo de 24 meses en todos los pacientes, excepto en 7
pacientes, de los cuales 5 fallecieron de recidiva antes de los dos años de seguimiento,
y 2 fallecieron durante el seguimiento por otras causas sin presentar recidiva. Por lo
tanto el seguimiento fue de entre 6 y 295 meses, hasta final de 2011, la pérdida del
seguimiento, la aparición de recidiva o la muerte del paciente. Durante el seguimiento
se detectaron 25 recidivas en el grupo estudiado, lo que supone una incidencia
acumulada de recidiva de casi un 18% de los casos.
1.2 VARIABLES DEMOGRÁFICAS
La cohorte está compuesta por 141 pacientes con diagnóstico de adenocarcinoma de
colon (T2-4 N0), con una media de edad al diagnóstico de 67,2 años, y un rango de 26 a
92 años. Sin embargo, la edad fue ligeramente superior en el grupo de recidiva
respecto al grupo con ausencia de enfermedad (69,2 años vs. 66,9).
La distribución de la cohorte por sexos fue de un 55,3 % de varones frente a un 44,7 %
de mujeres, similar en los dos grupos.
118 RESULTADOS
GRÁFICO 1. Distribución por sexo y aparición de recidiva.
1.3 CARACTERÍSTICAS DE LOS PACIENTES QUE PRESENTARON RECIDIVA
Se identificaron 25 pacientes con diagnóstico de CC en estadio T2-T4 N0 que durante el
seguimiento presentaron recidiva tumoral. De los casos de recidiva, 9 fueron mujeres y
16 fueron hombres, con una edad media de70, 2 y desviación estándar de 9,3 para
mujeres y una edad media de 68,44 y una desviación estándar de 12,5 años para
hombres, similar para ambos sexos.
La localización del cáncer más frecuente fue el colon izquierdo/sigma, seguido de la
afectación colon derecho/ciego y por ultimo de colon transverso. En el anexo II se
encuentran recogidas las principales características tumorales en estos pacientes.
1.4 CARACTERÍSTICAS DE LOS PACIENTES CON AUSENCIA DE ENFERMEDAD
Se identificaron durante el estudio 116 pacientes con diagnóstico de CC en estadio T2-
T4 N0 confirmado por anatomía patológica sin evidencia de recidiva durante el
Hombres Mujeres
Ausencia de enfermedad
Recidiva
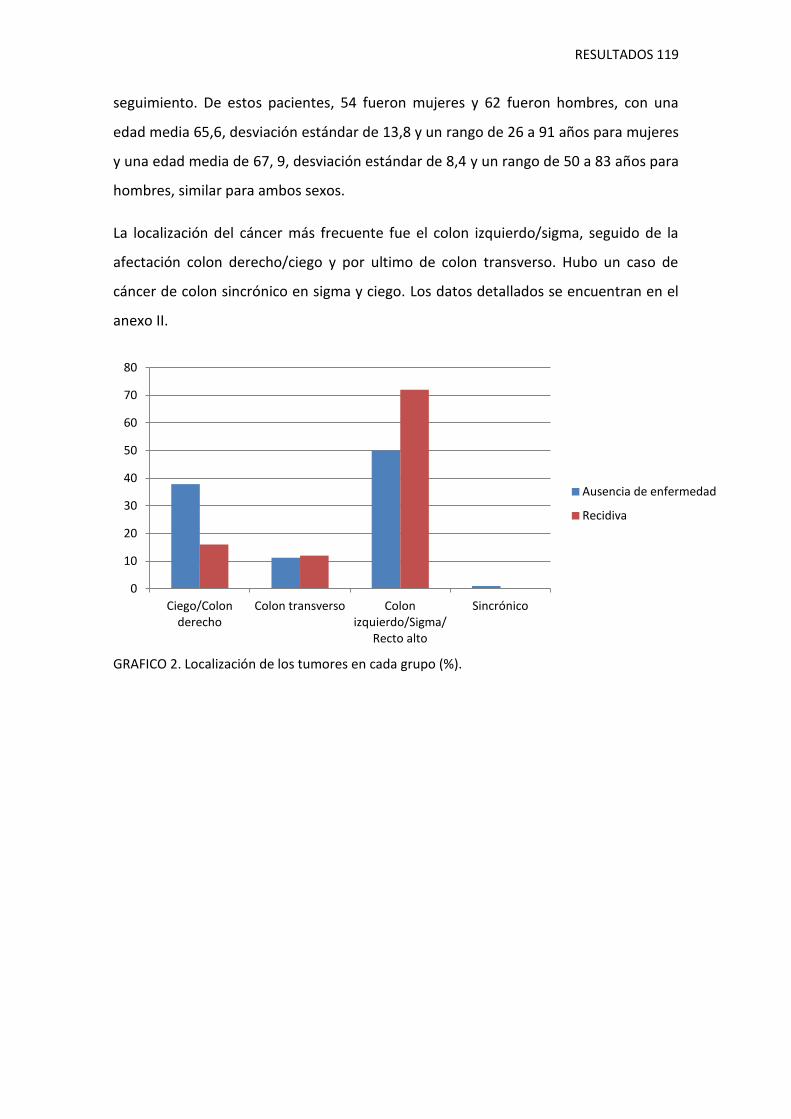
RESULTADOS 119
seguimiento. De estos pacientes, 54 fueron mujeres y 62 fueron hombres, con una
edad media 65,6, desviación estándar de 13,8 y un rango de 26 a 91 años para mujeres
y una edad media de 67, 9, desviación estándar de 8,4 y un rango de 50 a 83 años para
hombres, similar para ambos sexos.
La localización del cáncer más frecuente fue el colon izquierdo/sigma, seguido de la
afectación colon derecho/ciego y por ultimo de colon transverso. Hubo un caso de
cáncer de colon sincrónico en sigma y ciego. Los datos detallados se encuentran en el
anexo II.
GRAFICO 2. Localización de los tumores en cada grupo (%).
0
10
20
30
40
50
60
70
80
Ciego/Colon derecho
Colon transverso Colon izquierdo/Sigma/
Recto alto
Sincrónico
Ausencia de enfermedad
Recidiva

120 RESULTADOS
1.5 VARIABLES DE PRESENTACIÓN CLÍNICA
Tanto en los casos que presentaron recidiva como los que no la presentaron, el estadio
tumoral mas diagnosticado fue T3, siendo más frecuente en segundo lugar el T2 en los
pacientes sin recidiva que en los pacientes con recidiva.
La obstrucción colónica en la presentación clínica se produjo en 17 pacientes del global
de la muestra, lo que supone un 12,3%, siendo más frecuente en el grupo que
posteriormente desarrollaron recidiva con un 17,4% de presentaciones en forma de
obstrucción intestinal frente a un 10,2% en el grupo sin recidiva. La información no
estaba disponible en 4 pacientes.
GRÁFICO 3. Incidencia de obstrucción o perforación en la muestra (%).
La perforación colónica estaba presente en el momento de la intervención quirúrgica
en 7 pacientes, un 5,2% de los pacientes de forma global. No se encontró ningún caso
de perforación en el grupo de recidiva cuya información estaba disponible. La
información no estaba disponible en 7 pacientes.
Si 12%
No 88%
Obstrucción
5%
95%
Perforación
Si No

RESULTADOS 121
1.6 VARIABLES ANALÍTICAS: CEA SÉRICO PREQUIRÚRGICO
Los datos estaban disponibles en 113 pacientes del total de 141, con una distribución
no normal. La mediana de CEA en la muestra fue de 3,48 ng/mL con un rango de 0 a
203.
En el grupo recidiva, la mediana de CEA fue claramente superior en comparación con
el grupo ausencia de enfermedad (6,3 vs 3,2), aunque el rango de valores de CEA fue
muy amplio en ambos casos.
Posteriormente se dividieron los datos en mayor y menor de 5ng/m, punto de corte
habitual en los laboratorios. Los resultados se muestran a continuación.
GRÁFICO 4. Valores de CEA sérico prequirúrgico (%).
29,2 40
34
60,8
24 46
10
36
20
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Ausencia enfermedad Recidiva Global
Desconocido
CEA menor de 5
CEA mayor de 5

122 RESULTADOS
1.7 ESTADIAJE TUMORAL
1.7.1 TNM
Respecto a las características histológicas de los tumores, lo más frecuente fue el
estadio IIa (T3 N0) en ambos grupos.
GRÁFICO 5. Estadiaje TNM según grupos (%).
0
10
20
30
40
50
60
70
80
90
I IIA IIB IIC
Ausencia de enfermedad
Recidiva

RESULTADOS 123
1.7.2 Estadiaje modificado de Astler-Coller
Los pacientes fueron diagnosticados según el estadiaje modificado de Astler-Coller. La
mayor parte de los pacientes fueron diagnosticados en estadio B2 en ambos grupos,
siendo los diagnósticos B1 y B3 minoritarios en ambos grupos.
GRÁFICO 6. Estadiaje según la clasificación de Astler-Coller por grupos (%).
0
10
20
30
40
50
60
70
80
90
B1 B2 B3
Ausencia de enfermedad
Recidiva

124 RESULTADOS
1.7.3. Clasificación de Dukes
Los pacientes fueron diagnosticados según el estadiaje de Dukes, perteneciendo la
mayor parte de los pacientes al estadio B, y siendo muy escasos el porcentaje de los
pacientes del estadio A (menor del 10%, en ambos grupos).
GRÁFICO 7. Estadiaje según la clasificación de Dukes por grupos (%).
0
10
20
30
40
50
60
70
80
90
100
A B
Ausencia de enfermedad
Recidiva

RESULTADOS 125
1.8 VARIABLES ANATOMOPATOLÓGICAS
1.8.1 Diferenciación histológica
Lo más frecuente en ambos grupos fue el tumor bien diferenciado, encontrándose de
forma global en el 78,1% de los casos. En la muestra, el tumor fue moderadamente
diferenciado en 14,6 % de los pacientes y pobremente diferenciado en el 7,3%.
GRÁFICO 8. Diferenciación histológica tumoral por grupos (%).
1.8.2 Histología de células en anillo de sello
La histología en anillo de sello se encontró de forma anecdótica solo en 3 casos, es
decir, 2,6% del grupo con ausencia de enfermedad, y en ningún caso en los pacientes
con recidiva. Los datos estaban ausentes en 2 pacientes con recidiva y en un caso de
los pacientes con ausencia de enfermedad.
1.8.3 Componente mucinoso
La histología mucinosa, definida por más de un 50% de componente de mucina, se
encontró en el 22, 6 % de los pacientes con ausencia de enfermedad y en el 20% de los
0
10
20
30
40
50
60
70
80
90
Bien diferenciado Moderadamente diferenciado
Pobremente diferenciado
Ausencia de enfermedad
Recidiva
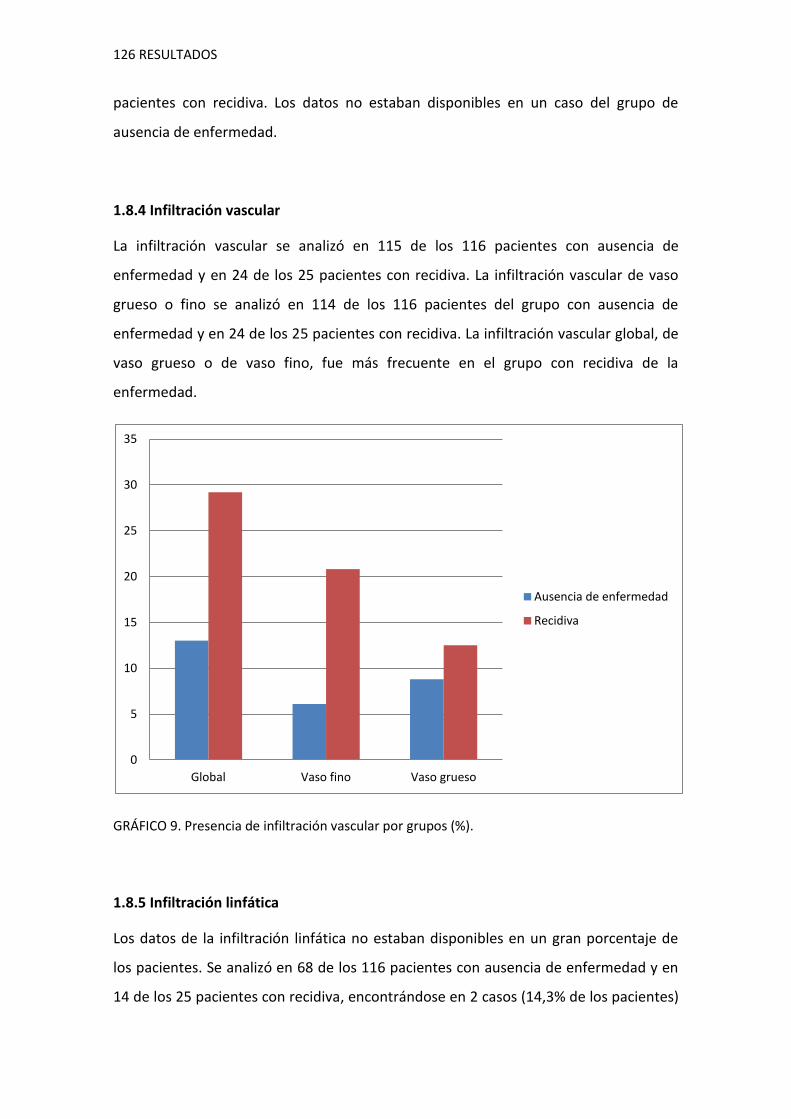
126 RESULTADOS
pacientes con recidiva. Los datos no estaban disponibles en un caso del grupo de
ausencia de enfermedad.
1.8.4 Infiltración vascular
La infiltración vascular se analizó en 115 de los 116 pacientes con ausencia de
enfermedad y en 24 de los 25 pacientes con recidiva. La infiltración vascular de vaso
grueso o fino se analizó en 114 de los 116 pacientes del grupo con ausencia de
enfermedad y en 24 de los 25 pacientes con recidiva. La infiltración vascular global, de
vaso grueso o de vaso fino, fue más frecuente en el grupo con recidiva de la
enfermedad.
GRÁFICO 9. Presencia de infiltración vascular por grupos (%).
1.8.5 Infiltración linfática
Los datos de la infiltración linfática no estaban disponibles en un gran porcentaje de
los pacientes. Se analizó en 68 de los 116 pacientes con ausencia de enfermedad y en
14 de los 25 pacientes con recidiva, encontrándose en 2 casos (14,3% de los pacientes)
0
5
10
15
20
25
30
35
Global Vaso fino Vaso grueso
Ausencia de enfermedad
Recidiva
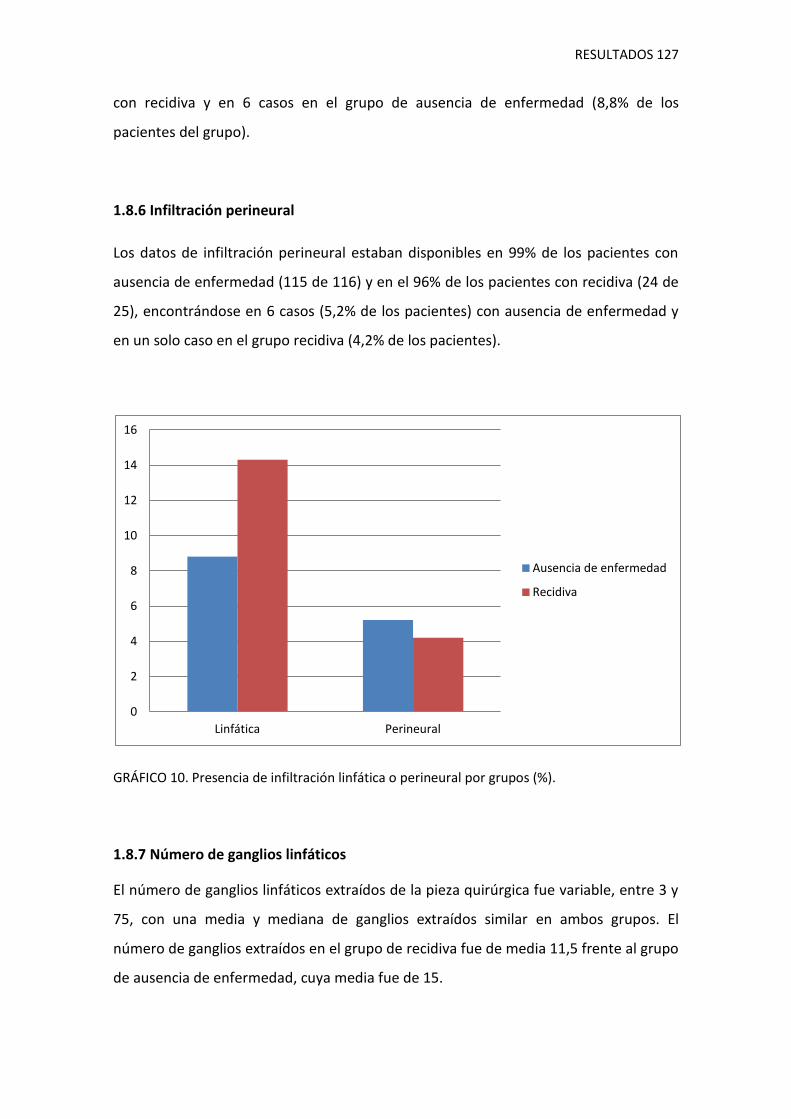
RESULTADOS 127
con recidiva y en 6 casos en el grupo de ausencia de enfermedad (8,8% de los
pacientes del grupo).
1.8.6 Infiltración perineural
Los datos de infiltración perineural estaban disponibles en 99% de los pacientes con
ausencia de enfermedad (115 de 116) y en el 96% de los pacientes con recidiva (24 de
25), encontrándose en 6 casos (5,2% de los pacientes) con ausencia de enfermedad y
en un solo caso en el grupo recidiva (4,2% de los pacientes).
GRÁFICO 10. Presencia de infiltración linfática o perineural por grupos (%).
1.8.7 Número de ganglios linfáticos
El número de ganglios linfáticos extraídos de la pieza quirúrgica fue variable, entre 3 y
75, con una media y mediana de ganglios extraídos similar en ambos grupos. El
número de ganglios extraídos en el grupo de recidiva fue de media 11,5 frente al grupo
de ausencia de enfermedad, cuya media fue de 15.
0
2
4
6
8
10
12
14
16
Linfática Perineural
Ausencia de enfermedad
Recidiva
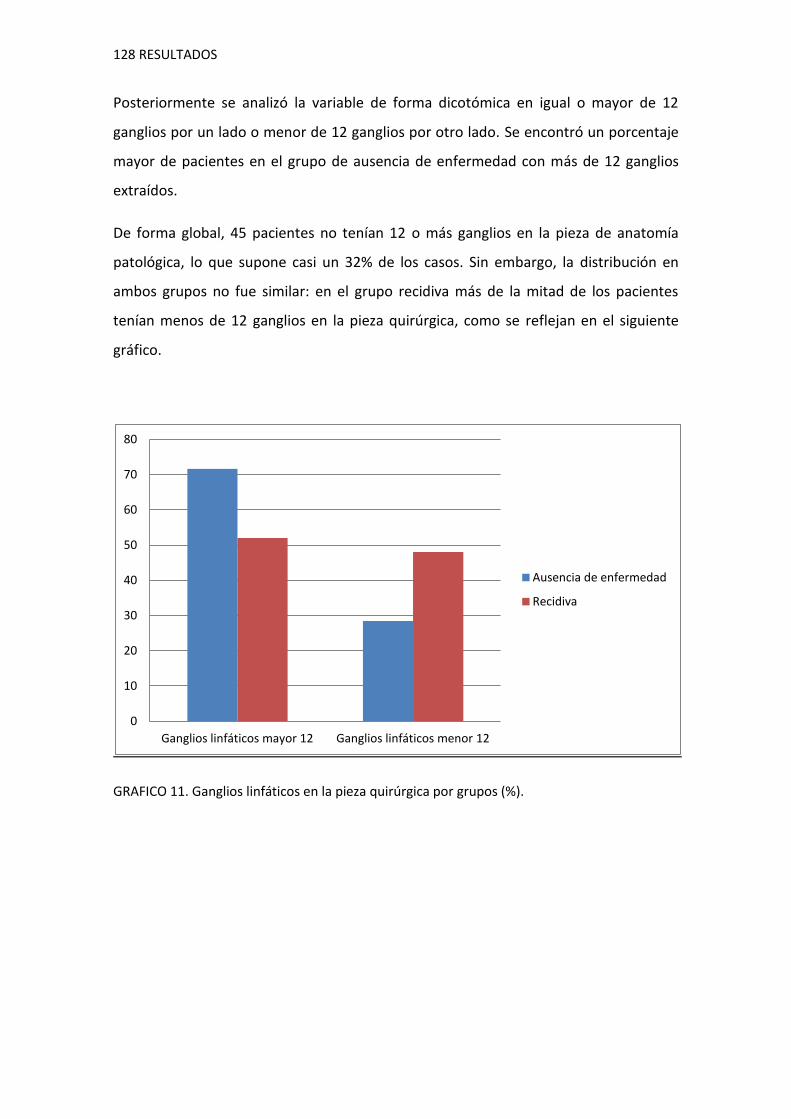
128 RESULTADOS
Posteriormente se analizó la variable de forma dicotómica en igual o mayor de 12
ganglios por un lado o menor de 12 ganglios por otro lado. Se encontró un porcentaje
mayor de pacientes en el grupo de ausencia de enfermedad con más de 12 ganglios
extraídos.
De forma global, 45 pacientes no tenían 12 o más ganglios en la pieza de anatomía
patológica, lo que supone casi un 32% de los casos. Sin embargo, la distribución en
ambos grupos no fue similar: en el grupo recidiva más de la mitad de los pacientes
tenían menos de 12 ganglios en la pieza quirúrgica, como se reflejan en el siguiente
gráfico.
GRAFICO 11. Ganglios linfáticos en la pieza quirúrgica por grupos (%).
0
10
20
30
40
50
60
70
80
Ganglios linfáticos mayor 12 Ganglios linfáticos menor 12
Ausencia de enfermedad
Recidiva

RESULTADOS 129
1.8.8 Características de riesgo agrupadas
Dado que la literatura describe variables anatomopatológicas reconocidas como
pronósticas, se clasificó a los pacientes en alto riesgo o bajo riesgo en función de si
presentaban alguna de las características de mal pronóstico reconocidas actualmente
como son: menos de 12 ganglios, estadio T4, presencia de perforación u obstrucción,
invasión linfovascular o diferenciación G3.
Más del 70% de los pacientes que presentaron recidiva, tenían algún factor de mal
pronóstico, frente a menos del 50% de los pacientes que no presentaron recidiva. Los
resultados se exponen en el gráfico siguiente:
GRÁFICO 12. Factores de riesgo por grupos (%).
0
10
20
30
40
50
60
70
80
Alto riesgo No alto riesgo
Ausencia de enfermedad
Recidiva

130 RESULTADOS
1.9 VARIABLES MOLECULARES
Las siguientes variables fueron excluidas del análisis del resto de las muestras, al
encontrarse en el análisis exploratorio de la primera placa de microarrays que no se
expresaban de manera diferencial en las muestras: Beta-catenina, C-Erb2, ciclina D1,
COX-2, EMA, N-Cadherina, neurotensina, NM-23, NTSR-1, osteonectina, osteopontina,
p16, p21, PTEN citoplasma, PTEN núcleo, SMADY y VEGFR.
En el anexo V se encuentran los resultados cuantitativos de todas las variables
moleculares que se analizaron en la muestra.
A continuación se muestran los gráficos de aquellas variables analizadas en toda la
muestra.
GRÁFICO 13. Expresión BCL-2 por grupos (%).
0
10
20
30
40
50
60
70
80
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
RESULTADOS 131
GRÁFICO 14. Expresión Caveolina por grupos (%).
GRÁFICO 15. Expresión CD133 en estroma por grupos (%).
GRÁFICO 16. Expresión CD133 total por grupos (%).
0
10
20
30
40
50
60
Pobreza de estroma
Moderada presencia de
estroma
Riqueza de estroma
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
70
80
90
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
Negativo Positivo débil
Positivo intenso
Positivo muy intenso
Ausencia de enfermedad
Recidiva

132 RESULTADOS
GRÁFICO 17. Expresión CEA por grupos (%).
GRÁFICO 18. Expresión CK-19 por grupos (%).
GRÁFICO 19. Expresión CK-20 por grupos (%).
0
10
20
30
40
50
60
70
80
90
100
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
70
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
5
10
15
20
25
30
35
40
45
50
Negativo Positivo débil
Positivo intenso
Positivo muy intenso
Ausencia de enfermedad
Recidiva

RESULTADOS 133
GRÁFICO 20. Expresión C-myc por grupos (%).
GRÁFICO 21. Expresión E-Cadherina por grupos (%).
GRÁFICO 21. Expresión EGFR por grupos (%).
0
10
20
30
40
50
60
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
70
80
90
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
70
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
134 RESULTADOS
GRÁFICO 22. Expresión MLH1 por grupos (%).
GRÁFICO23. Expresión MSH2 por grupos (%).
GRÁFICO 24. Expresión MSH6 por grupos (%).
0
10
20
30
40
50
60
70
80
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
70
80
90
100
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
70
Negativo Positivo débil
Positivo intenso
Positivo muy intenso
Ausencia de enfermedad
Recidiva

RESULTADOS 135
GRÁFICO 25. Estado de los microsatélites por grupos (%).
GRÁFICO 26. Expresión Nanog por grupos (%).
GRÁFICO 27. Expresión p27 por grupos (%).
0
10
20
30
40
50
60
70
80
90
Inestable Estable
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
Negativo <25% células
positivas
25-75% células
positivas
>75% células
positivas
Ausencia de enfermedad
Recidiva
0
10
20
30
40
50
60
Negativo <10% células
positivas
10-70% células
positivas
>70% células
positivas
Ausencia de enfermedad
Recidiva
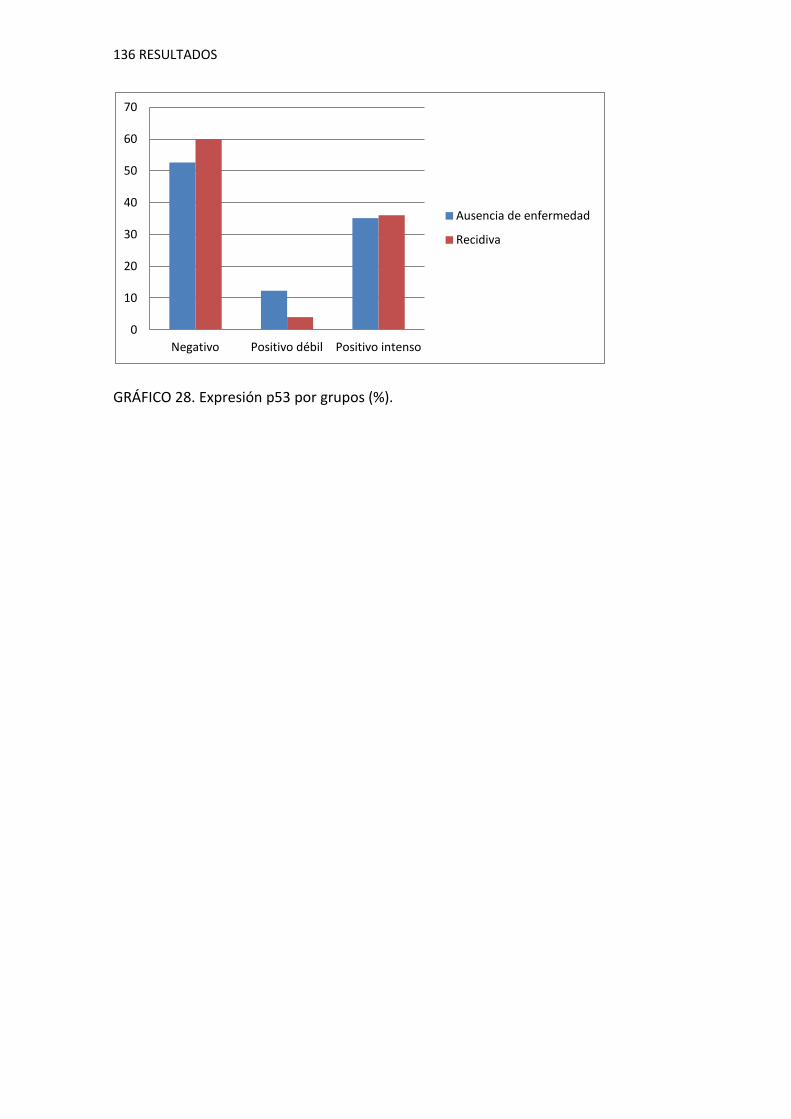
136 RESULTADOS
GRÁFICO 28. Expresión p53 por grupos (%).
0
10
20
30
40
50
60
70
Negativo Positivo débil Positivo intenso
Ausencia de enfermedad
Recidiva

RESULTADOS 137
2. Estadística analítica
2.1 SEGUIMIENTO Y SUPERVIVENCIA
De la cohorte de 141 pacientes, 25 pacientes desarrollaron una recidiva local o a
distancia durante el seguimiento. La mediana de seguimiento de la cohorte fue de 72
meses con un rango de 6 a 258 meses y un rango intercuatílico de 46 a 93 meses. Sólo
8 pacientes tuvieron un seguimiento inferior a 24 meses, 7 de ellos por fallecimiento
antes de los 24 meses y uno por pérdida de seguimiento.
TABLA 6. Pacientes con seguimiento menor a 24 meses.
La mediana de supervivencia libre de enfermedad fue de 19 meses y la mediana de
supervivencia global fue de 45 meses en el grupo que presentó recidiva.
La mediana de supervivencia global fue de 78 meses para el grupo con ausencia de
enfermedad.
En el postoperatorio falleció un paciente, que se excluyó posteriormente del estudio
dada la imposibilidad para incluirle en ninguno de los grupos al no haber podido
comprobarse la presencia o ausencia de enfermedad durante el seguimiento.
Paciente Recidiva Tiempo de
seguimiento (meses) Causa
1 No 6 Éxitus por cáncer de esófago
2 No 21 Perdida de seguimiento
3 No 22 Éxitus por hemorragia subaracnoidea
4 Si 8 Éxitus por recidiva pélvica
5 Si 21 Recidiva pélvica con exenteración y exitus post-quirúrgico.
6 Si 21 Éxitus por metástasis hepáticas irresecables
7 Si 18 Éxitus por metástasis pulmonares
8 Si 18 Éxitus por recidiva pélvica

138 RESULTADOS
GRAFICO 29. Comparativa supervivencia global a 5 años en el CC entre los datos de la literatura
y el trabajo actual. 10
En el gráfico siguiente aparece reflejada la supervivencia global de los pacientes en
función de la aparición o no de recidiva, mostrándose una menor supervivencia en
aquellos pacientes de la cohorte que durante el seguimiento presentaron recidiva.
GRÁFICO 30. Supervivencia global en función de la aparición de recidiva.
0
10
20
30
40
50
60
70
80
90
100
Estadio I Estadio II a Estadio II b Estadio II c
Supervivencia global a 5 años
SEER
Estudio actual
Supervivencia global (meses)
300250200150100500
Supe
rviv
enci
a ac
umul
ada
1,0
0,8
0,6
0,4
0,2
0,0
Grupo Recidiva-
censurado
Grupo Ausencia
Enfermedad-
censurado
Grupo Recidiva
Grupo Ausencia de
Enfermedad

RESULTADOS 139
Grupo de ausencia de
enfermedad Grupo Recidiva
Número de pacientes 116 25
Supervivencia global. Me (rango) 78 (6-295) 44,5 (8-107)
Supervivencia libre de enfermedad. Me (rango) - 19 (7-72)
Supervivencia tras la detección de recidiva. Me (rango)
- 8,5 (0-69)
Supervivencia global a un año (%) 99,1 95,8
Supervivencia global a dos años (%) 97,4 75,0
Supervivencia global a cinco años (%) 87,4 18,5
TABLA 7. Supervivencia por grupos

140 RESULTADOS
2.2 ANÁLISIS POR REGRESIÓN DE COX
2.2.1 Variables clínicas y analíticas
El análisis univariante de las variables clínicas y analíticas para la supervivencia libre de
enfermedad no fue significativo en ninguna de las variables clínicas ni analíticas
analizadas en este estudio. Los datos se reflejan en la siguiente tabla.
TABLA 8. Análisis univariante para la supervivencia libre de enfermedad en función de la
aparición de recidiva.
En cuanto a la supervivencia global en el análisis univariante, resultaron significativas:
La edad, como parece razonable, ya que a mayor edad, mayor mortalidad.
El sexo, como refleja la estadística general, en la que la esperanza de vida es
mayor para las mujeres que para los hombres.
P HR (IC 95%)
Edad mediana/rango(años) 0,559 1,011 (0,974-1,049)
Sexo (femenino vs. masculino) 0,254 0,788 (0,523-1,187)
Existencia de obstrucción 0,429 1,553 (0,521-4,623)
Existencia de perforación 0,467 0,045(0,00-189,552)
Localización
Ciego/colon derecho 0,954 1446 (0-3,6E+110)
Transverso 0,948 3615 (0-8,9E+110)
Colon izquierdo/sigma 0,947 4584 (0-1,1E+111)
CEA 0,634 1,004 (0,989-1,018)
CEA mayor de 5 0,106 2,315 (0,836-6,410)
p: probabilidad; HR: hazard ratio; IC: intervalo de confianza

RESULTADOS 141
P HR (IC 95%)
Edad mediana/rango(años) 0,004 1,041 (1,013-1,070)
Sexo (femenino vs. masculino) 0,027 0,726 (0,546-0,964)
Presencia de obstrucción sí 0,978 0,989 (0,459-2,132)
Presencia de perforación 0,355 0,392 (0,054-2,852)
Localización
Ciego/colon derecho
Transverso
Colon izquierdo/sigma
CEA 0,880 0,999 (0,988-1,011)
CEA mayor de 5ng/mL 0,538 1,223 (0,645-2,319)
SG: Supervivencia global; CEA: antígeno carcinoembrionario; p: probabilidad; HR: hazard ratio; IC:
intervalo de confianza.
TABLA 9. Análisis univariante para la supervivencia global en función de la aparición de
recidiva.
A continuación aparece reflejada la curva de supervivencia global en función de sexo:
GRÁFICO 31.Supervivencia global en función del sexo
Supervivencia global (meses)
300250200150100500
Sup
ervi
venc
ia a
cum
ulad
a
1,0
0,8
0,6
0,4
0,2
0,0
varón-
censurado
mujer-
censurado
varón
mujer
Sexo

142 RESULTADOS
2.2.2 Variables anatomopatológicas
En la tabla siguiente se encuentra reflejado el análisis univariante de las variables
anatomopatológicas en función de la supervivencia libre de enfermedad.
Análisis univariante para SLE
P HR (IC 95%)
Profundidad invasión T2-T3 vs. T4 0,013 3,913 (1,33-11,51)
Bien o moderadamente vs. Pobremente o
indiferenciado
0,336 0,043 (0,00-26,02)
Células en anillo de sello 0,635 0,048 (0,00-13187,3)
Numero de ganglios 0,917 1,003 (0,955-1,052)
Nº ganglios linfáticos < 12 0,067 2,087 (0,951-4,579)
Infiltración vascular 0,009 3,427 (1,363-8,619)
Infiltración vascular gruesa 0,004 4,316 (1,594-11,687)
Infiltración vascular fina 0,531 1,475 (0,438-4,967)
Infiltración linfática 0,427 1,842 (0,408-8,320)
Infiltración perineural 0,788 0,759 (0,102-5,633)
Componente mucinoso 0,784 0,871 (0,325-2,334)
SLE: Supervivencia libre de enfermedad; p: probabilidad; HR: hazard ratio; IC: intervalo de confianza.
TABLA 10. Supervivencia libre de enfermedad de las variables anatomopatológicas.
En el análisis univariante para la supervivencia libre de enfermedad, se encontró que
las siguientes variables fueron significativas o cercanas a la significación:
La posibilidad de recidiva se multiplica por 3,9 si el tumor era un T4 en vez de
T2 o T3.
Aunque no fue significativo, la posibilidad de recidiva se multiplica por 2,1 en
aquellos tumores con menos de 12 ganglios recuperados en la pieza.
En los casos en los que se encontró infiltración vascular, la posibilidad de
recidiva aumentaba en 3,4 veces.

RESULTADOS 143
Cuando la infiltración era de un vaso grueso, esta posibilidad de recidiva era
mayor aun, multiplicándose por 4,3.
En el análisis multivariante de la supervivencia libre de enfermedad con las variables
que previamente habían sido significativas o cercanas a la significación (profundidad
de invasión T2-3 frente a T4, número de ganglios menor de 12 y presencia de
infiltración vascular) solo se encontraron diferencias significativas en la invasión
vascular.
Análisis multivariante para SLE
P HR (IC 95%)
Profundidad invasión T2-T3 vs. T4 0,158 1,882 (0,782-4,527)
Nº ganglios linfáticos < 12 0,071 2,389 (0,927-6,16)
Infiltración vascular 0,012 3,824 (1,346-10,869)
SLE: Supervivencia libre de enfermedad; p: probabilidad; HR: hazard ratio; IC: intervalo de confianza.
TABLA 11. Análisis multivariante en función de la SLE de las variables previamente significativas
en el análisis univariante.

144 RESULTADOS
A continuación se encuentran los gráficos de supervivencia de las variables
significativas en el análisis univariante:
GRÁFICO 32. Supervivencia libre de enfermedad en función de la profundidad de invasión del
tumor.
GRÁFICO 33. Supervivencia libre de enfermedad en función del número de ganglios.
SLE (meses)
300250200150100500
Su
per
vive
nci
a ac
um
ula
da
1,0
0,8
0,6
0,4
0,2
0,0
T4-censurado
T2-T3 -censurado
T4
T2-T3
TNM dicotómica
Supervivencia libre de enfermedad (meses)
300250200150100500
Sup
ervi
venc
ia a
cum
ulad
a
1,0
0,8
0,6
0,4
0,2
0,0
Igual o mayor de 12 -
censurado
Menor de 12-
censurado
Igual o mayor de 12
Menor de 12
Número de ganglios

RESULTADOS 145
GRÁFICO 34. Supervivencia libre de enfermedad en función de la invasión vascular.
Respecto a la variable riesgo (definida como la aparición de alguno de los siguientes
factores: menos de 12 ganglio linfáticos analizados en la pieza quirúrgica, T4, presencia
de perforación u obstrucción, infiltración linfovascular o diferenciación G3), se
encontró significativa la presencia de factores de mal pronóstico, tanto para la SLE
como para la SG, aumentando el riesgo 2,6 veces y 2,5 veces respectivamente para la
SLE y la SG frente a los pacientes que no presentaban ninguno de dichos factores de
riesgo.
.
Supervivencia libre de
enfermedad
Supervivencia Global
P HR (95% IC) p HR (95% IC)
Riesgo 0,032 2,604 (1,050-6,806) 0,004 2,536 (1,345-4,781)
p: probabilidad; HR: hazard ratio; IC: intervalo de confianza.
TABLA 12. Análisis univariante en función de la SLE y la SG de la variable riesgo.
Supervivencia libre de enfermedad (meses)
300250200150100500
Su
perv
iven
cia
acu
mu
lad
a1,0
0,8
0,6
0,4
0,2
0,0
sí-censurado
no-censurado
sí
no
Infiltración vascular
Funciones de supervivencia
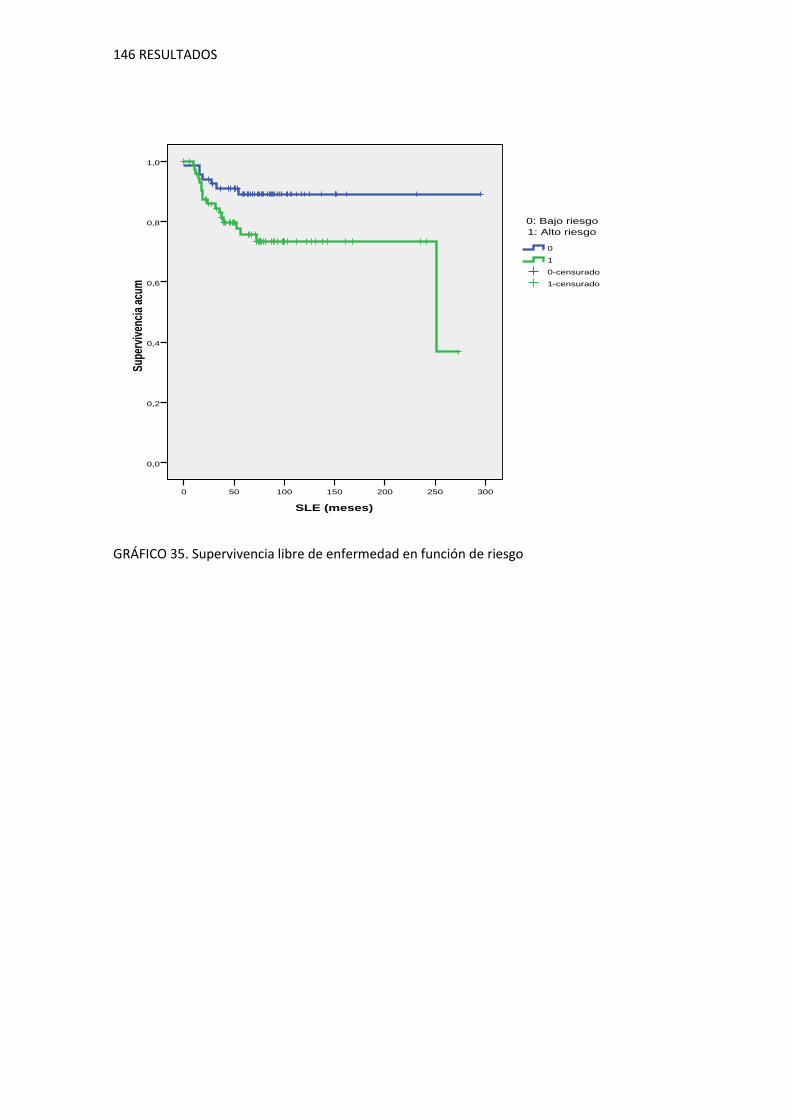
146 RESULTADOS
GRÁFICO 35. Supervivencia libre de enfermedad en función de riesgo
SLE (meses)
300250200150100500
Sup
ervi
venc
ia a
cum
1,0
0,8
0,6
0,4
0,2
0,0
1-censurado
0-censurado
1
0
0: Bajo riesgo
1: Alto riesgo

RESULTADOS 147
Se realizó el análisis univariante y multivariante en función de la supervivencia global,
encontrándose significativos y de peor pronóstico la presencia de T4 frente a T2-3 y la
presencia de invasión vascular, en ambos análisis. Los resultados aparecen reflejados
en la siguiente tabla:
Análisis univariante
para SG
Análisis multivariante para SG
P HR (IC 95%) P HR (IC 95%)
Profundidad invasión
T2-T3 vs. T4 0,030 2,597 (1,096-6,152) 0,005 3,588 (1,464-8,792)
Bien o moderadamente vs.
Pobremente o indiferenciado
0,124 0,212 (0,029-1,533)
Células en anillo de sello 0,558 0,048 (0,00-1228,1)
Numero de ganglios 0,990 1,00 (0,969-1,032)
Ganglios linfáticos >12 0,274 0,739 (0,430-1,270)
Infiltración vascular 0,000 3,308 (1,758-6,188) 0,000 3,727 (1,965-7,069)
Infiltración vascular grueso 0,000 4,287 (2,109-8,716)
Infiltración vascular fino 0,186 1,722 (0,770-3,848)
Infiltración linfática 0,377 1,612 (0,559-4,652)
Infiltración perineural 0,733 1,226 (0,380-3,959)
Componente mucinoso 0,697 0,871 (0,435-1,746)
Riesgo 0,004 2,536 (1,345-4,781)
SG: Supervivencia global; p: probabilidad; HR: hazard ratio; IC: intervalo de confianza del 95%.
TABLA 10. Análisis uni y mulitvariante de la SG en función de la aparición de recidiva en las
variables anatomopatológicas.
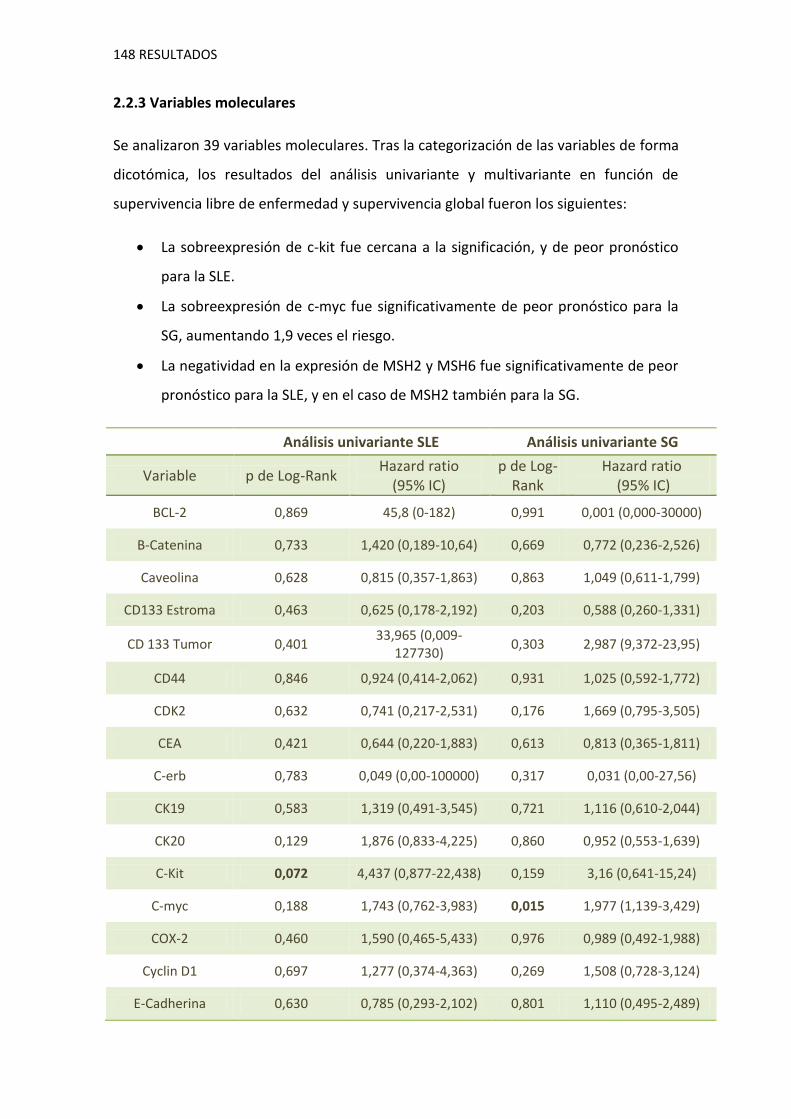
148 RESULTADOS
2.2.3 Variables moleculares
Se analizaron 39 variables moleculares. Tras la categorización de las variables de forma
dicotómica, los resultados del análisis univariante y multivariante en función de
supervivencia libre de enfermedad y supervivencia global fueron los siguientes:
La sobreexpresión de c-kit fue cercana a la significación, y de peor pronóstico
para la SLE.
La sobreexpresión de c-myc fue significativamente de peor pronóstico para la
SG, aumentando 1,9 veces el riesgo.
La negatividad en la expresión de MSH2 y MSH6 fue significativamente de peor
pronóstico para la SLE, y en el caso de MSH2 también para la SG.
Análisis univariante SLE Análisis univariante SG
Variable p de Log-Rank Hazard ratio
(95% IC) p de Log-
Rank Hazard ratio
(95% IC)
BCL-2 0,869 45,8 (0-182) 0,991 0,001 (0,000-30000)
B-Catenina 0,733 1,420 (0,189-10,64) 0,669 0,772 (0,236-2,526)
Caveolina 0,628 0,815 (0,357-1,863) 0,863 1,049 (0,611-1,799)
CD133 Estroma 0,463 0,625 (0,178-2,192) 0,203 0,588 (0,260-1,331)
CD 133 Tumor 0,401 33,965 (0,009-
127730) 0,303 2,987 (9,372-23,95)
CD44 0,846 0,924 (0,414-2,062) 0,931 1,025 (0,592-1,772)
CDK2 0,632 0,741 (0,217-2,531) 0,176 1,669 (0,795-3,505)
CEA 0,421 0,644 (0,220-1,883) 0,613 0,813 (0,365-1,811)
C-erb 0,783 0,049 (0,00-100000) 0,317 0,031 (0,00-27,56)
CK19 0,583 1,319 (0,491-3,545) 0,721 1,116 (0,610-2,044)
CK20 0,129 1,876 (0,833-4,225) 0,860 0,952 (0,553-1,639)
C-Kit 0,072 4,437 (0,877-22,438) 0,159 3,16 (0,641-15,24)
C-myc 0,188 1,743 (0,762-3,983) 0,015 1,977 (1,139-3,429)
COX-2 0,460 1,590 (0,465-5,433) 0,976 0,989 (0,492-1,988)
Cyclin D1 0,697 1,277 (0,374-4,363) 0,269 1,508 (0,728-3,124)
E-Cadherina 0,630 0,785 (0,293-2,102) 0,801 1,110 (0,495-2,489)

RESULTADOS 149
EFGR 0,857 1,095 (0,408-2,942) 0,267 1,407 (0,77-2,57)
EMA 0,688 0,784 (0,239-2,570) 0,742 0,887 (0,434-1,813)
HES 1 No se puede calcular No se puede calcular
H-TERT 0,696 0,663 (0,85-5,187) 0,403 1,514 (0,573-4,004)
KI 67 0,696 0,663 (0,085-5,187)
MLH1 0,419 0,715 (0,318-1,611) 0,686 0,891 (0,509-1,560)
MSH2 0,060 0,389 (0,145-1,042) 0,033 0,437 (0,204-0,937)
MSH6 0,022 0,216 (0,118-0,848) 0,186 0,561 (0,239-1,320)
NANOG 0,472 21,6 (0,005-3837) 0,845 0,868 (0,209-3,597)
N-Cadherina No se puede calcular No se puede calcular
Neurotensina 0,674 0,752 (0,199-2,839) 0,382 1,382 (0,669-2,855)
NM 23 No se puede calcular No se puede calcular
NTSR 1 0,733 0,699 (0,089-5,465) 0,896 1,073 (0,373-3,092)
Osteonectina No se puede calcular No se puede calcular
Osteopontina 0,267 0,420 (0,91.1,944) 0,759 0,892 (0,428-1,858)
P16 0,163 0,417 (0,133-1,426) 0,411 0,743 (0,367-1,508)
P21 0,942 1,047 (0,306-3,577) 0,209 1,606 (0,767-3,365)
P27 0,991 0,995 (0,446-2,222) 0,582 1,163 (0,680-1,989)
P53 0,446 1,371 (0,609-3,088) 0,343 1,304 (0,753-2,256)
PTEN citoplasma No se puede calcular NO SE PUEDE CALCULAR
PTEN núcleo 0,479 0,43 (0,00-264) 0,258 0,430 (0,100-1,857)
RBP 0,447 1,585 (0,484-5,195) 0,767 1,120 (0,529-2,373)
SMADY 0,521 1,545 (0,410-5,826) 0,440 1,358 (0,625-2,953)
VEGRF No se puede calcular No se puede calcular
P: probabilidad; HR: hazard ratio; IC: intervalo de confianza del 95%.
TABLA 13. Análisis supervivencia de las variables moleculares en función de la aparición de
recidiva.

150 RESULTADOS
A continuación se reflejan las curvas de supervivencia de las variables moleculares que
resultaron significativas en el análisis univariante para la SLE.
GRÁFICO 36. Supervivencia libre de enfermedad en función de la expresión de MSH2.
GRÁFICO 37. Supervivencia libre de enfermedad en función de la expresión de MSH2.
Supervivencia libre de enfermedad (meses)
300250200150100500
Su
perv
iven
cia
acu
mu
lad
a
1,0
0,8
0,6
0,4
0,2
0,0
Positivo-censurado
Negativo-censurado
Positivo
Negativo
Expresión MSH2
Funciones de supervivencia
Supervivencia libre de enfermedad (meses)
300250200150100500
Su
pe
rviv
en
cia
ac
um
ula
da
1,0
0,8
0,6
0,4
0,2
0,0
1-censurado
0-censurado
Positivo
Negativo
Expresión MSH6
Funciones de supervivencia
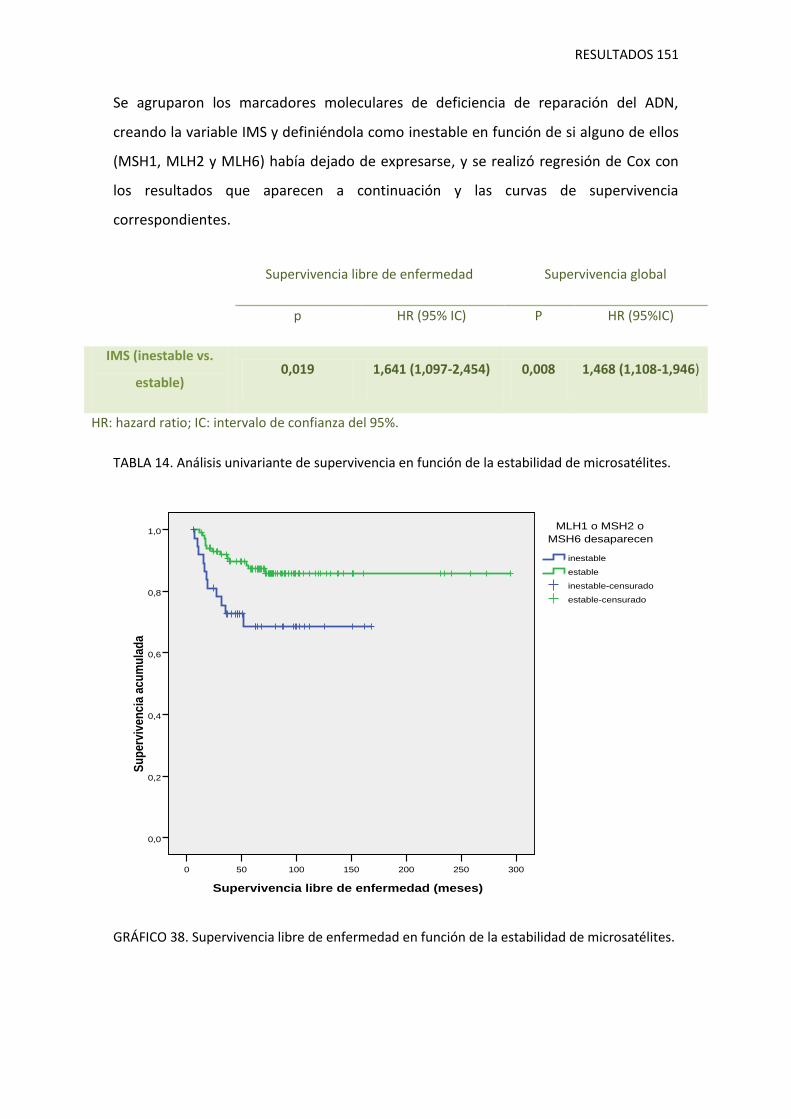
RESULTADOS 151
Se agruparon los marcadores moleculares de deficiencia de reparación del ADN,
creando la variable IMS y definiéndola como inestable en función de si alguno de ellos
(MSH1, MLH2 y MLH6) había dejado de expresarse, y se realizó regresión de Cox con
los resultados que aparecen a continuación y las curvas de supervivencia
correspondientes.
TABLA 14. Análisis univariante de supervivencia en función de la estabilidad de microsatélites.
GRÁFICO 38. Supervivencia libre de enfermedad en función de la estabilidad de microsatélites.
Supervivencia libre de enfermedad (meses)
300250200150100500
Su
pe
rviv
en
cia
ac
um
ula
da
1,0
0,8
0,6
0,4
0,2
0,0
estable-censurado
inestable-censurado
estable
inestable
MLH1 o MSH2 o
MSH6 desaparecen
Funciones de supervivencia
Supervivencia libre de enfermedad Supervivencia global
p HR (95% IC) P HR (95%IC)
IMS (inestable vs.
estable) 0,019 1,641 (1,097-2,454) 0,008 1,468 (1,108-1,946)
HR: hazard ratio; IC: intervalo de confianza del 95%.

152 RESULTADOS
GRÁFICO 39. Supervivencia global en función de la estabilidad de microsatélites.
Supervivencia global (meses)
300250200150100500
Su
per
vive
nci
a ac
um
ula
da
1,0
0,8
0,6
0,4
0,2
0,0
estable-censurado
inestable-censurado
estable
inestable
MLH1 o MSH2 o
MSH6 desaparecen
Funciones de supervivencia

DISCUSIÓN


DISCUSIÓN 155
1.- La situación actual de cáncer de colon en estadio II
Aproximadamente un tercio de los pacientes con CC no extendido a distancia en el
momento del diagnóstico, tendrán afectación de los ganglios linfáticos (estadio III) y
una cuarta parte de los pacientes invasión de la pared intestinal sin afectación de los
ganglios linfáticos (estadios I y II).13
El CC en estadio II representa una enfermedad de amplio espectro y pronóstico,
modulado por características del tumor, del paciente y de la presentación clínica 60. La
nueva clasificación de la AJCC de 2010 divide el estadio II en función de la profundidad
de la invasión del tumor, y nos muestra como es uno de los factores pronósticos más
importantes en estos pacientes.9
Según el estudio de la AJCC basado en registro norteamericano SEER, con más de
120.000 pacientes evaluados entre los años 1991 y 2000, la supervivencia a los cinco
años de los pacientes con estadiaje IIa (T3 N0) se sitúa en 84,7%, mientras que los
pacientes con estadiaje IIb-c (T4 N0) se encuentra en 72,2%. Las cifras de este último
grupo son inferiores a la supervivencia a los cinco años de los pacientes estadio IIIa
(T1-2 N+) de cáncer de colon, que reciben adyuvancia tras el tratamiento quirúrgico, y
cuya supervivencia a los cinco años es del 83,4%. 4,10
Por lo tanto, dentro del grupo de pacientes con estadio II, existe un subgrupo de
pacientes que deberían ser seleccionados para quimioterapia adyuvante tras el
tratamiento quirúrgico, por lo que es necesario determinar qué factores pronósticos
nos permiten detectar aquellos pacientes de alto riesgo de recidiva.
El objetivo de la quimioterapia adyuvante es mejorar la supervivencia global y libre de
enfermedad, actuando a nivel de la enfermedad micrometastásica. Actualmente se ha
demostrado el beneficio que produce la quimioterapia adyuvante en el estadio III del
CC, disminuyendo la posibilidad de recurrencia local o a distancia. Sin embargo, en el
estadio II este beneficio no ha podido ser establecido. La probabilidad de curación sólo
con cirugía en pacientes con CC estadio II se sitúa en el 82,5% a los cinco años según la
5ª edición de la AJCC. Sin embargo, algunos pacientes con factores de riesgo tienen
una probabilidad mayor de recidiva y su pronóstico puede situarse más cerca de los

156 DISCUSIÓN
datos de supervivencia del estadio III. En estos casos, estaría indicado el tratamiento
adyuvante. Diversos estudios actuales parecen mostrar que ciertas características
tumorales en pacientes con CC en estadio II, pueden conferir un mayor riesgo de
recidiva en algunos pacientes.13
Se han publicado múltiples trabajos que han estudiado factores clínicos, analíticos y
anatomopatológicos que puedan ser pronósticos de supervivencia en el CC estadio II,
al ser datos disponibles de manera habitual en el estudio de dichos pacientes con CC.
Sin embargo, son menos los trabajos que han estudiado factores moleculares
pronósticos de supervivencia, aunque en los últimos años, se están investigando
diversos marcadores moleculares para determinar que pacientes en estadio II se
pueden beneficiar del tratamiento adyuvante.
Actualmente la NCCN identifica como factores pronósticos de riesgo de recidiva los
siguientes factores anatomopatológicos en estadio II, y por tanto, susceptibles de
plantear adyuvancia post-quirúrgica de forma selectiva:
Pobre diferenciación celular (grado 3-4), exclusivamente en tumores con IMS
alta.
Invasión linfática o vascular.
Menos de 12 ganglios linfáticos examinados.
Invasión perineural.
Perforación localizada.
Márgenes positivos, cercanos o indeterminados en la pieza quirúrgica.
El estudio de la supervivencia libre de enfermedad en el CC ha demostrado ser un valor
objetivo para la estimación de la supervivencia global como se muestra en el
metaanálisis de Sargent et al. 61 Es por este motivo, que en este trabajo, aunque
también hablaremos de supervivencia global, el análisis se dirigirá primariamente a la
supervivencia libre de enfermedad de los enfermos de CC T2-T4 N0 tras cirugía
curativa.
En el metaanálisis de Sargent et al. 62 sobre la supervivencia de los pacientes con CC en
estadio II se extraen tres conclusiones fundamentales: la primera es que la terapia

DISCUSIÓN 157
adyuvante basada en 5-FU es capaz de erradicar las células cancerígenas de colon; la
segunda es que las recurrencias tardías pueden ocurrir, pero tras ocho años se puede
decir que el paciente está curado; y la tercera es que la mayor parte de las recurrencias
ocurren en los dos primeros años tras la cirugía, por lo que es prioritario el
seguimiento estrecho durante este periodo. En este mismo trabajo se ve que la mejora
global tras terapia adyuvante se sitúa en un 5% en los pacientes en estadio II.
Por estos motivos, el seguimiento en esta cohorte se realizó por un periodo mínimo de
dos años, o hasta el fallecimiento del paciente si era anterior a este periodo.
La supervivencia tras la aparición de recidiva fue 8,5 meses de mediana, aunque con
un rango amplio de hasta 69 meses. Estos datos nos indican por un lado que la
aparición de recidiva, afecta claramente a la supervivencia global de los pacientes,
aunque en alguno de estos pacientes, se llegó a supervivencias altas gracias al
tratamiento de dichas recidivas en los casos que fue posible.

158 DISCUSIÓN
2.- Factores clínicos: obstruccion/perforación
En diversos estudios se ha demostrado que la presencia de obstrucción o perforación
en el momento del diagnóstico tiene un peor pronóstico de supervivencia y mayor
mortalidad a largo plazo 63. De hecho, en las últimas recomendaciones terapéuticas de
la NCCN, la presencia de perforación en la pieza quirúrgica se considera como un factor
de riesgo adverso para la aparición de recidiva 12.
En el estudio de factores pronósticos en CC estadio B de Dukes de Saha et al.64 con
180 pacientes, la presencia de perforación intestinal fue un factor pronóstico adverso
con un HR de 2,9 y dentro de la significación estadística.
Sin embargo, en otros trabajos similares como el de Gertler et al. 65 o el de Quah et
al.60 en los que se analizó entre otros factores la presencia de perforación o de
obstrucción intestinal no se encontraron diferencias significativas respecto a estos dos
factores.
En nuestro estudio, la presencia de perforación u obstrucción colónica no demostró
ser factor de mal pronóstico significativo, probablemente debido al pequeño tamaño
de la cohorte en el caso de obstrucción intestinal y a que no se produjo ningún caso de
perforación en el grupo de recidiva.
La presencia de obstrucción o perforación intestinal debe considerarse como un factor
de mal pronóstico para la aparición de recidiva en los pacientes con CC en estadio II. El
hecho de que algunos trabajos no sean capaces de demostrarlo probablemente se
deba a que la aparición de obstrucción o perforación colónica no es muy habitual en
este estadio, y por tanto, es necesario que la cohorte de pacientes sea grande para
conseguir llegar a la significación estadística.

DISCUSIÓN 159
3.- Factores analíticos: CEA.
La medición del CEA preoperatorio se ha convertido en la actualidad en un
procedimiento de rutina dentro del estudio de extensión del CC. La ausencia de
normalización de sus valores tras la cirugía con intención curativa, nos tiene que hacer
pensar en la presencia de enfermedad tumoral residual.
Diversos estudios, incluyendo el trabajo de Chen et al. 66, han encontrado un peor
pronóstico asociado a la elevación preoperatoria del CEA por encima de sus niveles
normales (es decir, por encima de 5ng/mL) en pacientes con CC sin metástasis
ganglionares ni a distancia 67.
En el trabajo de Wolmark et al. 68 se demostró que la elevación preoperatoria del CEA
estaba asociada con un aumento relativo de fallos en el tratamiento. Los pacientes en
estadio Dukes B con niveles preoperatorios de CEA superiores a 10 ng/mL presentaban
un aumento del riesgo de recurrencia 3,24 veces mayor a pacientes similares con
niveles de CEA inferiores a 2,4ng/mL. En el trabajo de Sener et al 69 con 533 pacientes
en estadio de Dukes B, encontraron que la elevación marcada del CEA preoperatorio
(por encima de 15 ng/mL) tenía un pronóstico de supervivencia significativamente
peor.
En nuestro trabajo, la elevación del CEA preoperatorio por encima de 5ng/ml no
resultó significativo en relación con la supervivencia libre de enfermedad ni con la
supervivencia global. La ausencia de significación probablemente se deba al pequeño
tamaño de la muestra, pero el CEA sérico preoperatorio debe considerarse como
factor pronóstico adverso. Tal vez el nivel de corte de los niveles de CEA a partir de los
cuales podamos considerarlo como factor adverso deba ser superior a 5 ng/mL,
aunque el punto no ha quedado claramente establecido, por lo que se sigue utilizando
el valor de laboratorio de referencia.

160 DISCUSIÓN
4.- Factores anatomopatológicos
4.1 PROFUNDIDAD DE INVASIÓN (ESTADIO T)
La profundidad de invasión del tumor es un parámetro fundamental para la
determinación del estadio del paciente con CC tras la resección quirúrgica, y confiere
pronóstico diferente según su menor o su mayor graduación, desde T1 en el que solo
afecta a la muscular de la mucosa hasta T4 que sobrepasa los límites de la afectación
de la pared del colon.
De hecho, uno de los cambios fundamentales de la nueva edición del estadiaje TNM de
la AJCC tras el análisis de los datos de supervivencia del estudio SEER con más de
28.000 pacientes con adenocarcinoma de colon, se basa en la creación de subgrupos
dentro del estadio II en función de la profundidad de invasión (T), por encontrarse
diferencias en la supervivencia.10
En el estudio de Uen et al. 70 sobre factores pronósticos de CC estadio II, se encontró
que la mayor profundidad de invasión (T4 vs. T3) era pronóstico de recidiva de CC con
un HR de 4 (p=0.013) 70.
En el estudio sobre factores pronósticos en cáncer de colon de Mroczkowski71
encontraron que la profundidad de invasión era el factor pronóstico más importante
para la supervivencia global.
En nuestro estudio se comparó la SLE en función de la profundidad de invasión de T2 a
T4, no encontrando diferencias significativas entre cada grupo en el análisis
univariante. Sin embargo, al agrupar la variable comparando T2-T3 frente a T4, se
encontraron diferencias significativas en la SLE en el análisis univariante, aunque no el
multivariante. En el análisis de la supervivencia global, se encontró un peor pronóstico
en los pacientes con T4, con un aumento de riesgo de 3,9 veces respecto a los
pacientes con estadiaje T2 o T3 (p =0,013).
Estos resultados son concordantes con publicaciones previas, en las que la
estadificación T4 se asocia a un peor pronóstico. De hecho, la 7ª edición de la AJCC
subdivide el estadio T4 en función de si el tumor invade el peritoneo visceral u otros

DISCUSIÓN 161
órganos, teniendo peor pronóstico este ultimo. Actualmente, las guías oncológicas
para el tratamiento del CC reconocen el estadio T4 como factor de mal pronóstico para
plantear quimioterapia adyuvante tras cirugía curativa en el estadio II9,12,13.

162 DISCUSIÓN
4.2 GRADO HISTOLÓGICO
El grado histológico refleja el grado de diferenciación tumoral, y que se ha mostrado
ser factor pronóstico independiente del estadio tumoral. Sin embargo, el grado
histológico es un parámetro subjetivo, con una variabilidad interobservador y sin un
sistema uniforme aceptado de forma general. La interpretación del grado se basa en la
valoración del tumor por completo o de la peor área, por la valoración de la cantidad
de formación glandular únicamente o la valoración combinada de las glándulas y de
otras características citológicas o estructurales6,8,10. Tanto la AJCC como el CAP
recomiendan la adopción de un sistema dicotómico (bajo y alto grado histológico) con
un punto de corte bien definido para reducir las diferencias interobservador y
preservar o mejorar el poder pronóstico del grado histológico10.
El grado histológico se ha descrito en varios trabajos como factor pronóstico adverso
para la aparición de recidiva. El análisis multivariante de factores pronósticos en CC se
ha demostrado ser factor predictivo de supervivencia, de forma que los tumores de
alto grado (pobremente diferenciados o indiferenciados), están asociados con una
mayor penetración de la pared intestinal, y la presencia de metástasis ganglionares y a
distancia.72
La NCCN recomienda la administración de quimioterapia adyuvante en el CC en estadio
II en aquellos pacientes con grado histológico G3-G4, pero sólo en pacientes con IMS
alta12.
En nuestro trabajo, el grado histológico, expresado como variable dicotómica, no
resultó significativo en el análisis univariante de SLE ni de SG. Es posible que dado que
el análisis inicial se realizó en cuatro grupos (bien, moderado, pobremente
diferenciado e indiferenciado), y que este valor es subjetivo, aunque posteriormente
se agrupe de forma dicotómica, no refleje la realidad del grado histológico.
Por otro lado, el grado histológico ha demostrado en la literatura ser factor pronóstico
adverso principalmente en el análisis global de CC, y sólo en algún caso en el caso del
subgrupo del CC en estadio II, lo que hace que encontrar diferencias significativas sea

DISCUSIÓN 163
más difícil, y probablemente por este motivo requiera de una muestra mucho mayor,
aunque se trate probablemente de un factor pronóstico adverso en el CC.

164 DISCUSIÓN
4.3 INVASIÓN VASCULAR
No todos los estudios reconocen la invasión vascular como factor pronóstico en el CC,
aunque hay diversos estudios que lo han encontrado significativo.
En el estudio de Uen et al. 70 sobre factores pronósticos de CC estadio II, se encontró
que la presencia de invasión vascular era pronóstico de recidiva de CC con un HR de
3.5 (p=0.032).
En el estudio de Betge et al. 14 analizaron la presencia o ausencia de invasión vascular
como factor pronóstico en el CC. Dicho estudio mostró que la invasión vascular,
especialmente en el caso de ser extramural fue un factor pronóstico adverso, y se
relacionaba también con la presencia de mayor estadiaje T y de afectación ganglionar
linfática. También analizaron selectivamente dicha afectación en pacientes del estadio
II, y concluyeron que la invasión venosa, pero no la linfática, constituía un factor
pronóstico independiente en este subgrupo, con un aumento de riesgo cinco veces
mayor que en los que no tenían afectación venosa.
Sin embargo, en el trabajo sobre factores pronósticos en CC de Mroczkowski et al. 71
no encontraron que la invasión vascular fuera factor pronóstico de supervivencia,
aunque su resultado fue cercano a la significación.
En nuestro trabajo la invasión vascular, especialmente si se trataba de un vaso grueso,
se ha mostrado de nuevo como factor de mal pronóstico tanto en la supervivencia
libre de enfermedad, como en la supervivencia global, con significación estadística en
ambos casos y un aumento de riesgo de más de tres veces.
Estos datos son concordantes con los hallazgos de la literatura, y deberían estar
incluidos en el informe anatomopatológico de las piezas quirúrgicas de CC de forma
rutinaria.

DISCUSIÓN 165
4.4 INVASIÓN LINFÁTICA
La invasión linfática constituye el paso previo a la extensión de las células tumorales a
los ganglios linfáticos regionales. El CAP incluye dentro de sus recomendaciones
anatomopatológicas de las piezas quirúrgicas de CC el análisis de la presencia de
invasión linfática en cada bloque y que debe reflejarse en el informe patológico 10.
En el trabajo de Betge et al. 14 encontraron que los pacientes con presencia de invasión
linfática tenían más riesgo de desarrollar una progresión de la enfermedad, aunque
dicha diferencia alcanzaba la significación estadística.
En el trabajo alemán de Gertler et al. 65 encontraron que la presencia de invasión
linfática era factor pronóstico adverso para la supervivencia libre de enfermedad en el
análisis multivariante.
Hay diversos trabajos que hablan de la invasión linfovascular de forma conjunta,
debido a que a veces es difícil diferenciar a nivel capilar si se trata de un capilar venoso
o linfático, lo que dificulta analizar la importancia pronóstica de cada factor de forma
independiente.
En nuestro trabajo, el análisis de la invasión linfática solo estuvo disponible en el 70%
de los pacientes, y aunque fue mucho más frecuente en aquellos pacientes que
presentaban recidiva, dicha diferencia no tuvo significación estadística.
Aunque hay diversos trabajos que hablan del significado pronóstico de la invasión
linfática en el análisis anatomopatológico de las piezas de CC, sin embargo, el pequeño
tamaño muestral de este estudio y la ausencia de datos de un número importante de
muestras hacen que este factor no sea valorable en este trabajo. Sin embargo, parece
que pueda tener un papel como factor pronóstico adverso en el CC en estadio II,
aunque probablemente sea de mayor importancia la invasión vascular que la linfática
con los datos actualmente disponibles en la literatura.

166 DISCUSIÓN
4.5 INVASION PERINEURAL
La invasión perineural es poco frecuente, y se caracteriza por la invasión por parte de
las células tumorales de estructuras nerviosas, y la extensión del tumor a través de las
vainas tendinosas.
Aunque hay diversos estudios que identifican la invasión perineural como factor
pronóstico, todavía no hay evidencia científica suficiente para considerarlo un factor
de evidencia IIA.10
En la última edición de la NCCN (2012), la invasión perineural se añade a la valoración
de factores pronósticos adversos en el CC en estadio II, en relación con la necesidad de
tratamiento adyuvante en estos pacientes12.
Probablemente la invasión perineural esté actualmente infravalorada debido a que son
muchos los análisis de las piezas de CC que no reflejan la presencia o ausencia de este
factor.
En el estudio de Liebig et al.15 en el que se analizó específicamente la invasión
perineural en el CC en diferentes estadios, se encontró que aunque inicialmente sólo
se identificó invasión en un 0,5% de los pacientes, tras el estudio selectivo, un 22% de
los pacientes tuvieron invasión perineural. Analizados los pacientes en estadio I y II
(con N negativo), encontraron que la SLE para el grupo sin invasión perineural fue tres
veces superior a los pacientes con invasión, siendo dicha diferencia significativa
estadísticamente. Incluso proponen que la supervivencia de pacientes N0 con invasión
perineural tienen peor SLE que los pacientes con ganglios positivos.
En el trabajo de Quah et al.60 se encontró que un 14% de los pacientes con CC en
estadio II presentaron afectación perineural, siendo este factor pronóstico adverso en
el análisis univariante con el doble de riesgo de recidiva.
En nuestro trabajo, la presencia de invasión perineural fue del 5% de forma global,
únicamente en 6 pacientes, y en porcentaje muy similar en ambos grupos, por lo que
el análisis estadístico no mostró diferencias significativas.

DISCUSIÓN 167
Sin embargo, consideramos que se debe continuar analizando este factor dado que su
baja aparición y la ausencia de la búsqueda sistemática en las piezas quirúrgicas de CC
hace que sea más difícil demostrar si constituye un factor pronóstico en los pacientes
con CC en estadios iniciales.

168 DISCUSIÓN
4.6 NÚMERO DE GANGLIOS
Los factores que influyen en una adecuada extirpación y evaluación de los ganglios
linfáticos regionales y un estadiaje más preciso en los pacientes con CC no se
entienden completamente y dependen de muchos factores, entre los que está la
calidad de la resección quirúrgica, la calidad de la evaluación patológica, factores
tumorales y factores intrínsecos del paciente. La calidad de la resección quirúrgica es
importante porque el cirujano debe extirpar un segmento de colon con márgenes
adecuados y con el mesenterio correspondiente hasta la raíz de los vasos.
Posteriormente, el patólogo debe analizar el mesenterio para identificar los ganglios
linfáticos. Cuando se encuentran menos de 12 ganglios, el CAP recomienda la
utilización de técnicas adicionales para la identificación de ganglios. De hecho, la
presencia de 12 o menos ganglios analizados está incluido dentro de los criterios de la
NCCN para plantear adyuvancia en pacientes con cáncer de colon y ganglios negativos
(N0).3,12,73
El número total de ganglios extirpados y analizados en la pieza quirúrgica está
relacionado con el pronóstico tanto en el CC estadio II como en estadio III. En el
metaanálisis de Chang et al. 3 encontraron que un aumento del número de ganglios
examinados en la pieza quirúrgica se asociaba con un mejor pronóstico en el CC en
estadio II.
En el trabajo de Vather et al.73 en el que se analizaba la relación entre la supervivencia
y el número de ganglios examinados en CC estadio II y III, encontraron que la
mortalidad a los 5 años disminuía cuantos más ganglios se examinaban. Esto era más
marcado en los pacientes con menos de 13 ganglios, a partir de lo cual, la
supervivencia era similar en los grupos pese a aumentar el número de ganglios
examinados.
En nuestro trabajo, el número de ganglios extirpados no se demostró factor pronóstico
de recidiva. Sin embargo, al realizar el análisis dividiendo los pacientes en menos o más
de 12 ganglios, se encontraron diferencias en el pronóstico de cada grupo en relación
con la aparición de recidiva; dichas diferencias no llegaron a la significación estadística
(p=0,067) pero mostraban un aumento del riesgo de recidiva del doble que aquellos

DISCUSIÓN 169
pacientes que tenían menos de 12 ganglios linfáticos en la pieza quirúrgica. Esta
tendencia al aumento de riesgo en los pacientes con menos de 12 ganglios analizados,
concuerda con los datos publicados en la literatura. El trabajo actual probablemente
no tenga la suficiente potencia para detectar dicha diferencia debido al tamaño
muestral.

170 DISCUSIÓN
5.- Marcadores moleculares
Los trabajos de investigación en CC más actuales, intentan detectar características y
marcadores moleculares en los tumores, que permitan determinar la agresividad y el
pronóstico de cada paciente de forma independiente.
Actualmente se han desarrollado baterías de pruebas moleculares en el CC en estadio
II como es el caso de Oncotype DX para cáncer de colon o el ColoPrint, aunque los
resultados de dichos análisis no están suficientemente comprobados en estudios
prospectivos como para recomendar su uso de forma habitual en la práctica clínica.
En este trabajo hemos analizado múltiples factores moleculares de forma exploratoria;
los hallazgos de la literatura en torno a dichos factores han sido expuestos en la
introducción, por lo la discusión se centrará en aquellos que han mostrado
significación estadística o próxima a ella.

DISCUSIÓN 171
5.1 C-KIT
Muy pocos trabajos han analizado la importancia de c-Kit como factor pronóstico en el
cáncer de colon. C- kit se sobreexpresa en otros tumores como las leucemias
mieloides, los melanomas y los tumores de células germinales. 74
Los pacientes con CC en los que se expresa KIT/SCF, son candidatos al uso de Imatinib
mesilato, un inhibidor selectivo de tirosin quinasa (incluyendo KIT) en combinación con
la terapia estándar.
En el único trabajo publicado sobre la importancia de c-kit en el CC, Bellone G et al.34
analizan c-kit como factor pronóstico en el CC, encontrando que la expresión de c-kit
se asocia a un peor pronóstico y se expresa en mayor medida en estadios avanzados.
Los autores no ven claro el significado biológico de su expresión en el proceso de
carcinogénesis de colon, aunque se sospecha que c-kit puede tener un papel de
regulación autocrina en el desarrollo del CC.
En nuestro trabajo la sobreexpresión de c-kit resultó cercano a la significación,y de
peor pronóstico para la SLE pero no para la SG, aunque estos resultados deben ser
valorados con cautela dado que solo se analizó en un 16,4 % de los pacientes (23 de los
141).

172 DISCUSIÓN
5.2 C-MYC
La sobreexpresión de c-myc como factor pronóstico adverso puede tener relación con
el mecanismo biológico de este oncogen, que promueve el inicio del ciclo celular
mediante varios mecanismos superpuestos, además de tener un efecto sobre genes
que aumentan el crecimiento celular.
En diversos trabajos se ha estudiado la expresión de c-myc como factor pronóstico en
el CC con resultados dispares:
En el trabajo Intergroup study EST2284, el 32% de los pacientes presentaban
positividad para la expresión de c-myc y se encontraron diferencias significativas en la
supervivencia libre de enfermedad cuando se realizaba adyuvancia con levamisole y 5-
FU, lo que puede sugerir un papel la determinación de c-myc para la valoración de
quimioterapia adyuvante.31
Dos trabajos analizaron la expresión inmunohistoquímica de c-myc en los tumores de
colon y recto. En el estudio de Miller et al (1992)75 con 118 pacientes y el Bhatavdekar
et al (1997)76 con 48 pacientes encontraron un 71 y 65% respectivamente de pacientes
con tinción positiva para c-myc. No encontraron asociación al estadio tumoral ni al
grado histológico en ninguno de los estudios. Se encontró una mejor supervivencia
global en los pacientes tinción negativa para c-myc al compararlo con los de tinción
positiva en el estudio de Bhatavdekar76. Por el contrario, c-myc no fue factor
pronóstico de importancia en el estudio de Miller75.
Sin embargo, en el trabajo de Smith and Goh (1996) 77 en el que se estudió la
sobreexpresión de c-myc, presente en el 60% de los pacientes, mostraba que los
pacientes que presentaban sobreexpresión de c-myc tenían un mejor pronóstico que
aquellos que no lo tenían (P = 0.02).
En nuestro trabajo la expresión de c-myc de forma intensa frente a la expresión débil o
a la ausencia de expresión fue un factor pronóstico adverso en relación con la
supervivencia global, pero no con la supervivencia libre de enfermedad. Estos
hallazgos son concordantes con los de otros estudios previos como el de Bhatavdekar
et al, pero contrario a los resultados de otros autores como de Smith and Goh (1996).
DISCUSIÓN 173
El análisis de la expresión de c-myc en los tumores de colon es una vía de estudio que
nos puede aportar información pronóstica, tanto para la determinación del riesgo del
paciente como para la administración de quimioterapia adyuvante, aunque todavía no
está claro el papel que representa la sobreexpresión de este marcador.

174 DISCUSIÓN
5.3 INESTABILIDAD DE MICROSATÉLITES
La inestabilidad de microsatélites consiste en la presencia de mutaciones esporádicas o
heredadas, que hacen que las células pierdan la capacidad de reparar los defectos de
emparejamiento del ADN. Esta alteración se detecta en el 15% de los pacientes con
cáncer colorrectal, correspondiendo un 3% a síndromes hereditarios y un 12% a la
forma esporádica16.
En la revisión publicada por Boland16 en 2010 se ha encontrado que la inestabilidad de
microsatélites, se asocia a un ligero mejor pronóstico y a una peor respuesta al
tratamiento con 5-FU y cisplatino.27 Así mismo, la IMS tiene un fenotipo que se asocia
a una histología característica ya que presentan con más frecuencia histología
mucinosa, células en anillo de sello y pobre diferenciación celular 16.
En la revisión de Graziano del 200332 sobre factores pronósticos en CC estadio B de
Dukes se analizaron 17 estudios retrospectivos, encontrando una asociación
significativa entre la presencia de IMS y un mejor pronóstico. La mayor parte de estos
estudios mostraba una frecuencia de inestabilidad de microsatélites de entre 5-20% en
tumores esporádicos. La mayoría de estos estudios se realizó en pequeñas muestras,
por lo que no se pudo analizar de forma independiente el subgrupo de estadio B de
Dukes. Sólo el estudio de Gryfe et al. 78 confirmó de forma consistente e independiente
la asociación entre la presencia de inestabilidad de microsatélites y un mejor
pronóstico en pacientes con CC estadio B de Dukes.
En nuestro trabajo, la presencia de IMS fue factor de mal pronóstico para la
supervivencia libre de enfermedad y para la supervivencia global. Estos resultados
concuerdan con que fenotípicamente los tumores con inestabilidad de microsatélites
presentan baja diferenciación celular, lo que los hace más agresivos.
Sin embargo, estos resultados contradicen lo publicado en la literatura, por lo que han
de ser tomados con precaución, y precisan de estudios más amplios para determinar el
verdadero papel de la inestabilidad de microsatélites en el cáncer de colon.
Es posible que la inestabilidad de microsatélites nos sirva para crear grupos de riesgo, y
dentro del grupo de riesgo deban valorarse otros factores que modulen el riesgo de

DISCUSIÓN 175
recidiva a nivel individual. Sin embargo, en nuestro trabajo, dado lo reducido de la
muestra, no fue posible encontrar datos concluyentes a este respecto.


CONCLUSIONES


CONCLUSIONES 179
Tras el análisis retrospectivo de la evolución de un grupo de 141 pacientes
intervenidos de cáncer de colon en estadio T2-T4 N0 sin quimioterapia adyuvante, en
los que se analizaron una serie de factores analíticos, anatomopatológicos y genético-
moleculares, las conclusiones que podemos deducir de nuestro estudio en relación con
el riesgo de recidiva, supervivencia libre de enfermedad y supervivencia global son las
que se exponen a continuación:
1. Entre los pacientes que fueron intervenidos de cáncer de colon estadio T2-T4 N0
(estadio B de Astler-Coller) sin quimioterapia adyuvante, entre los años 1975 y
2007, el 17,73% presentó recidiva de su enfermedad tras cirugía curativa a lo largo
del seguimiento en un período de tiempo que abarcó entre los 6 meses y 24 años.
2. Entre los factores anatomopatológicos, resultaron de peor pronóstico en relación
con la supervivencia libre de enfermedad los siguientes:
a. El estadio T4 frente a T2 o T3.
b. Menos de 12 ganglios analizados en la pieza quirúrgica.
c. La presencia de invasión vascular de vaso de pared gruesa.
3. La presencia de inestabilidad de microsatélites resultó factor pronóstico adverso
tanto para la supervivencia libre de enfermedad como para la supervivencia global.
4. Entre de los factores moleculares estudiados resultaron con pronóstico
desfavorable:
a. La sobreexpresión de c-kit como factor adverso para la supervivencia libre
de enfermedad.
b. La sobreexpresión de c-myc como factor adverso para la supervivencia
global.
5. En nuestro estudio, no resultaron significativos en relación al pronóstico los
factores clínicos (obstrucción y perforación tumoral) ni los analíticos (CEA sérico
prequirúrgico):

180 CONCLUSIONES
6. En nuestro estudio, no resultaron significativos en relación al pronóstico los
siguientes factores anatomopatológicos:
a. Grado de diferenciación tumoral
b. Presencia de células en anillo de sello
c. Presencia de invasión linfática
d. Presencia de invasión perineural
e. Grado de componente mucinoso en el tumor.
7. En nuestro estudio, no resultaron significativos en relación al pronóstico los
siguientes factores moleculares divididos en función de su implicación en el
proceso de carcinogénesis:
a. Oncogenes: cERB-B2, EGFR y NTSR-1.
b. Genes supresores tumorales: p53, p21, p27, Nm23, SMAD 4, caveolina, p16,
pRB, N-cadherina y PTEN.
c. Proteínas de la vía de la apoptosis: BCL-2.
d. Proliferación celular: ciclina D1, beta-catenina y CDK-2.
e. Angiogénesis, metástasis e invasión tisular: VEGF, CD44, E-cadherina,
osteonectina y osteopontina.
f. Estructura celular: CEA, CK19, CK20 y EMA.
g. Diferenciación celular: CD133, HES-1 y NANOG.
8. Por el tamaño de la muestra en el grupo recidiva, hay varios factores que rozan
la significación estadística pero no la cumplen, pero según nuestro trabajo, la
presencia de cualquiera de los factores de mal pronóstico analizados con
significación estadística (estadio T4, menos de 12 ganglios linfáticos en la pieza
quirúrgica, presencia de invasión vascular, inestabilidad de microsatélites,
sobreexpresión de c-kit o sobreexpresión de c-myc), permiten establecer un
subgrupo de riesgo de recidiva en los pacientes con cáncer de colon
intervenidos con estadiaje T3-T4 N0, estadio B de la clasificación de Astler-Coller
y estadio II de la clasificación TNM, que nos debe hacer considerar la
conveniencia de tratamiento adyuvante y un seguimiento intensivo para el
diagnóstico precoz de la recidiva.

BIBLIOGRAFÍA


BIBLIOGRAFÍA 183
1. López-Abente Ortega G et al. La situación del cáncer en España. Ministerio de Sanidad y Consumo. 2005.
2. Siegel R, Desantis C, Virgo K, et al. Cancer Treatment and Survivorship Statistics , 2012. Am Cancer Soc. 2013. doi:10.3322/caac.21149.
3. Chang GJ, Rodriguez-Bigas M a, Skibber JM, Moyer V a. Lymph node evaluation and survival after curative resection of colon cancer: systematic review. J Natl Cancer Inst. 2007;99(6):433-41. doi:10.1093/jnci/djk092.
4. O’Connell JB, Maggard M a, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96(19):1420-5. doi:10.1093/jnci/djh275.
5. Ahnen DJ, Macrae FA, Bendell J, Tanabe KK, Savarese DM, Grover S. Clinical manifestations, diagnosis, and staging of colorectal cancer. UP TO DATE. 2012:1-27.
6. Compton C, Hawk E, Grochow L, Lee F, Ritter M, Niederhuber JE. Colon Cancer. In: Abeloff’s Clinical Oncology,. 4TH ed. Elsevier Inc.; 2008:1477-1534.
7. Fry RD, Mahmoud NN, Maron DJ, Bleier JIS. COLON AND RECTUM. In: Sabiston Textbook of Surgery. Nineteenth. Elsevier Inc.; 2012:1294-1380. doi:10.1016/B978-1-4377-1560-6.00052-4.
8. Bresalier RS. Colorectal Cancer. In: Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Ninth Edit. Copyright © 2010, 2006, 2002, 1998, 1993, 1989, 1983, 1978, 1973 by Saunders, an imprint of Elsevier Inc.; 2010:2191-2238. doi:10.1016/B978-1-4160-6189-2.00123-2.
9. American Joint Committee of Cancer 7th Edition, Edge S, Byrd D, Compton C, (Eds) et al (Eds). Colon and Rectum Cancer Staging. 2010:143.
10. Compton C, Tanabe KK, Savarese DM. Pathology and prognostic determinants of colorectal cancer. UP TO DATE. 2012:1-41.
11. Sanoff H, Goldberg R, Savarese DM. Adjuvant chemotherapy for resected stage II colon cancer. UP TO DATE. 2012:1-19.
12. NCCN GUIDELINES Version 3.2012. Practice Guidelines in Oncology - Colon Cancer. 2012.
13. Dotan E, Cohen SJ. Challenges in the management of stage II colon cancer. Semin Oncol. 2011;38(4):511-20. doi:10.1053/j.seminoncol.2011.05.005.
14. Betge J, Pollheimer MJ, Lindtner R a, et al. Intramural and extramural vascular invasion in colorectal cancer: prognostic significance and quality of pathology reporting. Cancer. 2012;118(3):628-38. doi:10.1002/cncr.26310.

184 BIBLIOGRAFÍA
15. Liebig C, Ayala G, Wilks J, et al. Perineural Invasion Is an Independent Predictor of Outcome in Colorectal Cancer. J Clin Oncol. 2009;27(31):5131-5137. doi:10.1200/JCO.2009.22.4949.
16. Boland CR, Goel A. Microsatellite Instability in Colorectal Cancer. Gastroenterology. 2010;138(6):2073-2087. doi:10.1053/j.gastro.2009.12.064.Boland.
17. Alberts B, Bray D, A J. Introducción a la biología celular.; 1999.
18. Ko TC. MOLECULAR AND CELL BIOLOGY. Nineteenth. Elsevier Inc.; 24-39. doi:10.1016/B978-1-4377-1560-6.00003-2.
19. Jacobs T. Control of the cell cycle. In: Abeloff’s Clinical Oncology,.; 1992:49-66. Available at: http://www.sciencedirect.com/science/article/pii/001216069290087W.
20. Alberts B, Johnson A, J L, et al. Molecular Biology of the Cell.; 2002.
21. Fearon E, Bommer G. Progressing from gene mutations to cancer. Abeloff MD, al Abeloff’s Clin Oncol 4th ed …. 2008:207-222. Available at: http://scholar.google.com/scholar?hl=en&btnG=Search&q=intitle:Progressing+from+Gene+Mutations+to+Cancer#1.
22. Alberts B, Johnson A, Lewis J et al. Programmed Cell Death (Apoptosis). In: Molecular Biology of the Cell. 4th editio. New York: Garland Science; 2002. Available at: Available from: http://www.ncbi.nlm.nih.gov/books/NBK26873/.
23. Elstrom RL, Thompson CB. Cell Life and Death. In: Abeloff’s Clinical Oncology,.; 2002:67-76.
24. Alberts B, Johnson A, Lewis J et al. Cancer as a Microevolutionary Process. In: Molecular Biology of the Cell. 4th Editio. New York: Garland Science; 2002. Available at: Available from: http://www.ncbi.nlm.nih.gov/books/NBK26891/.
25. Jass JR. Pathogenesis of colorectal cancer. In: Surgical Clinics of North America.Vol 82.; 2002:891-904. Available at: http://www.sciencedirect.com/science/article/pii/S1590865800803618.
26. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479-507. doi:10.1146/annurev-pathol-011110-130235.
27. Al-Sohaily S, Biankin A, Leong R, Kohonen-Corish M, Warusavitarne J. Molecular pathways in colorectal cancer. J Gastroenterol Hepatol. 2012. doi:10.1111/j.1440-1746.2012.07200.x.
28. Itzkowitz SH, Potack J. Colonic Polyps and Polyposis Syndromes. In: Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Ninth Edit. Copyright © 2010,

BIBLIOGRAFÍA 185
2006, 2002, 1998, 1993, 1989, 1983, 1978, 1973 by Saunders, an imprint of Elsevier Inc.; 2010:2155-2190. doi:10.1016/B978-1-4160-6189-2.00122-0.
29. Markowitz SD, Bertagnolli MM. Molecular Origins of cancer. N Engl J Med. 2010;361(25):2449-2460. doi:10.1056/NEJMra0804588.Molecular.
30. Saijo N. Critical comments for roles of biomarkers in the diagnosis and treatment of cancer. Cancer Treat Rev. 2012;38(1):63-7. doi:10.1016/j.ctrv.2011.02.004.
31. McLeod HL, Murray GI. Tumour markers of prognosis in colorectal cancer. Br J Cancer. 1999;79(2):191-203. doi:10.1038/sj.bjc.6690033.
32. Graziano F. Prognostic molecular markers for planning adjuvant chemotherapy trials in Dukes’ B colorectal cancer patients: how much evidence is enough? Ann Oncol. 2003;14(7):1026-1038. doi:10.1093/annonc/mdg284.
33. Alberts B, Johnson A, Lewis J et al. Finding the Cancer-Critical Genes. In: Molecular Biology of the Cell. 4th editio. New York: Garland Science; 2002. Available at: Available from: http://www.ncbi.nlm.nih.gov/books/NBK26816/.
34. Bellone G, Smirne C, Carbone A, et al. KIT/stem cell factor expression in premalignant and malignant lesions of the colon mucosa in relationship to disease progression and outcomes. Int J Oncol. 2006;29(4):851-9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16964380.
35. Dupouy S, Mourra N, Doan VK, Gompel A, Alifano M, Forgez P. The potential use of the neurotensin high affinity receptor 1 as a biomarker for cancer progression and as a component of personalized medicine in selective cancers. Biochimie. 2011;93(9):1369-78. doi:10.1016/j.biochi.2011.04.024.
36. Gui X, Guzman G, Dobner PR, Kadkol SS. Increased neurotensin receptor-1 expression during progression of colonic adenocarcinoma. Peptides. 2008;29(9):1609-15. doi:10.1016/j.peptides.2008.04.014.
37. Ogino S, Nosho K, Shima K, et al. p21 Expression in Colon Cancer and Modifying Effects of Patient Age and Body Mass Index on Prognosis. 2010;18(9):617-632. doi:10.1158/1055-9965.EPI-09-0451.p21.
38. Zhang H, Sun XF. Loss of p27 expression predicts poor prognosis in patients with Dukes’ B stage or proximal colorectal cancer. Int J Oncol. 2001;19(1):49-52. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11408921.
39. Demirbaş S, Sücüllü I, Yildirim S, Celenk T. Influence of the c-erb B-2, nm23, bcl-2 and p53 protein markers on colorectal cancer. Turk J Gastroenterol. 2006;17(1):13-9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16830272.

186 BIBLIOGRAFÍA
40. Mesker WE, Liefers G-J, Junggeburt JMC, et al. Presence of a high amount of stroma and downregulation of SMAD4 predict for worse survival for stage I-II colon cancer patients. Cell Oncol. 2009;31(3):169-78. doi:10.3233/CLO-2009-0478.
41. Patlolla JMR, Swamy M V, Raju J, Rao C V. Overexpression of caveolin-1 in experimental colon adenocarcinomas and human colon cancer cell lines. Oncol Rep. 2004;11(5):957-63. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15069532.
42. Hehlgans S, Cordes N. Caveolin-1 : an essential modulator of cancer cell radio- and chemoresistance. Am J Cancer Res. 2011;1(4):521-530.
43. Kaori Shima1, Katsuhiko Nosho1, Yoshifumi Baba1, Mami Cantor1 JAM, Edward L. Giovannucci2, 3, Charles S. Fuchs1, 2 and SO. Prognostic Significance of CDKN2A (p16) Promoter Methylation and Loss of Expression in 902 Colorectal Cancers: Cohort Study and Literature Review. 2012;128(5):1080-1094. doi:10.1002/ijc.25432.Prognostic.
44. De Wever O, Westbroek W, Verloes A, et al. Critical role of N-cadherin in myofibroblast invasion and migration in vitro stimulated by colon-cancer-cell-derived TGF-beta or wounding. J Cell Sci. 2004;117(Pt 20):4691-703. doi:10.1242/jcs.01322.
45. Ogino S, Nosho K, Irahara N, et al. A cohort study of cyclin D1 expression and prognosis in 602 colon cancer cases. Clin Cancer Res. 2009;15(13):4431-8. doi:10.1158/1078-0432.CCR-08-3330.
46. Chew A, Salama P, Robbshaw A, et al. SPARC, FOXP3, CD8 and CD45 correlation with disease recurrence and long-term disease-free survival in colorectal cancer. PLoS One. 2011;6(7):e22047. doi:10.1371/journal.pone.0022047.
47. Topak N, Nuri O. β -Catenin and its relation to VEGF and cyclin D1 expression in pT3 rectosigmoid cancers. Turkish J Gastroenterol. 2010;2010(4):365-371. doi:10.4318/tjg.2010.0122.
48. Pritchard C c, Grady WM. Colorectal Cancer Molecular Biology Moves into Clinical Practice. Gut. 2012;60(1):116-129. doi:10.1136/gut.2009.206250.Colorectal.
49. Mao Y, Li Z, Lou C, Zhang Y. Expression of phosphorylated Stat5 predicts expression of cyclin D1 and correlates with poor prognosis of colonic adenocarcinoma. Int J Colorectal Dis. 2011;26(1):29-35. doi:10.1007/s00384-010-1090-7.
50. Galizia G, Gemei M, Del Vecchio L, et al. Combined CD133/CD44 expression as a prognostic indicator of disease-free survival in patients with colorectal cancer. Arch Surg. 2012;147(1):18-24. doi:10.1001/archsurg.2011.795.

BIBLIOGRAFÍA 187
51. Agrawal D, Chen T, Irby R, et al. Osteopontin identified as lead marker of colon cancer progression, using pooled sample expression profiling. J Natl Cancer Inst. 2002;94(7):513-21. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11929952.
52. Weber GF. The cancer biomarker osteopontin: combination with other markers. Cancer Genomics Proteomics. 2011;8(6):263-88. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22086896.
53. Yeatman TJ, Chambers AF. Osteopontin and colon cancer progression. Clin Exp Metastasis. 2003;20(1):85-90. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12650611.
54. Nazato M, Luongo L, Waisberg DR, Martins R, Concei L, Waisberg J. Prognostic value of carcinoembryonic antigen distribution in tumor tissue of colorectal carcinoma. Arq gastroenterol. 2009;46(1):26-31.
55. Jain R, Fischer S, Serra S, Chetty R. The use of Cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl Immunohistochem Mol Morphol. 2010;18(1):9-15. doi:10.1097/PAI.0b013e3181ad36ea.
56. Bayrak R, Yenidünya S, Haltas H. Cytokeratin 7 and cytokeratin 20 expression in colorectal adenocarcinomas. Pathol Res Pract. 2011;207(3):156-60. doi:10.1016/j.prp.2010.12.005.
57. Davidson BR, Sams VR, Styles J, Dean C, Boulos PB. Comparative study of carcinoembryonic antigen and epithelial membrane antigen expression in normal colon, adenomas and adenocarcinomas of the colon and rectum. Gut. 1989;30(9):1260-5. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1434233&tool=pmcentrez&rendertype=abstract.
58. Ishiguro T, Sato A, Ohata H, Sakai H, Nakagama H, Okamoto K. Differential expression of nanog1 and nanogp8 in colon cancer cells. Biochem Biophys Res Commun. 2012;418(2):199-204. doi:10.1016/j.bbrc.2011.10.123.
59. Benson AB, Schrag D, Somerfield MR, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol. 2004;22(16):3408-19. doi:10.1200/JCO.2004.05.063.
60. Quah H-M, Chou JF, Gonen M, et al. Identification of patients with high-risk stage II colon cancer for adjuvant therapy. Dis Colon Rectum. 2008;51(5):503-7. doi:10.1007/s10350-008-9246-z.
61. Shi Q, Sargent DJ. Meta-analysis for the evaluation of surrogate endpoints in cancer clinical trials. Int J Clin Oncol. 2009;14(2):102-11. doi:10.1007/s10147-009-0885-4.

188 BIBLIOGRAFÍA
62. Sargent D, Sobrero A, Grothey A, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27(6):872-7. doi:10.1200/JCO.2008.19.5362.
63. Mulcahy HE, Toner M, Patchett SE, Daly L, O’Donoghue DP. Identifying stage B colorectal cancer patients at high risk of tumor recurrence and death. Dis Colon Rectum. 1997;40(3):326-31. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9118749.
64. Saha a K, Smith KJE, Sue-Ling H, Sagar PM, Burke D, Finan PJ. Prognostic factors for survival after curative resection of Dukes’ B colonic cancer. Colorectal Dis. 2011;13(12):1390-4. doi:10.1111/j.1463-1318.2010.02507.x.
65. Gertler R, Rosenberg R, Schuster T, Friess H. Defining a high-risk subgroup with colon cancer stages I and II for possible adjuvant therapy. Eur J Cancer. 2009;45(17):2992-9. doi:10.1016/j.ejca.2009.07.008.
66. Chen C-C, Yang S-H, Lin J-K, et al. Is it reasonable to add preoperative serum level of CEA and CA19-9 to staging for colorectal cancer? J Surg Res. 2005;124(2):169-74. doi:10.1016/j.jss.2004.08.013.
67. Harrison LE, Guillem JG, Paty P, Cohen AM. Preoperative Carcinoembryonic Antigen Predicts Outcomes in Node-Negative Colon Cancer Patients : A Multivariate Analysis of 572 Patients. J Am Coll Surg. 1997;185:55-59.
68. Wolmark N, Fisher B, Wieand HS, et al. The prognostic significance of preoperative carcinoembryonic antigen levels in colorectal cancer. Results from NSABP (National Surgical Adjuvant Breast and Bowel Project) clinical trials. Ann Surg. 1984;199(4):375-82. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1353353&tool=pmcentrez&rendertype=abstract.
69. Sener SF, Imperato JP, Chmiel J, Fremgen A, Sylvester J. The use of cancer registry data to study preoperative carcinoembryonic antigen level as an indicator of survival in colorectal cancer. CA Cancer J Clin. 39(1):50-7. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2492877.
70. Uen Y-H, Lin S-R, Wu D-C, et al. Prognostic significance of multiple molecular markers for patients with stage II colorectal cancer undergoing curative resection. Ann Surg. 2007;246(6):1040-6. doi:10.1097/SLA.0b013e318142d918.
71. Mroczkowski P, Schmidt U, Sahm M, Gastinger I, Lippert H, Kube R. Prognostic Factors Assessed for 15,096 Patients with Colon Cancer in Stages I and II. World J Surg. 2012:6-11. doi:10.1007/s00268-012-1531-2.

BIBLIOGRAFÍA 189
72. Marzouk O, Schofield J. Review of histopathological and molecular prognostic features in colorectal cancer. Cancers (Basel). 2011;3(2):2767-810. doi:10.3390/cancers3022767.
73. Vather R, Sammour T, Kahokehr A, Connolly AB, Hill AG. Lymph node evaluation and long-term survival in Stage II and Stage III colon cancer: a national study. Ann Surg Oncol. 2009;16(3):585-93. doi:10.1245/s10434-008-0265-8.
74. Bellone G, Silvestri S, Artusio E, et al. Growth stimulation of colorectal carcinoma cells via the c-kit receptor is inhibited by TGF-beta 1. J Cell Physiol. 1997;172(1):1-11. doi:10.1002/(SICI)1097-4652(199707)172:1<1::AID-JCP1>3.0.CO;2-S.
75. Miller F, Heimann TM, Quish A, et al. ras and c-myc protein expression in colorectal carcinoma. Study of cancer-prone patients. Dis Colon Rectum. 1992;35(5):430-5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1568393.
76. Bhatavdekar JM, Patel DD, Ghosh N, et al. Coexpression of Bcl-2, c-Myc, and p53 oncoproteins as prognostic discriminants in patients with colorectal carcinoma. Dis Colon Rectum. 1997;40(7):785-90. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9221853.
77. Smith DR, Goh HS. Overexpression of the c-myc proto-oncogene in colorectal carcinoma is associated with a reduced mortality that is abrogated by point mutation of the p53 tumor suppressor gene. Clin Cancer Res. 1996;2(6):1049-53. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9816266.
78. Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342(2):69-77. doi:10.1056/NEJM200001133420201.


ANEXOS

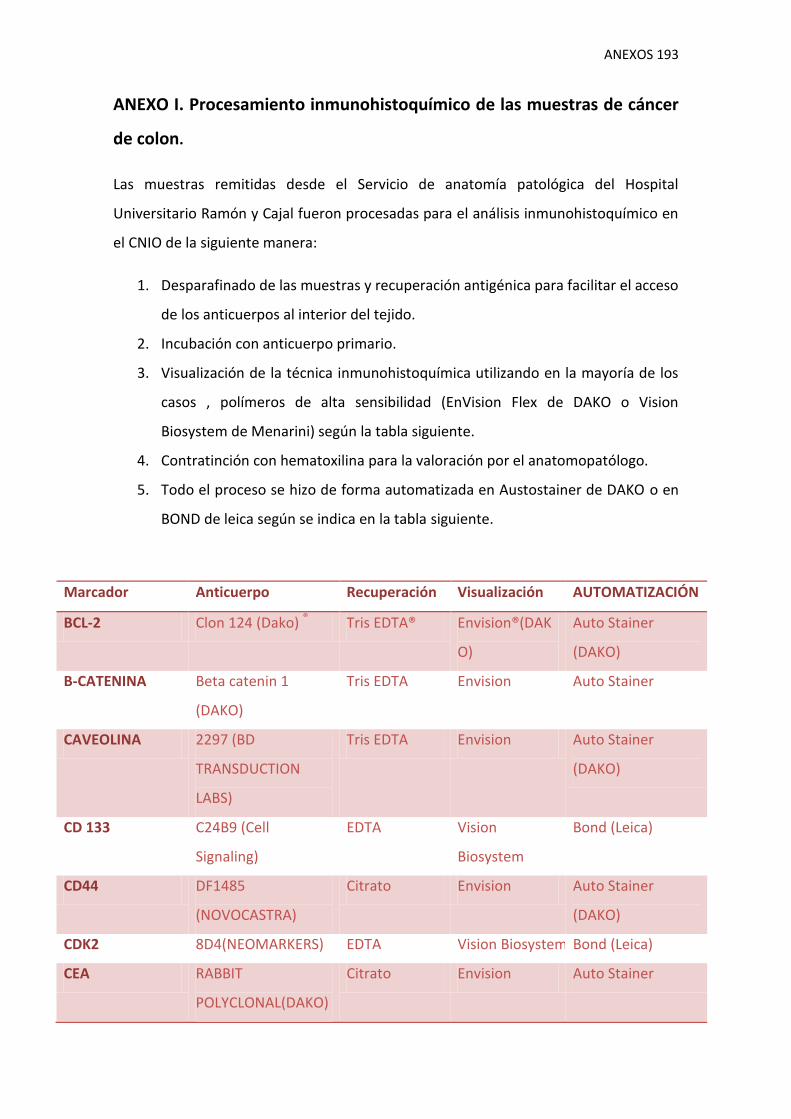
ANEXOS 193
ANEXO I. Procesamiento inmunohistoquímico de las muestras de cáncer
de colon.
Las muestras remitidas desde el Servicio de anatomía patológica del Hospital
Universitario Ramón y Cajal fueron procesadas para el análisis inmunohistoquímico en
el CNIO de la siguiente manera:
1. Desparafinado de las muestras y recuperación antigénica para facilitar el acceso
de los anticuerpos al interior del tejido.
2. Incubación con anticuerpo primario.
3. Visualización de la técnica inmunohistoquímica utilizando en la mayoría de los
casos , polímeros de alta sensibilidad (EnVision Flex de DAKO o Vision
Biosystem de Menarini) según la tabla siguiente.
4. Contratinción con hematoxilina para la valoración por el anatomopatólogo.
5. Todo el proceso se hizo de forma automatizada en Austostainer de DAKO o en
BOND de leica según se indica en la tabla siguiente.
Marcador Anticuerpo Recuperación Visualización AUTOMATIZACIÓN
BCL-2 Clon 124 (Dako) ® Tris EDTA® Envision®(DAK
O)
Auto Stainer
(DAKO)
B-CATENINA Beta catenin 1
(DAKO)
Tris EDTA Envision Auto Stainer
CAVEOLINA 2297 (BD
TRANSDUCTION
LABS)
Tris EDTA Envision Auto Stainer
(DAKO)
CD 133 C24B9 (Cell
Signaling)
EDTA Vision
Biosystem
Bond (Leica)
CD44 DF1485
(NOVOCASTRA)
Citrato Envision Auto Stainer
(DAKO)
CDK2 8D4(NEOMARKERS) EDTA Vision Biosystem Bond (Leica)
CEA RABBIT
POLYCLONAL(DAKO)
Citrato Envision Auto Stainer

194 ANEXOS
C-ERBB2 RABBIT
POLYCLONAL(DAKO)
Citrato LSAB TM500 (DAKO)
CK-19 RCK108(DAKO) Citrato LSAB TM500 (DAKO)
CK-20 KS20.8 (DAKO) Citrato LSAB TM500 (DAKO)
C-KIT RABBIT
POLYCLONAL(DAKO)
Citrato Envision Auto Stainer
C-MYC Y 69 (EPITOMICS) Tris EDTA Envision Auto Stainer
COX2 SP21 RABBIT
MONOCLONAL
Neomarkers
Tris EDTA Envision Auto Stainer
CYCLIN D1 SP4 RABBIT
MONOCLONAL
DAKO
Tris EDTA Envision Auto Stainer
E-CADHERINA NCH-38 (DAKO) Tris EDTA® Envision® Auto Stainer®
EGFR EGFR.113(NOVOCAS
TRA)
Citrato Envision® Auto Stainer®
EMA E29 (DAKO) Tris EDTA® Envision® Auto Stainer®
HES1 Diseñado por Dr.
Maraver (CNIO)
EDTA Vision
biosystem
Bond
H-TERT 44F12(NOVOCASTRA
)
Citrato Envision Auto Stainer
KI67 DAKO (clon MIB-1) Citrato Envision Auto Stainer
MLH1 G168-15(BD
PHARMINGEN)
Tris EDTA® Envision® Auto Stainer®
MSH2 FE11(ONCOGENE) Tris EDTA® Envision® Auto Stainer®
MSH6 44(BD
TRANSDUCTION
LABS)
Citrato Envision Auto Stainer
NANOG GOAT
POLYCLONAL(R&D
SYSTEMS)
Citrato Envision Auto Stainer

ANEXOS 195
N-CADHERINA 3B9 (ZYMED) EDTA® Vision
Biosystem
Bond
NEUROTENISINA RABBIT
POLYCLONAL(ENZO)
Citrato +
Proteinasa K
Envision Auto Stainer
NM23 RABBIT
POLYCLONAL(SANTA
CRUZ)
EDTA Vision
Biosystem
Bond
NTSR1 RABBIT
POLYCLONAL(GENW
AY)
EDTA Vision
Biosystem
Bond
OSTEONECTINA 15G12(NOVOCASTR
A)
Citrato Envision Auto Stainer
OSTEOPONTINA Polyclonal Rabbit
(Abcam)
Citrato Envision Auto Stainer
P16 E6H4(MTM) Tris EDTA Envision Auto Stainer
P21 EA10(CALBIOCHEM) Citrato Envision Auto Stainer
P27 57(BD
TRANSDUCTION
LABS)
Citrato Envision Auto Stainer
P53 DO-7(NOVOCASTRA) Citrato Envision Auto Stainer
PTEN Tris EDTA Envision Auto Stainer
RBP RABBIT
POLYCLONAL(SANTA
CRUZ)
Tris EDTA Envision Auto Stainer
SMADY B8 (SANTA CRUZ) Citrato Envision Auto Stainer
VEGRF NOVOCASTRA CITRATO LSAB TM500
196 ANEXOS
ANEXO II. Tablas de variables demográficas.
Edad (años) Ausencia de enfermedad
N (%)
Recidiva
N (%)
Media 66,9 69,6
DE 11,2 11,2
Sexo Ausencia de enfermedad
N (%)
Recidiva
N (%)
Hombres 62 (53,4) 16 (64,0)
Mujeres 54 (46,6) 9 (36,0)
TOTAL 116 25
Localización Ausencia de enfermedad
N (%)
Recidiva
N (%)
Colon derecho/ciego 44 (37,9) 4 (16,0)
Colon transverso 13 (11,2) 3 (12,0)
Colon izquierdo/recto-sigma 58 (50,0) 18 (72,0)
Colon izquierdo/ciego 1 (0,9) 0
TOTAL 116 25

ANEXOS 197
Recidiva Ausencia de Enfermedad
Mediana Rango Mediana Rango
CEA (ng/ml) 7,0 0,99-78 3,2 0-203
Ausencia de enfermedad
N (%)
Recidiva
N (%)
CEA mayor de 5 38 (29,2) 10 (40)
CEA menor de 5 59 (60,8) 6 (24)
TOTAL 97 16
Presentación Ausencia de enfermedad
N (%)
Recidiva
N (%) P (IC 95%)
Obstrucción sí 12 (10,6) 4 (17,4)
Perforación sí 7 (6,3) 0
Los datos sobre perforación estaban ausentes en 2 controles y en 4 casos. Los datos sobre
perforación estaban ausentes en 3 controles y en 5 casos.
TNM 2009 Ausencia de enfermedad
N (%)
Recidiva
N (%)
T2 N0 (I) 11 (10) 2 (10)
T3N0 (II A) 91 (82,7) 15 (75)
T4aN0 (II B) 3 (2,7) 1 (5)
T4bN0 (II C) 5 (4,5) 2 (10)
TOTAL 110 20
5 valores perdidos en el grupo de ausencia de enfermedad y 6 valores perdidos en el grupo de
recidiva.

198 ANEXOS
ANEXO III. Tablas de variables del estadiaje tumoral.
TNM 2009 Ausencia de enfermedad
N (%)
Recidiva
N (%)
T2 N0 (I) 11 (10) 2 (10)
T3N0 (II A) 91 (82,7) 15 (75)
T4aN0 (II B) 3 (2,7) 1 (5)
T4bN0 (II C) 5 (4,5) 2 (10)
TOTAL 110 20
5 valores perdidos en el grupo de ausencia de enfermedad y 6 valores perdidos en el grupo de recidiva.
Astler-Coller Ausencia de enfermedad
N (%)
Recidiva
N (%) P (IC 95%)
B1 11 ( 9,9) 2 (8,3)
B2 95 (85,6) 17 (70,8)
B3 5 (4,5) 2 (8,3)
TOTAL 111 21
4 valores perdidos en el grupo de ausencia de enfermedad y 5 valores perdidos en el grupo recidiva.
Dukes Ausencia de enfermedad
N (%)
Recidiva
N (%) P (IC 95%)
A 10 (9,1) 2 (9,1)
B 100 (90,9) 20 (90,9)
TOTAL 110 22
5 valores perdidos en el grupo de ausencia de enfermedad y 4 valores perdidos en el grupo recidiva.
ANEXOS 199
ANEXO IV. Tablas de variables histológicas.
Ausencia de Enfermedad Recidiva
Mediana Rango Mediana Rango
Número de Ganglios 15 3-46 11,5 3-75
Diferenciación Ausencia de enfermedad
N (%)
Recidiva
N (%) P (IC 95%)
Bien diferenciado 89 (78,8) 18 (75,0)
Moderadamente
diferenciado 14 (12,4) 6 (24,0)
Pobremente diferenciado 10 (8,8) 0
TOTAL 113 24
2 valores perdidos en el grupo de ausencia de enfermedad y 1 valor perdidos en el grupo recidiva.
Infiltración vascular
presente
Ausencia de enfermedad
N (%)
Recidiva
N (%)
Global 15 (13,0) 7 (29,2)
Vaso grueso 7 (6,1) 5 (20,8)
Vaso fino 10 (8,8) 3 (12,5)
Los datos están disponibles en 138 de los 141 pacientes.
Infiltración presente Ausencia de enfermedad
N (%)
Recidiva
N (%)
Linfática 6 (8,8) 2 (14,3)
Perineural 6 (5,2) 1 (4,2)

200 ANEXOS
Ganglios linfáticos
Ausencia de
enfermedad
N (%)
Recidiva
N (%)
Mayor o igual a 12 83 (71,6) 13 (52,0)
Menor de 12 33 (28,4) 12 (48,0)
TOTAL 116 25
Riesgo Ausencia de enfermedad
N (%)
Recidiva
N (%)
Alto riesgo 56 (48,3) 18 (72)
No alto riesgo 60 (51,7) 7 (28)
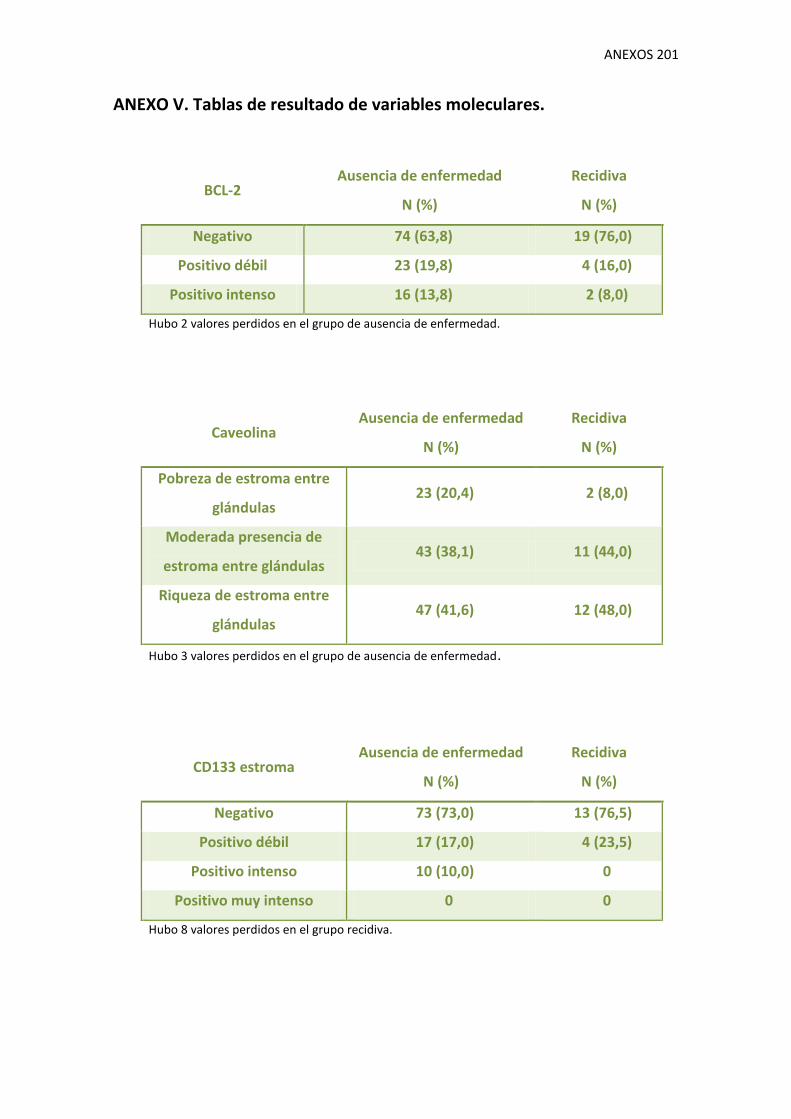
ANEXOS 201
ANEXO V. Tablas de resultado de variables moleculares.
BCL-2 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 74 (63,8) 19 (76,0)
Positivo débil 23 (19,8) 4 (16,0)
Positivo intenso 16 (13,8) 2 (8,0)
Hubo 2 valores perdidos en el grupo de ausencia de enfermedad.
Caveolina Ausencia de enfermedad
N (%)
Recidiva
N (%)
Pobreza de estroma entre
glándulas 23 (20,4) 2 (8,0)
Moderada presencia de
estroma entre glándulas 43 (38,1) 11 (44,0)
Riqueza de estroma entre
glándulas 47 (41,6) 12 (48,0)
Hubo 3 valores perdidos en el grupo de ausencia de enfermedad.
CD133 estroma Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 73 (73,0) 13 (76,5)
Positivo débil 17 (17,0) 4 (23,5)
Positivo intenso 10 (10,0) 0
Positivo muy intenso 0 0
Hubo 8 valores perdidos en el grupo recidiva.

202 ANEXOS
CD133 total Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 15 (18,5) 4 (28,6)
Positivo débil 41 (50,6) 3 (21,4)
Positivo intenso 15 (18,5) 4 (28,6)
Positivo muy intenso 3 (3,7) 2 (14,3)
Hubo 8 valores perdidos en el grupo recidiva.
CEA Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 3 (2,6) 1 (4,0)
Positivo débil 11 (9,6) 2 (8,0)
Positivo intenso 100 (87,7) 22 (88,0)
Hubo 2 valores perdidos en el grupo ausencia de enfermedad.
CK19 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 2 (2,0) 2 (11,8)
Positivo débil 48 (48,0) 4 (23,5)
Positivo intenso 50 (50,0) 11 (64,7)
Hubo 16 casos perdidos en el grupo ausencia de enfermedad y 8 perdidos en el grupo
recidiva.
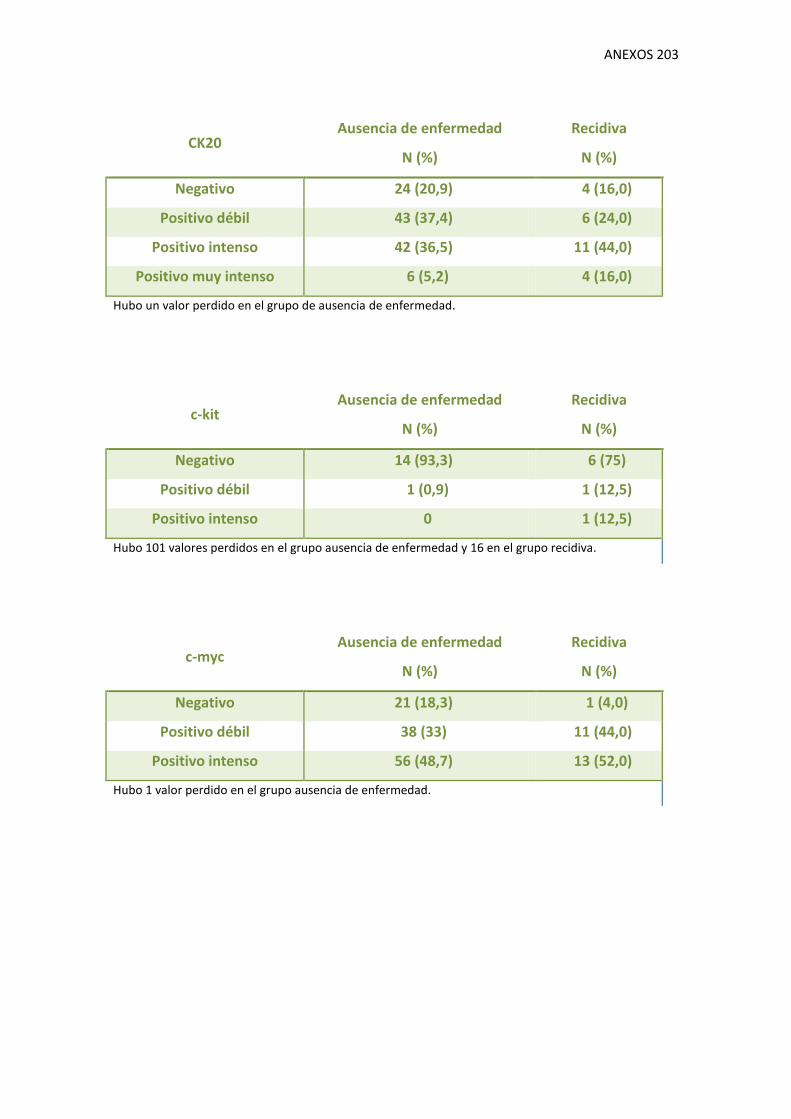
ANEXOS 203
CK20 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 24 (20,9) 4 (16,0)
Positivo débil 43 (37,4) 6 (24,0)
Positivo intenso 42 (36,5) 11 (44,0)
Positivo muy intenso 6 (5,2) 4 (16,0)
Hubo un valor perdido en el grupo de ausencia de enfermedad.
c-kit Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 14 (93,3) 6 (75)
Positivo débil 1 (0,9) 1 (12,5)
Positivo intenso 0 1 (12,5)
Hubo 101 valores perdidos en el grupo ausencia de enfermedad y 16 en el grupo recidiva.
c-myc Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 21 (18,3) 1 (4,0)
Positivo débil 38 (33) 11 (44,0)
Positivo intenso 56 (48,7) 13 (52,0)
Hubo 1 valor perdido en el grupo ausencia de enfermedad.

204 ANEXOS
E-Cadherina Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 6 (5,3) 0
Positivo débil 14 (12,3) 4 (16,0)
Positivo intenso 94 (82,5) 21 (84,0)
Hubo 2 valores perdidos en el grupo ausencia de enfermedad.
EGFR Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 59 (59,0) 9 (52,9)
Positivo débil 39 (39,0) 8 (47,1)
Positivo intenso 2 (2,0) 0
Hubo 2 valores perdidos en el grupo ausencia de enfermedad.
MLH1 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 25 (21,7) 10 (40,0)
Positivo débil 13 (11,3) 1 (4,0)
Positivo intenso 77 ( 67,0) 14 (56,0)
Hubo un valor perdido en el grupo ausencia de enfermedad.

ANEXOS 205
MSH2 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 3 (2,6) 2 (8,0)
Positivo débil 5 (4,4) 3 (12,0)
Positivo intenso 106 (93,0) 20 (80,0)
Hubo 2 valores perdidos en el grupo ausencia de enfermedad.
MSH6 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 6 (5,3) 5 (20,0)
Positivo débil 69 (60,5) 14 (56,0)
Positivo intenso 24 (21,1) 6 (24,0)
Positivo muy intenso 15 (13,2) 0
Hubo 2 valores perdidos en el grupo de ausencia de enfermedad.
MSI Ausencia de enfermedad
N (%)
Recidiva
N (%)
Inestable 27 (23,5) 11 (44,0)
Estable 88 (76,5) 14 (56,0)
Hubo un valor perdido en el grupo ausencia de enfermedad.

206 ANEXOS
NANOG Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 0
<25% de células positivas 8 (7,0) 1 (4,0)
25-75% de células positivas 51 (44,7) 12 (48,0)
>75% de células positivas 55 (48,2) 12 (48,0)
Hubo un valor perdido en el grupo ausencia de enfermedad.
P27 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 50 (43,5) 12 (48,0)
<10% de células positivas 56 (48,7) 11 (44,0)
10-70% de células positivas 9 (7,8) 2 (8,0)
>70% de células positivas 0 0
Hubo un valor perdido en el grupo ausencia de enfermedad.
P53 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 60 (52,6) 15 (60,0)
Positivo débil 14 (12,3) 1 (4,0)
Positivo intenso 40 (35,1) 9 (36,0)
Hubo 2 valores perdidos en el grupo ausencia de enfermedad.
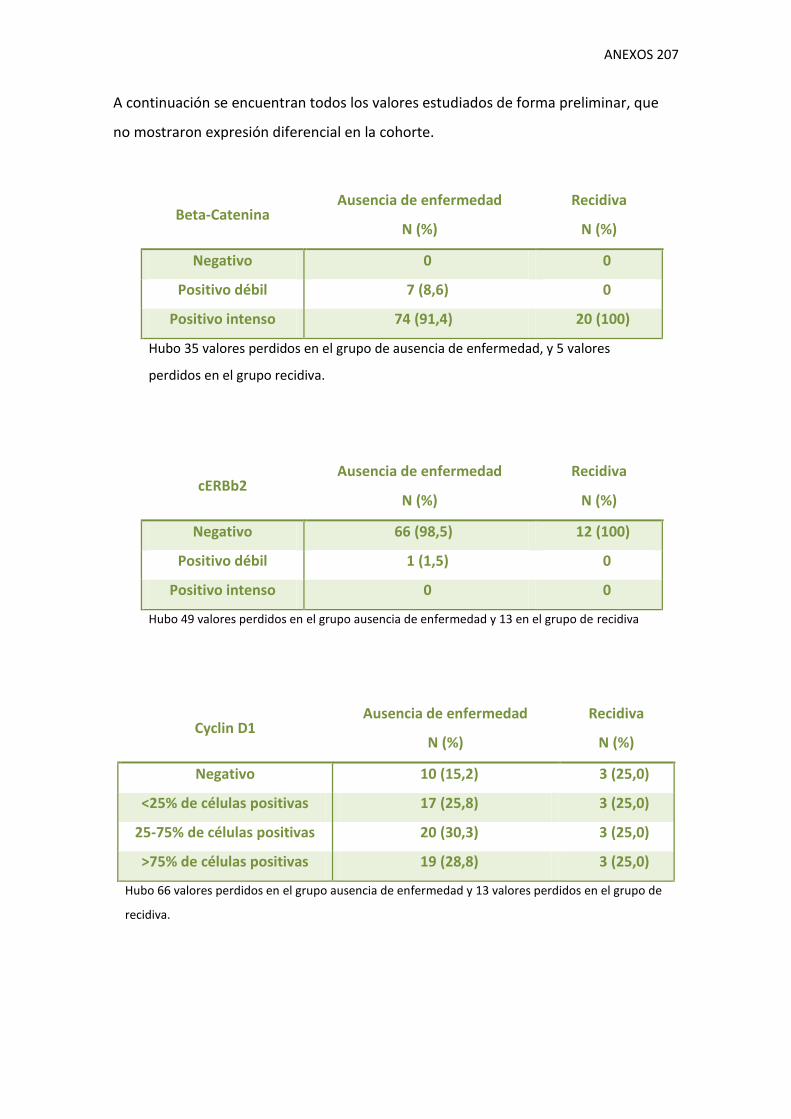
ANEXOS 207
A continuación se encuentran todos los valores estudiados de forma preliminar, que
no mostraron expresión diferencial en la cohorte.
Beta-Catenina Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 0 0
Positivo débil 7 (8,6) 0
Positivo intenso 74 (91,4) 20 (100)
Hubo 35 valores perdidos en el grupo de ausencia de enfermedad, y 5 valores
perdidos en el grupo recidiva.
cERBb2 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 66 (98,5) 12 (100)
Positivo débil 1 (1,5) 0
Positivo intenso 0 0
Hubo 49 valores perdidos en el grupo ausencia de enfermedad y 13 en el grupo de recidiva
Cyclin D1 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 10 (15,2) 3 (25,0)
<25% de células positivas 17 (25,8) 3 (25,0)
25-75% de células positivas 20 (30,3) 3 (25,0)
>75% de células positivas 19 (28,8) 3 (25,0)
Hubo 66 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo de
recidiva.

208 ANEXOS
N-Cadherina: hubo 49 valores perdidos en el grupo de ausencia de enfermedad y 13
valores perdidos en el grupo recidiva. Todos los casos analizados fueron negativos en
ambos grupos.
NM-23: hubo 49 valores perdidos en el grupo de ausencia de enfermedad y 13 valores
perdidos en el grupo recidiva. Todos los casos analizados fueron positivos intensos en
ambos grupos.
COX-2 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 17 (25,4) 4 (33,3)
<25% de células positivas 14 (20,9) 2 (16,7)
25-75% de células positivas 9 (13,4) 4 (33,3)
>75% de células positivas 27 (40,3) 2 (16,7)
Hubo 49 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.
EMA Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 9 (13,6) 0
<25% de células positivas 10 (15,2) 1 (8,3)
25-75% de células positivas 6 (9,1) 4 (33,3)
>75% de células positivas 41 (62,1) 7 (58,3)
Hubo 50 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.

ANEXOS 209
Osteonectina: hubo 49 valores perdidos en el grupo de ausencia de enfermedad y 14
valores perdidos en el grupo recidiva. Todos los casos analizados fueron negativos en
ambos grupos.
NEUROTENSINA Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 44 (65,7) 8 (66,7)
<25% de células positivas 15 (22,4) 4 (33,3)
25-75% de células positivas 8 (11,9) 0
>75% de células positivas 0 0
Hubo 49 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.
NTSR-1 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 59 ( 88,1) 11 (91,7)
<25% de células positivas 5 (7,5) 0
25-75% de células positivas 3 (4,5) 1 (8,3)
>75% de células positivas 0 0
Hubo 49 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.
OSTEOPONTINA Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 45 (67,2) 8 (66,7)
Positivo débil 20 (29,9) 4 (33,3)
Positivo intenso 2 (3,0) 0
Hubo 49 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.

210 ANEXOS
PTEN citoplasma: hubo 49 valores perdidos en el grupo de ausencia de enfermedad y
13 valores perdidos en el grupo recidiva. Todos los casos analizados fueron negativos
en ambos grupos.
PTEN núcleo: hubo 49 valores perdidos en el grupo de ausencia de enfermedad y 13
valores perdidos en el grupo recidiva. Todos los casos analizados fueron negativos en
el grupo recidiva. En el grupo ausencia de enfermedad, se encontraron 8 valores
positivos con menos del 25% de células positivas (un 11,9%), y el resto negativos.
P16 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 12 (18,2) 3 (25,0)
<25% de células positivas 16 (24,2) 3 (25,0)
25-75% de células positivas 19 (28,8) 3 (25,0)
>75% de células positivas 19 (28,8) 3 (25,0)
Hubo 50 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.
P21 Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 25 (38,5) 2 (16,7)
<25% de células positivas 17 (26,2) 6 (50,0)
25-75% de células positivas 16 (24,6) 3 (25,0)
>75% de células positivas 7 (10,8) 1 (8,3)
Hubo 51 valores perdidos en el grupo ausencia de enfermedad y 13 valores
perdidos en el grupo recidiva.

ANEXOS 211
VEGFR: hubo 49 valores perdidos en el grupo de ausencia de enfermedad y 13 valores
perdidos en el grupo recidiva. Todos los casos analizados fueron negativos en ambos
grupos.
pRB Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 28 (42,4) 6 (50,0)
Positivo débil 10 (15,2) 1 (8,3)
Positivo intenso 11 (16,7) 1 (8,3)
Positivo muy intenso 17 (25,8) 4 (33,3)
Hubo 50 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.
SMADY Ausencia de enfermedad
N (%)
Recidiva
N (%)
Negativo 14 (20,9) 1 (8,3)
<25% de células positivas 10 (14,9) 3 (25,0)
25-75% de células positivas 6 (9,0) 2 (16,7)
>75% de células positivas 37 (55,2) 6 (50,0)
Hubo 49 valores perdidos en el grupo ausencia de enfermedad y 13 valores perdidos en el grupo
recidiva.

212 ANEXOS
ANEXO VI. Agrupación variables moleculares de forma dicotómica
I. BCL2: NEGATIVO vs. POSITIVO DEBIL o POSITIVO INTENSO
II. B-CATENINA: NEGATIVO vs. POSITIVO DEBIL o POSITIVO INTENSO
III. CAVEOLINA: POBREZA DE ESTROMA ENTRE GLÁNDULAS vs. MODERADA
PRESENCIA DE ESTROMA ENTRE GLÁNDULAS o RIQUEZA DE ESTROMA ENTRE
GLÁNDULAS
IV. CD133 (VALORADO EN TOTAL, EN ESTROMA Y EN TUMOR): NEGATIVO o
POSITIVO DEBIL vs. POSITIVO INTENSO o POSITIVO MUY INTENSO
V. CD44: NEGATIVO o POSITIVO DÉBIL vs. POSITIVO INTENSO
VI. CDK2 (CITOPLASMA): NEGATIVO o POSITIVO DÉBIL vs. POSITIVO INTENSO.
VII. CDK2 (NUCLEO): NEGATIVO o MENOR DEL 25% o ENTRE EL 25-75% vs. MAYOR
DEL 75%.
VIII. CEA: NEGATIVO o POSITIVO DÉBIL vs. POSITIVO INTENSO
IX. C-ERB: NEGATIVO vs. POSITIVO DÉBIL o POSITIVO INTENSO
X. CK19: NEGATIVO o POSITIVO DÉBIL vs. POSITIVO INTENSO
XI. CK20: NEGATIVO o POSITIVO DEBIL vs. POSITIVO INTENSO o POSITIVO MUY
INTENSO
XII. C-KIT: NEGATIVO vs. POSITIVO DÉBIL o POSITIVO INTENSO
XIII. C-MYC: NEGATIVO o POSITIVO DÉBIL vs. POSITIVO INTENSO
XIV. COX2: NEGATIVO o MENOR DEL 25% o ENTRE EL 25-75% vs. MAYOR DEL 75%
XV. CYCLIN D1: NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XVI. E-CADHERINA NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XVII. EGFR NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XVIII. EMA NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XIX. HES1 NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XX. HTERT NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%

ANEXOS 213
XXI. KI67 NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXII. MLH1 NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXIII. MSH2 NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXIV. MSH6 NEGATIVO POSITIVO DEBILPOSITIVO INTENSO POSITIVO MUY INTENSO
XXV. MSI (MLH1 o MSH2 o MSH6 desaparecen) INESTABLE vs. ESTABLE
XXVI. NANOG NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XXVII. NCADHERINA NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXVIII. NEUROTENSINA NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL
75%
XXIX. NM23 NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXX. NTSR1 NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XXXI. OSTEONECTINA NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXXII. OSTEOPONTINA NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXXIII. P16 NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XXXIV. P21 NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XXXV. P27 NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XXXVI. P53 NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO
XXXVII. PTEN citoplasma NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL
75%
XXXVIII. PTEN nucleo NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XXXIX. RBP NEGATIVO POSITIVO DEBIL POSITIVO INTENSO POSITIVO MUY INTENSO

214 ANEXOS
XL. SMADY NEGATIVO MENOR DEL 25% ENTRE EL 25-75% MAYOR DEL 75%
XLI. VEGRF NEGATIVO POSITIVO DÉBIL POSITIVO INTENSO