explanation for excessive dna single-strand breaks and endogenous repair foci in pluripotent mouse...
TRANSCRIPT

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r. com/ loca te /yexc r
Research Article
Explanation for excessive DNA single-strand breaks andendogenous repair foci in pluripotent mouse embryonicstem cells
J.P. Banáth, C.A. Bañuelos, D. Klokov1, S.M. MacPhail, P.M. Lansdorp2, P.L. Olive⁎
British Columbia Cancer Research Centre, Vancouver, B.C., Canada, V5Z 1L3
A R T I C L E I N F O R M A T I O N
⁎ Corresponding author. Medical Biophysics DeCanada. Fax: +1 604 6758049.
E-mail address: [email protected] (P.L. Olive).1 Current address: Radiation Biology and Heal2 Terry Fox Laboratory, BCCRC.
0014-4827/$ – see front matter © 2008 Elseviedoi:10.1016/j.yexcr.2008.12.007
A B S T R A C T
Article Chronology:
Received 28 October 2008Revised version received
4 December 2008Accepted 5 December 2008Available online 24 December 2008
Pluripotent mouse embryonic stem cells (mES cells) exhibit ∼100 large γH2AX repair foci in theabsence of measurable numbers of DNA double-strand breaks. Many of these cells also showexcessive numbers of DNA single-strand breaks (N10,000 per cell) when analyzed using the
alkaline comet assay. To understand the reasons for these unexpected observations, variousmethods for detecting DNA strand breaks were applied to wild-type mES cells and to mES cellslacking H2AX, ATM, or DNA-PKcs. H2AX phosphorylation and expression of other repair complexeswere measured using flow and image analysis of antibody-stained cells. Results indicate that highnumbers of endogenous γH2AX foci and single-strand breaks in pluripotent mES cells do notrequire ATM or DNA-PK kinase activity and appear to be associated with global chromatindecondensation rather than pre-existing DNA damage. This will limit applications of γH2AX focianalysis in mES cells to relatively high levels of initial or residual DNA damage. Excessive numbersof single-strand breaks in the alkaline comet assay can be explained by the vulnerability ofreplicating chromatin in mES cells to osmotic shock. This suggests that caution is needed ininterpreting results with the alkaline comet assay when applied to certain cell types or after
treatment with agents that make chromatin vulnerable to osmotic changes. Differentiation of mEScells caused a reduction in histone acetylation, γH2AX foci intensity, and DNA single-strandbreakage, providing a link between chromatin structural organization, excessive γH2AX foci, andsensitivity of replicating mES cell chromatin to osmotic shock.
© 2008 Elsevier Inc. All rights reserved.
Keywords:
Mouse embryonic stem cellsDNA repair fociGamma-H2AXDNA single-strand breaksComet assay
Introduction
Pluripotent mouse embryonic stem cells (mES cells) are char-acterized by a unique higher order chromatin architecture.Features include an open heterochromatic structure, nuclear
partment, British Columbia
th Physics, Chalk River Lab
r Inc. All rights reserved.
“plasticity”, and an abundance of chromatin-remodeling factors[1,2]. Mobility of chromatin in pluripotent ES cells is thought to bemuch greater than in differentiated cells [3], and post-translationalhistone modifications play a critical role in maintaining pluripo-tency and poising mES cells for differentiation [4]. In spite of the
Cancer Research Centre, 675 W. 10th Avenue Vancouver, B.C., V5Z 1L3
oratories, AELC, Chalk River, ON, K0J 1PO, Canada.

1506 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
unique organization of chromatin in pluripotent mES cells, thegenome of these cells is highly stable due to an enhanced ability toaccurately repair DNA damage or to undergo apoptosis whendamage is excessive [5–7].
Replicating mammalian cells typically exhibit fewer than 2000endogenous DNA single-strand breaks when analyzed using analkaline gel electrophoresis method called the comet assay [8]. Itwas therefore surprising to observe as many as 40,000 single-strand breaks (ssb) per cell in untreated mES cells when analyzedusing this method. Because these breaks are associated with cellsin S phase, we initially considered the idea that theymay representa large number or an unusual clustering of replication origins thatare known to behave as ssb in the alkaline comet assay [8].However, it became apparent during the course of these experi-ments that other methods used to detect DNA single-strand breaks(but based on different physical principles) failed to revealexcessive DNA damage. We then examined several potentialreasons for the unexpected formation of breaks in mES cellswhen analyzed using the alkaline comet assay.
The unusual chromatin organization in mES cells may alsoexplain a second unexpected observation. Histone H2AX becomesphosphorylated in response to the presence of DNA double-strand breaks resulting in microscopically visible γH2AX foci.However, pluripotent mES cells can contain 100 or more largeendogenous γH2AX foci, equivalent to the number of foci that areproduced after exposure to about 4 Gy X-rays [9]. In comparison,most other untreated mammalian cells exhibit fewer than 5 largeγH2AX foci per cell [10,11]. We have recently examined double-strand break induction and rejoining in mES cells, and there wasno evidence to suggest the presence of excessive DNA double-strand breaks that could explain these foci [12]. However,endogenous foci can also indicate the presence of unrepaired ormisrepaired DNA double-strand breaks, telomere dysfunction,replication stress, genomic instability, or senescence [10,11,13]. Inaddition, hundreds of much smaller γH2AX foci have also beenobserved in differentiated cell lines, although these small fociappear to be unrelated to the DNA damage response [14,15].Importantly, the size of γH2AX foci is related to chromatinstructure; it is substantially reduced in condensed heterochro-matic regions [16,17] and increased when chromatin is hyper-acetylated [18] or deficient in histone H1 [19]. Since chromatin inpluripotent mES cells exhibits global histone hyperacetylationand is organized as euchromatin, it is possible that these cellssimply contain larger versions of the small endogenous γH2AXfoci observed in differentiated cells. In other words, endogenousfoci in mES cells are unrelated to the DNA damage response.However, excessive γH2AX phosphorylation has also beenreported in precancerous lesions [20,21], cells undergoingmeiosis [22], and proliferating germ cap cells in mouse hairfollicles [23]. Since mES cells proliferate rapidly and lack a G1checkpoint [24], perhaps replication stress in these cells leads toexcessive γH2AX foci formation.
The anomalous expression of γH2AX foci and the expla-nation for the appearance of ssb in the comet assay were exa-mined in pluripotent wild-type mES cells as well as mEScells deficient in H2AX and the kinases that phosphorylateH2AX. The effects of inhibiting replicon initiation and promotingdifferentiation were also examined. To gain insight into thenature of the DNA damage, several methods for detecting DNAssb were applied.
Materials and methods
Cell lines and X-irradiation
Two cultures of undifferentiated murine embryonic stem cellswere used. The R1 wild-type mES cell line derived from 129/Sv EScellswas obtained fromDr. A. Nagyat the University of Toronto [25]and was used between passages 8 and 16. Confirmatory experi-ments were performed using a second wild-type mES cell line,C1+/+ of the same genetic background [26]. H2AX−/−, ATM−/−
and DNA-PKcs−/− mES were generated in 129/Sv/Ev ES cells [27]
and were kindly provided by Dr. Fred Alt, Harvard [28,29]. Mouseembryonic stem cellsweremaintained as previously described [30]in Dulbecco's Modified Eagle's medium containing 15% fetal calfserum (StemCell Technologies) supplemented with 10 mM non-essential amino acids, 200 mM L-glutamine, 100 mM Na-pyruvate,10 ng/ml leukemia inhibitor factor and 100 μM monothioglycerol(StemCell Technologies) [30]. All cells were grown at 37 °C underambient air and 5% CO2. Mouse ES cells were maintained inexponential growth in the absence of feeders by subcultivation 3times per week on gelatin coated dishes seeded at 5×104 cells/60 mm dish. The differentiation status of the mES cells wasconfirmed using intact cells stained with antibodies against SSEA1,a cell surface marker of the undifferentiated mES phenotype [31]and by immunoblotting of nuclear proteins using antibodiesto lamin A/C that are not expressed in undifferentiated stem cells[32]. To promote differentiation of mES cells, LIF was omitted fromthe medium and cells were grown either as monolayers in theabsence of gelatin-coating or in spinner culture vessels for 2 weeks.
For comparison with mES cells, we used mouse embryofibroblasts (MEFs) purchased from StemCell Technologies andobtained from day 12 mouse embryos from CD1 mice; MEFs werecultured in the same medium as mES cells but lacking LIF. Toemploy cell lines with similar cell cycle time as mES cells, we usedhamster V79 fibroblasts and mouse L5178Y-R lymphoblastoid cells(designated LYR cells). V79 cells were originally obtained fromDr. Warren Sinclair and maintained in MEM with 10% fetal bovineserum. Mouse LYR cells were obtained from ATCC and grown inMEM with 10% heat-inactivated bovine calf serum. Differentiatedcell lines were maintained in exponential growth by twice weeklysub-cultivation in the appropriate medium.
For ionizing radiation exposures, cells were exposed on ice to300 kV X-rays at a dose rate of 5.1 Gy/min. Cells were thenincubated at 37 °C for up to 24 h after radiation to allow time forrepair. After trypsin digestion (0.1% in PBS) to produce a single cellsuspension, cells were centrifuged and fixed in 70% ethanol forsubsequent antibody staining and flow cytometry analysis.
Flow cytometry analysis for γH2AX
Antibody staining was performed as previously described usingmouse monoclonal anti-phosphoserine-139 H2AX anti-body (Upstate Biotechnology, 1:500 dilution or Abcam 2F3,1:5000 dilution) [9]. After secondary antibody labeling withAlexa 488-labeled goat anti-mouse antibody, cells were rinsedand resuspended in 1 μg/ml ml 4′,6-diamidino-2-phenylindoledihydrochloride hydrate (DAPI; Sigma) to stain DNA. Sampleswere analyzed using a Coulter Elite flow cytometer using UVand 488 nm laser excitation. In order to account for differences in

1507E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
cell cycle distribution that affect average γH2AX intensitymeasurements, γH2AX expression was first corrected for DNAcontent of each cell and results were expressed as a ratio of thesignal intensity for treated versus untreated cells.
Immunoblotting
Whole cell extracts were used for H2AX, γH2AX, and acetylatedhistone H3 and H4. Nuclear extracts were used tomeasure PP2A andPP1α, and were prepared as previously described [12]. Cells weretrypsinized, rinsed and lysed in lysis buffer (150 mM NaCl, 50 mMTrisHCl, 0.5%NP-40,withNaVa,NaF, aprotinin, pepstatin, andPMSF).Proteins were separated using SDS-PAGE with 15% polyacrylamidegels. All antibodies were used at given dilutions in 5% non-fat drymilk in phosphate buffer or Tris-buffered saline containing 0.1%Tween-20. After transferring proteins, overnight incubation wasconducted at 4 °C with primary anti-histone antibodies or 2 h atroom temperature for other proteins. The following primaryantibodies and dilutions were used: anti-acetylated histone H3rabbit polyclonal, (Upstate, #06-599, 0.1 μg/ml), anti-acetylatedhistone H4 (lys12) rabbit polyclonal (Upstate, #06-761-MN, 1 μg/ml), anti-α-tubulin mouse monoclonal antibody (Calbiochem,#CP06 1:4000 dilution), anti-acetylated tubulin mouse monoclonalantibody (Sigma, Clone 6-11B-1, 1:1000 dilution), anti-PP2A mousemonoclonal antibody (Upstate, #05-421, 1:1000 dilution), and anti-PP1α rabbit polyclonal antibody (Upstate, #07-273, 1:500 dilution).This was followed by rinsing and 1 h incubation with secondaryantibody, typically horseradish peroxidase conjugated goat anti-mouse or anti-rabbit IgG, antibody (Santa Cruz, 1:2000 dilution atroom temperature). To visualize proteins, a luminescence detectionreagent was prepared immediately before use and was applied forabout 4 min. Membranes were imaged using a chemiluminescenceimager (Alpha Innotech Corp, San Leandro, CA).
Immunohistochemistry
Mouse ES cells were grown attached to gelatin-coated coverslips.Cells on coverslips were fixed for 30 min in 2% paraformaldehydeand processed as previously described [12]. Antibodies used were:mouse monoclonal anti-RPA antibody (Oncogene, AB-3, 1:100dilution), rabbit polyclonal anti-53PB1 antibody (Novus NB100-2279, 1:200 dilution), rabbit polyclonal anti-RAD51 antibody(Calbiochem, PC130, 1:500 dilution), mouse monoclonal anti-γH2AX antibody (Upstate, 1:200 or AbCam, 1:1000 dilution), ratmonoclonal anti-BrdU antibody (Becton-Dickinson, BU1/75, 1:150dilution), and mouse monoclonal anti-SSEA1 antibody (ChemiconInternational, 1:100 dilution). Slides were viewed using a Zeissepifluorescence microscope using a 100× Neofluor objective and aQ-Imaging 1350 EX digital camera. For counting foci numbers pernucleus, foci images were obtained using Northern Eclipsedeconvolution software [9]. Approximately 50–100 cells wereanalyzed for average number of γH2AX or RAD51 foci per cell. Forcomparisons between cell lines, a constant exposure timewas usedfor image acquisition for each antibody.
Cell synchrony
Isoleucine deprivation and cycloheximide treatment were used toinhibit replicon initiation.Mouse ES cells were incubated for 24 h inisoleucine free minimal essential medium (Gibco) supplemented
with stem cell factors and 10% dialyzed FBS. After 24 h, completeregular growth medium with LIF was added to some of the dishesfor further incubation at 37 °C. For treatment with cycloheximide,mES cells were incubated for 2 h in complete medium containing10–100 μM cycloheximide (Sigma). Cells were then rinsed free ofdrug and incubated in fresh medium for up to 24 h.
Counterflow centrifugal elutriation separates cells largely onthe basis of cell size [33] and has been used to enrich for cells invarious phases of the cell cycle. Mouse ES cells were separated byelutriation as previously described [12]. Separated cells wereanalyzed for DNA ssb using the alkaline comet assay.
The fraction of cells in S phase was determined using ModFitsoftware (Verity Software House). Analysis was applied to DNAhistograms from DAPI-stained cells that were analyzed by flowcytometry using a Coulter Elite cell sorter.
Alkaline and neutral comet assays
The alkaline comet assay was performed as previously described[34] using a 1 h lysis method. Agarose embedded cells onmicroscope slides were incubated for 1 h in 1.2 M NaCl, 0.03 MNaOHwith 0.1% sarkosyl. After rinsing for 1 h to remove salt, slidesunderwent electrophoresis for 25 min at 0.6 V/cm in a solutioncontaining 0.03 M NaOH and 2 mM Na2EDTA. DNA was stainedwith 2.5 μg/ml propidium iodide. Modifications to the lysismethod were also examined, including omission of sarkosyl andinclusion of 2% DMSO or 0.5 mg/ml proteinase K. Analysis ofendogenous DNA double-strand breaks was conducted using theneutral version of the comet assay as previously described [12].
Alkali-unwinding assay
The alkali unwinding method was employed to measure therelative number of endogenous DNA ssb as well as the rate ofelongation of nascent single-stranded DNA strands into double-stranded DNA [35]. Cells radiolabeled for 24 h with 0.1 μCi/ml 14C-thymidine were incubated for 15 minwith 1 μCi/ml 3H-thymidine.At various times after uptake of 3H-thymidine, cells were lysed in0.03 M NaOH, 1 M NaCl for 1 h, then neutralized, sonicated, andanalyzed for the percentage of DNA in double-stranded form usinghydroxyapatite chromatography as previously described [36].The percentage of DNA in double-stranded form was determinedfor 3–6 replicates.
Alkaline DNA precipitation assay
The alkaline DNA precipitation assay [37] was also used tomeasureendogenous and radiation-induced DNA ssb inwild-type mES cellsand LYR cells. Cells were incubated for 24–48 h in completemedium containing 0.1 μCi/ml 14C-thymidine, allowed 2 h in freshmedium, then lysed in 0.5% SDS, 10 mM EDTA and 10 mM Tris towhich 0.05 M NaOH was added just before use (pH 12.4). Then0.12 M KCl was gently added and the tubes were incubated at 65 °Cfor 10 min. After cooling on ice for 5 min, cells were centrifugedand the supernatant containing small DNA fragments wasdecanted into a liquid scintillation vial. The pellet containingprotein-DNA aggregates was resuspended and decanted into asecond vial. Samples were counted on a liquid scintillation counterand the percentage of DNA in the pellet was determined. Resultsfrom 3 samples per radiation dose are reported.

1508 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
Halo assay
A modified version of the fluorescent halo assay [38] wasemployed to examine the association of DNA with the nuclearprotein matrix and to measure the ability of radiation-inducedDNA breaks to cause relaxation of DNA supercoils. Single cellsuspensions (mES and LYR) were lysed with an equal volume of2MNaCl, 20mMTris pH 7.8, 0.01% Triton X-100, 20mMEDTA, 0.1%DMSO and 1 mM PMFS. The fluorescent DNA stain propidiumiodide (PI) was included at final concentrations of 5 to 30 μg/ml.Cells were lysed at room temperature for 10 min and halos wereviewed within 15 min using an inverted Zeiss fluorescencemicroscope. Digitized images were analyzed with Image J softwareto calculate the diameter of the resulting halos. Results wereaveraged for at least 20 cells per sample.
Alkaline halo-gel assay
To simulate lysis conditions of the comet assay while avoidingusing electrophoresis or creating osmotic shock, we applied thealkaline halo-gel assay of Sestili and Cantoni [39]. Cells embeddedin agarose were lysed in 0.03 M NaOH, 1.2 M NaCl, 0.1% sarkosylfor 2 h or cells were lysed in this solution for 1 h followed by a 1–2 h rinse in 0.03 M NaOH and 2 mM Na2EDTA. Slides in their lysisor rinse solution were stained with 2.5 μg/ml propidium iodidebefore viewing using an inverted fluorescence microscope.Digitized images were examined for halo diameter using ImageJ software, and 18–25 nuclei were scored for each sample for 3independent experiments.
Results
Excessive endogenous γH2AX foci are associated with highlevels of histone acetylation in mES cells
Phosphorylation of histone H2AX is associated with the DNAdamage response after exposure of cells to genotoxic agents [40]. Itwas therefore surprising to observe excessive γH2AX foci inuntreated mES cells. No γH2AX antibody staining was observed inH2AX null mES cells, confirming the lack of non-specific staining(Figs. 1a, f). The number of microscopically visible endogenous fociwas high in untreated mES cells (Figs. 1b, e) but negligible in MEFs(Fig. 1c). However, radiation-induced γH2AX foci were easilydetected inMEFs 1 h after exposure to 1 Gy (Fig.1d).Mouse ES cellsshowed on average 100 large γH2AX foci per nucleus compared tofewer than 5 in V79 cells (Fig. 1e). Exposure to 4 Gy X-rays isrequired to produce about 100 foci per cell and is also consistentwith an average γH2AX signal that is 3–4 times background whenmeasured by flow cytometry [9]. Flow cytometry results confirmeda three-fold higher average γH2AX expression in wild-type mEScells relative to MEFs (Fig. 1f). As a result of the higher endogenousexpression of γH2AX, saturation of the radiation-induced γH2AXsignal occurred at a much lower radiation dose for mES cells(10 Gy) than V79 cells (30 Gy) (Fig. 1g).
An increase in histone acetylation has been associated withenlargement of γH2AX foci [18]. The level of acetylated histonesH3 and H4 was significantly higher in pluripotent mES cells (Figs.1h, i) although acetylated tubulin was lower in mES cell linescompared to MEFs. Increased histone acetylation was observed for
wild-typemES cells as well as mES cells lacking H2AX or DNA-PKcs.Results are consistent with the fact that chromatin is moredecondensed in pluripotent mES cells, and this may be responsiblefor the larger size of endogenous γH2AX foci.
Both DNA-PKcs−/− and ATM−/− mES cells showed similar
numbers of γH2AX foci as wild-type cells (Figs. 2a–c), indicatingthat neither of these DNA damage related kinases is required forendogenous H2AX phosphorylation in mES cells. The remainingkinase implicated in H2AX phosphorylation, ATR, is associatedwith phosphorylation that occurs in association with damagedreplication forks [41]. In an effort to reduce endogenous γH2AXexpression, mES cells were treated with the PI-3 kinase inhibitors,caffeine and wortmannin. We previously reported that mES cellstreated for 1 h with 15 μM wortmannin showed a modestreduction in endogenous γH2AX from 1 to 0.75±0.12 [12].However, treatment for 4 h with 5–20 mM caffeine increasedrather than decreased the expression of endogenous γH2AX by afactor of 1.7–2.2 (Figs. 2d–g). A small population with excessiveγH2AX staining was also observed, and this population increasedfrom 2% to 10% as caffeine concentration was raised from 5 to20 mM. Since excessive γH2AX staining is associated withapoptosis [42], it is likely that caffeine triggered apoptosis whichhas been reported to occur when ATR is inhibited in mES cells [43].Moreover, although caffeine typically inhibits ATR kinase, stimula-tion has also been reported [44].
High γH2AX is not explained by high H2AX, low phosphataseactivity, or excessive double-strand breaks
It is possible that the increased expression of γH2AX inmES cells isa result of an increase in expression of H2AX itself. However,immunoblotting revealed no significant differences between H2AXexpression in wild-type mES cells, DNA-PKcs
−/− mES cells, anddifferentiatedMEFs (Figs. 2h, i). Note that the increased expressionof γH2AX in mES cells was also detected using immunoblottingafter cell lysis (Fig. 2h); this indicates that the increased size ofγH2AX foci is not a result of better access of the antibody to thedecondensed DNA in pluripotent mES cells.
Low expression of phosphatases that remove γH2AX mightalso explain higher than normal expression of γH2AX. However,levels of PP2A and PP1α, phosphatases, known to de-phosphor-ylate γH2AX [45,46], were similar for mES and differentiatedmouse cells (Suppl. Figs. 1a,b). More importantly, the rate ofloss of γH2AX after irradiation was rapid and consistent withother rodent cell lines indicating that phosphatase activity wasnot compromised (Suppl. Fig. 1c). The average rate of loss ofradiation-induced γH2AX for 13 rodent cell lines measuredbetween 1 and 4 h after X-irradiation was 3±1.0 h (mean±SD,n=13). This was not significantly different from the loss half-time of 2.7±0.4 h measured for irradiated wild-type mES cells(Suppl. Fig. 1d).
Expression of γH2AX is also associated with DNA fragmenta-tion that results during apoptosis [42,47]. Although radiationeventually caused the appearance of cells with sub-G1 content(Suppl. Fig. 1e), fewer than 5% of mES cells were positive foractivated caspase 3 when measured 4 h after exposure to 20 Gy(Suppl. Fig. 2d). Moreover, there was no evidence that double-strand breaks in untreated mES cells were any higher than thoseobserved in MEFs when measured using the neutral comet assay(Suppl. Figs. 2a,b).

Fig. 1 – Endogenous expression of γH2AX foci in pluripotent mES cells is excessive. Panel a: confirmation of lack of γH2AX foci inH2AX−/−mES cells. Panel b: deconvolution image showing large numbers of endogenous γH2AX foci present inwild-typemES cells.Panel c: low endogenous expression of γH2AX in mouse embryo fibroblasts (MEFs). Panel d: radiation-induced foci are easilydetected in MEF 1 h after 1 Gy. Panel e: the number of large γH2AX foci per cell is shown for untreated wild-type mES cells (filledbars) in comparison to hamster V79 cells (hatched bars). Panel f: flow cytometry confirms higher expression of γH2AX in mES cellsrelative to differentiated MEF and V79 cells (Mean±SD for 3–4 measurements). Panel g: expression of radiation-induced γH2AXrelative to background γH2AX shows saturation at a lower radiation dose for mES cells than V79 cells. Panel h: immunoblot ofacetylated histones H3 and H4, acetylated tubulin and α-tubulin in mES wild-type cells, H2AX−/− mES cells, DNA-PKcs−/− mES cellsand MEFs. Panel i: quantitation of acetylation of histone H4 in various mES cell lines relative to MEF (mean and SD, n=4).
1509E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0

Fig. 2 – Endogenous H2AX phosphorylation does not require ATM or DNA-PK kinase activity. Panels a–c: appearance of endogenousγH2AX foci inwild-type, ATM−/−, or DNA-PKcs−/−mES cells. Panels d–g: effect of a 4 h treatment of wild-typemES cells with 0, 5, 10or 20 mM caffeine on expression of endogenous γH2AX. Arrows point to possible apoptotic populations expressing 10 times moreγH2AX. Panel h: immunoblot of H2AX and γH2AX in extracts from mES cell lines compared to MEFs and V79 cells. Panel i:quantitation of H2AX in 3–4 blots showing mean and SD for mES wild-type and null cells relative to MEFs.
1510 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
Endogenous RPA and 53BP1 foci appear normal in mES cells;RAD51 is high
Many proteins involved in the DNAdamage response can aggregateas microscopically visible foci at sites of DNA double-strand breaks.Mouse ES cells were examined for three of these proteins bothbefore and after exposure to X-rays. Although RPA associates withsingle-stranded DNA, aggregations of thousands of RPA moleculesinto foci is associated with sites of double-strand breaks [48]. In allcases, the number of DNA repair foci increased after exposure to X-rays (Suppl. Figs. 3a–h). However, the large size of endogenous
γH2AX foci (Suppl. Fig. 3a) preventedmicroscopic identification ofradiation-induced foci in mES cells (Suppl. Fig. 3b). Endogenousexpression of 53BP1was low (Suppl. Fig. 3c), and RPA foci were notvisible before irradiation (Suppl. Fig. 3e). However, higher thannormal numbers of endogenous RAD51 foci were observed inwild-type mES cells (Suppl. Fig. 3g) with an average of 13.4±1.7(mean±SEM, n=50) RAD51 foci per nucleus. H2AX−/− mESnuclei showed fewer endogenous RAD51 foci than wild-type mEScells (Suppl. Figs. 3i,j) with an average of 4.5±0.5 RAD51 foci pernucleus. This was significantly higher than the value obtained forMEFs of 1.6±0.5 RAD51 foci per nucleus. Only occasional co-

1511E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
localization was observed between γH2AX foci and RAD51 foci inmES cells (Suppl. Fig. 3k). Endogenous γH2AX foci and RAD51 fociwere present in cells lacking BrdU foci, and there was no obviousco-localization between BrdU foci (green) with either RAD51 foci(Suppl. Fig. 3l) orγH2AX foci (Fig. 3m) (red). Results of foci analysisare summarized in Table 1.
The alkaline comet assay reveals excessive DNA ssb inmES cells
Although the numbers of double-strand breaks in mES cells wereconsistent with those observed in other cell lines (Suppl. Fig. 2b),excessive numbers of DNA ssbwere observed inmES cells analyzedusing the alkaline comet assay. Tail moment, a measure of thenumber of ssb per cell, averaged 1–2 in differentiated mouse LYRlymphoblasts, (Figs. 3b,h) and 15–20 for wild-type mES cells (Figs.3a,g). The large range in tail moments in mES cells included valuesfrom 1 to 48, the maximum tail moment observed with our comet
Fig. 3 – Mouse ES cells exhibit excessive numbers of ssb using theDNA in the comet tail and distance of DNAmigration, is proportionashow awide range in ssb. Panels b–f: mouse LYR lymphoblasts showto dose of X-rays. Panel g–h: relation between tail moment and DNshows a tail moment of 5, typical of the maximum response observ
software. When compared to initial DNA ssb produced by X-rays inLYR cells (Figs. 3c–f), mouse ES cells exhibited on average about8 Gy or 8000 DNA ssb, but the range extended tomore than 20,000ssb in some cells. Similar results were obtained for all undiffer-entiated mES cell lines examined, including mES cells lackingH2AX, ATM or DNA-PKcs (data not shown).
Excessive DNA strand breakage is associated withreplicating DNA
Replication forks present in S phase cells are known to behave asssb in the alkaline comet assay [8]. This is responsible for thetypical pattern of DNA damage in relation to DNA content shown inFig. 3h for untreated LYR cells. Damage in mES cells was alsohighest in cells with S phase DNA content (Fig. 3g). The associationof DNA damage with DNA content is less apparent for mES cellssince excessive DNA damage also reduces the efficiency of DNAbinding by propidium iodide [49]. Consistent with these results,
alkaline comet assay. Tail moment, a measure of the amount ofl to the number of ssb. Panel a: untreated pluripotent mES cellsa response typical for differentiated cells with ssb proportionalA content for wild-type mES cells and LYR cells. The dotted lineed in differentiated S phase cells.

Table 1 – Summary of endogenous repair foci in mES cells
Cell Line EndogenousγH2AX Foci
RAD51foci
RPAfoci
53BP1foci
Wild-type mES cells +++ (+++) ++ (++) − (+) − (++)H2AX−/− mES cells − + (++) − (+) − (++)DNA-PKcs
−/− mES cells +++ (+++) + (++) − (+) − (++)MEFs + (++) − (++) − (+) − (++)Wild-type,differentiatedmES cells
+(+) (++) − (++) − (+) − (++)
−, fewer than 3 foci/nucleus.+ 3–10 foci/nucleus.++, 11–25 foci/nucleus.+++, N25 foci/nucleus.Symbols in brackets indicate the response to 1 Gy measured 1 h afterirradiation.
1512 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
when centrifugal elutriation was used to enrich for cells indifferent phases of the cell cycle, the comet tail moment averaged22±11 for enriched S phase cells compared to 10.6±2.3 for G2phase cells (data not shown).
Growth of wild-type mES cells in isoleucine-free medium for24 h reduced cell growth rate and caused a significant reduction intail moment. This reduction was reversible upon return of mEScells to completemedium (Figs. 4a–d,i). Isoleucine deprivation hadno effect on endogenous ssb in MEFs. A shorter 2 h treatment withcycloheximide, which is also known to inhibit replicon initiation[50], caused a similar decrease in ssb in S phase cells (Figs. 4e–h, j).Tail moment continued to decrease for 2 h after cycloheximideremoval and was partially reversible only after exposure to thelowest drug dose. For both isoleucine and cycloheximide-treatedcells, the decrease in tail moment occurred in cells with S phaseDNA content, and could not be explained by redistribution into G1or G2 phases. Therefore the presence of ssb in mES cells isassociated with actively replicating DNA.
No evidence for differences in mES replicon size or DNAelongation rate in mES cells
Subnuclear replication factories can be visualized by staining cellsfor proliferating cell nuclear antigen (PCNA). If organization ofreplicons in mES cells differs from that in differentiated cells, onemight expect to observe differences in the patterns of PCNAstaining. However, the pattern of PCNA staining in S phase mEScells was similar to that of differentiated V79 cells (Suppl. Fig. 4).There was no apparent increase in the number of PCNA foci in mEScells that might account for the unusual response of these cells inthe alkaline comet assay. This result is consistent with results byPanning and Gilbert who examined BrdUrd staining duringdifferentiation of mES cells and found no change in pattern in Sphase cells relative to other cell lines [51].
The fluorescent halo method [38,52] measures the frequencyand strength of DNA attachment sites to the nuclear proteinmatrix, and nuclear matrix attachment sites have been associatedwith sites of DNA replication [52]. When cells are lysed in highsalt and mild detergent, the resulting supercoiled DNA loopsappear as a halo surrounding the nuclear matrix core. The widthof the halo can be related to the number of matrix associatedreplication sites with a smaller loop size associated with morefrequent replication sites [53]. To determine if DNA loop sizes
might be smaller for mES cells, maximum halo diameter wasmeasured in cells exposed to increasing concentrations of theDNA intercalating dye propidium iodide. Loop sizes in mES cellswere compared to those of mouse LYR lymphoblast cells thathave a similar cell cycle time and similar S phase fraction as mEScells. These cells were also chosen for comparison because theyshow the same sensitivity to detergent lysis as mES cells. Therewas no difference in maximum loop size for undifferentiated mEScells compared to differentiated LYR cells suggesting that bothcell lines show similar numbers of matrix attachment sites andsimilar numbers of replicons (Suppl. Fig. 5a).
It is also possible that the rate of elongation of newly initiatedreplicons might be faster for mES cells. The alkali unwindingmethod of Ahnstrom and Rydberg [35,54] allows measurement ofDNA replication initiation and elongation rates. However, the rateof movement of newly incorporated 3H-thymidine into double-stranded DNA was the same for undifferentiated mES cells andmouse LYR cells, indicating similar rates of DNA chain elongation(Suppl. Fig. 5b)
Other methods that measure ssb do not detect excessive DNAbreakage in mES cells
Results using the halo assay did not indicate excessive numbers ofendogenous ssb in mES cells. Since the halo method detects loss ofDNA supercoiling for cells exposed to high salt lysis under neutralpH conditions, we questioned whether the breaks we observedusing the alkaline comet assay might be caused by the presence ofalkali-labile sites in mES cells. However, this was not the case sincethe alkali unwinding assay uses the same lysis solution as thecomet assay but also failed to detect the presence of excessive ssb(Suppl. Fig. 5c). Similarly, the alkaline DNA precipitation assay alsomeasures ssb, but in the absence of high salt, did not detectexcessive breaks in mES cells relative to LYR cells (Suppl. Fig. 5d).Therefore, of the 4 methods that are capable of detecting ssb withsimilar sensitivities, only the alkaline comet assay showedexcessive ssb in untreated mES cells.
The alkaline comet assay was then modified to: 1) excludedetergent or include proteinase K in the lysis buffer, 2) reduce freeradicals by adding DMSO to the lysis buffer, 3) alter the strengthand duration of alkali during lysis, or 4) reduce voltage duringelectrophoresis. None of these changes decreased the fraction ofcells with excessive ssb (Table 2). However, the comet assay differsfrom the other ssb detection methods in twoways. First, it requiresexposure of lysed cells to electrophoresis which may be respon-sible for DNA breakage. Second, the high salt concentration in thelysis buffer is removed by rinsing before the cells undergoelectrophoresis. This rinse step is necessary to avoid salt gradientsduring electrophoresis since they will affect the rate of DNAmigration. However, rapid removal of NaCl will also create anosmotic shock.
The alkaline halo-gel method detects ssb in mES cells onlyafter osmotic shock
To examine the role of electrophoresis and osmotic shock, amodification of the alkaline halo-gel method of Sestili and Cantoni[39] was employedwhich does not require electrophoresis and canbe performed with or without a rinse step after alkali lysis. Cellswere embedded in agarose and lysed in a high salt and alkaline
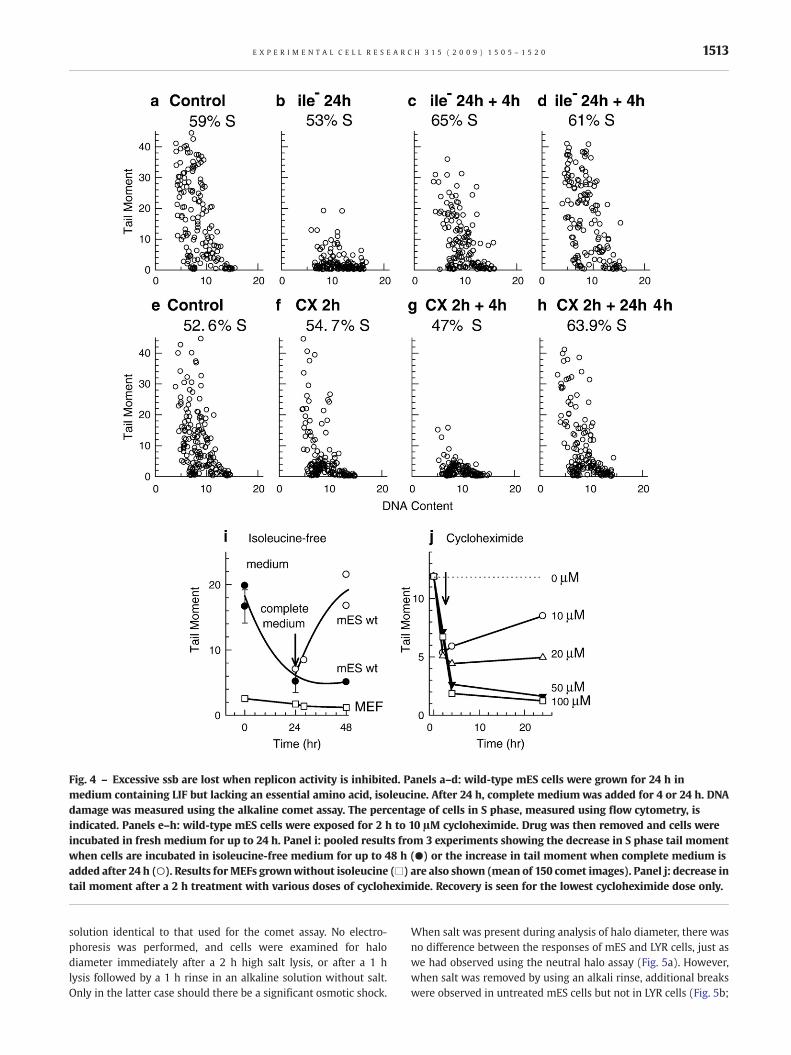
Fig. 4 – Excessive ssb are lost when replicon activity is inhibited. Panels a–d: wild-type mES cells were grown for 24 h inmedium containing LIF but lacking an essential amino acid, isoleucine. After 24 h, complete medium was added for 4 or 24 h. DNAdamage was measured using the alkaline comet assay. The percentage of cells in S phase, measured using flow cytometry, isindicated. Panels e–h: wild-type mES cells were exposed for 2 h to 10 μM cycloheximide. Drug was then removed and cells wereincubated in fresh medium for up to 24 h. Panel i: pooled results from 3 experiments showing the decrease in S phase tail momentwhen cells are incubated in isoleucine-free medium for up to 48 h (●) or the increase in tail moment when complete medium isadded after 24 h (○). Results forMEFs grownwithout isoleucine (□) are also shown (mean of 150 comet images). Panel j: decrease intail moment after a 2 h treatment with various doses of cycloheximide. Recovery is seen for the lowest cycloheximide dose only.
1513E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
solution identical to that used for the comet assay. No electro-phoresis was performed, and cells were examined for halodiameter immediately after a 2 h high salt lysis, or after a 1 hlysis followed by a 1 h rinse in an alkaline solution without salt.Only in the latter case should there be a significant osmotic shock.
When salt was present during analysis of halo diameter, there wasno difference between the responses of mES and LYR cells, just aswe had observed using the neutral halo assay (Fig. 5a). However,when salt was removed by using an alkali rinse, additional breakswere observed in untreated mES cells but not in LYR cells (Fig. 5b;

Table 2 – Summary of results using different methods for detecting DNA ssb in mES cells
Method High salt lysis Alkaline lysis High salt presentduring entire method
Excessive ssb
Alkaline comet assay• 0.03 M NaOH Yes Yes No Yes• 0.28 M NaOH for 16 h Yes Yes No Yes• Minus detergent Yes Yes No Yes• Plus proteinase K Yes Yes No Yes• Low voltage electrophoresis Yes Yes No YesAlkaline unwinding assay Yes Yes Yes NoHalo assay Yes No Yes NoAlkaline DNA precipitation assay No Yes – NoAlkaline halo-gel assay, no rinse Yes Yes Yes NoAlkaline halo-gel assay, with rinse Yes Yes No Yes
1514 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
Table 2), similar to results we obtained using the alkaline cometassay. The average halo width of mES cells was significantly higherthan the average halo width of LYR cells, but only for mES cells thatreceived the rinse (Figs. 5c, d). Representative halo-gel images areshown for the samples that underwent a rinse before analysis(Figs. 5e–h). Heterogeneity was observed only for untreated mEScells after the removal of salt (Fig. 5g). Since the alkaline halo-gelmethod with the rinse but no electrophoresis gives similar resultsas the alkaline comet assay, electrophoresis does not appear to beresponsible for the appearance of ssb using the comet assay. Theseresults provide direct support for the hypothesis that replicatingmES cell DNA is unusually sensitive to ssb caused by osmotic shock.
Modifying chromatin structure through differentiationof mES cells causes decreases in γH2AX, ssb, and histoneacetylation
Chromatin compaction occurs upon differentiation of pluripotentmES cells [19,55]. Differentiation was therefore used to examinethe importance of chromatin structure in excessive H2AXphosphorylation and sensitivity of replicating DNA to osmoticshock. Differentiation was accomplished by growth for 2 weeks inthe absence of LIF, either as monolayers on non-gelatin-coateddishes or in spinner culture as spheroids. Differentiation statuswas confirmed by a significant decrease in the expression of theSSEA1 cell surface antigen, a marker of the undifferentiatedphenotype (Suppl. Figs. 6a,b). γH2AX foci in pluripotent mEScells were larger than those in differentiated cells (Figs. 6a, b),and flow cytometry confirmed a two-fold reduction in averageγH2AX intensity per cell (Fig. 6f). The number of ssb revealedusing the comet assay was also significantly reduced indifferentiated mES cells especially when grown as spheroids(Figs. 6c–e). The fraction of S phase cells decreased duringdifferentiation, but those with S phase DNA content still showed areduction in ssb. Histone H4 acetylation was 3–6 times lower indifferentiated relative to pluripotent mES cells (Fig. 6g). Smallereffects were seen for acetylated histone H3. Therefore chromatin-induced changes that occur upon differentiation and areconfirmed by reduction in histone acetylation are associatedwith a decrease in sensitivity of mES cell DNA to breakage duringthe comet assay and a decrease in phosphorylation of H2AX.
Our results do not explain why the range in apparent ssb is sogreat in pluripotent mES cells, or why some differentiated mEScells continue to exhibit sensitivity to osmotic shock. Hetero-
geneity in expression of various differentiation markers andreversible changes in histone markings have been reported inpluripotent mES cells [56]. When pluripotent and differentiatedmES cells were sorted on the basis of expression of the SSEA1 cellsurface antigen (Suppl. Fig. 6c) there was a decrease in sensitivityto breakage in differentiated compared to pluripotent mES cellsbut there was no difference in the range of ssb in the cellsexpressing the most SSEA1 versus the least SSEA1 (Suppl. Figs. 6d–g). Therefore heterogeneity in SSEA1 antigen expression does notexplain heterogeneity in ssb in the comet assay.
Discussion
The unusual organization of chromatin in mES cells appears tounderlie two observations in this study: excessive numbers of ssbthat appear during performance of the alkaline comet assay, andlarge numbers of endogenous γH2AX foci. Mouse ES cellsexpressed ∼100 large γH2AX foci per cell that could not bedistinguished from radiation-induced foci on the basis of foci sizeor intensity. The high expression of γH2AX in mES cells was notexplained by an increase in double-strand breaks, DNA degrada-tion, or apoptosis. Two recent papers have also reported excessivenumbers of γH2AX foci inmESwhen compared toMEF cells. In onecase, the authors believed this to be an indication that double-strand break repair is important for ES cells [57], and in the secondpaper, the excessive ssb detected using the comet assay werepresumed to be responsible for producing occasional double-strand breaks and thus γH2AX foci [58]. As shown here, there is noevidence that single- or double-strand breaks actually exist insufficient numbers in intact mES cells to explain the γH2AX foci,and no evidence from this study or another [58] that theendogenous foci mark sites of active double-strand break repair.Rather, the euchromatic chromatin structure of pluripotent mEScells, assisted by global histone acetylation and abundant chro-matin remodeling complexes [2,55], is more likely to allow thedevelopment of larger γH2AX foci.
Chromatin structure is affected by chromatin remodelingprocesses which also influence the extent of H2AX phosphoryla-tion. Loss of the histone remodeling complex, SWI/SNF, results in adecrease in H2AX phosphorylation after irradiation [59]. H2AX inheterochromatin, which is largely absent from undifferentiatedmES cells, is known to be resistant to phosphorylation [16].Relaxation of chromatin organization, such as that caused by
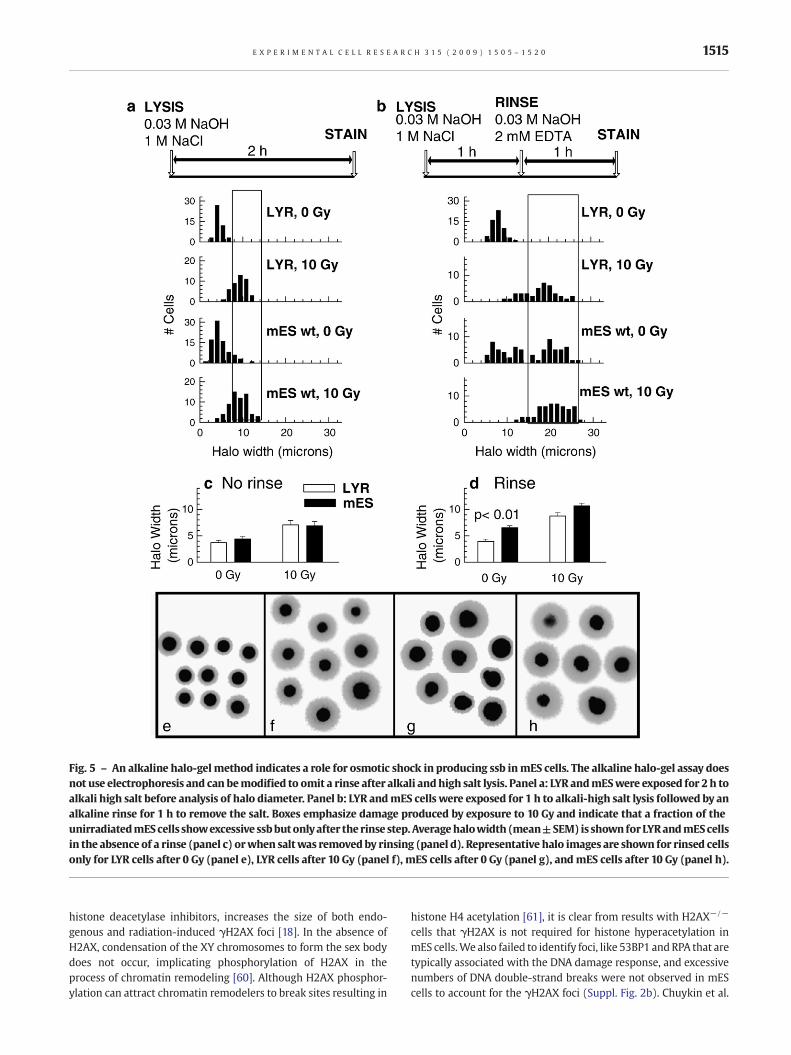
Fig. 5 – An alkaline halo-gelmethod indicates a role for osmotic shock in producing ssb inmES cells. The alkaline halo-gel assay doesnot use electrophoresis and can bemodified to omit a rinse after alkali andhigh salt lysis. Panel a: LYR andmESwere exposed for 2 h toalkali high salt before analysis of halo diameter. Panel b: LYR andmES cells were exposed for 1 h to alkali-high salt lysis followed by analkaline rinse for 1 h to remove the salt. Boxes emphasize damage produced by exposure to 10 Gy and indicate that a fraction of theunirradiatedmEScells showexcessive ssbbutonlyafter the rinse step. Averagehalowidth (mean±SEM) is shown forLYRandmEScellsin the absence of a rinse (panel c) orwhen saltwas removedby rinsing (panel d). Representative halo images are shown for rinsed cellsonly for LYR cells after 0 Gy (panel e), LYR cells after 10 Gy (panel f), mES cells after 0 Gy (panel g), and mES cells after 10 Gy (panel h).
1515E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
histone deacetylase inhibitors, increases the size of both endo-genous and radiation-induced γH2AX foci [18]. In the absence ofH2AX, condensation of the XY chromosomes to form the sex bodydoes not occur, implicating phosphorylation of H2AX in theprocess of chromatin remodeling [60]. Although H2AX phosphor-ylation can attract chromatin remodelers to break sites resulting in
histone H4 acetylation [61], it is clear from results with H2AX−/−
cells that γH2AX is not required for histone hyperacetylation inmES cells.We also failed to identify foci, like 53BP1 andRPA that aretypically associated with the DNA damage response, and excessivenumbers of DNA double-strand breaks were not observed in mEScells to account for the γH2AX foci (Suppl. Fig. 2b). Chuykin et al.

Fig. 6 – Differentiation of mES cells reduces γH2AX expression, ssb, and global histone H4 acetylation. Panels a: foci appearancein pluripotent mES cells. Panel b: decrease in γH2AX foci size after mES cells are grown as spheroids for twoweeks in the absence ofLIF. Panel c–e: decrease in ssb detected using the comet assay when mES cells are grown for 2 weeks as spheroids in the absenceof LIF. Panel f: differentiation causes a decrease in γH2AX expression in mES cells as measured using flow cytometry. Panel g:differentiation causes a significant decrease in histone H4 acetylation. Diff 1 represents mES cells allowed to differentiate asmonolayers in the absence of LIF. Diff 2 represents mES cells allowed to differentiate as spheroids in the absence of LIF.
1516 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
[58] also concluded that endogenous γH2AX foci in mES cells didnot constitute part of the DNA repair system based on apparentlack of phospho-ATM foci; this is consistent with our resultsshowing no decrease in γH2AX foci in ATM−/− mES cells. Itseems most likely that the endogenous foci that develop in mEScells simply represent much larger versions of small focipreviously characterized by McManus and Hendzel [15] and
whose function remains unknown. Although these authors havereported the presence of 200–300 sites of γH2AX accumulationthroughout the genome of normally growing mammalian cells,these are typically much smaller and thus easily distinguishedfrom foci induced by ionizing radiation. This is not the case withmES cells where endogenous and radiation-induced γH2AX fociare similar in size (Suppl. Figs. 3a,b). If maintenance of

1517E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
pluripotency requires a more open chromatin organization [55], aconsequence appears to be an increase in the size of endogenousγH2AX foci. Whether these endogenous foci serve a function interms of structure or signaling is not known. Mouse ES cells thatlack H2AX show a lower plating efficiency and exhibit genomicinstability but have a similar cell doubling time as wild-type cells[12,29]. Our results have implications for the use of γH2AX as amarker of DNA breaks in mES cells since the high backgroundreduces the sensitivity for detecting exogenously produced γH2AXfoci, at least microscopically.
Higher than normal numbers of RAD51 foci were alsoobserved in mES cells. RAD51 foci are associated with post-replicative chromatin [62], consistent with their role in homo-logous recombination. Unlike mES cells where ∼80% of the cellsare positive for RAD51 foci, fewer than 10% of differentiated,untreated cells contain RAD51 foci [63], often associated withreplication fork collapse and subsequent recombination [64]. Apart of the explanation for high RAD51 foci numbers is the highpercentage of S/G2 phase cells in mES cultures. The lack ofendogenous RPA foci in mES cells questions the relation betweenRAD51 focus formation and recombination in mES cells sincethese two molecules generally co-localize at sites of replicationfork arrest [65]. RAD51 foci may be higher as a result of the highexpression of γH2AX which is known to retain RAD51 at sites ofDNA damage [66]. The decrease in RAD51 foci observed inH2AX−/− mES cells (Suppl. Fig. 3i) supports this possibility, butthe modest co-localization between γH2AX and RAD51 (SupplFig. 3k) questions it. A more likely explanation is that RAD51protein expression has been reported to be 20 fold higher in mEScells than MEFs [57] so that foci size becomes sufficiently large tobe visible microscopically. It has been suggested that RAD51 focimay serve as a storage site for preformed repair proteincomplexes [62] which would be an advantage in mES cells thatlack a G1 checkpoint [7].
ATR kinase is responsible for phosphorylation of H2AX that isassociated with damaged replication forks [41], and this kinaseseems most likely to be involved in the endogenous phosphor-ylation in mES cells. Expression of γH2AX in both ATM−/− andDNA-PKcs−/− mES cells was similar to wild-type (Figs. 2b, c).Endogenous γH2AX could be reduced by treatment withwortmannin but not caffeine. Interestingly, mouse hair folliclecells exhibit a hair cycle dependent change in the expression ofendogenous γH2AX that has also been associated with ATRkinase [23].
Vulnerability to ssb in the alkaline comet assay appears to beassociated with decondensed chromatin in replicating mES cells.The observed decrease in ssb with differentiation (Fig. 6) isconsistent with the decrease in global histone acetylation which isassociated with an increase in chromatin condensation. Therehave been three previous reports of excessive ssb in mES cellsmeasured using the alkaline comet assay [58,67,68], and onenegative report using the alkaline unwinding assay [69]. Ourresults are in agreement with all of these studies but point to theimportance of osmotic shock in creating ssb when mES nucleiexposed to high salt lysis solutions are rapidly rinsed free of salt.The alkaline halo gel method was modified to directly test theimportance of osmotic shock. No ssb were detected when highsalt was maintained throughout the procedure, but breaksoccurred when osmotic shock was created by rinsing slides in asalt-free alkaline solution (Fig. 5b). The heterogeneity in ssb
remains unexplained, however, it could be related to theheterogeneity in epigenetic states that are known to be highlydynamic in pluripotent mES cells [56].
In the alkaline halo-gel assay of Sestili and Cantoni [39], haloformation is thought to be caused by the switch from high salt lysisto hypotonic solution; the authors suggested that the osmoticgradient produced by removing the salt drives the radial diffusionof DNA fragments through the agarose. In their experiments, onlycells containing DNA breaks reacted to the decrease in saltconcentration while undamaged control cells showed no changein halo diameter as a result of osmotic shock. Our results using LY-Rcells confirm this finding, but untreated mES cells clearlyresponded differently. Cook and Brazell discussed the importanceof salt in the stabilization of nucleoid structures [70], and it hasbeen suggested that as the ionic strength is decreased from high tolow salt, repulsive interactions within the chromatin core particlemay be able to break ionic bonds [71]. It would seem that osmoticforces have little effect on DNA integrity for the majority ofdifferentiated cell lines, but the unusual open chromatin structurein mES cells makes them particularly vulnerable in the alkalinecomet assay. Therefore, although there are no ssb present in intactpluripotent mES cells, the comet assay revealed the uniquesensitivity of mES chromatin to breakage caused by osmoticshock. It will be interesting to determine whether other mousestem cells, including tumor stem cells, share this property.
The presence of excessive DNA ssb in the comet assay has beenreported for a few specific differentiated cell types and only inrelation to the alkaline version of the comet assay. Evidence for thepresence of true breaks in any of these cell types is controversial.Kidney cells [72,73], chicken erythrocytes [74], and mouse andhuman sperm cell DNA [74] have been reported to contain highnumbers of ssb when examined using the alkaline comet assay.Breaks in spermhave been attributed to alkali-labile sites that occuras a result of chromatin compaction [75]. Although not directlyrelevant to mES cells where chromatin is decondensed rather thancondensed, it does point to the importance of higher orderchromatin structure in the response of cells in the alkaline cometassay. Breaks in mouse renal medulla cells remain largelyunexplained, and like mES cells, significant heterogeneity incomet tail moment has been observed [73]; it is possible thatosmotic shock also contributes to the excessive ssb seen in thesecells. Chicken erythrocyte DNA is more heavily damaged probablyas a result of the presence of heme which creates ssb during alkalilysis [76].
In our preliminary analysis of human embryonic stem cells, wefound no evidence for the presence of excessive ssb using thecomet assay or γH2AX foci. Similarly, Maynard et al. [5] applied thealkaline comet assay to human ES cells and did not report excessivessb. However, mouse and human ES cells differ in many importantways that include differences in chromatin organization [12,77,78].Moreover, human ES cells are believed to be further along in thedifferentiation program than mES cells [79,80]. Analysis of ssb andγH2AX foci in cells from mouse epiblasts which more closelyresemble human ES cells would be informative.
Acknowledgments
This work was supported by a grant from the Canadian CancerSociety. Flow cytometry expertise was provided by Denise

1518 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
McDougal. The authors thank Dr. Alt and Dr. Nagy for providing thepluripotent mES cell lines used in this study.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.yexcr.2008.12.007.
R E F E R E N C E S
[1] E. Meshorer, T. Misteli, Chromatin in pluripotent embryonic stemcells and differentiation, Nat. Rev., Mol. Cell Biol. 7 (2006)540–546.
[2] J.D. Pajerowski, K.N. Dahl, F.L. Zhong, P.J. Sammak, D.E. Discher,Physical plasticity of the nucleus in stem cell differentiation, Proc.Natl. Acad. Sci. U. S. A. 104 (2007) 15619–15624.
[3] S.M. Gasser, Visualizing chromatin dynamics in interphase nuclei,Science 296 (2002) 1412–1416.
[4] M. Spivakov, A.G. Fisher, Epigenetic signatures of stem-cellidentity, Nat. Rev. Genet. 8 (2007) 263–271.
[5] S. Maynard, A.M. Swistowska, J.W. Lee, Y. Liu, S.T. Liu, A.B. Da Cruz,M. Rao, N.C. de Souza-Pinto, X. Zeng, V.A. Bohr, Human embryonicstem cells have enhanced repair of multiple forms of DNAdamage, Stem Cells 26 (2008) 2266–2274.
[6] Y. Park, S.L. Gerson, DNA repair defects in stem cell function andaging, Annu. Rev. Med. 56 (2005) 495–508.
[7] P.J. Stambrook, An ageing question: do embryonic stem cellsprotect their genomes? Mech. Ageing Dev. 128 (2007) 31–35.
[8] P.L. Olive, J.P. Banáth, Induction and rejoining of radiation-inducedDNA single-strand breaks: “tail moment” as a function of positionin the cell cycle, Mutat. Res. 294 (1993) 275–283.
[9] S.H. MacPhail, J.P. Banath, T.Y. Yu, E.H. Chu, H. Lambur, P.L. Olive,Expression of phosphorylated histone H2AX in cultured celllines following exposure to X-rays, Int. J. Radiat. Biol. 79(2003) 351–358.
[10] T. Yu, S.H. MacPhail, J.P. Banath, D. Klokov, P.L. Olive, Endogenousexpression of phosphorylated histone H2AX in tumors in relationto DNA double-strand breaks and genomic instability, DNA Repair(Amst) 5 (2006) 935–946.
[11] O.A. Sedelnikova, I. Horikawa, D.B. Zimonjic, N.C. Popescu, W.M.Bonner, J.C. Barrett, Senescing human cells and ageing miceaccumulate DNA lesions with unrepairable double-strand breaks,Nat. Cell Biol. 6 (2004) 168–170.
[12] C.A. Banuelos, J.P. Banath, S.H. MacPhail, J. Zhao, C.A. Eaves, M.D. O,Connor, P.M. Lansdorp, P.L. Olive, Mouse but not humanembryonic stem cells are deficient in rejoining of ionizingradiation-induced DNA double-strand breaks, DNA Repair (Amst)7 (2008) 1471–1483.
[13] R.L. Warters, P.J. Adamson, C.D. Pond, S.A. Leachman, Melanomacells express elevated levels of phosphorylated histone H2AX foci,J. Invest. Dermatol. 124 (2005) 807–817.
[14] J. Han, M.J. Hendzel, J. lalunis-Turner, Quantitative analysis revealsasynchronous and more than DSB-associated histone H2AXphosphorylation after exposure to ionizing radiation, Radiat. Res.165 (2006) 283–292.
[15] K.J. McManus, M.J. Hendzel, ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normallygrowing mammalian cells, Mol. Biol. Cell 16 (2006) 5013–5025.
[16] J.A. Kim, M. Kruhlak, F. Dotiwala, A. Nussenzweig, J.E. Haber,Heterochromatin is refractory to gamma-H2AX modification inyeast and mammals, J. Cell Biol. 178 (2007) 209–218.
[17] I.G. Cowell, N.J. Sunter, P.B. Singh, C.A. Austin, B.W. Durkacz, M.J.Tilby, GammaH2AX foci form preferentially in euchromatin afterionising-radiation, PLoS ONE 2 (2007) e1057.
[18] C.A. Banuelos, J.P. Banath, S.H. MacPhail, J. Zhao, T. Reitsema, P.L.Olive, Radiosensitization by the histone deacetylase inhibitorPCI-24781, Clin. Cancer Res. 13 (2007) 6816–6826.
[19] M. Murga, I. Jaco, Y. Fan, R. Soria, B. Martinez-Pastor, M. Cuadrado,S.M. Yang, M.A. Blasco, A.I. Skoultchi, O. Fernandez-Capetillo,Global chromatin compaction limits the strength of the DNAdamage response, J. Cell Biol. 178 (2007) 1101–1108.
[20] J. Bartkova, Z. Horejsi, K. Koed, A. Kramer, F. Tort, K. Zieger, P.Guldberg, M. Sehested, J.M. Nesland, C. Lukas, T. Orntoft, J. Lukas, J.Bartek, DNA damage response as a candidate anti-cancer barrierin early human tumorigenesis, Nature 434 (2005) 864–870.
[21] V.G. Gorgoulis, L.V. Vassiliou, P. Karakaidos, P. Zacharatos, A.Kotsinas, T. Liloglou, M. Venere, R.A. Ditullio Jr., N.G. Kastrinakis, B.Levy, D. Kletsas, A. Yoneta, M. Herlyn, C. Kittas, T.D. Halazonetis,Activation of the DNA damage checkpoint and genomic instabilityin human precancerous lesions, Nature 434 (2005) 907–913.
[22] G. Hamer, H.L. Roepers-Gajadien, A. van Duyn-Goedhart, I.S.Gademan, H.B. Kal, P.P. van Buul, D.G. de Rooij, DNA double-strandbreaks and gamma-H2AX signaling in the testis, Biol. Reprod. 68(2003) 628–634.
[23] M. Koike, M. Mashino, J. Sugasawa, A. Koike, Dynamic change ofhistone H2AX phosphorylation independent of ATM and DNA-PKin mouse skin in situ, Biochem. Biophys. Res. Commun. 363(2007) 1009–1012.
[24] M.I. Aladjem, B.T. Spike, L.W. Rodewald, T.J. Hope, M. Klemm, R.Jaenisch, G.M.Wahl, ES cells do not activate p53-dependent stressresponses and undergo p53-independent apoptosis in responseto DNA damage, Curr. Biol. 8 (1998) 145–155.
[25] A. Nagy, J. Rossant, R. Nagy, W. bramow-Newerly, J.C. Roder,Derivation of completely cell culture-derived mice fromearly-passage embryonic stem cells, Proc. Natl. Acad. Sci. U. S. A.90 (1993) 8424–8428.
[26] H. Ding, M. Schertzer, X. Wu, M. Gertsenstein, S. Selig, M.Kammori, R. Pourvali, S. Poon, I. Vulto, E. Chavez, P.P. Tam, A. Nagy,P.M. Lansdorp, Regulation of murine telomere length by Rtel: anessential gene encoding a helicase-like protein, Cell 117 (2004)873–886.
[27] C. Deng, A. Wynshaw-Boris, F. Zhou, A. Kuo, P. Leder, Fibroblastgrowth factor receptor 3 is a negative regulator of bone growth,Cell 84 (1996) 911–921.
[28] Y. Gao, J. Chaudhuri, C. Zhu, L. Davidson, D.T. Weaver, F.W. Alt, Atargeted DNA-PKcs-null mutation reveals DNA-PK-independentfunctions for KU in V(D)J recombination, Immunity 9 (1998)367–376.
[29] C.H. Bassing, K.F. Chua, J. Sekiguchi, H. Suh, S.R. Whitlow, J.C.Fleming, B.C. Monroe, D.N. Ciccone, C. Yan, K. Vlasakova, D.M.Livingston, D.O. Ferguson, R. Scully, F.W. Alt, Increased ionizingradiation sensitivity and genomic instability in the absence ofhistone H2AX, Proc. Natl. Acad. Sci. U. S. A. 99 (2002)8173–8178.
[30] C.H. Glover, M. Marin, C.J. Eaves, C.D. Helgason, J.M. Piret, J. Bryan,Meta-analysis of differentiating mouse embryonic stem cell geneexpression kinetics reveals early change of a small gene set, PLoSComput. Biol. 2 (2006) e158.
[31] B.A. Fenderson, E.M. Eddy, S. Hakomori, Glycoconjugateexpression during embryogenesis and its biological significance,Bioessays 12 (1990) 173–179.
[32] D. Constantinescu, H.L. Gray, P.J. Sammak, G.P. Schatten, A.B.Csoka, Lamin A/C expression is a marker of mouse and humanembryonic stem cell differentiation, Stem Cells 24 (2006)177–185.
[33] J. Bauer, Advances in cell separation: recent developments incounterflow centrifugal elutriation and continuous flow cellseparation, J. Chromatogr., B, Biomed. Sci. Appl. 722 (1999)55–69.
[34] P.L. Olive, J.P. Banath, The comet assay: a method to measure DNAdamage in individual cells, Nat. Protoc. 1 (2006) 23–29.
[35] B. Rydberg, The rate of strand separation in alkali of DNA ofirradiated mammalian cells, Radiat. Res. 61 (1975) 274–287.

1519E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
[36] P.L. Olive, Inhibition of DNA synthesis by nitroheterocycles. II.Mechanisms of cytotoxicity, Br. J. Cancer 40 (1979) 94–104.
[37] P.L. Olive, DNA precipitation assay: a rapid and simple method fordetecting DNA damage in mammalian cells, Environ. Mol.Mutagen. 11 (1988) 487–495.
[38] J.L. Roti Roti, W.D. Wright, Visualization of DNA loops in nucleoidsfrom HeLa cells: assays for DNA damage and repair, Cytometry 8(1987) 461–467.
[39] P. Sestili, O. Cantoni, Osmotically driven radial diffusion ofsingle-stranded DNA fragments on an agarose bed as aconvenient measure of DNA strand scission, Free Radic. Biol. Med.26 (1999) 1019–1026.
[40] E.P. Rogakou, C. Boon, C. Redon, W.M. Bonner, Megabasechromatin domains involved in DNA double-strand breaks Invivo, J. Cell Biol. 146 (1999) 905–916.
[41] I.M. Ward, J. Chen, Histone H2AX is phosphorylated in anATR-dependent manner in response to replicational stress, J. Biol.Chem. 276 (2001) 47759–47762.
[42] X. Huang, H.D. Halicka, F. Traganos, T. Tanaka, A. Kurose, Z.Darzynkiewicz, Cytometric assessment of DNA damage inrelation to cell cycle phase and apoptosis, Cell Prolif. 38 (2005)223–243.
[43] L. Jirmanova, D.V. Bulavin, A.J. Fornace Jr., Inhibition of the ATR/Chk1 pathway induces a p38-dependent S-phase delay in mouseembryonic stem cells, Cell Cycle 4 (2005) 1428–1434.
[44] D. Cortez, Caffeine inhibits checkpoint responses withoutinhibiting the ataxia-telangiectasia-mutated (ATM) and ATM-and Rad3-related (ATR) protein kinases, J. Biol. Chem. 278 (2003)37139–37145.
[45] D. Chowdhury, M.C. Keogh, H. Ishii, C.L. Peterson, S. Buratowski, J.Lieberman, gamma-H2AX dephosphorylation by proteinphosphatase 2A facilitates DNA double-strand break repair, Mol.Cell 20 (2005) 801–809.
[46] J.S. Siino, I.B. Nazarov, M.P. Svetlova, L.V. Solovjeva, R.H. Adamson,I.A. Zalenskaya, P.M. Yau, E.M. Bradbury, N.V. Tomilin,Photobleaching of GFP-labeled H2AX in chromatin: H2AX has lowdiffusional mobility in the nucleus, Biochem. Biophys. Res.Commun. 297 (2002) 1318–1323.
[47] E.P. Rogakou, W. Nieves-Neira, C. Boon, Y. Pommier, W.M. Bonner,Initiation of DNA fragmentation during apoptosis inducesphosphorylation of H2AX histone at serine 139, J. Biol. Chem. 275(2000) 9390–9395.
[48] E.I. Golub, R.C. Gupta, T. Haaf, M.S. Wold, C.M. Radding,Interaction of human rad51 recombination protein withsingle-stranded DNA binding protein, RPA, Nucleic Acids Res. 26(1998) 5388–5393.
[49] P.L. Olive, J.P. Banáth, C.D. Fjell, DNA strand breakage and DNAstructure influence staining with propidium iodide using thealkaline comet assay, Cytometry 16 (1994) 305–312.
[50] R. Hand, DNA replication in mammalian cells. Altered patterns ofinitiation during inhibition of protein synthesis, J. Cell Biol. 67(1975) 761–773.
[51] M.M. Panning, D.M. Gilbert, Spatio-temporal organization of DNAreplication in murine embryonic stem, primary, andimmortalized cells, J. Cell. Biochem. 95 (2005) 74–82.
[52] B. Vogelstein, D.M. Pardoll, D.S. Coffey, Supercoiled loops andeucaryotic DNA replication, Cell 22 (1980) 79–85.
[53] J.M. Lemaitre, E. Danis, P. Pasero, Y. Vassetzky, M. Mechali, Mitoticremodeling of the replicon and chromosome structure, Cell 123(2005) 787–801.
[54] G. Ahnstrom, Techniques to measure DNA single-strand breaks incells: a review, Int. J. Radiat. Biol. 54 (1988) 695–707.
[55] E. Meshorer, D. Yellajoshula, E. George, P.J. Scambler, D.T. Brown, T.Misteli, Hyperdynamic plasticity of chromatin proteins inpluripotent embryonic stem cells, Dev. Cell 10 (2006) 105–116.
[56] K. Hayashi, S.M. Chuva de Sousa Lopes, F. Tang, M.A. Surani,Dynamic equilibrium and heterogeneity of mouse pluripotentstem cells with distinct functional and epigenetic states, Cell 3(2008) 391–401.
[57] E.D. Tichy, P.J. Stambrook, DNA repair in murine embryonic stemcells and differentiated cells, Exp. Cell Res. 314 (2008) 1929–1936.
[58] I.A. Chuykin, M.S. Lianguzova, T.V. Pospelova, V.A. Pospelov,Activation of DNA damage response signaling in mouseembryonic stem cells, Cell Cycle 7 (2008) 2922–2928.
[59] J.H. Park, E.J. Park, H.S. Lee, S.J. Kim, S.K. Hur, A.N. Imbalzano, J.Kwon, Mammalian SWI/SNF complexes facilitate DNAdouble-strand break repair by promoting gamma-H2AXinduction, EMBO J. 25 (2006) 3986–3997.
[60] O. Fernandez-Capetillo, S.K. Mahadevaiah, A. Celeste, P.J.Romanienko, R.D. Camerini-Otero, W.M. Bonner, K. Manova, P.Burgoyne, A. Nussenzweig, H2AX is required for chromatinremodeling and inactivation of sex chromosomes in male mousemeiosis, Dev. Cell 4 (2003) 497–508.
[61] C. Thiriet, J.J. Hayes, Chromatin in need of a fix: phosphorylationof H2AX connects chromatin to DNA repair, Mol. Cell 18 (2005)617–622.
[62] S. Tashiro, J. Walter, A. Shinohara, N. Kamada, T. Cremer, Rad51accumulation at sites of DNA damage and in postreplicativechromatin, J. Cell Biol. 150 (2000) 283–292.
[63] L.R. van Veelen, T. Cervelli, M.W. van de Rakt, A.F. Theil, J. Essers, R.Kanaar, Analysis of ionizing radiation-induced foci of DNAdamage repair proteins, Mutat. Res. 574 (2005) 22–33.
[64] M. Tarsounas, D. Davies, S.C. West, BRCA2-dependent andindependent formation of RAD51 nuclear foci, Oncogene 22(2003) 1115–1123.
[65] K.M. Sleeth, C.S. Sorensen, N. Issaeva, J. Dziegielewski, J. Bartek, T.Helleday, RPA mediates recombination repair during replicationstress and is displaced from DNA by checkpoint signalling inhuman cells, J. Mol. Biol. 373 (2007) 38–47.
[66] T.T. Paull, E.P. Rogakou, V. Yamazaki, C.U. Kirchgessner, M. Gellert,W.M. Bonner, A critical role for histone H2AX in recruitment ofrepair factors to nuclear foci after DNA damage, Curr. Biol. 10(2000) 886–895.
[67] R.S. Tebbs, J.E. Cleaver, R.A. Pedersen, A. Hartmann, Modificationof the comet assay for the detection of DNA strand breaks inextremely small tissue samples, Mutagenesis 14 (1999) 437–438.
[68] S.Y. Vatolin, E.V. Okhapkina, N.M. Matveeva, A.G. Shilov, S.I.Baiborodin, V.V. Philimonenko, N.S. Zhdanova, O.L. Serov,Scheduled perturbation in DNA during in vitro differentiation ofmouse embryo-derived cells, Mol. Reprod. Dev. 47 (1997) 1–10.
[69] G. Saretzki, L. Armstrong, A. Leake, M. Lako, Z.T. von, Stressdefense in murine embryonic stem cells is superior to that ofvarious differentiated murine cells, Stem Cells 22 (2004)962–971.
[70] P.R. Cook, I.A. Brazell, Supercoils in human DNA, J. Cell. Sci. 19(1975) 261–279.
[71] L.J. Libertini, E.W. Small, Reversibility of the low-salt transition ofchromatin core particles, Nucleic Acids Res. 15 (1987)6655–6664.
[72] D.W. Fairbairn, W.A. Reyes, K.L. O, Neill, Alkali-labile sites areprevalent in kidney tissue DNA, Cancer Lett. 81 (1994) 67–76.
[73] N.I. Dmitrieva, Q. Cai, M.B. Burg, Cells adapted to high NaClhave many DNA breaks and impaired DNA repair both in cellculture and in vivo, Proc. Natl. Acad. Sci. U. S. A. 101 (2004)2317–2322.
[74] N.P. Singh, D.B. Danner, R.R. Tice, M.T. McCoy, G.D. Collins, E.L.Schneider, Abundant alkali-sensitive sites in DNA of human andmouse sperm, Exp. Cell Res. 184 (1989) 461–470.
[75] L. Muriel, E. Segrelles, V. Goyanes, J. Gosalvez, J.L. Fernandez,Structure of human sperm DNA and background damage,analysed by in situ enzymatic treatment and digital imageanalysis, Mol. Hum. Reprod. 10 (2004) 203–209.
[76] M. Glei, S. Klenow, J. Sauer, U. Wegewitz, K. Richter, B.L.Pool-Zobel, Hemoglobin and hemin induce DNA damage inhuman colon tumor cells HT29 clone 19A and in primary humancolonocytes, Mutat. Res. 594 (2006) 162–171.
[77] J. Yu, J.A. Thomson, Pluripotent stem cell lines, Genes Dev. 22(2008) 1987–1997.

1520 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 5 ( 2 0 0 9 ) 1 5 0 5 – 1 5 2 0
[78] S. Koestenbauer, N.H. Zech, H. Juch, P. Vanderzwalmen, L.Schoonjans, G. Dohr, Embryonic stem cells: similarities anddifferences between human and murine embryonic stem cells,Am. J. Reprod. Immunol. 55 (2006) 169–180.
[79] P.J. Tesar, J.G. Chenoweth, F.A. Brook, T.J. Davies, E.P. Evans, D.L.Mack, R.L. Gardner, R.D. McKay, New cell lines from mouse
epiblast share defining features with human embryonic stemcells, Nature 448 (2007) 196–199.
[80] I.G. Brons, L.E. Smithers, M.W. Trotter, P. Rugg-Gunn, B. Sun, S.M.Chuva de Sousa Lopes, S.K. Howlett, A. Clarkson, L. hrlund-Richter,R.A. Pedersen, L. Vallier, Derivation of pluripotent epiblast stemcells from mammalian embryos, Nature 448 (2007) 191–195.