ethics of drug research - semmelweis
TRANSCRIPT

Kerpel-Fronius S. 1
Ethics of drug research
History, Declaration of Helsinki.
Risks, benefits and burden of study participation.
Ethical review process.
Safety and human dignity of research subjects
Informed consent.
Sándor Kerpel-Fronius, M.D., D.Sc.
Semmelweis University
Department of Pharmacology and Pharmacotherapy
Budapest, Hungary
Email: [email protected]

KERPEL-FRONIUS S. 2
Patient, physician and industry
An uneasy ethical relationship

Differences in ethical opinions
KERPEL-FRONIUS S. 3
Society
Healthy population
Sick persons
Health care
Health insurance Health care
specialists
Opposing
ethical
values
The ethical values of the different groups are influenced by
opposing financial and social interests
Bridging the gaps between the different ethical values is of
outmost social interest

KERPEL-FRONIUS S. 4
Claude Bernard (1813-1878)
Study of Experimental Medicine (1927)
Session at the Vivisection Laboratory, 1889 by Léon Augustin L'hermitte
Claude Bernard postulated that according to medical ethics
no experiemnts can be performed in humans which can
couse any harm even when the results could provide great
benefit for science or for others.

KERPEL-FRONIUS S: ICH-GCP09/eload09 5
Porosz humán vizsgálati rendelet, 1900

KERPEL-FRONIUS S: ICH-GCP09/eload09 6
Reichsrichtlinien zur Forschung an
Menschen (1931)
Reichsrundschreiben des Reichsminsters des Inneren, 28 Februar, 1931;
Reichsgesundheitsblatt 6(55) 174f
1. (1-3) The definition of difference between clinical trials and human experiments
2. The therapeutic investigational plan must be in line with medical knowledge. If possible it should be based on the results of animal experiements. Finally the risks should be in acceptable relation to the expected benefits
4. The enclosed subjects must provide informed consent.
5. The enclosure of patients below 18 years of age must be done with increased care
6. It is unethical to make advantage of the social vulnerability of patients

KERPEL-FRONIUS S. 7
Reichsrichtlinien zur Forschung an
Menschen (1931)
Reichsrundschreiben des Reichsminsters des Inneren, 28 Februar, 1931;
Reichsgesundheitsblatt 6(55) 174f
8. Human studies can be done only by the chief physician or his/her authorised medical coworker. The entire responsibility is carried by the chief physician.
The investigation must be adequately documented describing the goal and performance of the study, as well as the information provided together with the informed consent given by the patient megadását

KERPEL-FRONIUS S. 8
Main medical experts of the
Nurenberg trial of physicians
Werner Leibbrand
1896-1974
A. C. Ivy 1893-1978

KERPEL-FRONIUS S. 9
Summary of the principles of the
Nurenberg Code
Three basic principles laid down by Drs. Leo Alexander and Andrew Ivy described in 10 paragraphs
Human subjects can be entered into experiemnts only following adequate information and obtaining informed consent.
The experiments must be based on reliable animal experiments.
The experiments must be performed by educated professionals. The risks and the physical and psychic suffering must be kept on the possible lowest level. Human subjects cannot be entered into experiments which will lead to predermined permanent remaining damage or death

KERPEL-FRONIUS S: 10
Principles of the Nurenberg
Code
The 9th paragraph most probably entered by
the presiding judge
The human subjects must be free to
immediately quit the experiment at any
point when they feel physically or mentally
unable to go on

KERPEL-FRONIUS S: ICH-GCP09/eload09 11
REMARKS BY PRESIDENT CLINTON
IN APOLOGY FOR STUDY DONE IN
TUSKEGEE (May 16, 1997)
Today, all we can do is apologize. But you have
the power, for only you -- Mr. Shaw, the others who
are here, the family members who are with us in
Tuskegee -- only you have the power to forgive.

KERPEL-FRONIUS S. 12
Important documents dealing
with the ethics of human studies
1947 - Nurenberg Code
1948 (revised 1968, 1983, 1994) - Declaration of Geneva
(Svájc) (Általános etikai maximák)
1949 (revised 1968, 1983, 1994) - World Medical
Association International Code of Medical Ethics
1954 - World Medical Association Principles for Those in
Research and Experimentation
1964 (revised 1975, 1983, 1989, 1996, 2000, 2002,
2004, 2008, 2013) - Declaration of Helsinki. (WMA)
1979 - Belmont Report (USA)
1997 - Convention of the European Council, Oviedo,

Convention of the European
Council, Oviedo, 1997.
Convention for the Protection of Human
Beings with regard to the Application of
Biology and Medicine: Convention on
Human Rights and Biomedicine
KERPEL-FRONIUS S. 13

KERPEL-FRONIUS S. 14
The scientific, social, ethical and legal
background in the time of globalized
medicines development
ICH-GCP
Scientifically
adequate
methods
Health care research
in different political and
cultural environment
Correct
medical
behaviour
Declaration of
Helsinki
Health care research in
in differently developed
economic environment
Social control:
IEC, audit
Human and
patient rights
Reliable
Documentation
Report of AEs
Evaluation of
benefit/riskr
ratio
World Medical Association Drug Regulatory Agency
Pharmaceutical Industry
Clinical Investigators

KERPEL-FRONIUS S.
Good Clinical Practice (ICH-GCP)
Good clinical practice of drug development
ICH: International Committee for Harmonization unified
the guidelines for clinical trials of Europe, USA and Japan
It specifies exactly the tasks of the sponsor, the
investigator and the ethics committee
GCP is a standard of procedures, the aim of which is to
ensure the scientific quality, reliability and authenticity of
the design, conduct and documentation of clinical trials as
well as the rights and safety of the investigated subjects
and the confidentiality of their personal data
Final result of GCP: the establishment of an authentic
„paper trail“ that may be well followed by the authorities
15

INTERNATIONAL CONFERENCE ON
HARMONISATION OF TECHNICAL
REQUIREMENTS FOR REGISTRATION OF
PHARMACEUTICALS FOR HUMAN USE
ICH HARMONISED TRIPARTITE GUIDELINE
GUIDELINE FOR GOOD CLINICAL PRACTICE
E6(R1)
Current Step 4 version
dated 10 June 1996
KERPEL-FRONIUS S: ICH-GCP09/eload09 16

KERPEL-FRONIUS S. 17
EU convention, regulation on the research
and investigation of medicines on human
subjects
Directive 2001/20/EC of the European Parliament and of the
Council of 4 April 2001 on the approximation of the laws, regulations
and administrative provisions of the Member States relating to the
implementation of good clinical practice in the conduct of clinical trials on
medicinal products for human use
REGULATION (EU) No 536/2014 OF THE EUROPEAN
PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on
clinical trials on medicinal products for human use, and
repealing Directive 2001/20/EC
X

KERPEL-FRONIUS S. 18
The goal of ICH-GCP is to harmonize different
interests for the benefit odf the society
Pharmaceutical
industry
Demonstration of efficacy
and safety
Rapid and cheap
development
Competent Authority
Accurate determination
of benefit/risk ratio
Safety of the society
Independent Ethical Committee
ICH
-GC
P
ICH
-GC
P

KERPEL-FRONIUS S.
The distribution of the tasks of human
clinical trials (ICH-GCP)
The clinical trial may endanger the health and social status of the subjects
In lieu of the society the Independent Ethics Committee determines the
socially acceptable measure of the risk/benefit ratio and continuously
controls its development during the trials
Independent Ethics Committee
Sponsor Investigator
Human
trial
Administrative organization
Drug documentation
Drug supply
Collection and reporting
of adverse events
Data processing
Trial report
Archiving
Trial conduct
Observation, treatment and
reporting of adverse events
Archiving
Opinion Control
19

KERPEL-FRONIUS S.
The Independent Ethics Committee controls the safety of the subject,
asserts his/her rights and the confidentiality of his/her personal data in
the course of the trial
Independent Ethics Committe
Investigator
Health
institute Sponsor Social
rela-
tions
Sponsor
Subject
Tasks of the Independent Ethics
Committee
20

KERPEL-FRONIUS S. 21
Informed consent
BMJ 1998;316:1000-1005
Obtaining fully informed consent can be needlessly cruel

KERPEL-FRONIUS S.
Legal aspects of clinical trials
The conditions determined in the trial approval
and the prescriptions of the accepted protocol
as well as the requirements laid down in this
law concerning the conduct of clinical trials are considered occupational rules.
1997 Act Nr. CLIV of the Ministry of Health
Detailed regulations of medical investigations performed in human
subjects 22

KERPEL-FRONIUS S. 23
New challenges for clinical drug
development
Pharmaceutical industry
+
Clinical investigators
+
Competent Authority
+
Society
Medical need
for treating
vulnerable
patients Children
Elderly
Mentally
handicapped
Unconcious
patients, etc.
New scientific
possibilities Genomic research
New targets
New biological
products
Gene therapy
Advanced therapies
Medical device+drug
combinations

KERPEL-FRONIUS S. 24
Economical interests in clinical
drug development
Pharmaceutical
industry
Healt care provider
Out patient units
Hospitals
Research sites
Reesearch support
Joint ventures
Companies founded by
health care providers
Pharmaceutical
industry
Healt care provider
Out patient units
Hospitals
Research sites

KERPEL-FRONIUS S. 25
Social challenges created by the globalization of
the clinical drug development
Needs of the society
Many new, more effective and
safer medicinal products
Pharmaceutical industry
Increasing costs of
drug production
The number of patients
enrolled into trials increses
rapidly and significantly
The placement of “hightech”
studies into developing
countries leads frequently to
ethical, cultural and
social tensions
Strategic Initiative for
Developing Capacity of
Ethical Reviews 2002
(SIDCER)
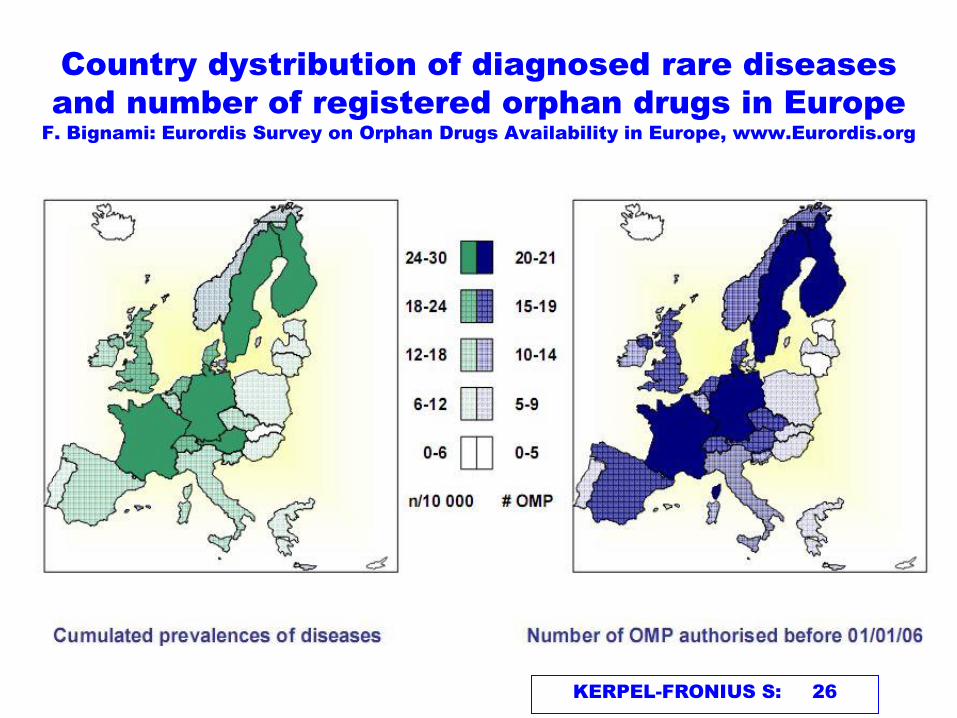
KERPEL-FRONIUS S: 26
Country dystribution of diagnosed rare diseases
and number of registered orphan drugs in Europe
F. Bignami: Eurordis Survey on Orphan Drugs Availability in Europe, www.Eurordis.org

Kerpel-Fronius S. 27
Equity
Each group of patients, irrespective of their social status,
should receive all drugs essential for their treatment with
appropriate level of reimbursement
Solidarity Very expensive treatments are fully reimbursed
Co-payments for cheaper and/or not essential drugs are fixed
at relatively higher levels to equilibrate drug budget
Ethical issues in drug
application

RAS–BRAF pathway
RTK = receptor tyrosine kinase; GTP = guanosine triphosphate; ERK =
extracellular signal-related kinase; MEK = MAP (mitogen-activated protein).
39. Garnett MJ, et al. Cancer Cell 2004;6:313–9.
41. Wan PTC, et al. Cell 2004;116:855–67.
BRAF
Activated RAS
MEK
ERK
P
Oncogen BRAF pathway41
Normal RAS–RAF pathway39
Growth factors
RTK
Mutated BRAF
Excesszív sejtproliferáció és
antiapoptosis
MEK
ERK
P
P P
RAS–GTP
Normal cell proliferation and survival
Normal RAS activation
PLX 4032
Vemurafenib
Inhibited cell
proliferation
Cell death
With the kind permission of A. Nagy
KERPEL-FRONIUS S: 28
KERPEL-FRONIUS S: 28

Inhibition of mutated, activated BRAF
in patients with pretreated melanoma
Flaherty et al., NEJM, 363. 2010
KERPEL-FRONIUS S: 29
-100
-75
-50
-25
0
25
50
75
100
%C
han
ge
Fro
m B
asel
ine
(Su
m o
f Les
ion
Siz
e)
RECIST 30% decrease)
ORR = 81%
Phase I study in pretreated patients
With the kind permission of A. Nagy

Confirmatory phase III trial Chapman PB et al. NEJM 364: 2507-2516_2011
KERPEL-FRONIUS S: 30
Stage IIIc/IV, AJCC)
BRAFV600 mutation
Diagnostic mutation test:
cobas® 4800 BRAF V600
Primary treatment 680
patients
No crossover at PD (was
permitted only after
survival end point was
reached
R 1:1
Dacarbazine
(1000 mg/m2
i.v. q3w)
Vemurafenib
(2x960 mg p.o.)
Response rate:
(> 30% RECIST) : 48% vs 5%
RRR 63% halálozás kockázata
RRR 74% halálozás és progresszió
The ethical problem
Are confirmatory phase III
trials always needed?

Vemurafenib vs DTIC
Chapman PB et al. NEJM 364: 2507-2516_2011
KERPEL-FRONIUS S. 31

Could we develop alternative approaches
for rapidly approving targeted medicines in
patients with precisely diagnosed target?
Chabner B: NEJM 364: 1087-1089, 2011
Suggestions of B. Chabner:
„High response rates (>50%), high disease-control rates
(>75%), and an acceptable toxicity profile in a biomarker-
defined population of 75 to 100 subjects should be
sufficient for accelerated approval if there is a clear unmet
need.
Randomized comparisons with minimally effective
treatments or placebo should not be required.
Specific end points for early approval should be maximally
flexible and adjusted according to the targeted disease
and the effectiveness and toxicity of alternative therapies.”
KERPEL-FRONIUS S: 32

Translational genomics
Kerpel-Fronius S.: 33

Identifiability of biological
materials of human origin
Steering Committee on Bioethics (CDBI) Recommendaqtion of the Committee of
Ministers to member states on biological materials of human origin.
Identifiable biological materials: the person concerned
can be identified alone or in combination with associated
data
In the case of coded material the investigator has
direct access to the code
Linked anonymised (pseudoanonymised)
material: the investigator has no access to the code
Non identifiable (unlinked anonymised) biological
material: the identification of the person is made
permanently impossible
KERPEL-FRONIUS S. 34

The harmonization of contradictory
scientific aims, social needs and
ethical principles
KERPEL-FRONIUS S: 35
Freedom of research
The value of the
scientific results
for the society
Safety and
the protection of
personal rights
and dignity
New medicines, techniques and processes will lead
to new ethical challenges

The harmonization of ethical
requirements associated with the
collection of human biological material
for genetic research within clinical trials
KERPEL-FRONIUS S. 36
Ethical guidelines
for
research on
human tissues
Ethical guidelines
for
Clinical research
„Combined research” Clinical drug trial
+
Genetic research
How to handle ethical issues satisfying both types of research?

Information and consent for the
primary and secondary use of
biological material and related
personal data
The information should be specific and as detailed as
possible with regard to any foreseen research uses and
the choices available in this respect
The consent should specify the options:
A general consent is recommended when the future
uses cannot be clearly defined
Possibility of refusing or future opting out
Consent limited to only unlinked anonymised use, or
limited only to specific research projects
KERPEL-FRONIUS S. 37

EU Data Protection Regulation
Critical comments
Article 81: Processing of personal data concerning
health which is necessary for historical, statistical or
scientific research purposes, such as patient registries
set up for improving diagnoses and differentiating
between similar types of diseases and preparing
studies for therapies, is shall be permitted only with
the consent of the data subject, and shall be subject to
the conditions and safeguards referred to in Article 83.
This amendment makes the exemption from consent for
the use of data concerning health in research very
narrow. This will prevent valuable health research that is
currently legal and already tightly regulated under
European and Member State law.
KERPEL-FRONIUS S. 38

EU Data Protection Regulation
Critical comments
Article 81: (Am 86) The processing of personal data
concerning health, as a special category of data, may
be necessary for reasons of historical, statistical or
scientific research. Therefore this Regulation
foresees an exemption from the requirement of
consent in cases of research that serves a high
public interest.
The concept of “high public interest” is problematic.
“High public interest” suggests the exemption is to be
used only in a very limited set of circumstances. This is
likely to be problematic for many studies, particularly
because the results and impact of the study are not
known at the outset.
KERPEL-FRONIUS S. 39

Article 83: In accordance with the rules set out in this
Regulation, personal data may be processed for historical,
statistical or scientific research purposes only if:
(a) these purposes cannot be otherwise fulfilled by
processing data which does not permit or not any longer
permit the identification of the data subject;
(b) data enabling the attribution of information to an identified
or identifiable data subject is kept separately from the other
information as long as these purposes can be fulfilled in
this manner under the highest technical standards, and
all necessary measures are taken to prevent
unwarranted re-identification of the data subjects.
This amendment makes the exemption from consent for the
use of health data in research very narrow, which will prevent
valuable research that is currently legal. KERPEL-FRONIUS S:. 40
EU Data Protection Regulation
Critical comments