effects of mechanical stress and carvedilol in lamin a/c...
TRANSCRIPT

Effects of Mechanical Stress and Carvedilol in LaminA/C–Deficient Dilated Cardiomyopathy
Suchitra Chandar,* Li Sze Yeo,* Christiana Leimena, Ju-Chiat Tan, Xiao-Hui Xiao,Vesna Nikolova-Krstevski, Yoshinori Yasuoka, Margaret Gardiner-Garden, Jianxin Wu, Scott Kesteven,Lina Karlsdotter, Shweta Natarajan, Arthur Carlton, Stephen Rainer, Michael P. Feneley, Diane Fatkin
Rationale: Mutations in the LMNA gene, which encodes the nuclear lamina proteins lamin A and lamin C, are themost common cause of familial dilated cardiomyopathy (DCM). Mechanical stress-induced apoptosis has beenproposed as the mechanism underpinning DCM in lamin A/C–deficient hearts, but supporting in vivo evidencehas been lacking.
Objective: Our aim was to study interventions to modify mechanical stress in heterozygous Lmna knockout(Lmna�/�) mice.
Methods and Results: Cardiac structure and function were evaluated before and after exercise training, thoracicaortic constriction, and carvedilol treatment. Lmna�/� mice develop adult-onset DCM with relatively moresevere disease in males. Lmna�/� cardiomyocytes show altered nuclear morphology and perinuclear desminorganization, with enhanced responses to hypo-osmotic stress indicative of cytoskeletal instability. Despite thesestructural defects that provide a template for mechanical stress-induced damage, young Lmna�/� mice subjectedto 6 weeks of moderate or strenuous exercise training did not show induction of apoptosis or accelerated DCM.In contrast, regular moderate exercise attenuated DCM development in male Lmna�/� mice. Sustained pressureoverload generated by thoracic aortic constriction depressed ventricular contraction in young wild-type andLmna�/� mice with no sex or genotype differences in the time-course or severity of response. Treatment of maleLmna�/� mice from 12 to 40 weeks with the �-blocker, carvedilol, prevented the dilatation and contractiledysfunction that was observed in placebo-treated mice.
Conclusions: These data suggest that factors other than mechanical stress-induced apoptosis contribute to DCM andprovide the first demonstration that regular moderate exercise and carvedilol can modify disease progression inlamin A/C–deficient hearts. (Circ Res. 2010;106:573-582.)
Key Words: familial dilated cardiomyopathy � lamin A/C � mechanical stress � exercise � carvedilol
Mutations in the LMNA gene that encodes the nuclearlamina proteins lamin A and lamin C are the most
common cause of familial dilated cardiomyopathy (DCM)identified to date,1 accounting for 5% to 10% familial DCMoverall and 30% to 45% families with DCM and conductionsystem disease (CD).2–5 Affected individuals frequently havea rapidly progressive downhill clinical course, requiringpacemaker implantation or heart transplantation, with anincreased risk of sudden death.2–5 Despite the clinical impor-tance of LMNA mutations, very little is known about mech-anisms of disease pathogenesis and strategies to preventDCM have not been investigated.
Because one-third of DCM-causing LMNA mutations arestop codons, splice site variants or insertions/deletions that
reduce lamin A/C protein levels,1,5 Lmna knockout mice area useful and clinically relevant model to study DCM mech-anisms.6 We have previously reported that homozygous Lmnaknockout (Lmna�/�) mice exhibit severe DCM by 4 to 6weeks.7 Heterozygous Lmna knockout (Lmna�/�) mice showCD at 10 weeks and DCM in later adult life.8 A detailedanalysis of the cardiac conduction-system in these mice hasrecently been performed.8 The basis for DCM in Lmna�/�
mice remains unexplained and is the major focus of thisstudy.
Lamins are intermediate filament proteins present in thenuclear lamina and matrix that are critical determinants ofnuclear architecture and function. A fundamental and unan-swered question is how defects in these nuclear proteins
Original received July 2, 2009; revision received November 30, 2009; accepted December 3, 2009.From the Molecular Cardiology Division (S.C., L.S.Y., C.L., V.N.-K., Y.Y., J.W., L.K., S.N., D.F.) and Cardiac Physiology and Transplantation
Division (J.-C.T., X.-H.X., S.K., M.P.F.), Victor Chang Cardiac Research Institute; Cancer Program (M.G.-G.), Garvan Institute of Medical Research;Synapse Technology Pty Ltd (A.C.); Division of Anatomical Pathology (S.R.) and Cardiology Department (M.P.F., D.F.), St Vincent’s Hospital; andFaculties of Medicine and Life Sciences (M.P.F., D.F.), University of New South Wales, Sydney, Australia.
*Both authors contributed equally to this work.Correspondence to Diane Fatkin, Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool St, PO Box 699, Darlinghurst NSW
2010, Australia. E-mail [email protected]© 2010 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.109.204388
573
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on M
ay 12, 2018http://circres.ahajournals.org/
Dow
nloaded from

cause cardiac contractile dysfunction. Cells lacking laminA/C have altered nuclear shape and chromatin organizationand show increased deformability and reduced viability inresponse to biaxial strain in vitro.9,10 Because of these nuclearstructural defects, it has been proposed that cardiomyocyteloss attributable to mechanical stress-induced apoptosis mightbe an important determinant of impaired contraction in laminA/C–deficient hearts (“structural hypothesis”).11,12 Myocar-dial apoptosis is an attractive disease mechanism because it isseen in failing hearts and “wear-and-tear” effects of repeatedcardiac contractions and hemodynamic load could accountfor age-related DCM in individuals with LMNA mutations. Inaddition to intrinsic nuclear defects, altered interactionsbetween the nucleus and the cytoskeleton may further pre-dispose to mechanical stress-induced damage. Desmin fila-ments form an intricate web that links myofibrils with thenucleus, intercalated discs and costameres. We found alteredperinuclear desmin organization in Lmna�/� cardiomyocytesand proposed a model in which loss of nuclear anchoringattributable to lack of lamin A/C destabilizes the desminscaffolding and promotes altered force transduction.7 Despitethe compelling rationale for the “structural hypothesis,” invivo data to validate mechanical stress as a determinant ofDCM in lamin A/C–deficient hearts are lacking.
The central hypothesis underpinning our study is thatinterventions which increase mechanical stress will promoteDCM and conversely, that reduction of mechanical stress willattenuate DCM in Lmna�/� mice. We first performed adetailed characterization of cardiac structure and function inmale and female Lmna�/� mice. Although a trend toward ahigher prevalence of LMNA mutations in females has beensuggested,4 the effects of sex on disease severity have notbeen evaluated. To determine the “wear and tear” effects ofenhanced cardiac contractile activity, young wild-type (WT)and Lmna�/� mice without DCM were subjected to periodsof moderate and strenuous exercise treadmill training. Wehypothesized that exercise would induce apoptosis and accel-erate the onset of DCM. We also evaluated the effects ofthoracic aortic constriction (TAC) in WT and Lmna�/� mice.TAC is a widely used intervention to induce left ventricular(LV) pressure overload and is known to increase cardiomyo-cyte apoptosis.13 We hypothesized that Lmna�/� mice would
have increased apoptotic vulnerability and contractile impair-ment after TAC.
�-Adrenergic receptor blocking drugs reduce myocardialchronotropic and inotropic activity and are widely used in thetreatment of symptomatic heart failure.14 Individuals withDCM caused by LMNA mutations are generally treated withstandard heart failure therapies, including �-blockers, oncesymptoms develop. Although genotype-positive family mem-bers can be identified preclinically, no preventative interven-tions have been studied. To determine whether �-blockertherapy would mitigate against the development of DCM, wealso evaluated the effects of carvedilol in young Lmna�/�
mice. These data provide the first comprehensive in vivoanalysis of mechanical stress in lamin A/C–deficient heartsand the first evaluation of therapeutic interventions to modifydisease progression.
MethodsAnimalsLmna knockout mice in a C57Bl6�129Sv genetic backgroundwere generated as described.6 Mice were genotyped by PCRamplification of tail DNA. Mutant mice and WT littermates werestudied according to protocols approved by the institutionalAnimal Ethics Committee.
Cardiac ProceduresEchocardiography was performed as described.7 Mice were venti-lated and anesthetized for surgical procedures with ketamine (75mg/kg), xylazine (20 mg/kg), and atropine (0.6 mg/kg). Heart rateswere obtained using telemetry (Data Sciences International, ArdenHill, Minn) and were analyzed using AcqKnowledge version 3.1software (Biopac Systems Inc, Goleta, Calif). TAC was achieved byplacement of a ligature (6.0 silk) between the innominate and leftcommon carotid arteries. Aortic pressure gradients were determinedin anesthetized mice (isoflurane 1% to 3%) using 1.4F and 1.0Fmicrotip catheter pressure transducers (Millar Instruments Inc, Hous-ton, Tex).
Exercise TrainingExercise performance was assessed using an Eco 3/6 rodent treadmill(Columbus Instruments, Columbus, Ohio) and an exercise tolerancetest comprised of graded increments of running speed (7 to 25m/min) at 3-minute intervals with a fixed 50 incline. Exercisetolerance test end points were: final stage achieved and number ofstimuli received from a grid at the base of each lane if running speedwas not maintained. For moderate exercise training, mice ran at 17m/min for 40 minutes at a 50 incline, 5 sessions/wk for 6 weeks.15
For strenuous exercise training, mice ran at the highest toleratedspeed (titrated from 15 m/min to 22 m/min over a 3-week period) for40 minutes at a 150 incline, 2 sessions/wk for 6 weeks.16 Eachregimen included 2 days of acclimatization and 10 minutes ofwarm-up and cool-down periods.
Drug StudiesCarvedilol (Roche, Mannheim, Germany) was mixed with 1.5%Triton X-100 and administered at a dose of 10 mg/kg per day indrinking water.17 Control mice received 1.5% Triton X-100 indrinking water.
Myocyte StudiesCardiomyocyte morphology, shortening, and Ca2� transients wereevaluated as described.7
For osmotic studies, isolated cardiomyocytes were placed in basicTyrode solution, then sequentially perfused at 37°C in isotonic (1T)and hypotonic (0.5T) solutions (80 mmol/L NaCl in basic Tyrodesolution replaced with 160 mmol/L and 22.5 mmol/L mannitol,
Non-standard Abbreviations and Acronyms
Ab antibody
CD conduction system disease
DCM dilated cardiomyopathy
ERK extracellular signal-regulated kinase
LV left ventricular
LVDD left ventricular end-diastolic diameter
LVFS left ventricular fractional shortening
MAPK mitogen-activated protein kinase
TAC thoracic aortic constriction
WT wild type
574 Circulation Research February 19, 2010
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
respectively), followed by a washout in 1T solution.18 Myocyteimages were captured (MyoCam; IonOptix Corp, Milton, Mass) anddimensions obtained using Image Tool software.
HistopathologySections (4 �m) of paraffin-embedded hearts stained with hematox-ylin/eosin were examined using light microscopy. Frozen tissuesections were fixed then incubated with anti-desmin antibody (Ab)(1:100 dilution, Novocastra Laboratories Ltd, Newcastle, UnitedKingdom) and a fluorescein isothiocyanate–conjugated secondaryAb. DNA fragmentation detected by TUNEL assay7 was quantifiedas the apoptotic index (number of TUNEL-positive nuclei divided bythe total number of cardiomyocytes). To detect caspase activation,deparaffinized sections were rehydrated and incubated with cleavedcaspase-3 Ab (1:200 dilution, Cell Signaling Technology Inc, Dan-vers, Mass) and a fluorescein isothiocyanate–conjugated secondaryAb. Sections were counterstained with DAPI and examined usingfluorescence microscopy at �40 magnification. Immunogold elec-tron microscopy was performed as described.7 The distribution ofgold-labeled desmin in standardized regions of interest was evalu-ated quantitatively using nearest-neighbor analysis.19
Protein AnalysisTotal protein extracts were separated by SDS-PAGE and hybridizedwith primary anti-desmin Ab (1:100 dilution, Novocastra) or totaland phosphorylated anti-p44/42 MAPK Ab, anti–stress-activatedprotein kinase/c-Jun N-terminal kinase (anti-SAPK/JNK) Ab, anti-p38 MAPK Ab (all 1:1000 dilution, Cell Signaling); or anti–�-actin
Ab (1:400 dilution, Sigma-Aldrich, St Louis, Mo) with horseradishperoxidase–conjugated or Alexa Fluor 680/750 secondary Abs.Hybridization signals were quantified and normalized to �-actin.
Statistical AnalysisDifferences between groups were assessed using ANOVA andStudent t test. Data are expressed as means�SD, and probabilityvalues of �0.05 were considered statistically significant.
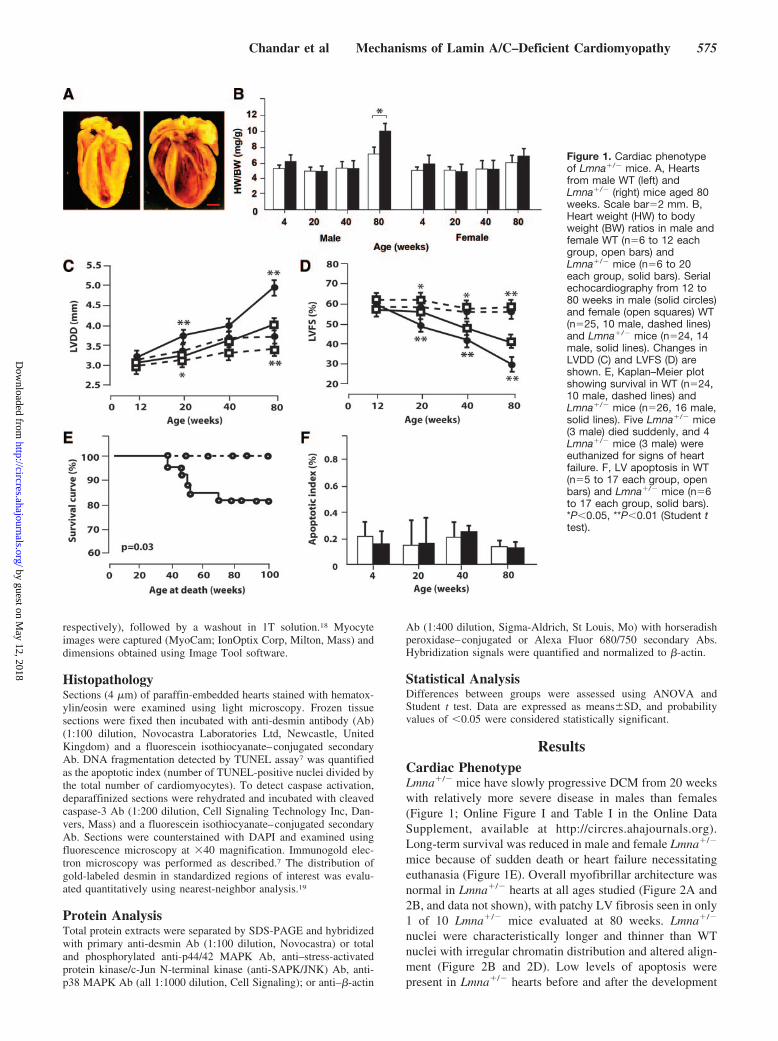
ResultsCardiac PhenotypeLmna�/� mice have slowly progressive DCM from 20 weekswith relatively more severe disease in males than females(Figure 1; Online Figure I and Table I in the Online DataSupplement, available at http://circres.ahajournals.org).Long-term survival was reduced in male and female Lmna�/�
mice because of sudden death or heart failure necessitatingeuthanasia (Figure 1E). Overall myofibrillar architecture wasnormal in Lmna�/� hearts at all ages studied (Figure 2A and2B, and data not shown), with patchy LV fibrosis seen in only1 of 10 Lmna�/� mice evaluated at 80 weeks. Lmna�/�
nuclei were characteristically longer and thinner than WTnuclei with irregular chromatin distribution and altered align-ment (Figure 2B and 2D). Low levels of apoptosis werepresent in Lmna�/� hearts before and after the development
Figure 1. Cardiac phenotypeof Lmna�/� mice. A, Heartsfrom male WT (left) andLmna�/� (right) mice aged 80weeks. Scale bar�2 mm. B,Heart weight (HW) to bodyweight (BW) ratios in male andfemale WT (n�6 to 12 eachgroup, open bars) andLmna�/� mice (n�6 to 20each group, solid bars). Serialechocardiography from 12 to80 weeks in male (solid circles)and female (open squares) WT(n�25, 10 male, dashed lines)and Lmna�/� mice (n�24, 14male, solid lines). Changes inLVDD (C) and LVFS (D) areshown. E, Kaplan–Meier plotshowing survival in WT (n�24,10 male, dashed lines) andLmna�/� mice (n�26, 16 male,solid lines). Five Lmna�/� mice(3 male) died suddenly, and 4Lmna�/� mice (3 male) wereeuthanized for signs of heartfailure. F, LV apoptosis in WT(n�5 to 17 each group, openbars) and Lmna�/� mice (n�6to 17 each group, solid bars).*P�0.05, **P�0.01 (Student ttest).
Chandar et al Mechanisms of Lamin A/C–Deficient Cardiomyopathy 575
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
of DCM with no age or genotype differences (Figure 1F).Isolated cardiomyocytes from male and female Lmna�/�
mice aged 40 weeks showed normal morphology, shortening,and Ca2� transients (Table 1).
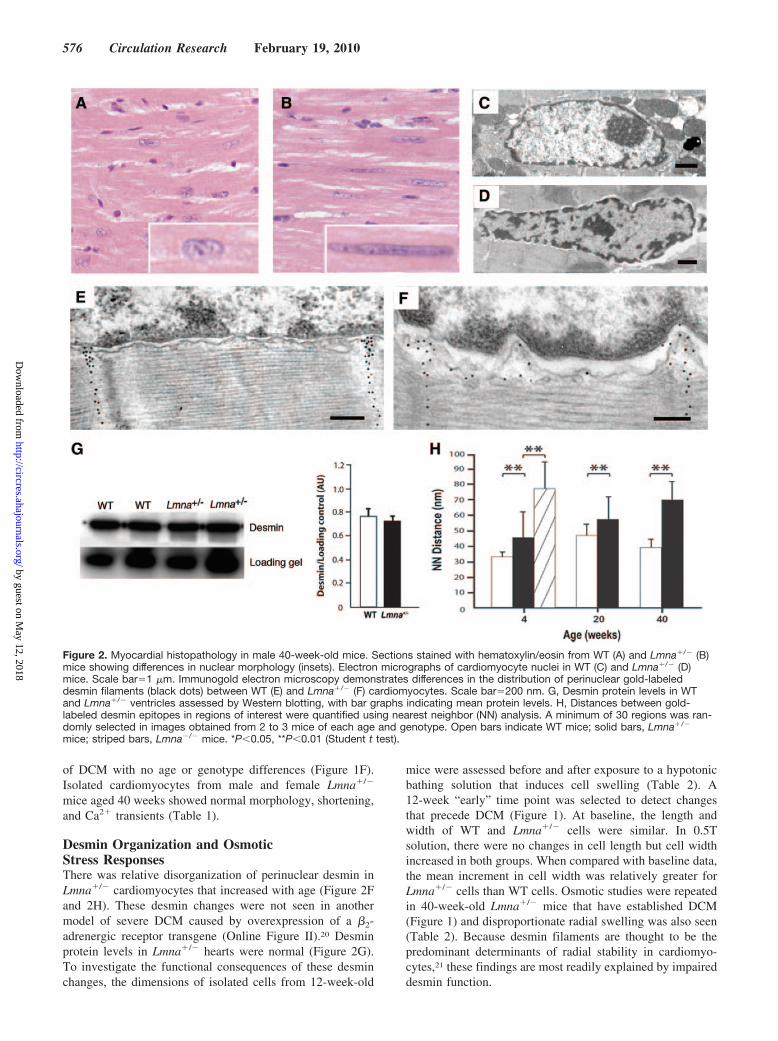
Desmin Organization and OsmoticStress ResponsesThere was relative disorganization of perinuclear desmin inLmna�/� cardiomyocytes that increased with age (Figure 2Fand 2H). These desmin changes were not seen in anothermodel of severe DCM caused by overexpression of a �2-adrenergic receptor transgene (Online Figure II).20 Desminprotein levels in Lmna�/� hearts were normal (Figure 2G).To investigate the functional consequences of these desminchanges, the dimensions of isolated cells from 12-week-old
mice were assessed before and after exposure to a hypotonicbathing solution that induces cell swelling (Table 2). A12-week “early” time point was selected to detect changesthat precede DCM (Figure 1). At baseline, the length andwidth of WT and Lmna�/� cells were similar. In 0.5Tsolution, there were no changes in cell length but cell widthincreased in both groups. When compared with baseline data,the mean increment in cell width was relatively greater forLmna�/� cells than WT cells. Osmotic studies were repeatedin 40-week-old Lmna�/� mice that have established DCM(Figure 1) and disproportionate radial swelling was also seen(Table 2). Because desmin filaments are thought to be thepredominant determinants of radial stability in cardiomyo-cytes,21 these findings are most readily explained by impaireddesmin function.
Figure 2. Myocardial histopathology in male 40-week-old mice. Sections stained with hematoxylin/eosin from WT (A) and Lmna�/� (B)mice showing differences in nuclear morphology (insets). Electron micrographs of cardiomyocyte nuclei in WT (C) and Lmna�/� (D)mice. Scale bar�1 �m. Immunogold electron microscopy demonstrates differences in the distribution of perinuclear gold-labeleddesmin filaments (black dots) between WT (E) and Lmna�/� (F) cardiomyocytes. Scale bar�200 nm. G, Desmin protein levels in WTand Lmna�/� ventricles assessed by Western blotting, with bar graphs indicating mean protein levels. H, Distances between gold-labeled desmin epitopes in regions of interest were quantified using nearest neighbor (NN) analysis. A minimum of 30 regions was ran-domly selected in images obtained from 2 to 3 mice of each age and genotype. Open bars indicate WT mice; solid bars, Lmna�/�
mice; striped bars, Lmna�/� mice. *P�0.05, **P�0.01 (Student t test).
576 Circulation Research February 19, 2010
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from

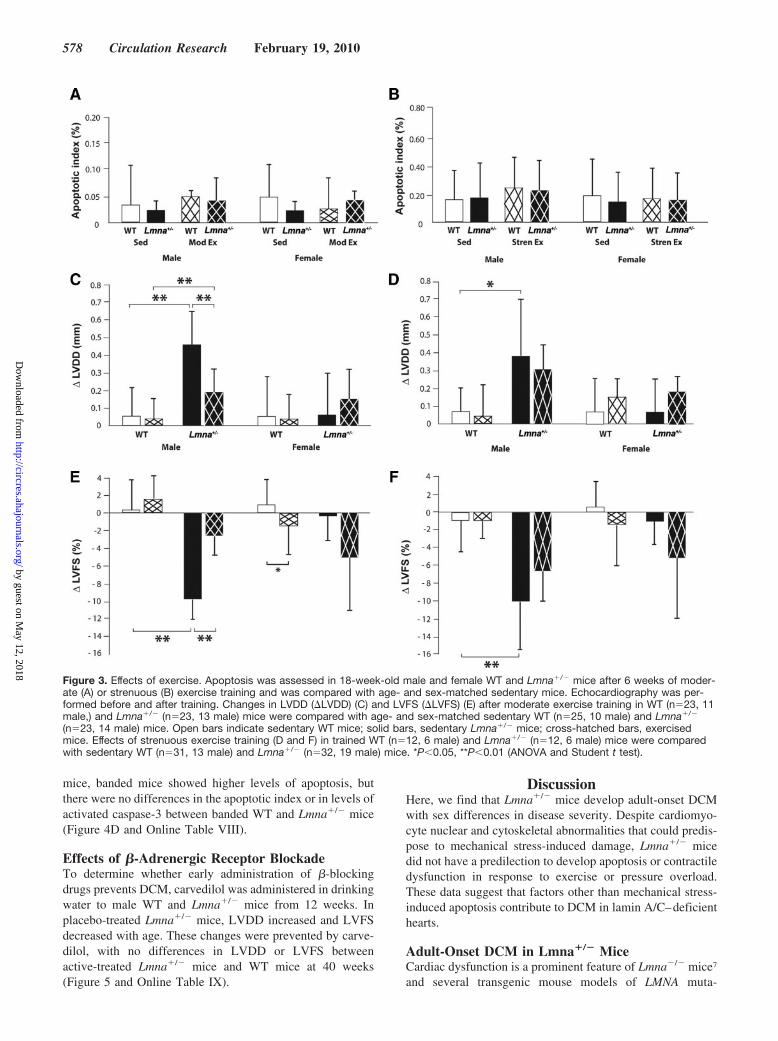
Effects of Exercise TrainingThere were no differences in baseline exercise tolerance testor echocardiographic parameters in WT and Lmna�/� miceaged 12 weeks. After 6 weeks of moderate exercise training,exercise performance improved in both groups with anincrease in the proportion of mice reaching the final stage ofthe exercise tolerance test and a reduction in the number ofstimuli received (Online Table II). Cardiomyocyte nuclearmorphology was qualitatively similar in sedentary and trainedLmna�/� hearts with no evidence of an exercise-inducedincrease in apoptosis, assessed by TUNEL assay or levels ofactivated caspase-3 (Figure 3A and Online Table III). LVend-diastolic diameter (LVDD) and fractional shortening(LVFS) were similar in trained and sedentary male WT mice.Trained male Lmna�/� mice had relatively higher LVDD andlower LVFS than trained WT mice. However, LV size andfunction in trained Lmna�/� mice were not significantlyworse than in sedentary Lmna�/� mice (Figure 3C and 3E;Online Table II). When baseline and postexercise data werecompared, trained male Lmna�/� mice had relatively lesschange in LVDD (�LVDD) and LVFS (�LVFS) than sed-entary male Lmna�/� mice over the same time period. Therewere only small differences in LV size and between sedentaryfemale WT and Lmna�/� mice, and these were unchanged bymoderate exercise.
To determine the effects of exercise intensity, a separatecohort of mice underwent a regimen of twice-weekly stren-uous exercise, in which the degree of difficulty was up-titrated to maximal tolerated levels at weekly intervals aspeak performance improved. A similar exercise protocolexacerbates myocardial histopathology in mdx mice that lackthe cytoskeletal protein, dystrophin.16 This regimen proved tobe initially more difficult for Lmna�/� mice; however,
exercise performance improved with training (Online TableIV). There were no differences in apoptosis between seden-tary and trained WT and Lmna�/� mice (Figure 3B andOnline Table V). LVDD and LVFS in trained male andfemale Lmna�/� mice remained similar to sedentary mice(Figure 3D and 3F; Online Table IV). The beneficial effectsof regular moderate-intensity exercise training on the�LVDD and �LVFS seen in male Lmna�/� mice were notrecapitulated with the intermittent high-intensity exerciseregimen.
Effects of TACTAC was performed in 12-week-old mice. All male micedeveloped myocardial hypertrophy with an increase in theheart weight/body weight ratio (Figure 4C). Male WT andLmna�/� mice also showed LV dilatation and reducedcontraction with no genotype differences at 7, 14 or 21 dayspost-TAC (Figure 4A and 4B; Online Table VI). Responsesto TAC in female WT and Lmna�/� mice were similar tomale mice (Figure 4A and 4B; Online Table VII).
Mitogen-activated protein kinase (MAPK) signaling path-ways regulate myocyte growth and survival in response tomechanical stress22 and have recently been implicated incardiac dysfunction in laminopathies.23 Western blot analysisshowed increased levels of phosphorylated extracellularsignal-regulated kinase (ERK)1/2 in LV tissue at 14 daysafter TAC, with no change in JNK or p38 proteins. Therewere no differences in indices of MAPK activation atbaseline or after TAC between WT and Lmna�/� mice(Figure 4E through 4H). When compared with sham-operated
Table 2. Osmotic Studies in Ventricular Cardiomyocytes
12 Weeks 40 Weeks
WT Lmna�/� WT Lmna�/�
No. cells 120 120 79 80
Baseline
Length (�m) 122.3�11.4 120.7�7.3 137.4�6.0 141.9�4.3
Width (�m) 21.4�1.6 20.3�1.6 26.4�0.9 25.6�1.3
1T
Length (�m) 121.8�12.3 120.1�8.2 132.0�9.0 137.6�9.1
Width (�m) 21.5�1.7 20.0�1.9 26.8�1.5 26.5�2.2
0.5T
Length (�m) 124.1�13.1 122.9�8.9 133.8�8.3 138.9�8.6
Width (�m) 23.6�1.8 24.4�2.0 29.9�0.7 30.6�1.4
1T (washout)
Length (�m) 121.9�12.1 120.4�8.4 128.9�12.8 133.0�13.0
Width (�m) 21.3�1.8 20.8�2.2 27.0�1.3 26.8�1.9
Change(baseline �0.5T)
Length (%) 1.4�1.4 1.8�2.3 2.5�4.0 2.0�3.5
Width (%) 10.4�0.8 20.2�1.7* 13.4�1.6 19.8�0.7*
Ventricular cardiomyocytes were isolated from 12- and 40-week-old maleWT and Lmna�/� mice (n�4–6 each group) and placed in basic Tyrodesolution. Cell dimensions were measured at baseline and 8 minutes aftersequential bathing in isotonic (1T) and hypotonic (0.5T) Tyrode solution.*P�0.05 vs WT (Student t test).
Table 1. Functional Studies of Ventricular Cardiomyocytes
Male Female
WT Lmna�/� WT Lmna�/�
Myocyte length (�m) 121�10 126�8 118�8 122�5
Myocyte width (�m) 28�4 27�3 26�3 24�2
Sarcomere length (�m) 1.61�0.03 1.62�0.04 1.57�0.06 1.60�0.05
Shortening (%) 4.4�1.2 4.2�1.6 4.9�1.7 3.8�1.1
Time to peak shortening(ms)
67�8 66�16 65�5 60�7
Time to 50% relaxation(ms)
70�18 64�22 62�12 64�12
Baseline Ca2� transient 0.80�0.07 0.78�0.08 NA 0.76�0.06
Peak transient amplitude 0.23�0.03 0.25�0.08 NA 0.24�0.04
Time to peak amplitude(ms)
48�10 41�8 NA 34�8
Time to 50% decay (ms) 120�38 144�29 NA 164�29
Ventricular cardiomyocytes (n�80–84 each group) were isolated from WT(n�17, 11 male) and Lmna�/� (n�17, 10 male) mice aged 40 weeks.Contractile function was assessed using edge detection. Cells were loaded withthe Ca2�-sensitive fluorescent dye Indo-1/AM, and intracellular �Ca2� wasmeasured as the change in the 405:485 nm emission ratio. Values are reportedas relative change in fluorescence ratio units following subtraction of back-ground values. NA indicates not available.
Chandar et al Mechanisms of Lamin A/C–Deficient Cardiomyopathy 577
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
mice, banded mice showed higher levels of apoptosis, butthere were no differences in the apoptotic index or in levels ofactivated caspase-3 between banded WT and Lmna�/� mice(Figure 4D and Online Table VIII).
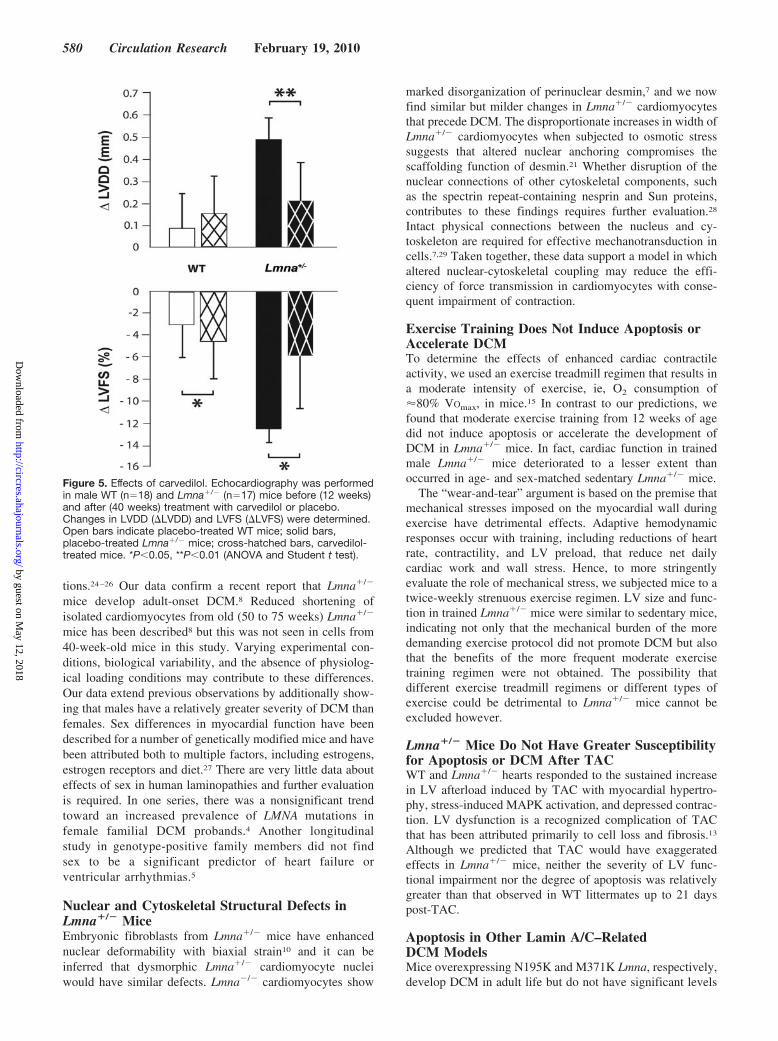
Effects of �-Adrenergic Receptor BlockadeTo determine whether early administration of �-blockingdrugs prevents DCM, carvedilol was administered in drinkingwater to male WT and Lmna�/� mice from 12 weeks. Inplacebo-treated Lmna�/� mice, LVDD increased and LVFSdecreased with age. These changes were prevented by carve-dilol, with no differences in LVDD or LVFS betweenactive-treated Lmna�/� mice and WT mice at 40 weeks(Figure 5 and Online Table IX).
DiscussionHere, we find that Lmna�/� mice develop adult-onset DCMwith sex differences in disease severity. Despite cardiomyo-cyte nuclear and cytoskeletal abnormalities that could predis-pose to mechanical stress-induced damage, Lmna�/� micedid not have a predilection to develop apoptosis or contractiledysfunction in response to exercise or pressure overload.These data suggest that factors other than mechanical stress-induced apoptosis contribute to DCM in lamin A/C–deficienthearts.
Adult-Onset DCM in Lmna�/� MiceCardiac dysfunction is a prominent feature of Lmna�/� mice7
and several transgenic mouse models of LMNA muta-
Figure 3. Effects of exercise. Apoptosis was assessed in 18-week-old male and female WT and Lmna�/� mice after 6 weeks of moder-ate (A) or strenuous (B) exercise training and was compared with age- and sex-matched sedentary mice. Echocardiography was per-formed before and after training. Changes in LVDD (�LVDD) (C) and LVFS (�LVFS) (E) after moderate exercise training in WT (n�23, 11male,) and Lmna�/� (n�23, 13 male) mice were compared with age- and sex-matched sedentary WT (n�25, 10 male) and Lmna�/�
(n�23, 14 male) mice. Open bars indicate sedentary WT mice; solid bars, sedentary Lmna�/� mice; cross-hatched bars, exercisedmice. Effects of strenuous exercise training (D and F) in trained WT (n�12, 6 male) and Lmna�/� (n�12, 6 male) mice were comparedwith sedentary WT (n�31, 13 male) and Lmna�/� (n�32, 19 male) mice. *P�0.05, **P�0.01 (ANOVA and Student t test).
578 Circulation Research February 19, 2010
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from

Figure 4. Effects of TAC. TAC was performed in 12-week-old WT (n�14, 7 male) and Lmna�/� (n�15, 7 male) mice. LVDD (A), LVFS(B), heart weight/body weight ratio (HW/BW) (C), and apoptosis (D) at 14 days post-TAC were compared with sham-operated WT(n�10, 5 male) and Lmna�/� (n�9, 5 male) mice. E through H, MAPK pathway activation was evaluated in male WT and Lmna�/� micebefore and at 14 days after TAC. Levels of phosphorylated ERK1/2, JNK, and p38 were assessed after normalization to �-actin andexpressed as a ratio with total ERK1/2, JNK, and p38. Representative gels (E) and quantification of replicates (n�3) (F through H) areshown. I, Mean aortic gradients at 14 days in sham-operated and TAC mice. Open bars indicate WT mice; solid bars, Lmna�/� mice.*P�0.05, **P�0.01 (ANOVA and Student t test).
Chandar et al Mechanisms of Lamin A/C–Deficient Cardiomyopathy 579
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from
tions.24–26 Our data confirm a recent report that Lmna�/�
mice develop adult-onset DCM.8 Reduced shortening ofisolated cardiomyocytes from old (50 to 75 weeks) Lmna�/�
mice has been described8 but this was not seen in cells from40-week-old mice in this study. Varying experimental con-ditions, biological variability, and the absence of physiolog-ical loading conditions may contribute to these differences.Our data extend previous observations by additionally show-ing that males have a relatively greater severity of DCM thanfemales. Sex differences in myocardial function have beendescribed for a number of genetically modified mice and havebeen attributed both to multiple factors, including estrogens,estrogen receptors and diet.27 There are very little data abouteffects of sex in human laminopathies and further evaluationis required. In one series, there was a nonsignificant trendtoward an increased prevalence of LMNA mutations infemale familial DCM probands.4 Another longitudinalstudy in genotype-positive family members did not findsex to be a significant predictor of heart failure orventricular arrhythmias.5
Nuclear and Cytoskeletal Structural Defects inLmna�/� MiceEmbryonic fibroblasts from Lmna�/� mice have enhancednuclear deformability with biaxial strain10 and it can beinferred that dysmorphic Lmna�/� cardiomyocyte nucleiwould have similar defects. Lmna�/� cardiomyocytes show
marked disorganization of perinuclear desmin,7 and we nowfind similar but milder changes in Lmna�/� cardiomyocytesthat precede DCM. The disproportionate increases in width ofLmna�/� cardiomyocytes when subjected to osmotic stresssuggests that altered nuclear anchoring compromises thescaffolding function of desmin.21 Whether disruption of thenuclear connections of other cytoskeletal components, suchas the spectrin repeat-containing nesprin and Sun proteins,contributes to these findings requires further evaluation.28
Intact physical connections between the nucleus and cy-toskeleton are required for effective mechanotransduction incells.7,29 Taken together, these data support a model in whichaltered nuclear-cytoskeletal coupling may reduce the effi-ciency of force transmission in cardiomyocytes with conse-quent impairment of contraction.
Exercise Training Does Not Induce Apoptosis orAccelerate DCMTo determine the effects of enhanced cardiac contractileactivity, we used an exercise treadmill regimen that results ina moderate intensity of exercise, ie, O2 consumption of80% VOmax, in mice.15 In contrast to our predictions, wefound that moderate exercise training from 12 weeks of agedid not induce apoptosis or accelerate the development ofDCM in Lmna�/� mice. In fact, cardiac function in trainedmale Lmna�/� mice deteriorated to a lesser extent thanoccurred in age- and sex-matched sedentary Lmna�/� mice.
The “wear-and-tear” argument is based on the premise thatmechanical stresses imposed on the myocardial wall duringexercise have detrimental effects. Adaptive hemodynamicresponses occur with training, including reductions of heartrate, contractility, and LV preload, that reduce net dailycardiac work and wall stress. Hence, to more stringentlyevaluate the role of mechanical stress, we subjected mice to atwice-weekly strenuous exercise regimen. LV size and func-tion in trained Lmna�/� mice were similar to sedentary mice,indicating not only that the mechanical burden of the moredemanding exercise protocol did not promote DCM but alsothat the benefits of the more frequent moderate exercisetraining regimen were not obtained. The possibility thatdifferent exercise treadmill regimens or different types ofexercise could be detrimental to Lmna�/� mice cannot beexcluded however.
Lmna�/� Mice Do Not Have Greater Susceptibilityfor Apoptosis or DCM After TACWT and Lmna�/� hearts responded to the sustained increasein LV afterload induced by TAC with myocardial hypertro-phy, stress-induced MAPK activation, and depressed contrac-tion. LV dysfunction is a recognized complication of TACthat has been attributed primarily to cell loss and fibrosis.13
Although we predicted that TAC would have exaggeratedeffects in Lmna�/� mice, neither the severity of LV func-tional impairment nor the degree of apoptosis was relativelygreater than that observed in WT littermates up to 21 dayspost-TAC.
Apoptosis in Other Lamin A/C–RelatedDCM ModelsMice overexpressing N195K and M371K Lmna, respectively,develop DCM in adult life but do not have significant levels
Figure 5. Effects of carvedilol. Echocardiography was performedin male WT (n�18) and Lmna�/� (n�17) mice before (12 weeks)and after (40 weeks) treatment with carvedilol or placebo.Changes in LVDD (�LVDD) and LVFS (�LVFS) were determined.Open bars indicate placebo-treated WT mice; solid bars,placebo-treated Lmna�/� mice; cross-hatched bars, carvedilol-treated mice. *P�0.05, **P�0.01 (ANOVA and Student t test).
580 Circulation Research February 19, 2010
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from

of LV apoptosis under baseline conditions.25,26 Young (4-week-old) Lmna�/� mice lack LV free wall apoptosis but doshow CD and apoptosis in atrioventricular node cells.8 Takentogether, these observations indicate that myocardial apopto-sis is unlikely to be a primary cause of DCM in any of thesemodels, and further suggest that CD and LV dysfunction canoccur as independent pathologies.
Exercise and Carvedilol Favorably ModifyDisease ProgressionExercise training at moderate intensity improves peak oxygenconsumption increases exercise time and improves quality oflife in patients with heart failure.30 These benefits have beenattributed to factors such as reduced resting heart rate andblood pressure, increased coronary blood flow, improvedvascular endothelial function, reduced platelet aggregation,reduced oxidative stress, improved lipid and blood glucoselevels, and modification of obesity and mental stress.30
Although it is generally assumed that exercise in heart failureis beneficial, studies in murine models suggest that the effectsmay vary according to the cause of DCM. For example,cytoskeletal protein models such as desmin-null, dysferlin-null, and dystrophin-deficient mdx mice, have a worse out-come with exercise.16,31,32
Carvedilol also has negative inotropic and chronotropicactions, as well as potent vasodilation, antiischemic, antiapo-ptotic and antioxidant effects.33 Given that mechanical stress-induced apoptosis does not explain lamin A/C–related DCM,it seems likely that the hemodynamic and antiapoptoticbenefits of exercise and carvedilol are relatively less impor-tant than other direct myocardial effects. Antioxidant actionsof exercise and carvedilol are of particular interest in view ofrecent data implicating oxidative stress in heart failure and inother laminopathy phenotypes, including lipodystrophy, pre-mature ageing, and amyotrophic quadricipital syndrome withcardiac involvement.34,35 Further studies of the role of oxida-tive stress and changes induced by exercise training andcarvedilol in Lmna�/� hearts are warranted.
Clinical ImplicationsBecause individuals at risk of DCM can be recognized early bygenetic testing or by presentation with CD, identification ofpreventive strategies is imperative. The “structural hypothesis”predicts that enhanced cardiac contractile activity would accel-erate DCM in patients with LMNA mutations and thus exerciseshould be avoided. Although our study was designed to deter-mine the role of mechanical stress as a primary pathogenicfactor, potential adverse effects once DCM is manifest cannot beexcluded. Genotype-positive individuals who engage in highlevel competitive sport for prolonged periods (10 years orlonger) have recently been shown to have a 3- to 4-foldincreased risk of adverse cardiac events.5 Although the numberswere relatively small, these data urge caution in exercise recom-mendations to families. An exciting finding in young maleLmna�/� mice was that regular moderate exercise and carvedilolappeared to protect against DCM development. Whether thesebenefits are also seen in older female mice and patients withestablished DCM will be important to ascertain.
AcknowledgmentsWe thank Colin L. Stewart for providing the Lmna knockout miceand Xiao-Jun Du for �2-adrenergic receptor transgenic mice. Wealso thank Mark Hicks for pharmacological supplies; Matt Wand forstatistical advice; Robert Graham and Peter Macdonald for helpfuldiscussions; and Aisling McMahon, Jan Michalicek, and IshtiaqAhmed for assistance with physiological studies.
Sources of FundingThis work was supported by the National Health and MedicalResearch Council of Australia, the Sylvia and Charles ViertelCharitable Foundation, and the Cardiac Society of Australia andNew Zealand.
DisclosuresNone.
References1. Fatkin D, Otway R, Richmond Z. Genetics of dilated cardiomyopathy.
Heart Fail Clin. In press.2. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M,
Atherton J, Vidaillet HJ, Spudich S, De Girolami U, Seidman JG,Seidman CE. Missense mutations in the rod domain of the lamin A/Cgene as causes of dilated cardiomyopathy and conduction-system disease.N Engl J Med. 1999;341:1715–1724.
3. Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, LiD, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/Cmutation analysis in a cohort of 324 unrelated patients with idiopathic orfamilial dilated cardiomyopathy. Am Heart J. 2008;156:161–169.
4. Taylor MRG, Fain PR, Sinagra G, Robinson ML, Robertson AD, CarnielE, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, BoucekMM, Lascor J, Moss AC, Li WLP, Stetler GL, Muntoni F, Bristow MR,Mestroni L. Natural history of dilated cardiomyopathy due to lamin A/Cgene mutations. J Am Coll Cardiol. 2003;4:771–780.
5. Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, MannarinoS, Gambarin F, Favalli V, Grasso M, Agozzino M, Campana C, Gavazzi A,Febo O, Marini M, Landolina M, Mortara A, Piccolo G, Vigano M, TavazziL, Arbustini E. Long-term outcome and risk stratification in dilated cardio-laminopathies. J Am Coll Cardiol. 2008;52:1250–1260.
6. Sullivan T, Escalante-Alcade D, Bhatt H, Anver M, Bhat N, NagashimaK, Stewart CL, Burke B. Loss of A-type lamin expression compromisesnuclear envelope integrity leading to muscular dystrophy. J Cell Biol.1999;147:913–919.
7. Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D,Kesteven S, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL,Martin D, Feneley MP, Fatkin D. Defects in nuclear structure andfunction promote dilated cardiomyopathy in lamin A/C–deficient mice.J Clin Invest. 2004;113:357–369.
8. Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, SherwoodM, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, ConnerDA, Berul CI, Seidman CE, Seidman JG. Lamin A/C haploinsufficiencycauses dilated cardiomyopathy and apoptosis-triggered cardiac con-duction system disease. J Mol Cell Cardiol. 2008;44:293–303.
9. Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, KammRD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclearmechanics and mechanotransduction. J Clin Invest. 2004;113:370–378.
10. Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT.Lamins A and C but not lamin B1 regulate nuclear mechanics. J BiolChem. 2006;35:25768–25780.
11. Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease: whydo ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J Cell Sci. 2001;114:9–19.
12. Burke B, Stewart CL. Life at the edge: the nuclear envelope and humandisease. Nat Rev Mol Cell Biol. 2002;3:575–585.
13. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z,Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor-�mediates cardiac remodelling and ventricular dysfunction after pressureoverload state. Circulation. 2007;115:1398–1407.
14. Packer M, Bristow MR, Cohn JN, et al. The effect of carvedilol onmorbidity and mortality in patients with chronic heart failure. N EnglJ Med. 1996;334:1349–1355.
Chandar et al Mechanisms of Lamin A/C–Deficient Cardiomyopathy 581
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from

15. Fernando P, Bonen A, Hoffman-Goetz L. Predicting submaximal oxygenconsumption during treadmill running in mice. Can J Physiol Pharmacol.1993;71:854–857.
16. Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S. Progression ofdystrophic features and activation of mitogen-activated protein kinasesand calcineurin by physical exercise in hearts of mdx mice. FEBS Lett.2002;520:18–24.
17. Nishio R, Shioi T, Sasayama S, Matsumori A. Carvedilol increases theproduction of interleukin-12 and interferon-� and improves the survivalof mice infected with the encephalomyocarditis virus. J Am Coll Cardiol.2003;41:340–345.
18. Suleymanian MA, Baumgarten CM. Osmotic gradient-induced waterpermeation across the sarcolemma of rabbit ventricular myocytes. J GenPhysiol. 1996;107:503–514.
19. Seul M, O’Gorman L, Sammon M. Analysis of point patterns. In: Seul M,O’Gorman L, Sammon M, eds. Practical Algorithms for Image Analysis.Cambridge, United Kingdom: Cambridge University Press; 2000:221–245.
20. Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age-dependent cardiomyopathy and heart failure phenotype in mice overex-pressing �2-adrenergic receptors in the heart. Cardiovasc Res. 2000;48:448–454.
21. Roos KP. Length, width, and volume changes in osmotically stressedmyocytes. Am J Physiol Heart Circ Physiol. 1986;251:H1373–H1378.
22. Wang Y. Mitogen-activated protein kinases in heart development anddiseases. Circulation. 2007;116:1413–1423.
23. Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G,Worman HJ. Activation of MAPK pathways links LMNA mutations tocardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest.2007;117:1282–1293.
24. Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E,Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, MalissenM, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutationdevelops muscular dystrophy and dilated cardiomyopathy similar to humanstriated muscle laminopathies. Hum Mol Genet. 2005;14:155–169.
25. Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of anLMNA-N195K variant of A-type lamins results in cardiac conductiondefects and death in mice. Hum Mol Genet. 2005;14:2167–2180.
26. Wang Y, Herron AJ, Worman HJ. Pathology and nuclear abnormalities inhearts of transgenic mice expressing M371K lamin A encoded by anLMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum MolGenet. 2006;15:2479–2489.
27. Konhilas JP, Leinwand LA. The effects of biological sex and diet on thedevelopment of heart failure. Circulation. 2007;116:2747–2759.
28. Warren DT, Zhang Q, Weissberg PL, Shanahan CM. Nesprins: intra-cellular scaffolds that maintain cell architecture and coordinate cellfunction? Expert Rev Mol Med. 2005;7:1–14.
29. Lee JSH, Hale CM, Panorchan P, Khatau SB, George JP, Tseng Y,Stewart CL, Hodzic D, Wirtz D. Nuclear lamin A/C deficiency inducesdefects in cell mechanics, polarization, and migration. Biophys J. 2007;93:2542–2552.
30. Pina IL, Apstein CS, Balady GJ, Belardinelli R, Chaitman BR, DuschaBD, Fletcher BJ, Fleg JL, Myers JN, Sullivan MJ. Exercise and heartfailure. A statement from the American Heart Association Committee onexercise, rehabilitation, and prevention. Circulation. 2003;107:1210–1225.
31. Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, GerdesAM, Capetanaki Y. The absence of desmin leads to cardiomyocytehypertrophy and cardiac dilation with compromised systolic function.J Mol Cell Cardiol. 1999;31:2063–2076.
32. Han R, Bansal D, Miyake K, Muniz VP, Weiss RM, McNeill PL,Campbell KP. Dysferlin-mediated membrane repair protects the heartfrom stress-induced left ventricular injury. J Clin Invest. 2007;117:1805–1813.
33. Cheng J, Kamiya K, Kodama I. Carvedilol: molecular and cellular basisfor its multifaceted therapeutic potential. Cardiovasc Drug Rev. 2001;19:152–171.
34. Caron M, Auclair M, Donadille B, et al. Human lipodystrophies linked tomutations in A-type lamins and to HIV protease inhibitor therapy are bothassociated with prelamin A accumulation, oxidative stress and prematurecellular senescence. Cell Death Differ. 2007;14:1759–1767.
35. Charniot JC, Bonnefont-Rousselot D, Marchand C, Zerhouni K, VignatN, Peynet J, Plotkine M, Legrand A, Artigou JY. Oxidative stress impli-cation in a new phenotype of amyotrophic quadricipital syndrome withcardiac involvement due to lamin A/C mutation. Free Radic Res.2007;41:424–431.
Novelty and Significance
What Is Known?
● Mutations in the LMNA gene, which encodes the nuclear laminaproteins lamin A and lamin C, are the most common cause offamilial DCM.
● Lmna�/� mice develop adult-onset DCM.● Lamin A/C deficiency in humans and in mice alters nuclear morphology
and nuclear mechanical properties.
What New Information Does This Article Contribute?
● In vivo evidence that mechanical stress-induced apoptosis is not aprimary determinant of DCM in Lmna�/� mice.
● Support for a model in which changes in cytoskeletal properties causedby loss of normal nuclear anchoring impair force transmission inLmna�/� cardiomyocytes.
● First demonstration that regular moderate exercise training and earlyadministration of carvedilol can modify disease progression in laminA/C–deficient hearts.
LMNA mutations are the most common cause of familial dilatedcardiomyopathy (DCM), but the mechanisms linking nuclear defects
to contractile dysfunction are unresolved. Lamin A/C–deficientnuclei have altered cytoarchitecture and structural properties, and,hence, mechanical stress-induced apoptosis has been widelyproposed as a key factor in DCM pathogenesis. We evaluated this“structural hypothesis” in heterozygous Lmna knockout (Lmna�/�)mice and found that despite cardiomyocyte nuclear abnormalities,young Lmna�/� mice subjected to exercise training or thoracicaortic constriction did not have increased vulnerability to apoptosisor accelerated DCM. In addition to nuclear defects, Lmna�/�
cardiomyocytes show disorganization of perinuclear desmin andexaggerated responses to hypo-osmotic stress. These observationssupport an alternative disease model in which loss of nuclear–cytoskeletal connections destabilizes the cytoskeletal scaffoldingand impairs force transmission in cardiomyocytes. Early recognitionof individuals at risk in families with LMNA mutations provides anopportunity for intervention to prevent DCM, but this has not beeninvestigated previously. Here we find, for the first time, that regularmoderate exercise and administration of carvedilol to young maleLmna�/� mice can attenuate the development of DCM. Thesefindings have implications for exercise recommendations andprovide a basis for clinical trials in presymptomatic genotype-positive family members.
582 Circulation Research February 19, 2010
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from

Feneley and Diane FatkinKesteven, Lina Karlsdotter, Shweta Natarajan, Arthur Carlton, Stephen Rainer, Michael P.
Nikolova-Krstevski, Yoshinori Yasuoka, Margaret Gardiner-Garden, Jianxin Wu, Scott Suchitra Chandar, Li Sze Yeo, Christiana Leimena, Ju-Chiat Tan, Xiao-Hui Xiao, Vesna
CardiomyopathyDeficient Dilated−Effects of Mechanical Stress and Carvedilol in Lamin A/C
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2009 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.109.2043882010;106:573-582; originally published online December 17, 2009;Circ Res.
http://circres.ahajournals.org/content/106/3/573World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2009/12/17/CIRCRESAHA.109.204388.DC1Data Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 12, 2018
http://circres.ahajournals.org/D
ownloaded from

SUPPLEMENT MATERIAL 1
EFFECTS OF MECHANICAL STRESS AND CARVEDILOL IN
LAMIN A/C-DEFICIENT DILATED CARDIOMYOPATHY
SUPPLEMENT MATERIAL 2
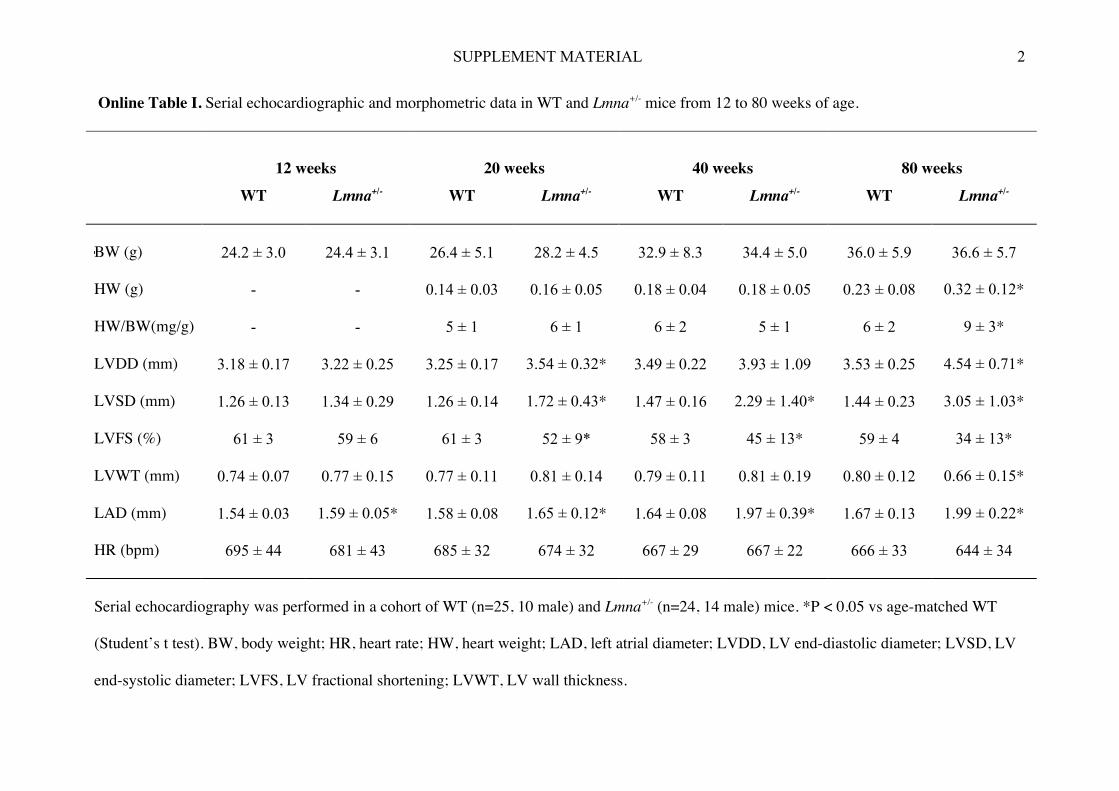
Online Table I. Serial echocardiographic and morphometric data in WT and Lmna+/- mice from 12 to 80 weeks of age.
12 weeks
20 weeks
40 weeks
80 weeks
WT Lmna+/- WT Lmna+/- WT Lmna+/- WT Lmna+/-
BW (g)
24.2 ± 3.0
24.4 ± 3.1
26.4 ± 5.1
28.2 ± 4.5
32.9 ± 8.3
34.4 ± 5.0
36.0 ± 5.9
36.6 ± 5.7
HW (g) - - 0.14 ± 0.03 0.16 ± 0.05 0.18 ± 0.04 0.18 ± 0.05 0.23 ± 0.08 0.32 ± 0.12*
HW/BW(mg/g) - - 5 ± 1 6 ± 1 6 ± 2 5 ± 1 6 ± 2 9 ± 3*
LVDD (mm) 3.18 ± 0.17 3.22 ± 0.25 3.25 ± 0.17 3.54 ± 0.32* 3.49 ± 0.22 3.93 ± 1.09 3.53 ± 0.25 4.54 ± 0.71*
LVSD (mm) 1.26 ± 0.13 1.34 ± 0.29 1.26 ± 0.14 1.72 ± 0.43* 1.47 ± 0.16 2.29 ± 1.40* 1.44 ± 0.23 3.05 ± 1.03*
LVFS (%) 61 ± 3 59 ± 6 61 ± 3 52 ± 9* 58 ± 3 45 ± 13* 59 ± 4 34 ± 13*
LVWT (mm) 0.74 ± 0.07 0.77 ± 0.15 0.77 ± 0.11 0.81 ± 0.14 0.79 ± 0.11 0.81 ± 0.19 0.80 ± 0.12 0.66 ± 0.15*
LAD (mm) 1.54 ± 0.03 1.59 ± 0.05* 1.58 ± 0.08 1.65 ± 0.12* 1.64 ± 0.08 1.97 ± 0.39* 1.67 ± 0.13 1.99 ± 0.22*
HR (bpm) 695 ± 44 681 ± 43 685 ± 32 674 ± 32 667 ± 29 667 ± 22 666 ± 33 644 ± 34
Serial echocardiography was performed in a cohort of WT (n=25, 10 male) and Lmna+/- (n=24, 14 male) mice. *P < 0.05 vs age-matched WT
(Student’s t test). BW, body weight; HR, heart rate; HW, heart weight; LAD, left atrial diameter; LVDD, LV end-diastolic diameter; LVSD, LV
end-systolic diameter; LVFS, LV fractional shortening; LVWT, LV wall thickness.

3
Online Table II. Echocardiographic data in 18-week-old mice with and without 6 weeks of moderate exercise training.
Male
Female
Sedentary Trained Sedentary Trained
WT Lmna+/- WT Lmna+/- WT Lmna+/- WT Lmna+/-
Final stage achieved:
- Baseline ETT
- 6-week ETT
- -
- -
3 (43%)
7 (100%)
8 (89%)
9 (100%)
- -
- -
6 (60%)
10 (100%)
7 (88%)
8 (100%)
No. stimuli: - Baseline ETT
- 6-week ETT
- -
- -
6.1 ± 8.8
0.1 ± 0.4
7.4 ± 7.0
1.2 ± 1.6
- -
- -
2.1 ± 3.2
0.8 ± 0.8
2.6 ± 1.2
1.0 ± 1.1 BW (g)
37.6 ± 5.1
35.1 ± 3.1
29.1 ± 1.7†
28.5 ± 1.9†
26.8 ± 3.5
25.3 ± 1.8
24.1 ± 2.1†
23.8 ± 1.7
LVDD (mm) 3.36 ± 0.14 3.70 ± 0.31* 3.41 ± 0.09 3.56 ± 0.17* 3.17 ± 0.15 3.29 ± 0.11* 3.28 ± 0.15 3.32 ± 0.15
LVSD (mm) 1.37 ± 0.13 1.91 ± 0.37* 1.41 ± 0.13 1.63±0.21*† 1.19 ± 0.10 1.43 ± 0.37* 1.30 ± 0.15† 1.45 ± 0.28
LVFS (%) 59 ± 3 49 ± 7* 59 ± 3 54 ± 4*† 62 ± 2 57 ± 10* 60 ± 3 57 ± 7
LAD (mm) 1.65 ± 0.09 1.73 ± 0.08 1.67 ± 0.09 1.69 ± 0.11 1.54 ± 0.04 1.53 ± 0.04 1.62 ± 0.07† 1.68 ± 0.12†

4
HR (bpm) 705 ± 28 687 ± 23 671 ± 57 677 ± 50 672 ± 28 654 ± 35 693 ± 50 666 ± 49
Cardiac function in trained WT (n=23, 11 male) and Lmna+/- (n=23, 13 male) mice was compared with age-matched sedentary WT (n=25, 10
male) and Lmna+/- (n=23, 14 male) mice. *P<0.05 vs WT, †P<0.05 vs sedentary mice (ANOVA and Student’s t test).
.

5
Online Table III. Evaluation of apoptosis after moderate exercise training.
Male
Female
Sedentary Trained Sedentary Trained
WT Lmna+/- WT Lmna+/- WT Lmna+/- WT Lmna+/-
TUNEL assay
TUNEL +ve cells 0.8 ± 1.7 1.0 ± 0.8 1.8 ± 1.3 1.8 ± 1.7 1.3 ± 1.5 0.8 ± 0.7 0.8 ± 0.9 1.6 ± 1.1
Total nuclei 3384 ± 565 3705 ± 1420 3123 ± 1450 3737 ± 1179 2730 ± 801 3166 ± 602 2887 ± 719 3625 ± 887
Apoptotic index (%) 0.03 ± 0.06 0.02 ± 0.02 0.05 ± 0.01 0.04 ± 0.03 0.05 ± 0.06 0.02 ± 0.02 0.03 ± 0.04 0.04 ± 0.02
Activated Caspase-3
Caspase-3 +ve cells 0 0.2 ± 0.4 0.5 ± 0.8 0.3 ± 0.4 0.3 ± 0.5 0.3 ± 0.5 0 0
Total nuclei 1977 ± 82 1988 ± 162 2059 ± 293 1934 ± 419 2045 ± 177 1988 ± 146 2316 ± 471 1889 ± 140
Sequential images from 14-20 random fields in LV sections from sedentary and trained WT (n=21, 11 male) and Lmna+/- (n=25, 14 male) mouse
hearts were evaluated. No statistically significant differences were observed.

6
Online Table IV. Echocardiographic data in 18-week-old mice with and without 6 weeks of strenuous exercise training.
Male
Female
Sedentary Trained Sedentary Trained
WT Lmna+/- WT Lmna+/- WT Lmna+/- WT Lmna+/-
Final stage achieved:
- Baseline ETT
- 6-week ETT
-
-
-
-
4 (67%)
4 (67%)
3 (50%)
5 (83%)
-
-
-
-
6 (100%)
6 (100%)
1 (17%)
6 (100%)
No. stimuli: - Baseline ETT
- 6-week ETT
-
-
-
-
13.5 ± 11.3
5.5 ± 9.2
12.0 ± 10.2
9.0 ± 13.2
-
-
-
-
7.3 ± 4.3
4.2 ± 9.7
15.0 ± 10.9
7.3 ± 13.2
BW (g) 35.5 ± 6.1 33.0 ± 4.8 27.5 ± 1.9† 29.6 ± 4.2 26.7 ± 3.3 24.8 ± 1.8 26.0 ± 2.3 24.9 ± 1.8
LVDD (mm) 3.35 ± 0.13 3.60 ± 0.32* 3.17 ± 0.13† 3.51 ± 0.26* 3.18 ± 0.14 3.22 ± 0.15 3.24 ± 0.15 3.29 ± 0.18
LVSD (mm) 1.37 ± 0.12 1.85 ± 0.34* 1.21 ± 0.10† 1.64 ± 0.32* 1.22 ± 0.11 1.37 ± 0.32 1.33 ± 0.11 1.42 ± 0.27
LVFS (%) 59 ± 3 49 ± 7* 62 ± 3 54 ± 6* 62 ± 3 58 ± 8 59 ± 3 57 ± 7
LAD (mm) 1.65 ± 0.08 1.79 ± 0.12* 1.65 ± 0.10 1.95±0.15*† 1.55 ± 0.04 1.55 ± 0.05 1.58 ± 0.04 1.67±0.09*†

7
HR (bpm) 706 ± 30 685 ± 36 654 ± 47† 694 ± 42 667 ± 30 665 ± 43 669 ± 9 666 ± 40
Cardiac function after strenuous exercise training in WT (n=12, 6 male) and Lmna+/- (n=12, 6 male) mice was compared with age-matched
sedentary WT (n=31, 13 male) and Lmna+/- (n=32, 19 male) mice. ETT indicates exercise tolerance test. *P<0.05 vs WT, †P<0.05 vs sedentary
mice (ANOVA and Student’s t test).
8
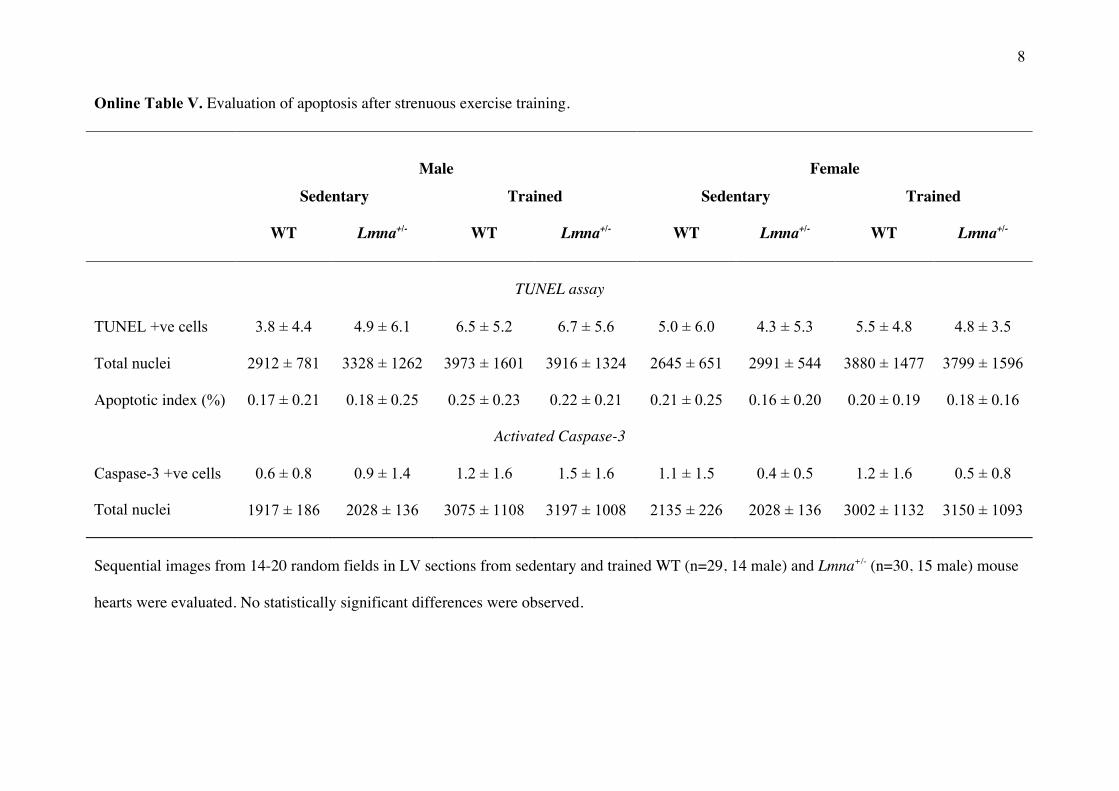
Online Table V. Evaluation of apoptosis after strenuous exercise training.
Male
Female
Sedentary Trained Sedentary Trained
WT Lmna+/- WT Lmna+/- WT Lmna+/- WT Lmna+/-
TUNEL assay
TUNEL +ve cells 3.8 ± 4.4 4.9 ± 6.1 6.5 ± 5.2 6.7 ± 5.6 5.0 ± 6.0 4.3 ± 5.3 5.5 ± 4.8 4.8 ± 3.5
Total nuclei 2912 ± 781 3328 ± 1262 3973 ± 1601 3916 ± 1324 2645 ± 651 2991 ± 544 3880 ± 1477 3799 ± 1596
Apoptotic index (%) 0.17 ± 0.21 0.18 ± 0.25 0.25 ± 0.23 0.22 ± 0.21 0.21 ± 0.25 0.16 ± 0.20 0.20 ± 0.19 0.18 ± 0.16
Activated Caspase-3
Caspase-3 +ve cells 0.6 ± 0.8 0.9 ± 1.4 1.2 ± 1.6 1.5 ± 1.6 1.1 ± 1.5 0.4 ± 0.5 1.2 ± 1.6 0.5 ± 0.8
Total nuclei 1917 ± 186 2028 ± 136 3075 ± 1108 3197 ± 1008 2135 ± 226 2028 ± 136 3002 ± 1132 3150 ± 1093
Sequential images from 14-20 random fields in LV sections from sedentary and trained WT (n=29, 14 male) and Lmna+/- (n=30, 15 male) mouse
hearts were evaluated. No statistically significant differences were observed.
9
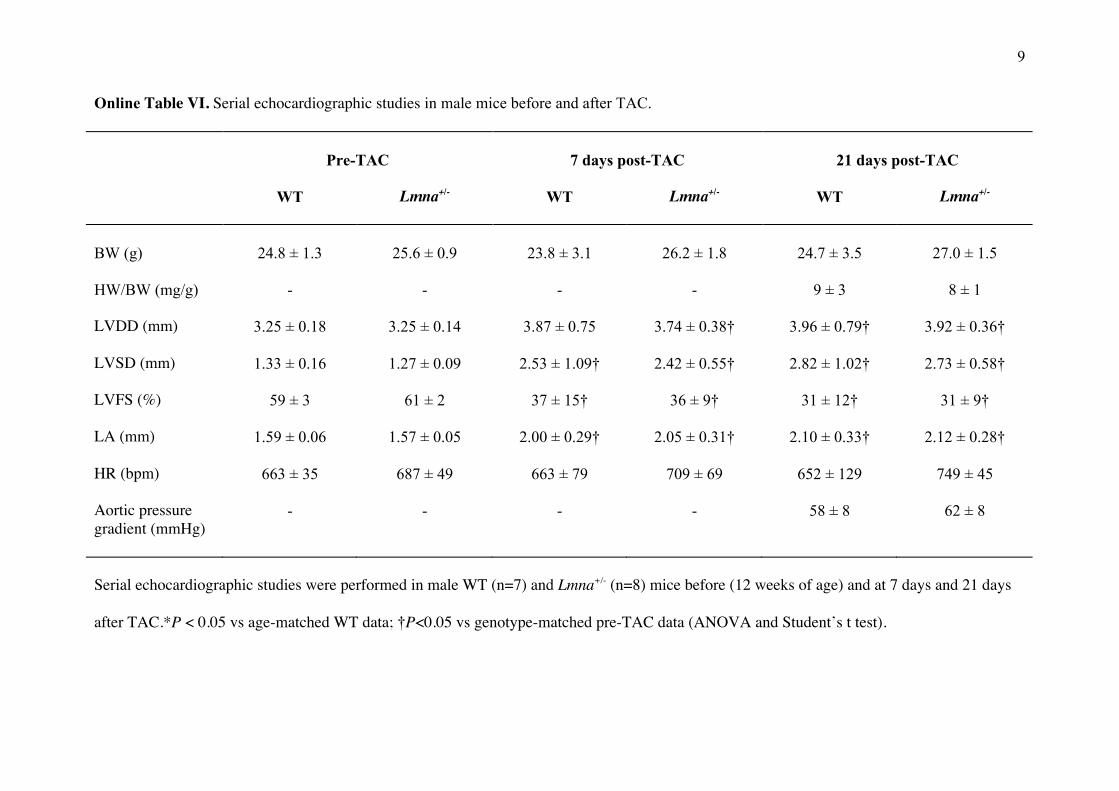
Online Table VI. Serial echocardiographic studies in male mice before and after TAC.
Pre-TAC
7 days post-TAC
21 days post-TAC
WT Lmna+/- WT Lmna+/- WT Lmna+/-
BW (g)
24.8 ± 1.3
25.6 ± 0.9
23.8 ± 3.1
26.2 ± 1.8
24.7 ± 3.5
27.0 ± 1.5
HW/BW (mg/g) - - - - 9 ± 3 8 ± 1
LVDD (mm) 3.25 ± 0.18 3.25 ± 0.14 3.87 ± 0.75 3.74 ± 0.38† 3.96 ± 0.79† 3.92 ± 0.36†
LVSD (mm) 1.33 ± 0.16 1.27 ± 0.09 2.53 ± 1.09† 2.42 ± 0.55† 2.82 ± 1.02† 2.73 ± 0.58†
LVFS (%) 59 ± 3 61 ± 2 37 ± 15† 36 ± 9† 31 ± 12† 31 ± 9†
LA (mm) 1.59 ± 0.06 1.57 ± 0.05 2.00 ± 0.29† 2.05 ± 0.31† 2.10 ± 0.33† 2.12 ± 0.28†
HR (bpm) 663 ± 35 687 ± 49 663 ± 79 709 ± 69 652 ± 129 749 ± 45
Aortic pressure gradient (mmHg)
- - - - 58 ± 8 62 ± 8
Serial echocardiographic studies were performed in male WT (n=7) and Lmna+/- (n=8) mice before (12 weeks of age) and at 7 days and 21 days
after TAC.*P < 0.05 vs age-matched WT data; †P<0.05 vs genotype-matched pre-TAC data (ANOVA and Student’s t test).

10
Online Table VII. Cardiac size and function at 14 days after TAC.
Male
Female
Sham TAC Sham TAC
WT Lmna+/- WT Lmna+/- WT Lmna+/- WT Lmna+/-
BW (g)
25.6 ± 1.4
25.8 ± 2.0
25.1 ± 2.1
26.4 ± 2.5
21.7 ± 1.5
23.0 ± 2.3
21.0 ± 1.5
22.7 ± 2.3
HW/BW(mg/g) 5 ± 1 6 ± 1 8 ± 1† 7 ±1 5 ± 1 6 ± 0 9 ± 2† 8 ± 1†
LVDD (mm) 3.32 ± 0.19 3.24 ± 0.07 3.83 ± 0.41† 3.88 ± 0.42† 3.19 ± 0.09 3.23 ± 0.11 3.94 ± 0.47† 3.75 ± 0.29†
LVSD (mm) 1.36 ± 0.12 1.36 ± 0.14 2.38 ± 0.65† 2.72 ± 0.66† 1.29 ± 0.11 1.33 ± 0.07 2.72 ± 0.77† 2.52 ± 0.48†
LVFS (%) 59 ± 2 58 ± 4 38 ± 12† 31 ± 10† 60 ± 5 59 ± 2 32 ± 13† 33 ± 8†
LAD (mm) 1.64 ± 0.04 1.63 ± 0.08 2.10 ± 0.09† 2.12 ± 0.38† 1.59 ± 0.03 1.61 ± 0.08 2.13 ± 0.14† 2.14 ± 0.30†
HR (bpm) 669 ± 52 736 ± 10 639 ± 88 671 ±65 690 ± 29 613 ± 86 612 ± 80 672 ± 62
Aortic pressure gradient (mmHg)
1 ± 1 1 ± 2 50 ± 6† 53 ± 6† 0 ± 3 1 ± 2 56 ± 13† 60 ± 13†
Data after TAC in WT (n=14, 7 male) and Lmna+/- (n=15, 7 male) mice are compared with sham-operated WT (n=10, 5 male) and Lmna+/- (n=9,
5 male) mice. *P < 0.05 vs age-matched WT; †P<0.05 vs genotype-matched sham group (ANOVA and Student’s t test).

11
Online Table VIII. Evaluation of apoptosis at 14 days post-TAC.
Sham
TAC
WT Lmna+/- WT Lmna+/-
TUNEL assay
TUNEL +ve cells 5.8 ± 2.1 6.5 ± 2.4 15.3 ± 4.4 13.7 ± 4.6
Total nuclei 2799 ± 427 3059 ± 872 2748 ± 482 2668 ± 472
Apoptotic index (%) 0.21 ± 0.11 0.21 ± 0.10 0.56 ± 0.15† 0.51 ± 0.13†
Activated Caspase-3
Caspase-3 +ve cells 0.8 ± 1.0 1.0 ± 0.8 9.0 ± 4.8† 11.8 ± 6.9†
Total nuclei 2309 ± 677 2479 ± 601 2988 ± 581 3006 ± 757
Apoptosis was evaluated after TAC or sham surgery in WT (n=10, 5 male) and Lmna+/-
(n=10, 5 male) mouse hearts. *P < 0.05 vs age-matched WT; †P<0.05 vs genotype-matched
sham group (ANOVA and Student’s t test).

12
Online Table IX. Cardiac function in 40 week-old mice after carvedilol or placebo treatment.
Placebo
Carvedilol
WT Lmna+/- WT Lmna+/-
BW (g)
33.8 ± 3.5
32.2± 3.2
30.3 ± 3.1
30.8 ± 2.7
LVDD (mm) 3.43 ± 0.17 3.73 ± 0.18* 3.42 ± 0.18 3.45 ± 0.18†
LVSD (mm) 1.44 ± 0.10 1.91 ± 0.18* 1.43 ± 0.13 1.58 ± 0.23†
LVFS (%) 58 ± 2 49 ± 3* 58 ± 2 55 ± 5†
LA (mm) 1.68 ± 0.04 1.85 ± 0.12* 1.69 ± 0.05 1.70 ± 0.12†
HR (bpm) ‡ 629 ± 47 622 ± 58 580 ± 62 581 ± 41
Male WT (n=18) and Lmna+/- (n=17) mice received oral carvedilol or placebo from 12 weeks
of age. Three WT mice (2 placebo-treated) and 2 Lmna+/- mice (1 placebo-treated) had
unexplained deaths. *P < 0.05 vs age-matched WT; †P<0.05 vs genotype-matched placebo
group (ANOVA and Student’s t test); ‡ Heart rates determined in unanesthetized mice by
telemetry.

SUPPLEMENT MATERIAL
Online Figure Legends
Online Figure I
Natural history of cardiac function in Lmna+/- mice. Serial echocardiography performed at 12,
20, 40 and 80 weeks in male and female wild-type (WT) and Lmna+/- mice demonstrates
temporal changes in left ventricular end-diastolic diameter (LVDD, A) and fractional
shortening (LVFS, B). The effects of genotype and sex on the rate of change of LVDD and
LVFS with age were assessed by the coefficients corresponding to the interaction between
genotype and age (β4), and sex and age (β5), respectively, using the nlme package in R1 and a
mixed effects model: log LVDDij (or LVFSij) = β0 + Ui + (β1 + Vi) ageij + β2genotypei + β3
sexi + β4ageijgenotypei + β5ageijgenotypei + εij, where i indexes animal, j indexes
measurement, Ui ~ N (0,σU2) and Vi ~ N(0, σv
2) represent random intercept and slope effects
respectively, age is measured as weeks since birth, genotype and sex are indicator variables
(WT = 0, Lmna+/- = 1; male = 0, female = 1), and εij ~ N(0,σe2) is within-subject error. There
was a significant influence of genotype on the rate of change of LVDD (P<0.001) and LVFS
(P<0.001). There was a trend towards an effect of sex in the full data-set (LVDD, P=0.07;
LVFS, P=016), with a significantly greater slope for LVDD (P=0.008) and LVFS (P=0.045)
when two severely-affected outliers were excluded.
Online Figure II
Desmin distribution in a non-lamin A/C-related DCM model. Immunogold electron
microscopy performed in β2-adrenergic receptor transgenic mice with severe DCM2 showed
normal cardiomyocyte nuclear morphology (A) and a normal perinuclear pattern of gold-
labelled desmin (black dots, B). Scale bars = (A) 2mm, (B) 0.25 mm.

SUPPLEMENT MATERIAL
References
1. Pinheiro J, Bates D, DebRoy S, Sarkar D. Linear and nonlinear mixed effects models. R
package version 2005;3:1-65.
2. Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age-dependent
cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic
receptors in the heart. Cardiovasc Res. 2000;48:448-454.

