effect of solvent quality on the polymer adsorption from bulk solution onto planar surfaces
TRANSCRIPT

Dynamic Article LinksC<Soft Matter
Cite this: Soft Matter, 2012, 8, 5140
www.rsc.org/softmatter PAPER
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online / Journal Homepage / Table of Contents for this issue
Effect of solvent quality on the polymer adsorption from bulk solution ontoplanar surfaces†
Per Linse
Received 11th January 2012, Accepted 5th March 2012
DOI: 10.1039/c2sm25074h
Adsorption of uncharged homopolymers in good and theta solvents onto planar surfaces at various
chain flexibility and polymer–surface attraction strengths was investigated by using a coarse-grained
bead–spring polymer model and simulation techniques. Equilibrium properties of the interfacial
systems were obtained from Monte Carlo simulations by monitoring the bead and polymer density
profiles, the number of adsorbed beads and polymers, the components of the radius of gyration
perpendicular and parallel to the surface as well as tail, loop, and train characteristics. The adsorption
process starting with a polymer-free zone adjacent to the surface was examined by Brownian dynamic
simulations. At equilibrium, the adsorbed amount increased upon increasing chain stiffness and in
poorer solvent conditions, and the structural characteristics depended also on the chain stiffness and
solvent condition. The initial adsorption was diffusion controlled, but soon it became governed by the
probability of a polymer to be captured by the surface attraction. Flexible polymers became flattened
after attaching, but their final relaxation mechanism involved an increase in perpendicular extension.
There were fewer adsorbed beads and longer tails, which was driven by the surface pressure originating
from the surrounding adsorbed polymers. This structural rearrangement became more prominent in
poorer solvent conditions. Finally, the integration time, which denotes the adsorption time for
adsorbed polymers to become fully integrated into the adsorbed layer, and the residence times of
integrated polymers were analyzed. In particular, the latter became longer with increasing chain
stiffness and shorter in poorer solvent conditions.
1 Introduction
Polymers in solution may adsorb onto surfaces if the effective
polymer–surface attraction exceeds the conformational entropy
loss of the polymer upon adsorption.1 The effective attraction
depends both on the direct polymer–surface and solvent–surface
interactions as well as on the polymer–solvent interaction.
Generally, poor solvent conditions promote adsorption.
The adsorption of polymers onto surfaces, whether desired or
not, has huge implications in many areas of research. Moreover,
understanding and controlling such processes is of great impor-
tance and is essential in many different technological aspects
ranging from the paper industry and paint formulation to
pharmaceutical applications,2 biophysics,3–5 and nanocomposite
materials.6
The adsorption of polymers onto surfaces is largely governed
by the prevailing conditions under which the polymer, solvent,
Physical Chemistry, Department of Chemistry, Lund University, P.O. Box124, S-221 00 Lund, Sweden
† Electronic supplementary information (ESI) available: Graphicalinformation displaying in more detail the adsorption process withflexible and rod-like polymers at the two solvent conditions areavailable. See DOI: 10.1039/c2sm25074h
5140 | Soft Matter, 2012, 8, 5140–5150
and surface interact. The equilibrium adsorbed layer in terms of
surface coverage and layer thickness is often of interest from
a technical point of view, where a surface is physically or
mechanically modified to meet specific requirements. Due to the
immense number of applications for polymer adsorption, there
has historically been a large interest in characterizing layers of
adsorbed polymers.7–11 However, often kinetics is so slow that
true equilibrium of the adsorbed polymers never is achieved
during realistic time scales. Resolving the different time scales
involved during the entire adsorption process, from diffusional
transport to a surface followed by subsequent attachment and
spreading on it, thus remains a significant task and a large
challenge within the field.
A number of different theoretical methods have been
employed to study the adsorption of uncharged polymers onto
uncharged surfaces from bulk solution. Such adsorption profiles
near adsorbing surfaces have been characterized by using mean-
field approaches12,13 as well as by using various simulation
techniques.14–17 The dynamics of polymer adsorption has also
been studied using dynamic mean-field schemes,18 as well as
dynamic Monte-Carlo,19–25 molecular dynamics,26–30 and Brow-
nian dynamics31,32 techniques. Some of the dynamic studies were
conducted on single polymers at a surface,22,25,31 while others
This journal is ª The Royal Society of Chemistry 2012

Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
were comprised of adsorption from solutions ranging from semi-
dilute conditions20,19,21,23,24,30,32 to polymer melts.26–29 Various
static and dynamic properties of polymer adsorption have been
examined as a function of strength of the polymer–surface
interaction,14,17,21,23,31 and only a few have been conducted on
polymer models with varying intrinsic stiffness.14,16,31 Further-
more, some attention has been given to diffusion and exchange in
an adsorbed layer.21,26,27,23,33
In a previous work,34 adsorption of flexible polymers in a good
solvent onto a planar and solid surface for different polymer
flexibility and surface attraction was examined. Both equilibrium
structure and adsorption dynamics starting from a polymer-free
zone adjacent to the surface were examined. In this study, we
extend these simulation studies to comprise adsorption from
a theta solvent using both Monte Carlo and Brownian dynamics
techniques. Beside an increase in the adsorbed amount, other
equilibrium and dynamical properties were found to depend on
the solvent quality.
2 Model
The adsorption of polymers from solution onto a planar surface
is studied using a simple coarse-grained model; basically the same
model as previously described is used.31–34 The solution contains
Np polymers and each polymer is represented by a sequence ofNb
spherical beads connected via harmonic potentials. The total
number of beads in the system N is thus given by N ¼ NpNb. The
polymers are confined in a rectangular simulation box with the
box lengths Lx, Ly, and Lz. At z ¼ �(Lz/2) we have adsorbing
surfaces, whereas periodic boundary conditions are applied in
the x- and y-directions. The length of the box edges are Lx¼ Ly¼200 �A and Lz ¼ 240 �A. Our focus is on structures and events
occurring near the surfaces. Therefore, a coordinate system with
a z-axis starting at z ¼ Lz/2 and directed into the solution is
adopted, and averages over both equivalent surfaces are made.
The total potential energy U of the system can be expressed as
a sum of four different terms according to
U ¼ Unonbond + Ubond + Uangle + Usurf (1)
The nonbonded bead–bead potential energy Unonbond is assumed
to be pairwise additive according to
Unonbond ¼XNi\j
u�rij�
(2)
where the Lennard-Jones (LJ) potential energy is given by
u�rij� ¼
(43
���s
rij
�6
þ�s
rij
�12
þ3shift
�; rij # scut
0; rij . scut
(3)
is used for the interaction between beads i and j, where rij is the
distance between the two beads, s ¼ 3.405 �A the diameter of the
bead, and 3 ¼ 0.9961 kJ mol�1 the interaction strength. The
standard (untruncated and unshifted) attractive LJ potential
with (scut, 3shift) ¼ (N, 0) was used to represent the theta solvent
condition, whereas the truncated and shifted version of the LJ
potential obtained with (scut, 3shift) ¼ (21/6s, 1/4), yielding a soft
This journal is ª The Royal Society of Chemistry 2012
repulsive potential, was used to represent the good solvent
condition. It should be remembered that eqn (3) represent
a solvent-averaged interaction between polymer beads, and is as
such an approximation. Its usefulness has to be assessed by
performing simulations with explicit solvent; however, that is
outside the scope of the current investigation.
The bond potential energy Ubond is given by
Ubond ¼ 1
2kbond
XNp
p¼1
XNb�1
i¼1
�ri;p � req
�2(4)
where ri,p is the length of the bond between bead i and i + 1 of the
p:th polymer, kbond ¼ 2.4088 kJ mol�1 �A�2 the bond force
constant, and req ¼ 5.0 �A the equilibrium bond length.
Furthermore, the angular potential energy Uangle is given by
Uangle ¼ 1
2kangle
XNp
p¼1
XNb�1
i¼2
�qi;p � qeq
�(5)
where qi,p is the angle formed by beads i � 1, i, and i + 1 of the
p:th polymer, kangle the angular force constant that determines
the stiffness of the polymer, and qeq ¼ 180� the equilibrium bond
angle. In the presence of all interactions, the root-mean-square
(rms) bead–bead separation of bonded beads along the polymers
becomes hR2bbi1/2 z 5.5 �A.
The polymer–surface interaction is taken as a sum of bead–
surface interactions according to
Usurf ¼XNi¼1
�usurfðziÞ þ usurfðLz � ziÞ
(6)
where an attractive 3–9 LJ potential35
usurfðziÞ ¼ 2p
3rss
3s 3s
"��ss
zi
�3
þ 2
15
�ss
zi
�9#
(7)
is used for the interaction between bead i and a surface. In eqn
(7), rs is the density of the (hypothetical) particles forming the
surface, ss the mean diameter of a bead and a surface particle,
3s a potential energy parameter describing the bead–surface
interaction, and zi the z-coordinate of bead i with respect to
surface. With this attractive 3–9 LJ potential, the potential
minimum appears at zmin ¼ (2/5)1/6ss and amounts to usurf(zmin)
¼ �[2p(10)1/2/9]rss3s3s. For simplicity, ss ¼ 3.5 �A and rss
3s ¼ 1
were chosen, giving zmin z 3.0 �A and usurf(zmin) z �2.23s.
In this work, Np ¼ 1056 polymers with chain length Nb ¼ 20
have been considered for (i) good and theta solvent conditions at
(ii) variable flexibility regulated by kangle and (iii) variable bead–
surface interaction regulated by 3s. With the radius of gyration
hR2gi1/2 ¼ 12.50 �A for a flexible 20-mer at infinite dilution in
a good solvent,32 one obtains (4p/3)hR2gi3/2Np/(LxLyLz) ¼ 0.9;
thus, the bulk density is below but near the overlap density.
In total 12 systems involving eight different chain flexibilities
and three different bead–surface interaction strengths have been
examined. The different angular force constants were kangle ¼0 (flexible), 0.3, 1.2 (semi-flexible), 3, 6, 10 (stiff), 20, and 30 (rod-
like polymer) J mol�1 deg�2. The different bead–surface interac-
tion strengths were 3s ¼ 1.5 (weak), 2.5 (intermediate), and 3.5
(strong bead–surface attraction) kJ mol�1. These systems were
divided into three sets: (i) Set I comprising systems characterized
by the intermediate bead–surface interaction strength (3s¼ 2.5 kJ
Soft Matter, 2012, 8, 5140–5150 | 5141

Table 1 Model parameters
Box length (x- and y-direction) Lx ¼ Ly ¼ 200 �ABox length (z-direction) Lz ¼ 240 �ATemperature T ¼ 298 KNumber of beads in a polymer Nb ¼ 20Number of polymers Np ¼ 1056Bead–bead LJ parameter s ¼ 3.405 �ABead–bead LJ parameter 3 ¼ 0.9961 kJ mol�1
Cutoff of s scut ¼ N and 21/6sShift of 3 3shift ¼ 0 and 1/4Force constant of bond potential kbond ¼ 2.4088 kJ mol�1 �A�2
Equilibrium separation of bond potential req ¼ 5.0 �AForce constant of angle potential kangle ¼ 0, 0.3, 1.2, 3, 6, 10, 20, and 30 J mol�1 deg�2
Equilibrium angle of angle potential qeq ¼ 180�Bead–surface LJ parameter ss ¼ 3.5 �ABead–surface LJ parameter 3s ¼ 1.5, 2.5, and 3.5 kJ mol�1
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
mol�1) at variable angular force constant kangle, (ii) Set II
comprising systems with flexible polymers (kangle ¼ 0) at variable
bead–surface interaction strength 3s, and (iii) Set III comprising
systems with rod-like polymers (kangle ¼ 30 J mol�1 deg�2)) at
variable bead–surface interaction strength 3s. General model
parameters are compiled in Table 1, and systems investigated and
their labels are found in Table 2.
Fig. 1 provides the rms radius of gyration hR2gi1/2 of single and
flexible chains versus Nb ranging fromNb ¼ 10 to 640 at different
solvent conditions. An analysis of the data leads to the scaling
relation hR2gi1/2 �Nn
b with n ¼ 0.61 � 0.01 for (scut, 3shift)¼ (21/6s,
1/4) and n ¼ 0.49 � 0.01 for (scut, 3shift) ¼ (N, 0), cf., e.g., ref. 36.
The intrinsic flexibility of the polymers was characterized by the
calculated persistence length lp based on the local folding of
a single polymer according to lp ¼ hR2bbi1/2/(1 + h cos qi)37,38 at
infinite dilution. At good solvent conditions, the persistence
length of the single polymers for the different angular force
constants became lp ¼ 6.5, 7.8, 13, 26, 47, 77, 149, and 221 �A,
respectively. The contour length is L ¼ (Nb � 1)hR2bbi1/2 and
becomes Lz 105 �A forNb ¼ 20. At theta solvent conditions, the
corresponding persistence lengths are 10% (flexible polymer) to
3% (rod-like polymer) smaller.
3 Method
3.1 Simulation details
Monte Carlo (MC) simulations were used to (i) obtain equilib-
rium properties and (ii) to prepare the initial configurations of
Table 2 Overview of investigated systemsa
kangle/J mol�1 deg�2
3s/kJ mol�1 0b 0.3 1.2c 3 6 10d 20 30e
1.5f II III2.5g I, II I I I I I I I, III3.5h II III
a I, II, and III denote that the system belongs to Set I, Set II, and Set III,respectively. b Referred to as flexible polymer. c Referred to as semi-flexible polymer. d Referred to as stiff polymer. e Referred to as rod-like polymer. f Referred to as weak bead–surface attraction. g Referredto as intermediate bead–surface attraction. h Referred to as strongbead–surface attraction.
5142 | Soft Matter, 2012, 8, 5140–5150
the Brownian dynamics (BD) simulations, whereas the BD
simulations were used to examine the adsorption dynamics and
the associated change of the internal structure of the polymers.
The canonical ensemble, characterized by a constant number of
particles, volume, and temperature was used throughout. The
variable adsorption among the systems lead to a variation of the
bulk polymer density (density far from the surfaces) up to 10% at
good solvent conditions and up to 50% at the theta solvent
conditions. Additional simulations have been made to examine
the consequences of the variation of the bulk polymer density.
All simulations were performed using the integrated Monte
Carlo/molecular dynamics/Brownian dynamics simulation
package MOLSIM.39
In more detail, the MC simulations were performed according
to the Metropolis algorithm40 using three types of trial moves: (i)
translation of individual beads, (ii) reptation of polymers, and
(iii) translation of polymers. The translational displacement
parameter of single-bead trial moves was 3 �A, the probability of
a reptation and of a polymer translation was 1/Nb of that of
a single-bead trial move, and the polymer translational
displacement parameter was 5�A. TheMC simulations comprised
1 � 105 trial moves per bead after equilibration.
The dynamic adsorption simulation studies were carried out as
follows: (i) First, preparative MC stimulation of polymer solu-
tions confined in a box with hard walls at the edges in the
z-direction with Lz ¼ 200 �A and periodic boundary conditions in
the x- and y-direction were performed. (ii) Second, the hard walls
Fig. 1 Rms radius of gyration hR2gi1/2 of single and flexible chains as
a function of chain length Nb with (scut, 3shift) ¼ (21/6s, 1/4) (open
symbols) referred to as the good solvent and (scut, 3shift) ¼ (N, 0) (solid
symbols) referred to as the theta solvent.
This journal is ª The Royal Society of Chemistry 2012

Fig. 2 Snapshots displaying (left) initial and (right) final equilibrium
polymer conformation at (top to bottom) kangle ¼ 0 (flexible polymers) in
a good solvent, kangle ¼ 30 J mol�1 deg�2 (rod-like polymers) in a good
solvent, kangle ¼ 0 (flexible polymers) in a theta solvent, and kangle ¼ 30 J
mol�1 deg�2 (rod-like polymers) in a theta solvent; all at 3s ¼ 2.5 kJ mol�1
Beads residing in adsorbed polymers are given in red.
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
were removed, the box length in the z-direction was increased to
Lz ¼ 240 �A, and attractive surfaces, whose potential is described
by eqn (7), were invoked. (iii) Finally, the BD simulations were
initiated. Hence, the initial configurations of the BD simulations
involved a z20 �A thick polymer-free zone adjacent to each
attractive surface. Beads located 20 �A from a surface experience
the negligible bead–surface potential usurf z 10�3 kbT, which is
only 0.05% of the value at zmin; hence, the initial polymer
adsorption ought to be controlled by translational diffusion.
The motion of the polymer beads in the BD simulations was
described by Ermak41
riðtþ DtÞ ¼ riðtÞ þD0Dt
kbTFiðtÞ þ Riðt;DtÞ (8)
where ri(t + Dt) is the location of bead i at the time t + Dt, ri(t)
the location of bead i at the time t, D0 is the bead self-diffusion
coefficient in the absence of systematic forces, kb is Boltz-
mann’s constant, T is the temperature, and Fi(t) is the
systematic force on bead i at time t arising from the potential
energy U given by eqn (1). Furthermore, Ri(t;Dt) is a random
displacement of bead i representing the effect of collisions with
solvent molecules at time t and is sampled from a Gaussian
distribution with the mean hRi(t;Dt)i ¼ 0 and the variance
hRi(t;Dt)$Rj(t0;Dt)i ¼ 6D0Dtdijd(t � t0) as obtained from the
fluctuation–dissipation theorem. In this work, hydrodynamic
interactions were neglected.
A bead self-diffusion coefficientD0¼ 0.1�A2 ps�1 was used, and
an integration time step Dt ¼ 0.025 ps was employed. The BD
simulations involved 3.2� 107 (good solvent) and 16� 107 (theta
solvent) time steps, providing a nominal simulation time of
800 ns and 4000 ns, respectively. Using sBD ¼ s2/D0 ¼ 116 ps as
the conventional unit of time, the integration time step becomes
Dt ¼ 2.2 � 10�4sBD and the total simulation time 6.9 � 103sBDand 34 � 103sBD, respectively. For a single flexible polymer with
20 beads in an infinite dilute solution in a good solvent,
a previous investigation31 gave the polymer self-diffusion coef-
ficient D ¼ 0.005 �A2 ps�1 and the relaxation time sR ¼ 55 ns
characterizing the end-to-end vector time correlation function.
The statistical uncertainties of the equilibrium properties given
in the Figures are based on block averaging and are negligible,
whereas those of the dynamic properties are comparable to the
symbol size unless otherwise stated.
4 Results and discussion
4.1 Overview
The BD simulations were started with nonequilibrium configu-
rations comprising polymer-free zones between an adsorbing
surface and the bulk polymer solution, and the BD simulations
were continued until equilibrium was achieved. Fig. 2 displays
the initial and final equilibrium configurations in good and theta
solvents for flexible and rod-like polymers at the intermediate
bead–surface attraction. Series of snapshots displaying the
adsorption process of these four systems are available in the
Electronic Supplementary Information (Fig. S1–S4).† The initial
configurations (Fig. 2, left) are characterized by a ca. 20 �A wide
polymer-free zone, and next to this zone there is a depletion
region of approximately a radius of gyration in the good solvent
and a more extended and laterally heterogeneous depletion
This journal is ª The Royal Society of Chemistry 2012
region in the theta solvent. At equilibrium (Fig. 2, right), also
here the bulk solution is more heterogeneous in the theta solvent.
The beads residing in adsorbed polymers (discussed further
below) are depicted in red. The different chain configurations of
flexible and rod-like polymers are also noticeable.
4.2 Equilibrium properties
4.2.1 Density distributions. The z-distribution of the reduced
bead density r*b(z) h rb(z)s3 and of the reduced center-of-mass
(com) density of polymers r*com(z)h rcom(z)s3 near the surface at
the two solvent conditions for flexible and rod-like polymers at
the intermediate bead–surface attraction are displayed in Fig. 3.
In the good solvent (dashed curves), the bead density distri-
butions given show one layer of beads that are in direct contact
with the surface (Fig. 3a). The number density of this layer is
three-fold larger for rod-like polymers than for the flexible ones.
A weak second bead layer ca. 3 �A further away is found for
flexible polymers but is absent for rod-like polymers. The bead
distributions in the theta solvent (solid curves) are characterized
by (i) a higher number density in the first bead layer, (ii)
a pronounced second bead layer, (iii) even a third bead layer for
rod-like polymers, and (iv) furthermore a gradual decay
approaching bulk density ca. 40 �A from the surface.
Soft Matter, 2012, 8, 5140–5150 | 5143

Fig. 3 (a) Reduced bead density r*b(z)h rb(z)s3 and (b) reduced polymer
density r*com(z)h rcom(z)s3 as a function of z-coordinate near a surface in
a good solvent (dashed curves) and a theta solvent (solid curves) at indi-
cated values of kangle in Jmol�1 deg�2 and 3s¼ 2.5 kJmol�1. The location of
the adsorption threshold is also given (dotted vertical lines).
Fig. 4 (a) Average number of adsorbed beads hNadsb i as a function of the
angular force constant kangle in a good solvent (open circles, dashed
curves) and a theta solvent (solid circles, solid curves) at 3s¼ 2.5 kJ mol�1.
(b) Corresponding results with ca. 10% (good solvent) and ca. 50% (theta
solvent) larger number of polymers, giving similar bulk densities, are also
given (crosses). The crosses partly overlap corresponding circles.
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
The com density distributions display a single maximum near
the surface; however, both the locations and the values vary
noticeably upon the solvent conditions examined here (Fig. 3b).
The maximum of the com distribution for flexible polymers
appears at z z 5–6 �A, whereas for rod-like polymers the
maximum shifts to z z 3.3 �A. As for the bead density distri-
butions, in the theta solvent (i) the maxima are larger and (ii)
tails of enhanced density stretch further into bulk solution.
A further analysis involving all systems in Set I shows that the
variations of the bead and com density distributions upon
increasing chain stiffness are nonmonotonic in different aspects
(data not shown). For example, upon an increase of chain stiff-
ness we found: (i) in the good solvent the bead density of the first
layer increases and the density of the second layer decreases, (ii)
in the theta solvent the magnitude of the second peak of the bead
distribution first decreases before it increases, and (iii) at both
solvent conditions the single maxima of the com density distri-
bution first decreases and thereafter increases. Later, we will
frequently encounter properties having nonmonotonic variation
upon an increase of the chain stiffness. Finally, at increasing
bead–surface interaction strength (Set II and Set III), the bead
and com density distributions display a regular behavior
involving larger bead and com density maxima appearing at
higher interaction strength.
In summary, the bead–surface attraction gives rise to adsorbed
polymer layers encompassing one to three distinct bead layers
and extending up to 10 �A (good solvent) and 40 �A (theta solvent)
from the surface. The single peak of the com density distribution
is broader and differs markedly between flexible and rod-like
polymers. Fig. 3 also displays the geometrical adsorption
threshold zads (dotted vertical line), later to be used. We see that
the beads selected by zads belongs essentially to the first layer of
adsorbed beads, and polymers with one or several adsorbed
beads will be referred to as adsorbed polymers.
5144 | Soft Matter, 2012, 8, 5140–5150
4.2.2 Adsorbed amount. The average number of adsorbed
beads hNadsb i and of adsorbed polymers hNads
p i in the first
adsorption layer as a function of the angle force constant for the
intermediate bead–surface attraction are given in Fig. 4.
In the theta solvent, the adsorbed amounts are 50 to 100%
larger than in the good solvent—consistent with the distributions
given in Fig. 3. At both solvent conditions, the average number
of adsorbed beads basically increases with polymer stiffness
(Fig. 4a); however, a small decrease in hNadsb i is initially observed.
The number of adsorbed polymers displays the opposite
behavior, displaying an initial increase as flexible polymers
become semi-flexible, thereafter a decrease, and finally a plateau
as the polymers become even stiffer (Fig. 4b). The location of the
maximum of hNadsp i appears at kangle ¼ 2–3 J mol�1 deg�2 (cor-
responding to a persistence length of lp ¼ 20–30 �A), which is
larger than the location of the minimum of hNadsb i.
Both hNadsb i and hNads
p i display monotonic increases at
increasing bead–surface interaction strength for all polymer
stiffnesses and at both solvent conditions (data not shown).
Generally, the variation of the average number adsorbed beads is
stronger (nearly 5-fold) than the variation of the average number
of adsorbed polymers (less than 3-fold) across the variation of
polymer flexibility and bead–surface interaction strength.
The use of a canonical ensemble implies that the polymer
bulk density decreases at increasing adsorption. In the good
solvent, the bulk density is reduced by z10% and in the theta
solvent it is reduced by z50% through the adsorption. Fig. 4
also shows results with an enhanced number of polymers to
compensate for those adsorbed (crosses), giving a bulk density
r*b(z) z N/(LxLyLz)s3 ¼ 0.087. The variation of the bulk
density across the conditions only mildly affect the adsorbed
amount, signifying that we are on the plateau of the adsorption
isotherm (as regarding the adsorbed amount within zads). In the
This journal is ª The Royal Society of Chemistry 2012
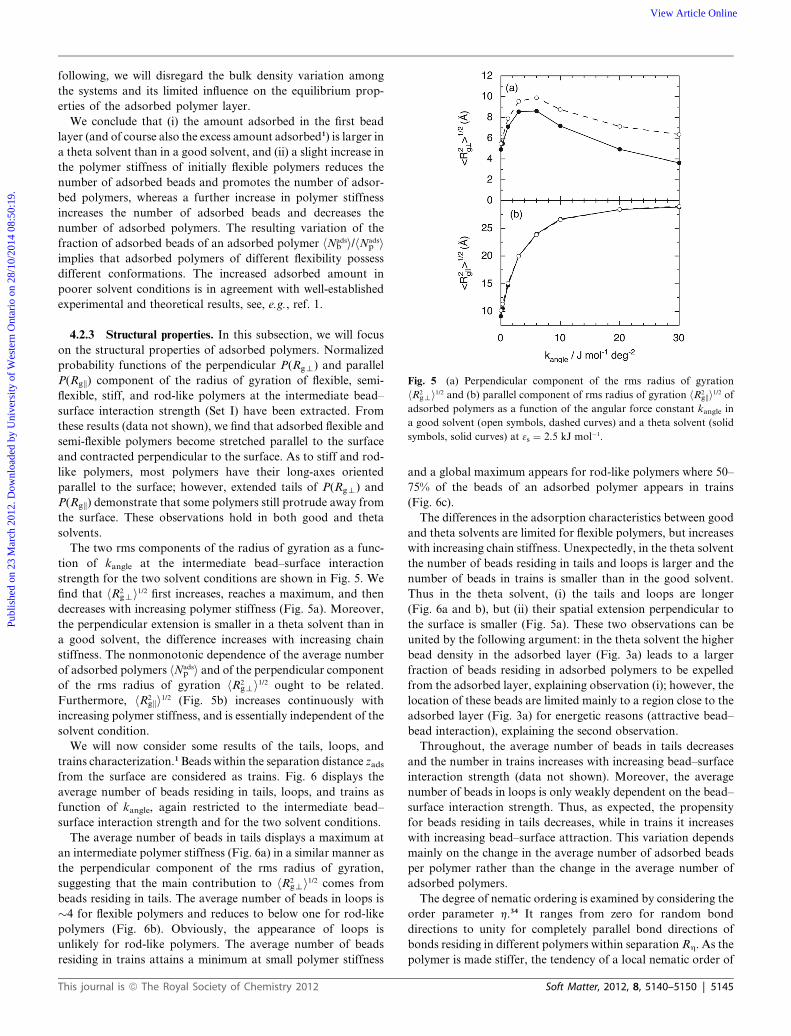
Fig. 5 (a) Perpendicular component of the rms radius of gyration
hR2gti1/2 and (b) parallel component of rms radius of gyration hR2
gki1/2 ofadsorbed polymers as a function of the angular force constant kangle in
a good solvent (open symbols, dashed curves) and a theta solvent (solid
symbols, solid curves) at 3s ¼ 2.5 kJ mol�1.
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
following, we will disregard the bulk density variation among
the systems and its limited influence on the equilibrium prop-
erties of the adsorbed polymer layer.
We conclude that (i) the amount adsorbed in the first bead
layer (and of course also the excess amount adsorbed1) is larger in
a theta solvent than in a good solvent, and (ii) a slight increase in
the polymer stiffness of initially flexible polymers reduces the
number of adsorbed beads and promotes the number of adsor-
bed polymers, whereas a further increase in polymer stiffness
increases the number of adsorbed beads and decreases the
number of adsorbed polymers. The resulting variation of the
fraction of adsorbed beads of an adsorbed polymer hNadsb i/hNads
p iimplies that adsorbed polymers of different flexibility possess
different conformations. The increased adsorbed amount in
poorer solvent conditions is in agreement with well-established
experimental and theoretical results, see, e.g., ref. 1.
4.2.3 Structural properties. In this subsection, we will focus
on the structural properties of adsorbed polymers. Normalized
probability functions of the perpendicular P(Rgt) and parallel
P(Rgk) component of the radius of gyration of flexible, semi-
flexible, stiff, and rod-like polymers at the intermediate bead–
surface interaction strength (Set I) have been extracted. From
these results (data not shown), we find that adsorbed flexible and
semi-flexible polymers become stretched parallel to the surface
and contracted perpendicular to the surface. As to stiff and rod-
like polymers, most polymers have their long-axes oriented
parallel to the surface; however, extended tails of P(Rgt) and
P(Rgk) demonstrate that some polymers still protrude away from
the surface. These observations hold in both good and theta
solvents.
The two rms components of the radius of gyration as a func-
tion of kangle at the intermediate bead–surface interaction
strength for the two solvent conditions are shown in Fig. 5. We
find that hR2gti1/2 first increases, reaches a maximum, and then
decreases with increasing polymer stiffness (Fig. 5a). Moreover,
the perpendicular extension is smaller in a theta solvent than in
a good solvent, the difference increases with increasing chain
stiffness. The nonmonotonic dependence of the average number
of adsorbed polymers hNadsp i and of the perpendicular component
of the rms radius of gyration hR2gti1/2 ought to be related.
Furthermore, hR2gki1/2 (Fig. 5b) increases continuously with
increasing polymer stiffness, and is essentially independent of the
solvent condition.
We will now consider some results of the tails, loops, and
trains characterization.1Beads within the separation distance zadsfrom the surface are considered as trains. Fig. 6 displays the
average number of beads residing in tails, loops, and trains as
function of kangle, again restricted to the intermediate bead–
surface interaction strength and for the two solvent conditions.
The average number of beads in tails displays a maximum at
an intermediate polymer stiffness (Fig. 6a) in a similar manner as
the perpendicular component of the rms radius of gyration,
suggesting that the main contribution to hR2gti1/2 comes from
beads residing in tails. The average number of beads in loops is
�4 for flexible polymers and reduces to below one for rod-like
polymers (Fig. 6b). Obviously, the appearance of loops is
unlikely for rod-like polymers. The average number of beads
residing in trains attains a minimum at small polymer stiffness
This journal is ª The Royal Society of Chemistry 2012
and a global maximum appears for rod-like polymers where 50–
75% of the beads of an adsorbed polymer appears in trains
(Fig. 6c).
The differences in the adsorption characteristics between good
and theta solvents are limited for flexible polymers, but increases
with increasing chain stiffness. Unexpectedly, in the theta solvent
the number of beads residing in tails and loops is larger and the
number of beads in trains is smaller than in the good solvent.
Thus in the theta solvent, (i) the tails and loops are longer
(Fig. 6a and b), but (ii) their spatial extension perpendicular to
the surface is smaller (Fig. 5a). These two observations can be
united by the following argument: in the theta solvent the higher
bead density in the adsorbed layer (Fig. 3a) leads to a larger
fraction of beads residing in adsorbed polymers to be expelled
from the adsorbed layer, explaining observation (i); however, the
location of these beads are limited mainly to a region close to the
adsorbed layer (Fig. 3a) for energetic reasons (attractive bead–
bead interaction), explaining the second observation.
Throughout, the average number of beads in tails decreases
and the number in trains increases with increasing bead–surface
interaction strength (data not shown). Moreover, the average
number of beads in loops is only weakly dependent on the bead–
surface interaction strength. Thus, as expected, the propensity
for beads residing in tails decreases, while in trains it increases
with increasing bead–surface attraction. This variation depends
mainly on the change in the average number of adsorbed beads
per polymer rather than the change in the average number of
adsorbed polymers.
The degree of nematic ordering is examined by considering the
order parameter h.34 It ranges from zero for random bond
directions to unity for completely parallel bond directions of
bonds residing in different polymers within separationRh. As the
polymer is made stiffer, the tendency of a local nematic order of
Soft Matter, 2012, 8, 5140–5150 | 5145

Fig. 6 Average number of beads residing in (a) tails hNtaili, (b) loopshNloopi, and (c) trains hNtraini of adsorbed polymers as a function of
the angular force constant kangle in a good solvent (open symbols,
dashed curves) and a theta solvent (solid symbols, solid curves) at 3s ¼2.5 kJ mol�1.
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
bonds in the adsorbed polymer layer increases. Fig. 7 shows the
average bond order hhi for Rh ¼ 20 �A as a function of kangle at
the intermediate bead–surface interactions strength. First, the
bond-order data show virtually no bond order (h z 0.1) for
adsorbed flexible polymers, whereas there is a large bond order
(hz 0.8–0.9) for adsorbed rod-like polymers. The bond order of
adsorbed bonds is basically insensitive to the solvent condition.
Fig. 7 Average bond order hhi as a function of the angular force
constant kangle in a good solvent (open symbols, dashed curves) and
a theta solvent (solid symbols, solid curves) for Rh ¼ 20 �A and at 3s ¼2.5 kJ mol�1.
5146 | Soft Matter, 2012, 8, 5140–5150
Snapshots of the final equilibrium configurations of the MC
simulations for adsorbed flexible and rod-like polymers were
given in Fig. 2 and illustrate a number of properties previously
quantified. For the rod-like polymers, it is obvious that some
polymers strongly protrude into the solution, giving rise to the
tails in P(Rgt) and P(Rgk) discussed above. Similar observations
of ‘‘hairpins’’ extending into solution for adsorbed semi-flexible
polymers in a good solvent have previously been discussed by
Kramarenko et al.14 Furthermore, the change from a disordered
to a nematic-like order of adsorbed polymers at increasing
polymer stiffness was quantified in Fig. 7. Snapshots in the ESI†
(Fig. S5) further illustrate this change of bond order with
increasing stiffness as well as the appearance of a few semi-flex-
ible and rod-like polymers protruding into the solution.
4.3 Dynamic properties
In the BD simulation, polymers diffuse to the surface, become
physically attached, and undergo various structural relaxations.
In the following, t0 will denote the time of the onset of the first
polymer attachments and t0 0 the time at which a quantity has
relaxed to its equilibrium value. As we will see, t0 0 is generally
property dependent. The equilibrium values given below are
taken from the separate MC simulations. Mostly, there is very
good agreement between the equilibrium values obtained from
the MC simulations and the values of the corresponding prop-
erties at the end of the BD simulations.
4.3.1 Adsorbed amount. The adsorption process will first be
characterized by considering the time dependence of the number
of adsorbed beads and of the number of adsorbed polymers.
Fig. 8a shows Nadsb (t) and Fig. 8b shows Nads
p (t) as a function of
the simulation time t in a logarithmic representation for flexible
and rod-like polymers in good and theta solvents at the inter-
mediate bead–surface attraction.
Though the first polymers adsorbed somewhat earlier, Fig. 8
shows that a more substantial rise ofNadsb (t) andNads
p (t) appears at
t0 z 2 ns in the good solvent and t0 z 10 ns in the theta solvent.
There are likely two reasons for this difference: (i) different initial
structures from the MC simulations with a wider region of
reduced polymer density in the theta solvent (see Fig. 2) and
hence a longer distance for the polymers to diffuse and (ii)
a smaller mutual diffusion coefficient in the theta solvent as
compared to that in the good solvent. To discriminate between
these two explanations, shorter and supplementary BD simula-
tions were performed for the two solvent conditions using the
MC structure of the other condition as the initial structures.
These results are also given in Fig. 8 (solid and dashed curves),
and they display that the dominant contribution comes from the
different mutual diffusion coefficient but that the contribution
from the different initial structures is not negligible.
The appearance of a common onset time t0 z 2 ns in the good
solvent and different from t0 z 10 ns in the theta solvent (i) is
consistent with the initial part of the adsorption being diffusion
controlled without any significant drift contribution from the
bead–surface attraction and (ii) the mutual diffusion is at most
marginally affected by the polymer stiffness at the conditions of
this study. As the adsorption process proceeds further, it
becomes faster for rod-like polymers than flexible ones in the
This journal is ª The Royal Society of Chemistry 2012
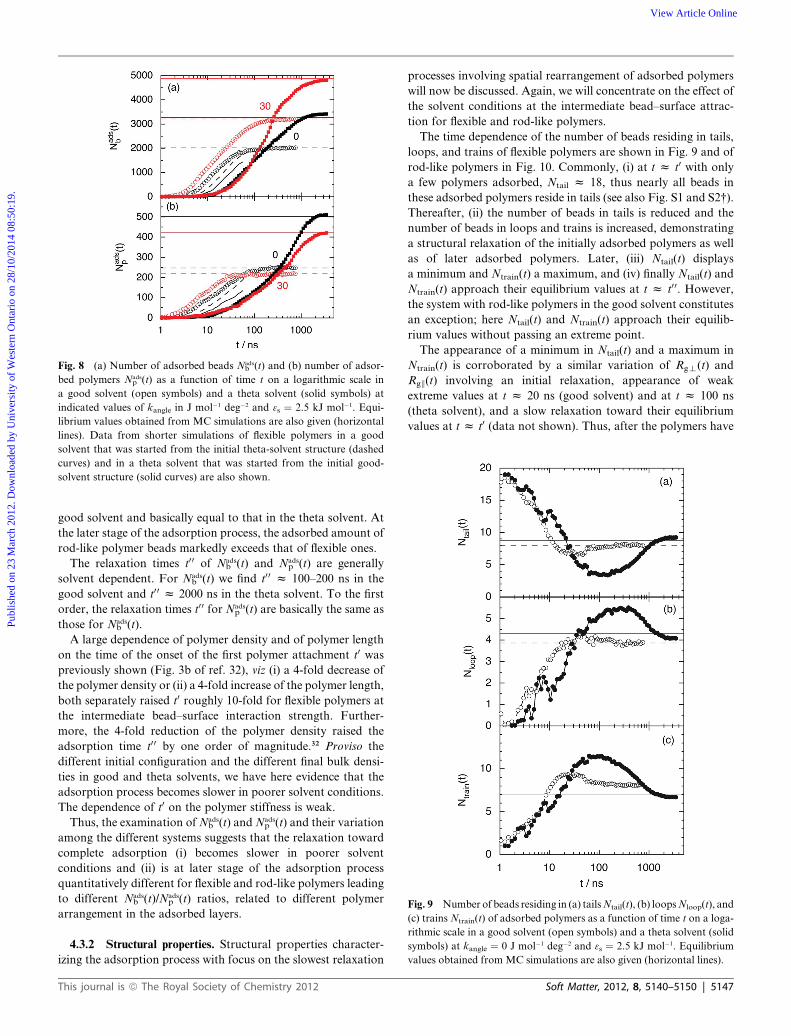
Fig. 8 (a) Number of adsorbed beads Nadsb (t) and (b) number of adsor-
bed polymers Nadsp (t) as a function of time t on a logarithmic scale in
a good solvent (open symbols) and a theta solvent (solid symbols) at
indicated values of kangle in J mol�1 deg�2 and 3s ¼ 2.5 kJ mol�1. Equi-
librium values obtained from MC simulations are also given (horizontal
lines). Data from shorter simulations of flexible polymers in a good
solvent that was started from the initial theta-solvent structure (dashed
curves) and in a theta solvent that was started from the initial good-
solvent structure (solid curves) are also shown.
Fig. 9 Number of beads residing in (a) tailsNtail(t), (b) loopsNloop(t), and
(c) trains Ntrain(t) of adsorbed polymers as a function of time t on a loga-
rithmic scale in a good solvent (open symbols) and a theta solvent (solid
symbols) at kangle ¼ 0 J mol�1 deg�2 and 3s ¼ 2.5 kJ mol�1. Equilibrium
values obtained from MC simulations are also given (horizontal lines).
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
good solvent and basically equal to that in the theta solvent. At
the later stage of the adsorption process, the adsorbed amount of
rod-like polymer beads markedly exceeds that of flexible ones.
The relaxation times t00 of Nadsb (t) and Nads
p (t) are generally
solvent dependent. For Nadsb (t) we find t00 z 100–200 ns in the
good solvent and t0 0 z 2000 ns in the theta solvent. To the first
order, the relaxation times t00 for Nadsp (t) are basically the same as
those for Nadsb (t).
A large dependence of polymer density and of polymer length
on the time of the onset of the first polymer attachment t0 waspreviously shown (Fig. 3b of ref. 32), viz (i) a 4-fold decrease of
the polymer density or (ii) a 4-fold increase of the polymer length,
both separately raised t0 roughly 10-fold for flexible polymers at
the intermediate bead–surface interaction strength. Further-
more, the 4-fold reduction of the polymer density raised the
adsorption time t0 0 by one order of magnitude.32 Proviso the
different initial configuration and the different final bulk densi-
ties in good and theta solvents, we have here evidence that the
adsorption process becomes slower in poorer solvent conditions.
The dependence of t0 on the polymer stiffness is weak.
Thus, the examination ofNadsb (t) andNads
p (t) and their variation
among the different systems suggests that the relaxation toward
complete adsorption (i) becomes slower in poorer solvent
conditions and (ii) is at later stage of the adsorption process
quantitatively different for flexible and rod-like polymers leading
to different Nadsb (t)/Nads
p (t) ratios, related to different polymer
arrangement in the adsorbed layers.
4.3.2 Structural properties. Structural properties character-
izing the adsorption process with focus on the slowest relaxation
This journal is ª The Royal Society of Chemistry 2012
processes involving spatial rearrangement of adsorbed polymers
will now be discussed. Again, we will concentrate on the effect of
the solvent conditions at the intermediate bead–surface attrac-
tion for flexible and rod-like polymers.
The time dependence of the number of beads residing in tails,
loops, and trains of flexible polymers are shown in Fig. 9 and of
rod-like polymers in Fig. 10. Commonly, (i) at t z t0 with only
a few polymers adsorbed, Ntail z 18, thus nearly all beads in
these adsorbed polymers reside in tails (see also Fig. S1 and S2†).
Thereafter, (ii) the number of beads in tails is reduced and the
number of beads in loops and trains is increased, demonstrating
a structural relaxation of the initially adsorbed polymers as well
as of later adsorbed polymers. Later, (iii) Ntail(t) displays
a minimum and Ntrain(t) a maximum, and (iv) finally Ntail(t) and
Ntrain(t) approach their equilibrium values at t z t0 0. However,
the system with rod-like polymers in the good solvent constitutes
an exception; here Ntail(t) and Ntrain(t) approach their equilib-
rium values without passing an extreme point.
The appearance of a minimum in Ntail(t) and a maximum in
Ntrain(t) is corroborated by a similar variation of Rgt(t) and
Rgk(t) involving an initial relaxation, appearance of weak
extreme values at t z 20 ns (good solvent) and at t z 100 ns
(theta solvent), and a slow relaxation toward their equilibrium
values at t z t0 (data not shown). Thus, after the polymers have
Soft Matter, 2012, 8, 5140–5150 | 5147

Fig. 10 Number of beads residing in (a) tails Ntail(t), (b) loops Nloop(t),
and (c) trains Ntrain(t) of adsorbed polymers as a function of time t on
a logarithmic scale in a good solvent (open symbols) and a theta solvent
(solid symbols) at kangle ¼ 30 J mol�1 deg�2 and 3s ¼ 2.5 kJ mol�1.
Equilibrium values obtained from MC simulations are also given (hori-
zontal lines).
Fig. 11 Probability distribution of the adsorption time P(tads) on a log-
arithmic scale in a good solvent (open symbols) and a theta solvent (solid
symbols) conditions (a) at kangle ¼ 0 (flexible polymers) for early (dotted
curves) and late (solid curves) adsorption and (b) for late adsorption at
kangle¼ 0 (flexible polymers, black) and kangle¼ 30 J mol�1 deg�2 (rod-like
polymers, red); all at 3s ¼ 2.5 kJ mol�1. Adsorption appearing before t ¼t1/2 ¼ 20 ns (good solvent) and 200 ns (poor solvent) were considered as
early, otherwise late.
Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
attained a two-dimensional shape and oriented parallel to the
surface (leading to larger Ntrain), they become laterally
compressed, resulting in fewer anchoring points (leading to
smaller Ntrain) and longer tails (leading to larger Ntail). For
flexible polymers in a good solvent, it was previously shown that
this final lateral compression becomes accentuated at increasing
bead–surface attraction.34 As compared to the good solvent, in
the theta solvent we find here that such a spatial restructuring is
much stronger and also appears for rod-like polymers.
The reason for this larger final spatial restructuring of
adsorbed flexible polymers at poorer solvent conditions (Fig. 9)
is mainly that the adsorbed polymers at intermediate time (t z100 ns) are more strongly attracted to the surface, not through
the direct bead–surface interaction, but indirectly through the
attractive bead–bead interaction. As at this stage the polymers
are individually adsorbed and have only limited contact with
bulk polymers (see Electronic Supplementary Information†),
the polymers coils are two-dimensional and localized parallel to
and at the surface. There is also a secondary effect that the
number of beads in tails at equilibrium is enhanced at poorer
solvent conditions. Here, we anticipated that the thicker
adsorbed layer appearing in the theta solvent (see Fig. 3)
energetically facilitates the tail formation. Turning to rod-like
5148 | Soft Matter, 2012, 8, 5140–5150
polymers (Fig. 10), the final relaxation of Ntail(t) to its equi-
librium values in the theta solvent dominates by the energetic
contribution as the difference between the equilibrium values of
Ntail(t) in good and theta solvents is larger than the differences
of Ntail(t) in good and theta solvents at the intermediate time.
The same holds for Ntrain.
Thus, after the polymers have attained a conformation pref-
erential parallel to the surface, they become laterally compressed,
resulting in fewer anchoring points (smaller Ntrain) and longer
tails (larger Ntail). This compression accentuates at increasing
polymer flexibly and at poorer solvent conditions.
4.3.3 Integration time and residence time. The dynamics of
the adsorption process will now be considered. For that reason,
let the distribution function P(tads) denote the probability that
a polymer remains (continuously) adsorbed during the time tadsafter it has become adsorbed. Obviously, P(0)¼ 1 and P(N)¼ 0.
Furthermore, let t1/2 denote the time at which half of the
maximum adsorbed amount is achieved. In our analysis, we will
separate polymers that adsorbed before t ¼ t1/2 ¼ 20 ns in the
good solvent and t¼ t1/2 ¼ 200 ns in the theta solvent (cf. Fig. 8).
It is anticipated that adsorption properties of polymers adsorb-
ing on a bare surface are different from those of polymers
adsorbing on a surface with adsorbed polymers.
Fig. 11 displays probability distributions of the adsorption
time on a logarithmic scale versus time for different cases.
Generally, lnP(tads) vs tads shows (i) a fast initial decay down to
P(tads) z 10�2 to 10�4 and (ii) thereafter a basically linear
dependence on the adsorption time tads. The time of the onset at
which lnP(tads) starts to become linearly dependent on tads will be
referred to as the integration time sint. Observation (i) implies
This journal is ª The Royal Society of Chemistry 2012

Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
that polymers with an adsorption time tads < sint have a larger
probability (per unit time) of becoming desorbed than those
polymers with an adsorption time tads > sint, whereas observation(ii) implies that for adsorption times tads > sint the desorption
process becomes a first order process and thus all adsorbed
polymers with tads > sint display the same probability of being
desorbed. Polymers with an adsorption time tads < sint are
considered as not yet being fully integrated in the adsorbed layer,
whereas those with tads > sint are considered as fully integrated.
The slope sres of lnP(tads) vs tads, tads > sint, provides the residencetime (average adsorption time) of fully integrated polymers. The
extracted integration sint and residence sres times are compiled in
Table 3.
In more detail, Fig. 11a shows P(tads) for adsorbed flexible
polymers in the good (open symbols) and the theta (solid
symbols) solvent separated for polymers being adsorbed early
(t < t1/2) and late (t > t1/2). The quasi-linear regimes after ca. 25 ns
falls off with different slopes, and obviously the residence time of
adsorbed polymers is longer for those being adsorbed at an early
stage (dotted curves). The difference is significant in the good
solvent and large in the theta solvent. Hence, polymers adsorbing
on a bare surface have a longer residence times than polymers
adsorbing on a polymer covered surface.
Fig. 11b displays also P(tads) but now for flexible and rod-
like polymers at the two solvent conditions. We find that the
integration times, sint ¼ 15–30 ns, do not strongly depend on
the polymer flexibility or the solvent conditions but are in
most cases significantly shorter than the residence times sres ¼30–100 ns. The residence times are longer for the rod-like
polymers as compared to the flexible ones—an observation
that could be understood by their larger contact and hence
adsorption energy with the surface. Furthermore, the residence
times in the theta solvent is smaller than those in the good
solvent. Here, the larger number density of beads outside the
layer comprising the primary adsorbed beads most likely
facilitate the exchange of polymers. Thus, the presence of
polymers available for an exchange appears more important
than any obstruction effect.
Hence, we have defined the time for a polymer to become fully
integrated into the adsorbed layer. Moreover, it was found that
the residence time of fully integrated polymers (i) is smaller for
flexible polymers as compared to rod-like ones and (ii) is smaller
in a theta solvent as compared to a good solvent.
Table 3 Integration time and residence time of integrated polymersa
Solvent kangle/J mol�1 deg�2 sint/ns sres/ns
Good 0 20 45Good 30 30 100Theta 0 15 30Theta 30 25 65
a The integration time sint is taken as the adsorption time at the onsetwhere the adsorption probability distribution P(tads) displays an
exponential behavior according to PðtadsÞ ¼ Ce�ðtads=sresÞ. The timeconstant sres of the exponential behavior is taken as the residence timeof fully integrated polymers. Estimated uncertainties are 50% (sint) and10% (sres).
This journal is ª The Royal Society of Chemistry 2012
5 Summary
The adsorption of uncharged 20-mers onto planar surfaces from
good and theta solvents has been studied for different intrinsic
stiffnesses of the polymers and various bead–surface attractions.
Equilibrium adsorption properties of 12 systems were deter-
mined by Monte Carlo simulations, and adsorption processes of
these were determined by Brownian dynamic simulations.
The layer of adsorbed polymers at the surface increased when
changing from good to theta solvents. At theta conditions, the
distribution of polymer beads becomes oscillatory, and the
individual polymers in the first adsorption layers became less
anchored to the surface.
The adsorption process started from a polymer-free surface
and the initial adsorption was diffusion controlled, but soon
became governed by the bead–surface attraction. The adsorption
was slower in the theta solvent mainly due to a slower mutual
diffusion. Flexible polymers became flattened after attaching,
but the final relaxation mechanism involved an increased
perpendicular extension with fewer adsorbed beads and longer
tails driven by the surface pressure originating from the
surrounding adsorbed polymers. This structural rearrangement
becomes stronger in poorer solvent conditions. The residence
times of adsorbed polymers became longer with increasing
polymer stiffness and smaller in poorer solvent conditions, the
latter effect consistent with a weakening of the anchoring of
adsorbed polymers.
Acknowledgements
Generous allocation of computer resources by Center for
Scientific and Technical Computing at Lund University
(LUNARC) and financial support by the Swedish Research
Council (VR), grant number 621-2007-5251, are gratefully
acknowledged.
References
1 G. J. Fleer and J. Lyklema, in Adsorption From Solution at the Solid/Liquid interface, Academic Press, New York, 1983.
2 D. F. Evans and H. Wennerstr€om, The Colloidal Domain: WherePhysics, Chemistry, Biology and Technology Meet, Wiley-VCH, NewYork, 2nd edn, 1999.
3 W. Norde, Colloids and Interfaces in Life Sciences, Marcell DekkerInc., New York, 2003.
4 J. J. Gray, Curr. Opin. Struct. Biol., 2004, 14, 110.5 M. Yaseen, H. J. Salacinski, A. M. Seifalian and J. R. Lu, Biomed.Mater., 2008, 3, 034123.
6 T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin,M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp,X. Chen and R. S. Ruoff, et al., Nat. Nanotechnol., 2008, 3, 327.
7 G. J. Fleer, M. A. Cohen Stuart, J. H. M. H. Scheutjens, T. Cosgroveand B. Vincent, Polymers at interfaces, Chapman & Hall, London,1993.
8 M. A. Cohen Stuart and G. J. Fleer, Annu. Rev. Mater. Sci., 1996, 26,463.
9 A. Yethiraj, Adv. Chem. Phys., 2002, 121, 89.10 M. A. Cohen Stuart, Surf. Sci. Ser., 2003, 110, 1.11 B. O’Shaughnessy and D. Vavylonis, J. Phys.: Condens. Matter, 2005,
17, R63.12 J. M. H. M. Scheutjens and G. J. Fleer, J. Phys. Chem., 1980, 84, 178.13 P. G. de Gennes, Macromolecules, 1981, 14, 1637.14 E. Yu. Kramarenko, R. G. Winkler, P. G. Khalatur, A. R. Khokhlov
and P. Reineker, J. Chem. Phys., 1996, 104, 4806.15 A. Striolo and J. M. Prausnitz, J. Chem. Phys., 2001, 114, 8565.
Soft Matter, 2012, 8, 5140–5150 | 5149

Publ
ishe
d on
23
Mar
ch 2
012.
Dow
nloa
ded
by U
nive
rsity
of
Wes
tern
Ont
ario
on
28/1
0/20
14 0
8:50
:19.
View Article Online
16 T. Sintes, K. Sumithra and E. Straube, Macromolecules, 2001, 34,1352.
17 A. Striolo, A. Jayaraman, J. Genzer and C. K. Hall, J. Chem. Phys.,2005, 123, 064710.
18 R. Hasegawa and M. Doi, Macromolecules, 1997, 30, 3086.19 L.-C. R. Jia and P. Y. Lai, J. Chem. Phys., 1996, 105, 11319.20 R. Zajec and A. Chakrabarti, J. Chem. Phys., 1996, 104, 2418.21 H. Takeuchi, Macromol. Theory Simul., 1999, 8, 391.22 A. L. Ponomarev, T. D. Sewell and C. J. Durning, Macromolecules,
2000, 33, 2662.23 J. K. Wolterink, G. T. Barkema and M. A. Cohen Stuart,
Macromolecules, 2005, 38, 2009.24 G. D. Smith, Y. Zhang, F. Yin and D. Bedrov, Langmuir, 2006, 22,
664.25 R. Descas, J.-U. Sommer and A. Blumen, J. Chem. Phys., 2006, 124,
094701.26 K. A. Smith, M. Vladkov and J.-L. Barrat,Macromolecules, 2005, 38,
571.27 V. A. Harmandaris, K. Ch Daoulas and V. G. Mavrantzas,
Macromolecules, 2005, 38, 5796.28 K. Ch Daoulas, V. A. Harmandaris and V. G. Mavrantzas,
Macromolecules, 2005, 38, 5780.
5150 | Soft Matter, 2012, 8, 5140–5150
29 Y. Li, D. Wei, C. C. Han and Q. Liao, J. Chem. Phys., 2007, 126,204907.
30 A. Chremos, E. Glynos, V. Koutsos and P. J. Camp, Soft Matter,2009, 5, 637.
31 N. K€allrot and P. Linse, Macromolecules, 2007, 40, 4669.32 N. K€allrot, M. Dahlqvist and P. Linse, Macromolecules, 2009, 42,
3641.33 N. K€allrot and P. Linse, J. Phys. Chem. B, 2010, 114, 3741.34 N. K€allrot and P. Linse, Macromolecules, 2010, 43, 2055.35 J. Baschnagel, H. Mayer, F. Varnik, S. Metzger, M. Aichele,
M. M€uller and K. Binder, Interface Sci., 2003, 11, 159.36 J. P. de Gennes, Scaling Concepts in Polymer Physics, Cornell
University Press, Ithaca, 1979.37 M. Ullner, B. J€onsson, C. Peterson, O. Sommelius and B. S€oderberg,
J. Chem. Phys., 1997, 107, 1279.38 A. Akinchina and P. Linse, Macromolecules, 2002, 35, 5183.39 MOLSIM, Version 4.7, P. Linse, Lund University, Sweden,
2009.40 M. P. Allen and D. J. Tildesley, Computer Simulations of Liquids,
Oxford University Press, Oxford, 1987.41 D. L. Ermak and J. A. McCammon, J. Chem. Phys., 1978, 69(4),
1352.
This journal is ª The Royal Society of Chemistry 2012