effect of c–h-s and c-h-cl interactions on the conformational preference of inhibitors of tibo...
TRANSCRIPT

8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 1/7
Effect of C–H Á Á Á S and C–H Á Á Á Cl interactions on theconformational preference of inhibitors of TIBO family
Renato F. Freitas, Sergio E. Galembeck *
Departamento de Quımica, Faculdade de Filosofia, Cie ncias e Letras de Ribeira o Preto, Universidade de Sa o Paulo, Aveinda Banderiantes
3900, 14040-901 Ribeirao Preto-SP, Brazil
Received 7 December 2005; in final form 13 March 2006Available online 22 March 2006
Abstract
The non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) are an important class of drugs employed in antiviral ther-apy. The coordinates of three inhibitors, derived from TIBO, tetrahydroimidazo-(4,5,1-1- jk )(1,4)-benzodi-azepin-2(1H )-one, (whichbelongs to the NNRTIs class), were taken from PDB database and the electronic structure were investigated by using the B3LYP/6-31+G(d,p) model. Results obtained by means of the natural bond orbital (NBO) and atoms in molecules (AIM) methods indicatedthe presence of weak hydrogen bonds of the C–H Á Á Á S and C–H Á Á Á Cl type, which are partially responsible for the conformational dif-ferences observed between the inhibitors 8 Cl-TIBO and 9 Cl-TIBO.Ó 2006 Elsevier B.V. All rights reserved.
1. Introduction
HIV-1 reverse transcriptase enzyme (HIV-1 RT) is a keytarget of anti-AIDS therapy because it catalyzes an essen-tial step in virus replication: the conversion of viral geno-mic RNA into proviral DNA, which later integrates thehost genome [1]. There are two classes of HIV-1 RT inhib-itors: the nucleoside (NRTIs) and the non-nucleoside ones(NNRTIs). NRTIs act on the catalytic site of the reversetranscriptase enzyme, preventing DNA synthesis, whereasNNRTIs bind non-competitively to a hydrophobic siteclose to the catalytic site, forcing the enzyme to adopt an
inactive conformation. [2].Three NNRTIs are currently being used in anti-HIVtreatment, namely nevirapine, delavirdine, and efavirenz[2]. The ‘first-generation’ inhibitors a-APA, TIBO 86183,9 Cl-TIBO, and nevirapine promote fast mutations of theenzyme. These mutations make the virus resistant to theseinhibitors because the interaction of these NNRTIs withaminoacids from the allosteric site of HIV-1 RT is similar.
Two of the commonest mutations that make HIV-1 resis-tant to NNRTIs are Lys103Asp and Tyr181Cys [1]. Manyefforts have been made toward novel NNRTIs that areresilient to such drug resistance mutations. To this end,new compounds such as capravirine, DPC and TMC 125have been obtained. These compounds have led to satisfac-tory results regarding inhibition of HIV-1 RT exhibitingone or more punctual mutations [3–5].
Compounds of TIBO family were the first to be shownto have high power and specificity toward the inhibitionof HIV-1 replication [6]. Since then, these compounds havebeen extensively studied with a view to increasing their
power and understanding the mechanism of action of NNRTIs [7,8]. Conformational analysis of compounds of TIBO family has been carried out in order to assess the roleplayed by the structure of these compounds in the inhibi-tion of HIV-1 RT [9,10]. These studies have shown thatthe bioactive conformation is not a global minimum, andthey have indicated that the compounds of TIBO familypresent high flexibility, which allows them to adjust tothe allosteric site of the enzyme [9,10].
In this Letter, an analysis of the intramolecular interac-tions taking place in 8 Cl-TIBO and 9 Cl-TIBO originally
0009-2614/$ - see front matter Ó 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.cplett.2006.03.041
* Corresponding author. Fax: +55 16 3602 4838/633 8151.E-mail address: [email protected] (S.E. Galembeck).
www.elsevier.com/locate/cplett
Chemical Physics Letters 423 (2006) 131–137
8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 2/7
complexed with wild-type HIV-1 RT, and in 8 Cl-TIBOcomplexed with the Tyr181Cys HIV-1 RT mutant will bepresented, by means of the natural bond orbital (NBO)[11] and atoms in molecules (AIM) [12] methods. Thisstudy was carried out aiming at understanding the differentconformational preferences exhibited by these molecules.
Our results indicate that interactions of the C–H Á Á Á Sand C–H Á Á Á Cl type are partially responsible for the diver-sity in the observed conformations.
2. Computational methods
In this work, three crystalline structures of the HIV-1RT enzyme, deposited in the Protein Data Bank (PDB)[13] and complexed with inhibitors of TIBO family wereused. The first structure contained the inhibitor 8 Cl-TIBO(R86183) complexed with wild-type HIV-1 RT (1HNV),the second structure contained 8 Cl-TIBO complexed withthe enzyme exhibiting two punctual mutations, Tyr181Cys
in subunit p66 and Cys280Ser in subunit p51 (1UWB) [8],and the third consisted of the inhibitor 9 Cl-TIBO(R82913) complexed with wild-type HIV-1 RT (1REV) [9].
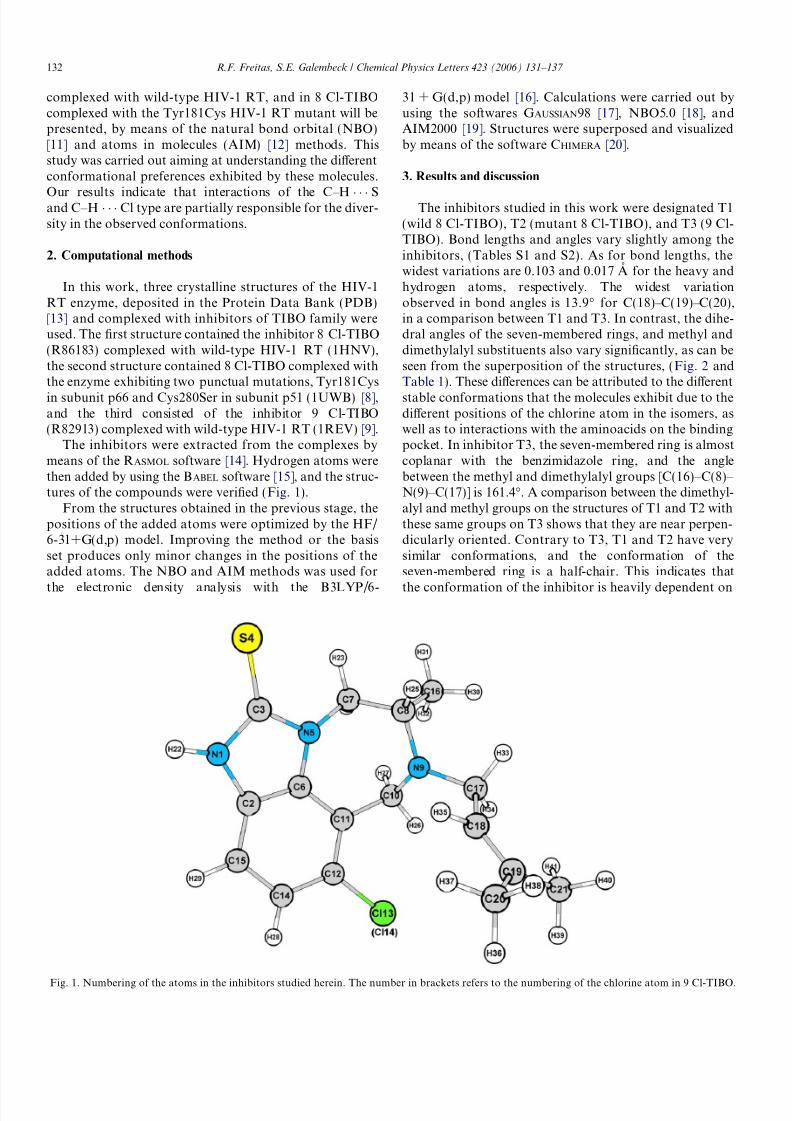
The inhibitors were extracted from the complexes bymeans of the RASMOL software [14]. Hydrogen atoms werethen added by using the BABEL software [15], and the struc-tures of the compounds were verified (Fig. 1).
From the structures obtained in the previous stage, thepositions of the added atoms were optimized by the HF/6-31+G(d,p) model. Improving the method or the basisset produces only minor changes in the positions of theadded atoms. The NBO and AIM methods was used for
the electronic density analysis with the B3LYP/6-
31 + G(d,p) model [16]. Calculations were carried out byusing the softwares GAUSSIAN98 [17], NBO5.0 [18], andAIM2000 [19]. Structures were superposed and visualizedby means of the software CHIMERA [20].
3. Results and discussion
The inhibitors studied in this work were designated T1(wild 8 Cl-TIBO), T2 (mutant 8 Cl-TIBO), and T3 (9 Cl-TIBO). Bond lengths and angles vary slightly among theinhibitors, (Tables S1 and S2). As for bond lengths, thewidest variations are 0.103 and 0.017 A for the heavy andhydrogen atoms, respectively. The widest variationobserved in bond angles is 13.9° for C(18)–C(19)–C(20),in a comparison between T1 and T3. In contrast, the dihe-dral angles of the seven-membered rings, and methyl anddimethylalyl substituents also vary significantly, as can beseen from the superposition of the structures, (Fig. 2 andTable 1). These differences can be attributed to the different
stable conformations that the molecules exhibit due to thedifferent positions of the chlorine atom in the isomers, aswell as to interactions with the aminoacids on the bindingpocket. In inhibitor T3, the seven-membered ring is almostcoplanar with the benzimidazole ring, and the anglebetween the methyl and dimethylalyl groups [C(16)–C(8)– N(9)–C(17)] is 161.4°. A comparison between the dimethyl-alyl and methyl groups on the structures of T1 and T2 withthese same groups on T3 shows that they are near perpen-dicularly oriented. Contrary to T3, T1 and T2 have verysimilar conformations, and the conformation of theseven-membered ring is a half-chair. This indicates that
the conformation of the inhibitor is heavily dependent on
Fig. 1. Numbering of the atoms in the inhibitors studied herein. The number in brackets refers to the numbering of the chlorine atom in 9 Cl-TIBO.
132 R.F. Freitas, S.E. Galembeck / Chemical Physics Letters 423 (2006) 131–137

8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 3/7
the position of the chlorine atom, making it also dependenton intramolecular interactions. Interactions between theinhibitor and aminoacids on the binding pocket should
alter the preferential conformation only slightly.The conformational analysis of a TIBO derivative
(R82913) was carried out in this laboratory by means of the pseudosystematic method SUMM [9]. The conformerswere grouped by using the clustering technique, accordingto the foldingof the seven-membered ring and to the positionof the dimethylalyl group. Results showed that the com-plexed structure of the inhibitor TIBO R82913 is equivalentto that of the conformer SP5. The inhibitors T1 and T2 canalso be classified as SP5, while T3 does not fit into any of the classes obtained in the clustering analysis.
4. NBO Analysis
The second-order perturbation energies, (DE (2)), of equiv-alent interactions between NBOs in the three inhibitors aresimilar, (Tables 2 and S4). In all cases, interactions of
p ! p* n ! p*, and n ! r* type take place. The p ! p*,np Nð1Þ ! pÃ
Cð2ÞÀCð6Þ, and np Nð5Þ ! pÃCð2ÞÀCð6Þ interactions
indicate the resonance in the benzimidazole ring. It can beseen that there is p backdonation from the chlorine atomto the benzoimidazole ring, np Clð13Þ ! pÃ
Cð11ÞÀCð12Þ, as wellas conjugation of the thiourea group, np Nð1Þ !
p
Ã
Cð3ÞÀSð4Þ and np Nð5Þ ! p
Ã
Cð3ÞÀSð4Þ. These are the most intenseinteractions encountered in these compounds, specially inT3. Although the electronegativityof the sulfur atom is mod-erate, the resonance of the thiourea group increases the effec-tive electronegativity of this atom significantly, making it agood proton acceptor [21]. This leads to stabilization of theinteractions between non-chemically bonded atoms and alsoof the intramolecular interactions np Sð4Þ ! r
ÃCð7ÞÀHð23Þ in T1
and T2, and np Sð4Þ ! rÃCð7ÞÀHð24Þ in T3, (Fig. 3A). Despite
being relatively little studied, several papers indicate thatC–H Á Á Á S interactions are weak hydrogen bonds that havegreat importance in the design of supramolecular organicsystems and in molecular stacking [22,23].
There is another intramolecular interaction np Clð13Þ !r
ÃCð10ÞÀHð26Þ in T1 and T2, (Fig. 3B). To date, there is no con-
sensus on whether interactions of the C–H Á Á Á Cl type arehydrogen bonds. Apparently, interactions of the C– H Á Á Á ClÀ and C–H Á Á Á Cl–M, (M = metal), type can beconsidered hydrogen bonds, whereas C–H Á Á Á Cl–C inter-actions would hardly be taken as such without activationof the acceptor via formation of an anion or by coordina-tion to a metal [24]. Nevertheless, C–H Á Á Á Cl interactionshave shown to be of great importance in several areas suchas molecular recognition, reactivity and structure of bio-chemical species, stability of metal complexes, crystal engi-
neering, and determination of molecular conformations[25–29].
In the case of inhibitor T3, there is no evidence that aninteraction takes place between the lone electron pair of the chlorine atom and the hydrogen atom present on theseven-membered ring, because the position of the chlorineatom hinder such interaction. Furthermore, the np Sð4Þ !r
ÃCð7ÞÀHð24Þ interaction in T3 is weaker than that in T1 and
T2. These results indicate that T3 experiences more confor-mational freedom, allowing the seven-membered ring toadopt a quasiplanar conformation in relation to the benz-imidazole ring.
5. Natural atomic charges
The natural atomic charges obtained by the NPAmethod are very similar in the three inhibitors, (Table S5).
Fig. 2. Superposition of the inhibitors from the TIBO family: T1 (blue),T2 (green), and T3 (yellow), using benzimidazole ring as reference.
Table 1Dihedral angles presenting the widest variations (°)
Dihedral angle T1 T2 T3
C(7)–C(8)–N(9)–C(10) 30.3 40.4 93.0C(8)–N(9)–C(10)–C(11) À90.9 À85.5 À91.1N(5)–C(7)–C(8)–C(16) 159.4 144.8 68.3C(7)–C(8)–N(9)–C(17) 180.0 À168.6 À73.9C(8)–N(9)–C(17)–C(18) 111.9 121.8 À174.4
Table 2Second-order perturbation energy (DE (2)) of the most relevant interactions taking place in inhibitors from the TIBO family, (kcal/mol)
T1 T2 T3
Interaction DE (2) Interaction DE (2) Interaction DE (2)
np Nð1Þ ! pÃCð3ÞÀSð4Þ 46.40 np Nð1Þ ! pÃ
Cð3ÞÀSð4Þ 56.68 np Nð1Þ ! pÃCð3ÞÀSð4Þ 71.54
np Nð5Þ ! pÃCð3ÞÀSð4Þ 53.97 np Nð5Þ ! p
ÃCð3ÞÀSð4Þ 71.08 np Nð5Þ ! p
ÃCð3ÞÀSð4Þ 75.48
np Sð4Þ ! rÃCð7ÞÀHð23Þ 3.77 np Sð4Þ ! r
ÃCð7ÞÀHð23Þ 3.65 np Sð4Þ ! r
ÃCð7ÞÀHð24Þ 1.03
np Clð13Þ ! r
Ã
Cð10ÞÀHð26Þ 2.49n
p Clð13Þ ! r
Ã
Cð10ÞÀHð26Þ 2.47
R.F. Freitas, S.E. Galembeck / Chemical Physics Letters 423 (2006) 131–137 133

8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 4/7
The largest differences are related to the different locationsof the chlorine atom in the isomers and they can beobserved between the carbon atoms and the chlorine atomin T1 and T3, (Table 3). The hydrogen atoms involved inhydrogen bonding, H(23) and H(26) in T1 and T2, and
H(24) in T3, are the atoms with the highest positive charges.
6. AIM analysis
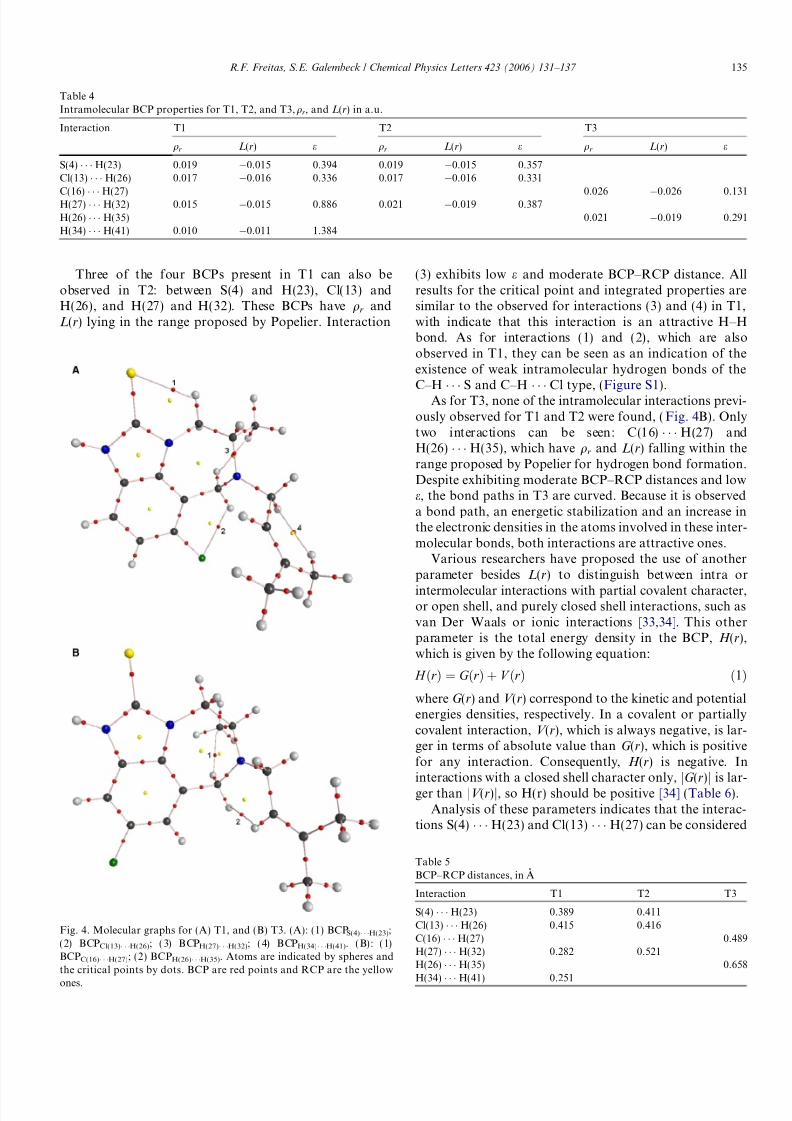
The various properties of BCPs are similar, (Tables 4and S6). The molecular graphs for T1 show four bondpaths that indicate the occurrence of the intramolecular
interactions S(4) Á Á Á H(23), (1); Cl(13) Á Á Á H(26), (2);H(27) Á Á Á H(32), (3); and H(34) Á Á Á H(41), (4), (Fig. 4A).For all the four BCPs qr and L(r) fall in the range proposedby Popelier to characterize a hydrogen bond [30]. BothH Á Á Á H interactions are traditionally considered as stericrepulsions, but Matta et al. demonstrated that these H–H
bonds substantially stabilize the system. [31] These authorspropose several criteria for these bonds. The nature of allintramolecular interactions can be understood by e andthe distance between the BCP and the RCP, (Tables 4and 5). A high e indicates that the interaction is structurallyunstable since the BCP may be destroyed upon its bindingto the RCP [32]. Therefore, the e value shows how stable asystem is in the face of a geometry variation in the system[30]. By analyzing the results for (3) and (4) it is possible toobserve that e is higher than the suggested threshold,qb = qr and BCP–RCP distances are small. By comparingthe integrated properties for the atoms involved inH Á Á Á H interaction with similar hydrogens it is possible
to observe that the energetic stabilization and the chargevariations are very small, around 2.0 kcal/mol and 0.001,but G b is larger than jV bj. It can be concluded that, despitethe disagreement of some parameters with the criteria pro-posed by Matta et al., the observed H Á Á Á H interactions areattractive H–H bonds, but the associated bond paths arevery close to instability. As for interactions (1) and (2), theyhave low e and moderate BCP–RCP distance. Domagalaet al. have observed the existence of intramolecular interac-tions of the C–H Á Á Á S type in some thiazolidinic com-pounds, where the qr and L(r) values are close to thosefound in this work [33]. According to these authors, the
C–H Á Á Á S interaction may be regarded as a weak hydrogenbond. The presence of the interactions C(7)–H(23) Á Á Á S(4)and C(10)–H(26) Á Á Á Cl(13) as confirmed by the AIM anal-ysis are in agreement with the results obtained during theNBO analysis, which showed that these interactions couldbe partially responsible for the conformational preferenceobserved in T1 and T2.
Fig. 3. Interactions in inhibitor T1: (A) np Sð4Þ ! rÃCð7ÞÀHð23Þ; (B)
np Clð13Þ ! rÃCð10ÞÀHð26Þ.
Table 3NPA charges exhibiting the widest variations among the monomers T1, T2, and T3
T1 T2 T3
Atom Charge Atom Charge Atom Charge
N(1) À0.611 N(1) À0.606 N(1) À0.574S(4) À0.212 S(4) À0.223 S(4) À0.263C(6) 0.144 C(6) 0.153 C(6) 0.177C(11) À0.033 C(11) À0.041 C(11) À0.072C(12) À0.062 C(12) À0.072 C(12) À0.248Cl(13) 0.011 Cl(13) 0.020 C(13) À0.086C(14) À0.248 C(14) À0.251 Cl(14) 0.036C(15) À0.254 C(15) À0.254 C(15) À0.282C(16) À0.700 C(16) À0.698 C(16) À0.720C(19) À0.037 C(19) À0.035 C(19) À0.010H(23) 0.300 H(23) 0.300 H(23) 0.256H(24) 0.245 H(24) 0.244 H(24) 0.275H(26) 0.261 H(26) 0.263 H(26) 0.239H(27) 0.224 H(27) 0.227 H(27) 0.261
H(31) 0.236 H(31) 0.237 H(31) 0.259
134 R.F. Freitas, S.E. Galembeck / Chemical Physics Letters 423 (2006) 131–137
8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 5/7
Three of the four BCPs present in T1 can also beobserved in T2: between S(4) and H(23), Cl(13) andH(26), and H(27) and H(32). These BCPs have qr andL(r) lying in the range proposed by Popelier. Interaction
(3) exhibits low e and moderate BCP–RCP distance. Allresults for the critical point and integrated properties aresimilar to the observed for interactions (3) and (4) in T1,with indicate that this interaction is an attractive H–Hbond. As for interactions (1) and (2), which are alsoobserved in T1, they can be seen as an indication of theexistence of weak intramolecular hydrogen bonds of theC–H Á Á Á S and C–H Á Á Á Cl type, (Figure S1).
As for T3, none of the intramolecular interactions previ-ously observed for T1 and T2 were found, (Fig. 4B). Onlytwo interactions can be seen: C(16) Á Á Á H(27) andH(26) Á Á Á H(35), which have qr and L(r) falling within therange proposed by Popelier for hydrogen bond formation.Despite exhibiting moderate BCP–RCP distances and lowe, the bond paths in T3 are curved. Because it is observeda bond path, an energetic stabilization and an increase inthe electronic densities in the atoms involved in these inter-molecular bonds, both interactions are attractive ones.
Various researchers have proposed the use of anotherparameter besides L(r) to distinguish between intra or
intermolecular interactions with partial covalent character,or open shell, and purely closed shell interactions, such asvan Der Waals or ionic interactions [33,34]. This otherparameter is the total energy density in the BCP, H (r),which is given by the following equation:
H ðr Þ ¼ G ðr Þ þ V ðr Þ ð1Þ
where G (r) and V (r) correspond to the kinetic and potentialenergies densities, respectively. In a covalent or partiallycovalent interaction, V (r), which is always negative, is lar-ger in terms of absolute value than G (r), which is positivefor any interaction. Consequently, H (r) is negative. Ininteractions with a closed shell character only, jG (r)j is lar-ger than jV (r)j, so H(r) should be positive [34] (Table 6).
Analysis of these parameters indicates that the interac-tions S(4) Á Á Á H(23) and Cl(13) Á Á Á H(27) can be considered
Table 4Intramolecular BCP properties for T1, T2, and T3, qr, and L(r) in a.u.
Interaction T1 T2 T3
qr L(r) e qr L(r) e qr L(r) e
S(4) Á Á Á H(23) 0.019 À0.015 0.394 0.019 À0.015 0.357Cl(13) Á Á Á H(26) 0.017 À0.016 0.336 0.017 À0.016 0.331
C(16) Á Á Á H(27) 0.026 À0.026 0.131H(27) Á Á Á H(32) 0.015 À0.015 0.886 0.021 À0.019 0.387H(26) Á Á Á H(35) 0.021 À0.019 0.291H(34) Á Á Á H(41) 0.010 À0.011 1.384
Fig. 4. Molecular graphs for (A) T1, and (B) T3. (A): (1) BCPS(4)Á Á ÁH(23);(2) BCPCl(13)Á Á ÁH(26); (3) BCPH(27)Á Á ÁH(32); (4) BCPH(34)Á Á ÁH(41). (B): (1)BCPC(16)Á Á ÁH(27); (2) BCPH(26)Á Á ÁH(35). Atoms are indicated by spheres andthe critical points by dots. BCP are red points and RCP are the yellow
ones.
Table 5BCP–RCP distances, in A
Interaction T1 T2 T3
S(4) Á Á Á H(23) 0.389 0.411Cl(13) Á Á Á H(26) 0.415 0.416C(16) Á Á Á H(27) 0.489H(27) Á Á Á H(32) 0.282 0.521H(26) Á Á Á H(35) 0.658
H(34) Á Á Á H(41) 0.251
R.F. Freitas, S.E. Galembeck / Chemical Physics Letters 423 (2006) 131–137 135
8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 6/7
weak hydrogen bonds, since L(r) < 0 and H (r) > 0. Theseresults are in agreement with the work of Rozas et al , whopropose that weak hydrogen bonds exhibit L(r) < 0 andH (r) > 0, intermediate hydrogen bonds present L(r) < 0and H (r) < 0, and strong hydrogen bonds have L(r) > 0and H (r) < 0 [35]. However, H (r) > 0 indicates that all theintramolecular interactions observed in the compoundsstudied herein are purely of the closed shell type, with nocovalent character. This fact reinforces the conclusion thatthe hydrogen bond of the C–H Á Á Á S type is weak, since thestrongest hydrogen bonds present small covalent character.
7. Conclusions
Analysis of the intramolecular interactions of two inhib-itors of TIBO family (8 Cl-TIBO and 9 Cl-TIBO) extractedfrom three crystalline structures of complexes formedbetween these inhibitors and HIV1 RT was carried outby means of the NBO and AIM methods. It can be
observed that 8 Cl-TIBO and 9 Cl-TIBO present differentconformations. This indicates that the intramolecular inter-actions determine the conformation of these molecules,while the intermolecular interactions act by only distortingthe preferential conformation. The presence of interactionsof the C–H Á Á Á S and C–H Á Á Á Cl type was observed in 8Cl-TIBO, T1 and T2. The former interaction may beconsidered a weak hydrogen bond, and both interactionscontribute to the stabilization of the observed conforma-tions. 9 Cl-TIBO does not display the C–H Á Á Á Cl interac-tion because of the position of the chlorine atom, and theNBO and AIM analyses showed that the hydrogen bond
C–H Á Á Á S is relatively weaker in this inhibitor than in 8Cl-TIBO. The absence of the former interaction and thelow intensity of the latter, if compared with the same inT1 and T2, render 9 Cl-TIBO greater conformational free-dom, which allows the seven-membered ring of this inhib-itor to adopt a conformation that is approximatelycoplanar with the benzimidazole ring.
Appendix A. Supplementary data
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/
j.cplett.2006.03.041.
References
[1] J. Ren, R. Esnouf, A. Hopkins, C. Ross, Y. Jones, D. Stammers, D.Stuart, Structure 3 (1995) 915.
[2] E. De Clercq, J. Med. Chem. 48 (2005) 1297.[3] J. Ren, C. Nichols, L.E. Bird, T. Fujiwara, H. Sugimoto, D.I. Stuart,
D.K. Stammers, J. Biol. Chem. 275 (2000) 14316.[4] J.W. Corbett, K.J. Kresge, S. Pan, B.C. Cordova, R.M. Klabe, J.D.
Rodgers, S.K.E. Viitanen, Antimicrob. Agents 43 (1999) 2893.[5] J. Guillemont, E. Pasquier, P. Palandjian, D. Vernier, S. Gaurrand,
P.J. Lewi, J. Heeres, M.R. de Jonge, L.M.H. Koymans, F.F.D.Daeyaert, M.H. Vinkers, E. Arnold, K. Das, R. Pauwels, K. Andries,M.P. de Bethune, E. Bettens, K. Hertogs, P. Wigerinck, P. Timm-erman, P.A.J. Janssen, J. Med. Chem. 48 (2005) 2072.
[6] R. Pauwels, K. Andries, J. Desmyter, D. Schols, M. Kukla, H.Breslin, A. Raeymaekers, J.V. Gelder, R. Woestenborghs, J. Heyk-ants, K. Schellenkens, M.A.C. Janssen, E. De Clercq, P. Janssen,Nature 343 (1990) 470.
[7] W. Ho, M. Kukla, H. Breslin, D. Ludovici, P. Grous, C. Diamond,M. Miranda, J. Rodgers, C. Ho, E. De Clercq, R. Pauwels, K.Andries, M.A.C. Janssen, P. Janssen, J. Med. Chem 38 (1995) 794.
[8] K. Das, J. Ding, Y. Hsiou, A.D. Clark, H. Moereels, L. Koymans, K.
Andries, R. Pauwels, P.A. Janssen, P.L. Boyer, P. Clark, R.H. Smith,M.B.K. Smith, c.J. Michejda, S.H. Hughes, E. Arnold, J. Mol. Biol.264 (1996) 1085.
[9] O.J. Abrahao, P.G.B.D. Nascimento, S.E. Galembeck, J. Comput.Chem. 22 (2001) 1817.
[10] S. Saen-oon, S. Hannonguba, P. Wolschann, J. Chem. Inf. Comput.Sci. 43 (2003) 1412.
[11] A.E. Reed, R.B. Weinstock, F.J. Weinhold, J. Chem. Phys. 83 (1985)735.
[12] R.F.W. Bader, Atomns in Molecules – A Quantum Theory, Inter-national Series of Monographs on Chemistry, vol. 22, OxfordUniversity Press, Oxford, 1990.
[13] H.M. Berman, J. Westbrook, Z. Feng, Z.G. Gilliland, T.N. Bhat, H.Weissig, I.N. Shindyalov, P.E. Bourne, Nucl. Acid Res. 28 (2000) 235.
[14] R. Sayle, Molecular Graphics Visualization Tool, Glaxo Research
and Development, Greeford, Middlesex, UK, 1994.[15] J.J. Gosper, BabelWin A Molecular Structure Information Inter-change Hub, Brunel University, 1998.
[16] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.[17] M.J. Frisch et al., GAUSSIAN 98, Revision A.9, Gaussian Inc.,
Pittsburgh PA, 1998.[18] E.D. Glendening, J.K. Badenhoop, A.E. Reed, J.E. Carpenter, J.A.
Bohmann, C.M. Morales, F. Weinhold, NBO 5.0, TheoreticalChemistry Institute, University of Wisconsin, Madison, WI, 2001.
[19] F. Biegler-Konig, J. Schonbohm, D. Bayles, J. Comput. Chem. 22(2001) 545.
[20] E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M.Greenblatt, E.C. Meng, T.E. Ferrin, J. Comput. Chem. 25 (2004)1605.
[21] F. Allen, C.M. Bird, R.S. Rowland, P.R. Raithby, Acta Cryst. B53
(1997) 680.
Table 6
G (r), V (r), and H (r), (·10À3 a.u.), for T1, T2, and T3
Interaction T1 T2 T3
G (r) V (r) H (r) G (r) V (r) H (r) G (r) V (r) H (r)
S(4) Á Á Á H(23) 13.777 À12.329 1.448 13.390 À12.013 1.377Cl(l3) Á Á Á H(26) 13.712 À11.312 2.399 13.860 À11.458 2.402
C(16) Á Á Á H(27) 24.084 À21.776 2.309H(27) Á Á Á H(32) 12.598 À9.757 2.841 16.996 À15.022 1.974H(26) Á Á Á H(35) 17.120 À15.214 1.906H(34) Á Á Á H(41) 8.169 À5.634 2.534
136 R.F. Freitas, S.E. Galembeck / Chemical Physics Letters 423 (2006) 131–137

8/7/2019 Effect of C–H-S and C-H-Cl interactions on the conformational preference of inhibitors of TIBO family
http://slidepdf.com/reader/full/effect-of-ch-s-and-c-h-cl-interactions-on-the-conformational-preference 7/7
[22] S.J. Narayanan, B. Sridevi, T.K. Chandrashekar, A. Vij, R. Roy, R.Angew, Chem. Int. Ed. 37 (1998) 3394.
[23] M.J. Potrzebowski, M. Michalska, A.E. Koziol, S. Kazmierski, T.Lis, J. Pluskowski, W. Ciesielski, J. Org. Chem. 63 (1998) 4209.
[24] C.B. Aakeroy, T.A. Evans, K.R. Seddon, I. Palinko, New J. Chem. 23(1999) 145.
[25] E.M.D. Keegstra, A.L. Spek, J.W. Zwikker, L.W. Jenneskens, J.Chem. Soc. Chem. Commun. 14 (1994) 1633.
[26] Z.S. Derewenda, L. Lee, U. Derewenda, J. Mol. Biol. 252 (1995) 248.[27] T. Steiner, W. Saenger, J. Chem. Soc. Chem. Commun. 20 (1995)
2087.[28] V. Balamurugan, W. Jacob, J. Mukherjee, R. Mukherjee, Cryst. Eng.
Commun. 6 (2004) 396.
[29] G. Muller, M. Lutz, S. Harder, Acta Crystallog. 52 (1996) 1014.[30] P. Popelier, Atoms in Molecules: An Introduction, Prentice Hall,
England, 2000.[31] C.F. Matta, J. Hernandez-Trujillo, T.-H. Tang, R.F.W. Bader,
Chem. Eur. J. 9 (2003) 1940.[32] D. Cremer, E. Kraka, T.S. Slee, R.F.W. Bader, C.D.H. Lau,
T.T.N. Ding, P.J. MacDougall, J. Am. Chem. Soc. 105 (1983)5069.
[33] M. Domagala, S.J. Grabowski, K. Urbaniak, G. Mloston, J. Phys.Chem. A 107 (2003) 2730.
[34] S.J. Grabowski, J. Phys. Org. Chem. 17 (2004) 18.[35] I. Rozas, I. Alkorta, J. Elguero, J. Am. Chem. Soc. 122 (2000)
11154.
R.F. Freitas, S.E. Galembeck / Chemical Physics Letters 423 (2006) 131–137 137