
REPUBLICA BOLIVARIANA DE VENEZUELA UNIVERSIDAD DEL ZULIA
FACULTAD EXPERIMENTAL DE CIENCIAS DIVISIÓN DE ESTUDIOS PARA GRADUADOS
MAESTRIA EN BIOLOGÍA MENCIÓN INMUNOLOGÍA BÁSICA
PAPEL DE LA MICROGLIA Y LA SEROTONINA EN LA HIPERALGESIA INDUCIDA POR ESTRÉS REPETIDO
Trabajo presentado para optar al Grado de Magíster Scientiarum en Biología, Mención Inmunología Básica.
Autor: Shirley Medina de Leendertz, Licda.
C.I.: 6913159
Tutor: Dr. Heberto Suárez Roca
C.I.: 4.533.204
Maracaibo, Septiembre de 2007

12
PAPEL DE LA MICROGLIA Y LA SEROTONINA EN LA HIPERALGESIA INDUCIDA POR ESTRÉS REPETIDO
Trabajo presentado para optar al Grado de Magíster Scientiarum en Biología, Mención Inmunología Básica.
Autor: Shirley Medina de Leendertz, Licda. C.I.: 6913159
E-mail: [email protected] Tutor: Dr. Heberto Suárez Roca
C.I.: 4.533.204 E-mail: [email protected]

13
ACTA VEREDICTO
Este jurado aprueba el Trabajo de Grado titulado ¨PAPEL DE LA MICROGLIA Y LA SEROTONINA EN LA HIPERALGESIA INDUCIDA POR ESTRÉS REPETIDO¨, que la Licenciada en Biología Shirley Medina de Leendertz C.I.: 6.913.159, presenta ante el Consejo Técnico de Estudios para Graduados de la Facultad Experimental de Ciencias, en cumplimiento de los requisitos señalados en los Artículos del Reglamento de Estudios para Graduados para optar al titulo de MAGISTER SCIENTIARUM EN BIOLOGÍA, MENCIÓN INMUNOLOGÍA BÁSICA.
Jurado:
MSc. Sandra Azuero de G. Dra. María Zabala (Coordinadora) (Jurado Principal) C.I.7.885.249 ׃ C.I.7.492.506 ׃
Dr. Hector Pons P. (Jurado Principal) C.I.4.521.139 ׃
Maracaibo, Septiembre 2007

14
AGRADECIMIENTOS:
• A mis padres, a mi esposo e hijas, a mis hermanos y sus familias todos
un hermoso regalo de Dios.
• Mis más sincero agradecimiento a mi tutor Dr. Heberto Suarez y al MSc .
Luis Quintero hombres pacientes, espectaculares personas, muy buenos
amigos y maestros, a todos los compañeros del Laboratorio de
Farmacología del Instituto de Investigaciones Clínicas Edixon,
Jacqueline, Ricardo y Heisher todos personas inigualables muy buenos
amigos.
• A los que laboran conmigo y el Dr. Ernesto Bonilla por permitirme seguir
creciendo profesionalmente, a todo el personal del Instituto de
Investigaciones Clínicas todas personas muy especiales y de las que en
momentos difíciles recibí todo su apoyo.
• A todos mis amigos, maravillosas personas que siempre están a mí
alrededor brindándome su amistad, su amor, su paciencia y sus
conocimientos a todos gracias.

15
DEDICATORIA: A Dios espíritu renovador de vida, creación vibrante y sabiduría infinita que esta
en mí y en todos seres del mundo. A mis padres los amo, a mi esposo e hijas
fuente de amor, comprensión y paciencia son mi vida, a mis hermanos y a
todos mis familiares doy gracias por ser tan afortunada de tenerlos a todos a mi
lado. A Norma y King por su apoyo. A mi madre y padre espiritual Gloria y
Rubén, y a todos mis queridos amigos ellos saben quienes son.

16
INDICE DE CONTENIDO Pág.
Índice de figuras vii
Resumen ix
Abstract x
1. Introducción 11
2. Revisión Bibliográfica 20
3. Objetivos 36
3.1. Objetivo General 36
3.2. Objetivos Específicos 36
4. Metodología 38
4.1. Animales de experimentación 38
4.1.1. Modelo Experimental 1 38
4.1.1.2.- Nocicepción Química 39
4.1.1.3. Protocolo de Inmunohistoquímica para microglia 41
4.1.2. Modelo experimental 2 44
4.1.3. Modelo experimental 3 45
4.1.3.1. Protocolo de Inmunohistoquímica para c-Fos 46
4.2. Análisis estadístico 49
4.3. Análisis de las Imágenes 50
5. Resultados 51
6. Discusión 63
7. Conclusión 77
8. Referencias Bibliográficas 81

17
ÍNDICE DE FIGURAS
FIGURA Pág
1 Curva de intensidad del estímulo vs. sensación dolorosa 29
2 Diseño Experimental 1 44
3 Diseño Experimental 2 45
4 Diseño Experimental 3 49
5 Cambios morfológicos de la Microglia. A microglia en reposo, B microglia activada y C microglia totalmente activa como macrófago. 52
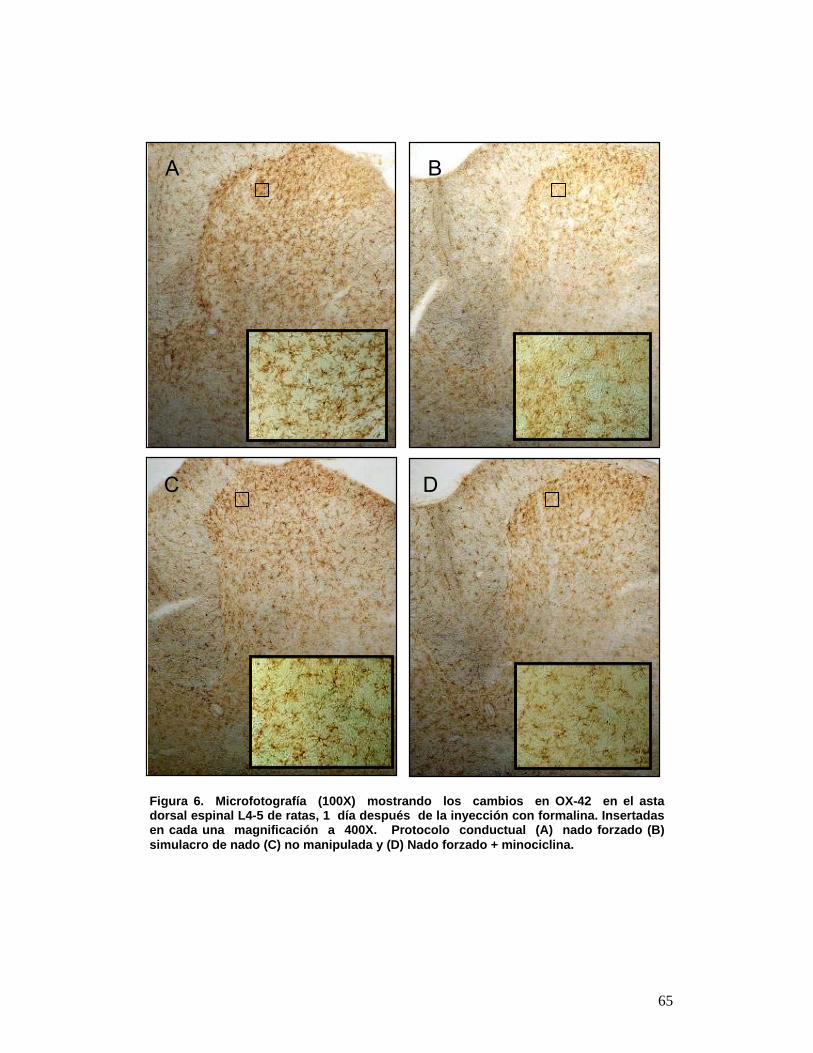
6 Microfotografía (100X) mostrando los cambios en OX-42 en el asta dorsal espinal L4-5 de ratas, 1 día después de la inyección con formalina. Insertadas en cada una magnificación a 400X. Protocolo conductual (A) nado forzado (B) simulacro de nado (C) no manipulada y (D) Nado forzado + minociclina. 55
7 Curvas de respuesta nociceptiva. Todas las ratas fueron sometidas a la prueba de formalina. A) Ratas sometidas a los procedimientos condicionantes, mostrando respuestas hiperalgésicas en ratas estresadas, * representa una diferencia significativa con p< 0,001 de las ratas salinas estresadas al compararlas con ratas no manipuladas, B) Las ratas sometidas a nado forzado mostraron una respuesta hiperalgésica frente a la prueba de formalina, inhibida al pretratarlas con minociclina,
representa una diferencia significativa con p<0,001 de las ratas salinas estresadas al compararla con ratas estresadas y pretratadas con minociclina, C) simulacro de nado, D) no manipuladas. Las barras representan 2 períodos de no observación conductual (15 min. Cada unos). 56
8 Declive progresivo del tiempo de lucha después de la sección de nado forzado repetido una dosis baja de un inhibidor de la recaptación de serotonina clomipramina (2,5 mg/Kg, i.p. por día) y Minociclina una tetraciclina (40 mg/Kg)*
vii

18
representa una diferencia significativa con p<0,0001 al compararla con minociclina. 57 9 Curvas de respuesta nociceptiva. Todas las ratas sometidas a la prueba de formalina. A) Las ratas sometidas a nado
forzado mostraron una respuesta hiperalgésica frente a la prueba de formalina, conductual (15 min cada unos). Cada punto representa la media ± el error estándar obtenido de 6 ratas representa una diferencia significativa de p< 0.01 en todos los intervalos marcados, excepto el 8 y el 18 con un p<0,001 con respecto a control salino (A). 58 10 Expresión laminar de la proteína c-Fos en ratas seguida de la
inyección subcutánea de formalina en la pata trasera derecha. A) Las ratas pretratadas con solución salina
sometidas a nado forzado mostraron un incremento significativo en el marcaje de los núcleos inmunoreactivos en la lámina I-II y V-VI. B) simulacro de nado, C) no manipuladas. * representa una diferencia significativa con p<0,0001 de las ratas salinas estresadas al compararla con ratas estresadas y pretratadas con clomipramina. 59
11 Microfotografía (100X) mostrando la expresión de la
proteína c-Fos inducida por formalina en el asta dorsal espinal de ratas. Protocolo conductual (A) Salina con Nado
forzado (B) Clomipramina con nado forzado (C) Salina Simulacro, (D) Clomipramina Simulacro, (E) Salina No manipulada y (F) Clomipramina No manipulada. 62
viii

19
SHIRLEY MEDINA DE LEENDERTZ. PAPEL DE LA MICROGLIA Y LA SEROTONINA EN LA HIPERALGESIA INDUCIDA POR ESTRÉS REPETIDO. Trabajo de Grado para optar al titulo de Magister Scientiarum en Biología, Mención Inmunología Básica. División de Estudios para Graduados. Facultad Experimental de Ciencias. Universidad del Zulia. Maracaibo, Venezuela, 2007. p 91.
RESUMEN
El propósito de este estudio fue determinar si la microglia y la serotonina están involucradas en la hiperalgesia provocada por exposición repetida a estrés. Ratas machos Sprague-Dawley fueron sometidos a estrés psicofísico (nado forzado) o psicológico (simulacro de nado) sin condiciones de escape, por 10-20 min, durante 3 días consecutivos. Los animales controles no fueron manipulados y permanecieron en sus jaulas. Cada uno de estos tres grupos de ratas fueron subdivididos en: (1) aquellos que no recibían ningún pretratamiento farmacológico; (2) pretratados con 40 mg/Kg i.p. de minociclina (un inhibidor selectivo de la microglia) ó con vehículo (NaCl 0.9%, i.p.), 1 hora antes de cada nado, simulacro o enjaulamiento; y (3) pretratados con 2.5 mg/Kg i.p. de clomipramina (un bien conocido antidepresivo e inhibidor selectivo de la recaptación de serotonina) ó con vehículo (NaCl 0.9%, i.p.), 7 dias antes y durante cada nado, simulacro o enjaulamiento. Todas las ratas fueron luego inyectadas en la pata trasera con formalina 1% para producir un dolor inflamatorio agudo. Posteriormente las ratas fueron sacrificadas y perfundidas para removerles quirúrgicamente el segmento lumbar de la médula espinal. Los segmentos medulares de los grupos no tratados y tratados con minociclina fueron procesados inmunohistoquímicamente para observar la microglia (usando un anticuerpo anti-CR3, OX-42). A los tejidos provenientes de ratas tratadas con clomipramina se procesaron inmunohistoquímicamente para el marcador neuronal c-fos. El estrés de nado incrementó la activación microglial inducida por la inyección subcutánea de formalina. La minociclina inhibió la activación microglial central observada en la medula espinal de ratas estresadas con nado forzado. Al mismo tiempo, la administración de minociclina evitó el desarrollo de la hiperalgesia cutánea post-estrés de nado. La hiperalgesia cutánea post-estrés de nado, la disminución de los intentos de escape a lo largo de los nados consecutivos y la sobre-expresión de c-Fos inducida por un dolor inflamatorio agudo fueron bloqueadas por el pretratamiento con clomipramina. En conclusión, la hiperalgesia inducida por estrés de nado forzado estuvo vinculada a una sobre-activación de la microglia en el sistema nervioso central y a una deficiencia en la transmisión central serotoninérgica. Así, el estrés de nado pareciera sensibilizar los circuitos neuronales del dolor y la inmunidad intrínseca de la medula espinal para una respuesta incrementada a un estímulo inmunológicos y nocivos periféricos subsiguientes.
Palabras claves: activación microglial, estrés, dolor, serotonina, inflamación. Correo electrónico del autor: [email protected]
ix

20
SHIRLEY MEDINA DE LEENDERTZ. ROLE OF MICROGLIA AND SEROTONIN IN HIPERALGESIA INDUCED BY REPEATED STRESS. Trabajo de Grado para optar al titulo de Magister Scientiarum en Biología, Mención Inmunología Básica. División de Estudios para Graduados. Facultad Experimental de Ciencias. Universidad del Zulia. Maracaibo, Venezuela, 2007. p 91.
ABSTRACT
The purpose of this study was to determine if microglia and serotonin are involved in hyperalgesia induced by repeated exposure to stress. Male Sprague-Dawley rats were submitted to psychophysical (forced swimming) or psychological stress (swimming simulacrum) without escape conditions, for 10-20 min, for 3 consecutive days. The control animals were not manipulated and remained in their cages. Each one of these three rat groups were subdivided in: (1) no pharmacologic pretreatment; (2) pretreatment with minocicline 40 mg/Kg i.p. or with vehicle (NaCl 0.9%, i.p.), 1 hour before each swim, simulacrum or encaged and (3) pretreatment with clomipramine 2.5 mg/Kg i.p. (a well known antidepressant and selective inhibitor of serotonin uptake) or with vehicle (NaCl 0.9%, i.p.), 7 days before and during each swim, simulacrum or while caged. All rats were injected afterwards on the hind paw with 1% formalin to produce acute inflammatory pain. The rats were sacrificed and perfused to surgically remove the lumbar segment of the spinal cord. The medullary segments of the minocicline treated and not treated groups were treated immunohystochemically for microglia observation (using an AntiCR3 antibody, OX-42). The tissues obtained from rats treated with clomipramine were processed immunohystochemically for the neuronal marker c-fos. Swimming stress increased microglial activation induced by the subcutaneous injection of formalin. Minocicline inhibited central microglial activation observed in the spinal cord of rats under swimming stress. At the same time, the administration of minocicline avoided the development of cutaneous hyperalgesia post swimming stress. Cutaneous hiperalgesia post swimming stress, reduced escape attempts in consecutive swims and the overexpression of c-Fos induced by an acute inflammatory pain were blocked by pretreatment with clomipramine. In conclusion, hyperalgesia induced by swimming stress could be linked to an overactivation of microglia in the central nervous system and a deficiency in central serotoninergic transmission. Thus, swimming stress seems to sensibilize the pain neuronal circuits and the intrinsic immunity of the spinal cord for an increased answer to a subsequent immunological and nocive peripheral stimulus.
Key Words: microglial activation, stress, pain, serotonin, inflammation. Authors Electronic Mail: [email protected]
x

21
1.- INTRODUCCIÓN
Las microglias son células de la inmunidad innata, que están residenciadas
en el SNC, representan en adultos aproximadamente el 10 % de la población
celular del sistema nervioso central (Town y col. 2005). El papel principal de la
microglia es mediar respuestas inmunitarias intrínsecas innatas frente a injurias
tisulares en una gran variedad de condiciones (Stence y col., 2001). Diversas
evidencias sugieren que la microglía juega un papel doble de neuroprotector ó
citotóxico dependiendo de la severidad de injuria sobre el tejido (Benati y col.,
1993).
La adhesión específica (no aleatoria) entre diferentes células o entre
células o matrices extracelulares es un componente esencial de la migración y
el reconocimiento celular y subyace a muchos procesos biológicos. El receptor
para el complemento tipo 3 (CD11b), es una integrina con cadenas αM de 165
kD y cadenas β2 de 95 kD. Las integrinas como su nombre lo indica integran la
unión del ligando extracelular con la motilidad dependiente del citoesqueleto, el
cambio de forma y la respuesta fagocítica. La integrina CD11b en humanos y
CR3 en ratas cuyo ligando es la proteína del complemento iC3b en humanos,
es expresado por monocitos/macrófagos y la microglía, permitiendo la
visualización de dichas células mediante inmunohistoquímica con el anticuerpo
monoclonal OX-42 (Abbas y col., 2003; Draskovic-Pavlovic y col., 1999; Choi

22
y col., 2003). También la microglia expresa el receptor CD200R principalmente
expresado por células mieloides (Wright y col., 2003).
Además de los receptores antes mencionados la microglia posee diferentes
tipos de receptores implicados en la modulación de la transmisión nociceptiva,
tales como, receptores adrenérgicos, serotoninérgico tipo 5-HT2a, purinérgicos,
GABAérgicos, glutamatérgicos y glicinérgicos. De esta manera, la actividad de
la neuroglia pudiera ser modulada o regulada por diferentes neurotransmisores,
entre ellos la 5-HT (Lee y col., 2001; Synowitz y col., 2004). Las células gliales
(microglia y astrocitos) de la médula espinal, son críticamente importantes en la
mediación de estos procesos nociceptivos (Watkins y col., 2003). Aunque las
células gliales no son eléctricamente excitables ni establecen sinapsis con las
neuronas, mantienen un diálogo continuo entre ellas y con las neuronas de un
modo bastante complejo, asegurándose el soporte estructural, metabólico y
trófico durante toda la vida (Aschner y col., 2002).
Las respuestas inmunitarias están inhibidas en el cerebro en condiciones
normales. Existen evidencias de que las neuronas controlan el estado de
activación de la microglia, contribuyendo a su estado quiescente. Uno de los
mecanismos sugeridos de cómo las neuronas controlan la activación microglial
es la interacción con el ligando CD200, una glicoproteína de membrana
presente en la neurona, con sus receptores presentes en la microglia (Neuman,

23
2001). También existen antibióticos con una acción potente y preferencial
inhibitoria sobre microglia y no sobre los astrocitos o neuronas, como es la
minociclina una tetraciclina semisintética de segunda generación que penetra
bien en SNC a través de la barrera hematoencefalica (Alonso, 1980). Además
de su acción como antibiótico, la minociclina tiene efecto neuroprotector y
antiinflamatorio en el SNC (Tikka y col., 2001; Yrjanheikki y col 1998).
Recientes estudios han demostrado que las células gliales del cordon
espinal, tales como los astrocitos y la microglia, juegan un papel importante en
la creación y mantenimiento del dolor patológico inducido por virus y bacterias,
elevando las citoquinas proinflamatorias. (Coyle 1998; Watkins y col. 1997;
Watkins y col. 2001a, b; Watkins y Maier 1999, 2000; Tsuda y col, 2003). La
microglia también puede sensibilizarse respondiendo a un estimulo nocivo,
expresando el receptor del complemento tipo CD11b en humanos y CR3 en
ratas cuyo ligando es la proteína del complemento iC3b. Se ha demostrado que
las células gliales pudieran ser activadas por sustancias relevantes para el
mecanismo del dolor como la sustancia P, bradykinina, péptidos, genes
relacionados con calcitonina (Fu y col, 1999; Watkins y col, 1997, 2001a, b).
Desde la sensación hasta la percepción del dolor y su modulación son vistos
clásicamente mediados solamente por neuronas. Recientes hallazgos implican
a la glía del cordón espinal con un papel importante en la creación y

24
mantenimiento del dolor patológico. Existen evidencias cada vez mayores de
que la glía puede ser la clave para crear estados de dolor exagerado en
diversas patologías, esto sugiere que pueden estar ampliamente involucrados
en la creación del dolor patológico y tal vez en otros fenómenos sensoriales
(Watkins y col., 2001). La inyección subcutanea de formalina en la superficie
plantar (prueba de formalina) nos provee de un valioso modelo de dolor
inflamatorio (Cho y col., 2006), la hiperalgesia inducida por formalina se ha
sugerido que involucra la glia espinal (Teng y col., 1998; Cho y col., 2006).
Los astrocitos y las microglías de las astas dorsales son activados en
respuesta a una amplia gama de condiciones que producen hiperalgesia. Estos
incluyen, inflamación subcutánea (Sweitzer y col., 1999; Fu y col., 1999),
bacterias intraperitoneales (Sweitzer y col., 1999), trauma de nervio
periférico (DeLeón y col., 1999), cáncer de hueso (Schwel y col., 1999),
constricción de raíces raquídeas lumbares (Watkins y col., 2000) y activación
inmunitaria dentro del cordón (Milligan y col. 2001).
La microglia y los astrocitos son atractivos candidatos como mediadores de
hiperalgesia, ya que: primero, la glia activada libera sustancias que excitan las
neuronas espinales que responden al dolor, tales como: especies de oxígeno
reactivo y Oxido Nítrico, prostaglandinas y otros productos del ácido
araquidónico, aminoácidos excitadores y factores de crecimiento (Watkins y

25
col., 2000). Segundo, sustancias liberadas de la glia activada originan
liberación exagerada de transmisores del dolor a partir de neuronas sensoriales
que forman sinapsis en el asta dorsal (Inoue y col., 1999; Southall y col., 1998).
Tercero, la microglia y los astrocitos forman asas de control positivo, creando
una liberación perseverante de mediadores del dolor (Watkins y col., 2000).
Cuarto, las sustancias derivadas de la glia ejercen funciones autocrinas y
paracrinas por lo cual están bien posicionadas para afectar globalmente la
actividad en el cordón espinal (Aldskogius y col., 1998). Finalmente, la glia es
activada por neurotransmisores del dolor en el cordón dorsal (Watkins y col.,
2001).
Entonces, es importante destacar que la función inmunológica de la
microglia y los astrocitos guardan cierto paralelismo con los mecanismos de
percepción del dolor, ya que ambas son células indispensables para la
protección del organismo al alertar de daño tisular frente a la exposición de
estímulos potencialmente nocivos. La percepción del dolor es regulada por el
organismo a través de diferentes mecanismos a diferentes niveles. Un nivel de
control está ubicado en las astas posteriores o dorsales de la médula espinal.
Varias teorías han surgido de datos experimentales que explican la regulación
espinal de la nocicepción. Una de ellas es la teoría de la puerta de control del
dolor (Melzack y Loeser, 1978; Loeser y Melzack, 1999; Ribera y col., 2003).
En esta teoría se considera que la transmisión de impulsos de dolor,

26
conducidos hasta la médula espinal por fibras aferentes nociceptoras que
provienen de tejidos periféricos, pueden ser inhibidas pre- o post-
sinápticamente, a nivel del asta dorsal espinal por: (1) impulsos conducidos por
fibras aferentes no nociceptoras (ej., táctiles, de temperatura no nociva, etc.)
que inervan tejidos periféricos, o (2) por fibras descendentes que se originan en
los niveles centrales del sistema nervioso, como la sustancia periacueductal
gris y que liberan serotonina o 5-hidroxitriptamina (5HT), como uno de sus
neurotransmisores mayores (Cabana y col., 2004).
La 5-HT regula la nocicepción a nivel de la medula espinal a través de
diferentes subtipos de receptores 5-HT y cambios funcionales de larga duración
en los mismos (Eide y col., 1993). Algunos estudios han evidenciado que la
estimulación de los receptores 5-HT1, 5-HT2 y 5-HT3 parece reducir la
nocicepción mientras que otros han reportado que la activación de 5-HT2 y 5-
HT3 parece incrementar la percepción del dolor. Además, el incremento o la
reducción adaptativa del número de los receptores pueden originar cambios de
larga duración o plasticidad en el sistema 5-HT. La lesión de neuronas
serotoninérgicas induce supersensibilidad por denervación a la 5-HT, por el
contrario, la estimulación prolongada de los receptores 5-HT puede producir
subsensibilidad a la 5-HT. El tratamiento a largo plazo con antidepresivos
utilizados en el manejo clínico del dolor parecen incrementar los receptores 5-
HT1 y disminuir los receptores 5-HT2 (Eide y col., 1993)

27
La liberación de 5HT dentro del SNC puede ser modificada por situaciones
estresantes que conducen a estados fisiológicos o emocionales que
interrumpen el balance homeostático del individuo o alostasis, equilibrio
dinámico del organismo (Ribera y col., 2003). Estudios con microdiálisis
intracerebral en ratas con libertad de movimientos han mostrado un incremento
en la liberación de 5HT en varias regiones del cerebro, especialmente en el
rafe magnus después de una exposición de 5 – 15 min a estrés de nado
forzado (Adell y col., 1997; Hellhammer y col., 1983; Ikeda y col., 1985). Por lo
contrario, un nado forzado prolongado por 30 min. disminuye la liberación de
5HT en algunas estructuras cerebrales tales como la amígdala y septum lateral
(Kirby y col, 1995, 1997). Una correlación entre la inhibición de la liberación de
5-HT y hacer frente activo en la prueba de estrés por nado (tratar de escapar)
involucra a este efecto neuroquímico en la respuesta conductual al estrés de
nado (Kirby y col., 1997).
Cambios en la actividad del sistema serotoninérgico central, pueden
explicar, en parte, los cambios bidireccionales en la nocicepción (analgesia e
hiperalgesia) observados después de diferentes condiciones de estrés. De
forma interesante, la actividad de las fibras serotoninérgicas descendentes del
rafe magno inhibe tónicamente la transmisión del dolor en la médula espinal y
la inhibición de la síntesis de serotonina a nivel espinal produce hiperalgesia en
ratas (Adell y col., 1997)

28
Estímulos y señales externas como estrés prolongado y el dolor pueden
activar a nivel celular numerosos genes con el fin de generar una gama de
factores transcripcionales o proteínas accesorias que interaccionan con
factores transcripcionales necesarios para responder adecuadamente al
ambiente y garantizar la supervivencia del organismo (Griffiths y col., 1995). El
prototipo de estos genes inmediatos es el c-fos, cuya proteína forma
heterodímeros específicos con otras proteínas nucleares como c-jun, a través
de una interacción ¨leucine-zipper¨ y de esta forma se fija al DNA de los genes
cuya expresión regula (Hughes y col., 1985). Por ello, la expresión de c-Fos ha
sido empleada como un índice marcador de la actividad neuronal (Hughes y
col., 1985; Hsu y col., 1981).
De la evidencia experimental señalada anteriormente pueden extraerse
varias conclusiones: 1) que la microglia posee receptores para 5-HT, y por lo
tanto es susceptible a un control regulatorio por fibras inhibitorias descendentes
que liberan 5-HT como neurotransmisor, 2) que la exposición aguda al estrés
induce la secreción de serotonina mientras que la exposición repetida al estrés
produciría una disminución de la liberación de 5-HT por mecanismos no
conocidos, probablemente por agotamiento del neurotransmisor almacenado
en los terminales sinápticos; 3) la serotonina es un modulador inhibitorio de la
nocicepción, por lo cual, aumentos de sus niveles llevarían a una analgesia
(disminución de la percepción del dolor) y una disminución de sus

29
concentraciones extracelulares provocarían un estado hiperalgésico (aumento
de la percepción del dolor).
De esta manera, es posible que el estrés repetido en la rata, como el
producido por el sometimiento a nado forzado, produzca una disminución de
los niveles de 5-HT en la medula espinal, lo cual disminuiría el control
inhibitorio de la nocicepción, y ocasionaría la hiperalgesia observada 24 horas
después del último episodio de estrés. Esta disminución del control inhibitorio
de la serotonina se reflejaría como una mayor activación de poblaciones de
neuronas y microglias en la medula espinal de la rata. El propósito de este
estudio fue determinar si bajo condiciones de estrés repetido la serotonina y la
microglia están involucradas en la hiperalgesia existiendo 1) cambios en
incremento de la percepción del dolor inflamatorio provocado por el estrés de
nado repetido al administrar la clomipramina un inhibidor de la recaptación de
serotonina, 2) cambios en la expresión de la proteína c-Fos indicador de
activación neuronal espinal sensorial al administrar clomipramina en ratas
estresadas con dolor inflamatorio, 3) cambios en activación microglial en ratas
estresadas con dolor inflamatorio inducido por la inyección subcutánea de
formalina indicado por la expresión del receptor CR3 y por último la 4) cambios
sobre el incremento de la percepción del dolor inflamatorio producido por estrés
repetido al administrar minociclina un inhibidor de la activación microglial.

30
2.- REVISIÓN BIBLIOGRÁFICA
Activación de microglia
Después de lesión o de la infección, la microglia se activa, esto se
manifiesta mediante un proceso de contracción, proliferación e incremento de
la expresión de varios factores en la superficie de la célula. El nivel de
activación de la microglia y la expresión de sus receptores depende del tipo y la
severidad de lesión del cerebro (Raivich y col., 1999; Streit y col., 1988).
Después de una axonotomía reversible, la microglia prolifera y rodea los
nervios secretando factores solubles tales como factor de crecimiento del
fibroblasto (FGF) y factor de crecimiento del nervio (FCN) (Heumann y col.,
1987; Gomez-Pinilla y col., 1990; Araujo y Cotman, 1992). También ocurre un
incremento en la expresión de varios receptores como los de complemento, Fc,
Trombina, ¨scavenger¨, citoquinas como la IL-6 (que estimula el crecimiento de
los linfocitos B productores de anticuerpos) y la IL-18 (inflamatoria glia),
quimiocinas (que inician y promueven las reacciones inflamatorias, ya que
regulan el tráfico y afluencia al sitio de la inflamación de varios tipos celulares y
determinan un incremento de su adhesión a las células endoteliales y/o su
activación) y de co-receptores de CD4 (CMH clase II) y CD8 (CMH clase I). Así,
la microglia parece mostrar un efecto neuroprotector en su estado activado o
esferoide, y colabora en la recuperación de neuronas con daños reversibles.

31
Inversamente, la degeneración neuronal inducida por una proteína toxica
llamada ricina (acontecimiento irreversible y letal) da lugar a que la microglia se
active completamente como fagocito. Esta etapa de la activación está
caracterizada por un aumento significativo en la expresión de los marcadores
observados en la etapa fagocitaria incluyendo varias integrinas (α5β1, α6β1 y
αMβ2), y antígenos del complejo principal de histocompatibilidad de clase I y II.
Así, la microglia muestra una plasticidad "funcional" notable dependiendo de la
severidad de lesión (Kitamura y col., 1977; Dickson y col., 1991; Lee y col.,
2001).
Por otra parte, es importante mencionar la acción de los nucleótidos y
nucleósidos extracelulares en situaciones de daño porque pueden estimular la
síntesis de ADN y la proliferación de la microglia, astrocitos y células
endoteliales. Cabe destacar que los nucleótidos y nucleósidos junto con el ATP
y otros componentes inducen a que haya un aumento de calcio en esas
células, siendo los responsables de su activación, e incluso podrían actuar
como sus reguladores endógenos además de serlo también para los
oligodendrocitos (Nearty y col., 1996).
Además, la microglia activada libera factores de crecimiento polipeptídicos y
citoquinas que estimulan la proliferación de células endoteliales capilares del
cerebro y cambios reactivos en los astrocitos. Por un lado, la glia juega un

32
papel positivo en los eventos regenerativos post-lesión, pero por el otro, la
reacción glial tiene un efecto negativo que se opone a la recuperación de
funciones porque desafortunadamente las reacciones gliales a la lesión
constituyen también un impedimento a la recuperación posterior, ya que la
cicatriz glial hecha por los astrocitos suele considerarse como un obstáculo
físico que limita la regeneración axonal (Streit, 2002).
Ahora bien, existe una gran cantidad de reportes que implican la activación
y la proliferación excesiva de la microglia en el desarrollo de la muerte neuronal
en varios estados patológicos de ciertas enfermedades del SNC. Los ejemplos
incluyen: el síndrome de Wernicke-Korsakoff, Enfermedad de Parkinson,
Enfermedad de Alzheimer, isquemia, y varios HIV-1 (Todd y Butterworth, 1999;
McGeer y McGeer, 1998; Walton y col., 1999; Akiyama y col., 2000; Xiong y
col., 2000). Una concordancia en los mecanismos observados en estas
enfermedades es la producción de una variedad de neurotoxinas en exceso por
la microglia, incluyendo el óxido nítrico (ON), factor de la necrosis tumoral alpha
(TNFα) y especies de oxigeno reactivo como el peróxido. La producción
excesiva de estos factores conduce a una cascada de efectos incluyendo la
activación de astrocitos, activación adicional de la microglia, y finalmente la
muerte neuronal (Lee y col., 2001).

33
Serotonina
Por otra parte, el oxido nítrico (ON) y la serotonina (5-hidroxitriptamina; 5-
HT) son importantes neuromoduladores que están involucrados en
innumerables reacciones bioquímicas. Linden y col. (2002) describieron un
nuevo sistema de co-cultivo para estudiar las interacciones entre el ON y la 5-
HT. El óxido nítrico derivado de las células microgliales Bv2 (una línea celular
de microglia de murido modificada con el oncogen v-raf/v-myc transportado por
el retrovirus J2, estas células expresan una actividad no específica de la
esterasa, tienen habilidad fagocítica y la peroxidasa carece de actividad)
estimuladas por citoquinas depletan la 5-HT en ratas con células de leucemia
basofílica (RBL-2H3) (Linden y col., 2002).
Se conoce que el sistema serotoninérgico modula el humor, la emoción, el
sueño y el apetito, así como también está implicado en el control de numerosas
funciones fisiológicas y del comportamiento. Se ha propuesto que la
neurotransmisión serotoninérgica disminuida pudiera desempeñar un papel
clave en la etiología de la depresión. La concentración sináptica de serotonina
es controlada directamente por su recaptación en la terminal pre-sináptica, así,
las drogas que bloquean la recaptación de serotonina se han utilizado con éxito
para el tratamiento de la depresión. Por ello, se han desarrollado los
antidepresivos tricíclicos tales como clomipramina, un inhibidor no selectivo de

34
la recaptación de serotonina, fluoxetina y paroxetine inhibidores selectivos de
serotonina, los cuales son usualmente prescritos en pacientes con depresión
(Basbaum y col., 1978; Ardid y col., 1995).
Estrés
El estrés ha estado implicado en muchos desórdenes médicos y
psiquiátricos (Elenkov y Chrousos, 1999; Chrousos, 2000; Habib y col., 2001).
Un modelo de estrés experimental que resulta en cambios de comportamiento
a largo plazo es el estrés por nado, y este fenómeno es el elemento clave de la
prueba de nado forzado, un protocolo válido para la actividad de antidepresivos
(Porsolt y col., 1977). En la exposición inicial al estrés del nado forzado, las
ratas exhiben ambos comportamientos activo (nadar o luchar) y pasivo (el
flotar). Durante una segunda exposición (24 horas a 2 semanas más tarde), los
patrones cambian tal que el comportamiento activo disminuye y predomina el
comportamiento pasivo. Como otras estrategias experimentales que modelan
aspectos de depresión, este modelo inductor de estrés se inclina hacia el
comportamiento pasivo que es sensible a los tratamientos del antidepresivo
(Maier y Jackson, 1979; Weiss y col., 1981, Roche y col., 2003).
Estudios efectuados por Kirby y col. (1995), compararon el efecto de
diferentes estresores (nado, inmovilización, locomoción forzada y frío) sobre los

35
niveles extracelulares de 5-HT en estriado y hipocampo ventral usando
microdiálisis y midiendo corticosterona en plasma como indicador de estrés
(Kirby y col., 1994). De los cuatro estresantes estudiados: nado, inmovilización,
locomoción forzada y frío, el nado forzado produjo un gran efecto neuroquímico
y neuroendocrino (Kirby y col., 1995). Cambios bidireccionales en la actividad
serotoninérgica de núcleos cerebrales conocidos, que se relacionan en
respuesta al estrés han sido observados, por ejemplo, después de la
exposición aguda a diferentes tipos de estímulos adversos tanto físicos como
psicológicos aparece un incremento en la concentración extracelular de
serotonina en varias regiones del cerebro, especialmente en el rafe magnus.
Controversialmente, el estrés prolongado disminuye la concentración de
serotonina en algunas estructuras cerebrales conocidas que son activadas por
el estrés como son la amígdala y el septum lateral. La magnitud de la inhibición
tónica de la transmisión del dolor en las astas dorsales de la médula espinal
depende del estado conductual del organismo (deprimido, ansiedad, miedo).
El concepto de que el estrés puede contribuir a la generación y
mantenimiento del dolor no esta aún sistemáticamente explorado. Quizás, esto
se deba al hecho de que el interés científico durante varias décadas se ha
dirigido, a estudiar la disminución en la percepción del dolor (analgesia)
transitoria que se observa cuando la nocicepción es medida inmediatamente al
final de una exposición aguda o crónica de agentes estresores (Lewis y col.,

36
1980; Amit y Galina, 1986; Pignatiello y col., 1989). Varios estudios han
reportado que bajo algunas condiciones experimentales como exposiciones
simples o repetidas a situaciones de estrés incontrolable e inescapable pueden
provocar una hiperalgésia transitoria o persistente, en lugar de analgesia.
Usualmente, el aumento de la percepción del dolor que sigue a la exposición
de estresores ha sido interpretado erróneamente como un indicador del
desarrollo de tolerancia al efecto de estresores repetido o ha sido pasado por
alto a pesar de su significado clínico (Madden y col., 1977).
Se sabe que las neuronas responden al estímulo que les llega a la
membrana con cambios a corto plazo, que vienen mediados por canales
iónicos, o por segundos mensajeros, y con cambios a largo plazo, involucrando
nuevos patrones de expresión genética como son los proto-oncogenes, los
cuales no necesitan síntesis proteica para su inducción. La mayoría de estos
proto-oncogenes nucleares o genes tempranos o inmediatos codifican
proteínas reguladoras que controlan la expresión de otros genes.
El c-fos esta implicado en la codificación de una proteína nuclear que
interviene en la regulación de los genes de transcripción (Draisci y Ladarola,
1989). Las exposiciones a estímulos nocivos externos provocan la expresión
del proto-oncogen c-fos. En la médula espinal se ha demostrado que la
estimulación dolorosa de la pata trasera de la rata produce un aumento de la

37
expresión de c-fos, observándose un intenso marcaje en los segmentos
medulares L3 y L4 (ensanchamiento lumbar de la médula espinal) a nivel de las
láminas I, II, V y VI del asta posterior, siendo en estas láminas donde llegan las
ramas centrales de los nervios periféricos (Abbadie y Benson., 1993). En forma
similar, la inflamación de los tejidos puede inducir una rápida expresión de c-fos
y otros genes inmediatos tempranos, paralelamente con el desarrollo de una
conducta hiperalgésica. La estimulación nociceptiva periférica por inflamación
puede provocar la aparición de la inmunoreactividad de c-fos en las láminas I y
II así como en las láminas V y VI de la médula espinal. Muchos estudios han
demostrado que la síntesis de la proteína c-Fos inducida por inflamación tisular
muestra un curso variable, dependiendo del origen del estímulo inflamatorio
(Noguchi y col., 1991).
Nocicepción
El dolor es una sensación heterogénea que puede subdividirse en tres
categorías desde el punto de vista fisiopatológico: el dolor fisiológico,
inflamatorio y neuropático. El dolor normal o fisiológico es una sensación
protectora que nos alerta acerca de la presencia en el ambiente de estímulos
lasivos. A su vez, después de un proceso inflamatorio o injuria se suceden una
serie de alteraciones en el sistema somato sensorial, que amplifica las
respuestas e incrementa la sensibilidad a estímulos periféricos, de tal manera

38
que el dolor puede ser activado por estímulos normalmente inocuos o de baja
intensidad. Este tipo de dolor denominado clínico o patológico, es una
expresión de la plasticidad del sistema somatosensorial, definida como la
capacidad de estas neuronas de cambiar sus funciones, perfil bioquímico y su
estructura. Estos cambios operan en múltiples sitios y se producen por diversos
mecanismos. La hipersensibilidad que acompaña al dolor inflamatorio
usualmente retorna a lo normal si el proceso o enfermedad causante es
controlado, mientras que el dolor neuropático persiste por tiempo prolongado,
aún cuando haya ocurrido un proceso de cicatrización (Woolf y Costean, 1999;
Woolf y Salter, 2000).
Desde la sensación hasta la percepción del dolor y su modulación son
vistas clásicamente mediados solamente por neuronas. Recientes hallazgos
implican a la glia del cordón espinal con un papel importante en la creación y
mantenimiento del dolor patológico. Existen evidencias cada vez mayores de
que la glia puede ser la clave para crear estados de dolor exagerado en
diversas patologías, esto sugiere que pueden estar ampliamente involucrados
en la creación del dolor patológico y tal vez en otros fenómenos sensoriales
(Watkins y col., 2001). Subcutánea inyección de formalina en la superficie
plantar (prueba de formalina) nos provee de un valioso modelo de dolor
inflamatorio (Cho y col., 2006), la hiperalgesia inducida por formalina se ha
sugerido que involucra la glia espinal (Teng y col., 1998 y Cho y col., 2006).

39
Desde el punto de vista psicofisiológico, el término hiperalgesia se refiere a
un desplazamiento hacia la izquierda en la curva que relaciona la intensidad del
estímulo con la sensación dolorosa percibida e incluye la adolinia cuando el
estímulo es de baja intensidad (Garcia y col, 2001) Figura 1.
Figura 1. Curva de intensidad del estímulo vs. sensación dolorosa percibida.
La nocicepción es la actividad neurológica desencadenada por estímulos
potencialmente dañinos para los tejidos. Está modulada por factores
psicobiológicos y puede percibirse o no como dolor. Sin embargo, dolor y
nocicepción no son conceptos sinónimos. Nocicepción implica la excitación de

40
los nociceptores (receptores periféricos del dolor), y, si bien esta excitación
puede conducir a la percepción del dolor, éste también se puede originar en
ausencia de nocicepción. Actualmente, gracias a las técnicas de biología
molecular y a las técnicas de imagen se ha comprobado que son muchas las
estructuras nerviosas a nivel periférico, medular, subcortical y cortical que
intervienen en la percepción del dolor, con lo que esta percepción afecta a
nuestros pensamientos, memoria, actitudes, emociones, movimientos y
conducta, y a la vez se ve afectada por cada uno de estos procesos (Ribera y
col., 2003).
Un receptor sensitivo es una estructura anatómica capaz de responder ante
determinados estímulos convirtiendo la energía de estos estímulos en un
potencial eléctrico que se transmite por una vía periférica hasta los niveles
centrales del sistema nervioso. Habitualmente se trata de estructuras
especializadas de las propias neuronas o de terminaciones libres. Aunque la
mayor parte de receptores responden preferentemente a un estímulo
determinado (mecánico, químico, térmico), también existen receptores
polimodales (Ribera y col., 2003).
Una característica diferencial de los nociceptores en comparación con
los demás receptores radica en la posibilidad de que otras estructuras puedan
modular sus propiedades receptoras. Según el tipo de estímulo los

41
nociceptores se pueden dividir en tres grupos mecánicos, térmicos y
polimodales (activados por estímulos tanto mecánicos como térmicos)
(Basbaum y Jessell, 2000).
Uno de los avances más importantes en la investigación sobre el dolor
ha sido el descubrimiento de la existencia de varios circuitos neuronales de tipo
modulador cuya función consiste en regular la percepción del dolor. Estos
circuitos se encuentran a varios niveles del sistema nervioso, de tal forma que
las vías nociceptivas aferentes son permanentemente moduladas por sistemas
reguladores situados a distintos niveles del SNC (Ribera y col., 2003).
El primer nivel de modulación se encuentra en la médula espinal, donde las
conexiones que se establecen entre las fibras aferentes nociceptivas (de
pequeño diámetro) y las fibras aferentes no nociceptivas (de gran diámetro)
controlan la información nociceptiva que se transmite hacia los centros
nerviosos superiores, de tal forma que ésta es regulada por la actividad de las
fibras aferentes mielinizadas (Aβ) no relacionadas directamente con la
transmisión del dolor. Este primer circuito modulador se conoce como la Teoría
del Umbral del Dolor o Teoría de la Puerta de Control del Dolor (Wall y
Meltzack, 1994), según la cual el dolor es la resultante del equilibrio de la
actividad de las fibras aferentes nociceptivas y no nociceptivas.

42
La teoría de la puerta de control del dolor ha sufrido varias modificaciones
(Melzack y Loeser, 1978; Loeser y Melzack, 1999), y actualmente se considera
que la inhibición de la transmisión del dolor se produce tanto por fibras
aferentes presinápticas como postsinápticas periféricas, y también por fibras
descendentes que se originan en los niveles centrales del sistema nervioso. La
teoría de la puerta de control del dolor ha constituido la base para la utilización
de la Estimulación Nerviosa Eléctrica Transcutánea (ENET) y de la
estimulación la columna dorsal medular, para el alivio del dolor (Melzack y
Loeser, 1978; Loeser y Melzack, 1999; Ribera y col., 2003).
Dolor, inflamación, estrés y microglia.
Varios estudios recientes han demostrado el efecto de la estimulación
nociceptiva periférica en la respuesta de las neuronas de las astas dorsales.
Diversos estudios han evaluado los efectos de la lesión tisular en la respuesta
neuronal. Pocos estudios han evaluado los efectos de la estimulación
nociceptiva en la respuesta glial. Sin embargo parece que la glia juega un papel
muy significativo en la función del sistema nervioso central de lo que se
pensaba anteriormente, por ejemplo la microglia y los astrocitos pueden
sintetizar y secretar citoquinas, como la IL-1β (Kronfol y Remick, 2000), TNF-α
(Bethea y col., 1992), IL-6 (Fu y col 1998, Le y col 1989) y factor de crecimiento
neuronal (NGF) (Gilad y Gilad , 1995) las cuales juegan un papel crítico en el

43
mantenimiento de la homeostasis del SNC (Pousset 1994; Nakajima y
Kohsaka, 2004).
Además de la liberación de citoquinas la glia puede liberar otras sustancias
neuromoduladoras como los productos de la ciclooxigenasa y el óxido nítrico.
Los productos de la ciclooxigenasa y el óxido nítrico se cree que son
mediadores de la hiperalgesia. El comportamiento de dolor puede bloquearse
1) por antagonista de un receptor recombinante de la IL-1 humana, 2) un
anticuerpo dirigido contra el factor de crecimiento neural ó 3) inhibidores
metabólicos de la función de la glia. Dadas estas observaciones es razonable
postular que las células gliales (astrocitos y microglias) son fuentes importantes
de citoquinas, productos de ciclooxigenasa y óxido nítrico que contribuyen al
desarrollo del dolor persistente luego de un estimulo nociceptivo periférico (Fu y
col 1999).
Fu y col 1999 efectuaron el primer estudio que reportaba que la inyección
subcutánea de formalina en la superficie plantar de la pata trasera de rata
induce una activación de microglia en el cordón espinal lumbar. Aunque no
pudieron determinar si esta respuesta era resultado de la actividad nociceptiva,
de la inflamación periférica ó de la degeneración de las neuronas aferentes
primarias o centrales. Sin importar, que mecanismo determinen como el estrés
conlleva a la activación microglial existen diversas evidencias de que la

44
microglia puede prepararse por un número de estímulos diferentes incluyendo
neurodegeneración, envejecimiento y estrés para que un segundo estímulo
inflamatorio sistémico que pueda conllevar a un cambio en el fenotipo o un
fenotipo pro-inflamatorio exagerado. Esta respuesta exagerada a las citoquinas
puede llevar a cambios agudos en el comportamiento o exacerbar patologías.
Por lo tanto las microglias son tanto sensores de patología como también
juegan un papel crítico en la comunicación entre estresores de medio
ambiente, patógenos sistémicos, lesión, injuria ó toxinas en el cerebro. No es
difícil ver que esto tendrá consecuencias importantes para nuestro
entendimiento de las influencias ambientales en la salud mental y
enfermedades del cerebro (Perry 2007).
Para finalizar, por otra parte, la función inmunitaria intracerebral está
sometida a un estricto control y el privilegio inmunitario del cerebro viene
determinado no sólo por la barrera hematoencefálica sino también por la
existencia de señales de freno que hacen que la microglia, el macrófago del
SNC, se mantenga en un estado quiescente cuando el tejido nervioso no se
encuentra en una situación de estrés, a través de la interacción local del
receptor CD200 (CD200R) presente en la microglia y de su ligando CD200
(OX2) una glicoproteína de membrana que se expresa en neuronas, endotelio,
células del estroma y linfocitos; y proporciona una señal inhibitoria a células de
la línea mieloide como la microglia. Cuando se produce una lesión inflamatoria

45
en el cerebro, la microglia se activa y se produce un descenso en la expresión
del CD200R, para hacer frente a la situación de daño, sin embargo esta señal
debe ser controlada para que no sobrepase un umbral de tolerabilidad y
contribuya al daño (Wright y col, 2003; M. Hernangómez y col 2006).

46
3.- OBJETIVOS
3.1.- Objetivo General:
Determinar si la serotonina y la microglia están involucradas en la
hiperalgesia inducida por estrés repetido en ratas.
3.2.- Objetivos Específicos:
3.2.1.- Determinar la activación microglial midiendo el incremento de la
expresión del receptor CR3 del complemento en ratas estresadas con dolor
inflamatorio inducido por la inyección subcutánea de formalina.
3.2.2.- Determinar el efecto de la minociclina, inhibidor de la activación
microglial, sobre el incremento de la percepción del dolor inflamatorio producido
por estrés repetido, evaluando el efecto de este agente sobre la prueba de
formalina en ratas.
3.2.3.- Determinar el efecto del inhibidor de la recaptación de serotonina
clomipramina sobre el incremento en la percepción del dolor inflamatorio
provocado por estrés de nado repetido.

47
3.2.4.- Determinar el efecto del inhibidor de la recaptación de serotonina
clomipramina sobre la activación de las neuronas espinales sensoriales en
ratas estresadas con dolor inflamatorio, evaluando la expresión del factor de
trascripción temprana c-Fos.

48
4.- METODOLOGÍA:
4.1.- Animales de experimentación:
4.1.1- Modelo Experimental 1
Ratas machos Sprague-Dawley (250 – 300 gramos de peso) fueron
usadas para este estudio. En un experimento típico 12 ratas fueron enjauladas
individualmente y llevadas al lugar de las pruebas 3 días antes del
procedimiento conductual con agua y comida disponibles ad libitum. Cada
animal fue usado una sola vez. Los observadores desconocieron a qué
procedimiento conductual condicionante fueron sometidos los grupos
experimentales.
4.1.1.1.- Estrés de Nado
Un grupo de ratas (estrés de nado) fueron sometidas a
nado forzado en un cilindro con agua a 25 ºC y una profundidad de 30
centímetros por 10 minutos. Durante la sección de nado fueron medidos los
tiempos de inmovilidad y lucha siguiendo el método descritos por Porsolt y col.,
1978. La inmovilidad ocurre cuando la rata se mantiene sobre la superficie del

49
agua con movimientos mínimos para mantener la cabeza sobresaliendo de
esta. La lucha ocurre cuando la rata nada activamente, brinca, se mueve
vigorosamente con sus cuatro patas rompiendo la superficie del agua o cuando
intenta escalar la pared del cilindro buscando una escapatoria. Otro grupo de
ratas fueron sometidas a un simulacro de nado, colocándolas en un cilindro con
agua (25 ºC) de solo una profundidad de 2-3 centímetros, solo lo suficiente
para mojar parcialmente el animal, pero no para obligaralo a nadar. Finalmente,
un grupo de ratas no fueron manipuladas y permanecieron en sus jaulas. En
los días 2 y 3 las ratas fueron sometidas nuevamente a 20 minutos de nado
forzado, a simulacro de nado o a permanencia en sus jaulas (controles).
4.1.1.2.- Nocicepción Química
Nocicepción química se evaluó mediante la prueba de
formalina, la cual se realizó 48 horas después de la última sesión de nado
forzado o simulacro de nado y consistió en inyectar 0,1 cc de formalina al 1%
(37,5% de formaldehído en 0,9% de solución salina) en la superficie plantar de
la pata trasera derecha. La inyección de formalina indujo un comportamiento
característico de la rata ante un dolor inflamatorio agudo, el cual fue observado
por 2 horas a intervalos de 180 segundos. La primera hora de observación se
dividió en dos períodos: un primer período de 30 minutos durante el cual se
registró la conducta nociceptiva y un segundo período de 30 minutos durante el

50
cual no se registró la conducta. La segunda hora se dividió en cuatro períodos
de 15 minutos cada uno, alternando con 15 minutos de registro de la
nocicepción y 15 minutos donde no se registró. En total, fueron medidos 20
intervalos de 3 min. de duración por 2 horas. La intensidad del dolor fue
registrada de acuerdo a la siguiente escala basada en 4 categorías de
comportamientos básicos de la rata: 0 = el peso del cuerpo del animal estaba
igualmente distribuido en todas las patas, ya sea que esta estuviera quieta o en
movimiento; T1 = poco peso corporal apoyado sobre la planta de la pata
inyectada; T2 = pata inyectada elevada; T3 = la pata inyectada es lamida,
mordida o sacudida. El valor numérico fue calculado mediante la siguiente
formula (T1 + 2T2 + 3T3) /180, donde T1, T2 y T3 fueron el tiempo (en
segundos) gastado por categoría 1, 2 ó 3 durante cada intervalo de
observación de 180 segundos. Estas observaciones se realizaron con la
asistencia de un programa de computadora diseñado para esta prueba.
Un día después del estimulo, las ratas fueron sacrificadas, induciéndoles un
coma profundo al introducirlas por varios minutos en una cámara sellada
conteniendo CO2 a una presión controlada entre 4 y 6 libras. Los animales
fueron perfundidos vía intracardíaca, con una solución de Buffer Fosfato Salino
(PBS) 0,1 M pH 7,4 para limpiar los tejidos. Después se perfundió la misma
cantidad de paraformaldehido al 4% en PBS para fijar los tejidos. Una vez que
se aplicaron ambas soluciones, se procedió a la extracción de la médula

51
espinal del animal mediante disección quirúrgica. El segmento lumbar de la
médula espinal extraída se sumergió en 30 ml de formaldehído al 4 % en PBS
por 2 horas, para una post-fijación del tejido, posteriormente fue sumergido en
una solución crioprotectora de sacarosa al 30% por toda la noche. Los tejidos
fueron guardados a 4°C para posteriores estudios por inmunohistoquímica con
marcadores selectivos para la identificación de la microglia.
4.1.1.3.- Protocolo de Inmunohistoquímica para microglia:
Selección del tejido y cortes:
Se realizó la localización final del ensanchamiento lumbar, a
través de la lupa estereoscópica y se uso una hojilla de bisturí para seccionar la
región de interés. El trozo de tejido se puso sobre una placa metálica, con
previa adición de Histo-PrepTM, cuidando de que no quedaran burbujas, y se
colocó en la zona de congelamiento del micrótomo (Modelo HM505N)
temperatura de trabajo (-25°C). Una vez congelados los bloques, se procedió
a realizar cortes o rebanadas de 40 μm de grosor, cuidando la integridad del
tejido medular. Luego fueron tomadas las rebanadas con un pincel fino y
colectadas en PBS 0,1 M. Se seleccionaron morfológicamente los cortes
correspondientes a los segmentos lumbares L4 y L5.

52
Inmunotinción de microglias: El procedimiento duró tres días y
consistió en:
Día # 1: Los cortes fueron transferidos a un recipiente con malla que
contiene PBS. Se realizaron dos lavados de 10 minutos y dos lavados de 5
minutos. Se incubaron los tejidos en una solución bloqueadora (suero normal
de caballo al 4 % en PBS) por 1 hora. Luego, se incubaron los cortes en placas
serológica con anticuerpo primario de ratón, monoclonal anti-CR3 de rata (OX-
42; Serotec, USA) en una dilución de (1:1200 en 4% de suero normal de
caballo), por 36 horas a 4°C. Un grupo de cortes no se incubaron en anticuerpo
primario con el fin de determinar la especificidad de reacción cromática.
Día # 2: Se lavaron los cortes con PBS dos veces por 10 minutos y dos
veces por 5 minutos, seguidamente se realizo un incubación de bloqueo con
suero normal de caballo al 4% en PBS por 30 minutos, y se incubaron con
anticuerpo secundario biotinilado de caballo anti-IgG de ratón (absorbido en
ratas) en una dilución de 1:200 en NHS al 4% por 90 min. Se procedió a lavar
nuevamente con PBS 2 lavados por 5 min y 2 lavados por 10 min.
Aproximadamente 1 hora antes de comenzar el lavado se procedió a la
preparación del complejo reactivo ABC (Estreptavidina – Biotina – HRP) según
instrucciones del fabricante (Vector Laboratories, Burlingame, C.A, U.S.A).

53
Terminados los lavados se incubaron los cortes con ABC por 90 minutos, para
luego lavarlos con PBS, por el mismo tiempo anterior. Finalmente se incubaron
con diaminobencidina (DAB) [20 mg en una mezcla de 33 μl H2O2 al 30% y 100
ml de Buffer Tris (0,05 M pH 7,6)] para revelar las proteínas marcadas, con una
reacción que produce un color negro-marrón. Los cortes se lavaron con buffer
Tris por 5 min y luego con PBS por 30 min. y se transfirieron a portaobjetos
precubiertos con gelatina. Los cortes se dejaron secar toda la noche.
Día # 3: Los cortes se lavaron sumergiéndolos en agua de grifo por 20
segundos y se procedió a la deshidratación de los mismos sumergiéndolos en
forma progresiva en etanol al 50 %, 70%, 95 %, 100 % por 1 minuto cada uno,
y en xileno por 4 minutos. Los cortes se montaron de forma permanente
cubiertos con Permount® y cubreobjetos, para la observación bajo microscopio
de luz.
Figura 2. Diseño Experimental 1.
• Procedimiento condicionante: Nado, simulacro y sin manipulación • • •
∗ Prueba de evaluación nociceptiva: Prueba de Formalina ∗
° Cirugía y extracción de la medula espinal °
Días 1 2 3 4 5 6

54
4.1.2- Modelo Experimental 2
Ratas machos Sprague-Dawley (250 – 300 gramos de peso) fueron
usadas para este estudio. En un experimento típico 12 ratas fueron enjauladas
individualmente y llevadas al lugar de las pruebas 3 días antes del
procedimiento conductual con agua y comida disponibles ad libitum. Cada
animal fue usado una sola vez. Las ratas fueron sometidas a nado forzado
(n=4) o simulacro de nado (n=4) 10 min el primer día, el segundo y el tercer día
se sometieron a 20 min. Un grupo de ratas permaneció en sus jaulas no
manipuladas (n=4). La mitad de las ratas de cada grupo fueron tratadas: con 40
mg/Kg de minociclina intraperitonealmente (i.p.) o con vehículo (NaCl 0.9%,
i.p.), 1 hora antes de cada nado, simulacro o enjaulamiento.
La evaluación de la nocicepción química mediante la prueba de
formalina), realizada 48 horas después de la última sesión de nado forzado
como se describe en el Modelo Experimental 1. Un día después del estimulo,
las ratas fueron sacrificadas y prefundidas para luego removerle
quirúrgicamente el segmento lumbar de la médula espinal como se describe
en el Modelo Experimental 1.

55
♦ Tratamiento farmacológico: Minociclina ó vehículo ♦ ♦ ♦
• Procedimiento condicionante: Nado, Simulacro y no manipuladas • • •
∗ Prueba de evaluación nociceptiva: Prueba de Formalina ∗
° Cirugía y Extracción medular °
Figura 3. Diseño experimental 2
4.1.3.- Modelo experimental 3:
Ratas machos Sprague-Dawley (250 – 300 gramos de peso) fueron
usadas para este estudio. En un experimento típico 12 ratas fueron enjauladas
individualmente y llevadas al lugar de las pruebas 3 días antes del
procedimiento conductual con agua y comida disponibles ad libitum. Cada
animal fue usado una sola vez. Los observadores desconocían a qué
tratamiento farmacológico y a cuál procedimiento conductual serán sometidos
los grupos experimentales
Las ratas fueron sometidas a nado forzado (n=4) o simulacro de nado
(n=4) 10 min el primer día, y 20 min el segundo y el tercer día. Un grupo de
ratas permaneció en sus jaulas no manipuladas (n=4). La mitad de las ratas de
Días 1 2 3 4 5 6

56
cada grupo experimental fueron tratadas: 2.5 mg/Kg i.p. de clomipramina o con
vehículo (NaCl 0.9%, i.p.), desde el día 1 hasta el final del protocolo conductual
el día 10.
La evaluación de la nocicepción química mediante la prueba de
formalina), realizada 48 horas después de la última sesión de nado forzado
como se describe en el Modelo Experimental 1. Un día después del estimulo,
las ratas fueron sacrificadas y perfundidas para luego removerle
quirúrgicamente el segmento lumbar de la médula espinal como se describe
en el Modelo Experimental 1.
4.1.3.1. Protocolo de Inmunohistoquímica para c-Fos:
Selección del tejido y cortes:
Se localizo el ensanchamiento lumbar de la medula
espinal a través de la lupa estereoscópica y se uso una hojilla de bisturí para
seccionar dicha región de interés. El trozo de tejido se puso sobre una placa
metálica, con previa adición de Histo-prepTM, cuidando de que no quedaran
burbujas, y colocándose luego en la zona de congelamiento del micrótomo
(Modelo HM505N). Una vez congelados los bloques, se procedió a realizar
cortes o rebanadas de 40 μm de grosor, cuidando la integridad del tejido

57
medular. Luego fueron tomadas las rebanadas con un pincel fino y colectadas
en buffer PBS/TX; (buffer fosfato alino 0,1 M con 4% Triton X-100). Se
seleccionaron morfológicamente los cortes correspondientes a los segmentos
lumbares L4 y L5.
Inmunotinción para c-Fos:
Se realizo en tres días y consistió en:
Día # 1: Los cortes fueron transferidos a un recipiente con malla que
contenían PBS/TX. Se realizaron dos lavados de 10 minutos y dos lavados de
5 minutos. Una vez culminado este lapso, se procedió a reducir la actividad de
la peroxidasa endógena exponiendo los cortes a H2O2 al 0.3 % por 30 minutos.
Luego se repitieron los lavados en PBS/TX, igual que la vez anterior, y se
procedió a bloquear con suero normal de cabra al 4 % en PBS/TX por 1 hora.
Por último se incubaron los cortes en placas serológicas con anticuerpo
primario policlonal anti-c-Fos antisuero levantado en conejo en una dilución de
1:2000 en suero normal de cabra (NGS), por 36 horas a 4°C. (Vector
Laboratories, Burlingame, C.A, U.S.A). Un grupo de cortes no se incubaron en
anticuerpo primario con el fin de determinar la especificidad de reacción
cromática.

58
Día # 2: Se lavaron los cortes con PBS/TX dos veces por 10 minutos y dos
veces por 5 minutos, seguidamente se bloquearon con NGS al 4% en PBS/TX
por 30 minutos, y se incubaron con biotinilado de cabra anti-IgG de conejo
(absorbido en ratas) a una dilución de 1:2000 en NGS. Se procedió a lavar
nuevamente con PBS/TX con los mismos tiempos estipulados.
Aproximadamente 1 hora antes de comenzar el lavado se preparo el complejo
reactivo ABC (Estreptavidina - Biotina- HRP) según instrucciones del fabricante
(Vector Laboratories, Burlingame, C.A, U.S.A). Terminados los lavados se
incubaron los cortes con ABC por 90 minutos, luego fueron lavados con
PBS/TX, por el mismo tiempo anterior. Finalmente se incubaron con
diaminobencidina (DAB) para revelar las proteínas marcadas, con una reacción
que produce un color negro-marrón. Los cortes se lavaron con TBS/TX, por el
mismo período de tiempo y se transfirieron a portaobjetos precubiertos con
gelatina. Los cortes se dejaron secar toda la noche.
Día # 3: Los cortes se lavaron sumergiéndolos en agua de grifo por 20
segundos y se procedió a la deshidratación de los mismos sumergiéndolos en
forma progresiva en etanol al 50 %, 70%, 95 %, 100 % por 1 minuto cada uno,
y en xileno por 4 minutos. Los cortes se montaron de forma permanente
cubiertos con Permount® y cubreobjetos, para luego observarlos bajo
microscopio de luz.

59
Figura 4. Diseño experimental 3
4.2. Análisis estadístico:
Para procesar los datos obtenidos, se utilizó un análisis de varianza
(ANOVA), para determinar si hubo diferencias globales entre los promedios de
más de dos grupos. ANOVA de una vía fue aplicada para una variable
independiente, y ANOVA de dos vías, para dos variables independientes. Para
las diferencias globales significativas, se aplicó la prueba de rangos múltiples
de Duncan´s para determinar las diferencias de los promedios entre sí. La
significancia estadística fue asumida a una p< 0,05.
♦ Tratamiento farmacológico: Clomipramina ó vehículo ♦ ♦ ♦ ♦ ♦ ♦ ♦ ♦ ♦ ♦ ♦
• Procedimiento condicionante: Nado, Simulacro y no manipuladas • • •
∗ Prueba de evaluación nociceptiva: Prueba de Formalina ∗
° Cirugía y Extracción de la médula °
Días 1 2 3 4 5 6 7 8 9 10 11 12 13

60
4.2.1 Análisis de las Imágenes
El análisis cuantitativo de la inmunoreactividad al anticuerpo OX-
42 fue realizado en la lámina I – II de las astas dorsales de la médula espinal.
Fueron seleccionados al azar 4 cortes por rata. Las imágenes fueron
capturadas por un microscopio Axioskop 2 plus Zeiss® acoplado a una cámara
de video cuyas señales fueron enviadas a un computador. Estas fueron
adquiridas primero a una amplificación de 100 X, luego fueron ubicadas las
láminas I - II para tomar una amplificación de 400 X, con la finalidad de hacer
conteo de las microglias, las láminas I - II del asta dorsal recibe inervaciones de
la superficie plantar de la pata trasera que es estimulada durante la prueba
nociceptiva de formalina. La imagen fue capturada mediante el Imagen-pro®
Plus versión 4.5.1 de Media Cybernetics®. Posteriormente la imagen original
fue transferida a una plataforma Mac OS 10.4 y para el conteo individual de las
microglias se utilizó el programa Photo Shop CS3 extended.

61
5.- RESULTADOS El estrés incrementó la activación microglial inducida por la inyección
subcutánea de formalina. Las micrografías observadas en la Figura 6 muestran
que la inyección subcutánea de formalina (prueba de dolor inflamatorio) causa
activación microglial en todos los grupos estudiados aunque pudo observarse
que la activación de la microglia fue substancialmente mayor en las ratas que
fueron sometidas a estrés de nado repetido en comparación con aquellas
sometidas a simulacro de nado o no manipuladas.
Todos los grupos sometidos a la prueba de formalina, mostraron microglias
en reposo y activadas OX-42 positivas, tanto en sustancia gris como en blanca.
En el simulacro de nado y no manipuladas (microfotografía Figura 6B y 6C) se
pueden observar microglias OX-42 positivas con ramificaciones extendidas en
todas las direcciones y con un citoplasma perinuclear (microglias en reposo ver
Figura 5A), además de observarse escasas microglia OX-42 positivas
mostrando acortamiento de las ramificaciones y engrosamiento en su centro
(Figura 5B). La distribución se concentra en mayor número hacia la parte
posterior del asta dorsal.
En los animales que fueron sometidos a estrés de nado forzado repetido
durante 3 días consecutivos e inyectados subcutáneamente con formalina

62
mostraron abundantes microglias OX-42 positivas fuertemente teñidas con un
significativo acortamiento de las ramificaciones y engrosamiento en su centro,
aunque sin llegar al estado final de activación cuando las células toman una
morfología ameboide, estas se observan distribuidas homogéneamente en el
asta dorsal lumbar lamina I y II (Figura 6A).
Figura 5. Cambios morfológicos de la Microglia. A microglia en reposo, B microglia activada y C microglia totalmente
activa como macrófago.
La minociclina inhibió la activación microglial en ratas estresadas y
sometidas a la prueba de formalina además produjo una disminución en el
incremento de la percepción del dolor inflamatorio provocado por estrés de
nado forzado repetido. En los animales que fueron pretratados con minociclina
una hora antes de cada prueba de estrés por nado forzado, durante 3 días
consecutivos, y luego inyectados subcutáneamente con formalina en la pata
trasera derecha, mostraron patrones similares a los exhibidos por las ratas no
manipuladas o por aquellas sometidas a simulacro de nado, esto es, microglias
en reposo OX-42 positivas, tanto en sustancia gris como en blanca. De esta
A C B

63
manera, se puede observar el efecto inhibitorio de la sobre-activación microglial
provocada por exposición concomitante a estrés de nado y a la prueba de
formalina al comparar las ratas tratadas con minociclina con aquellas tratadas
con solución salina (Figura 6 D y A, respectivamente).
La cuantificación de las microglias OX-42 positivas observadas por campo
a 400X reveló que las ratas sometidas a nado forzado tenían un numero
significativamente mayor de dichas células/campo (104,3 ± 5,3) que las ratas
pretratadas con minociclina y sometidas a estrés (49 ± 2,51) (p<0,0001). Las
ratas simulacro de nado (68,3 ± 2,5) y no manipuladas (58,66 ± 3,752)
mostraron una diferencia significativa compararlas con las ratas pretratadas
con minociclina y sometidas a estrés (p<0,001).
Se pudo observar que las ratas pre-tratadas con solución salina (vehículo) y
sometidas a nado forzado exhibieron una mayor respuesta nociceptiva
(hiperalgesia) durante los intervalos de observación del 16 al 20 que los
animales no manipuladas (p<0,001) (Figura 7A). En la Figura 7B se muestra
que las ratas estresadas y pretratadas con minociclina mostraron una menor
respuesta nociceptiva que las ratas estresadas tratadas o no con solución
salina (vehículo) (p<0,001). No se observó diferencia significativa en las ratas
simulacro y no manipuladas al comparar dentro el mismo grupo las pretratadas
con minociclina o con solución salina (vehículo) (Figura 7C y D).

64
Las ratas pretratadas con clomipramina, varios días antes y durante las
exposiciones a las situaciones de estrés de nado forzado, tuvieron el típico
patrón bifásico de comportamiento nociceptivo frente a la prueba de formalina;
la magnitud de dicho comportamiento fue significativamente menor, durante la
fase II de la curva de respuesta (intervalos 18 – 19) en comparación con el
observado en las ratas pretratadas con solución salina (p<0,01) (Figura 9A). En
las Figuras 9B y 9C, se muestra que no hubo diferencias significativas en la
conducta nociceptiva de los animales sometidos a simulacro de nado o los no
manipuladas pretratados con clomipramina ó con solución salina. De esta
manera, un tratamiento crónico y a bajas dosis con un inhibidor de la
recaptación de serotonina como la clomipramina produjo un bloqueo de la
hiperalgesia inducida por exposición repetida a estrés de nado forzado.
65
Figura 6. Microfotografía (100X) mostrando los cambios en OX-42 en el asta dorsal espinal L4-5 de ratas, 1 día después de la inyección con formalina. Insertadas en cada una magnificación a 400X. Protocolo conductual (A) nado forzado (B) simulacro de nado (C) no manipulada y (D) Nado forzado + minociclina.
A B
C D

66
0 5 10 15 200
1
2
3SalinaMinociclina
** ****
**** *
Periodo de 15 min. sin observación
Nado Forzado Repetido
Intervalos min.
Punt
aje
de D
olor
Simulacro de Nado Forzado
0 5 10 15 200
1
2
3SalinaMinociclina
Periodo de 15 min. sin observación
Intervalos min.
Punt
aje
de D
olor
No Manipuladas
0 5 10 15 200
1
2
3SalinaMinociclina
Periodo de 15 min. sin observación
Intervalos min.
Punt
aje
de D
olor
A B
C D
Periodo de 15 min.sin observación
*
**
*** *
0 5 10 15 200
1
2
3 Salina nadoSalina simulacroSalina no manipulada
Intervalos min
Punt
aje
de d
olor *
*
*
Figura 7. Curvas de respuesta nociceptiva. Todas las ratas fueron sometidas a la prueba de formalina. A) Ratas salinas sometidas a los 3 procedimientos condicionantes, mostrando respuesta hiperalgésica en ratas estresadas. A) Ratas salinas sometidas a los 3 procedimientos condicionantes, mostrando respuesta hiperalgésica en ratas estresadas, * representa una diferencia significativa con p<0,001 de las ratas salinas estresadas al compararla con ratas no manipuladas, B) ratas sometidas a nado forzado mostraron una respuesta hiperalgésica frente a la prueba de formalina, inhibida al pretratarlas con minociclina, * representa una diferencia significativa con p<0,001 de las ratas salinas estresadas al compararla con ratas estresadas y pretratadas con minociclina, C) simulacro de nado, D) no manipuladas. Las barras representan 2 períodos de no observación conductual (15 min. cada unos).

67
Nado Forzado Clomipramina-Minociclina
1. 2. 3.0
50
100
150
200SalinaClomipraminaMinociclina
*
*
Sesiones de Nado Forzado
Tiem
po d
e Lu
cha
(seg
)
Nado forzado Porcentual Clomipramina-Minociclina
1. 2. 3.0
10
20
30
40SalinaClomipraminaMinociclina
**
Sesiones de Nado Forzado
Tiem
po d
e Lu
cha
(%)
Figura 8. Declive progresivo del tiempo de lucha después de la sección de nado forzado repetido una dosis baja de un inhibidor de la recaptación de serotonina clomipramina (2,5 mg/Kg, i.p. por día) y Minociclina una tetraciclina (40 mg/Kg). * representa una diferencia significativa con p<0,0001 observada en las ratas pretratadas con clomipramina al compararla con minociclina.

68
Nado Forzado Repetido
0 5 10 15 200
1
2
3SalinaClomipramina
* *
*
*
***
**
* *
Periodo de 15 min. sin observación
Intervalos min.
Punt
aje
de D
olor
Simulacro de Nado Forzado
0 5 10 15 200
1
2
3SalinaClomipramina
Periodo de 15 min. sin observación
Intervalos min.
Punt
aje
de D
olor
No Manipuladas
0 5 10 15 200
1
2
3SalinaClomipramina
Periodo de 15 min. sin observación
Intervalos min.
Punt
aje
de D
olor
A
B
C
Figura 9. Curvas de respuesta nociceptiva. Todas las ratas fueron sometidas a la prueba de formalina. A) Las ratas sometidas a nado forzado mostraron una respuesta hiperalgésica frente a la prueba de formalina, B) simulacro de nado, C) no manipuladas. Las barras negras representan 2 períodos de no observación conductual (15 min cada unos). Cada punto representa la media ± el error estándar obtenido de 6 ratas. * representa una diferencia significativa de p< 0.01 en todos los intervalos marcados, excepto el 8 y el 18 con un p<0,001 con respecto a control salino (A).

69
Nado Forzado Repetido
I-II III-IV V-VI VII VIII IX X0
10
20
30
40
50SalinaClomipramina
**
*
Laminas
Núm
ero
de c
élul
as c
-Fos
+
Simulacro de Nado Forzado
I-II III-IV V-VI VII VIII IX X0
10
20
30
40
50SalinaClomipramina
Laminas
Núm
ero
de c
élul
as c
-Fos
+
No Manipuladas
I-II III-IV V-VI VII VIII IX X0
10
20
30
40
50SalinaClomipramina
Laminas
Núm
ero
de c
élul
as c
-Fos
+
A
B
C
Figura 10. Expresión laminar de la proteína c-Fos en ratas seguida de la inyección subcutánea de formalina en la pata trasera derecha. A) Las ratas pretratadas con solución salina sometidas a nado forzado mostraron un incremento significativo en el marcaje de los núcleos inmunoreactivos en la lámina I-II y V-VI. B) simulacro de nado, C) no manipuladas. * representa una diferencia significativa con p<0,0001 de las ratas salinas estresadas al compararla con ratas estresadas y pretratadas con clomipramina.

70
En la Figura 8 A y B muestran que las ratas sometidas a nado forzado
repetido, pretratadas con solución salinas ó con minociclina, tienen una
disminución progresiva en el tiempo de lucha o intentos de escape durante las
tres secciones consecutivas de nado, sin una diferencia significativa entre ellas.
Esta falta de deseo de escape (lo que es denominado “inmovilidad” en la
prueba de nado forzado), no se observó en las ratas que fueron tratadas con
clomipramina, un inhibidor de la recaptura de serotonina. Una diferencia
significativa entre las ratas pretratadas con clomipramina y minociclina fue
observada durante el segundo y tercer día de las secciones de nado forzado
(p<0,0001).
En las ratas sometidas a estrés, pretratadas con clomipramina, tuvieron una
disminución altamente significativa en el número de núcleos inmunoreactivos al
anticuerpo contra la proteina c-Fos (un marcador de activación de neuronas
sensoriales) en comparación con aquellas pretratadas con solución salina. En
las ratas salinas sometidas a nado forzado, la prueba de formalina indujo una
significativa activación neuronal demostrada por la presencia de núcleos c-Fos
inmunoreactivos en las láminas I-VI (sensoriales) del asta dorsal de la medula
espinal lumbar (Figuras 10 y 11). Las ratas sometidas a nado forzado y
pretratadas con clomipramina tuvieron un número significativamente menor de
núcleos c-Fos inmunoreactivos en las láminas I-VI (sensoriales) del asta dorsal
que las ratas tratadas con solución salina (p<0,0001) (Figuras 10A, 11A-B). La

71
mayoría de los núcleos estuvieron localizados en las laminas superficiales
(láminas I-II) y en la coyuntura (laminas V-VI) del asta dorsal de la medula
espinal lumbar, lo cual coincide con la proyección topográfica de las aferentes
en médula espinas de las patas traseras (Molander y Grant 1985, 1986). En las
ratas sometidas a simulacro de nado, y en aquellas no manipuladas, no se
observó diferencia significativa en el número de núcleos c-Fos inmunoreactivos
entre aquellas pretratadas con salina o con clomipramina (Figura 10 B-C y
Figura 11 C-F).

72
B
C D
E F
Figura 11. Microfotografía (100X) mostrando la expresión de la proteína c-Fos inducida por formalina en el asta dorsal espinal de ratas. Protocolo conductual (A) Salina con Nado forzado (B) Clomipramina con nado forzado (C) Salina Simulacro, (D) Clomipramina Simulacro, (E) Salina No manipulada y (F) Clomipramina No manipulada.
I-II
I-II
I-II
I-II
I-II
III-IV
III-IV III-IV
III-IV III-IV
V-VI
V-VI V-VI
V-VI V-VI
V-VI
III-IV
I-II A B I-II
III-IV
V-VI

73
6.- DISCUSION
En este estudio se observo que existe una relación entre el dolor
inflamatorio inducido por formalina y la activación de microglia. Los animales
sometidos a estrés de nado, simulacro de nado y no manipulados, a los cuales
se les produjo un dolor inflamatorio agudo con la inyección subcutánea de
formalina, mostraron microglia tanto en reposo como activadas marcadas con
OX-42. En los animales que fueron sometidos a estrés de nado forzado
repetido durante 3 días consecutivos y luego a dolor cutáneo inflamatorio
agudo mostraron un aumento significativo de las células microgliales OX-42
positivas, tanto aquellas que parecían en reposo como aquellas que lucían
activas desde el punto de vista morfológico. La microglia activada estuvo
fuertemente teñida con un significativo acortamiento de las ramificaciones y
engrosamiento en su centro, aunque sin llegar al estado final de activación
cuando las células toman una morfología ameboide. La microglia activada
estaba distribuida homogéneamente en el asta dorsal lumbar, observándose un
gran número de ellas en láminas I y II, la cual coincide con la proyección
topográfica de las aferentes en médula espinal de las patas traseras (Molander
y Grant 1985, 1986).
El anticuerpo monoclonal OX-42 en roedores (en humanos clon 25f9 y en
monos clon M1/70.15.1) se une al receptor para el complemento tipo 3 CR3,

74
también llamado Integrina αM [CD11b], Mac-1, Mo-1 ó integrina αM, cuyo
ligando natural endógeno es la proteína del complemento iC3b. El CR3 es
expresado constitutivamente en la microglia y por ello la microglia tanto en
reposo como activa se tiñen con el procedimiento inmunohistoquímico que usa
OX-42. Por este motivo, el criterio de activación para la evaluación de la
microglia se basa en cambios morfológicos que pueda sufrir la célula. Se uso el
OX-42 que marca el receptor constitutivo CR3, en vez de un anticuerpo que
inmunoreaccionara con receptores microgliales inducibles, porque se ha
establecido una estrecha relación entre la activación microglial, el aumento de
la expresión de este receptor CR3 y dos represores que mantienen la microglia
en reposo: la proteína CD200 expresada en neurona y el receptor CD200R
expresado en microglia.
Existen evidencias de que las neuronas controlan el estado de activación
de la microglia, contribuyendo a su estado quiescente. Uno de los mecanismos
sugeridos de cómo las neuronas controlan la activación microglial es la
interacción con el ligando CD200, una glicoproteína de membrana presente en
las neuronas, con su receptor CD200R que esta expresado abundantemente
en las células mieloides, entre ellas la microglia (Neuman, 2001; Wright y col.,
2003). Hoek y col. (2000) crearon un ratón homocigoto (- / -) para la expresión
de la glicoproteína CD200. En ratones con una expresión normal de la
glicoproteína CD200, las microglias que estaban quiescentes, expresaban en

75
su superficie celular muy bajos niveles de MHC II, CR3 y CD45, mostrando una
morfología muy ramificada. En contraste, las microglias de los animales
homocigoto (- / -), sin expresión detectable de la glicoproteína CD200, exhibían
muchas características de activación, incluyendo pocas ramificaciones,
engrosamiento en su centro, incremento en la expresión de MHC II, CR3 y
CD45. El CR3 es el receptor del iC3b, fragmento generado por la proteolisis de
C3b; y desempeña un papel importante en la fagocitosis de partículas
recubiertas por iC3b. El CD45 en su dominio citoplasmático contiene una
región con actividad proteica tirosina fosfatasa intrínseca que cataliza la
eliminación de fosfatos de los residuos de tirosina en varios sustratos. Uno de
esos sustratos es la Lck (tirosina quinasa específica de linfocitos, no receptora,
de la familia Src), la cual está asociada de forma no covalente (aunque
firmemente) a las colas citoplasmáticas de las moléculas CD4 y CD8, y es
necesaria para la maduración y activación de las células T. Además, el CD45
desempeña un papel esencial en la señalización mediada por receptores de las
células T y B para antígenos. Frank y col., 2007 demostraron que el estrés
origina una regulación corriente abajo de la expresión hipocámpica de la
proteína CD200 24 horas después del estrés. El efecto del estrés provocado
por el skock inescapable en CD200 estuvo limitado sólo a el análisis del ARN
mensajero, por lo tanto hasta que no se establezca que el skock inescapable
modula el CD200 a nivel de proteínas, estos resultado debe tratarse con
cuidado (Frank y col., 2007). Sin embargo, el efecto del estrés en el CD200

76
sugiere que la sensibilización inducida por estrés de la microglia puede
mediarse en parte por atenuación del control neuronal de microglia, el cual
resulta en un microambiente permisivo proinflamatorio en el sistema nervioso
central. Es de hacer notar que la expresión de CD200 también está reducido en
animales ancianos sugiriendo que el control neuronal reducido de la microglia
puede ser una característica general que contribuya a la activación microglial.
La señal neuroquímica o neurohormonal que es inducida por el estrés y que
actúa sobre el CD200 todavía no es bien conocida (Hernangómez y col.,
2006).
La exposición previa a una fuente de estrés puede potenciar la respuesta
inmunológica proinflamatoria del sistema nervioso central a un estímulo
inmunológico periférico, sin embargo los sustratos neuroinmunológicos que
median este efecto no se han determinado (Frank y col., 2007). Tanto la
exposición al estrés como la activación inmunológica inducen la producción de
citoquinas en el sistema nerviosos central. Jonson y col. (2004) demostraron
que la IL1β exógena simula los efectos del estrés, estos resultados sugieren
que la IL1β central media la potenciación inducida por estrés de la respuesta
inmunológica en el SNC (O´Connor y col., 2003), el estrés pareciera
sensibilizar o preparar el sistema nervioso para una respuesta incrementada a
un estímulo inmunológico periférico subsiguiente. Debido a que las microglias
son las células efectoras inmunológicas primarias en el sistema nervioso

77
central y son una fuente principal de citoquinas proinflamatorias en este
sistema, la microglia pudiera servir como sustrato neuroinmunológico. Los
astrocitos pueden jugar un papel importante, pero las microglias son más
reactivas que los astrocitos y la inducción de IL1β por activación inmunológica
periférica es más prominente en las microglias que en los astrocitos (Aloisi,
2001).
La prueba de formalina produce inflamación, la cual se acompaña de
acidosis tisular (Sawynok y col., 2003; Mogil y col., 2005), lo que es uno de los
factores que pueden contribuir a los mecanismos periféricos centrales de la
hiperalgesia. Mamet y col. (2005) demostraron que los niveles
transcripcionales de los canales iónicos ácido sensibles (ASICs) se
incrementaron in vitro en condiciones inflamatorias, observando que este
incremento era causado por NGF, serotonina, IL-1 y bradikinina. Una mezcla
de estos mediadores incrementaría el número de neuronas que expresan
ASICs llevando a una excitabilidad mayor de las neuronas sensoriales,
pudiendo especular que esta mayor excitabilidad pudiera de alguna manera
estar asociada a los cambios transcripcionales que pudieran modificar por
mecanismos no establecidos la expresión de la glicoproteína CD200 y a su vez
influir en la regulación del estado de quiescencia de las microglias, así como
también el efecto de la clomipramina sobre los niveles de serotonina pudiera a

78
través de esta vía aunque en menor grado que el NGF disminuir la
hiperalgesia.
Por otra parte, existen antibióticos con una acción potente y preferencial
inhibitoria sobre microglia y no sobre los astrocitos o neuronas como la
minociclina, que es una tetraciclina semisintética de segunda generación que
penetra bien en SNC a través de la barrera hematoencefálica (Cho y col.,
2006). Además de su acción como antibiótico, la minociclina tiene efecto
neuroprotector y antiinflamatorio en el SNC (Tikka y col., 2001; Yrjanheikki y col
1998). En el presente estudio se pudo observar que el tratamiento con
minociclina disminuyo considerablemente el número de microglias activas que
aparecían teñidas intensamente en el segmento lumbar de la medula espinal
de animales estresados sin minociclina (ratas controles tratadas con solución
salina), observándose un mayor número de microglia con morfología
característica de microglias en reposo, poco teñidas, con muchas
ramificaciones y con un centro citoplasmático perinuclear, adquiriendo una
apariencia muy similar a la observada en las ratas que fueron sometidas a
simulacro de nado y no manipulada, las cuales mostrando más microglias en
reposo que activadas.
El típico patrón bifásico de comportamiento nociceptivo frente al dolor
inflamatorio agudo, inducido por la inyección subcutánea de formalina en la

79
pata trasera de la rata fue observado en los animales pretratados con
minociclina sometidos a estrés, simulacro de nado y no manipuladas, un
patrón similar fue observado en las ratas pretratadas con solución salina
sometidas a simulacro de nado y no manipuladas. Observándose la máxima
actividad de comportamiento nociceptivo (dolor) en el inicio de la fase II con un
decrecimiento progresivo a través del tiempo.
Las ratas a las que se les inyectó solución salina y fueron sometidas a
estrés de nado mostraron una mayor respuesta nociceptiva (hiperalgesia) en
los 15 últimos minutos de observación de la fase II al compararlas con las ratas
estresadas y pretratadas con minociclina.
Las ratas pretratadas con minociclina y sometidas a estrés, mostraron un
bloqueo de la hiperalgesia observada durante la fase II de la prueba de
formalina en comparación con las ratas estresadas pretratadas con solución
salina. La fase I de la prueba de formalina se cree refleja dolor nociceptivo
agudo originado por la acción irritante que la formalina ejerce directamente
sobre las terminales nerviosas activadas por estímulos dolorosos, mientras que
la fase II se atribuye a la combinación de impulsos aferentes de nervios
sensoriales estimulados por el proceso inflamatorio del tejido subcutáneo
periférico (en la pata trasera) y cambios funcionales en la neurotransmisión

80
moduladora de la información dolorosa en el asta dorsal de la médula espinal
que conducen a una facilitación o sensibilización central a la transmisión y
procesamiento de los impulsos dolorosos. Debido a que la minociclina penetra
bien a través de la barrera hematoencefálica puede producir efecto
antinociceptivo mediante la regulación periférica y/o mecanismos centrales
relacionados con el dolor inflamatorio inducido por formalina e incrementados
por el estrés previo. Cho y col. (2006) reportaron resultados similares sobre el
efecto inhibitorio de la minociclina sobre el dolor inflamatorio en el modelo de
formalina; aunque sin someter los animales a estrés, ellos además observaron
que la minociclina tenía un efecto antiedematoso en el tejido periférico. A pesar
de que Cho y col (2006) no observaron cambio en las propiedades eléctricas
de la membrana o en la excitabilidad de las neuronas en el ganglion dorsal,
plantean que no se puede excluir la posibilidad de que la minociclina pueda
inhibir la respuesta inmune periférica inducida por la inyección de formalina,
inhibiendo por lo tanto de manera indirecta la actividad neuronal sensorial.
Los antidepresivos son el grupo coadyuvante más ampliamente utilizado en
el tratamiento del dolor, especialmente en el dolor crónico, unas veces como
complemento de los analgésicos antipiréticos (AAP)/antiinflamatorio no
esteroideos (AINE), opioides y otros como fármacos de primera elección
(McQuay y col., 1997; Yerro y col., 2000; Lynch 2001; Flórez, 2004). La acción
analgésica de los antidepresivos es independiente de su acción antidepresiva

81
(Cruciani y col. 2006), aunque existan situaciones de dolor crónico con
componente depresivo en las que el alivio de este factor contribuye a mejorar la
actividad general del paciente, dolor incluido (Flórez, 2004). El mecanismo de
su acción analgésica sigue siendo objeto de controversia. Suele afirmarse que
es el resultado del incremento de la actividad conjunta de los sistemas
endógenos descendentes noradrenérgicos y serotonérgicos que proyectan al
asta posterior de la médula espinal y controlan las vías aferentes del dolor,
como consecuencia de su acción bioquímica que consiste en la inhibición de la
recaptación de noradrenalina y serotonina en las respectivas terminaciones
nerviosas (Atkinson y col. 1999). Pero esta explicación dista de ser aceptada
por todos, y se proponen otras teorías como son la acción neurotrófica, la
acción reguladora de la plasticidad sináptica, los cambios a largo plazo en la
actividad y equilibrio entre diversos tipos de receptores, e incluso acciones
cerebrales supraespinales que contribuyan a reducir la percepción del dolor
(Eschalier y col., 1988; Flórez, 2004; Marchand y col., 2006).
Los fármacos con los que se tiene una experiencia más amplia son la
amitriptilina, la clomipramina, la desipramina y la doxepina (Flórez, 2004;
Veldhuijzen y col. 2006). En ocasiones, y en función del tipo de dolor que se
instaura en un determinado paciente, el antidepresivo será el primer fármaco
que se utilice: es el caso, por ejemplo, de una neuralgia postherpética o de una
neuralgia diabética (Vidal y col., 2004; Neira y col., 1998). En otras, en cambio,

82
el antidepresivo se suma al analgésico opioide o AAP/AINE que se estaba ya
utilizando: es el caso de un dolor oncológico en el que aparece un componente
neuropático nuevo, o un componente depresivo (Almanza-Muñoz y col., 2000).
La clomipramina es menos sedante y más euforizante que la amitriptilina, lo
que puede ser ventajoso para algunos pacientes, pero en conjunto, suele ser
peor tolerada, produciéndose un mayor número de abandonos del tratamiento
(Flórez, 2004).
El típico patrón bifásico de comportamiento nociceptivo frente a la prueba
de formalina fue observado en los animales pretratados con clomipramina ó
solución salina sometidos a simulacro de nado y no manipuladas.
Las ratas pretratadas con clomipramina, varios días antes y durante las
exposiciones a las situaciones de estrés, mostraron un bloqueo de la
hiperalgesia observada durante la fase II de la curva de respuesta nociceptiva
en comparación con las ratas pretratadas con solución salinas y sometidas a
estrés. Un tratamiento crónico y a bajas dosis con un inhibidor de la
recaptación de serotonina como la clomipramina produjo un bloqueo de la
hiperalgesia observada en las ratas salinas sometidas a nado forzado, para
verificar los efectos nociceptivos de la clomipramina en el dolor inflamatorio
inducido por formalina se observo inmunohistoquímicamente cambios en la
expresión de la proteína c-Fos inducida por dolor en el asta dorsal espinal. En

83
el presente estudio se demostró que la regulación corriente arriba de la
expresión de c-Fos originada por la inyección de formalina fue inhibida por la
clomipramina. Debido a que el c-Fos es un marcador que puede regularse
corriente arriba por muchos tipos de estímulos nocivos periféricos (Curran y
Morgan, 1995; Vergnolle y col, 2001; Watkins y col 1997), la reducción de su
expresión en el asta dorsal de la medula indica claramente la acción
antinociceptiva de la clomipramina.
Estudios en microdiálisis con ratas con movimiento libre mostraron un
incremento en los niveles de serotonina en varias regiones del cerebro,
específicamente el rafe magno, después de 15 minutos de nado forzado (Ikeda
y col., 1985; Adell y col., 1997). Por el contrario, el nado forzado prolongado por
30 minutos, disminuye el flujo de serotonina en ciertas estructuras cerebrales
como la amigdala y el septum lateral, por lo tanto especulamos que el estrés
de nado crónico produce un incremento inicial en la actividad de los sistemas
de serotonina central, el cual es seguido por una reducción en la liberación
como respuesta al estrés prolongado.
El presente estudio sugiere que el incremento en la percepción del dolor
inflamatorio cutáneo agudo que ocurre luego de la exposición repetida estrés
de nado forzado pudieran estar mediados por cambios en la actividad del
sistema central serotoninérgico, la serotonina es un modulador inhibitorio de la

84
nocicepción, por lo tanto, aumentos en sus niveles llevarían a una analgesia
(disminución de la percepción del dolor) y una disminución de sus
concentraciones extracelulares provocarían un estado hiperalgésico (aumento
de la percepción del dolor). En el presente estudio se encontró que la
hiperalgesia por exposición repetida a nado forzado disminuye
considerablemente por administración, antes de la exposición a estrés, de
bajas dosis de clomipramina, un inhibidor selectivo de la recaptación de
serotonina. Los sitios de acción de la clomipramina todavía no han sido bien
aclarados, debido a que su efecto antihiperalgesico puede ser llevado a cabo
en diferentes áreas inervadas por el sistema serotoninérgico central. Un
componente principal del efecto antihiperalgesico de este agente
serotoninérgico puede deberse a su acción en neuronas serotononérgicas en el
rafe magno que envía fibras descendentes a las capas superficiales de las asta
dorsales medulares. De hecho, se ha encontrado que las neuronas del rafe
magno inhiben la respuesta al dolor provocada por formalina (Abbott y col.,
1982). El estrés por nado agudo reduce significativamente y por largo plazo los
niveles extracelulares de 5-HT en el septum lateral y amigdala (Kirby y col.,
1995, 1997). Hallazgos experimentales convergentes implican en este efecto
inhibitorio al factor liberador de corticotropina (CRF), un neuropeptido liberado
rápida y masivamente durante una situación de estrés. La administración de
CRF en el núcleo dorsal del rafe imita el efecto inhibitorio del nado forzado
sobre el incremento de 5-HT (Price y col. 1998, 2002). Además el incremento

85
de la inhibición de 5-HT por el estrés de nado puede prevenirse por la previa
administración de antagonistas del CRF (Xiang y col., 2003).
El dolor evocado por formalina puede inhibirse por estimulación eléctrica de
la sustancia gris periacueductal, por lo tanto, el estrés de nado forzado repetido
puede producir hiperalgesia mediante el bloqueo del control inhibitorio de la
sustancia gris periacueductal y esto puede prevenirse con la administración de
agentes serotoninérgicos antes de la exposición a estrés (Dubuisson y col.,
1977). El bloqueo de varias estructuras en el circuito límbico activadas durante
la respuesta fisiológica de estrés, como el núcleo paraventricular del
hipotálamo y el hipocampo, también reduce el comportamiento de dolor
inducido por formalina (Vaccarino y col., 1992). De esta manera, el estrés
crónico puede incrementar la nocicepción al aumentar la actividad de las
estructuras límbicas del hipotálamo e hipocampo para producir hiperalgesia y
agentes serotoninérgicos pueden actuar en estas regiones para inhibir la
hiperalgesia inducida por estrés mediada por el sistema límbico (Akil y col.,
1995).
En el presente estudio también se observo, en las ratas controles tratadas
solo con solución salina, que a medida que se exponía repetidamente al estrés
de nado inescapable, había una reducción progresiva en el tiempo dedicado a
tratar de escaparse del recipiente con agua. Las ratas expuestas al estrés de

86
nado y tratadas con clomipramina antes del estrés mostraron un incremento
significativo en el tiempo dedicado a tratar de escapar del estresor. La
disminución del comportamiento de escape inducido por el estrés de nado
forzado inescapable se ha denominado comportamiento de desesperación o
invalidez aprendida y se cree que representa en los roedores un equivalente de
la depresión clínica en humanos. Es interesante el hecho de que el estrés
crónico produce cambios funcionales y bioquímicos en el eje límbico
hipotalámico pituitario que son muy similares a los observados durante la
depresión severa, siendo dichos cambios considerados una característica
necesaria de este síndrome (Akil y col., 1995). La sensibilidad al dolor se
incrementa en individuos con depresión menor o mayor (Merskey, 1965) y los
individuos deprimidos reportan una prevalencía mayor de problemas de dolor
clínico (Merskey, 1965; Piñerua-Shuhaibar, 1999). En el presente estudio, las
ratas mostraron una falta exagerada de deseo de escape (lo que es
denominado “inmovilidad” en la prueba de nado forzado) y una respuesta
hiperalgesica a la inflamación aguda, siendo dichos cambios revertidos por la
clomipramina, un inhibidor de la recaptura de serotonina (Izumi R, 1983;
Quintero y col., 2000). Esto nos podría llevar a especular que la falta exagerada
al deseo de escape y la hiperalgesia podría resultar de una deficiencia en la
transmisión central serotoninérgica, lo que produce tanto depresión como
sensibilización a las vías centrales del dolor.

87
7.- CONCLUSIÓN
1) El estrés incrementó la activación microglial inducida por la inyección
subcutánea de formalina. El estrés pareciera sensibilizar el sistema nervioso
para una respuesta incrementada a un estímulo inmunológico periférico
subsiguiente. La activación microglial esta condicionada por varios factores,
uno de los más importantes es la permanencia en un estado de quiescencia o
reposo provocada por la unión del receptor CD200R en la microglia con la
glicoproteína CD200 presente en las neuronas. La exposición repetida a estrés
pudiera de alguna manera estar asociada a cambios transcripcionales y de
señalización que causarían modificaciones en la expresión de la glicoproteína
CD200 en las neuronas.
2) La minociclina inhibió la activación microglial central observada en la
medula espinal de ratas estresadas y con un proceso inflamatorio agudo
doloroso. Al mismo tiempo, la administración de minociclina antes de cada
estrés de nado forzado evitó el desarrollo de la hiperalgesia cutánea, la cual si
se desarrolló en ratas estresadas pero tratadas solo con solución salina. Por lo
tanto, la hiperalgesia inducida por estrés de nado forzado podría estar
vinculada a una sobre-activación de la microglia en el sistema nervioso central.

88
3) El presente estudio sugiere que el incremento en la percepción del
dolor inflamatorio cutáneo agudo que ocurre luego de la exposición repetida a
estrés de nado forzado pudieran estar mediados por cambios en la actividad
del sistema central serotoninérgico. En el presente estudio, la hiperalgesia
cutánea, la falta exagerada del deseo de escape de un ambiente estresante
(“inmovilidad”) y la sobre-activación de las neuronas espinales sensoriales
inducida por un dolor inflamatorio agudo (estimada como un expresión
incrementada de la proteína c-Fos) fueron considerablemente bloqueadas por
administración de clomipramina antes de la exposición a estrés. Esto nos
podría llevar a especular que una deficiencia en la transmisión central
serotoninérgica produce tanto una conducta depresiva como una
sensibilización a las vías centrales del dolor.

89
8.- REFERENCIAS BIBLIOGRAFÍCAS ABBAS A, LICHTMAN A, POBER J. (2003). Inmunología Celular y Molecular. 4ta Edición. McGraw-Hill-Interamericana. Madrid. España. ADELL A, CASANOVAS J, ARTIGAS F. (1997). Comparative study in the rat of the actions different types of stress on the release of 5-HT in raphe nuclei and forebrain areas. Neuropharmacology. 36. AKIL HA, MORANO MI. Stress. In: Bloom, F.E.; Kupter: 735 -741.. DJ. Editors. (1995). Psychopharmacology: The fourth generation of progress. New York: Raven Press. 67: 773-785.
AKIYAMA H, ARAI T, KONDO H, TANNO E, HAGA C, IKEDA K. (2000). Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Discord. 1:S47–S53.
ALMANZA-MUÑOZ JJ, JIMMIE C HOLLAND JC. Psico-oncología: estado actual y perspectivas futuras Revista del Instituto Nacional de Carcinología. 46(3):196-206. ALOISI F. (2001). Immune function of microglia. Glia 36:165–179. AMIT Z, GALINA ZH. (1986). Strees-induced analgesia: adaptative pain suppression. Physiol Rev. 66(4):1091-120.
ASCHNER M, SONNEWALD U, TAN KH. (2002). Astrocyte modulation of neurotoxic injury. Brain Pathol. 12(4):475-81. ARAUJO DM, COTMAN CW. (1992). Basic FGF in astroglial, microglial and neuronal cultures: characterization of binding sites and modulation of release by lymphokines and trophic factors. J Neurosci. 12:1668-1678.
ARDID D, JOURDAN D, MESTRE C, VILLANUEVA L, LE BARS D, ESCHALIER A. (1995). Involvement of bulbospinal pathways in the antinociceptive effect of clomipramine in the rat. Brain Res. 695:253-256.
ATKINSON JH, SLATER MA, WAHLGREN DR, WILLIAMS RA, ZISOOK S, PRUITT SD, EPPING-JORDAN JE, PATTERSON TL, GRANT I, ABRAMSON I, GARFIN SR. 1999 Effects of noradrenergic and serotonergic antidepressants on chronic low back pain intensity. Pain. 83(2):137-45. BASBAUM AI, FIELDS HL. (1978). Endogenous pain control mechanisms: review and hypothesis. Ann Neurol. 4:451-462.

90
BASBAUM AI, JESSELL TM. The percepcion of pain. Kandel ER, Schwartz Jessell TM, Eds. (2000). Principles of neural science, 4a ed. McGraw-Hill, New York. pp 472-491
BAUER J, BRADL M, HICKEY WF, FORSS-PETTER S, BREITSCHOPF H, LININGTON C, WEKERLE H, LASSMANN H. (1998). T-cell apoptosis in inflammatory brain lesions: destruction of T cells does not depend on antigen recognition. Am. J. Pathol. 153:715.
BETHEA JR, GILLESPIE GY, BENVENISTE EN. (1992). Interleukin-1 beta induction of TNF-alpha gene expression: involvement of protein kinase C. J Cell Physiol. 152(2):264–273. CABANA SALAZAR JA, RUIZ REYES CR. (2004). Analgesia por acupuntura. Rev Cubana Med Milit. 33(1):1-6.
CHOI SH, JOE EH, KIM SU, JIN BK. (2003). Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. The Journal of Neuroscience. 23(13):5877–5886. CHO IH, CHENG YM, KYU PARK C, PARK SH, YING LI H, KIM D, PIAO ZG, CHOI SY, LEE SJ PARK K, KIM JS, JUNG SJ, OH SB. (2006) Systenic administration of minocycline inhibits formalin-induced inflammatory pain in rat. Brain Research. 1072 (910):208-214 CHROUSOS GP. (2000). Stress, chronic inflammation, and emotional and physical well-being: concurrent effects and chronic sequelae. J Allergy Clin Immunol. 106:S275–S291.
CRUCIANI RA, NIETO MJ. (2006). Fisiopatología y tratamiento del dolor neuropático:avances más recientes. Rev. Soc. Esp. Dolor. 5: 312-327. DALLMAN MF, AKANA SF, SCRIBER KA, BRADBURY MJ, WALKER CD, STRACK AM, CASCIO CS. (1992). Stress, feedback, and facilitation in the hypothalamopituitary- adrenal axis. J Neuroendocrinol. 4:517–526.
DE GOEIJ DCE, JEZOVA D, TILDERS FJH. (1992). Repeated stress enhances vasopressin synthesis in corticotropin releasing factor neurons in the paraventricular nucleus. Brain Res. 557:265–268
DE LA TORRE A, NÚÑEZ MX. (2002). Inmunología ocular: síndromes de ojo seco. Coloma Med. 33(3):113-122.

91
DELEO JA, COLBURN RW. (1999) Proinflammatory cytokines and glial cells: their role in neuropathic pain. In Cytokines and Pain (Watkins, L., ed.). pp. 159–182. Birkhauser
DICKSON DW, MATTIACE LA, KURE K, HUTCHINS K, LYMAN WD, BROSNAN CF. (1991). Microglia in human disease, with an emphasis on acquired immune deficiency syndrome. Lab Invest. 64:135–156
DRAGUNOW M. (1999). Neuronal death and survival in two models of hypoxicischemic brain damage. Brain Res Rev. 29:137–168.
DUBUISSON D, DENNIS SG. (1977). The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 4:161-174. ESCHALIER A, FIALIP J, VAROQUAUX O, MAKAMBILA M.C, MARTY H. ASTIDE P. (1988) Pharmacokinetic patterns of repeated administrations of antidepressants in animals. I. Implications for the antinociceptive action of clomipramine in mice. J. Pharmacol. Exp. Ther. 245: 963−968. ELENKOV IJ, CHROUSOS GP. (1999). Stress, cytokine patterns, and susceptibility to disease. Ballieres Best Pract Res Clin Endocrinol Metab. 13:583–595.
FISHER HG, REICHMANN G. (2001). Brain Dendritic Cells and Macrophages/Microglia in Central Nervous System Inflammation. The Journal of Immunology. 166: 2717-2726. FLORES J. (2004). FARMACOS Y DOLOR. Edita: Ergon. C/ Arboleda, 1 - 28220 Majadahonda (Madrid). p. 141. FRANCIS K, BEEK JV., CANOVA C, NEAL JW, GASQUE P. (2003). Innate immunity and brain inflammation: the key role of complement. Expert Reviews in Molecular Medicine. 5:1-19. FRANK MG, BARATTA MV, SPRUNGER DV, WATKINS LR, MAIER SF. (2007) Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inXammatory cytokine responses. Brain, Behavior, and Immunity. 21:47–59 FREI K, MALIPIERO UV, LEIST YP, ZINKERNAGEL RM, SCHWAB MS, FONTANA A. (1989). On the cellular source and function of interleukin 6 PRODUCED BY DE CENTRAL NERVOUS SYSTEM. Eur. J. Immunol.19:689-694.

92
FU K-Y, LIGHT AR, MATSUSHIMA GK, MAIXNER W. (1999). Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 825: 59–67 GARCIA LE, MARTINEZ MP, GONZALEZ HL. (2001). Inflamación y dolor: cambios en el sistema nervioso periférico y central. MEDUNAB-España. 4(10):1-14 GILAD GM, GILAD VH. BRUCE R. (1995). Chemotaxis and accumulation of nerve growth factor by microglia and macrophages J Neurosci Res. 41(5):594-602 GOMEZ-PINILLA F, CUMMINGS BJ, COTMAN CW. (1990). Induction of basic fibroblast growth factor in Alzheimer’s disease pathology. Neuroreport. 1:211–214.
GRIFFITHS, MD. (1995). Adolescent Gambling. London : Routledge. HABIB KE, GOLD PW, CHROUSOS GP. (2001). Neuroendocrinology of stress. Endocrinol Metab Clin North Am. 30:695–728.
HELLHAMMER DH, HINGTGEN JN, WADE SE, SHEN PA, APRISON MH. (1983). Serotoninergic changes in specific areas of rat brain associated with activity stress gastric lesions. Psychosom. Med. 45:115-122.
HEUMANN R, LINDHOLM D, BANDTLOW C, MEYER M, RADEKE MJ, MISKO TP, SHOOTER E, THOENEN H. (1987). Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration and regeneration: role of macrophages. Proc Natl Acad Sci USA. 84:8735–8739.
HOEK R, RUULS S, MURPHY C, WRIGHT G, GODDARD R, ZURAWSKI S, BLOM B, HOMOLA M, STREIT W, BROWN M, BARCLAY N, SEDGWICK J. (2000). Down-Regulation of the macrophage lineage through interaction with OX2 (CD200). Science. 290:1768-1771. HSU SM, RAINE L, FANGER H. (1981). Use of avidin-biotin-peroxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem Cytochem. 29:577-580.
HUGHES P, DRAGUNOW M. (1985). Induction of immediate-early genes and the control of neurotransmitter- regulated gene expression within the nervous system. Pharmacological Rew. 47:133-178.

93
IKEDA M, NAGATSU T. (1985). Effect of short-term swimming stress and diazepam on 3,4-dihydroxyphenylacetic acid (DOPAC) and 5-hydroxyindolacetic acid (5-HIAA) levels in the caudate nucleus: an in vivo voltammetric study. Naunyn-Schmiedebrg's Archive of Pharmacology. 331:23-26.
INOUE A, IKOMA K, MORIOKA N, KUMAGAI K, HASHIMOTO T, HIDE I, NAKATA Y. (1999). Interleukin-1beta induces substance P release from primary afferent neurons through the cyclooxygenase-2 system. J. Neurochem. 73:2206–2213. IZUMI R, TAKAHASHI M, KANETO H. (1983). Involvement of different mechanisms, opioid and non-opioid forms, in the analgesia induced by footshock (FS) and immobilized-water immersion (IW) stress. Jpn J Pharmacol. 33:1104-1106.
JANEWAY CA, TRAVERS P, WALPORT M, CAPRA D. (2000). Inmunobiología. El Sistema Inmunitario en condiciones de salud y enfermedad. Editorial II Masson. España. Pág. 642.
JOHNSON JD, O’CONNOR KA, WATKINS LR, MAIER SF. (2004). The role of IL-1beta in stress-inducd sensitization of proinflammatory cytokine and corticosterone responses. Neuroscience. 127:569–577. KIRBY LG, ALLEN AR, LUCKI I. (1995). Regional differences in the effects of forced swimming on extracellular levels of 5-hydroxytryptamine and 5-hydroxyindoleacetic acid. Brain Res. 682. KIRBY LG, ALLEN AR, LUCKI I. (1995). Regional differences in the effects of forced swimming on extracellular levels of 5-hydroxytryptamine and 5-hydroxyindoleacetic acid. Brain Res. 682:189–196.
KIRBY LG, CHOU-GREEN JM, DAVIS K, LUCKI I. (1997). The effects of different stressors on extracellular 5-hydroxytryptamine and 5-hydroxyindoleacetic acid. Brain Res. 760:218–230.
KIRBY LG, LUCKI I. (1997). Interaction between the forced swimming test and fluoxetin treatment on extracellular 5-HT and 5-HIAA in the rat. J. Pharmacol Exp Ther. 282:967-976.
KITAMURA T, TSUCHIHASHI Y, TATEBE A, FUJITA S. (1977). Electron microscopic features of the resting microglia in the rabbit hippocampus, identified by silver carbonate staining. Acta Neuropathol. 38:195–201.

94
KRONFOL Z, REMICK DG. (2000). Cytokines and the brain: implications for clinical psychiatry. M J Psychiatry. 157(5):683-94 LE JM, VILCEK J. (1989). Interleukin 6: a multifunctional cytokine regulating immune reactions and the acute phase protein response. 61(6):588-602. LEE G, DALLAS S, HONG M, BENDAYAN R. (2001). Drug Transporters in the Central Nervous System: Brain Barriers and Brain Parenchyma Considerations. Pharmacol Rev. 53:569–596.
LEWIS J, CANNON JT, LIEBESKIND JC. (1980). Opioid and nonopioid mechanisms of stress analgesia. Science. 208:623-625.
LYNCH ME. (2001) Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 26(1):30-6. LOESER JD, MELZACK R. (1999). Pain: an overview. Lancet. 353:1607-1609.
MADDEN J, AKIL H, PATRICK RL, BARCHAS JD. (1977). Stress-induced parallel changes in central opioid levels and pain responsiveness in the rat. Nature. 265:358-60.
MAMET J, BARON A, LAZDUNSKI M, VOILLEY N. (2002). Proinflammatory mediators, stimulators of sensory neuron excitability via the expression of acid-sensing ion channels. The Journal of Neurocience. 22(24):10662-70. MCGEER PL, MCGEER EG. (1998). Mechanisms of cell death in Alzheimer disease–immunopathology. J Neural Transm Suppl. 54:159–166.
MCQUAY HJ, MOORE RA. (1997). Antidepressants and chronic pain. Br. Med. J. 414:763-764. MAGISTRETTI PJ, PELLERIN L, MARTIN JL. (1995). Brain energy metabolism: An integrated cellular perspective. In: Bloom FE, Kupfer DJ, Edt. Psychopharmacology: The Fourth Generation of Progress. New York: Raven, 921-932.
MAIER SF, JACKSON R. (1979). Learned helplessness: all of us were right (and wrong): inescapable shock has multiple effects. Psychol Learn Motiv. 13:155-218.
F. MARCHAND, D. ARDID, E. CHAPUY, A. ALLOUI, D. JOURDAN, A. ESCHALIER . 2006. Evidence for an involvement of supraspinal delta and

95
spinal mu opioid receptors in the antihyperalgesic effect of chronically administered clomipramine in mononeuropathic rats. JPET Fast Forward Published on September 3. MASSON P, LEUNG CG. (1996). Physiological functions of pontomedullary raphe and medial reticular neurons. Prog Brain Res. 107:269-282.
MELZACK R., LOESER JD. (1978). Phantom body pain in paraplegics: evidence for a central “pattern generating mechanism” for pain. Pain. 4:195-210. MERSKEY H. (1965). The effect of chronic pain upon the response to noxious stimuli by psychiatric patients. J Psychiatr Res.8:405-419. MILLIGAN ED., MEHMERT KK., HINDE JL., HARVEY LO., MARTIN D., TRACEY KJ., MAIER SF., WATKINS LR. (2000). Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the Human Immunodeficiency Virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 861:105–116.
MOLANDER C, GRANT G. (1985). Cutaneous projections from the rat hindlimb foot to the substantia gelatinosa of the spinal cord studied by transganglionic transport of WGA-HRP conjugate. J. Comp. Neurol. 237: 476–484. MOLANDER C, GRANT G, Laminar distribution and somatotropic organization of primary afferent fibers from hindlimb nerves in the dorsal horn. A study by transganglionic transport of horseradish peroxidase in the rat, Neuroscience 19 (1986) 297–312. NEARY JT, RATHBONE MP, CATTABENI F, ABBRACCHIO MP, BURNSTOCK G. (1996). Trophic actions of extracellular nucleotides and nucleosides on glial and neuronal cells. Trends in Neuroscience. 19(1): 13-18. NEUMANN H. (2001) Control of glial immune function by neurons. Glia. 6(2):191-9. NEIRA F, ORTEGA JL. (1998). La neuralgia postherpética. ¿Un problema sin resolver? Rev. Soc. Esp. Dolor. 5: 128-143. O’CONNOR KA, JOHNSON JD, HANSEN MK, WIESELER FRANK JL, MAKSIMOVA E, WATKINS LR, MAIER SF. 2003. Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res. 991, 123–132.

96
PEREIRA CF, NOTTET HSLM. (2000). The Blood-Brain Barrier in HIV-associated Dementia. NeuroAids. 3(2):1-8. PERRY VH. (2007). Stress primes microglia to the presence of systemic inflammation: Implications for environmental influences on the brain. Brain, Behavior, and Immunity. 21:45–46 PETERSEN MA., DAILEY ME. (2004). Diverse Microglial motility behaviors during clearance of dead cells in hippocampal slices. Glia. 46(2):195-206.
PIGNATIELLO MF., OLSON GA., KASTIN AJ., EHRENSING RH., MCLEAN JH., OLSON RD. (1989). MIF-1 is active in a chronic stress animal model of depression. Pharmacol Biochem Behav. 32:737-742.
PIÑERUA-SHUHAIBAR L, PRIETO-RINCON D, FERRER A, BONILLA E, MAIXNER, H, SUAREZ-ROCA H. Reduced tolerance and cardiovascular response to ischemic pain in minor depression. J Affective Dis. 56:119-26. PORSOLT RD, LE PINCHON M, JALFRE M. (1977). Depression: a new animal model sensitive to antidepressant treatments. Nature. 266:730–732. POUSSET F. (1994). Developmental expression of cytokine genes in the cortex and hippocampus of the rat central nervous system Brain Res Dev Brain Res. 81(1):143-6.
PRIETO YERRO C, VARGAS CASTRILLON E. (2000). Problemas e uso de los antiinflamatorios no esteroideos (AINES) en pacientes con patología cronica asociada. Información del sistema nacional de salud (Espana). 24(4):85-91.
QUINTERO L, MORENO M, AVILA C, ARCAYA J, MAIXNER W, SUAREZ-ROCA H. (2000). Long-lasting delayed hyperalgesia after subchronic swim stress. Pharmacology, Biochemistry and Behavior. 67(3):449-458.
RAIVICH G., BOHATSCHEK M., KLOSS CU., WERNER A., JONES LL., KREUTZBERG GW. (1999). Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Rev. 30:77–105.
REXED B. (1952). The cytoarchitectonic organization of the spinal cord of the cat. J. Comp. Neurol. 96:415-495.
RIBERA MV., BUSQUETS C. (2003). Unidades del Dolor. Realidad Hoy, reto para el futuro. Clínica del Dolor. Hospital Valle de Hebrón. Capitulo XIX. Vilallonga JR. Neuroanatomía del dolor: bases anatómicas de la

97
percepción del dolor. Hospital Valle del Hebrón. Barcelona. España. pp. 217-250
ROCHE M, COMMONS KG, PEOPLES A, VALENTINO RJ. 2003. Circuitry underlying regulation of the serotonergic system by swim stress. The Journal of Neuroscience. 23(3):970 –977. SAWYNOK J, XUE JUN L. (2003) The formalin test: Characteristics and usefulness of the model. Reviews in Analgesia. 7(2):145-163 SCHWEI MJ, HONORE P, ROGERS SD, SALAK-JOHNSON JL, FINKE MP, RAMNARAINE ML, CLOHISY DR, MANTYH PW. (1999). Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. J. Neurosci. 19: 10886–10897.
SORKIN LS, STEINMAN JL, HUGHES MG, WILLIS WD, MCADOO DJ. (1988). Microdialysis recovery of serotonin released in spinal cord dorsal horn. J Neurosci Methods. 23(2):131-8.
SOUTHALL, D. (1998). Manual of acute pain management in Children. Archives of Disease in childhood. 79(4):380. STENCE N, WAITE M, DAILEY ME. (2001). Dynamics of microglial activation: a confocal time-lapse análisis in hippocampal slices. GLIA. 33:256-266.
STORKSON RV, KJORSVIK A, TJOLSEN A, HOLE K. (1996). Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 65(2):167-72.
STREIT WJ. MICROGLIAL CELLS. KETTENMANN H, RANSOM B. (1995). Neuroglia. Oxford University Press. New York. pp. 85-96. STREIT WJ, GRAEBER MB, KREUTZBERG GW. (1988). Functional plasticity of microglia: a review. Glia. 1:301–307. STREIT, WJ. (2002). Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 40(2):133-9. STREIT WJ, MRAK RE, GRIFFIN WS. (2004). Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 1(1):14.
SWEITZER SM, COLBURN RW, RUTKOWSKI M, DELEO JA. (1999). Acute peripheral inflammation induces moderate glial activation and spinal IL-1 beta

98
expression that correlates with pain behavior in the rat. Brain Res. 829, 209–221. TODD K, BUTTERWORTH RF. (1999). Mechanisms of selective neuronal cell death due to thiamine deficiency. Ann NY Acad Sci. 893:404–411. TOWN T, NIKOLIC V, TAN J. (2005). The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation. 31(2):24. VACCARINO AL, MELZACK R. (1992).Temporal processes of formalin pain: differential role of the cingulum bundle, fornix pathway and medial bulboreticular formation. Pain. 49:257-271. VELDHUIJZEN DS,ALBERT WIJCK AJM, VERSTER JC, KENEMANS JL, KALKMAN CJ, BEREND OLIVIER, VOLKERTS ER. (2006). Acute and subchronic effects of amitriptyline 25 mg on actual driving in chronic neuropathic pain patients. Journal of Psychopharmacology. 20(6):782–788 VIDAL MA, MARTÍNEZ-FERNÁNDEZ E, MARTÍNEZ-VÁZQUEZ DE CASTRO J, TORRES LM. (2004). Eficacia de la gabapentina y de la amitriptilina en el dolor neuropático del diabético. Rev. Soc. Esp. Dolor. 11: 292-305. WALL PD, MELZACK R editors. (1994). Textbook of Pain. Myofascial Pain Syndrome. Churchill Livingstone 263-267. WATKINS LR, MAIER SF. (2000). The Pain of Being Sick: Implications of Immune-to-Brain Communication for Understanding Pain. Annual Review of Psychology. 51: 29-57.
WATKINS LR, MAIER SF. (2000). The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 51: 29–57.
WATKINS LR, MILLIGAN ED, MAIER SF. (2001). Glial activation: a driving force for pathological pain. TRENDS in Neurosciences. 24(8):450-455.
WALTON M, CONNOR B, LAWLOR P, YOUNG D, SIRIMANNE E, GLUCKMAN P, COLE G, DRAGUNOW M. (1999). Neuronal death and survival in two models of hypoxicischemic brain damage. Brain Res Rev. 29:137–168.
WEISS JM., GOODMAN PA., LOSITO BG., CORRIGAN S., CHARRY JM., BAILEY WH. (1981). Behavioral depression produced by an uncontrollable stressor: relationship to norepinephrine, dopamine, and serotonin levels in various regions of rat brain. Brain Res Rev. 1981; 3:167–205.

99
WOOLF CJ, COSTIGAN M. (1999). Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci U S A. 96(14):7723-30.
WOOLF CJ. (2000). Pain. Neurobiol Dis. 7(5):504-10. WRIGHT G, CHERWINSKI H, FOSTER-CUEVAS M, BROOKE G, PUKLAVEC M, BIGLER M, SONG Y, JENMALM M, GORMAN D, MCCLANAHAN T, LIU M, BROWN MH, SEDGWICK JD, PHILLIPS JH, BARCLAY N. (2003). Characterization of CD200 receptor family in mice and humans and their interactions with CD200. The journal of Inmmunology. 171(6):3034-46.
XIONG H., ZENG YC., LEWIS T., ZHENG J., PERSIDSKY Y., GENDELMAN HE. (2000). HIV-1infected mononuclear phagocyte secretory products affect neuronal physiology leading to cellular demise: relevance for HIV-1- associated dementia. J Neurovirol. 6:S14–23. YRJANHEIKKI J, KEINANEN R, PELLIKKA M, HOKFELT T, KOISTINAHO J. (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 95(26):15769-74.
XIANG Z, PRINCE DA. (2003). Heterogeneous Actions of Serotonin on Interneurons in Rat Visual Cortex. J Neurophysiol 89: 1278-1287







