
Monoclonal antibodies (mAbs) have
emerged as important therapeutics
for the treatment of life-threatening
diseases, including cancer and
autoimmune diseases (1,2,3). Today,
more than 40 mAbs are marketed
in the United States and Europe, of
which 18 display blockbuster status;
six have sales greater than $6 billion
(Humira, Remicade, Enbrel, Rituxan,
Avastin, and Herceptin) and over 50
are in late-stage clinical development.
mAbs are currently considered
to be the fastest growing class of
therapeutics with sales more than
doubled since 2008 (1,2,3).
The successes of mAbs have
triggered the development of various
next generation formats, such as
antibody–drug conjugates (ADC),
bispecific mAbs, mAb mixtures,
polyclonal antibodies, antibody
fragments (Fab, Fc, nanobodies),
Fc fusion proteins, brain penetrant
mAbs, and glyco-engineered
mAbs. In oncology, ADCs are
particularly promising because they
synergistically combine a specific
mAb linked to a biologically active
cytotoxic drug using a stable linker
(4–6). The promise of ADCs is that
highly toxic drugs can be selectively
delivered to tumour cells thereby
substantially lowering side effects
as typically experienced with
classical chemotherapy. Two ADCs
are currently marketed, namely
brentuximab vedotin (Adcetris)
and ado-trastuzumab emtansine
(Kadcyla), and over 30 are in clinical
trials (4–6).
With the top-selling mAbs evolving
out of patent there has been a
growing interest in the development of
biosimilars (7). In 2013, we witnessed
the European approval of the first
two mAb biosimilars (Remsima and
Inflectra), which both contain the
same active substance, infliximab (8).
In April 2016, Inflectra also reached
marketing authorization in the US and
a third infliximab biosimilar (Flixabi)
was recently approved in Europe.
Remicade, infliximab’s blockbuster
originator, reached global sales of
$8.9 billion in 2013 (3).
Structural ComplexityTogether with a huge therapeutic
potential, these products come with
a structural complexity that drives
state-of-the-art chromatography and
mass spectrometry to its limits and
this is exactly where two-dimensional
liquid chromatography (2D-LC)
comes into play. mAbs are tetrameric
immunoglobulin G (IgG) molecules
with a molecular weight (MW) of
150 kDa (± 1300 amino acids)
composed of two light (Lc – 25 kDa)
and two heavy (Hc – 50 kDa)
polypeptide chains connected
through interchain disulphide
bridges (Figure 1). Twelve intrachain
disulphide bridges, four within
each Hc and two within each Lc,
furthermore guarantee its structural
integrity. The structure can also
be divided in the antigen binding
fragment (Fab) and the crystallizable
fragment (Fc). Antigen binding is
mediated by the Fab fragment,
while the Fc fragment is responsible
for the effector function, that is,
antibody dependent cell-mediated
cytotoxicity (ADCC) and complement
dependent cytotoxicity (CDC). All
mAbs are glycoproteins and have two
conserved N -glycosylation sites in
the Fc region that can be occupied
with complex and high mannose type
N -glycans.
These glycan structures are known
to play a role, amongst others, in the
effector function and are the subject
of extensive research towards the
development of glyco-engineered
mAbs with improved effector
functions. A variety of other chemical
and enzymatic modifications
(wanted and unwanted) taking place
during expression, purification, and
long-term storage further shape the
mAb and give rise to a substantial
heterogeneity. Modifications and
Characterizing Monoclonal Antibodies and Antibody–Drug Conjugates Using 2D-LC–MSKoen Sandra, Isabel Vandenheede, Mieke Steenbeke, Gerd Vanhoenacker, and Pat Sandra, Research Institute for
Chromatography, Kortrijk, Belgium
Two-dimensional liquid chromatography (2D-LC) has in recent years seen an enormous evolution, and with the introduction of commercial instrumentation, the technique is no longer considered a specialist tool. One of the fi elds where 2D-LC is being widely adopted is in the analysis of biopharmaceuticals, including monoclonal antibodies (mAbs) and antibody–drug conjugates (ADCs). These molecules come with a structural complexity that drives state-of-the-art chromatography and mass spectrometry (MS) to its limits. Using practical examples from the authors’ laboratory complemented with background literature, the possibilities of on-line 2D-LC for the characterization of mAbs and ADCs are presented and discussed.
149www.chromatographyonline.com
BIOPHARMACEUTICAL PERSPECTIVES

variants often encountered include
(but are not limited to) asparagine
deamidation, aspartate isomerization,
succinimide formation, N -terminal
pyroglutamate formation, C-terminal
lysine truncation, methionine or
tryptophan oxidation, glycation,
cysteine variants, and sequence
variants (9). Even though only a single
molecule is cloned, thousands of
possible variant combinations may
exist for one given mAb and they all
contribute to the safety and efficacy
of the product.
Compared to naked mAbs,
ADCs further add to the complexity
because the heterogeneity of the
initial antibody is superimposed with
the variability associated with the
conjugation strategy. Conjugation
typically takes place on the amino
groups of lysine residues or on the
sulphydryl groups of interchain
cysteine residues as is the case
in, respectively, ado-trastuzumab
emtansine and brentuximab vedotin
(b)(a)
1D-Column 1D-Column
2D-Column
2D-Column
2D-Pump
2D-Pump
Waste
Waste
OUT
OUT
Fill-direction
Fill-direction
Analyze-direction
Analyze-direction
Loop
Loop
Figure 2: (a) Modern modulators for comprehensive LC×LC and (b) for multiple heart-cutting LC (mLC–LC).
(a)
(b) (c)
FabF(ab’)2
N-ter
C-ter
DM1 Thioether linker
Trastuzumab Brentuximab
Proteasecleavable linker MMAE
Light chain (Lc)
Heavy chain (Lc)
C-ter
G0F
n = 3.5
G0F G0F G0Fn = 4
G0F
G0F
N-ter
Hc
Lc
FcFc/2
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYN(glycan)STYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG
Figure 1: Schematic representation of mAb and ADC. (a) Anatomy of humanized IgG1 trastuzumab (Herceptin). Primary sequence of the light and heavy chain and schematic representation of a functional monoclonal antibody. Subunits that can be formed following chemical or enzymatic treatment are shown: light chain (Lc), heavy chain (Hc), crystallizable fragment (Fc), fragment antigen binding (Fab), Fc/2, and F(ab’)2. Asparagine at position 300 in the heavy chain is occupied with bi-antennary N-glycans. (b) Anatomy of the ADC ado-trastuzumab emtansine (Kadcyla), which combines the trastuzumab (a) with the cytotoxic microtubule-inhibiting maytansine derivative DM1 conjugated to lysine residues via a non-reducible thioether linker. DM1 is conjugated with an average of 3.5 drugs per antibody. (c) Anatomy of the ADC brentuximab-vedotin (Adcetris), which combines the antibody brentuximab with the antimitotic drug monomethylauristatin E (MMAE) conjugated to interchain cysteine residues via a cathepsin cleavable valine-citrulline linker. MMAE is conjugated with an average of 4 drugs per antibody.
-$r($�&VSPQF March 2017150
BIOPHARMACEUTICAL 1&341&$5*7&4

(Figure 1). With 80–100 lysine and only eight interchain
cysteine residues available, lysine conjugation yields a
more heterogeneous mixture of species compared to
cysteine conjugation.
On top of the described primary structure, mAbs and
ADCs also have distinct higher order structures dictating
their function, which might be influenced by the above
described modifications, and can appear as dimers or
aggregates, which have the potential to induce immune
responses.
Two-Dimensional Liquid ChromatographyAn emerging tool to tackle this complexity is 2D-LC
(10–13). In 2D-LC, two different chromatographic
separation mechanisms are combined and peaks eluting
from the first column are further separated on a second
column with an orthogonal separation mechanism. The
aim is to substantially increase the separation power
for complex samples or to make the first-dimension
separation, in case it makes use of non-volatile salts,
compatible with mass spectrometry (MS). Orthogonal
behaviour is typically governed when combining the
following chromatographic modes: affinity chromatography
(Protein A), anion or cation exchange chromatography
(AEX, CEX), hydrophilic interaction liquid chromatography
(HILIC), hydrophobic interaction chromatography
(HIC), reversed-phase liquid chromatography (LC), and
size-exclusion chromatography (SEC).
Peaks can be transferred from one dimension to
the other dimension in either an on-line or an off-line
approach. Historically, off-line transfer has been the
method of choice and is still widely applied. With robust,
on-line 2D-LC instrumentation made commercially
available in recent years, however, on-line 2D-LC should
no longer be considered a specialist tool and is finding
its way to mainstream laboratories. This article focuses on
on-line 2D-LC.
In on-line 2D-LC one makes use of a first-dimension
pump, injector, column, and, optionally, a first-dimension
detector, which is combined with a second pump, a
second injector—now termed modulator, a second
column, and a detector. The aim of the modulator is to
efficiently transfer peaks from one dimension to the other
without compromising the separation (Figure 2). In cases
where the modulator transfers all first-dimension peaks to
the second dimension, this is known as comprehensive
2D-LC or LC×LC. The modulator is composed of a valve
with two loops installed (Figure 2[a]). While one loop
is being filled with peaks from the first dimension, the
second loop is being analyzed on the second dimension
column. This second-dimension separation is typically
very fast (in the order of 30 s) to facilitate a high sampling
frequency and maintain the first-dimension separation. In
cases where the modulator is programmed to transfer one
or a couple of peaks from the first dimension to the second
dimension, the method is termed (multiple) heart-cutting
2D-LC or (m)LC–LC. A modulator that allows one to store
up to 12 first dimension fractions is pictured in Figure 2(b).
Loops are then analyzed on the second dimension one
by one and the first and second dimension run times are
decoupled, which means that there is no requirement
to perform fast separations. The following section will
highlight how both modes are being applied in the analysis
of mAbs and ADCs.
(Multiple) Heart-Cutting LC–LC(m)LC–LC is typically applied for the analysis of intact
mAbs or large fragments thereof (for example, Lc, Hc, Fab,
Fc, F[ab’]2, Fc/2, Fd), which can be generated following
chemical reduction or enzymatic digestion, with the aim of
increasing resolution (for mAbs and ADCs, peaks typically
contain many variants) and allowing an in-depth product
characterization and to make separations compatible with
MS (11,12).
Birdsall et al. showed the potential of an on-line
HIC–reversed-phase LC–UV–MS setup for the
characterization of interchain cysteine conjugated ADCs
(14). HIC separates the ADC based on the number of
drugs conjugated for each antibody and as such allows
the drug-to-antibody ratio (DAR) and drug distribution
to be determined. HIC maintains mAbs in their native
state, so it is especially well suited for interchain cysteine
conjugated ADCs. The inherent problem of HIC is its
incompatibility with MS detection as a result of the use
of non-volatile salts. However, applying heart-cutting
2D-LC facilitates the combination of HIC and MS using
a reversed-phase LC desalting step prior to MS. The
denaturing reversed-phase conditions give rise to
dissociation of the ADC into the respective subunits and
as such this heart-cutting setup provides unambiguous
identification of positional isomers. The downside of the
described approach is that the setup only allows the
151www.chromatographyonline.com
BIOPHARMACEUTICAL PERSPECTIVES
Don’t buy any more your gas, you can produce it yourself !
F-DGSi is �UVW�)UHQFK�SURGXFHU�RI�WKH�ODERUDWRU\�JDV�JHQHUDWRUV��
)�'*6L��VLWXDWHG�LQ�WKH�3DULV�UHJLRQ��KDV�RSHQHG�LWV�PDQXIDFWXULQJ�SODQW�,*60�WR�
VDWLVI\�LWV�FOLHQWV�LQ�WHUPV�RI�TXDOLW\�DQG�LQQRYDWLRQ�RI�SURGXFWV�
7KH�YDOXHV�RI�)�'*6L
´�7KH�H[SHUWLVH��D�SDVVLRQ�WKDW�ZH�VKDUH
´�7KH�FOLHQWÊV�VDWLVIDFWLRQ��DQ�DEVROXWH�SULRULW\
´�7KH�LQQRYDWLRQ�LWV�SDUW�RI�RXU�'1$
´�7KH�TXDOLW\��DQ�HQJDJHPHQW�WKDW�SHUVLVW
´�7KH�GHVLJQ��EHFDXVH�WKH�SHUIRUPDQFHV�DUH�QRW�VXIILFLHQW
´�7KH�VHFXULW\��EHFDXVH�LW�EHORQJV�WR�DOO�RI�XV
&20(�72�',6&29(57KH�DOWHUQDWLYH�WR�WKH�+HOLXP�IRU�*&�DQG�*&06�
2XU�QLWURJHQ�JHQHUDWRUV�ZLWK�DQ�LQQRYDWLYH�
GHVLJQ�DQG�XQLTXH�FKDUDFWHULVWLFV
One step ahead !
ARABLAB DUBAI STAND 1006
ANALYTICA VIETNAM STAND C26
ES899870_LCE0317_151.pgs 03.06.2017 18:37 ADV blackyellowmagentacyan

second dimension reversed-phase
LC–MS analysis of one heart-cut,
even though the HIC chromatogram
is heavily populated with peaks.
Characterizing all peaks, therefore,
requires several re-injections.
This issue is alleviated in multiple
heart-cutting LC–LC where the
modulator design allows the storage
of multiple first dimension fractions.
Figures 3 and 4 show the mLC–LC
analysis of cetuximab (Erbitux).
Compared to other mAbs, which
have one N -glycosylation site in
the conserved region of the Hc,
cetuximab contains an additional
glycosylation site in the variable
region of the heavy chain (15).
Both the conserved and second
N -glycosylation site are occupied
with different types of sugars. While
the former contains the typical
mAb bi-antennary complex glycans
G0F, G1F, and G2F, the latter site is
occupied with a diverse mixture of
glycans containing a large amount
of α-(1J3) linked galactose residues
next to N -glycolylneuraminic (NGNA)
acid terminating glycans. This unique
glycosylation pattern stems from
the fact that these antibodies are
produced in murine SP2/0 opposed
to CHO cell lines. In this mLC–LC
example (Figures 3 and 4), IdeZ
digested cetuximab was analyzed
on the orthogonal combination
CEX–reversed-phase LC hyphenated
to UV and QTOF-MS detection.
An immunoglobulin-degrading
enzyme from Streptococcus equi
ssp zooepidemicus (IdeZ) cleaves
mAbs underneath the hinge region
thereby generating a F(ab’)2
and Fc/2 fragment and as such
separates out the two glycosylation
sites. Six CEX heart-cuts were
transferred to reversed-phase LC–
MS. Chromatographic conditions
are described in Sandra et al. (16).
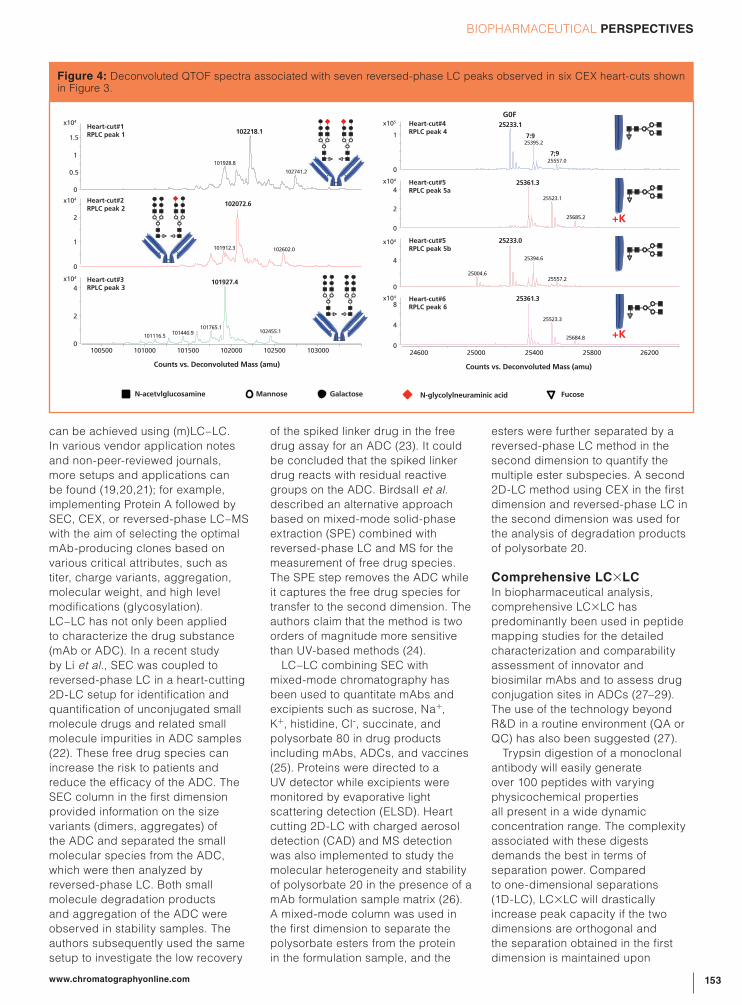
CEX peaks 1, 2, and 3 correspond
to the 100 kDa F(ab’)2 fragment and
from the corresponding MS data it is
shown that these peaks differ in their
NGNA degree with peak 1 carrying
NGNA terminating N -glycans on
both F(ab’)2 arms, peak 2 carrying
one NGNA terminating glycan on
one F(ab’)2 arm and a neutral glycan
on the other F(ab’)2 arm, and peak
3 carrying neutral glycans on both
arms. N -glycolylneuraminic acid
renders the protein more acidic,
which explains the earlier elution on
CEX. Peaks 4, 5, and 6 correspond
to the Fc/2 fragments and differ in
C-terminal lysine (K) truncation with
peak 6 carrying two lysine residues,
peak 5 one, and peak 4 being fully
truncated. Lysine is a basic amino
acid explaining the increased
retention time on CEX. Both Fc/2
fragments, which are no longer
connected via interchain disulphide
bridges, remain associated
under the native CEX conditions.
Denaturation, however, occurs during
reversed-phase LC–MS analysis,
which explains the detection of
25 kDa fragments using MS (Figure 4)
and the double peaks (5a: Fc/2 + K
and 5b: Fc/2 without K) observed
on reversed-phase LC (heart-cut 5,
Figure 3[b]).
An identical CEX–reversed-phase
LC(–UV)–MS setup has recently been
used to identify the main isoforms
of the mAb rituximab (17) and to
characterize the antibody–drug
conjugate (ADC) ado-trastuzumab
emtansine (16). Instead of storing
first dimension fractions in loops,
Alvarez et al. designed a setup that
allowed six peaks of interest from
a CEX or SEC separation to be
analyzed and identified by trapping
and desalting the fractions onto a
series of reversed-phase cartridges
with subsequent MS analysis (18).
An on-line disulphide reduction step
was furthermore incorporated into
the workflow, allowing more detailed
characterization of mAbs.
The examples described in this
section offer a small taste of what
2.0 3.0 4.0
0
100
200
300
400
500
2
3
15b
5a
64
15 17.5 20 22.5 25 27.5
Time (min)
(a)
(b)
Fc/2
F(ab’)2
Time (min)
mA
Um
AU
30 32.5 35 37.5
0
10
20
30
40
50
60
70
1
2
3
4
65
Figure 3: mLC–LC data of IdeZ treated cetuximab. CEX–UV (214 nm) chromatograms with (a) the heart-cuts and (b) the representative reversed-phase LC–UV (214 nm) chromatograms. Deconvoluted QTOF-MS spectra associated with the annotated peaks are shown in Figure 4. Fractions were transferred from one dimension to the other using the modulator shown in Figure 2(b). Experimental conditions can be obtained from reference 16.
-$r($�&VSPQF March 2017152
BIOPHARMACEUTICAL 1&341&$5*7&4
can be achieved using (m)LC–LC.
In various vendor application notes
and non-peer-reviewed journals,
more setups and applications can
be found (19,20,21); for example,
implementing Protein A followed by
SEC, CEX, or reversed-phase LC–MS
with the aim of selecting the optimal
mAb-producing clones based on
various critical attributes, such as
titer, charge variants, aggregation,
molecular weight, and high level
modifications (glycosylation).
LC–LC has not only been applied
to characterize the drug substance
(mAb or ADC). In a recent study
by Li et al., SEC was coupled to
reversed-phase LC in a heart-cutting
2D-LC setup for identification and
quantification of unconjugated small
molecule drugs and related small
molecule impurities in ADC samples
(22). These free drug species can
increase the risk to patients and
reduce the efficacy of the ADC. The
SEC column in the first dimension
provided information on the size
variants (dimers, aggregates) of
the ADC and separated the small
molecular species from the ADC,
which were then analyzed by
reversed-phase LC. Both small
molecule degradation products
and aggregation of the ADC were
observed in stability samples. The
authors subsequently used the same
setup to investigate the low recovery
of the spiked linker drug in the free
drug assay for an ADC (23). It could
be concluded that the spiked linker
drug reacts with residual reactive
groups on the ADC. Birdsall et al.
described an alternative approach
based on mixed-mode solid-phase
extraction (SPE) combined with
reversed-phase LC and MS for the
measurement of free drug species.
The SPE step removes the ADC while
it captures the free drug species for
transfer to the second dimension. The
authors claim that the method is two
orders of magnitude more sensitive
than UV-based methods (24).
LC–LC combining SEC with
mixed-mode chromatography has
been used to quantitate mAbs and
excipients such as sucrose, Na+,
K+, histidine, Cl-, succinate, and
polysorbate 80 in drug products
including mAbs, ADCs, and vaccines
(25). Proteins were directed to a
UV detector while excipients were
monitored by evaporative light
scattering detection (ELSD). Heart
cutting 2D-LC with charged aerosol
detection (CAD) and MS detection
was also implemented to study the
molecular heterogeneity and stability
of polysorbate 20 in the presence of a
mAb formulation sample matrix (26).
A mixed-mode column was used in
the first dimension to separate the
polysorbate esters from the protein
in the formulation sample, and the
esters were further separated by a
reversed-phase LC method in the
second dimension to quantify the
multiple ester subspecies. A second
2D-LC method using CEX in the first
dimension and reversed-phase LC in
the second dimension was used for
the analysis of degradation products
of polysorbate 20.
Comprehensive LC×LCIn biopharmaceutical analysis,
comprehensive LC×LC has
predominantly been used in peptide
mapping studies for the detailed
characterization and comparability
assessment of innovator and
biosimilar mAbs and to assess drug
conjugation sites in ADCs (27–29).
The use of the technology beyond
R&D in a routine environment (QA or
QC) has also been suggested (27).
Trypsin digestion of a monoclonal
antibody will easily generate
over 100 peptides with varying
physicochemical properties
all present in a wide dynamic
concentration range. The complexity
associated with these digests
demands the best in terms of
separation power. Compared
to one-dimensional separations
(1D-LC), LC×LC will drastically
increase peak capacity if the two
dimensions are orthogonal and
the separation obtained in the first
dimension is maintained upon
x104 x105
x104
x104
x104
x104
x104
0
0.5
1
1.5102218.1
101928.8
102741.2
0
1
2
102072.6
101912.3 102602.0
0
2
4101927.4
101765.1102455.1101440.9
101116.5
Counts vs. Deconvoluted Mass (amu) Counts vs. Deconvoluted Mass (amu)
N-acetvlglucosamine N-glycolylneuraminic acidMannose Galactose Fucose
100500 101000 101500 102000 102500 103000
Heart-cut#1RPLC peak 1
Heart-cut#2RPLC peak 2
Heart-cut#3RPLC peak 3
Heart-cut#4RPLC peak 4
Heart-cut#5RPLC peak 5a
Heart-cut#5RPLC peak 5b
Heart-cut#6RPLC peak 6
0
1
25233.1
25395.2
25557.0
0
2
425361.3
25523.1
25685.2
0
4
25233.0
25394.6
25004.625557.2
0
4
825361.3
25523.3
25684.8
24600 25000 25400 25800 26200
+K
+K
G0F
7:9
7;9
Figure 4: Deconvoluted QTOF spectra associated with seven reversed-phase LC peaks observed in six CEX heart-cuts shown in Figure 3.
153www.chromatographyonline.com
BIOPHARMACEUTICAL 1&341&$5*7&4

transfer to the second dimension.
Orthogonal combinations for
LC×LC-based peptide mapping
are strong cation exchange (SCX)
× reversed-phase LC, hydrophilic
interaction liquid chromatography
(HILIC) × reversed-phase LC, and
reversed-phase LC × reversed-phase
LC at different pHs in the two
dimensions (27). Note that in the
above combinations, reversed-phase
LC is always used in the second
dimension. The reasons for that
are the commercial availability of
high-quality, “fast” reversed-phase
LC second dimension columns and
their compatibility with MS. The
highest orthogonality is obtained
with SCX × reversed phase LC
and HILIC × reversed phase LC
because the separation mechanisms
are completely different. However,
reversed-phase LC × reversed-phase
LC is the preferred approach
because (i) it offers the highest
ruggedness from the excellent solvent
compatibility in both dimensions,
(ii) the peak capacity is very high as
a result of the high plate numbers
in the individual dimensions, and
(iii) orthogonality is acceptable as
a result of the zwitterionic nature of
peptides resulting in major selectivity
differences when performing
separations using reversed-phase LC
at pH extremes (pH 2 versus pH 10).
Vanhoenacker et al. recently
described the potential
of reversed-phase LC ×
reversed-phase LC combined with
UV and MS detection for the detailed
characterization and comparability
assessment of a trastuzumab innovator
(Herceptin) and candidate biosimilar
(27). A wealth of information was
provided in the peptide map allowing
identity, purity, and comparability to
be assessed. Striking similarities were
noticed when comparing the peptide
maps, but when looking in more detail
differences could be found and these
were shown to reside in modifications
(C-terminal lysine truncation,
asparagine deamidation).
To further illustrate the
attractiveness of LC×LC for detailed
characterization and comparability
assessment of mAbs, Figure 5
compares the reversed-phase LC
× reversed-phase LC tryptic peptide
maps of an infliximab originator
and candidate biosimilar for which
apparently, the recombinant
expression in Chinese Hamster
Ovary (CHO) cells was going
wrong. Both plots are very similar
but some striking differences are
noted when the MS data is taken into
consideration. The spots SLSLSPG
and SLSLSPGK clearly present in
the originator are replaced by one
spot SLSLSPGI in the biosimilar,
which according to the mass spectral
data corresponds to the addition
of an isoleucine (I) to SLSLSPG
or the replacement of lysine (K)
by isoleucine in SLSLSPGK at the
C -termini of the heavy chain.
The origin of the two spots,
SLSLSPG and SLSLSPGK, in the
originator mAb can be explained
by the knowledge that heavy
chains are historically cloned with
a C -terminal lysine but during
cell culture production, host cell
carboxypeptidases act on the
antibody resulting in the partial
removal of these lysine residues.
A very small retention shift was
also noted in another spot that
could be attributed to a threonine
to serine substitution in heavy
chain peptide NYYGS(TJS)
Figure 5: Reversed-phase LC × reversed-phase LC–QTOF-MS peptide mapping of infliximab originator and candidate biosimilar and drawing of the mAb with annotation of the modifications. First and second dimension separation consist of reversed-phase LC operated at high and low pH, respectively. Fractions were transferred from one dimension to the other using the modulator shown in Figure 2(a). First dimension peaks were sampled 3–4 times. Experimental conditions can be obtained from reference 16.
-$r($�&VSPQF March 2017154
BIOPHARMACEUTICAL 1&341&$5*7&4

YDYWGQGTTLTVSSASTK. Two
point mutations are at the origin of
this wrong recombinant expression
(Figure 5). These mutations were
confirmed with reverse transcription
polymerase chain reaction (RT-PCR)
and DNA sequencing. According
to US and European regulatory
authorities, identical primary
sequence is primordial to similarity,
thereby ruling out this candidate
biosimilar from further development.
Very recently, the possibilities of
LC×LC-based peptide mapping
in the characterization of ADCs
have been demonstrated (16). The
methodology was shown to be
particularly valuable to assess drug
conjugation sites. Figures 6(a) and
6(b) display the reversed-phase LC
× reversed-phase LC–MS tryptic
peptide maps of naked trastuzumab
(Herceptin) and ado-trastuzumab
emtansine (Kadcyla). The latter
combines trastuzumab with the
cytotoxic microtubule-inhibiting
maytansine derivative DM1
conjugated to lysine residues via
a non-reducible thioether linker.
Since Herceptin and Kadcyla have
the same amino acid sequence,
most of the peptide map is
identical. Differentiating spots are,
nevertheless, observed and are in
Figure 6: Reversed-phase LC × reversed-phase LC–MS peptide maps of (a) Herceptin and (b) Kadcyla and reversed-phase LC × reversed-phase LC all ions MS/MS peptide maps of (c,d) Herceptin and (d,f) Kadcyla showing extracted ion at m/z 547.22056 (extracted at 20 ppm mass accuracy). Collision energy was, respectively, 20 (c,d) and 40 eV (e,f). The identity of the annotated spots is shown in Table 1. First and second dimension separation consist of reversed-phase LC operated at high and low pH, respectively. Fractions were transferred from one dimension to the other using the modulator shown in Figure 2(a). First dimension peaks were sampled 3–4 times. Experimental conditions can be obtained from reference 16.
(a)
(c)
(e)
(b)
(d)
(f)
Table 1: A selection of conjugated peptides corresponding to the annotated spots in
the LC×LC peptide map shown in Figures 6(d) and (f). The conjugated lysine residues
are marked.
ChainSequence Location
Sequence
1 Lc 184–190 ADYEKHK
2 Hc 292–295 TKPR
3 Hc 413–419 LTVDKSR
4 Hc 222–251 SCDKTHTCPPCPAPELLGGPSVFLFPPKPK(1)
5 Hc 20–38 LSCAASGFNIKDTYIHWVR
6 Hc 305–325 VVSVLTVLHQDWLNGKEYKCK(1)
7 Hc 259–291 TPRVTCVVVDVSHEDPEVKFNWYVDGVEVHNAK
(1) Two potential conjugation sites.
155www.chromatographyonline.com
BIOPHARMACEUTICAL 1&341&$5*7&4

the upper right corner of the plot of
Kadcyla, which corresponds to the
most hydrophobic part. Since the
conjugation of the drugs makes the
peptides more hydrophobic, these
spots are expected to correspond
to the conjugated peptides. The
fragmentation spectra associated
with DM1 conjugated peptides are
heavily populated by fragments
originating from the drug, for
example, at m/z 547.2206. This
ion can be used to recognize
conjugated peptides in LC×LC–
MS maps. One can operate the
QTOF-MS system in the all ions MS/
MS mode, which means that the
quadrupole is operated in the RF
only mode thereby transferring all
peptides to the collision cell where
collision-induced dissociation takes
place.
As illustrated in Figures 6(c–f),
when extracting from the data
the DM1 specific fragment ion at
m/z 547.2206 at high mass accuracy,
the upper right corner of the 2D
peptide map lights up for Kadcyla
in comparison to Herceptin, making
this LC×LC methodology extremely
powerful to highlight conjugated
peptides. The latter plot is virtually
empty illustrating the selectivity that
is offered by this all ions MS/MS
functionality. Figures 6(c) and (d)
display the all ions MS/MS data
obtained at a collision energy of
20 eV, Figures 6(e) and (f) the data
at a higher collision energy of 40 eV.
Complementary information is
obtained when acquiring the data at
two different collision energy values.
A selection of identified conjugated
peptides is shown in Table 1. Since
the cytotoxic drug has a maximum
UV absorbance at 252 nm, this
wavelength can also be used to
confirm the positioning of conjugated
peptides in the peptide map as an
alternative to the all ions MS/MS
approach—evidently with a lower
specificity.
A similar strategy was applied
to reveal the conjugation sites on
brentuximab-vedotin (Adcetris),
the interchain cysteine conjugated
ADC. With only eight residues (four
on each half antibody) available for
conjugation, Adcetris is much simpler
compared to Kadcyla. Figure 7(a)
displays the reversed-phase LC
× reversed-phase LC–MS tryptic
peptide map of Adcetris. In analogy
to DM1 conjugated peptides,
MMAE conjugated peptides also
contain specific ions originating
from the cytotoxic molecule, that
is, at m/z 718.5113. As shown in
Figures 7(b) and (c), these ions can
be extracted from all ions MS/MS
data allowing conjugated peptides
in the 2D plots to be recognized. A
selection of identified conjugated
peptides is shown in Table 2. Of
particular interest is the detection
of conjugated intrachain cysteine
residues here exemplified with heavy
chain peptide NQVSLTCLVK (peak 3
in Figure 7[c]). These intrachain
residues are not targeted during
1
23
4
5
1
23
4
5
2D
, R
PLC
, 2
4s
2D
, R
PLC
, 2
4s
1D, RPLC, 45 min
(a)
(b)
(c)
1D, RPLC, 45 min
2D
, R
PLC
Figure 7: Reversed-phase LC × reversed-phase LC–MS peptide map of (a) Adcetris and (b,c) reversed-phase LC × reversed-phase LC all ions MS/MS peptide map showing extracted ion at m/z 718.5113 (extracted at 20 ppm mass accuracy). Collision energy was at 20 eV. (b) Full plot, (c) Zoomed intensified plot. The identity of the annotated spots is shown in Table 2. First and second dimension separation consist of reversed-phase LC operated at high and low pH, respectively. Fractions were transferred from one dimension to the other using the modulator shown in Figure 2(a). First dimension peaks were sampled 3–4 times. Experimental conditions can be obtained from reference 16.
-$r($�&VSPQF March 2017156
BIOPHARMACEUTICAL 1&341&$5*7&4

ADC manufacturing. Apparently the
TCEP reduction step applied to the
mAb prior to conjugation results
in collateral intrachain disulphide
bridge reduction next to interchain
reduction (the latter links are more
labile). The resulting free thiol groups
can then react with the protease
cleavable linker-MMAE intermediate.
These unwanted side products
are only detected at trace levels
and illustrate the sensitivity of the
presented methodology.
It is clear that LC×LC holds
great potential in peptide mapping.
Recently, this 2D-LC mode has been
used at the protein level to resolve
intact mAbs or large fragments
thereof. Sorensen et al. demonstrated
the use of CEX × reversed-phase
LC coupled to MS to compare mAb
originators and biosimilars following
IdeS digestion thereby generating
F(ab’)2 and Fc/2 fragments and
subsequent reduction generating Lc,
Fd’, and Fc/2 (30). 2D-LC enabled
direct identification of charge variants
separated by the CEX column by
indirect coupling of the separation
to MS detection through the second
dimension reversed-phase LC
separation. The authors concluded
that the richness of the data enables
facile assessment of the degree of
similarity between mAbs. In a series
of papers, Sarrut et al. described the
analysis of the interchain cysteine
conjugated ADC brentuximab-vedotin
using HIC × reversed-phase
LC combined with UV and MS
detection (31,32). This setup
simultaneously allowed the DAR and
the predominant positional isomers
to be determined. The authors
furthermore suggested the use of this
methodology in quality control when
operated in UV-only mode.
AcknowledgementThe authors acknowledge Sonja
Schneider and Udo Huber
(Agilent Technologies, Waldbronn,
Germany), Maureen Joseph
(Agilent Technologies, Wilmington,
USA), and our colleagues from the
biopharmaceutical industry.
3FGFSFODFT(1) K. Sandra, I. Vandenheede, and P.
Sandra, J. Chromatogr. A 1335, 81–103
(2014).
(2) S. Fekete, D. Guillarme, P. Sandra, and
K. Sandra, Anal. Chem. 88, 480–507
(2016).
(3) G. Walsh, Nat. Biotechnol. 32,
992–1000 (2014).
(4) S. Panowski, S. Bhakta, H. Raab, P.
Polakis, and J.R. Junutula, mAbs 6,
34–45 (2014).
(5) A. Wakankar, Y. Chen, Y. Gokarn, and
F.S. Jacobson, mAbs 3, 161–172 (2011).
(6) A. Beck and J.M. Reichert, mAbs 6,
15–17 (2014).
(7) K. Sandra, I. Vandenheede, E.
Dumont, and P. Sandra, “Advances in
Biopharmaceutical Analysis” supplement
to LCGC Europe, Volume 28, Number
s10, October 2015.
(8) A. Beck and J.M. Reichert, mAbs 5,
621–623 (2013).
(9) A. Beck, E. Wagner-Rousset, D.
Ayoub, A. Van Dorsselaer, and S.
Sanglier-Cianférani, Anal. Chem. 85,
715–736 (2013).
(10) I. François, K. Sandra, and P. Sandra,
Anal. Chim. Acta 641, 14–31 (2009).
(11) K. Sandra and P. Sandra, Bioanalysis 7,
2843–2847 (2015).
(12) D. Stoll, J. Danforth, K. Zhang, and A.
Beck, J. Chromatogr. B 1032, 51–60
(2016).
(13) D. Stoll and P.W. Carr, Anal. Chem. 89,
519–531 (2017).
(14) R.E. Birdsall, H. Shion, F.W. Kotch, A.
Xu, T.J. Porter, and W. Chen, mAbs 7,
1036–1044 (2015).
(15) J. Qian, T. Liu, L. Yang, A. Daus, R.
Crowley, and Q. Zhou, Anal. Biochem.
364, 8–18 (2007).
(16) K. Sandra, G. Vanhoenacker, I.
Vandenheede, M. Steenbeke, M.
Joseph, and P. Sandra, J. Chromatogr. B
1032, 119–130 (2016).
(17) D.R. Stoll, D.C. Harmes, J. Danforth, E.
Wagner, D. Guillarme, S. Fekete, and
A. Beck, Anal. Chem. 87, 8307–8315
(2015).
(18) M. Alvarez, G. Tremintin, J. Wang, M.
Eng, Y.H. Kao, J. Jeong, V.T. Ling, and
O.V. Borisov, Anal. Biochem. 419, 17–25
(2011).
(19) S.M. McCarthy, T.E. Wheat, Y.Q. Yu, and
J.R. Mazzeo, Waters Application Note;
720004304EN; (2012).
(20) S. Schneider, Agilent Technologies
Application Note; 5991-6673EN; (2016).
(21) E. Largy, A. Catrain, G. Van Vyncht,
and A. Delobel, Current Trends in Mass
Spectroscopy 14(2), 29–35 (2016).
(22) Y. Li, C. Gu, J. Gruenhagen, K. Zhang,
P. Yehl, N.P. Chetwyn, and C.D. Medley,
J. Chromatogr. A 1393, 81–88 (2015).
(23) Y. Li, C. Stella, L. Zheng, C. Bechtel,
J. Gruenhagen, F. Jacobson, and C.D.
Medley, J. Chromatogr. B 1032, 112–118
(2016).
(24) R.E. Birdsall, S.M. McCarthy, M.C.
Janin-Bussat, M. Perez, J.F. Haeuw, W.
Chen, and A. Beck, mAbs 8, 306–317
(2016).
(25) Y. He, O.V. Friese, M.R. Schlittler, Q.
Wang, X. Yang, L.A. Bass, and M.T.
Jones, J. Chromatogr. A 1262, 122–129
(2012).
(26) Y. Li, D. Hewitt, Y. Lentz, J. Ji, T. Zhang,
and K. Zhang, Anal. Chem. 86,
5150–5157 (2014).
(27) G. Vanhoenacker, I. Vandenheede, F.
David, P. Sandra, and K. Sandra, Anal.
Bioanal. Chem. 407, 355–366 (2015).
(28) G. Vanhoenacker, K. Sandra, I.
Vandenheede, F. David, P. Sandra,
U. Huber, and E. Naegele, Agilent
Technologies Application Note;
5991-2880EN; (2013).
(29) G. Vanhoenacker, K. Sandra , I.
Vandenheede, F. David, P. Sandra,
and U. Huber, Agilent Technologies
Application Note; 5991-4530EN;
(2014).
(30) M. Sorensen, D.C. Harmes, D.R. Stoll,
G.O. Staples, S. Fekete, D. Guillarme,
and A. Beck, mAbs 8, 1224–1234 (2016).
(31) M. Sarrut, A. Corgier, S. Fekete,
D. Guillarme, D. Lascoux, M.C.
Janin-Bussat, A. Beck, and S.
Heinisch, J. Chromatogr. B 1032,
103–111 (2016).
(32) M. Sarrut, S. Fekete, M.C. Janin-Bussat,
O. Colas, D. Guillarme, A. Beck, and S.
Heinisch, J. Chromatogr. B 1032, 91–102
(2016).
Koen Sandra is Scientific Director
at the Research Institute for
Chromatography (RIC, Kortrijk,
Belgium).
Isabel Vandenheede is a Protein
Analyst at RIC.
Mieke Steenbeke is LC Lab Technician
at RIC.
Gerd Vanhoenacker is LC Product
Manager at RIC.
Pat Sandra is President of the RIC and
Emiritus Professor at Ghent University
(Ghent, Belgium).
Table 2: A selection of conjugated peptides corresponding to the annotated spots
in the LC×LC peptide map shown in Figure 7(b) and (c). The conjugated cysteine
residues are underlined. Payload: MMAE + linker. Carbamidomethylation results from
alkylation of free cysteine residues with iodoacetamide.
ChainSequence Location
Sequence
1 Lc 216-218 GEC + payload
2 Hc 219-222 SCDK + payload
3 Hc 361-370 NQVSLTCLVK + payload
4 Hc 223-248THTCPPCPAPELLGGPSVFLFPPKPK + 1* payload + 1*
carbamidomethylation
5 Hc 223-248 THTCPPCPAPELLGGPSVFLFPPKPK + 2* payload
157www.chromatographyonline.com
BIOPHARMACEUTICAL 1&341&$5*7&4

![Monoclonal Antibodies - Copy [Autosaved]](https://cdn.vdocuments.site/doc/165x107/577c7e6a1a28abe054a109e9/monoclonal-antibodies-copy-autosaved.jpg)





