docket # fda-2007-d-0369 - informa/media/images/publications/archi… · docket # fda-2007-d-0369...
TRANSCRIPT

13 September 2010
Division of Dockets Management (HFA-305) RLD Application Number: 21346 Docket # FDA-2007-D-0369 5630 Fishers Lane Room 1061 Rockville, MD 20852 Dear Sir or Madam: On behalf of Ortho-McNeil-Janssen Pharmaceutical, Inc., Johnson and Johnson Pharmaceutical Research and Development (J&JPRD) is providing the following recommendations and comments with regard to the FDA draft guidance entitled, “Guidance on Risperidone,” dated February 2010, for the intramuscular injection. J&JPRD appreciates the opportunity to submit these comments as appropriate studies and metrics to evaluate bioequivalence will be critical to ensuring the safety and efficacy of substitutes for the currently approved intramuscular risperidone formulation, submitted under an ANDA. The comments below focus on overall issues relating to the assessment of bioequivalence for intramuscular (i.m.) long-acting injectable drug products (LAIs), in general, and risperidone in particular. Included are eleven detailed comments, which address specific sections within the draft Guidance on risperidone or provide recommendations for addressing additional issues that are not covered in the draft Guidance. 1. BACKGROUND: THE IMPORTANCE OF GUIDELINES FOR EVALUATING
BIOEQUIVALENCE OF IM LAIs Bioequivalence (BE) studies have, of course, been routinely performed for oral drug products to evaluate new formulations during the development phase or to implement post-approval changes. BE trials are used primarily to infer therapeutic equivalence of different formulations in terms of safety and efficacy and, therefore, to guarantee switchability between these formulations. For two orally administered drug products to be bioequivalent, the active drug ingredient or active moiety in the Test drug product must exhibit comparable rate and extent of absorption as the Reference drug product. Most BE trials rely on the standard metrics of Cmax and AUC to evaluate different formulations, but both FDA and the broader scientific and medical community have recognized the importance of relying on additional metrics and studies to evaluate bioequivalence for particular types of products. In contrast to oral drug products, guidelines outlining the design of bioequivalence programs for intramuscular (i.m.) long-acting injectables (LAIs) are not available. Nevertheless, specific recommendations to govern the design of a suitable bioequivalence program are critical for LAI antipsychotics because of their complex release and pharmacokinetic characteristics; 1,2 subject

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 2 of 15
to a complex interplay between the drug, the excipients and the injection site; and the significant patient safety and tolerability issues related directly to the early or immediate release of drug product. In the absence of formal guidelines for i.m. LAIs, the following regulatory references are used as a basis to comment on the draft FDA Guideline on intramuscular risperidone.3
• FDA Guidance for Industry. Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. March 2003.4
• FDA bioequivalence recommendations for Specific Products (released from 2007 onwards).
• FDA Guidance for Industry Bioequivalence Recommendations for Specific Products, June 2010.5
• Minutes and transcript for the April 13, 2010 Meeting of the Pharmaceutical Science and Clinical Pharmacology Advisory Committee.6
• CPMP/EWP/QWP/1401/98 Rev 1 (January 20, 2010): Guideline on the investigation of bioequivalence.7
• CPMP/EWP/280/96 Corr*, July 28 1999. Note for guidance on quality of modified release oral and transdermal dosage forms: Section II (pharmacokinetic and clinical evaluation).8
2. RECOMMENDATIONS FOR THE “GUIDANCE ON RISPERIDONE,” DATED
FEBRUARY 2010 2.1. Study Design: Fasting The draft Guidance for i.m. risperidone recommends drug administration under fasting conditions.3 The Company does not consider the requirement to administer the drug under fasting conditions to be essential because of the i.m administration route. Proposed Change The Company proposes to delete this requirement from the Guidance. 2.2. Study Design: Single-dose The draft Guidance for i.m. risperidone proposes a single-dose two-way crossover study design. 3 Single-dose (SD) BE study designs are generally recommended for both immediate-release and modified-release drug products because they are generally more sensitive in assessing the release of the drug substance from the drug product in the systemic circulation and in detecting differences in release/absorption rate.4 A thorough assessment of the release is especially important for LAI risperidone for at least two reasons. First, because of the high drug load, there are risks related to early or immediate release of risperidone intended to be released over several weeks (dose dumping). Second, the drug release rate governs both the initial PK profile and the apparent half-life (flip-flop kinetics) and, as a consequence, determines the onset of action as

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 3 of 15
well as key steady-state PK characteristics (e.g. Cmin,ss, Cmax,ss and the peak/trough ratio). Therefore, conducting a single-dose study is essential to assure bioequivalence. Specifically, the release of risperidone from RISPERDAL® CONSTA® is characterized by a lag time (Tlag) before the main release occurs after a single i.m. (gluteal) injection of RISPERDAL® CONSTA®. Thus, there is only a small, initial release of the drug (< 1% of the dose) followed by a lag time of 3 weeks before the main release of the drug starting from the end of Week 3 onwards. Therefore, oral supplementation is needed during the first 3 weeks to ensure sufficient antipsychotic coverage. That main release is maintained from weeks 4 to 6 and then subsides fairly rapidly by 7 weeks after the i.m. injection. RISPERDAL® CONSTA® has an apparent half-life of 3-6 days.1 Due to the unique PK profile of RISPERDAL® CONSTA® with the 3-week lag time (i.e. at least as long as the duration of the main release phase) and a relatively short (relative to the 2-week dosing interval) apparent half-life of 3-6 days, small changes in initial release or a slightly earlier start of the main release phase may cause significant changes in the duration of the oral supplementation required and on the PK parameters at steady-state (Cmin,ss, Cmax,ss and/or peak/trough ratio). Therefore, reliance solely on Cmax and AUClast in a SD study to ensure bioequivalence would not allow one to infer bioequivalence at steady-state and to ensure switchability. For instance, if a patient was receiving RISPERDAL® CONSTA® and was switched to a new risperidone long acting injection (LAI) that has a lag time of less than 3 weeks, then there would be additive exposure from the previous RISPERDAL® CONSTA® injections and the new risperidone injection resulting in potential adverse events. Similarly, if a patient was receiving RISPERDAL® CONSTA® and was switched to a new risperidone long acting injection that has a lag time of more than 3 weeks, then the minimal plasma exposure, approximately 6-7 weeks after the last RISPERDAL® CONSTA® dose, would be lower than the minimal plasma concentration during treatment with RISPERDAL® CONSTA®, potentially resulting in a higher risk for relapse. To address this problem, the guidelines should call for an assessment of the PK profile during the initial release phase; that is, the plasma concentration-time profile prior to Tmax should be comparable for the Test and Reference drug products (see section 2.4. PK Parameters to be Assessed by Single Dose: Tlag, Tmax, AUClast,, Partial AUC). Proposed Change No change. The Company agrees that conducting a single-dose study should always be required. 2.3. Study Design: Multiple-dose The draft Guidance for i.m. risperidone does not propose a cross-over multiple-dose (MD) bioequivalence or switching study design. Due to the unique PK profile of RISPERDAL® CONSTA® with the 3-week lag time and the relatively short apparent half-life (see above), small changes in initial release or a slightly earlier start of the main release phase may cause significant changes in the PK parameters at steady-state (Cmin,ss, Cmax,ss and/or peak/trough ratio). Therefore, reliance solely on Cmax and AUClast in a SD study to ensure bioequivalence would not allow one

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 4 of 15
to infer bioequivalence at steady-state and to ensure switchability. In order to ensure therapeutic equivalence and infer similar safety and efficacy between such formulations at steady-state, a multiple dose (MD) study is considered essential in addition to the single-dose (SD). Because of the complex PK profile (i.e. initial release, lag time, main burst, rapid decline) and because small changes in initial release or a slightly earlier start of the main release phase may cause significant changes in the PK parameters (Cmin,ss, Cmax,ss and/or peak/trough ratio) upon switching from Reference to Test drug product or vice versa, a cross-over multiple-dose switching study demonstrating switchability (test-to-reference and reference-to-test) at steady-state should be required in addition to the single-dose (SD) in order to ensure similarity in exposure and equivalence in terms of safety and efficacy profile upon switching from Reference to Test drug product or vice versa. This is recognized in the general FDA Bioequivalence Guidance, 4 stating that, for a modified-release drug product submitted as an ANDA, it must be demonstrated that the Test drug product’s steady-state performance is equivalent to the currently marketed non-controlled release or controlled-release drug product that contains the same active drug ingredient or therapeutic moiety and that is subject to an approved full NDA. In the European Union, for demonstration of bioequivalence of suspensions intended to delay or prolong the release of the active substance for i.m. or s.c. administration, the rules for extra vascular modified release (MR) formulations7, e.g. transdermal drug delivery systems (TDDS) apply and bioequivalence is recommended to be assessed after both SD and MD administration. Since antipsychotic medications are typically administered for chronic treatment, it is important to completely characterize the Test drug product’s safety and tolerability profile over a period of time. Not all treatment emergent adverse events occur in the first few days of treatment; therefore conducting a MD study will allow evaluation of safety and tolerability (including local tolerability and injection site reactions) over several injection cycles, thereby enabling a better understanding of the safety and tolerability profile of the Test drug product. A complex interplay exists between the drug (product), excipients, and injection site. Based on our experience with other long acting injectable antipsychotics, parameters such as site of injection (e.g. gluteal vs deltoid muscles), particle size, volume, and concentration can affect the active substance release rate. When altering the composition and/or manufacturing process or equipment, small changes may result in an altered initial release as observed in pilot formulations of long acting injectable risperidone9, potentially increasing the risk of adverse events or relapse. 2.3.1. Adverse Events Associated with Early Release of Medication: Adverse events that can be worsened if early or immediate release occurs include but may not be limited to:
a) Extrapyramidal symptoms (EPS). EPS such as acute dystonia, parkinsonism, and akasthisia are important safety and tolerability issues that are related to both higher and

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 5 of 15
fluctuating plasma concentrations of typical and, to a lesser extent, atypical antipsychotic drugs.10 These symptoms may occur acutely in response to higher plasma concentrations, and there is evidence to suggest that acute EPS is a risk factor for tardive dyskinesia (a syndrome of involuntary dyskinetic movements, which are potentially irreversible).11,12 While it is difficult to predict the occurrence of EPS symptoms for any individual patient, on a population level, D2 receptor occupancy levels >80% have been associated with higher rates of EPS. 13,14,15,16 Studies have shown that EPS symptoms have a steep concentration-response profile for risperidone: around an EC50 in plasma of approximately 32 ng/mL for the active moiety (sum of risperidone and active metabolite), the risk to develop EPS increases significantly, which is comparable with the data observed for paliperidone showing an increase in EPS incidence at concentrations between 20 and 40 ng/mL.17 In comparison with daily immediate-release oral therapy, long-acting injectable risperidone is associated with reduced peak blood levels and decreased plasma peak-to-trough ratios, and it is believed that these sustained release characteristics help to maintain optimal dopamine receptor occupancy, thereby reducing associated side effects.9,18 However, even with these sustained release characteristics, a dose-related increase in the incidence of EPS-related adverse events has been observed with up to 75 mg long-acting injectable risperidone over a 12-week double-blind period.19 Therefore, early release of risperidone over multiple injection cycles, leading to unexpectedly high and fluctuating plasma concentrations and D2 receptor occupancy, could potentially increase the risk of EPS-related adverse events and the complications associated with these events. (b) Sedation and somnolence. These events are often observed early in the course of antipsychotic treatment and tend to be dose-related. 20,21 Sedation and somnolence can be significant issues leading to a potential for cognitive and motor impairment, falls (with resultant injuries), sleep cycle disruption, and inability to participate in normal activities, including impairments in work, study, or social functioning. These events are of particular concern in elderly patients who are more prone to adverse consequences from falls, such as bone fractures, injuries, functional decline, dependency, and death.22 In addition, these effects can be dose limiting, and can also be a cause of poor treatment adherence, which is the most common cause of relapse for patients with schizophrenia and bipolar disorder.19,20 Risperidone has high affinity for α1 adrenoceptors, which may partially regulate somnolence and sedation.23 The incidence of somnolence with long-acting injectable risperidone over a 12-week double-blind period was similar for the 25 mg (5%) and 50 mg (6%) dose groups, and further increased in the 75 mg dose group (10%) versus placebo (3%).19 Therefore, early release of risperidone over multiple injection cycles, leading to unexpectedly high plasma concentrations, could potentially increase the risk of sedation or somnolence and the complications associated with these events. (c) Orthostatic hypotension. Long-acting injectable risperidone may induce orthostatic hypotension associated with dizziness, tachycardia, and in some patients, syncope, especially during the initial dose-titration period with oral risperidone, probably reflecting its alpha-adrenergic antagonistic properties.1 These events are also of particular

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 6 of 15
concern in elderly patients.22 If managed with careful dose adjustment, patients can become partially or fully tolerant to this adverse effect.21 Therefore, early release of risperidone over multiple injection cycles, leading to unexpectedly high and fluctuating plasma concentrations, could potentially increase the risk of orthostatic hypotension and the complications associated with these events. d) QTc Prolongation. An undesirable property of some non-antiarrhythmic drugs is their ability to delay cardiac repolarization, an effect that can be measured as prolongation of the QT interval on the surface ECG. A delay in cardiac repolarization creates an electrophysiological environment that favors the development of cardiac arrhythmias, most clearly Torsade de Pointes. Torsade de Pointes can degenerate into ventricular fibrillation, potentially leading to sudden death.24 QTc prolongation has been identified as a potential safety concern with several atypical antipsychotics.19,20 QTc prolongation has been observed in overdoses of both oral risperidone and paliperidone. 1,25 One case study of risperidone overdose describes correlation of serum drug concentration with QTc prolongation, which corrected as exposure to the drug fell.26 There is a theoretical risk that early release of risperidone over multiple injection cycles, leading to unexpectedly high plasma concentrations, could potentially increase the risk of QTc prolongation. This risk factor is of particular importance for patients with a history or presence of other risk factors for a prolongation of the QTc interval, such as specific cardiac disorders (sick sinus syndrome, complete AV block, congestive heart failure, polymorphic ventricular tachycardia and congenital prolongation of the QT interval), clinically relevant hypocalcaemia, hypokaelemia, or hypomagnesaemia, or concomitant use of other medications that prolong the QTc interval (particularly Class I or Class III antiarrhythmics).20,21 2.3.2 Adverse Events Associated with Delayed Release of Medication:
In case of a longer lag time for the Test drug product compared to the Reference product, a decreased Cmin,ss would be observed upon switching, potentially increasing the risk of relapse. Relapse in schizophrenia is common and often extremely serious, with significant consequences for the patient and his/her support network. Episodes of relapse or even exacerbation of symptomatology, which can occur abruptly, are often the result of poor treatment adherence leading to lower plasma concentrations of the medication. There may be a recurrence of both positive and negative symptoms of the disorder. Positive symptoms may include but are not limited to hallucinations, delusions, disorganization of thought and behavior, and aggressive or even violent outbursts, Negative symptoms are generally characterized by increasing social withdrawal, flattening of affect, impairments in attention and anhedonia, or a loss of interest in usual activities. There may also be an accompanying deterioration in cognitive functioning. Obviously, all of these symptoms can impair social and vocational functioning to a significant degree, lead to increased caregiver burden, as well as an increased risk of suicidal and violent behaviors. Successive relapses can result in reduced periods of subsequent remission, with many patients unable to regain their previous level of functionality and cognitive capacity after each

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 7 of 15
episode.27,28 With each successive relapse, patients experience an increase in the severity of their symptoms and the progression of their illness moves them further away from recovery.29 About 60% to 75% of patients with schizophrenia relapse within 1 to 2 years without antipsychotic medication; even with oral medications this rate is still over 40% due in large part to intermittent treatment adherence.30 Continuous medication, with maintenance of stable concentration of drug, has been shown to reduce this risk further to between 20% and 30%. This is an important factor to be considered when switching a patient’s treatment from RISPERDAL® CONSTA® to another long acting injectable risperidone medication. Bipolar disorder affects up to 5% of the population.31,32 Typically, functioning and well being are impaired even after symptomatic recovery, meaning bipolar disorder is a cause of significant disability, morbidity, and mortality and is one of the leading causes of disability in young adults. Education and career development are impeded. Relationships with family and friends are adversely affected. Self-image can be devastated. Direct and indirect health care costs of bipolar disorder are considerable—estimated to be $45 billion per year in the U.S.33 The natural history of bipolar disorder is characterized by relapses and recurrences. More than 90% of individuals who have a single manic episode will have future episodes, and 10% to 15% of patients will have more than 10 episodes in their lifetime.34 Consequently, bipolar disorder is increasingly recognized as a chronic recurrent illness that requires long-term maintenance treatment following alleviation of the acute symptoms of the disease. Much as with the diagnosis of schizophrenia, as described above, there is a high rate of poor treatment adherence, hence the potential benefits of treatment with RISPERDAL® CONSTA®, the long-acting injectable formulation, have been significant in reducing the rates of recurrent episodes. These relapses, whether into acute mania, depression, or mixed mania, are often associated with severe impairments in social and occupational functioning. Particularly if patients are well stabilized on a given medication, any change of treatment, with consequent change in the plasma levels of active medication, can increase the risk of such relapses. A change from RISPERDAL® CONSTA® to an alternative long acting injectable risperidone could have the potential to precipitate such relapses during the transition period, and there is of course no guarantee that with an alternative formulation, the same degree of efficacy and safety can be maintained. For these reasons, it would be critical to establish bioequivalence in the evaluation of any potential alternative version formulation of long acting injectable risperidone, prior to approving its use as a substitute for RISPERDAL® CONSTA® in the treatment of either schizophrenia or as a maintenance treatment for bipolar disorder. A MD BE or switching study provides the opportunity to establish the tolerability and safety profile of the Test drug product, which is considered especially important when differences in lag time between Reference and Test drug product can result in sudden increases or decreases in systemic exposure upon switching.

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 8 of 15
Proposed Change In addition to a SD study, the Company considers a MD (BE or switching) study essential to guarantee bioequivalence and infer therapeutic equivalence in terms of safety/tolerability and efficacy for i.m. risperidone LAIs. 2.4. PK Parameters to be Assessed by Single Dose: Tlag, Tmax, AUClast, Partial AUC The draft Guidance for i.m. risperidone does not specify the PK parameters to be evaluated. To demonstrate bioequivalence in terms of rate and extent of absorption, Cmax and AUClast have generally been considered appropriate PK parameters. However, due to the importance of the 3-week lag time in view of the steady-state PK profile, it is critical to define maximum criteria for early exposure measures to demonstrate similarity of the pharmacokinetics during the initial release phase. Accordingly, the guidance should add criteria based on the lag time recognized in the approved labeling, Tlag, and time to peak exposure, Tmax. The use of statistical criteria based on Tlag and Tmax in a single dose study is essential to ensuring bioequivalence at steady-state. The partial AUC is also considered an essential and adequate PK parameter to ensure that the early release of risperidone does not exceed maximum criteria. This approach is becoming more accepted as a means to demonstrate early onset of action6, and would be equally valuable in defining maximum criteria in terms of early exposure and ensuring an adequate safety profile of substitutes for the currently approved i.m. risperidone formulation, submitted under an ANDA.
At the recent FDA Advisory Committee meeting, it was stated that partial AUCs would be required by FDA for multiphasic modified-release (MR) drug products for which the Test and Reference drug products may not be therapeutically equivalent (switchable), despite being deemed bioequivalent when the traditional metrics (Cmax and AUC) were compared. As described above, this is considered to be the case for i.m. risperidone.
Criteria for the partial AUC measure can be derived based on the historical information obtained for different LAI risperidone formulations.9 Because of the complex interplay between the drug, the excipients and the injection site environment, the drug release rate can be significantly affected by small changes in residual solvent or excipients, mediated by physiological reactions (see below). Although such an increase in release rate, mediated by a physiological response is unpredictable by in vitro dissolution experiments, the currently approved in vitro dissolution specifications are associated with partial AUC values of ≤ 2.5% on Day 1 and ≤ 40% on Day 24 and defining these values as maximum criteria for the partial AUC values at these critical time points would ensure equivalent initial and early release by any proposed substitute for the currently approved i.m. risperidone formulation, submitted under an ANDA.
When comparing the bioavailability of immediate-release (IR) Test and Reference formulations, the 90% confidence interval for the ratio of AUCt (or truncated AUC72h for compounds with a long half-life) and Cmax for the Test and Reference drug products should be contained within the 80 to 125% acceptance interval. 4 However, the terminal half-life of LAI antipsychotics is not driven by their elimination rate but driven by the slow release rate of drug from the formulation (flip-flop kinetics). The absorption phase continues for a long period of time (several months for RISPERDAL® CONSTA®) after injection. 1,2 Truncation to a time point when the drug release is not complete would not allow characterization of the entire release/absorption phase in spite of

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 9 of 15
the objectives of a BE study to demonstrate bioequivalence in terms of rate and extent of drug release. Moreover, because the drug release of i.m. risperidone depends on aspects such as polymer composition of the formulation and the excipients and residual solvents, or may be affected (dependent on the manufacturing technology) by particle size, particle size distribution or injection site, as observed for other LAI antipsychotics, these active pharmaceutical ingredient (API) and drug product related characteristics may affect the drug release throughout the entire release period and, therefore, truncation to a time point when the drug release is not complete is considered unacceptable. For risperidone LAI, AUCt should capture at least a period of 85 days.35,36,37.
Proposed Change The Company recommends the Guidance specify Cmax, AUClast, Tlag, and Tmax and partial AUC (until Day 1 and Day 24) as the PK parameters to be evaluated in the single-dose study. The Company also proposes to recommend in the Guidance that, for a risperidone LAI, AUCt should capture at least a period of 85 days. 2.5. PK Parameters to be Assessed via Multiple-Dose: Cmin,ss, Cmax,ss, AUCss The company considers a MD BE study (a BE study at steady-state and/or a cross-over switching study at steady-state) essential in order to guarantee equivalence of Reference and Test drug products at steady-state and assure switchability between formulations as previously discussed. The key PK parameters to be assessed in a MD study are well defined: Cmin,ss, Cmax,ss, AUCss. Because of the complex PK profile and because small changes in initial release or a slightly earlier start of the main release phase may cause significant changes in the PK parameters (Cmin,ss, Cmax,ss and/or peak/trough ratio) upon switching from Reference to Test drug product or vice versa, a cross-over multiple-dose switching study demonstrating switchability (test-to-reference and reference-to-test) at steady-state should be required in order to ensure similarity in exposure and equivalence in terms of safety and efficacy profile upon switching from Reference to Test drug product or vice versa. The key PK parameters to be assessed in a MD switching study are the same: Cmin,ss, Cmax,ss, AUCss.
Proposed Change The key PK parameters to be assessed in a MD study are well defined: Cmin,ss, Cmax,ss, AUCss. The company recommends FDA specify the requirement to demonstrate BE for the steady-state PK parameters Cmin,ss, Cmax,ss, and AUCss in the Guidance. Safety and tolerability must also be carefully assessed. 2.6. Strengths: 25 mg and 50 mg The draft Guidance for i.m. risperidone proposes a single-dose study at 25 mg, accepting, conditionally, waivers for 12.5, 37.5 and 50 mg strengths. 3 The guidelines on the Investigation of Bioequivalence, 4 state that it may be sufficient to establish bioequivalence at only one or two strengths, provided that the strengths are proportionally

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 10 of 15
similar in composition, and appropriate comparative dissolution data are available. In the case of linear PK (AUC-based) a biowaiver can be conditionally granted for all lower strengths (criteria specified in the applicable guideline). A waiver for the 12.5, 37.5 and 50 mg doses based on the BE data obtained for the 25 mg strength conflicts with other countries’ guidance for modified release products8 and is not considered a valid approach as the dissolution rate (and therefore the apparent half-life) may depend on the dose as observed for other LAIs.34 For drugs, such as LAIs, with non-linear pharmacokinetics characterized by a less than dose-proportional increase in PK parameters, caused by limited solubility, BE should be demonstrated at the most critical dose, i.e. the highest strength. The kinetics of risperidone, after oral administration of risperidone and after i.m. administration of RISPERDAL® CONSTA® are dose-proportional in the 12.5-50 mg range. However, because of the complex interplay between the drug (product), excipients and injection site, the dose-proportionality may be dependent on characteristics other than the intrinsic physicochemical properties of the API, e.g. chemical composition of the polymer, excipients and residual solvents or particle size (distribution) and injection site, which may affect the PK of the drug (dependent on the applied manufacturing technology) as observed for other antipsychotics. Therefore, the draft Guidance for i.m. risperidone should also call for a demonstration of BE at least at the lowest and the highest strength. Biowaivers may be acceptable for the intermediate strengths of LAI antipsychotics provided that both: BE is minimally demonstrated at the highest and the lowest strength and dose-proportionality for the Test Formulation is demonstrated for all strengths. Proposed Change The Company recommends requiring demonstration of BE at least at the lowest and the highest strength. 2.7. Waiver Request of In-vivo Testing subsection (i) Due to the difference in adipose tissue between the deltoid and gluteal muscle, the drug release rate from the Test drug product may depend (based on the manufacturing technology) on the injection site as observed for other antipsychotics. Although this was not observed for RISPERDAL® CONSTA®, similar effects may apply for other LAI risperidone formulations, dependent on the manufacturing technology. Proposed Change The Company proposes that the Guidance recommends the evaluation of bioequivalence of Reference and Test drug product for both deltoid and gluteal injection sites. 2.8. Waiver Request of In-vivo Testing subsection (ii) In the case of a Test drug product demonstrating a dose-proportional increase in AUC but a less than dose-proportional increase in Cmax, reflecting a dose-dependent release rate, the draft

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 11 of 15
Guidance for i.m. risperidone does not recommend at which dose(s) bioequivalence should be demonstrated. Proposed Change In this situation the Company recommends requiring demonstration of BE at the lowest and the highest strength, provided that the different strengths are proportionally similar. 2.9. Waiver Request of In-vivo Testing subsection (iii) The draft Guidance for i.m. risperidone proposes a single-dose study at 25 mg, accepting waivers of 12.5, 37.5 and 50 mg strengths: a waiver is considered conditionally acceptable, one of the conditions being to establish an acceptable in vitro dissolution testing. 3 In addition to the intrinsic release characteristics of the drug product, physiological aspects may mediate the in vivo release. Based on the observations with pilot formulations of LAI risperidone that small changes in amounts of residual solvents or excipients can significantly impact the in vivo release and cause an early release of drug, the biorelevance of a dissolution test may depend on these excipients or residual solvents. Early or immediate release of a large dose of medication intended for a period of 2 weeks may potentially be an important safety issue for patients. Because an increase in release rate, mediated by a physiological response, is unlikely to be predicted by most in vitro dissolution methods, the Company strongly recommends specifying in the Guidance how the biorelevance of the in vitro dissolution test should be demonstrated. When changes to the composition of the drug product or to the manufacturing process/equipment are implemented compared to the original drug product, waivers based on in vitro dissolution testing are not considered acceptable unless the biorelevance of the testing is documented, especially for waivers of higher strengths, containing the highest amount of residual solvent and excipients. Proposed Change The Company recommends the Guidance specifies that the bio-relevance of the used in vitro release test has been demonstrated at all clinically relevant time points, for example Day 1 and Day 24. 2.10. Analytes to Measure Consistently, guidelines recommend demonstration of bioequivalence based on parent drug plasma concentrations, as the most sensitive approach to detect differences in rate and/or extent of absorption. This approach is considered appropriate because risperidone LAI exhibits flip-flop kinetics (apparent half-life and steady-state PK profile reflects the rate of absorption). Proposed Change No change. The Company agrees with including this approach in the Guidance.

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 12 of 15
2.11. Safety The draft Guidance for i.m. risperidone proposes a single-dose two-way crossover study design at one strength. 3 The proposed requirements to demonstrate BE for an i.m. risperidone LAI (SD BE study at 25 mg strength) are inconsistent with the concepts (BE program including BE and safety study) applied in the draft Guidances for transdermal application forms38,39,40,41,42 and described in the corresponding CPMP Note for Guidance8, in spite of the fact that both types of drug products have similar characteristics:
• the complex pharmacokinetic characteristics are driven by the release of drug (or prodrug), often subject to complex interactions between drug (product), excipients and the (status of the) injection site; e.g. in the original program of risperidone LAI it was experienced that small changes in amounts of residual solvents or excipients can cause a significant early release upon injection.
• the drug release and absorption characteristics are potentially dependent on the status of the local environment of skin/muscle and mediated by physiological reactions (pH, inflammation, irritation etc.).
• both types of drug products have a potential for interaction between the excipients and drug release rate, e.g. for early LAI risperidone formulations, the drug load for these types of drug products, providing drug load for several days or weeks, is similarly high, LAIs providing even less opportunity to interfere with the drug release after administration, requiring a conservative bioequivalence approach.
It was noted that a draft guidance was released by FDA for Doxorubicin, stating that a generic doxorubicin HCl liposome injection, being a parenteral drug product, must be qualitatively and quantitatively the same as the Reference Listed Drug except differences in buffers, preservatives and antioxidants provided that the applicant identifies and characterizes these differences and demonstrates that the differences do not impact the safety/efficacy profile of the drug product. The current FDA Guideline on risperidone LAI does not reflect similar thinking, despite the fact that the development program of RISPERDAL® CONSTA® demonstrated for the early pilot formulations, that small changes in excipients or solvent could cause a significant early release in some subjects. It is unclear how FDA recommends assessing the safety profile of the new Test drug product. A cross-over BE study for LAI AP’s is typically feasible with a sample size of 75-100 patients/dose/study. However, as observed with early pilot formulations of LAI risperidone, an increased proportion of patients may encounter increased release immediately upon injection of drug, e.g. due to excipient-mediated inflammatory reactions. Early or immediate release of a large dose of medication intended for a period of 2 weeks may potentially be an important safety issue for patients. Because such an increase in release rate, mediated by a physiological response, is unpredictable by in vitro dissolution experiments and potentially dependent on the administered volume/dose, generating PK data in a sufficient number of subjects after multiple injections at the highest dose is considered appropriate. Testing the highest dose would also provide important safety and tolerability data at the highest proposed dose. The bioequivalence program, as currently proposed by FDA does not allow reliable detection of an increase in early exposure nor does it allow evaluation of the incidence of related AE’s from the highest dose.

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 13 of 15
Due to the difference in adipose tissue between the deltoid and gluteal muscle, the drug release rate of the drug may depend on the injection site as observed for other antipsychotics. Although this was not observed for RISPERDAL® CONSTA®, similar effects may apply for other LAI risperidone formulations, dependent on the manufacturing technology. In addition, such a study design will allow documentation of the comparative local tolerability profile of deltoid versus gluteal injection for the Test drug product. Proposed Change The Company recommends the guidance specify the minimal sample size to adequately evaluate the safety profile of a new Test drug product prior to considering approval that the Test drug product can substitute for the Reference product. 2. CONCLUSION
Considering all of the above, the Company believes that the bioequivalence program, as currently proposed by FDA would not lead to adequate BE testing of new risperidone intramuscular injections and therefore strongly recommends the Guidance for i.m. risperidone include the following requirements:
• Conduct a cross-over multiple-dose bioequivalence and/or switching study design in addition to the SD study
• Demonstrate BE at least at the lowest and the highest strength • Demonstrate BE for both deltoid and gluteal injection sites • Specify the critical design elements and outcome measures:
o Cmax, AUClast, Tlag, and Tmax and partial AUC (until Day 1 and Day 24) as the PK parameters to be evaluated in the single-dose study
o AUCt should capture at least a period of 85 days o Demonstrate BE for the steady-state PK parameters Cmin,ss, Cmax,ss, and AUCss. o Safety and tolerability must also be carefully assessed o Specify the minimal sample size to adequately evaluate the safety profile of the
Test drug product. • Demonstrate the bio-relevance of the used in vitro release test • A new i.m. risperidone injection must be qualitatively and quantitatively the same as the
Reference Listed Drug, including residual solvents and excipients and the applicant must identify and characterize any differences and demonstrate that the differences do not impact the safety and efficacy profile of the drug product.
Thank you for the opportunity to review and comment on this draft guidance. Should you have any questions or comments, please contact me directly at (609) 730-7028. Sincerely, Heddie Martynowicz, M.S. Senior Director, Regulatory Affairs Johnson and Johnson Pharmaceutical Research and Development, L.L.C.

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 14 of 15
1 USPI RISPERDAL® CONSTA® 2 USPI INVEGA® SUSTENNA® 3 FDA Draft Guideline on Risperidone, dated February 2010 4 FDA Guidance for Industry - Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations, issued March 2003 5 FDA Guidance for Industry Bioequivalence Recommendations for Specific Products, June 2010. 6 FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting, April 13, 2010 7 CPMP/EWP/QWP/1401/98 Rev 1 (January 20, 2010): Guideline on the investigation of bioequivalence. 8 CPMP/EWP/280/96 Corr*, July 28 1999. Note for guidance on quality of modified release oral and transdermal dosage forms: Section II (pharmacokinetic and clinical evaluation). 9 RISPERDAL® CONSTA® NDA Clinical Pharmacology and Biopharmaceutics review 10 Keith S (2006). Advances in psychotropic formulations. Prog Neuropsychopharmacol Biol Psychiatry 2006; 30: 996-1008. 11 Tenback DE (2006), van Harten PN, Slooff CJ, van Os J, SOHO Study Group. Evidence that early extrapyramidal symptoms predict later tardive dyskinesia: a prospective analysis of 10,000 patients in the European schizophrenia outpatient health outcomes (SOHO) study. Am J Psychiatry 2006; 163: 1438-1440. 12 Remington G (2007). Tardive dyskinesia: eliminated, forgotten, or overshadowed? Curr Opin Psychiatry 2007; 20: 131-137. 13 Farde L (1992), Nordstron A-L, Wiesel F-A, et al. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry 1992; 49: 538-544. 14 Nyberg S (1999), Eriksson B, Oxenstierna G, Halldin C, Farde L. Suggested minimal effective dose of risperidone based on PET-measured D2 and 5-HT2A receptor occupancy in schizophrenic patients. Am J Psychiatry 1999; 156: 869-875. 15 Kapur S (2000), Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry (2000); 157: 514-520. 16 Remington G (2006), Mamo D, Labelle A, et al. A PET study evaluating dopamine D2 receptor occupancy for long-acting injectable risperidone. Am J Psychiatry 2006; 163: 396-401. 17 De Ridder F, Vermeulen A, Cleton, A, Kramer M, Eerdekens M. Evaluation of the clinical relevance of food effect observed with paliperidone ER, using a pharmacokinetic/pharmacodynamic modeling approach. Poster at the 3rd Pharmaceutical Sciences World Congress (PSWC), Amsterdam, The Netherlands, April 22-25, 2007. (Attached) 18 Yoshimura R (2009), Ueda N, Ikenough-Sugita A, et al. Fluctuating plasma levels of the active moiety of risperidone is related to occurrence of extrapyramidal symptoms. Int J Psychiatry Clin Practive 2009; 13: 21-24. 19 Kane JM (2003), Eerdekens M, Lindenmayer J-P, et al. Long-acting injectable risperidone: efficacy and safety of the first long-acting atypical antipsychotic. Am J Psychiatry 2003; 160: 1125-1132. 20 Muench J (2010), Hamer AM. Adverse effects of antipsychotic medications. Am Fam Physician 2010; 81: 617-622. 21 Haddad PM (2007), Sharma SG. Adverse effects of atypical antipsychotics. Differential risk and clinical implications. CNS Drugs 2007; 21: 911-936. 22 Maixner SM (1999), Mellow AM, Tandon R. The efficacy, safety, and tolerability of antipsychotics in the elderly. J Clin Psychiatry 1999; 60 (Suppl 8): 29-41. 23 Mauri MC (2007), Volonteri LS, Colasanti A, et al. Clinical pharmacokinetics of atypical antipsychotics. A critical review of the relationship between plasma concentrations and clinical response. Clin Pharmacokinet 2007; 46: 359-388. 24 ICH Guidance for Industry E14. Clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-aniarrhythmic drugs. http://www.ich.org/LOB/media/MEDIA1476.pdf Accessed 07 September 2010. 25 USPI INVEGA® 26 Pollack PT (2010), Lyon AW, Verjee ZH. Risperidone induced QT prolongation following overdose correlates with serum drug concentration and resolves rapidly with no evidence of altered pharmacokinetics. Clin Pharm Ther 2010; 7 (Suppl 1): PII-6.

Docket # FDA-2007-D-0369
J&J PRD Comments: Docket # FDA-2007-D-0369 Page 15 of 15
27 Johnson DA, et al (1983). The discontinuance of maintenance neuroleptic therapy in chronic schizophrenic patients: drug and social consequences. Acta Psychiatr Scand 1983;67:339-352 28 Molina V, et al (2004). Lower prefrontal gray matter volume in schizophrenia in chronic but not in first episode schizophrenia patients. Psychiatry Res 2004;131:45-56 29 Wyatt R. (1991) Neuroleptics and the natural course of schizophrenia. Schizophr Bull 1991;17:325-351. 30 Kane JM, et al. (2003) Expert opinion paper and literature review on the need for a long-acting injectable formulation of a second-generation antipsychotic for the treatment of schizophrenia. White paper (not published); 2003. (Attached) 31 Akiskal HS (2000), Bourgeois ML, Angst J, Post R, Moller HJ, Hirschfeld R. Re-evaluating the prevalence of and diagnostic composition within the broad clinical spectrum of Bipolar disorders. J Affect Disord 2000; 59(suppl 1):S5-30 32 Benazzi F. (2007) Bipolar II disorder: epidemiology, diagnosis and management. CNS Drugs 2007; 21(9):727-740 33 Wyatt RJ, (1995) Henter I. An economic evaluation of manic-depressive illness: 1991. Soc Psychiatry Psychiatr Epidemiol 1995; 30:213-219. 34 Müller-Oerlinghausen B, (2002) Berghöfer A, Bauer M. Bipolar Disorder. Lancet 2002; 359(9302):241-247 35 Eerdekens M, (2004) Van Hove I, Remmerie B, Mannaert E. Pharmacokinetics and Tolerability of Long Acting Risperidone in Schizophrenia. Schizophrenia Research 70 (1), P.91-100, 2004 36 Mannaert E, (2005) Vermeulen A, Remmerie B, Bouhours P, Levron JC: Pharmacokinetic Profile of Long acting injectable risperidone at steady state, comparison with oral administration. L’Encephale 31 (1) P.609-615, 2005 37 Thyssen A, (2010) Rusch S, Herben V, Quiroz J, Mannaert E, Risperidone long Acting Injection: Pharmacokinetis Following Administartion in deltoid versus Gluteal Muscle in schizophrenic patients. Journal of Clinical Pharmacology, Epub, ahead of print, 2010 38 Draft Guideline on Clonidine, released by FDA Nov 2009 39 Draft Guideline on ethinyl estradiol/norelgestromin, released by FDA May 2009, revised Jul 2009 40 Draft Guideline on Scopolamine, released by FDA Aug 2009, revised 2009 41 Draft Guideline on Selegiline, released by FDA Aug 2009 42 Draft Guideline on Fentanyl, released by FDA May 2009, Feb 2010

Attachments

EVALUATION OF THE CLINICAL RELEVANCE OF THEFOOD EFFECT OBSERVED WITH PALIPERIDONE ER,USING A PHARMACOKINETIC/PHARMACODYNAMIC
MODELLING APPROACHF. de Ridder, A. Vermeulen, A. Cleton, M. Kramer, M. Eerdekens
Johnson & Johnson Pharmaceutical Research & Development, Beerse, Belgium.

BACKGROUND■ Paliperidone extended-release tablets (paliperidone ER; INVEGA™) is a psychotropic agent with
demonstrated consistent efficacy and favourable tolerability in the treatment of patients withschizophrenia [1–3]
■ Paliperidone is a monoaminergic antagonist that exhibits the characteristic dopamine type 2 andserotonin type 2A antagonism of the newer, second-generation, antipsychotic drugs
■ Paliperidone ER uses OROS® delivery technology, resulting in a controlled, gradually ascendingrelease of the drug and, therefore, only small 24-hour peak-to-trough fluctuations in paliperidoneplasma concentrations at steady state compared with immediate-release oral formulations [4, 5]
■ This formulation was designed to permit initiation of treatment at an effective dose without the needfor dose titration for initial tolerability reasons
■ Changes in the pharmacokinetic (PK) profile and subsequent changes in exposure to a drug, such asmight be observed with changes in timing of administration with respect to food intake, may impactthe safety and adverse event (AE) profile associated with a drug
OBJECTIVES■ To establish a model-based characterization of the relationship between paliperidone exposure (steady
state concentration using a population PK model [6]) and the risk of developing an extrapyramidal (EPS)related AE, a side effect observed in some individuals with some antipsychotic medications
■ To apply the PK/pharmacodynamic (PD)-model to investigate the impact of food intake with increasingdose of paliperidone ER on EPS incidence
METHODOLOGYStudy design and data■ Pooled data from the safety analysis set (all patients who received at least one dose of study medication
during the double-blind phase) from three Phase III, 6 week, randomized, double-blind, parallel-group,placebo-controlled studies in patients with schizophrenia treated with fixed doses of paliperidone ERwere included in the analyses
■ Paliperidone ER 3–15 mg once daily in the morning was dosed without regard to food. Those treatedwith paliperidone ER 15 mg received paliperidone ER 12 mg for the first 7 days followed bypaliperidone ER 15 mg for the remainder of the study
■ The occurrence of treatment-emergent EPS related-AEs (defined as ≥1 EPS-related AE according toWorld Health Orgainisation Adverse Reaction Terminology preferred terms) were recorded in allstudies across five main groups: dyskinesia, dystonia, hyperkinesia, Parkinsonism and tremor
■ Plasma concentrations of paliperidone were to be measured for all subjects following a sparsesampling scheme (planned number of samples was 3 at each of 3 visits)
■ A total of 1318 patients were included in the analysis; the demographics of the population in eachgroup was similar [7]
Exposure model■ The average steady state paliperidone plasma concentration was considered as the explanatory
variable for the PK/PD-model. For each patient the average steady state concentration (Css, i ) wascalculated as:
■ For 91 patients, no estimate of individual clearance was available and these were excluded from theanalysis. However, a sensitivity analysis (not shown) illustrated that the exclusion of these individualswould not bias the results of the PK/PD modelling presented here
ABSTRACTPurpose: Evaluation of the clinical relevance of the increase in exposure observed with food intakeof paliperidone extended-release (ER) tablets using a population PK/PD-modelling approach.
Methods: Fixed doses (3–15 mg) of paliperidone ER were administered for 6 weeks, once daily inthe morning with or without food. The PK/PD-model related steady state plasma concentrations ofpaliperidone ER with the risk of developing adverse events related to extrapyramidal symptoms(EPS), the most common type of adverse event reported with antipsychotics. For a range of doses,the population distribution of plasma concentrations in the fed and fasted state was simulated usinga population pharmacokinetic model. These simulated concentrations were used as input to thePK/PD-model in order to predict the incidence of EPS in the fed and fasted state.
Results: The relationship between EPS-risk and plasma concentrations was adequately described bya steep, sigmoidal curve, with an EC50 estimated at 24 ng/mL and a Hill factor (steepness) of 5.7.The Emax value corresponded to approximately a 2.8-fold increase of the placebo-risk. A slightlyhigher EPS incidence was predicted in the fed state at all dose levels (3–15 mg), consistent with theincreased plasma exposure in the fed state, after administration of paliperidone ER. An increase of1.6% was predicted for a dose of 3 mg and 4% for doses of 6 to 15 mg.
Conclusions: Although a high fat breakfast increases plasma concentrations by approximately 50%compared to the fasted state, the impact on EPS incidence according to the PK/PD simulations isminimal at any given dose level.
Css,i=D/CLi/τ
Where:• D is the total dose (3, 6, 9, 12 or 15 mg paliperidone ER)• CLi is the individual apparent clearance (CL/F). Individual CL/F were the empirical Bayesian estimates for the subjects in the studies, obtained from the population PK model that was previously developed [6]• τ is the dosing interval=1 day (24 hours)

Model for extrapyramidal symptom incidence■ The outcome of interest is the incidence of EPS, defined as the number of subjects with at least one
EPS-related treatment-emergent AE
■ The hazard of developing EPS at time t (number of days on treatment) was modelled (Equation 2)
■ The choice of function f was based on a graphical data exploration using Kaplan-Meier plots of the time toEPS for the placebo patients only
■ The choice of function g was based on an exploratory data analysis, which suggested a sigmoidal Emax
(maximum drug-induced effect) model (Hill-model) (Equation 3)
■ Model parameters were estimated through maximum likelihood using the SAS-procedure NLMIXED
Model qualification■ The final model was qualified by its ability to simulate the clinical trial data from which it was derived
(posterior predictive check)
■ The Phase III studies were simulated with the following settings for the exposure model:
■ The subject-specific CLi/F was assumed to follow a log normal distribution in the populationcharacterized by:
– Mean=log (13.7 L/h)
– Standard deviation=0.5 (inter-individual variability)
■ Individual values were sampled from this distribution from which the subjects’ average steady statepaliperidone plasma concentration was calculated
Food effect on extrapyramidal symptom incidence■ A population dose-response for EPS incidence in a 6-week clinical study was predicted using the simulation
model where parameters of the exposure model depended on fed conditions
■ Empirical distributions for individual clearance values for the fasted and fed condition were derived fromthe population PK model, assuming an absolute oral bioavailability factor of 28% for the fasted state and42% for the fed state
CONCLUSIONS■ The PK/PD-relationship between the risk of EPS-related AE and paliperidone steady state plasma
concentrations was described by a steep, sigmoidal curve
■ For plasma concentrations below 20 ng/ml, the EPS risk was similar to placebo. For paliperidone ER, aplasma concentration of 20 ng/ml corresponds to a central D2-receptor occupancy of 80%, which is thegenerally accepted threshold for the occurrence of EPS-related side effects with D2 antagonists
■ A PK/PD simulation predicted an increase of the EPS incidence in a 6-week clinical study in fed compared with fasted conditions. Thus, it can be inferred from this simulation exercise that at any given dose level, thePD implications of a fed versus fasted state will be minimal
■ The predicted increase was 1.6% for 3 mg and around 4% for doses between 6 mg and 15 mg
■ This difference in EPS incidence at any given dose level between the fed and fasted state is not clinically relevant
ACKNOWLEDGEMENTS■ Supported by funding from Johnson & Johnson Pharmaceutical Services, LLC., and Johnson & Johnson
Pharmaceutical Research & Development. The authors would like to thank Samantha Kew, PhD,Medicus International, for her editorial assistance
REFERENCES1. Kane J, et al. Schizophr Res 2007; 90:147–61.
2. Marder SR, et al. Biol Psychiatry 2007; in press.
3. Davidson M, et al. Schizophr Res 2007; in press.
4. Karlsson P, et al. Schizophr Res 2006; 81:85–6.
5. Conley R, et al. Curr Med Res Opin 2006; 22:1879–92.
6. Vermeulen A, et al. Clin Pharm Ther 2007; [Abstract].
7. Meltzer H, et al. Int J Neuropsychopharmacol 2006; 9(Suppl 1): S225.
8. Vermeulen A, et al. Johnson & Johnson, data on file.
Equation 1 logλ (t,ci) = f(t) + g (ci)
• λ = Hazard, modelled as a function of time and of covariates at the subject level [7]• t = Time since start of trial (days)• ci = Patient’s individual average steady state concentration• f = Function that describes how hazard changes over time in patients treated with placebo. f (t) was assumed to be a linear function of time (see Equation 2)• g = Function that describes the drug effect as a function of ci
Equation 2 f (t) = β0 + β1t f (t) = β0 + β1 log (t)
(ci)
g (ci) =
13θ2
θ1θ θ1:Emax
(maximal effect)θ2:EC
50 (half effect concentration)
θ3:Hill-factor (steepness)
Equation 3
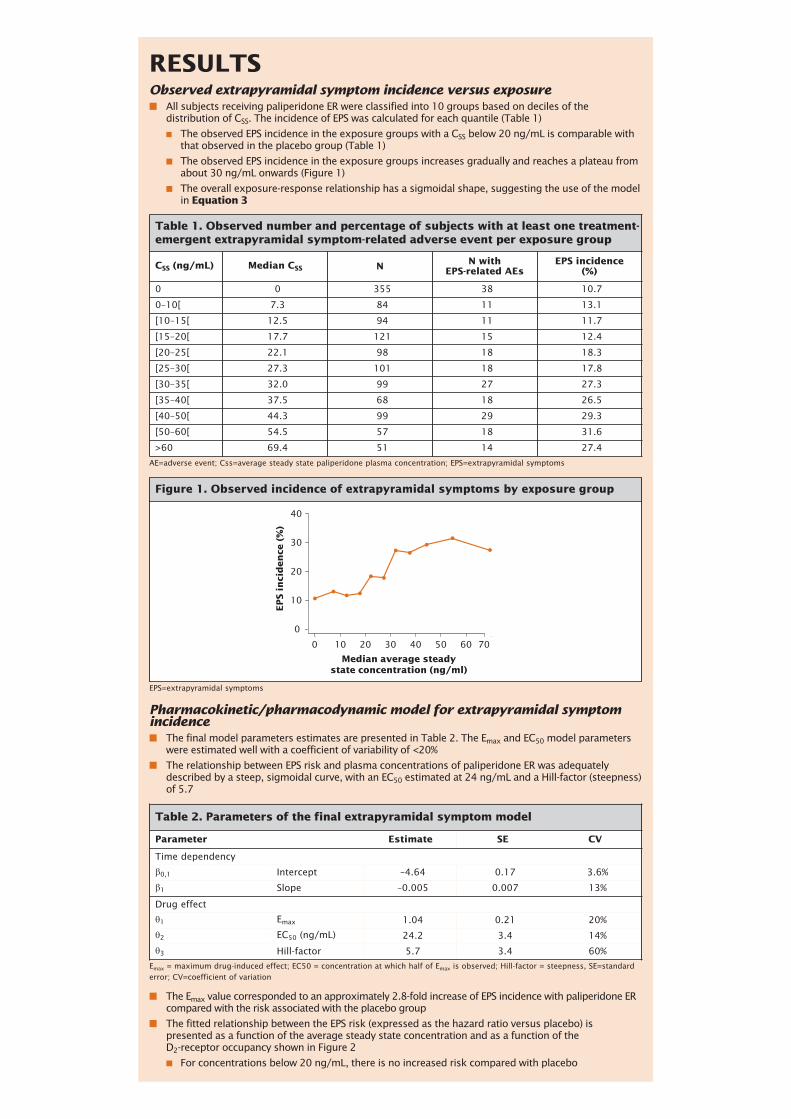
RESULTSObserved extrapyramidal symptom incidence versus exposure■ All subjects receiving paliperidone ER were classified into 10 groups based on deciles of the
distribution of CSS. The incidence of EPS was calculated for each quantile (Table 1)
■ The observed EPS incidence in the exposure groups with a CSS below 20 ng/mL is comparable withthat observed in the placebo group (Table 1)
■ The observed EPS incidence in the exposure groups increases gradually and reaches a plateau fromabout 30 ng/mL onwards (Figure 1)
■ The overall exposure-response relationship has a sigmoidal shape, suggesting the use of the modelin Equation 3
Table 1. Observed number and percentage of subjects with at least one treatment-emergent extrapyramidal symptom-related adverse event per exposure group
CSS (ng/mL) Median CSS N N withEPS-related AEs
EPS incidence(%)
0 0 355 38 10.7
0–10[ 7.3 84 11 13.1
[10–15[ 12.5 94 11 11.7
[15–20[ 17.7 121 15 12.4
[20–25[ 22.1 98 18 18.3
[25–30[ 27.3 101 18 17.8
[30–35[ 32.0 99 27 27.3
[35–40[ 37.5 68 18 26.5
[40–50[ 44.3 99 29 29.3
[50–60[ 54.5 57 18 31.6
>60 69.4 51 14 27.4
AE=adverse event; Css=average steady state paliperidone plasma concentration; EPS=extrapyramidal symptoms
EPS=extrapyramidal symptoms
Emax = maximum drug-induced effect; EC50 = concentration at which half of Emax is observed; Hill-factor = steepness, SE=standarderror; CV=coefficient of variation
Pharmacokinetic/pharmacodynamic model for extrapyramidal symptomincidence ■ The final model parameters estimates are presented in Table 2. The Emax and EC50 model parameters
were estimated well with a coefficient of variability of <20%
■ The relationship between EPS risk and plasma concentrations of paliperidone ER was adequatelydescribed by a steep, sigmoidal curve, with an EC50 estimated at 24 ng/mL and a Hill-factor (steepness)of 5.7
■ The Emax value corresponded to an approximately 2.8-fold increase of EPS incidence with paliperidone ERcompared with the risk associated with the placebo group
■ The fitted relationship between the EPS risk (expressed as the hazard ratio versus placebo) ispresented as a function of the average steady state concentration and as a function of the D2-receptor occupancy shown in Figure 2
■ For concentrations below 20 ng/mL, there is no increased risk compared with placebo
Median average steadystate concentration (ng/ml)
EP
S i
nci
de
nce
(%
)
10 20 30 40 50 60 70
0
10
20
30
40
0
Figure 1. Observed incidence of extrapyramidal symptoms by exposure group
Table 2. Parameters of the final extrapyramidal symptom model
Parameter Estimate SE CV
Time dependency
β0,1 Intercept –4.64 0.17 3.6%
β1 Slope –0.005 0.007 13%
Drug effect
θ1 Emax 1.04 0.21 20%
θ2 EC50 (ng/mL) 24.2 3.4 14%
θ3 Hill-factor 5.7 3.4 60%
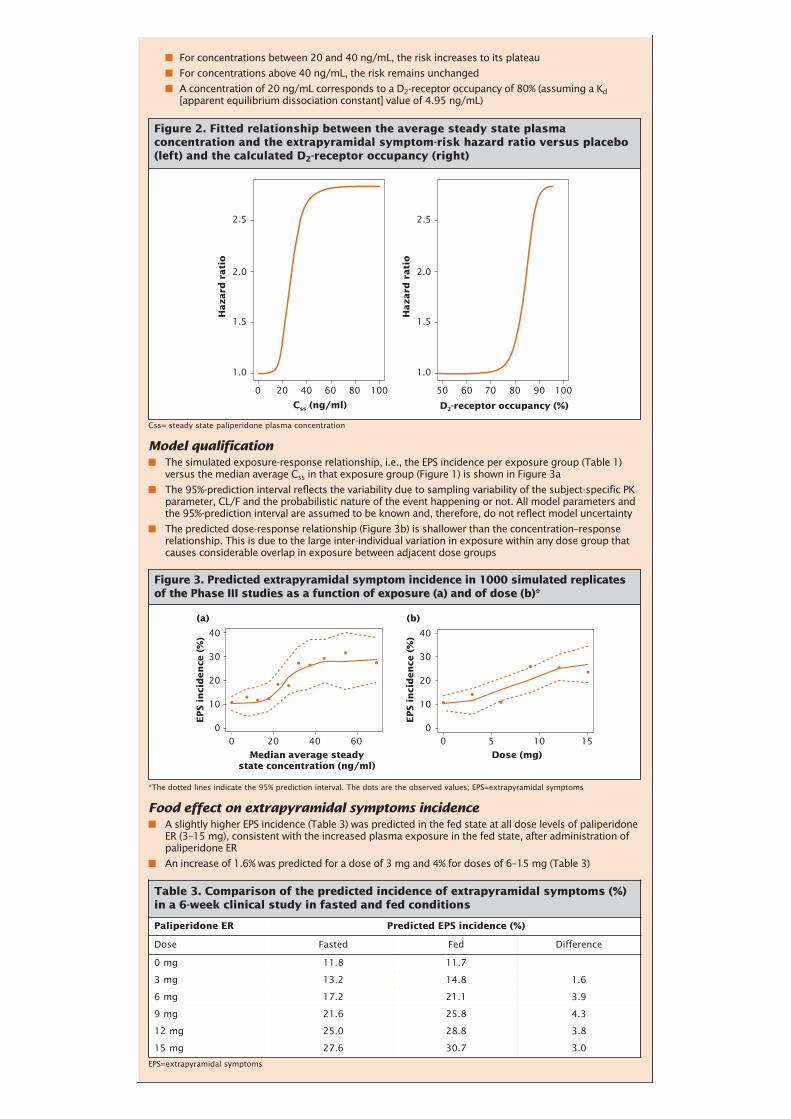
■ For concentrations between 20 and 40 ng/mL, the risk increases to its plateau
■ For concentrations above 40 ng/mL, the risk remains unchanged
■ A concentration of 20 ng/mL corresponds to a D2-receptor occupancy of 80% (assuming a Kd[apparent equilibrium dissociation constant] value of 4.95 ng/mL)
Model qualification■ The simulated exposure-response relationship, i.e., the EPS incidence per exposure group (Table 1)
versus the median average Css in that exposure group (Figure 1) is shown in Figure 3a
■ The 95%-prediction interval reflects the variability due to sampling variability of the subject-specific PKparameter, CL/F and the probabilistic nature of the event happening or not. All model parameters andthe 95%-prediction interval are assumed to be known and, therefore, do not reflect model uncertainty
■ The predicted dose-response relationship (Figure 3b) is shallower than the concentration–responserelationship. This is due to the large inter-individual variation in exposure within any dose group thatcauses considerable overlap in exposure between adjacent dose groups
Food effect on extrapyramidal symptoms incidence■ A slightly higher EPS incidence (Table 3) was predicted in the fed state at all dose levels of paliperidone
ER (3–15 mg), consistent with the increased plasma exposure in the fed state, after administration ofpaliperidone ER
■ An increase of 1.6% was predicted for a dose of 3 mg and 4% for doses of 6–15 mg (Table 3)
Css= steady state paliperidone plasma concentration
Ha
za
rd r
ati
o
1.0
0 20 40 60 80 100 50 60 70 80 90 100
1.5
2.0
2.5
Ha
za
rd r
ati
o
1.0
1.5
2.0
2.5
Css (ng/ml) D2-receptor occupancy (%)
Figure 2. Fitted relationship between the average steady state plasmaconcentration and the extrapyramidal symptom-risk hazard ratio versus placebo(left) and the calculated D2-receptor occupancy (right)
*The dotted lines indicate the 95% prediction interval. The dots are the observed values; EPS=extrapyramidal symptoms
EP
S i
nci
de
nce
(%
) 40
30
20
10
00 20 40 60
Median average steadystate concentration (ng/ml)
EP
S i
nci
de
nce
(%
)
(a) (b)
40
30
20
10
00 5 10 15
Dose (mg)
Figure 3. Predicted extrapyramidal symptom incidence in 1000 simulated replicatesof the Phase III studies as a function of exposure (a) and of dose (b)*
EPS=extrapyramidal symptoms
Table 3. Comparison of the predicted incidence of extrapyramidal symptoms (%)in a 6-week clinical study in fasted and fed conditions
Paliperidone ER Predicted EPS incidence (%)
Dose Fasted Fed Difference
0 mg 11.8 11.7
3 mg 13.2 14.8 1.6
6 mg 17.2 21.1 3.9
9 mg 21.6 25.8 4.3
12 mg 25.0 28.8 3.8
15 mg 27.6 30.7 3.0











