development of ni-catalyzed alkene
TRANSCRIPT

University of New MexicoUNM Digital Repository
Chemistry ETDs Electronic Theses and Dissertations
Summer 7-15-2019
Development of Ni-Catalyzed AlkeneDicarbofunctionalization ReactionsShekhar KCUniversity of New Mexico - Main Campus
Follow this and additional works at: https://digitalrepository.unm.edu/chem_etdsPart of the Inorganic Chemistry Commons, Medicinal-Pharmaceutical Chemistry Commons,
and the Organic Chemistry Commons
This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has beenaccepted for inclusion in Chemistry ETDs by an authorized administrator of UNM Digital Repository. For more information, please [email protected].
Recommended CitationKC, Shekhar. "Development of Ni-Catalyzed Alkene Dicarbofunctionalization Reactions." (2019). https://digitalrepository.unm.edu/chem_etds/156

i
Shekhar KC
Candidate
Department of Chemistry and Chemical Biology
Department
This dissertation is approved, and it is acceptable in quality and form for publication:
Approved by the Dissertation Committee:
Prof. Ramesh Giri, Chairperson
Prof. Yang Qin
Prof. Mark Chalfant Walker
Prof. Rodolfo Tello-Aburto

ii
Development of Ni-Catalyzed Alkene Dicarbofunctionalization
Reactions
by
Shekhar KC
M.Sc. Chemistry, Tribhuvan University, 2009
DISSERTATION
Submitted in Partial Fulfillment of the
Requirements for the Degree of
Doctor of Philosophy
Chemistry
The University of New Mexico
Albuquerque, New Mexico
July 2019

iii
DEDICATION
To my parents and my lovely wife!

iv
ACKNOWLEDGEMENTS
First, I would like to express my sincere thanks to my advisor, Professor Ramesh
Giri for his encouragement, guidance and financial support during my Ph. D. study. I am
fortunate to have him as my mentor in organic chemistry and of my life. Thanks for opening
my eyes to a new stage of opportunity and strength. I will be forever grateful for your
guidance.
I wish to extend my thanks to my committee members, Professor Yang Qin,
Professor Mark Chalfant Walker, Professor Rodolfo Tello-Aburto for their time and
attention in regard to my work. Also, I am thankful to my research proposal committee
member Prof. Richard Kemp for his time and insightful comments.
Many thanks to the past and present group members of the Giri research group,
especially Prakash, Roshan, Surendra, Bijay, Lucas, Vivek, Robert, Namrata, Arjun,
Santosh, Rajani, Sangita and Ryan for the assistance and friendship. I am especially
grateful to Lucas Chesley for his relentless help in proofreading and giving feedback on
my dissertation.
My special thank goes to my friends Umesh, Zhen, Brad, Griffin, Khadanand, Amrit,
Ranjana, Bijesh and Tefera. I am also grateful to all the professors and staffs of the
Chemistry Department at UNM for their help and co-operation throughout my Ph.D. study.
Finally, and most importantly, I would like to thank my family, my father Khem B.
KC, my mother Maya Devi KC, my brother Santosh, and sisters Sanju and Sangrila, for
their unconditional support, motivation and encouragement. I would like to thank my
lovely wife Sheela Thapa for her sacrifice, love, encouragement and continual support.
This work would not have been possible without these people.

v
Development of Ni-Catalyzed Alkene Dicarbofunctionalization
Reactions
by
Shekhar KC
M.Sc. Chemistry, Tribhuvan University, 2009
Ph.D., Chemistry, University of New Mexico, USA, 2019
Abstract
Alkenes serve as one of the most important feedstocks for organic synthesis, having two
vicinal sites for bond formation. In alkenes, both vicinal sites can be functionalized with
two reagents in a process commonly known as alkene difunctionalization, which results in
the formation of two new bonds. A number of alkenes difunctionalization reactions, such
as diamination, dioxygenation, carboamination and carbooxygenation, are known.
However, difunctionalization of alkenes with two carbon-based entities, termed alkene
dicarbofunctionalization, is relatively less common. Development of such a process could
provide a powerful method to introduce two different carbon fragments across an alkene
in a regioselective manner, enabling a modular, convergent and expedient synthesis of
complex structural cores prevalent in pharmaceutical and natural products. In this
dissertation, we describe the discovery and development of two novel Ni-catalyzed alkene
dicarbofunctionalization reactions.

vi
The first part of my dissertation focuses on the development of three-component Ni-
catalyzed regioselective alkylarylation of vinylarenes with alkyl halides and arylzinc
reagents. This reaction enables the successful addition of primary, secondary and tertiary
alkyl halides, and arylzinc reagents across the alkenes in vinylarenes in a highly
regioselective manner. The reaction also shows a high degree of functional group tolerance.
Detailed mechanistic investigations by quantitative kinetics, competition studies, and
radical probes indicate that this reaction proceeds by a single electron transfer (SET)
process with the direct halogen atom abstraction from alkyl halides by a Ni-catalyst being
the rate limiting step. We have also demonstrated the application of this novel reaction in
the synthesis of a precursor of Zoloft (an antidepression drug) and in the late-state synthesis
of a potential FLAP inhibitor and its analogs.
The second part of my dissertation describes a Ni/terpyridine-catalyzed two-component
cyclization/coupling reaction of alkene tethered to alkyl halides with arylzinc reagents.
This reaction enabled us to synthesize a large number of complex carbo- and N and O-
based heterocycles, which are prevalent in bioactive natural products and pharmaceutically
relevant molecules. We further applied this new cyclization/coupling method to the concise
synthesis of six bioactive lignan natural products containing three different structural
frameworks. The synthesis of these natural products can also be performed in gram-scale
quantities. We also conducted mechanistic investigations through competition studies and
radical probes, which indicated that the current reaction proceeds via Ni(I)/Ni(III) catalytic
cycle in an analogous manner to the well-known Ni/terpyridine-catalyzed Negishi cross-
coupling reaction.

vii
Table of Contents
Dedication………………………………………………………………………….……..iii
Acknowledgement………………………………………………………………………..iv
Abstract………………………………………………………...………………...………..v
Table of Contents……………………………………………………………………..…..vi
List of Schemes………………………………………………………………………..…ix
List of Tables………………………………………………………………...…………..xiv
List of Figures………………………………………………………………….….……..xv
List of Abbreviations…………………………………………………………...……..…xvi
Chapter 1. Chapter 1. Alkene Dicarbofunctionalization Reaction.............................1
1.1 Introduction .......................................................................................................... 1
1.2 Alkene Dicarbofunctionalization ......................................................................... 3
1.2.1 Cyclization/Coupling Reactions ................................................................... 8
1.2.2 Three-Component Reactions ...................................................................... 23
1.3 Conclusion .......................................................................................................... 37
Chapter 2. Three-Component Alkylarylation of Vinyl Arenes ................................39
2.1 Introduction ........................................................................................................ 39
2.2 Ni-Catalyzed Alkylarylation of Vinylarenes...................................................... 39

viii
2.3 Mechanistic Study .............................................................................................. 50
2.4 Conclusion .......................................................................................................... 60
Chapter 3. Cyclization/Coupling Reaction .................................................................62
3.1 Introduction ........................................................................................................ 62
3.2 Ni-Catalyzed Cyclization/Coupling of Alkene tethered to Alkyl Halides ......... 66
3.3 Application to the concise synthesis of natural products ................................... 73
3.4 Mechanistic Study .............................................................................................. 77
3.5 Conclusion .......................................................................................................... 85
Chapter 4. Experimental Section .................................................................................86
4.1 Ni-Catalyzed Alkylarylation of Vinylarenes...................................................... 86
4.1.1 General Information .................................................................................... 86
4.1.2 Experimental Procedure .............................................................................. 87
4.1.3 Mechanistic studies ..................................................................................... 91
4.1.4 Characterization data for new compounds ................................................ 113
4.2 Ni-Catalyzed Cyclization/Coupling ................................................................. 147
4.2.1 General Information .................................................................................. 147
4.2.2 Experimental Procedure ............................................................................ 148
4.2.3 Mechanistic Study ..................................................................................... 153
4.2.4 Characterization data for new compounds ................................................ 161

ix
List of Schemes
Chapter 1
Scheme 1.1. Application of Heck reaction in the synthesis of estradiol………………. ….2
Scheme 1.2. Three-component dicarbofunctionalization and cyclization/coupling reaction
…………………………………………………….………………………...…….……….5
Scheme 1.3. Possible problems in dicarbofunctionalization reactions…………………….7
Scheme 1.4. Problems in alkene dicarbofunctionalization by cyclization/coupling……....9
Scheme 1.5. Ligands used in cross-coupling reaction…………………………………….10
Scheme 1.6. Pd-catalyzed cyclization/coupling of alkenes tethered to enolates………...11
Scheme 1.7. Pd-catalyzed cyclization/coupling of unactivated alkenes tethered to enolates
……….…………………………………………………………………….……….…….12
Scheme 1.8. Pd-catalyzed cyclization/coupling of alkenes tethered to aryl bromides with
organotin reagents…………………………………………….…………….……………13
Scheme 1.9. Pd-catalyzed cyclization/coupling of activated alkenes tethered to aryl iodides
with organotin reagents…………………………………………………….…………….13
Scheme 1.10. Pd-catalyzed aryl C-H bond cyclization/cyanomethylation……………….14
Scheme 1.11. Pd-catalyzed cyclization/coupling of alkenes tethered to alkyl iodides with
CO……………………………………………………………………………………......15
Scheme 1.12. Pd-catalyzed cyclization/coupling of alkenes tethered to alkyl iodides with
organoboron reagents…………………………………………………………………….16

x
Scheme 1.13. Pd-catalyzed aryl C-H cyclization/carbonylation………………………….16
Scheme 1.14. Co-catalyzed cyclization/coupling of tethered alkenes with arylmagnesium
reagents…………………………………………………………………………….…….18
Scheme 1.15. Ni-catalyzed cyclization/coupling of tethered alkenes with alkylzinc
reagents………………………………………………………………...……………...…19
Scheme 1.16. Ni-catalyzed reductive cyclization/coupling of tethered alkenes with aryl
iodides………………………...………………………………………………………….19
Scheme 1.17. Ni-catalyzed reductive cyclization/coupling of tethered alkenes with aryl
iodides…………………………………………...……………………………………….20
Scheme 1.18. Pd-catalyzed, Cu-mediated sequential cyclization/coupling of alkylzinc
reagents with different electrophiles…………………………………….……………….20
Scheme 1.19. Cyclization/coupling of aryl-9-BBN reagents with aryl iodides…………...21
Scheme 1.20. Enantioselective Ni-catalyzed cyclization/coupling with alkyl bromides....21
Scheme 1.21. Cu-catalyzed cyclization/coupling reaction of with alkenes tethered to
alkyl/arylzinc reagents with aryl iodides…………………………...…………………….22
Scheme 1.22. Ni-catalyzed carboacylation of tethered alkenes with pinacol arylboronates
…………….………………………………………………………………………..…….22
Scheme 1.23. Rh-catalyzed intramolecular arylacylation………………………………...23
Scheme 1.24. Pd-catalyzed dicarbofunctionlization of norbornene and norbornadiene….25
Scheme 1.25. Co-catalyzed dicarbofunctionalization reaction of 1,3-diene…………….25

xi
Scheme 1.26. Pd-catalyzed dicarbofunctionalization reaction of 1,3-diene with arylboronic
acids and vinyl triflates…………………………………………………………………...26
Scheme 1.27. Pd-catalyzed dicarbofunctionalization reaction of styrenes with arylboronic
acids and vinyl triflates…………………………………………………………………...26
Scheme 1.28. Cu-catalyzed trifluoromethylarylation of styrenes………………………...27
Scheme 1.29. Ti-catalyzed reductive dialkylation of styrenes with alkyl bromides………28
Scheme 1.30. Ag mediated Fe-catalyzed difunctionalization reaction of styrenes……….28
Scheme 1.31. Ni-catalyzed diarylation of vinylarene………………………………….…29
Scheme 1.32. Copper-catalyzed asymmetric conjugate addition/silylation………………30
Scheme 1.33. Nickel-catalyzed three-component reaction of benzylacrylates..…………30
Scheme 1.34. Palladium catalyzed decarboxylation of unactivated alkenes……………...31
Scheme 1.35. Palladium catalyzed 1,1-difunctionalization of simple alkene…………….32
Scheme 1.36. Palladium catalyzed coordination assisted diarylation of vinyl ethers…….32
Scheme 1.37. Ni-catalyzed dicarbofunctionalization of enamide………………………...33
Scheme 1.38. Nickel-catalyzed 1,2-diarylation of vinylarenes using coordinating group.33
Scheme 1.39. Pyridine assisted Nickel-catalyzed 1,2-diarylation of vinylsilanes……….34
Scheme 1.40. Nickel-catalyzed 1,3-diarylation of unactivated alkenes in ketimines…….34
Scheme 1.41. Nickel-catalyzed 1,2-diarylation of unactivated alkenes in ketimines…….35
Scheme 1.42. Nickel-catalyzed alkylarylation of 8-aminoquinolinamide……………….36

xii
Scheme 1.43. Nickel-catalyzed functionalization reaction of N-Allyl aminopyrimidines.36
Scheme 1.44. Nickel-catalyzed reductive alkylarylation of alkenes……………………...37
Scheme 1.45. Nickel-catalyzed carboacylation of alkene………………………………...37
Chapter 2
Scheme 2.1. Plan A for the synthesis of potential FLAP inhibitor…………………….…48
Scheme 2.2. Plan B for the synthesis of potential FLAP inhibitor……………………….49
Scheme 2.3. Reduction experiment of Ni(II) to Ni(0) by ArZnI………………………….51
Scheme 2.4. Radical clock experiment…………………………………………………...52
Scheme 2.5. Formation of radically dimerized product………………………………….52
Scheme 2.6. Competition experiments with 1° and 2° R-X……………………………....53
Scheme 2.7. Competition experiments with 3° and 2° R-X………………………………54
Scheme 2.8. Competition experiments with RI, RBr and RCl……………………………54
Scheme 2.9. Competition experiments with ArZnI………………………………………55
Scheme 2.10. Proposed catalytic cycle…………………………………………………...59
Chapter 3
Scheme 3.1. Pathways for alkene dicarbofunctionalization and problems……………….64
Scheme 3.2. Initial optimization of this reaction using FeCl2 catalyst……….………….65

xiii
Scheme 3.3. Concise synthesis of dimethylretrodendrin, kusunokinin and dimethylmetair-
esinol……………………………………………………………………………….…….74
Scheme 3.4. Concise synthesis of yatein and bursehernin……………………………….75
Scheme 3.5. Concise synthesis of collinusin…………………………………………….76
Scheme 3.6. Proposed catalytic cycle……………………………………………………78
Scheme 3.7. Selectivity study in Negishi cross-coupling reaction with electronically bias-
ed arylzinc reagents……...……………………………………………………………….79
Scheme 3.8. Selectivity in cyclization/coupling with electronically biased arylzinc reage-
nts……………………………………………………………….……………………….80
Scheme 3.8. Diastereoselectivity studies with cis- and trans-1-(allyloxy)-2 bromocycloh-
exane…………………………………………….…………………………………….…81
Scheme 3.9. Diastereoselectivity in the known radical cyclization and the current
cyclization/coupling reactions…………………………………………………………...83

xiv
List of Tables
Chapter 2
Table 2.1. Optimization of reaction conditions…………………………………………...41
Table 2.2. Scope with RX and ArZnI………………………………………….…………42
Table 2.3. Scope with vinylarenes, RX and ArZnI……………………………………….43
Table 2.4. Scope with vinylarenes, RX and ArZnI……………………………………….45
Table 2.5. Scope of ArZnI with vinylarenes and -bromo esters…………………………47
Chapter 3
Table 3.1. Optimization of reaction conditions for cyclization/coupling.….…….……...67
Table 3.2. Cyclization/coupling of alkenes tethered to alkyl halides…………………....69
Table 3.3. Diastereoselective cyclization/coupling………………………………………70
Table 3.4. Tolerance of base-sensitive and racemizable stereocenters…………………...72

xv
List of Figures
Chapter 1
Figure 1-1. Figure showing different reactions in alkene functionalization……………….1
Figure 1-2. Graph showing the no. of publications in dicarbofunctionalization by the year
…...…………………………………………………………………………………...……5
Chapter 2
Figure 2-1. Application of dicarbofunctionalization in the synthesis of drug molecule….39
Figure 2-2. A typical reaction kinetic profile…………………………………………….56
Figure 2-3. a) A plot of product yields Vs cyclohexyl iodide concentrations. b) A plot of kin
vs. cyclohexyl iodide concentrations…………………………………………………….56
Figure 2-4. a) A plot of product yields Vs NiBr2 concentrations. b) A plot of kin vs. NiBr2
concentrations……………………………………………………………………………57
Figure 2-5. a) A plot of product yields Vs PhZnI concentrations. b) A plot of kin vs. PhZnI
concentrations……………………………………………………………………………58
Figure 2-6. A plot of product yields vs. alkene concentrations…………………………...58
Chapter 3
Figure 3-1. Structure of heterocyclic cores……………………………………………….61
Figure 3-2. Lignan natural products and bioactive molecules containing (arylmethyl)
heterocyclic cores………………………………………………………………………...63
Figure 3-3. Crystal structure of Fe(II)Cl2(HMPA)2 complex……………………………66

xvi
List of Abbreviations
Å angstrom
AIBN azobisisobutyronitrile
APPI atmospheric pressure photoionization
aq. aqueous
BCl3 boron trichloride
BOC tert-butyloxycarbonyl
Bipy bipyridine
cat. catalyst
cbz carboxybenzyl group
CDCl3 deuterated chloroform
CG coordinating group
CHCl3 chloroform
CH2Cl2 methylene chloride
CO carbon monoxide
COSY COrelated SpectroscopY
CsF caesium fluoride
d doublet
dd doublet of doublet
DCE 1,2-dichloroethane
DFT density functional theory
DMF dimethylformamide

xvii
DMSO dimethyl sulfoxide
DPPE 1,2-Bis(diphenylphosphino) ethane
FLAP 5-Lipoxygenase Acting Protein
δ chemical shift
dt doublet of triplet
dr diastereomeric ratio
ee enantiomeric excess
ESI electrospray ionization
EtOAc ethyl acetate
Et2Zn diethylzinc
er enantiomeric ratio
equiv equivalent
g gram(s)
GC gas chromatography
GC-MS gas chromatography-mass spectrometry
h hour(s)
HMPA hexamethylphosphoramide
HPLC high-performance liquid chromatography
HRMS high resolution mass spectrometry
Hz hertz
J coupling constants
L liter
LDA lithium diisopropylamide

xviii
λ wavelength
m multiplet
m meta
M molar
MeCN acetonitrile
Me3Si trimethyl silane
mg milligram(s)
MHz megahertz
min minute(s)
L microliter
mL milliter
mm millimeter
mmol millimole
Mol mole
mp melting point
NCbz N- protected carboxybenzyl group
NaH sodium fluoride
NaNO2 sodium nitrite
NHC N-heterocyclic carbene
NiBr2 nickel(II) bromide
NiBr2.dme nickel(II) bromide ethylene glycol dimethyl ether
NiCl2(PPh3)2 bis(triphenylphosphine)nickel(II) dichloride
Ni(cod)2 bis(1,5-Cyclooctadiene)nickel(0)

xix
NMR nuclear magnetic resonance
NMP N-methyl-2-pyrrolidinone
o ortho
OTf trifluoromethanesulfonate
p para
PhI(OPiv)2 di-(pivaloyloxy)iodobenzene
PdCl2(dppf) 1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)
Pd(dba)2 bis(dibenzylideneacetone)palladium(0)
Pd(PPh3)4 tetrakis(triphenylphosphine)palladium
Pd(OAc)2 palladium(II)acetate
Ph phenyl
ppm parts per millions
Rf retention factor
RDS rate determining step
rt room temperature
s singlet
SET single electron transfer
SIMes HCl 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride
SOCl2 thionyl chloride
SnCl2 tin (II) chloride
TBS tert-butyldimethylsilyl
TEMPO 2,2,6,6-Tetramethyl-1-piperidinyloxy
Terpy terpyridine

xx
THF tetrahydrofuran
TMEDA N,N′,N′-Tetramethylethylenediamine
TMs transition metals
TMSCl chlorotrimethyl silane
UV ultra violet
1°,2°, 3° primary, secondary, tertiary

1
Chapter 1. Chapter 1. Alkene Dicarbofunctionalization Reaction
1.1 Introduction
Alkenes constitute a large class of industrial petrochemicals and are among the most
common chemicals used for making complex molecules in both academia and industry.
Functionalization of alkenes to synthesize valuable products has been extensively growing
in the last fifty years. Typically, the functionalization of an alkene can be categorized into
monofunctionalization and difunctionalization. Some of the most important alkene
monofunctionalization and difunctionalization processes are listed below (Figure 1-1).
Figure 1-1. Figure showing different reactions in alkene functionalization
Alkene monofunctionalization reactions have been widely used for the carbon-carbon (C-
C) or C-heteroatom bond formation. One of the most well-known alkene
monofunctionalization reactions that generates a new C-C bond is hydroformylation,1-2
where there is an addition of carbon monoxide (CO) and H2 into the double bond. The
aldehyde product formed from this reaction can be used to form other useful industrial
products. Usually, transition metals (TMs) like Co,3-4 and Rh,5 and in some cases

2
heterogeneous catalysts,6 have been used to catalyze these reactions. Another prominent
example of alkene monofunctionalization that generates a new C-C bond is the Mizoroki-
Heck reaction. This Nobel prize-wining reaction (2010),7-9 developed by Mizoroki10 and
Heck,11 combines organic halides with alkenes to generate a new C-C bond along with the
regeneration of the C=C bond. This reaction has been widely utilized in the synthesis and
manufacturing of building blocks in pharmaceuticals and materials.12-14 A well-known
example for the application of the Heck reaction is the synthesis of the natural steroid
estradiol15 1.1 (Scheme 1.1).
Scheme 1.1. Application of Heck reaction in the synthesis of estradiol
Adding heteroatoms to monofunctionalize alkenes has also been developed as an important
method for making complex molecules. One such example is hydrosilylation,16 which is
used for making organosilicon compounds. These organosilicon compounds can further be
utilized in stereospecific oxidation and cross-coupling reactions. Another prominent
example of heteroatom-based alkene monofunctionalization is hydroamination,17-18 a
reaction that provides a wide variety of nitrogen-based structures commonly found in
bioactive molecules.

3
Functionalization of alkenes with two moieties, often called alkene difunctionalization, is
also pervasive in organic synthesis. This class of reactions can be either be non-TM or TM
catalyzed.19 Non-TM based alkene difunctionalization reactions20 are usually initiated by
the addition of carbon-centered or heteroatom-centered radicals to the unsaturated bond.
The difunctionalized products are subsequently formed after the oxidation of radical
intermediates to carbocations followed by their trapping with nucleophiles. TM-catalyzed
difunctionalization involves a TM catalyst, where a nucleophile is typically added to
alkenes to form alkylmetal intermediates. These intermediates are then further intercepted
with another reagent to form difunctionalized products.21
One of the most useful alkene difunctionalization reactions is the Sharpless
dihydroxylation reaction.22 In this reaction, alkenes are converted to diols with peroxide
oxidants in the presence of osmium catalysts. This process has shaped the art of asymmetric
catalysis and total synthesis.22 More recently, several variants of this alkene
heterodifunctionalization have been reported. Some of these examples are alkene
dioxygenation,23-24 diamination,25 dihalogenation,26-27 carboamination,28
carbooxygenation,29 aminooxygenation.30
1.2 Alkene Dicarbofunctionalization
More recently, difunctionalization of alkenes with carbon-based entities, termed alkene
dicarbofunctionalization, is also gaining momentum in the field of method development.
In this process, carbon-based reagents are added across the double bond to generate two
new C-C bonds. Although the reports of this type of reaction can be traced back to the
1980s, their development as a useful synthetic method has been very slow. The slow

4
development can be attributed to the difficulty of adding two different reagents across an
alkene in a single step. Despite such difficulties, several notable examples of alkene
dicarbofunctionalization reactions have been developed. Among them, the Diels-alder
reaction31 and cyclopropanation32 are classic examples in a broad perspective. Although
these reactions proceed with mechanistic scenarios completely different from those of the
reactions this thesis outlines and treats in detail in the subsequent sections and chapters,
the Diels-Alder reaction and cyclopropanation are the most successful examples that
demonstrate the power of generating two C-C bonds in synthetic step to form complex
molecules. While the exhaustive treatment of use of these reactions is beyond the scope of
this thesis, several notable examples of natural product synthesis,33 pharmaceutical
synthesis,13 and material synthesis34 highlight the synthetic significance of reactions that
generate two C-C bonds in one step.
TM-catalyzed dicarbofunctionalization of alkenes35-38 that proceeds via stepwise addition
of two carbon-based entities and involves the formation of organotransition metal
intermediates (as opposed to the concerted processes in the Diels-Alder and
cyclopropanation reactions) is one of the powerful methods to construct two C-C bonds
across the alkene. While reductive alkene dicarbofunctionalization also has been emerging
more recently, the most common alkene dicarbofunctionalization utilizes organic halides
and organometallic reagents as carbon sources. These reactions are typically conducted
either through cyclization/coupling or in a three-component process (Scheme 1.2). The
historical reports and the progress over several years in developing these reactions are
described below with pertinent examples.
5
Scheme 1.2. Three-component dicarbofunctionalization and cyclization/coupling reaction
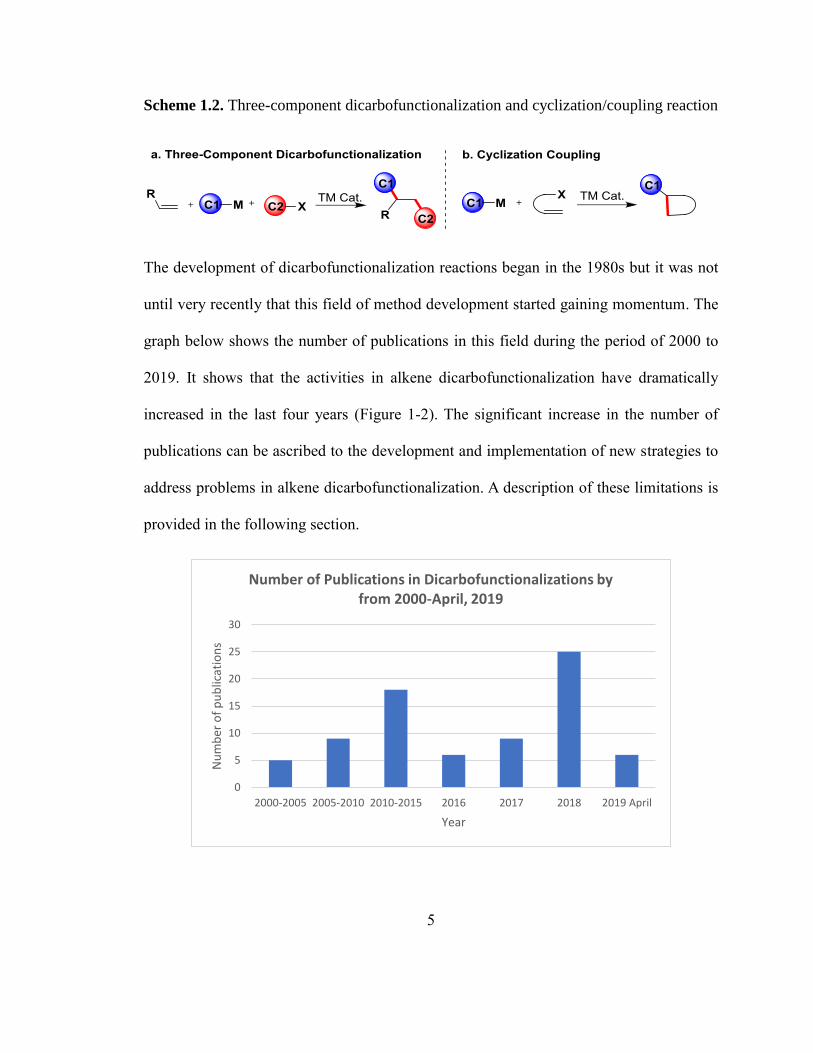
The development of dicarbofunctionalization reactions began in the 1980s but it was not
until very recently that this field of method development started gaining momentum. The
graph below shows the number of publications in this field during the period of 2000 to
2019. It shows that the activities in alkene dicarbofunctionalization have dramatically
increased in the last four years (Figure 1-2). The significant increase in the number of
publications can be ascribed to the development and implementation of new strategies to
address problems in alkene dicarbofunctionalization. A description of these limitations is
provided in the following section.
0
5
10
15
20
25
30
2000-2005 2005-2010 2010-2015 2016 2017 2018 2019 April
Nu
mb
er o
f p
ub
licat
ion
s
Year
Number of Publications in Dicarbofunctionalizations by from 2000-April, 2019

6
Figure 1-2. Graph showing the no. of publications in dicarbofunctionalization by the year
(Source; Scifinder and ISI citation index at Web of Science; search criteria:
“dicarbofunctionalization” and other related terms; duplicates removed; refined by year)
Catalytic Cycle. The catalytic cycle of dicarbofunctionalization reactions is mainly
divided into four basic steps – oxidative addition, migratory insertion, transmetalation and
reductive elimination (Scheme 1.3). First, organic halides undergo oxidative addition or
single electron transfer (SET) to form a new organometallic species 1.2. This is followed
by an alkene insertion to an organometallic species (RM-X) or organic halide (R-X)
through migratory insertion or radical addition generating a new alkylmetal species 1.3.
These alkylmetal species are intercepted by an organometallic reagent and undergoes
transmetalation 1.4 which is followed by reductive elimination to furnish the
dicarbofunctionalized product 1.5. However, there are two challenges in these reactions,
firstly the alkylmetal intermediates have a higher tendency to undergo β-hydride
elimination and leads to the formation of Heck products 1.7. Secondly, the oxidatively
added intermediate can transmetalate with organometallic reagent and reductively
eliminate to give the cross-coupling product 1.6 (Scheme 1.3).

7
Scheme 1.3. Possible problems in dicarbofunctionalization reactions
These two processes need to be avoided in order to perform a dicarbofunctionalization
reaction successfully. This is the most challenging problem in such chemistry. Different
strategies were used to difunctionalize alkenes in three-component and
cyclization/coupling reaction. In three-component reaction, substrates that can form
geometrically constrained alkylmetal intermediates, -allyl and -benzyl intermediates are
used and, in some reports, the alkylmetal species are intercepted by CO and coordinating
group (CG). While several strategies such as stabilizing alkylpalladium with ligands,
alkylmetal species lacking β-Hs, intercepting alkylpalladium species with CO, alkylmetal
species of 1st row transition metals and other reactions involving acylmetal species are used
in cyclization/coupling. These strategies are very important to suppress the β-hydride
elimination from the alkylmetal intermediates. In the subsequent sections, the early reports

8
and recently developed strategies to address existing problems in the context of
cyclization/coupling and three-component reactions will be discussed.
1.2.1 Cyclization/Coupling Reactions
Cyclization/coupling is one of the most well-studied reactions in the difunctionalization of
alkenes. In this method, two C-C bonds are easily constructed across the alkene forming
complex molecular structures using simple and readily available substrates.
Cyclization/coupling reactions are important for making carbocycles and heterocycles in
drugs, drug targets and natural products.39-40 These two-component reactions are more
favorable than the three-component reactions due to the intramolecular nature of the
reactants41-42 and therefore suffer less from β-hydride elimination. In these processes, the
tethered alkene undergoes cyclization by a radical or a nonradical migratory insertion
process43-44 to form a five-membered or a six-membered ring. These reactions proceed by
initial cyclization upon tethered alkenes generating cyclized alkylmetal intermediates,
which are subsequently intercepted by organic halides, enolates, CO, or organometallic
reagents to furnish dicarbofunctionalized products.
The catalytic cycle for cyclization/coupling involves four elementary steps with the
involvement of three organometallic species 1.8-1.10 prone to undergo -hydride
elimination. The cycle starts with the oxidative addition of organic halides to give the
alkylmetal complex 1.8 which is susceptible to undergo β-hydride elimination (Scheme
1.4). Then the migratory insertion of the alkylmetal species 1.8 into the alkene results in
the formation of cyclized alkylmetal species 1.9, another species that is also susceptible to

9
decompose by β-hydride elimination. Transmetalation of species 1.9 with organometallic
reagent gives species 1.10, a third species prone to undergo -hydride elimination. Finally,
the cyclized/cross-coupled product is formed after reductive elimination of complex 1.10.
Scheme 1.4. Problems in alkene dicarbofunctionalization by cyclization/coupling
In this catalytic cycle of cyclization/coupling reaction, there are three alkylmetal
intermediates which are potential for β-hydride elimination and results Heck product. Over
the years, several strategies have been implemented to address the issue of -hydride
elimination in cyclization/coupling reactions. These strategies are discussed in the
subsequent sections in detail with pertinent examples.
Scheme 1.5. Ligands used in cross-coupling reaction

10
1.2.1.1 Stabilizing Alkylpalladium with ligands
The addition of extraneous ligands (Scheme 1.5) is known to address the problem of β-
hydride elimination from alkylmetal intermediates in direct cross-coupling reactions. In
these reactions, the ligands not only block additional coordination sites and create high
coordination metal complexes but also prevent the C-H bonds in these complexes from
attaining syn-coplanarity with the metal and slow down β-hydride elimination process. A
similar strategy could also be expected to address the problem of -hydride elimination in
alkene dicarbofunctionalization. However, this strategy has been successful in alkene
difunctionalization reactions only in limited cases.
In one such example, bidentate phosphine ligands are used to intercept alkylpalladium
species in alkene tethered to dicarbonyl compounds with enolizable -hydrogens. In 1987,
Balme and coworkers45 developed a dicarbofunctionalization reaction of an alkene tethered
to dicarbonyl compounds bearing enolizable - hydrogen (DMSO pKa ~13) with aryl
iodides (Scheme 1.6).46-47 The reaction shows successful formation of cyclopentyl rings
containing exocyclic benzyl groups in presence of palladium catalyst. This transformation
was possible after electrophilic activation of alkenes by Ar-Pd(II)-X species followed by
anti-carbopalladation and stabilization of alkylpalladium species 1.11 with bidentate

11
phosphine ligands. The final product was obtained after the reductive elimination of species
1.11.
Scheme 1.6. Pd-catalyzed cyclization/coupling of alkenes tethered to enolates
Later, Balme and coworkers further expanded the idea of intercepting alkylpalladium
species by the use of bidentate ligands in ,-unsaturated dicarbonyl compounds for the
synthesis of 3-benzyltetrahydrofuran derivatives.67 In a similar page, Waser and coworkers
showed that the dicarbofunctionalization reaction of alkene tethered to enolates can be
possible using different type of ligands and electrophiles like bromoalkynylsilane.48 Not
only in the formation of five membered rings, Balme and coworkers further expanded their
work using the same strategy for the formation of six membered ring.49 This way they were
able to construct 5/5 or 5/6 bicyclic systems50-52 which is applicable to the synthesis of the
monounsaturated sesquiterpene, (±)-Δ9(12)-capnellene.53
Recently our group also developed a similar Pd-catalyzed cyclization/coupling reaction
(Scheme 1.7).54 Herein, the unactivated alkenes in N-allylarylacetamides are
difunctionalized, which furnish cyclized/arylated products by employing aryl halides and
enolates as carbon sources to create new C-C bonds. The formation of cyclization/coupling
product would be surprising if the reaction involved alkylpalladium intermediates 1.12
given the widespread understanding that the alkylpalladium intermediates are highly prone

12
to undergo -hydride elimination. Detailed mechanistic studies indeed revealed that the
reaction did not involve alkylpalladium intermediates 1.12, which would have formed by
migratory insertion of the tethered alkenes. The reaction proceeded through the formation
of expected Heck products, which subsequently underwent base-promoted conjugate
addition on the styryl moieties55 with the enolate 1.13.
Scheme 1.7. Pd-catalyzed cyclization/coupling of unactivated alkene tethered to enolates
1.2.1.2 Alkylmetal Species Lacking a -H
Another strategy to avoid β-hydride elimination is to utilize 2,2-disubstituted tethered
alkenes, which generate alkylmetal intermediates lacking -H’s. In 1988, Grigg and
coworkers explored this idea in alkenes tethered to aryl halides by intercepting an
alkylmetal species 1.14 that did not have a -H through the use of organotin reagents to
form different five and six-membered cyclized products (Scheme 1.8).30, 31 These types of
reactions with intermediates lacking -H’s enable transmetalation and reductive
elimination to form the cyclized products. In some cases, the use of 1,2-disubstituted
alkenes tethered to organic halides was also possible due to the unfavorable geometry for

13
-hydride elimination. This strategy was applied to derivatize complex molecules such as
sugars, aminoacids, nucleotides and purines.56-57
Scheme 1.8. Pd-catalyzed cyclization/coupling of alkenes tethered to aryl bromides with
organotin reagents
A similar approach was also applied in functionalizing activated alkenes tethered to aryl
halides. Grigg and coworkers demonstrated this by performing a cyclization/coupling
reaction of activated tethered alkenes to make five-membered cyclic products,58-59 and
Wilson was able to use this method to make six-membered cyclic products.60 Grigg and
coworkers were able to prove that this reaction proceeded via the formation of species 1.15,
an intermediate that lacks a -H (Scheme 1.9).
Scheme 1.9. Pd-catalyzed cyclization/coupling of activated alkenes tethered to aryl iodides
with organotin reagents
In 1989, Negishi and Zhang introduced a method where an alkylmetal species can be
intercepted by a variety of intramolecular carbon sources such as C-H bonds, alkynes and

14
benzynes. In this method, they reported a Pd-catalyzed cyclization cascade of alkynyl
alkenes tethered to vinyl iodides.61 Similarly, de Meijere and coworkers reported a Pd-
catalyzed cascade reaction for the sequential formation of four 6/5/6/3-membered rings in
one step.62
Additionally, Grigg and several other groups reported that a Pd-catalyzed cyclization of
aryl halides tethered to disubstituted alkenes could be intercepted intramolecularly by the
activated aryl C-H bonds of arenes.63-65 The authors further expanded this idea by
intercepting an alkylpalladium species with CN anions.66 Similarly, Xu and coworkers
trapped the similar alkylpalladium intermediates by C-H bonds of fluorinated arenes.67 Liu
and coworkers also developed a PhI(OPiv)2/CsF-promoted Pd-catalyzed
dicarbofunctionalization reaction of N-allylaniline by activating the ortho-C-H bond of
aniline followed by the cyclization and interception of an alkylpalladium species 1.16 with
acetonitrile (Scheme 1.10).68
Scheme 1.10. Pd-catalyzed aryl C-H bond cyclization/cyanomethylation
Moreover, Sodeoka and coworkers developed a Cu-catalyzed trifluoromethylation reaction
through the cyclization/coupling of alkenes tethered to arenes via ortho-C-H bond
activation. In this reaction, there is an interception of an alkylmetal species by Togni’s
reagent to form 5-and 6-membered carbo- and heterocycles.69

15
1.2.1.3 Interception of Alkylpalladium Species with CO
Out of the different strategies that are employed to intercept the alkylmetal species prior to
β-hydride elimination, use of CO to intercept an alkylpalladium species is also well-known.
In this method, alkylmetal species are intercepted prior to β-hydride elimination resulting
in an acylmetal species, which does not undergo β-hydride elimination. Negishi and Tour
developed a Pd-catalyzed double carbonylative cyclization/coupling reaction of alkenes
tethered to organic halides by intercepting an alkylpalladium species 1.17 (Scheme 1.11)
with CO. 70-73 They further expanded this reaction with the addition of one molecule of CO
into the alkenes tethered to organic halides.74
Scheme 1.11. Pd-catalyzed cyclization/coupling of alkenes tethered to alkyl iodides with
CO
Alexanian,75 and Ryu and coworkers76 also independently described a Mn and a Pd-
catalyzed dicarbofunctionalization reaction of unactivated alkenes tethered to alkyl halides
in the presence of CO. Furthermore, Suzuki and Miyaura developed a carbonylative
cyclization/coupling reaction of 9-alkyl-9-borabicyclo[3.3.1]nonane (9-alkyl-9-BBN)
reagent under CO atmosphere in the presence of UV light (Scheme 1.12).77, 78 The reaction
was believed to proceed through the presence of a radical intermediate, which cyclize and
inserts CO to form an acylpalladium species 1.18.
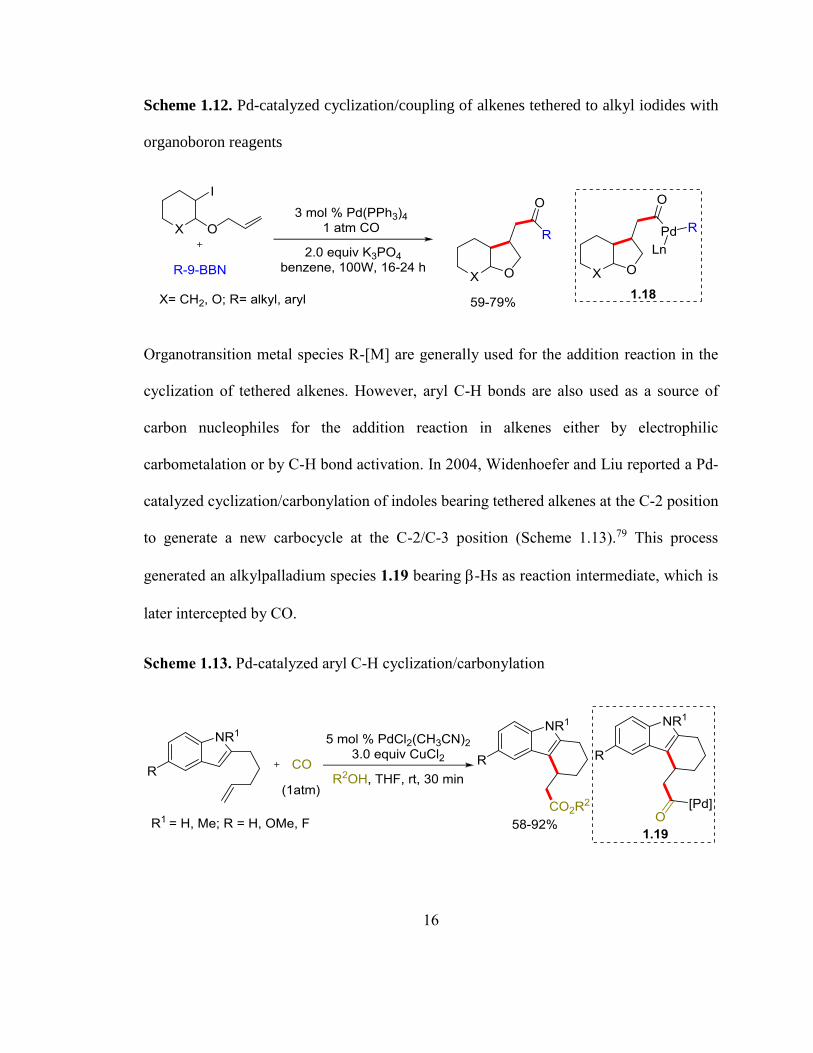
16
Scheme 1.12. Pd-catalyzed cyclization/coupling of alkenes tethered to alkyl iodides with
organoboron reagents
Organotransition metal species R-[M] are generally used for the addition reaction in the
cyclization of tethered alkenes. However, aryl C-H bonds are also used as a source of
carbon nucleophiles for the addition reaction in alkenes either by electrophilic
carbometalation or by C-H bond activation. In 2004, Widenhoefer and Liu reported a Pd-
catalyzed cyclization/carbonylation of indoles bearing tethered alkenes at the C-2 position
to generate a new carbocycle at the C-2/C-3 position (Scheme 1.13).79 This process
generated an alkylpalladium species 1.19 bearing -Hs as reaction intermediate, which is
later intercepted by CO.
Scheme 1.13. Pd-catalyzed aryl C-H cyclization/carbonylation

17
1.2.1.4 Alkylmetal Species of First Row Transition Metals
In recent years the use of first row transition metals such as manganese, iron, cobalt, nickel,
and copper have attracted attention due to their natural abundance, unique reactivity and
bio-compatibility.80 These metals have afforded a general reaction for alkene
dicarbofunctionalization in substrates that generate alkylmetal intermediates having -Hs.
There is no need for special consideration in substrate design, reaction condition, or
heteroatom coordination. The ability of these TMs to perform such transformations without
complications from -hydride elimination is because of their potential ability to reduce
organic halides by a single electron transfer,81 which form carbon-centered radicals.
Addition of carbon-centered radicals to alkenes generate alkylmetal intermediates. These
intermediates when compared to analogous alkylpalladium intermediates, have a higher
energy barrier for -hydride elimination and a lower energy barrier for reductive
elimination.82
Dicarbofunctionalization reactions of alkenes tethered to alkyl halides using first row TMs
that generate alkylmetal intermediates bearing -H’s are well-known processes in
cyclization/coupling. In 1994, this idea was first reported by Delgado and coworkers for
the stoichiometric Ni-mediated cyclization/carbonylation and cyclization/cyanation of
alkenes tethered to N-benzylated vinyl bromides.83-84 In 2001, Oshima and coworkers
developed a Co-catalyzed dicarbofunctionalization reaction of tethered alkene with aryl
Grignard reagent (Scheme 1.14).85 This transformation was proposed to proceed through a
radical pathway by the formation of an alkyl[Co] complex 1.20. Later, they further
expanded this chemistry using vinylalkylsilyl ethers bearing tethered alkyl iodides as

18
substrates.86 Similarly, Kang and coworkers reported an Fe-catalyzed cyclization/coupling
reaction of alkenes tethered to alkyl iodides with aryl Grignard reagents.87
Scheme 1.14. Co-catalyzed cyclization/coupling of alkenes tethered to alkyl halides with
arylmagnesium reagents
In 2007, Cárdenas and coworkers published a Ni-Catalyzed cyclization/coupling reaction
of alkenes tethered to alkyl halides with alkylzinc reagents (Scheme 1.15).88 The
dicarbofunctionalized product was formed after reductive elimination of an alkylnickel
intermediate 1.21. Mechanistic investigations based on a ring-opening experiment and
TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) trapping experiment showed that there is
generation of an alkyl radical in the reaction. Later, the same author expanded this
chemistry with aryl Grignard reagents using Ni/TMEDA (N,N,N′,N′-
tetramethylethylenediamine) as a catalyst.89
Scheme 1.15. Ni-catalyzed cyclization/coupling of alkenes tethered to alkyl bromides with
alkylzinc reagents

19
The use of first row TMs for the reductive dicarbofunctionalization reaction of tethered
alkenes with various electrophiles is another important area which has been developed
recently. Peng and coworkers reported a Ni-catalyzed cyclization/coupling reaction of
tethered alkenes with aryl iodides (Scheme 1.16). Here, ethyl crotonate is used as a ligand
and zinc powder as a stoichiometric reductant to regenerate the catalyst.90-93 The formation
of the dicarbofunctionalized product is possible after reductive elimination of an alkyl[Ni]
species 1.22. In 2018, Diao and coworkers also reported a Ni-catalyzed reductive
dicarbofunctionalization of tethered alkenes with aryl bromides.94-95 Later, the same
authors also developed an enantioselective reductive dicarbofunctionalization reaction of
activated alkenes.96
Scheme 1.16. Ni-catalyzed reductive cyclization/coupling of alkenes with aryl iodides
Additionally, Wang and coworkers also developed a Ni-catalyzed reductive
dicarbofunctionalization of alkenes with alkyl bromides.97 The same authors later
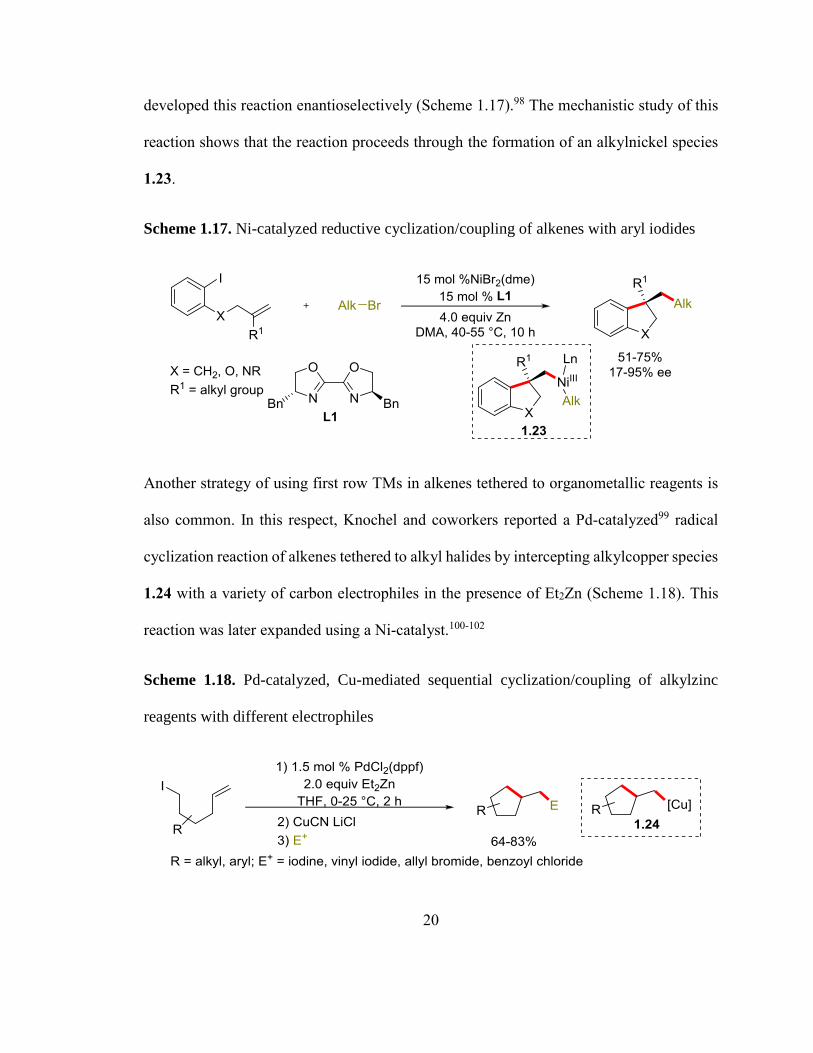
20
developed this reaction enantioselectively (Scheme 1.17).98 The mechanistic study of this
reaction shows that the reaction proceeds through the formation of an alkylnickel species
1.23.
Scheme 1.17. Ni-catalyzed reductive cyclization/coupling of alkenes with aryl iodides
Another strategy of using first row TMs in alkenes tethered to organometallic reagents is
also common. In this respect, Knochel and coworkers reported a Pd-catalyzed99 radical
cyclization reaction of alkenes tethered to alkyl halides by intercepting alkylcopper species
1.24 with a variety of carbon electrophiles in the presence of Et2Zn (Scheme 1.18). This
reaction was later expanded using a Ni-catalyst.100-102
Scheme 1.18. Pd-catalyzed, Cu-mediated sequential cyclization/coupling of alkylzinc
reagents with different electrophiles

21
Brown and coworkers reported a cyclization/coupling reaction of tethered alkenes with aryl
iodides by intercepting an alkylcopper species 1.25 (Scheme 1.19).103 Later, the same
authors extended this work enantioselectively by using CuBr/BenzP* as a chiral catalyst.104
Scheme 1.19. Cyclization/coupling of aryl-9-BBN reagents with aryl iodides
Similar work in enantioselective cyclization/coupling of tethered alkenes with primary and
secondary alkyl halides was developed by Fu and Cong using a Ni/diamine*-catalyst
(Scheme 1.20).105 The catalytic cycle of the reaction shows the formation of the product
from an alkylnickel species 1.26.
Scheme 1.20. Enantioselective Ni-catalyzed cyclization/coupling with alkyl bromides
In this respect, our group also disclosed a Cu-catalyzed cyclization/coupling of tethered
alkenes with aryl iodides (Scheme 1.21).106 The mechanism shows that the reaction
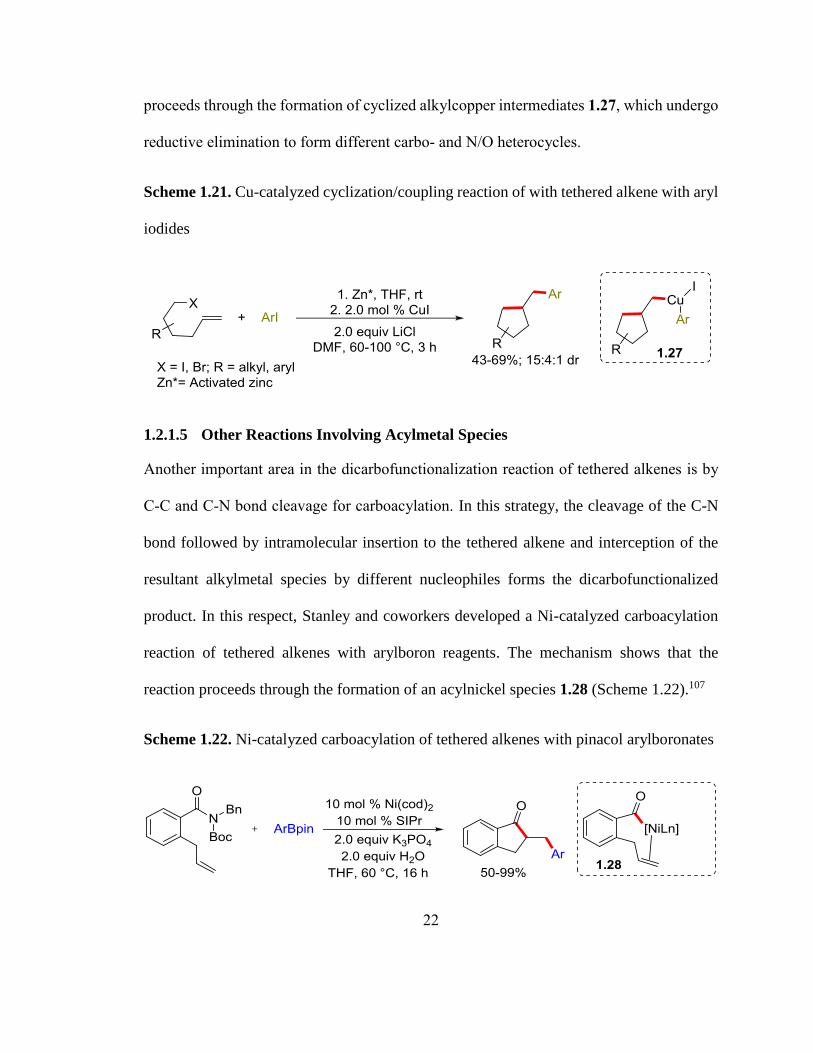
22
proceeds through the formation of cyclized alkylcopper intermediates 1.27, which undergo
reductive elimination to form different carbo- and N/O heterocycles.
Scheme 1.21. Cu-catalyzed cyclization/coupling reaction of with tethered alkene with aryl
iodides
1.2.1.5 Other Reactions Involving Acylmetal Species
Another important area in the dicarbofunctionalization reaction of tethered alkenes is by
C-C and C-N bond cleavage for carboacylation. In this strategy, the cleavage of the C-N
bond followed by intramolecular insertion to the tethered alkene and interception of the
resultant alkylmetal species by different nucleophiles forms the dicarbofunctionalized
product. In this respect, Stanley and coworkers developed a Ni-catalyzed carboacylation
reaction of tethered alkenes with arylboron reagents. The mechanism shows that the
reaction proceeds through the formation of an acylnickel species 1.28 (Scheme 1.22).107
Scheme 1.22. Ni-catalyzed carboacylation of tethered alkenes with pinacol arylboronates

23
Douglas and Dreis also reported a Rh-catalyzed carboacylation reaction of 8-quinolinyl
ketones bearing an ortho-allylphenyl ether via C-C bond cleavage (Scheme 1.23).108 The
mechanism of this reaction shows that there is the formation of an acyl[Rh] species 1.29,
an intermediate which avoids -hydride elimination.
Scheme 1.23. Rh-catalyzed intramolecular arylacylation
Similarly, Takemoto and coworkers,109 Murakami and coworkers,110 Cramer and
coworkers,111-112 and Dong and coworkers113 disclosed a cyclization reaction of a tethered
alkene via C-C bond cleavage.
1.2.2 Three-Component Reactions
The three-component reaction is a well-known reaction where an alkene, electrophile and
a nucleophile are involved to construct two C-C bonds across an alkene. These reactions
are very important for making complex carbon skeletons. Different types of readily
available electrophiles like organic halides, triflates, and nucleophiles like organometallic
reagents and enolates can be utilized in three-component reactions. This method is very
challenging due to the intermolecular nature of the reactants. In this process, -hydride
elimination and cross-coupling are more pronounced due to inefficient alkene binding

24
which leads to the undesired products i.e. Heck products and cross-coupling products. To
this date, three-component reactions in activated alkenes and alkenes bearing coordination
groups are well developed but the three-component reaction of unactivated alkenes are not
fully developed. Also, three-component reductive and oxidative dicarbofunctionalization
reactions114 are also known. Different strategies used in three-component alkene
dicarbofunctionalization reactions are discussed below.
1.2.2.1 Geometrically Constrained Alkylmetal Intermediates
The complications from -hydride elimination was avoided using geometrically strained
molecules like norbornene and norborndiene. These bicyclic molecules suppress -hydride
elimination from the alkylmetal species by restricting bond rotations that prevent the
alkylpalladium species 1.30 in attaining the syn-coplanarity with a -H required for -
hydride elimination. The earliest discovery was made in 1982 by Chiusoli and Catellani115-
116 for a Pd-catalyzed dicarbofunctionalization reaction of norbornene and norbornadiene
with arylbromide and sodium tetrafluoroborate (Scheme 1.24). This is a modular example
of three-component dicarbofunctionalization reaction, which formed cis exo-products.
Later, this reaction was further extended using a Ni-catalyst.117 Also, norbornadiene
substrates were used by Torii,39 Kang,118 and Goodson119 for the three-component alkene
dicarbofunctionalization reactions.
Scheme 1.24. Pd-catalyzed dicarbofunctionlization of norbornene and norbornadiene

25
1.2.2.2 -Allyl and -Benzyl Intermediates
Another approach to overcome -hydride elimination is by forming -allyl/benzyl-[M]
intermediates in the reaction. Conjugated dienes were used as a substrate where an
additional alkene of the diene forms a -allyl-[M] species after Heck carbometallation
process. Takai and coworkers120 first reported a 1,2-dicarbofunctionalization of 1,3-dienes
with alkyl halides and benzaldehyde using stoichiometric amount of chromium catalyst.
Later, Oshima and coworkers reported Co-catalyzed regioselective
dicarbofunctionalization of conjugated dienes with Me3SiCH2MgCl and alkyl halides
(Scheme 1.25).121 This reaction is believed to proceed through the formation of alkyl
radicals and alkylcobalt species, which later stabilized by -allyl-[Co] species 1.31.
Similarly, Kambe and coworkers developed a 1,4-difunctionalization reaction of
conjugated dienes with alkyl halides and PhMgBr/PhZnCl.122-123
Scheme 1.25. Co-catalyzed dicarbofunctionalization reaction of 1,3-diene

26
Sigman and coworkers also reported a Pd-catalyzed dicarbofunctionalization reaction of 1-
3-dienes with vinyl triflates and aryl boronic acids (Scheme 1.26).124-127 The mechanistic
studies show that there is the formation of a -allylpalladium species 1.32, a stable
intermediate which prevent -hydride elimination and enables the pathway for the
dicarbofunctionalization reaction.
Scheme 1.26. Pd-catalyzed dicarbofunctionalization reaction of 1,3-diene with arylboronic
acids and vinyl triflates
Alike dienes, styrenes were also used as a substrate for the Pd-catalyzed three-component
reaction. The mechanism of the reaction shows the formation of a -benzylpalladium 1.33
species.128 In 2017, Song and coworker reported a dicarbofunctionalization reaction of
styrenes with aryl boronic acids and vinyl triflates (Scheme 1.27).129
Scheme 1.27. Pd-catalyzed dicarbofunctionalization reaction of styrenes with arylboronic
acids and vinyl triflates

27
Carbon-centered radicals can be used for the dicarbofunctionalization reaction of styrenes.
In 2013, Liu and coworkers developed a Cu-catalyzed trifluoromethylarylation of both
unactivated alkenes and styrenes with Tognis reagents and arylboronic acids (Scheme
1.28).130 The mechanistic insight of this reaction shows that Togni’s reagent generates a
trifluoromethyl radical which would be added to the alkenes to form alkylArCu(II) species
1.34 and reductively eliminate to form the product.
Scheme 1.28. Cu-catalyzed trifluoromethylarylation of styrenes
This trifluoromethylation was later developed enantioselectively131 by the same authors
and this area was further shed into light form several authors like Szabó,132 Liu,133-134 and
Liang135 for the trifluoromethylcyanation reaction in styrenes.
In 1998, Kambe and coworkers reported a Cp2TiCl2 catalyzed dicarbofunctionalization of
styrenes with two different types of alkyl halides in the presence of stoichiometric amount
of nBuMgCl (Scheme 1.30).136 The reaction is proposed to proceed by the addition of alkyl
radicals to styrenes followed by transmetalation of alkyl-[Ti] intermediates 1.35 to
nBuMgCl and reaction of the resultant alkyl-MgCl with alkyl halides.
Scheme 1.29. Ti-catalyzed reductive dialkylation of styrenes with alkyl bromides
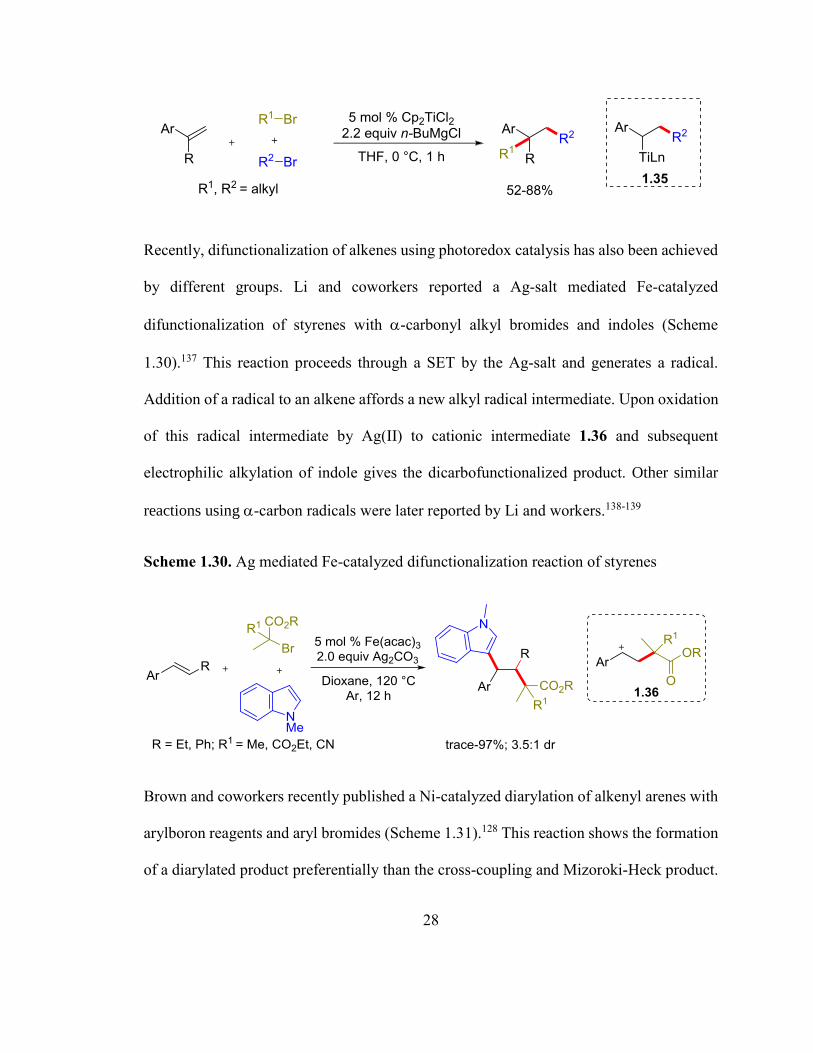
28
Recently, difunctionalization of alkenes using photoredox catalysis has also been achieved
by different groups. Li and coworkers reported a Ag-salt mediated Fe-catalyzed
difunctionalization of styrenes with -carbonyl alkyl bromides and indoles (Scheme
1.30).137 This reaction proceeds through a SET by the Ag-salt and generates a radical.
Addition of a radical to an alkene affords a new alkyl radical intermediate. Upon oxidation
of this radical intermediate by Ag(II) to cationic intermediate 1.36 and subsequent
electrophilic alkylation of indole gives the dicarbofunctionalized product. Other similar
reactions using -carbon radicals were later reported by Li and workers.138-139
Scheme 1.30. Ag mediated Fe-catalyzed difunctionalization reaction of styrenes
Brown and coworkers recently published a Ni-catalyzed diarylation of alkenyl arenes with
arylboron reagents and aryl bromides (Scheme 1.31).128 This reaction shows the formation
of a diarylated product preferentially than the cross-coupling and Mizoroki-Heck product.

29
The proposed reaction mechanism in the catalytic cycle shows the formation of an
alkylnickel intermediates 1.37 to furnish the diarylated product. Also a visible light
mediated three-component dicarbofunctionalization reaction using benzylic radicals has
recently been developed by Glorius and coworkers.140
Scheme 1.31. Ni-catalyzed diarylation of vinylarene
1.2.2.3 Stabilizing Alkylmetal Species as Enolates
Conjugate addition reactions141 of organometallic nucleophiles or nucleophilic radicals to
,-unsaturated substrates are also an important and well-known method for building two
C-C bonds.142 In this reaction, the nucleophile adds to the -carbon of the electron
deficient-alkene giving the carbanion followed by trapping with the electrophile to give -
substituted product. This method is very potential for the formation of various bioactive
molecules by employing varieties of donor and acceptor substrates. The very first example
of conjugate addition was reported in 1883 by Komemnos.143 TM-catalyzed and TM-free
organocatalytic asymmetric synthesis144 has significantly developed over the past decades
to enable the molecular complexity from readily available starting materials. Alexakis and
coworkers developed a Lewis acid activated copper-catalyzed tandem conjugate addition
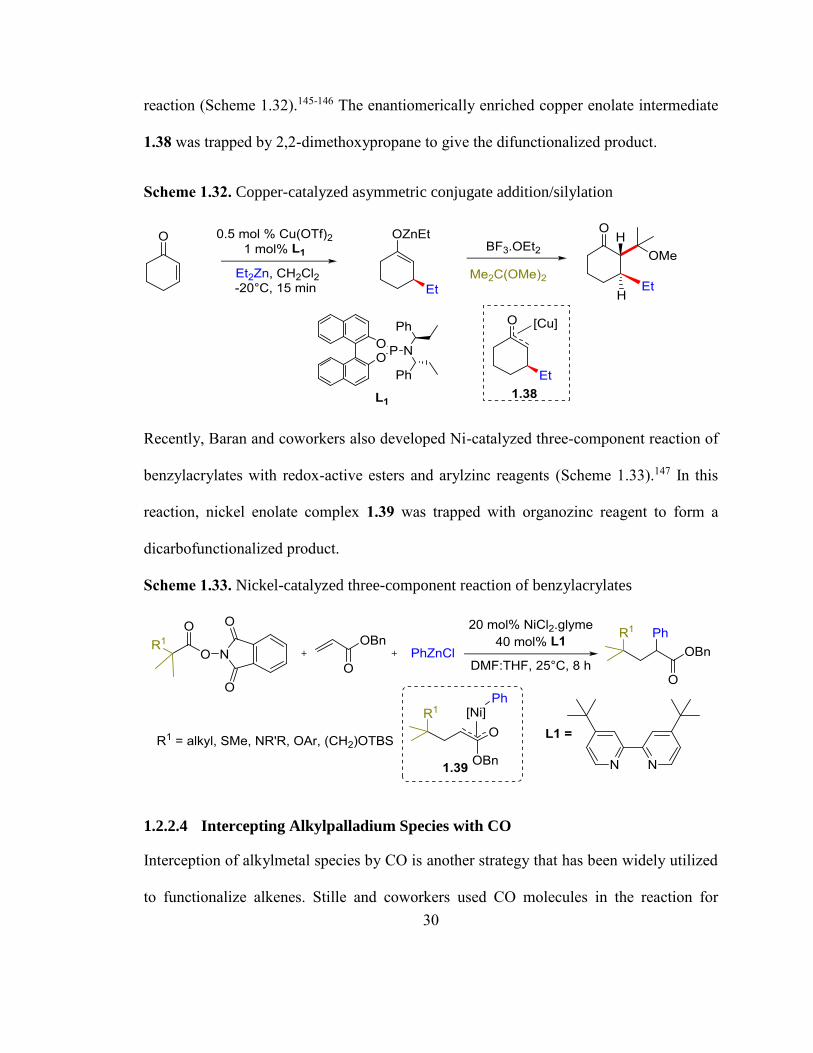
30
reaction (Scheme 1.32).145-146 The enantiomerically enriched copper enolate intermediate
1.38 was trapped by 2,2-dimethoxypropane to give the difunctionalized product.
Scheme 1.32. Copper-catalyzed asymmetric conjugate addition/silylation
Recently, Baran and coworkers also developed Ni-catalyzed three-component reaction of
benzylacrylates with redox-active esters and arylzinc reagents (Scheme 1.33).147 In this
reaction, nickel enolate complex 1.39 was trapped with organozinc reagent to form a
dicarbofunctionalized product.
Scheme 1.33. Nickel-catalyzed three-component reaction of benzylacrylates
1.2.2.4 Intercepting Alkylpalladium Species with CO
Interception of alkylmetal species by CO is another strategy that has been widely utilized
to functionalize alkenes. Stille and coworkers used CO molecules in the reaction for

31
intercepting alkylmetal species to generate alkylacylmetal intermediates 1.40, which are
not susceptible to undergo β-hydride elimination. They reported the addition of two alkoxy
groups in unactivated alkenes using carbon monoxide and methanol in presence of
palladium chloride and copper chloride (Scheme 1.34).148 Later, Ishii and coworkers
developed the similar dicarboalkoxylation reaction of unactivated alkenes using CO and
methanol in the presence of palladium catalyst, and molybdovanadophosphate as an
oxidant.149
Scheme 1.34. Palladium catalyzed decarboxylation of unactivated alkenes
The dicarbofunctionalization reactions of completely unbiased alkenes that do not have
any means to stabilize the alkylmetal species have also been developed. Usually reaction
without any stabilizing factor lead to the formation of a Heck product. However, some
report shows the formation of 1,1 difunctionalized product instead of 1,2-difunctionalized
product. Sigman and coworkers reported a Pd-catalyzed 1, 1-dicarbofunctionalization
reaction of unactivated alkenes with vinyl triflates and arylboronic acids.124 The reaction
is possible due to β-hydride elimination and re-insertion of metal-hydrides (Pd-H) into
alkenes forms an allylpalladium species 1.41, which undergoes transmetalation followed
by reductive elimination to result 1,1-dicarbofunctionalized products (Scheme 1.35).150

32
Scheme 1.35. Palladium catalyzed 1,1-difunctionalization of simple alkene
1.2.2.5 Stabilizing Alkylmetal Species by Coordination Group
Intramolecular coordination using heteroatom with TMs are one of the well-known
examples for the installment of two different carbon entities across alkenes. Due to this,
stable transient metallacycles are formed,151 which slow down the -hydride elimination.112
The rate of decomposition of metallacyles by -hydride elimination is slower by almost
four orders than their acyclic variants due to restricted bond rotations, which prevent
attainment of favorable geometry for -hydride elimination.152-157
The history for the use of coordination in dicarbofunctionalization started from Larhed and
coworkers. They reported a Pd-catalyzed oxidative diarylation of alkenes tethered to
dialkylated amines with arylboronic acids (Scheme 1.36).158 The mechanistic study shows
that β-hydride elimination from the alkylpalladium species was prevented due to the
formation of N-coordinated palladacycle 1.42.
Scheme 1.36. Palladium catalyzed coordination assisted diarylation of vinyl ethers

33
The use of a coordinating group was further explored in a Ni-catalyzed
difluoroalkylarylation of enamides by Zhang and coworkers in 2016. They proposed that
the oxygen in the enamide acts as a coordinating group to form a transient nickellacycle
1.43, which prevent β-hydride elimination (Scheme 1.37).159
Scheme 1.37. Ni-catalyzed dicarbofunctionalization of enamide
Our group has been broadly involved in the regioselective difunctionalization of mildly
activated and unactivated alkenes using coordination groups. Recently, we reported Ni-
catalyzed diarylation of mildly activated 2-vinyl aldimines with arylzinc reagents and aryl
iodides. The proposed mechanism shows that imines as a coordinating group are
instrumental for the formation of 6-membered nickellacycle 1.44, which would stabilize
the alkylmetal species from undergoing β-hydride elimination (Scheme 1.38).160
Scheme 1.38. Nickel-catalyzed 1,2-diarylation of vinylarenes using coordinating group

34
Similarly, our group also reported a dicarbofunctionalization reaction of pyridylvinyl
silanes where pyridine acts as a coordination group for the formation of 5-membered
transient nickellacycle 1.45 to prevent β-hydride elimination (Scheme 1.39).161
Scheme 1.39. Pyridine assisted Nickel-catalyzed 1,2-diarylation of vinylsilanes
Our group was not limited in the functionalization of mildly activated alkenes, we also
explored the coordination approach for the functionalization of unactivated alkenes. We
reported a Ni-catalyzed 1,3-diarylation of N-phenylhex-5-en-2-imine where an imine is
used as a coordinating group (Scheme 1.42). The 1,3-dicarbofunctionalized product is
possible due to ring contraction from fluxional six-membered nickellacycle to five-
membered nickellacycle 1.46 through β-hydride elimination and re-insertion into the
alkene (Scheme 1.40).162
Scheme 1.40. Nickel-catalyzed 1,3-diarylation of unactivated alkenes in ketimines

35
Later, our group further disclosed the chemistry of 1,2-difunctionalization using alkenyl
ketimines in the presence of a bimetallic catalyst.163 The mechanistic study further opens
the door of the formation of cationic nickel species 1.47 (Scheme 1.41), which speeds up
the alkene binding and transmetalation, and slows down the pathway for β-hydride
elimination. All these reactions using coordinating group can be easily removed under mild
condition to give the difunctionalized products.
Scheme 1.41. Nickel-catalyzed 1,2-diarylation of unactivated alkenes in ketimines
Similarly, Engle and coworkers also reported a dicarbofunctionalization reaction by using
8-aminoquinolinamide as a coordinating group. They reported a Ni-catalyzed
alkylarylation reaction of 8-aminoquinolinamide using aryl iodides and alkylzinc reagents.
The reaction forms a transient metallacycle 1.48 to prevent the β-hydride elimination
(Scheme 1.42).164 They further expanded this work for the dialkylation reaction165 of 8-
aminoquinolinamide and diarylation of alkenyl amides using a Ni-catalyst.166
Scheme 1.42. Nickel-catalyzed alkylarylation of 8-aminoquinolinamide
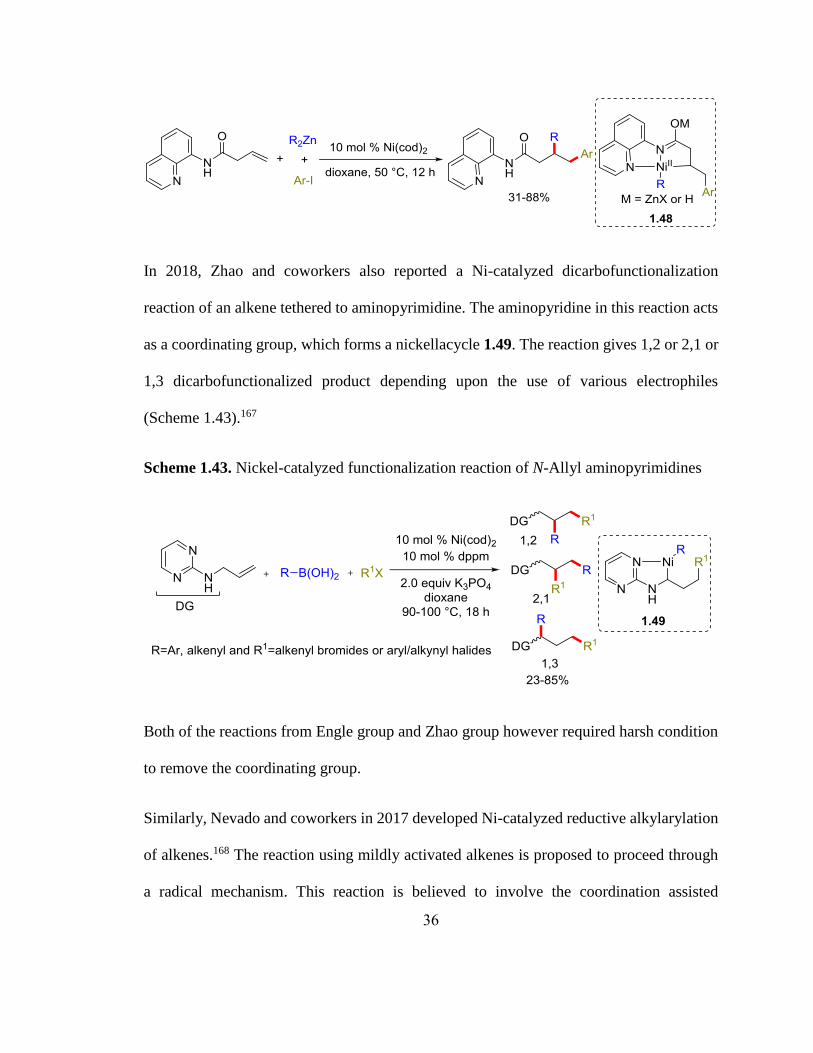
36
In 2018, Zhao and coworkers also reported a Ni-catalyzed dicarbofunctionalization
reaction of an alkene tethered to aminopyrimidine. The aminopyridine in this reaction acts
as a coordinating group, which forms a nickellacycle 1.49. The reaction gives 1,2 or 2,1 or
1,3 dicarbofunctionalized product depending upon the use of various electrophiles
(Scheme 1.43).167
Scheme 1.43. Nickel-catalyzed functionalization reaction of N-Allyl aminopyrimidines
Both of the reactions from Engle group and Zhao group however required harsh condition
to remove the coordinating group.
Similarly, Nevado and coworkers in 2017 developed Ni-catalyzed reductive alkylarylation
of alkenes.168 The reaction using mildly activated alkenes is proposed to proceed through
a radical mechanism. This reaction is believed to involve the coordination assisted
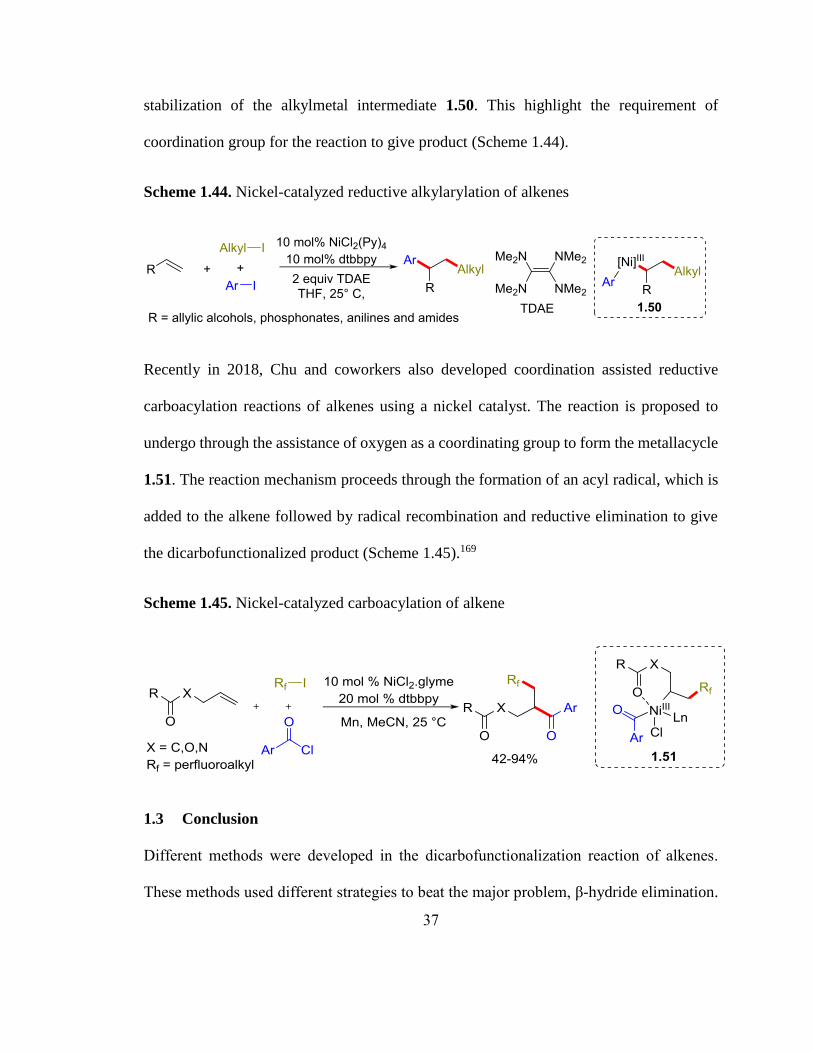
37
stabilization of the alkylmetal intermediate 1.50. This highlight the requirement of
coordination group for the reaction to give product (Scheme 1.44).
Scheme 1.44. Nickel-catalyzed reductive alkylarylation of alkenes
Recently in 2018, Chu and coworkers also developed coordination assisted reductive
carboacylation reactions of alkenes using a nickel catalyst. The reaction is proposed to
undergo through the assistance of oxygen as a coordinating group to form the metallacycle
1.51. The reaction mechanism proceeds through the formation of an acyl radical, which is
added to the alkene followed by radical recombination and reductive elimination to give
the dicarbofunctionalized product (Scheme 1.45).169
Scheme 1.45. Nickel-catalyzed carboacylation of alkene
1.3 Conclusion
Different methods were developed in the dicarbofunctionalization reaction of alkenes.
These methods used different strategies to beat the major problem, β-hydride elimination.

38
The strategies used to address the major problem in three-component and
cyclization/coupling reaction were discussed.

39
Chapter 2. Three-Component Alkylarylation of Vinyl Arenes
2.1 Introduction
Vinylarenes serve as one of the most synthetically valuable sources of alkenes which could
attenuate the effects of -hydride elimination by in situ formation of -benzyl-[M]
species.114, 129, 170 Dicarbofunctionalization of alkenes in vinylarenes could afford a concise
synthetic process to construct a 1,1-diarylalkane scaffold, an important motif that is widely
present in various bioactive molecules against breast cancer (MCF-7), lung cancer (H-460),
brain cancer (SF-268), and membrane protein FLAP (5-Lipoxygenase Acting Protein)
(Figure 2-1).171-173
Figure 2-1. Application of dicarbofunctionalization in the synthesis of drug molecules
Although there are some examples in the functionalization of vinylarenes, which proceed
for homodicarboalkoxylation,148, 174 homodiarylation/homodivinylation,114
trifluoromethylarylation,130-131 trifluoromethylcyanation132, 134-135 and vinylarylation,129
there are not any methods for three-component catalytic functionalization of alkenes in
vinylarenes with alkyl halides.175
2.2 Ni-Catalyzed Alkylarylation of Vinylarenes

40
In this respect, we developed a Ni-catalyzed alkylarylation of vinylarenes with primary,
secondary and tertiary alkyl halides, and arylzinc reagents that furnishes diversely
substituted 1,1-diarylalkanes via the formation of two C(sp3)-C(sp3) and C(sp3)-C(sp2)
bonds in one step. We started our attempts to alkylarylate 2-vinylnaphthanlene 2.1 with
cyclohexyl iodide and phenylzinc iodide with different TM catalyst. After screening and
optimization, we were pleased to find that the reaction was efficiently catalyzed by 5 mol%
NiBr2 in NMP at room temperature affording the alkylarylated product 2.2 in 81% yield
with 2 equiv. each of cyclohexyl iodide and PhZnI (entry 1). Lowering the amount of
cyclohexyl iodide or PhZnI decreased the yield (entries 2-3). Neither alkyl chloride nor
alkyl fluoride give the product (entry 4). The reaction is also catalyzed by Ni(0) sources
such as Ni(cod)2 and (Ph3P)4Ni in similar yields (entries 5-6). Fe, Cu and Pd-catalysts did
not form any product (entry 8) while CoCl2 also catalyzed the reaction in moderate yields
(entry 7). There is no reaction without the presence of NiBr2 (entry 9). The reaction also
works well in DMA with similar yield and only a small amount of the product 2.1 was
formed when THF or toluene is used as a solvent (entries 10-11).
Table 2.1. Optimization of reaction conditionsa

41
a0.1 mmol scale reactions. bIsolated yield (0.5 mmol scale) in parenthesis
After having the optimized conditions, we examined the scope of the reaction of 2-
vinylnaphthalene with different alkyl halides and arylzinc reagents (Table 2.2). The
reaction proceeds well with primary and secondary alkyl halides (I, Br) by generating
secondary and tertiary carbon centers. The reaction also tolerates various functional groups
such as OTBS (2.5), phthalimide (2.6), CF3 (2.7-2.8) and OMe (2.4, 2.10), and ArZnI
bearing ortho-OMe (2.4).
Table 2.2. Scope with RX and ArZnIa
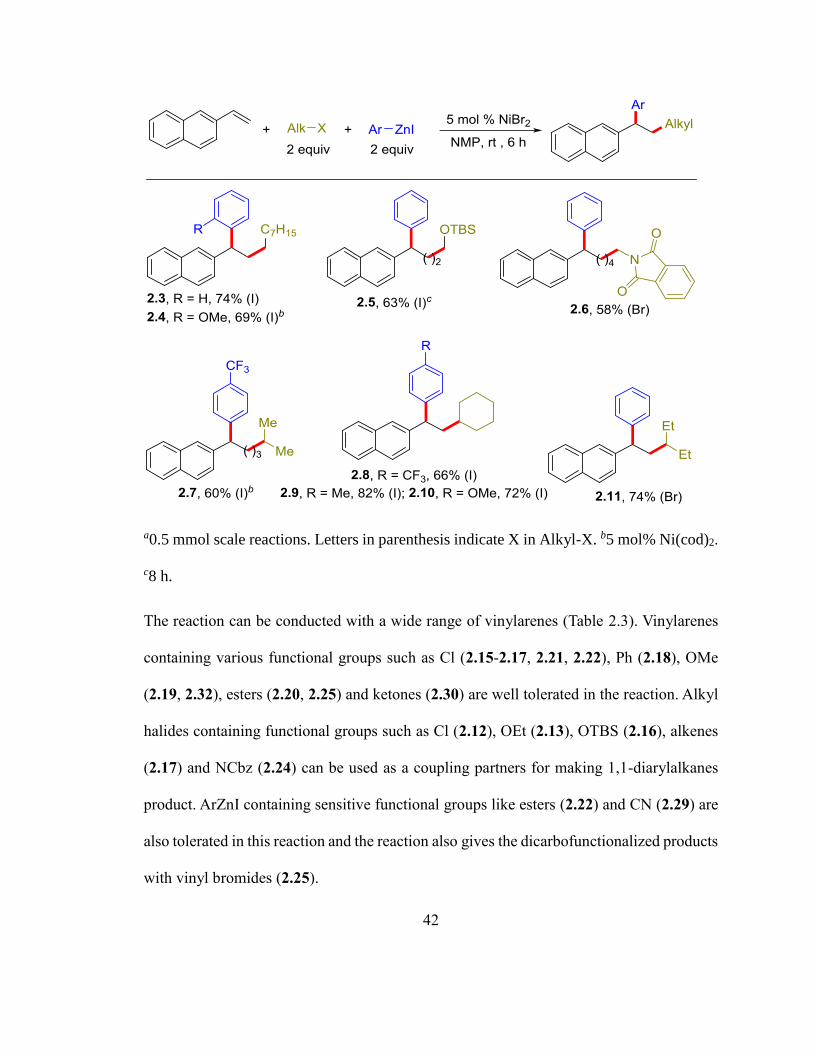
42
a0.5 mmol scale reactions. Letters in parenthesis indicate X in Alkyl-X. b5 mol% Ni(cod)2.
c8 h.
The reaction can be conducted with a wide range of vinylarenes (Table 2.3). Vinylarenes
containing various functional groups such as Cl (2.15-2.17, 2.21, 2.22), Ph (2.18), OMe
(2.19, 2.32), esters (2.20, 2.25) and ketones (2.30) are well tolerated in the reaction. Alkyl
halides containing functional groups such as Cl (2.12), OEt (2.13), OTBS (2.16), alkenes
(2.17) and NCbz (2.24) can be used as a coupling partners for making 1,1-diarylalkanes
product. ArZnI containing sensitive functional groups like esters (2.22) and CN (2.29) are
also tolerated in this reaction and the reaction also gives the dicarbofunctionalized products
with vinyl bromides (2.25).
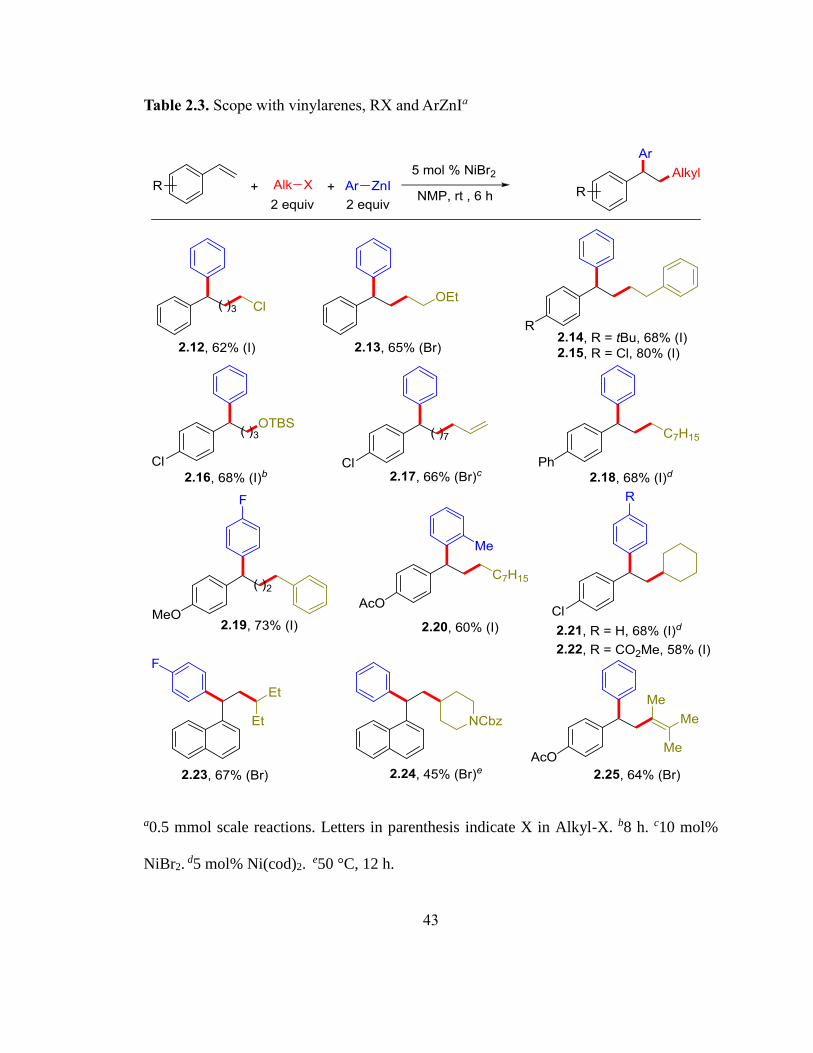
43
Table 2.3. Scope with vinylarenes, RX and ArZnIa
a0.5 mmol scale reactions. Letters in parenthesis indicate X in Alkyl-X. b8 h. c10 mol%
NiBr2. d5 mol% Ni(cod)2.
e50 °C, 12 h.

44
The reactions of tert-alkyl halides were examined and proved to be more challenging than
those of the primary and secondary alkyl halides due to the increased difficulty to form
quaternary carbon centers. After catalyst optimization, we found that (Ph3P)2NiCl2 was an
excellent catalyst for the coupling with tert-alkyl halides, which afforded products in good
to excellent yields (2.26-2.32).
To demonstrate the application of this reaction in the context of complex molecules, two
styryl scaffolds were installed in non-steroidal anti-inflammatory drugs (NSAIDs)
indometacin and tolmetin. These derivatized substrates were alkylarylated efficiently with
ArZnI and primary and secondary alkyl halides, which afforded the corresponding products
(2.34-2.36) in 46-67% yields.
Table 2.4. Scope with vinylarenes, RX and ArZnIa
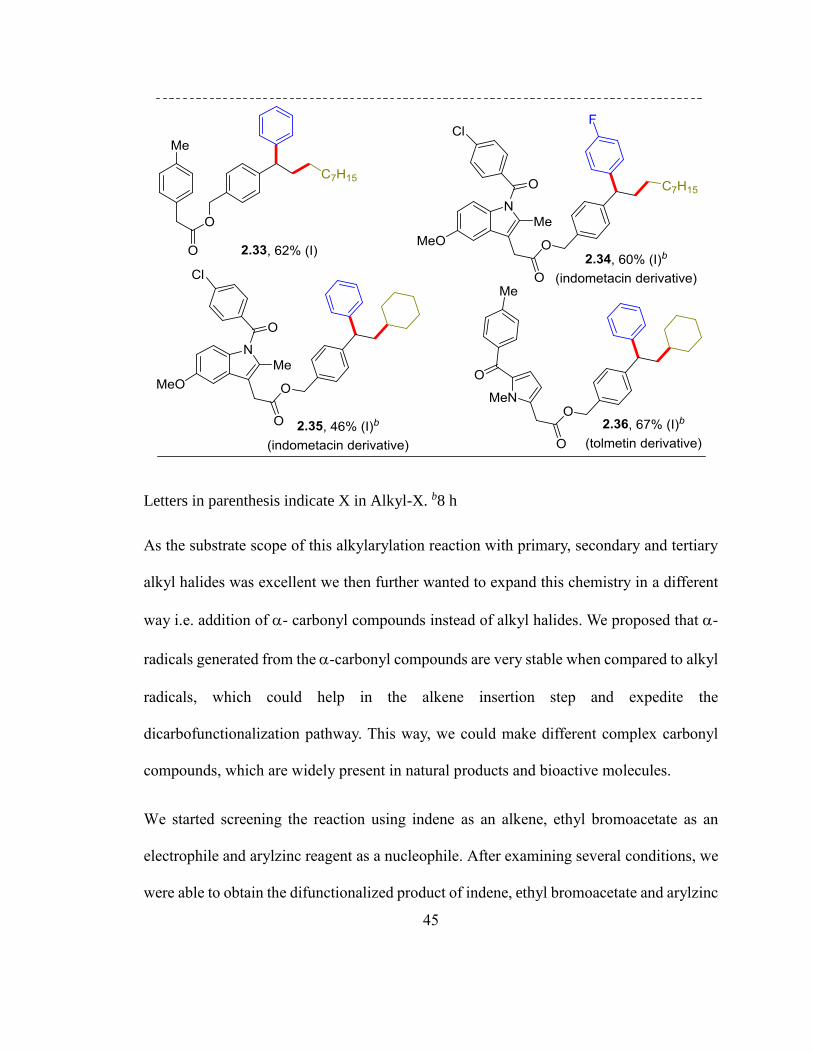
45
Letters in parenthesis indicate X in Alkyl-X. b8 h
As the substrate scope of this alkylarylation reaction with primary, secondary and tertiary
alkyl halides was excellent we then further wanted to expand this chemistry in a different
way i.e. addition of - carbonyl compounds instead of alkyl halides. We proposed that -
radicals generated from the -carbonyl compounds are very stable when compared to alkyl
radicals, which could help in the alkene insertion step and expedite the
dicarbofunctionalization pathway. This way, we could make different complex carbonyl
compounds, which are widely present in natural products and bioactive molecules.
We started screening the reaction using indene as an alkene, ethyl bromoacetate as an
electrophile and arylzinc reagent as a nucleophile. After examining several conditions, we
were able to obtain the difunctionalized product of indene, ethyl bromoacetate and arylzinc
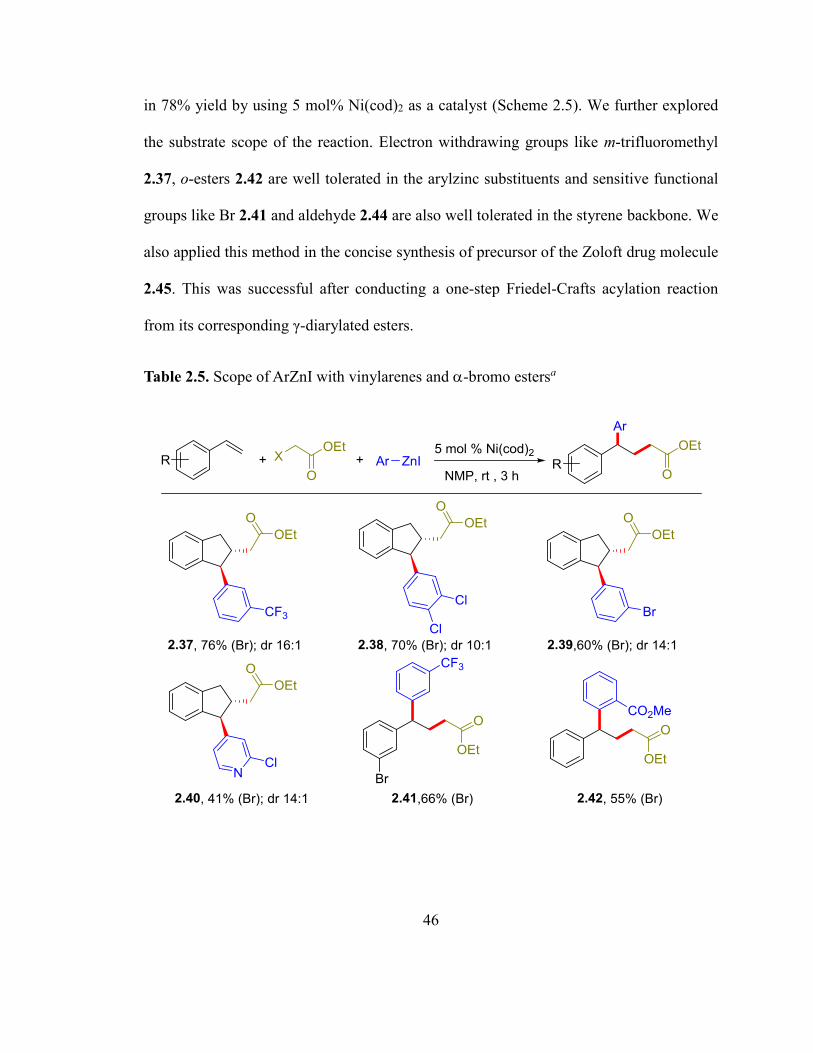
46
in 78% yield by using 5 mol% Ni(cod)2 as a catalyst (Scheme 2.5). We further explored
the substrate scope of the reaction. Electron withdrawing groups like m-trifluoromethyl
2.37, o-esters 2.42 are well tolerated in the arylzinc substituents and sensitive functional
groups like Br 2.41 and aldehyde 2.44 are also well tolerated in the styrene backbone. We
also applied this method in the concise synthesis of precursor of the Zoloft drug molecule
2.45. This was successful after conducting a one-step Friedel-Crafts acylation reaction
from its corresponding γ-diarylated esters.
Table 2.5. Scope of ArZnI with vinylarenes and -bromo estersa

47
a 0.5 mmol scale reactions. Letters in parenthesis indicate X in Alkyl-X. b Reaction in DCM,
cThis was prepared after Friedel-Crafts acylation reaction of ethyl 4-(3,4-dichlorophenyl)-
4-phenyl butanoate
The area of dicarbofunctionalization reactions have grown interest in recent years but its
application in the synthesis of biologically important target molecules largely remains
unknown. In this respect, we applied the current alkylarylation reaction of alkenes as a new
retrosynthetic disconnection to concisely synthesize a potential 5-lipoxygenase activating
protein (FLAP) inhibitor 56. We first proposed our reaction scheme for the synthesis of
potential FLAP inhibitor 56 (Scheme 2.1) starting from methyl 4-methoxy-2-
hydroxybenzoate, a cheap (0.6$/1g) and commercially available starting material. The
starting material was reacted with triflic anhydride to provide the methyl 4-methoxy-2-
(((trifluoromethyl)sulfonyl)oxy)benzoate 2.46. Then the styryl backbone in the compound
2.46 was installed by conducting a Pd-catalyzed vinylation reaction to give compound 2.47.
The dicarbofunctionalization reaction of methyl 4-methoxy-2-vinylbenzoate 2.47, t-BuI
and PhZnI was performed using NiCl2(PPh3)2 as a catalyst and successfully gives the
product 2.48 in 67% yield. Then, selective demethylation of -OMe group over the ester -
OMe 2.48 was performed using 2 equiv. of BCl3 at very low temperature. However, the
reaction didn’t furnish the desired demethylated product 2.49. Instead of forming the
48
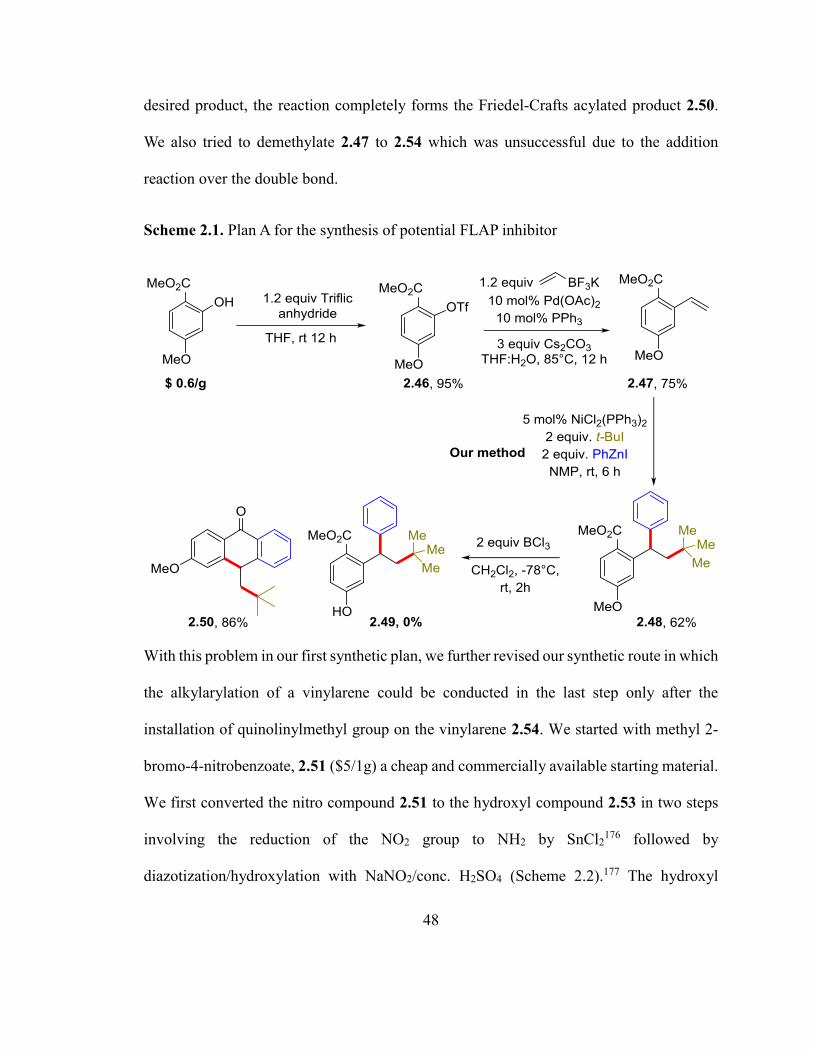
desired product, the reaction completely forms the Friedel-Crafts acylated product 2.50.
We also tried to demethylate 2.47 to 2.54 which was unsuccessful due to the addition
reaction over the double bond.
Scheme 2.1. Plan A for the synthesis of potential FLAP inhibitor
With this problem in our first synthetic plan, we further revised our synthetic route in which
the alkylarylation of a vinylarene could be conducted in the last step only after the
installation of quinolinylmethyl group on the vinylarene 2.54. We started with methyl 2-
bromo-4-nitrobenzoate, 2.51 ($5/1g) a cheap and commercially available starting material.
We first converted the nitro compound 2.51 to the hydroxyl compound 2.53 in two steps
involving the reduction of the NO2 group to NH2 by SnCl2176 followed by
diazotization/hydroxylation with NaNO2/conc. H2SO4 (Scheme 2.2).177 The hydroxyl

49
compound 2.53 was then vinylated by a Pd-catalyzed Suzuki-Miyaura coupling with the
Molander’s reagent to form the hydroxyvinylarene compound 2.54.178 The
hydroxyvinylarene 2.54 was then arylmethylated with 2-(chloromethyl)quinoline
hydrochloride179 to generate the vinylarene intermediate 2.55. The vinylarene 2.55 was
then subjected to Ni-catalyzed alkylarylation with tBuI and PhZnI. Despite the presence of
the ortho-ester group and the acidic benzylic methylene group activated by both the oxygen
and quinoline, the alkylarylation reaction proceeded smoothly to furnish the potential
FLAP inhibitor 2.56 in 63% yield. The reaction can also be conducted in a gram-scale
quantity (5.0 mmol, 1.47 g) without compromising the product yield (65%) (Scheme 2.2).
Previously, Chu and coworkers from the Merck synthetic lab reported the synthesis of this
compound in 12 synthetic steps,172 while we were able to synthesize this compound in just
5 synthetic steps. This shows that our current method is concise and economical for the
synthesis of potential FLAP inhibitor 2.56.
Scheme 2.2. Plan B for the synthesis of potential FLAP inhibitor
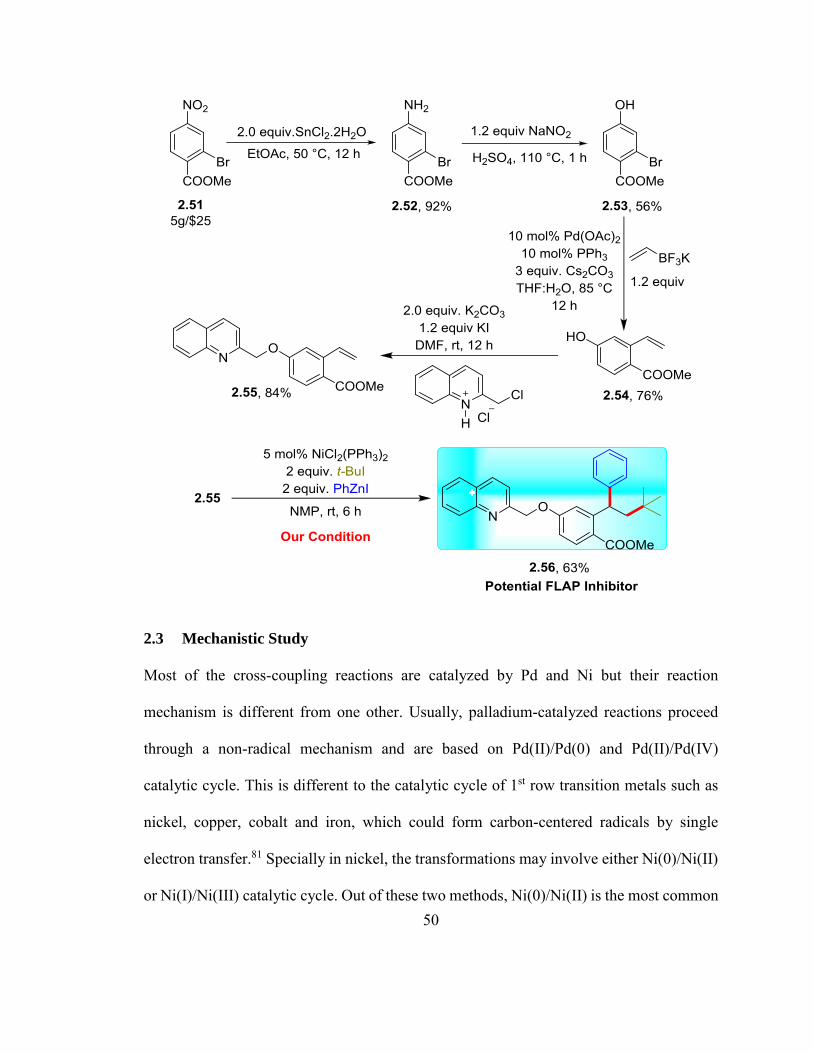
50
2.3 Mechanistic Study
Most of the cross-coupling reactions are catalyzed by Pd and Ni but their reaction
mechanism is different from one other. Usually, palladium-catalyzed reactions proceed
through a non-radical mechanism and are based on Pd(II)/Pd(0) and Pd(II)/Pd(IV)
catalytic cycle. This is different to the catalytic cycle of 1st row transition metals such as
nickel, copper, cobalt and iron, which could form carbon-centered radicals by single
electron transfer.81 Specially in nickel, the transformations may involve either Ni(0)/Ni(II)
or Ni(I)/Ni(III) catalytic cycle. Out of these two methods, Ni(0)/Ni(II) is the most common

51
catalytic cycle. We performed several competitive experiments and quantitative kinetics
experiments to understand the mechanism of the reaction Ni(I)/Ni(III). Our reaction is also
catalyzed by Ni(cod)2 and (Ph3P)4Ni similar to NiBr2 (Table 2.1), from there we believe
that Ni(0) is likely the active catalyst in our reaction system. This could be possible after
in situ reduction of NiBr2 to Ni(0) by ArZnI in our reaction. So we performed a reduction
experiment of the precatalyst NiBr2 to Ni(0) by using organozinc reagents. Experimentally,
we reacted NiBr2•DME with an excess of 4-FC6H5ZnI reagent and it was followed by in
situ 19F NMR (Scheme 2.3). The reaction formed a dark solution as soon as we mixed
NiBr2•DME and 4-FC6H5ZnI which is likely due to the formation of Ni(0) species after
reduction of Ni(II) catalyst by organozinc reagent. We found the formation of 4,4’-
difluorobiphenyl in nearly stoichiometric ratio to NiBr2•DME and the concentration of the
biaryl product does not changes with time (Scheme 2.3). This experiment indicates that
NiBr2 is instantaneously reduced to Ni(0) and the biaryl product would arise by reductive
elimination from (4-FC6H5)2Ni generated from double transmetalation of the two
equivalent 4-FC6H5ZnI to NiBr2•DME.
Scheme 2.3. Reduction experiment of Ni(II) to Ni(0) by ArZnI
Also it is well-known that the Ni-catalyzed cross-coupling reaction of alkyl halides
proceeds through single-electron transfer (SET)81 radical process. Ni(0) reduces organic
halides by two single-electron transfer process, generating an organic radical as an

52
intermediate. In order to examine the possibility of a radical intermediate in our reaction,
we conducted a radical clock experiment using iodoethoxypropene as a radical probe
(Scheme 2.4). It is reported that the radical derived from iodoethoxypropene undergoes
cyclization at the rate of 4.0 × 109 s-1 in THF/HMPA at room temperature.180 When
iodoethoxypropene was reacted with 4-chlorostyrene and PhZnI under the standard
reaction condition, cyclized product 2.57 was generated in 38% yield. The product was
formed with the presence of an alkyl radical which is formed after SET of alkyl halides by
Ni-catalyst. The alkyl radical then undergoes cyclization/coupling and form cyclized
methyl radical which recombines with the Ni-catalyst and forms alkylnickel intermediate.
The cyclized product is finally formed after the alkylnickel intermediate undergoes
subsequent transmetalation and reductive elimination.
Scheme 2.4. Radical clock experiment
In addition, a dimerized product from our reaction was isolated, which was formed through
the formation of a benzylic radical intermediate. When 2-vinylnaphthalene was reacted
with tBuBr and PhZnI under our standard reaction condition, dimerized product 2.58 was
isolated in 14% yield (Scheme 2.5). The formation of dimerized product is possible from
the intermediate 2.59, which is formed after the addition of the tertiary butyl radical to the
styrene. These two experiments indicate that our reaction likely proceeds through the
involvement of a radical intermediate.

53
Scheme 2.5. Formation of radically dimerized product
Reactions involving radical chemistry can happen either through an inner sphere
mechanism or an outer sphere mechanism. In inner sphere electron transfer, a bridging
atom takes a significant role in the study of reaction mechanism. In our reaction system, if
the halogen atom abstraction happens through a bridging process then the inner sphere
reaction rate is significantly dependent on the strength of C-X bond. As the bond strength
of halides follows this order C-F > C-Cl > C-Br > C-I, we expect to see the rate difference
of our reaction from competitive experiments. In contrast, the rate constants for the outer-
sphere electron transfer process exhibit a dependence on the solvent effect and negligible
dependence on different types of halides.181 To understand whether this radical process
proceeds through an inner sphere or outer sphere electron transfer mechanism, we
conducted competition experiments using different types of alkyl halides.
We performed a competition experiment by reacting 2-vinyl naphthalene with excess of n-
octyl iodide and cyclohexyl iodide under our standard condition (Scheme 2.6). We found
that the rate of formation of the difunctionalized product from cyclohexyl iodide to n-octyl
iodide is in the ratio of 3:1.
Scheme 2.6. Competition experiments with 1° and 2° R-X
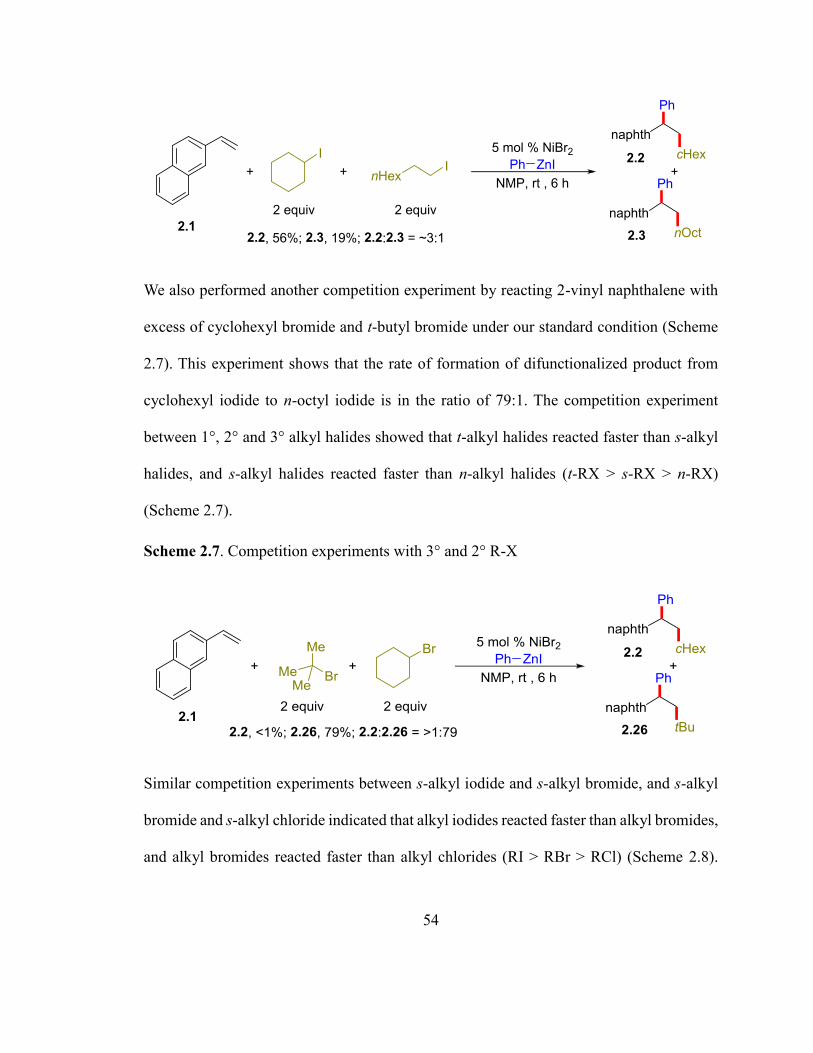
54
We also performed another competition experiment by reacting 2-vinyl naphthalene with
excess of cyclohexyl bromide and t-butyl bromide under our standard condition (Scheme
2.7). This experiment shows that the rate of formation of difunctionalized product from
cyclohexyl iodide to n-octyl iodide is in the ratio of 79:1. The competition experiment
between 1°, 2° and 3° alkyl halides showed that t-alkyl halides reacted faster than s-alkyl
halides, and s-alkyl halides reacted faster than n-alkyl halides (t-RX > s-RX > n-RX)
(Scheme 2.7).
Scheme 2.7. Competition experiments with 3° and 2° R-X
Similar competition experiments between s-alkyl iodide and s-alkyl bromide, and s-alkyl
bromide and s-alkyl chloride indicated that alkyl iodides reacted faster than alkyl bromides,
and alkyl bromides reacted faster than alkyl chlorides (RI > RBr > RCl) (Scheme 2.8).
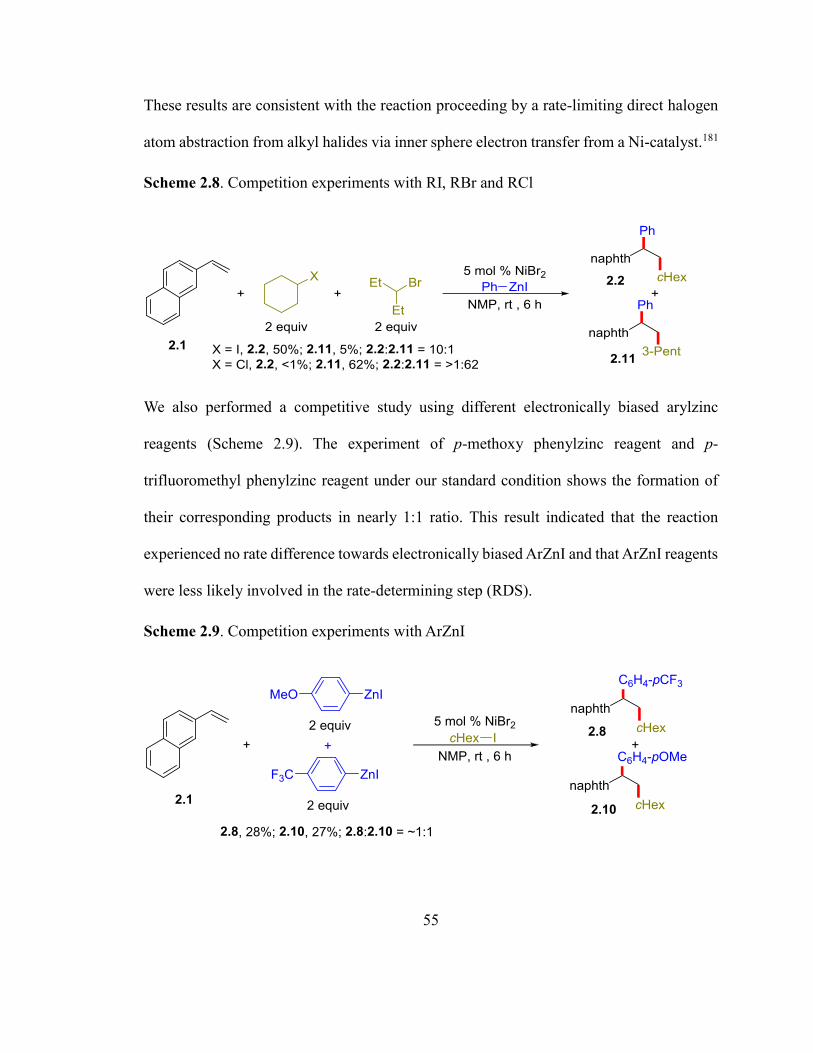
55
These results are consistent with the reaction proceeding by a rate-limiting direct halogen
atom abstraction from alkyl halides via inner sphere electron transfer from a Ni-catalyst.181
Scheme 2.8. Competition experiments with RI, RBr and RCl
We also performed a competitive study using different electronically biased arylzinc
reagents (Scheme 2.9). The experiment of p-methoxy phenylzinc reagent and p-
trifluoromethyl phenylzinc reagent under our standard condition shows the formation of
their corresponding products in nearly 1:1 ratio. This result indicated that the reaction
experienced no rate difference towards electronically biased ArZnI and that ArZnI reagents
were less likely involved in the rate-determining step (RDS).
Scheme 2.9. Competition experiments with ArZnI
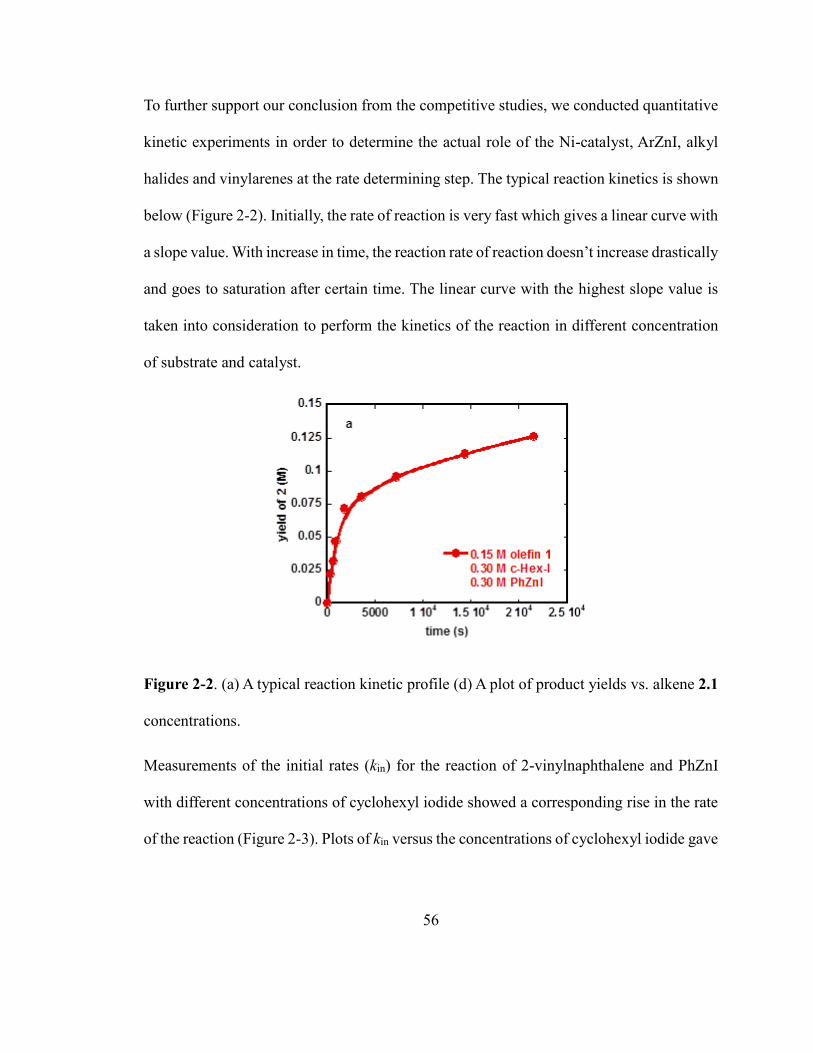
56
To further support our conclusion from the competitive studies, we conducted quantitative
kinetic experiments in order to determine the actual role of the Ni-catalyst, ArZnI, alkyl
halides and vinylarenes at the rate determining step. The typical reaction kinetics is shown
below (Figure 2-2). Initially, the rate of reaction is very fast which gives a linear curve with
a slope value. With increase in time, the reaction rate of reaction doesn’t increase drastically
and goes to saturation after certain time. The linear curve with the highest slope value is
taken into consideration to perform the kinetics of the reaction in different concentration
of substrate and catalyst.
Figure 2-2. (a) A typical reaction kinetic profile (d) A plot of product yields vs. alkene 2.1
concentrations.
Measurements of the initial rates (kin) for the reaction of 2-vinylnaphthalene and PhZnI
with different concentrations of cyclohexyl iodide showed a corresponding rise in the rate
of the reaction (Figure 2-3). Plots of kin versus the concentrations of cyclohexyl iodide gave
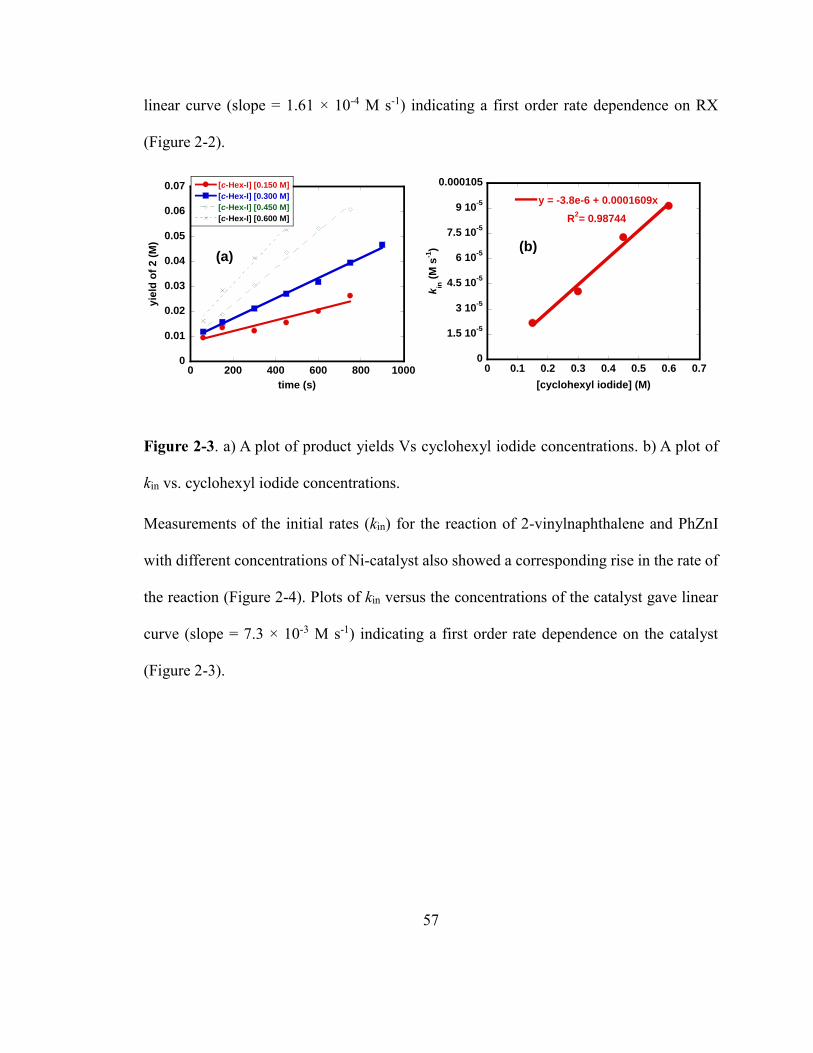
57
linear curve (slope = 1.61 × 10-4 M s-1) indicating a first order rate dependence on RX
(Figure 2-2).
Figure 2-3. a) A plot of product yields Vs cyclohexyl iodide concentrations. b) A plot of
kin vs. cyclohexyl iodide concentrations.
Measurements of the initial rates (kin) for the reaction of 2-vinylnaphthalene and PhZnI
with different concentrations of Ni-catalyst also showed a corresponding rise in the rate of
the reaction (Figure 2-4). Plots of kin versus the concentrations of the catalyst gave linear
curve (slope = 7.3 × 10-3 M s-1) indicating a first order rate dependence on the catalyst
(Figure 2-3).
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0 200 400 600 800 1000
[c-Hex-I] [0.150 M]
[c-Hex-I] [0.300 M]
[c-Hex-I] [0.450 M]
[c-Hex-I] [0.600 M]
yie
ld o
f 2
(M
)
time (s)
(a)
0
1.5 10-5
3 10-5
4.5 10-5
6 10-5
7.5 10-5
9 10-5
0.000105
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
y = -3.8e-6 + 0.0001609x
R2= 0.98744
kin
(M
s-1
)
[cyclohexyl iodide] (M)
(b)
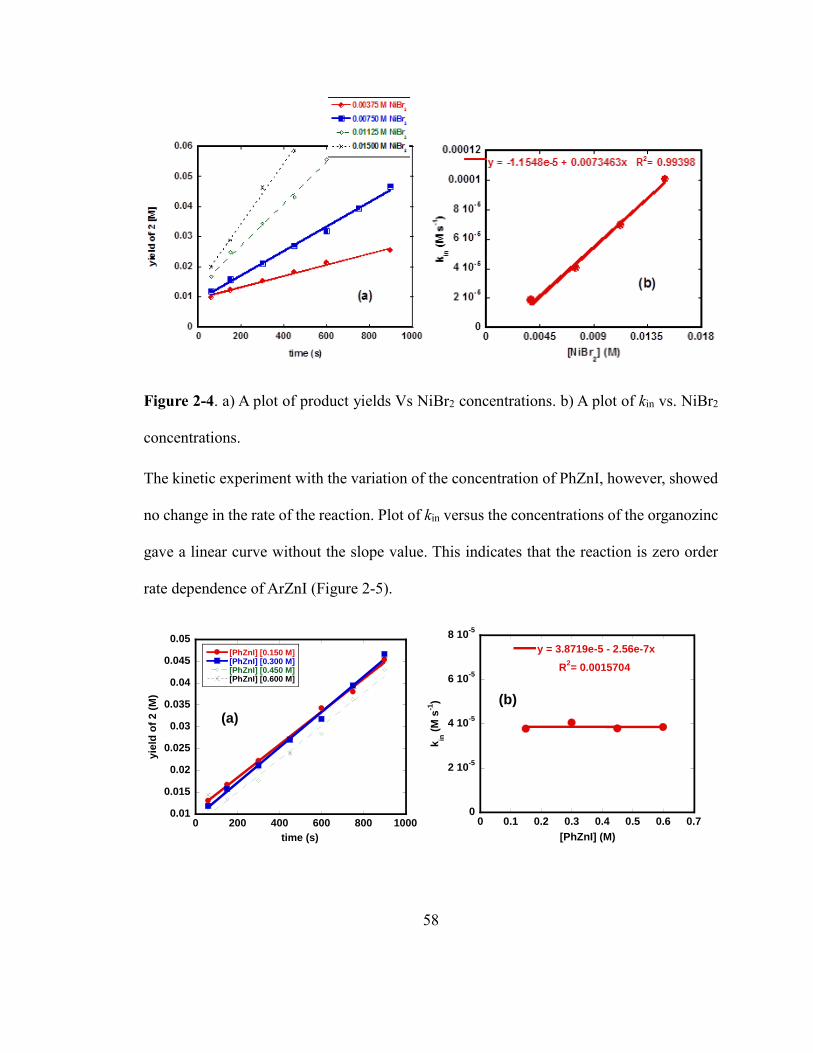
58
Figure 2-4. a) A plot of product yields Vs NiBr2 concentrations. b) A plot of kin vs. NiBr2
concentrations.
The kinetic experiment with the variation of the concentration of PhZnI, however, showed
no change in the rate of the reaction. Plot of kin versus the concentrations of the organozinc
gave a linear curve without the slope value. This indicates that the reaction is zero order
rate dependence of ArZnI (Figure 2-5).
0.01
0.015
0.02
0.025
0.03
0.035
0.04
0.045
0.05
0 200 400 600 800 1000
[PhZnI] [0.150 M][PhZnI] [0.300 M][PhZnI] [0.450 M][PhZnI] [0.600 M]
yie
ld o
f 2 (
M)
time (s)
(a)
0
2 10-5
4 10-5
6 10-5
8 10-5
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
y = 3.8719e-5 - 2.56e-7x
R2= 0.0015704
kin
(M
s-1
)
[PhZnI] (M)
(b)
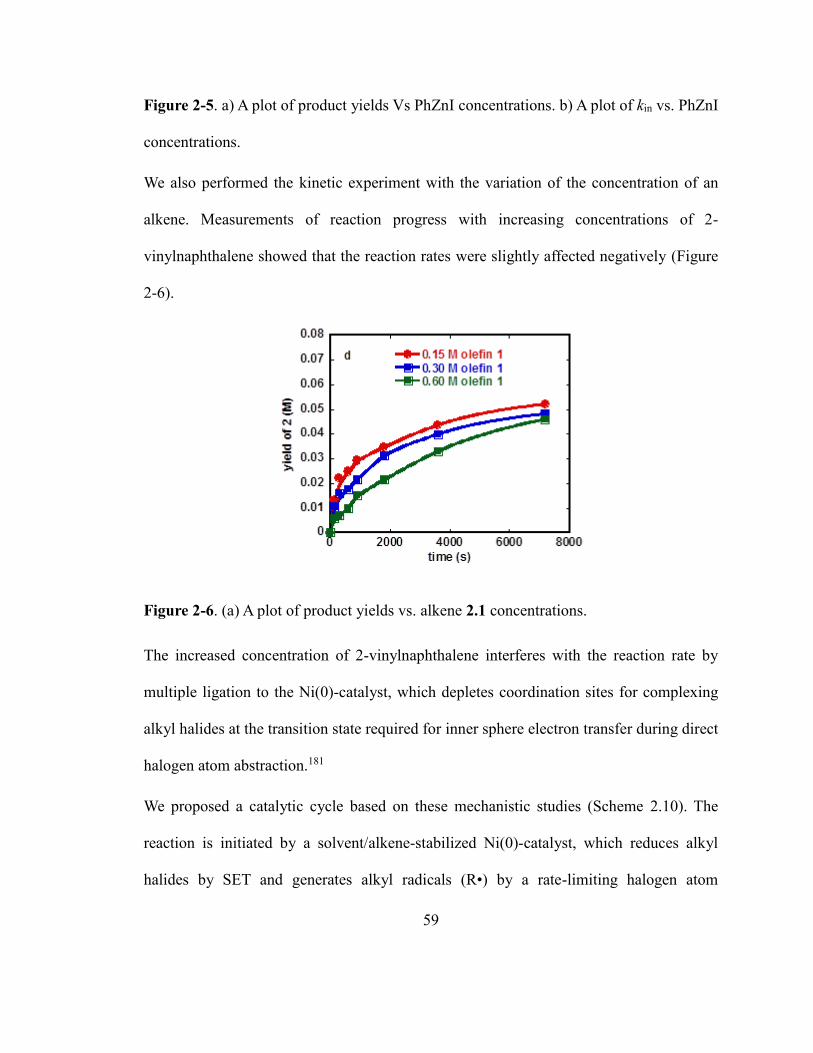
59
Figure 2-5. a) A plot of product yields Vs PhZnI concentrations. b) A plot of kin vs. PhZnI
concentrations.
We also performed the kinetic experiment with the variation of the concentration of an
alkene. Measurements of reaction progress with increasing concentrations of 2-
vinylnaphthalene showed that the reaction rates were slightly affected negatively (Figure
2-6).
Figure 2-6. (a) A plot of product yields vs. alkene 2.1 concentrations.
The increased concentration of 2-vinylnaphthalene interferes with the reaction rate by
multiple ligation to the Ni(0)-catalyst, which depletes coordination sites for complexing
alkyl halides at the transition state required for inner sphere electron transfer during direct
halogen atom abstraction.181
We proposed a catalytic cycle based on these mechanistic studies (Scheme 2.10). The
reaction is initiated by a solvent/alkene-stabilized Ni(0)-catalyst, which reduces alkyl
halides by SET and generates alkyl radicals (R•) by a rate-limiting halogen atom

60
abstraction process. The alkyl radicals then undergo addition to vinylarenes to form
benzylic radicals. The benzylic radicals will recombine with the [NiI-X] species which
undergoes transmetalation with ArZnX followed by reductive elimination to form the
alkylarylated products and regenerate the active Ni(0)-catalyst.
Scheme 2.10: Proposed catalytic cycle
2.4 Conclusion
We developed a catalyst-controlled Ni-catalyzed regioselective alkylarylation of
vinylarenes with alkyl halides and arylzinc reagents. This reaction shows successful
coupling of primary, secondary, and tertiary alkyl halides as well as tolerating a wide
variety of functional groups. The mechanistic investigations by quantitative kinetics,
competition studies, and radical probes show that this reaction proceeds through a single
electron transfer (SET) with the rate limiting step being the direct halogen atom abstraction
from alkyl halides by a Ni-catalyst. This method is useful for the synthesis of 1,1-
diarylalkanes complex structural cores which are found in many biologically active
compounds.

61
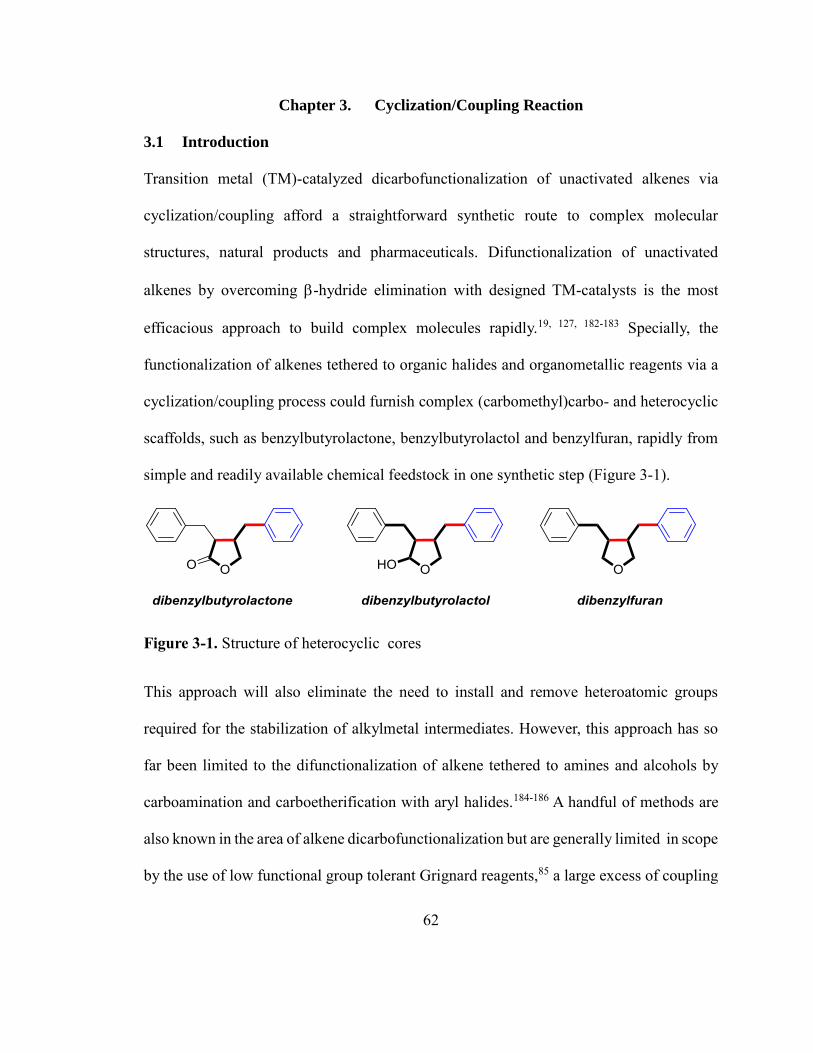
62
Chapter 3. Cyclization/Coupling Reaction
3.1 Introduction
Transition metal (TM)-catalyzed dicarbofunctionalization of unactivated alkenes via
cyclization/coupling afford a straightforward synthetic route to complex molecular
structures, natural products and pharmaceuticals. Difunctionalization of unactivated
alkenes by overcoming -hydride elimination with designed TM-catalysts is the most
efficacious approach to build complex molecules rapidly.19, 127, 182-183 Specially, the
functionalization of alkenes tethered to organic halides and organometallic reagents via a
cyclization/coupling process could furnish complex (carbomethyl)carbo- and heterocyclic
scaffolds, such as benzylbutyrolactone, benzylbutyrolactol and benzylfuran, rapidly from
simple and readily available chemical feedstock in one synthetic step (Figure 3-1).
Figure 3-1. Structure of heterocyclic cores
This approach will also eliminate the need to install and remove heteroatomic groups
required for the stabilization of alkylmetal intermediates. However, this approach has so
far been limited to the difunctionalization of alkene tethered to amines and alcohols by
carboamination and carboetherification with aryl halides.184-186 A handful of methods are
also known in the area of alkene dicarbofunctionalization but are generally limited in scope
by the use of low functional group tolerant Grignard reagents,85 a large excess of coupling

63
reagents,85, 88, 103-105 or substrates that lack in -H’s.60, 187 Therefore, the objective is to
design and develop novel catalyst in combination with ligands that will enable us to
overcome this limitation and to intercept alkylmetal intermediates with organometallic
reagents prior to -hydride elimination. Successful development of this aim will enable us
to utilize tethered alkene to synthesize complex carbocycles, heterocycles and acyclic
motifs relevant to natural products and bioactive molecules from simple and readily
available chemical feedstock in one synthetic step. These important targets that are
otherwise difficult to access rapidly from a known synthetic method. Such cyclic
frameworks are profusely imbedded as structural cores in a variety of natural products and
biologically active molecules such as lignans (Figure 3.2).188-191 Lignans are an extremely
large group of polyphenol natural products which are generally extracted from plants.
Molecules containing these structural scaffolds generally display a wide range of biological
activities including antiviral, antitumor and anti-HIV.188-191
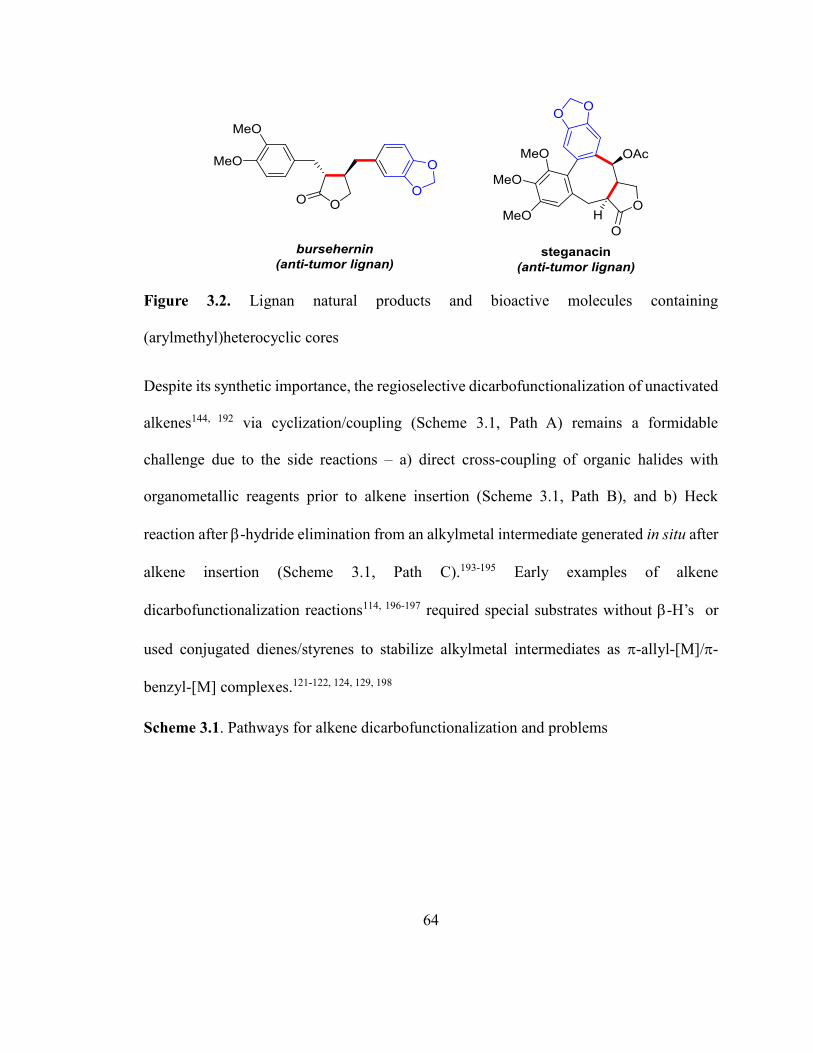
64
Figure 3.2. Lignan natural products and bioactive molecules containing
(arylmethyl)heterocyclic cores
Despite its synthetic importance, the regioselective dicarbofunctionalization of unactivated
alkenes144, 192 via cyclization/coupling (Scheme 3.1, Path A) remains a formidable
challenge due to the side reactions – a) direct cross-coupling of organic halides with
organometallic reagents prior to alkene insertion (Scheme 3.1, Path B), and b) Heck
reaction after -hydride elimination from an alkylmetal intermediate generated in situ after
alkene insertion (Scheme 3.1, Path C).193-195 Early examples of alkene
dicarbofunctionalization reactions114, 196-197 required special substrates without -H’s or
used conjugated dienes/styrenes to stabilize alkylmetal intermediates as -allyl-[M]/-
benzyl-[M] complexes.121-122, 124, 129, 198
Scheme 3.1. Pathways for alkene dicarbofunctionalization and problems

65
While no general solution has emerged yet108, 199-200 a limited number of reactions have
demonstrated that the use of base metal catalysts enables the cyclization/coupling, a
reaction that proceeds by a radical process90, 100, 125, 159, 201-203 thus expediting alkene
insertion and avoiding complications by -hydride elimination.46, 160-161, 164, 167, 204-205 In this
respect, Oshima,87 and Cárdenas147, 206-207 developed a dicarbofunctionalization reaction of
an alkene tethered to alkyl halides with organometallic reagent using first row transition
metal i.e. Ni as a catalyst. However, the scope and synthetic utility of the
cyclization/coupling reactions is still limited especially for their application to the synthesis
of natural products.
Our group recently reported a Cu-catalyzed cyclization/coupling of alkyl and aryl
organometallic reagents with aryl halides.106 The functional group tolerance of this reaction
was excellent in terms of substrate scope, but this reaction does not tolerate electron-rich
aryl halides due to the low reactivity of Cu-catalyst to the electron-rich aryl halides. This
limitation restricts the application of these reactions broadly for the synthesis of natural
products such as lignans (Figure 3-2) that contain benzylbutyrolactone frameworks with
electron-rich polyphenol derivatives. All these early examples represent some progress in

66
the area of cyclization/coupling but their application has never been explored to the
synthesis of natural products.
3.2 Ni-Catalyzed Cyclization/Coupling of Alkene tethered to Alkyl Halides
First, we hypothesized that the use of alkene tethered to alkyl halides in different ways can
be useful for making these carbocycles and heterocycles in shorter synthetic routes. We
planned to use alkene tethered to alkyl halides, which can undergo SET to form alkyl
radicals in presence of Ni-catalyst and recombine with the catalyst to result an alkylmetal
species. These alkylmetal species could be intercepted with organometallic reagents before
undergoing -hydride elimination.
We started screening the cyclization/coupling reaction of unactivated alkenes by using 6-
bromo hexene as an alkene tethered to alkyl halide and diphenylzinc as an organometallic
reagent with 5 mol% of FeCl2, 10 mol% of HMPA as a ligand in dioxane at 100°C for 12
h (Scheme 3.3). After several screening, we found that the cyclized coupled product 3.1
was formed in 45% yield. To understand the role of ligand and mechanism of the reaction,
we synthesized a Fe(II)Cl2(HMPA)2 complex (Figure 3-2). Despite exploring several other
conditions, the substrate scope of the reaction was limited, and the yield of product
remained low.
Scheme 3.2. Initial optimization of this reaction using FeCl2 catalyst

67
Figure 3-3. Crystal structure of Fe(II)Cl2(HMPA)2 complex
After screening the reaction different catalyst, we found that Ni-catalysts and terpyridine
ligand works well for this chemistry. The reaction of diethyl 2-allyl-2-(2-
bromoethyl)malonate 3.13 with (4-cyanophenyl)zinc iodide using 3 mol% of NiBr2, 4
mol% of terpy in NMP at 50 °C gives cyclized cross-coupling product 3.14 in 87% yield
(Table 3.1). The different substituents in terpy. like tBu, Me and MeO- 3.3-3.5 were further
examined out of which only the tBu-substituted terpy 3.3 furnished the expected product
in comparable yield (entries 2-3). A similar tridentate ligand, pybox 3.6, afforded the
product only in 12% yield (entry 4). The phosphorus and nitrogen based bidentate ligands
such as phen 3.7, bipy 3.8, bis-amines 3.9-3.10, dppbz 3.11 and xantphos 3.12 also gave
the product in low to moderate yields (entries 5-6). Only a trace amount of product was
formed in the absence of terpy or catalyst which shows that the current reaction is strongly
ligand and catalyst-controlled (entry 7 and 8). The yield of the product 3.14 decreases with
decrease in the reaction time and temperature (entries 9-10). The reaction gave moderate
yields when NiBr2 was replaced with Ni(0) catalyst like Ni(cod)2 or Ni(PPh3)4 (entry 11).
The reaction was completely dependent on the polarity of the solvents. Solvents like THF,
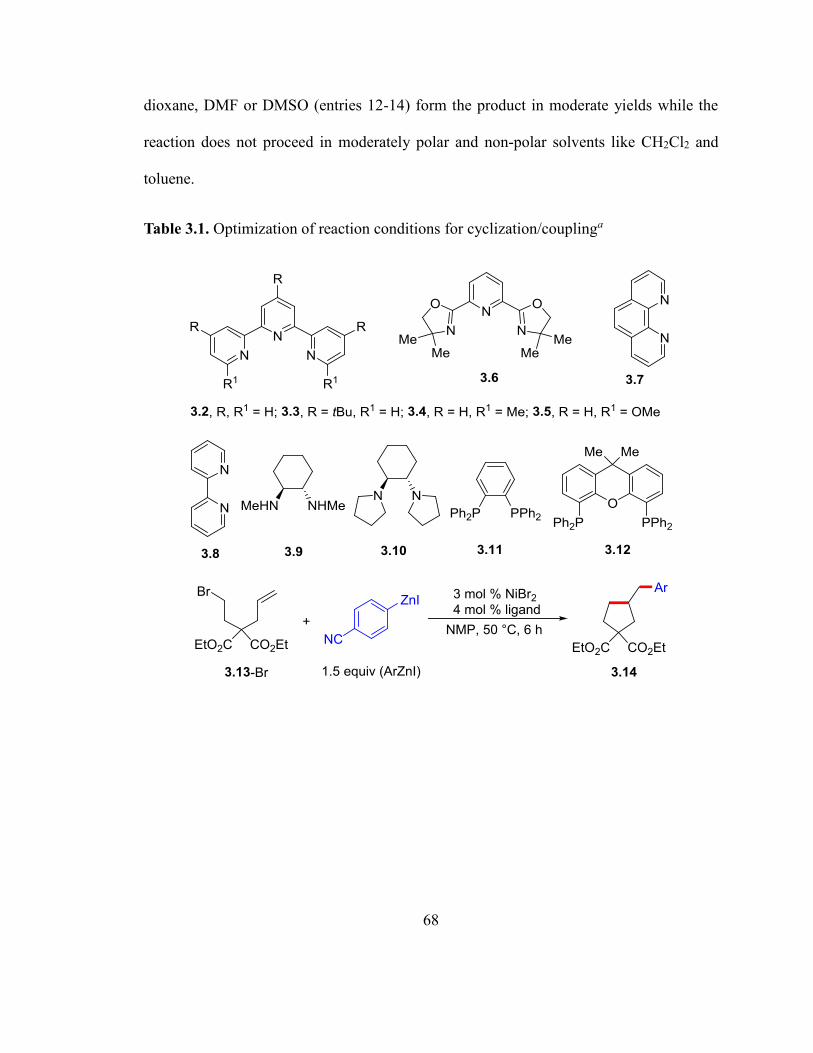
68
dioxane, DMF or DMSO (entries 12-14) form the product in moderate yields while the
reaction does not proceed in moderately polar and non-polar solvents like CH2Cl2 and
toluene.
Table 3.1. Optimization of reaction conditions for cyclization/couplinga

69
aYields were determined by 1H NMR using pyrene as an internal standard. Value in
parenthesis is the isolated yield from a 0.5 mmol scale reaction.
Substrate Scope. We further studied the scope of the current cyclization/coupling reaction
with respect to both reagents, the alkene tethered to alkyl halides and arylzinc reagents
(Table 3.2). Different types of alkene tethered to alkyl halides can be cyclized to form
carbocycles (3.15-3.18), and N- (3.19-3.21) and O-heterocycles (3.22-3.26) in good yields.
Both electron-rich and deficient arylzinc reagents proceeds well in the reaction forming
good to excellent yields of products. Functional groups which are challenging in
organometallic chemistry such as nitriles (3.18 and 3.24), esters (3.19), ketones (3.24) and
halides (F, Cl, Br) (3.21, 3.22 and 3.26) are also well tolerated in our reaction.
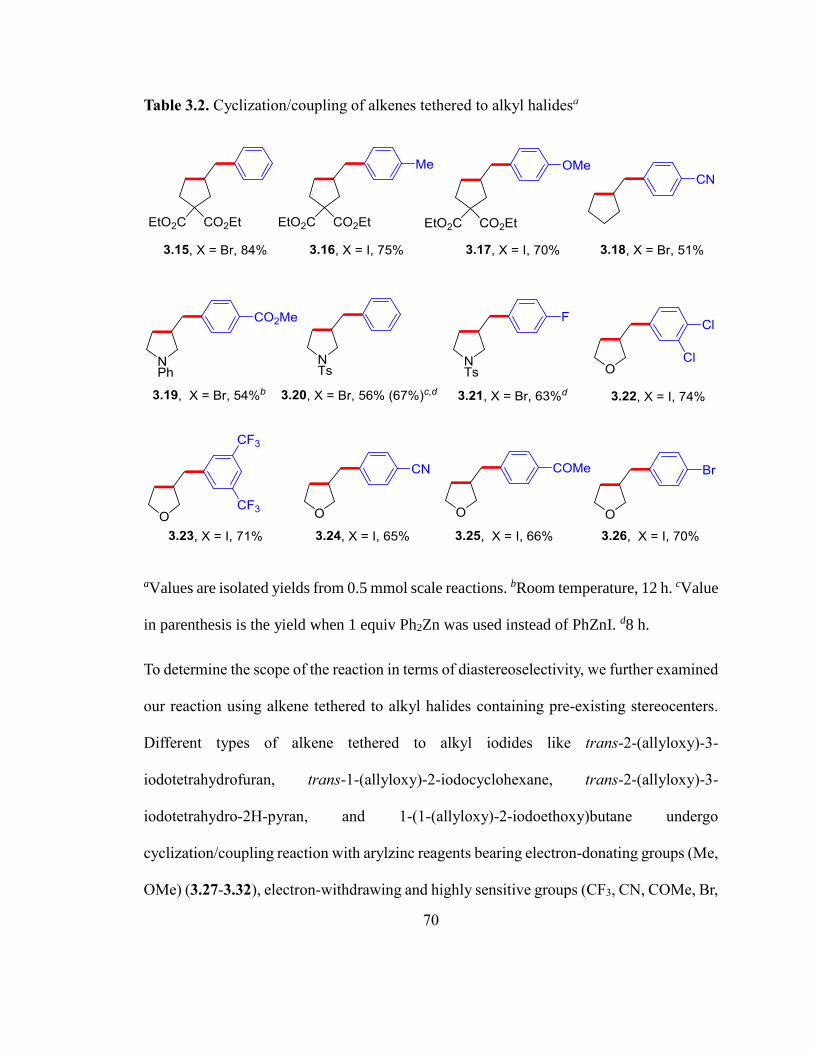
70
Table 3.2. Cyclization/coupling of alkenes tethered to alkyl halidesa
aValues are isolated yields from 0.5 mmol scale reactions. bRoom temperature, 12 h. cValue
in parenthesis is the yield when 1 equiv Ph2Zn was used instead of PhZnI. d8 h.
To determine the scope of the reaction in terms of diastereoselectivity, we further examined
our reaction using alkene tethered to alkyl halides containing pre-existing stereocenters.
Different types of alkene tethered to alkyl iodides like trans-2-(allyloxy)-3-
iodotetrahydrofuran, trans-1-(allyloxy)-2-iodocyclohexane, trans-2-(allyloxy)-3-
iodotetrahydro-2H-pyran, and 1-(1-(allyloxy)-2-iodoethoxy)butane undergo
cyclization/coupling reaction with arylzinc reagents bearing electron-donating groups (Me,
OMe) (3.27-3.32), electron-withdrawing and highly sensitive groups (CF3, CN, COMe, Br,

71
Cl) (3.34-3.38 and 3.43-3.46), and ortho-substituents (3.28, 3.29, 3.32) to furnish bicyclic
heterocycles in good to excellent yields (Table 3.3). These products were formed in
moderate to good levels of diastereoselectivity with the major diastereoisomers containing
three contiguous stereocenters in all cis-configuration. Also heteroarylzinc reagents, such
as (2-chloropyridin-4-yl)zinc iodide, thiophen-2-ylzinc iodide and di(furan-2-yl)zinc
(3.39-3.42) were well-tolerated in good to excellent yields with moderate to good degrees
of diastereoselectivities.
Table 3.3. Diastereoselective cyclization/coupling

72
aValues are isolated yields from 0.5 mmol scale reactions. dr was determined by 1H NMR
and GC. b1 equiv di(furan-2-yl)zinc was used. 10 h.
Furthermore, we examined the tolerance of molecules containing racemizable
stereocenters under our reaction conditions. Under our standard reaction condition, we used
two commercially available enantiomerically enriched compounds containing one chiral
center – N-Boc-D-proline methyl ester (3.47, R-enantiomer) and (R)-
dimethylmethylsuccinate (3.48) – as stoichiometric additives (Table 3.4). We isolated the
compound 3.47 and 3.48 after performing our standard reaction and the ratio of
enantiomers of the isolated compounds were determined by chiral high-performance liquid
chromatography (HPLC). The results showed that there is no change in the er’s ratio when

73
compared to the prior er’s ratio. This experiment confirms that our cyclization/coupling
reaction tolerates base sensitive and racemizable stereocenters. It shows that our current
reaction is very important for the tolerance of challenging molecules, which contain base
sensitive enantioenriched chiral centers.
Table 3.4. Tolerance of base-sensitive and racemizable stereocentersa
aReactions were run in 0.4 mmol scale in 2.0 mL NMP with 1.0 equiv of chiral additives.
The additives were isolated by column chromatography and their er’s were determined by
chiral HPLC.
3.3 Application to the concise synthesis of natural products
Our current method is also applied to the concise synthesis of six lignan natural products –
(±)-dimethylretrodendrin, (±)-kusunokinin, (±)-dimethylmatairesinol, (±)-bursehernin,
(±)-yatein and (±)-collinusin– with three different structural frameworks that contain
benzylbutyrolactone backbones (Scheme 3.4-3.6). These lignan natural products

74
containing dibenzylbutyrolactone and aryltetralin structural cores display a wide range of
biological properties such as fungicidal, antibiotic, antiviral, antitumor and anti-HIV.208-209
(±)-dimethylretrodendrin and (±)-collinusin belong to the class of aryltetralin lignans,
which show antitumor activities by functioning as a potent inhibitors of human DNA
topoisomerase II.210 Collinusin was isolated from the leaves of Cleistanthus collinus
(Roxb.) that has insecticidal and piscicidal activities211 and kusunokinin shows insecticidal
and antitrypanosomal activities.212-213 Bursehernin shows potent cytotoxic activities in
colon, prostate and breast cancer cell lines among others.214 Recent study shows that yatein
exhibits antiproliferative activity215 and suppresses herpes simplex virus type 1 replication
in HeLa cells.216
Prior synthesis of these lignan natural products required a multi-step process for the
construction the benzylbutyrolactone skeleton.217-223 This new method allowed us to
synthesize the benzylbutyrolactone structure in one-pot two steps in gram-scale quantities.
The reaction of 1-(1-(allyloxy)-2-iodoethoxy)butane (3.49) with (3,4-
dimethoxyphenyl)zinc iodide followed by oxidation of the crude product with the Jones
reagent (Scheme 3.3) gives the intermediate lactone 3.50 in just one-pot two steps. To see
the synthetic utility, the reaction was conducted in large scale, 10 mmol scale, which
afforded the lactone 3.50 in 62% isolated yield (1.464 g). (±)-dimethylretrodendrin (3.51)
was synthesized from the lactone 3.50 after deprotonation with lithium diisoproylamide
(LDA) at -78 °C and treated with 3,4-dimethoxybenzaldehyde followed by Friedel-Crafts
alkylation in presence of trifluoroacetic acid to furnish as a 19:1 diastereomeric mixture in
73% isolated yield (Scheme 3.4). The intermediate lactone 3.50 further used for the
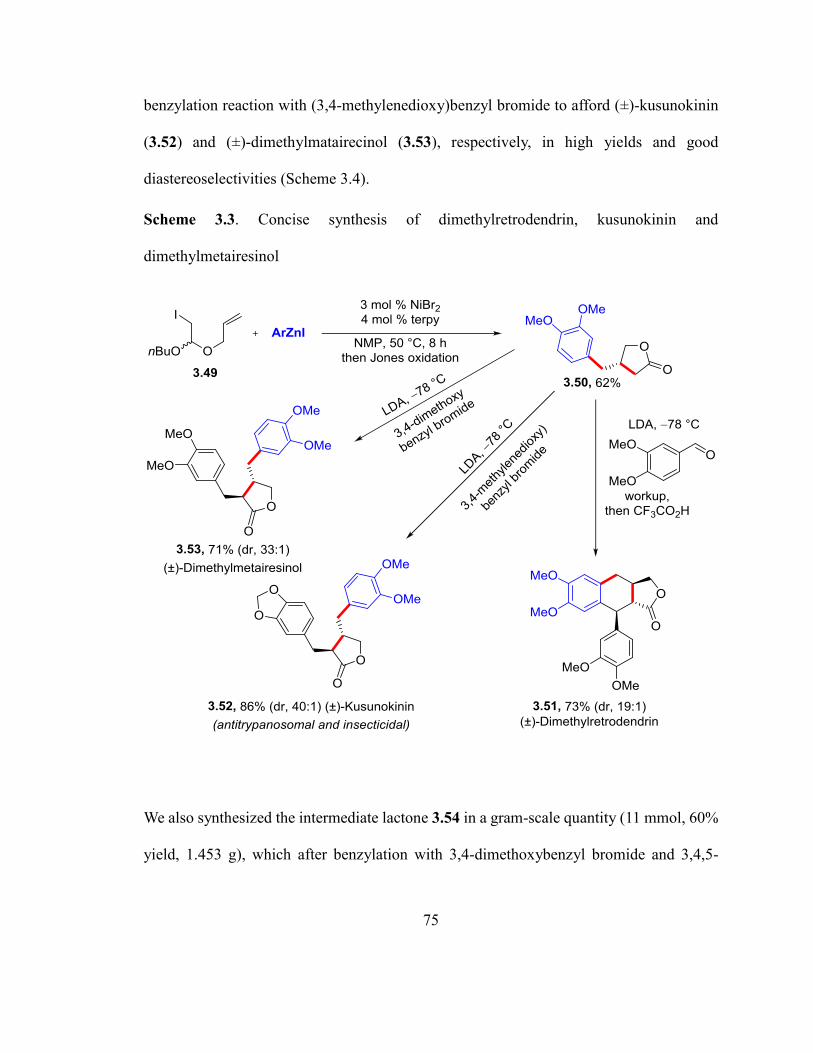
75
benzylation reaction with (3,4-methylenedioxy)benzyl bromide to afford (±)-kusunokinin
(3.52) and (±)-dimethylmatairecinol (3.53), respectively, in high yields and good
diastereoselectivities (Scheme 3.4).
Scheme 3.3. Concise synthesis of dimethylretrodendrin, kusunokinin and
dimethylmetairesinol
We also synthesized the intermediate lactone 3.54 in a gram-scale quantity (11 mmol, 60%
yield, 1.453 g), which after benzylation with 3,4-dimethoxybenzyl bromide and 3,4,5-
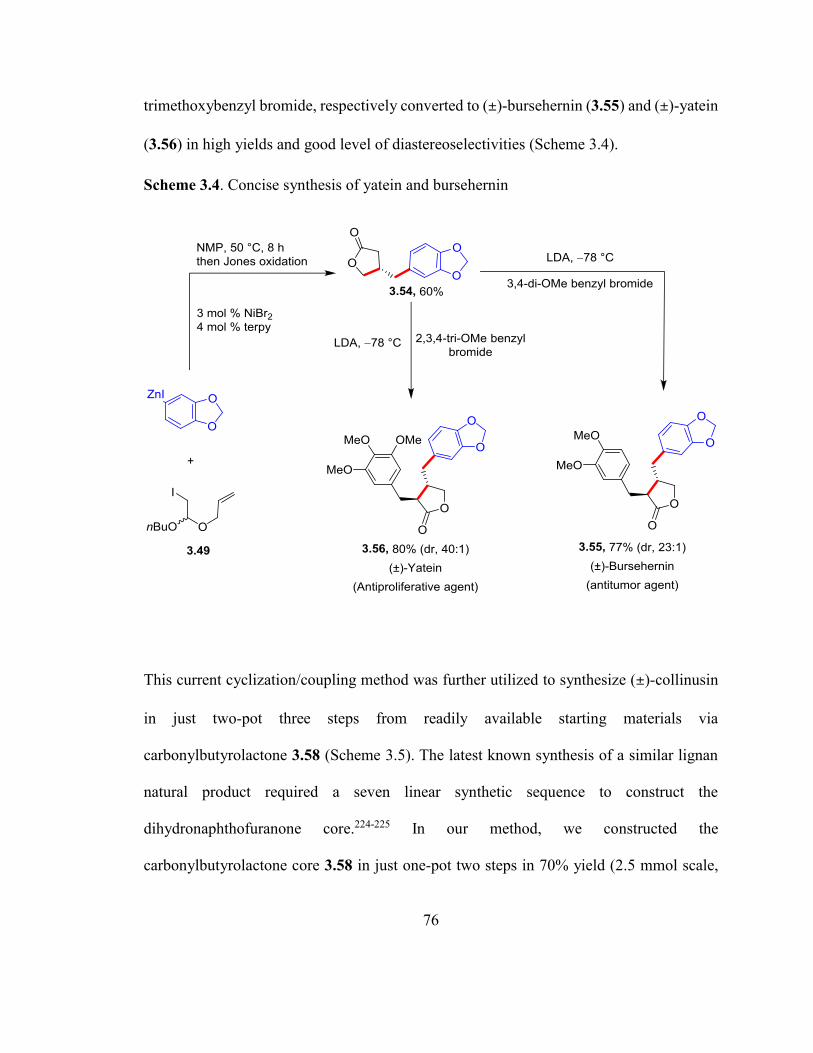
76
trimethoxybenzyl bromide, respectively converted to (±)-bursehernin (3.55) and (±)-yatein
(3.56) in high yields and good level of diastereoselectivities (Scheme 3.4).
Scheme 3.4. Concise synthesis of yatein and bursehernin
This current cyclization/coupling method was further utilized to synthesize (±)-collinusin
in just two-pot three steps from readily available starting materials via
carbonylbutyrolactone 3.58 (Scheme 3.5). The latest known synthesis of a similar lignan
natural product required a seven linear synthetic sequence to construct the
dihydronaphthofuranone core.224-225 In our method, we constructed the
carbonylbutyrolactone core 3.58 in just one-pot two steps in 70% yield (2.5 mmol scale,
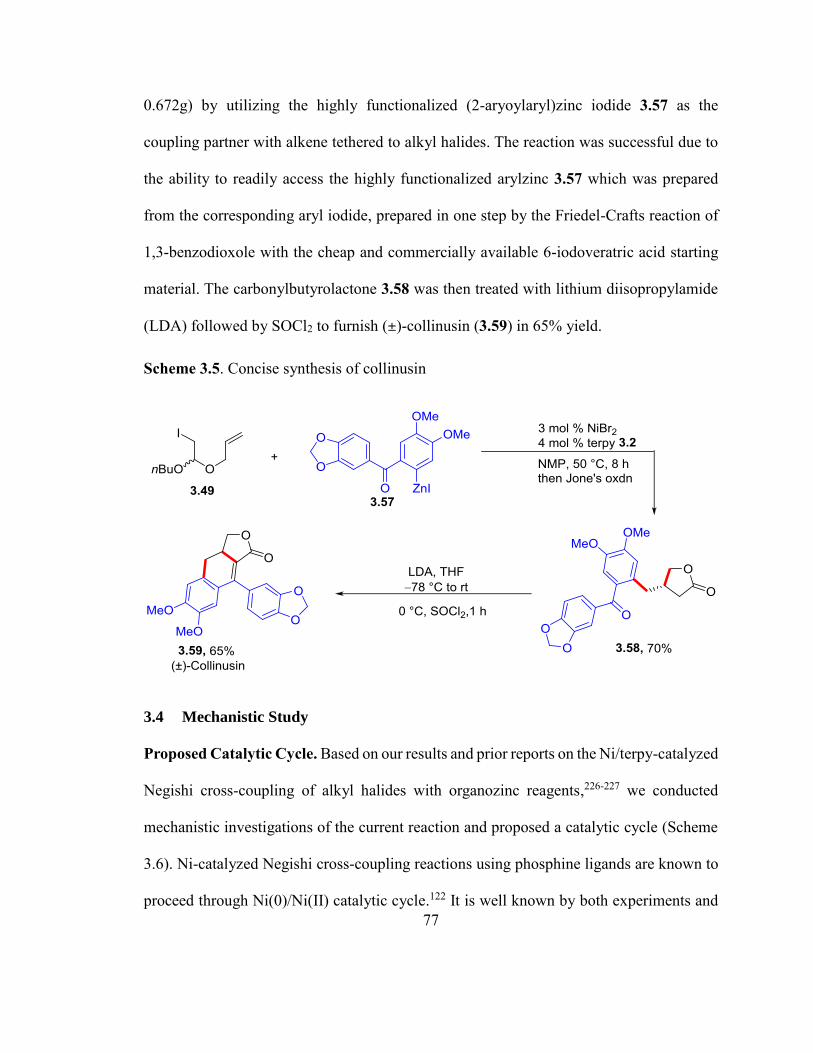
77
0.672g) by utilizing the highly functionalized (2-aryoylaryl)zinc iodide 3.57 as the
coupling partner with alkene tethered to alkyl halides. The reaction was successful due to
the ability to readily access the highly functionalized arylzinc 3.57 which was prepared
from the corresponding aryl iodide, prepared in one step by the Friedel-Crafts reaction of
1,3-benzodioxole with the cheap and commercially available 6-iodoveratric acid starting
material. The carbonylbutyrolactone 3.58 was then treated with lithium diisopropylamide
(LDA) followed by SOCl2 to furnish (±)-collinusin (3.59) in 65% yield.
Scheme 3.5. Concise synthesis of collinusin
3.4 Mechanistic Study
Proposed Catalytic Cycle. Based on our results and prior reports on the Ni/terpy-catalyzed
Negishi cross-coupling of alkyl halides with organozinc reagents,226-227 we conducted
mechanistic investigations of the current reaction and proposed a catalytic cycle (Scheme
3.6). Ni-catalyzed Negishi cross-coupling reactions using phosphine ligands are known to
proceed through Ni(0)/Ni(II) catalytic cycle.122 It is well known by both experiments and

78
density functional theory (DFT) calculations that Ni-catalyzed Negishi cross-coupling
reactions using terpyridine ligands proceed through Ni(I)/Ni(III) catalytic cycle with the
involvement of alkyl radical intermediates. 84, 227-234 The different oxidation state of the
metal is attained because of terpyridine being a non-innocent ligand and excellent electron
acceptor through its extended system, can often involve in redox chemistry. This makes
a Ni/terpy complex a ligand-centered radical or metal-centered radical.228
Usually the catalytic cycle Ni-catalyzed Negishi cross-coupling reactions starts with the
combination of NiX2 and terpyridine ligand with the generation of a low valent
(terpy)Ni(I)X catalyst (3.60) that undergoes transmetalation with organozinc reagents to
form a (terpy)NiR species.82, 229-236 The alkyl halides are then reduced by a single electron
transfer (SET) from (terpy)NiR species followed by recombination of the resultant alkyl
radicals with (terpy)Ni(X)(R) and reductive elimination to generate the cross-coupled
products. Similarly, under our catalytic conditions, the alkyl radicals are generated by
single electron transfer from a (terpy)NiAr species (3.61). The alkyl radicals generated can
undergo cyclization onto the tethered alkene to generate a cyclized primary alkyl radical
prior to recombination with the (terpy)Ni(X)(Ar) species (3.63) to generate
(terpy)Ni(R)(Ar) species (3.64). The cyclized/cross-coupled product and the regeneration
of the active (terpy)NiX catalyst (3.60) is possible after the reductive elimination of the
species 3.64.
Scheme 3.6. Proposed catalytic cycle
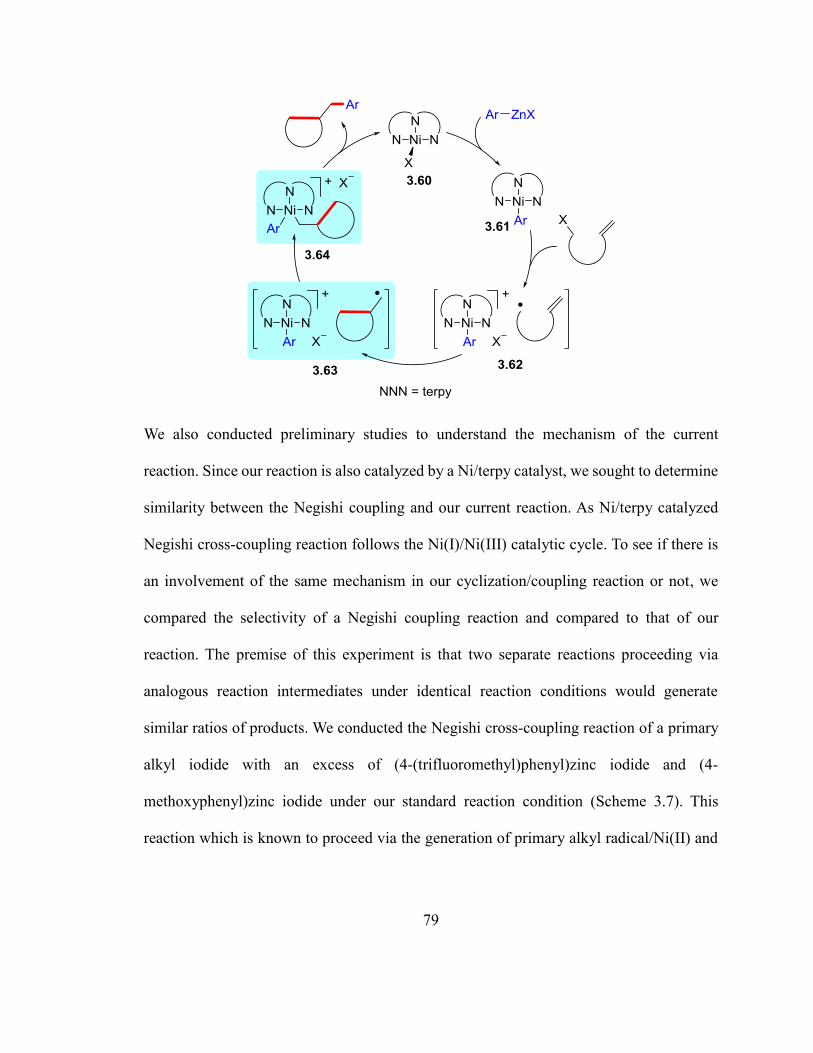
79
We also conducted preliminary studies to understand the mechanism of the current
reaction. Since our reaction is also catalyzed by a Ni/terpy catalyst, we sought to determine
similarity between the Negishi coupling and our current reaction. As Ni/terpy catalyzed
Negishi cross-coupling reaction follows the Ni(I)/Ni(III) catalytic cycle. To see if there is
an involvement of the same mechanism in our cyclization/coupling reaction or not, we
compared the selectivity of a Negishi coupling reaction and compared to that of our
reaction. The premise of this experiment is that two separate reactions proceeding via
analogous reaction intermediates under identical reaction conditions would generate
similar ratios of products. We conducted the Negishi cross-coupling reaction of a primary
alkyl iodide with an excess of (4-(trifluoromethyl)phenyl)zinc iodide and (4-
methoxyphenyl)zinc iodide under our standard reaction condition (Scheme 3.7). This
reaction which is known to proceed via the generation of primary alkyl radical/Ni(II) and
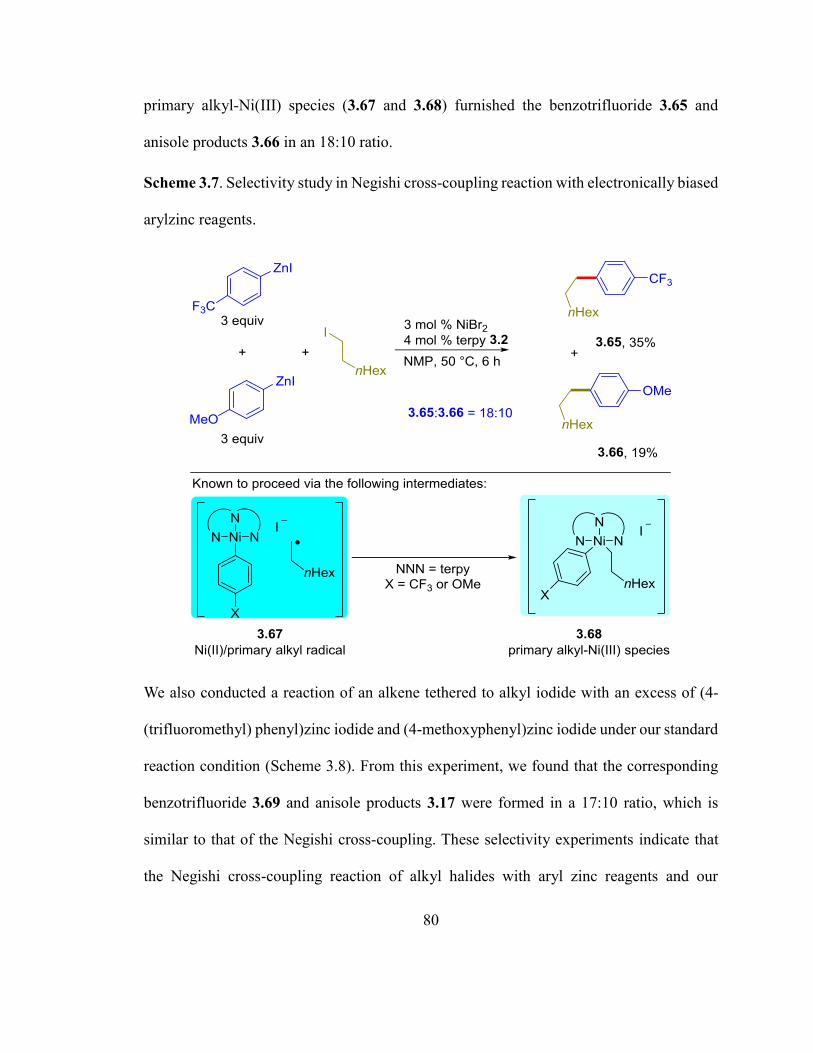
80
primary alkyl-Ni(III) species (3.67 and 3.68) furnished the benzotrifluoride 3.65 and
anisole products 3.66 in an 18:10 ratio.
Scheme 3.7. Selectivity study in Negishi cross-coupling reaction with electronically biased
arylzinc reagents.
We also conducted a reaction of an alkene tethered to alkyl iodide with an excess of (4-
(trifluoromethyl) phenyl)zinc iodide and (4-methoxyphenyl)zinc iodide under our standard
reaction condition (Scheme 3.8). From this experiment, we found that the corresponding
benzotrifluoride 3.69 and anisole products 3.17 were formed in a 17:10 ratio, which is
similar to that of the Negishi cross-coupling. These selectivity experiments indicate that
the Negishi cross-coupling reaction of alkyl halides with aryl zinc reagents and our
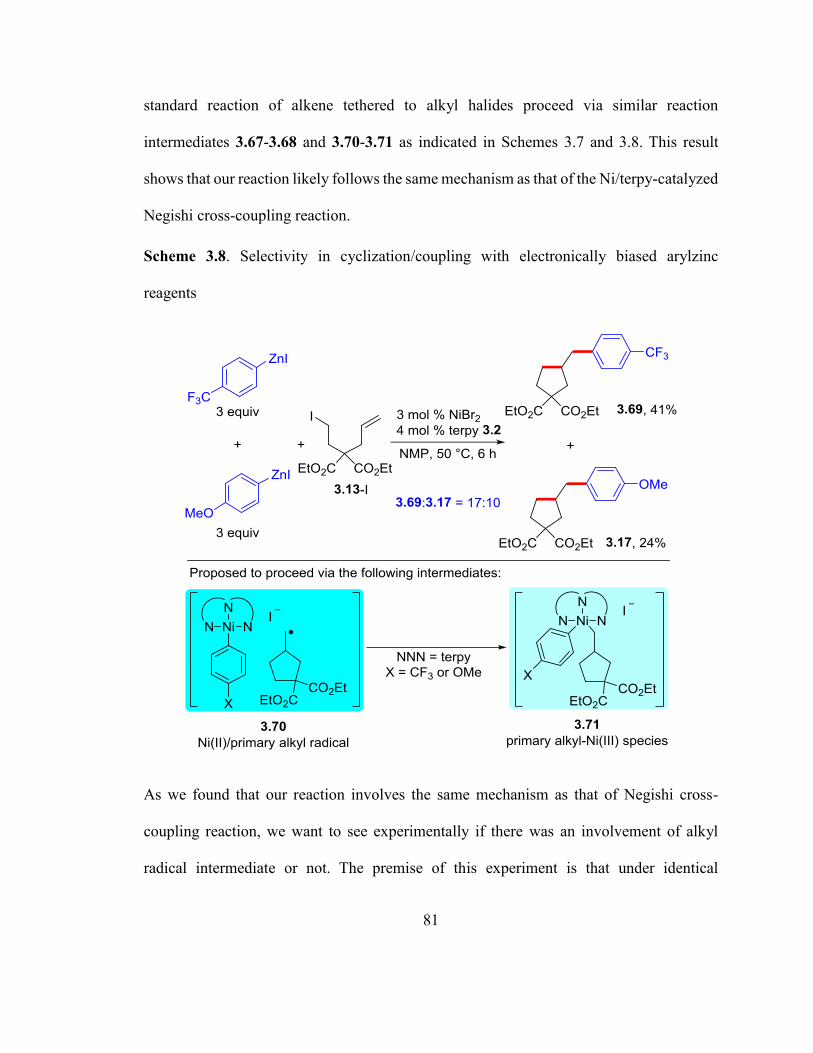
81
standard reaction of alkene tethered to alkyl halides proceed via similar reaction
intermediates 3.67-3.68 and 3.70-3.71 as indicated in Schemes 3.7 and 3.8. This result
shows that our reaction likely follows the same mechanism as that of the Ni/terpy-catalyzed
Negishi cross-coupling reaction.
Scheme 3.8. Selectivity in cyclization/coupling with electronically biased arylzinc
reagents
As we found that our reaction involves the same mechanism as that of Negishi cross-
coupling reaction, we want to see experimentally if there was an involvement of alkyl
radical intermediate or not. The premise of this experiment is that under identical

82
conditions, two different stereoisomers must form the same ratio of product if there is an
involvement of a radical intermediate. With the formation of a radical intermediate, there
is the loss of a stereocenter and the selectivity of the product no longer depends upon the
stereochemistry of the starting materials. To examine the presence of an alkyl radical
intermediate, we conducted several experiments. In the first experiment, the stereochemical
outcomes of a reaction involving the cis- and trans-isomers of 1-(allyloxy)-2-
bromocyclohexane (3.72) were analyzed (Scheme 3.9). We conducted an experiment of
cis- and trans-1-(allyloxy)-2-bromocyclohexane (3.72) separately with PhZnI under our
standard reaction condition. Both cis- and trans-isomers generated the cyclization/coupling
product 3.73 in 63% and 74% yields, respectively, with the same degree of
diastereoselectivity (1.3:1). These results indicate that the reactions of both cis- and trans-
isomers of 1-(allyloxy)-2-bromocyclohexane (3.72) proceed via the formation of the same
radical intermediate 3.74 after the loss of stereochemistry at the C-Br chiral center.
Scheme 3.9. Diastereoselectivity studies with cis- and trans-1-(allyloxy)-2-
bromocyclohexane

83
To further support the presence of an alkyl radical intermediate, we also performed an
experiment using a racemic chiral alkene tethered to alkyl bromide 3.75 under our reaction
condition and compared the diastereoselectivity of our reaction with a separate reaction
using the same alkyl bromide 3.75 under a condition which is known to proceed through a
radical cyclization pathway (Scheme 3.10). The premise of the experiment is that if there
is an involvement of a radical intermediate, a chiral-racemic substrate must form the same
ratio of product as that of product obtained from a well-known standard radical cyclization
reaction. We performed an experiment by reacting the racemic chiral alkyl bromide 3.75
with Bu3SnH in the presence of 10 mol % a 2,2′-Azobis(2-methylpropionitrile) (AIBN) as
a radical initiator237 under UV light (300 nm). This reaction is known to proceed with the
formation of uncyclized and cyclized alkyl radicals 3.78 and 3.79.237 We found a trans-
isomer of the cyclization/H-atom abstraction product 3.76 as a single diastereomer from
this reaction in 70% yield.
We also conducted our standard cyclization/coupling reaction using the same racemic
chiral alkene tethered to alkyl bromide 3.75 and PhZnI under our standard condition. This
experiment also furnished a trans-isomer of the cyclization/coupling product 3.77 as a
single diastereomer in 58% yield. The results of these experiments indicate that our
standard cyclization/coupling reaction proceeds via the same diastereoselectivity-
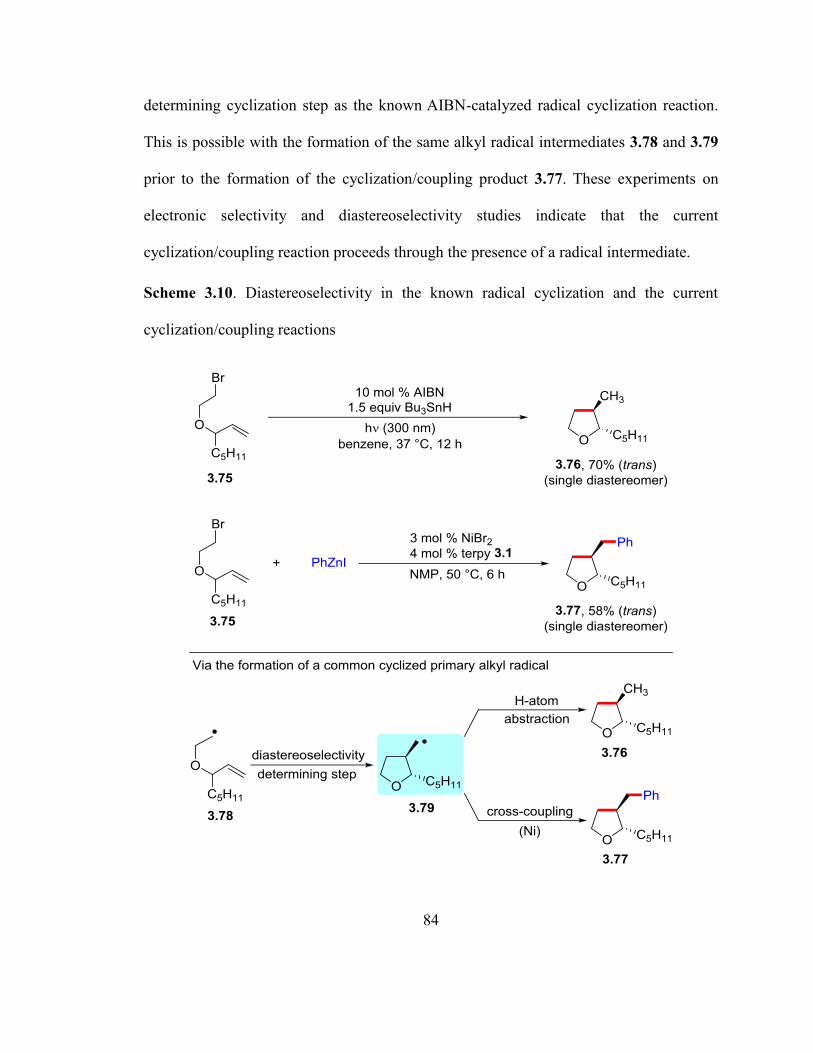
84
determining cyclization step as the known AIBN-catalyzed radical cyclization reaction.
This is possible with the formation of the same alkyl radical intermediates 3.78 and 3.79
prior to the formation of the cyclization/coupling product 3.77. These experiments on
electronic selectivity and diastereoselectivity studies indicate that the current
cyclization/coupling reaction proceeds through the presence of a radical intermediate.
Scheme 3.10. Diastereoselectivity in the known radical cyclization and the current
cyclization/coupling reactions

85
3.5 Conclusion
We developed an efficient (terpy)NiBr2 catalytic system for the cyclization/coupling of
alkenes tethered to alkyl halides with arylzinc reagents regioselectively. This reaction
tolerates a wide variety of functional groups and base-sensitive racemizable stereocenters.
This reaction protocol also provides rapid access to (arylmethyl)carbo- and heterocyclic
scaffolds, which occur widely as structural cores in various natural products and bioactive
molecules. We further applied this new method for the concise synthesis of six lignan
natural products containing three different structural frameworks in gram-scale quantities.
Mechanistic studies with radical probes and product selectivities show that the current
cyclization/coupling reaction proceeds via a single electron transfer (SET) process.

86
Chapter 4. Experimental Section
4.1 Ni-Catalyzed Alkylarylation of Vinylarenes
4.1.1 General Information
Unless otherwise noted, all the reactions were carried out under an atmosphere of nitrogen
and all the chemicals were handled under the nitrogen atmosphere. All the glassware
including the 4-dram and 1-dram borosilicate (Kimble-Chase) vials, and pressure vessels
were properly dried in an oven before use. Bulk solvents were obtained from EMD and
anhydrous solvents (DMF, DMA, DMSO, NMP, dioxane, toluene) were obtained from
Sigma-Aldrich, and were used directly without further purification. Deuterated solvents
were purchased from Sigma-Aldrich. NiBr2 and ZnCl2 (99.95%) was purchased from Alfa-
Aesar. Aryl halides were purchased from Acros, Sigma-Aldrich, Oakwood, TCI-America,
Matrix and Alfa-Aesar. 1H, 13C, and 19F NMR spectra were recorded on a Bruker
instrument (300 & 500, 75 & 126, and 282 & 470 MHz respectively) at Department of
Chemistry and Chemical Biology, University of New Mexico (UNM) and internally
referenced to the residual solvent signals of CDCl3 for 1H, 13C NMR at 7.26 and 77.16
ppm, and externally referenced to C6F6 for 19F NMR at −164.9 ppm. The chemical shifts
of NMR and the coupling constants (J) for 1H, 13C, and 19F NMR are reported in δ parts
per millions (ppm) and in Hertz, respectively. The following conventions are used for
multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of
doublet; br, broad. High resolution mass spectra for new compounds were recorded at the
Mass Spectrometry facilities at the Department of Chemistry and Chemical Biology, UNM,
and at the University of Texas Austin.

87
4.1.2 Experimental Procedure
General procedure for the preparation of arylzinc reagent
Organozinc reagents were prepared according to a literature procedure.238 In a glovebox to
a Schlenk flask , anhydrous LiCl (420 mg, 10 mmol) and zinc powder (984 mg, 15 mmol)
was added and the mixture was dried under high vacuum at 150 °C to 170 °C for 2 h outside
the glovebox. It was cooled down to room temperature after 2 h and the reaction flask was
flushed with nitrogen. Then it was again taken to a glovebox and anhydrous THF (10 mL)
was added with stirring the solution at room temperature. Later, with the addition of 5
mol% of BrCH2CH2Br and 3 mol% of TMSCl to the zinc/THF suspension zinc was
activated and the mixture was stirred for 5 min at room temperature. To this stirred solution
was added corresponding aryl iodides (10 mmol) (neat) dropwise or portionwise and the
reaction mixture was refluxed for electron-deficient (also iodobenzene) for 24 h and
electron rich aryl iodides for 48-96 h. The final concentration of the arylzinc reagent was
determined by titration with molecular iodine in THF.239
General procedure for screening reaction conditions
In a glovebox, a solution of phenylzinc iodide (0.20 mmol, 240 µL of 0.833 M stock
solution in THF) was taken in a 1-dram vial and the solvent was removed under vacuum.
To the PhZnI residue was added catalyst (0.005 mmol), 2-vinylnaphthalene (15.4 mg, 0.10
mmol) and iodocyclohexane (42.0 mg, 0.20 mmol). The mixture was then dissolved in 0.5
mL solvent. The vial was tightly capped and removed from the glovebox. It was vigorously
stirred at room temperature for 6 h. After 6 h, 50 µL of pyrene (0.010 mmol, 0.2 M stock

88
solution) as an internal standard was added in the reaction, diluted with EtOAc (2 mL) and
filtered through a short pad of silica gel in a pipette. The filtrate was then analyzed by 1H
NMR. The percentage yields of the product 2.2 was calculated by integrating against
pyrene as an internal standard.
General procedure for product isolation
In a glovebox, a solution of arylzinc reagents (1.0 mmol, 1.20 mL of 0.833 M stock solution
in THF) was taken in a 15 mL sealed tube and the solvent was removed under vacuum. To
the residue of ArZnI, NiBr2 (5.5 mg, 0.025 mmol), vinylarenes (0.5 mmol) and alkyl halides
(1.0 mmol). 10 mol% NiBr2 was used for 2.17. For tert-butyl halides, 5 mol% (Ph3P)2NiCl
was used instead of NiBr2 unless stated otherwise. 5 mol% Ni(cod)2 was used for 2.4, 2.7,
2.18 and 2.21 instead of NiBr2. The mixture was then dissolved in 2.5 mL NMP. The vial
was tightly capped and removed from glove box. It was vigorously stirred at room
temperature. After 6 h (or 8 h for 2.5, 2.6, 2.34, 2.35 and 2.36, and 12 h at 50 °C for 2.24),

89
the reaction mixture diluted with EtOAc (Et2O was used for hydrocarbons without any
functional group – compounds 2.2, 2.3, 2.9, 2.11, 2.14, 2.18 and 2.26) (10 mL) and washed
with H2O (5 mL × 3). The aqueous fraction was extracted back with EtOAc (5 mL × 3)
and combined with the first ethyl acetate fraction. The combined ethyl acetate fraction was
dried over Na2SO4 and the solvent was removed in a rotary evaporator. The product was
purified by silica gel column chromatography using different percentage of solvent as
eluent.
Preparation of substrates
Procedure A
Preparation of 4-vinylbenzyl 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-
yl)acetate: In a dry RB flask, indometacin (357.78 mg, 1 mmol) was weigh out and DMF
(5 mL) was added. Later K2CO3 (207.3 mg, 1.5 mmol) and KI (249 mg, 1.5 mmol) was
added and stirred. To this stirring suspension 4-vinylbenzyl chloride (167.8 mg, 1.1 mmol)
was added let it stirred for 12 h at room temperature. To the reaction EtOAc (10 mL) was
added and water (6 mL) was added then it was extracted and washed three times with water.
The organic layer was collected and dried. The product was purified by flash column
chromatography using 30% EtOAc/hex as eluent and white solid of 4-vinylbenzyl 2-(1-(4-

90
chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetate was obtained, 95% yield. 1H
NMR (300 MHz, CDCl3): 2.35 (s, 3H), 3.70 (s, 2H), 3.74 (s, 3H), 5.11 (s, 2H), 5.26 (d,
J = 12.0 Hz, 1H), 5.74 (d, J = 18.0 Hz, 1H), 6.69 (dd, J = 18.0 Hz, J = 12.0 Hz, 2H), 6.90
(dd, J = 12.0 Hz, J = 9.0 Hz, 2H), 7.23 (s, 1H), 7.35 (d, J = 9.0 Hz, 2H), 7.43 (d, J = 9.0
Hz, 2H), 7.62 (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 13.5, 30.5, 55.7, 66.6,
101.3, 111.9, 112.6, 114.5, 115.1, 126.4, 128.6, 129.2, 130.7, 130.9, 131.3, 134.0, 135.3,
136.0, 136.4, 137.7, 139.3, 156.1, 168.4, 170.7; IR (neat) cm-1 : 2980, 1733, 1473, 1091,
914; HRMS (ESI): Calcd for C28H24ClNO4 (M+H)+ 474.1472, found 474.1453.
Preparation of 4-vinylbenzyl 2-(1-methyl-5-(4-methylbenzoyl)-1H-pyrrol-2-yl)acetate:
Above procedure A was used to prepare 4-vinylbenzyl 2-(1-methyl-5-(4-methylbenzoyl)-
1H-pyrrol-2-yl)acetate. Here, instead of indometacin and K2CO3, tolmetin sodium salt
dihydrate (315.3 mg, 1 mmol) was used. The product 4-vinylbenzyl 2-(1-methyl-5-(4-
methylbenzoyl)-1H-pyrrol-2-yl)acetate was purified by flash column chromatography
using 25% EtOAc/hex as eluent and white solid of 4-vinylbenzyl 2-(p-tolyl)acetate was
obtained, 96% yield. 1H NMR (300 MHz, CDCl3): 2.40 (s, 3H), 3.73 (s, 2H), 3.89 (s,
3H), 5.14 (s, 2H), 5.25 (d, J = 12.0 Hz, 1H), 5.73 (d, J = 9.0 Hz, 1H), 6.08 (d, J = 3.0 Hz,
1H), 6.64 (d, J = 3.0 Hz, 1H), 6.69 (dd, J = 18.0 Hz, J = 12.0 Hz, 1H), 7.21 (d, J = 6.0 Hz,
2H), 7.27 (d, J = 9.0 Hz, 2H), 7.38 (d, J = 6.0 Hz, 2H), 7.68 (d, J = 6.0 Hz, 2H); 13C NMR

91
(75 MHz, CDCl3): 21.6, 33.0, 33.3, 66.9, 109.6, 122.3, 126.5, 128.7, 128.8, 129.5, 131.5,
134.3, 134.9, 136.3, 137.4, 137.9, 141.9, 169.2, 185.9; IR (neat) cm-1 : 2958, 1735, 1608,
1480, 1174, 1052, 919; HRMS (ESI): Calcd for C24H24NO3 (M+H)+ 374.1756, found
374.1760.
Preparation of 4-vinylbenzyl 2-(p-tolyl)acetate: Above procedure A was used to prepare 4-
vinylbenzyl 2-(p-tolyl) acetate. Here, instead of Indometacin, p-tolylacetic acid (150.1 mg,
1 mmol) was used to get 4-vinylbenzyl 2-(p-tolyl)acetate. The product 2-(p-tolyl)acetate
was purified by flash column chromatography using 5% EtOAc/hex as eluent and a
colorless liquid of 4-vinylbenzyl 2-(p-tolyl)acetate was obtained, 98% yield. 1H NMR (300
MHz, CDCl3): 2.33 (s, 3H), 3.63 (s, 2H), 5.11 (s, 2H), 5.26 (d, J = 12.0 Hz, 1H), 5.75
(d, J = 15.0 Hz, 1H), 6.71 (dd, J = 18.0 Hz, J = 12.0 Hz, 1H), 7.13 (d, J = 9.0 Hz, 2H), 7.18
(d, J = 9.0 Hz, 2H), 7.27 (d, J = 9.0 Hz, 2H), 7.38 (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz,
CDCl3): 21.2, 41.0, 66.4, 141.4, 126.4, 128.5, 129.2, 129.3, 130.9, 135.5, 136.4, 136.8,
137.6, 171.7; IR (neat) cm-1 : 2980, 1731, 1514, 1239, 1138, 988, 824; HRMS (APPI):
Calcd for C18H19O2 (M+H)+ 267.1385, found 267.1384.
4.1.3 Mechanistic studies
Experiment for the reduction of Ni(II) to Ni(0) by ArZnX

92
In a 1 dram vial, NiBr2.DME (3.08 mg, 0.010 mmol) and 2-vinyl naphthalene (15.4 mg,
0.10 mmol) were weighed and was dissolved in 0.4 mL NMP solution. The homogeneous
solution was carefully transferred to a clean NMR tube with septum. The vial was further
washed with extra 0.3 mL of NMP and was completely transferred to NMR tube. The
internal standard, benzotrifluoride (20 µL of 1.0 M stock solution) was then added to the
NMR tube and tightly capped. To this solution in the NMR tube, 4-(fluorophenyl)zinc
iodide (0.10 mmol, 133 µL of 0.750 M ) was added. The solution instantaneously turned
black upon mixing. The resulting dark solution was then monitored my 19F NMR at < 0.5
min and at 20 min. The amount of homocoupling was obtained by integrating the 19F signal
of 4,4’-difluorobiphenyl against that of benzotrifluoride. The amount of 4,4’-
difluorobiphenyl was found to be 0.009 mmol both at < 0.5 min and 20 min.
Competition Experiments
a. Competition experiments with 1°, 2° and 3° R-X

93
To a dry 1 dram vial, PhZnI (0.20 mmol, 240 µL of 0.833 M stock solution in THF) was
taken and the solvent was removed under vacuum. To the residue, NiBr2 (1.1 mg, 0.005
mmol), 2-vinyl naphthalene (2.1) (15.4 mg, 0.10 mmol), iodocyclohexane (42.0 mg, 0.20
mmol) and iodooctane (48 mg, 0.20 mmol) were added respectively. The mixture was then
dissolved in NMP (0.5 mL) and stirred vigorously at room temperature. After 6 h, the
reaction mixture was quenched with H2O and was diluted with EtOAc (4 mL) and filtered
through silica pad. The clear solution was run in GC using pyrene as an internal standard.
The product peak of 2.2 and 2.3 were analyzed by GC and GC/MS using pyrene as an
internal standard. The ratio of 2.2:2.3 was found as ~3:1 after correction for the response
factors of the products against pyrene as an internal standard.
To a dry 1 dram vial, PhZnI (0.20 mmol, 240 µL of 0.833 M stock solution in THF) was
taken and the solvent was removed under vacuum. To the residue, NiBr2 (1.1 mg, 0.005
mmol), 2-vinyl naphthalene (2.1) (15.4 mg, 0.1 mmol), t-butyl bromide (27.4 mg, 0.2
mmol) and bromo cyclohexane (32.6 mg, 0.2 mmol) were added respectively. The mixture
was then dissolved in NMP (0.5 mL) and stirred vigorously at room temperature. After 6
h, the reaction mixture was quenched with H2O and was diluted with EtOAc (4 mL) and
filtered through silica pad. The clear solution was run in GC using pyrene as an internal

94
standard. The product peak of 2.2 and 2.26 were analyzed by GC and GC/MS using pyrene
as an internal standard. The ratio of 2.2:2.26 was found as >1: 79 after correction for the
response factors of the products against pyrene as an internal standard.
b. Competition experiments with RI, RBr and RCl
To a dry 1-dram vial, PhZnI (0.20 mmol, 240 µL of 0.833 M stock solution in THF) was
taken and the solvent was removed under vacuum. To the residue, NiBr2 (1.1 mg, 0.005
mmol), 2-vinyl naphthalene (2.1) (15.4 mg, 0.1 mmol), iodo cyclohexane (42.0 mg, 0.2
mmol) and 3-bromopentane (30.2 mg, 0.2 mmol) were added respectively. The mixture
was dissolved in NMP (0.5 mL) and stirred vigorously at room temperature. After 6 h, the
reaction mixture was quenched with H2O and was diluted with EtOAc (4 mL) and filtered
through silica pad. The clear solution was run in GC using pyrene as an internal standard.
The product peak of 2.2 and 2.11 were analyzed by GC and GC/MS using pyrene as an
internal standard. The ratio of 2.2:2.11 was found as 10: 1 after correction for the response
factors of the products against pyrene as an internal standard.
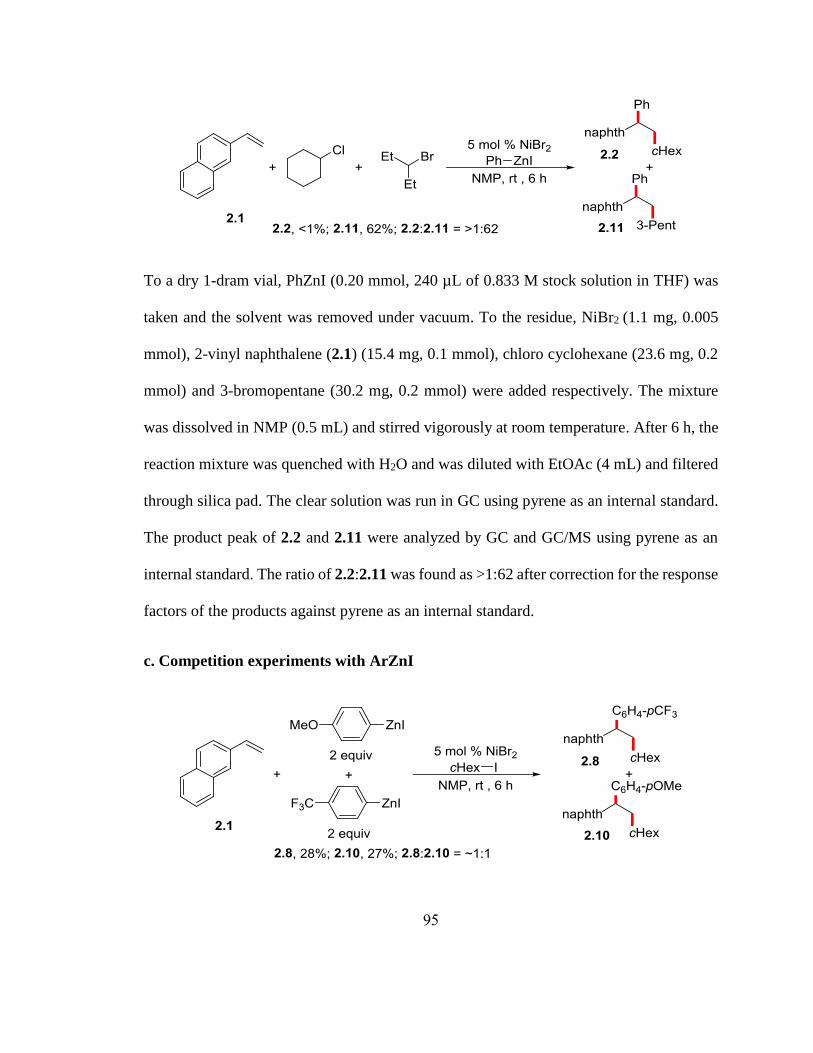
95
To a dry 1-dram vial, PhZnI (0.20 mmol, 240 µL of 0.833 M stock solution in THF) was
taken and the solvent was removed under vacuum. To the residue, NiBr2 (1.1 mg, 0.005
mmol), 2-vinyl naphthalene (2.1) (15.4 mg, 0.1 mmol), chloro cyclohexane (23.6 mg, 0.2
mmol) and 3-bromopentane (30.2 mg, 0.2 mmol) were added respectively. The mixture
was dissolved in NMP (0.5 mL) and stirred vigorously at room temperature. After 6 h, the
reaction mixture was quenched with H2O and was diluted with EtOAc (4 mL) and filtered
through silica pad. The clear solution was run in GC using pyrene as an internal standard.
The product peak of 2.2 and 2.11 were analyzed by GC and GC/MS using pyrene as an
internal standard. The ratio of 2.2:2.11 was found as >1:62 after correction for the response
factors of the products against pyrene as an internal standard.
c. Competition experiments with ArZnI

96
To a dry 1 dram vial, (4-(trifluoromethyl)phenyl)zinc iodide (0.2 mmol) and (4-
methoxyphenyl)zinc iodide (0.2 mmol) stock solution in THF was taken and the solvent
was removed under vacuum. To the residue, NiBr2 (1.1 mg, 0.005 mmol), 2-vinyl
naphthalene (2.1) (15.4 mg, 0.1 mmol), iodo cyclohexane (42.0 mg, 0.2 mmol) were added
respectively. The mixture was dissolved in NMP (0.5 mL) and stirred vigorously at room
temperature. After 6 h, the reaction mixture quenched with H2O and was diluted with
EtOAc (4 mL) and filtered through silica pad. The clear solution was run in GC using
pyrene as an internal standard. The product peak of 2.8 and 2.10 were analyzed by GC and
GC/MS using pyrene as an internal standard. The ratio of 2.8:2.10 was found as ~1:1 after
correction for the response factors of the products against pyrene as an internal standard.
Kinetic Studies
Preparation of stock solutions. 2-Vinylnaphthalene (1.00 M): The stock solution of 2-
vinyl naphthalene was prepared in 1.0 mL volumetric flask by dissolving 154.2 mg of 2-
vinyl naphthalene in NMP.
PhZnI (0.830 M): 5 mL of PhZnI solution (0.830 M) in THF was transferred to a 4-dram
vial and THF was removed under vacuum. The residue was then transferred to a 5.0 mL
volumetric flask by dissolving in NMP and the total volume was made up to 5.0 mL.
Iodocyclohexane (2.00 M): The stock solution of iodocyclohexane was prepared in 1.0 mL
volumetric flask by dissolving 420 mg of 2-vinylnaphthalene in NMP.
a. General procedure for typical reaction kinetics

97
For the reaction of 2-vinylnaphthalene (0.150 M), PhZnI (0.30 M) with
iodocyclohexane (0.30 M): In a glove box, NiBr2 (0.015 mmol, 3.30 mg) was weigh out
into a 2.0 mL volumetric flask and 300 µL of iodocyclohexane (2.00 M), 300 µL of 2-
vinylnaphthalene (1.00 M) and 720 µL of PhZnI (0.830 M) stock solutions were added,
respectively. The mixture was diluted to 2.0 mL to obtain a homogeneous solution
containing final concentrations of 0.30 M iodocyclohexane, 0.30 M PhZnI and 0.15 M 2-
vinylnaphthalene. The solution was then transferred to a 1dram vial containing a stirring
bar and stirred vigorously at room temperature. 200 µL of the reaction mixture was taken
out at 5, 10, 15, 30, 60, 120, 240, 360 minutes and quenched with H2O immediately. To
the quenched solution was added EtOAc (2.0 mL), pyrene (15 µL of 0.20 M) as an internal
standard and the organic layer was washed with H2O (0.5 mL × 3). The organic fraction
was dried over Na2SO4, the solvent was removed in a rotary evaporator and the sample was
analyzed by 1H NMR. The percentage yields of the product 2.2 were calculated by
integrating against pyrene as an internal standard, which were then converted to molar
concentrations. A duplicate reaction was also run under otherwise identical conditions and
an average value was taken for each time point. The yields in molar concentrations are
presented in Table 1. The molar concentrations of the product 2.2 were plotted against the
reaction time to obtain a typical reaction kinetic profile.
Table 1. The molar concentration of product 2.2 at different time interval
Time (s) Yield of 2 (M)
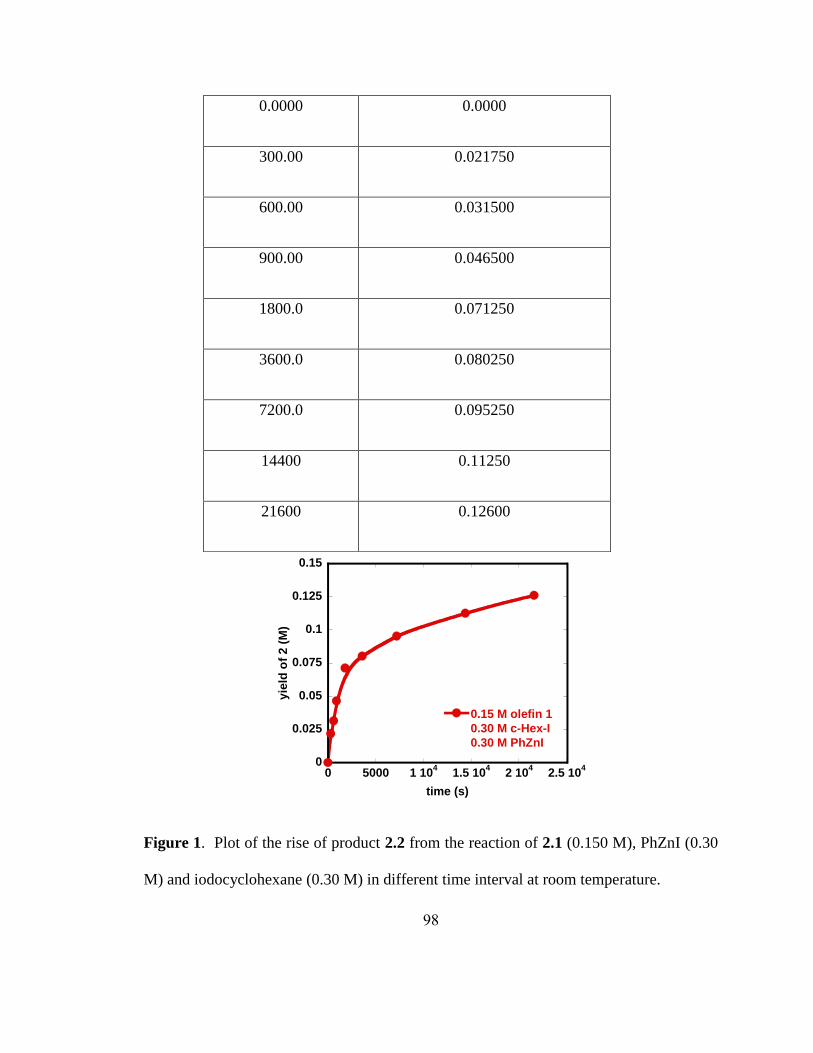
98
0
0.025
0.05
0.075
0.1
0.125
0.15
0 5000 1 104
1.5 104
2 104
2.5 104
0.15 M olefin 1
0.30 M c-Hex-I
0.30 M PhZnI
yie
ld o
f 2 (
M)
time (s)
Figure 1. Plot of the rise of product 2.2 from the reaction of 2.1 (0.150 M), PhZnI (0.30
M) and iodocyclohexane (0.30 M) in different time interval at room temperature.
0.0000 0.0000
300.00 0.021750
600.00 0.031500
900.00 0.046500
1800.0 0.071250
3600.0 0.080250
7200.0 0.095250
14400 0.11250
21600 0.12600

99
b. General procedure to determine the dependence of reaction rate on the
concentration of iodocyclohexane
For the reaction of 2-vinlylnaphthalene (0.15 M), PhZnI (0.30 M) with
iodocyclohexane (0.15 M): In a glove box, NiBr2 (0.015 mmol, 3.3 mg) was weigh out
into a 2.0 mL volumetric flask and 150 µL of iodocyclohexane (2.00 M), 300 µL of 2-
vinylnaphthalene (1.00 M) and 720 µL of PhZnI (0.830 M) stock solutions were added,
respectively. The mixture was diluted to 2.0 mL to obtain a homogeneous solution
containing final concentrations of 0.15 M iodocyclohexane, 0.30 M PhZnI and 0.15 M 2-
vinylnaphthalene. The solution was then transferred to a 1-dram vial containing a stirring
bar and stirred vigorously at room temperature. 200 µL of the reaction mixture was taken
out at 1, 2.5, 5, 7.5, 10, 12.5, 15 min and quenched with H2O immediately. To the quenched
solution was added EtOAc (2 mL), pyrene (15 µL of 0.20 M) as an internal standard and
the organic layer was washed with H2O (0.5 mL × 3). The organic fraction was dried over
Na2SO4, the solvent was removed in a rotary evaporator and the sample was analyzed by
1H NMR.
For the reaction of 2-vinylnaphthalene (0.15 M), PhZnI (0.30 M) with
iodocyclohexane (0.30 M): The procedure for this reaction was the same as above but
instead of 150 µL of iodocyclohexane (2.0 M), 300 µL of iodocyclohexane (2.0 M) was
added in the reaction.
For the reaction of 2-vinylnaphthalene (0.15 M), PhZnI (0.30 M) with
iodocyclohexane (0.45 M): The procedure for this reaction was the same as above but

100
instead of 150 µL of iodocyclohexane (2.0 M), 450 µL of iodocyclohexane (2.0 M) was
added in the reaction.
For the reaction of 2-vinylnaphthalene (0.15 M), PhZnI (0.30 M) with
iodocyclohexane (0.60 M): The procedure for this reaction was the same as above but
instead of 150 µL of iodocyclohexane (2.0 M), 600 µL of iodocyclohexane (2.0 M) was
added in the reaction.
The percentage yields of the product 2.2 were calculated by integrating against pyrene as
an internal standard, which were then converted to molar concentrations. A duplicate
reaction was also run under otherwise identical conditions and an average value was taken
for each time point. The molar concentration of product 2.2 (only the data corresponding
to the linear portion of the graph, typically < 40% yield, was used) was plotted against the
reaction time and the slope of linear portion of the curve was used to determine the initial
rates of the reaction. The table showing molar concentration of product 2.2 in different
concentration of iodocyclohexane, graph showing the rate at different concentration of
iodocyclohexane, table with kin value and the graph showing kin versus [Cyhex-I] are shown
below.
Table 2. The molar concentration of product 2.2 in different concentration of
iodocyclohexane at different time interval
Time
(s)
0.15 M [Cyhex-
I]
0.30 M [Cyhex-
I]
0.45 M [Cyhex-
I]
0.60 M [Cyhex-
I]
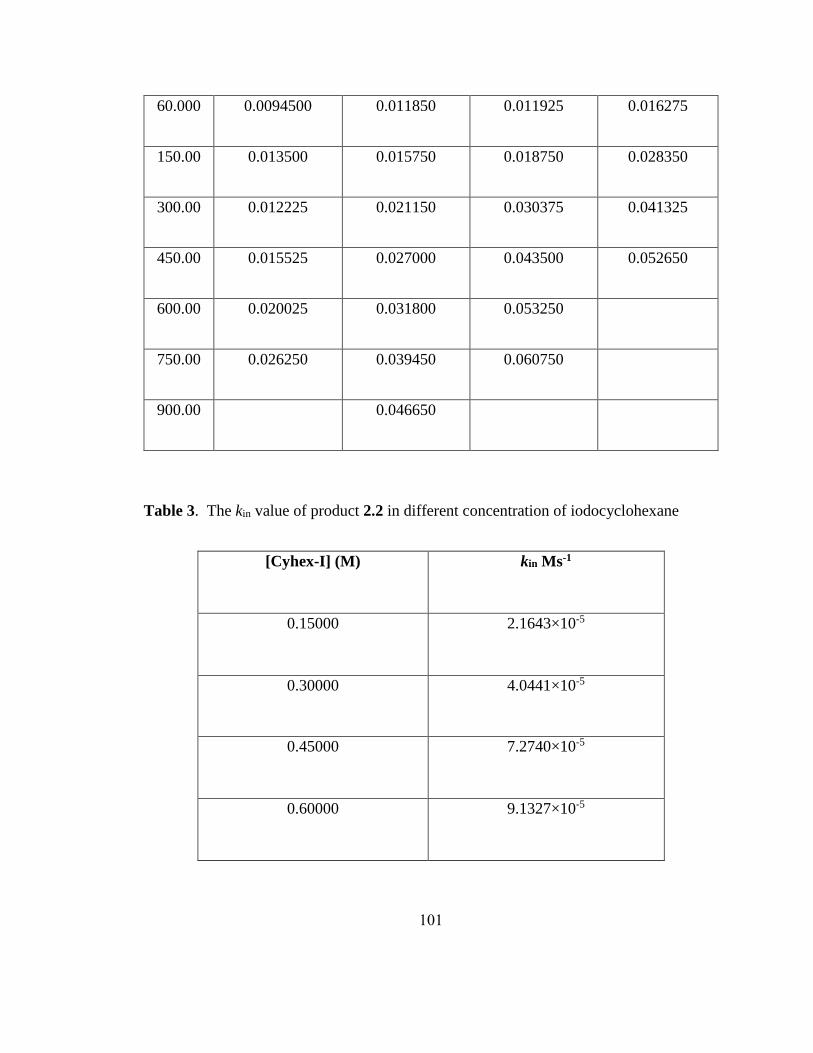
101
60.000 0.0094500 0.011850 0.011925 0.016275
150.00 0.013500 0.015750 0.018750 0.028350
300.00 0.012225 0.021150 0.030375 0.041325
450.00 0.015525 0.027000 0.043500 0.052650
600.00 0.020025 0.031800 0.053250
750.00 0.026250 0.039450 0.060750
900.00 0.046650
Table 3. The kin value of product 2.2 in different concentration of iodocyclohexane
[Cyhex-I] (M) kin Ms-1
0.15000 2.1643×10-5
0.30000 4.0441×10-5
0.45000 7.2740×10-5
0.60000 9.1327×10-5
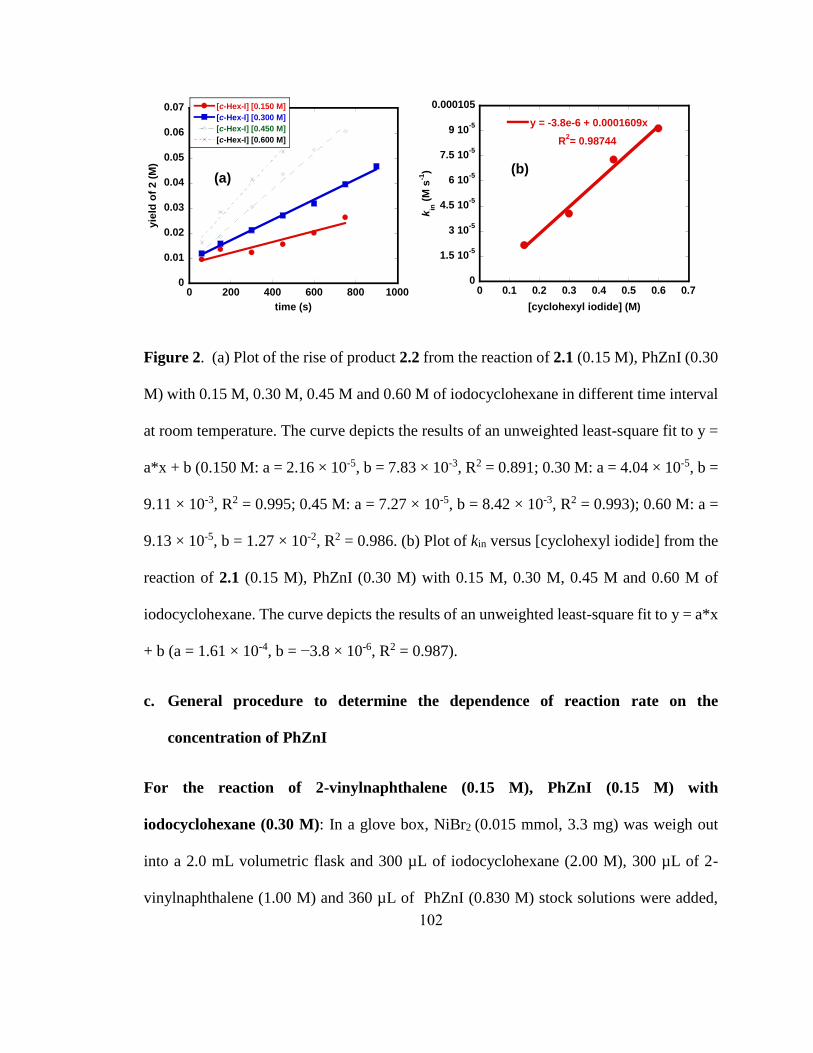
102
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0 200 400 600 800 1000
[c-Hex-I] [0.150 M]
[c-Hex-I] [0.300 M]
[c-Hex-I] [0.450 M]
[c-Hex-I] [0.600 M]
yie
ld o
f 2
(M
)
time (s)
(a)
0
1.5 10-5
3 10-5
4.5 10-5
6 10-5
7.5 10-5
9 10-5
0.000105
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
y = -3.8e-6 + 0.0001609x
R2= 0.98744
kin
(M
s-1
)
[cyclohexyl iodide] (M)
(b)
Figure 2. (a) Plot of the rise of product 2.2 from the reaction of 2.1 (0.15 M), PhZnI (0.30
M) with 0.15 M, 0.30 M, 0.45 M and 0.60 M of iodocyclohexane in different time interval
at room temperature. The curve depicts the results of an unweighted least-square fit to y =
a*x + b (0.150 M: a = 2.16 × 10-5, b = 7.83 × 10-3, R2 = 0.891; 0.30 M: a = 4.04 × 10-5, b =
9.11 × 10-3, R2 = 0.995; 0.45 M: a = 7.27 × 10-5, b = 8.42 × 10-3, R2 = 0.993); 0.60 M: a =
9.13 × 10-5, b = 1.27 × 10-2, R2 = 0.986. (b) Plot of kin versus [cyclohexyl iodide] from the
reaction of 2.1 (0.15 M), PhZnI (0.30 M) with 0.15 M, 0.30 M, 0.45 M and 0.60 M of
iodocyclohexane. The curve depicts the results of an unweighted least-square fit to y = a*x
+ b (a = 1.61 × 10-4, b = −3.8 × 10-6, R2 = 0.987).
c. General procedure to determine the dependence of reaction rate on the
concentration of PhZnI
For the reaction of 2-vinylnaphthalene (0.15 M), PhZnI (0.15 M) with
iodocyclohexane (0.30 M): In a glove box, NiBr2 (0.015 mmol, 3.3 mg) was weigh out
into a 2.0 mL volumetric flask and 300 µL of iodocyclohexane (2.00 M), 300 µL of 2-
vinylnaphthalene (1.00 M) and 360 µL of PhZnI (0.830 M) stock solutions were added,

103
respectively. The mixture was diluted to 2.0 mL to obtain a homogeneous solution
containing final concentrations of 0.30 M iodocyclohexane, 0.15 M PhZnI and 0.15 M 2-
vinylnaphthalene. The solution was then transferred to a 1 dram vial containing a stirring
bar and stirred vigorously at room temperature. 200 µL of the reaction mixture was taken
out at 1, 2.5, 5, 7.5, 10, 12.5, 15 min and quenched with H2O immediately. To the quenched
solution was added EtOAc (2 mL), pyrene (15 µL of 0.20 M) as an internal standard and
the organic layer was washed with H2O (0.5 mL × 3). The organic fraction was dried over
Na2SO4, the solvent was removed in a rotary evaporator and the sample was analyzed by
1H NMR.
For the reaction of 2-vinylnaphthalene (0.15 M), iodocyclohexane (0.30 M) with
PhZnI (0.30 M): The procedure for this reaction was same as above but instead of 360 µL
of PhZnI (0.830 M stock solution, 720 µL of PhZnI (0.830 M stock solution) was added in
the reaction.
For the reaction of 2-vinylnaphthalene (0.15 M), iodocyclohexane (0.30 M) with
PhZnI (0.45 M): The procedure for this reaction was same as above but instead of 360 µL
of PhZnI (0.830 M stock solution, 900 µL of PhZnI (1.0 M stock solution) was added in
the reaction.
For the reaction of 2-vinylnaphthalene (0.15 M), iodocyclohexane (0.30 M) with
PhZnI (0.60 M): The procedure for this reaction was same as above but instead of 360 µL
of PhZnI (0.830 M stock solution, 1200 µL of PhZnI (1.0 M stock solution) was added in
the reaction.
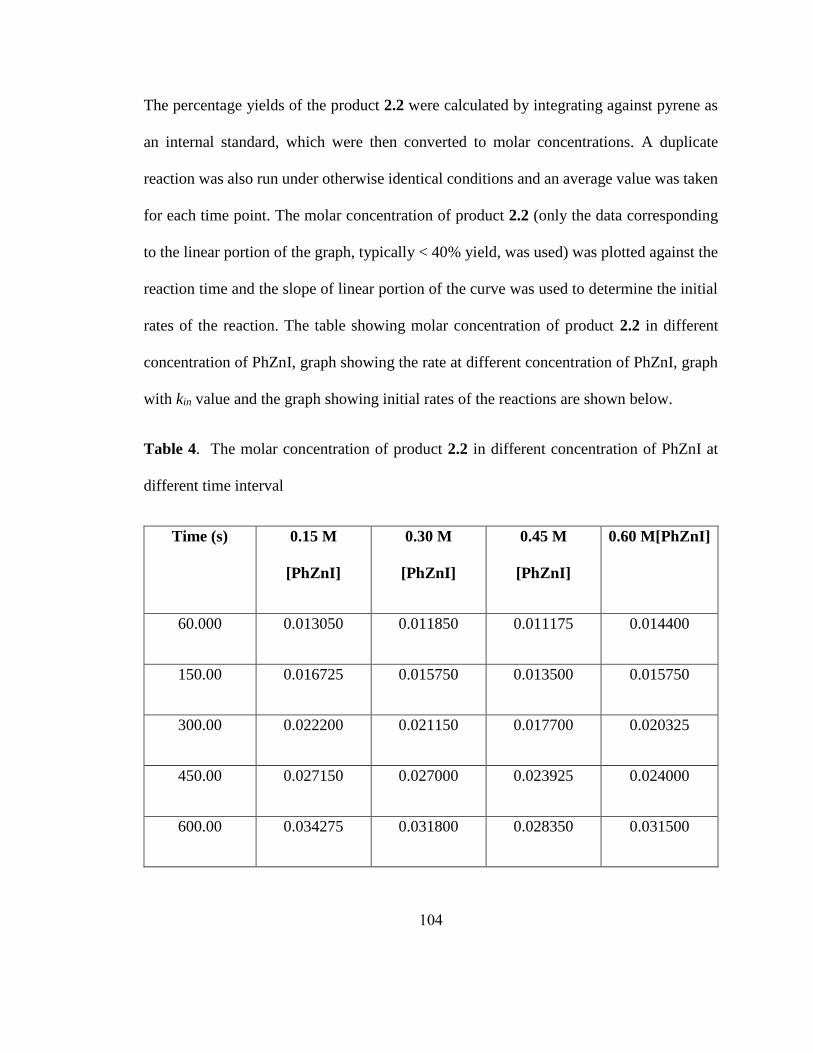
104
The percentage yields of the product 2.2 were calculated by integrating against pyrene as
an internal standard, which were then converted to molar concentrations. A duplicate
reaction was also run under otherwise identical conditions and an average value was taken
for each time point. The molar concentration of product 2.2 (only the data corresponding
to the linear portion of the graph, typically < 40% yield, was used) was plotted against the
reaction time and the slope of linear portion of the curve was used to determine the initial
rates of the reaction. The table showing molar concentration of product 2.2 in different
concentration of PhZnI, graph showing the rate at different concentration of PhZnI, graph
with kin value and the graph showing initial rates of the reactions are shown below.
Table 4. The molar concentration of product 2.2 in different concentration of PhZnI at
different time interval
Time (s) 0.15 M
[PhZnI]
0.30 M
[PhZnI]
0.45 M
[PhZnI]
0.60 M[PhZnI]
60.000 0.013050 0.011850 0.011175 0.014400
150.00 0.016725 0.015750 0.013500 0.015750
300.00 0.022200 0.021150 0.017700 0.020325
450.00 0.027150 0.027000 0.023925 0.024000
600.00 0.034275 0.031800 0.028350 0.031500
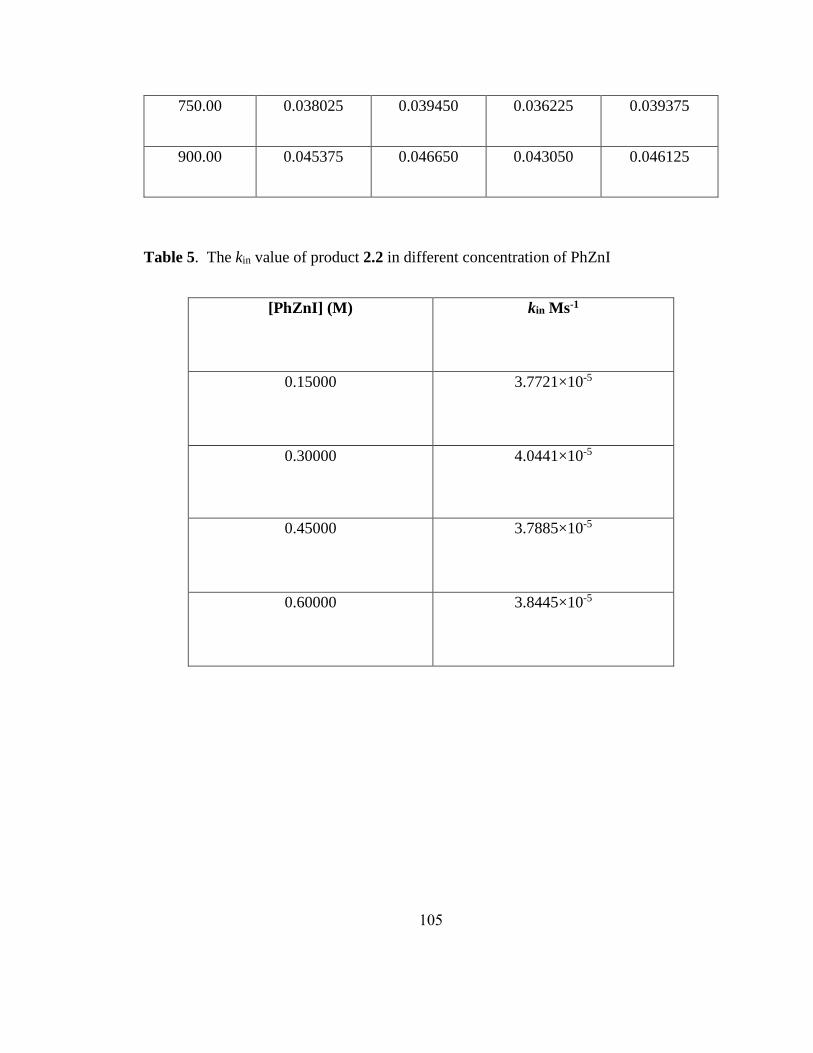
105
750.00 0.038025 0.039450 0.036225 0.039375
900.00 0.045375 0.046650 0.043050 0.046125
Table 5. The kin value of product 2.2 in different concentration of PhZnI
[PhZnI] (M) kin Ms-1
0.15000 3.7721×10-5
0.30000 4.0441×10-5
0.45000 3.7885×10-5
0.60000 3.8445×10-5

106
0.01
0.015
0.02
0.025
0.03
0.035
0.04
0.045
0.05
0 200 400 600 800 1000
[PhZnI] [0.150 M][PhZnI] [0.300 M][PhZnI] [0.450 M][PhZnI] [0.600 M]
yie
ld o
f 2
(M
)
time (s)
(a)
0
2 10-5
4 10-5
6 10-5
8 10-5
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
y = 3.8719e-5 - 2.56e-7x
R2= 0.0015704
kin
(M
s-1
)
[PhZnI] (M)
(b)
Figure 3. (a) Plot of the rise of product 2.2 from the reaction of 2.1 (0.15 M),
iodocyclohexane (0.30 M) with 0.15 M, 0.30 M, 0.45 M and 0.60 M of PhZnI in different
time interval at room temperature. The curve depicts the results of an unweighted least-
square fit to y = a*x + b (0.150 M: a = 3.772 × 10-5, b = 1.08 × 10-2, R2 = 0.996; 0.30 M: a
= 4.044 × 10-5, b = 9.11 × 10-3, R2 = 0.995; 0.45 M: a = 3.78 × 10-5, b = 7.47 × 10-3, R2 =
0.988); 0.60 M: a = 3.84 × 10-5, b = 9.72 × 10-3, R2 = 0.975. (b) Plot of kin versus [PhZnI]
from the reaction of 2.1 (0.15 M), iodocyclohexane (0.30 M) with 0.15 M, 0.30 M, 0.45 M
and 0.60 M of PhZnI. The curve depicts the results of an unweighted least-square fit to y
= a*x + b (a = −2.56 × 10-7, b = 3.87 × 10-5, R2 = 0.0015).
d. General procedure to determine the dependence of reaction rate on the
concentration of (2-vinylnaphthalene)
For the reaction of iodocyclohexane (0.30 M), PhZnI (0.15 M) with 2-
vinylnaphthalene (0.15 M): In a glove box, NiBr2 (0.015 mmol, 3.3 mg) was weigh out
into a 2.0 mL volumetric flask and 300 µL of iodocyclohexane (2.00 M), 300 µL of 2-

107
vinylnaphthalene (1.00 M) and 360 µL of PhZnI (0.830 M) stock solutions were added,
respectively. The mixture was diluted to 2.0 mL to obtain a homogeneous solution
containing final concentrations of 0.30 M iodocyclohexane, 0.15 M PhZnI and 0.15 M 2-
vinylnaphthalene. The solution was then transferred to a 1dram vial containing a stirring
bar and stirred vigorously at room temperature. 200 µL of the reaction mixture was taken
out at 2.5, 5, 10, 15, 30, 60, 120 min and quenched with H2O immediately. To the quenched
solution was added EtOAc (2 mL), pyrene (15 µL of 0.20 M) as an internal standard and
the organic layer was washed with H2O (0.5 mL × 3). The organic fraction was dried over
Na2SO4, the solvent was removed in a rotary evaporator and the sample was analyzed by
1H NMR.
For the reaction of iodocyclohexane (0.30 M), PhZnI (0.15 M) with 2-
vinylnaphthalene (0.30 M): The procedure for this reaction was same as above but instead
of 300 µL of 2-vinyl naphthalene (1.0 M stock solution), 600 µL of 2-vinylnaphthalene
(1.0 M stock solution) was added in the reaction.
For the reaction of iodocyclohexane (0.30 M), PhZnI (0.15 M) with 2-
vinylnaphthalene (0.60 M): The procedure for this reaction was same as above but instead
of 300 µL of 2-vinylnaphthalene (1.0 M stock solution), 600 µL of 2-vinylnaphthalene (2.0
M stock solution) was added in the reaction.
The percentage yields of the product 2.2 were calculated by integrating against pyrene as
an internal standard, which were then converted to molar concentrations. A duplicate
reaction was also run under otherwise identical conditions and an average value was taken
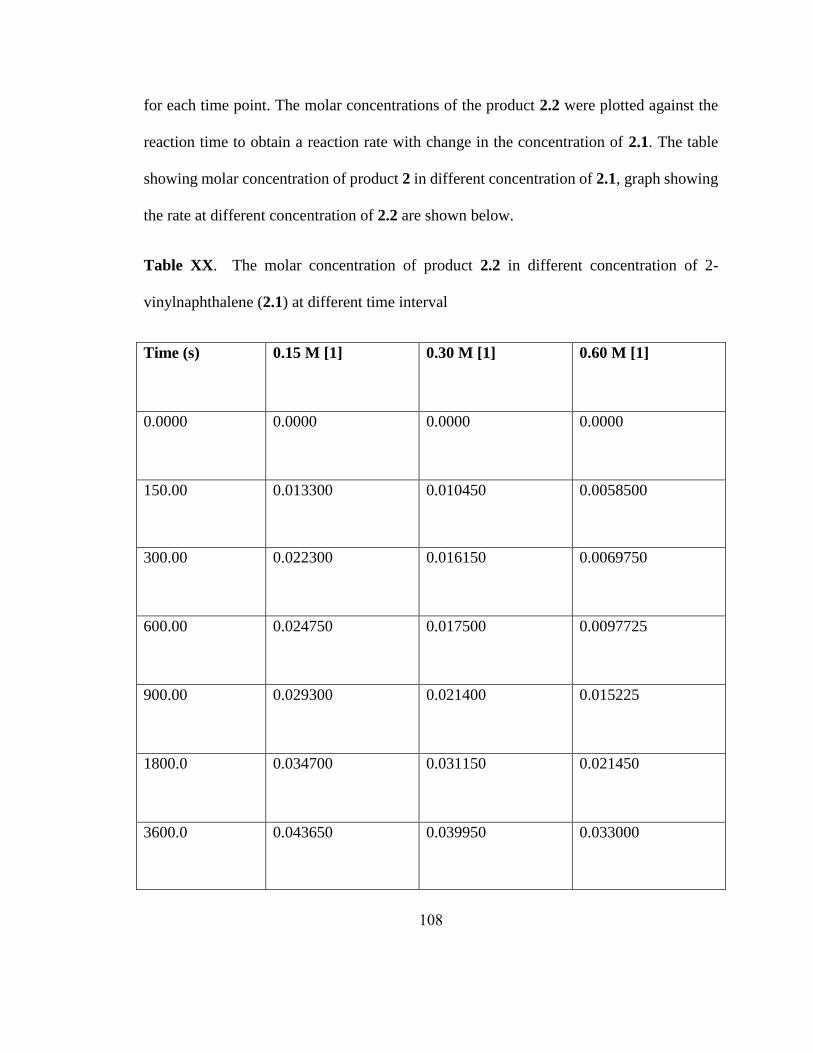
108
for each time point. The molar concentrations of the product 2.2 were plotted against the
reaction time to obtain a reaction rate with change in the concentration of 2.1. The table
showing molar concentration of product 2 in different concentration of 2.1, graph showing
the rate at different concentration of 2.2 are shown below.
Table XX. The molar concentration of product 2.2 in different concentration of 2-
vinylnaphthalene (2.1) at different time interval
Time (s) 0.15 M [1] 0.30 M [1] 0.60 M [1]
0.0000 0.0000 0.0000 0.0000
150.00 0.013300 0.010450 0.0058500
300.00 0.022300 0.016150 0.0069750
600.00 0.024750 0.017500 0.0097725
900.00 0.029300 0.021400 0.015225
1800.0 0.034700 0.031150 0.021450
3600.0 0.043650 0.039950 0.033000
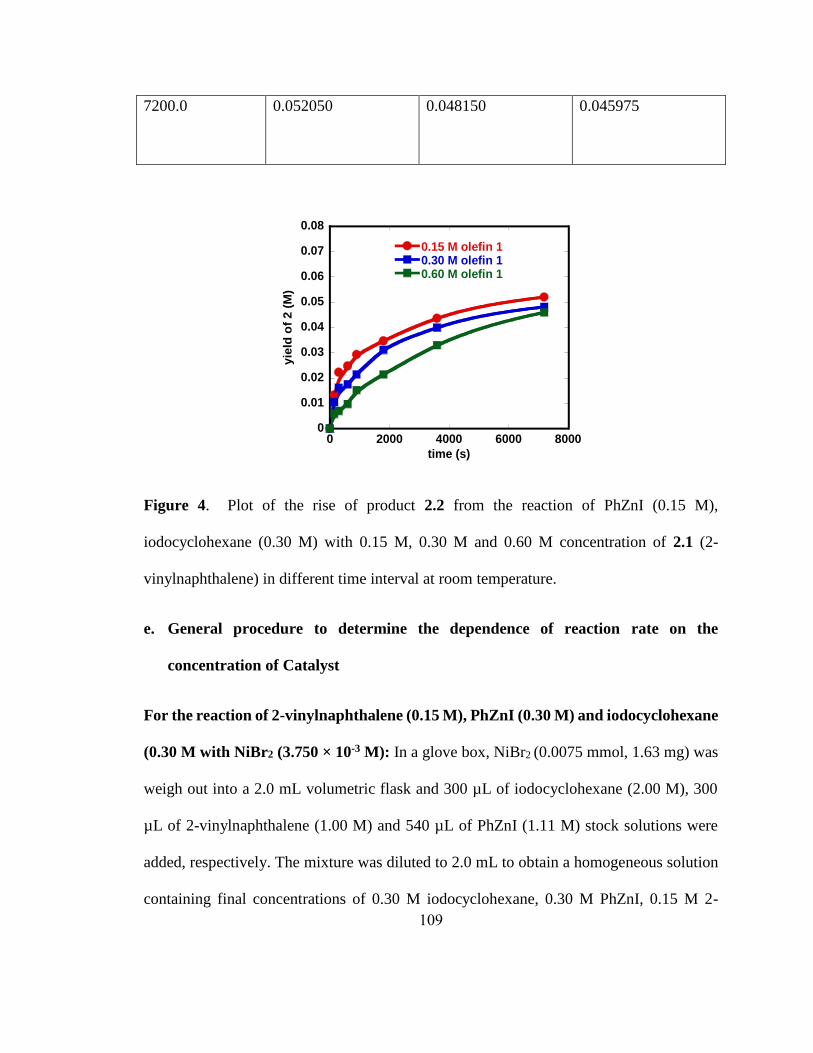
109
7200.0 0.052050 0.048150 0.045975
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0 2000 4000 6000 8000
0.15 M olefin 10.30 M olefin 10.60 M olefin 1
yie
ld o
f 2
(M
)
time (s)
Figure 4. Plot of the rise of product 2.2 from the reaction of PhZnI (0.15 M),
iodocyclohexane (0.30 M) with 0.15 M, 0.30 M and 0.60 M concentration of 2.1 (2-
vinylnaphthalene) in different time interval at room temperature.
e. General procedure to determine the dependence of reaction rate on the
concentration of Catalyst
For the reaction of 2-vinylnaphthalene (0.15 M), PhZnI (0.30 M) and iodocyclohexane
(0.30 M with NiBr2 (3.750 × 10-3 M): In a glove box, NiBr2 (0.0075 mmol, 1.63 mg) was
weigh out into a 2.0 mL volumetric flask and 300 µL of iodocyclohexane (2.00 M), 300
µL of 2-vinylnaphthalene (1.00 M) and 540 µL of PhZnI (1.11 M) stock solutions were
added, respectively. The mixture was diluted to 2.0 mL to obtain a homogeneous solution
containing final concentrations of 0.30 M iodocyclohexane, 0.30 M PhZnI, 0.15 M 2-

110
vinylnaphthalene and 3.750 × 10-3 M of NiBr2. The solution was then transferred to a 1
dram vial containing a stirring bar and stirred vigorously at room temperature. 200 µL of
the reaction mixture was taken out at 1, 2.5, 5, 7.5, 10, 12.5, 15 min and quenched with
H2O immediately. To the quenched solution was added EtOAc (2 mL), pyrene (15 µL of
0.20 M) as an internal standard and the organic layer was washed with H2O (0.5 mL × 3).
The organic fraction was dried over Na2SO4, the solvent was removed in a rotary
evaporator and the sample was analyzed by 1H NMR.
For the reaction of 2-vinylnaphthalene (0.15 M), iodocyclohexane (0.30 M) and PhZnI
(0.30 M) with NiBr2 (7.50 × 10-3 M): The procedure for this reaction was same as above
but instead of NiBr2 (0.0075 mmol, 1.63 mg), NiBr2 (0.015 mmol, 3.30 mg) was added in
the reaction.
For the reaction of 2-vinylnaphthalene (0.15 M), iodocyclohexane (0.30 M) and PhZnI
(0.30 M) with NiBr2 (11.250 × 10-3 M): The procedure for this reaction was same as above
but instead of NiBr2 (0.0075 mmol, 1.63 mg), NiBr2 (0.0225 mmol, 4.90 mg) was added in
the reaction.
For the reaction of 2-vinylnaphthalene (0.15 M), iodocyclohexane (0.30 M) and PhZnI
(0.30 M) with NiBr2 (15.0 × 10-3 M): The procedure for this reaction was same as above
but instead of NiBr2 (0.0075 mmol, 1.63 mg), NiBr2 (0.030 mmol, 6.540 mg) was added in
the reaction.
The percentage yields of the product 2.2 were calculated by integrating against pyrene as
an internal standard, which were then converted to molar concentrations. A duplicate
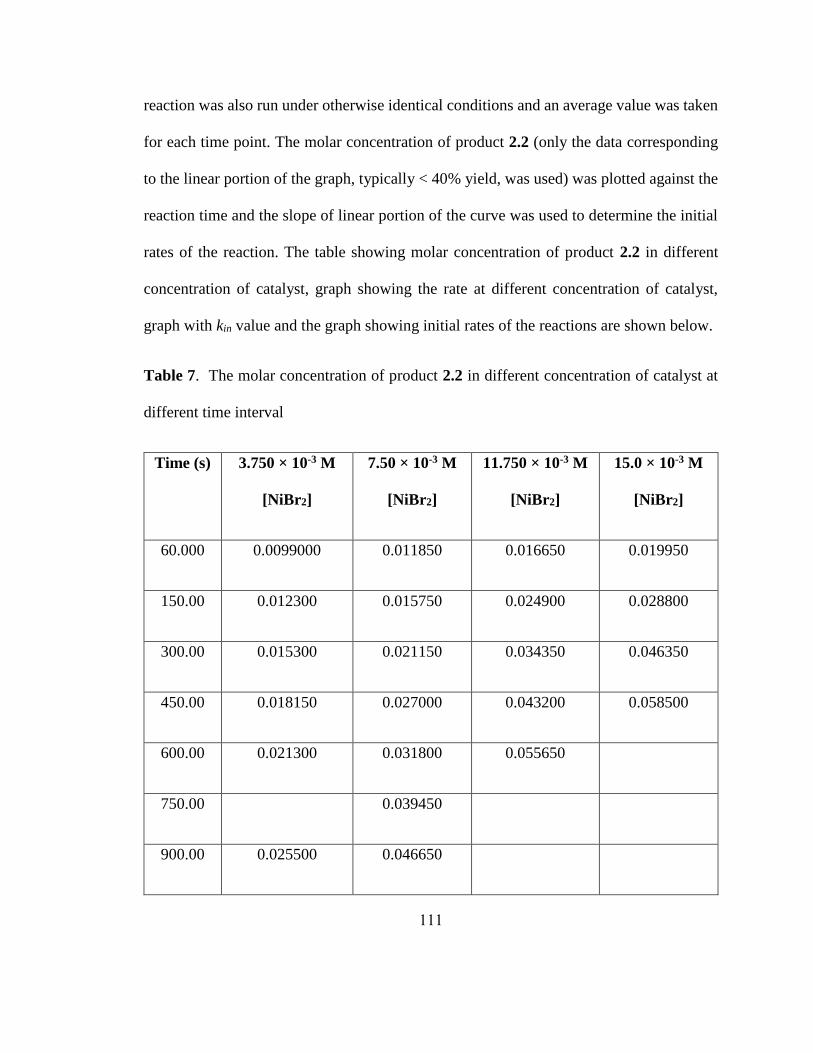
111
reaction was also run under otherwise identical conditions and an average value was taken
for each time point. The molar concentration of product 2.2 (only the data corresponding
to the linear portion of the graph, typically < 40% yield, was used) was plotted against the
reaction time and the slope of linear portion of the curve was used to determine the initial
rates of the reaction. The table showing molar concentration of product 2.2 in different
concentration of catalyst, graph showing the rate at different concentration of catalyst,
graph with kin value and the graph showing initial rates of the reactions are shown below.
Table 7. The molar concentration of product 2.2 in different concentration of catalyst at
different time interval
Time (s) 3.750 × 10-3 M
[NiBr2]
7.50 × 10-3 M
[NiBr2]
11.750 × 10-3 M
[NiBr2]
15.0 × 10-3 M
[NiBr2]
60.000 0.0099000 0.011850 0.016650 0.019950
150.00 0.012300 0.015750 0.024900 0.028800
300.00 0.015300 0.021150 0.034350 0.046350
450.00 0.018150 0.027000 0.043200 0.058500
600.00 0.021300 0.031800 0.055650
750.00 0.039450
900.00 0.025500 0.046650
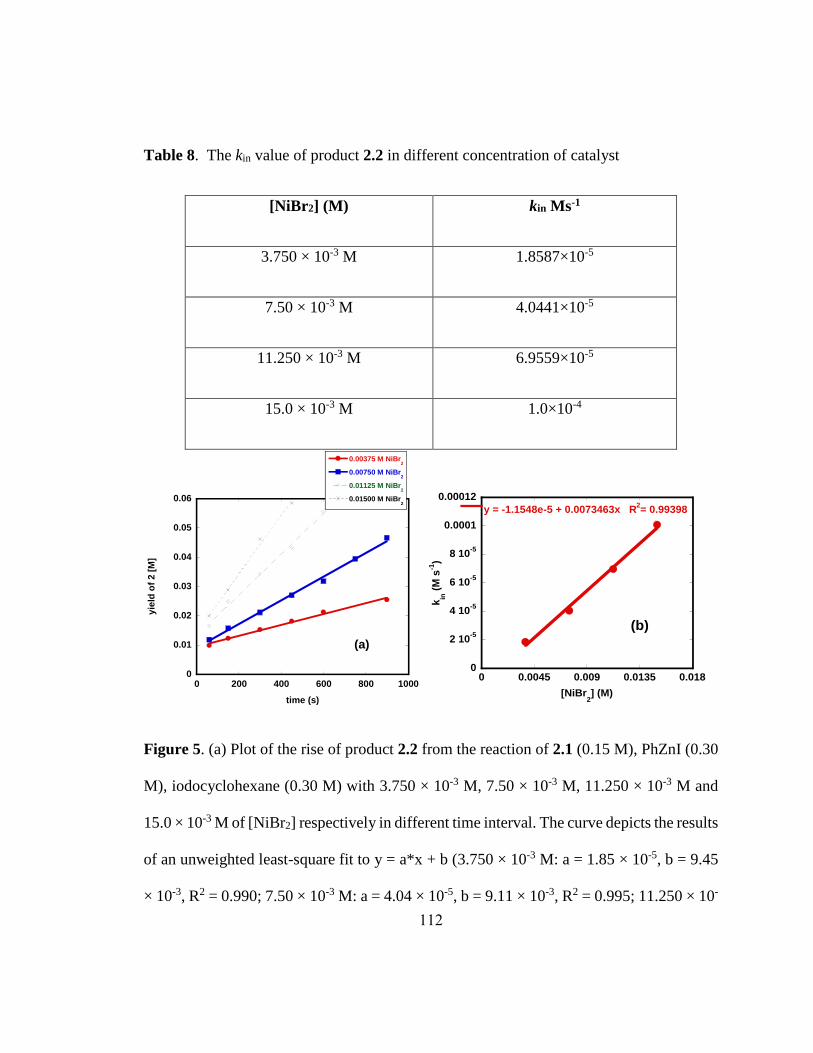
112
Table 8. The kin value of product 2.2 in different concentration of catalyst
[NiBr2] (M) kin Ms-1
3.750 × 10-3 M 1.8587×10-5
7.50 × 10-3 M 4.0441×10-5
11.250 × 10-3 M 6.9559×10-5
15.0 × 10-3 M 1.0×10-4
0
0.01
0.02
0.03
0.04
0.05
0.06
0 200 400 600 800 1000
0.00375 M NiBr2
0.00750 M NiBr2
0.01125 M NiBr2
0.01500 M NiBr2
yie
ld o
f 2
[M
]
time (s)
(a)
0
2 10-5
4 10-5
6 10-5
8 10-5
0.0001
0.00012
0 0.0045 0.009 0.0135 0.018
(b)
y = -1.1548e-5 + 0.0073463x R2= 0.99398
kin
(M
s-1
)
[NiBr2] (M)
Figure 5. (a) Plot of the rise of product 2.2 from the reaction of 2.1 (0.15 M), PhZnI (0.30
M), iodocyclohexane (0.30 M) with 3.750 × 10-3 M, 7.50 × 10-3 M, 11.250 × 10-3 M and
15.0 × 10-3 M of [NiBr2] respectively in different time interval. The curve depicts the results
of an unweighted least-square fit to y = a*x + b (3.750 × 10-3 M: a = 1.85 × 10-5, b = 9.45
× 10-3, R2 = 0.990; 7.50 × 10-3 M: a = 4.04 × 10-5, b = 9.11 × 10-3, R2 = 0.995; 11.250 × 10-

113
3 M: a = 6.95 × 10-5, b = 1.32 × 10-2, R2 = 0.995); 15.0 × 10-3 M: a = 1.00 × 10-4, b = 1.42
× 10-2, R2 = 0.994. (b) Plot of kin versus [NiBr2] from the reaction of 2.1 (0.15 M), PhZnI
(0.30 M), iodocyclohexane (0.30 M) with 3.750 × 10-3 M, 7.50 × 10-3 M, 11.250 × 10-3 M
and 15.0 × 10-3 M of [NiBr2]. The curve depicts the results of an unweighted least-square
fit to y = a*x + b (a = 7.34 × 10-4, b = −1.15 × 10-5, R2 = 0.993).
4.1.4 Characterization data for new compounds
2-(2-Cyclohexyl-1-phenylethyl)naphthalene (2.2): The title compound 2.2 was obtained as
a white solid (127.2 mg, 81% yield) after purification by silica gel column chromatography
in hexane. 1H NMR (300 MHz, CDCl3): 0.98-1.28 (m, 6H), 1.60-1.83 (m, 5H), 1.96-
2.12 (m, 2H), 4.25 (t, J = 7.5 Hz, 1H), 7.16-7.20 (m, 1H), 7.23-7.30 (m, 4H), 7.35-7.38
(dd, J = 9.0 Hz, J = 1.8 Hz, 1H), 7.41-7.48 (m, 2H), 7.72 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H),
7.78-7.82 (m, 2H); 13C NMR (75 MHz, CDCl3): 26.3, 26.7, 33.5, 33.7, 35.0, 43.5, 48.1,
125.4, 126.0, 126.1, 127.0, 127.6, 127.8, 128.1, 128.5, 132.2, 133.7, 143.0, 145.4; IR
(neat) cm-1 : 2913, 1597, 1445, 819, 741, 698; HRMS (APPI): Calcd for C24H26 (M)+
314.2035, found 314.2027.

114
2-(1-Phenyldecyl)naphthalene (2.3): The title compound 2.3 was obtained as a colorless
oil (127.3 mg, 74% yield) after purification by silica gel column chromatography in
hexane. 1H NMR (300 MHz, CDCl3): 0.90 (t, J = 6.0 Hz, 3H), 1.27-1.36 (m, 14H),
2.10-2.23 (m, 2H), 4.08 (t, J = 7.5 Hz, 1H), 7.17-7.22 (m, 1H), 7.27-7.32 (m, 4H), 7.35-
7.39 (dd, J = 9.0 Hz, J = 3.0 Hz, 1H), 7.41-7.50 (m, 2H), 7.74 (s, 1H), 7.76 (d, J = 9.0 Hz,
1H), 7.79-7.84 (m, 2H); 13C NMR (75 MHz, CDCl3): 14.2, 22.8, 28.2, 29.4, 29.6, 29.7,
29.8, 32.0, 35.6, 51.5, 125.4, 126.0, 126.1, 126.9, 127.6, 127.8, 128.1, 128.5, 132.2, 133.6,
142.9, 145.3; IR (neat) cm-1 : 2922, 1493, 1452, 812, 742, 698; HRMS (APPI): Calcd for
C26H32 (M)+ 344.2504, found 344.2502.
2-(1-(2-Methoxyphenyl)decyl)naphthalene (2.4): The title compound 2.4 was obtained as
a colorless oil (129.0 mg, 69% yield) after purification by silica gel column
chromatography in 2% Et2O/Hex. 1H NMR (300 MHz, CDCl3): 0.86 (t, J = 9.0 Hz, 3H),
1.23-1.31 (m, 14H), 2.03-2.13 (m, 2H), 3.78 (s, 3H), 4.54 (t, J = 6.0 Hz, 1H), 6.83 (d, J =
3.0 Hz, 1H), 6.92 (t, J = 4.5 Hz, 1H), 7.16 (t, J = 4.5 Hz, 1H), 7.27 (d, J = 4.5 Hz, 1H),

115
7.38-7.44 (m, 3H), 7.71 (d, J = 6.0 Hz, 2H), 7.77 (t, J = 4.5 Hz, 2H); 13C NMR (75 MHz,
CDCl3): 14.2, 22.8, 28.1, 29.4, 29.6, 29.7, 29.8, 32.0, 35.0, 43.3, 55.6, 110.8, 120.6,
125.1, 125.7, 126.2, 127.1, 127.5, 127.6, 127.8, 132.1, 133.6, 133.9, 143.0, 157.2; IR
(neat) cm-1 : 2920, 1465, 1448, 1127, 950, 815, 698; HRMS (APPI): Calcd for C27H34O
(M)+ 374.2610, found 374.2607.
tert-Butyldimethyl(4-(naphthalen-2-yl)-4-phenylbutoxy)silane (2.5): The title compound
2.5 was obtained as a colorless oil (122.8 mg, 63% yield) after purification by silica gel
column chromatography in 10% DCM/Hex. 1H NMR (300 MHz, CDCl3): 0.04 (s, 6H),
0.90 (s, 9H), 1.50-1.60 (m, 2H), 2.16-2.29 (m, 2H), 3.66 (t, J = 6.0 Hz, 2H), 4.09 (t, J =
7.5 Hz, 1H), 7.15-7.22 (m, 1H), 7.26-7.30 (m, 4H), 7.35 (dd, J = 9.0 Hz, J = 3.0 Hz, 1H),
7.39-7.49 (m, 2H), 7.73 (s, 1H), 7.74-7.82 (m, 3H); 13C NMR (75 MHz, CDCl3): -5.1,
18.5, 26.1, 31.4, 31.8, 51.2, 63.1, 125.4, 126.0, 126.2, 126.9, 127.6, 127.8, 128.1, 128.5,
132.2, 133.6, 142.7, 145.1; IR (neat) cm-1 : 2915, 1450, 1245, 1098, 833, 773; HRMS
(ESI): Calcd for C26H35OSiNa (M+Na)+ 413.2277, found 413.2266.

116
2-(6-(Naphthalen-2-yl)-6-phenylhexyl)isoindoline-1,3-dione (2.6): The title compound 2.6
was obtained as a colorless oil (125.0 mg, 58% yield) after purification by silica gel column
chromatography in 10% EtOAc/Hex. 1H NMR (300 MHz, CDCl3): 1.27-1.46 (m, 4H),
1.65 (m, 2H), 2.10-2.21 (m, 2H), 3.65 (t, J = 6.0 Hz, 2H), 4.04 (t, J = 7.5 Hz, 1H), 7.13-
7.20 (m, 1H), 7.23-7.28 (m, 4H), 7.32 (dd, J = 9.0 Hz, J = 1.8 Hz, 1H), 7.37-7.47 (m, 2H),
7.65-7.74 (m, 4H), 7.75-7.84 (m, 4H); 13C NMR (75 MHz, CDCl3): 27.0, 27.7, 28.5,
35.4, 38.1, 51.4, 123.2, 125.4, 125.9, 126.0, 126.2, 126.8, 127.6, 127.8, 128.0, 128.1, 128.5,
132.2, 133.6, 133.9, 142.6, 145.1, 168.5; IR (neat) cm-1 : 2931, 1704, 1393, 1087, 814,
716; HRMS (ESI): Calcd for C30H27NNaO2 (M+Na)+ 456.1939, found 456.1940.
2-(5-Methyl-1-(4-(trifluoromethyl)phenyl)hexyl)naphthalene (2.7): The title compound 2.7
was obtained as a white solid (114.7 mg, 62% yield) after purification by silica gel column
chromatography in 5% Et2O/Hex. 1H NMR (500 MHz, CDCl3): 0.85 (s, 3H), 0.86 (s,
3H), 1.25-1.35 (m, 4H), 1.49-1.57 (m, 1H), 2.09-2.33 (m, 2H), 4.14 (t, J = 7.5 Hz, 1H),
7.33 (dd, J = 5.0 Hz, J = 1.0 Hz, 1H), 7.41 (d, J = 10.0 Hz, 2H), 7.44-7.50 (m, 2H), 7.55
(d, J = 10.0 Hz, 2H), 7.72 (s, 1H), 7.78 (d, J = 10.0 Hz, 1H), 7.82 (t, J = 10.0 Hz, 2H); 13C
NMR (126 MHz, CDCl3): 22.7, 22.7, 25.8, 27.9, 35.6, 39.0, 51.3, 124.4 (q, J = 222.5
Hz), 125.5 (q, J = 3.75 Hz), 125.7, 126.1, 126.2, 126.6, 127.7, 127.8, 128.4, 128.5 (q, J =

117
28.7 Hz), 132.3, 133.6, 141.8, 149.4; 19F NMR (470 MHz, CDCl3) -62.5; IR (neat) cm-
1 : 2930, 1322, 1160, 1113, 1017, 842; HRMS (APPI): Calcd for C24H25F3 (M)+ 370.1908,
found 370.1899.
2-(2-Cyclohexyl-1-(4-(trifluoromethyl)phenyl)ethyl)naphthalene (2.8): The title
compound 2.8 was obtained as a white solid (126.0 mg, 66% yield) after purification by
silica gel column chromatography in hexane. 1H NMR (300 MHz, CDCl3): 0.94-1.27
(m, 6H), 1.61-1.85 (m, 5H), 1.95-2.14 (m, 2H), 4.30 (t, J = 7.5 Hz, 1H), 7.32 (dd, J = 9.0
Hz, J = 1.8 Hz, 1H), 7.39 (d, J = 9.0 Hz, 2H), 7.44-7.51 (m, 2H), 7.53 (d, J = 9.0 Hz, 2H),
7.70 (s, 1H), 7.76 (d, J = 9.0 Hz, 1H), 7.79-7.83 (m, 2H); 13C NMR (75 MHz, CDCl3):
26.2, 26.7, 33.5, 33.6, 35.0, 43.3, 48.0, 124.3 (q, J = 262.5 Hz), 125.5 (q, J = 3.75 Hz),
125.7, 126.1, 126.2, 126.7, 127.7, 127.8, 128.4, 128.5 (q, J = 31.5 Hz), 132.3, 133.6, 141.8,
149.6; 19F NMR (282 MHz, CDCl3) -60.8; IR (neat) cm-1 : 2923, 1653, 1418, 1321,
1115, 823; HRMS (APPI): Calcd for C25H25F3 (M)+ 382.1908, found 382.1904.

118
2-(1-(p-Tolyl)decyl)naphthalene (2.9): The title compound 2.9 was obtained as a colorless
oil (134.5 mg, 82% yield) after purification by silica gel column chromatography in
hexane. 1H NMR (300 MHz, CDCl3): 0.91-1.16 (m, 6H), 1.60-1.83 (m, 5H), 1.94-2.10
(m, 2H), 2.31 (s, 3H), 4.22 (t, J = 7.5 Hz, 1H), 7.10 (d, J = 6.0 Hz, 2H), 7.19 (d, J = 6.0
Hz, 2H), 7.36 (dd, J = 9.0 Hz, J = 3.0 Hz, 1H), 7.39-7.49 (m, 2H), 7.71 (s, 1H), 7.74 (d, J
= 9.0 Hz, 1H), 7.77-7.83 (m, 2H); 13C NMR (75 MHz, CDCl3): 21.1, 26.2, 26.8, 33.5,
33.6, 35.0, 43.6, 47.7, 125.3, 125.8, 125.9, 127.0, 127.6, 127.8, 127.9, 128.1, 129.2, 132.2,
133.7, 135.6, 142.4, 143.2; IR (neat) cm-1 : 2917, 1633, 1507, 1446, 811, 742; HRMS
(APPI): Calcd for C25H28 (M)+ 328.2191, found 328.2184.
2-(2-Cyclohexyl-1-(4-methoxyphenyl)ethyl)naphthalene(2.10): The title compound 2.10
was obtained as a colorless oil (134.5 mg, 82% yield) after purification by silica gel column
chromatography in hexane. 1H NMR (300 MHz, CDCl3): 0.95-1.14 (m, 6H), 1.59-1.82
(m, 5H), 1.90-2.06 (m, 2H), 3.77 (s, 3H), 4.18 (t, J = 7.5 Hz, 1H), 6.81 (d, J = 15.0 Hz,
2H), 7.19 (d, J = 15.0 Hz, 2H), 7.33 (d, J = 6.0 Hz, 1H), 7.38-7.47 (m, 2H), 7.67 (s, 1H),
7.72-7.80 (m, 3H); 13C NMR (75 MHz, CDCl3): 26.3, 26.8, 33.6, 35.0, 43.7, 47.2, 55.3,
113.9, 125.3, 125.8, 125.9, 126.9, 127.6, 127.8, 128.1, 129.0, 132.2, 133.7, 137.6, 143.4,

119
157.9; IR (neat) cm-1 : 2918, 1608, 1508, 1244, 1035, 889; HRMS (ESI): Calcd for
C25H32NO (M+NH4)+ 362.2484, found 362.2489.
2-(3-Ethyl-1-phenylpentyl)naphthalene (2.11): The title compound 2.11 was obtained as a
colorless oil (111.8 mg, 74% yield) after purification by silica gel column chromatography
in hexane. 1H NMR (300 MHz, CDCl3): 0.88 (t, J = 7.5 Hz, 6H), 1.19-1.27 (m, 1H),
1.36-1.48 (m, 4H), 2.02-2.19 (m, 2H), 4.24 (t, J = 7.5 Hz, 1H), 7.18-7.12 (m, 1H), 7.28-
7.35 (m, 4H), 7.42-7.50 (m, 3H), 7.76 (s, 1H), 7.78 (d, J = 9.0 Hz, 1H), 7.79-7.84 (m, 2H);
13C NMR (75 MHz, CDCl3): 10.5, 10.6, 25.2, 25.3, 37.4, 39.1, 48.7, 125.4, 126.0, 126.1,
127.0, 127.6, 127.8, 128.1, 128.5, 132.2, 133.7, 142.9, 145.4; IR (neat) cm-1 : 2919, 1493,
1451, 855, 717; HRMS (CI): Calcd for C23H26 (M)+ 302.2035, found 302.2028.
(5-Chloropentane-1,1-diyl)dibenzene (2.12): The title compound 2.12 was obtained as a
colorless oil (79.9 mg, 62% yield) after purification by silica gel column chromatography
in 2% Et2O/Hex . 1H NMR (500 MHz, CDCl3): 1.40-1.46 (m, 2H), 1.80-1.85 (m, 2H),
2.07-2.11 (m, 2H), 3.51 (t, J = 7.5 Hz, 2H), 3.92 (t, J = 7.5 Hz, 1H), 7.19 (t, J = 7.5 Hz,

120
2H), 7.25-7.31 (m, 8H); 13C NMR (126 MHz, CDCl3): 25.5, 32.7, 35.1, 44.9, 51.3,
126.3, 127.9, 128.5, 128.6, 144.9; IR (neat): 2935, 1599, 1449, 1030, 744, 696; HRMS
(CI) cm-1 : Calcd for C17H19Cl (M)+ 258.1175, found 258.1172.
(4-Ethoxybutane-1,1-diyl)dibenzene (2.13): The title compound 2.13 was obtained as a
colorless oil (82.5 mg, 65% yield) after purification by silica gel column chromatography
in 2% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 1.18 (t, J = 7.5 Hz, 3H), 1.51-1.60 (m,
2H), 2.08-2.16 (m, 2H), 3.40-3.47 (m, 4H), 3.91 (t, J = 9.0 Hz, 1H), 7.13-7.30 (m, 10H);
13C NMR (75 MHz, CDCl3): 15.3, 28.4, 32.4, 51.3, 66.2, 70.6, 126.2, 128.0, 128.5,
145.1; IR (neat) cm-1 : 2930, 1493, 1394, 1108, 744; HRMS (ESI): Calcd for C18H22ONa
(M+Na)+ 277.1568, found 277.1564.
(1-(4-(tert-Butyl)phenyl)butane-1,4-diyl)dibenzene (2.14): The title compound 2.14 was
obtained as a colorless oil (116.2 mg, 68% yield) after purification by silica gel column
chromatography in 2% Et2O:Hex . 1H NMR (500 MHz, CDCl3): 1.31 (s, 9H), 1.60-1.69

121
(m, 2H), 2.08-2.13 (m, 2H), 2.66 (t, J = 7.5 Hz, 2H), 3.91 (t, J = 7.5 Hz, 1H), 7.14-7.20 (m,
6H), 7.25-7.31 (m, 8H); 13C NMR (126 MHz, CDCl3): 29.9, 31.5, 34.4, 35.4, 36.0, 51.0,
125.3, 125.7, 126.1, 127.4, 128.0, 128.3, 128.5, 128.6, 142.1, 142.5, 145.3, 148.8; IR
(neat) cm-1 : 2960, 1508, 1473, 1029, 745, 696; HRMS (CI): Calcd for C26H30 (M)+
342.2348, found 342.2347.
(1-(4-Chlorophenyl)butane-1,4-diyl)dibenzene (2.15): The title compound 2.15 was
obtained as a colorless oil (128.0 mg, 80% yield) after purification by silica gel column
chromatography in 5% Et2O:Hex. 1H NMR (500 MHz, CDCl3): 1.58-1.64 (m, 2H),
2.04-2.12 (m, 2H), 2.66 (t, J = 7.5 Hz, 2H), 3.91 (t, J = 7.5 Hz, 1H), 7.14-7.16 (m, 4H),
7.19-7.31 (m, 10H); 13C NMR (126 MHz, CDCl3): 29.8, 35.2, 35.9, 50.7, 125.9, 126.4,
127.8, 128.4, 128.5, 128.6, 129.3, 131.9, 142.2, 143.6, 144.6; IR (neat) cm-1 : 2937, 1652,
1489, 746, 696; HRMS (CI): Calcd for C22H21Cl (M)+ 320.1332, found 320.1329.

122
tert-Butyl(3-(4-chlorophenyl)-3-phenylpropoxy)dimethylsilane (2.16): The title compound
2.16 was obtained as a colorless oil (127.1 mg, 68% yield) after purification by silica gel
column chromatography in 10% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 0.05 (s, 6H),
0.91 (s, 9H), 1.45-1.57 (m, 2H), 2.06-2.13 (m, 2H), 3.63 (t, J = 6.0 Hz, 2H), 3.90 (t, J =
7.5 Hz, 1H), 7.17-7.32 (m, 9H); 13C NMR (75 MHz, CDCl3): -5.1, 18.4, 26.1, 31.2,
32.0, 50.5, 63.0, 126.4, 127.9, 128.6, 129.3, 131.8, 143.8, 144.7; IR (neat) cm-1 : 2951,
1489, 1252, 1091, 1013, 832; HRMS (CI): Calcd for C22H32ClOSi (M+H)+ 375.1911,
found 375.1910.
1-Chloro-4-(1-phenylnon-8-en-1-yl)benzene (2.17): The title compound 2.17 was obtained
as a colorless oil (102.9 mg, 66% yield) after purification by silica gel column
chromatography in 10% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 1.19-1.39 (m, 8H),
1.96-2.04 (m, 4H), 3.85 (t, J = 7.5 Hz, 1H), 4.90-5.01 (m, 2H), 5.72-5.86 (m, 1H), 7.14-
7.28 (m, 9H); 13C NMR (75 MHz, CDCl3): 28.0, 29.0, 29.1, 29.5, 33.9, 35.7, 114.3,
126.3, 127.8, 128.6, 129.3, 131.8, 139.2, 143.9, 144.9; IR (neat) cm-1 : 2926, 1488, 1451,
1091, 1013, 908, 815; HRMS (CI): Calcd for C21H25Cl (M)+ 312.1645, found 312.1645.

123
4-(1-Phenyldecyl)-1,1'-biphenyl (2.18): The title compound 2.18 was obtained as a white
solid (125.8 mg, 68% yield) after purification by silica gel column chromatography in
hexane. 1H NMR (300 MHz, CDCl3): 0.89 (t, J = 6.0 Hz, 3H), 1.26-1.33 (m, 14H), 2.05-
2.12 (m, 2H), 3.95 (t, J = 7.5 Hz, 1H), 7.28-7.35 (m, 7H), 7.40-7.46 (m, 3H), 7.50-7.59 (m,
4H); 13C NMR (75 MHz, CDCl3): 14.2, 22.8, 28.2, 29.4, 29.6, 29.7, 29.8, 32.0, 35.9,
51.2, 126.2, 127.1, 127.2, 128.0, 128.3, 128.5, 128.8, 139.0, 141.1, 144.6, 145.4; IR (neat)
cm-1 : 2922, 1486, 1451, 1007, 830; HRMS (APPI): Calcd for C28H34 (M)+ 370.2661,
found 370.2661.
1-Fluoro-4-(1-(4-methoxyphenyl)-4-phenylbutyl)benzene (2.19): The title compound 2.19
was obtained as a colorless oil (121.9 mg, 73% yield) after purification by silica gel column
chromatography in 5% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 1.61-1.68 (m, 2H), 2.09
(q, J = 7.9 Hz, 2H), 2.67 (t, J = 7.5 Hz, 2H), 3.79 (s, 3H), 3.90 (t, J = 7.5 Hz, 1H), 6.84 (d,
J = 9.0 Hz, 2H), 7.15-7.30 (m, 11H); 13C NMR (75 MHz, CDCl3): 29.9, 35.5, 35.9, 50.5,

124
55.3, 113.8, 125.8 (d, J = 22.5 Hz), 127.8, 128.4 (d, J = 10.5 Hz), 128.8, 137.3, 142.4,
145.6, 157.9; 19F NMR (282 MHz, CDCl3) -116.1; IR (neat) cm-1 : 2922, 1607, 1508,
1451, 1244, 1032. HRMS (CI): Calcd for C23H23FO (M)+ 334.1733, found 334.1733.
4-(1-(o-Tolyl)decyl)phenyl acetate (2.20): The title compound 2.20 was obtained as a
colorless oil (109.8 mg, 60% yield) after purification by silica gel column chromatography
in 10% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 0.88 (t, J = 6.0 Hz, 3H), 1.25-1.31 (m,
14H), 1.95-2.06 (m, 2H), 2.26 (s, 3H), 2.27 (s, 3H), 4.09 (t, J = 7.5 Hz, 1H), 6.97 (d, J =
9.0 Hz, 2H), 7.11 (d, J = 3.0 Hz, 2H), 7.16-7.23 (m, 3H), 7.30-7.36 (m, 1H); 13C NMR (75
MHz, CDCl3): 14.2, 20.0, 21.2, 22.8, 28.1, 29.4, 29.6, 29.7, 29.8, 32.0, 36.4, 46.4, 121.2,
126.1, 126.7, 129.1, 130.6, 136.4, 142.6, 142.8, 148.7, 169.6; IR (neat) cm-1 : 2923, 1762,
1505, 1458, 1194, 910; HRMS (CI): Calcd for C25H34O2 (M)+ 366.2559, found 366.2558.
1-Chloro-4-(2-cyclohexyl-1-phenylethyl)benzene (2.21): The title compound 2.21 was
obtained as a colorless oil (121.9 mg, 68% yield) after purification by silica gel column

125
chromatography in 5% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 0.89-1.03 (m, 2H),
1.12-1.15 (m, 4H), 1.63-1.80 (m, 5H), 1.92 (t, J = 7.5 Hz, 2H), 4.07 (t, J = 7.5 Hz, 1H),
7.17-7.33 (m, 9H); 13C NMR (75 MHz, CDCl3): 26.2, 26.7, 33.4, 33.5, 34.9, 43.6, 47.4,
126.3, 127.9, 128.6, 129.3, 131.7, 144.0, 145.0; IR (neat) cm-1 : 2919, 1488, 10292, 1013,
815, 716; HRMS (CI): Calcd for C20H23Cl (M)+ 298.1488, found 298.1487.
Methyl 4-(1-(4-chlorophenyl)-2-cyclohexylethyl)benzoate (2.22): The title compound 2.22
was obtained as a colorless oil (103.2 mg, 58% yield) after purification by silica gel column
chromatography in 5% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 0.89-0.99 (m, 2H),
1.08-1.14 (m, 4H), 1.60-1.76 (m, 5H), 1.90 (t, J = 7.5 Hz, 2H), 3.89 (s, 3H), 4.09 (t, J = 7.5
Hz, 1H), 7.14 (d, J = 9.0 Hz, 2H), 7.23-7.29 (m, 4H), 7.95 (d, J = 6.0 Hz, 2H); 13C NMR
(75 MHz, CDCl3): 26.2, 26.6. 33.4, 33.5, 35.0, 43.3, 47.5, 52.1, 127.9, 128.3, 128.7, 129.3,
130.0, 132.1, 143.0, 150.3, 167.0; IR (neat) cm-1 : 2920, 1717, 1609, 1489, 1274, 1103,
1013, 773; HRMS (APPI): Calcd for C22H26ClO2 (M+H)+ 357.1621, found 357.1618.

126
1-(3-Ethyl-1-(4-fluorophenyl)pentyl)naphthalene (2.23): The title compound 2.23 was
obtained as a colorless oil (107.2 mg, 67% yield) after purification by silica gel column
chromatography in 2% EtOAc:Hex. 1H NMR (500 MHz, CDCl3): 0.88 (t, J = 7.5 Hz,
3H), 0.87 (t, J = 7.5 Hz, 3H), 1.24-1.34 (m, 3H), 1.36-1.52 (m, 2H), 1.97-2.03 (m, 1H),
2.07-2.13 (m, 1H), 4.85 (t, J = 7.5 Hz, 1H), 6.93 (t, J = 7.5 Hz, 2H), 7.24 (dd, J = 7.5 Hz,
J = 2.5 Hz, 2H), 7.42-7.48 (m, 4H), 7.73 (t, J = 7.5 Hz, 1H), 7.84 (d, J = 2.5 Hz, 1H), 8.11
(d, J = 2.5 Hz, 1H); 13C NMR (126 MHz, CDCl3): 10.5, 10.7, 25.3, 25.4, 37.6, 40.3,
42.7, 115.1, 115.3, 123.5, 124.5, 125.4 (d, J = 6.2 Hz), 126.1, 127.1, 129.0, 129.5 (d, J =
7.5 Hz), 132.0, 134.2, 140.5, 141.1, 161.3 (d, J = 242.5 Hz); 19F NMR (470 MHz, CDCl3)
-116.0; IR (neat) cm-1 : 2921, 1505, 1458, 1221, 1157, 797; HRMS (APPI): Calcd for
C23H25F (M)+ 320.1940, found 320.1940.
Benzyl 4-(2-(naphthalen-1-yl)-2-phenylethyl)piperidine-1-carboxylate (2.24): The title
compound 2.24 was obtained as a viscous colorless oil (101.0 mg, 45% yield) after
purification by silica gel column chromatography in 15% Et2O:Hex. 1H NMR (300 MHz,
CDCl3): 1.21-1.28 (m, 2H), 1.45-1.57 (m, 1H), 1.66-1.70 (m, 1H), 1.89-1.92 (m, 1H),
2.03-2.21 (m, 2H), 2.59-2.75 (m, 2H), 4.15 (br.s, 2H), 4.91 (t, J = 7.5 Hz, 1H), 5.13 (s,
2H), 7.14-7.20 (m, 1H), 7.27-7.37 (m, 9H), 7.44-7.50 (m, 4H), 7.74 (dd, J = 6.0 Hz, J =
3.0 Hz, 1H), 7.85 (dd, J = 6.0 Hz, J = 3.0 Hz, 1H), 8.13 (dd, J = 6.0 Hz, J = 3.0 Hz, 1H);

127
13C NMR (75 MHz, CDCl3): 32.4, 33.6, 42.7, 43.3, 44.1, 67.0, 123.4, 124.5, 125.5,
126.1, 126.3, 127.1, 127.9, 128.0, 128.1, 128.5, 128.6, 129.0, 131.9, 134.2, 137.0, 140.0,
144.7, 155.3; IR (neat) cm-1 : 2923, 1687, 1429, 1233, 1176, 906; HRMS (ESI): Calcd
for C31H32NO2 (M+H)+ 450.2433, found 450.2425.
4-(3,4-Dimethyl-1-phenylpent-3-en-1-yl)phenyl acetate (2.25): The title compound 2.25
was obtained as a colorless oil (98.5 mg, 64% yield) after purification by silica gel column
chromatography in 10% EtOAc:Hex. 1H NMR (300 MHz, CDCl3): 1.37 (s, 3H), 1.53
(s, 3H), 1.55 (s, 3H), 2.27 (s, 3H), 2.76 (d, J = 6.0 Hz, 2H), 3.07 (t, J = 7.5 Hz, 1H), 6.96-
7.00 (m, 2H), 7.17-7.35 (m, 7H); 13C NMR (75 MHz, CDCl3): 121.5, 125.1, 126.2,
128.2, 129.1, 142.7, 144.8, 148.9, 169.6; IR (neat) cm-1 : 2980, 1760, 1504, 1190, 1015.
HRMS (ESI): Calcd for C21H28NO2 (M+NH4)+ 326.2120, found 326.2118.

128
2-(3,3-Dimethyl-1-phenylbutyl)naphthalene (2.26): The title compound 2.26 was obtained
as a white solid (125.2 mg, 87% yield) after purification by silica gel column
chromatography in hexane. 1H NMR (300 MHz, CDCl3): 0.93 (s, 9H), 2.23 (dd, J =
12.0 Hz, , J = 6.0 Hz, 1H), 2.30 (dd, J = 15.0 Hz, , J = 6.0 Hz, 1H), 4.29 (t, J = 7.5 Hz,
1H), 7.19 (t, J = 7.5 Hz, 1H), 7.26-7.33 (m, 2H), 7.39-7.51 (m, 5H), 7.77-7.85 (m, 4H); 13C
NMR (75 MHz, CDCl3): 30.4, 31.7, 48.5, 49.3, 125.3, 125.8, 126.0, 126.9, 127.6, 127.8,
128.0, 128.1, 128.5, 132.1, 133.7, 144.2, 146.7; IR (neat) cm-1 : 2950, 1596, 1506, 1362,
1240, 813; HRMS (CI): Calcd for C22H24 (M)+ 288.1878, found 288.1883.
2-(1-(4-Fluorophenyl)-3,3-dimethylbutyl)naphthalene (2.27): The title compound 2.27
was obtained as a white solid (126.9 mg, 83% yield) after purification by silica gel column
chromatography in hexane. 1H NMR (500 MHz, CDCl3): 0.89 (s, 9H), 2.16 (dd, J = 15.0
Hz, , J = 5.0 Hz, 1H), 2.24 (dd, J = 15.0 Hz, , J = 5.0 Hz, 1H), 4.25 (t, J = 5.0 Hz, 1H),
6.98 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.42-7.48 (m, 3H), 7.73 (s, 1H), 7.79 (dd,
J = 20.0 Hz, J = 10.0 Hz, 3H); 13C NMR (126 MHz, CDCl3): 30.4, 31.7, 47.7, 49.4,
115.2, 115.3, 125.6 (d, J = 30.0 Hz), 126.1, 126.7, 127.7, 127.8, 128.3, 129.3 (d, J = 6.25
Hz), 132.1, 133.7, 142.3 (d, J = 3.75 Hz), 144.1, 161.3 (d, J = 242.5 Hz); 19F NMR (470

129
MHz, CDCl3) -116.2; IR (neat) cm-1 : 2952, 1599, 1505, 1325, 1095, 836; HRMS
(APPI): Calcd for C22H23F (M)+ 306.1784, found 306.1782.
1-(1-(4-Methoxyphenyl)-3,3-dimethylbutyl)naphthalene (2.28): The title compound 2.28
was obtained as a white solid (114.4 mg, 72% yield) after purification by silica gel column
chromatography in 25% DCM:Hex. 1H NMR (300 MHz, CDCl3): 0.90 (s, 9H), 2.18
(dd, J = 15.0 Hz, , J = 6.0 Hz, 1H), 2.27 (dd, J = 15.0 Hz, , J = 6.0 Hz, 1H), 3.75 (s, 3H),
4.93 (t, J = 6.0 Hz, 1H), 6.80 (d, J = 9.0 Hz, 2H), 7.30 (d, J = 9.0 Hz, 2H), 7.43-7.59 (m,
4H), 7.70 (d, J = 9.0 Hz, 1H), 7.85 (d, J = 9.0 Hz, 1H), 8.31 (d, J = 6.0 Hz, 1H); 13C NMR
(75 MHz, CDCl3): 30.5, 31.8, 41.5, 50.0, 55.3, 113.8, 123.6, 124.8, 125.3, 125.5, 125.9,
126.5, 129.1, 131.5, 134.3, 138.4, 142.5, 157.7; IR (neat) cm-1 : 2950, 1608, 1508, 1440,
1245, 1030; HRMS (ESI): Calcd for C23H30NO (M+NH4)+ 336.2327, found 336.2328.
4-(3,3-Dimethyl-1-phenylbutyl)benzonitrile (2.29): The title compound 2.29 was obtained
as a white solid (85.4 mg, 65% yield) after purification by silica gel column

130
chromatography in 20% DCM:Hex. 1H NMR (300 MHz, CDCl3): 0.81 (s, 9H), 2.04
(dd, J = 15.0 Hz, J = 6.0 Hz, 1H), 2.11 (dd, J = 15.0 Hz, , J = 6.0 Hz, 1H), 4.08 (t, J = 7.5
Hz, 1H), 7.15-7.29 (m, 5H), 7.38 (d, J = 6.0 Hz, 2H), 7.53 (d, J = 6.0 Hz, 2H); 13C NMR
(75 MHz, CDCl3): 30.3, 31.6, 48.6, 49.1, 109.8, 119.1, 126.6, 127.8, 128.6, 128.8, 132.4,
145.2, 152.4; IR (neat) cm-1 : 2969, 2226, 1378, 1159, 1127, 950 ; HRMS (CI): Calcd for
C19H21N (M)+ 263.1674, found 263.1678.
1-(4-(3,3-Dimethyl-1-phenylbutyl)phenyl)ethan-1-one (2.30): The title compound 2.30
was obtained as a white solid (116.2 mg, 83% yield) after purification by silica gel column
chromatography in 25% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 0.85 (s, 9H), 2.13 (d,
J = 6.0 Hz, 2H), 2.55 (s, 3H), 4.13 (t, J = 6.0 Hz, 1H), 7.14-7.30 (m, 5H), 7.40 (d, J = 9.0
Hz, 2H), 7.86 (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 26.6, 30.3, 31.6, 48.5,
49.2, 126.3, 127.8, 128.1, 128.7, 135.1, 145.8, 152.5, 197.8; IR (neat) cm-1 : 2950, 1674,
1602, 1362, 1265, 957; HRMS (ESI): Calcd for C20H25O (M+H)+ 281.1905, found
281.1905.

131
1-(3,3-Dimethyl-1-phenylpentyl)-4-(trifluoromethyl)benzene (2.31): The title compound
2.31 was obtained as a colorless oil (112.0 mg, 70% yield) after purification by silica gel
column chromatography in hexane. 1H NMR (300 MHz, CDCl3): 0.76 (s, 9H), 1.23 (q,
J = 6.9 Hz, 2H), 2.06 (dd, J = 15.0 Hz, J = 6.0 Hz, 1H), 2.13 (dd, J = 15.0 Hz, J = 6.0 Hz,
1H), 4.10 (t, J = 6.0 Hz, 1H), 7.15-7.28 (m, 5H), 7.41 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 9.0
Hz, 2H); 13C NMR (75 MHz, CDCl3): 8.5, 27.4, 34.2, 35.0, 46.9, 48.0, 125.5 (q, J = 2.5
Hz), 126.3, 127.3, 127.8, 128.1 (q, J = 8.2 Hz), 128.75, 145.9, 151.1; 19F NMR (282 MHz,
CDCl3) -62.9; IR (neat) cm-1 : 2960, 1465, 1323, 1162, 1067; HRMS (CI): Calcd for
C20H23F3 (M)+ 320.1752, found 320.1750.
1-(3,3-Dimethyl-1-phenylpentyl)-4-methoxybenzene (2.32): The title compound 2.32 was
obtained as a colorless oil (121.9 mg, 78% yield) after purification by silica gel column
chromatography in 10% DCM:Hex. 1H NMR (300 MHz, CDCl3): 0.77 (s, 9H), 1.25 (q,
J = 7.9 Hz, 2H), 2.07 (d, J = 6.0 Hz, 2H), 3.77 (s, 3H), 4.02 (t, J = 7.5 Hz, 1H), 6.81 (d, J
= 9.0 Hz, 2H), 7.12-7.31 (m, 7H); 13C NMR (75 MHz, CDCl3): 8.5, 27.4, 34.1, 35.0,
47.1, 47.3, 55.3, 113.8, 125.8, 127.8, 128.5, 128.7, 139.2, 147.4, 157.8; IR (neat) cm-1 :
2958, 1508, 1462, 1246, 1176, 821; HRMS (ESI): Calcd for C20H30NO (M+NH4)+
300.2327, found 300.2329.
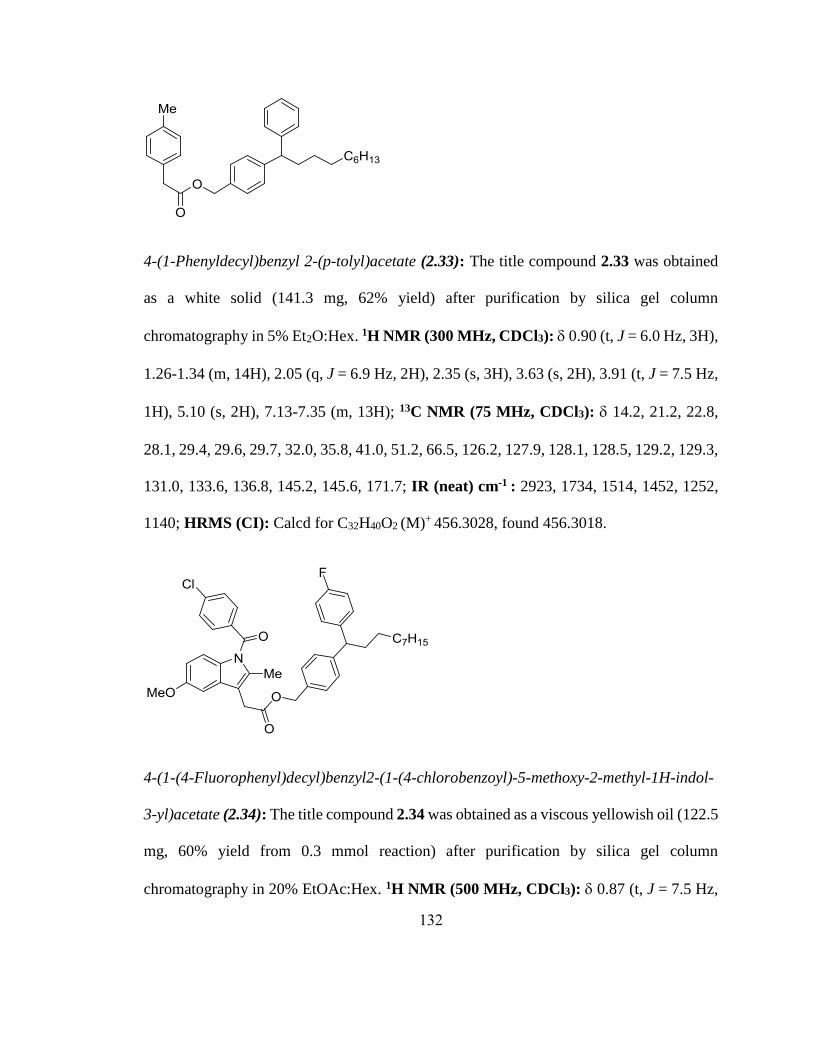
132
4-(1-Phenyldecyl)benzyl 2-(p-tolyl)acetate (2.33): The title compound 2.33 was obtained
as a white solid (141.3 mg, 62% yield) after purification by silica gel column
chromatography in 5% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 0.90 (t, J = 6.0 Hz, 3H),
1.26-1.34 (m, 14H), 2.05 (q, J = 6.9 Hz, 2H), 2.35 (s, 3H), 3.63 (s, 2H), 3.91 (t, J = 7.5 Hz,
1H), 5.10 (s, 2H), 7.13-7.35 (m, 13H); 13C NMR (75 MHz, CDCl3): 14.2, 21.2, 22.8,
28.1, 29.4, 29.6, 29.7, 32.0, 35.8, 41.0, 51.2, 66.5, 126.2, 127.9, 128.1, 128.5, 129.2, 129.3,
131.0, 133.6, 136.8, 145.2, 145.6, 171.7; IR (neat) cm-1 : 2923, 1734, 1514, 1452, 1252,
1140; HRMS (CI): Calcd for C32H40O2 (M)+ 456.3028, found 456.3018.
4-(1-(4-Fluorophenyl)decyl)benzyl2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-
3-yl)acetate (2.34): The title compound 2.34 was obtained as a viscous yellowish oil (122.5
mg, 60% yield from 0.3 mmol reaction) after purification by silica gel column
chromatography in 20% EtOAc:Hex. 1H NMR (500 MHz, CDCl3): 0.87 (t, J = 7.5 Hz,

133
3H), 1.23-1.30 (m, 14H), 1.98 (q, J = 8.3 Hz, 2H), 2.36 (s, 3H), 3.70 (s, 2H), 3.73 (s, 3H),
3.86 (t, J = 7.5 Hz, 1H), 5.09 (s, 2H), 6.67 (dd, J = 10.0 Hz, J = 5.0 Hz, 1H), 6.88 (d, J =
6.0 Hz, 1H), 6.92-6.98 (m, 3H), 7.15-7.21 (m, 4H), 7.22 (d, J = 6.0 Hz, 2H), 7.45 (d, J =
3.0 Hz, 2H), 7.64 (d, J = 3.0 Hz, 2H); 13C NMR (126 MHz, CDCl3): 13.5, 14.2, 22.8,
28.0, 29.4, 29.6, 29.7, 30.5, 32.0, 35.9, 50.4, 55.7, 66.7, 101.3, 111.9, 112.6, 115.0, 115.2,
115.4, 126.1, 128.0, 128.5, 129.2, 129.3, 130.8 (d, J = 26.2 Hz), 131.3, 133.8 (d, J = 50.0
Hz), 136.0, 139.3, 140.8, 145.6, 156.1, 161.4 (d, J = 242.5 Hz), 168.4, 170.8; 19F NMR
(470 MHz, CDCl3) -117.2; IR (neat) cm-1 : 2924, 1734, 1682, 1476, 1313, 1220, 1088;
HRMS (APPI): Calcd for C42H46ClFNO4 (M+H)+ 682.3099, found 682.3032.
4-(2-Cyclohexyl-1-phenylethyl)benzyl2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-
indol-3-yl)acetate (2.35): The title compound 2.35 was obtained as a viscous colorless oil
(87.3 mg, 46% yield from 0.3 mmol reaction) after purification by silica gel column
chromatography in 20% EtOAc:Hex. 1H NMR (300 MHz, CDCl3): 0.83-0.98 (m, 2H),
1.08-1.14 (m, 4H), 1.58-1.77 (m, 5H), 1.90 (t, J = 6.0 Hz, 2H), 2.40 (s, 3H), 3.71 (s, 5H),
4.05 (t, J = 7.5 Hz, 1H), 5.10 (s, 2H), 6.66 (dd, J = 9.0 Hz, J = 3.0 Hz, 1H), 6.93 (d, J = 3.0
Hz, 1H), 6.96 (d, J = 9.0 Hz, 1H), 7.18-7.28 (m, 6H), 7.42-7.52 (m, 3H), 7.65-7.72 (m,

134
3H), 7.79 (d, J = 6.0 Hz, 1H); 13C NMR (126 MHz, CDCl3): 13.5, 26.2, 26.7, 30.5, 33.5,
34.9, 43.6, 47.8, 55.7, 66.7, 101.2, 112.0, 112.6, 115.0, 126.2, 127.4, 127.9, 128.1, 128.4,
128.5, 129.2, 130.7, 130.9, 131.3, 133.4, 134.0, 136.0, 139.3, 145.2, 145.9, 156.1, 168.4,
170.8; IR (neat) cm-1 : 2980, 1733, 1716, 1558, 1456, 1374; HRMS (ESI): Calcd for
C40H41ClNO4 (M+H)+ 634.2724, found 634.2728.
4-(2-Cyclohexyl-1-phenylethyl)benzyl2-(1-methyl-5-(4-methylbenzoyl)-1H-pyrrol-2-
yl)acetate (2.36): The title compound 2.36 was obtained as a viscous colorless liquid (107.1
mg, 67% yield) after purification by silica gel column chromatography in 20% EtOAc:Hex.
1H NMR (300 MHz, CDCl3): 0.91-1.02 (m, 2H), 1.11-1.19 (m, 4H), 1.62-1.81 (m, 5H),
1.94 (t, J = 7.5 Hz, 2H), 2.44 (s, 3H), 3.74 (s, 2H), 3.92 (s, 3H), 4.09 (t, J = 7.5 Hz, 1H),
5.14 (s, 2H), 6.11 (d, J = 3.0 Hz, 1H), 6.68 (d, J = 3.0 Hz, 1H), 7.18-7.37 (m, 11H), 7.73
(d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 21.6, 26.2, 26.7, 33.0, 33.2, 33.4, 34.9,
43.5, 47.8, 67.0, 109.5, 122.3, 126.1, 127.9, 128.2, 128.5, 128.6, 128.7, 129.5, 131.5, 133.0,
134.4, 137.4, 141.9, 145.1, 146.0, 169.3, 185.9; IR (neat) cm-1 : 2919, 1735, 1622, 1371,
1261, 1163; HRMS (ESI): Calcd for C36H40NO3 (M+H)+ 534.3008, found 534.3008.

135
3-(3-(4-Chlorophenyl)-3-phenylpropyl)tetrahydrofuran (2.57): The product was obtained
using 1.5 equiv of PhZnI and 1.5 equiv of 3-(2-iodoethoxy)prop-1-ene in our standard
condition. The title compound 2.57 (inseparable diastereomeric mixture) was obtained as
a colorless oil (57 mg, 38% yield) after purification by silica gel column chromatography
in 5% Et2O:Hex. 1H NMR (300 MHz, CDCl3): 1.26-1.51 (m, 3H), 1.97-2.23 (m, 4H),
3.28 (t, J = 7.5 Hz, 1H), 3.68-3.91 (m, 4H), 7.15-7.31 (m, 9H); 13C NMR (75 MHz,
CDCl3): 31.8, 32.5, 34.7, 39.5, 51.0, 68.0, 73.4, 126.5, 127.7, 128.7, 129.2, 132.0, 143.5,
144.4; IR (neat) cm-1 : 2929, 1488, 1451, 1372, 1090, 820; HRMS (ESI): Calcd for
C19H22ClO (M+H)+ 301.1359, found 301.1380.
2,2'-(2,2,7,7-Tetramethyloctane-4,5-diyl)dinaphthalene (2.58): 5 mol% NiBr2 was used as
a catalyst for this reaction (see Scheme 4 for reaction). The title compound 2.58 was
obtained as a white solid (29.5 mg, 14% yield; 1:1 dr) along with 2.26 (79.2 mg, 55% yield)
after purification by silica gel column chromatography in 3% Et2O:Hex. 1H NMR (300
MHz, CDCl3): 0.56 (s, 9H), 0.76 (s, 9H), 1.50 (d, J = 15.0 Hz, 1H), 1.70-1.78 (m, 1H),

136
1.80-1.89 (m, 2H), 3.05 (dd, J = 15.0 Hz, J = 6.0 Hz, 1H), 3.15 (dd, J = 12.0 Hz, J = 3.0
Hz, 1H), 7.12 (dd, J = 9.0 Hz, J = 3.0 Hz, 1H), 7.30 (dd, J = 9.0 Hz, J = 3.0 Hz, 1H), 7.38-
7.48 (m, 5H), 7.61 (d, J = 6.0 Hz, 2H), 7.68-7.85 (m, 5H); 13C NMR (75 MHz, CDCl3):
30.2, 30.3, 31.2, 31.4, 46.4, 47.0, 50.0, 50.2, 125.0, 125.1, 125.5, 125.7, 126.6, 127.5,
127.7, 128.2, 128.8, 132.1, 132.3, 133.1, 133.5, 142.1, 143.8; IR (neat) cm-1 : 2924, 1394,
1363, 1047, 717; HRMS (CI): Calcd for C32H38 (M)+ 422.2974, found 422.2968.
ethyl 2-((1S,2R)-1-(3-(trifluoromethyl)phenyl)-2,3-dihydro-1H-inden-2-yl)acetate (2.37):
The title compound 2.37 was obtained as a colorless liquid (132.2 mg, 76% yield; 16:1 dr)
after purification by silica gel column chromatography in ether: hexane = 1:10. 1H NMR
(300 MHz, CDCl3): 1.19 (t, J = 7.5 Hz, 3H), 2.47-2.62 (m, 2H), 2.71-2.85 (m, 2H), 3.27-
3.34 (m, 1H), 3.98-4.07 (m, 3H), 6.81 (d, J = 6.0 Hz, 1H), 7.11-7.28 (m, 3H), 7.36 (d, J =
9.0 Hz, 1H), 7.41-7.45 (m, 2H), 7.52 (d, J = 6.0 Hz, 1H); 13C NMR (126 MHz, CDCl3):
14.2, 38.1, 38.3, 47.2, 57.3, 60.5, 124.3 (q, J = 273.4 Hz), 123.8 (q, J = 3.7 Hz), 124.5,
124.8, 125.5 (q, J = 3.7 Hz), 126.9, 127.3, 129.1, 130.9 (q, J = 32.3 Hz), 132.2, 142.8,
144.4, 145.2, 172.4; 19F NMR (282 MHz, CDCl3) -60.9; IR (neat) cm-1: 1730, 1446,
1324, 1160, 1120, 1072; HRMS (ESI): Calcd for C20H20F3O2 (M+H)+ 349.1415, found
349.1425.
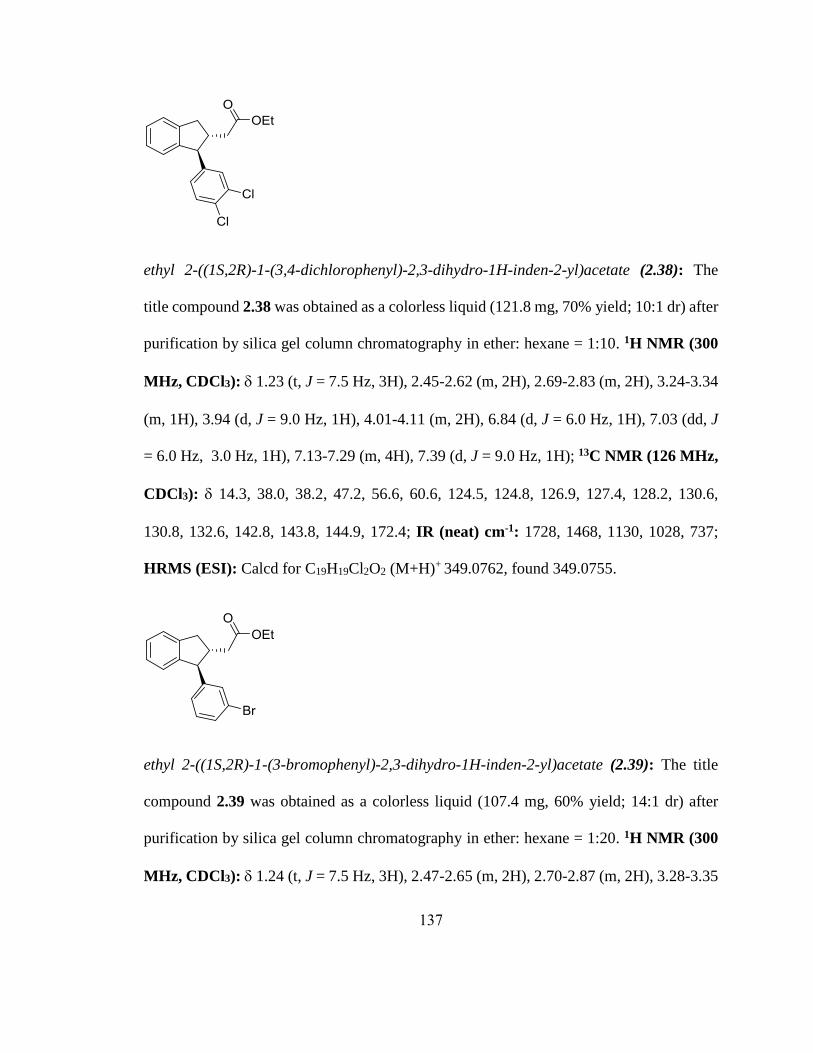
137
ethyl 2-((1S,2R)-1-(3,4-dichlorophenyl)-2,3-dihydro-1H-inden-2-yl)acetate (2.38): The
title compound 2.38 was obtained as a colorless liquid (121.8 mg, 70% yield; 10:1 dr) after
purification by silica gel column chromatography in ether: hexane = 1:10. 1H NMR (300
MHz, CDCl3): 1.23 (t, J = 7.5 Hz, 3H), 2.45-2.62 (m, 2H), 2.69-2.83 (m, 2H), 3.24-3.34
(m, 1H), 3.94 (d, J = 9.0 Hz, 1H), 4.01-4.11 (m, 2H), 6.84 (d, J = 6.0 Hz, 1H), 7.03 (dd, J
= 6.0 Hz, 3.0 Hz, 1H), 7.13-7.29 (m, 4H), 7.39 (d, J = 9.0 Hz, 1H); 13C NMR (126 MHz,
CDCl3): 14.3, 38.0, 38.2, 47.2, 56.6, 60.6, 124.5, 124.8, 126.9, 127.4, 128.2, 130.6,
130.8, 132.6, 142.8, 143.8, 144.9, 172.4; IR (neat) cm-1: 1728, 1468, 1130, 1028, 737;
HRMS (ESI): Calcd for C19H19Cl2O2 (M+H)+ 349.0762, found 349.0755.
ethyl 2-((1S,2R)-1-(3-bromophenyl)-2,3-dihydro-1H-inden-2-yl)acetate (2.39): The title
compound 2.39 was obtained as a colorless liquid (107.4 mg, 60% yield; 14:1 dr) after
purification by silica gel column chromatography in ether: hexane = 1:20. 1H NMR (300
MHz, CDCl3): 1.24 (t, J = 7.5 Hz, 3H), 2.47-2.65 (m, 2H), 2.70-2.87 (m, 2H), 3.28-3.35

138
(m, 1H), 3.95 (d, J = 12.0 Hz, 1H), 4.01-4.12 (m, 2H), 6.86 (d, J = 9.0 Hz, 1H), 7.12-7.29
(m, 5H), 7.40 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 14.3, 38.0, 38.3, 47.2,
57.1, 60.5, 122.7, 124.4, 124.8, 126.8, 127.2, 127.5, 130.0, 130.2, 131.7, 142.8, 145.3,
145.8, 172.4; IR (neat) cm-1: 1728, 1473, 1158, 1025, 745; HRMS (ESI): Calcd for
C19H20BrO2 (M+H)+ 359.0647, found 359.0641.
ethyl 2-((1S,2R)-1-(2-chloropyridin-4-yl)-2,3-dihydro-1H-inden-2-yl)acetate (2.40): The
title compound 2.40 was obtained as a colorless liquid (64.5 mg, 41% yield; 14:1 dr) after
purification by silica gel column chromatography in ether: hexane = 1:5. 1H NMR (500
MHz, CDCl3): 1.23 (t, J = 7.5 Hz, 3H), 2.50-2.60 (m, 2H), 2.73-2.84 (m, 2H), 3.29-3.33
(m, 1H), 4.01 (d, J = 10.0 Hz, 1H), 4.03-4.10 (m, 2H), 6.85 (d, J = 10.0 Hz, 1H), 7.05 (d,
J = 5.0 Hz, 1H), 7.16 (s, 1H), 7.17 (d, J = 5.0 Hz, 1H), 7.23-7.31 (m, 2H), 8.32 (d, J = 5.0
Hz, 1H); 13C NMR (126 MHz, CDCl3): 14.3, 38.1, 38.4, 46.5, 56.3, 60.7, 122.7, 124.3,
124.7, 124.8, 127.1, 127.8, 142.8, 143.5, 149.9, 152.0; IR (neat) cm-1: 1726, 1589, 1387,
1122, 1024, 736; HRMS (ESI): Calcd for C18H19ClNO2 (M+H)+ 316.1104, found
316.1110.

139
ethyl 4-(3-bromophenyl)-4-(3-(trifluoromethyl)phenyl)butanoate (2.41): The title
compound 2.41 was obtained as a colorless liquid (136.6 mg, 66% yield) after purification
by silica gel column chromatography in ether: hexane = 1:20. 1H NMR (300 MHz,
CDCl3): 1.24 (t, J = 7.5 Hz, 3H), 2.23-2.41 (m, 4H), 3.98 (t, J = 7.5 Hz, 1H), 4.11 (q, J
= 6.9 Hz, 2H), 7.16-7.21 (m, 2H), 7.34-7.47 (m, 6H); 13C NMR (126 MHz, CDCl3):
14.3, 30.3, 32.5, 50.0, 60.6, 123.0, 123.8 (q, J = 3.7 Hz), 124.1 (q, J = 273.4 Hz), 124.6 (q,
J = 3.7 Hz), 126.5, 129.3, 130.1, 130.4, 131.0, 131.1 (q, J = 49.1 Hz), 131.2, 144.4, 145.2,
173.0; 19F NMR (282 MHz, CDCl3) -61.0; IR (neat) cm-1: 1728, 325, 1120, 1072, 782;
HRMS (ESI): Calcd for C19H22BrF3O2N (M+NH4)+ 432.0786, found 432.0795.
methyl 2-(4-ethoxy-4-oxo-1-phenylbutyl)benzoate (2.42): The title compound 2.42 was
obtained as a colorless liquid (89.6 mg, 55% yield) after purification by silica gel column
chromatography in ether: hexane = 1:10. 1H NMR (500 MHz, CDCl3): 1.21 (t, J = 7.5
Hz, 3H), 2.23-2.42 (m, 4H), 3.86 (s, 3H), 4.08 (q, J = 6.6 Hz, 2H), 5.01 (t, J = 7.5 Hz, 1H),
7.14-7.19 (m, 1H), 7.21-7.26 (m, 5H), 7.37-7.44 (m, 2H), 7.76 (dd, J = 1.5 Hz, J = 10.0

140
Hz, 1H); 13C NMR (126 MHz, CDCl3): 14.3, 31.0, 32.9, 44.6, 52.2, 60.4, 126.1, 126.3,
128.3, 128.5, 130.3, 130.6, 132.0, 143.9, 145.3, 168.6, 173.5; IR (neat) cm-1: 1717, 1256,
1127, 1074; HRMS (ESI): Calcd for C20H23O4 (M+H)+ 327.1596, found 327.1609.
ethyl 1-(2-phenyl-2-(3-(trifluoromethyl)phenyl)ethyl)cyclobutane-1-carboxylate (2.43):
The title compound 2.43 was obtained as a colorless liquid (105.2 mg, 56% yield) after
purification by silica gel column chromatography in ether: hexane = 1:20. 1H NMR (300
MHz, CDCl3): 1.10 (t, J = 7.5 Hz, 3H), 1.77-1.88 (m, 4H), 2.23-2.37 (m, 2H), 2.62 (d, J
= 9.0 Hz, 2H), 3.68 (q, J = 7.0 Hz, 2H), 3.95 (t, J = 7.5 Hz, 1H), 7.14-7.28 (m, 5H), 7.32-
7.41 (m, 3H), 7.49 (s, 1H); 13C NMR (75 MHz, CDCl3): 14.0, 16.0, 30.8, 31.1, 43.9,
47.7, 48.2, 60.3, 123.2 (q, J = 3.0 Hz), 124.3 (q, J = 270.7 Hz), 124.6 (q, J = 3.7 Hz), 126.7,
128.0, 128.6, 128.9, 130.7 (q, J = 32.2 Hz), 131.7, 144.0, 145.9, 176.5; 19F NMR (282
MHz, CDCl3) -61.0; IR (neat) cm-1: 1721, 1325, 1159, 1025; HRMS (ESI): Calcd for
C22H24F3O2 (M+H)+ 377.1729, found 377.1742.

141
ethyl 4-(2-formylphenyl)-2,2-dimethyl-4-(3-(trifluoromethyl)phenyl)butanoate (2.44): The
title compound 2.44 was obtained as a colorless liquid (121.5 mg, 62% yield) after
purification by silica gel column chromatography in ether: hexane = 1:10. 1H NMR (300
MHz, CDCl3): 1.08 (t, J = 7.5 Hz, 3H), 1.17 (s, 6H), 2.45 (d, J = 6.0 Hz, 2H), 3.63 (q, J
= 6.9 Hz, 2H), 5.61 (t, J = 7.5 Hz, 1H), 7.32-7.41 (m, 3H), 7.53-7.61 (m, 4H), 7.73 (d, J =
9.0 Hz, 1H), 10.22 (s, 1H); 13C NMR (126 MHz, CDCl3): 14.0, 26.0, 26.3, 39.9, 42.2,
46.4, 60.4, 123.3 (q, J = 3.7 Hz), 124.2 (q, J = 272.1 Hz), 124.6 (q, J = 3.7 Hz), 127.0,
128.8, 128.9, 130.8 (q, J = 32.3 Hz), 131.9, 133.3, 133.8, 134.7, 145.8, 146.2, 177.0, 193.3;
19F NMR (282 MHz, CDCl3) -61.0; IR (neat) cm-1: 1692, 1447, 1325, 1119, 1073;
HRMS (ESI): Calcd for C22H24F3O3 (M+H)+ 393.1678, found 393.1659.
4-(3,4-dichlorophenyl)-3,4-dihydronaphthalen-1(2H)-one (2.45): The title compound 2.45
was obtained as a white solid (37.1 mg, 64% yield from 0.2 mmol) after purification by
silica gel column chromatography in ether: hexane = 1:5. 1H NMR (300 MHz, CDCl3):
2.20-2.31 (m, 1H), 2.39-2.52 (m, 1H), 2.58-2.77 (s, 2H), 4.27 (dd, J = 9.0 Hz, J = 6.0 Hz,
1H), 6.93-6.96 (m, 2H), 7.22 (d, J = 3.0 Hz, 1H), 7.36-7.50 (m, 3H), 8.12 (d, J = 6.0 Hz,
1H); 13C NMR (75 MHz, CDCl3): 31.8, 36.6, 44.6, 127.5, 127.6, 128.0, 129.4, 130.6,
130.7, 131.1, 132.8, 132.9, 133.9, 144.1, 144.9, 197.4; IR (neat) cm-1: 1672, 1590, 1469,
1284, 1028; HRMS (ESI): Calcd for C16H13Cl2O (M+H)+ 291.0343, found 291.0347.

142
methyl 4-methoxy-2-(((trifluoromethyl)sulfonyl)oxy)benzoate (2.46): The title compound
2.46 was obtained as a brown solid (95% yield) after purification by silica gel column
chromatography in ethyl acetate: hexane = 1:5. 1H NMR (500 MHz, CDCl3): 3.88 (s,
3H), 3.93 (s, 3H), 6.77 (s, 1H), 6.95 (d, J = 6.0 Hz, 1H), 8.06 (d, J = 6.0 Hz, 1H); 13C NMR
(126 MHz, CDCl3): 52.4, 56.1, 109.0, 113.5, 116.4, 118.8 (q, J = 320.8 Hz), 134.2,
149.7, 164.1; 19F NMR (282 MHz, CDCl3) -73.2; IR (neat) cm-1: 1731, 1614, 1420,
1128, 1068; HRMS (ESI): Calcd for C10H10F3O6S (M+H)+ 315.0150, found 315.0147.
methyl 4-methoxy-2-vinylbenzoate (2.47): The title compound 2.47 was obtained as a
colorless liquid (75% yield) after purification by silica gel column chromatography in
ether: hexane = 1:10. 1H NMR (500 MHz, CDCl3): 3.86 (s, 6H), 5.35 (d, J = 10.0 Hz,
1H), 5.62 (d, J = 15.0 Hz, 1H), 6.82 (dd, J = 10.0 Hz, 3.0 Hz, 1H), 7.03 (d, J = 5.0 Hz, 1H),
7.55 (dd, J = 20.0 Hz, 10.0 Hz, 1H), 7.91 (d, J = 10.0 Hz, 1H); 13C NMR (126 MHz,
CDCl3): 51.9, 55.4, 112.5, 112.9, 116.4, 120.8, 132.8, 136.5, 142.4, 162.6, 167.3; IR
(neat) cm-1: 1708, 1598, 1433, 1232, 1125; HRMS (ESI): Calcd for C11H13O3 (M+H)+
193.0865, found 193.0866.
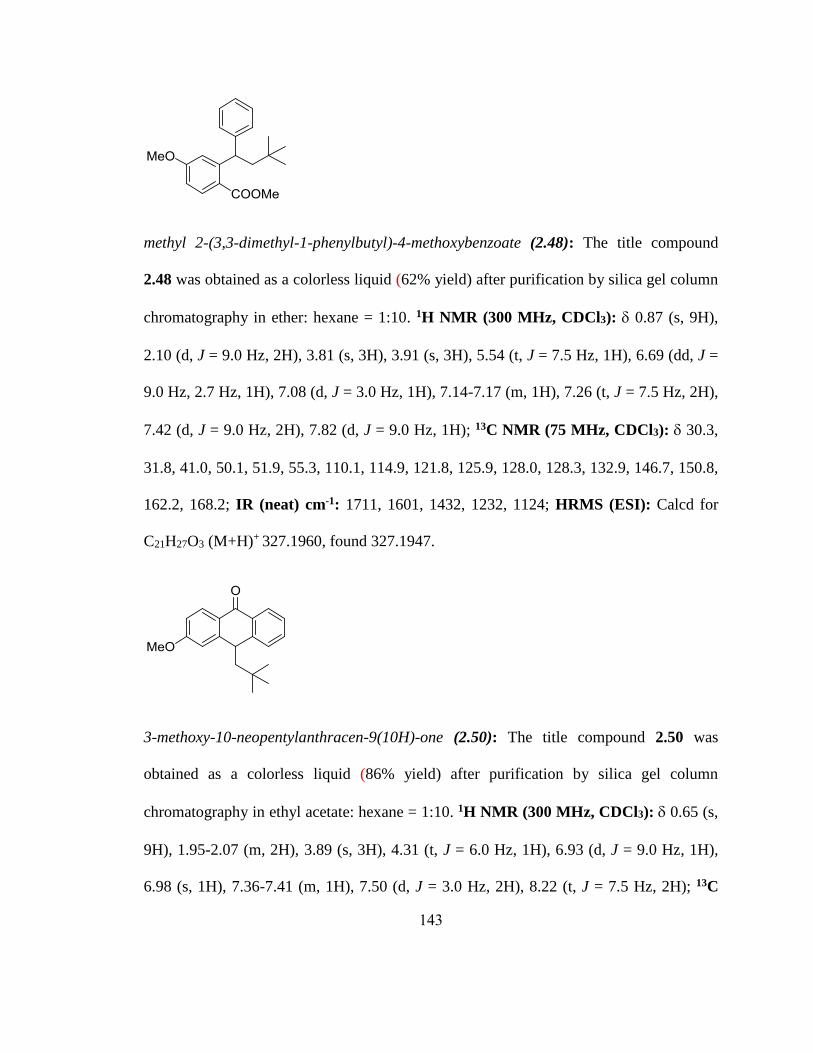
143
methyl 2-(3,3-dimethyl-1-phenylbutyl)-4-methoxybenzoate (2.48): The title compound
2.48 was obtained as a colorless liquid (62% yield) after purification by silica gel column
chromatography in ether: hexane = 1:10. 1H NMR (300 MHz, CDCl3): 0.87 (s, 9H),
2.10 (d, J = 9.0 Hz, 2H), 3.81 (s, 3H), 3.91 (s, 3H), 5.54 (t, J = 7.5 Hz, 1H), 6.69 (dd, J =
9.0 Hz, 2.7 Hz, 1H), 7.08 (d, J = 3.0 Hz, 1H), 7.14-7.17 (m, 1H), 7.26 (t, J = 7.5 Hz, 2H),
7.42 (d, J = 9.0 Hz, 2H), 7.82 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 30.3,
31.8, 41.0, 50.1, 51.9, 55.3, 110.1, 114.9, 121.8, 125.9, 128.0, 128.3, 132.9, 146.7, 150.8,
162.2, 168.2; IR (neat) cm-1: 1711, 1601, 1432, 1232, 1124; HRMS (ESI): Calcd for
C21H27O3 (M+H)+ 327.1960, found 327.1947.
3-methoxy-10-neopentylanthracen-9(10H)-one (2.50): The title compound 2.50 was
obtained as a colorless liquid (86% yield) after purification by silica gel column
chromatography in ethyl acetate: hexane = 1:10. 1H NMR (300 MHz, CDCl3): 0.65 (s,
9H), 1.95-2.07 (m, 2H), 3.89 (s, 3H), 4.31 (t, J = 6.0 Hz, 1H), 6.93 (d, J = 9.0 Hz, 1H),
6.98 (s, 1H), 7.36-7.41 (m, 1H), 7.50 (d, J = 3.0 Hz, 2H), 8.22 (t, J = 7.5 Hz, 2H); 13C

144
NMR (75 MHz, CDCl3): 30.7, 32.5, 41.6, 55.2, 55.5, 112.8, 113.1, 126.1, 126.8, 127.5,
128.3, 130.1, 132.1, 132.6, 146.5, 149.3, 162.9, 184.1; IR (neat) cm-1: 1651, 1598, 1456,
1274, 1241, 1091, 931; HRMS (ESI): Calcd for C20H23O2 (M+H)+ 295.1698, found
295.1702.
methyl 4-amino-2-bromobenzoate (2.52): The title compound 2.52 was obtained as a light-
yellow solid (92%) yield and it used for next step without further purification. 1H NMR
(500 MHz, CDCl3): 3.84 (s, 3H), 4.13 (br.s, 2H), 6.54 (dd, J = 10.0 Hz, 2.5 Hz, 1H),
6.91 (d, J = 2.0 Hz, 1H), 7.73 (d, J = 2.0 Hz, 1H); 13C NMR (126 MHz, CDCl3): 51.9,
112.9, 119.5, 119.8, 124.1, 133.7, 150.7, 166.1; IR (neat) cm-1: 3324, 1704, 1586, 1428,
1240, 1032; HRMS (ESI): Calcd for C8H9BrNO2 (M+H)+ 229.2817, found 229.9809.
methyl 2-bromo-4-hydroxybenzoate (2.53): The title compound 2.53 was obtained as a
white solid (56%) yield after purification by silica gel column chromatography in ethyl
acetate: hexane = 1:10. 1H NMR (300 MHz, CDCl3): 3.87 (s, 3H), 4.94 (br.s, 1H), 6.82
(d, J = 9.0 Hz, 1H), 7.13 (s, 1H), 7.77 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3):
52.4, 115.3, 122.2, 123.0, 124.1, 134.4, 162.5, 167.6; IR (neat) cm-1: 3333, 1684, 1261,
1031; HRMS (ESI): Calcd for C8H8BrO3 (M+H)+ 230.9657, found 230.9663.

145
methyl 4-hydroxy-2-vinylbenzoate (2.54): The title compound 2.54 was obtained as a white
solid (76%) yield after purification by silica gel column chromatography in ether: hexane
= 1:5. 1H NMR (300 MHz, CDCl3): 3.88 (s, 3H), 5.32 (d, J = 9.0 Hz, 1H), 5.57 (d, J =
15.0 Hz, 1H), 5.89 (s, 1H), 6.78 (dd, J = 9.0 Hz, 3.0 Hz, 1H), 7.00 (d, J = 3.0 Hz, 1H), 7.50
(dd, J = 18.0 Hz, 12.0 Hz, 1H), 7.86 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3):
52.2, 114.2, 114.7, 116.8, 120.4, 133.2, 136.0, 142.8, 159.6, 168.2; IR (neat) cm-1: 3291,
1677, 1560, 1438, 1138; HRMS (ESI): Calcd for C10H11O3 (M+H)+ 179.0708, found
179.0713.
methyl 4-(quinolin-2-ylmethoxy)-2-vinylbenzoate (2.55): The title compound 2.55 was
obtained as a white solid (84% yield) after purification by silica gel column
chromatography in ethyl acetate: hexane = 1:5. 1H NMR (300 MHz, CDCl3): 3.86 (s,
3H), 5.34 (d, J = 12.0 Hz, 1H), 5.45 (s, 2H), 5.60 (d, J = 18.0 Hz, 1H), 6.95 (dd, J = 9.0
Hz, 3.0 Hz, 1H), 7.23 (d, J = 2.4 Hz, 1H), 7.48-7.59 (m, 2H), 7.65 (d, J = 12.0 Hz, 1H),
7.75 (t, J = 7.5 Hz, 1H), 7.87 (dd, J = 18.0 Hz, 9.0 Hz, 2H), 8.08 (d, J = 9.0 Hz, 1H), 8.20
(d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 51.9, 71.4, 113.6, 113.7, 116.7, 119.1,
121.4, 126.7, 127.7, 127.8, 129.0, 130.0, 132.9, 136.2, 137.2, 142.4, 147.6, 157.1, 161.3,

146
167.2; IR (neat) cm-1: 1709, 1564, 1426, 1258, 1138, 1091; HRMS (ESI): Calcd for
C20H18NO3 (M+H)+ 320.1287, found 320.1293.
methyl 2-(3,3-dimethyl-1-phenylbutyl)-4-(quinolin-2-ylmethoxy)benzoate (2.56): The title
compound 2.56 was obtained as a colorless viscous liquid (63% yield from 0.5 mmol
reaction) after purification by silica gel column chromatography in THF: hexane = 1:20.
1H NMR (500 MHz, CDCl3): 0.75 (s, 9H), 1.98 (dd, J = 6.0 Hz, 3.0 Hz, 2H), 3.88 (s,
3H), 5.42 (s, 2H), 5.45 (t, J = 4.5 Hz, 1H), 6.82 (dd, J = 3.0 Hz, 1.5 Hz, 1H), 7.05-7.11 (m,
3H), 7.18 (d, J = 3.0 Hz, 1H), 7.29 (d, J = 3.0 Hz, 2H), 7.57-7.61 (m, 2H), 7.76-7.78 (m,
2H), 7.83 (d, J = 6.0 Hz, 1H), 8.14 (dd, J = 12.0 Hz, 6.0 Hz, 2H); 13C NMR (126 MHz,
CDCl3): 30.2, 31.7, 41.0, 50.0, 52.0, 71.4, 112.3, 114.9, 119.2, 122.2, 125.8, 126.8,
127.7, 127.8, 127.9, 128.3, 129.1, 130.0, 132.8, 137.2, 146.6, 147.7, 151.0, 157.4, 160.9,
168.2; IR (neat) cm-1: 1711, 1598, 1430, 1234, 1124, 1041; HRMS (ESI): Calcd for
C30H32NO3 (M+H)+ 454.2382, found 454.2397.

147
4.2 Ni-Catalyzed Cyclization/Coupling
4.2.1 General Information
All the reactions were set up inside a nitrogen-filled glovebox and all the chemicals were
handled under nitrogen atmosphere unless stated otherwise. All the glassware including
the 4-dram and 1-dram borosilicate (Kimble-Chase) vials, and pressure vessels were
properly dried in an oven before use. Bulk solvents were obtained from EMD and
anhydrous solvents (DMF, DMA, DMSO, NMP, dioxane, toluene) were obtained from
Sigma-Aldrich, and were used directly without further purification. Deuterated solvents
were purchased from Sigma-Aldrich. NiBr2 was purchased from Alfa-Aesar. Aryl halides
were purchased from Acros, Sigma-Aldrich, Oakwood, TCI-America, Matrix and Alfa-
Aesar. ZnCl2 (99.95%) was obtained from Alfa-Aesar was used as received. 1H, 13C, and
19F NMR spectra were recorded on a Bruker instrument (300, 75, and 282 MHz
respectively) and internally referenced to the residual solvent signals of CDCl3 for 1H and
13C NMR, and 19F NMR at 7.26, 77.16 ppm, −164.9 ppm respectively. The chemical shifts
of NMR and the coupling constants (J) for 1H, 13C, and 19F NMR are reported in δ parts
per millions (ppm) and in Hertz, respectively. The following conventions are used for
multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of
doublet; br, broad. High resolution mass spectra for new compounds were recorded at the
Mass Spectrometry, Department of Chemistry and Chemical Biology, University of New
Mexico (UNM), University of Texas Austin and University of California, Riverside. All
NMR spectra were collected at Department of Chemistry and Chemical Biology,
University of New Mexico (UNM). The HPLC used was Waters e2695 separations module

148
and Waters 2489 UV/Vis detector. Infrared (IR) spectra were recorded on Bruker Alpha-P
ATR-IR at UNM and νmax is reported in cm-1. The starting materials diethyl-2-allyl-2-(2-
bromoethyl)malonate240(12-Br), 3-(2-iodoethoxy)prop-1-ene, N-allyl-N-(2-bromoethyl)-
4-methylbenzenesulfonamide241, N-allyl-N-(2-bromoethyl)aniline242, trans-2-(allyloxy)-3-
iodotetrahydrofuran and trans-2-(allyloxy)-3-iodotetrahydro-pyran100, 243, trans-1-
(allyloxy)-2-iodocyclohexane244, (1-(allyloxy)-2-iodoethyl)benzene245, 1-(1-(allyloxy)-2-
iodoeth-oxy)butane100, 1-(allyloxy)-1-ethoxy-2-iodobutane100, 3-(2-bromoethoxy)oct-1-
ene203, trans-1-(Allyloxy)-2-bromocyclohexane244, cis-1-(allyloxy)-2-
bromocyclohexane246 and 3-(2-bromoethoxy)oct-1-ene203 were prepared according to the
given literature procedure.
4.2.2 Experimental Procedure
General procedure for the preparation of arylzinc reagent. Procedure A.238 To a
Schlenk flask in a glovebox, anhydrous LiCl (210 mg, 5 mmol) and zinc powder (492 mg,
7.5 mmol) was added and the mixture was dried under high vacuum at 150 °C to 170 °C
for 2 h outside the glovebox. After 2 h, it was cooled down to room temperature and the
reaction flask was flushed with nitrogen. Then it was again taken to a glovebox and
anhydrous THF (5mL) was added with stirring the solution at room temperature. Later,
zinc was activated with the addition of 5 mol% of BrCH2CH2Br and 3 mol% of TMSCl to
the zinc/THF suspension and the mixture was stirred for 5 minutes at room temperature.
To this stirred solution was added corresponding aryl iodides (5 mmol) (neat) dropwise
(liquid) or portion-wise (solid) and the reaction mixture was either heated at 50 °C for

149
heteroaryl iodides for 6 h or refluxed for electron-deficient and electron rich aryl iodides
for 12-96 h. The final concentration of the arylzinc reagent was determined by titration
with molecular iodine in THF.239
Procedure B. Under nitrogen atmosphere, naphthalene (563.9 mg, 4.4 mmol) was
dissolved in anhydrous THF (4 ml) in 15 mL pressure vessel, potassium (156.4 mg, 4
mmol) was added to the solution and stirred overnight at room temperature. The solution
turned dark green immediately indicating the generation of potassium naphthalenide.
Anhydrous ZnCl2 (272.6 mg, 2.0 mmol) was then suspended in dry THF (4 mL) in a
separate vial, which was then added dropwise to the potassium naphtalenide solution. The
resultant solution was then stirred for 12 h at room temperature. Aryl iodide (1.0 mmol,
neat) was added to the stirred solution and stirred again overnight at room temperature.
Thus, formed organozinc was filtered through Celite upon completion of the reaction
(monitored by GC for the remaining starting aryl iodides as well as the protonation product
by quenching the organozinc reagents with acetic acid). The final concentration was
determined by titration with molecular iodine in THF.239
Procedure C.247 Under nitrogen atmosphere, LiCl (63 mg, 0.3 mmol), InCl3 (30mg, 0.03
mmol) and Zn powder (983 mg, 15 mmol) were weigh out in a dry schlenk-tube equipped
with a stir bar. The mixture was heated at 170°C under high vaccum for 3 h. After cooling
down to room temperature, the tube was flushed with nitrogen and freshly distilled THF
(5mL) and DMPU (5ml) was added and stirred at room temperature. Later TMSCl (3
mol%) and aryl iodide (5 mmol) was added to the suspension. The mixture was further

150
stirred at room temperature for 1 h. The completion of reaction was monitored by GC
analysis of acetic acid-quenched aliquots. The excess zinc dust was allowed to settle down
and filtered through celite pad. Finally, the concentration of resulting organozinc was
determined by titration with molecular iodine in THF.239
General procedure for screening reaction conditions. In a glovebox, THF solution of
(4-cyanophenyl)zinc iodide (0.150 mmol) was taken in a 1-dram vial and the solvent was
removed under vacuum. To the ArZnI residue was added NiBr2 (0.65 mg, 0.003 mmol),
terpyridine (0.9 mg, 0.004 mmol), and diethyl-2-allyl-2-(2-bromoethyl)malonate (30.6 mg,
0.10 mmol). The mixture was then dissolved in 0.5 mL of NMP. The vial was tightly
capped and removed from the glovebox. It was placed in a hotplate preheated to 50 °C with
vigorous stirring. After 6 h, the reaction mixture was cooled to room temperature and 50
µL of pyrene (0.010 mmol, 0.2 M stock solution) as an internal standard was added, diluted
with EtOAc (2 mL) and filtered through a short pad of silica gel in a pipette. The filtrate
was then analyzed by GC, GC-MS and 1H NMR.
General procedure for large scale reaction. In a glovebox, arylzinc stock solution in
THF (0.750 mmol) was taken in a 15mL sealed tube and the solvent was removed under
vacuum. To the residue of ArZnI was added NiBr2 (3.3 mg, 0.015 mmol), terpyridine (4.7
mg, 0.020 mmol), and alkene tethered to alkyl halides (0.5 mmol). The mixture was then
dissolved in NMP (2.5 mL). The sealed tube was tightly capped and removed from the
glovebox. It was then placed in an oil-bath preheated to 50 °C with vigorous stirring. After
6 h, the reaction mixture was cooled to room temperature, diluted with EtOAc (10 mL) and
washed with H2O (5 mL × 3). The aqueous fraction was extracted back with ethyl acetate

151
(5 mL × 3) and combined with the first ethyl acetate fraction. The combined ethyl acetate
fraction was dried over Na2SO4 and the solvent was removed in a rotary evaporator. The
product was purified by silica gel column chromatography using ethyl acetate/hexane as
eluent.
Determination of diastereoselectivity and identification of major diastereoisomers.
The diastereoselectivity was determined based on crude 1H NMR and crude GC trace. The
corresponding GC traces are provided in the respective places. The dr’s in the reported 1H
NMR spectra of analytically pure compounds do not reflect the actual dr’s of the reaction.
The diastereomer either contained the other diastereomer in analytically pure samples, or
a small fraction of the separated minor diastereomer was contaminated with some other
impurities, which are not included in the reported yields. Therefore, the reported 1H NMR
show different dr’s than those that are actually formed in the reaction. The structures of
major diastereomers were determined by comparing 1H NMR spectra and the coupling
constants with the same or similar compounds in the literature.85, 88-89, 100, 106
Test for the tolerance of base-sensitive racemizable stereocenter in N-Boc D-proline
methyl ester (3.47). In a sealed tube, (4-cyanophenyl)zinc iodide stock solution in THF
(0.60 mmol) was taken and the solvent was removed under vacuum. To the residue of
ArZnI was added NiBr2 (2.5 mg, 0.012), terpyridine (3.6 mg, 0.016 mmol), and diethyl-2-
allyl-2-(2-bromoethyl)malonate (3.13-Br) (0.4 mmol) respectively. The chiral additives N-
Boc D-Proline methyl ester (0.4 mmol) was then added in the mixture. Then the mixture
was dissolved in NMP (2.0 mL). The sealed tube was tightly capped and placed in an oil-
bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction mixture was cooled

152
to room temperature, diluted with EtOAc (10 mL) and washed with H2O (5 mL × 3). The
aqueous fraction was extracted back with ethyl acetate (5 mL × 3) and combined with the
first ethyl acetate fraction. The combined ethyl acetate fraction was dried over Na2SO4 and
the solvent was removed in a rotary evaporator. The product was purified by column
chromatography using 20% EtOAc in hexane. The compound diethyl 3-(4-
cyanobenzyl)cyclopentane-1,1-dicarboxylate (3.14) was obtained in 76% yield with the
recovery of chiral additives N-Boc D-proline methyl ester in 93% yield. After the reaction,
the racemization was checked using chiral HPLC. At first, the racemic mixture of N-Boc
proline methyl ester in chiral HPLC were separated by chiral pak IA-3 in 1% IPA in hexane
with the flow of 1.0 mL/min with detection by UV detector at 210 nm. The two peaks of
(S)- and (R)-compounds were observed at 11.1 min and 12.8 min. Then pure N-Boc D-
proline methyl ester [(R)-enantiomer)] appeared as a single peak at 12.8 min with 100:0
enantiomeric ratio (er). Later, N-Boc D-proline methyl ester recovered from our reaction
was analyzed with the chiral HPLC which showed a single peak at 12.8 min with 100:0 er.
For complete picture of both enantiomer, their % area and retention time see SI.
Test for the tolerance of base-sensitive racemizable stereocenter in (R)-
Dimethylmethylsuccinate (3.48). To a sealed tube, (4-cyanophenyl)zinc iodide stock
solution in THF (0.6 mmol) was taken and the solvent was removed under vacuum. To the
residue of ArZnI, NiBr2 (2.5 mg, 0.012 mmol), terpyridine (3.6 mg, 0.016 mmol), and
diethyl-2-allyl-2-(2-bromoethyl)malonate (3.13-Br) (0.4 mmol) were added respectively.
The chiral additives (R)-dimethylmethylsuccinate (0.5 mmol) was then added in the
mixture. Then the mixture was dissolved in NMP (2.0 mL). Later, the tube was sealed and

153
placed in an oil-bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction
mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with
H2O (5 mL × 3). The aqueous fraction was extracted back with ethyl acetate (5 mL × 3)
and combined with the first ethyl acetate fraction. The combined ethyl acetate fraction was
dried over Na2SO4 and the solvent was removed in a rotary evaporator. The product was
purified by column chromatography using 15% EtOAc in hexane. The compound diethyl
3-(4-cyanobenzyl)cyclopentane-1,1-dicarboxylate (3.14) was obtained in 78% yield with
the recovery of chiral additives (R)-dimethylmethylsuccinate in 89% yield. After this
reaction, the epimerization of chiral additives was checked using chiral HPLC. At first,
racemic mixture of (R,S)-dimethylmethylsuccinate were separated in HPLC by using
Chiral-Pak-IB in 5% IPA in hexane with the flow of 0.5ml/min at 210 nm wavelength. The
two peaks of (R)- and (S)- configuration were observed at 10.9 and 13.9 min. Then pure
(R)-dimethylmethylsuccinate appeared as a peak at 10.9 and minor at 13.9 min with
90.5:9.5 er. Later, (R)-dimethylmethylsuccinate which is recovered from our reaction was
analyzed through chiral HPLC. It shows two peaks at 10.9 and 13.9 min with 90.5:9.5 er
which is exactly same with the value of (R)-dimethylmethylsuccinate before reaction. For
complete picture of both enantiomer, their % area and retention time see SI.
4.2.3 Mechanistic Study
Scheme 3.7. Selectivity study in Negishi cross-coupling reaction with electronically biased
arylzinc reagents.
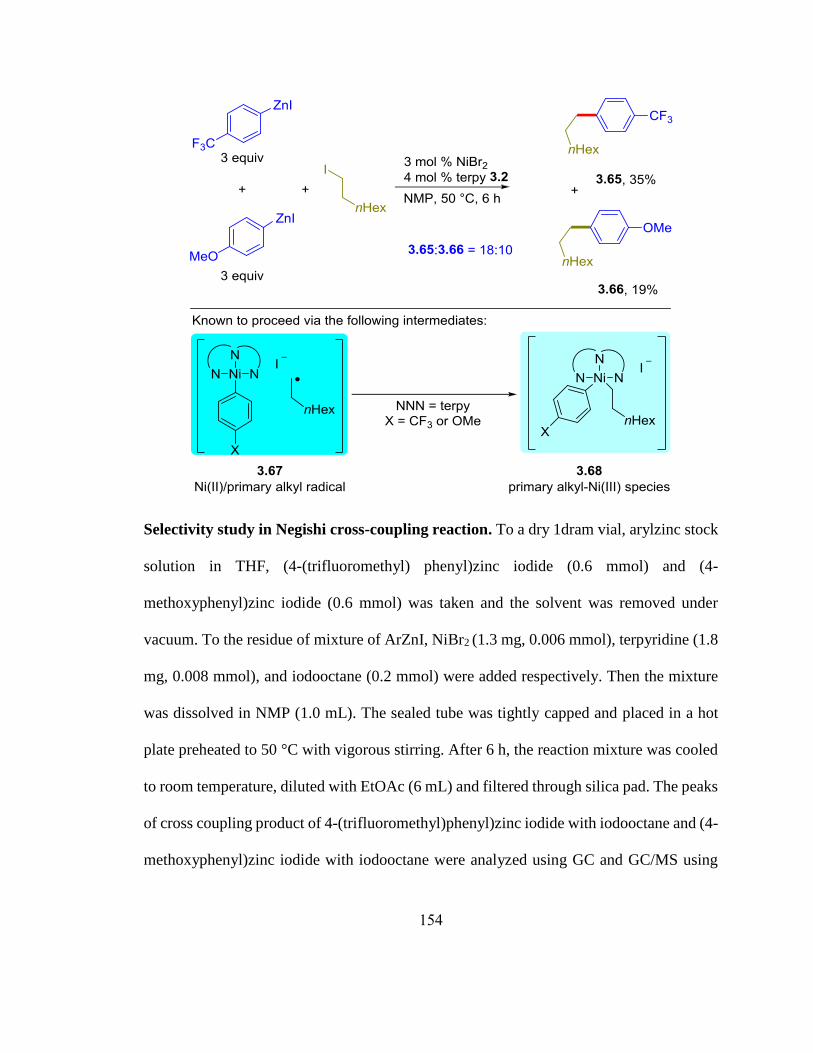
154
Selectivity study in Negishi cross-coupling reaction. To a dry 1dram vial, arylzinc stock
solution in THF, (4-(trifluoromethyl) phenyl)zinc iodide (0.6 mmol) and (4-
methoxyphenyl)zinc iodide (0.6 mmol) was taken and the solvent was removed under
vacuum. To the residue of mixture of ArZnI, NiBr2 (1.3 mg, 0.006 mmol), terpyridine (1.8
mg, 0.008 mmol), and iodooctane (0.2 mmol) were added respectively. Then the mixture
was dissolved in NMP (1.0 mL). The sealed tube was tightly capped and placed in a hot
plate preheated to 50 °C with vigorous stirring. After 6 h, the reaction mixture was cooled
to room temperature, diluted with EtOAc (6 mL) and filtered through silica pad. The peaks
of cross coupling product of 4-(trifluoromethyl)phenyl)zinc iodide with iodooctane and (4-
methoxyphenyl)zinc iodide with iodooctane were analyzed using GC and GC/MS using
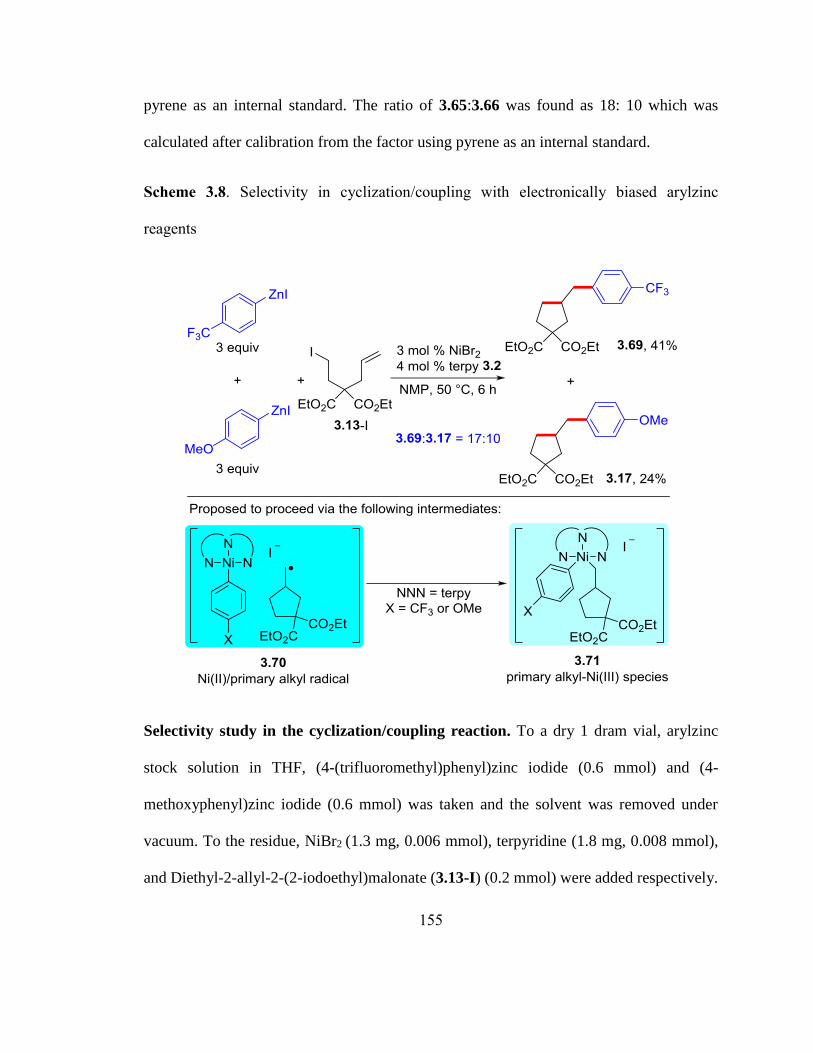
155
pyrene as an internal standard. The ratio of 3.65:3.66 was found as 18: 10 which was
calculated after calibration from the factor using pyrene as an internal standard.
Scheme 3.8. Selectivity in cyclization/coupling with electronically biased arylzinc
reagents
Selectivity study in the cyclization/coupling reaction. To a dry 1 dram vial, arylzinc
stock solution in THF, (4-(trifluoromethyl)phenyl)zinc iodide (0.6 mmol) and (4-
methoxyphenyl)zinc iodide (0.6 mmol) was taken and the solvent was removed under
vacuum. To the residue, NiBr2 (1.3 mg, 0.006 mmol), terpyridine (1.8 mg, 0.008 mmol),
and Diethyl-2-allyl-2-(2-iodoethyl)malonate (3.13-I) (0.2 mmol) were added respectively.

156
The mixture was dissolved in NMP (1.0 mL) as a solvent then sealed tube was tightly
capped and placed in a hot plate preheated to 50 °C with vigorous stirring. After 6 h, the
reaction mixture was cooled to room temperature, diluted with EtOAc (10 mL) and filtered
through silica pad. The clear solution was run in GC using pyrene as an internal standard.
The product peak of cyclization/cross coupled product of 4-(trifluoromethyl)phenyl)zinc
iodide with 3.13-I and (4-methoxyphenyl)zinc iodide with 3.13-I were analyzed by GC
and GC/MS using pyrene as an internal standard. The ratio of 3.69:3.17 was found as 17:
10 which was calculated after calibration of their value from the factor using pyrene as an
internal standard.
Scheme 3.9. Diastereoselectivity studies with cis- and trans-1-(allyloxy)-2-
bromocyclohexane

157
Diastereoselectivity study of trans-1-(allyloxy)-2-bromocyclohexane. The organozinc
prepared for this reaction was according to procedure A. In a sealed tube, phenylzinc iodide
stock solution in THF (0.750 mmol) was taken and the solvent was removed under vacuum.
To the residue of PhZnI was added NiBr2 (3.3 mg, 0.015 mmol), terpyridine (4.7 mg, 0.020
mmol), and trans-1-(allyloxy)-2-bromocyclohexane (3.72) (0.5 mmol) respectively. Then
the mixture was dissolved in NMP (2.5 mL). The sealed tube was tightly capped and placed
in an oil-bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction mixture was
cooled to room temperature, diluted with EtOAc (10 mL) extracted with ethyl acetate. The
combined ethyl acetate fraction was dried over Na2SO4 and the solvent was removed in a
rotary evaporator. The crude reaction solution was analyzed in GC and GC/MS and their
diastereomeric ratio was calculated. The diastereomeric ratio of the product from this
reaction was found to be 1.3:1. The product (3R,3aS,7aS)-3-benzyloctahydrobenzofuran
(3.73) was purified by column chromatography in 63% yield using 5% EtOAc in hexane.
1H NMR (300 MHz, CDCl3): 1.06-1.39 (m, 3H), 1.44-1.62 (m, 4H), 1.71-1.87 (m, 2H),
1.95-2.05 (m, 1x0.43H), 2.23-2.34 (m, 1x0.57H), 2.55-2.63 (m, 1H), 2.69-2.80 (m, 1H),
3.52 (dd, J = 6.0, 3.0 Hz, 1x0.57H), 3.65 (t, J = 8.5 Hz, 1x0.43H), 3.86-4.10 (m, 2H), 7.14-
7.30 (m, 5H); 13C NMR (75 MHz, CDCl3): 20.5, 21.2, 22.3, 23.7, 24.6, 27.6, 28.5, 28.7,
33.7, 39.9, 40.1, 43.1, 45.6, 45.8, 71.2, 72.0, 76.3, 78.4, 126.0, 126.1, 128.5, 128.8, 140.7,
141.0; IR (neat): 2926, 1452, 1022, 749, 698; HRMS (CI): (M)+ Calcd for C15H20O
216.1514; found 216.1503.
Diastereoselectivity study of cis-1-(allyloxy)-2-bromocyclohexane. In a sealed tube,
phenylzinc iodide stock solution in THF (0.750 mmol) was taken and the solvent was

158
removed under vacuum. To the residue of PhZnI was added NiBr2 (3.3 mg, 0.015 mmol),
terpyridine (4.7 mg, 0.020 mmol), and cis-1-(allyloxy)-2-bromocyclohexane (3.72) (0.5
mmol) respectively. Then the mixture was dissolved in NMP (2.5 mL). The sealed tube
was tightly capped and placed in an oil-bath preheated to 50 °C with vigorous stirring.
After 6 h, the reaction mixture was cooled to room temperature, diluted with EtOAc (10
mL) extracted with ethyl acetate. The combined ethyl acetate fraction was dried over
Na2SO4 and the solvent was removed in a rotary evaporator. The clear solution was
analyzed in GCand GC/MS and their diastereomeric ratio was calculated. The
diastereomeric ratio of the product from this reaction was found to be 1.3:1. The product
(3R,3aS,7aS)-3-benzyloctahydrobenzofuran (3.73) was purified by column
chromatography in 74% yield using 5% EtOAc in hexane.
Scheme 3.10. Diastereoselectivity in the known radical cyclization and the current
cyclization/coupling reactions
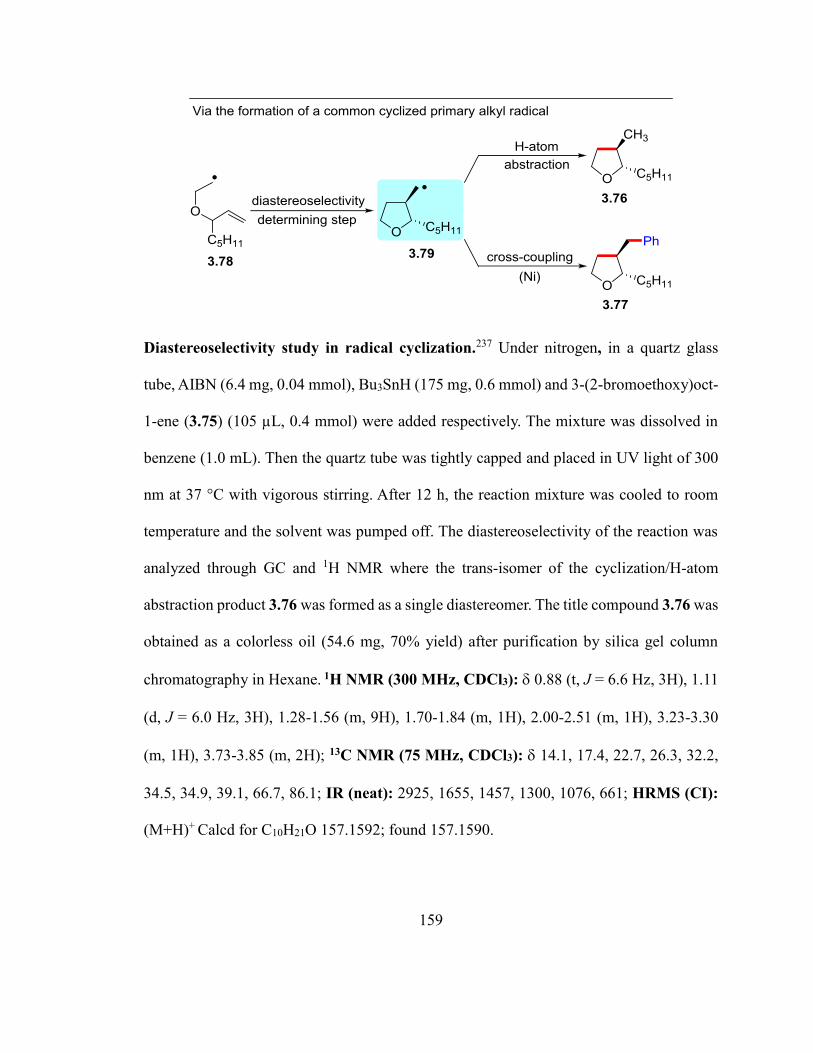
159
Diastereoselectivity study in radical cyclization.237 Under nitrogen, in a quartz glass
tube, AIBN (6.4 mg, 0.04 mmol), Bu3SnH (175 mg, 0.6 mmol) and 3-(2-bromoethoxy)oct-
1-ene (3.75) (105 µL, 0.4 mmol) were added respectively. The mixture was dissolved in
benzene (1.0 mL). Then the quartz tube was tightly capped and placed in UV light of 300
nm at 37 °C with vigorous stirring. After 12 h, the reaction mixture was cooled to room
temperature and the solvent was pumped off. The diastereoselectivity of the reaction was
analyzed through GC and 1H NMR where the trans-isomer of the cyclization/H-atom
abstraction product 3.76 was formed as a single diastereomer. The title compound 3.76 was
obtained as a colorless oil (54.6 mg, 70% yield) after purification by silica gel column
chromatography in Hexane. 1H NMR (300 MHz, CDCl3): 0.88 (t, J = 6.6 Hz, 3H), 1.11
(d, J = 6.0 Hz, 3H), 1.28-1.56 (m, 9H), 1.70-1.84 (m, 1H), 2.00-2.51 (m, 1H), 3.23-3.30
(m, 1H), 3.73-3.85 (m, 2H); 13C NMR (75 MHz, CDCl3): 14.1, 17.4, 22.7, 26.3, 32.2,
34.5, 34.9, 39.1, 66.7, 86.1; IR (neat): 2925, 1655, 1457, 1300, 1076, 661; HRMS (CI):
(M+H)+ Calcd for C10H21O 157.1592; found 157.1590.

160
Diastereoselectivity study in cyclization/coupling reaction. The organozinc prepared for
this reaction was according to procedure A. To a sealed tube, phenylzinc iodide stock
solution in THF (0.750 mmol) was taken and the solvent was removed under vacuum. To
the residue of PhZnI, NiBr2 (3.3 mg, 0.015 mmol), terpyridine (4.7 mg, 0.02 mmol), and
3-(2-bromoethoxy)oct-1-ene (3.75) (0.5 mmol) were added respectively. Then the mixture
was dissolved in NMP (2.5 mL). The sealed tube was tightly capped and placed in an oil-
bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction mixture was cooled
to room temperature and extracted with ethyl acetate (15 mL) and H2O (10 mL). The
combined ethyl acetate fraction was dried over Na2SO4 and the solvent was removed in a
rotary evaporator. The diastereomer was analyzed through 1H NMR and GC. The
diastereoselectivity of the reaction was analyzed through GC and 1H NMR where the trans-
isomer of the cyclization/coupling product 3.77 was formed as a single diastereomer. The
title compound 3.77 was obtained as a colorless oil (67.2 mg, 58% yield) after purification
by silica gel column chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3):
0.87 (t, J = 7.5 Hz, 3H), 1.25-1.44 (m, 8H), 1.57-1.68 (m, 1H), 1.89-2.11 (m, 2H), 2.54
(dd, J = 12.0, 9.0 Hz, 1H), 2.77 (dd, J = 12.0 Hz, J = 9.0 Hz, 1H), 3.49-3.55 (m, 1H), 3.80
(t, J = 6.0 Hz, 2H), 7.15-7.31 (m, 5H); 13C NMR (75 MHz, CDCl3): 14.1, 22.7, 26.1,
32.0, 32.6, 34.8, 39.5, 46.32, 66.8, 84.3, 126.1, 128.4, 128.9, 140.8; IR (neat): 2926, 1453,
1079, 754, 689; HRMS (ESI-TOF): Calcd for C16H24ONa (M+Na)+ 255.1725; found
255.1720.

161
4.2.4 Characterization data for new compounds
Ethyl 2-acetyl-2-(2-bromoethyl)pent-4-enoate. The title compound was prepared
according to the modified procedure described in the given literature248. To a dry flask,
NaH (864 mg, 36 mmol) was weigh out and dry THF (30 mL) was added. The flask was
stirred and cooled to 0 °C. To the stirring suspension, ethyl acetoacetate (30 mmol) was
added dropwise for 5 minutes. The resulting mixture was stirred at room temperature for
an hour. Later, the reaction mixture was again cooled down to 0 °C and allylbromide (2.83
mL, 33 mmol) was added dropwise in the mixture. After the complete addition of
allylbromide, the reaction mixture was stirred at room temperature for 12 h. It was diluted
with EtOAc (30 mL) and washed with H2O (15 mL). The ethyl acetate fraction was dried
over Na2SO4 and the solvent was removed in a rotary evaporator. The product was purified
by silica gel column chromatography using ethyl acetate/hexane as eluent and colorless oil
of ethyl 2-acetyl-2-(2-bromoethyl)pent-4-enoate was obtained in 90% yield.
To a dry flask, NaH (720 mg, 30 mmol) was weigh out and dry DMF (20 mL) was added.
The flask was stirred and cooled to 0 °C. To the stirring suspension, ethyl 2-acetylpent-4-
enoate (20 mmol) was added dropwise for 5 minutes. The resulting mixture was stirred at
room temperature for an hour. Later, the reaction mixture was again cooled down to 0 °C
and dibromoethane ( 3.5 ml, 40 mmol) was added dropwise in the mixture. After the

162
complete addition of dibromoethane, the reaction mixture was stirred at room temperature
for 12 h. It was then diluted with EtOAc (20 mL) and washed with H2O (10 mL). The ethyl
acetate fraction was dried over Na2SO4 and the solvent was removed in a rotary evaporator.
The title compound was obtained as a colorless oil (3.03 ml, 55% yield) after purification
by silica gel column chromatography (Hex : Et2O = 8:1). 1H NMR (300 MHz, CDCl3):
1.28 (t, J = 7.5 Hz, 3H), 2.16 (s, 3H), 2.30-2.40 (m, 1H), 2.45-2.56 (m, 1H), 2.64 (d, J =
9.0 Hz, 2H), 3.20-3.34 (m, 2H), 4.23 (q, J = 7.0 Hz, 2H), 5.07-5.17 (m, 2H), 5.51-5.64 (m,
1H); 13C NMR (75 MHz, CDCl3): 14.1, 26.9 27.0, 35.3, 36.6, 61.9, 63.4, 119.8, 131.6,
171.0, 203.4.
Diethyl 3-(4-cyanobenzyl)cyclopentane-1,1-dicarboxylate (3.14):106 This reaction was
performed by using organozinc prepared according to procedure A in our standard
condition. The title compound 3.14 was obtained as a yellow oil (133.2 mg, 81% yield)
after purification by silica gel column chromatography (Hex: EtOAc = 19:1). 1H NMR
(300 MHz, CDCl3): 1.22 (q, J = 7.9 Hz, 6H), 1.31-1.41 (m, 1H), 1.76-1.84 (m, 2H),
2.11-2.39 (m, 4H), 2.64-2.76 (m, 2H), 4.11-4.21 (m, 4H), 7.25 (d, J = 9.0 Hz, 2H), 7.55 (d,
J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 14.1, 32.0, 33.7, 40.2, 41.2, 41.4, 59.9,
61.5, 109.9, 119.1, 129.5, 132.2, 146.9, 172.5, 172.6; IR (neat): 2980, 2937, 2227, 1723,

163
1607,1249, 1156, 1024; HRMS (ESI-TOF) m/z: (M+H)+ Calcd for C19H24NO4 330.1705;
found 330.1694.
Diethyl 3-benzylcyclopentane-1,1-dicarboxylate (3.15):249 This reaction was performed by
using organozinc prepared according to procedure A in our standard condition. The title
compound 3.15 was obtained as a colorless oil (127.6 mg, 84% yield) after purification by
silica gel column chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3):
1.23 (q, J = 7.9 Hz, 6H), 1.34-1.44 (m, 1H), 1.80-1.87 (m, 2H), 2.04-2.44 (m, 4H), 2.58-
2.72 (m, 2H), 4.12-4.22 (m, 4H), 7.14-7.29 (m, 5H); 13C NMR (75 MHz, CDCl3): 14.1,
32.0, 33.8, 40.4, 41.3, 41.6, 60.0, 61.3, 61.4, 125.9, 128.3, 128.8, 141.3, 172.7, 172.8; IR
(neat): 2980, 1725, 1453, 1247, 1155, 1096 ; HRMS (ESI-TOF) m/z: (M+H)+ Calcd for
C18H25O4 305.1753; found 305.1737.
Diethyl 3-(4-methylbenzyl)cyclopentane-1,1-dicarboxylate (3.16). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.16 was
obtained as a colorless oil (119.2 mg, 75% yield) after purification by silica gel column
chromatography (Hex: EtOAc = 19:1) 1H NMR (300 MHz, CDCl3): 1.23 (q, J = 6.9

164
Hz, 6H), 1.29-1.43 (m, 1H), 1.76-1.86 (m, 2H), 2.08-2.43 (m, 4H), 2.31 (s, 3H), 2.54-2.68
(m, 2H), 4.12-4.22 (m, 4H), 7.03-7.12 (m, 4H); 13C NMR (75 MHz, CDCl3): 14.1, 21.1,
32.0, 33.8, 40.4, 40.9, 41.7, 60.0, 61.3, 128.7, 129.0, 135.3, 138.2, 172.7, 172.8; IR (neat):
2980, 1725, 1445, 1366, 1247, 1096; HRMS (ESI-TOF) m/z: (M+H)+ Calcd for C19H27O4
319.1909; found 319.1910.
Diethyl 3-(4-methoxybenzyl)cyclopentane-1,1-dicarboxylate (3.17). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.17 was
obtained as a colorless oil (116.9 mg, 70% yield) after purification by silica gel column
chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3): 1.23 (q, J = 6.9
Hz, 6H), 1.28-1.45 (m, 1H), 1.77-1.84 (m, 2H), 2.08-2.42 (m, 4H), 2.52-2.65 (m, 2H), 3.78
(s, 3H), 4.12-4.21 (m, 4H), 6.81 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H); 13C NMR
(75 MHz, CDCl3): 14.1, 31.9, 33.7, 40.3, 41.8, 55.2, 60.0, 61.3, 113.7, 129.6, 133.3,
157.8, 172.7, 172.8; IR (neat): 2980, 1724, 1611, 1443, 1242, 1095; HRMS (ESI-TOF)
m/z: (M+H)+ Calcd for C19H27O5 335.1858; found 335.1858.

165
4-(cyclopentylmethyl)benzonitrile (3.18).106 The organozinc prepared for this reaction was
according to procedure A. The title compound 3.18 was obtained as a colorless oil (47 mg,
51% yield) after purification by silica gel column chromatography (Hex: EtOAc = 19:1).
1H NMR (300 MHz, CDCl3: 1.11-1.27 (m, 2H), 1.49-1.72 (m, 6H), 2.07 (app. septet, J
= 7.5 Hz, 1H), 2.65 (d, J = 9.0 Hz, 2H), 7.26 (d, J = 9.0 Hz, 2H), 7.55 (d, J = 9.0 Hz, 2H);
13C NMR (75 MHz, CDCl3): 25.0, 32.5, 41.7, 42.3, 109.6, 119.3, 129.6, 132.1, 148.1 ;
IR (neat): 2948, 2865, 2226, 1606, 1506.
Methyl 4-((1-phenylpyrrolidin-3-yl)methyl)benzoate (3.19). The organozinc prepared for
this reaction was according to procedure A. The reaction was done at room temperature for
12 h using standard condition. The title compound 3.19 was obtained as a white solid (79.6
mg, 54% yield) after purification by silica gel column chromatography (Hex: EtOAc =
19:1). 1H NMR (300 MHz, CDCl3): 1.69-1.82 (m, 1H), 2.06-2.16 (m, 1H), 2.62 (app.
septet, J = 7.5 Hz, 1H), 2.82 (d, J = 9 Hz, 2H), 3.02 (t, J = 9 Hz, 1H), 3.26-3.45 (m, 3H),
3.93 (s, 3H), 6.54 (d, J = 9 Hz, 2H), 6.68 (t, J = 9.0 Hz, 1H), 7.21-7.31 (m, 4H), 8.01 (d, J
= 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 31.4, 40.0, 40.2, 47.2, 52.1, 53.0, 111.5,
115.6, 128.3, 128.8, 129.2, 129.9, 146.2, 147.8, 167.1; IR (neat): 2959, 2822, 1713, 1599,
1505, 1308, 1276, 1107; HRMS (ESI-TOF) m/z: (M+H)+ Calcd for C19H22NO2 296.1651;
found 296.1648.

166
3-Benzyl-1-tosylpyrrolidine (3.20). The reaction was conducted using one equiv
diphenylzinc (115.5 mg, 67% yield) instead of PhZnI (88.2mg, 56%) in our standard
condition for 8 h. The title compound 3.20 was obtained as a colorless oil after purification
by silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3):
1.40-1.55 (m, 1H), 1.82-1.92 (m, 1H), 2.32 (app. septet, J = 7.5 Hz, 1H), 2.25-2.40 (m,
1H), 2.44 (s, 3H), 2.54 (d, J = 9 Hz, 2H), 2.92 (t, J = 9 Hz, 1H), 3.19 (q, J = 7.9 Hz 1H),
3.31-3.42 (m, 2H), 7.05 (d, J = 6 Hz, 2H), ), 7.16-7.33 (m, 5H), 7.69 (d, J = 9 Hz, 2H) ;
13C NMR (75 MHz, CDCl3): 21.6, 31.1, 39.1, 40.4, 47.4, 52.8, 126.3, 127.5, 128.5,
128.6, 129.7, 133.9, 139.8, 143.4; IR (neat): 2949, 1733, 1339, 1157, 1027, 1014; HRMS
(ESI-TOF) m/z: (M+H)+ Calcd for C18H22NO2S 316.1371; found 316.1363.
3-(4-Fluorobenzyl)-1-tosylpyrrolidine (3.21). The organozinc prepared for this reaction
was according to procedure A. This reaction was completed in 8 h using our standard
condition. The title compound 3.21 was obtained as a colorless oil (104.8 mg, 63% yield)
after purification by silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300
MHz, CDCl3): 1.40-1.53 (m, 1H), 1.80-1.91 (m, 1H), 2.28 (app. septet, J = 6.9 Hz, 1H),
2.43 (s, 3H), 2.52 (d, J = 9 Hz, 2H), 2.89 (q, J = 6 Hz, 1H), 3.14-3.22 (m, 1H), 3.28-3.42

167
(m, 2H), 6.90-7.03 (m, 4H), ), 7.32 (d, J = 9 Hz, 2H), 7.68 (d, J = 9 Hz, 2H); 13C NMR
(75 MHz, CDCl3): 21.6, 31.0, 38.3, 40.5, 47.4, 52.7, 115.3 (d, JCF = 21.0 Hz), 127.6,
129.7, 130.0 (d, JCF = 8.2 Hz), 133.9, 135.4 (d, JCF = 3.0 Hz), 143.4, 161.4 (d, JCF = 243
Hz); 19F NMR (282 MHz, CDCl3) -115.2 IR (neat): 1598, 1508, 1448, 1155, 1090,
1014 ; HRMS (CI) m/z: (M+H)+ Calcd for C18H21FNO2S 334.1277; found 334.1284.
3-(3,4-Dichlorobenzyl)tetrahydrofuran (3.22). The organozinc prepared for this reaction
was according to procedure A. The title compound 3.22 was obtained as a colorless oil
(85.1 mg, 74% yield) after purification by silica gel column chromatography (Hex: EtOAc
= 9:1). 1H NMR (300 MHz, CDCl3): 1.51-1.63 (m, 1H), 1.93-2.04 (m, 1H), 2.46 (app.
septet, J = 7.5 Hz, 1H), 2.57-2.70 (m, 2H), 3.42 (t, J = 7.5 Hz, 1H), 3.71-3.93 (m, 3H),
6.99 (d, J = 6 Hz, 1H), 7.26 (s, 1H), 7.34 (d, J = 6 Hz, 1H); 13C NMR (75 MHz, CDCl3):
32.0, 38.5, 40.7, 67.9, 72.8, 128.2, 130.1, 130.4, 130.6, 132.4, 141.1; IR (neat): 2966,
2856, 1470, 1372, 1240, 1130, 1044, 1029; HRMS (CI) m/z: (M)+ Calcd for C11H12Cl2O
230.0265; found 230.0273.

168
3-(3,5-Bis(trifluoromethyl)benzyl)tetrahydrofuran (3.23). The organozinc prepared for this
reaction was according to procedure A. The title compound 3.23 was obtained as a
colorless oil (105.7 mg, 71% yield) after purification by silica gel column chromatography
(Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3): 1.56-1.67 (m, 1H), 1.97-2.08 (m,
1H), 2.55 (app. septet, J = 7.5 Hz, 1H), 2.76-2.90 (m, 2H), 3.47 (t, J = 7.5 Hz, 1H), 3.74-
3.97 (m, 3H), 7.63 (s, 2H), ), 7.73 (s, 1H); 13C NMR (75 MHz, CDCl3): 32.0, 39.0, 40.6,
67.9, 72.7, 120.4, 123.4 (q, JCF = 270.9 Hz), 128.9, 131.9 (q, JCF = 32.7 Hz), 143.3 ; 19F
NMR (282 MHz, CDCl3) -61.5 IR (neat): 2936, 2864, 1622, 1456, 1378, 1274, 1166,
1002; HRMS (CI) m/z: (M+H)+ Calcd for C13H13F6O 299.0871; found 299.0864.
4-((Tetrahydrofuran-3-yl)methyl)benzonitrile (3.24). The organozinc prepared for this
reaction was according to procedure A. The title compound 3.24 was obtained as a
colorless oil (60.7 mg, 65% yield) after purification by silica gel column chromatography
(Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3): 1.53-1.65 (m, 1H), 1.94-2.05 (m,
1H), 2.51 (app. septet, J = 7.5 Hz, 1H), 2.68-2.81 (m, 2H), 3.44 (t, J = 7.5 Hz, 1H), 3.72-
3.94 (m, 3H), 7.28 (d, J = 6.0 Hz, 2H), 7.58 (d, J = 6 Hz, 2H); 13C NMR (75 MHz,
CDCl3): 32.0, 39.4, 40.5, 67.8, 72.7, 110.1, 119.0, 129.5, 132.3, 146.4 ; IR (neat): 2965,
2856, 2226, 1606, 1506, 1415, 1177, 1042; HRMS (ESI-TOF) m/z: (M+H)+ Calcd for
C12H14NO 188.1075; found 188.1081.

169
1-(4-((tetrahydrofuran-3-yl)methyl)phenyl)ethan-1-one (3.25). The organozinc prepared
for this reaction was according to procedure C. The title compound 3.25 was obtained as a
colorless oil (67.3mg, 66% yield) after purification by silica gel column chromatography
(Hex: EtOAc = 4:1). 1H NMR (300 MHz, CDCl3): 1.55-1.67 (m, 1H), 1.94-2.05 (m,
1H), 2.48-2.69 (m, 4H), 2.68-2.75 (d, J = 6.0 Hz, 2H), 3.45 (t, J = 7.5 Hz, 1H), 3.74-3.94
(m, 3H), 7.26 (d, J = 6.0 Hz, 2H), 7.89 (d, J = 6 Hz, 2H); 13C NMR (75 MHz, CDCl3):
26.6, 32.2, 39.4, 40.7, 67.9, 72.9,128.7, 129.0, 135.4, 146.6, 197.8 ; IR (neat): 2855,
1677, 1605, 1413, 1265, 956; HRMS (APCI) m/z: (M+H)+ Calcd for C13H17O2 205.1229;
found 205.1217.
3-(4-bromobenzyl)tetrahydrofuran (3.26). The organozinc prepared for this reaction was
according to procedure A. The title compound 3.26 was obtained as a colorless oil (84.0
mg, 70% yield) after purification by silica gel column chromatography (Hex: EtOAc =
19:1). 1H NMR (300 MHz, CDCl3): 1.53-1.65 (m, 1H), 1.92-2.04 (m, 1H), 2.47 (app.
septet, J = 7.0 Hz, 1H), 2.61-2.70 (m, 2H), 3.43 (t, J = 7.5 Hz, 1H), 3.71-3.93 (m, 3H),
7.04 (d, J = 9.0 Hz, 2H), 7.41 (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 32.1,
38.8, 40.9, 67.9, 72.9, 119.9, 130.5, 131.6, 139.8 ; IR (neat): 2965, 2856, 2226, 1606,

170
1506, 1415, 1177, 1042; HRMS (CI) m/z: (M)+ Calcd for C11H13BrO 240.0150; found
240.0152.
(±)-(3R,3aS,6aR)-3-(4-Methoxybenzyl)hexahydrofuro[2,3-b]furan (3.27).250 The
organozinc prepared for this reaction was according to procedure A. The title compound
3.27 was obtained as a colorless oil (90.0 mg, 77% yield; 8:1 dr) after purification by silica
gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3): 1.77-
2.03 (m, 2H), 2.55-2.79 (m, 4H), 3.52 (t, J = 10.5 Hz, 1H), 3.76 (s, 3H), 3.81-3.97 (m, 3H),
5.69 (d, J = 6 Hz, 1H), 6.81 (d, J=9.0 Hz, 1H), 7.07 (d, J=6.0 Hz, 1H); 13C NMR (75 MHz,
CDCl3): 25.0, 32.8, 43.9, 45.4, 55.2, 69.0, 72.1, 109.8, 113.9, 129.1, 131.9, 158.0; IR
(neat): 2952, 1735, 1611, 1511, 1372, 1242, 1178; HRMS (ESI-TOF) m/z: (M+H)+ Calcd
for C14H19O3 235.1334; found 235.1330. The actual dr (8:1) of this compound was
calculated using crude 1H NMR by integrating peaks at 5.69 ppm (major isomer) and
5.72 ppm (minor isomer). The characterization data given above correspond to the major
isomer isolated by column chromatography.

171
(±)-(3R,3aS,7aS)-3-(2-Methoxybenzyl)octahydrobenzofuran (3.28). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.28 was
obtained as a colorless oil (78.7 mg, 64% yield; 1.7:1 dr) after purification by silica gel
column chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3): 1.14-1.40
(m, 3H), 1.44-1.64 (m, 4H), 1.71-1.89 (m, 2H), 1.96-2.00 (m, 1x0.37H), 2.26-2.33 (m,
1x0.63H), 2.53-2.62 (m, 1H), 2.67-2.78 (m, 1H), 3.52 (dd, J = 9.0, 6.0 Hz, 1x0.63H), 3.64
(t, J = 7.5 Hz, 1x0.37H), 3.80 (s, 3H), 3.88-4.11 (m, 2H), 6.73-6.79 (m, 3H), 7.17-7.23 (m,
1H); 13C NMR (75 MHz, CDCl3): 20.5, 21.2, 22.2, 23.7, 24.6, 27.5, 28.4, 28.7, 33.7,
39.9, 40.1, 43.1, 45.4, 45.7, 55.2, 70.9, 72.0, 76.2, 78.4, 111.1, 111.2, 114.4, 114.7, 120.9,
121.2, 129.4, 142.3, 142.7, 159.7; IR (neat): 2930, 1737, 1584, 1239, 1152, 1043; HRMS
(ESI-TOF) m/z: (M+H)+ Calcd for C16H23O2 247.1698; found 247.1691. The actual dr
(1.7:1) was calculated using GC. See SI for GC trace.
(±)-(3R,3aS,6aR)-3-(2-Methylbenzyl)hexahydrofuro[2,3-b]furan (3.29). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.29 was
obtained as a colorless oil (65.4 mg, 60% yield; 5:1 dr) after purification by silica gel
column chromatography (Hex: EtOAc = 9:1;). 1H NMR (300 MHz, CDCl3): 1.87-2.08
(m, 2H), 2.32 (s, 3H), 2.62-2.86 (m, 4H), 3.56-3.64 (m, 1H), 3.83-4.00 (m, 3H), 5.73 (d, J
= 6 Hz, 1H), 7.10-7-16 (m, 4H); 13C NMR (75 MHz, CDCl3): 19.5, 25.3, 31.0, 42.3,

172
45.8, 69.2, 72.2, 109.8, 126.1, 126.4, 128.6, 130.5, 135.8, 138.2 ; IR (neat): 2949, 2867,
1603, 1490, 1371, 1180, 1071; HRMS (ESI-TOF) m/z: (M+H)+ Calcd for C14H19O2
219.1385; found 219.1377. The actual dr (5:1) of this compound was calculated using
crude 1H NMR by integrating peaks at 5.73 ppm (major isomer) and 5.74 ppm (minor
isomer). The characterization data given above correspond to the major isomer isolated by
column chromatography.
(±)-(3R,3aS,7aR)-3-(4-Methylbenzyl)hexahydro-4H-furo[2,3-b]pyran (3.30). The
organozinc prepared for this reaction was according to procedure A. The title compound
3.30 was obtained as colorless oil (71.9 mg, 62% yield; 5:1 dr) after purification by silica
gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3): 1.49-
1.63 (m, 3H), 1.72-1.79 (m, 1H), 1.91-1.99 (m, 1H), 2.32(s, 3H), 2.53-2.74 (m, 3H), 3.63-
3.67 (m, 1H), 3.74-3.79 (m, 2H), 3.88 (t, J = 7.5 Hz, 1H), 5.27 (d, J = 3.0 Hz, 1H), 7.04-
7.11 (m, 4H); 13C NMR (75 MHz, CDCl3): 19.5, 21.0, 23.2, 32.9, 36.5, 42.6, 61.0, 69.9,
102.0, 128.2, 129.2, 135.6, 137.0; IR (neat): 2939, 1736, 1515, 1144, 1109, 1018; HRMS
(ESI-TOF) m/z: (M+H)+ Calcd for C15H21O2 233.1542; found 233.1537. The actual dr
(5:1) of this compound was calculated using crude 1H NMR by integrating peaks at 5.27
ppm (major isomer) and 5.01 ppm (minor isomer). The characterization data given above
correspond to the major isomer isolated by column chromatography.

173
(±)-(3R,3aS,7aS)-3-(4-Methylbenzyl)octahydrobenzofuran (3.31). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.31 was
obtained as a colorless oil (79.3 mg, 69% yield; 1.7:1 dr) after purification by silica gel
column chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3): 1.18-1.42
(m, 3H), 1.47-1.65 (m, 4H), 1.73-1.89 (m, 2H), 1.98-2.02 (m, 1x0.37H), 2.24-2.31 (m,
1x0.63H), 2.34 (s, 3H), 2.54-2.63 (m, 1H), 2.67-2.79 (m, 1H), 3.54 (dd, J = 9.0, 6.0 Hz,
1x0.63H), 3.67 (t, J = 7.5 Hz, 1x0.37H), 3.89-4.12 (m, 2H), 7.06-7.13 (m, 4H); 13C NMR
(75 MHz, CDCl3): 20.5, 21.0, 21.2, 22.2, 23.7, 24.5, 27.5, 28.4, 28.6, 33.1, 39.4, 40.0,
43.0, 45.6, 45.8, 70.9, 72.0, 76.2, 78.4, 128.2, 128.6, 129.1, 135.4, 137.5, 137.9; IR (neat):
2926, 2853, 1514, 1446, 1061, 1022; HRMS (CI) m/z: (M)+ Calcd for C16H22O 230.1671;
found 230.1669. The actual dr (1.7:1) was calculated using GC. See SI for GC trace.
(±)-(2S,4R)-2-Butoxy-4-(2,4-dimethoxybenzyl)tetrahydrofuran (3.32). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.32 was
obtained as a colorless oil (98.4 mg, 67% yield; 1.4:1 dr) after purification by silica gel
column chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3): 0.88-0.96

174
(m, 3H), 1.30-1.46 (m, 2H), 1.50-1.65 (m, 3H), 1.92-1.98 (m, 1x0.42H), 2.10-2.20 (m,
1x0.58H), 2.45-2.55 (m, 1x0.58H), 2.59 (d, J = 6.0 Hz, 1x0.42H), 2.69 (d, J = 9.0 Hz, 2H),
3.34-3.42 (m, 1H), 3.54-3.73 (m, 2H), 3.79 (s, 6H), 3.83-3.96 (m, 1H), 5.08-5.12 (m, 1H),
6.38-6.43 (m, 2H), 7.00 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 14.0, 19.5,
32.0, 33.2, 33.7, 37.5, 38.6, 38.9, 39.2, 55.2, 55.4, 67.2, 67.5, 72.0, 98.5, 103.7, 104.3,
104.7, 121.7, 130.5, 158.4, 159.4; IR (neat): 2934, 1738, 1612, 1506, 1456, 1286, 1207,
1155, 1063; HRMS (CI) m/z: (M)+ Calcd for C17H26O4 294.1831 found 294.1832. The
actual dr (1.4:1) was calculated using GC. See SI for GC trace.
(±)-(2S,4R)-4-Benzyl-2-phenyltetrahydrofuran (3.33). The organozinc prepared for this
reaction was according to procedure A. The title compound 3.33 was obtained as a
colorless oil (94.0 mg, 79% yield; 6.5:1 dr) after purification by silica gel column
chromatography (Hex: EtOAc = 19:1).1H NMR (300 MHz, CDCl3): 1.97-2.07 (m, 1H),
2.11-2.20 (m, 1H), 2.45 (app. septet, J = 7.1 Hz, 1x0.13H), 2.71 (app. septet, J = 7.5 Hz,
1x0.87H), 2.79 (d, J = 9.0 Hz, 2H), 3.70 (t, J = 7.5 Hz, 1x0.87H), 3.82 (t, J = 7.5 Hz,
1x0.13H), 4.09 (t, J = 7.5 Hz, 1x0.13H), 4.20 (t, J = 7.5 Hz, 1x0.87H), 4.93 (t, J = 7.5 Hz,
1x0.13H), 5.12 (t, J = 7.5 Hz, 1x0.87H), 7.20-7.39 (m, 10H); 13C NMR (75 MHz, CDCl3):
39.2, 39.5, 40.6, 40.7, 41.9, 42.3, 73.7, 73.8, 80.1, 81.4, 125.5, 125.6, 126.2, 127.1,
128.3, 128.4, 128.5, 128.7, 140.6, 143.8; IR (neat): 3061, 2933, 1602, 1494, 1067, 1028;

175
HRMS (CI) m/z: (M)+ Calcd for C17H18O 238.1358; found 238.1357. The actual dr (6.5:1)
was calculated using GC. See SI for GC trace.
(±)-(2S,4R)-2-Phenyl-4-(4-(trifluoromethyl)benzyl)tetrahydrofuran (3.34). The
organozinc prepared for this reaction was according to procedure A. The title compound
3.34 was obtained as colorless oil (114.7 mg, 75% yield; 10:1 dr) after purification by silica
gel column chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3): 1.95-
2.05 (m, 1H), 2.08-2.16 (m, 1H), 2.42 (app. septet, J = 6.75 Hz, 1x0.10H), 2.66 (app. septet,
J = 6.90 Hz, 1x0.90H), 2.83 (d, J = 9.0 Hz, 2H), 3.65 (t, J = 7.5 Hz, 1x0.90H), 3.79 (t, J =
7.5 Hz, 1x0.10H), 4.06 (t, J = 7.5 Hz, 1x0.10 H), 4.18 (t, J = 7.5 Hz, 1x0.90H), 4.92 (t, J =
7.5 Hz, 1x0.10H), 5.10 (t, J = 7.5 Hz, 1x0.90H), 7.24-7.36 (m, 7H), 7.55 (d, J = 9.0 Hz,
2H),; 13C NMR (75 MHz, CDCl3): 39.0, 39.4, 40.5, 40.6, 41.7, 42.0, 73.5, 73.6, 80.1,
81.4, 124.4 (q, JCF = 159.0 Hz), 125.5 (q, , JCF = 12.6 Hz ), 127.3, 127.4, 128.4 (q, JCF = 5.3
Hz ), 128.9, 129.1, 143.0, 143.6, 144.7; 19F NMR (282 MHz, CDCl3): -61.1; IR (neat):
2973, 2867, 1614, 1321, 1151, 1108, 1062; HRMS (CI) m/z: (M-H)+ Calcd for C18H16F3O
305.1153; found 305.1145. The actual dr (10:1) of this compound was calculated using
crude 1H NMR by integrating peaks at 5.10 ppm (major isomer) and 4.92 ppm (minor
isomer).

176
(±)-(3R,3aS,6aR)-3-(4-(Trifluoromethyl)benzyl)hexahydrofuro[2,3-b]furan (3.35). The
organozinc prepared for this reaction was according to procedure A. The title compound
3.35 was obtained as a colorless oil (93.8 mg, 69% yield; 10:1 dr) after purification by
silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3):
1.79-2.03 (m, 2H), 2.60-2.84 (m, 4H), 3.55 (t, J = 9.0 Hz, 1H), 3.82-3.99 (m, 3H), 5.70 (d,
J = 6.0 Hz, 1H), 7.28 (d, J = 6.0 Hz, 2H), 7.54 (d, J = 6.0 Hz, 2H); 13C NMR (75 MHz,
CDCl3): 25.1, 33.6, 43.5, 45.4, 69.0, 71.9, 109.7, 124.2 (q, JCF= 270.4 Hz), 125.5(q,
JCF= 3.9 Hz), 128.0, 128.6 (q, JCF= 10.9 Hz), 144.1; 19F NMR (282 MHz, CDCl3) -60.8
IR (neat): 2951, 2871, 1321, 1160, 1108, 1065, 997; HRMS (ESI-TOF): (M+H)+ Calcd
for C14H16F3O2 273.1102; found 273.1093. The actual dr (10:1) of this compound was
calculated using crude 1H NMR by integrating peaks at 5.70 ppm (major isomer) and
5.74 ppm (minor isomer). The characterization data given above correspond to the major
isomer isolated by column chromatography.
(±)-4-(((3R,3aS,7aR)-Hexahydro-4H-furo[2,3-b]pyran-3-yl)methyl)benzonitrile (3.36).
The organozinc prepared for this reaction was according to procedure A. The title

177
compound 3.36 was obtained as colorless oil (93.5 mg, 77% yield; 10:1 dr) after
purification by silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300
MHz, CDCl3): 1.48-1.64 (m, 3H), 1.67-1.73 (m, 1x0.90H), 1.81-1.85 (m, 1x0.10H),
1.90-1.98 (m, 1H), 2.62-2.82 (m, 3x0.90H), 2.82-2.85 (m, 3x0.10H), 3.37-3.45 (m,
1x0.10H), 3.60-3.66 (m, 1x0.90H), 3.71-3.79 (m, 2H), 3.85 (t, J = 7.5 Hz, 1x0.90H), 4.13
(t, J = 7.5 Hz, 1x0.10H), 5.01 (d, J = 3.0 Hz, 1x0.10H), 5.25 (d, J = 3.0 Hz, 1x0.90H), 7.27
(d, J = 6.0 Hz, 2H), 7.57 (d, J = 6.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 19.6, 23.0,
33.7, 36.5, 42.0, 61.0, 69.6, 101.8, 110.2, 118.8, 129.2, 132.4, 145.8; IR (neat): 2940,
2869, 2226, 1716, 1606, 1252, 1177, 1049; HRMS (ESI-TOF): (M+H)+ Calcd for
C15H18NO2 244.1338; found 244.1320. The actual dr (10:1) of this compound was
calculated using crude 1H NMR by integrating peaks at 5.27 ppm (major isomer) and
5.03 ppm (minor isomer). The characterization data given above correspond to the major
isomer isolated by column chromatography.
(±)-(2S,4R)-2-Butoxy-4-(3-chlorobenzyl)tetrahydrofuran (3.37). The organozinc prepared
for this reaction was according to procedure A. The title compound 3.37 was obtained as a
colorless oil (101.8 mg, 76% yield; 4.1:1 dr) after purification by silica gel column
chromatography (Hex: EtOAc = 19:1). 1H NMR (300 MHz, CDCl3): 0.88-0.96 (m, 3H),
1.30-1.43 (m, 2H), 1.50-1.69 (m, 3H), 1.95-2.02 (m, 1x0.20H), 2.11-2.20 (m, 1x0.80H),

178
2.46 (app.septet, J = 7.9 Hz, 1x0.80H), 2.62-2.65 (m, 1x0.20H), 2.75 (d, J = 9.0 Hz, 2H),
3.31-3.42 (m, 1H), 3.51-3.73 (m, 2H), 3.88-4.00 (m, 1H), 5.09-5.13 (m, 1H), 7.03-7.06 (m,
1H), 7.15-7.26 (m, 3H); 13C NMR (75 MHz, CDCl3): 14.0, 19.5, 32.0, 38.5, 39.2, 39.7,
67.5, 71.6, 104.5, 126.3, 126.9, 128.8, 129.7, 134.2, 143.0; IR (neat): 2956, 2931, 2869,
1598, 1474, 1430, 1080, 1011; HRMS (CI) m/z: (M)+ Calcd for C15H21ClO2 268.1230;
found 268.1221. The actual dr (4.1:1) was calculated using GC. See SI for GC trace.
(±)-4-(3,4-Dichlorobenzyl)-2-ethoxy-3-ethyltetrahydrofuran (3.38). The organozinc
prepared for this reaction was according to procedure A. The. title compound 3.38 was
obtained as a colorless oil (104.2 mg, 69% yield; 6: 4.5: 3.4: 1 dr) after purification by
silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3):
0.85-0.90 (m, 1H), 0.93-1.02 (m, 2H), 1.16-1.23 (m, 3H), 1.32-1.73 (m, 2H), 1.99-2.11 (m,
1H), 2.36-2.86 (m, 3H), 3.39-3.56 (m, 1H), 3.59-3.90 (m, 3H), 4.78 (d, J = 2.1 Hz,
1x0.40H), 4.85 (d, J = 2.6 Hz, 1x0.23H), 4.95 (d, J = 4.8 Hz, 1x0.30H), 4.98 (d, J = 4.5
Hz, 1x0.07H), 6.98 (d, J = 9.0 Hz, 1H), 7.23-7.34 (m, 2H); 13C NMR (75 MHz, CDCl3):
12.2, 12.8, 13.0, 15.3, 15.4, 15.5, 18.0, 19.3, 20.8, 25.5, 32.8, 35.1, 38.5, 40.4, 41.9, 45.8,
48.8, 50.0, 52.9, 63.0, 63.3, 70.6, 71.6, 71.7, 104.1, 107.9, 109.1, 128.1, 128.4, 129.8,
130.1, 130.3, 130.4, 130.6, 130.8, 132.3, 132.4, 140.9, 141.1, 141.2, 142.2; IR (neat):
2965, 2874, 1472, 1131, 1044, 1028, 999; HRMS (CI) m/z: (M)+ C15H20Cl2O2 302.0840;

179
found 302.0840. The actual dr (6: 4.5: 3.4: 1) was calculated using GC. See SI for GC
trace.
(±)-(2S,4R)-4-((5-Butoxytetrahydrofuran-3-yl)methyl)-2-chloropyridine (3.39). The
organozinc prepared for this reaction was according to procedure A. The title compound
3.39 was obtained as a colorless oil (108.9 mg, 81% yield; 2.2:1 dr) by silica gel column
chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3): 0.87-0.96 (m, 3H),
1.30-1.44 (m, 2H), 1.47-1.64 (m, 3H), 1.97-2.04 (m, 1x0.31H), 2.10-2.19 (m, 1x0.69H),
2.48 (app.septet, J = 6.9 Hz, 1x0.69H), 2.64-2.67 (m, 1x0.31H), 2.79 (d, J = 9.0 Hz, 2H),
3.33-3.41 (m, 1H), 3.54-3.72 (m, 2H), 3.91-4.01 (m, 1H), 5.09-5.12 (m, 1H), 7.03 (t, J =
6.0 Hz, 1H), 7.15 (s, 1H), 8.28 (d, J = 6.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 13.9,
19.4, 19.5, 31.8, 31.9, 37.8, 38.3, 38.6, 38.7, 39.0, 67.2, 67.5, 71.3, 71.5, 103.9, 104.3,
122.9, 123.0, 124.5, 149.7, 151.8, 152.9, 153.3; IR (neat): 2955, 2869, 1592, 1465, 1385,
1085, 1013; HRMS (ESI-TOF): (M+H)+ Calcd for C14H21ClNO2 270.1261; found
270.1247. The actual dr (2.2:1) was calculated using GC. See SI for GC trace.

180
(±)-(3R,3aS,7aR)-3-(Furan-2-ylmethyl)hexahydro-4H-furo[2,3-b]pyran (3.40). This
reaction was performed by using di(furan-2-yl)zinc (1 eqvt.) in our standard condition. The
title compound 3.40 was obtained as colorless oil (84.2 mg, 81% yield; 8:1 dr) after
purification by silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300
MHz, CDCl3): 1.45-1.64 (m, 3H), 1.72-1.80 (m, 1H), 1.96-2.05 (m, 1H), 2.58-2.79 (m,
3H), 3.60-3.67 (m, 1H), 3.72-3.82 (m, 2H), 3.97 (t, J = 7.5 Hz, 1H), 5.28 (d, J = 3.0 Hz,
1H), 5.99 (d, J = 3.0 Hz, 1H), 6.27 (t, J = 3.0 Hz, 1H), 7.29 (d, J = 3.0 Hz, 1H); 13C NMR
(75 MHz, CDCl3): 19.5, 23.2, 26.1, 36.7, 39.9, 61.2, 69.9, 102.0, 105.5, 110.2, 141.3,
154.1; IR (neat): 2936, 2871, 1769, 1436, 1143, 1047, 1015; HRMS (ESI-TOF): (M+H)+
Calcd for C12H17O3 209.1178; found 209.1174. The actual dr (8:1) of this compound was
calculated using crude 1H NMR by integrating peaks at 5.30 ppm (major isomer) and
5.01 ppm (minor isomer). The characterization data given above correspond to the major
isomer isolated by column chromatography.
(±)-(3R,3aS,6aR)-3-(Furan-2-ylmethyl)hexahydrofuro[2,3-b]furan (3.41). This reaction
was performed by using di(furan-2-yl)zinc (1 eqvt.) in our standard condition. The title
compound 3.41 was obtained as colorless oil (63.0 mg, 65% yield; 9.3:1 dr) after
purification by silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300
MHz, CDCl3): 1.82-2.01 (m, 2H), 2.65-2.89 (m, 4H), 3.54 (t, J = 9.0 Hz, 1H), 3.84-3.99

181
(m, 3H), 5.74 (d, J = 6.0 Hz, 1H), 6.01 (d, J = 3.0 Hz, 1H), 6.28 (t, J = 1.5 Hz, 1H), 7.31
(d, J = 0.72 Hz, 1H); 13C NMR (75 MHz, CDCl3): 25.2, 26.3, 41.1, 45.6, 69.2, 72.2,
105.6, 109.8, 110.3, 141.3, 153.8; IR (neat): 2949, 2873, 1716, 1254, 1106, 1072, 996;
HRMS (ESI-TOF): (M+H)+ Calcd for C11H15O3 195.1021; found 195.1015. The actual
dr (9.3:1) of this compound was calculated using crude 1H NMR by integrating peaks at
5.70 ppm (major isomer) and 5.81 ppm (minor isomer). The characterization data given
above correspond to the major isomer isolated by column chromatography.
(±)-(3R,3aS,6aR)-3-(Thiophen-2-ylmethyl)hexahydrofuro[2,3-b]furan (3.42). The
organozinc prepared for this reaction was according to procedure A. The title compound
3.42 was obtained as a colorless oil (76.6 mg, 73% yield; 10:1 dr) after purification by
silica gel column chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3):
1.82-2.04 (m, 2H), 2.64-3.01 (m, 4H), 3.54 (t, J = 10.5 Hz, 1H), 3.84-3.99 (m, 3H), 5.74
(d, J = 3.0 Hz, 1H), 6.80 (d, J = 3.0 Hz, 1H), 6.93 (t, J = 4.5 Hz, 1H), 7.14 (d, J = 6.0 Hz,
1H); 13C NMR (75 MHz, CDCl3): 25.2, 28.0, 44.1, 45.4, 69.2, 72.2, 109.9, 123.6, 124.8,
126.9, 142.5; IR (neat): 2948, 1438, 1252, 1107, 997, 921; HRMS (ESI-TOF): (M+H)+
Calcd for C11H15O2S 211.0793; found 211.0792. The actual dr (10:1) of this compound
was calculated using crude 1H NMR by integrating peaks at 5.70 ppm (major isomer)

182
and 5.80 ppm (minor isomer). The characterization data given above correspond to the
major isomer isolated by column chromatography.
1-(4-(((3R,3aS,7aR)-hexahydro-4H-furo[2,3-b]pyran-3-yl)methyl)phenyl)ethan-1-one
(3.43). This reaction was performed by using organozinc prepared according to procedure
C in our standard condition. The title compound 3.43 was obtained as white solid (101.4
mg, 78% yield; 10:1 dr) after purification by flash column chromatography (Hex: EtOAc
= 2:1). 1H NMR (300 MHz, CDCl3): 1.51-1.61 (m, 3H), 1.72-1.76 (m, 1H), 1.92-1.98
(m, 1H), 2.58 (s, 3H), 2.64-2.82 (m, 3H), 3.64 (t, J = 7.5 Hz, 1H), 3.74-3.81 (m, 2H), 3.87
(t, J = 7.5 Hz, 1H), 5.27 (d, J = 6.0 Hz, 1H), 7.26 (d, J = 8.1 Hz, 2H), 7.88 (d, J = 8.1 Hz,
2H); 13C NMR (75 MHz, CDCl3): 19.6, 23.1, 26.6, 33.6, 36.6, 42.2, 61.0, 69.7, 101.9,
128.6, 128.7, 135.5, 145.9, 197.7; IR (neat): 2932, 1676, 1355, 1138, 1014, 866; HRMS
(APCI): (M+H)+ Calcd for C16H21O3 261.1491; found 261.1481 The actual dr (10:1) of
this compound was calculated using crude 1H NMR by integrating peaks at 5.25 ppm
(major isomer) and 5.03 ppm (minor isomer). The characterization data given above
correspond to the major isomer isolated by column chromatography.

183
1-(4-(((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)methyl)phenyl)ethan-1-one (3.44).
This reaction was performed by using organozinc prepared according to procedure C in
our standard condition. The title compound 3.44 was obtained as a white solid (86.1 mg,
70% yield; 10:1 dr) after purification by flash column chromatography (Hex: EtOAc =
2:1). 1H NMR (300 MHz, CDCl3): 1.78-2.02 (m, 2H), 2.57 (s, 3H), 2.62-2.84 (m, 4H),
3.54 (t, J = 10.5 Hz, 1H), 3.81-3.98 (m, 3H), 5.69 (d, J = 6 Hz, 1H), 7.25 (d, J = 9 Hz, 2H),
), 7.87 (d, J = 9 Hz, 2H); 13C NMR (75 MHz, CDCl3): 25.2, 26.6, 33.9, 43.5, 45.4, 69.1,
72.0, 109.8, 128.6, 128.8, 135.5, 145.7, 197.6; IR (neat): 1669, 1415, 1267, 1107, 1003,
921; HRMS (APCI): (M+H)+ Calcd for C15H19O3 247.1334; found 247.1323. The actual
dr (10:1) of this compound was calculated using crude 1H NMR by integrating peaks at
5.70 ppm (major isomer) and 5.80 ppm (minor isomer). The characterization data given
above correspond to the major isomer isolated by column chromatography.
(2S,4R)-4-(4-bromobenzyl)-2-phenyltetrahydrofuran (3.45). This reaction was performed
by using organozinc prepared according to procedure A in our standard condition. The title
compound 3.45 was obtained as colorless oil (98.0mg, 62% yield; 10:1 dr) after
purification by silica gel column chromatography (Hex: EtOAc = 19:1). 1H NMR (300
MHz, CDCl3): 1.93-2.02 (m, 1H), 2.05-2.14 (m, 1H), 2.41 (app. septet, J = 6.0 Hz,
1x0.10H), 2.63 (app. septet, J = 6.75 Hz, 1x0.90H), 2.72 (d, J = 9.0 Hz, 2H), 3.63 (t, J =

184
7.5 Hz, 1x0.90H), 3.76 (t, J = 7.5 Hz, 1x0.10H), 4.04 (t, J = 7.5 Hz, 1x0.10H), 4.16 (t, J =
7.5 Hz, 1x0.90H), 4.90 (t, J = 7.5 Hz, 1x0.10H), 5.07 (t, J = 7.5 Hz, 1x0.90H), 7.05 (d, J
= 8.1 Hz, 2H), 7.25-7.35 (m, 5H), 7.41 (d, J = 8.1 Hz, 2H),; 13C NMR (75 MHz, CDCl3):
38.6, 38.9, 40.5, 41.7, 42.1, 73.5, 73.6, 80.0, 81.3, 120.0, 125.5, 125.6, 127.2, 127.4,
128.4 (br, s), 131.6 (br, s), 139.5, 143.6; IR (neat): 1558, 1161, 1008, 760, 701, 528;
HRMS (CI): (M)+ Calcd for : C17H17BrO 316.0463; found 316.0461. The actual dr (10:1)
of this compound was calculated using crude 1H NMR by integrating peaks at 5.07 ppm
(major isomer) and 4.90 ppm (minor isomer).
ethyl (1R)-1-acetyl-3-(4-cyanobenzyl)cyclopentane-1-carboxylate (3.46). The organozinc
prepared for this reaction was according to procedure A. The title compound 3.46 was
obtained as a colorless oil (89.7 mg, 60 % yield; 3:2 dr) after purification by silica gel
column chromatography (Hex: Et2O = 8:1). 1H NMR (500 MHz, CDCl3): 1.21-1.23
(m, 3H), 1.30-1.40 (m, 1H), 1.99-2.13 (m, 4H), 2.22-2.34 (m, 3H), 2.65-2.74 (m, 2H), 4.14-
4.21 (m, 2H), 7.26 (d, J =7.5 Hz, 2H), 7.55 (d, J = 7.5 Hz, 2H); 13C NMR (125 MHz,
CDCl3): 14.1, 26.2, 26.5, 32.0, 32.1, 32.2, 32.3, 38.5, 38.7, 41.2, 41.3, 41.4, 61.6, 66.3,
66.5, 109.9, 119.1, 129.5, 132.2, 146.8, 146.9, 173.3, 173.4, 203.4; HRMS (CI): (M+H)+
Calcd for for C18H22NO3 300.1600; found 300.1590. The actual dr (3:2) of this compound

185
was calculated using crude 1H NMR by integrating peaks at 2.10 ppm (major isomer)
and 2.08 ppm (minor isomer).
(±)-4-(3,4-Dimethoxybenzyl)dihydrofuran-2(3H)-one (3.50). The dicarbofunctionalization
reaction was conducted in 10.0 mmol scale in 50 mL NMP in 8 h using the standard
procedure. Organozinc ((3,4-dimethoxyphenyl)zinc iodide) for this reaction was prepared
according to above procedure B. Under nitrogen atmosphere, (3,4-dimethoxyphenyl)zinc
iodide stock solution in THF (15 mmol) was taken in a 150 mL sealed tube and the solvent
was removed under vacuum. To the residue of ArZnI was added NiBr2 (30 mg, 0.3 mmol),
terpyridine (45 mg, 0.4 mmol), and 1-(1-(allyloxy)-2-iodoethoxy)butane (3.49) (10 mmol).
The mixture was then dissolved in NMP (50 mL). The sealed tube was tightly capped, and
placed in an oil-bath preheated to 50 °C with vigorous stirring. The resultant cyclized cross
coupled product was oxidized without further purification as follow: The crude reaction
mixture (brown color) was diluted with ethyl acetate (30 mL) and washed three times with
10 mL water. The combined EtOAc layer was dried with Na2SO4 and solvent was removed.
For oxidation, the crude reaction mixture was taken in a reaction flask and 200 mL acetone
was added to the flask. 30 mL of Jones reagent (prepared by dissolving 1 gm CrO3 in 1 mL
conc. H2SO4 and 3 mL H2O) was added dropwise to the reaction mixture at 0 °C. The
reaction was stirred for 1 h at 0 °C. Then the reaction was quenched with isopropyl alcohol
(30 mL) and stirred for a while and filtered. The filtrate was neutralized with saturated

186
NaHCO3 solution (10 mL) and extracted with EtOAc (30 mL). The combined EtOAc layer
was dried with Na2SO4 and the solvent was pumped off. The title compound 3.50 was
obtained as a colorless oil (1463.2 mg, 62% yield) after purification by silica gel column
chromatography (Hex: EtOAc = 4:1). 1H NMR (300 MHz, CDCl3): 2.21 (dd, J = 18.0,
6.0 Hz 1H), 2.52 (dd, J = 18.0, 9.0 Hz 1H), 2.63-2.84 (m, 3H), 3.78 (d, J = 3.0 Hz, 6H),
3.95 (dd, J = 9.0, 6.0 Hz, 1H), 4.25 (dd, J = 9.0, 6.0 Hz, 1H), 6.62-6.65 (m, 2H), 6.75 (d, J
= 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 34.0, 37.1, 38.3, 55.7, 72.5, 111.2, 111.7,
120.5, 130.7, 147.6, 148.9, 176.8; IR (neat): 2935, 2835, 1767, 1512, 1261, 1138, 1011;
HRMS (ESI-TOF): (M+H)+ Calcd for C13H17O4 237.1127; found 237.1116.
(3aR,9S,9aR)-9-(3,4-dimethoxyphenyl)-6,7-dimethoxy-3a,4,9,9a-tetrahydronaphtho[2,3-
c]furan-1(3H)-one (3.51).220 Compound 3.50 (94.4 mg, 0.4 mmol) was dissolved in dry
THF (2.5 mL) then the solution was cooled to -78 °C. To the cooled solution, LDA (0.8
mmol, 1.6 ml of 0.5M solution in THF) was added dropwise for 5 minutes. The solution
was stirred for 1h at -78 °C which turns the solution to yellow. To the reaction mixture,
solution 3,4-dimethoxybenzaldehyde (99.6 mg, 0.6 mmol) in 1.5 mL THF was added
dropwise at -50 °C and and stirred at room temperature for 6 h. Then solvent was removed,
and the reaction crude was dissolved in 1 mL CH2Cl2. To the stirring reaction mixture at

187
room temperature, Trifluoroacetic acid (309 µl, 4 mmol) was added dropwise. It was stirred
overnight at room temperature and quenched with saturated NaHCO3 (2.0 ml). The reaction
mixture was extracted with CH2Cl2 (8 mL). The organic layer was dried with Na2SO4 and
the solvent was removed. The title compound 3.51 was obtained as a white solid (112.1
mg, 73% yield; 19:1 dr) after purification by flash column chromatography (Hex: EtOAc
= 2:1). 1H NMR (500 MHz, CDCl3): 2.50 (dd, J = 13.8, 10.8 Hz, 1H), 2.58-2.68 (m,
1H), 2.93 (t, J = 13.2 Hz, 1H), 3.00 (dd, J = 15.0, 4.5 Hz, 1H), 3.60 (s, 3H), 3.82 (s, 3H),
3.87 (s, 6H), 3.99 (dd, J = 10.0, 9.5 Hz, 1H), 4.11 (d, J = 11.0 Hz, 1H), 4.52 (dd, J = 8.0,
7.5 Hz, 1H), 6.33 (s, 1H), 6.61 (s, 1H), 6.70 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.83 (d, J =
8.5 Hz, 1H); 13C NMR (75 MHz, CDCl3): 32.6, 40.1, 45.8, 48.9, 55.8, 55.9, 56.0, 71.0,
111.0, 111.4, 112.4, 112.9, 121.9, 126.9, 131.4, 135.6, 147.7, 147.8, 147.9, 148.8, 175.6, ;
IR (neat): 1759, 1652, 1514, 1246, 1217, 1102, 981 ; HRMS (ESI-TOF): (M+H)+ Calcd
for C22H25O6 385.1651; found 385.1653. The stereochemistry of the compound was
assigned by comparing the spectral data and the J-coupling values with the known
compound in the literature.220 The actual dr (19:1) was calculated using GC and
observation of single isomer in crude 1H NMR. See SI for GC trace.

188
(±)-(3R,4R)-3-(Benzo[d][1,3]dioxol-5-ylmethyl)-4-(3,4-dimethoxybenzyl)dihydrofuran-
2(3H)-one (3.52).251 In a 4 dram vial, compound 3.50 (94.4 mg, 0.4 mmol) was dissolved
in dry THF (4 mL) . The solution was cooled to -78 °C. To the cooled solution, LDA (0.48
mmol, 0.96 ml of 0.5M) solution was added dropwise for 5 minutes. The solution was
stirred for 1 h at -78 °C which turns the solution to yellow. To the reaction mixture, solution
5-(bromomethyl)benzo[d][1,3]dioxole252 (127.8 mg, 0.6 mmol) in 1.5 mL THF was added
dropwise at -50 °C followed by addition of HMPA ( 72 µl, 0.4 mmol) and stirred at room
temperature 6 h. The reaction mixture was quenched with NH4Cl (1 mL) and extracted
with EtOAc (10 mL). The organic layer was dried with Na2SO4 and the solvent was
removed. The title compound 3.52 was obtained as a colorless viscous oil (127.2 mg, 86%
yield; 40:1 dr) after purification by silica gel column chromatography (Hex: EtOAc = 3:1).
1H NMR (300 MHz, CDCl3): 2.46-2.62 (m, 4H), 2.84 (dd, J = 15.0, 9.0 Hz, 1H), 2.96
(dd, J = 15.0, 6.0 Hz, 1H), 3.82 (s, 3H), 3.85 (s, 3H), ), 3.87-3.90 (m, 1H), 4.14 (dd, J =
9.0, 6.8 Hz, 1H), 5.92 (dd, J = 3.2, 1.4 Hz, 1H), 6.47 (d, J = 3.0 Hz, 1H), 6.55-6.59 (m,
3H), 6.73 (dd, J = 15.0, 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): 34.8, 38.3, 41.3,
46.5, 55.8, 56.0, 71.3, 101.1, 108.2, 109.5, 111.4, 111.8, 120.7, 122.3, 130.5, 131.4, 146.5,
147.9, 149.1, 178.5; IR (neat): 1767, 1732, 1514, 1442, 1236, 1155, 1027; HRMS (ESI-
TOF): (M+H)+ Calcd for C21H23O6 371.1495, found 371.1490. The actual dr (40:1) was
calculated using GC. See SI for GC trace.

189
(±)-(3R,4R)-3,4-Bis(3,4-dimethoxybenzyl)dihydrofuran-2(3H)-one (3.53).251 In a 4 dram
vial, compound 3.50 (94.4 mg, 0.4 mmol) was dissolved in dry THF (4 mL) . The solution
was cooled to -78 °C. To the solution, LDA (0.48 mmol, 0.96 ml of 0.5M) solution was
added dropwise for 5 minutes. The solution was stirred for 1 h at -78 °C which turns the
solution to yellow. To the reaction mixture, solution of 4-(bromomethyl)-1,2-
dimethoxybenzene253 (, 138 mg, 0.6 mmol) in 1.5 mL THF was added dropwise at -50 °C
followed by addition of HMPA (72 µl, 0.4 mmol) and stirred at room temperature 6 h. The
reaction mixture was quenched with NH4Cl (1 mL) and extracted with EtOAc 10 (mL).
The organic layer was dried with Na2SO4 and the solvent was removed. The title compound
3.53 was obtained as a white solid (109.6 mg, 71% yield; 33:1 dr) after purification by
silica gel column chromatography (Hex: EtOAc = 3:1). 1H NMR (300 MHz, CDCl3);
2.47-2.65 (m, 4H), 2.88-3.00 (m, 2H), 3.81-3.89 (m, 13H), 4.12 (dd, J = 9.0, 6.0 Hz, 1H),
6.48 (s, 1H), 6.54 (d, J = 9.0 Hz, 1H), 6.63-6.67 (m, 2H), 6.75 (dd, J = 9.0, 6.0 Hz, 2H);
13C NMR (75 MHz, CDCl3): 34.6, 38.3, 41.1, 46.6, 55.9, 71.3, 111.1, 111.4, 111.9,
112.4, 120.6, 121.4, 130.3, 130.5, 147.9, 148.0, 149.1, 178.8; IR (neat): 2935, 1765, 1512,
1452, 1234, 1155, 1014; IR (neat): 1765, 1512, 1452, 1234, 1081, 974; HRMS (ESI-

190
TOF): (M+H)+ Calcd for C22H27O6 387.1808; found 387.1818. The actual dr (33:1) was
calculated using GC. See SI for GC trace.
(±)-4-(Benzo[d][1,3]dioxol-5-ylmethyl)dihydrofuran-2(3H)-one (3.54). The
dicarbofunctionaliz-ation reaction was conducted in 10.0 mmol scale in 50 mL NMP in 8
h using the standard procedure. Organozinc (benzo[d][1,3]dioxol-5-ylzinc iodide) for this
reaction was prepared according to procedure B. Under nitrogen atmosphere,
benzo[d][1,3]dioxol-5-ylzinc iodide stock solution in THF (15 mmol) was taken in a 150
mL sealed tube and the solvent was removed under vacuum. To the residue of ArZnI was
added NiBr2 (30 mg, 0.3 mmol), terpyridine (45 mg, 0.4 mmol), and 1-(1-(allyloxy)-2-
iodoethoxy)butane (3.49) (10 mmol). The mixture was then dissolved in NMP (50 mL).
The sealed tube was tightly capped and placed in an oil-bath preheated to 50 °C with
vigorous stirring. The resultant cyclized cross coupled product was oxidized without
further purification as follow: The crude reaction mixture (brown color) was diluted with
ethyl acetate (30 mL) and washed three times with 10 mL of water. The combined EtOAc
layer was dried with Na2SO4 and solvent was removed. For oxidation, the crude reaction
mixture was taken in a reaction flask and 220 mL acetone was added to the flask. 33 mL
of Jones reagent (prepared by dissolving 1 gm CrO3 in 1 mL conc. H2SO4 and 3 mL H2O)
was added dropwise to the reaction mixture at 0°C. The reaction was stirred for 1 h at 0°C.
Then the reaction was quenched with isopropyl alcohol (33 ml) and stirred for a while and

191
filtered. The filtrate was neutralized with saturated NaHCO3 solution (10 mL) and extracted
with EtOAc (30 mL). The combined EtOAc layer was dried with Na2SO4 and the solvent
was pumped off. The title compound 3.54 was obtained as a colorless oil (1452 mg, 60%
yield) after purification by silica gel column chromatography (Hex: EtOAc = 4:1). 1H
NMR (300 MHz, CDCl3): 2.19 (dd, J = 18.0, 9.0 Hz 1H), 2.52 (dd, J = 18.0, 9.0 Hz 1H),
2.60-2.78 (m, 3H), 3.93 (dd, J = 9.0, 6.0 Hz, 1H), 4.25 (dd, J = 12.0, 9.0 Hz, 1H), 5.86 (s,
2H), 6.54 (d, J = 6.0 Hz, 1H), 6.58 (s, 1H), 6.68 (d, J = 6.0 Hz, 1H); 13C NMR (75 MHz,
CDCl3): 33.8, 37.0, 38.3, 72.3, 100.8, 108.2, 108.7, 121.4, 131.9, 146.1, 147.7, 176.7;
IR (neat): 2906, 1769, 1488, 1238, 1166, 1011; HRMS (ESI-TOF): (M+H)+ Calcd for
C12H13O4 221.0814; found 221.0806.
(±)-(3R,4R)-4-(Benzo[d][1,3]dioxol-5-ylmethyl)-3-(3,4-dimethoxybenzyl)dihydrofuran-
2(3H)-one (3.55).223 In a 4 dram vial, compound 3.54 (88.0 mg, 0.4 mmol) was dissolved
in dry THF (4 mL) . The solution was cooled to -78 °C. To the solution, LDA (0.48 mmol,
0.96 ml of 0.5M) solution was added dropwise for 5 minutes. The solution was stirred for
1 h at -78 °C which turns the solution to yellow. To the reaction mixture, solution of 4-
(bromomethyl)-1,2-dimethoxybenzene253 ( 138 mg, 0.6 mmol) in 1.5 mL THF was added
dropwise at -50 °C followed by addition of HMPA ( 72 µl, 0.4 mmol) and stirred at room

192
temperature 6 h. The reaction mixture was quenched with NH4Cl (1 mL) and extracted
with EtOAc (10 mL). The organic layer was dried with Na2SO4 and the solvent was
removed. The title compound 3.55 was obtained as a colorless viscous oil (113.9 mg, 77%
yield; 23:1 dr) after purification by silica gel column chromatography (Hex: EtOAc = 3:1).
The actual dr ratio is calculated using GC trace. See SI for GC trace. 1H NMR (300 MHz,
CDCl3): 2.42-2.60 (m, 4H), 2.88 (dd, J = 15.0, 9.0 Hz, 1H), 2.96 (dd, J = 15.0, 6.0 Hz,
1H), 3.83 (s, 3H), 3.86 (s, 3H), 3.86-3.88 (m, 1H), 4.11 (dd, J = 9.0, 6.0 Hz, 1H), 5.92 (dd,
J = 3.3, 1.5 Hz, 1H), 6.42-6.47 (m, 2H), 6.66-6.70 (m, 3H), 6.79 (d, J = 9.0 Hz, 1H); 13C
NMR (75 MHz, CDCl3): 34.7, 38.4, 41.1, 46.6, 55.9, 71.2, 101.1, 108.3, 108.8, 111.2,
112.2, 121.4, 121.6, 130.2, 131.7, 146.4, 148.0, 149.1, 178.7; IR (neat): 1766, 1514, 1464,
1236, 1140, 1025; HRMS (ESI-TOF): Calcd for (M+H)+ C21H23O6 371.1495; found
371.1529. The actual dr (23:1) was calculated using GC. See SI for GC trace.
(±)-(3R,4R)-4-(Benzo[d][1,3]dioxol-5-ylmethyl)-3-(3,4,5-trimethoxybenzyl)dihydrofuran-
2(3H)-one (3.56).217 In a 4 dram vial, compound 3.54 (88 mg, 0.4 mmol) was dissolved in
dry THF (4 mL) . The solution was cooled to -78 °C. To the solution, LDA (0.48 mmol,
0.96 mL of 0.5M) solution was added dropwise for 5 minutes. The solution was stirred for
1 h at -78 °C which turns the solution to yellow. To the reaction mixture, solution of 5-

193
(bromomethyl)-1,2,3-trimethoxybenzene254-255 (165.6 mg, 0.6 mmol,) in 1.5 mL THF was
added dropwise at -50 °C followed by addition of HMPA (72 µl, 0.4 mmol) and stirred at
room temperature 6 hrs. The reaction mixture was quenched with NH4Cl (1 mL) and
extracted with EtOAc (10 mL). The organic layer was dried with Na2SO4 and the solvent
was removed. The title compound 3.56 was obtained as a colorless oil (128.0 mg, 80%
yield; 40:1 dr) after purification by silica gel column chromatography (Hex: EtOAc = 3:1).
1H NMR (300 MHz, CDCl3): 2.44-2.65 (m, 4H), 2.84-2.96 (m, 2H), 3.82 (s, 9H), 3.84-
3.90 (m, 1H), 4.17 (dd, J = 9.0, 6.0 Hz, 1H), 5.93 (dd, J = 2.8, 1.3 Hz, 1H), 6.35 (s, 2H),
6.45-6.48 (m, 2H), 6.69 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 35.3, 38.4,
41.1, 46.5, 56.1, 60.9, 71.2, 101.1, 106.2, 108.3, 108.8, 121.6, 131.6, 133.4, 136.9, 146.4,
147.9, 153.3, 178.5; IR (neat): 1766, 1589, 1489, 1237, 1123, 1010; HRMS (ESI-TOF):
(M+H)+ Calcd for C22H25O7 401.1600; found 401.1594. The actual dr (40:1) was calculated
using GC. See SI for GC trace.
Benzo[d][1,3]dioxol-5-yl(2-iodo-4,5-dimethoxyphenyl)methanone . The compound was
prepared by Friedel-Crafts acylation of 1,3-benzodioxole with 6-iodoveratric acid
according to the literature procedure.256 The title compound benzo[d][1,3]dioxol-5-yl(2-
iodo-4,5-dimethoxyphenyl)methan-one was obtained as a white solid (94% yield) after
purification by silica gel column chromatography (Hex : EtOAc = 9:1). 1H NMR (300

194
MHz, CDCl3): 3.80 (s, 3H), 3.88 (s, 3H), 6.03 (s, 2H), 6.78 (d, J = 6.0 Hz, 2H), 7.24-
7.26 (m, 2H), 7.33 (s, 1H); 13C NMR (75 MHz, CDCl3): 56.1, 56.3, 81.5, 102.0, 107.9,
109.4, 111.7, 121.8, 127.9, 130.6, 136.8, 148.3, 148.9, 150.4, 152.3, 195.2; IR (neat):
2900, 1635, 1499, 1323, 1179, 1033; HRMS (ESI-TOF): (M+H)+ Calcd for C16H14IO5
412.9886, found 412.9879. The title compound was used directly for the preparation of
organozinc 3.57 using the standard procedure A.
(±)-4-(2-(Benzo[d][1,3]dioxole-5-carbonyl)-4,5-dimethoxybenzyl)dihydrofuran-2(3H)-
one (3.58). The dicarbofunctionalization reaction was conducted in 2.50 mmol scale in
12.5 mL NMP using the standard procedure for 8 h. For this reaction, the organozinc (3.57)
(2-(benzo[d][1,3]dioxole-5-carbonyl)-4,5-dimethoxyphenyl)zinc iodide was prepared
according to the standard procedure A. The resultant product was oxidized without further
purification as follows: The crude reaction mixture (brown color) was diluted with ethyl
acetate (15 mL) and washed three times with 5 mL water. The aqueous layer was extracted
back with EtOAc (10 mL) and all the EtOAc layer were combined and dried with Na2SO4
and solvent was removed. For oxidation, the crude reaction mixture was taken in a reaction
flask and acetone (70 mL) was added to the flask. 7.5 mL of Jones reagent (prepared by
dissolving 1 gm CrO3 in 1 mL conc. H2SO4 and 3 mL H2O) was added dropwise to the

195
reaction mixture at 0 °C. The reaction was stirred for 1 h at 0 °C. Then the reaction was
quenched with isopropyl alcohol (6 mL) and stirred for a while and filtered. The filtrate
was neutralized with NaHCO3 (2 mL) and extracted with EtOAc (15 mL). The combined
EtOAc layer was dried with Na2SO4 and the solvent was pumped off. The title compound
3.58 was obtained as a white solid (672.0 mg, 70% yield) after purification by silica gel
column chromatography (Hex: EtOAc = 3:1). 1H NMR (300 MHz, CDCl3): 2.14 (dd, J
= 18.0, 6.0 Hz 1H), 2.37 (dd, J = 18.0, 9.0 Hz 1H), 2.66-2.80 (m, 3H), 3.67 (s, 3H), 3.80
(s, 3H), 3.86 (dd, J = 9.0, 6.0 Hz, 1H), 4.13 (dd, J = 9.0, 6.0 Hz, 1H), 5.91 (s, 2H), 6.64-
6.69 (m, 2H), 6.73 (s, 1H), 7.13 (d, J = 6.0 Hz, 1H), 7.17 (s, 1H); 13C NMR (75 MHz,
CDCl3): 33.8, 35.3, 37.1, 55.7, 72.3, 101.8, 107.4, 108.9, 112.4, 112.9, 126.8, 130.2,
131.1, 132.2, 146.3, 147.9, 150.2, 151.7, 176.7, 195.3; IR (neat): 2935, 1771, 1646, 1436,
1225, 1032; HRMS (ESI-TOF): (M+H)+ Calcd for C21H21O7 385.1287; found 385.1276.
(±)9-(Benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-3a,4-dihydronaphtho[2,3-c]furan-1(3H)-
one (3.59).257 Compound 3.58 (76.8 mg, 0.25 mmol) was dissolved in dry THF (2.5 mL)
and the solution was cooled to -78 °C. To the cooled solution, LDA (0.5 mmol, 1.0 mL of
0.5M solution in THF) was added dropwise for 5 minutes. The solution was stirred for 1 h
at -78 °C which turns the solution to yellow. To the same reaction mixture, HMPA (45 µl,

196
0.25 mmol) was added dropwise and stirred at room temperature for additional 3 h. Then
the solvent was removed, and the crude reaction was dissolved in 2.4 mL pyridine solution.
The reaction mixture was cooled back to 0 °C and SOCl2 (182 µl, 2.5 mmol,) was added
dropwise. It was stirred for 1 h at 0 °C and was diluted with CH2Cl2 (10 mL) and quenched
with 1M HCl (2.5 ml). The reaction mixture was extracted with additional 10 mL CH2Cl2.
The organic layer was dried with Na2SO4 and the solvent was removed. The title compound
3.59 was obtained as a white solid (59.4 mg, 65% yield) after purification by silica gel
column chromatography (Hex: EtOAc = 1:1). 1H NMR (300 MHz, CDCl3): 2.79 (t, J
= 9.9 Hz, 1H), 2.93 (dd, J = 15.0, 6.0 Hz, 1H), 3.33-3.46 (m, 1H), 3.67 (s, 3H), 3.92 (s,
3H), 4.00 (t, J = 9.0 Hz, 1H), 4.69 (t, J = 9.0 Hz, 1H), 6.02 (d, J = 3.0 Hz, 2H), 6.54 (s,
1H), 6.78 (br.s, 3H), 6.86 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): 33.0, 35.8,
56.1, 71.05, 101.3, 107.9, 110.6, 111.2, 112.5, 119.6, 123.9, 127.9, 128.6, 129.3, 147.1,
147.2, 147.7, 147.9, 150.3, 168.4; IR (neat): 2915, 2849, 1745, 1683, 1540, 1378, 1063 ;
HRMS (ESI): (M+H)+ Calcd for C21H19O6 367.1182; found 367.1190.
1-octyl-4-(trifluoromethyl)benzene (3.65).258 The organozinc prepared for this reaction was
according to procedure A. Under nitrogen atmosphere, in a sealed tube, (4-
(trifluoromethyl)phenyl)zinc iodide stock solution in THF (0.750 mmol) was taken and the
solvent was removed under vacuum. To the residue of ArZnI, NiBr2 (3.3 mg, 0.015 mmol),
terpyridine (4.7 mg, 0.02 mmol), and iodooctane (0.5 mmol) were added successively.

197
Then the mixture was dissolved in NMP (2.5 mL). Later, sealed tube was tightly capped,
and placed in an oil-bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction
mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with
H2O (5 mL × 3). The aqueous fraction was extracted back with ethyl acetate (5 mL × 3)
and combined with the first ethyl acetate fraction. The combined ethyl acetate fraction was
dried over Na2SO4 and the solvent was removed in a rotary evaporator. The title compound
3.65 was obtained as a colorless oil (78.1 mg, 88% yield) after purification by silica gel
column chromatography in hexane. 1H NMR (300 MHz, CDCl3): 0.87 (t, J = 6.0 Hz,
3H), 1.25-1.33 (m, 10H), 1.56-1.63 (m, 2H), 2.64 (t, J = 7.8 Hz, 2H), 7.26 (d, J = 6.0 Hz,
2H), 7.45 (d, J = 7.8 Hz, 2H); 13C NMR (75 MHz, CDCl3): 14.2, 22.8, 29.4, 29.5, 31.3,
32.0, 35.98, 123.5 (q, J = 72.0 Hz), 125.3 (q, J = 4.1 Hz), 126.8 (q, J = 63.7 Hz), 128.8,
147.1; 19F NMR (282 MHz, CDCl3) -62.3. The NMR data are consistent with the
reported values.
1-methoxy-4-octylbenzene (3.66).258 The organozinc prepared for this reaction was
according to procedure A. Under nitrogen atmosphere, in a sealed tube, (4-((4-
methoxyphenyl)zinc iodide stock solution in THF (0.750 mmol) was taken and the solvent
was removed under vacuum. To the residue of ArZnI, NiBr2 (3.3 mg, 0.015 mmol),
terpyridine (4.7 mg, 0.020 mmol), and iodooctane (0.5 mmol) were added successively.
Then the mixture was dissolved in NMP (2.5 mL). Later, sealed tube was tightly capped

198
and placed in an oil-bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction
mixture was cooled to room temperature, diluted with EtOAc (10 mL) and washed with
H2O (5 mL × 3). The aqueous fraction was extracted back with ethyl acetate (5 mL × 3)
and combined with the first ethyl acetate fraction. The combined ethyl acetate fraction was
dried over Na2SO4 and the solvent was removed in a rotary evaporator. The title compound
3.66 was obtained as a colorless oil (46.5 mg, 69% yield) after purification by silica gel
column chromatography (Hex: EtOAc = 32:1). 1H NMR (300 MHz, CDCl3): 0.90 (t, J
= 6.3 Hz, 3H), 1.28-1.35 (m, 10H), 1.54-1.64 (m, 2H), 2.56 (t, J = 7.8 Hz, 2H), 3.80 (s,
3H), 6.82 (d, J = 9.0 Hz, 2H), 7.10 (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3):
14.2, 22.8, 29.4, 29.6, 31.9, 32.0, 35.2, 55.3, 113.7, 129.3, 135.2, 157.7. The NMR data are
consistent with the reported values.
diethyl 3-(4-(trifluoromethyl)benzyl)cyclopentane-1,1-dicarboxylate (3.69).106 The
organozinc prepared for this reaction was according to procedure A. Under nitrogen
atmosphere, in a sealed tube, (4-(trifluoromethyl)phenyl)zinc iodide stock solution in THF
(0.750 mmol) was taken and the solvent was removed under vacuum. To the residue of
ArZnI, NiBr2 (3.3 mg, 0.015 mmol), terpyridine (4.7 mg, 0.02 mmol), and diethyl-2-allyl-
2-(2-Iodoethyl)malonate (3.13-I) (0.5 mmol) were added successively. Then the mixture
was dissolved in NMP (2.5 mL). Later, sealed tube was tightly capped, and placed in an
oil-bath preheated to 50 °C with vigorous stirring. After 6 h, the reaction mixture was

199
cooled to room temperature, diluted with EtOAc (10 mL) and washed with H2O (5 mL ×
3). The aqueous fraction was extracted back with ethyl acetate (5 mL × 3) and combined
with the first ethyl acetate fraction. The combined ethyl acetate fraction was dried over
Na2SO4 and the solvent was removed in a rotary evaporator. The title compound 3.69 was
obtained as a colorless oil (148.8 mg, 80% yield) after purification by silica gel column
chromatography (Hex: EtOAc = 9:1). 1H NMR (300 MHz, CDCl3): 1.17-1.25 (m, 6H),
1.28-1.41 (m, 1H), 1.76-1.84 (m, 2H), 2.07-2.40 (m, 4H), 2.62-2.75 (m, 2H), 4.10-4.20 (m,
4H), 7.26 (d, J = 6.0 Hz, 2H), 7.51 (d, J = 6.0 Hz, 2H); 13C NMR (75 MHz, CDCl3):
14.1, 32.1, 33.7, 40.3, 41.1, 41.4, 60.0, 61.5, 122.6, 125.4(q, JCF= 3.8 Hz), 126.0 (q, JCF=
31.5 Hz), 128.4 (q, JCF= 34.1 Hz), 129.1, 145.4, 172.6, 172.7; 19F NMR (282 MHz, CDCl3)
-60.8; IR (neat): 2981, 2944, 2855, 1725, 1617.
References
1. Hebrard, F.; Kalck, P., Cobalt-Catalyzed Hydroformylation of Alkenes: Generation
and Recycling of the Carbonyl Species, and Catalytic Cycle. Chemical Reviews 2009, 109
(9), 4272-4282.
2. Franke, R.; Selent, D.; Börner, A., Applied Hydroformylation. Chemical Reviews
2012, 112 (11), 5675-5732.
3. Slaugh, L. H.; Mullineaux, R. D., Novel Hydroformylation catalysts. Journal of
Organometallic Chemistry 1968, 13 (2), 469-477.
4. Breit, B.; Seiche, W., Recent Advances on Chemo-, Regio- and Stereoselective
Hydroformylation. Synthesis 2001, 2001 (01), 0001-0036.

200
5. Daubignard, J.; Detz, R. J.; Jans, A. C. H.; de Bruin, B.; Reek, J. N. H., Rational
Optimization of Supramolecular Catalysts for the Rhodium-Catalyzed Asymmetric
Hydrogenation Reaction. Angewandte Chemie International Edition 2017, 56 (42), 13056-
13060.
6. Hou, C.; Zhao, G.; Ji, Y.; Niu, Z.; Wang, D.; Li, Y., Hydroformylation of alkenes
over rhodium supported on the metal-organic framework ZIF-8. Nano Research 2014, 7
(9), 1364-1369.
7. Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V.,
Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010
Nobel Prize. Angewandte Chemie International Edition 2012, 51 (21), 5062-5085.
8. Negishi, E.-i., Magical Power of Transition Metals: Past, Present, and Future
(Nobel Lecture). Angewandte Chemie International Edition 2011, 50 (30), 6738-6764.
9. Suzuki, A., Cross-Coupling Reactions Of Organoboranes: An Easy Way To
Construct C C Bonds (Nobel Lecture). Angewandte Chemie International Edition 2011,
50 (30), 6722-6737.
10. Tsutomu, M.; Kunio, M.; Atsumu, O., Arylation of Olefin with Aryl Iodide
Catalyzed by Palladium. Bulletin of the Chemical Society of Japan 1971, 44 (2), 581-581.
11. Heck, R. F.; Nolley, J. P., Palladium-catalyzed vinylic hydrogen substitution
reactions with aryl, benzyl, and styryl halides. The Journal of Organic Chemistry 1972, 37
(14), 2320-2322.
12. Corbet, J.-P.; Mignani, G., Selected Patented Cross-Coupling Reaction
Technologies. Chemical Reviews 2006, 106 (7), 2651-2710.

201
13. Nicolaou, K. C.; Bulger, P. G.; Sarlah, D., Palladium-Catalyzed Cross-Coupling
Reactions in Total Synthesis. Angewandte Chemie International Edition 2005, 44 (29),
4442-4489.
14. Trost, B. M.; Crawley, M. L., Asymmetric Transition-Metal-Catalyzed Allylic
Alkylations: Applications in Total Synthesis. Chemical Reviews 2003, 103 (8), 2921-2944.
15. Tietze Lutz, F.; Düfert, A., Multiple Pd-catalyzed reactions in the synthesis of
natural products, drugs, and materials. In Pure and Applied Chemistry, 2010; Vol. 82, p
1375.
16. Nakajima, Y.; Shimada, S., Hydrosilylation reaction of olefins: recent advances and
perspectives. RSC Advances 2015, 5 (26), 20603-20616.
17. Shigehisa, H.; Koseki, N.; Shimizu, N.; Fujisawa, M.; Niitsu, M.; Hiroya, K.,
Catalytic Hydroamination of Unactivated Olefins Using a Co Catalyst for Complex
Molecule Synthesis. Journal of the American Chemical Society 2014, 136 (39), 13534-
13537.
18. Gui, J.; Pan, C.-M.; Jin, Y.; Qin, T.; Lo, J. C.; Lee, B. J.; Spergel, S. H.; Mertzman,
M. E.; Pitts, W. J.; La Cruz, T. E.; Schmidt, M. A.; Darvatkar, N.; Natarajan, S. R.; Baran,
P. S., Practical olefin hydroamination with nitroarenes. Science 2015, 348 (6237), 886.
19. Yin, G.; Mu, X.; Liu, G., Palladium(II)-Catalyzed Oxidative Difunctionalization of
Alkenes: Bond Forming at a High-Valent Palladium Center. Accounts of Chemical
Research 2016, 49 (11), 2413-2423.

202
20. Lan, X.-W.; Wang, N.-X.; Xing, Y., Recent Advances in Radical
Difunctionalization of Simple Alkenes. European Journal of Organic Chemistry 2017,
2017 (39), 5821-5851.
21. Mai, D. N.; Wolfe, J. P., Asymmetric Palladium-Catalyzed Carboamination
Reactions for the Synthesis of Enantiomerically Enriched 2-(Arylmethyl)- and 2-
(Alkenylmethyl)pyrrolidines. Journal of the American Chemical Society 2010, 132 (35),
12157-12159.
22. Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B., Catalytic Asymmetric
Dihydroxylation. Chemical Reviews 1994, 94 (8), 2483-2547.
23. Giglio, B. C.; Schmidt, V. A.; Alexanian, E. J., Metal-Free, Aerobic Dioxygenation
of Alkenes Using Simple Hydroxamic Acid Derivatives. Journal of the American
Chemical Society 2011, 133 (34), 13320-13322.
24. Beller, M.; Seayad, J.; Tillack, A.; Jiao, H., Catalytic Markovnikov and anti-
Markovnikov Functionalization of Alkenes and Alkynes: Recent Developments and
Trends. Angewandte Chemie International Edition 2004, 43 (26), 3368-3398.
25. Bar, G. L. J.; Lloyd-Jones, G. C.; Booker-Milburn, K. I., Pd(II)-Catalyzed
Intermolecular 1,2-Diamination of Conjugated Dienes. Journal of the American Chemical
Society 2005, 127 (20), 7308-7309.
26. Banik, S. M.; Medley, J. W.; Jacobsen, E. N., Catalytic, Diastereoselective 1,2-
Difluorination of Alkenes. Journal of the American Chemical Society 2016, 138 (15),
5000-5003.

203
27. Molnár, I. G.; Gilmour, R., Catalytic Difluorination of Olefins. Journal of the
American Chemical Society 2016, 138 (15), 5004-5007.
28. Liu, G.; Stahl, S. S., Highly Regioselective Pd-Catalyzed Intermolecular
Aminoacetoxylation of Alkenes and Evidence for cis-Aminopalladation and SN2 C−O
Bond Formation. Journal of the American Chemical Society 2006, 128 (22), 7179-7181.
29. Liu, W.; Li, Y.; Liu, K.; Li, Z., Iron-Catalyzed Carbonylation-Peroxidation of
Alkenes with Aldehydes and Hydroperoxides. Journal of the American Chemical Society
2011, 133 (28), 10756-10759.
30. Sun, X.; Li, X.; Song, S.; Zhu, Y.; Liang, Y.-F.; Jiao, N., Mn-Catalyzed Highly
Efficient Aerobic Oxidative Hydroxyazidation of Olefins: A Direct Approach to β-Azido
Alcohols. Journal of the American Chemical Society 2015, 137 (18), 6059-6066.
31. Whiting, A.; Windsor, C. M., What makes a neutral imino dieneophile undergo a
thermal, non-catalysed, Diels-Alder reaction? Tetrahedron 1998, 54 (22), 6035-6050.
32. Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A. B., Stereoselective
Cyclopropanation Reactions. Chemical Reviews 2003, 103 (4), 977-1050.
33. Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G., The Diels–
Alder Reaction in Total Synthesis. Angewandte Chemie International Edition 2002, 41
(10), 1668-1698.
34. Tasdelen, M. A., Diels–Alder “click” reactions: recent applications in polymer and
material science. Polymer Chemistry 2011, 2 (10), 2133-2145.
35. Derosa, J. T., V. T.; van der Puyl; V. A.; Engle, K. M. , Carbon–Carbon π-Bonds
as Conjunctive Reagents in Cross-Coupling. Aldrichimica Acta 2018, 51, 21-32.

204
36. Giri, R.; Kc, S., Strategies toward Dicarbofunctionalization of Unactivated Olefins
by Combined Heck Carbometalation and Cross-Coupling. The Journal of Organic
Chemistry 2018, 83 (6), 3013-3022.
37. Dhungana, R. K.; Kc, S.; Basnet, P.; Giri, R., Transition Metal-Catalyzed
Dicarbofunctionalization of Unactivated Olefins. The Chemical Record 2018, 18 (9), 1314-
1340.
38. Zhang, J.-S.; Liu, L.; Chen, T.; Han, L.-B., Transition-Metal-Catalyzed Three-
Component Difunctionalizations of Alkenes. Chemistry – An Asian Journal 2018, 13 (17),
2277-2291.
39. Zweig, J. E.; Kim, D. E.; Newhouse, T. R., Methods Utilizing First-Row Transition
Metals in Natural Product Total Synthesis. Chemical Reviews 2017, 117 (18), 11680-
11752.
40. Mason, J. S.; Morize, I.; Menard, P. R.; Cheney, D. L.; Hulme, C.; Labaudiniere,
R. F., New 4-Point Pharmacophore Method for Molecular Similarity and Diversity
Applications: Overview of the Method and Applications, Including a Novel Approach to
the Design of Combinatorial Libraries Containing Privileged Substructures. Journal of
Medicinal Chemistry 1999, 42 (17), 3251-3264.
41. Baldwin, J. E., Rules for ring closure. Journal of the Chemical Society, Chemical
Communications 1976, (18), 734-736.
42. Martí-Centelles, V.; Pandey, M. D.; Burguete, M. I.; Luis, S. V., Macrocyclization
Reactions: The Importance of Conformational, Configurational, and Template-Induced
Preorganization. Chemical Reviews 2015, 115 (16), 8736-8834.

205
43. Cavell, K. J., Recent fundamental studies on migratory insertion into metal-carbon
bonds. Coord. Chem. Rev. 1996, 155, 209-243.
44. Iqbal, J.; Bhatia, B.; Nayyar, N. K., Transition Metal-Promoted Free-Radical
Reactions in Organic Synthesis: The Formation of Carbon-Carbon Bonds. Chemical
Reviews 1994, 94 (2), 519-564.
45. Fournet, G.; Balme, G.; Gore, J., Attaque nucleophile intramoleculaire
d'organopalladiques issus de la carbopalladation d'alkylidenecyclopropanes. Tetrahedron
Letters 1987, 28 (39), 4533-4536.
46. Balme, G.; Bouyssi, D.; Lomberget, T.; Monteiro, N., Cyclisations Involving
Attack of Carbo- and Heteronucleophiles on Carbon-Carbon π-Bonds Activated by
Organopalladium Complexes. Synthesis 2003, 2003 (14), 2115-2134.
47. Dénès, F.; Pérez-Luna, A.; Chemla, F., Addition of Metal Enolate Derivatives to
Unactivated Carbon−Carbon Multiple Bonds. Chemical Reviews 2010, 110 (4), 2366-
2447.
48. Nicolai, S.; Swallow, P.; Waser, J., Intramolecular palladium-catalyzed alkene
carboalkynylation. Tetrahedron 2015, 71 (35), 5959-5964.
49. Bouyssi, D.; Coudanne, I.; Uriot, H.; Goré, J.; Balme, G., Formation of
cyclohexanes in the palladium catalyzed reaction of ɛ-unsaturated cyano esters and
congeners. Tetrahedron Letters 1995, 36 (44), 8019-8022.
50. Vittoz, P.; Bouyssi, D.; Traversa, C.; Goré, J.; Balme, G., A general solution to the
synthesis of triquinanes by a palladium catalyzed process. Tetrahedron Letters 1994, 35
(12), 1871-1874.

206
51. Bruyère, D.; Gaignard, G.; Bouyssi, D.; Balme, G.; Lancelin, J.-M., A steric control
of regioselectivity in palladium-catalyzed cyclizations of alkenes bearing arylbromides and
nucleophiles. Tetrahedron Letters 1997, 38 (5), 827-830.
52. Bruyère, D.; Bouyssi, D.; Balme, G., A study on the regio- and stereoselectivity in
palladium-catalyzed cyclizations of alkenes and alkynes bearing bromoaryl and
nucleophilic groups. Tetrahedron 2004, 60 (18), 4007-4017.
53. Balme, G.; Bouyssi, D., Total synthesis of the triquinane marine sesquiterpene
(±)Δ9(12) capnellene using a palladium-catalyzed bis-cyclization step. Tetrahedron 1994,
50 (2), 403-414.
54. Dhungana, R. K.; Shrestha, B.; Thapa-Magar, R.; Basnet, P.; Giri, R., Pd-Catalyzed
Regioselective 1,2-Dicarbofunctionalization of Unactivated Olefins by a Heck
Reaction/Enolate Cyclization Cascade. Organic Letters 2017, 19 (8), 2154-2157.
55. Rodriguez, A. L.; Bunlaksananusorn, T.; Knochel, P., Potassium tert-Butoxide
Catalyzed Addition of Carbonyl Derivatives to Styrenes. Organic Letters 2000, 2 (21),
3285-3287.
56. Casaschi, A.; Grigg, R.; Sansano, J. M., Palladium catalysed tandem cyclisation–
anion capture. Part 7:1 Synthesis of derivatives of α-amino esters, nitrogen heterocycles
and β-aryl/heteroaryl ethylamines via in situ generated vinylstannanes. Tetrahedron 2001,
57 (3), 607-615.
57. Casaschi, A.; Grigg, R.; Sansano, J. M., Palladium Catalysed Tandem Cyclisation–
Anion Capture. Part 6:1 Synthesis of Sugar, Nucleoside, Purine, Benzodiazepinone and β-

207
lactam Analogues via Capture of in situ Generated Vinylstannanes. Tetrahedron 2000, 56
(38), 7553-7560.
58. Grigg, R.; Sansano, J.; Santhakumar, V.; Sridharan, V.; Thangavelanthum, R.;
Thornton-Pett, M.; Wilson, D., Palladium catalysed tandem cyclisation-anion capture
processes. Part 3. Organoboron anion transfer agents. Tetrahedron 1997, 53 (34), 11803-
11826.
59. Grigg, R.; Mariani, E.; Sridharan, V., Palladium-catalysed in situ zipper
generation–cyclisation–anion capture. Synthesis of 3,3-disubstituted indolines and 2,3-
dihydrobenzofurans. Tetrahedron Letters 2001, 42 (49), 8677-8680.
60. Wilson, J. E., Diastereoselective synthesis of tetrahydroquinolines via a palladium-
catalyzed Heck–Suzuki cascade reaction. Tetrahedron Letters 2012, 53 (18), 2308-2311.
61. Zhang, Y.; Negishi, E., Metal-promoted cyclization. 25. Palladium-catalyzed
cascade carbometalation of alkynes and alkenes as an efficient route to cyclic and
polycyclic structures. Journal of the American Chemical Society 1989, 111 (9), 3454-3456.
62. Schweizer, S.; Song, Z.-Z.; Meyer, F. E.; Parsons, P. J.; de Meijere, A., Two New
Modes of Pd-Catalyzed Domino-Tetracyclization of Bromodienynes—5-exo-trig
Cyclization Wins over β-Hydride Elimination. Angewandte Chemie International Edition
1999, 38 (10), 1452-1454.
63. Grigg, R.; Fretwell, P.; Meerholtz, C.; Sridharan, V., Palladium catalysed synthesis
of spiroindolines. Tetrahedron 1994, 50 (2), 359-370.

208
64. Brown, D.; Grigg, K.; Sridharan, V.; Tambyrajah, V., a palladium catalysed
cascade cyclisation-friedel-crafts alkylation approach to angularly fused ring systems.
Tetrahedron Letters 1995, 36 (44), 8137-8140.
65. Satyanarayana, G.; Maichle-Mossmer, C.; Maier, M. E., Formation of pentacyclic
structures by a domino sequence on cyclic enamides. Chemical Communications 2009,
(12), 1571-1573.
66. Grigg, R.; Santhakumar, V.; Sridharan, V., Palladium catalysed cascade cyclisation
- cyanide ion capture. Tetrahedron Letters 1993, 34 (19), 3163-3164.
67. Wu, X.-X.; Chen, W.-L.; Shen, Y.; Chen, S.; Xu, P.-F.; Liang, Y.-M., Palladium-
Catalyzed Domino Heck/Intermolecular C–H Bond Functionalization: Efficient Synthesis
of Alkylated Polyfluoroarene Derivatives. Organic Letters 2016, 18 (8), 1784-1787.
68. Zhang, H.; Chen, P.; Liu, G., Palladium-Catalyzed Oxidative Arylalkylation of
Unactivated Alkenes: Dual C–H Bond Cleavage of Anilines and Acetonitrile. Synlett 2012,
23 (19), 2749-2752.
69. Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M., Alkene Trifluoromethylation
Coupled with C C Bond Formation: Construction of Trifluoromethylated Carbocycles
and Heterocycles. Angewandte Chemie International Edition 2013, 52 (14), 4000-4003.
70. Tour, J. M.; Negishi, E., Metal promoted cyclization. 9. Controlled and catalytic
acylpalladation. A novel route to cyclopentenone and cyclohexenone derivatives. Journal
of the American Chemical Society 1985, 107 (26), 8289-8291.

209
71. Negishi, E.-i.; Copéret, C.; Ma, S.; Mita, T.; Sugihara, T.; Tour, J. M., Palladium-
Catalyzed Carbonylative Cyclization of 1-Iodo-2-alkenylbenzenes. Journal of the
American Chemical Society 1996, 118 (25), 5904-5918.
72. Negishi, E.-i.; Ma, S.; Amanfu, J.; Copéret, C.; Miller, J. A.; Tour, J. M., Palladium-
Catalyzed Cyclization of 1-Iodo-Substituted 1,4-, 1,5-, and 1,6-Dienes as Well as of 5-
Iodo-1,5-dienes in the Presence of Carbon Monoxide. Journal of the American Chemical
Society 1996, 118 (25), 5919-5931.
73. Negishi, E.; Sawada, H.; Tour, J. M.; Wei, Y., Metal-promoted cyclization. 17. A
novel bicyclization methodology via cyclialkylation of .omega.-halo-1-metallo-1-alkynes
containing aluminum and zinc. The Journal of Organic Chemistry 1988, 53 (4), 913-915.
74. Copéret, C.; Negishi, E.-i., Palladium-Catalyzed Highly Diastereoselective Cyclic
Carbopalladation−Carbonylative Esterification Tandem Reaction of Iododienes and
Iodoarylalkenes. Organic Letters 1999, 1 (1), 165-168.
75. McMahon, C. M.; Renn, M. S.; Alexanian, E. J., Manganese-Catalyzed
Carboacylations of Alkenes with Alkyl Iodides. Organic Letters 2016, 18 (16), 4148-4150.
76. Ryu, I.; Kreimerman, S.; Araki, F.; Nishitani, S.; Oderaotoshi, Y.; Minakata, S.;
Komatsu, M., Cascade Radical Reactions Catalyzed by a Pd/Light System: Cyclizative
Multiple Carbonylation of 4-Alkenyl Iodides. Journal of the American Chemical Society
2002, 124 (15), 3812-3813.
77. Ishiyama, T.; Miyaura, N.; Suzuki*, A., Palladium-catalyzed carbonylative cross-
coupling reaction of iodoalkanes with 9-alkyl-9-BBN derivatives. A direct and selective
synthesis of ketones. Tetrahedron Letters 1991, 32 (47), 6923-6926.

210
78. Ishiyama, T.; Murata, M.; Suzuki, A.; Miyaura, N., Synthesis of ketones from
iodoalkenes, carbon monoxide and 9-alkyl-9-borabicyclo[3.3.1]nonane derivatives via a
radical cyclization and palladium-catalysed carbonylative cross-coupling sequence.
Journal of the Chemical Society, Chemical Communications 1995, (3), 295-296.
79. Liu, C.; Widenhoefer, R. A., Palladium-Catalyzed Cyclization/Carboalkoxylation
of Alkenyl Indoles. Journal of the American Chemical Society 2004, 126 (33), 10250-
10251.
80. Bullck, M., Catalysis without Precious Metals. Wiley-VCH, Weinheim 2011.
81. Jana, R.; Pathak, T. P.; Sigman, M. S., Advances in Transition Metal (Pd,Ni,Fe)-
Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners.
Chemical Reviews 2011, 111 (3), 1417-1492.
82. Menezes da Silva, V. H.; Braga, A. A. C.; Cundari, T. R., N-Heterocyclic Carbene
Based Nickel and Palladium Complexes: A DFT Comparison of the Mizoroki–Heck
Catalytic Cycles. Organometallics 2016, 35 (18), 3170-3181.
83. Sole, D.; Cancho, Y.; Llebaria, A.; Moreto, J. M.; Delgado, A., Intramolecular
Nitrogen Assistance in the Nickel-Promoted Tandem Cyclization-Capture of Amino-
Tethered Vinyl Bromides and Alkenes. Journal of the American Chemical Society 1994,
116 (26), 12133-12134.
84. Solé, D.; Cancho, Y.; Llebaria, A.; Moretó, J. M.; Delgado, A., Nitrogen Assistance
in Intramolecular Nickel-Promoted Tandem Cyclization−Quenching Processes. The
Journal of Organic Chemistry 1996, 61 (17), 5895-5904.

211
85. Wakabayashi, K.; Yorimitsu, H.; Oshima, K., Cobalt-Catalyzed Tandem Radical
Cyclization and Cross-Coupling Reaction: Its Application to Benzyl-Substituted
Heterocycles. Journal of the American Chemical Society 2001, 123 (22), 5374-5375.
86. Someya, H.; Kondoh, A.; Sato, A.; Ohmiya, H.; Yorimitsu, H.; Oshima, K., A New
Approach to 4-Aryl-1,3-butanediols by Cobalt-Catalyzed Sequential Radical Cyclization-
Arylation Reaction of Silicon-Tethered 6-Iodo-1-hexene Derivatives. Synlett 2006, 2006
(18), 3061-3064.
87. Kim, J. G.; Son, Y. H.; Seo, J. W.; Kang, E. J., Iron-Catalyzed Tandem Cyclization
and Cross-Coupling Reactions of Iodoalkanes and Aryl Grignard Reagents. European
Journal of Organic Chemistry 2015, 2015 (8), 1781-1789.
88. Phapale, V. B.; Buñuel, E.; García-Iglesias, M.; Cárdenas, D. J., Ni-Catalyzed
Cascade Formation of C(sp3) C(sp3) Bonds by Cyclization and Cross-Coupling
Reactions of Iodoalkanes with Alkyl Zinc Halides. Angewandte Chemie International
Edition 2007, 46 (46), 8790-8795.
89. Guisán-Ceinos, M.; Soler-Yanes, R.; Collado-Sanz, D.; Phapale, V. B.; Buñuel, E.;
Cárdenas, D. J., Ni-Catalyzed Cascade Cyclization–Kumada Alkyl–Alkyl Cross-Coupling.
Chemistry – A European Journal 2013, 19 (26), 8405-8410.
90. Yan, C.-S.; Peng, Y.; Xu, X.-B.; Wang, Y.-W., Nickel-Mediated Inter- and
Intramolecular Reductive Cross-Coupling of Unactivated Alkyl Bromides and Aryl
Iodides at Room Temperature. Chemistry – A European Journal 2012, 18 (19), 6039-6048.

212
91. Peng, Y.; Xu, X.-B.; Xiao, J.; Wang, Y.-W., Nickel-mediated stereocontrolled
synthesis of spiroketals via tandem cyclization-coupling of [small beta]-bromo ketals and
aryl iodides. Chemical Communications 2014, 50 (4), 472-474.
92. Peng, Y.; Xiao, J.; Xu, X.-B.; Duan, S.-M.; Ren, L.; Shao, Y.-L.; Wang, Y.-W.,
Stereospecific Synthesis of Tetrahydronaphtho[2,3-b]furans Enabled by a Nickel-
Promoted Tandem Reductive Cyclization. Organic Letters 2016, 18 (19), 5170-5173.
93. Xiao, J.; Wang, Y.-W.; Peng, Y., Nickel-Promoted Reductive Cyclization Cascade:
A Short Synthesis of a New Aromatic Strigolactone Analogue. Synthesis 2017, 49 (16),
3576-3581.
94. Kuang, Y.; Wang, X.; Anthony, D.; Diao, T., Ni-catalyzed two-component
reductive dicarbofunctionalization of alkenes via radical cyclization. Chemical
Communications 2018, 54 (20), 2558-2561.
95. Anthony, D.; Lin, Q.; Baudet, J.; Diao, T., Nickel-Catalyzed Asymmetric
Reductive Diarylation of Vinylarenes. Angewandte Chemie International Edition 2019, 58
(10), 3198-3202.
96. Wang, K.; Ding, Z.; Zhou, Z.; Kong, W., Ni-Catalyzed Enantioselective Reductive
Diarylation of Activated Alkenes by Domino Cyclization/Cross-Coupling. Journal of the
American Chemical Society 2018, 140 (39), 12364-12368.
97. Jin, Y.; Wang, C., Ni-catalysed reductive arylalkylation of unactivated alkenes.
Chemical Science 2019, 10 (6), 1780-1785.

213
98. Jin, Y.; Wang, C., Nickel-Catalyzed Asymmetric Reductive Arylalkylation of
Unactivated Alkenes. Angewandte Chemie International Edition 2019, 58 (20), 6722-
6726.
99. Stadtmueller, H.; Lentz, R.; Tucker, C. E.; Stuedemann, T.; Doerner, W.; Knochel,
P., Palladium-catalyzed iodine-zinc exchange reactions. A new palladium-mediated
intramolecular carbozincation of alkenes. Journal of the American Chemical Society 1993,
115 (15), 7027-7028.
100. Vaupel, A.; Knochel, P., Stereoselective Synthesis of Heterocyclic Zinc Reagents
via a Nickel-Catalyzed Radical Cyclization. The Journal of Organic Chemistry 1996, 61
(17), 5743-5753.
101. Stadtmüller, H.; Vaupel, A.; Tucker, C. E.; Stüdemann, T.; Knochel, P.,
Stereoselective Preparation of Polyfunctional Cyclopentane Derivatives by Radical
Nickel- or Palladium-Catalyzed Carbozincations. Chemistry – A European Journal 1996,
2 (10), 1204-1220.
102. Logan, K. M.; Sardini, S. R.; White, S. D.; Brown, M. K., Nickel-Catalyzed
Stereoselective Arylboration of Unactivated Alkenes. Journal of the American Chemical
Society 2018, 140 (1), 159-162.
103. You, W.; Brown, M. K., Diarylation of Alkenes by a Cu-Catalyzed Migratory
Insertion/Cross-Coupling Cascade. Journal of the American Chemical Society 2014, 136
(42), 14730-14733.
104. You, W.; Brown, M. K., Catalytic Enantioselective Diarylation of Alkenes. Journal
of the American Chemical Society 2015, 137 (46), 14578-14581.

214
105. Cong, H.; Fu, G. C., Catalytic Enantioselective Cyclization/Cross-Coupling with
Alkyl Electrophiles. J. Am. Chem. Soc. 2014, 136 (10), 3788-3791.
106. Thapa, S.; Basnet, P.; Giri, R., Copper-Catalyzed Dicarbofunctionalization of
Unactivated Olefins by Tandem Cyclization/Cross-Coupling. Journal of the American
Chemical Society 2017, 139 (16), 5700-5703.
107. Walker, J. A.; Vickerman, K. L.; Humke, J. N.; Stanley, L. M., Ni-Catalyzed
Alkene Carboacylation via Amide C–N Bond Activation. Journal of the American
Chemical Society 2017, 139 (30), 10228-10231.
108. Dreis, A. M.; Douglas, C. J., Catalytic Carbon−Carbon σ Bond Activation: An
Intramolecular Carbo-Acylation Reaction with Acylquinolines. Journal of the American
Chemical Society 2009, 131 (2), 412-413.
109. Yasui, Y.; Kamisaki, H.; Takemoto, Y., Enantioselective Synthesis of 3,3-
Disubstituted Oxindoles through Pd-Catalyzed Cyanoamidation. Organic Letters 2008, 10
(15), 3303-3306.
110. Liu, L.; Ishida, N.; Murakami, M., Atom- and Step-Economical Pathway to Chiral
Benzobicyclo[2.2.2]octenones through Carbon–Carbon Bond Cleavage. Angewandte
Chemie International Edition 2012, 51 (10), 2485-2488.
111. Souillart, L.; Parker, E.; Cramer, N., Highly Enantioselective Rhodium(I)-
Catalyzed Activation of Enantiotopic Cyclobutanone C C Bonds. Angewandte Chemie
International Edition 2014, 53 (11), 3001-3005.
112. Souillart, L.; Cramer, N., Enantioselective Rhodium-catalyzed C–C Bond
Activation of Cyclobutanones. Chimia 2015, 69 (4), 187-190.

215
113. Xu, T.; Dong, G., Rhodium-Catalyzed Regioselective Carboacylation of Olefins:
A C C Bond Activation Approach for Accessing Fused-Ring Systems. Angewandte
Chemie International Edition 2012, 51 (30), 7567-7571.
114. Urkalan, K. B.; Sigman, M. S., Palladium-Catalyzed Oxidative Intermolecular
Difunctionalization of Terminal Alkenes with Organostannanes and Molecular Oxygen.
Angewandte Chemie International Edition 2009, 48 (17), 3146-3149.
115. Catellani, M.; Paolo Chiusoli, G., One-pot palladium-catalyzed synthesis of 2,3-
disubstituted bicyclo[2.2.1]heptanes and bicyclo[2.2.1]hept-5-enes. Tetrahedron Letters
1982, 23 (43), 4517-4520.
116. Catellani, M.; Chiusoli, G. P.; Concari, S., A new palladium catalyzed synthesis of
cis,exo-2,3-diarylsubstituted bicyclo[2.2.1]heptanes or bicyclo [2.2.1] hept-2-enes.
Tetrahedron 1989, 45 (16), 5263-5268.
117. Catellani, M.; Chiusoli, G. P.; Mari, A., Palladium- or nickel-catalyzed sequential
reaction of organic bromides, bicyclo[2.2.1]hept-2-ene or bicyclo[2.2.1]hepta-2,5-diene
and alkynes. Journal of Organometallic Chemistry 1984, 275 (1), 129-138.
118. Kang, S.-K.; Kim, J.-S.; Choi, S.-C.; Lim, K.-H., Three-Component Coupling
Reaction: Palladium-Catalyzed Coupling of Norbornadiene and Iodonium Salts or
Diazonium Salts with Organostannanes, Alkynes, and Sodium Tetraphenylborate.
Synthesis 1998, 1998 (09), 1249-1251.
119. Shaulis, K. M.; Hoskin, B. L.; Townsend, J. R.; Goodson, F. E.; Incarvito, C. D.;
Rheingold, A. L., Tandem Suzuki Coupling−Norbornadiene Insertion Reactions. A

216
Convenient Route to 5,6-Diarylnorbornene Compounds. The Journal of Organic
Chemistry 2002, 67 (16), 5860-5863.
120. Takai, K.; Matsukawa, N.; Takahashi, A.; Fujii, T., Three-Component Coupling
Reactions of Alkyl Iodides, 1,3-Dienes, and Carbonyl Compounds by Sequential
Generation of Radical and Anionic Species with CrCl2. Angewandte Chemie International
Edition 1998, 37 (1-2), 152-155.
121. Mizutani, K.; Shinokubo, H.; Oshima, K., Cobalt-Catalyzed Three-Component
Coupling Reaction of Alkyl Halides, 1,3-Dienes, and Trimethylsilylmethylmagnesium
Chloride. Organic Letters 2003, 5 (21), 3959-3961.
122. Terao, J.; Nii, S.; Chowdhury, F. A.; Nakamura, A.; Kambe, N., Nickel-Catalyzed
Regioselective Three Component Coupling Reaction of Alkyl Halides, Butadienes, and
Ar-M (M=MgX, ZnX). Advanced Synthesis & Catalysis 2004, 346 (8), 905-908.
123. Iwasaki, T.; Fukuoka, A.; Yokoyama, W.; Min, X.; Hisaki, I.; Yang, T.; Ehara, M.;
Kuniyasu, H.; Kambe, N., Nickel-catalyzed coupling reaction of alkyl halides with aryl
Grignard reagents in the presence of 1,3-butadiene: mechanistic studies of four-component
coupling and competing cross-coupling reactions. Chemical Science 2018, 9 (8), 2195-
2211.
124. Liao, L.; Jana, R.; Urkalan, K. B.; Sigman, M. S., A Palladium-Catalyzed Three-
Component Cross-Coupling of Conjugated Dienes or Terminal Alkenes with Vinyl
Triflates and Boronic Acids. Journal of the American Chemical Society 2011, 133 (15),
5784-5787.

217
125. Stokes, B. J.; Liao, L.; de Andrade, A. M.; Wang, Q.; Sigman, M. S., A Palladium-
Catalyzed Three-Component-Coupling Strategy for the Differential Vicinal Diarylation of
Terminal 1,3-Dienes. Organic Letters 2014, 16 (17), 4666-4669.
126. DeLuca Ryan, J.; Stokes Benjamin, J.; Sigman Matthew, S., The strategic
generation and interception of palladium-hydrides for use in alkene functionalization
reactions. In Pure and Applied Chemistry, 2014; Vol. 86, p 395.
127. Jensen, K. H.; Sigman, M. S., Mechanistic approaches to palladium-catalyzed
alkene difunctionalization reactions. Org. Biomol. Chem. 2008, 6 (22), 4083-4088.
128. Gao, P.; Chen, L.-A.; Brown, M. K., Nickel-Catalyzed Stereoselective Diarylation
of Alkenylarenes. Journal of the American Chemical Society 2018, 140 (34), 10653-10657.
129. Kuang, Z.; Yang, K.; Song, Q., Pd-Catalyzed Regioselective 1,2-
Difunctionalization of Vinylarenes with Alkenyl Triflates and Aryl Boronic Acids at
Ambient Temperature. Organic Letters 2017, 19 (10), 2702-2705.
130. Wang, F.; Wang, D.; Mu, X.; Chen, P.; Liu, G., Copper-Catalyzed Intermolecular
Trifluoromethylarylation of Alkenes: Mutual Activation of Arylboronic Acid and CF3+
Reagent. Journal of the American Chemical Society 2014, 136 (29), 10202-10205.
131. Wu, L.; Wang, F.; Wan, X.; Wang, D.; Chen, P.; Liu, G., Asymmetric Cu-
Catalyzed Intermolecular Trifluoromethylarylation of Styrenes: Enantioselective
Arylation of Benzylic Radicals. Journal of the American Chemical Society 2017, 139 (8),
2904-2907.

218
132. Ilchenko, N. O.; Janson, P. G.; Szabó, K. J., Copper-Mediated
Cyanotrifluoromethylation of Styrenes Using the Togni Reagent. The Journal of Organic
Chemistry 2013, 78 (21), 11087-11091.
133. Liang, Z.; Wang, F.; Chen, P.; Liu, G., Copper-catalyzed intermolecular
cyanotrifluoromethylation of alkenes: Convenient synthesis of CF3-containing alkyl
nitriles. Journal of Fluorine Chemistry 2014, 167 (Supplement C), 55-60.
134. Wang, F.; Wang, D.; Wan, X.; Wu, L.; Chen, P.; Liu, G., Enantioselective Copper-
Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes via Radical Process.
Journal of the American Chemical Society 2016, 138 (48), 15547-15550.
135. He, Y.-T.; Li, L.-H.; Yang, Y.-F.; Zhou, Z.-Z.; Hua, H.-L.; Liu, X.-Y.; Liang, Y.-
M., Copper-Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes. Organic
Letters 2014, 16 (1), 270-273.
136. Terao, J.; Saito, K.; Nii, S.; Kambe, N.; Sonoda, N., Regioselective Double
Alkylation of Styrenes with Alkyl Halides Using a Titanocene Catalyst. Journal of the
American Chemical Society 1998, 120 (45), 11822-11823.
137. Ouyang, X.-H.; Song, R.-J.; Hu, M.; Yang, Y.; Li, J.-H., Silver-Mediated
Intermolecular 1,2-Alkylarylation of Styrenes with α-Carbonyl Alkyl Bromides and
Indoles. Angewandte Chemie International Edition 2016, 55 (9), 3187-3191.
138. Yong, X.; Han, Y.-F.; Li, Y.; Song, R.-J.; Li, J.-H., Alkylarylation of styrenes via
direct C(sp3)–Br/C(sp2)–H functionalization mediated by photoredox and copper
cooperative catalysis. Chemical Communications 2018, 54 (91), 12816-12819.

219
139. Ouyang, X.-H.; Cheng, J.; Li, J.-H., 1,2-Diarylation of alkenes with aryldiazonium
salts and arenes enabled by visible light photoredox catalysis. Chemical Communications
2018, 54 (63), 8745-8748.
140. Klauck, F. J. R.; Yoon, H.; James, M. J.; Lautens, M.; Glorius, F., Visible-Light-
Mediated Deaminative Three-Component Dicarbofunctionalization of Styrenes with
Benzylic Radicals. ACS Catalysis 2019, 9 (1), 236-241.
141. Rossiter, B. E.; Swingle, N. M., Asymmetric conjugate addition. Chemical Reviews
1992, 92 (5), 771-806.
142. Conjugate Addition Reactions in Organic Synthesis. In Tetrahedron Organic
Chemistry Series, Perlmutter, P., Ed. Elsevier: 1992; Vol. 9, pp 1-373.
143. Komemnos, T. L., 1883, 218, 145.
144. Guo, H.-C.; Ma, J.-A., Catalytic Asymmetric Tandem Transformations Triggered
by Conjugate Additions. Angewandte Chemie International Edition 2006, 45 (3), 354-366.
145. Alexakis, A.; Trevitt, G. P.; Bernardinelli, G., Tandem Enantioselective Conjugate
Addition: Electrophile Trapping Reactions. Application in the Formation of Syn or Anti
Aldols. Journal of the American Chemical Society 2001, 123 (18), 4358-4359.
146. Hayashi, Y.; Umekubo, N., Direct Asymmetric Michael Reaction of α,β-
Unsaturated Aldehydes and Ketones Catalyzed by Two Secondary Amine Catalysts.
Angewandte Chemie International Edition 2018, 57 (7), 1958-1962.
147. Qin, T.; Cornella, J.; Li, C.; Malins, L. R.; Edwards, J. T.; Kawamura, S.; Maxwell,
B. D.; Eastgate, M. D.; Baran, P. S., A general alkyl-alkyl cross-coupling enabled by redox-
active esters and alkylzinc reagents. Science 2016, 352 (6287), 801.

220
148. James, D. E.; Stille, J. K., The palladium(II) catalyzed olefin carbonylation
reaction. Mechanisms and synthetic utility. Journal of the American Chemical Society
1976, 98 (7), 1810-1823.
149. Yokota, T.; Sakaguchi, S.; Ishii, Y., Oxidative Carbomethoxylation of Alkenes
Using a Pd(II)/Molybdovanadophosphate (NPMoV) System under Carbon Monoxide and
Air. The Journal of Organic Chemistry 2002, 67 (14), 5005-5008.
150. Orlandi, M.; Hilton, M. J.; Yamamoto, E.; Toste, F. D.; Sigman, M. S., Mechanistic
Investigations of the Pd(0)-Catalyzed Enantioselective 1,1-Diarylation of Benzyl
Acrylates. J. Am. Chem. Soc. 2017, 139 (36), 12688-12695.
151. Albrecht, M., Cyclometalation Using d-Block Transition Metals: Fundamental
Aspects and Recent Trends. Chemical Reviews 2010, 110 (2), 576-623.
152. McDermott, J. X.; White, J. F.; Whitesides, G. M., Thermal decomposition of
bis(phosphine)platinum(II) metallocycles. Journal of the American Chemical Society
1976, 98 (21), 6521-6528.
153. Burke, B. J.; Overman, L. E., Enantioselective Synthesis of Six-Membered
Palladacycles Having Metal-Bound Stereogenic Carbons: Isolation and Reactivity of
Palladacycles Containing Readily Accessible β-Hydrogens. Journal of the American
Chemical Society 2004, 126 (51), 16820-16833.
154. Tanaka, D.; Romeril, S. P.; Myers, A. G., On the Mechanism of the Palladium(II)-
Catalyzed Decarboxylative Olefination of Arene Carboxylic Acids. Crystallographic
Characterization of Non-Phosphine Palladium(II) Intermediates and Observation of Their

221
Stepwise Transformation in Heck-like Processes. Journal of the American Chemical
Society 2005, 127 (29), 10323-10333.
155. Hoberg, H.; Peres, Y.; Krüger, C.; Tsay, Y.-H., A 1-Oxa-2-nickela-5-
cyclopentanone from Ethene and Carbon Dioxide: Preparation, Structure, and Reactivity.
Angewandte Chemie International Edition in English 1987, 26 (8), 771-773.
156. Hoberg, H.; Jenni, K.; Angermund, K.; Krüger, C., CC-Linkages of Ethene with
CO2 on an Iron(0) Complex—Synthesis and Crystal Structure Analysis of
[(PEt3)2Fe(C2H4)2]. Angewandte Chemie International Edition in English 1987, 26 (2),
153-155.
157. Cohen, S. A.; Bercaw, J. E., Titanacycles derived from reductive coupling of
nitriles, alkynes, acetaldehyde, and carbon dioxide with
bis(pentamethylcyclopentadienyl)(ethylene)titanium(II). Organometallics 1985, 4 (6),
1006-1014.
158. Trejos, A.; Fardost, A.; Yahiaoui, S.; Larhed, M., Palladium(ii)-catalyzed coupling
reactions with a chelating vinyl ether and arylboronic acids: a new Heck/Suzuki domino
diarylation reaction. Chemical Communications 2009, (48), 7587-7589.
159. Gu, J.-W.; Min, Q.-Q.; Yu, L.-C.; Zhang, X., Tandem Difluoroalkylation-Arylation
of Enamides Catalyzed by Nickel. Angewandte Chemie International Edition 2016, 55
(40), 12270-12274.
160. Shrestha, B.; Basnet, P.; Dhungana, R. K.; Kc, S.; Thapa, S.; Sears, J. M.; Giri, R.,
Ni-Catalyzed Regioselective 1,2-Dicarbofunctionalization of Olefins by Intercepting Heck

222
Intermediates as Imine-Stabilized Transient Metallacycles. Journal of the American
Chemical Society 2017, 139 (31), 10653-10656.
161. Thapa, S.; Dhungana, R. K.; Magar, R. T.; Shrestha, B.; Kc, S.; Giri, R., Ni-
catalysed regioselective 1,2-diarylation of unactivated olefins by stabilizing Heck
intermediates as pyridylsilyl-coordinated transient metallacycles. Chemical Science 2018,
9 (4), 904-909.
162. Basnet, P.; Dhungana, R. K.; Thapa, S.; Shrestha, B.; Kc, S.; Sears, J. M.; Giri, R.,
Ni-Catalyzed Regioselective β,δ-Diarylation of Unactivated Olefins in Ketimines via
Ligand-Enabled Contraction of Transient Nickellacycles: Rapid Access to Remotely
Diarylated Ketones. Journal of the American Chemical Society 2018, 140 (25), 7782-7786.
163. Basnet, P.; Kc, S.; Dhungana, R. K.; Shrestha, B.; Boyle, T. J.; Giri, R., Synergistic
Bimetallic Ni/Ag and Ni/Cu Catalysis for Regioselective γ,δ-Diarylation of Alkenyl
Ketimines: Addressing β-H Elimination by in Situ Generation of Cationic Ni(II) Catalysts.
Journal of the American Chemical Society 2018, 140 (46), 15586-15590.
164. Derosa, J.; Tran, V. T.; Boulous, M. N.; Chen, J. S.; Engle, K. M., Nickel-Catalyzed
β,γ-Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Conjunctive Cross-
Coupling. Journal of the American Chemical Society 2017, 139 (31), 10657-10660.
165. Derosa, J.; van der Puyl, V. A.; Tran, V. T.; Liu, M.; Engle, Keary M., Directed
nickel-catalyzed 1,2-dialkylation of alkenyl carbonyl compounds. Chemical Science 2018,
9 (23), 5278-5283.

223
166. Derosa, J.; Kleinmans, R.; Tran, V. T.; Karunananda, M. K.; Wisniewski, S. R.;
Eastgate, M. D.; Engle, K. M., Nickel-Catalyzed 1,2-Diarylation of Simple Alkenyl
Amides. Journal of the American Chemical Society 2018, 140 (51), 17878-17883.
167. Li, W.; Boon, J. K.; Zhao, Y., Nickel-catalyzed difunctionalization of allyl moieties
using organoboronic acids and halides with divergent regioselectivities. Chemical Science
2018, 9 (3), 600-607.
168. García-Domínguez, A.; Li, Z.; Nevado, C., Nickel-Catalyzed Reductive
Dicarbofunctionalization of Alkenes. Journal of the American Chemical Society 2017, 139
(20), 6835-6838.
169. Zhao, X.; Tu, H.-Y.; Guo, L.; Zhu, S.; Qing, F.-L.; Chu, L., Intermolecular selective
carboacylation of alkenes via nickel-catalyzed reductive radical relay. Nature
Communications 2018, 9 (1), 3488.
170. Jun, T.; Yuichiro, K.; Nobuaki, K., Titanocene‐Catalyzed Regioselective
Alkylation of Styrenes with Grignard Reagents Using β‐Bromoethyl Ethers, Thioethers, or
Amines. Chem. Asian J. 2008, 3 (8‐9), 1472-1478.
171. Sanchez, L. A.; Olmedo, D.; Lopez-Perez, J. L.; Williams, T. D.; Gupta, M. P.,
Two new alkylresorcinols from Homalomena wendlandii and their cytotoxic activity. Nat.
Prod. Commun. 2012, 7 (8), 1043-1046.
172. Chu, L.; Armstrong, H. M.; Chang, L. L.; Cheng, A. F.; Colwell, L.; Cui, J.; Evans,
J.; Galka, A.; Goulet, M. T.; Hayes, N.; Lo, J.; Menke, J.; Ok, H. O.; Ondeyka, D. L.; Patel,
M.; Quaker, G. M.; Sings, H.; Witkin, S. L.; Zhao, A.; Ujjainwalla, F., Evaluation of endo-
and exo-aryl-substitutions and central scaffold modifications on diphenyl substituted

224
alkanes as 5-lipoxygenase activating protein inhibitors. Bioorg. Med. Chem. Lett. 2012, 22
(12), 4133-4138.
173. Yonova, I. M.; Johnson, A. G.; Osborne, C. A.; Moore, C. E.; Morrissette, N. S.;
Jarvo, E. R., Stereospecific Nickel‐Catalyzed Cross‐Coupling Reactions of Alkyl Grignard
Reagents and Identification of Selective Anti‐Breast‐Cancer Agents. Angew. Chem. Int.
Ed. 2014, 53 (9), 2422-2427.
174. Heck, R. F., Dicarboalkoxylation of olefins and acetylenes. Journal of the
American Chemical Society 1972, 94 (8), 2712-2716.
175. (a) For a stoichiometric Ag-mediated a-alkyl-indolation of styrenes, s. O., X.-H.;
Song, R.-J.; Hu, M.; Yang, Y.; Li, J.-H. Angew. Chem. Int. Ed. 2016, 55, 3187; (b) For a
visible light/Ir-catalyzed a-alkyl-indolation of styrenes, see: Li, M.; Yang, J.; Ouyang, X.-
H.; Yang, Y.; Hu, M.; Song, R.-J.; Li, J.-H. J. Org. Chem. 2016, 81, 7148; (c) For
alkylarylation of dienes, see: Takanori, I.; Xin, M.; Asuka, F.; Hitoshi, K.; Nobuaki, K.
Angew. Chem. Int. Ed. 2016, 55, 5550.
176. Jung, K.-Y.; Vanommeslaeghe, K.; Lanning, M. E.; Yap, J. L.; Gordon, C.; Wilder,
P. T.; MacKerell, A. D.; Fletcher, S., Amphipathic α-Helix Mimetics Based on a 1,2-
Diphenylacetylene Scaffold. Org. Lett. 2013, 15 (13), 3234-3237.
177. Ueberschaar, N.; Xu, Z.; Scherlach, K.; Metsä-Ketelä, M.; Bretschneider, T.;
Dahse, H.-M.; Görls, H.; Hertweck, C., Synthetic Remodeling of the Chartreusin Pathway
to Tune Antiproliferative and Antibacterial Activities. J. Am. Chem. Soc. 2013, 135 (46),
17408-17416.

225
178. Molander, G. A.; Brown, A. R., Suzuki−Miyaura Cross-Coupling Reactions of
Potassium Vinyltrifluoroborate with Aryl and Heteroaryl Electrophiles. J. Org. Chem.
2006, 71 (26), 9681-9686.
179. Kolasa, T.; Gunn, D. E.; Bhatia, P.; Woods, K. W.; Gane, T.; Stewart, A. O.;
Bouska, J. B.; Harris, R. R.; Hulkower, K. I.; Malo, P. E.; Bell, R. L.; Carter, G. W.;
Brooks, C. D. W., Heteroarylmethoxyphenylalkoxyiminoalkylcarboxylic Acids as
Leukotriene Biosynthesis Inhibitors. J. Med. Chem. 2000, 43 (4), 690-705.
180. Molander, G. A.; Harring, L. S., Reductive radical cyclizations of haloalkenes
promoted by samarium diiodide. Sequential cyclization/intermolecular carbonyl addition
reactions. The Journal of Organic Chemistry 1990, 55 (25), 6171-6176.
181. Huber, T. A.; Macartney, D. H.; Baird, M. C., Kinetics and Mechanisms of Halogen
Abstraction Reactions of the 17-Electron, Metal-Centered Radical CpCr(CO)3 with
Organic Halides. Organometallics 1995, 14 (2), 592-602.
182. Chemler, S. R.; Fuller, P. H., Heterocycle synthesis by copper facilitated addition
of heteroatoms to alkenes, alkynes and arenes. Chem. Soc. Rev. 2007, 36 (7), 1153-1160.
183. Zhang, L.; Lovinger, G. J.; Edelstein, E. K.; Szymaniak, A. A.; Chierchia, M. P.;
Morken, J. P., Catalytic conjunctive cross-coupling enabled by metal-induced metallate
rearrangement. Science 2016, 351 (6268), 70-74.
184. Lira, R.; Wolfe, J. P., Palladium-Catalyzed Synthesis of N-Aryl-2-benzylindolines
via Tandem Arylation of 2-Allylaniline: Control of Selectivity through in Situ Catalyst
Modification. J. Am. Chem. Soc. 2004, 126 (43), 13906-13907.

226
185. Hay, M. B.; Wolfe, J. P., Palladium-Catalyzed Synthesis of 2,1‘-Disubstituted
Tetrahydrofurans from γ-Hydroxy Internal Alkenes. Evidence for Alkene Insertion into a
Pd−O Bond and Stereochemical Scrambling via β-Hydride Elimination. J. Am. Chem. Soc.
2005, 127 (47), 16468-16476.
186. Borrajo-Calleja, G. M.; Bizet, V.; Mazet, C., Palladium-Catalyzed Enantioselective
Intermolecular Carboetherification of Dihydrofurans. J. Am. Chem. Soc. 2016, 138 (12),
4014-4017.
187. Seashore-Ludlow, B.; Somfai, P., Domino Carbopalladation–Cross-Coupling for
the Synthesis of 3,3-Disubstituted Oxindoles. Org. Lett. 2012, 14 (15), 3858-3861.
188. Timple, J. M. V.; Magalhães, L. G.; Souza Rezende, K. C.; Pereira, A. C.; Cunha,
W. R.; Andrade e Silva, M. L.; Mortensen, O. V.; Fontana, A. C. K., The Lignan (−)-
Hinokinin Displays Modulatory Effects on Human Monoamine and GABA Transporter
Activities. Journal of Natural Products 2013, 76 (10), 1889-1895.
189. Heinonen, S.; Nurmi, T.; Liukkonen, K.; Poutanen, K.; Wähälä, K.; Deyama, T.;
Nishibe, S.; Adlercreutz, H., In Vitro Metabolism of Plant Lignans: New Precursors of
Mammalian Lignans Enterolactone and Enterodiol. Journal of Agricultural and Food
Chemistry 2001, 49 (7), 3178-3186.
190. Barbosa, L. C.; Furtado, R. A.; Bertanha, H. C. C.; Tomazella, I. M.; Costa, E. S.;
Bastos, J. K.; Silva, M. L. A. e.; Tavares, D. C., Chemopreventive Effects of (−)-Hinokinin
against 1,2-Dimethylhydrazine-Induced Genotoxicity and Preneoplastic Lesions in Rat
Colon. Journal of Natural Products 2014, 77 (10), 2312-2315.

227
191. Borne, R. F.; Aboul-Enein, H. Y.; Waters, I. W.; Hicks, J., Synthesis and
cholinergic activity of some structural analogs of pilocarpine. Journal of Medicinal
Chemistry 1973, 16 (3), 245-247.
192. Qin, T.; Cornella, J.; Li, C.; Malins, L. R.; Edwards, J. T.; Kawamura, S.; Maxwell,
B. D.; Eastgate, M. D.; Baran, P. S., A general alkyl-alkyl cross-coupling enabled by redox-
active esters and alkylzinc reagents. Science 2016, 352, 801-805.
193. Saini, V.; Sigman, M. S., Palladium-Catalyzed 1,1-Difunctionalization of Ethylene.
Journal of the American Chemical Society 2012, 134 (28), 11372-11375.
194. Werner, E. W.; Urkalan, K. B.; Sigman, M. S., PdII-Catalyzed Oxidative 1,1-
Diarylation of Terminal Olefins. Organic Letters 2010, 12 (12), 2848-2851.
195. Saini, V.; Liao, L.; Wang, Q.; Jana, R.; Sigman, M. S., Pd(0)-Catalyzed 1,1-
Diarylation of Ethylene and Allylic Carbonates. Organic Letters 2013, 15 (19), 5008-5011.
196. Yahiaoui, S.; Fardost, A.; Trejos, A.; Larhed, M., Chelation-Mediated
Palladium(II)-Catalyzed Domino Heck−Mizoroki/Suzuki−Miyaura Reactions Using
Arylboronic Acids: Increasing Scope and Mechanistic Understanding. The Journal of
Organic Chemistry 2011, 76 (8), 2433-2438.
197. Trejos, A.; Odell, L. R.; Larhed, M., Development of Stereocontrolled
Palladium(II)-Catalyzed Domino Heck/Suzuki β,α-Diarylation Reactions with Chelating
Vinyl Ethers and Arylboronic Acids. ChemistryOpen 2012, 1 (1), 49-56.
198. Wu, X.; Lin, H.-C.; Li, M.-L.; Li, L.-L.; Han, Z.-Y.; Gong, L.-Z., Enantioselective
1,2-Difunctionalization of Dienes Enabled by Chiral Palladium Complex-Catalyzed

228
Cascade Arylation/Allylic Alkylation Reaction. Journal of the American Chemical Society
2015, 137 (42), 13476-13479.
199. Liu, X.; Li, B.; Gu, Z., Palladium-Catalyzed Heck-type Domino Cyclization and
Carboxylation to Synthesize Carboxylic Acids by Utilizing Chloroform as the Carbon
Monoxide Source. The Journal of Organic Chemistry 2015, 80 (15), 7547-7554.
200. Xu, T.; Ko, H. M.; Savage, N. A.; Dong, G., Highly Enantioselective Rh-Catalyzed
Carboacylation of Olefins: Efficient Syntheses of Chiral Poly-Fused Rings. Journal of the
American Chemical Society 2012, 134 (49), 20005-20008.
201. García-Domínguez, A.; Li, Z.; Nevado, C., Nickel-Catalyzed Reductive
Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 2017, 139 ( 20), 6835–6838.
202. Xue, W.; Qu, Z.-W.; Grimme, S.; Oestreich, M., Copper-Catalyzed Cross-Coupling
of Silicon Pronucleophiles with Unactivated Alkyl Electrophiles Coupled with Radical
Cyclization. Journal of the American Chemical Society 2016, 138 (43), 14222-14225.
203. Iwamoto, H.; Akiyama, S.; Hayama, K.; Ito, H., Copper(I)-Catalyzed Stereo- and
Chemoselective Borylative Radical Cyclization of Alkyl Halides Bearing an Alkene
Moiety. Organic Letters 2017, 19 (10), 2614-2617.
204. Liu, Z.; Zeng, T.; Yang, K. S.; Engle, K. M., β,γ-Vicinal Dicarbofunctionalization
of Alkenyl Carbonyl Compounds via Directed Nucleopalladation. Journal of the American
Chemical Society 2016, 138 (46), 15122-15125.
205. Fournet, G.; Balme, G.; Gore, J., Intramolecular nucleophilic attack of
organopalladium compounds resulting from carbopalladation of alkylidenecyclopropanes.
Tetrahedron Lett. 1987, 28 (39), 4533-6.

229
206. González-Bobes, F.; Fu, G. C., Amino Alcohols as Ligands for Nickel-Catalyzed
Suzuki Reactions of Unactivated Alkyl Halides, Including Secondary Alkyl Chlorides,
with Arylboronic Acids. Journal of the American Chemical Society 2006, 128 (16), 5360-
5361.
207. Powell, D. A.; Maki, T.; Fu, G. C., Stille Cross-Couplings of Unactivated
Secondary Alkyl Halides Using Monoorganotin Reagents. Journal of the American
Chemical Society 2005, 127 (2), 510-511.
208. Davin, L. B.; Lewis, N. G., An historical perspective on lignan biosynthesis:
Monolignol, allylphenol and hydroxycinnamic acid coupling and downstream metabolism.
Phytochem. Rev. 2003, 2 (3), 257.
209. Saleem, M.; Kim, H. J.; Ali, M. S.; Lee, Y. S., An update on bioactive plant lignans.
Natural Product Reports 2005, 22 (6), 696-716.
210. Lee, K. H.; Beers, S. A.; Mori, M.; Wang, Z. Q.; Kuo, Y. H.; Li, L.; Liu, S. Y.;
Chang, J. Y.; Han, F. S.; Cheng, Y. C., Antitumor agents. 111. New 4-hydroxylated and 4-
halogenated anilino derivatives of 4'-demethylepipodophyllotoxin as potent inhibitors of
human DNA topoisomerase II. J. Med. Chem. 1990, 33 (5), 1364-1368.
211. Stevenson, R.; Block, E., Lignan lactones. Synthesis of (+-)-collinusin and
justicidin B. J. Org. Chem. 1971, 36 (22), 3453-3455.
212. Messiano, G. B.; Vieira, L.; Machado, M. B.; Lopes, L. M. X.; de Bortoli, S. A.;
Zukerman-Schpector, J., Evaluation of Insecticidal Activity of Diterpenes and Lignans
from Aristolochia malmeana against Anticarsia gemmatalis. J. Agr. Food Chem. 2008, 56
(8), 2655-2659.

230
213. Sartorelli, P.; Salomone Carvalho, C.; Quero Reimão, J.; Lorenzi, H.; Tempone, A.
G., Antitrypanosomal Activity of a Diterpene and Lignans Isolated from Aristolochia
cymbifera. Planta Med. 2010, 76 (13), 1454-1456.
214. Rojas-Sepúlveda, A. M.; Mendieta-Serrano, M.; Mojica, M. Y. A.; Salas-Vidal, E.;
Marquina, S.; Villarreal, M. L.; Puebla, A. M.; Delgado, J. I.; Alvarez, L., Cytotoxic
Podophyllotoxin Type-Lignans from the Steam Bark of Bursera fagaroides var. fagaroides.
Molecules 2012, 17 (8), 9506.
215. Donoso-Fierro, C.; Tiezzi, A.; Ovidi, E.; Ceccarelli, D.; Triggiani, D.;
Mastrogiovanni, F.; Taddei, A. R.; Pérez, C.; Becerra, J.; Silva, M.; Passarella, D.,
Antiproliferative activity of yatein isolated from Austrocedrus chilensis against murine
myeloma cells: Cytological studies and chemical investigations. Pharm. Biol. 2015, 53 (3),
378-385.
216. Kuo, Y.-C.; Kuo, Y.-H.; Lin, Y.-L.; Tsai, W.-J., Yatein from Chamaecyparis obtusa
suppresses herpes simplex virus type 1 replication in HeLa cells by interruption the
immediate-early gene expression. Antiviral Res. 2006, 70 (3), 112-120.
217. Itoh, T.; Chika, J.; Takagi, Y.; Nishiyama, S., An efficient enantioselective total
synthesis of antitumor lignans: synthesis of enantiomerically pure 4-hydroxyalkanenitriles
via an enzymic reaction. J. Org. Chem. 1993, 58 (21), 5717-5723.
218. Alizadeh, B. H.; Foroumadi, A.; Emami, S.; Khoobi, M.; Panah, F.; Ardestani, S.
K.; Shafiee, A., Isochaihulactone analogues: Synthesis and anti-proliferative activity of
novel dibenzylbutyrolactones. Eur. J. Med. Chem. 2010, 45 (12), 5979-5984.

231
219. Fukuyama, T.; Nishitani, S.; Inouye, T.; Morimoto, K.; Ryu, I., Effective
Acceleration of Atom Transfer Carbonylation of Alkyl Iodides by Metal Complexes.
Application to the Synthesis of the Hinokinin Precursor and Dihydrocapsaicin. Org. Lett.
2006, 8 (7), 1383-1386.
220. Maddaford, S. P.; Charlton, J. L., A general asymmetric synthesis of (-)-.alpha.-
dimethylretrodendrin and its diastereomers. J. Org. Chem. 1993, 58 (15), 4132-4138.
221. DeMartino, M. P.; Chen, K.; Baran, P. S., Intermolecular Enolate Heterocoupling:
Scope, Mechanism, and Application. J. Am. Chem. Soc. 2008, 130 (34), 11546-11560.
222. Ting, C. P.; Maimone, T. J., C H Bond Arylation in the Synthesis of Aryltetralin
Lignans: A Short Total Synthesis of Podophyllotoxin. Angew. Chem. Int. Ed. 2014, 53
(12), 3115-3119.
223. Welin, E. R.; Warkentin, A. A.; Conrad, J. C.; MacMillan, D. W. C.,
Enantioselective α-Alkylation of Aldehydes by Photoredox Organocatalysis: Rapid Access
to Pharmacophore Fragments from β-Cyanoaldehydes. Angewandte Chemie International
Edition 2015, 54 (33), 9668-9672.
224. Park, J.-E.; Lee, J.; Seo, S.-Y.; Shin, D., Regioselective route for arylnaphthalene
lactones: convenient synthesis of taiwanin C, justicidin E, and daurinol. Tetrahedron Lett.
2014, 55 (4), 818-820.
225. Morimoto, T.; Chiba, M.; Achiwa, K., EFFICIENT SYNTHESIS OF NATURAL
(+)-COLLINUSIN USING CATALYTIC ASYMMETRIC HYDROGENATION WITH
A CHIRAL BISPHOSPHINE-RHODIUM(I) COMPLEX. Chem. Pharm. Bull. 1989, 37
(11), 3161-3163.

232
226. Hu, X., Nickel-catalyzed cross coupling of non-activated alkyl halides: a
mechanistic perspective. Chem. Sci. 2011, 2 (10), 1867-1886.
227. Pérez García, P. M.; Ren, P.; Scopelliti, R.; Hu, X., Nickel-Catalyzed Direct
Alkylation of Terminal Alkynes at Room Temperature: A Hemilabile Pincer Ligand
Enhances Catalytic Activity. ACS Catal. 2015, 5 (2), 1164-1171.
228. Budnikova, H. Y.; Vicic, A. D.; Klein, A., Exploring Mechanisms in Ni Terpyridine
Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics 2018, 6 (1).
229. Anderson, T. J.; Jones, G. D.; Vicic, D. A., Evidence for a NiI Active Species in
the Catalytic Cross-Coupling of Alkyl Electrophiles. J. Am. Chem. Soc. 2004, 126 (26),
8100-8101.
230. Jones, G. D.; Martin, J. L.; McFarland, C.; Allen, O. R.; Hall, R. E.; Haley, A. D.;
Brandon, R. J.; Konovalova, T.; Desrochers, P. J.; Pulay, P.; Vicic, D. A., Ligand Redox
Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl−Alkyl Cross-
Coupling Catalyst. J. Am. Chem. Soc. 2006, 128 (40), 13175-13183.
231. Jones, G. D.; McFarland, C.; Anderson, T. J.; Vicic, D. A., Analysis of key steps in
the catalytic cross-coupling of alkyl electrophiles under Negishi-like conditions. Chemical
Communications 2005, (33), 4211-4213.
232. Zhou, J.; Fu, G. C., Cross-Couplings of Unactivated Secondary Alkyl Halides:
Room-Temperature Nickel-Catalyzed Negishi Reactions of Alkyl Bromides and Iodides.
J. Am. Chem. Soc. 2003, 125 (48), 14726-14727.
233. Son, S.; Fu, G. C., Nickel-Catalyzed Asymmetric Negishi Cross-Couplings of
Secondary Allylic Chlorides with Alkylzincs. J. Am. Chem. Soc. 2008, 130 (9), 2756-2757.

233
234. Smith, S. W.; Fu, G. C., Nickel-Catalyzed Negishi Cross-Couplings of Secondary
Nucleophiles with Secondary Propargylic Electrophiles at Room Temperature. Angew.
Chem. Int. Ed. 2008, 47 (48), 9334-9336.
235. Lin, X.; Phillips, D. L., Density Functional Theory Studies of Negishi Alkyl–Alkyl
Cross-Coupling Reactions Catalyzed by a Methylterpyridyl-Ni(I) Complex. The Journal
of Organic Chemistry 2008, 73 (10), 3680-3688.
236. Lin, X.; Sun, J.; Xi, Y.; Lin, D., How Racemic Secondary Alkyl Electrophiles
Proceed to Enantioselective Products in Negishi Cross-Coupling Reactions.
Organometallics 2011, 30 (12), 3284-3292.
237. Stork, G.; Mook, R.; Biller, S. A.; Rychnovsky, S. D., Free-radical cyclization of
bromo acetals. Use in the construction of bicyclic acetals and lactones. J. Am. Chem. Soc.
1983, 105 (11), 3741-3742.
238. Krasovskiy, A.; Malakhov, V.; Gavryushin, A.; Knochel, P., Efficient Synthesis of
Functionalized Organozinc Compounds by the Direct Insertion of Zinc into Organic
Iodides and Bromides. Angew. Chem. Int. Ed. 2006, 45 (36), 6040-6044.
239. Krasovskiy, A.; Knochel, P., Convenient Titration Method for Organometallic
Zinc, Magnesium, and Lanthanide Reagents. Synthesis 2006, 2006 (05), 0890-0891.
240. Kuehne, M. E.; He, L.; Jokiel, P. A.; Pace, C. J.; Fleck, M. W.; Maisonneuve, I. M.;
Glick, S. D.; Bidlack, J. M., Synthesis and Biological Evaluation of 18-
Methoxycoronaridine Congeners. Potential Antiaddiction Agents. Journal of Medicinal
Chemistry 2003, 46 (13), 2716-2730.

234
241. Lokantha Rai, K. M.; Hassner, A., Synthetic methods. 33. Versatile methods of
synthesis of 5- to 8-membered ring N-heterocycles via intramolecular cycloadditions of
allylamines. Heterocycles 1990, 30 (2), 817-830.
242. Alcaide, B.; Almendros, P.; Alonso, J. M., Ruthenium-Catalyzed Chemoselective
N-Allyl Cleavage: Novel Grubbs Carbene Mediated Deprotection of Allylic Amines.
Chemistry – A European Journal 2003, 9 (23), 5793-5799.
243. Middleton, D. S.; Simpkins, N. S., A Mild Method for the Preparation of
Functionalised, Unsymmetrical Ketene Acetals. Synth. Commun. 1989, 19 (1-2), 21-29.
244. Seel, S.; Dagousset, G.; Thaler, T.; Frischmuth, A.; Karaghiosoff, K.; Zipse, H.;
Knochel, P., Preparation of Stereodefined Secondary Alkyllithium Compounds. Chemistry
– A European Journal 2013, 19 (14), 4614-4622.
245. Moriya, O.; Kakihana, M.; Urata, Y.; Sugizaki, T.; Kageyama, T.; Ueno, Y.; Endo,
T., A new synthetic route to alkenyl-substituted tetrahydrofurans via successive intra- and
inter-molecular radical reactions. Journal of the Chemical Society, Chemical
Communications 1985, (20), 1401-1402.
246. Jaseer, E. A.; Naidu, A. B.; Kumar, S. S.; Rao, R. K.; Thakur, K. G.; Sekar, G.,
Highly stereoselective chlorination of [small beta]-substituted cyclic alcohols using PPh3-
NCS: factors that control the stereoselectivity. Chemical Communications 2007, (8), 867-
869.
247. Benischke, A. D.; Le Corre, G.; Knochel, P., Preparation of Polyfunctional
Organozinc Halides by an InX3- and LiCl-Catalyzed Zinc Insertion to Aryl and Heteroaryl
Iodides and Bromides. Chemistry – A European Journal 2017, 23 (4), 778-782.

235
248. Bouissane, L., Synthesis and 1,3-dipolar cycloaddition reactions of new
pyrazolo[1,5,4-ef][1,5]benzodiazepines. Heterocycles 2004, 63 (7), 1651-1658.
249. Kim, S., Facile generation of alkyl, aminyl, and alkoxy radicals from Se-phenyl
benzoselenohydroximate derivatives. Synlett 1997, (8), 950-952.
250. Yan, C.-S., Nickel-Mediated Inter- and Intramolecular Reductive Cross-Coupling
of Unactivated Alkyl Bromides and Aryl Iodides at Room Temperature. Chemistry - A
European Journal 2012, 18 (19), 6039-6048.
251. Brown, E., Lignans. 10. Preparation of (R)-(+)- and (S)-(-)-b-piperonyl- and -b-
veratryl-g-butyrolactones and their use in the total synthesis of optically active lignans.
Tetrahedron 1989, 45 (1), 141-154.
252. Coyle, E. E.; Doonan, B. J.; Holohan, A. J.; Walsh, K. A.; Lavigne, F.; Krenske, E.
H.; O'Brien, C. J., Catalytic Wittig Reactions of Semi- and Nonstabilized Ylides Enabled
by Ylide Tuning. Angewandte Chemie International Edition 2014, 53 (47), 12907-12911.
253. Wu, W.-B.; Huang, J.-M., Electrochemical Cleavage of Aryl Ethers Promoted by
Sodium Borohydride. The Journal of Organic Chemistry 2014, 79 (21), 10189-10195.
254. Wang, M.; Mickens, J.; Gao, M.; Miller, K. D.; Sledge, G. W.; Hutchins, G. D.;
Zheng, Q.-H., Design and synthesis of carbon-11-labeled dual aromatase–steroid sulfatase
inhibitors as new potential PET agents for imaging of aromatase and steroid sulfatase
expression in breast cancer. Steroids 2009, 74 (12), 896-905.
255. Chen, P.-C.; Tsai, W.-J.; Ueng, Y.-F.; Tzeng, T.-T.; Chen, H.-L.; Zhu, P.-R.;
Huang, C.-H.; Shiao, Y.-J.; Li, W.-T., Neuroprotective and Antineuroinflammatory Effects

236
of Hydroxyl-Functionalized Stilbenes and 2-Arylbenzo[b]furans. Journal of Medicinal
Chemistry 2017, 60 (9), 4062-4073.
256. Elger, B.; Schneider, M.; Winter, E.; Carvelli, L.; Bonomi, M.; Fracasso, C.; Guiso,
G.; Colovic, M.; Caccia, S.; Mennini, T., Optimized Synthesis of AMPA Receptor
Antagonist ZK 187638 and Neurobehavioral Activity in a Mouse Model of Neuronal
Ceroid Lipofuscinosis. ChemMedChem 2006, 1 (10), 1142-1148.
257. Morimoto, T., Asymmetric reactions catalyzed by chiral metal complexes. LV.
Efficient asymmetric syntheses of naturally occurring lignan lactones using catalytic
asymmetric hydrogenation as a key reaction. Tetrahedron 1993, 49 (9), 1793-1806.
258. Everson, D. A.; Shrestha, R.; Weix, D. J., Nickel-Catalyzed Reductive Cross-
Coupling of Aryl Halides with Alkyl Halides. Journal of the American Chemical Society
2010, 132 (3), 920-921.