crescentic glomerulonephritis: an update on pauci … neeraja kambham, md, department of pathology,...
TRANSCRIPT

Crescentic Glomerulonephritis: An Updateon Pauci-immune and Anti-GBM Diseases
Neeraja Kambham, MD
Abstract: Crescentic glomerulonephritis (GN) in a renal biopsy is awidely accepted “critical diagnosis” in Anatomic Pathology prac-tice. Prompt biopsy evaluation and notification of the referringphysician is essential to facilitate rapid therapeutic intervention.The differential diagnostic categories of crescentic GN includepauci-immune GN, anti-glomerular basement membrane (GBM)nephritis and immune complex-mediated GN, distinguished fromone another by immunofluorescence and electron microscopicstudy of the renal biopsy. Immune complex-mediated GN ischaracterized by abundant glomerular deposits and encompassesseveral diseases including but not limited to lupus nephritis,cryoglobulinemic GN and immunoglobulin A nephropathy. Pauci-immune GN, with paucity of deposits, correlates closely with an-tineutrophil cytoplasmic antibody disease due to the identifiablecirculating pathogenic antineutrophil cytoplasmic antibody in mostpatients. Recent studies have identified other antibodies in pauci-immune GN and implicated infectious organisms in triggeringautoimmunity in a susceptible host by molecular mimicry of hostantigens. Anti-GBM nephritis is a rare but potentially life-threat-ening autoimmune disease with circulating antibodies againstGBM epitopes in a3 chain of type IV collagen. It is characterizedby a linear immunoglobulin G deposition along GBM on im-munofluorescence microscopy. Environmental triggers includinginfections and solvent exposure seem to change the tertiary struc-ture of the type IV collagen a345 hexamer in GBM, expose neo-epitopes, and initiate autoimmunity. Even in light of advancesin understanding of pathophysiology and serologic testing, renalbiopsy remains the mainstay of diagnosis of crescentic GN.
Key Words: crescentic glomerulonephritis, pauci-immune,
anti-GBM, ANCA
(Adv Anat Pathol 2012;19:111–124)
Glomerular crescents in a renal biopsy evoke certaindifferential diagnostic considerations and often, the
need for aggressive therapeutic interventions. Extraca-pillary proliferation within a glomerulus, either partially orcompletely filling up the Bowman’s space defines a crescent(Fig. 1). It is composed of proliferating parietal epithelialcells, podocytes, macrophages, and fibroblasts.1 Crescentformation is stimulated by the entry of fibrin and otherplasma proteins from the capillary lumens and indicatesglomerular basement membrane (GBM) rupture.2 Recrui-tment of T cells, macrophages, and other inflammatory cellsto the site of injury follows, with cytokine release and tissueinjury. These “cellular” crescents may evolve into a chronicphase of fibrocellular or fibrous crescents with increased
extracellular matrix and collagen deposition.2 Identifying adefinite site of GBM rupture in a sclerosed glomerulusshould raise suspicion for previous necrotizing or crescenticglomerular injury.
Crescentic glomerulonephritis (GN) is defined as suchin the presence of crescents in >50% of glomeruli sampledfor renal biopsy.2 This is a critical diagnosis with clinicalimplications that ideally is communicated to the referringphysician immediately. As per the guidelines set forth byAssociation of Directors of Anatomic and Surgical Patho-logy, this represents a practice improvement and patientsafety initiative.3 In most practices, the turnaround time forrenal biopsy results is significantly shorter than con-firmatory serological tests for anti-GBM nephritis and an-tineutrophil cytoplasmic antibodies (ANCA)-associatedGN; thus, prompt reporting is crucial. It cannot be over-emphasized that it is also important to document suchnotification of critical diagnoses. Although not a “criticaldiagnosis,” the presence of even a single crescent in a renalbiopsy can indicate potential rapid progression of disease.This finding may fall under the category of “urgent orsignificant, unexpected diagnosis” and rapid institution oftherapy may be beneficial in such cases. It is recommendedthat each institution develop their own policies and proce-dures regarding effective communication of such importantdiagnoses.4
Typically, a patient with crescentic GN presents withnephritic syndrome and rapid deterioration of renal functionwithin 3 months or less. This clinical syndrome is referred to as“rapidly progressive glomerulonephritis” (RPGN) or rapidlyprogressive renal failure and is often considered synonymouswith histological evidence of crescent GN.2 The proteinuria increscentic GN is usually subnephrotic (<3.5g/d) and is ac-companied by hematuria, oliguria, and hypertension, all fea-tures of nephritic syndrome. Urinalysis may reveal dysmorphicred blood cells (RBC) or RBC casts, supportive of a glomer-ular origin of bleed.2 On occasion, other diseases that promptvastly different therapy such as thrombotic microangiopathycan present with RPGN, underscoring the importance ofa renal biopsy-proven diagnosis. Serological testing in asuspected case of RPGN includes screening for relevant anti-bodies including but limited to, anti-GBM, ANCA and anti-DNA antibodies.
The finding of crescentic GN on light microscopicevaluation prompts a differential diagnosis encompassing 3distinct categories of diseases: pauci-immune GN, anti-GBMantibody-associated GN, and immune complex-mediatedGN.2 Most cases of crescentic immune complex-mediatedGN, but not pauci-immune or anti-GBM nephritis, havevariable mesangial and/or endocapillary proliferation in un-affected (ie, without crescents) glomeruli. The extent andseverity of crescentic process including widespread disruptionof Bowman’s capsules tend to be severe in anti-GBM neph-ritis and pauci-immune GN. However, these histological
From the Department of Pathology, Stanford University MedicalCenter, Stanford, CA.
The author has no funding or conflicts of interest to disclose.Reprints: Neeraja Kambham, MD, Department of Pathology,
Stanford University Medical Center, Rm H2110, 300 Pasteur Drive,Stanford, CA (e-mail: [email protected]).
Copyright r 2012 by Lippincott Williams & Wilkins
REVIEW ARTICLE
Adv Anat Pathol � Volume 19, Number 2, March 2012 www.anatomicpathology.com | 111

distinctions are not reliable indicators of subtype of crescenticGN. In addition to clinical and serological parameters, fur-ther classification of crescentic GN rests largely on im-munofluorescence microscopic (IF) evaluation of the renalbiopsy, with additional diagnostic electron microscopic (EM)findings in some cases. Although all 3 categories are indeed“immune-mediated,” the immune complex-mediated GNrefers to the presence of immune complex deposits visible byboth IF (granular deposits) and EM. As the name suggests,pauci-immune GN has relative absence of deposits by bothIF and EM. Anti-GBM nephritis is characterized by linearimmunoglobulin (Ig) G staining along GBM, but no depositsare visible by EM, possibly due to uniform binding ofcirculating autoantibodies to GBM epitopes rather thanformation of clumps of deposits.
In general, pauci-immune GN is the most frequentcause of crescentic GN in all age groups and among pa-tients older than 60 years of age, it accounts for up to 80%of cases.2,5,6 It is also one of the most common diagnosisrendered when necrotizing arteritis is encountered on renalbiopsy (accounts for >80% of arteritis).2,7 Although im-mune complex-mediated GN may reveal associated arteritison biopsy as in cryoglobulinemia, lupus nephritis, orHenoch-Schonlein purpura, it is rather infrequent.2,7 Isolatedanti-GBM nephritis does not cause necrotizing vasculitisand if present, should raise concern for concurrent pauci-immune disease.
This review will focus mainly on pauci-immune GNand anti-GBM nephritis, highlighting the recent insightsgained into their pathogenic mechanisms. Detailed dis-cussion about immune complex-mediated GN is beyond thescope of this review, but salient renal biopsy findings arelisted in Table 1 and Figure 2.
PAUCI-IMMUNE GLOMERULONEPHRITISMore than half of crescentic GN have complete lack of
or relative paucity of immune complex deposits as dem-onstrated by IF and/or EM, and thus belong to pauci-im-mune GN category.2,5 A vast majority of patients withpauci-immune GN have circulating ANCA and hence, thisentity is often termed as ANCA-mediated GN.2,8 Pauci-immune GN is a renal manifestation of ANCA-associatedsmall-vessel vasculitides with involvement of small arteries,arterioles, venules, and capillaries (and thus glomeruli).ANCA-associated vasculitides involving the kidney includemicroscopic polyangiitis (MPA), granulomatosis with poly-angiitis (GPA; formerly known as Wegener’s granulomatosis),Churg-Strauss syndrome (CSS) and a renal-limited form.5,9
Necrotizing vasculitides of medium and small-sized arteries inthe absence of GN or involvement of arterioles, venules, orcapillaries can be observed in the setting of polyarteritis no-dosa, an entity very distinct from (and not be confused with)MPA.5 Polyarteritis nodosa is linked to hepatitis B and
FIGURE 1. A, Glomerular cellular crescents with severely compressed capillary tufts preclude an adequate evaluation of mesangium andcapillary lumens [Jones methenamine silver stain (JMS); �400]. B, C, In addition to a crescent, foci of GBM rupture with extravasatedfibrin (long arrows) are evident on JMS (B) and trichrome (C) stain (�400). D, Crescentic GN is often associated with prominentinterstitial inflammation and acute tubular injury as seen here with loss of tubular brush borders and sloughed epithelial cells (*; �200).E, F, Fibrocellular (arrowhead) and fibrous crescents represent subacute and chronic glomerular changes. Focal capillary loops may beseen enmeshed in a crescent (short arrow) [periodic acid-Schiff stain (PAS), �400]. GBM indicates glomerular basement membrane;GN, glomerulonephritis.
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
112 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins
possibly hepatitis C infection, is ANCA negative and usuallydoes not demonstrate glomerular crescents.
Clinical PresentationWith a worldwide prevalence, the mean age at pre-
sentation for pauci-immune GN is 60 years and there is nosignificant sex predilection.2,5 Constitutional flu-likesymptoms such as fever, malaise, myalgias, and arthralgiasoften precede the onset of disease. The renal manifestationsinclude RPGN, and in one study, the mean serum crea-tinine at presentation was 6.5±4.0mg/dL.2 Urinalysis re-veals dysmorphic RBCs and frequently, RBC casts. Theoverall presentation with elevated serum creatinine can bevery nonspecific and a high index of suspicion is needed toinitiate further work up and confirm the diagnosis.
ANCA disease is usually a multisystem disorder andpulmonary renal syndrome is a common presentation.2,5,10
The pulmonary symptoms range from mild cough andblood tinged sputum to severe hemoptysis due to life-threatening massive pulmonary hemorrhage. A suddendrop in hemoglobin levels should raise concern for an in-ternal hemorrhage, even if not otherwise clinically evident.Bilateral nodular infiltrates and cavitary lesions may beseen on imaging studies, especially with GPA and CSS.
Head and neck involvement is quite common in GPA andmay take the form of recurrent sinusitis, nasal septumdamage, orbital disease, or otitis media. CSS is charac-terized by relatively less frequent renal involvement, andthese patients have allergic rhinitis, asthma, peripheraleosinophilia, and in some instances eosinophilic pneumoniaor gastroenteritis.2,5,10 Interestingly, the onset of asthma inCSS can precede small-vessel vasculitides by a few years oreven decades.
Localized symptoms may vary based on the organinvolvement by vasculitis such as skin purpura (leukocy-toclastic vasculitis), livedo reticularis, urtricaria, peripheralneuropathy (especially mononeuritis multiplex), meningitis,hemetemesis, melena, etc.11 Overall, signs of extrarenalvasculitis are observed in >75% of patients.2,5,10 Lesscommon localized forms of ANCA disease include renal-limited disease and conversely a limited form of GPA withonly upper and lower respiratory tract disease, withoutrenal or systemic involvement.10,12
Laboratory InvestigationsPauci-immune GN is associated with normal se-
rum complement levels and laboratory studies to excludecryoglobulinemia or lupus nephritis are often indicated.
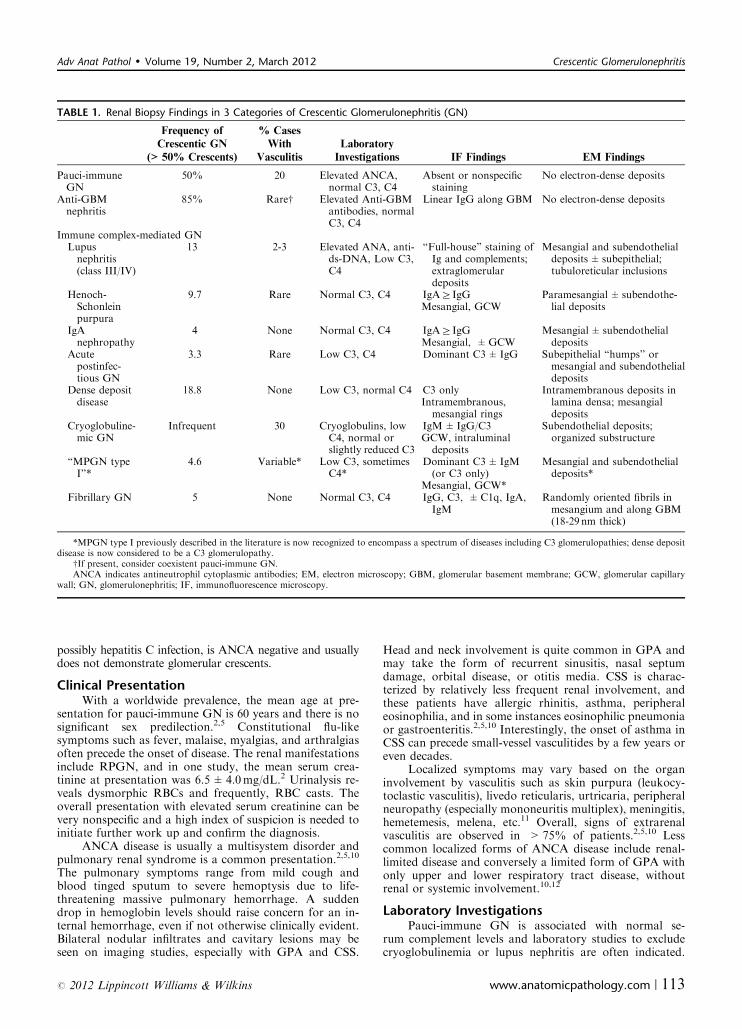
TABLE 1. Renal Biopsy Findings in 3 Categories of Crescentic Glomerulonephritis (GN)
Frequency of
Crescentic GN
(>50% Crescents)
% Cases
With
Vasculitis
Laboratory
Investigations IF Findings EM Findings
Pauci-immuneGN
50% 20 Elevated ANCA,normal C3, C4
Absent or nonspecificstaining
No electron-dense deposits
Anti-GBMnephritis
85% Rarew Elevated Anti-GBMantibodies, normalC3, C4
Linear IgG along GBM No electron-dense deposits
Immune complex-mediated GNLupusnephritis(class III/IV)
13 2-3 Elevated ANA, anti-ds-DNA, Low C3,C4
“Full-house” staining ofIg and complements;extraglomerulardeposits
Mesangial and subendothelialdeposits±subepithelial;tubuloreticular inclusions
Henoch-Schonleinpurpura
9.7 Rare Normal C3, C4 IgAZIgGMesangial, GCW
Paramesangial±subendothe-lial deposits
IgAnephropathy
4 None Normal C3, C4 IgAZIgGMesangial, ±GCW
Mesangial± subendothelialdeposits
Acutepostinfec-tious GN
3.3 Rare Low C3, C4 Dominant C3±IgG Subepithelial “humps” ormesangial and subendothelialdeposits
Dense depositdisease
18.8 None Low C3, normal C4 C3 onlyIntramembranous,mesangial rings
Intramembranous deposits inlamina densa; mesangialdeposits
Cryoglobuline-mic GN
Infrequent 30 Cryoglobulins, lowC4, normal orslightly reduced C3
IgM±IgG/C3GCW, intraluminaldeposits
Subendothelial deposits;organized substructure
“MPGN typeI”*
4.6 Variable* Low C3, sometimesC4*
Dominant C3±IgM(or C3 only)
Mesangial, GCW*
Mesangial and subendothelialdeposits*
Fibrillary GN 5 None Normal C3, C4 IgG, C3, ±C1q, IgA,IgM
Randomly oriented fibrils inmesangium and along GBM(18-29 nm thick)
*MPGN type I previously described in the literature is now recognized to encompass a spectrum of diseases including C3 glomerulopathies; dense depositdisease is now considered to be a C3 glomerulopathy.
wIf present, consider coexistent pauci-immune GN.ANCA indicates antineutrophil cytoplasmic antibodies; EM, electron microscopy; GBM, glomerular basement membrane; GCW, glomerular capillary
wall; GN, glomerulonephritis; IF, immunofluorescence microscopy.
Adv Anat Pathol � Volume 19, Number 2, March 2012 Crescentic Glomerulonephritis
r 2012 Lippincott Williams & Wilkins www.anatomicpathology.com | 113

FIGURE 2. Immune complex-mediated crescentic GN is often associated with mesangial and/or endocapillary proliferation. A–C, Biopsyfindings of diffuse proliferative lupus nephritis (A: PAS, �400) with mesangial and capillary wall “full-house” staining including IgG (B),IgA, IgM, C3, and C1q. Corresponding mesangial (*) and subendothelial (arrowhead) electron-dense deposits are seen (�4000) alongwith tubuloreticular inclusions (inset C, �25,000). D–F, Renal features of Henoch-Schonlein purpura. The glomerulus shows crescentand mesangial sclerosis along with increased endocapillary cellularity (JMS, �400); IF demonstrates dominant IgA (E) staining andsubendothelial deposits (arrowhead) are observed on EM (F, �10,000). G–I, Postinfectious GN with diffuse glomerular proliferation andnumerous neutrophils (exudative pattern) seen on H&E stain (G, �400). The deposits on IF are granular and strongly positive for C3 (H)and characteristic subepithelial “humps” (arrow) are identified on EM (I, �12,000). J–L, Cryoglobulinemic GN shows characteristicmembranoproliferative features (JMS, �400) and, on occasion, necrotizing arteritis ( ). IF demonstrates IgM and C3 staining (notshown) and EM shows reduplicated GBMs ( > ) along with subendothelial deposits. EM indicates electron microscopy; GBM, glomerularbasement membrane; GN, glomerulonephritis; H&E, hematoxylin-eosin; IF, immunofluorescence microscopy; Ig, immunoglobulin;JMS, Jones methenamine silver stain; PAS, periodic acid-Schiff.
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
114 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins

Davies and colleagues first reported ANCA in pauci-immuneGN patients and its significance has been confirmed bysubsequent studies.13–15 ANCA testing can be performedeither by indirect IF or by enzyme-linked immunosorbentassay (ELISA) using purified specific antigens proteinase-3(PR3) and myeloperoxidase (MPO).10 Both PR3 and MPOare components of neutrophil and monocyte lysosomes.Traditionally, the IF testing involves incubation of patient’sserum with ethanol fixed human neutrophils as a substrate todetect the presence of circulating antibodies. Two patterns ofANCA staining observed on indirect IF include perinuclearwith nuclear extension (p-ANCA) and cytoplasmic granularwith interlobular accentuation pattern (c-ANCA).16 Classicc-ANCA is almost always directed against PR3, but p-ANCAmay be directed against MPO or PR3. Hence, indirect IF andELISA should test for both subcategories of IF patterns andspecific antigens. The frequency of ANCA subtypes is dif-ferent in various ANCA vasculitides (Table 2). In addition toclassic IF patterns, atypical ANCA patterns have been rec-ognized and these include cytoplasmic homogenous pattern(c-ANCA atypical) or perinuclear without nuclear extension(p-ANCA atypical).
In general, indirect IF is more sensitive while ELISAtest is more specific, but certain caveats need to be con-sidered.8,16 Nonspecific or atypical ANCA (positive by IF,but ELISA for PR3 and MPO negative) can be detected innonvasculitic autoimmune or inflammatory conditions (eg,ulcerative colitis, autoimmune hepatitis, sclerosing chol-angitis), and reportedly with cocaine abuse.17 The antigentargets responsible for such pattern include lactoferrin,elastase, cathepsin-G, lysozyme, and other minor antigensnot routinely tested in the laboratories.8,16 Factors such asformalin fixation of neutrophil substrate, in-house prepa-ration of substrate, or different commercial substrates andELISA kits seemingly contribute to variable sensitivitiesand specificities of indirect IF and ELISA reported in theliterature. The International Consensus Statement for ANCAtesting recommends that all sera for ANCA be screened firstby indirect IF followed by a positive result confirmation byELISA.16,18 However, on rare occasion, ELISA may be
positive in the context of negative indirect IF test and in thatlight, there may be a role for simultaneous IF and ELISAtesting.17 In general, testing for ANCA by indirect IF orELISA alone is not be ideal.
The positive and negative predictive values of thesetests are closely linked to the clinical presentation.19,20 Inthe setting of RPGN, the positive predictive value is ap-proximately 99%, but negative predictive value is only 80%(and 65% in patients older than 50 years), indicating theneed for pursuing further studies such as a renal biopsydespite a negative ANCA test. It is now well documentedthat approximately 10% to 20% of biopsy-proven pauci-immune GN patients lack indirect IF ANCA and ELISAsupport. CSS patients have higher frequency of false-neg-ative ANCA, especially in the absence of renal disease, asdo patients with limited form of GPA.17,19,20 On the otherend of the spectrum, if the patient has minimally increasedserum creatinine with hematuria and mild proteinuria, thenegative predictive value of ANCA test is 99%, essentiallyruling out ANCA disease. The positive predictive value inthis clinical scenario is also low (47% to 66% depending onthe age of patient). Thus, an ANCA serology result shouldbe interpreted with caution and always in the context ofclinical presentation. Although active pauci-immune dis-ease usually has high ANCA levels, there are no standardANCA units and the levels of antibody reported are labo-ratory specific. Of note, several medications and infectionscan induce ANCA, but this does not necessarily representactive disease with vasculitis.16
PathologyGiven the long turnaround times for ELISA results, a
renal biopsy is of immense value in the diagnosis of clin-ically suspected pauci-immune GN. Even in patients withpredominantly pulmonary symptoms, relative to lungsampling, a renal biopsy is associated with ease of IF in-terpretation, higher yield of definitive diagnosis, and alower procedural risk.21 The biopsy findings of pauci-immune GN are represented in Figure 3.
TABLE 2. Clinicopathological Subtypes of Pauci-immune Glomerulonephritis
Granulomatosis With
Polyangiitis (GPA)
Microscopic
Polyangiitis (MPA)
Churg-Strauss Syndrome
(CSS)
Renal-limited
Disease
Frequency of renalinvolvement (%)
80 90 45w 100
Other frequentlyinvolved organs
Pulmonary, head andneck (90%)
Pulmonary (50%),musculoskeletal(60%)
Pulmonary, neurologic(70%)
None
Other manifestations Constitutionalsymptoms
Constitutionalsymptoms
Constitutional symptoms,asthma, allergic rhinitis,blood eosinophilia
Constitutionalsymptoms
Frequency of ANCApositivity (%)
95 90 70 80
ANCA subtype(MPO:PR3%)
25:75 50:50 60:40 60:40
Additional renalbiopsy findings
Interstitialgranulomatousinflammation rare*
None Eosinophil-rich infiltrate;interstitial granulomatousinflammation rare*
None
*Granulomatous inflammation in GPA and CSS is more often described in respiratory tract and in the case of GPA also in head and neck lesions.wCan be even lower, B25% in the absence of circulating ANCA.ANCA indicates antineutrophil cytoplasmic antibody; PR3, Proteinase 3; MPO, myeloperoxidase.
Adv Anat Pathol � Volume 19, Number 2, March 2012 Crescentic Glomerulonephritis
r 2012 Lippincott Williams & Wilkins www.anatomicpathology.com | 115
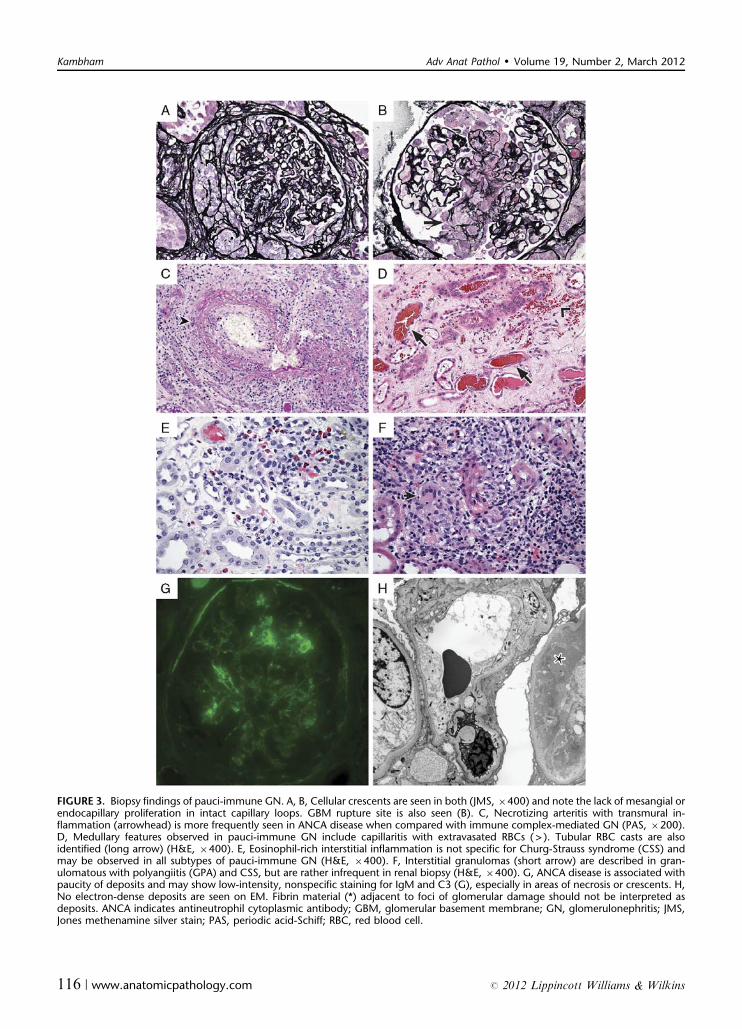
FIGURE 3. Biopsy findings of pauci-immune GN. A, B, Cellular crescents are seen in both (JMS, �400) and note the lack of mesangial orendocapillary proliferation in intact capillary loops. GBM rupture site is also seen (B). C, Necrotizing arteritis with transmural in-flammation (arrowhead) is more frequently seen in ANCA disease when compared with immune complex-mediated GN (PAS, �200).D, Medullary features observed in pauci-immune GN include capillaritis with extravasated RBCs ( > ). Tubular RBC casts are alsoidentified (long arrow) (H&E, �400). E, Eosinophil-rich interstitial inflammation is not specific for Churg-Strauss syndrome (CSS) andmay be observed in all subtypes of pauci-immune GN (H&E, �400). F, Interstitial granulomas (short arrow) are described in gran-ulomatous with polyangiitis (GPA) and CSS, but are rather infrequent in renal biopsy (H&E, �400). G, ANCA disease is associated withpaucity of deposits and may show low-intensity, nonspecific staining for IgM and C3 (G), especially in areas of necrosis or crescents. H,No electron-dense deposits are seen on EM. Fibrin material (*) adjacent to foci of glomerular damage should not be interpreted asdeposits. ANCA indicates antineutrophil cytoplasmic antibody; GBM, glomerular basement membrane; GN, glomerulonephritis; JMS,Jones methenamine silver stain; PAS, periodic acid-Schiff; RBC, red blood cell.
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
116 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins

Light MicroscopyThe cellular crescents can be segmental or circum-
ferential, often with disrupted Bowman’s capsules. In addi-tion, a variable number of glomeruli have areas of necrosis,karyorrhexis, or fibrin extravasation.2,10,15 A GBM rupturesite may be evident on periodic acid-Schiff or silver stain andfibrin is brightly fuschsinophilic (red) on trichrome stain. Fociof necrosis and crescents often have associated inflammatorycells, but there is usually no evidence of mesangial or endo-capillary proliferation in uninvolved segments of glomeruli.The biopsy may demonstrate uniformly acute lesions or mayhave variable combination of subacute or chronic lesionssuch as fibrocellular/fibrous crescents and Bowman’s capsu-lar synechiae. These changes correspond to the rapidity ofonset and duration of ANCA disease. Global glomerulo-sclerosis and chronic tubulointerstitial damage identified canbe attributed to either progression of ANCA GN or preex-isting disease such as hypertensive nephrosclerosis and carefulexamination of sclerosed glomeruli is helpful in distinguishingbetween the two possibilities. Acute tubular injury is oftenobserved with loss of brush borders and sloughed epithelialcells. Occasionally, no necrotizing or crescentic lesions areidentified despite a clinical scenario of RPGN and positiveANCA serology. If the biopsy sample in such cases is limitedby glomerular paucity, consideration should be given to un-sampled ANCA-related crescents, especially if RBC casts areidentified in the biopsy or urine microscopy.
Tubulointerstitial and periglomerular inflammationtends to be prominent in ANCA disease and is likely withinthe spectrum of disease rather than just a secondary phe-nomenon due to glomerular damage.2,10,15 Multinucleatedgiant cells seen on occasion within periglomerular in-flammation may be misinterpreted as interstitial gran-ulomas. Although described in GPA and CSS, interstitialgranulomas are rather infrequent in kidney (2% to 4% ofcases).22 In contrast, granulomatous inflammation in GPAis relatively common in pulmonary and head and neck le-sions. Although eosinophil-rich interstitial inflammation isconsidered as a hallmark of CSS, it is not a helpful featureas eosinophils can be quite prominent in all ANCA dis-eases.23 In essence, the subtype of ANCA vasculitis is oftendifficult to discern on renal biopsy alone. However, thereseem to be differences in the pathology seen in MPO versusPR3-ANCA-associated disease. Although extrarenal dis-ease is more frequent, the extent of glomerular disease andchronic renal damage appears to be less in PR3-ANCAdisease when compared with MPO-ANCA disease.24
Vasculitis can be observed in up to a third of renalbiopsies, characterized by fibrinoid necrosis and transmuralinfiltrates of lymphocytes and neutrophils. The disruptedelastic lamina in arteries may be evident on silver stain. Theaffected blood vessels are usually of small caliber and in-clude interlobular arteries, arterioles and medullary vasarecta.7 When present, medullary capillaritis results in in-terstitial hemorrhage and RBC extravasation admixed withapoptotic bodies. Other medullary-specific lesions describedin ANCA disease include necrotizing arteriolitis, gran-ulomas and papillary tip necrosis.25
Immunofluorescence MicroscopyBy definition, there is a paucity of immune deposits on
IF. Necrotic foci are highlighted by fibrinogen and may dem-onstrate entrapped C3 and IgM. However, no linear or gran-ular deposits of Ig or complements of Z2+ (scale of 0 to 4)intensity should be observed in unaffected glomeruli.2,10,15
Although occasional cases have non-specific–associated im-mune staining,26 prominent deposits in “normal” glomerulishould raise concern for superimposed immune complex-mediated disease.
Electron MicroscopySimilar to IF, no electron-dense deposits are observed
on ultrastructural examination. Electron-dense fibrin ma-terial in the vicinity of necrosis should not be misinterpretedas deposits. On occasion, the ruptured site of GBM may bevisible in the area of fibrinoid necrosis or cellular crescent.The adjacent endothelial cell usually has features of injuryand cytoplasmic swelling.2,10
Pathological ClassificationA recently proposed histological classification27 catego-
rizes the glomerular lesions as focal (>50% normal glomer-uli), crescentic (>50% glomeruli with cellular or fibrocellularcrescents), sclerotic (>50% glomeruli are globally sclerosed),and mixed (heterogenous lesions, none involving >50% ofglomeruli). A normal glomerulus is defined as one with nosegmental sclerosis, GBM double contours, or extensive is-chemic change. This classification is progonstically relevant inthat the 5-year renal survival rates for focal, crescentic, mixed,and sclerotic categories are 93%, 76%, 61%, and 50%, re-spectively.27 The tubulointerstitial parameters did not provideindependent predictive value. The study results reinforce thesignificance of describing the extent and distribution of activeand chronic glomerular lesions in the pathology report.
PathogenesisDespite absence of visible immune deposits, the
pathogenic mechanisms in ANCA disease are evidentlyimmune mediated with development of autoantibodies.14,15
ANCA have been extensively studied in various in vitroexperiments and animal models. Passive transfer of MPO-ANCA IgG induces pauci-immune GN and vasculitis inmice and as per one report, transplacental transfer of thesematernal antibodies caused human neonatal renal pulmo-nary syndrome, confirming the pivotal role of ANCA.14,28
Although abundant in vitro evidence exists for pathogenicrole of MPO-ANCA, convincing animal models are notavailable for PR3-ANCA GN.14,15,29
Upon binding with the target antigens PR3 and MPO,ANCAs activate neutrophils, promote neutrophil-endo-thelial cell binding, and cause release of reactive oxygenspecies and tissue-degrading proteolytic enzymes fromneutrophils with resultant vessel wall injury. In vitro studieshave demonstrated that ANCA-mediated neutrophil acti-vation is mediated by binding of Fab02 and Fc receptorswith subsequent initiation of signal induction pathways andactivation of alternative complement pathway.9,30
PR3 and MPO are intracellular enzymes within neu-trophil and monocyte granules. For the ANCA-mediatedinjury mechanisms to kick in, these antigens have to beexposed on the cell surfaces as in activated state of neu-trophils and monocytes. The milieu of proinflammatorycytokines in the setting of infection is very conducive to thiscell priming.11,14,15 The cytokines released by degranulatingneutrophils and monocytes also, in turn, exacerbate thisautoimmune injury. The fenestrated endothelium in glomer-ulus and negatively charged GBMmay facilitate trapping ofactivated neutrophils and promote the renal predilectionfor injury.20
Adv Anat Pathol � Volume 19, Number 2, March 2012 Crescentic Glomerulonephritis
r 2012 Lippincott Williams & Wilkins www.anatomicpathology.com | 117

Antibodies other than ANCA have been studied in pa-tients with pauci-immune GN. Recently, antibodies to lyso-somal membrane protein-2 (LAMP-2) were detected inpatients with pauci-immune GN at prevalence rates sig-nificantly greater than that of MPO-ANCA and PR3-ANCAcombined.31 The pathogenicity of LAMP-2 antibodies wasconfirmed in in vitro and in vivo studies. LAMP-2 is amembrane surface protein expressed on neutrophils and en-dothelial cells and unlike ANCA, is readily accessible to thecirculating antibodies. LAMP-2 is involved in cell adhesionand homeostasis. Although it is an attractive alternative tointracellular ANCA antigens, a subsequent study could notconfirm the significance of LAMP-2 antibodies.32 Anti-plas-minogen antibodies were detected in approximately 25% ofANCA patients, possibly triggered by sequence homologybetween plasminogen and complementary PR3 (see below)and thus, contributing to deep vein thrombosis.33
What triggers the formation of these autoantibodies?Several lines of evidence point to infectious organisms andtheir successful mimicry of host molecules in initiating au-toimmunity.9 Previous observations that GPA patientsoften are chronic nasal carriers of Staphylococcus aureusand relapses in them can be reduced with antimicrobialtherapy support this hypothesis.34–36 Seasonal variation inthe incidence of pauci-immune GN also suggests a corre-lation with microbial infection.37 Toll-like receptors thatrecognize infectious agents can exacerbate anti-MPO GN inexperimental models.38 ANCA-positive serology has beenreported in many infections including suppurative lungdisease, Pseudomonas infection in cystic fibrosis and sub-acute bacterial endocarditis.16 More recent evidence in-cludes identification of a LAMP-2 epitope with 100%homology to fimbrial adhesin (FimH) seen in gram-negative bacteria.31 It has been hypothesized that immuneresponse to FimH as in urosepsis triggers anti-LAMP-2 antibodies due to molecular mimicry.31,39
Neutrophils can kill bacterial organisms such as S. aureusextracellularly by undergoing respiratory burst and forming aweb of released DNA called “neutrophil extracellular traps.”40
These traps, demonstrated in ANCA vasculitis, contain PR3and MPO antigens, and are thus capable of initiating and ex-acerbating autoimmune disease.41 Ross river virus, S. aureus,and Entamoeba histolytica have sequences similar to com-plementary PR3 peptide.42 Experimental evidence showsthat complementary PR3 peptide (generated by antisense DNAstrand of PR3) is capable of inducing antibodies that inturn generate a second round of antibodies (anti-idiotypic anti-bodies) that have affinity to PR3 antigen.42,43 This cascade ofantibody production continues when a third generation ofantibodies are produced by these anti-idiotypic antibodies withbinding properties to complementary PR3. Although this theoryof autoantigen complementarity generated much enthusiasm,other laboratories are yet to confirm these observations.44
In addition to infections, other environmental factorscan induce ANCA formation. These include silica ex-posure, drugs such as propothiouracil, hydralazine, pen-icillamine, cefotaxime, clozapine, indomethacin, isoniazid,rifampicin, and minocycline.45,46 Patients respond to dis-continuation of the offending drug, but ANCA levels maypersist despite disease remission. There seems to be an as-sociation between development of CSS and use of leuko-triene receptor antagonist in treatment of bronchialasthma.47 Recent reports have implicated antitumor ne-crosis factor a (anti-TNFa) used in the treatment of rheu-matoid arthritis in inducing ANCA48,49 and in a smaller
fraction of patients, ANCA-associated renal disease. Thedetection of circulating MPO or PR3-ANCA was tempo-rally related to anti-TNFa treatment and at least in somecases, the patients responded to drug withdrawal.
Obviously, not all patients exposed to bacterial infections,drugs, or other environmental factors develop pauci-immuneGN. There seems to be a role for genetic susceptibility. Differ-ences in surface expression of PR3/MPO-ANCA on neutro-phils due to loss of epigenetic silencing, gene polymorphismssuch as defective a-1 antitrypsin allele (a physiological inhibitorof PR3), leptin receptor gene, CTLA-4 and CD226 are someexamples accounting for increased risk.9,14,50,51 The presence ofMHC class II gene HLA-DRB1*0401 is associated with higherrisk of developing GPA and HLA-DRB4 with susceptibilityto CSS.
In addition to autoantibodies, T-cell–mediated immuneresponses also have a significant role in pauci-immune GN,especially CSS.52 As is widely known, CD4+ helper T cellswith interferon-g secretion are involved in granuloma for-mation. Patients with pauci-immune GN have T-cell ab-normalities such as dysregulated T-helper cells, defectiveT-regulatory cells52,53 and a preferential Th17 response.Th17 cells are increasingly being implicated in auto-immunity including ANCA disease.54,55 Neutrophil activa-tion by cytokines synthesized by Th17 cells may contributeto exacerbation of GN. Studies have demonstrated thatTh17 inducing toll-like receptors may be a potential linkbetween infection and autoimmunity.38 In addition to au-toantibody responses, B cells can present antigens to T cellsand may modulate T-cell responses in small-vessel vasculi-tis.52 Eosinophil activation with subsequent tissue damagehas been shown to be an important component of patho-genesis in CSS.
Treatment and PrognosisIn the setting of highly suspicious and life-threatening
clinical picture of ANCA, empiric therapy may be initiateddespite pending serological or biopsy results. The treatment issimilar irrespective of the subtype of pauci-immune GN andincludes induction and maintenance immunosuppression.9
Induction of remission is achieved with steroids and cyclo-phosphamide. Plasmapharesis to remove the circulating an-tibody is beneficial for patients with pulmonary hemorrhageand severe kidney disease. Mycophenolate mofetil and aza-thioprine are usually used in maintenance of remission. Theefficacy and safety of B-cell depletion therapies are also beinginvestigated. Anti-TNFa therapy has been considered be-cause of its ability to interfere with immunological pathwaysinvolved in ANCA disease. In light of reports of ANCAdisease in rheumatoid arthritis patients treated with anti-TNFa, the role of this agent is unclear. Approximately athird of patients with ANCA disease relapse and overall, 5-year patient and renal survival rates are in the range of 60%to 70%.2,15 The subtype of ANCA (MPO vs. PR3) is a betterpredictor of outcomes and relapse rates than the specificclinicopathological subtype such as GPA versus MPA. Forexample, PR3-ANCA and lung disease are associated withhigher relapse rates.56,57 A repeat biopsy may be justified insome to determine the extent of chronic changes and toevaluate the options for therapeutic modulation.
ANCA serological titers do correlate with disease ac-tivity and a new negative result in a patient with ANCAportends complete remission. It is generally suggested thatthe patient be in clinical remission before transplantation.58
However, persistently positive ANCA is not predictive of
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
118 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins

relapse and renal transplantation may be successful despitea positive test.59 There are no good predictors of diseaserecurrence after transplantation, which, based on pooledanalysis of multiple studies, is estimated to be 17%.60 Inmost instances, such relapse in a renal allograft occurs morethan 1 year after transplantation.61
ANTIGLOMERULAR BASEMENT MEMBRANENEPHRITIS
Anti-GBM nephritis is a rare autoimmune disordercharacterized by autoantibodies to noncollagenous (NC1)domain of a3 chain of type IV collagen,2 an integral com-ponent of GBM. The clinical presentation is RPGN withbiopsy findings of severe crescentic GN and a linear dep-osition of IgG along the GBM as evidenced by IF. When itis accompanied by pulmonary involvement, it is referred toas anti-GBM disease or “Goodpasture syndrome,” afterErnest Goodpasture who first described the index case withpulmonary renal syndrome in 1919. Anti-GBM disease isby far the most severe form of crescentic GN both in termsof clinical presentation and extent of glomerular involve-ment by crescents.2,62
Clinical PresentationFrequently seen in whites, the incidence of anti-GBM
disease shows a bimodal age distribution, the first peakbeing at 20 to 30 years of age with a male preponderanceand a second peak at 50 to 70 years, mainly in females.2,63
The overall incidence in the United States is <1 case permillion population per year.63 It is quite rare in children,presumably due to structural transition that occurs frompediatric to adult GBM. Renal pulmonary syndrome is afrequent presentation, especially in younger individuals.About a third of patients, primarily older individuals,present with isolated GN. Isolated pulmonary hemorrhageis, however, uncommon in anti-GBM nephritis.2,62,64,65
General malaise and weakness are usual systemic manifes-tations, likely related to anemia and renal disease. Manypatients develop flu-like illness before the onset of diseaseand some report exposure to hydrocarbons.65–67 AlthoughRPGN is the most frequent presentation, older patientsmay present with milder kidney damage and less frequentpulmonary disease.68 On rare occasion, a patient may havenormal renal function despite severe pulmonary hemo-rrhage with the only evidence of anti-GBM nephritis beinglinear IgG along GBM.69 RBC casts on urine microscopyconfirm the glomerular origin of bleeding and the protei-nuria is usually subnephrotic.
Laboratory InvestigationsThe diagnosis of anti-GBM nephritis requires the
demonstration of anti-GBM antibodies, usually IgG class,in the patient’s serum or tissue. The circulating antibodiesare detected by an ELISA test using human or animalGBM or recombinant antigen as a substrate. Althoughcommercially available kits vary in their sensitivity, ELISAtesting is considered most sensitive and specific method ofanti-GBM antibody detection. The anti-GBM antibodylevels also seem to correlate with renal disease activity.70
However, the standard ELISA test can be negative in ap-proximately 10% to 15% of biopsy-proven anti-GBMnephritis, and in some of these cases, the autoantibodies aredetectable only by specialized biosensor analysis.71,72 Aproportion of these cases may have circulating antibodieswith specificity to a3 chain determinants not routinely de-
tected by ELISA substrate or have specificity to other GBMcomponents such as entactin.73,74 Rarely, the circulatingautoantibodies belong to IgA class (rather than IgG) andcan be directed against novel epitopes within GBM in-cluding a1/2 chains or NC1 domains of a5 and a6 chains oftype IV collagen.75–77
Pathology
Light MicroscopyAnti-GBM nephritis is a severe form of crescentic GN
with widespread crescent formation evident on biopsy inmost patients (Fig. 4). More than 80% of the patients haveZ50% glomerular crescents at the time of diagnosis.2,62 Onoccasion, the renal biopsy may demonstrate only focal andsegmental glomerular necrosis. The crescents range fromsegmental to circumferential and tend to be similar inacuity, reflecting the abrupt onset of GN. The Bowman’scapsules are often disrupted extensively and periglomerularinflammation may appear granuloma-like with occasionalmultinucleated giant cells. Although infiltrating leukocytesand granulocytes accompany the crescents and fibrinoidnecrosis, the unaffected segments of glomeruli are normo-cellular, similar to pauci-immune GN. But in some cases,the extensive circumferential crescents with compressedcapillary loops preclude evaluation of mesangium orcapillary lumens. The GBM rupture may be evident onperiodic acid-Schiff or silver stain and the extravasated fi-brin within the urinary space and crescent is highlighted bytrichrome stain. Extensive tubulointerstitial inflammation,interstitial edema, and acute tubular injury often accom-pany the glomerular change. The interstitial infiltrate isusually mixed and may contain several eosinophils. Vas-culitis, if present, should raise suspicion for combined anti-GBM and ANCA disease and a serological confirmation isrecommended. Based on duration of disease before biopsy,subacute and chronic changes can be observed withglomerular fibrocellular and fibrous crescents and chronictubulointerstitial damage.
Immunofluorescence MicroscopyDiffuse linear IgG staining, intensity Z2 (on a scale of
0 to 4), along the GBM is diagnostic of anti-GBM neph-ritis, especially in the setting of clinical RPGN or crescentson biopsy. Nonspecific or low-intensity IgG staining maybe observed in elderly individuals, patients with diabetesmellitus and obesity, allograft biopsies, and autopsy speci-mens. Hence, clinicopathological correlation is of greatrelevance in interpreting these IF results. Also, when indoubt, comparing the intensity of linear staining of IgGwith that of albumin is helpful as anti-GBM nephritis isassociated with IgG intensity greater than that of albumin.If the crescents are extensive, linear IgG staining may bedifficult to identify or may be limited to a few fragments ofremnant GBM in the glomerulus. Linear, but less-intense kand l staining (and on occasion IgA and IgM) is also ob-served in anti-GBM nephritis, whereas C3, if present, tendsto be patchy and granular. Distal tubular basement mem-branes and Bowman’s capsules that express a3 chain oftype IV collagen occasionally have linear IgG staining too.Antisera to fibrin highlight foci of necrosis and cellularcrescents. In rare patients with circulating IgA anti-GBMantibodies, the linear GBM staining is observed with anti-sera to IgA rather than IgG. Linear Ig heavy and/orlight chains may be noted in patients with diabetic
Adv Anat Pathol � Volume 19, Number 2, March 2012 Crescentic Glomerulonephritis
r 2012 Lippincott Williams & Wilkins www.anatomicpathology.com | 119

FIGURE 4. Anti-GBM nephritis is usually a severe form of crescentic GN. A, Unaffected capillary loops, if present, are devoid ofproliferation (JMS, �400). B, Severe destruction of glomerulus and Bowman’s capsule is frequent. Multinucleated cells (arrowhead) andperiglomerular palisading of inflammatory cells (long arrow) should not be misinterpreted as granulomatous inflammation (H&E,�400). C, The IF is diagnostic with linear IgG staining of GBM. D, Fibrinogen stain highlights the cellular crescent of a differentglomerulus. E, F, On EM, the GBM of the affected glomerulus is collapsed (*) and fibrin (short arrow) is seen admixed with the cellularcomponent of crescent. But preserved capillary loops (F) are entirely normal (�4000). EM indicates electron microscopy; GBM,glomerular basement membrane; GN, glomerulonephritis; IF, immunofluorescence microscopy; Ig, immunoglobulin; JMS, Jonesmethenamine silver stain;
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
120 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins

glomerulosclerosis, idiopathic nodular sclerosis, and mon-oclonal Ig deposition disease, but light microscopy is dis-tinctly different in these entities with nodular mesangialsclerosis rather than glomerular crescents. In rare cases ofanti-GBM nephritis with coexistent membranous nephr-opathy, granular capillary wall IgG staining may be barelydiscernable in the presence of linear pattern and ultra-structural confirmation is needed.
Electron MicroscopyOther than confirming the lack of electron-dense de-
posits, ultrastructural features in anti-GBM nephritis arediagnostically nonspecific and show crescents and GBMbreaks in the acute phase. Some of the changes observedadjacent to necrotic foci include subendothelial expansionby lucent material, margination of neutrophils and mono-cytes in capillary lumens, and fibrin tactoids in crescents.Increasing fibrosis and collagen deposition is seen as thelesions evolve into chronic phase with fibrous crescents.These EM findings are similar to pauci-immune GN. In thesetting of concurrent membranous nephropathy, sub-epithelial electron-dense deposits are observed.
A renal biopsy is invaluable in the diagnosis of anti-GBM nephritis due to the ease of interpretation and a shortturnaround time. In contrast, confirmatory serological teststypically take a few days. Even in cases with predominantpulmonary involvement, a renal biopsy is preferred overtransbronchial biopsies that have a significant rate of false-negative IF results. Lung biopsy IF interpretation is ratherdifficult due to high background, autofluorescence, andirregular poorly localized pattern of IgG staining in anti-GBM disease.
PathogenesisThe pathogenicity of anti-GBM antibodies was first
demonstrated by Lerner et al,78 by passive transfer of dis-ease in monkeys by injecting antibodies from patients withlinear IgG-positive glomerulonephritis. Subsequent animalstudies have confirmed causative role of anti-GBM anti-bodies.79,80 Temporal associations between relapse and re-emergence of antibodies and prognostic value of anti-GBMlevels also support this hypothesis.70 The susceptibility forautoantibody generation may be genetically determined assupported by strong association between HLA DR and DQantigens and anti-GBM disease.81 Despite geographic var-iations, patients with HLA-DRB1*1501 alleles seem proneto develop anti-GBM nephritis.81,82 Environmental triggerssuch as viral infections, exposure to organic solvents (glueand solvent sniffing), and cigarette smoke probably are“second hits” to developing autoimmunity. It is well knownthat respiratory infections precede anti-GBM nephritiscausing flu-like symptoms and miniepidemics of anti-GBMnephritis have been reported.65,67
In vast majority of cases, anti-GBM antibodies targetthe NC1 domain, specifically epitopes within EA and EB
regions of a3 chain of type IV collagen.14,63,83 This antigen,also referred to as “Goodpasture antigen,” is an importantconstituent of the kidney GBM as well as alveolar capillarybasement membranes, choroid plexus, and eye.84 NormalGBM is composed of a3, 4, and 5 chains of type IV col-lagen assembled as triple helical protomers. Two triplehelical protomers are associated together at the C-terminalNC1 domain forming a hexamer that is in turn stabilized bysulfilimine bonds.85 In the presence of intact sulfiliminebonds, anti-GBM antibodies cannot bind to its hidden
target antigen. It is postulated that triggering events maychange the conformation of these hexamers, expose neo-epitopes, elicit autoantibody production, and facilitate an-tigen-antibody binding.83 Some possible triggers includeenzymatic and nonenzymatic modifications (oxidation, ni-trosylation, glycation), rise in body temperature, and pro-teolytic cleavage. Environmental exposure to cigarettesmoke or organic solvents may inhibit the enzymes thatcatalyze the sulfilimine bond formation and alter the qua-ternary structure of the hexamer.86 These events may alsoinitiate epitope spreading and in fact, many patients withanti-GBM nephritis have circulating autoantibodies to a5NCI monomer in addition to expected anti-a3 NCI anti-bodies.83,85 The specificity of these antibodies is similarirrespective of involvement of either lung or kidney.
In addition to autoantibodies, T cells and macrophageshave a role in exacerbating the inflammatory process andglomerular injury in anti-GBM disease.2,87 Antigen-anti-body binding activates complement and recruitment ofneutrophils and monocytes which facilitate GBM ruptureand crescent formation.
Treatment and PrognosisEarly diagnosis and treatment is of paramount im-
portance for improved outcomes in anti-GBM nephritis.High-dose corticosteroids and cyclophosphamide therapyneed to be instituted rapidly and plasmapharesis is useful inremoval of circulating antibodies. Severe renal disease re-quiring dialysis at presentation and serum creatinine>5mg/dL is associated with poor prognosis. In general,anti-GBM nephritis has worse prognosis than pauci-immune GN or immune complex-mediated GN. Relapse ofanti-GBM nephritis as an active disease is quite infrequent,but can occur even after several years. Anti-GBM nephritisrecurrence in renal allografts is uncommon, especially if theantibody titers remain low.
De novo anti-GBM nephritis can occur in approx-imately 3% to 5% of patients with Alport syndrome whoundergo renal transplantation. It is most frequently seen inmales with X-linked mutation in genes encoding a5 chain oftype IV collagen. In the absence of a5 chain, these patientsfail to assemble the a345 triple helix in the GBM. Sub-sequent to renal transplantation, they develop antibodiesagainst a5 NC1 of the intact hexamer in the allograft,a novel protein in the recipient.83,85 Unlike native anti-GBM nephritis, the antigenic epitope appears to be on thesurface of the hexamer and thus can elicit antibody re-sponse without the triggering factors needed for hexamerdissociation.83,85,86
Concurrent DiseasePositive ANCA serology, especially anti-MPO has
been identified in approximately a third of the patients withanti-GBM disease. ANCA can be detected either before orafter the appearance of anti-GBM antibodies. In additionto glomerular crescents, the renal biopsies from these pa-tients likely demonstrate vasculitis. Despite previous re-ports,88,89 the prognosis of dual-positive patients iscomparable to patients with isolated anti-GBM nephritis.90
However, similar to isolated ANCA disease patients, thesedual-positive cases also have higher frequency of activerelapses.90 This necessitates close clinical follow-up of suchpatients for early detection of relapses. It is presumed that de-velopment of one disease, either anti-GBM or pauci-immune
Adv Anat Pathol � Volume 19, Number 2, March 2012 Crescentic Glomerulonephritis
r 2012 Lippincott Williams & Wilkins www.anatomicpathology.com | 121

GN causes exposure of hidden antigens, thus precipitatingthe concurrent disease process.88,91,92
A recent study identified naturally occurring low-titeranti-MPO and anti-GBM antibodies in healthy individualsthat are evidently nonpathogenic.93 Although such natu-rally occurring low-avidity antibodies suggest incompleteimmune tolerance, they may have an important role indefense against microbial infections, removal of apoptoticor neoplastic cells. Geographic variation in the prevalenceof such antibodies may be related to frequency of exposureto infectious pathogens.93 It is speculated that dysregulatedimmunological mechanisms result in higher pathogenictiters of antibody. Although the discussion of immuno-logical theories is beyond the scope of this review, it seemsthat idiotypic and anti-idiotypic antibodies may play a rolein both anti-GBM and ANCA diseases.43,94
Rarely, anti-GBM nephritis and membranous nephr-opathy can occur simultaneously or one can precede theother.95 Although this concurrence can be entirely co-incidental, epitope spreading or release of damaged GBMantigens into circulation can be triggered by one disease,precipitating the occurrence of a second glomerular dis-ease.96,97 IF can be challenging to interpret, but EM con-firms the presence of subepithelial membranous deposits.
CONCLUSIONSDespite advances in understanding the pathophysio-
logy of ANCA GN and anti-GBM nephritis, and theavailability of serologic laboratory studies with reasonablesensitivity and specificity, renal biopsy remains the main-stay of diagnosis for patients presenting with the clinicalsyndrome of RPGN. IF, together with light microscopyand EM, allow distinction of ANCA GN, anti-GBM dis-ease, and immune complex-mediated GN, including over-lap syndromes and mimics. Clinical correlation and closecommunication with the clinical team remains critical forrapid initiation of appropriate therapy in these critically illpatients.
ACKNOWLEDGMENT
The author thanks Megan Troxell MD, PhD at OregonHealth and Science University for reviewing the manuscript.
REFERENCES
1. Thorner PS, Ho M, Eremina V, et al. Podocytes contribute tothe formation of glomerular crescents. J Am Soc Nephrol.2008;19:495–502.
2. Jennette JC. Rapidly progressive crescentic glomerulonephritis.Kidney Int. 2003;63:1164–1177.
3. Association of Directors of Anatomic and Surgical Pathology.Critical diagnoses (critical values) in anatomic pathology. Am JSurg Pathol. 2006;30:897–899.
4. Nakhleh RE, Myer JL, Allen TC, et al. Consensus statementon effective communication of urgent diagnoses and signifi-cant, unexpected diagnoses in surgical pathology and cyto-pathology from the College of American Pathologists andAssociation of Directors of Anatomic and Surgical Pathology.Arch Pathol Lab Med. 2011;135:1–7.
5. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med.1997;337:1512–1523.
6. Bomback AS, Appel GB, Radhakrishnan J, et al. ANCA-associated glomerulonephritis in the very elderly. Kidney Int.2010.
7. Jennette JC, Falk RJ. The pathology of vasculitis involving thekidney. Am J Kidney Dis. 1994;24:130–141.
8. Savige J, Davies D, Falk RJ, et al. Antineutrophil cytoplasmicantibodies and associated diseases: a review of the clinical andlaboratory features. Kidney Int. 2000;57:846–862.
9. Falk RJ, Jennette JC. ANCA disease: where is this fieldheading? J Am Soc Nephrol. 2010;21:745–752.
10. Jennette JCTD. Pauci-immune and antineutrophil cytoplasmicautoantibody-mediated crescentic glomerulonephritis and vas-culitis. In: Jennette JCOJ, Schwartz MM, Silva FG, eds.Pathology of the Kidney. Philadelphia: Lippincott Williams &Wilkins; 2007:664–673.
11. Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med. 2004;117:39–50.
12. Holle JU, Laudien M, Gross WL. Clinical manifestations andtreatment of Wegener’s granulomatosis. Rheum Dis Clin NorthAm. 2010;36:507–526.
13. Davies DJ, Moran JE, Niall JF, et al. Segmental necrotisingglomerulonephritis with antineutrophil antibody: possiblearbovirus aetiology? Br Med J (Clin Res Ed). 1982;285:606.
14. Lionaki S, Jennette JC, Falk RJ. Anti-neutrophil cytoplasmic(ANCA) and anti-glomerular basement membrane (GBM)autoantibodies in necrotizing and crescentic glomeruloneph-ritis. Semin Immunopathol. 2007;29:459–474.
15. Morgan MD, Harper L, Williams J, et al. Anti-neutrophilcytoplasm-associated glomerulonephritis. J Am Soc Nephrol.2006;17:1224–1234.
16. Savige J, Pollock W, Trevisin M. What do antineutrophilcytoplasmic antibodies (ANCA) tell us? Best Pract Res ClinRheumatol. 2005;19:263–276.
17. Gaffo AL. Diagnostic approach to ANCA-associated vasculi-tides. Rheum Dis Clin North Am. 2010;36:491–506.
18. Savige J, Gillis D, Benson E, et al. International ConsensusStatement on Testing and Reporting of AntineutrophilCytoplasmic Antibodies (ANCA). Am J Clin Pathol. 1999;111:507–513.
19. Jennette JC, Wilkman AS, Falk RJ. Diagnostic predictivevalue of ANCA serology. Kidney Int. 1998;53:796–798.
20. Rutgers A, Sanders JS, Stegeman CA, et al. Pauci-immunenecrotizing glomerulonephritis. Rheum Dis Clin North Am.2010;36:559–572.
21. Schnabel A, Holl-Ulrich K, Dalhoff K, et al. Efficacy oftransbronchial biopsy in pulmonary vaculitides. Eur Respir J.1997;10:2738–2743.
22. Hauer HA, Bajema IM, van Houwelingen HC, et al. Renalhistology in ANCA-associated vasculitis: differences betweendiagnostic and serologic subgroups. Kidney Int. 2002;61:80–89.
23. Sinico RA, Di Toma L, Maggiore U, et al. Renal involvementin Churg-Strauss syndrome. Am J Kidney Dis. 2006;47:770–779.
24. Vizjak A, Rott T, Koselj-Kajtna M, et al. Histologic andimmunohistologic study and clinical presentation of ANCA-associated glomerulonephritis with correlation to ANCAantigen specificity. Am J Kidney Dis. 2003;41:539–549.
25. Bonsib SM, Goeken JA, Fandel T, et al. Necrotizing medullarylesions in patients with ANCA associated renal disease. ModPathol. 1994;7:181–185.
26. Neumann I, Regele H, Kain R, et al. Glomerular immunedeposits are associated with increased proteinuria in patientswith ANCA-associated crescentic nephritis. Nephrol DialTransplant. 2003;18:524–531.
27. Berden AE, Ferrario F, Hagen EC, et al. Histopathologicclassification of ANCA-associated glomerulonephritis. J AmSoc Nephrol. 2010;21:1628–1636.
28. Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmicautoantibodies specific for myeloperoxidase cause glomerulo-nephritis and vasculitis in mice. J Clin Invest. 2002;110:955–963.
29. Kallenberg CG. Pathogenesis of PR3-ANCA associatedvasculitis. J Autoimmun. 2008;30:29–36.
30. Xiao H, Schreiber A, Heeringa P, et al. Alternative comple-ment pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64.
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
122 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins

31. Kain R, Exner M, Brandes R, et al. Molecular mimicry inpauci-immune focal necrotizing glomerulonephritis. Nat Med.2008;14:1088–1096.
32. Roth AJ, Brown MC, Smith RN, et al. Anti-LAMP-2antibodies are not prevalent in patients with antineutrophilcytoplasmic autoantibody glomerulonephritis. J Am SocNephrol. 2011.
33. Bautz DJ, Preston GA, Lionaki S, et al. Antibodies with dualreactivity to plasminogen and complementary PR3 in PR3-ANCA vasculitis. J Am Soc Nephrol. 2008;19:2421–2429.
34. Bell EK, Chugh SS, Cook WJ. A case of infection-associatedantiproteinase-3-negative cytoplasmic antineutrophil cytoplas-mic antibody pauci-immune focal necrotizing glomeruloneph-ritis. Nephrol Dial Transplant. 2010;25:3119–3123.
35. Stegeman CA, Tervaert JW, de Jong PE, et al. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention ofrelapses of Wegener’s granulomatosis. Dutch Co-TrimoxazoleWegener Study Group. N Engl J Med. 1996;335:16–20.
36. Stegeman CA, Tervaert JW, Sluiter WJ, et al. Association ofchronic nasal carriage of Staphylococcus aureus and higherrelapse rates in Wegener granulomatosis. Ann Intern Med.1994;120:12–17.
37. Tidman M, Olander R, Svalander C, et al. Patients hospi-talized because of small vessel vasculitides with renal involve-ment in the period 1975–95: organ involvement, anti-neutrophilcytoplasmic antibodies patterns, seasonal attack rates andfluctuation of annual frequencies. J Intern Med. 1998;244:133–141.
38. Summers SA, van der Veen BS, O’Sullivan KM, et al. Intrinsicrenal cell and leukocyte-derived TLR4 aggravate experimentalanti-MPO glomerulonephritis. Kidney Int. 2011;78:1263–1274.
39. Bosch X. LAMPs and NETs in the pathogenesis of ANCAvasculitis. J Am Soc Nephrol . 2009;20:1654–1656.
40. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophilextracellular traps kill bacteria. Science. 2004;303:1532–1535.
41. Kessenbrock K, Krumbholz M, Schonermarck U, et al.Netting neutrophils in autoimmune small-vessel vasculitis.Nat Med. 2009;15:623–625.
42. Pendergraft WF III, Preston GA, Shah RR, et al. Auto-immunity is triggered by cPR-3(105-201), a protein comple-mentary to human autoantigen proteinase-3. Nat Med.2004;10:72–79.
43. Pendergraft WF III, Pressler BM, Jennette JC, et al.Autoantigen complementarity: a new theory implicatingcomplementary proteins as initiators of autoimmune disease.J Mol Med. 2005;83:12–25.
44. Tadema H, Kallenberg CG, Stegeman CA, et al. Reactivityagainst complementary proteinase-3 is not increased in patientswith PR3-ANCA-associated vasculitis. PLoS One. 2011;6:e17972.
45. Gao Y, Zhao MH. Review article: drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Neph-rology (Carlton). 2009;14:33–41.
46. Wiik A. Drug-induced vasculitis. Curr Opin Rheumatol. 2008;20:35–39.
47. Kanda T, Tanio H, Wu C, et al. Churg-Strauss syndrome withsevere granulomatous angiitis and crescentic glomeruloneph-ritis, which developed during therapy with a leukotrienereceptor antagonist. Clin Exp Nephrol. 2010;14:602–607.
48. Hirohama D, Hoshino J, Hasegawa E, et al. Development ofmyeloperoxidase-antineutrophil cytoplasmic antibody-associ-ated renal vasculitis in a patient receiving treatment withanti-tumor necrosis factor-alpha. Mod Rheumatol. 2010;20:602–605.
49. Stokes MB, Foster K, Markowitz GS, et al. Development ofglomerulonephritis during anti-TNF-alpha therapy for rheu-matoid arthritis. Nephrol Dial Transplant. 2005;20:1400–1406.
50. Borgmann S, Haubitz M. Genetic impact of pathogenesis andprognosis of ANCA-associated vasculitides. Clin Exp Rheu-matol. 2004;22:S79–S86.
51. Ciavatta DJ, Yang J, Preston GA, et al. Epigenetic basis foraberrant upregulation of autoantigen genes in humans withANCA vasculitis. J Clin Invest. 2010;120:3209–3219.
52. Lepse N, Abdulahad WH, Kallenberg CG, et al. Immuneregulatory mechanisms in ANCA-associated vasculitides.Autoimmun Rev. 2011.
53. Rimbert M, Hamidou M, Braudeau C, et al. Decreasednumbers of blood dendritic cells and defective function ofregulatory T cells in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS One. 2011;6:e18734.
54. Gan PY, Steinmetz OM, Tan DS, et al. Th17 cells promoteautoimmune anti-myeloperoxidase glomerulonephritis. J AmSoc Nephrol. 2010;21:925–931.
55. Ooi JD, Kitching AR, Holdsworth SR. Review: T helper 17cells: their role in glomerulonephritis. Nephrology (Carlton).2010;15:513–521.
56. Hogan SL, Falk RJ, Chin H, et al. Predictors of relapse andtreatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann InternMed. 2005;143:621–631.
57. Pagnoux C, Hogan SL, Chin H, et al. Predictors of treatmentresistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: comparison of two independ-ent cohorts. Arthritis Rheum. 2008;58:2908–2918.
58. Little MA, Hassan B, Jacques S, et al. Renal transplantation insystemic vasculitis: when is it safe? Nephrol Dial Transplant.2009;24:3219–3225.
59. Geetha D, Seo P. Renal transplantation in the ANCA-associated vasculitides. Am J Transplant. 2007;7:2657–2662.
60. Nachman PH, Segelmark M, Westman K, et al. RecurrentANCA-associated small vessel vasculitis after transplantation:a pooled analysis. Kidney Int. 1999;56:1544–1550.
61. Moroni G, Torri A, Gallelli B, et al. The long-term prognosisof renal transplant in patients with systemic vasculitis. Am JTransplant. 2007;7:2133–2139.
62. Daly C, Conlon PJ, Medwar W, et al. Characteristics andoutcome of anti-glomerular basement membrane disease: asingle-center experience. Ren Fail. 1996;18:105–112.
63. Bolton WK. Goodpasture’s syndrome. Kidney Int. 1996;50:1753–1766.
64. Saraf P, Berger HW, Thung SN. Goodpasture’s syndrome withno overt renal disease. Mt Sinai J Med. 1978;45:451–454.
65. Wilson CB, Dixon FJ. Anti-glomerular basement membraneantibody-induced glomerulonephritis. Kidney Int. 1973;3:74–89.
66. Bombassei GJ, Kaplan AA. The association between hydro-carbon exposure and anti-glomerular basement membraneantibody-mediated disease (Goodpasture’s syndrome). Am JInd Med. 1992;21:141–153.
67. Proskey AJ, Weatherbee L, Easterling RE, et al. Good-pasture’s syndrome. A report of five cases and review of theliterature. Am J Med. 1970;48:162–173.
68. Cui Z, Zhao J, Jia XY, et al. Clinical features and outcomes ofanti-glomerular basement membrane disease in older patients.Am J Kidney Dis. 2011;57:575–582.
69. Cui Z, Zhao MH, Singh AK, et al. Antiglomerular basementmembrane disease with normal renal function. Kidney Int.2007;72:1403–1408.
70. Yang R, Hellmark T, Zhao J, et al. Levels of epitope-specificautoantibodies correlate with renal damage in anti-GBMdisease. Nephrol Dial Transplant. 2009;24:1838–1844.
71. Hellmark T, Segelmark M, Unger C, et al. Identification of aclinically relevant immunodominant region of collagen IV inGoodpasture disease. Kidney Int. 1999;55:936–944.
72. Salama AD, Dougan T, Levy JB, et al. Goodpasture’s diseasein the absence of circulating anti-glomerular basementmembrane antibodies as detected by standard techniques. AmJ Kidney Dis. 2002;39:1162–1167.
73. Saxena R, Bygren P, Cederholm B, et al. Circulating anti-entactin antibodies in patients with glomerulonephritis. KidneyInt. 1991;39:996–1004.
74. Saxena R, Bygren P, Rasmussen N, et al. Circulatingautoantibodies in patients with extracapillary glomeruloneph-ritis. Nephrol Dial Transplant. 1991;6:389–397.
75. Border WA, Baehler RW, Bhathena D, et al. IgA antibasementmembrane nephritis with pulmonary hemorrhage. Ann InternMed. 1979;91:21–25.
Adv Anat Pathol � Volume 19, Number 2, March 2012 Crescentic Glomerulonephritis
r 2012 Lippincott Williams & Wilkins www.anatomicpathology.com | 123

76. Ho J, Gibson IW, Zacharias J, et al. Antigenic heterogeneity ofIgA anti-GBM disease: new renal targets of IgA autoanti-bodies. Am J Kidney Dis. 2008;52:761–765.
77. Shaer AJ, Stewart LR, Cheek DE, et al. IgA antiglomerularbasement membrane nephritis associated with Crohn’s disease:a case report and review of glomerulonephritis in inflammatorybowel disease. Am J Kidney Dis. 2003;41:1097–1109.
78. Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesisof human glomerulonephritis. J Exp Med. 1967;126:989–1004.
79. Germuth FG Jr., Choi IJ, Taylor JJ, et al. Antibasementmembrane disease. I. The glomerular lesions of Goodpasture’sdisease and experimental disease in sheep. Johns Hopkins MedJ. 1972;131:367–384.
80. Ohnuki T. Crescentic glomerulonephritis induced in the goatby immunization with homologous or heterologous glomerularbasement membrane antigen. Acta Pathol Jpn. 1975;25:319–331.
81. Huey B, McCormick K, Capper J, et al. Associations of HLA-DR and HLA-DQ types with anti-GBM nephritis by sequence-specific oligonucleotide probe hybridization. Kidney Int. 1993;44:307–312.
82. Yang R, Cui Z, Zhao J, et al. The role of HLA-DRB1 alleleson susceptibility of Chinese patients with anti-GBM disease.Clin Immunol. 2009;133:245–250.
83. Pedchenko V, Bondar O, Fogo AB, et al. Moleculararchitecture of the Goodpasture autoantigen in anti-GBMnephritis. N Engl J Med. 2010;363:343–354.
84. Cashman SJ, Pusey CD, Evans DJ. Extraglomerular distributionof immunoreactive Goodpasture antigen. J Pathol. 1988;155:61–70.
85. Salant DJ. Goodpasture’s disease—new secrets revealed.N Engl J Med. 2010;363:388–391.
86. Borza DB, Neilson EG, Hudson BG. Pathogenesis of Goodpasturesyndrome: a molecular perspective. Semin Nephrol. 2003;23:522–531.
87. Bolton WK, Innes DJ Jr., Sturgill BC, et al. T-cells andmacrophages in rapidly progressive glomerulonephritis: clin-icopathologic correlations. Kidney Int. 1987;32:869–876.
88. Bosch X, Mirapeix E, Font J, et al. Prognostic implication ofanti-neutrophil cytoplasmic autoantibodies with myeloperox-idase specificity in anti-glomerular basement membranedisease. Clin Nephrol. 1991;36:107–113.
89. Clyne S, Frederick C, Arndt F, et al. Concurrent and discreteclinicopathological presentations of Wegener granulomatosisand anti-glomerular basement membrane disease. Am J KidneyDis. 2009;54:1116–1120.
90. Rutgers A, Slot M, van Paassen P, et al. Coexistence of anti-glomerular basement membrane antibodies and myeloperox-idase-ANCAs in crescentic glomerulonephritis. Am J KidneyDis. 2005;46:253–262.
91. Hellmark T, Niles JL, Collins AB, et al. Comparison of anti-GBM antibodies in sera with or without ANCA. J Am SocNephrol. 1997;8:376–385.
92. Peces R, Rodriguez M, Pobes A, et al. Sequential developmentof pulmonary hemorrhage with MPO-ANCA complicatinganti-glomerular basement membrane antibody-mediated glo-merulonephritis. Am J Kidney Dis. 2000;35:954–957.
93. Cui Z, Zhao MH, Segelmark M, et al. Natural autoantibodiesto myeloperoxidase, proteinase 3, and the glomerular basementmembrane are present in normal individuals. Kidney Int.2010;78:590–597.
94. Shoenfeld Y. The idiotypic network in autoimmunity: anti-bodies that bind antibodies that bind antibodies. Nat Med.2004;10:17–18.
95. Troxell ML, Saxena AB, Kambham N. Concurrent anti-glomerular basement membrane disease and membranousglomerulonephritis: a case report and literature review. ClinNephrol. 2006;66:120–127.
96. Fukatsu A, Brentjens JR, Killen PD, et al. Studies on theformation of glomerular immune deposits in brown Norwayrats injected with mercuric chloride. Clin Immunol Immuno-pathol. 1987;45:35–47.
97. Basford AW, Lewis J, Dwyer JP, et al. Membranous nephro-pathy with crescents. J Am Soc Nephrol. 2011:22:1804–1808.
Kambham Adv Anat Pathol � Volume 19, Number 2, March 2012
124 | www.anatomicpathology.com r 2012 Lippincott Williams & Wilkins