copyright by kenneth stanley matthews 2005
TRANSCRIPT

Copyright
by
Kenneth Stanley Matthews
2005

The Dissertation Committee for Kenneth Stanley Matthews Certifies that this is the
approved version of the following dissertation:
THE TOTAL SYNTHESIS OF (±)-RENIERAMYCIN G AND
STUDIES TOWARD THE SYNTHESIS OF (±)-LEMONOMYCIN
AND (±)-SAFRAMYCIN B
Committee:
Philip D. Magnus
Stephen F. Martin
Eric V. Anslyn
Sean M. Kerwin
Hung-wen Liu

THE TOTAL SYNTHESIS OF (±)-RENIERAMYCIN G AND
STUDIES TOWARD THE SYNTHESIS OF (±)-LEMONOMYCIN
AND (±)-SAFRAMYCIN B
by
Kenneth Stanley Matthews, B.S.
Dissertation
Presented to the Faculty of the Graduate School of
The University of Texas at Austin
in Partial Fulfillment
of the Requirements
for the Degree of
Doctor of Philosophy
The University of Texas at Austin
August, 2005

Dedication
To my father, mother, and brother, for all your love and support.

v
Acknowledgements
Ms. Robin Spalty: you got the ball rolling and I am forever grateful that you
taught chemistry long enough to teach its wonders.
Dr. Christopher Creighton: thank you for giving me a chance, getting me into
organic chemistry, and all the advice that has taken me so far.
Dr. Jamie Reuter: your patience and generosity were exactly what I needed, and
your passionate management style still can not be beat.
Dr. Konrad Fiechtinger: you taught me so much chemistry I could not begin to
thank you. You are a great boss and an inspiring chemist.
Prof. Murray Goodman: you let me in your group with so little experience and I
will forever thank you for giving me that first chance.
Dr. Gary Snyder: the only organic chemistry teacher that was clear, concise, and
thorough. Thank you for giving the best foundation to be a researcher.
Prof. John C. Wheeler: your excitement for chemistry and demand of students
helped me focus and achieve far above my own expectations.
Dr. Luca Araldi: your training has allowed me to understand so much of the
chemistry process and your patience and happy spirit were the best to work with.
Dr. Jean Cui: your drive to do great things is unmatched. I am very grateful to
have learned not only chemistry from you but how to work in the midst of circumstance.

vi
Dr. Odile Levy: a truly great boss, you were so very keen on keeping me focused,
and your leadership showed me how it all gets done.
Dr. Scott Kemp: I can not thank you enough for teaching me the ways of the Jedi.
Without your insight and vigilant teaching I would have been lost in graduate school.
Mr. Maxwell Lawrence: you gave me so much advice on how to be successful in
chemistry and taught me the essential lesson of doing it right the first time.
Dr. Jennifer Kreisburg: you helped me get into a great research group and gave
me great advice throughout graduate school. I am very grateful.
Prof. Cyril Olivier: I owe you for all the helpful discussions and advice. Your
knowledge and passion for chemistry were an inspiration.
Dr. Trevor Rainey: anyone who can stand five years sitting next to me deserves
praise, and thank you for the countless discussions and sharing your knowledge.
Dr. Rachel Turnbull: thank you for all your writing advice and time editing. I
would have been lost without it.
Mr. Vincent Lynch: your skills as an X-ray crystallographer saved the day many a
time, and I thank you for all the helpful discussions.
Mr. Steve Sorey and Dr. Ben Shoulders: thank you for the countless discussions
about NMR interpretation and for dealing with my nagging.
Prof. Philip D. Magnus: you gave me a chance in your group and a wonderful
project. I enjoyed all the guidance and support that you have provided.

vii
THE TOTAL SYNTHESIS OF (±)-RENIERAMYCIN G AND
STUDIES TOWARD THE SYNTHESIS OF (±)-LEMONOMYCIN
AND (±)-SAFRAMYCIN B
Publication No._____________
Kenneth Stanley Matthews, Ph.D.
The University of Texas at Austin, 2005
Supervisor: Philip Douglas Magnus
Herein is described our synthetic approach to the tetrahydroisoquinoline alkaloids.
The first chapter describes relevant background related to the biological significance of
these alkaloids. The analysis of various syntheses of saframycins, renieramycins, and
lemonomycin is also discussed. Chapter 2 describes the development of a new synthesis
of 1,3-cis-substituted tetrahydroisoquinolines and a novel lactam formation in our
approach to (±)-saframycin B. Chapter 3 applies the new tetrahydroisoquinoline
formation toward the synthesis of (±)-lemonomycin. The bicyclo[3.2.1] system is
constructed from an intramolecular N-acyliminium cyclization and leads to the synthesis
(±)-lemonomycinone amide. Chapter 4 reports the total synthesis of (±)-renieramycin G
from an advanced intermediate used in our approach to (±)-lemonomycin, demonstrating
a divergent approach to the tetrahydroisoquinoline alkaloids. Chapter 5 contains the
experimental details and characterization data for all new reported compounds.

viii
Table of Contents
Chapter 1: Tetrahydroisoquinoline Alkaloids..........................................................1 1.0. Introduction ................................................................................................1 1.1. Biological Activity .....................................................................................4 1.2. Mechanism of Action .................................................................................5 1.3. Biosynthesis................................................................................................7 1.4. Synthetic Approaches to Tetrahydroisoquinoline Alkaloids......................9
1.4.1. The Saframycins..............................................................................10 1.4.1.1. Fukuyama’s Total Synthesis of (±)-Saframycin B.................10 1.4.1.2. Kubo’s Total Synthesis of (±)-Saframycin B.........................13 1.4.1.3. Fukuyama’s Total Synthesis of (±)-Saframycin A.................15 1.4.1.4. William’s Studies Toward the Saframycins...........................17 1.4.1.5. Corey’s Enantioselective Synthesis of (–)-Saframycin A......19
1.4.2. The Renieramycins..........................................................................21 1.4.2.1. Fukuyama’s Synthesis of (±)-Renieramycin A ......................21 1.4.2.2. Danishefsky’s Synthesis of Cribrostatin IV ...........................23
1.4.3. Lemonomycin..................................................................................28 1.4.3.1. Stoltz’s Synthesis of (–)-Lemonomycin.................................28 1.4.3.2. Fukuyama’s Studies Toward (–)-Lemonomycin....................31
1.5. Retrosynthetic Analysis ............................................................................35 1.6. Conclusion ................................................................................................37 1.7. References ................................................................................................38
Chapter 2: Toward the Synthesis of Saframycins B ..............................................42 2.0. Introduction ..............................................................................................42 2.1. Background: Enantioselective Alkylation of Isoquinolines.....................43 2.2. Results and Discussion.............................................................................51
2.2.1. Investigation of the Enantioselective Alkylation of Isoquionline ...51 2.2.2. Formation of 1,3-cis-Substituted Tetrahydroisoquinoline ..............55 2.2.3. 3-(Aminomethyl)-Isoquinoline .......................................................59

ix
2.2.4. Oxygen Analogue............................................................................67 2.2.5. Nitrogen Analogue for Pummerer Cyclization ...............................72 2.2.6. Formation of the Real System.........................................................76 2.2.7. Alkylation, Reduction, and Cyclization of the Real System...........80
2.3. Conclusion ................................................................................................85 2.4. References ................................................................................................87
Chapter 3: Toward the Synthesis of Lemonomycin ..............................................89 3.0. Introduction ..............................................................................................89 3.1. Results and Discussion.............................................................................91
3.1.1. Alkylation with Benzyloxymethyllithium.......................................91 3.1.2. Formation 1,3-cis-Substituted Tetrahydroisoquinoline...................97 3.1.3. Amide Coupling ............................................................................103 3.1.4. Stereoselective Incorporation of C14-C15 ....................................107 3.1.5. Formation of the Bicyclo[3.2.1] System .......................................117
3.2. Conclusion ..............................................................................................123 3.3. References ..............................................................................................125
Chapter 4: The Total Synthesis of (±)-Renieramycin G......................................126 4.0. Introduction ............................................................................................126 4.1. Results and Discussion...........................................................................128
4.1.1. Benzyl Chloride Formation ...........................................................128 4.1.2. Formation the Bicyclo[3.3.1] System............................................131 4.1.3. Completion of the Synthesis..........................................................137
4.2. Conclusion ..............................................................................................142 4.3. References ..............................................................................................143
Chapter 5: Experimentals.....................................................................................144 5.0. General Information ...............................................................................144 5.1. Experimental Conditions and Characterization......................................145 5.2. References ..............................................................................................244

x
Appendix A: X-Ray Data for the Chloride 198 .................................................252
Appendix B: X-Ray Data for the Bicyclo[3.3.1] 213 ........................................256
Appendix C: X-Ray Data for the Lactam 229 ...................................................260
Appendix D: X-Ray Data for the Amino Alcohol 283 ......................................264
Appendix E: X-Ray Data for Thioaminal 300....................................................268
Appendix F: X-Ray Data for the Allylated Product 301 ...................................273
Appendix G: X-Ray Data for Oxime 314 ..........................................................278
Appendix H: X-Ray Data for the Diol 334 ........................................................283
Abbreviations ......................................................................................................288
Vita .....................................................................................................................290

1
Saframycins A (1a) R1=CN,R2=H B (1b) R1=R2=H C (1c) R1=H,R2=OMe G (1d) R1=CN,R2=OH S (1e) R1=OH,R2=H
Renieramycins
A (2a) R1=R2=H,R3=OH B (2b) R1=R2=H,R3=OMe C (2c) R1=R2=O,R3=OH D (2d) R1=R2=O,R3=OEt E (2e) R1=OH,R2=R3=H F (2f) R1=OH,R2=H,R3=OMe G (2g) R1=R2=O,R3=H
Safracins A (3a) R1=R2=H B (3b) R1=H,R2=OH (3c) R1=CN,R2=H
Ecteinascidin
Et743 (4a) R1=OH,R2=Me Et729 (4b) R1=OH,R2=H Et745 (4c) R1=H,R2=Me Et770 (4d) R1=CN,R2=Me
Chapter 1: Tetrahydroisoquinoline Alkaloids
1.0. INTRODUCTION
The tetrahydroisoquinoline alkaloids are a broad family of natural products with
nearly 60 members isolated and more than a hundred derivatives. Members of this family
include those with a bis-tetrahydroisoquinoline core; the saframycins, renieramycins,
safracins, and ecteinascidins (Figure 1.01), and those with a mono-tetrahydroisoquinoline
core; naphthyridinomycin, cyanocycline, the bioxalomycins, the quinocarcins,
tetrazomine, and lemonomycin (Figure 1.02).1-10
Figure 1.01. Bis-Tetrahydroisoquinolines
NN
Me
O
OOMe
O
O
Me
MeO
HH
H
NH
H
1
3
OO
Me
Me
R2
R1
NN
Me
O
OOMe
O
O
Me
MeO
HH
H
O
H
1
3
OMe
MeR3
R1
H
MeR2
NN
Me
R2
HOOMe
O
O
Me
MeO
HH
H
NH
H
1
3
ONH2
Me
Me
R1
NN
MeHOOMe
OAcMe
HH
HR1
OHS
NHMeO
HO
O
13 R2
OO

2
naphthridinomycin (5) R1=OH,R2=Me,R3=Mecyanocycline A (6a) R1=CN,R2=Me,R3=Me
B (6b) R1=CN,R2=Me,R3=H F (6c) R1=CN,R2=H,R3=Me
bioxalomycin β1 (7a) R=H bioxalomycin β2 (7b) R=Me
quinocarcin (8) tetrazomine (9) lemonomycin (10)
Figure 1.02. Mono-Tetrahydroisoquinolines
The first tetrahydroisoquinoline alkaloid structure elucidated was in 1974 with the
X-ray analysis of the unstable ruby red crystal of naphthyridinomycin.11 This compound
was isolated from an unknown Streptomycete strain, Streptomyces lusitanus AYB-1026,
found in an Easter Island soil sample.12 Within the next 10 years cyanocyclines,13,14
quinocarcin,15 safracins16 and numerous saframcyins9 were isolated and characterized
from strains of Streptomyces. During the 1990’s further fermentation studies led to the
discovery of tetrazomine17 and bioxalomycins.18 One of the most recent additions to the
family was lemonomycin which was first isolated in 196019 from Streptomyces candidus
however the structure was not elucidated until 2000.20 Lemonomycin is the only
tetrahydroisoquinoline alkaloid to possess a glycidic linkage in the C1 side-chain and the
rare aldehyde hydrate functionality.
N
OMe
HH
HO
H
CO2H
N3
1
Me
H
NNR
O
O
Me
MeO
H
H
1
3
N
O
HH
OH
N
OMe
HH
HO
HN3
1
Me
HNH
O
NH
OH
H
H
OH
NNR3
O
O
Me
R2O
H
R1
OH
H
1
3
N
O
HH
N
O
Me
MeO
HH
HOH
O
H
OOH
NMe2
HOOH
NH3
1
O

3
Tetrahydroisoquinoline alkaloids are also found as marine natural products. All of
the renieramycins were isolated from marine sources: A-F from the bright blue sea
sponge Reniera sp. found in Mexico,21,22 G from the Fijian sponge Xestospongia
caycedoi23 and most recently H and I from the sponge Haliclona cribricutis found off the
coast of India.24 The renieramycins possess a similar bis-isoquinolinequinone core to the
saframycins. The main difference is an angelate C1 side-chain in place of a pyruvamide.
The most structurally complex tetrahydroisoquinoline alkaloids, the ecteinascidins which
are actually tris-tetrahydroisoquinolines, were isolated from a Caribbean tunicate
Ecteinascidia turbinata in 1990.25,26

4
1.1. BIOLOGICAL ACTIVITY
The tetrahydroisoquinoline alkaloids possess a range of biological activity as
potent cytotoxic agents. This includes antitumor, antibiotic and antimicrobial activity
through the inhibition of RNA, DNA, and protein synthesis. A recent review by
Williams1 gives a comprehensive account of the biology of tetrahydroisoquinoline
antitumor antibiotics. While many of the tetrahydroisoquinoline alkaloids have well
investigated in vitro activity, saframycin and the ecteinascidins have the most well
studied in vivo biological activity in this family. However none of these alkaloids or
derivatives are currently on the market as therapeutics.
Ecteinascidin 743 (Et743, 4a) is currently in Phase III clinical trials for the
treatment of ovarian, endometrial and breast cancer.27 The antiproliferative activity of
Et743 is greater than that of taxol, mitomycin C and cisplatin (Table 1.01). Furthermore,
the unique mode of action of Et743 is thought to be responsible for its high level of
activity in advanced sarcomas that had relapsed or were resistant to conventional therapy.
cell line drug
P388 leukemia
L1210 leukemia
A549 lung cancer
Et743 0.34 nM28 0.66 nM28 0.26 nM28
saframycin A N/A N/A 133 nM29
taxol 16.9 nM30 41.0 nM31 2.0 nM32
mitomycin C 14.0 nM30 4.2 μM33 0.23 μM34
cisplatin 0.11 μM35 7.0 μM35 18.1 μM36
Table 1.01. Comparison of Inhibitory Concentrations (IC50)

5
1.2. MECHANISM OF ACTION1,9,37
Multiple theories have been proposed for the mechanism of action leading to the
observed biological activity of the tetrahydroisoquinoline alkaloids. The current accepted
theory for Et743 involves binding to the minor groove of DNA through hydrogen
bonding interactions between the A and E-ring of Et743 (4a) and three base pairs, leading
to unique sequence specificities (Scheme 1.01). Protonation at N12 and subsequent acid
catalyzed dehydration of carbinolamine 11 leads to formation of iminium 12. The
exocyclic 2-amino group of guanine 13 attacks the C21 iminium to form a covalently
bonded adduct 15. The initial Et743/DNA complex is formed reversibly under non-
denaturing conditions, migrating from a non-favored binding sequence to the favored
DNA target site (5′-AGC). The Et743/DNA covalent adduct 15 has been shown to form
reversibly with DNA denaturization.38
NN
MeHOOMe
AcO
O
Me
O
HH
HOH
OHS
NHMeO
HO
O
13 MeA
E18
2113
11
87
6
5
14
15
16
17
B
H
GF
C
NN
MeHOOMe
OH
MeH
5´-AGC
NN
MeHOOMe
Me
HN
N
N
NH2N
O
5´-AGC
H
OH
NN
MeHOOMe
Me
HN
N
N
NHN
O
5´-AGC
H
OHH
NN
MeHOOMe
Me
HN
N
N
NHN
O
5´-AGC
H
Scheme 1.01. Et743 DNA Alkylation
4a
14
13
15
12
11

6
The saframycins possess a similar mechanism of binding as Et743, involving
reversible formation of a noncovalent complex (hydrogen bonded) and an acid-promoted
covalent binding to guanine. In addition, the saframycins possess a quinone functionality
which allows for an alternative method of covalent bonding. Using saframycin A (1a) as
an example, reducing cofactors such as dithiothreitol can lead to the formation of a
hydroquinone 16 (Scheme 1.02). At this oxidation level a ring fragmentation and loss of
cyanide can occur, forming the electrophilic iminium 18 via intermediate 17. Alkylation
of the 2-amino group of guanine 13, as before, generates the covalently bonded adduct
19. Yet another possible mechanism for biological activity is through single strand
scission of DNA caused by reactive oxygen species (O2−·) produced by the redox cycling
of the quinone functionality present in saframcyins.39
NN
Me
O
OOMe
O
O
Me
MeO
HH
H
NH
H
1
3
OO
Me
Me
CN87
6
5 4
18 17
1615
142113
11
NN
Me
O
OOMe
OH
O
Me
MeO
HH
H
NH
H
OO
Me
Me
CN
[H]
H
NN
Me
O
OOMe
OH
O
Me
MeO
HH
H
NH
OO
Me
Me
H
NN
Me
O
OOMe
OH
OH
Me
MeO
HH
H
NH
OO
Me
Me
HN
N
N
NH2N
O
NN
Me
O
OOMe
OH
OH
Me
MeO
HH
H
HN
OO
Me
Me
HN
N
N
NH2N
O
Scheme 1.02. Saframycin A DNA Alkylation
1a
19 13
17 16
18

7
1.3. BIOSYNTHESIS1,9
The biosynthesis of some tetrahydroisoquinoline alkaloids, namely the
saframycins, safracins, and cyanocycline A is well investigated, while that of the more
structurally complex Et743 has not been fully realized. In 1985, 14C-labeled experiments
demonstrated that the biosynthesis of saframycin A involves the amino acids tyrosine,
glycine, alanine, and methionine.40 Two tyrosine residues 20 cyclize to form a dimeric
intermediate (Scheme 1.03), followed by incorporation of a glycine-alanine dipeptide 21
which becomes the pyruvamide side chain. The five methyl groups were shown to be
incorporated from methionine units 22.
NN
Me
O
OOMe
O
O
Me
MeO
HH
H
NH
H
OO
Me
Me
CN
CO2H
NH2HO
CO2H
NH2HO O
NH
ONH2
Me
OH
MeS NH2
CO2H
*
SMeH2N
CO2H
*
*
**
*
*
Scheme 1.03. Biosynthesis of Saframycin A.
The biosynthesis of unstable naphthyridinomycin was elucidated by analogy with
the more stable cyanocycline A (6a). In 1982, 14C-labeled and 15N-labeled experiments
showed the involvement of tyrosine, methionine, glycine, and ornithine (Scheme 1.04).41
The glycine unit 23 is converted to serine (24) which becomes the oxazolidine portion of
20
22 1a 21
22
20

8
6a.42 The ornithine unit 25 forms the bicyclo[3.2.1] system, while the three methyl
groups were shown to be incorporated from methionine units 22. Studies showed that
methylation of tyrosine (20) was followed by hydroxylation to form catechol 27 before
incorporation into cyanocycline A.43
NNMe
O
O
Me
MeO
H
CNOH
H
N
O
HH
HO2C 15NH2
*
HO2C 15NH2
OH
*
*
HO2C NH2
H2N
CO2H
NH2HO
CO2H
NH2HO
CO2H
NH2HO
Me
Me
OH
*
*
MeS NH2
CO2H
*
Scheme 1.04. Biosynthesis of Cyanocyline A
23
25 27
6a
22
20
26
24

9
29
1.4. SYNTHETIC APPROACHES TO TETRAHYDROISOQUINOLINE ALKALOIDS
Numerous syntheses of tetrahydroisoquinolines have been reported since the first
synthesis of saframycin B by Fukuyama in 1982.44 The emergence of Et743 as a
promising therapeutic agent and the discovery of new structures, including lemonomycin,
have kept a number of research groups publishing in this area. While a variety of
different synthetic approaches have been published there are fundamental similarities in
strategy to address the formation of key stereogenic centers. Formation of the 1,3-cis-
substituted tetrahydroisoquinoline ring system of the western fragment has proven to be
the primary focus of many strategies (Scheme 1.05). The most common strategy is
formation via a Pictet-Spengler cyclization late in the synthesis. This is used in the
syntheses of saframycins, renieramycins, and lemonomycin. The 1,3-cis-substituted
tetrahydroisoquinoline ring system of the eastern fragment is also often formed via a
Pictet-Spengler cyclization or by a Mannich reaction. These approaches and other
solutions to stereochemical control will be discussed in detail in the proceeding sections.
NN
Me
O
OOMe
O
O
Me
MeO
HH
H
X
H
1
3 R
R
HNN
Me
OMe
HOOMe
OMe
OH
Me
MeO
HH
H
R
R
1
3 HNN
Me
OMe
HOOMe
OMe
OH
Me
MeOH
R
R
1
3
1st 1,3-cis-substituted tetrahydroisoquinoline
2nd 1,3-cis-substituted tetrahydroisoquinoline
X
OHC+
H1
3
Scheme 1.05. Pictet-Spengler Approach to Tetrahydroisoquinoline Alkaloids
28
30
31

10
Saframycins
A (1a) R1=CN, R2=H B (1b) R1=R2=H C (1c) R1=H, R2=OMe G (1d) R1=CN, R2=OH S (1e) R1=OH,R2=H
1.4.1. THE SAFRAMYCINS
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
H
NH
OO
Me
R1
R21
11
13
1615
8
75
4
3
19
24
22
18
17
21
10
1.4.1.1. Fukuyama’s Total Synthesis of (±)-Saframycin B
Fukuyama and co-workers were the first to publish the total synthesis of a
saframycin, saframycin B.44 The synthesis starts with the formation of intermediates 32
and 34, each from aldehyde 33 (Scheme 1.06). The symmetric structure of the arene units
of the saframycins enable their synthesis from the same aldehyde 33. A condensation of
32 and 34 followed by acetate formation gave amide 35. Formation of aldehyde 36 was
performed in a three-step sequence; ozonolysis and treatment with dimethyl sulfide gave
the correct oxidation state at the C11 stereocenter (saframycin numbering), followed by
elimination of acetic acid with DBU. N-Acyliminium cyclization via a Pictet-Spengler
ring closure was promoted by heating 36 in formic acid to give the bicyclo[3.3.1] 37.
Fukuyama notes that a similar intermediate 38 does not undergo the analogous
cyclization to 39 (Scheme 1.07). The lack of reactivity was attributed to steric bulk
around the C3 stereocenter and the arylbenzyl ether.

11
MeOMe
MeOOBn
CHO MeOMe
MeOOBn
MeOMe
MeOOBn
NH2
PhOH
CO2H
NHCbz
4 steps
76% yield
6 steps
84% yield
MeOMe
MeOOBn
HN
PhOAc
O
NHCbz
OMeMe
OMeOBn
MeOMe
MeOOBn
HN
O
NHCbz
OMeMe
OMeOBn OMe
Me
MeOOBn
HN
O
N
BnOOMe
Me
OMeCbz
H
H
O
i, ii iii, iv
v11
Reaction Conditions: i) DCC, CH2Cl2 (83%); ii) Ac2O, pyr., 60 °C (98%); iii) O3, −78 °C; then Me2S; iv) DBU, CH2Cl2, 0 °C; v) HCO2H, 60 °C (74% over 3 steps).
Scheme 1.06. Fukuyama’s Synthesis of (±)-Saframycin B
OMeMe
MeOOBn
N
O
N
BnOOMe
Me
OMeCO2Me
H
H
MeOMe
MeOOBn
N
O
N
OMeMe
OMeOBn
H
HOH
CO2Me Hi3 3
Reaction Conditions: i) HCO2H, 60 °C, 20 min.
Scheme 1.07. Fukuyama’s Unsuccessful Pictet-Spengler Cyclization
32 33 34
32 + 34
35
36 37
38 39

12
41
The synthesis continued with catalytic hydrogenation of 37 which
stereoselectively formed the C3 center (Scheme 1.08). Reductive alkylation followed by
reduction of the lactam gave N-methylamine 40. Hydrogenolysis and a second Pictet-
Spengler cyclization with aldehyde 41 gave predominantly the desired cyclized product
42 in a ratio of 6:1 over the α-isomer. The final steps included removal of the Cbz group,
pyruvamide formation, and final oxidation to the bis-isoquinolinequinone to give
(±)-saframycin B (1b). The total synthesis was completed in 18 steps with an 8% overall
yield from 33.
CbzHN CHO
i-iiiiv
v-vii
OMeMe
MeOOH
HNN
HOOMe
Me
OMeMe
H
H
H
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
NHCbz
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
H
NHCOCOMe
3
Reaction Conditions: i) H2 (1000 psi), Raney Ni, 100 °C; ii) H2 (1000 psi), 37% aq. HCHO, Raney Ni; iii) AlH3, THF (75%, 3 steps); iv) 41, CH3CN, 70 °C (75%); v) H2, 10% Pd/C, AcOH vi) ClCOCOMe, PhNMe2 (72%, 2 steps); vii) CAN, 0 °C (37%).
Scheme 1.08. Fukuyama’s Synthesis (±)-Saframycin B
42
37
1b
40

13
1.4.1.2. Kubo’s Total Synthesis of (±)-Saframycin B
The next total synthesis of a saframycin was of (±)-saframycin B by Kubo and
co-workers.45 In this new approach, the C2-symmetrical diketopiperazine 44 (Scheme
1.09) was the key starting building block. A sequence of condensations with aldehyde 43
led to arylidenediketopiperazine 45. Benzyl protection of the amide followed by
conversion of the N-acetyl protected amide furnished the isopropylcarbamate 46. The
first Pictet-Spengler ring closure was carried out by selectively reducing the C11
carbonyl moiety (saframycin numbering) to form a hemi-aminal which cyclized, as in
Fukuyama’s work, upon heating in formic acid. The isopropylcarbamate was then
removed and the secondary amine methylated. Reduction of the amide and
stereoselective catalytic hydrogenation gave 47.
AcNNAc
O
O
MeOMe
MeOOMe
CHO
OMeMe
MeOOMe
HNN
MeOOMe
Me
OMeMe
H
H
+Me
OMe
MeOOMe
HN
O
NAc
OMeMe
OMeOMeO
i-iv
MeOMe
MeOOMe
BnN
O
N
OMeMe
OMeOMeO
CO2iPr
v-vii
viii-xiii11
Reaction Conditions: i) t-BuOK, t-BuOH, DMF, 25 °C (66%); ii) 10% Pd/C, EtOH, DMF; iii) Ac2O, 110 °C (80% yield 2 steps); iv) t-BuOK, t-BuOH, DMF, 25 °C (81%); v) BnBr, NaH; vi) NH2NH2·H2O (94%, 2 steps); vii) ClCO2
iPr, DMAP, TEA (94%); viii) LiAlH(OtBu)3; ix) HCO2H, 60 °C (52%, 2 steps); x) H2SO4, TFA xi) 37% aq. HCOH, HCO2H, 70 °C (96%, 2 steps); xii) AlH3, THF, 0 °C (93%); xiii) 20% Pd/C, H2 (4 atm), EtOH, 80 °C (99%).
Scheme 1.09. Kubo’s Synthesis of (±)-Saframycin B
47 46
4544 43

14
48 49
At this stage in the synthesis Kubo used an alternative substrate than Fukuyama to
install the side-chain group, butyl glyoxylate. Kubo used a two step procedure that
coupled the n-butyl glyoxylate to 47 and then a second Pictet-Spengler ring closure was
promoted by treatment with TFA (Scheme 1.10). In contrast to Fukuyama’s work, Kubo
only observed the opposite stereochemistry at the newly formed C1 stereocenter, giving
solely the 1,3-trans-substituted tetrahydroisoquinoline 48. Numerous attempts to
epimerize the stereocenter under basic conditions were unsuccessful. Complete inversion
of the stereocenter was achieved by oxidation to the iminium with mercury acetate and
reduction with sodium borohydride yielding solely 1,3-cis-49.
i, ii iii, iv
v-viii ix, xOMe
Me
MeOOMe
NN
MeOOMe
Me
OMeMe
H
H
H
H
NHCOCOMe
OMeMe
MeOOMe
NN
MeOOMe
Me
OMeMe
H
H
H
CO2nBu
OMeMe
MeOOMe
NN
MeOOMe
Me
OMeMe
H
H
H
CO2nBu
H1
Reaction Conditions: i) CHOCO2Bu, K2CO3, BuOH; ii) TFA, 25 °C (70%, 2 steps); iii) Hg(OAc)2, 5% AcOH/H2O, 90 °C; iv) NaBH4, EtOH/H2O (71%, 2 steps); v) LAH, THF, reflux (77%); vi) DEAD, PhtNH, PPh3, THF; vii) NH2NH2·H2O, EtOH, reflux; viii) ClCOCOMe, DMAP, TEA, CH2Cl2 (76%, 3 steps); ix) BBr3, CH2Cl2, −78 °C to 0 °C; x) 10 M HNO3, 25 °C (41%, 2 steps). Scheme 1.10. Kubo’s Synthesis of (±)-Saframycin B
50
1b
47

15
The side-chain pyruvamide was installed by reduction of the ester, Mitsunobu
displacement of the alcohol with phthalamide, hydrazinolysis and treatment with
pyruvoyl chloride to give 50. The final oxidation to the quinone was accomplished by
first deprotecting the ArOMe with BBr3. Direct oxidation of the trimethoxybenzene gave
only decomposition. Oxidation of the phenol with nitric acid gave (±)-saframycin B (1b).
The total synthesis was accomplished in 22 steps with a 3% overall yield.
1.4.1.3. Fukuyama’s Total Synthesis of (±)-Saframycin A
Soon after Kubo’s synthesis Fukuyama and co-workers reported a total synthesis
of (±)-saframycin A.46 Their approach utilized a similar initial strategy previously
reported by Kubo, using the C2-symmetrical diketopiperazine 44 and aldehyde 33
(Scheme 1.11). A similar sequence of 11 steps afforded amide 51. At this stage
Fukuyama addressed the key difference between saframycin A and B, the oxidation state
at C21. Boc protection of the phenols and amide of 51 allowed reduction of the amide
carbonyl to the ring opened alcohol 52. Boc deprotection followed by a Pictet-Spengler
with aldehyde 53, stereoselectively formed the 1,3-cis-substituted tetrahydroisoquinoline
54 with only trace amounts of the unwanted α-isomer. Swern oxidation formed an
unstable hemi-aminal which was stereoselectively converted to the amino nitrile 55
providing the correct stereochemistry at C21. The final steps involved deprotection of the
side-chain, pyruvamide formation, and oxidation with DDQ to give (±)-saframycin A
(1a). The total synthesis was completed in 20 steps with 9% overall yield.

16
OMeMe
MeOOH
HN
O
N
HOOMe
Me
OMeMe
H
H
H
OMeMe
MeOOBoc
NHBoc
H NH H
MeOH
OMeMe
MeO
BocO OMeMe
MeOOH
NH
H NH H
MeOH
OMeMe
MeO
HO
BocHN CHO
i, ii
iii
ivH
NHBoc
viiiv-vii
11 steps
41% yield
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
NHCOCOMeCN
21
Reaction Conditions: i) Boc2O, DMAP, DMF, 60 °C (81%); ii) NaBH4, EtOH, 0 °C (92%); iii) TFA; iv) 53, MeOH, 60 °C (82%, 2 steps); v) Swern oxid., then NaCN, MeOH (67%); vi) TFA; vii) ClCOCOMe, NaHCO3, CH2Cl2 (86%, 2 steps); viii) DDQ, 0 °C (60%).
Scheme 1.11. Fukuyama’s Synthesis of (±)-Saframycin A
33
52
53
54
51
1a
55
44 +

17
1.4.1.4. William’s Studies Toward the Saframycins
Another approach to the saframycins that involved Pictet-Spengler ring closure
was reported by Williams.47 This enantioselective approach started from the condensation
of aldehyde 33 (Scheme 1.12). Treatment of the resulting imine with the ketene, formed
from the reaction of acid chloride 56 with triethylamine at −78 °C, gave β-lactam 57. The
chiral auxiliary was removed under hydrogenolysis and the free amine underwent Pictet-
Spengler ring closure with methyl glyoxylate to give a single diastereomer 58, possessing
the unwanted α-orientation at C1. Contrary to Kubo’s inversion of C1, basic conditions
were sufficient to epimerize the C1 center as a 3:1 mixture in favor of the cis-isomer 59.
The side-chain and phenol were converted to benzyl ethers in four steps to arrive at 60.
MeMeO
MeOOBn
N
OPh
PhO
COCl
i, ii
BnNH
NH
O
OPh
Ph
O
MeMeO
MeOOH
NH
CO2Me
BnN OH
HMe
MeO
MeOOH
NH
CO2Me
BnN OH
HMe
MeO
MeOOBn
NH
BnN OH
H
OBn
H H
iii, iv
v vi-ix1
Reaction Conditions: i) BnNH2, PhH, reflux; ii) TEA, CH2Cl2, −78 °C to 0 °C (99%, 2 steps); iii) Pd(OH)2, H2 (60 psi), MeOH/THF; iv) CHOCO2Me, MeOH (84%, 2 steps); v) DBU, THF (75%); vi) Boc2O, EtOH; vii) LiBH4, MeOH, Et2O, reflux (78%, 2 steps); viii) NaH; BnBr, nBu4NI; ix) TMSOTf, 2,6-lutidine (55%, 2 steps).
Scheme 1.12. William’s Studies Toward Saframycins
58 59
57
60
56
33
18
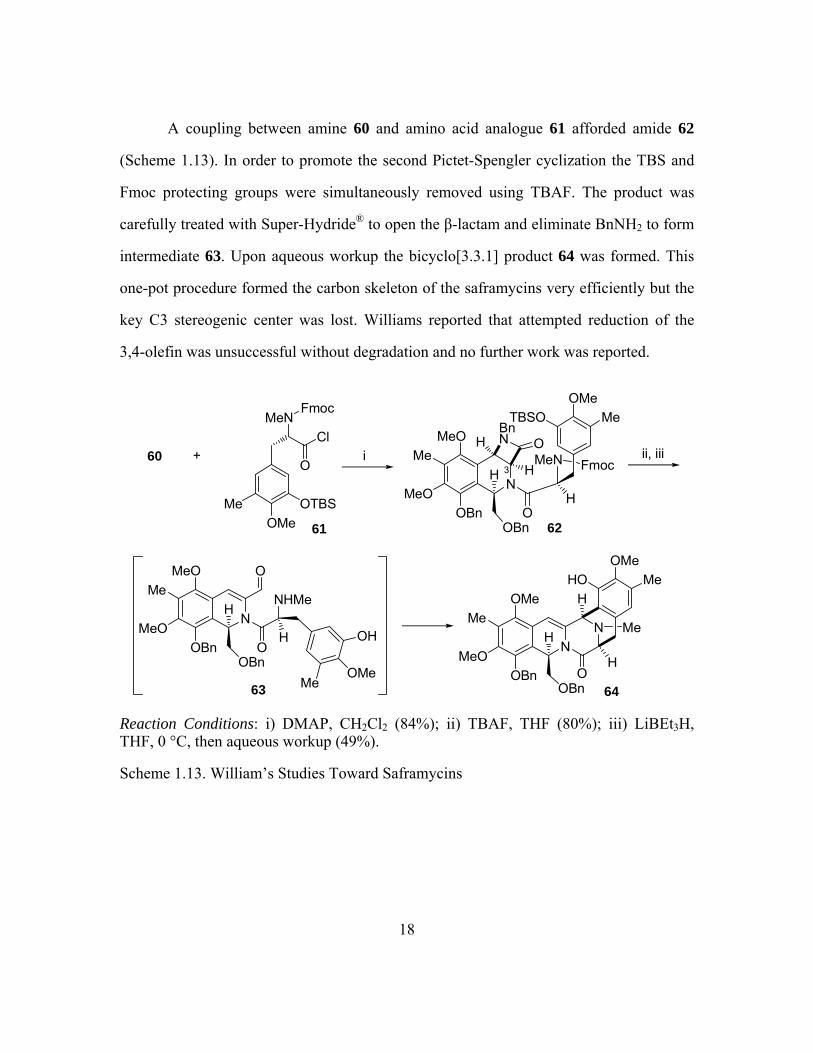
A coupling between amine 60 and amino acid analogue 61 afforded amide 62
(Scheme 1.13). In order to promote the second Pictet-Spengler cyclization the TBS and
Fmoc protecting groups were simultaneously removed using TBAF. The product was
carefully treated with Super-Hydride® to open the β-lactam and eliminate BnNH2 to form
intermediate 63. Upon aqueous workup the bicyclo[3.3.1] product 64 was formed. This
one-pot procedure formed the carbon skeleton of the saframycins very efficiently but the
key C3 stereogenic center was lost. Williams reported that attempted reduction of the
3,4-olefin was unsuccessful without degradation and no further work was reported.
MeNFmoc
Cl
O
OMeOTBSMe
+ MeMeO
MeOOBn
N
BnN OH
H
OBn
H
O
MeN
H
Fmoc
TBSOOMe
Me
MeMeO
MeOOBn
N
OBn
H
O
NHMe
O
H
MeOMe
OH
OMeMe
MeOOBn
NN
HOOMe
Me
Me
H
H
OBn
H
O
i ii, iii3
Reaction Conditions: i) DMAP, CH2Cl2 (84%); ii) TBAF, THF (80%); iii) LiBEt3H, THF, 0 °C, then aqueous workup (49%).
Scheme 1.13. William’s Studies Toward Saframycins
64
6261
60
63

19
1.4.1.5. Corey’s Enantioselective Synthesis of (–)-Saframycin A
In 1996 E. J. Corey and co-workers published the enantioselective synthesis of
ecteinascidin 743.48 Three years later Corey reported a divergent approach in this
synthesis that allowed access to the saframycins.49 Corey demonstrated this by
synthesizing (−)-saframycin A. The enantioselective synthesis was a convergent approach
utilizing amino acid analogues 67 and 68 (Scheme 1.14). The highlight of this strategy
employed dimethyl acetal 66, formed in a seven step sequence from phenol 65, for the
intramolecular Pictet-Spengler ring closure to form the 1,3-cis-substituted
tetrahydroisoquinoline 67. With the C3 stereocenter already established, formation of the
trans-isomer during the Pictet-Spengler was not a possibility, resulting in complete
control of diastereoselectivity. Amine 67 was condensed with aldehyde 68 in the
presence of acetic acid and KCN to stereoselectively form the aminonitrile 69. Formation
of the bicyclo[3.3.1] system required reduction of the lactone to the lactol, followed by
desilylation and acid catalyzed Pictet-Spengler, which gave solely the cis-isomer 70 due
to the established C13 stereocenter. At this stage the backbone structure of the
saframycins was established. A 16 step sequence of functional group interconversions
successfully completed the enantioselective synthesis of (−)-saframycin A (1a). From
aldehyde 65 the synthesis was completed in 31 steps with an overall yield of 0.4%.

20
MeOBn
HNO
O
OBn
O
OMe
OMe
H
NH
OO
H
HMe
OHOMe
OTBSTBSO
CHO
AllocHN N
OO
H
MeOAllyl
HN
TBSOOMe
OTBS
CN
AllocH
H
+
7 steps
42% yield
i-iii
iv, v
vi-viii 16 steps
4% yield
OH
OMeMe
NN
HOOMe
OH
Alloc
H
H
H
H
OHCN
OO
OO
OO
OO
OO
3
13
Reaction Conditions: i) BF3·OEt2, H2O; ii) BF3·OEt2, 4Å molecular sieves (73%, 2 steps); iii) 10% Pd/C, H2 (100%); iv) HOAc, KCN (61%); v) allyl bromide, Cs2CO3, DMF (87%); vi) DIBAL, PhMe, −78 °C; vii) KF·2H2O, MeOH; viii) CH3SO3H, 3Å molecular sieves, CH2Cl2 (55%, 3 steps).
Scheme 1.14. Corey’s Enantioselective Synthesis of (–)-Saframycin A
67
70
68 69
1a
6665

21
Renieramycins
A (2a) R1=R2=H, R3=OH B (2b) R1=R2=H, R3=OMe C (2c) R1=R2=O, R3=OH D (2d) R1=R2=O, R3=OEt E (2e) R1=OH, R2=R3=H F (2f) R1=OH, R2=H, R3=OMe G (2g) R1=R2=O, R3=H
1.4.2. THE RENIERAMYCINS
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
H
O
OMe
R1
R31
11
13
1615
8
75
4
3
19
24
22
18
17
21
10
R2
H
Me
26
1.4.2.1. FUKUYAMA’S SYNTHESIS OF (±)-RENIERAMYCIN A
The only total synthesis of a renieramycin before 2005 was a stereoselective
synthesis of (±)-renieramycin A (2a) completed by Fukuyama and co-workers in 1990.50
Although much of the strategy is similar to Fukuyama’s previous work in the area, the
synthesis was essential to unambiguously prove the stereochemistry at the C1 center. The
stereochemistry of the angelate side-chain of renieramycins was originally assigned as
the α-isomer,51 however further review of spectroscopic data revealed similarities
between saframycin C and renieramycin A, suggesting that both share the same
β-configuration. Faulkner revised the assignment,52 and Fukuyama proved this to be
correct upon completion of his total synthesis.
The synthesis began from readily available aldehyde 71 (Scheme 1.15). In
anticipation of installation of the C14 hydroxyl late in the synthesis, Fukuyama takes
great measures to synthesize differentially protected phenol 72, so that C17-OH can be
generated to facilitate benzylic oxidation. Conversion of 71 to 72 requires 10 steps,
followed by a condensation with diketopiperazine 44. Similar steps to the previous
synthesis of saframycin A led to phenol 73.

22
CHO
OMe
AcNNAc
O
OMe
OMe
10 steps
55% yield
OMOMOR
MeOMe
R = (CH2)3ODMTS
+
OMeMe
MeOOBn
HN
O
NR
BnOOH
Me
OMeH
H
H
11 steps
13% yield
OMeMe
MeOOBn
HN
O
N
BnOOMe
Me
OMeMe
H
H
H
OMeMe
MeOOH
HNN
HOOMe
Me
OMeMe
H
H
H
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
O
OMe
Me
OHOH
OH
v, vii-iv
vii viii
R = CO2(CH2)2CO2Me
OO
Me
Me
CHO
HH
Reaction Conditions: i) DDQ, THF/H2O (8:1); ii) MeI, K2CO3, DMF; iii) DBU, MeOH; iv) HCHO, NaBH3CN, TFA, MeOH (45%, 4 steps); v) AlH3, THF vi) H2, Pd/C, EtOH (64%, 2 steps) vii) glycolaldehyde angelate, CH3CN, 50 °C (66%) viii) DDQ, actone/H2O (20:1) (48%).
Scheme 1.15. Fukuyama’s Synthesis of (±)-Renieramycin A
The C18 phenol was protected as the benzyl ether, while the C17 phenol existed
as the free alcohol, which upon treatment with DDQ stereoselectively hydroxylates from
the exo-face. The phenol was alkylated with MeI and the carbamate converted to the
2a
4471 72
74
75
77
73
76

23
N-methylamine 74. Reduction of the amide with AlH3 and diastereoselective catalytic
hydrogenation gave amine 75. The angelate side-chain was installed by Pictet-Spengler
reaction with glycolaldehyde angelate 76 to furnish a 5:1 mixture of 77 and its α-isomer.
Fukuyama was able to establish the relative stereochemistry by a single crystal X-ray
analysis of 77. (±)-Renieramycin A (2a) was formed upon oxidation with DDQ giving a
structure identical to that of the authentic sample. The synthesis was completed in 29
steps with an overall yield of 0.7%.
1.4.2.2. Danishefsky’s Synthesis of Cribrostatin IV (Renieramycin H)
OMe
MeOO
NN
HOOMe
Me
OHMe
H
H
H
O
OMe
Me
O
O
H
1
11
13
1615
8
7
5
43
19
24
22
18
17
21
10
26
Danishefsky and co-workers have recently reported the synthesis of cribrostatin
IV (renieramycin H, 78).53 Pettit reported the isolation and structural elucidation of this
compound from a blue marine sponge, Cribrochalina.54 However, around the same time
Kubo had semi-synthetically derived the same compound and characterized it as
renieramycin H.55 The significant differences when compared with renieramycin A are
the 3,4-olefin, the C14 ketone and the existence of the hydroquinone, whose presence is
attributed to the adjacent ketone.
Cribrostatin IV (Renieramycin H, 78)

24
Danishefsky used a strategy he developed in the 1980’s in his approach to the
quinocarcins.56 The enantioselective synthesis began with the construction of
tetrahydroisoquinoline 81 derived from phenol 79 (Scheme 1.16). A 12 step sequence of
reactions, in which a Noyori protocol57 enantioselectively installed the C1 stereocenter,
gave dimethylacetal 80. The tetrahydroisoquinoline 81 was formed from a Bobbitt
modified Pomeranz-Fritsch cyclization.58 The amine of 81 was coupled with amino acid
analogue 82, then PMB removal followed by Dess-Martin oxidation formed the C4-keto-
benzaldehyde 83.
MeCHO
HOOH
MeOAllyl
MeOOMe
NH
OMe
OMe
OBn
MeAllylO
MeOOMe
NH
OBn
OH
H
BnOOMe
Me
BocHNHO
O
PMBO
Me
H
BnOOMe
Me
BocN
O
Me
H
MeAllylO
MeOOMe
N
OBn
O
H
O
12 steps
22% yield
i
+ii-iv
Reaction Conditions: i) 8.0 M HCl/dioxane (97%); ii) BOPCl, TEA, CH2Cl2 (89%); iii) DDQ, CH2Cl2, pH = 7 buffer (90%); iv) DMP, 2,6-lutidine, CH2Cl2 (84%).
Scheme 1.16. Danishefsky’s Synthesis of Cribrostatin IV
79
83
80 81
81
82

25
Formation of the bicyclo[3.3.1] system was achieved by treatment of aldehyde 83
with formic acid at 100 °C (Scheme 1.17). This led to loss of the N-Boc protecting group,
imine formation and Mannich closure to form 84. The diastereoselective ring closure
formed solely the 1,3-trans-substituted tetrahydroisoquinoline, which is normally the
undesired orientation. However, the target molecule possesses unsaturation at the C3-C4
position making the stereochemistry at the newly formed C3 stereocenter irrelevant.
Reduction of the C4 ketone and elimination was facilitated by first deprotecting the
epi-allylether to generate the phenol. The phenol was reprotected as the TBS ether and
selective debenzylation of the arylbenzyl ether over the alkylbenzyl ether afforded 85. In
order to install the C14 ketone the phenol was first oxidized to the quinone with Fremy’s
salt. Benzylic oxidation to the ketone was promoted with selenium dioxide followed by
oxidation with Dess-Martin periodinane. The quinone was reduced back to the
hydroquinone and the alkylbenzyl ether was hydrogenolyzed in the same step under
catalytic hydrogenation conditions, producing alcohol 86. The angelate ester was readily
formed by simply allowing a solution of alcohol 86 and angeloyl chloride (87) to stand
for 12 hours, followed by TBS removal to afford phenol 88. A three step sequence was
necessary to furnish the mono-quinone cribrostatin IV. Oxidation to the bis-
isoquinolinequinone was necessary to remove the arylmethyl ether. Reduction of the bis-
isoquinolinequinone and air oxidation selectively oxidized the A-ring hydroquinone to
furnish cribrostatin IV (78). The synthesis was completed in 31 steps with an overall
yield of 2%.

26
AllylOMe
MeOOMe
NN
BnOOMe
Me
Me
H
H
H
H
OBnO
O OTBSMe
MeOOMe
NN
HOOMe
Me
Me
H
H
H
OBnO
OTBSMe
MeOOMe
NN
HOOMe
Me
Me
H
H
H
OHO
OHMe
MeOOMe
NN
HOOMe
Me
OHMe
H
H
H
O
OMe
Me
O
O
OH
O
i ii-vi
vii-x
Cl
O
MeH
Me
xi, xii
xiii-xv
H
OMe
MeOO
NN
HOOMe
Me
OHMe
H
H
H
O
OMe
Me
O
O
H
Reaction Conditions: i) HCO2H, 100 °C (59%) ii) NaBH4, THF/H2O; iii) AcOH, Bu3SnH, (Ph3P)2PdCl2, CH2Cl2 (98%, 2 steps) iv) CSA, PhH, 80 °C (80%); v) TBSOTf, TEA, CH2Cl2 (90%); vi) 5% Pd/C, H2 (1 atm), EtOAc (90%); vii) Fremy’s salt, KH2PO4, CH3CN/H2O (84%); viii) SeO2, dioxane, 100 °C (87%) ix) DMP, CH2Cl2; x) 10% Pd/C H2 (1 atm), MeOH (89%, 2 steps); xi) 87, CH2Cl2; xii) AcOH, TBAF, THF (75%, 2 steps); xiii) PIFA, CH3CN/H2O; xiv) Zn, AcOH; xv) air, DMF (65%, 3 steps).
Scheme 1.17. Danishefsky’s Synthesis of Cribrostatin IV
88
86
87
85 84
83
78

27
This strategy was successful in the synthesis of Cribrostatin IV, due to the lack of
stereochemistry at the C3 center, but Danishefsky’s attempts to synthesize other
tetrahydroisoquinoline alkaloids employing the same methodology were not as
successful. In his approach to the saframycins59 a similar Mannich cyclization on a
slightly different substrate 89 yielded only the 1,3-cis-isomer 90 rather than the trans-
isomer observed above (Scheme 1.18). In further contrast, this same strategy was
employed in Danishefsky’s synthesis of quinocarcins.56 However, dimethylacetal 91
cyclized to give only the trans-isomer on formation of the bicyclo[3.2.1] 92 (Scheme
1.19).
MeOOMe
Me
BocN
O
Me
H
MeMeO
MeOOMe
N
OBn
O
H
OOMe OMe
Me
MeOOMe
NN
MeOOMe
Me
Me
H
H
H
OBnO
OMeH
OHCO2H, reflux
Scheme 1.18. Danishefsky’s Approach Toward Saframycins
MeOBocN
O
Me
HOMe
N
O
H
MeOCO2Me
CO2Me
HCO2H, reflux
OMe
N
O
HNMe
HH
CO2MeMeO2C
HO
Scheme 1.19. Danishefsky’s Approach Toward Quinocarcin
89 90
92 91

28
The strategy outlined by Danishefsky was the first to combine the two subclasses
of tetrahydroisoquinoline alkaloids, the mono- and bis-tetrahydroisoquinoline alkaloids.
Unfortunately, the stereoselectivity necessary for formation of the
1,3-cis-tetrahydroisoquinoline was substrate dependent such that in the bicyclo[3.2.1]
ring formation the undesired stereochemistry was observed.
1.4.3. LEMONOMYCIN
NNH
H
HOH
MeO
Me
O
H
H
O
O
Me
OHMe
NMe2
O
HOOH
1
11
13
16
15
8
75
4
3
22
21
10
1.4.3.1. Stoltz’s Synthesis of (–)-Lemonomycin
The structure of (–)-lemonomycin (10) is quite unique in that it is the only
tetrahydroisoquinoline alkaloid to possess a glycosidic linkage and the rare aldehyde
hydrate functionality. The enantioselective synthesis of (–)-lemonomycin by Stoltz
further confirmed the rare functional groups.60 The synthetic strategy proved to be not
nearly as unique but rather an efficient combination of known strategies in the synthesis
of tetrahydroisoquinoline alkaloids. The early enantioselective formation of the
bicyclo[3.2.1] system was a slight modification of the strategy used by Garner and
co-workers for the synthesis of (−)-quinocarcin.61 Garner assembles the bicyclo[3.2.1]
(–)-Lemonomycin (10)

29
system via a 1,3-dipolar addition of azomethine ylide 94, formed from irradiation of
aziridine 93, with Oppolzer’s chiral acryloyl sultam 95 to diastereoselectively form the
bridged ring system 96 (Scheme 1.20).
hν (2537 Å)
dioxane+
N
SO2
ON
O
OOMe
Me
OH
NMe
N
OMeOH
NMeO
O
Me
NN
H
HO
O
OMe
MeH
OH
ON
SO2Me
61% yield
Scheme 1.20. Garner’s Approach to Quinocarcin
The cycloaddition employed by Stoltz used the Joule oxidopyrazinium62 97 and
the same Oppolzer’s chiral acryloyl sultam 95 to arrive at bicycle 98 in 94% ee (Scheme
1.21). Conversion to vinyl iodide 99 was followed by a Suzuki coupling with boronate
100, to give eneamide 101. The diastereoselectivity of the 3,4-olefin reduction followed
known observations,61 giving solely amide 102. Formation of the 1,3-cis-substituted
tetrahydroisoquinoline used methodology developed by Fukuyama in his synthesis of
renieramycin A; employing a Pictet-Spengler with the fully constructed side-chain
tethered to an aldehyde.50 Unfortunately the Pictet-Spengler ring closure between 102 and
104 proved unsuccessful, as the nitrogen amide was not reactive. Amide 102 was then
converted to amine 103 in five steps.
9495 93
96

30
HNN
O
BnBr
N
O
SOO
HNNBn
H
HO
OH
HNNBn
H
HO
OTIPS
I
OMeBMe
MeOOTs
O
O
HNNBn
H
HO
OTIPS
MeO
MeOMe
OTs
HNNBn
H
HO
OTIPS
MeO
MeOMe
OTs
H
H2NN
H
HOH
OH
MeO
MeOMe
OH
HCbz
+i, ii iii, iv
+ v
vi vii-xi
Reaction Conditions: i) N-Me-morpholine, CH3CN, −20 °C; ii) NaBH4, EtOH (72%, 2 steps); iii) TIPSOTf, CH2Cl2, 2,6-lutidine (82%); iv) ICl, CH2Cl2, 0 °C (81%); v) Pd(PPh3)4, K2CO3, PhH, MeOH, 70 °C (69%); vi) Pd/C, H2 (1000 psi), TFA, EtOH (72%); vii) CbzCl, DMAP, CH3CN; viii) KOTMS, CH3CN (87%, 2 steps); ix) Boc2O, DMAP, CH3CN; x) NaBH4, EtOH; xi) HCl, MeOH (81%, 3 steps).
Scheme 1.21. Stoltz’s Synthesis of (–)-Lemonomycin
The Pictet-Spengler cyclization with the more reactive amine 103 and aldehyde
104 gave the 1,3-cis-substituted tetrahydroisoquinoline 105 with excellent
diastereoselectivity (Scheme 1.22). Completion of the synthesis followed with N-Cbz
removal, bis-Swern oxidation, equilibration to the bicyclo[3.2.1] ring system and
oxidation to give (−)-lemonomycin (10). This very efficient enantioselective synthesis
was completed in 15 steps from 97 with an overall yield of 3%.
95 98
101 10099
97
103102

31
NHN
H
HOH
OH
MeO
MeOMe
OH
HCbz
H
O
O
Me
OHMe
NMe2
CHO
O
O
Me
OHMe
NMe2
+i ii-iv
Reaction Conditions: i) EtOH (63%) ii) Pd/C, H2, EtOH iii) Swern, then aq. HCl (52%); iv) CAN, 0 °C (51%).
Scheme 1.22. Stoltz’s Synthesis of (–)-Lemonomycin
1.4.3.2. Fukuyama’s Approach Toward the Synthesis of (–)-Lemonomycin
Fukuyama reported a partial synthesis of (–)-lemonomycin in 2005 employing a
strategy his group had previously developed.63 In their work on Et743, a synthetic
strategy using the Ugi four-component condensation was successful in building the core
ring structure.64 The approach to (–)-lemonomycin began with phenol 106 (Scheme 1.23)
which undergoes a Mannich-type reaction with chiral template 107 to yield
phenylglycine derivative 108. A five-step sequence installed the requisite differential
protecting groups and side-chain oxidation state to form the phenylglycinol derivative
109.
The Ugi four-component condensation was effected by heating amine 109,
aldehyde 110, isonitrile 111, and amino acid 112 to provide in excellent yield amide 113
(Scheme 1.24). This efficient procedure installed in one step all of the necessary atoms
needed to form the core of lemonomycin. Unfortunately, arranging the atoms to the
correct connectivity proved tedious. Mild acid hydrolysis of the dimethyl acetate formed
cyclized product enecarbamate 114.
105
104
103 10

32
MeOMe
MeOOH
O
N
O
Ph MeOMe
MeOOH
O
HN
O
Ph+
i MeOMe
MeOOMs
OTBDPS
NH2
ii-vi
Reaction Conditions: i) TFA, CH2Cl2, −10 °C (93%); ii) MsCl, TEA, CH2Cl2, 0 °C; iii) NaBH4, MeOH; iv) TBDPSCl, imid., DMF (90%, 3 steps); v) Pb(OAc)4, CH3CN 0 °C; vi) NH2OH·HCl, NaOAc, EtOH, 0 °C (74%, 2 steps).
Scheme 1.23. Fukuyama’s Approach to (–)-Lemonomycin
MeO CHO
OMe
OCO2PhCN
HO2C
NHBoc i
MeMeO
MeOOMs
OTBDPS
NO
HNOMeMeO OCO2Ph
O
NHBocMe
MeO
MeOOMs
NNBoc
OOTBDPS
HN
OCO2Ph
Oii
MeOMe
MeOOMs
OTBDPS
NH2
Reaction Conditions: i) CF3CH2OH, 50 °C; ii) CSA, quinoline, PhMe, reflux (73%, 2 steps).
Scheme 1.24. Fukuyama’s Approach to (–)-Lemonomycin
108107
106 109
112
111109
110
114113

33
Prior to the essential formation of the bicyclo[3.2.1] system the C4 amide was
converted to the acetate protected alcohol, as was the TBDMS ether. A metathesis
reaction was used to install the allyltrimethylsilane moiety providing cyclization
precursor 115 (Scheme 1.25). Exposure of 115 to BF3·Et2O generated a reactive
N-acyliminium intermediate 116 which was trapped by the allylsilane to give cyclized
product 117. This cyclization proved diastereoselective in formation of the C15
stereocenter, with no discussion of the observed selectivity. To achieve the required
formation of the 1,3-cis-substituted tetrahydroisoquinoline Fukuyama had previously
developed a method that treated an analogous eneamide with DMDO in methanol,
followed by an acidic reduction to reduce the C3 stereocenter.65 Unfortunately in this
synthesis the protecting groups proved to be incompatible. The acid sensitive protecting
groups were converted to more stable protecting groups and treatment with DMDO
formed methyl-aminal 118. Reduction with NaCNBH3 in TFA/THF provided the
requisite stereochemistry at C3. The final ring closure required a more electron-donating
aromatic ring thus the mesyl group was replaced by the corresponding benzyl ether.
Oxidation of the primary alcohol generated the aldehyde which upon exposure to TFA
cyclized to 119. No further work was reported on this intermediate.

34
NN
H
HO
MeO
Me
OMs
BocH
OAc
MeMeO
MeOOMs
NNBoc
OOAc
TMSOAc
MeMeO
MeOOMs
NNBoc
OOAc
Me3Si
NN
HO
MeO
MeOMe
OMs
CbzH
OTIPS
OHOMe
NN
HO
MeO
MeOMe
OBn
CbzH
OTIPS
OHH
i-v vi
vii-xi
xii-xvi3
OMe 15
Reaction Conditions: i) t-BuOK, 4Å molecular sieves, THF, 0 °C; ii) NaBH4, THF; iii) TBAF, THF, 50 °C; iv) Ac2O, pyr. (68%, 4 steps); v) Grubbs 2nd generation catalyst (2 mol%), allyltrimethylsilane, CH2Cl2, reflux (51%); vi) BF3·Et2O, CH2Cl2, −78 °C (95%); vii) TMSOTf, CH2Cl2, 0 °C; viii) CbzCl, DMAP, CH3CN; ix) K2CO3, MeOH; x) TIPSOTf, 2,6-lutidine, CH2Cl2, 0 °C (55%, 4 steps); xi) DMDO, Na2SO4, MeOH, −78 °C; CSA; xii) NaBH3CN, TFA/THF, 0 °C; xiii) KOSiMe3, CH3CN, 0 °C; xiv) BnBr, 50 °C (53%, 4 steps); xv) DMP, CH2Cl2; xvi) TFA/CH2Cl2 (76%, 2 steps).
Scheme 1.25. Fukuyama’s Approach to (–)-Lemonomycin
114
115
116 117
119118

35
1.5. RETROSYNTHETIC ANALYSIS
In our approach to the tetrahydroisoquinoline alkaloids we envisioned an
alternative route to form the key 1,3-cis-substituted tetrahydroisoquinoline core 122
(Scheme 1.26). With the exception of Corey’s work, the tetrahydroisoquinoline ring
forming reaction via Pictet-Spengler or Mannich cyclization has been shown to give both
the desired 1,3-cis and undesired 1,3-trans product depending on the substrate, thus
planning a strategy which predicts the stereoselectivity of these cyclizations a priori is
not possible. To avoid this potential problem our aim was to develop an alternative
method for the synthesis of the 1,3-cis-substituted tetrahydroisoquinoline ring system.
Unlike most syntheses, we planned to enantioselectively install the C1
stereocenter first. This chiral center would then be used to direct the formation of
subsequent stereocenters. The C3 stereocenter could be obtained via a reduction where
the steric bulk of the C1 side-chain would provide means for diastereoselectivity. For
installation of the C1 substituent we envision an enantioselective alkylation of
3-substituted-isoquinoline 125 with a nucleophile, M-CH2-X, to generate the
1,2-dihydroisoquinoline 124 followed by an ionic hydrogenation to establish the
1,3-cis-substituted tetrahydroisoquinoline core 123.
By building the western fragment first and elaborating the eastern bicycle last we
will have a potentially divergent route that allows formation of both the bicyclo[3.3.1]
system 120 and the bicyclo[3.2.1] system 121 late in the synthesis. Construction of the
bridged ring system will be realized from tricycle 122 by a carbon-carbon bonding
forming reaction at either C11 or C13, or by a cycloaddition generating both bonds at the
same time. The diastereoselectivity of these reactions will be essential but difficult to

36
predict. Our strategy has great potential to provide an efficient approach to both mono-
and bis-tetrahydroisoquinoline alkaloids.
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
X
H
Y Z
NNH
H CHO
HMeO
MeO
O
H
H
XY Z
NNR3
MeO
MeOMe
OR1
H
H
XO
NR2Y
MeO
MeOMe
OR1
H
H
XNR2Y
MeO
MeOMe
OR1
H
X
NY
MeO
MeOMe
OR1
1
11
13
3
1
3 1
3
1
11
13
3
1
11
13
3
1
3
Scheme 1.26. Retrosynthetic Analysis
122
121 120
124
123
125

37
1.6. CONCLUSION
The tetrahydroisoquinoline alkaloids have been of biological and chemical
significance for over 25 years. Their potent biological properties make them promising
therapeutics, while their structural complexity provides challenging synthetic targets.
Numerous research groups, far more than have been mentioned above, have made
important contributions in the synthesis of both mono- and bis-tetrahydroisoquinoline
alkaloids. Fukuyama has been the most prolific researcher publishing the first synthesis
of a tetrahydroisoquinoline in 1982 and a partial synthesis of lemonomycin using an
altogether different route in 2005. The strategy employed by Danishefsky had the most
potential for efficient access to both mono- and bis-tetrahydroisoquinoline alkaloids from
a common advanced intermediate, but the requisite diastereoselectivity during the
formation of the 1,3-cis-substituted tetrahydroisoquinoline could not be attained.
The results of our efforts to combine an efficient strategy for the synthesis of
mono- (lemonomycin) and bis-tetrahydroisoquinolines (saframycin B and renieramycin
G) will be discussed in the proceeding chapters.

38
1.7. REFERENCES
1) The chemistry and biology of tetrahydroisoquinoline alkaloids has been extensively reviewed in following review and references 2 through 10: Williams, R. M.; Scott, J. D. Chem. Rev. 2002, 102, 1669. 2) Kubo, A.; Arai, T. The Alkaloids; Brossi. A., Ed.; Academic Press: New York, 1983; Vol. 21, p 55. 3) Remers, W. A. The Chemistry of Antitumor Antibiotics; Wiley: New York, 1988, Vol. 2, p 93. 4) Remers, W. A. The Chemistry of Antitumor Antibiotics; Wiley: New York, 1988, Vol. 2, p 120. 5) Arai, T. Journal of Chromatography Library: Natural Product Isolation; Wagman, G. H., Cooper, R., Eds.; Elsevier: New York, 1989; Vol. 43, p 191. 6) Kubo, A.; Saito, N. Studies in Natural Products Chemistry; Elsevier: New York, 1992; Vol. 10, p 77. 7) Fukuyama, T. Adv. Heterocycl. Nat. Prod. Synth. 1992, 2, 189. 8) Katoh, T.; Terashima, S. Studies in Natural Products Chemistry; Elseveir: New York, 1997; Vol. 19, p 289. 9) Ozturk, T. The Alkaloids; Brossi, A., Ed.; Academic Press: New York, 2000; Vol. 53, p 119. 10) Rinehart, K. L. Med. Res. Rev. 2000, 20, 1. 11) Sygusch, J.; Brisse, F.; Hanessian, S.; Kluepfel, D. Tetrahedron Lett. 1974, 15, 4021. 12) Kluepfel, D.; Baker, H. A.; Piattoni, G.; Sehgal, S. N.; Sidorowicz, A.; Singh, K.; Vezina, C. J. Antibiot. 1975, 28, 497. 13) Zmijewski, M. M., Jr.; Goebel, M. J. Antibiot. 1982, 35, 771. 14) Hayashi, T.; Noto, T.; Nawata, T.; Okazaki, H.; Sawada, M; Ando, K. J. Antibiot. 1982, 35, 771. 15) Tomita, F.; Takahashi, K.; Shimizu, K. J. Antibiot. 1983, 36, 463.

39
16) Ikeda, T.; Idemoto, H.; Hirayama, F.; Yamamoto, K.; Iwao, K. Asao, T.; Munakata, T. J. Antibiot. 1983, 36, 1279. 17) Suzuki, K.; Sato, T.; Morioka, M.; Nagai, K.; Abe, K.; Yamaguchi, H.; Saito, T. J. Antibiot. 1991, 44, 479. 18) Bernan, V. S.; Montenegro, D. A.; Korshalla, J. D.; Maiese, W. M.; Steinberg, D. A.; Greenstein, M. J. Antibiot. 1994, 47, 1417. 19) Whaley, H. A.; Patterson, E. L.; Dann, M.; Shay, A. J.; Porter, J. N. Antimicrob. Agents Chemother. 1964, 8, 83. 20) He, H.; Shen, B.; Carter, G. T. Tetrahedron Lett. 2000, 41, 2067. 21) Frincke, J. M.; Faulkner, D. J. J. Am. Chem. Soc. 1982, 104, 265. 22) He. H.; Faulkner, D. J. J. Org. Chem. 1989, 54, 5822. 23) Davidson, B. S. Tetrahedron Lett. 1992, 33, 3721. 24) Parameswaran, P. S.; Naik, C. G.; Kamat, S. Y.; Pramanik, B. N. Ind. J. Chem. 1998, 37B, 1258. 25) Original isolation: Rinehart, K. L.; Holt, T. G.; Fregeau, N. L.; Stroh, J. G.; Kieffer, P. A.; Sun, F.; Li, L. H.; Martin, D. G. J. Org. Chem. 1990, 55, 4512. 26) Structural revision: Rinehart, K. L.; Holt, T. G.; Fregeau, N. L.; Stroh, J. G.; Kieffer, P. A.; Sun, F.; Li, L. H.; Martin, D. G. J. Org. Chem. 1991, 56, 1676. 27) Phase I clinical data, see: Ryan, D. P.; Supko, J. G.; Eder, J. P.; Seiden, M. V.; Demetri, G.; Lynch, T. J.; Fischman, A. J.; Davis, J.; Jieno, J.; Clark, J. W. Clin. Cancer Res. 2001, 7, 231. 28) Rinehart, K. L. Med. Res. Rev. 2000, 20, 1. 29) Myers, A. G.; Plowright, A. T. J. Am. Chem. Soc. 2001, 123, 5114. 30) Pastrone, I.; Viale, M; Cafaggi, S.; Mariggio, M. A.; Parodi, A.; Esposito, M. Invest. New Drugs 1999, 16 (4), 297. 31) Martoli, M. H.; Boitard, M.; Fessi, H.; Beriel, H.; Devissaguet, J. P.; Picot, F.; Puisieux, F. J. Microencapsul. 1990, 7 (2), 191.

40
32) Martello, L. A.; McDaid, H. M.; Regl, D. L.; Yang, C. P.; Meng, D.; Pettus, T. R.; Kaufman, M. D.; Arimoto, H.; Danishefsky, S. J.; Smith, A. M.; Horwitz, A. B. Clin. Cancer Res. 2000, 6, 1978. 33) Dorr, R. T.; Liddil, J. D.; Trent, J. M.; Dalton, W. S. Biochem. Pharmacol. 1987, 36 (19), 3115. 34) Kohn, H.; Na, Y.; Li, V.-S.; Nakanishi, Y.; Bastow, K. F. J. Med. Chem. 2001, 44, 3453. 35) Viale, M.; Vannozzi, M. O.; Merlo, F.; Cafaggi, S.; Parodi, B.; Esposito, M. Eur. J. Cancer 1996, 32 (13), 2327. 36) Ying, Y.; Qiu-Jun, L.; Qing-You, D., Bing-Hu, Y.; Ru-Xian, L.; Sheng-Qi, W. World J. Gastroenterol 2005, 11 (16), 2491. 37) Aune, G. J.; Furuta, T.; Pommier, Y. Anti-Cancer Drugs 2002, 13, 545. 38) Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K. W. Biochemistry, 1996, 35, 13303. 39) Bernan, V. S.; Montenegro, D. A.; Korshalla, J. D.; Maiese, W. M.; Steinberg, D. A.; Greenstein, M. J. Antibiot. 1994, 47, 1417. 40) Mikami, Y.; Yazawa, K.; Takahashi, K.; Arai, T.; Namikoshi, M.; Iwasaki, S.; Okuda, S. J. Biol. Chem. 1985, 260, 334. 41) Zmijewski, M. J., Jr.; Mikolajczak, M.; Viswanatha, V.; Hruby, V. J. J. Am. Chem. Soc. 1982, 104, 4969. 42) Zmijewski, M. J., Jr.; Palaniswamy, V. A.; Gould, S. J. J. Chem. Soc., Chem. Comm. 1985, 1261. 43) Palaniswamy, V. A.; Gould, S. J. J. Am. Chem. Soc. 1986, 108, 5651. 44) Fukuyama, T.; Schleben, R. A. J. Am. Chem. Soc. 1982, 104, 4957. 45) Kubo, A.; Saito, N.; Yamato, H.; Masubuchi, K.; Nakamura, M. J. Org. Chem. 1988, 53, 4295. 46) Fukuyama, T.; Yang, L.; Ajeck, K. L.; Sachleben, R. A. J. Am. Chem. Soc. 1990, 112, 3712. 47) Williams, R. M.; Jin, W.; Metobo, S. Org. Lett. 2003, 5, 2095.

41
48) Corey, E. J.; Gin, D. Y.; Kania, R. S. J. Am. Chem. Soc. 1996, 118, 9202. 49) Corey, E. J; Martinez, E. J. Org. Lett. 1999, 1, 75. 50) Fukuyama, T.; Linton, S. D.; Tun, M. M. Tetrahedron Lett. 1990, 31, 5989. 51) Faulkner, D. J.; Frincke, J. M. J. Am. Chem. Soc. 1982, 104, 265. 52) Faulkner, D. J.; He, H. J. Org. Chem. 1989, 54, 5822. 53) Danishefsky, S. J.; Chan, C.; Heid, R.; Zheng, S.; Guo, J.; Zhou, B.; Furuuchi, T. J. Am. Chem. Soc. 2005, 127, 4596. 54) Pettit, G. R.; Knight, J. C.; Collins, J. C.; Herald, D. L.; Pettit, R. K.; Boyd, M. R.; Young, V. G. J. Nat. Prod. 2000, 63, 793. 55) Kubo, A.; Saito, B.; Sakai, H.; Suwanborirux, K.; Pummangura, S. Heterocycles 2001, 55, 21. 56) Danishefsky, S. J.; Harrison, P. J.; Webb, R. R. II; O’Neill, B. T. J. Am. Chem. Soc. 1985, 107, 1421. 57) Noyori, R.; Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T. J. Am. Chem. Soc. 1996, 118, 2521. 58) Bobbitt, J. M.; Moore, T. E. J. Org. Chem. 1968, 33, 2958. 59) Danishefsky, S. J.; Guo, J.; Zhou, B. Tetrahedron Lett. 2000, 41, 2043. 60) Stoltz, B. M.; Cruz, E. G.; Ashley, E. R. J. Am. Chem. Soc. 2003, 125, 15000. 61) Garner, P.; Ho, W. B.; Shin, H. J. Am. Chem. Soc. 1993, 115, 10742. 62) Joule, J. A.; Yates, N. D.; Peteres, D. A.; Allway, P. A.; Beddeoes, R. L.; Scopes, D. I. C. Heterocycles 1995, 40, 331. 63) Fukuyama, T.; Rikimaru, K.; Mori, K.; Kan, T. Chem. Comm. 2005, 394. 64) Fukuyama, T.; Endo, A.; Yanagisawa, A.; Abe, S.; Toma , T. K. J. Am. Chem. Soc. 2002, 142, 6552. 65) Fukuyama, T.; Mori, K.; Rikimaru, K.; Kan, T. Org. Lett. 2004, 6, 3095.

42
Chapter 2: Toward the Synthesis of Saframycin B
2.0. INTRODUCTION
In our retrosynthetic analysis of the tetrahydroisoquinoline alkaloids we plan to
form the first stereocenter at C1 early in the synthesis via an enantioselective alkylation
of an isoquinoline derivative 128 (Scheme 2.01). The stereoselective reduction of the
3,4-olefin 127 will be essential for obtaining the requisite 1,3-cis-substituted
tetrahydroisoquinoline core 126. We anticipate the C1 substituent will provide steric bulk
for facial selectivity during reduction. Saframycin B (1b) was chosen as our initial target
because of the simplicity of the structure, lacking oxidation at C20, C14, C4, and
possessing the pyruvamide side-chain. The alkylation and reduction chemistry will be
investigated with isoquinoline, a 3-substituted isoquinoline, and a substituted
isoquinoline representative of the real system.
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
NHCOCOMe
H
1
11
13
314
4
21NR2
YH
H
X
NR2Y
H
X
NY
1
3
1
3
1
3
R1
R1 R1
Scheme 2.01. Synthetic Approach Toward Saframycin B
126
127128
1b

43
2.1. BACKGROUND: ENANTIOSELECTIVE ALKYLATION OF ISOQUINOLINES
The alkylation of isoquinolines at C1 is a well documented reaction.1 When
treated with a strong organometallic nucleophile (130) isoquinoline (129) is alkylated
solely at the C1 position giving 131 (Scheme 2.02). In theory, if one shifts the aromatic
resonance to the pyridine portion of isoquinoline (132), one can envision alkylation at the
C3 position. However, this is not observed because the alkylated product 133 would no
longer possess an aromatic ring. The activation energy necessary to alkylate the C3 center
is significantly higher than alkylation at the C1 center.
N M X+ N
X
M
NM
N M XX+
Scheme 2.02. C1 Selectivity in Isoquinoline Alkylation
The stereoselective alkylation of an isoquinoline at C1 has been demonstrated on
few substrates and with limited success. In contrast, the enantioselective alkylation of
3,4-dihydroisoquinolines is much more well studied and will not be discussed here.2 One
of the oldest and well studied methods to alkylate isoquinolines is the Reissert reaction.3
Treatment of isoquinoline with an acid chloride in the presence of a cyanide source
generates the 1-cyano-1,2-dihydroisoquinoline 135 via the N-acylisoquinolinium
activated intermediate 134 (Scheme 2.03). Shibasaki and co-workers reported an
129 130
132 133
131
130

44
enantioselective Reissert reaction by catalyzing the alkylation in the presence of a chiral
lewis acid 137 (Scheme 2.03).4 The alkylation with 3-methylisoquinoline 136 under
optimized conditions gave the 1,2-dihydroisoquinoline 138 in 99% yield and 77% ee. The
Reissert reaction is a viable method for installation of the C1 stereocenter, and there is
potential for the formation of the requisite aminomethyl side-chain upon reduction.
However a drawback of this method is the use of an acid chloride which generates an
amide, a rather robust functional group, which could be potentially difficult to remove.
NKCN, AcCl
N Me
O
N Me
OCN
CN
N
Me TMSCN, AcCl
137, CH2Cl2N
MeO
N
Me
Me
OCN
OO
P(O)Ph2
P(O)Ph2
AlCl
Me3Si CN
Scheme 2.03. Variants of the Reissert Reaction
Reissert Reaction
Enantioselective Reissert Reaction
135
134
138 99% yield, 77% ee
137
136
129

45
Another method for the alkylation of activated isoquinolines involves the use of a
chiral acid chloride or chloroformate. Comins and co-workers have developed an
efficient method for the diastereoselective addition of Grignard reagents to homochiral 1-
acylpyridinium salts.5 Comins published one paper on the application of this
methodology to isoquinolines.6 Isoquinoline was treated with (−)-8-phenylmenthyl
chloroformate, forming a chiral N-acylisoquinolinium. Addition of MeMgI gave an 80%
yield of the alkylated product 139 with 60% de under optimized conditions (Scheme
2.04). This route is potentially attractive because the more readily cleaved carbamate is
formed rather than an amide. However, there is little potential for greater than 60% ee
and the alkylations are reported to work for Grignards but not alkyllithium nucleophiles.
Organolithium nucleophiles are reported to alkylate at the acyliminium carbonyl not at
the C1 imine carbon.7
N1) R*OCOCl, PhMe/THF, −23 °C
2) MeMgI
R* = (−)-8-phenylmenthyl
N
MeCO2R*
Scheme 2.04. Comins’ Diastereoselective Alkylations
139 80% yield, 60% de
129

46
The use of a chiral auxiliary is another potential option for the enantioselective
alkylation of isoquinolines. Marazano and co-workers reported modest
diastereoselectivity in the alkylation of the isoquinolinium salt 140 (Scheme 2.05).8 The
addition of RMgI formed the 1-substituted-1,2-dihydroisoquinoline 141 in 32-57% yield
and 38-90% de. The modest yield and diastereoselectivity make this route less attractive.
In addition, formation of the chiral salt 140 required a three step procedure from
isoquinoline.
NPhOH(CH2)11
OSO3
RMgI, THF
0 °CN
PhOH
HH
R
Scheme 2.05. Chiral Isoquinolinium Salts
A recent publication by the Alexakis group offered the most general method to
enantioselectively alkylate isoquinolines.9 Alexakis and Amiot applied known
methodology used for the enantioselective alkylation of acyclic and cyclic imines to
isoquinolines.2 They reported an alkylation of isoquinoline with an alkyllithium
nucleophile in the presence of a chiral bidentate ligand, however the initial alkylated
product could not be isolated. Alexakis observed that treatment of isoquinoline with
MeLi resulted in the rearomatised product 142 upon workup (Scheme 2.06) and not the
1,2-dihydroisoquinoline 143. In order to obtain the 1,2-dihydroisoquinoline the
intermediate alkylated product 146 was acylated with methyl chloroformate, however
acylation occurred at both C2 and C4 forming a mixture of 144 and 145. After MeLi
addition to isoquinoline the N2-lithio 146 is in equilibrium with the C4-lithio 147 which
141 32-57% yield, 38-90% de
140

47
142
is then acylated. The resulting nucleophilic nitrogen is acylated a second time to give
intermediate 148 which can be readily deprotonated to generate the diacylated product
145.
N N
MeLi
N
Me
Li
MeLi
Et2O25 °C
N
Me
air
ClCO2Me
N
MeCO2Me N
Me
CO2Me
+CO2Me
N
Me
MeCO2 H
CO2Me
NH
Me
Scheme 2.06. Alexakis Alkylation of Unactivated Isoquinoline
It was observed that the alkylation of isoquinoline with MeLi was catalyzed by
the addition of a bidentate ligand such as dimethoxyethane. Furthermore, the use of a
chiral bidentate ligand, namely (−)-sparteine (149) (Table 2.01), resulted in
enantioselective alkylation. The results are summarized in Table 2.1.
143
145144
147
148
146129

48
N
i) MeLi, solventtemp., 149
+ii) ClCO2Me
N N
entry solvent temp. (°C)
equiv. of 149 yield (%) ratio
144/145 ee (%)
144
1 PhMe −40 1 77 75/25 42
2 PhMe −40 0.25 89 70/30 36
3 Et2O −40 0.2 76 70/30 23
4 Et2O −78 0.2 7 70/30 21
5 PhMe −78 0.2 54 70/30 29
6 PhMe −10 0 45 80/20 N/A
Table 2.01. Alexakis’ Enantioselective Alkylation of Isoquinoline7,9
The shortcomings of this approach include modest ee’s, inseparable mixtures of
mono- and di-acylated products and only a limited number of nucleophiles were
investigated (MeLi, n-BuLi, and PhLi). Although the reaction proved catalytic for
enantioselective alkylation (entries 2-5), variance of solvent and temperature did very
little to improve the ee. The alkylations with PhLi and n-BuLi gave similar results to
MeLi. The Alexakis group was able to prove through further experiments that the
addition of MeLi occurred from the top face, producing the desired absolute
stereochemistry at C1 in agreement with the tetrahydroisoquinoline alkaloid side-chain.
We hoped to improve upon this methodology with the use of a more
functionalized nucleophile, improving the overall yield of mono-acylated product and
increasing the ee for alkylation of unactivated isoquinolines. The tetrahydroisoquinoline
144 145
149 129

49
alkaloids have a C1 side-chain with a methylene then a heteroatom (N or O), suggesting
that our organolithium nucleophile should have the general structure Li-CH2-X. An initial
search in the literature found that Li-CH2-SPh (phenylthiomethyllithium or
lithiothioanisole, 150) was a well investigated nucleophile suitable for our requirements.
Phenylthiomethyllithium (151) can be generated by treatment of thioanisole with
n-BuLi in diethyl ether with stirring for 15 hours (Table 2.02). The lithiation takes place
on the aromatic ring as well as the methyl group generating a mixture of products.10 The
aryl-lithiated products 152 slowly equilibrate to the more thermodynamically favored
methyl-lithiated product over a period of 15 h. Corey and Seebach reported that under
similar conditions in the presence of a tertiary diamine, 1,3-diazabicyclo[2.2.2]octane 153
(DABCO), phenylthiomethyllithium was formed in 45 minutes.11 Peterson observed
similar results when tetramethylethylenediamine (TMEDA) was employed instead of
DABCO.12
S n-BuLi
Et2O
S
Li
S Li+
observed product composition (mole%) time
(min.) 153
(equiv.) methyl ortho and meta para
5 0 63 37 trace
1 0 90 9 trace
15 0 96 4 trace
45 min. (in THF) 1 98.5 trace trace
Table 2.02. Thioanisole Metalation
NN
150 153 151 152
NN

50
129
A further literature search revealed, to the best of our knowledge, that
(−)-sparteine has never been used in the lithiation of thioanisole. It was our hope to
combine the isoquinoline alkylation work of Alexakis and lithiation results of Corey to
enantioselectively alkylate isoquinoline with phenylthiomethyllithium formed in the
presence of (−)-sparteine (Scheme 2.07).
S
n-BuLi, THFS Li
N NN
NN
N
PhS
Li
ClCO2MeN
PhS
CO2Me
Scheme 2.07. Planned Enantioselective Alkylation of Isoquinoline
149
150 154
156155

51
2.2. RESULTS AND DISCUSSION
2.2.1. INVESTIGATION OF THE ENANTIOSELECTIVE ALKYLATION OF ISOQUINOLINE
During our initial investigation into the alkylation of isoquinoline with
phenylthiomethyllithium we chose to use the known methods of DABCO and TMEDA to
generate the lithiated nucleophile. When a solution of isoquinoline in toluene at −78 °C
was treated with phenylthiomethyllithium the alkylation proceeded slowly (Table 2.03).
Stirring at −20 °C for more than 24 hours was needed for greater than 80% conversion,
when either DABCO or TMEDA were used. A methyl chloroformate quench gave a
mixture of mono-acylated product 157 and di-acylated product 158, as observed by
Alexakis. In contrast to Alexakis’ results, these products could be separated and
characterized. When (−)-sparteine was used in place of DABCO/TMEDA the results
were similar although the yield of diacylated product was slightly lower. In an effort to
minimize the reaction time the reaction was allowed to warm to room temperature. It was
observed that the starting isoquinoline was rapidly consumed when warmed to room
temperature and upon quenching with methyl chloroformate only the mono-acylated
product 157 was observed.
These results suggest that the intermediate alkylated product, which exists in an
equilibrium as the N2-lithio or C4-lithio adducts, is temperature dependent. The C4-litho
product is either less stable at room temperature or the N2-lithio adduct is significantly
more reactive at room temperature such that none of the C4 acylated product is observed.

52
N N
PhS
CO2MeN
PhS
CO2Me
CO2Me
+
1) PhSCH2Li (1.5 equiv.), PhMe, diamine, temp., time
2) ClCO2Me
diamine temp. (°C) time yield (%) 157 yield (%) 158
DABCO −78 to −20 1 day 33 52
TMEDA −78 to −20 2 days 39 28
(−)-sparteine −78 to −20 1 day 45 10
(−)-sparteine −78 to −40 3 days 56 12
(−)-sparteine −78 to 25 1 h 91 0
Table 2.03. Alkylation of Isoquinoline with Phenylthiomethyllithium
With the success of the alkylation of isoquinoline with phenylthiomethyllithium
in the presence of (−)-sparteine we next had to determine if the alkylation was
enantioselective. We first attempted to resolve enantiomers in the 1H NMR using a
europium based chiral shift reagent, but this was unsuccessful. Instead, analytical HPLC
with a chiral column (Diacel, Chiracel OD) was employed and the 1:1 mixture of
enantiomers was readily separable. The separation was also performed on the product
formed from the use of DABCO and identical results were observed. A variety of
conditions were employed; varying reaction temperature, equivalents of chiral ligand and
nucleophile, and solvent in an effort to achieve some level of enantioselectivity without
success.
157 158 129

53
In a different approach to the enantioselective alkylation of isoquinolines we
investigated the use of chiral sulfoxides. Sulfoxides are more readily lithiated at the
methyl position and chiral sulfoxides have shown diastereoselectivity in the alkylation of
imines.13 Methyl phenyl sulfoxide (159) was used as a model (Scheme 2.08), but the
reaction was not analogous to the phenylthiomethyllithium reaction. The starting
isoquinoline was consumed by TLC analysis and a variety of products were observed, but
upon methyl chloroformate quench none of the desired product 160 was isolated.
SO i) LDA, THF, −78 °C
ii) isoquinoline (129), −78 °C to 25 °C
iii) ClCO2Me
N
SPh
OCO2Me
Scheme 2.08. Isoquinoline Alkylation with Methyl Phenyl Sulfoxide
There are a number of potential side reactions stemming from use of the lithiated
methyl phenyl sulfoxide 161 that are less likely and hence not observed with thioanisole.
Assuming the initial alkylation was successful, the sulfoxide adduct 162 (Scheme 2.09)
would have α-methylene protons which are still quite acidic. This could lead to not only
an equilibrium with C4-lithiation but also lithiation at C22 (saframycin numbering) to
give 165. Once lithiation at C22 occurs α-elimination to form 166 would be possible.
Furthermore, sulfoxides are also known to β-eliminate14 and 164 could potentially form
from deprotonation at C1 of intermediate 163. All approaches to enantioselective
alkylation of isoquinoline with a sulfoxide were abandoned owing to the wide variety of
possible by-products.
159 160

54
129 N
SPh
OLi
N
SPh
O
Li
NH
SPh
OLi
HN
SPh
OLi
H
N
LiSO
LiN
22
Scheme 2.09. Potential Side-Reactions of Sulfoxide Alkylation
Our investigation into the enantioselective alkylation of isoquinoline with
phenylthiomethyllithium failed to show any facial selectivity, but we were able to
successfully install a functionalized C1 side-chain and minimize the amount of diacylated
product. At this point we decided further investigation into the formation of the
1,3-cis-substituted tetrahydroisoquinoline would prove more fruitful and a potential
reinvestigation of the enantioselective alkylation could be done at a later stage.
166165
162161
164
163

55
2.2.2. FORMATION OF 1,3-CIS-SUBSTITUTED TETRAHYDROISOQUINOLINE
To form the 1,3-cis-substituted tetrahydroisoquinoline we needed to establish a
method of reduction. As a model system we used the previously formed
1,2-dihydroisoquinoline 157. We anticipated potential difficulties with reduction of the
hindered di-substituted cyclic olefin (and tri-substituted for the real system). In addition it
was postulated that the presence of the sulfur could poison catalytic hydrogenation
methods.
Initial investigations found that palladium catalyzed hydrogenations were
unsuccessful for obtaining the reduced product. Wilkson’s catalyst has been used to
successfully reduce olefins in the presence of sulfides.15 For our substrate no reduction
was observed. We next turned our attention to ionic hydrogenation with the use of a
strong acid and a trialkylsilane. A recent publication by Molinski et al. gave promise for
the reduction of our substrate.16 It was reported that the reduction of enecarbamates 167
with TFA and triethylsilane, through the intermediate iminium 168, gave carbamates 169
in excellent yields (Scheme 2.10). The reaction is the acyclic equivalent to our substrate
and uses reagents that should be compatible with the sulfide side-chain.
HN
CO2Et
R
Et3SiH
TFA, −10 °C
HN
CO2Et
R
NCO2Et
R
HH
H SiEt3O2CCF3
94-99% yield
Scheme 2.10. Molinski’s Ionic Hydrogenation of Enecarbamates
169
168
167

56
Upon treatment of 1,2-dihydroisoquinoline 157 with triethylsilane and TFA clean
reduction of the 3,4-olefin was observed, to give the tetrahydroisoquinoline 170 in
excellent yield (Scheme 2.11).
NCO2Me
SPh
Et3SiH, TFA
CH2Cl2, 0 °C to 25 °C, 1 hN
CO2Me
SPh95% yield
Scheme 2.11. Reduction of 1,2-Dihydroisoquinoline 157
In order to establish the stereoselectivity for this reduction a 3-substituted
isoquinoline was necessary. The simple, commercially available 3-methyl isoquinoline
(171), was chosen for the model study. Treatment of 3-methyl isoquinoline with
phenylthiomethyllithium and methyl chloroformate quench as before, afforded the
desired product 172 in excellent yield (Scheme 2.12).
NCO2Me
SPh
N
90% yield
Me Me i) PhSCH2Li, (−)-sparteinePhMe, −78 °C to 25 °C, 1 h
ii) ClCO2Me
Scheme 2.12. Alkylation of 3-Methyl Isoquinoline
It should be noted that although enantioselectivity was not observed for the
alkylation of isoquinolines with phenylthiomethyllithium generated in the presence of
(−)-sparteine, these alkylations gave superior results than the DABCO or TMEDA
promoted alkylations. DABCO is a hygroscopic solid which made for tedious reaction
170
171
157
172

57
setup on a small scale and capricious reaction results were observed. With TMEDA more
by-products were observed during alkylation. (−)-Sparteine is readily purified by
distillation, it is not particularly expensive, and the best, reproducible results were
observed using (−)-sparteine as the diamine for phenylthiomethyllithium formation.
The reduction of 3-methyl-1,2-dihydroisoquinoline 172 was conducted with
Et3SiH/TFA as before, to give in excellent yield the reduced 173 as what appeared to be
one diastereomer by TLC (Scheme 2.13). Further confirmation of the structure by 1H
NMR analysis was difficult because strong carbamate resonance led to an observed
broadening of signals. However, upon performing a 1H NMR acquisition at 100 °C, the
signals sharpened and it was obvious that only one diastereomer was formed. A NOESY
experiment was conducted to determine the stereochemistry at the C3 center. Irradiation
of the C3 methyl protons showed diagnostic nuclear Overhauser enhancements of C22
methylene protons, as well as C3 and C4 protons, but no enhancement of the C1 proton
was observed. These results suggested that indeed we had formed the 1,3-cis-substituted
tetrahydroisoquinoline 173.
NCO2Me
NCO2Me
SPh
Me Et3SiH, TFA
CH2Cl2, 0 °C to 25 °C, 45 min.
87% yield
MeH
HH H
HPhSH
Scheme 2.13. Stereoselective Reduction of 3-Methyl-1,2-Dihydroisoquinoline 172
observed nOe’s of C3-methyl
172 173

58
The observed cis-relationship is due to the steric bulk of the C1 side-chain. The
reduction occurs on the iminium intermediate 172 (Scheme 2.14) which possesses four
sp2 hybridized carbons leading to a flat structure, as modeled by Chem3D (Figure 2.01).
The C1 side-chain exists in the axial orientation hindering hydride addition to the C3
iminium from the top face which would give the trans-isomer. Instead only the
cis-isomer is observed from hydride addition to the bottom face.
NCO2Me
SPh
Me
NCO2Me
SPh
MeHN
CO2Me
SPh
MeH SiEt3 H
H
Scheme 2.14. Ionic Hydrogenation of 172
Figure 2.01 Chem3D Representation of the N-Acyliminium Intermediate 174
172 173 174

59
We have successfully demonstrated an efficient route to 1,3-cis-substituted
tetrahydroisoquinolines from alkylation of unactivated isoquinolines with
phenylthiomethyllithium followed by methyl chloroformate quench to give the
1,2-dihydroisoquinoline. A stereoselective 3,4-olefin reduction by ionic hydrogenation
provides the 1,3-cis-substituted tetrahydroisoquinoline. Investigation of this chemistry
using a more functionalized 3-substituted isoquinoline which is representative of the real
system was required.
2.2.3. 3-(AMINOMETHYL)-ISOQUINOLINE
Moving to an isoquinoline system that closely resembles saframycin B we wanted
to investigate the alkylation and reduction of a 3-(aminomethyl)-isoquinoline 178. The
nitrogen is in the correct orientation to generate the piperazine ring system present in
saframycin B. In addition, we anticipated that the benzyl protecting groups would be
stable to the strong basic conditions used for alkylation and the strong acidic conditions
used for reduction.
The initial synthesis of 178 followed a literature preparation from 1994 (Scheme
2.15).17 The problems with this route included the high cost of starting material, the
number of steps involved, and inconsistent yields for reduction of the nitrile 176 were
observed. Improvements in the synthesis of 3-substituted isoquinolines were essential for
this route to be viable.

60
NOH
O1) ClCO2Et, TEA, THF
2) NH4OH3) TFAA, Et3N, CH2Cl2
95% yield
N
CN
H2, Pd/C
HCl, MeOH NH·HClNH2·HCl
48-78% yield
NNBn2
Scheme 2.15. Synthesis of 3-(Aminomethyl)-Isoquinoline
The solution came in a timely publication by Larock.18 He reported a very
efficient synthesis of 3-substituted isoquinolines 181 from ortho-iodo imines 179 and
various acetylenes 180 in a two step process (Scheme 2.16). The first step involved a
Sonagashira coupling of the aryl iodide 179 and acetylene 180 to form the coupled
product 182. After filtration and removal of triethylamine the coupled product was
redissolved in DMF and heated in the presence of catalytic CuI. The copper complex 183
cyclized to 184 and elimination formed the isoquinoline 181. Larock did not report the
synthesis of isoquinoline 178, nor did he demonstrate that this methodology could be
used with a nitrogenous acetylene.
176, $128/1
178 177
175, $78/1 g

61
I
N+ RH
1) 2% PdCl2(PPh3)2, 1% CuI Et3N, 50 °C
2) 10% CuI, DMF, 100 °CN
R
81-95% yield
N
R
N
RCuLn
N
RCu
H
HSonagashira
CuI
Scheme 2.16. Larock Isoquinoline Synthesis
To generate the required isoquinoline 178, ortho-iodo imine 179 was synthesized
from inexpensive ortho-iodo benzoic acid 185 (Scheme 2.17). Following published
procedures the acid was reduced to the corresponding alcohol with borane19 then oxidized
to the aldehyde 186 with PCC20 in 89% yield over the two steps. The Larock procedure
for imine formation was modified; the aldehyde was stirred with tert-butylamine in
pentane over 4Å molecular sieves for 12 hours at room temperature. Upon filtration and
concentration, a 92% yield of imine 179 was obtained with no aqueous workup or
distillation, as the excess tert-butylamine is removed under aspirator vacuum. The
standard Larock conditions were used with dibenzylpropargylamine21 (187) for the
Sonagashira coupling, to give in near quantitative yield the coupled product after two
hours at 55 °C. The copper catalyzed cyclization step proceeded slower than expected
and was not complete until after heating at 100 °C for 14 hours. Under optimized
conditions isoquinoline 178 was isolated in 84% from imine 179.
182
180181179
183 184

62
I
CO2H
I
O
H1) BH3·SMe2, THF, reflux
2) PCC, CH2Cl2, 25 °C
t-butylamine4Å molecular sieves
25 °C, 16 h
I
N
1) 2% PdCl2(PPh3)2, 1% CuI Et3N, 55 °C, 2 h
2) 10% CuI, DMF, 100 °C, 14 h NNBn2+
NBn2
89% yield92% yield
84% yield
Scheme 2.17. 3-(Aminomethyl)-Isoquinoline Formation
The alkylation of isoquinoline 178 with 1.5 equivalents of
phenylthiomethyllithium gave the desired 1,2-dihydroisoquinoline 188 in 86% yield upon
quenching with methyl chloroformate (Scheme 2.18). From ortho-iodo benzoic acid, a
six step sequence yielded the 1,2-dihydroisoquinoline 188 in 59% yield.
NNBn2
NCO2Me
SPh86% yield
NBn2
i) PhSCH2Li, (−)-sparteinePhMe, −78 °C to 25 °C, 30 min.
ii) ClCO2Me
Scheme 2.18. Alkylation of Isoquinoline 178
The ionic hydrogenation conditions which worked so effectively for the 3-methyl-
1,2-dihydroisoquinoline 172 gave only starting material when applied to 188 (Scheme
2.19). Treatment of 188 with Et3SiH and TFA, even under refluxing conditions, gave
mostly starting material with only slight degradation. This dramatic change in reactivity
can be attributed to the tertiary amine. The strong acidic conditions the nitrogen will be
185, $15/25 g 186
179 187 178
178 188

63
protonated such that in solution the starting material exists as the fully protonated
intermediate 189. In order for the reduction to occur a second protonation at the C4 center
must occur generating a di-cationic species 190. This di-cation may be too high in energy
to form under these conditions, hence reduction is not observed.
NCO2Me
SPh
NBn2 Et3SiH, TFA
CH2Cl2, reflux NCO2Me
SPh
NBn2H
H
NCO2Me
SPh
NBn2
H
NCO2Me
SPh
NBn2
H
H
H
H
Scheme 2.19. Attempted Ionic Hydrogenation of 1,2-Dihydroisoquinoline 188
In an attempt to circumvent this problem we planned to convert the tertiary amine
to a carbamate via a known modification of the Von Brun reaction.22 Benzyl amines can
be removed in a two step procedure by first reacting with a chloroformate to form a
carbamate, followed by hydrolysis to generate the debenzylated amine. It was anticipated
that treatment of amine 188 with phenylchloroformate in refluxing 1,2-dichloroethane
would promote debenzylation via N-acyliminium 192 to generate carbamate 193 which
we hoped would undergo ionic hydrogenation more readily.
190189
191 188

64
NCO2Me
SPh
NBn2 ClCO2Ph
DCE, reflux NCO2Me
SPh
NBn
CO2Ph
NN
PhSCO2Me
Cl
CO2PhPh
Ph
Scheme 2.20. Attempted Formation of Carbamate 193
Unfortunately the desired product was not observed. Initial inspection of
spectroscopic data revealed that one benzyl group had been removed and IR revealed a
new stretch consistent with a carbamate carbonyl. This seemingly correct product was
isolated after chromatography to separate it from unreacted starting material. Upon
reduction under ionic hydrogenation conditions the expected product could not be
verified. Difficulty with characterization ensued as a mixture of inseparable products was
isolated and carbamate resonance distorted room temperature NMR. An X-ray structure
analysis revealed the answer. The product isolated after reduction was a 6:1 mixture of
chloride 198 and methyl 173 (Scheme 2.22). The attempted debenzylation with
phenylchloroformate (Scheme 2.21) was not successful; chloride 194 and carbamate 195
were formed and could not be separated by chromatography. The apparent loss of the
benzyl methylene in the 1H NMR came from symmetrization as the starting material had
diastereotopic benzylic methylenes, and the new carbonyl IR stretch was from the
carbamate 195. The preference for allylic chlorination over benzylic chlorination is
potentially due to neighboring group participation. After acylation to generate the
188
192
193

65
N-acyliminium 196 the N2 carbamate can displace the quaternized amine. Chlorination of
the intermediate 197 would occur readily to give 194.
N
Bn2N CO2Ph
O
OMePhS
NCO2Me
SPh
NBn2
N
PhS
O
OMe
ClCO2Ph
DCE, reflux NCO2Me
SPh
Cl+ Bn2N
CO2Ph
Cl
58% yield
Scheme 2.21. Formation of Chloride 194
Reduction of chloride 194 with TFA and triethylsilane yielded the reduced
compound 198 and the over-reduced compound 173 (Scheme 2.22). The reduction
proceeds via the expected pathway of ionic hydrogenation and enecarbamate protonation
followed by hydride addition. The over reduced product was potentially formed from the
same oxazoline intermediate 197, derived as above from neighboring group participation
of the methyl carbamate. Hydride addition to oxazoline 197 forms the 3-methyl-1,2-
dihydroisoquinoline 172, and further reduction gives the previously synthesized
isoquinoline 173. The X-ray structure of 198 and 173 (appendix A) resolved the
confusion observed from analysis of spectroscopic data. In addition the X-ray data
proved unambiguously that the ionic hydrogenation stereoselectivity forms the
1,3-cis-substituted tetrahydroisoquinoline.
188 195 194
197196

66
NCO2Me
SPh
ClN
CO2Me
SPh
MeH
H
H
H
+
45% yield6:1, X-ray
Et3SiH, TFA
CH2Cl2
N
PhS
O
OMe
NCO2Me
SPh
Cl
N
PhS
Me
CO2Me
NCO2Me
SPh
Cl
H H
NCO2Me
SPh
MeH
H
H
Scheme 2.22. Possible Mechanism for Formation of Chloride 198
The reduction of 188 proved troublesome and an alternative route for the
synthesis of 1,3-cis-substituted tetrahydroisoquinoline 191 was sought. We turned our
attention to the synthesis of an oxygen analogue of 188 speculating that reduction would
occur more readily in the absence of the basic amine. It was anticipated that after
stereoselective reduction the oxygen atom could be converted to the requisite nitrogen
atom.
198
172
173194
197 174
199

67
2.2.4. OXYGEN ANALOGUE
The new route required synthesis of the requisite isoquinolines 201 and 203. The
preferred route would utilize isoquinoline 201 as it is anticipated that during the reduction
step the silyl protecting group will be lost to afford the reduced and deprotected alcohol,
ready for conversion to a nitrogen atom.
The isoquinoline synthesis proceeded well for both the triisopropylsilylpropargyl
ether (200) and the propargylbenzyl ether23 (202) to afford 201 and 203 in excellent yield
(Scheme 2.23). These isoquinolines were alkylated as before to give the
1,2-dihydroisoquinolines 204 and 205 in good yield (Scheme 2.24).
1) 2% PdCl2(PPh3)2, 1% CuI Et3N, 55 °C, 6 h
2) 10% CuI, DMF, 100 °C, 9 h NOBn
+OBn
84% yield
1) 2% PdCl2(PPh3)2, 1% CuI Et3N, 55 °C, 1 h
2) 10% CuI, DMF, 100 °C, 4 h NOTIPS
+OTIPS
94% yield
Scheme 2.23. Isoquinoline Synthesis
NOR
NCO2Me
SPh
i) PhSCH2Li, (−)-sparteinePhMe, −78 °C to 25 °C, 30 min. OR
ii) ClCO2Me
Scheme 2.24. 1,2-Dihydroisoquinoline Synthesis
203
200
202
179
201
179
204 R = TIPS, 89% yield205 R = Bn, 76% yield
201 R = TIPS 203 R = Bn

68
207, 29% yield
The ionic hydrogenation was first attempted with 204. Treatment of a solution of
204 and Et3SiH in CH2Cl2 with TFA at −10 °C led to immediate deprotection of the silyl
ether (Scheme 2.25). Upon warming to 25 °C reduction was observed as well as the
formation of a by-product. Reduction gave the desired 1,3-cis-substituted
tetrahydroisoquinoline 206 as a single diastereomer in 53% yield and isoquinoline 207 in
29% yield. After silyl ether deprotection to form 208 an acid catalyzed acyl migration can
lead to the formation of by-product 207. Protonation of the methyl carbamate 208 gives
209 which can undergo hydroxyl addition to form intermediate 210 followed by a ring
opening to generate 211. The 1,2-dihydroisoquionoline 211 can readily undergo air
oxidation to form isoquinoline 207.
NCO2Me
SPh
OTIPSEt3SiH, TFA
NCO2Me
SPh
OH
NH
H
+ OCO2Me
−10 °C to 0°C1 h
CH2Cl2
NCO2Me
SPh
OH
−TIPS
NOH
PhSO
OMe
H
N
PhS
O
OHOMe
NH
PhS
OCO2Me
H
PhS
[O]
Scheme 2.25. Reduction of Silyl Ether 204
211 208
209 210
204
206, 53% yield

69
In dramatic contrast to the triisopropylsilyl ether 204, the benzyl ether analogue
205 proved more stable during reduction. When enecarbarmate 205 was subjected to the
same ionic hydrogenation conditions clean reduction was observed to give a single
diastereomer 212 in excellent yield (Scheme 2.26). As with previous reductions the
1,3-cis relative stereochemistry was based on the analogy with earlier
tetrahydroisoquinoline formation.
NCO2Me
SPh
OBn Et3SiH, TFA
CH2Cl2, −10 °C to 0°C, 3 h NCO2Me
SPh
OBnH
H
97% yield
Scheme 2.26. Reduction of Benzyl Ether 205
We now wanted to investigate the reactivity of the C1 and C3 substituents toward
each other to confirm their 1,3-cis relationship. To explore this reactivity we chose to
form a Pummerer intermediate. Treatment of sulfide 212 with N-chloro-succinimide 214
forms the α-chloro-sulfide 218 (Scheme 2.27). Addition of SnCl4 generates the reactive
thionium ion 217, the Pummerer intermediate, which quickly cyclizes to 219 followed by
debenzylation to afford 213 as a single diastereomer in 74% yield. The cyclized product
213 was isolated as a crystalline white solid and an X-ray structure (appendix B)
confirmed the formation of the bicyclo[3.3.1] system.
212205

70
NCO2Me
SPh
OBnH
H
1) NCS, PhCl, 2 h, 25 °C
2) SnCl4, 0 °C, 5 min.
N
ClO
O
NCO2Me
SPh
OBnH
H
Cl
H
N
PhS
OBnH
H
NOO
NO
CO2Me
PhS
NO
CO2Me
SPh
SnCl5
Ph
74% yield
Cl
N
PhS
OBnH
H
Cl
CO2Me
CO2Me
N
PhS
OBnH
H
CO2Me
SnCl4
Scheme 2.27. The Pummerer Cyclization
The cyclization introduced another potential route to the tetrahydroisoquinolines.
The structure of the cyclization product is that of a bicyclo[3.3.1] system, which is very
similar to that found in the saframycins except for the presence of the oxygen atom
instead of a nitrogen atom (Figure 2.02). The pseudo-symmetry of the saframycins makes
our strategy to synthesize the 1,3-cis-substituted tetrahydroisoquinoline potentially
applicable to both the western and eastern fragments of saframycin B. Although we
initially planned to synthesize the western fragment first we hoped to apply this
cyclization methodology to the nitrogen analogue, to form the 1,4-diaza-
bicyclo[3.3.1]system found in saframycin B (Scheme 2.28).
212
218
219
217
214
213, X-ray
217215
216

71
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
NHCOCOMe
H
OMe
OMe
O
NN
OMeO
MeO Me
H
H
H
NHCOCOMe
H
NO
CO2Me
SPh
Figure 2.02. Pseudo-Symmetry of Saframycin B
NCO2Me
SPh
NBn2H
H
R
NNBn
CO2Me
SPh
R
Scheme 2.28. Nitrogen Analogue to Form Bicyclo[3.3.1] System
1b
213
Saframycin B (1b)
221220

72
2.2.5. NITROGEN ANALOGUE FOR PUMMERER CYCLIZATION
To investigate the Pummerer cyclization on the nitrogen analogue conversion of
the oxygen atom was required. Previously, alcohol 206 was successfully generated from
simultaneous reduction and deprotection of silyl ether 204. However, modest yields for
this product were observed due to by-product formation. The benzyl ether proved to be a
more efficient route to alcohol 206. Ionic hydrogenation gave tetrahydroisoquinoline 212,
and benzyl ether deprotection was promoted by treatment with BCl3 to give alcohol 206
in 97% yield (Scheme 2.29). This two step procedure to form alcohol 206 was more
efficient than the one step procedure from silyl ether 204. The alcohol was then converted
to the tosylate 222 in 86% yield using standard conditions.
NCO2Me
SPh
OBnH
HBCl3, CH2Cl2
−78 °C to 25 °C, 30 min.
0 °C to 25 °C, 4 h
95% yield
NCO2Me
SPh
OHH
H
Me
SO2Cl
pyridine, CH2Cl2
86% yield
NCO2Me
SPh
OTsH
H
Scheme 2.29. Tosylate Formation
222
212 206

73
Treatment of tosylate 222 with sodium azide in DMF at 55 °C gave only modest
yields of the desired azide 223 (Scheme 2.30) and the by-product 224 in 31% yield. The
formation of this by-product was likely to have come from a similar neighboring group
participation as previously observed. Heating the tosylate 222 in DMF could lead to an
intramolecular displacement of the tosylate generating intermediate 225. Azide can then
demethylate the activated intermediate to give the oxazolidinone 224. It should be noted
that without heating no reaction occurs between sodium azide and tosylate 222.
NCO2Me
SPh
OTsH
H
NCO2Me
SPh
N3H
H
N
SPh
H
HNaN3, DMF
55 °C, 12 hO
O
N
PhS
TsO
H
H
O
OMe
N
PhS
H
H
O
O Me
N3
+
Scheme 2.30. Azide Formation
A more efficient synthesis of the azide was found using modified Mitsunobu
conditions.24 Treatment of alcohol 206 with diethylazodicarboxylate (DEAD),
triphenylphosphine, and diphenylphosphorylazide gave azide 223 (Scheme 2.31). Under
these conditions, which did not require heating, the oxazolidinone product 224 was not
observed.
223, 65% yield
225
222 224, 31% yield

74
(PhO)2PON3, DEAD
PPh3, THF0 °C to 25 °C, 2 h
NCO2Me
SPh
OHH
H
NCO2Me
SPh
N3H
H
82% yield
Scheme 2.31. Efficient Azide 223 Formation
With azide 223 in hand the initial plan was to investigate the Pummerer
cyclization which involved reduction of the azide to the amine followed by dibenzylation.
However, a thorough investigation of the Pummerer cyclization directly with azide 223
gave interesting results.
Azide 223 was treated under identical Pummerer cyclization conditions as benzyl
ether 212, NCS followed by SnCl4 (Scheme 2.32). The evolution of gas was observed and
the reaction was complete upon initial TLC analysis. After aqueous workup and
chromatography the unstable iminosulfide 226 was isolated in 49% yield. The
spectroscopic data appeared consistent with the cyclized product. The mechanism for this
reaction is outlined in Scheme 2.32 and proceeds through an analogous Pummerer
intermediate 227 as before. The azide cyclizes to form intermediate 228 which can
undergo rapid and irreversible loss of nitrogen gas to generate the iminosulfide 226. It
was observed that iminosulfide 226 readily hydrolyzed to lactam 229 under acidic
conditions. Treatment of the crude cyclized product with 5% aq. sodium hydrogen sulfate
rapidly formed the lactam 229 (Scheme 2.33), which was characterized by X-ray
crystallography (appendix C).
206 223

75
NCO2Me
SPh
N3H
H
1) NCS, PhCl, 2 h, 25 °C
2) SnCl4, 0 °C, 5 min.
49% yield
NN
CO2Me
SPh
NCO2Me
S
NH
H
Ph
N NN
N
CO2Me
PhSH
N2
SnCl5
Scheme 2.32. Azide Cyclization
NN
CO2Me
SPh
5% aq. NaHSO4
Et2O, 5 min.
>95% yield
NNH
CO2Me
O
Scheme 2.33. Lactam Formation
This lactam formation was unprecedented and the cyclization of an azide with a
Pummerer intermediate has, to the best of our knowledge, never been reported. In
addition cyclization gave the desired 1,4-diazabicyclo[3.3.1] system. Application of this
methodology to the real system was necessary to determine its potential utility in the
synthesis of saframycin B.
229, X-ray 226
226
228227
223

76
2.2.6. FORMATION OF THE REAL SYSTEM
Moving away from a model system necessitated the formation of a substituted
analogue of the bicyclo[3.3.1] system 229. To achieve this goal we required the synthesis
of hexasubstituted benzene 234 (Scheme 2.34). The arylbenzyl ether was important as it
provides easy access to the phenol, prior to oxidation to the quinone in the final steps of
the synthesis of saframycin B. Oxidation to the quinone has been shown to proceed in
higher yields from a phenol than from an aryl ether.25
In our first approach to the formation of 234 we started with commercially
available aldehyde 230 which was converted to phenol 231 via a known method of
Baeyer-Villiger oxidation, followed by formate ester hydrolysis.26 Phenol 231 was
formylated under Duff conditions27 and protected as the benzyl ether to give aldehyde
233. The installation of the aryl iodide was attempted with an electrophilic iodine source,
iodine, NIS, or AgO2CCF3/I2, but only complex reaction mixtures were isolated.
Me
MeOOH
OMe
Me
MeOOBn
OMe
O
N
NN
N
AcOH, CH2Cl2, 100 °C
1)
2) BnBr, Na2CO3, DMF
Me
MeOOBn
OMe
O
I
Me
MeOCHO
OMe
1) m-CPBA, CH2Cl2
2) NaOH, MeOH
>95% yield 48% yield
Scheme 2.34. Attempted Formation of Hexasubstituted Benzene 234
233 234
230 231
232

77
Further examination of the literature showed that ortho-iodination of similar
aldehydes was not known. More common was a two step procedure utilized by Suzuki in
his work on a similar benzaldehyde.28 A solution of phenol 231 and paraformaldehyde in
CH2Cl2 was treated with neat diethylaluminum chloride to afford diol 235 (Scheme 2.35).
The hydroxymethylated product was formed very cleanly but found to be unstable at
room temperature. The crude phenol was immediately protected as the arylbenzyl ether to
give alcohol 236 in 89% yield over the two steps. The alcohol 236 underwent facile aryl
iodination with silver trifluoroacetate and iodine to give iodide 237 in 82% yield.
Oxidation of the primary alcohol to the aldehyde with PCC gave the desired
hexasubstituted benzene 234 in 98% yield. This procedure provided a high yielding and
scaleable route to the benzaldehyde required for isoquinoline formation.
Me
MeOOH
OMe
OH
Me
MeOOH
OMe
(CH2O)n, Et2AlCl
CH2Cl2, 0 °C to 25 °C
Me
MeOOBn
OMe
OH
BnBr, K2CO3
acetone, 60 °C
Me
MeOOBn
OMe
OH
IAgOOCCF3
I2, CHCl3
Me
MeOOBn
OMe
CHO
IPCC
CH2Cl2
89% yieldtwo steps
82% yield 98% yield
Scheme 2.35. Synthesis of Hexasubstituted Benzene 235
237236
235231
234

78
With aldehyde 234 in hand we turned our attention to the synthesis of
isoquinoline 240. The imine 238 was formed as before in quantitative yield (Scheme
2.36) and then treated with acetylene 202 using the Larock isoquinoline procedure.18
Unfortunately, the reaction did not proceed smoothly. A mixture of hydrolyzed starting
material 234, hydrolyzed coupled product 239, and desired isoquinoline 240 were
isolated after aqueous workup. In previous isoquinoline formation, upon addition of the
palladium and heating to promote the Sonagashira coupling the solution became pale
yellow in color with a fluffy precipitate forming (triethylamine hydroiodide). However
for the substituted imine 238 the reaction appeared black in color and a small amount of
black solid formed suggesting that palladium(0) was precipitating out of solution.
Me
MeOOBn
OMe
CHO
I Me
MeOOBn
OMeI
Nt-butylamine, PhMe4Å molecular sieves
25 °C, 3 h
OBn 2% PdCl2(PPh3)2, 1% CuI, Et3N, 55 °C, 3 h
2) 10% CuI, DMF, 100 °C, 5 h
1)
Me
MeOOBn
OMe
CHO
OBnMe
MeOOBn
OMe
CHO
I Me
MeOOBn
OMe
NOBn
+ +
Scheme 2.36. Larock Conditions for Isoquinoline Synthesis
240, 33% yield 239, 18% yield 234, 41% yield
238234
202

79
To avoid the problems seemingly associated with the use of palladium we decided
to leave the palladium out of the reaction mixture. In the absence of palladium a
stoichiometric amount of copper was used to conduct the Castro-Stevens reaction.29
Under optimized conditions it was found that stirring imine 238 with acetylene 202 in
triethylamine with 1.2 equivalents of CuI at room temperature for 24 hours gave the
cleanly coupled product in near quantitative yield (Scheme 2.37). A filtration step was no
longer necessary as the same reaction flask was heated to 85 °C, after complete coupling,
and stirred for one hour to promote cyclization to isoquinoline 240. Attempts to heat the
reaction before complete imine 238 coupling led to significant by-product formation. The
yield for the one-pot procedure was 91% yield.
Me
MeOOBn
OMeI
N
OBn1.2 equiv. CuI, Et3N, 25 °C, 24 h
then 85 °C, 1 h
Me
MeOOBn
OMe
NOBn
91% yield
Scheme 2.37. Castro-Stevens Conditions for Isoquinoline Synthesis
Castro-Stevens reactions often require heating for the coupling to occur. With the
imine substrate 238 simply stirring at room temperature generated the coupled product in
near quanitiative yield. However it was noted that room temperature coupling with
aldehyde 234 was unsuccessful and only unreacted starting material was observed after
stirring overnight. It appears the imine nitrogen coordinates with the CuI to lower the
activation energy for acetylene insertion into the aryl iodine bond.
238 240

80
2.2.7. ALKYLATION, REDUCTION, AND CYCLIZATION OF THE REAL SYSTEM
The substituted isoquinoline could now be obtained very cleanly and efficiently
using a modification of the Larock isoquinoline synthesis. Alkylation of isoquinoline 240
with phenylthiomethyllithium in the presence of (−)-sparteine gave
1,2-dihydroisoquinoline 241 in 83% yield (Scheme 2.38). Three equivalents of
phenylthiomethyllithium were used to hasten the alkylation of the much less reactive
electron rich isoquinoline 240. The enantioselectivity of this reaction was also
investigated, but unfortunately separation of the enantiomers by chiral HPLC revealed a
1:1 mixture.
Me
MeOOBn
OMe
NOBn
1) PhSCH2Li (3.0 equiv.) (−)-sparteine (3.0 equiv.) PhMe, −78 °C to 25 °C, 15 min.
2) ClCO2Me
83% yield
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
Scheme 2.38. Alkylation of Substituted Isoquinoline 240
The alkylated product 241 was then subjected to ionic hydrogenation conditions,
previously used with much success for the unsubstituted analogue 205. Treatment of a
solution of 241 and Et3SiH in CH2Cl2 with TFA gave a mixture of two products (Scheme
2.38). The over-reduced 3-methyl tetrahydroisoquinoline 242 was isolated in 61% yield
and the desired product 243 was isolated in 31% yield. The 3-methyl product 242 was
unexpected as no by-products were observed in the model system. This suggests that
neighboring group participation of the methyl carbamate is not likely as it would have
241 240

81
been observed in the model system. The unwanted product was attributed to the electron
rich aromatic ring.
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
Me
MeOOBn
OMe
N
Me
CO2Me
SPh
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
H
H
H
H
TFA, Et3SiH, CH2Cl2−10 °C to 25 °C, 3 h
+
Scheme 2.39. Ionic Hydrogenation on the Real System
The 3-methyl tetrahydroisoquinoline 242 could be formed via the pathway
outlined in Scheme 2.40. In the presence of a strong acid small amounts of the protonated
ether 245 may exist. The lone pair of electrons on either arylmethoxy of 245 can donate
into the ring system and eliminate benzyl alcohol, forming oxonium intermediate 246.
Hydride addition to the activated intermediate would give 247. Subsequent ionic
hydrogenation of 247 leads to the over-reduced product 242. The reaction mechanism
suggests that the pathway for elimination of benzyl alcohol remains reversible until the
first hydride addition generates 247. In order to minimize the amount of eliminated
benzyl alcohol intermediate we conducted the reaction in excess benzyl alcohol, hoping
to employ Le Chatelier’s principle. By conducting the reaction with benzyl alcohol as a
co-solvent the yield of the desired product 243 was increased to 71% (Scheme 2.41) and
by-product 242 was isolated in only 22% yield.
241
242, 61% yield
243, 31% yield

82
241
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
TFA, Et3SiH
−10 °C to 25 °C, 3 h
H
H
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
H
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
Me
MeOOBn
OMe
NCO2Me
SPh
Me
MeOOBn
OMe
N
Me
CO2Me
SPhH
H
Scheme 2.40. Ionic Hydrogenation on the Real System
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
TFA, Et3SiH, BnOH
−10 °C to 25 °C, 16 h
Scheme 2.41. Improved Reduction Conditions
243 +
247
246
245
242
243 + 242
71% yield 22% yield
244
241

83
With an improved synthesis of the desired 1,3-cis-substituted
tetrahydroisoquinoline 243 enough material was available to investigate the formation of
the requisite azide. Benzyl ether 243 required deprotection, however in the real system an
additional benzyl ether is present on the aryl ring. Fortunately the previous conditions
established for benzyl ether deprotection, BCl3, provided the required selectivity. Boron
trichloride can selectively deprotect alkylbenzyl ethers over arylbenzyl ethers because the
mechanism first involves coordination of the boron with oxygen to form an activated
intermediate 249 (Scheme 2.42), and the alkylbenzyl ether oxygen has the most
nucleophilic lone pairs. Treatment of benzyl ether 243 with BCl3 at −40 °C, with careful
monitoring of the reaction, gave the desired alcohol 248 in 88% yield.
Me
MeOOBn
OMe
NOBn
CO2Me
SPh
Me
MeOOBn
OMe
NOH
CO2Me
SPh
H
H
H
HBCl3, CH2Cl2
−40°C, 2 h
Me
MeOOBn
OMe
NO
CO2MeSPh
H
H
Ph
BCl2Cl
BCl2Cl
88% yield
Scheme 2.42. Alkylbenzyl Ether Deprotection
243 248
249

84
Alcohol 248 was then converted to azide 250 in 85% yield (Scheme 2.43) using
the conditions from the model system with diisopropyl azodicarboxylate (DIAD)
replacing DEAD. DIAD could be stored longer than DEAD and gave identical results.
Azide 250 was subjected to the Pummerer cyclization conditions previously used. The
desired cyclized product 251 was isolated in 51% yield. For this substrate the
iminosulfide could not be isolated nor observed by TLC analysis. Lactam 251 was
characterized and the spectroscopic data was consistent with the model lactam system.
Me
MeOOBn
OMe
NOH
CO2Me
SPh
H
H
1) NCS, PhCl, 2 h, 25 °C
2) SnCl4, 0 °C, 5 min.
51% yield
NNH
CO2Me
O
(PhO)2PON3, DIADPPh3, THF
0 °C to 25 °C, 1.5 h
85% yield
Me
MeOOBn
OMe
NN3
CO2Me
SPh
H
H
MeOMe
MeOOMe
Scheme 2.43. Cyclization of the Real System
248
251
250

85
2.3. CONCLUSION
The model conditions proved successful in their application to the real system
leading to the synthesis of the bicyclo[3.3.1] system present in saframycin B. Further
work was needed to install the second 1,3-cis-substituted tetrahydroisoquinoline which
would likely go through intermediate 252 (Scheme 2.44). However, after the change in
our strategy we found our approach resembled the diketopiperazine derived intermediates
used by Kubo and Fukuyama for their work in the saframycins.
NNH
CO2Me
O
MeOMe
MeOOBn
NNR
CO2MeMeOMe
MeOOBn
Ar
ArHN
O
N
BnOOMe
Me
OMeCbz
H
H
ArRN
N
BnOOMe
Me
OMeCO2Me
H
H
ArHN
N
MeOOMe
Me
OMeMe
H
H
Scheme 2.44. Other Intermediates in the Synthesis of Saframycin B
252
Fukuyama’s Intermediate
251
Kubo’s Intermediate

86
At this stage, the approach to saframycin B was terminated. We were successful
in developing a new stereoselective approach for the formation of the 1,3-cis-substituted
tetrahydroisoquinoline core of the tetrahydroisoquinoline alkaloids by alkylating
unactivated isoquinolines with phenylthiomethyllithium followed by ionic hydrogenation.
The lack of enantioselectivity was disappointing but we hoped to reinvestigate this issue.
In addition a new lactam synthesis was developed from the reaction of an azide with a
Pummerer intermediate to form the bicyclo[3.3.1] system found in saframycin B. The
project now turned toward the synthesis of an alternative tetrahydroisoquinoline alkaloid.

87
2.4. REFERENCES
1) Dyke, S. F.; Kinsman, R. G. Heterocyclic Compounds; Wiley-Interscience: New York, 1981; Vol. 38, Part 1-3. 2) Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069. 3) Blasko, G.; Kerekes, P.; Makleit, S. Alkaloids 1987, 31, 1. 4) Shibasaki, M.; Takamura, M.; Funabashi, K.; Kania, M. J. Am. Chem. Soc. 2000, 122, 6327. 5) Comins, D. L.; Goehring, R. R.; Joseph, S. P.; O’Connor, S. J. Org. Chem. 1990, 55, 2574. 6) Comins, D. L.; Badawi, M. M. Heterocycles 1991, 32, 1869. 7) Frank Amiot’s Dissertation, 2002, Universite de Geneve, Switzerland. 8) Marazano, C.; Barbier, D.; Riche, C.; Das, B. C.; Potier, P. J. Org. Chem. 1998, 63, 1767. 9) Alexakis, A.; Amiot, F. Tetrahedron: Asymmetry 2002, 13, 2117. 10) Shirley, D. A.; Reeves, B. J. J. Organometallic Chemistry 1969, 16, 1. 11) Corey, E. J.; Seebach, D. J. Org. Chem. 1966, 31, 4097. 12) Peterson, D. J. Org. Chem. 1967, 32, 1717 13) Pyne, S. G.; Dikic, B. J. Org. Chem. 1990, 55, 1932. 14) Solladié, G. Synthesis 1981, 185. 15) Birch, A. J.; Walker, K. A. M. Tetrahedron Lett. 1967, 8, 1935. 16) Molinski, T. F.; Masuno, M. N. Tetrahedron Lett. 2001, 42, 8263. 17) Langy, K. C. Org. Prep. Proc. Int. 1994, 26, 429. 18) Larock, R. C.; Roesch, K. R. J. Org. Chem. 2002, 67, 86. 19) Bacon, R. G. R.; Lindsay, W. S. J. Chem. Soc., 1958, 1375.

88
20) Larock, R. C.; Doty, M. J.; Cacchi, S. J. J. Org. Chem. 1993, 58, 4579. 21) Ireland, R. E.; Anderson, R. C.; Badoud, R.; Fitzsimmons, B. J.; McGarvey, G. J.; Thaisrivongs, S.; Wilcox, C. S. J. Am. Chem. Soc. 1983, 105, 1988. 22) Cooley, J.; Evain, E. J. Synthesis 1989, 1. 23) Turos, E.; Ren, X. F.; Lake, C. H.; Churchill, M. R. J. Org. Chem. 1995, 60, 6468. 24) Bose, A. K.; Lal, B.; Pramanik, B. N.; Manhas, M. S. Tetrahedron Lett. 1977, 18, 1977. 25) Kubo, A.; Saito, N.; Yamato, H.; Masubuchi, K.; Nakamura, M. J. Org. Chem. 1988, 53, 4295. 26) Godfrey, I. M.; Sargent, M. V.; Elix, J. A. J. Chem. Soc., Perkin Trans. 1 1974, 1353. 27) Kubo, A.; Kitahara, T.; Nakahara, S.; Numata, R. Chem. Pharm. Bull. 1985, 33, 2122. 28) Suzuki, K.; Saito, T.; Morimoto, M.; Akiyama, C.; Matsumoto, T. J. Am. Chem. Soc. 1995, 117, 1075. 29) Castro, C. E.; Havlin, R.; Honwad, V. K.; Malte, A.; Moje, S. J. Am. Chem. Soc. 1969, 91, 6464.

89
Chapter 3: Toward the Synthesis of Lemonomycin
3.0. INTRODUCTION
The new target tetrahydroisoquinoline alkaloid, lemonomycin (10, Scheme 3.01),
was chosen because at the beginning of the project the total synthesis of this unique
structure had not been reported. It was not until halfway through the project that Stoltz
reported his enantioselective total synthesis.1
In our retrosynthetic analysis of lemonomycin, the western fragment was to be
synthesized employing the chemistry previously developed. We anticipated the necessity
for an alternative nucleophile than phenylthiomethyllithium for the alkylation of
isoquinoline 257 (Scheme 3.01). The presence of the resulting sulfide side-chain would
require extra steps for conversion to the requisite ether. Once the 1,2-dihydroisoquinoline
256 was formed, finding appropriate conditions for reduction that would provide an
efficient yield of the 1,3-cis-substituted tetrahydroisoquinoline 255 would be important.
The current conditions for ionic hydrogenation of the electron rich system led to
elimination of benzyl alcohol. We anticipate that the elimination by-product would be
observed no matter which protecting group was used. Further elaboration to
lemonomycin (10) would proceed through intermediate 254 followed by formation of the
bicyclo[3.2.1]system 253.

90
NNH
H
HOH
MeO
MeO
O
H
H
O
O
Me
OHMe
NMe2
HOOH
NN
H
HO
MeOMe
OBn
H
H
OBn
CHO
R2
NNR2
OMeO
MeOMe
OBn
H
H
OBn
OR3
NHMeO
MeOMe
OBn
H
H
X
NMeO
MeOMe
OBn
H
X
OHN
MeO
MeOMe
OBn
OR1 OH
1
11
13
314
4
17
15
16
18
6 10
8
1
3
1
3
1
3
1
3
1
3
MeO
CO2Me
Scheme 3.01. Retrosynthesis of Lemonomycin (10)
10
255
253 254
256257

91
3.1. RESULTS AND DISCUSSION
3.1.1. ALKYLATION WITH BENZYLOXYMETHYLLITHIUM
The alkylation of an isoquinoline with a nucleophile other than
phenylthiomethyllithium would still require the nucleophile to have the general structure
Li-CH2-X. The preference for our approach to lemonomycin would be where X is an
oxygen atom. A literature search revealed that benzyloxymethyllithium2 (BOM-Li, 260,
Scheme 3.02) was a well investigated nucleophile and a suitable choice. The
organolithium nucleophile is formed from trans-metallation of the stannane 259 with
n-BuLi. Stannane 259 is formed from stannylation of benzyl chloromethyl ether3 (258)
with Bu3SnLi.
O
ClBu3Sn-Li, THF
O
SnBu3n-BuLi
O
Li
−78 °C to 25 °C, 30 min. THF, −78 °C
70% yield
Scheme 3.02. Formation of Benzyloxymethyllithium
The alkylation of isoquinoline with benzyloxymethyllithium was used as a model
system to determine the reactivity of the nucleophile. A solution of stannane 259 and 1.7
equivalents of (−)-sparteine in toluene at −78 °C was treated with one equivalent of
n-BuLi (Scheme 3.03). After 10 minutes a solution of isoquinoline in THF was added and
the reaction warmed to room temperature. The starting material was consumed and the
reaction was quenched with methyl chloroformate to give the 1,2-dihydroisoquinoline
261 in 66% yield. The enantiomers were separated by chiral HPLC (Diacel, Chiracel OD
column) in a ratio of 2:1, revealing an enantioselectivity of 33% for the conversion.
258 259 260

92
229
O
i) n-BuLi, (−)-sparteine PhMe, −78 °C, 10 min.
SnBu3ii)
N
−78 °C to 25 °C, 15 min.
, THF
iii) ClCO2Me
N
OBn
CO2Me
Scheme 3.03. Alkylation of Isoquinoline with BOM-Li
These promising results demonstrated the utility of the nucleophile and that under
unoptimized conditions some enantioselectivity could be achieved. The observed
enantioselectivity was unexpected because the analogous alkylation with
phenylthiomethyllithium showed no enantioselectivity under a variety of conditions. If
the steric differences of the nucleophiles are considered negligible, then the main
difference is the heteroatom; sulfur and oxygen have different electronic properties that
could effect the coordination of (−)-sparteine to the respective lithiated species. The
better coordinating sulfur lone pairs could be stabilizing the anion without assistance
from the nitrogen lone pairs of (−)-sparteine, while in the case of oxygen the nitrogen
atoms of (−)-sparteine provide stabilization and form a chiral complex that shows facial
selectivity in the alkylation of isoquinoline. The reaction was not optimized for the model
system because application of this alkylation to the real system was of more importance.
In our approach to lemonomycin we wanted to use an alternative substituted
isoquinoline to 240, used in our work toward saframycin B. Using the same isoquinoline
for the alkylation with BOM-Li the product would possess two alkylbenzyl ethers which
could prove difficult to differentiate at a later stage. Instead a triisopropylsilyl protected
66% yield, 33% ee
261
259

93
alcohol at the C3 position was preferred, which would provide differentiation as well as
stability to the alkylation conditions.
The synthesis of isoquinoline 262 (Scheme 3.04) was first attempted using a
modified one-pot procedure of the Larock conditions that worked well before. The
Castro-Stevens reaction between imine 238 and acetylene 200 appeared to be consistent
with prior substrates, but upon heating the reaction to promote cyclization degradation
was observed and the desired isoquinoline 262 was not isolated. The only difference from
the prior isoquinoline synthesis was the presence of the triisopropylpropargyl ether.
During the Castro-Stevens coupling one equivalent of triethylamine hydroiodide was
formed, which at room temperature did not appear to deprotect the silyl ether. However,
heating to 80 °C could lead to acid catalyzed deprotection of the silyl ether. Larock
reported that propargyl alcohol does not undergo isoquinoline formation.4
Me
MeOOBn
OMeI
N
OTIPSCuI (1.2 equiv.), Et3N, 25 °C, 24 h
then 80 °C, 1 h
Me
MeOOBn
OMe
NOTIPS
Scheme 3.04. Attempted Synthesis of Isoquinoline 262
To solve this problem a basic workup was conducted after the Castro-Stevens
room temperature coupling was complete. The coupled intermediate was then dissolved
in DMF with catalytic CuI and heated, now in the absence of triethylamine hydroiodide,
to afford the desired isoquinoline 262 in 76% yield (Scheme 3.05).
238
200
262

94
Me
MeOOBn
OMeI
N
OTIPSCuI (1.2 equiv.), Et3N, 25 °C, 24 h
2) CuI (0.2 equiv.), DMF 100 °C, 1 h
Me
MeOOBn
OMe
NOTIPS
1)
76% yield
Scheme 3.05. Synthesis of Isoquinoline 262
The alkylation of isoquinoline 262 with BOM-Li in the presence of (−)-sparteine
was performed under the conditions used for the previous model isoquinoline (Scheme
3.06). The starting material was consumed but a mixture of unidentified products was
isolated.
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBn
O
i) n-BuLi, (−)-sparteine PhMe, −78 °C, 10 min.
SnBu3ii)
−78 °C to 25 °C, 15 min.262, THF
iii) ClCO2Me
Scheme 3.06. Attempted Synthesis of 263
The reaction conditions were simplified by removing (−)-sparteine and using only
THF as solvent. A variety of conditions were employed varying the equivalents of
stannane 259, the equivalents of n-BuLi, and reaction temperature (Scheme 3.07). The
results are summarized in Table 3.01.
262 238
259
263
200

95
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBni) n-BuLi (equiv.), THF, −78 °C, 10 min.
ii)THF, temp.262 (1.0 equiv.),
iii) ClCO2Me (5.0 equiv.)
Me
MeOOBn
OMe
NOTIPS
CO2Me
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBn
CO2Me
Scheme 3.07. Alkylation of 262
yield (%) entry 259
(equiv.) n-BuLi (equiv.)
temp. (°C) 262 263 264 265
1 1.2 1.2 −78 to 25 68 0 8 12
2 3 1 −78 to 25 43 0 32 0
3 3 3 −78 to 25 31 0 38 8
4 3 1 −78 28 57 0 0
5 3 3 −78 0 45 0 33
6 4 3 −78 0 79 0 trace
Table 3.01. Alkylation of 262
The subtle reaction changes demonstrate the capricious reactivity of BOM-Li and
isoquinoline 262. The conditions for entries 1 to 3 allowed warming to room temperature
using either one equivalent of BOM-Li or an excess, but a large percentage of recovered
starting material was isolated. It is likely that BOM-Li is not stable at room temperature
and the isoquinoline 262 is not consumed before degradation. As observed in the
264
263
265
259

96
alkylations with phenylthiomethyllithium, the electron rich isoquinoline 262 is less
reactive than the unsubstituted isoquinoline. The isoquinoline 262 that was alkylated gave
mainly diacylated product 264 upon methyl chloroformate quench. Keeping the reaction
at −78 °C and generating one equivalent of BOM-Li (entry 4) gave better conversion and
57% yield of the desired mono-acylated product 263. The diacylated product 264 was not
observed, however, nearly 30% of the starting isoquinoline was isolated. In an attempt to
drive the reaction to completion, three equivalents of BOM-Li were used (entry 5) which
did succeed in consuming all of the starting isoquinoline. Upon quenching with methyl
chloroformate the n-butyl adduct 265 was isolated in 33% yield along with the desired
product 263 in 45% yield. The origin of 265 was initially thought to be due to excess
n-BuLi in the reaction arising from improper titration of the commercial n-BuLi solution
in hexanes. After multiple reactions using a one to one ratio of carefully titrated n-BuLi
and stannane 259 the n-butyl adduct 265 was always isolated in significant yield, which
suggested that an aggregate could be present. It was then observed that a slight excess of
stannane 259 over n-BuLi (entry 6) minimized the amount of 265 and gave the desired
1,2-dihydroisoquinoline 263 in 79% yield.

97
3.1.2. FORMATION 1,3-CIS-SUBSTITUTED TETRAHYDROISOQUINOLINE
When 263 was treated under ionic hydrogenation conditions none of the desired
1,3-cis-substituted tetrahydroisoquinoline 266 could be isolated or observed (Scheme
3.08). The ionic hydrogenation of 1,2-dihydroisoquinoline 263 was anticipated to be
problematic, as consistent with the observations made in our approach to saframycin B.
Two possible by-products were the acyl-migration adduct 269 and the over-reduced
3-methyl adduct 272. The former coming from loss of the silyl ether and acid catalyzed
migration of the methyl carbamate to form the methyl carbonate 269. The latter could
arise from elimination of H2O or HOTIPS and reduction of the oxonium intermediate 271
leading to C3-methyl tetrahydroisoquinoline 272.
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBn
TFA, Et3SiH, CH2Cl2−10 °C to 25 °C, 1 h
H
Me
MeOOBn
OMe
NO
CO2MeOBn
H Me
MeOOBn
OMe
NCO2Me
OBn
Me
MeOOBn
OMe
N
Me
CO2Me
OBn
Me
MeOOBn
OMe
NOH
CO2Me
OBn
H
H
Me
MeOOBn
OMe
NOH
OBn
Me
MeOOBn
OMe
N
OBn
O
OHOMe
OMe
O
Me
MeOOBn
OMe
N
OBn
OCO2Me
-TIPS
H
H
H
Scheme 3.08. Failed Ionic Hydrogenation
266263
269
272 271
268267
270

98
The acyl-migration product could readily be avoided by replacing the acid
sensitive protecting group on the alcohol. The elimination product could not be avoided
without changing the electronics of the electron rich isoquinoline ring system. The first
solution to the problem was to change the reduction conditions and not the substrate.
Attempts to reduce the 3,4-olefin under catalytic hydrogenation conditions were
unsuccessful (Scheme 3.09). The cyclic tri-substituted olefin is quite hindered and
literature suggested hydrogen pressures over 1000 psi would have been necessary.5 In
addition, deprotection of the benzyl ethers was observed.
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBn
Me
MeOOH
OMe
NOTIPS
CO2Me
OH
PtO2, EtOH
H2 (1 atm)
87% yield
Scheme 3.09. Attempted Hydrogenation
273263

99
An effort was made to utilize the proximity of the C3 hydroxymethyl substituent,
which, when the silyl ether was removed by treatment with TFA gave allylic alcohol 274
in 88% yield (Scheme 3.10). Catalytic hydrogenation conditions were employed with the
hopes that neighboring group assistance would promote reduction. Reduction was not
observed only benzyl ether deprotection resulting in 275.
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBn
TFAMe
MeOOBn
OMe
NOH
CO2Me
OBn88% yield
CH2Cl2, 0 °C
5% Pt/C, EtOH
H2 (1 atm), 16 h
87% yield
Me
MeOOH
OMe
NOH
CO2Me
OBn
Scheme 3.10. Allylic Alcohol Formation and Attempted Reduction
275
274263

100
Reduction of the allylic alcohol was then attempted with NaBH4, which has
shown to be successful in berberine alkaloids for the reduction of eneamines.6 When
alcohol 274 was treated with NaBH4 in MeOH a new compound was formed (Scheme
3.11). However it proved not to be the reduced compound 266 but rather the cyclized
product 276. NaBH4 was reacting as a base generating the alkoxide 277, which forms the
cyclized intermediate 278 then the oxazolidinone derivative 276.
Me
MeOOBn
OMe
NOH
CO2Me
OBn
NaBH4, MeOH
25 °C, 15 h
Me
MeOOBn
OMe
N
OBn
O
O86% yield
Me
MeOOBn
OMe
NO
OBn
OMe
O
Me
MeOOBn
OMe
N
OBn
O
OOMe
Scheme 3.11. Unexpected Formation of Oxazolidinone 276
274
277 278
276

101
276
The new compound 276 was subjected to ionic hydrogenation conditions and
clean reduction to the tetrahydroisoquinoline 279 as one diastereomer was observed
(Scheme 3.12). It was expected that reduction of the oxazolidinone derivative 276 was
not likely to give the acyl-migration product, but by-products from elimination were
anticipated. One could imagine protonation of the carbonyl moiety resulting in
intermediate 280, which could lead to the ring-opened intermediate 281 and
decarboxylated intermediate 282. However, none of the anticipated by-products from the
elimination pathway were observed.
TFA, Et3SiH
−10 °C to 25 °C, 3 h
Me
MeOOBn
OMe
N
OBn
O
O
Me
MeOOBn
OMe
N
OBn
O
O95% yield
H
H
Me
MeOOBn
OMe
N
OBn
O
O H
Me
MeOOBn
OMe
N
OBn
O
OH
H
Me
MeOOBn
OMe
N
OBn
Me
-CO2
H
Scheme 3.12. Successful Ionic Hydrogenation
279
280
281
282

102
Conversion to the oxazolidinone 279 was optimized by treating silyl ether 263
with TBAF to give 276 in 91% yield (Scheme 3.13). Reduction with Et3SiH and TFA in
CH2Cl2 gave the tetrahydroisoquinoline 279, which was hydrazinolyzed to yield the
amino alcohol 283 in 86% yield over the two steps. The amino alcohol 283 was
recrystallized and an X-ray structure (appendix D) proved that the relative
stereochemistry was the anticipated cis-isomer.
Me
MeOOBn
OMe
NOTIPS
CO2Me
OBn
Me
MeOOBn
OMe
NHOH
OBn
H
H
Me
MeOOBn
OMe
N
OBn
O
O−10 °C to 25 °C
TBAF, THF
91% yield
1) TFA, Et3SiH−10 °C to 25 °C, 2 h
2) H2NNH2·H2O, KOHethylene glycol150 °C, 3 h
86% yield
Scheme 3.13. Optimized Formation of Amino Alcohol 283
283, X-ray
276 263

103
3.1.3. AMIDE COUPLING
In our approach to lemonomycin we intended to construct the bicyclo[3.2.1]
system 284 from a hemi-aminal 285, which would come from an amide coupling of the
amino alcohol 283 (Scheme 3.14).
Me
MeOOBn
OMe
NHOH
OBn
H
H
NN
H
HO
MeOMe
OBn
H
H
OBn
R2
R1
NNR1
OMeO
MeOMe
OBn
H
H
OBn
OH
MeO
Scheme 3.14. Retrosynthesis of Bicyclo[3.2.1] System
A literature search showed precedence for performing amino acid couplings with
hindered amines using PyBOP® as an activating reagent.7 A solution of amino alcohol
283 and one equivalent of BocGlyOH in CH2Cl2 was treated with PyBOP® and Hünig’s
base (Scheme 3.15). The only product isolated was ester 286, none of the amide 287 was
observed.
Me
MeOOBn
OMe
NHOH
OBn
H
HMe
MeOOBn
OMe
NHO
OBn
H
HNHBoc
O1.0 equiv. BocGlyOH
1.0 equiv. PyBOP®
Hünig's base, CH2Cl20 °C to 25 °C, 4 h
82% yield
Scheme 3.15. Ester Formation
285284 283
286283

104
287
The amide 287 was predicted to be more thermodynamically stable than the ester,
so conditions were investigated to promote thermal acyl-migration (Scheme 3.16).
Heating ester 286 in dichlorobenzene at 150 °C gave only unreacted starting material.
Heating in the presence of thiopyridone at 100 °C was also unsuccessful in migrating the
glycine to the amine.
Me
MeOOBn
OMe
NHO
OBn
H
HNHBoc
O dichlorobenzene
150 °C Me
MeOOBn
OMe
NOH
OBn
H
H
ONHBoc
N SHPhMe, 100 °C
Scheme 3.16. Attempted Acyl-Migration
It was observed that the use of an excess of BocGlyOH and PyBOP® during the
coupling with amino alcohol 283 gave a dicoupled product 288 in 85% yield (Scheme
3.17). The ester was hydrolyzed by treatment with NaOH to afford amide 287 in 60%
yield. While this two step procedure succeeded in providing the desired amide product it
was not particularly efficient, so an alternative method was developed.
Me
MeOOBn
OMe
NO
OBn
H
HNHBoc
O3.0 equiv. Boc-GlyOH
3.0 equiv. PyBOP®
Hünig's base, CH2Cl20 °C to 25 °C, 15 h
85% yield
ONHBoc
NaOH
DMSO
60% yield
Scheme 3.17. Unoptimized Formation of Amide 287
283 287
286
288

105
283 287
289
The previous results demonstrated that the first coupling occurred at the alcohol to
give the ester, but the amine was not too hindered to undergo a second coupling to
provide the amide 288. If the alcohol was protected and treated under coupling conditions
then amide 287 could be isolated directly. A multiple step sequence involving selective
protection of the alcohol would prove tedious. Thus, the best approach would be an in
situ protection of the alcohol during amide coupling and subsequent removal in the
workup.
The answer came by the way of silyl-activated amide coupling conditions, taking
advantage of the known silyl activation of amines.8 Treatment of amino alcohol 283 with
two equivalents of chlorotrimethylsilane gave the disilylated intermediate 290 (Scheme
3.18). The silyl ether prevents ester formation, while silylation of the amine leads to
activation of the nitrogen.
Me
MeOOBn
OMe
NHOH
OBn
H
H 1) TMSCl, Et3N, THF 0 °C to 25 °C, 3 h
2)
THF, −65 °C to 25 °C, 13 h
O
O ONHBoc
Me
MeOOBn
OMe
NOH
OBn
H
H
ONHBoc
Me
MeOOBn
OMe
NTMSOTMS
OBn
H
H
TMSCl
Me
MeOOBn
OMe
NOTMS
OBn
H
H
ONHBoc
82% yield H
Scheme 3.18. Silyl-Activated Amide Coupling Conditions
290 291
289

106
292 21% yield
The reaction conditions were carefully optimized to improve the yield and prevent
by-product formation. The disilylated intermediate 290 was added with filtration to a
solution of the mixed anhydride 289 at −65 °C and then slowly warmed to room
temperature with stirring overnight. Addition of the disilylated 290 to mixed anhydride
289 at −20 °C or warmer gave the tert-butyl amide product 292 from addition at the
wrong carbonyl (Scheme 3.19).
THF, −20°C to 25 °C, 14 h
Me
MeOOBn
OMe
N
OBn
H
H
O
OH
Scheme 3.19. tert-Butyl Amide By-Product Formation
The primary alcohol of amide 287 was then oxidized under Swern conditions9 to
afford hemi-aminal 293 as a mixture of diastereomers (3:2) in 94% yield (Scheme 3.20).
Care was taken during the oxidation to avoid dehydration as this is known to occur with
prolonged stirring at room temperature or with the use of Dess-Martin periodinane.10
Me
MeOOBn
OMe
NOH
OBn
H
H
ONHBoc
(COCl)2, DMSOEt3N, CH2Cl2
−78 °C to 25 °C, 8 min.
87% yield
Me
MeOOBn
OMe
N
OBn
H
HNBoc
OH
O
11
Scheme 3.20. Hemi-Aminal Formation
287 293, 3:2 mixture
at C11
290 287 57% yield
289 +

107
3.1.4. STEREOSELECTIVE INCORPORATION OF C14-C15
The strategy for building the bicyclo[3.2.1] system 294 (Scheme 3.21) in the
eastern fragment of lemonomycin was not planned from the beginning. The biggest
challenge lies with stereoselectivity for the incorporation of the three carbon unit. The
1,3-cis-substituted tetrahydroisoquinoline 295 possesses most of the steric bulk on the top
face, suggesting potential problems with stereoselectivity during the carbon-carbon bond
forming reactions at either C11 or C13.
Me
MeOOBn
OMe
N
OBn
H
HNBoc
OH
O
NN
H
HO
MeOMe
OBn
H
H
OBn
R1
Boc
1
3
MeO1
3 11 11
1313
15
14
Scheme 3.21. Alkylation Across C11 and C13
A cycloaddition process was avoided as this strategy has been shown to work best
on substrates which cannot undergo β-elimination at the C3 center.11 Instead, alkylation
at C11 was investigated to determine the reactivity and stereoselectivity at that center.
The first approach involved N-acyliminium alkylation with allylsilane (Scheme 3.22),
however, the alkylated product 296 was not observed and only starting material was
recovered. The starting hemi-aminal is identical to the hydrolyzed iminium intermediate
so it was not possible to determine if the N-acyliminium ion had formed.
294 295

108
Me
MeOOBn
OMe
N
OBn
H
HNBoc
OH
O
Si(Me)3Tf2O, DABCOEt2O, −78 °C
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
Scheme 3.22. Attempted Allylation
The hemi-aminal 293 was converted to the methyl aminal 297 in 95% yield on
treatment with CSA and trimethylorthoformate in methanol (Scheme 3.23). The methyl
aminal was formed in a ratio of 6:1 but the relative stereochemistry at C11 was not
determined, nor could the diastereomers be separated by flash chromatography. The
product was then subjected to allylation under a variety of conditions.
Me
MeOOBn
OMe
N
OBn
H
HNBoc
OH
O
HC(OMe)3, CSA
MeOH, 0 °C, 30 min.
95% yield
Me
MeOOBn
OMe
N
OBn
H
HNBoc
OMe
O
11
RLA,
R = Si(Me)3 Sn(Me)3LA = BF3·OEt2 TiCl4 SnCl4
TMSOTf
OTBS
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
CHO
Scheme 3.23. Attempted Allylations using Methyl Aminal 297
293 296
293
297296
298
297, 6:1 mixture at C11
299

109
Alkylation was not observed under any conditions. The main product isolated was
always hemi-aminal 293, which suggests that the N-acyliminium intermediate was
forming but not reacting. A more reactive nucleophile than allylsilane and allylstannane,
the silyl enol ether of acetaldehyde 298,12 was employed but again without success. An
observation made under heating was the apparent loss of the benzyl ethers, which may
have been from reaction with the lewis acid catalyst. To avoid this we investigated an
alternative method for the incorporation of the three carbon unit.
The hemi-aminal 293 was converted to thioaminal 300 by treatment with
thiophenol and para-toluenesulfonic acid in 86% yield from alcohol 287, under
optimized conditions (Scheme 3.24). Only one diastereomer was observed, which upon
recrystallization and X-ray analysis (appendix E) proved to be the cis-isomer. This
stereochemical result was surprising because the X-ray suggests that the axial orientation
of the C1 substituent ought to provide enough steric bulk to destabilize the axial
orientation of the C11 thiophenol (Figure 3.01). The likely cause of this preferred
orientation is the planar N12-nitrogen. Strong carbamate resonance was observed in 1H
NMR, suggested the nitrogen-carbon bond to the N-Boc carbonyl has imine character. As
a result the N-Boc is coplanar with the N12-nitrogen making the neighboring equatorial
positions more sterically hindered than the axial positions.
1) (COCl)2, DMSO, Et3N, CH2Cl2 −78 °C to 25 °C, 8 min.
2) PhSH, pTSA 0 °C, 1.5 h
86% yield
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPhMe
MeOOBn
OMe
NOH
OBn
H
H
ONHBoc
Scheme 3.24. Optimized Thioaminal Formation
287 300, X-ray

110
Figure 3.01. ORTEP Representation of Thioaminal 300 X-ray Structure
With application of the thioaminal we hoped to use a thiophilic lewis acid that
could selectively generate the N-acyliminium ion without reacting with the benzyl ethers
or N-Boc protecting group. Silver tetrafluoroborate was chosen as the thiophile.
Unfortunately treatment of thioaminal 300 with silver tetrafluoroborate in the presence of
tributylallylstannane gave only hemi-aminal 293 after an aqueous quench, 296 was not
observed (Scheme 3.25). Benzyl ether or N-Boc deprotection were not observed and the
C11 position proved unreactive toward alkylation by N-acyliminium chemistry.
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
Sn(Bu)3
Et2O, 0 °C
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
AgBF4
11 11
Scheme 3.25. Attempted Allylation of Thioaminal 300
300 296

111
300 301, X-ray
Our next approach focused on alkylation at the C13 position by enolization of the
amide and alkylation with an electrophile. Hemi-aminal 293 and methyl aminal 297 were
not used for this alkylation as they were never isolated as a single diastereomer, however
the thioaminal 300 was isolated as the cis-isomer exclusively. Examination of the X-ray
structure showed the expected stereochemistry for alkylation at C13 would be from the
bottom face to give the undesired isomer. Thioaminal 300 was deprotonated with lithium
hexamethyldisilazide at −78 °C then treated with allyl bromide to furnish the alkylated
product 301 in 70% yield (Scheme 3.26). The alkylation was complete after 20 minutes
and only one diastereomer was isolated. NMR analysis did not provide conclusive
evidence to determine the relative stereochemistry at the C13 position. However, X-ray
analysis of the recrystallized product (appendix F) showed the stereochemistry at the C13
position to be that of the expected trans-isomer (Figure 3.02).
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPhi) LiHMDS, THF
−78 °C, 15 min.
ii) Br
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
70% yield
13 13
Scheme 3.26. Allylation of Thioaminal 300

112
Figure 3.02. ORTEP Representation of Allylated Product 301 X-ray Structure
Since the alkylation was completely diastereoselective it was anticipated that
deprotonation of the C13 hydrogen and reprotonation would occur from the bottom face.
The alkylated product 301 was treated with potassium tert-butoxide, but no reaction
occurred at room temperature. However, heating at 55 °C for 16 hours produced a 1:1
mixture of diastereomers (Scheme 3.27).
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
t-BuOK, t-BuOH
THF, 55 °C, 16 h
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
13
Scheme 3.27. Epimerization of C13 Center
301 301/302, 1:1 mixture at C13

113
As an alternative to the thermodynamic conditions applied above, a
diastereoselective reprotonation was performed with complete deprotonation at C13 and
reprotonation in a second step. This reaction initially proved difficult because the C13
hydrogen of 301 was not as easily deprotonated as 300. Examination of the X-ray
structure (Figure 3.02) revealed the C13 hydrogen was equatorial and coplanar with
respect to the C17 carbonyl. Such alignment makes enolization more difficult because of
poor orbital overlap with the carbonyl. Treatment of 301 with LiHMDS, KHMDS, or
LDA at −78 °C showed no signs of deprotonation. Attempted deprotonation at elevated
temperatures led to degradation. A stronger base was used in conjunction with n-BuLi but
multiple by-products were observed.
Success was found using a procedure developed by S. G. Davies13 involving
treatment with t-BuLi at −78 °C followed by immediate quench with a bulky proton
source, butylated hydroxytoluene (BHT, 303, Scheme 3.28). Inversion of the C13
stereocenter afforded 302 in 79% yield, but it was observed that at least two equivalents
of t-BuLi were necessary to obtain complete inversion. Efforts to elaborate 302 to the
bridged system were unsuccessful and attempts to proceed with the synthesis from
allylated product 301 were abandoned.
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh−78 °C, 30 sec.
i) 2.4 equiv. t-BuLi, THF
ii) OH
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
79% yield
Scheme 3.28. Inversion of C13 Stereocenter
301 302 303

114
An alternative electrophile to allylbromide was chosen that would require less
manipulation to form the bicyclo[3.2.1] system. Thioaminal 300 was alkylated with
bromide 304 (Scheme 3.29), but initial results were poor. The optimized conditions
included deoxygenation of the starting solution, the use of KHMDS instead of LiHMDS,
and the use of iodide 306 instead of the analogous bromide 304 (Scheme 3.30). Under
these conditions the alkylated product 305 was isolated in 78% yield, along with 10%
recovered starting material. The remaining starting material could not be minimized by
the use of excess base or iodide 306. It is likely that O-alkylation is concomitant with
C-alkylation and the O-alkylated intermediate would be readily hydrolyzed upon aqueous
workup to give starting material.
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPhi) LiHMDS, THF
−78 °C, 5 min.
ii)Br
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
48% yield
OTIPSOTIPS
Scheme 3.29. Initial C13 Alkylation
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPhi) KHMDS, THF
−78 °C, 5 min.
ii)I
78% yield
OTIPS
Scheme 3.30. Optimized C13 Alkylation
305
300
306
304
305300

115
Inversion of the C13 stereocenter was performed as before, with care taken to
deoxygenate the reaction solution. The inverted product 307 was isolated in 73% yield
after purification and characterized as the cis-isomer (Scheme 3.31). On scale-up, a two
step procedure starting from 305 inverted the C13 stereocenter, then treatment of the
crude product mixture with 48% aqueous HF in acetonitrile removed the triisopropylsilyl
protecting group to furnish alcohol 308 in 83% yield over the two steps.
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
OTIPS
−78 °C, 30 sec.i) 2.4 equiv. t-BuLi, THF
ii) BHT
73% yield
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
OH
−78 °C, 30 sec.1) i) 2.2 equiv. t-BuLi, THF
ii) BHT
2) 48% aq. HF, CH3CN0 °C, 30 min.
83% yield
Scheme 3.31. Optimized C13 Inversion
305
305
307
308

116
With the three carbon unit installed with correct C13 stereochemistry the focus
turned to the formation of the C11-C15 bond. The approach would be to form the silyl
enol ether of the side-chain then cyclize to form the bicyclo[3.2.1] system. Alcohol 308
was converted to the silyl enol ether by oxidation to aldehyde 309 under Swern
conditions (Scheme 3.32),14 then treatment with TIPSOTf and triethylamine in diethyl
ether. The two step procedure furnished the trans-silyl enol ether in 88% yield.
TIPSOTf, Et3N0 °C to 25 °C, 16 h
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
OTIPS
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh(COCl)2, DMSO
Et3N, CH2Cl2−78 °C to 25 °C, 9 min. O
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
OH
Scheme 3.32. Silyl Enol Ether Formation
308 309
31088% yield, two steps

117
3.1.5. FORMATION OF THE BICYCLO[3.2.1] SYSTEM
It was hoped that the silyl enol ether would be nucleophilic enough to cyclize and
form the desired bicyclo[3.2.1] system under the condition required for N-acyliminium
ion formation. Intramolecular cyclizations are well precedented in the literature15 and
specifically, the N-acyliminium cyclization of a silyl enol ether to a 5-membered ring has
been reported.16
The initial conditions utilized AgBF4 as the thiophile to promote formation of
N-acyliminium 312. Treatment of 310 with AgBF4 in dichloroethane gave a mixture of
unidentified products and only 12% yield of the cyclized product 311 (Scheme 3.33,
Table 3.02, entry 1). Changing the solvent system to THF made a dramatic difference and
the cyclized product 311 was formed in nearly 80% yield (entry 2). Stoichiometric
amounts of AgBF4 were necessary to consume all of the starting thioaminal 310. The
intermediate N-acyliminium ion 312 formed rapidly but cyclization appeared slow and
was complete after stirring for four hours at room temperature. However, at 40 °C the
cyclization was complete after 30 minutes (entry 2).

118
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
OTIPS NN
H
O
MeOMe
OBn
H
H
OBn
CHO
Boc
MeO
Table 3.2
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
OTIPS
-SPh
NN
H
O
MeOMe
OBn
H
H
OBn
Boc
MeO
O
SiF
H
H
15
Scheme 3.33. Cyclization to the Bicyclo[3.2.1] System
entry reagent solvent temp (°C) time yield (%)
1 AgBF4 DCE 25 2 h 12
2 AgBF4 THF 25 40
4 h 30 min.
78 74
3 AgBF4 Et2O 25 30 min. 0
4 AgBF4 PhCl 25 2 h 0
5 BF3·OEt2 CH2Cl2 25 2 h 0
6 Hg(O2CCF3)2 THF 25 30 min. 0
7 Δ PhCl 115 24 h 0
8 1) m-CPBA 2) Δ
CH2Cl2 PhCl
80
2 h 0
9 Bu3SnH, AIBN PhH 80 20 h 0
Table 3.02. Cyclization to the Bicyclo[3.2.1] System
310
312 313
311

119
Further investigation of the reaction proved surprising because under no other
conditions was the cyclized product observed. Changing the solvent to diethyl ether or
chlorobenzene led to the formation of a mixture of unidentified products (entries 3 and
4). Other lewis acids and thiophiles also gave poor results (entries 5 and 6). Heating to
promote elimination of thiophenoxide led to deprotection of the silyl enol ether leaving
the thioaminal untouched (entry 7). Conversion of the sulfide to the sulfoxide resulted in
a better leaving group but upon elimination with heating only unidentified products were
observed (entry 8). Radical cyclization conditions17 gave no reaction (entry 9). The
significance of the solvent selectivity was not determined until our work on renieramycin
G.
Minor impurities were observed during the cyclization in THF and after filtration
of the crude reaction mixture through a plug of silica gel only small amounts of
impurities were present. The cyclized product appeared to be a single diastereomer at the
C15 stereocenter. To determine the relative stereochemistry with certainty a crystalline
derivative was made. Success in obtaining a crystal structure was found with formation of
the oxime derivative 314, from treatment of aldehyde 311 in methanol with
hydroxylamine hydrochloride and potassium acetate (Scheme 3.34). A 1:1 mixture of cis-
and trans-isomers was isolated in 83% yield. Chromatography was employed to obtain
modest separation of the isomers. The more polar fractions gave a 4:1 mixture, cis:trans,
as a white solid which upon recrystallization produced X-ray quality needles. The X-ray
structure (appendix G) shows that the cyclization furnished the aldehyde on the outside
face giving the desired stereochemistry at C15 (Figure 3.03). The selectivity was likely
due to the axial orientation of the C1 side-chain, which provides steric hindrance for
cyclization with the silyl enol ether on the inside face. The X-ray structure shows that the
C1 side-chain remains in the axial orientation even with the bridged ring system present.

120
NN
H
O
MeOMe
OBn
H
H
OBn
CHO
Boc
MeO
HONH2·HCl
KOAc, EtOH25 °C , 2 h
NN
H
O
MeOMe
OBn
H
H
OBn
Boc
MeO
NHOH
83% yield
HH
15
Scheme 3.34. Oxime Formation
Figure 3.03. ORTEP Representation of Oxime 314 X-ray Structure
With an efficient yield of the bicyclo[3.2.1] system we focused our attention on
elaboration of the remainder of lemonomycin. N-Boc deprotection, amide reduction,
debenzylation, C1 glycol incorporation, and a final oxidation to the quinone were
311 314, X-ray

121
required. However, at this stage of the project Stoltz published his total synthesis of
(−)-lemonomycin.1 As described in Chapter 1 his strategy was distinctly different, in fact
if we were to synthesize lemonomycin we would not share a common intermediate. The
lack of overlap between the strategies meant that we would not be able to demonstrate, by
formal synthesis, the completion of lemonomycin. The enantioselective synthesis by
Stoltz was more efficient so with our strategy offering no further improvement
completion of the total synthesis of lemonomycin was abandoned. However, formation of
the lemonomycin aglycone amide (lemonomycinone amide, 317, Scheme 3.36) derivative
was investigated.
Aldehyde 311 was treated under hydrogenolysis conditions to give the diol 315 in
79% yield from the silyl enol ether 310 (Scheme 3.35). The C15 aldehyde did not exist as
the hydrated aldehyde present in (−)-lemonomycin. However, it was observed that when
deuterated methanol was used as an NMR solvent none of the aldehyde signals could be
observed in the proton and carbon NMR. Although the spectra did not show clean
formation of a new compound, it was evident that hydration of the aldehyde did occur.
Evaporation of this sample and then reinvestigation by 1H NMR analysis in deuterated
chloroform indicated reformation of aldehyde 315.
NN
H
O
MeOMe
OBn
H
H
OBn
CHO
Boc
MeO HN
N
H
O
MeOMe
OH
H
H
OH
CHO
Boc
MeO H
Pd(OH)2, MeOH
1 atm H2, 25 °C , 4 h
79% yield from cyclization
1515
Scheme 3.35. Hydrogenolysis
315 311

122
The N-Boc protecting group was removed under acidic conditions to afford amine
316 (Scheme 3.36). Monitoring the reaction by TLC proved difficult and reverse-phase
HPLC conditions were necessary to accurately characterize the deprotected product. 1H
NMR experiments verified the aldehyde existed solely in the hydrated form. In addition,
2D NMR experiments showed epimerization at the C15 stereocenter had not occurred.
An efficient route to quinone 317 involved deprotection of the N-Boc protecting
group of aldehyde 315 and oxidation of the crude product with ammonium cerium (IV)
nitrate to afford the quinone 317 in 35% yield over the two steps. Reverse-phase HPLC
solvents contained 0.1% TFA and hence the TFA salt of the product was isolated after
purification. Characterization of the final compound by 2D NMR showed consistent
stereochemistry at the C15 stereocenter, a distinct doublet for the C16 hydrogen, and
quinone shifts consistent with the reported spectra of (−)-lemonomycin.
3 N HCl
1:1 MeOH:H2O25 °C , 3 h
NN
H
O
MeOMe
OH
H
H
OH
CHO
Boc
MeO HN
NH
H
O
MeOMe
OH
H
H
OH
MeO H
OHHO
· HCl
(NH4)2Ce(NO3)6
H2O, 25 °C , 4 h NNH
H
O
MeO
O
H
H
OH
MeO H
OHHO
· TFA
35% yieldtwo steps
15
16
Scheme 3.36. Formation of Lemonomycinone Amide 317
316315
317

123
3.2. CONCLUSION
The approach to (−)-lemonomycin succeeded in developing new methodology for
the synthesis of tetrahydroisoquinoline alkaloids. The alkylation of isoquinoline with
benzyloxymethyllithium was performed in the presence of (−)-sparteine, showing modest
enantioselectivity. Unfortunately, the analogous reaction with the electron-rich
isoquinoline 262 gave multiple unidentified products. In the absence of (−)-sparteine, the
1,2-dihydroisoquinoline 263 could be formed. Elaboration of the 1,3-cis-substitution
required formation of oxazolidinone derivative 276 but stereoselectivity during ionic
hydrogenation was still maintained. Formation of the eastern fragment required
development of silyl-activated coupling conditions that protected the C3-hydroxyl while
activating the amine to give the amide 287 in excellent yield.
Construction of the bicyclo[3.2.1] system proceeded through the thioaminal 300
which was synthesized as one diastereomer from the corresponding hemi-aminal.
Alkylation via an enolate, generated from the amide, gave the wrong stereochemistry at
the C13 center, but proved completely diastereoselective. Thus, inversion of the center by
diastereoselective reprotonation conditions were very efficient. To close the three carbon
unit to the bicyclo[3.2.1] system, an intramolecular N-acyliminium cyclization with silyl
enol ether 310 was developed. The cyclization was best performed in THF with thiophile
AgBF4, other reaction conditions gave at best only trace amounts of 311. The C15
aldehyde was formed diastereoselectively and the X-ray structure of the oxime derivative
314 proved the desired stereochemistry had been achieved. The unique aldehyde was
eventually converted to the hydrated aldehyde 316 after N-Boc deprotection and a final
oxidation afforded quinone 317. The quinone represented lemonomycinone amide, a

124
317
congener of the initial target. The (±)-lemonomycinone amide 317 was synthesized from
aldehyde 230 in 26 steps with an overall yield of 3% (Scheme 3.37).
OMeMe
MeOCHO
26 steps
3% yield NNH
H
O
MeO
O
H
H
OH
MeO H
OHHO
· TFA
Scheme 3.37. Total Synthesis of (±)-Lemonomycinone Amide 317
Although the total synthesis of lemonomycin was abandoned, success was found
with our approach to this class of tetrahydroisoquinoline alkaloids. Application of this
new methodology to another tetrahydroisoquinoline alkaloid was investigated to
demonstrate its utility in a divergent strategy.
230

125
3.3. REFERENCES 1) Stoltz, B. M.; Cruz, E. G.; Ashley, E. R. J. Am. Chem. Soc. 2003, 125, 15000. 2) Kaufman, T. S. Synlett 1997, 1377. 3) Adams, J.; Plamondon, L.; McCormack, T. A.; Destree, A. T.; Behnke, M. L.; Grenier, L.; Soucy, F. J. Am. Chem. Soc. 1999, 121, 9967. 4) Larock, R. C.; Roesch, K. R. J. Org. Chem. 2002, 67, 86. 5) Williams, R. M.; Metobo, S.; Jin, W. Org. Lett. 2003, 5, 2095. In this publication the reduction of a similar 3,4-olefin of a tetrahydroisoquinoline intermediate could not be performed under high pressure without degradation of the molecule. The failure of reduction led to the termination of this approach. 6) Davies, S. G.; Blagg, J. J. Chem. Soc., Chem. Comm. 1986, 492. 7) Aldrich, J. V.; Murray, T. F.; Berman, F.; Maeda, D. Y. J. Med. Chem. 2000, 43, 5044.
8) Klebe, J. F. In Advances in Organic Chemistry; Taylor, E. C., Ed.; Wiley-Interscience: New York, 1972; Vol. 8, p 97. 9) Fukumoto, K.; Sukegawa, Y.; Shishido, K. Heterocycles 1986, 24, 641. 10) Hu, L.; Yu, C. Tetrahedron Lett. 2001, 42, 5167. 11) Williams, R. M.; Scott, J. D. J. Am. Chem. Soc. 2002, 124, 2951. 12) Jung, M. E.; Blum, R. B. Tetrahedron Lett. 1977, 18, 3794. 13) Davies, S. G.; Bull, S. D.; Epstein, S. W.; Ouzman, J. V. A. Tetrahedron: Asymmetry 1998, 9, 2795. 14) Swern, D.; Mancuso, A. J.; Huang, S. L. J. Org. Chem. 1978, 43, 2480 15) Maryanoff, B. E.; Zhang, H.-C.; Cohen, J. H.; Turchi, I. J.; Maryanoff, C. A. Chem. Rev. 2004, 104, 1431. 16) Speckamp, W. N.; Hiemstra, H.; Vijn, R. J. J. Org. Chem. 1988, 53, 3882. 17) Hart, D. J.; Atarashi, S.; Choi, J.-K.; Ha, D.-C.; Kuzmich, D.; Lee, C.-S.; Maesh, S.; Wu, S. C. J. Am. Chem. Soc. 1997, 119, 6226.

126
Chapter 4: The Total Synthesis of (±)-Renieramycin G
4.0. INTRODUCTION
Continuing our approach to the tetrahydroisoquinoline alkaloids we chose as our
new target renieramycin G (2g, Scheme 4.01). The total synthesis of renieramycin G had
not been previously reported. Furthermore, at the start of the project the only total
synthesis of a renieramycin was by Fukuyama,1 but Danishefsky recently published the
synthesis of cribrostatin IV (renieramycin H).2
We hoped to develop a divergent strategy for the synthesis of
tetrahydroisoquinoline alkaloids, something that has not been previously demonstrated.
To do this we wanted to use an advanced intermediate from our approach to
lemonomycin. The divergence between lemonomycin and renieramycin G occurs from
sulfide 300 (Scheme 4.01). Alkylation with iodide 306 installed the framework for the
bicyclo[3.2.1] system of lemonomycin. However alkylation with a benzyl halide 320
would install the framework for the bicyclo[3.3.1] system of renieramycin G. The choice
of protecting groups was important as stability to the alkylation and the diastereoselective
reprotonation conditions were required. It was anticipated that the reactivity would be
improved if the cyclization precursor 319 was a phenol (R1 = H). This would require a
differential protecting group (R1) to be present on the benzyl halide 320 which could be
readily removed in the presence of the benzyl ether and the N-Boc protecting groups.
After cyclization to 318, removal of the N-Boc protecting group and methylation were
required. The final steps include benzyl ether deprotection, oxidation to the
bis-isoquinolinequinone, and angelate ester formation. The angelate ester is known to
readily isomerize to the more stable cis-isomer.3 Thus, it was important to minimize the
number of steps with the angelate ester incorporated.

127
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
H
O
OMe
MeO
OMeMe
MeOOBn
NN
HOOMe
Me
OMeBoc
H
H
H
H
OBnO
1
3
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh
1
3
X
MeOMe
OMe
OR1+
1
31
11
13
3
4
21
6 10
8
22
19
17
15
14
26
24
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OR1
OMe
MeOMe
H
Scheme 4.01. Retrosynthesis of Renieramycin G (2g)
2g 318
319
320
300

128
4.1. RESULTS AND DISCUSSION
4.1.1. BENZYL CHLORIDE FORMATION
Our first objective in the approach to renieramycin G was the synthesis of a
suitable benzyl halide that would fit our reaction profile. For this we chose the benzyl
chloride 321 (Figure 4.01). We hoped that the benzyl chloride would be a reactive
enough electrophile, but formation of the benzyl bromide was possible if this was a
problem. The trityl ether would provide differentiation from the benzyl ethers and N-Boc
(which are more stable to acid than the aryl trityl ether). It was anticipated that the trityl
group would be stable to both the alkylation conditions and the diastereoselective
reprotonation conditions. No literature precedence for this compound existed, but we
utilized known chemistry to synthesis the benzyl halide from commercially available
benzaldehyde 230 (Scheme 4.02). The same benzaldehyde was used to form the
tetrahydroisoquinoline of the western fragment, demonstrating the pseudo-symmetry of
the renieramycins.
MeOMe
MeO
Cl
OTrt
Figure 4.01. Benzyl Chloride 321
321

129
A solution of benzaldehyde 230 in 37% aqueous formaldehyde was treated under
chloromethylation conditions4 (Scheme 4.02), gaseous HCl and zinc (II) chloride. After
chromatography the benzyl chloride 322 was isolated as a white solid in 73% yield.
MeOMe
MeO
O H
MeOMe
MeO
O H
Cl37% aq. formaldehyde
HCl (g), ZnCl2reflux, 90 min.
73% yield
Scheme 4.02. Chloromethylation
A Baeyer-Villiger reaction was performed with benzyl chloride 322 furnishing the
formate ester 323 in 95% yield (Scheme 4.03). This type of Baeyer-Villiger in the
presence of a benzyl chloride had not been previously reported. There was concern that
by-products could form from the addition of m-CPBA at the benzyl chloride rather than
the benzaldehyde; however no by-products from this pathway were observed.
MeOMe
MeO
O H
Cl MeOMe
MeO
Cl
O
O
H
m-CPBA, CH2Cl20 °C to 25 °C, 3 h
95% yield
Scheme 4.03. Formate Ester Formation
322230
322 323

130
In our previous studies we used NaOH in MeOH to hydrolyze the formate ester,
however the analogous procedure with benzyl chloride 323 gave significant amounts of a
by-product that appeared to be from methoxide addition at the benzyl chloride. To avoid
this side-reaction the formate ester was reduced with LiBH4, to furnish the phenol 324 as
an unstable orange oil (Scheme 4.04). The crude phenol was protected as the trityl ether
upon treatment with recrystallized trityl chloride and triethylamine in CH2Cl2. The trityl
ether 321 was unstable and although the reaction appeared clean by TLC analysis
isolation of the pure product proved difficult. Silica gel chromatography was employed to
isolate the semi-pure ether, which upon recrystallization afforded the pure trityl ether 321
in 51% yield from the formate ester 323. The trityl ether was also found to be unstable to
storage at room temperature under argon in the dark.
MeOMe
MeO
Cl
O
O
H
LiBH4, THF
0 °C, 10 min.
MeOMe
MeO
Cl
OH
trityl chloride
Et3N, CH2Cl225 °C, 2 h
MeOMe
MeO
Cl
OTrt51% yieldtwo steps
Scheme 4.04. Trityl Ether 321 Formation
323 324 321

131
4.1.2. FORMATION OF THE BICYCLO[3.3.1] SYSTEM
With benzyl chloride 321 in hand, alkylation of the common intermediate 300
was conducted under identical conditions to those used for the approach to lemonomycin
with iodide 306. Treatment of a degassed solution of thioaminal 300 with KHMDS at
−78 °C followed by addition of benzyl chloride 321 as a solution in THF gave unreacted
starting material (Scheme 4.05). No alkylation product was observed and it is likely that
the benzyl chloride was not reactive enough. O-Alkylation was ruled out because the
starting benzyl chloride was recovered. If O-alkylation had occurred then upon workup
hydrolysis would have given the benzyl alcohol.
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPhKHMDS, THF, −78 °C
−78 °C to 25 °C, 3 h
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OTrtOMe
MeOMe
then 321
Scheme 4.05. Attempted Benzylation of 300
The instability of the benzyl chloride 321 made conversion to a more reactive
benzyl bromide an unattractive route. Instead, the reactivity of the enolate was enhanced.
Conducting the reaction in the presence of 18-crown-6 produced a change in reactivity
and the alkylated product was formed. However, there was noticeable by-product
formation and the alkylated product was formed in approximately 40% yield. It was
noted that deoxygenation of the reaction solution was essential and a dramatic increase in
reaction efficiency was found by starting with thioaminal 300, 18-crown-6, and benzyl
chloride 321 as a solution in THF followed by the addition of KHMDS (Scheme 4.06).
300 325

132
Combining the reagents reduced the time between deprotonation and alkylation,
minimizing by-product formation. The reaction was complete within 20 minutes and near
quantitative formation of the alkylated product 325 was observed. The alkylation
appeared to be diastereoselective.
18-crown-6, KHMDS
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OTrtOMe
MeOMe
THF, −78 °C, 20 min
98% yield
Scheme 4.06. Optimized Benzylation of Thioaminal 300
It was noted that manipulation of 325 did not lead to degradation or loss of the
trityl protecting group. Whereas the benzyl chloride 321 was much more susceptible to
deprotection and degradation. However, as with most of the compounds in this series,
pronounced carbamate resonance was observed, and the trityl group was not stable to
heat during a variable temperature NMR experiment. We anticipated the trityl group to be
stable to diastereoselective reprotonation.
Initial attempts to invert the C13 stereocenter utilized the conditions employed in
our approach to lemonomycin. The benzylated product 325 was treated with 2.5
equivalents of tert-butyllithium then immediately reprotonated with butylated hydroxyl-
toluene (BHT) (Scheme 4.07). A 6:1 mixture of the desired product 326 and starting
material was isolated in 73% yield. Using in excess of three or four equivalents did not
affect the diastereomeric ratio.
321 300 +
325

133
−78 °C, 30 sec.i) 2.5 equiv. t-BuLi, THF
ii) BHT, THF NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OTrtOMe
MeOMe
Scheme 4.07. Diastereoselective Reprotonation
The isomers were difficult to separate until removal of the trityl group produced a
mixture of the corresponding phenols. The more polar phenols were then separated by
chromatography. A one-pot, two-step procedure was developed that inverted the C13
stereocenter and removed the trityl protecting group. The benzylated product 326 was
treated to the same reprotonation conditions as above (Scheme 4.08). The solution was
then treated with anhydrous HCl in diethyl ether, with warming to 0 °C and careful
monitoring of the reaction; small portions of HCl were added until the trityl ether was not
detected. After separation from the other isomer, phenol 327 was isolated in 73% yield
over the two steps.
−78 °C, 30 sec.ii) BHT, THF
1) i) 2.5 equiv. t-BuLi, THF
2) HCl, Et2O, 0 °C, 1 hN
NBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OHOMe
MeOMe
73% yield
Scheme 4.08. Optimized Formation of Phenol 327
325 + 325
326
73% combined yield 326:325 = 6:1
325
327

134
The phenol 327 represented the cyclization precursor to be used in the
N-acyliminium cyclization to form the bicyclo[3.3.1] system. The optimized conditions
employed in our approach to lemonomycin were employed first. Treatment of phenol
327, as a solution in THF, with AgBF4 in THF led to immediate starting material
consumption and the formation of more polar products (Scheme 4.09). However upon
aqueous workup and concentration of the organic fractions well over 300% mass
recovery was isolated.
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OHOMe
MeOMe
OMeMe
MeOOBn
NN
HOOMe
Me
OMeBoc
H
H
H
H
OBnO
AgBF4
THF, 25 °C
Scheme 4.09. Attempted N-Acyliminium Alkylation
The reaction conditions are fairly simple with only AgBF4 being added, and the
extra weight did not appear to be coming from this reagent. A volatile oil was present in
large amounts, which upon 1H NMR analysis appeared to be one of the by-products
observed in our N-acyliminium cyclization of silyl enol ether 310 during our studies on
lemonomycin. This by-product possessed what appeared to be an aldehyde signal and
was originally thought to come from degradation of the starting silyl enol ether 310.
However, observation of the same by-product in the cyclization of phenol 327 suggested
it was unlikely that an aldehyde had formed from the degradation of 327. In addition the
mass recovery could not be explained if degradation of the starting phenol 327 was
assumed.
327 328

135
The answer appeared to be polymerization of the solvent THF. Tests showed that
THF was stable to AgBF4, but polymerization in the presence of a catalyst, our phenol
327, was observed. The polymer could be forming by two possible mechanisms. The first
mechanism could be acid catalyzed polymerization promoted by the HBF4 formed after
cyclization. The strong acid HBF4 is not formed during the cyclization of silyl enol ether
310, which could explain why more polymer was observed during the cyclization of
phenol 327. A second mechanism is needed to rationalize the formation of the same
polymer in the absence of HBF4. We postulate that an N-acyliminium ion 329, formed
from either silyl enol ether 310 or phenol 327, can react with THF to form an oxonium
intermediate 330 (Scheme 4.10). This intermediate can then react with another molecule
of THF leading to polymerization.
NN
OMeO
MeOMe
OBn
H
H
OBn
Boc
NN
OMeO
MeOMe
OBn
H
H
OBn
BocOO
AgBF4
THF, 25 °C
NN
OMeO
MeOMe
OBn
H
H
OBn
BocO
O
O
R
RR
polymerization
Scheme 4.10. Observed Polymerization
327 or 310
329
331 330

136
When the solvent was changed to chlorobenzene the by-product was not observed
(Scheme 4.11). N-Acyliminium formation was rapid and a much more polar compound
was the major product. Isolation of this compound revealed no tert-butyl proton signals
were present in 1H NMR suggesting the absence of the N-Boc protecting group. Further
characterization revealed that the desired cyclization had occurred but loss of N-Boc had
furnished amine 332, presumably due to the presence of HBF4 formed in the reaction.
NNBoc
OMeO
MeOMe
OBn
H
H
OBn
SPh OHOMe
MeOMe
OMeMe
MeOOBn
NNH
HOOMe
Me
OMeH
H
H
H
OBnO
AgBF4, PhCl
25 °C, 10 min.
56% yield
Scheme 4.11. N-Acyliminium Cyclization
The cyclization to 332 was very rapid and none of the N-Boc cyclized
intermediate could be observed or isolated. The amine product 332 was formed within 10
minutes of AgBF4 addition. The cyclization of phenol 327 was significantly more rapid
than the cyclization of silyl enol ether 310, which required four hours at room
temperature after N-acyliminium formation. The relative rates are not necessarily
surprising because formation of the 6-membered bicyclo[3.3.1] system is less strained
than the 5-membered bicyclo[3.2.1] system. There is also a difference of nucleophilicity
between the phenol and the silyl enol ether.
Cyclization of the phenol 327 proceeded rapidly and cleanly in chlorobenzene.
The loss of the N-Boc protecting group was a fortuitous result as it would have been
removed in the next reaction. Under optimized conditions reductive methylation with
327 332

137
sodium cyanoborohydride, acetic acid and formaldehyde (Scheme 4.12) afforded
N-methylamine 333. After chromatography 333 was isolated in 65% yield over the two
steps. The spectroscopic data were consistent for formation of the desired cyclized
product. Attempts to form a crystalline derivative were unsuccessful for this substrate.
1) AgBF4, PhCl25 °C, 10 min. OMe
Me
MeOOBn
NN
HOOMe
Me
OMeMe
H
H
H
H
OBnO
2) NaBH3CN, AcOH, 37% aq. formaldehyde, MeOH, 25 °C, 1h
65% yield
Scheme 4.12. Optimized N-Acyliminium Cyclization
4.1.3. COMPLETION OF THE SYNTHESIS
The final reactions in our approach to renieramycin G required deprotection of the
benzyl ethers, oxidation to the bis-isoquinolinequinone, and angelate ester formation.
Treatment of N-methyl 333 with Pd(OH)2 under a balloon atmosphere of hydrogen led to
debenzylation (Scheme 4.13). The debenzylation was much slower than in our approach
to lemonomycin and under unoptimized conditions the mono-benzylated product 334 was
isolated in 85% yield. After purification by chromatography the mono-benzyl ether 334
was recrystallized from methanol and ethyl acetate yielding small amounts of X-ray
quality needles. An X-ray structure of the mono-benzyl ether 334 (appendix H)
confirmed that the cyclization had occurred as expected with the correct stereochemistry.
327
333

138
OMeMe
MeOOBn
NN
HOOMe
Me
OMeMe
H
H
H
H
OBnO
Pd(OH)2, MeOH
1 atm H2, 1 h
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
OBnO
85% yield
Scheme 4.13. Incomplete Hydrogenolysis
The hydrogenolysis was optimized by the use of at least 40% by weight catalyst
and stirred for six hours under an atmosphere of hydrogen (Scheme 4.14). The triol 335
was isolated in 94% yield. The triol was not very soluble and we were unable to find
appropriate recrystallization conditions.
OMeMe
MeOOBn
NN
HOOMe
Me
OMeMe
H
H
H
H
OBnO
Pd(OH)2, MeOH
1 atm H2, 6 h
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
OHO
94% yield
Scheme 4.14. Optimized Hydrogenolysis
The order in which to do the remaining reactions (phenolic oxidation and angelate
ester formation) was our next concern. Our initial approach was to oxidize to the di-
quinone and then esterify, however, in Danishefsky’s work on cribrostatin IV (78) he
reported that “despite extensive efforts” formation of the angelate ester of the quinone
336 was unsuccessful (Scheme 4.15).2 Their synthetic route was altered in order to go
334, X-ray 333
335 333

139
87
through the phenol 86, which underwent esterification and silyl deprotection to afford 88
in 75% yield over the two steps. Our phenol 335 was slightly different than
Danishefsky’s phenol 86 in that the phenolic hydroxyls present in our substrate were
likely to be more reactive. This could lead to multiple ester products, so we decided to
proceed through oxidation first followed by esterification.
OMe
MeOO
NN
HOOMe
Me
OHMe
H
H
H
OHO
O
OMe
MeOO
NN
HOOMe
Me
OHMe
H
H
H
OO
O
O
Me
Me
H
OTBSMe
MeOOMe
NN
HOOMe
Me
OHMe
H
H
H
OHO
O
OHMe
MeOOMe
NN
HOOMe
Me
OHMe
H
H
H
OO
O
O
Me
Me
H
Me
MeClO
H CH2Cl2
1)
2) AcOH, TBAF, THF
75% yield
Scheme 4.15. Danishefsky’s Angelate Ester Formation
336
86
88
cribrostatin IV (renieramycin H, 78)

140
The triol 335 was oxidized to the diquinone using Stoltz’s conditions5 by
treatment with ammonium cerium (IV) nitrate (Scheme 4.16). The oxidation was
complete within 15 minutes and the diquinone 337 was isolated in 55% yield after silica
gel chromatography.
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
OHO
(NH4)2Ce(NO3)6CH3CN:H2O 6:1
−5 °C, 10 min.
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
H
OHO
55% yield
Scheme 4.16. Bis-Isoquinolinequinone Formation
For esterification we followed the procedure utilized by Joseph-Nathan and
co-workers in their work on butanoic acid derivatives.6 Excess angeloyl chloride7 (87)
was added to a solution of 337 in CH2Cl2 (Scheme 4.17). After standing in the dark for
24 hours the solution was concentrated under vacuum and purified directly by
reverse-phase HPLC. The initial HPLC conditions chosen were normal-phase, with
elution in isopropanol and hexanes, however the product was not stable in these solvents.
Under reverse-phase conditions the solvent system was H2O and acetonitrile, and in
contrast to our purification of lemonomycinone amide 317 TFA was not present. The
product was very stable to these conditions and trailing was not observed during
separation. After purification (±)-renieramycin G (2g) was isolated in 74% yield.
335 337

141
OMe
MeOO
NN
OOMe
Me
OMe
H
H
H
H
OHO
N
MeO
MeOO
OO
NH
OOMe
Me
OMe
OMe
Me
H
HH
H
Me
MeClO
H
CH2Cl2
74% yield
26
Scheme 4.17. Angelate Ester Formation
We attempted to compare our sample with an authentic one. However, after
discourse with Prof. Davidson, the author responsible for the isolation,7 he informed us
that he no longer had an authentic sample in his possession. Furthermore he was unable
to provide copies of the original spectra. In the isolation paper a list of the observed
proton and carbon signals was published and based on the initial spectra we acquired of
our sample of renieramycin G in CD2Cl2 it appeared we had indeed formed the correct
structure. The key 1H NMR signal was the C26 hydrogen which appears around 5.9 ppm
for the angelate ester but around 6.6 ppm for the tiglate ester, the trans-isomer.8 Thus,
epimerization of the double bond had not occurred. Unfortunately we were not satisfied
with the spectra in CD2Cl2 because trace water, CH2Cl2, and acid led to signal broadening
and overlap. For an improvement in the resolution of our spectra we chose C6D6 as the
NMR solvent. The signals were much sharper, trace C6H6 and H2O appeared out of the
range of renieramycin G signals so there was no overlap. 2D NMR experiments including
COSY, NOESY, HSQC, and HMBC were conducted and all data appeared consistent
with previously published observations of similar structures.
87
337
(±)-renieramycin G (2g)

142
4.2. CONCLUSION
The total synthesis of (±)-renieramycin G successfully demonstrated a divergent
approach to the tetrahydroisoquinoline alkaloids. In chapter three we synthesized a
(±)-lemonomycinone amide (317) from thioaminal 300 and from that same intermediate
we synthesized (±)-renieramycin G (2g). The alkylation of thioaminal 300 with benzyl
chloride 321 proved more efficient than the alkylation with iodide 306. Construction of
the bicyclo[3.3.1] system via an N-acyliminium cyclization was also more efficient than
formation of the bicyclo[3.2.1] system of 317. This can be attributed to the formation of a
less strained 6-membered ring. The incomplete hydrogenolysis provided the mono-benzyl
ether 334 as a crystalline solid. The X-ray structure of which was important to determine
with certainty that the desired cyclization had occurred and the C13 stereocenter had been
inverted. The synthesis of (±)-renieramycin G was completed after oxidation to the
diquinone and angelate ester formation. Although Danishefsky reported problems with
esterification of the C1 side-chain alcohol of his diquinone, difficulties were not observed
for our substrate. The total synthesis of (±)-renieramycin G from commercially available
benzaldehyde 230 was completed in 25 steps with an overall yield of 4% (Scheme 4.18).
OMeMe
MeOCHO
25 steps
4% yield
Scheme 4.18. Total Synthesis of (±)-Renieramycin G (2g)
(±)-renieramycin G (2g)
230

143
4.3. REFERENCES
1) Fukuyama, T.; Linton, S. D.; Tun, M. M. Tetrahedron Lett. 1990, 31, 5989. 2) Danishefsky, S. J.; Chan, C.; Heid, R.; Zheng, S.; Guo, J.; Zhou, B.; Furuuchi, T. J. Am. Chem. Soc. 2005, 127, 4596. 3) Buckles, R. E.; Mock, G. V. J. Org. Chem. 1950, 15, 680. 4) Geissman, T. A.; Schlatter, M. J.; Webb, I. D.; Roberts, J. D. J. Org. Chem. 1946, 11, 741. 5) Stoltz, B. M.; Cruz, E. G.; Ashley, E. R. J. Am. Chem. Soc. 2003, 125, 15000. 6) Joseph-Nathan, P.; Torres-Valencia, J. M.; Cerda-Garcia-Rojas, C. M. Tetrahedron: Asymmetry 1998, 9, 757. 7) Davidson, B. S. Tetrahedron Lett. 1992, 33, 3721. 8) Beeby, P. J. Tetrahedron Lett. 1977, 38, 3379.

144
Chapter 5: Experimentals
5.0. GENERAL INFORMATION
Melting points were taken on a Thomas-Hoover capillary tube apparatus and are
uncorrected. Infrared spectra were recorded on a Nicolet FT-IR spectrophotometer neat
unless otherwise indicated. 1H NMR spectra were recorded on either a General Electric
QE-300 spectrometer at 300 MHz or a Varian spectrometer at 500 MHz in the indicated
solvent and are reported in ppm relative to tetramethylsilane and referenced internally to
the residually protonated solvent. 13C NMR spectra were recorded on a General Electric
QE-300 at 75 MHz or a Varian spectrometer at 125 MHz in the solvent indicated and are
reported in ppm relative to tetramethylsilane and referenced internally to the residually
protonated solvent. Mass spectra were obtained on a VG ZAB2E or a Finnigan TSQ70.
Routine monitoring of reactions was performed using Merck 60 F254 silica gel,
aluminum-backed TLC plates. Flash column chromatography was performed using EMD
silica gel (particle size 0.040-0.063 μm). Reverse-phase HPLC purification was done
with a WATERS 1525 Binary HPLC Pump and WATERS 2996 Photoiodide Array
Detector using C18 packed analytical (VYDAC, 10 μm packing, 4.6 x 250 mm), semi-
preparatory (WATERS, μBondapak, 7.8 x 300 mm), and/or preparatory column
(VYDAC, 10 μm packing, 22 x 250 mm). Optical purities were determined by HPLC
analysis using an analytical chiral column (Daicel, Chiralcel OD; eluent,
hexane/isopropyl alcohol; UV detector, 254 nm) with a Beckman Coulter System Gold
126P solvent module and Beckman Coulter System Gold 168 detector.
Solvents and commercial reagents were purified in accordance with Perrin and
Armarego or used without further purification. All reactions were setup under an
atmosphere of argon and only degassed when specified.

145
5.1. EXPERIMENTAL CONDITIONS
1-Phenylsulfanylmethyl-1H-isoquinoline-2-carboxylic acid methyl ester (157)
1-Phenylsulfanylmethyl-1H-isoquinoline-2,4-dicarboxylic acid dimethyl ester (158)
NCO2Me
PhS
NCO2Me
SPh
CO2Me
To a stirred solution of thioanisole (0.594 mL, 5.06 mmol) and TMEDA (0.764 mL, 5.06
mmol) in THF (8.0 mL) at 0 °C was added n-BuLi (2.30 M in hexanes, 2.20 mL, 5.06
mmol) dropwise within 10 min. The cloudy, yellow solution was allowed to warm to 25
°C and was stirred for 1 h. The solution of phenylthiomethyllithium and TMEDA was
added dropwise within 10 min. to a degassed solution of isoquinoline (0.30 mL, 2.20
mmol) in toluene (18 mL) at –78 °C under an atmosphere of argon. The resulting orange
solution was stirred for 5 min. at –78 °C. The solution was warmed to –20 °C and stirred
for two days. The brown solution was quenched with methyl chloroformate (0.510 mL,
6.60 mmol) and the color of the clear solution changed to pale orange. The solution
stirred for 15 min. and was then cooled to 0 °C, and diluted with sat. aq. NaHCO3 (40
mL). The reaction warmed to room temperature and the aqueous layer was extracted with
EtOAc (2 x 75 mL). The organic extracts were combined, dried (Na2SO4), filtered, and
concentrated under vacuum. The crude yellow oil was purified by flash column
chromatography (SiO2, 5-15% EtOAc in hexanes) to provide 157 (0.267 g, 39% yield) as
a colorless oil and 158 (0.228 g, 28% yield) as a white solid. 157: Rf = 0.37 (20% EtOAc
in hexanes); IR (thin film) 2947, 1716, 1635, 1456, 1439, 1354, 1330, 1232 cm-1; 1H

146
NMR (300 MHz, CDCl3) δ 7.36-7.04 (9 H, m), 6.91 (0.5 H, d, J = 7.7 Hz), 6.69 (0.5 H,
d, J = 7.7 Hz), 5.95 (0.5 H, d, J = 7.8 Hz), 5.85 (0.5 H, d, J = 7.8 Hz), 5.52 (0.5 H, t, J =
7.0 Hz), 5.32 (0.5 H, t, J = 7.0 Hz), 3.79 (2 H, s), 3.71 (1 H, s), 3.27-3.02 (2 H, m) ppm; 1H NMR (500 MHz, DMSO-d6, 120 °C) δ 7.33-7.16 (8 H, m), 7.12 (1 H, d, J = 7.7 Hz),
6.78 (1 H, d, J = 7.7 Hz), 5.97 (1 H, d, J = 7.8 Hz), 5.44 (1 H, t, J = 6.7 Hz), 3.72 (3 H, s),
3.18 (1 H, dd, J = 13.7, 6.8 Hz), 3.13 (1 H, dd, J = 13.7, 6.7 Hz) ppm; 13C NMR (500
MHz, DMSO-d6, 120 °C) δ 152.5, 135.3, 130.0, 129.4, 128.2, 128.1, 127.4, 126.2, 126.0,
125.3, 123.9, 123.8, 108.0, 53.7, 52.4, 36.9 ppm; HRMS calcd. for C18H18NO2S (MH+)
312.1058 Da. Found 312.1049 Da. HPLC conditions for separation of enantiomers using
analytical column Chiracel OD: loaded 15 μl of 0.5 mg/mL sample in 1%
isopropanol/hexanes, gradient 2%-5% over 15 min., enantiomers elute at 11.9 min. and
13.9 min. in a ratio of 1:1. 158: mp = 104-107 °C; Rf = 0.23 (20% EtOAc in hexanes); IR
(thin film) 2953, 1732, 1705, 1614, 1438, 1371, 1243, 1168 cm-1; 1H NMR (300 MHz,
CDCl3) δ 8.23 (1 H, brs), 8.07 (0.5 H, brs), 7.87 (0.5 H, brs), 7.33-7.10 (8 H, m), 5.48
(0.5 H, brs), 5.35 (0.5 H, brs), 3.84 (6 H, s), 3.15 (1 H, brs), 3.05 (1 H, dd, J = 13.7, 7.0
Hz) ppm; 1H NMR (500 MHz, DMSO-d6, 120 °C) δ 8.10 (1 H, d, J = 8.0 Hz), 7.82 (1 H,
s), 7.33- 7.18 (8 H, m), 5.47 (1 H, t, J = 6.5 Hz), 3.81 (3 H, s), 3.80 (3 H, s), 3.20 (2 H, d,
J = 6.5 Hz) ppm; 13C NMR (500 MHz, DMSO-d6, 120 °C) δ 164.5, 152.0, 134.9, 134.2,
129.9, 128.2, 127.4, 126.9, 126.5, 126.4, 125.5, 123.7, 108.9, 54.4, 53.3, 50.6, 37.3 ppm;
HRMS calcd. for C20H20NO4S (MH+) 370.1113 Da. Found 370.1115 Da.

147
Optimized Formation of 1-phenylsulfanylmethyl-1H-isoquinoline-2-carboxylic acid
methyl ester (157)
NCO2Me
PhS
To a stirred solution of thioanisole (60 μL, 0.51 mmol) and (−)-sparteine (117 μL, 0.51
mmol) in THF (1.0 mL) at 0 °C was added n-BuLi (1.45 M in hexanes, 350 μL, 0.51
mmol) dropwise within 10 min. The cloudy, yellow solution was allowed to warm to 25
°C and was stirred for 1 h. The solution of phenylthiomethyllithium and (−)-sparteine was
added dropwise within 10 min. to a degassed solution of isoquinoline (50 μL, 0.43 mmol)
in toluene (3.5 mL) at –78 °C under an atmosphere of argon. The resulting orange
solution was stirred for 5 min. at –78 °C. The solution was warmed to 25 °C and stirred
for 1.5 h. The orange/brown solution was quenched with methyl chloroformate (100 μL,
1.28 mmol) and the color of the clear solution changed to canary yellow. The solution
stirred for 15 min. and was then cooled to 0 °C, and diluted with 1.5 mL of sat. aq.
NaHCO3. The reaction warmed to room temperature and the aqueous layer was extracted
with toluene (3 x 5 mL). The organic extracts were combined, dried (Na2SO4), filtered,
and concentrated under vacuum. The crude yellow oil was purified by flash column
chromatography (SiO2, 3-7% EtOAc in hexanes) to provide 157 (121 mg, 91% yield) as a
colorless oil: characterized as above.

148
1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic acid methyl ester
(170)
NCO2Me
PhS
To a solution of 157 (44 mg, 0.14 mmol) in CH2Cl2 (0.20 mL) at 0 °C was added
triethylsilane (112 μL, 0.70 mmol), followed by dropwise addition within 10 min. of TFA
(55 μL, 0.72 mmol). The reaction was allowed to warm to 25 °C and stirred for 1 h. The
reaction was quenched with ice, diluted with sat. aq. NaHCO3 (5 mL) and extracted with
CH2Cl2 (3 x 10 mL). The combined extracts were washed with sat. aq. NaCl (5 mL),
dried (Na2SO4), filtered, and concentrated under vacuum. The crude oil was purified by
flash column chromatography (SiO2, 10% EtOAc in hexanes) to provide 170 (43 mg,
95% yield) as a colorless oil: Rf = 0.31 (20% EtOAc in hexanes). IR (thin film) 2949,
1700, 1446, 1331, 1223, 743 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.43-7.39 (2 H, m),
7.34-7.25 (2 H, m), 7.20-7.16 (5 H, m), 5.43 (0.45 H, t, J = 6.6 Hz), 5.25 (0.55 H, t, J =
6.6 Hz), 4.14-4.09 (0.55 H, m), 3.88-3.82 (0.45 H, m), 3.76 (1.4 H, s), 3.66 (1.6 H, s),
3.50-3.35 (1 H, m), 3.32-3.26 (2 H, m), 2.96-2.76 (2 H, m) ppm; 13C NMR (75 MHz,
CDCl3) δ 156.4, 156.2, 136.6, 136.3, 135.7, 134.6, 134.5, 129.7, 129.3, 129.0, 128.8,
127.9, 127.5, 127.4, 126.4, 126.2, 76.8, 54.5, 54.0, 52.9, 52.7, 40.4, 40.1, 39.3, 38.2, 28.7,
28.3 ppm; HRMS calcd. for C18H20NO2S (MH+) 314.1215 Da. Found 314.1215 Da.

149
3-methyl-1-phenylsulfanylmethyl-1H-isoquinoline-2-carboxylic acid methyl ester
(172)
N
PhS
CO2Me
Me
To a stirred solution of degassed thioanisole (60 μL, 0.51 mmol) and (−)-sparteine (117
μL, 0.51 mmol) in THF (1.0 mL) at 0 °C was added n-butyllithium (1.45 M in hexanes,
350 μL, 0.51 mmol) dropwise within 10 min. The cloudy, yellow solution was allowed to
warm to 25 °C and was stirred for 1 h. The solution of phenylthiomethyllithium and
(−)-sparteine was added dropwise within 10 min. to a degassed solution of 3-methyl
isoquinoline (30 mg, 0.21 mmol) in toluene (3.5 mL) at –78 °C under an atmosphere of
argon. The resulting orange solution was stirred for 5 min. at –78 °C, the reaction was
then warmed to 25 °C and was stirred for 1 h. The orange/brown solution was quenched
with methylchloroformate (100 μL, 1.28 mmol) and the color changed to pale yellow.
The solution was stirred for 15 min., cooled to 0°C, and added sat. aq. NaHCO3 (1.5 mL).
The reaction warmed to 25 °C and was extracted with toluene (3 x 5 mL). The organic
extracts were combined, dried (Na2SO4), filtered, and concentrated under vacuum. The
crude yellow oil was purified by flash column chromatography (SiO2, 3-7% EtOAc in
hexanes) to provide 172 (61 mg, 90% yield) as a colorless oil: Rf = 0.40 (25% EtOAc in
hexanes); IR (thin film) 2950, 1715, 1438, 1340, 1317, 1262, 739 cm-1; 1H NMR (300
MHz, C6D6) δ 7.33 (2 H, m), 7.05-6.99 (3 H, m), 6.92-6.77 (4 H, m), 5.71 (2 H, m), 3.30
(3 H, s), 3.12 (1 H, dd, J = 13.6, 8.9 Hz), 2.84 (1 H, dd, J = 13.6, 7.0 Hz), 2.19 (3 H, brs)
ppm; 13C NMR (75 MHz, CD6) δ 136.9, 132.7, 131.1, 129.4, 128.3, 127.1, 126.6, 126.4,

150
126.1, 125.9, 124.3, 113.9, 55.9, 52.3, 36.4, 21.5 ppm; HRMS calcd. for C19H20NO2S
(MH+) 326.1215 Da. Found 326.1209 Da.
3-methyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic acid
methyl ester (173)
NCO2Me
SPh
MeH
H
To a solution of 172 (61 mg, 0.19 mmol) in CH2Cl2 (0.35 mL) at 0 °C was added
triethylsilane (151 μL, 1.0 mmol), followed by dropwise addition within 10 min. of
trifluoroacetic acid (145 μL, 1.88 mmol). The reaction was stirred at 0 °C for 30 min. The
solution was warmed to 0 °C and was stirred 15 min. The reaction appeared complete by
TLC analysis and was quenched with ice, diluted with sat. aq. NaHCO3 (5 mL) until
basic and extracted with CH2Cl2 (3 x 10 mL). The combined extracts were washed with
brine (5 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude oil was
purified by flash column chromatography (SiO2, 3-7% EtOAc in hexanes) to afford 173
(52 mg, 87% yield) as a colorless oil: Rf = 0.27 (25% EtOAc in hexanes); IR (thin film)
2951, 1696, 1440, 1387, 1324, 1085, 739 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 7.38-
7.27 (4 H, m), 7.23-7.14 (5 H, m), 5.20 (1 H, br s), 3.98-3.93 (1 H, m), 3.50 (3 H, brs),
2.99 (1 H, dd, J = 16.0, 7.0 Hz), 2.85 (1 H, dd, J = 16.0, 9.9 Hz), 1.31 (3 H, d, J = 6.0
Hz); (300 MHz, DMSO-d6, 100 °C) 7.39-7.37 (2 H, m), 7.32-7.29 (2 H, m), 7.22-7.17 (5
H, m), 5.30 (1 H, dd, J = 7.5, 7.5 Hz), 4.11-4.04 (1 H, m), 3.59 (3 H, s), 3.36 (1 H, dd, J =

151
13.4, 7.5 Hz), 3.31 (1 H, dd, J = 13.4, 7.7 Hz), 3.03 (1 H, dd, J = 6.9, 16.0 Hz), 2.84 (1 H,
dd, J = 16.0, 9.2 Hz), 1.36 (3 H, d, J = 6.3 Hz) ppm; 13C NMR (75 MHz, C6D6) δ 156.5,
136.5, 136.3, 133.8, 129.4, 129.2, 129.1, 128.2, 127.9, 127.6, 127.5, 127.3, 126.5, 126.2,
55.7, 55.4, 52.73 48.1, 40.1, 40.0, 35.4, 23.5, 23.2, 23.0 ppm; HRMS calcd. for
C19H22NO2S (MH+) 328.1371 Da. Found 328.1369 Da.
tert-Butyl-[1-(2-iodo-phenyl)-meth-(E)-ylidene]-amine (179)
NtBu
I
To a solution of 2-iodobenzaldehyde2 (1.0 g, 4.31 mmol) in pentane (22 mL) was added
4Å molecular sieves and tert-butylamine (0.91 mL, 8.62 mmol). The reaction was stirred
at 25 °C for 12 h. The solution was filtered through Celite® and washed with benzene,
then concentrated under vacuum to afford 179 (1.12 g, 92% yield as a pale yellow oil: Rf
= 0.39 (10% EtOAc in hexanes); bp = 78 °C at 0.4 mmHg; IR (neat): 2967, 2928, 2903,
1633, 1464, 1434, 1370, 1271, 1221, 1202, 1012, 752 cm-1; 1H NMR (300 MHz, CDCl3):
δ 8.41 (1 H, s), 7.93 (1 H, d, J = 7.8 Hz), 7.84 (1 H, d, J = 7.9 Hz), 7.37 (1 H, t, J = 7.5
Hz), 7.08 (1 H, t, J = 7.8 Hz), 1.33 (9 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 159.2,
139.5, 138.0, 131.7, 128.8, 128.6, 100.47, 58.1, 29.9 ppm; HRMS calcd. for C11H15NI
(MH+) 288.0249 Da. Found 288.0247 Da.

152
Dibenzyl-isoquinolin-3-ylmethyl amine (178)
NNBn2
To a stirred solution of imine 179 (0.711 g, 2.48 mmol) and
1-(dibenzylamino)-2-propyne3 (187) (0.70 g, 2.97 mmol) in triethylamine (9.9 mL) was
added PdCl2(PPh3)2 (35 mg, 0.05 mmol) and CuI (5 mg, 0.03 mmol). The mixture was
heated to 55 °C and stirred for 2 h. The mixture was cooled to 25 °C, filtered through
Celite®, and the filtrate was concentrated under vacuum. The resulting residue was
dissolved in anhydrous DMF (25 mL), and CuI (50 mg, 0.30 mmol) was added. The
reaction mixture was flushed with argon, sealed, heated to 100 °C, and stirred for 14 h.
The solution was cooled to 25 °C, diluted with sat. aq. NH4Cl (50 mL), and extracted
with Et2O (2 x 50 mL). The combined extracts were dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting residue was purified by flash column
chromatography (SiO2, 3-20% EtOAc-hexane) to afford 178 (0.705 g, 84% yield) as a
yellow oil: Rf = 0.22 (25% EtOAc in hexanes); IR (thin film): 3059, 3027, 2797, 1629,
1588, 1494, 1454, 1366, 1128, 974, 887, 745 cm-1; 1H NMR (300 MHz, CDCl3): δ 9.22
(1 H, s), 7.96-7.94 (2 H, m), 7.85 (1 H, d, J = 8.2 Hz), 7.68 (1 H, t, J = 7.3 Hz), 7.55 (1 H,
t, J = 7.8 Hz), 7.52-7.48 (4 H, m), 7.38-7.33 (4 H, m), 7.23-7.28 (2 H, m), 3.97 (2 H, s),
3.74 (4 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 153.5, 152.0, 139.6, 136.6, 130.4,
128.9, 128.4, 127.9, 127.6, 127.1, 126.8, 126.7, 118.6, 59.7, 58.4 ppm; m/z (C.I.) 339
(100%, MH+).

153
[(Dibenzylamino)-methyl]-1-phenylsulfanylmethyl-1H-isoquinoline-2-carboxylic
acid methyl ester (188)
NNBn2
CO2Me
PhS
To a stirred solution of thioanisole (0.26 mL, 2.18 mmol) and (−)-sparteine (0.50 mL,
2.18 mmol) in anhydrous THF (2.6 mL) at 0 °C was added n-BuLi (2.10 M in hexanes,
1.0 mL, 2.18 mmol) dropwise within 10 min. The cloudy, yellow solution was allowed to
warm to 25 °C and was stirred for 1 h. The above solution of phenylthiomethyllithium
and (−)-sparteine was added dropwise within 10 min. to a degassed solution of 178
(0.489 g, 1.45 mmol) in toluene (12 mL) at –78 °C under an atmosphere of argon. The
resulting orange solution was allowed to warm to 25 °C over 15 min. The solution
became black in color and methylchloroformate (1.1 mL, 13.8 mmol) was added. The
pale brown and clear solution was cooled to 0 °C, treated with sat. aq. NaHCO3 (10 mL),
and then extracted with EtOAc (3 x 25 mL). The organic extracts were combined, dried
(Na2SO4), filtered, and concentrated under vacuum. The crude brown oil was purified by
flash column chromatography (SiO2, 5%-10% EtOAc in hexanes) to provide 188 (0.651
g, 86% yield) as a yellow oil: Rf = 0.22 (20% EtOAc in hexanes); IR (thin film): 3060,
3026, 2928, 2797, 1716, 1636, 1439, 1398, 1343, 1320, 1263, 1113, 1065, 1027 971 cm-
1; 1H NMR (300 MHz, CDCl3): δ 7.44-7.09 (19 H, m), 6.62 (1 H, s), 5.45-5.40 (1 H, m),
5.30 (2 H, s), 3.74-3.45 (9 H, m), 3.18 (1 H, dd, J = 13.3, 7.8 Hz), 2.98 (1 H, dd, J = 13.3,
7.1 Hz) ppm; 1H NMR (500 MHz, toluene-d8): δ 7.40-7.39 (4 H, m), 7.25-7.22 (3 H, m),
7.20-7.06 (4 H, m), 7.02-6.97 (4 H, m), 6.93-6.89 (3 H, m), 6.87-6.85 (1 H, m), 6.48 (1
H, s), 5.66 (1 H, brs), 3.89 (1 H, d, J = 15.4 Hz), 3.88 (2 H, d, J = 13.9 Hz), 3.54-3.47 (3

154
H, m), 3.21-3.18 (4 H, m), 2.84 (1 H, dd, J = 13.2, 7.1 Hz) ppm; 13C NMR (125 MHz,
toluene-d8): δ 154.1, 139.3, 136.9, 133.2, 130.7, 129.4, 129.1, 128.6, 128.0, 127.9, 127.7,
127.4, 127.0, 126.8, 126.5, 125.2, 125.0, 124.9, 124.6, 114.6, 58.1, 56.9, 56.2, 52.2, 37.0
ppm; 1H NMR (500 MHz, toluene-d8, 100 °C): δ 7.35-7.34 (4 H, m), 7.24-7.22 (2 H, m),
7.16-7.05 (4 H, m), 7.04-7.02 (1 H, m), 7.00-6.96 (4 H, m), 6.93-6.89 (4 H, m), 6.48 (1
H, s), 5.60 (1 H, t, J = 7.3 Hz), 3.86 (1 H, dd, J = 16.0, 1.1 Hz), 3.76 (2 H, d, J = 13.9
Hz), 3.63 (1 H, d, J = 16.0 Hz), 3.59 (2 H, d, J = 13.9 Hz), 3.29 (3 H, s), 3.18 (1 H, dd, J
= 13.2, 7.5 Hz), 2.91 (1 H, dd, J = 13.3, 7.0 Hz) ppm; m/z (C.I.) 521 (100%, MH+).
3-Chloromethyl-1-phenylsulfanylmethyl-1H-isoquinoline-2-carboxylic acid methyl
ester (194)
Dibenzyl-carbamic acid phenyl ester (195)
NCl
CO2Me
PhS
NCO2Ph
To a stirred solution of 188 (0.651 g, 1.25 mmol) in CH2Cl2 (5 mL) at 25 °C was added
phenyl chloroformate (1.57 mL, 12.5 mmol). The reaction was heated to reflux and
stirred for 11 h. The reaction was cooled to 25 °C, treated with sat. aq. NaHCO3 (10 mL)
and stirred vigorously for 15 min. The reaction mixture was extracted with CH2Cl2 (3 x
20 mL). The combined extracts were dried (Na2SO4), filtered, and concentrated under
vacuum. The resulting residue was purified by flash column chromatography (SiO2, 3%-
10% EtOAc in hexanes) to provide 188 (0.29 g, 45% yield) and an inseparable mixture of
chloride 194 and carbamate 195 as a pale yellow oil (0.495 g, 58% yield): (characterized

155
without separation), Rf = 0.40 (20% EtOAc in hexanes); IR (thin film): 3061, 2953, 1717,
1455, 1439, 1344, 1204, 1068 cm-1; 1H NMR (300 MHz, C6D6): δ 7.29-6.71 (ArH), 5.71
(1 H, s), 5.47 (1 H, brs), 4.41 (1 H, s), 4.28 (1 H, s), 3.84 and 3.80 (carbamate
methylenes), 3.30-3.22 (1 H, m), 3.21 (3 H, s), 2.89 (1 H, dd, J = 13.9, 7.3 Hz) ppm; 13C
NMR (75 MHz, C6D6): δ 154.8, 152.0, 137.5, 136.6, 133.3, 129.9, 129.6, 129.3, 129.1,
129.0, 128.7, 128.4, 127.9, 126.9, 126.6, 126.5, 126.3, 126.2, 126.1, 125.9, 125.4, 125.1,
121.9, 117.1, 115.1, 55.4, 52.6, 49.9, 49.5, 45.5, 36.4 ppm.
3-methyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic acid
methyl ester (173)
3-chloromethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic
acid methyl ester (198)
NCO2Me
SPh
MeH
H
NCO2Me
SPh
ClH
H
To a stirred solution of the inseparable mixture of chloride 194 and carbmate 195 (0.50 g)
in CH2Cl2 (0.50 mL) at 25 °C was added Et3SiH (1.5 mL, 9.1 mmol). The solution was
cooled to −15 °C and added trifluoroacetic acid (pre-cooled to −15 °C, 1.1 mL, 13.6
mmol). The solution was warmed to 25 °C and stirred 14 h. The solution was cooled to 0
°C and poured over ice. The aqueous solution was basified with sat. aq. NaHCO3 until
pH = 9, and extracted with CH2Cl2 (2 x 25 mL). The organic extracts were combined,
dried (Na2SO4), filtered, and concentrated under vacuum. The yellow oil was purified by

156
flash column chromatography (SiO2, 5-10% EtOAc in hexanes) to provide an inseparable
mixture of 173 and chloride 198 (0.25 g, 75% yield) as a colorless oil: Rf = 20 (20%
EtOAc in hexanes); recrystallized from toluene as 6:1 ratio of 198 to 173; X-ray (see
Appendix A); mp = 75-80 °C; 198: IR (thin film): 2953, 2925, 1698, 1441, 1387, 1324
cm-1; 1H NMR (300 MHz, CDCl3): δ 7.41-7.15 (9 H, m), 5.52 (0.4 H, brs), 5.25 (0.6 H,
brs), 4.28 (1.2 H, brs), 4.06 (0.8 H, brs), 3.85-3.65 (4 H, m), 3.42-3.36 (1 H, m), 3.25-
3.01 (3 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 153.2, 136.5, 133.8, 132.9, 129.4,
129.1, 128.3, 128.2, 127.5, 126.7, 126.5, 126.2, 121.9, 55.5, 53.8, 52.7, 48.1, 47.0, 40.1,
35.4, 30.7 ppm; HRMS calcd. for C19H21NO2SCl (MH+) 362.0982 Da. Found 362.0969
Da.
Triisopropyl-prop-2-ynyloxy-silane (200)
OTIPS
To a stirred solution of propargyl alcohol (2.08 mL, 35.7 mmol) and imidazole (2.67 g,
39.3 mmol) in DMF (16 mL) at 0 °C was added TIPS-Cl (8.83 mL, 41.0 mmol). The
reaction was warmed to 25 °C and stirred for 16 h. The reaction mixture was partitioned
between hexanes (125 mL) and sat. aq. NaHCO3 (75 mL). The organic layer was washed
with sat. aq. NaHCO3 (50 mL), sat. aq. NaCl (50 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting colorless liquid was purified by flash column
chromatography (SiO2, 2-4% EtOAc-hexane) then by distillation (110 °C, 20 mmHg) to
afford 200 (3.8 g, 54% yield) as a colorless liquid: Rf = 0.35 (2% EtOAc in hexanes); IR
(neat): 3313, 2944, 2867, 1464, 1370, 1263, 1101, 1001, 883, 774 cm-1; 1H NMR (300

157
MHz, CDCl3): δ 4.38 (2 H, d, J = 2.2 Hz), 2.39 (1 H, t, J = 2.3 Hz), 1.16-1.05 (21 H, m)
ppm; 13C NMR (75 MHz, CDCl3): δ 82.2, 72.4, 51.5, 17.6, 11.7 ppm; HRMS calcd. for
C12H23OSi (M-H+) 211.1518 Da. Found 211.1515 Da.
3-Triisopropylsilanyloxymethyl-isoquinoline (201)
NOTIPS
To a stirred solution of imine 179 (1.10 g, 3.83 mmol) and alkyne 200 (0.98 g, 4.60
mmol) in THF (15.3 mL) was added PdCl2(PPh3)2 (53 mg, 0.08 mmol) and CuI (7 mg,
0.04 mmol). The reaction was flushed with argon, sealed, heated to 55 °C and stirred for
1 h. The reaction was cooled to 25 °C, filtered through Celite®, and the filtrate was
concentrated under vacuum. The resulting residue was dissolved in anhydrous DMF (38
mL), and added CuI (73 mg, 0.10 mmol). The reaction mixture was flushed with argon,
sealed, heated to 100 °C, and stirred for 4 h. The reaction was cooled to 25 °C, diluted
with Et2O (120 mL), washed with sat. aq. NH4Cl (130 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting residue was purified by flash column
chromatography (SiO2, 5-10% EtOAc-hexane) to afford isoquinoline 201 (1.13 g, 94%
yield) as a yellow oil: Rf = 0.36 (20% EtOAc in hexanes); IR (thin film): 2954, 2911,
2875, 1630, 1595, 1457, 1373, 1128, 1096, 1015, 879, 816 cm-1; 1H NMR (300 MHz,
CDCl3): δ 9.15 (1 H, s), 7.94 (1 H, d, J = 7.9 Hz), 7.86 (1 H, s), 7.83 (1 H, d, J = 8.3 Hz),
7.66 (1 H, t, J = 8.1 Hz), 7.54 (1 H, t, J = 7.9 Hz) 5.09 (2 H, s), 1.29-1.20 (3 H, m), 1.12
(18 H, d, J = 6.4 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 136.7, 130.5, 127.9, 127.7,

158
126.8, 126.7, 154.8, 151.9, 115.8, 65.8, 17.7, 12.0 ppm; HRMS calcd. for C19H30NOSi
(MH+) 316.2097 Da. Found 316.2105 Da.
1-Phenylsulfanylmethyl-3-triisopropylsilanyloxymethyl-1H-isoquinoline-2-
carboxylic acid methyl ester (204)
NOTIPS
PhS
CO2Me
To a stirred solution of thioanisole (0.178 mL, 1.52 mmol) and (−)-sparteine (0.340 mL,
1.52 mmol) in anhydrous THF (1.9 mL) at 0 °C was added n-BuLi (2.40 M in hexanes,
0.633 mL, 1.52 mmol) dropwise within 10 min. The cloudy, yellow solution was allowed
to warm to 25 °C and was stirred for 1 h. The above solution of phenylthiomethyllithium
and (−)-sparteine was added dropwise within 10 min. to a degassed solution of
isoquinoline 201 (0.319 g, 1.01 mmol) in toluene (8.2 mL) at −78 °C under an
atmosphere of argon. The resulting orange solution was allowed to warm to 25 °C over
15 min. The solution was stirred for 30 min. and quenched with methylchloroformate
(0.235 mL, 3.03 mmol). The pale yellow solution was cooled to 0 °C, treated with 10 mL
of sat. aq. NaHCO3, and then extracted with EtOAc (3 x 25 mL). The organic extracts
were combined, dried (Na2SO4), filtered, and the solvent was removed under vacuum.
The crude brown oil was purified by flash column chromatography (SiO2, 1-5% EtOAc
in hexanes) to provide 204 (0.501 g, 95% yield) as a pale yellow oil: Rf = 0.35 (20%
EtOAc in hexanes); IR (thin film): 2943, 2865, 1717, 1637, 1456, 1439, 1401, 1321,
1264, 1129, 1109, 1049, 883 cm-1; 1H NMR (300 MHz, C6D6): δ 7.25 (2 H, brs), 7.09-

159
6.84 (6 H, m), 6.78 (1 H, d, J = 7.1 Hz), 6.56 (1 H, brs), 5.60 (1 H, brs), 5.20-4.80 (2 H,
brs), 3.25 (3 H, s), 3.22-3.17 (1 H, m), 2.89 (1 H, dd, J = 13.7, 6.4 Hz), 1.09 (9 H, s) ppm; 13C NMR (75 MHz, C6D6): δ 154.1, 138.8, 136.8, 133.2, 130.8, 129.1, 128.9, 128.5,
128.3, 127.1, 126.8, 126.3, 125.8, 125.2, 112.0, 63.8, 55.8, 52.3, 36.6, 18.1, 12.2 ppm;
HRMS calcd. for C28H40NO3SSi (MH+) 498.2498 Da. Found 498.2514 Da.
Carbonic acid methyl ester 1-phenylsulfanylmethyl-isoquinolin-3-ylmethyl ester
(207)
3-Hydroxymethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic
acid methyl ester (206)
NOCO2Me
PhS
N
PhS
CO2Me
OHH
H
To a stirred solution of 204 (0.508 g, 1.03 mmol) and triethylsilane (0.818 mL, 5.13
mmol) in CH2Cl2 (1.0 mL) at −10 °C was added TFA (pre-cooled to 0 °C, 0.70 mL, 9.0
mmol). TLC analysis showed complete deprotection of silylether but no reduction after
15 min. at −10 °C. The reaction was warmed to 25 °C, stirred for 1 h, and quenched with
sat. aq. NaHCO3 (30 mL). The solution was extracted with CH2Cl2 (3 x 25 mL). The
combined extracts were dried (Na2SO4), filtered, and concentrated under vacuum. The
resulting residue was purified by flash column chromatography (SiO2, 10-70% EtOAc in
hexanes) to provide 207 (0.103 g, 29% yield) as a yellow oil and 206 (0.187 g, 53%
yield) as a colorless oil. 207: Rf = 0.24 (20% EtOAc in hexanes); IR (thin film): 2954,

160
1750, 1699, 1439, 1287, 1268, 741 cm-1; 1H NMR (300 MHz, CDCl3): δ 8.16 (1 H, d, J =
8.0 Hz), 7.81 (1 H, d, J = 8.1 Hz), 7.67 (1 H, t, J = 8.0 Hz), 7.62 (1 H, s), 7.58 (1 H, t, J =
7.9), 7.41-7.37 (2 H, m), 7.26-7.17 (3 H, m), 5.34 (2 H, s), 4.73 (2 H, s), 3.83 (3 H, s)
ppm; m/z (C.I.) 349 (100%, MH+). 206: Rf = 0.19 (50% EtOAc in hexanes); IR (thin
film) 3446, 2923, 2850, 1684, 1445, 1393, 1329, 1264, 1073, 1026, 739, 690 cm-1; 1H
NMR (300 MHz, CDCl3) δ 7.41 (2 H, d, J = 10.2 Hz), 7.37-7.20 (7 H, m), 5.62 (0.4 H,
brs), 5.34 (0.6 H, brs), 4.21-4.05 (2 H, m), 3.66 (4 H, brs), 3.43-3.02 (4 H, m), 2.98-2.90
(1 H, m) ppm; 13C NMR (75 MHz, CDCl3) δ 157.2, 136.1, 135.1, 133.6, 129.3, 128.8,
128.1, 127.8, 126.4, 126.3, 95.1, 65.7, 55.1, 52.8, 40.4, 29.2 ppm; HRMS calcd. for
C19H22NO3S (MH+) 344.1320 Da. Found 344.1317 Da.
3-Benzyloxymethylisoquinoline (203)
NOBn
To a solution of 179 (3.0 g, 10.45 mmol) and propargyl benzyl ether4 (1.83 g, 12.54
mmol) in triethylamine (42 mL) was added PdCl2(Ph3)2 (0.147 g, 2 mol %) followed by
CuI (20 mg, 1 mol %) under an atmosphere of argon at 25 °C. The mixture was warmed
to 55 °C and stirred for 6 hours. The mixture was allowed to cool to 25 °C and filtered
through Celite® to remove the yellow precipitate which was washed with triethylamine.
The filtrate was concentrated under reduced pressure, the resulting residue was dissolved
in DMF (105 mL), and CuI (0.200 g, 10 mol %) was added under an atmosphere of argon
at 25 °C. The mixture was heated to 100 °C and stirred for 9 h. The dark brown solution
was allowed to cool to 25 °C and diluted with Et2O (500 mL), then washed with sat. aq.

161
NH4Cl (400 mL), dried (Na2SO4), and filtered. The solvent was removed under reduced
pressure to give a black oil, which was purified by flash column chromatography (SiO2,
20% to 50% EtOAc in hexanes) to afford 203 as a pale yellow oil (2.2 g, 84% yield) Rf =
0.60 (100% EtOAc); IR (thin film) 3059, 3030, 2857, 1630, 1592, 1496, 1454, 1359,
1127, 1098, 747 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.24 (1 H, s), 7.95 (1 H, d, J = 8.1),
7.70-7.65 (2 H, m), 7.60-7.55 (2 H, m), 7.47-7.30 (5 H, m), 4.88 (2 H, s), 4.75 (2 H, s)
ppm; 13C NMR (75 MHz, CDCl3) δ 152.3, 151.9, 138.3, 136.5, 130.6, 128.6, 128.0,
127.9, 127.7, 127.1, 126.8, 117.6, 73.2, 73.1 ppm; HRMS calcd. for C17H16NO (MH+)
250.1232 Da. Found 250.1239 Da.
3-Benzyloxymethyl-1-phenylsulfanylmethyl-1H-isoquinoline-2-carboxylic acid
methyl ester (205)
N
PhS
CO2Me
OBn
To a stirred, degassed solution thioanisole (0.756 mL, 6.46 mmol) and (−)-sparteine (1.48
mL, 6.46 mmol) in anhydrous THF (8.05 mL) at 0 °C was added n-BuLi (1.95 M in
hexanes, 3.25 mL, 6.46 mmol) dropwise within 10 min. The cloudy, yellow solution was
allowed to warm to 25 °C and was stirred for 1 h. The solution of
phenylthiomethyllithium and (−)-sparteine was added dropwise within 10 min. to a
degassed solution of 203 (1.16 g, 4.61 mmol) in toluene (37 mL) at –78 °C under an
atmosphere of argon. The resulting orange solution was allowed to warm to 25 °C over
15 min. The solution became black in color, stirred 30 min. and quenched with

162
methylchloroformate (1.1 mL, 13.8 mmol). Upon addition the solution became pale
orange. The solution was cooled to 0 °C, treated with 30 mL of sat. aq. NaHCO3, and
then extracted with EtOAc (2 x 75 mL). The organic extracts were combined, washed
with sat. aq. NaCl (30 mL), dried (Na2SO4), filtered, and concentrated under vacuum.
The crude yellow oil was purified by flash column chromatography (SiO2, 10%-20%
EtOAc in hexanes) to provide 205 (1.50 g, 75% yield) as a colorless oil: Rf = 0.23 (20%
EtOAc in hexanes); IR (thin film) 3028, 2928, 2854, 1716, 1638, 1481, 1454, 1439,
1401, 1321, 1264, 1113, 738, 696 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.41-7.11 (14 H,
m), 6.44 (1 H, s), 5.49 (1 H, brs), 4.60 (2 H, s), 4.51 (2 H, brs), 3.70 (3 H, s), 3.25 (1 H,
dd, J = 13.4, 8.1 Hz), 3.04 (1 H, dd, J = 13.7, 6.7 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ
154.0, 138.0, 135.9, 134.7, 132.7, 129.9, 129.0, 128.9, 128.8, 128.2, 128.0, 127.5, 127.0,
126.1, 126.0, 125.0, 113.8, 72.7, 72.3, 69.9, 55.5, 52.9, 36.4 ppm; HRMS calcd. for
C26H26NO3S (MH+) 432.1633 Da. Found 432.1619 Da. HPLC conditions for separation
of enantiomers using ananlytical column Chiracel OD: loaded 15 μL of 0.5 mg/mL
sample in 1% isopropanol/hexanes, gradient 2%-5% over 15 min., enantiomers elute at
11.7 min. and 13.8 min. in a ratio of 1:1.

163
3-Benzyloxymethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-
carboxylic acid methyl ester (212)
N
PhS
CO2Me
OBnH
H
To a solution of 205 (1.50 g, 3.48 mmol) and triethylsilane (5.60 mL, 34.8 mmol) in
CH2Cl2 (35.0 mL) at –20 °C was added TFA (pre-cooled to –10 °C, 2.70 mL, 34.8 mmol)
in one portion. The solution was allowed to warm to 25 °C and was stirred for 3 h at
which point starting material could not be detected by TLC. The solution was quenched
by pouring over ice, basified with 10% aq. NaOH, and extracted with CH2Cl2 (2 x 50
mL). The combined extracts were washed with sat. aq. NaCl (20 mL), dried (Na2SO4),
filtered, and the solvent was removed under vacuum. The crude oil was purified by flash
column chromatography (SiO2, 10%-20% EtOAc in hexanes) to afford 212 (1.47 g, 97%
yield) as a colorless oil: Rf = 0.18 (20% EtOAc in hexanes); IR (thin film) 3028, 2951,
2855, 1697, 1442, 1390, 1325, 1105, 1026, 738, 697 cm-1; 1H NMR (300 MHz, CDCl3) δ
7.55-7.25 (10 H, m), 7.22-7.18 (4 H, m), 5.55 (0.4 H, brs), 5.25 (0.6 H, brs), 4.57 (2 H, s),
4.38-4.18 (1 H, m), 3.86-3.62 (5 H, m), 3.37 (1 H, m), 3.22-3.13 (2 H, m), 3.01 (1 H, dd,
J = 16.2, 7.2 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 156.8, 156.0, 138.1, 136.6, 133.4,
128.7, 128.2, 127.5, 126.9, 126.0, 125.7, 73.2, 73.1, 73.0, 72.3, 71.1, 55.1, 54.7, 52.6,
52.4, 51.8, 51.5, 39.3, 39.0, 30.2, 29.6 ppm; HRMS calcd. for C26H28NO3S (MH+)
434.1790 Da. Found 434.1790 Da.

164
12-Phenylsulfanyl-11-oxa-13-aza-tricyclo[7.3.1.02,7]trideca-2,4,6-triene-13-
carboxylic acid methyl ester (213)
O
SPh
N
H
H CO2Me
To a solution of 212 (51 mg, 0.117 mmol) in chlorobenzene (0.5 mL) under an
atmosphere of argon at 0 °C was added N-chlorosuccinimide (16 mg, 0.117 mmol). The
mixture was allowed to warm to 25 °C and was stirred for 2 h, then filtered through a pad
of glass wool into a dry flask under argon and washed with chlorobenzene (0.5 mL). The
solution was cooled to 0 °C and SnCl4 (3.0 M in CH2Cl2, 8 μL, 20 mol%) was added
dropwise. The resulting yellow solution was stirred for 15 min. at 0 °C, quenched with
sat. aq. NaHCO3 (5 mL), and extracted with EtOAc (2 x 10 mL). The combined extracts
were dried (Na2SO4), filtered, and concentrated under vacuum. The crude residue was
purified by flash column chromatography (SiO2, 5%-10% EtOAc in hexanes) to afford
213 (30 mg, 76% yield) as a colorless oil: crystallized from methanol to yield a white
solid; X-ray (see Appendix B); mp = 131-133 °C; Rf = 0.19 (25% EtOAc in hexanes); IR
(thin film) 2953, 1701, 1449, 1409, 1339, 1320, 1310, 1117, 1104, 1065, 742, 692 cm-1; 1H NMR (500 MHz, toluene-d8, 100 °C) δ 8.61 (2 H, d, J = 7.2 Hz), 7.01-6.91 (4 H, m),
6.86 (1 H, t, J = 7.5 Hz), 6.82 (1 H, d, J = 7.6 Hz), 6.70 (1 H, d, J = 7.5 Hz), 5.32 (1 H,
brs), 5.23 (1 H, s), 4.56 (1 H, dd, J = 11.4, 3.1 Hz), 4.25 (1 H, brs), 3.49 (3 H, s), 3.28 (1
H, d, J = 11.4 Hz), 3.05 (1 H, dd, J = 17.3, 7.5 Hz), 2.44 (1 H, d, J = 17.3 Hz) ppm; 13C
NMR (125 MHz, toluene-d8, 100 °C) δ 155.5, 137.8, 136.4, 136.3, 131.7, 129.2, 128.9,

165
128.1, 128.0, 127.1, 126.7, 125.6, 125.4, 88.9, 65.7, 57.5, 52.5, 47.9, 32.0 ppm; HRMS
calcd. for C19H20NO3S (MH+) 342.1164 Da. Found 342.1159 Da.
Improved route to 3-Hydroxymethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-
isoquinoline-2-carboxylic acid methyl ester (206)
N
PhS
CO2Me
OHH
H
To a stirred solution of 212 (1.05 g, 2.42 mmol) in CH2Cl2 (24.0 mL) at –78 °C was
added BCl3 (1.0 M in CH2Cl2, 4.84 mL, 4.84 mmol) dropwise. Upon complete addition
the solution was allowed to warm to 0 °C and was stirred for 30 min., then treated with
methanol (3 mL), diluted with sat. aq. NaHCO3 (20 mL), and extracted with CH2Cl2 (2 x
50 mL). The organic extracts were combined, dried (Na2SO4), filtered, and the solvent
was removed under vacuum. The crude residue was purified by flash column
chromatography (SiO2, 20%-40% EtOAc in hexanes) to afford 206 (0.806 g, 95% yield)
as a colorless oil: characterized as above.

166
1-Phenylsulfanylmethyl-3-(toluene-4-sulfonyloxymethyl)-3,4-dihydro-1H-
isoquinoline-2-carboxylic acid methyl ester (222)
N
PhS
CO2Me
OTsH
H
To a stirred solution of alcohol 206 (0.215 g, 0.627 mmol) in pyridine (0.50 mL) and
CH2Cl2 (0.50 mL) at 0 °C was added p-toluenesulfonyl chloride (0.144 g, 0.752 mmol).
The reaction was warmed to 25 °C and stirred for 4 h. The solution was diluted with
EtOAc (25 mL) and washed with H2O (15 mL), 10% aq. CuSO4 (4 x 15 mL), H2O (15
mL), and sat. aq. NaCl (15 mL). The organic layer was dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting residue was purified by flash column
chromatography (SiO2, 10-50% EtOAc in hexanes) to provide 222 (0.286 g, 86% yield)
as a colorless oil: Rf = 0.36 (40% EtOAc in hexanes); IR (thin film): 3030, 2953, 1698,
1443, 1390, 1363, 1328, 1189, 1177, 1096, 919 cm-1; 1H NMR (300 MHz, CDCl3): δ
7.83 (2 H, d, J = 7.9 Hz), 7.38-7.14 (11 H, m), 5.47 (0.5 H, brs), 5.18 (0.5 H, brs), 4.54
(0.5 H, brs), 4.24-4.10 (2.5 H, m), 3.57 (3 H, s), 3.28 (1 H, dd, J = 13.4, 6.7 Hz), 3.14-
2.95 (3 H, m), 2.45 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 156.1, 145.1, 136.2,
136.0, 132.9, 132.7, 130.1, 129.2, 129.1, 128.2, 127.5, 126.7, 126.5, 126.2, 71.1, 70.1,
60.5, 55.5, 52.9, 51.9, 51.1, 39.3, 30.0, 29.4, 21.8, 14.4 ppm; HRMS calcd. for
C26H28NO5S2 (MH+) 498.1409 Da. Found 498.1418 Da.

167
3-Azidomethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic
acid methyl ester (223)
Phenylsulfanylmethyl-1,5,10,10a-tetrahydro-oxazolo[3,4-b]isoquinolin-3-one (224)
N
PhS
CO2Me
N3H
H
N
PhS
H
H
O
O
To a stirred solution of 222 (0.131 g, 0.262 mmol) in DMF (0.5 mL) at 25 °C was added
NaN3 (51 mg, 0.789 mmol). The reaction was heated to 55 °C for 12 h and then cooled to
25 °C, diluted with sat. aq NaHCO3 (5 mL) and extracted with EtOAc (3 x 15 mL). The
combined extracts were washed with sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting residue was purified by flash column
chromatography (SiO2, 5-40% EtOAc in hexanes) to afford azide 223 as a colorless oil
63 mg, 65% yield) and oxazolidinone 224 (25 mg, 31% yield) as a white solid. 223: Rf =
0.18 (20% EtOAc in hexanes); IR (thin film) 2952, 2103, 1698, 1481, 1441, 1388, 1326,
1289, 1248, 739, 690 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.39 (2 H, d, J = 7.6 Hz), 7.37-
7.17 (7 H, m), 5.53 (0.4 H, brs), 5.24 (0.6 H, brs), 4.21-3.95 (2 H, m), 3.85-3.56 (4 H, m),
3.41 (1 H, dd, J = 13.4, 6.8 Hz), 3.20 (1 H, dd, J = 13.4, 8.0 Hz), 3.15-2.91 (2 H, m) ppm; 13C NMR (75 MHz, CDCl3) δ 156.6, 136.3, 133.0, 129.4, 129.1, 128.2, 127.9, 127.5,
126.7, 126.5, 126.3, 55.6, 54.1, 53.9, 53.3, 53.2, 53.0, 52.7, 48.1, 39.7, 30.6 ppm; HRMS
calcd. For C19H21N4O2S (MH+) 369.1385 Da. Found 369.1380 Da. 224: Rf = 0.17 (30%
EtOAc in hexanes); mp = 130-132 °C; IR (thin film): 2901, 1748, 1404, 1346, 1280,
1227, 1063, 1025, 743 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.28-7.09 (9 H, m), 5.10 (1
H, dd, J = 4.9, 2.4 Hz), 4.44 (1 H, t, J = 7.6 Hz), 4.08 (1 H, dd, J = 13.6, 4.9 Hz), 3.97-

168
3.90 (1 H, m), 3.76 (1 H, dd, J = 11.2, 8.0 Hz), 3.44 (1 H, dd, J = 13.9, 2.3 Hz), 3.10 (1
H, dd, J = 14.3, 11.3 Hz), 2.90 (1 H, dd, J = 14.4, 3.0 Hz) ppm; 13C NMR (75 MHz,
CDCl3): δ 157.0, 136.3, 134.6, 133.9, 129.2, 128.9, 127.8, 127.4, 126.1, 68.7, 54.6, 54.3,
39.7, 34.1 ppm; HRMS calcd. for C18H18NO2S (MH+) 312.1058 Da. Found 312.1065 Da.
3-Azidomethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic
acid methyl ester (223)
N
PhS
CO2Me
N3H
H
To a solution of 206 (0.790 g, 2.30 mmol) in THF (7.7 mL) at 0 °C was added
triphenylphosphine (1.33 g, 5.06 mmol) and diethyl azodicarboxylate (0.797 mL, 5.06
mmol). After 2 min. a white precipitate formed and diphenylphosphoryl azide (1.09 mL,
5.06 mmol) was added. The mixture was allowed to warm to 25 °C and was stirred for 2
h, then concentrated under reduced pressure. The residue was purified by flash column
chromatography (SiO2, 5% to 7% to 10% EtOAc in hexanes) to afford azide 223 (0.694
g, 82% yield) as a colorless oil: characterized as above.

169
12-Phenylsulfanyl-11,13-diaza-tricyclo[7.3.1.02,7]trideca-2,4,6,11-tetraene-13-
carboxylic acid methyl ester (226)
N
H
H CO2Me
N
SPh
To a solution of 223 (45 mg, 0.122 mmol) in chlorobenzene (0.61 mL) under an
atmosphere of argon at 0 °C was added N-chlorosuccinimide (16 mg, 0.122 mmol). The
mixture was allowed to warm to 25 °C and was stirred for 2 h, then filtered through a pad
of glass wool into a dry flask under argon and washed with chlorobenzene (0.6 mL). The
solution was cooled to 0 °C and SnCl4 (3.0 M in CH2Cl2, 41 μL, 0.122 mmol) was added
dropwise. The solution immediately became yellow, a precipitate formed, and the
evolution of gas was observed. Upon complete addition no starting material could be
detected by TLC. The mixture was diluted with sat. aq. NaHCO3 until basic (5 mL) and
extracted with CH2Cl2 (2 x 10 mL). The combined extracts were dried (Na2SO4), filtered,
and the solvent was removed under vacuum. The crude residue was purified by flash
column chromatography (SiO2, 10%-40% EtOAc in hexanes) to afford 226 as a yellow
oil (22 mg, 49% yield). Rf = 0.16 (30% EtOAc in hexanes); IR (thin film) 2922, 1705,
1637, 1449, 1320, 1121, 741, 688 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.42-7.30 (5 H,
m), 7.25-7.15 (4 H, m), 5.49 (0.4 H, brs), 5.33 (0.6 H, brs), 4.79 (0.4 H, m), 4.67 (0.6 H,
m), 4.24-4.12 (1 H, m), 3.72 (3 H, s), 3.67 (1 H, d, J = 18.1 Hz), 3.41-3.33 (1 H, m), 2.71
(1 H, d, J = 17.5 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 145.6, 134.8, 133.3, 129.5,
129.3, 129.2, 128.9, 127.8, 127.4, 126.1, 105.0, 56.6, 54.6, 53.1, 43.8, 33.9 ppm; HRMS
calcd. for C19H19N2O2S (MH+) 339.1167 Da. Found 339.1163 Da.

170
12-Oxo-11,13-diaza-tricyclo[7.3.1.02,7]trideca-2,4,6-triene-13-carboxylic acid methyl
ester (229)
N
H
H CO2Me
NH
O
To a solution of 226 (22 mg, 0.060 mmol) in Et2O (1.0 mL) was added 5% aq. sodium
hydrogen sulfate (2.0 mL) with vigorous stirring. After 5 min. the layers were separated
and the aqueous layer was extracted with Et2O (2 x 3.0 mL). The organic extracts were
combined, washed with sat. aq. NaCl (1.0 mL), dried (Na2SO4), filtered, and the solvent
was removed under vacuum. The crude oil was purified by flash column chromatography
(SiO2, 100% EtOAc) to afford 229 (15 mg, >95% yield) as a colorless oil: crystallized
from EtOAc to give a white solid; X-ray (appendix C); mp = 211-213 °C; Rf = 0.17
(100% EtOAc); IR (thin film) 3266, 2955, 1701, 1685, 1489, 1452, 1411, 1323, 1251,
1119, 746 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.39 (1 H, d, J = 6.0 Hz), 7.25-7.12 (3 H,
m), 6.16-5.99 (1 H, m), 5.53-5.41 (1 H, m), 5.02-4.85 (1 H, m), 3.91-3.85 (1 H, m), 3.73
(3 H, s), 3.47 (1 H, dd, J = 17.6, 8.0 Hz), 3.23 (1 H, dd, J = 11.9, 2.6 Hz), 2.81 (1 H, d, J
= 17.7 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 169.0, 154.8, 132.2, 128.7, 128.0, 126.9,
126.4, 56.9, 52.9, 47.1, 42.4, 32.6 ppm; HRMS calcd. for C13H15N2O3 (MH+) 247.1083
Da. Found 247.1082 Da.

171
2-Benzyloxy-3,5-dimethoxy-4-methyl benzaldehyde (233)
OMe
Me
MeOOBn
CHO
To a stirred solution of phenol 2315 (1.12 g, 6.67 mmol) in acetic acid (33 mL) was added
hexamethylenetetramine (5.61 g, 40.0 mmol) in one portion. The solution was heated to
100 °C and stirred 2 h. The brown/red solution was cooled to 25 °C and poured into H2O
(75 mL). The aqeous solution was extracted with CH2Cl2 (2 x 75 mL). The organic
extracts were combined, washed with 5% aq. Na2CO3 (2 x 75 mL), sat. aq. NaCl (75
mL), dried (Na2SO4), filtered, and concentrated under vacuum. The brown oil was
purified by flash column chromatography (SiO2, 15% to 25% EtOAc in hexanes) to
afford the aldehyde (0.676 g, 52% yield) as an impure yellow solid. The aldehyde (3.45
mmol) was then dissolved in DMF (35 mL) and to the stirred solution was added
tetrabutylammonium iodide (25 mg, 0.070 mmol), Na2CO3 (0.731 g, 6.90 mmol), and
benzyl bromide (0.492 mL, 4.14 mmol). The yellow solution was stirred 14 h at 25 °C.
The reaction was diluted with sat. aq. NH4Cl (60 mL) and extracted with Et2O (2 x 100
mL). The combined extracts were washed with sat. aq. NaHCO3 (75 mL), sat. aq. NaCl
(75 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The residue was
purified by flash column chromatography (SiO2, 10% EtOAc in hexanes) to afford
benzaldehyde 233 (0.910 g, 48% yield over the two steps) as a colorless oil: Rf = 0.36
(20% EtOAc in hexanes); IR (thin film) 2939, 2859, 1683, 1598, 1464, 1412, 1372, 1332,
1283, 1205, 1133, 1081, 1023 cm-1; 1H NMR (300 MHz, CDCl3) δ 10.15 (1 H, s), 7.38 (5
H,s), 7.01 (1 H, s), 5.12 (2 H, s), 3.90 (3 H, s), 3.84 (3 H, s), 2.24 (3 H, s) ppm; 13C NMR

172
(75 MHz, CDCl3) δ 189.3, 154.4, 152.0, 149.3, 136.1, 129.5, 128.5, 128.4, 128.3, 127.4,
101.8, 76.7, 60.5, 55.6, 9.5 ppm; HRMS calcd. for C17H19O4 (MH+) 287.1283 Da. Found
287.1281 Da.
(2-Benzyloxy-3,5-dimethoxy-4-methyl-phenyl)-methanol (236)
OMe
MeOOBn
Me
OH
To a stirred solution of phenol 2315 (7.31g, 43.5 mmol) and paraformaldehyde (1.96 g) in
CH2Cl2 (89 mL) at 0 °C under an atmosphere of argon was added neat Et2AlCl (6.27 mL,
50.0 mmol) dropwise within 15 min. The solution was stirred for 30 min. at 0 °C and 25
°C for 21 h. The solution was quenched with aq. 2 M HCl (120 mL) and quickly
extracted with EtOAc (2 x 200 mL). The organic extracts were combined, washed with
sat. aq. NaCl (100 mL), dried (Na2SO4), filtered through Celite® to remove trace
precipitate, and concentrated under vacuum to afford 235 as an acid sensitive yellow
solid (8.6 g, 99% yield) which was shown to be free of impurities by spectrographic
analysis and used immediately without further purification.
To a solution of 235 (8.6 g, 43.4 mmol) in acetone (217 mL) at 25 °C was added
anhydrous K2CO3 (25.0 g, 178 mmol) and benzyl bromide (25.8 mL, 217 mmol). The
mixture was heated at reflux for 4 h and then cooled to 25 °C, diluted with saturate
aqueous NH4Cl (400 mL), and extracted with EtOAc (2 x 400 mL). The combined
extracts were dried (Na2SO4), filtered, and concentrated under vacuum. The resulting

173
brown oil was purified by flash column chromatography (SiO2, 20% to 50% EtOAc in
hexanes) to afford 236 (11.2 g, 89% yield) as a burnt orange oil: Rf = 0.31 (40% EtOAc
in hexanes); IR (thin film) 3416, 2936, 2873, 2837, 1606, 1586, 1484, 1455, 1410, 1375,
1328, 1223, 1129, 1089, 1026, 845, 755, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.46-
7.35 (5 H, m), 6.63 (1 H, s), 5.01 (2 H, s), 4.55 (2 H, brs), 3.87 (3 H, s), 3.79 (3 H, s),
2.60 (1 H, brs), 2.20 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3) δ 154.2, 151.6, 143.0,
137.3, 131.8, 128.4, 128.3, 128.2, 128.0, 120.0, 105.2, 75.2, 60.8, 60.3, 55.5, 8.7 ppm;
HRMS calcd. for C17H21O4 (MH+) 289.1440 Da. Found 289.1432 Da.
(2-Benzyloxy-6-iodo-3,5-dimethoxy-4-methyl-phenyl)-methanol (237)
OMe
MeOOBn
Me
OH
I
To a stirred suspension of 236 (3.60 g, 12.5 mmol) and silver trifluoroacetate (5.52 g,
25.0 mmol) at 0 °C in CHCl3 (55 mL) was added I2 (4.76 g, 18.8 mmol) as a solution in
CHCl3 (82 mL). The mixture was stirred for 1 h at 0 °C, filtered through Celite® and
washed with CHCl3. The organic solution was washed with 10% aq. sodium thiosulfate
(50 mL) and sat. aq. NaCl (50 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The resulting yellow oil was purified by flash column chromatography (SiO2,
10% to 25% EtOAc in hexanes) to afford 237 (4.20 g, 82% yield) as a pale yellow oil: Rf
= 0.46 (20% EtOAc in hexanes); IR (thin film) 3474, 2934, 2888, 2835, 1447, 1404,
1371, 1314, 1228, 1104, 1088, 1012, 965, 736, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ

174
7.50-7.36 (5 H, m), 5.04 (2 H, s), 4.78 (2 H, d, J = 5.6 Hz), 3.86 (3 H, s), 3.76 (3 H, s),
2.31 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3) δ 154.5, 152.5, 147.1, 136.8, 135.2,
128.5, 128.2, 126.1, 91.8, 76.0, 64.3, 60.3, 60.2, 10.6 ppm; HRMS calcd. for C17H19O4I
(M) 414.0328 Da. Found 414.0323 Da.
2-Benzyloxy-6-iodo-3,5-dimethoxy-4-methyl benzaldehyde (234)
OMe
MeOOBn
Me
O
I
To a stirred solution of 237 (12.9 g, 31.1 mmol) at 0 °C in CH2Cl2 (78.0 mL) was added
pyridinium chlorochromate (20.1 g, 93.2 mmol) in small portions. The solution was
allowed to warm to 25 °C and was stirred for 12 h. Silica gel was added to the mixture
such that upon removal of the solvent under vacuum a fine brown powder was obtained,
which was purifed by flash column chromatography (SiO2, 10% to 25% EtOAc in
hexanes) to afford 234 (12.8 g, 98% yield) as a pale yellow oil: Rf = 0.29 (20% EtOAc in
hexanes); IR (thin film) 3031, 2935, 2870, 1699, 1445, 1392, 1362, 1304, 1255, 1238,
1105, 1017, 962, 748, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ 10.05 (1 H, s), 7.49-7.35
(5 H, m), 5.05 (2 H, s), 3.87 (3 H, s), 3.78 (3 H, s), 2.36 (3 H, s) ppm; 13C NMR (75
MHz, CDCl3) δ 191.9, 155.2, 153.1, 150.6, 136.1, 131.9, 129.1, 128.5, 128.3, 87.4, 76.7,
60.5, 60.3, 11.1 ppm; HRMS calcd. for C17H17O4I (M) 412.0171 Da. Found 412.0165 Da.

175
[1-(2-Benzyloxy-6-iodo-3,5-dimethoxy-4-methyl-phenyl)-meth-(E)-ylidene]-tert-butyl
amine (238)
2-Benzyloxy-6-(3-benzyloxy-prop-1-ynyl)-3,5-dimethoxy-4-methyl benzaldehyde
(239)
8-Benzyloxy-3-benzyloxymethyl-5,7-dimethoxy-6-methyl isoquinoline (240)
OMe
Me
MeOOBn
N
I
MeOMe
MeOOBn
CHO
OBn
NOBn
OMeMe
MeOOBn
To a stirred solution of 234 (1.28 g, 3.10 mmol) in toluene (8 mL) was added 4 Å
molecular sieves and t-butyl amine (0.98 mL, 9.31 mmole) at 25 °C. The mixture was
stirred for 3 h and filtered through Celite®, washed with toluene, and the solvent
evaporated under vacuum to afford imine 238, which was used without further
purification.
To a degassed solution of 238 (1.45 g, 3.10 mmol) and progargyl benzyl ether (0.543 g,
3.72 mmol) in triethylamine (12.4 mL) at 25 °C under argon was added PdCl2(PPh3)2 (44
mg, 0.062 mmol) and CuI (6 mg, 0.031 mmol). The reaction flask was sealed under
argon, heated to 55 °C and stirred for 3 h. The solution became black in color and a
precipitate formed. The mixture was cooled to 25 °C, filtered through Celite®, and
concentrated filtrate under vacuum. The resulting residue was dissolved in anhydrous
DMF (30 mL), and to this solution was added CuI (50 mg, 0.30 mmole). The reaction
flask was flushed with argon, sealed, heated to 100 °C, and stirred for 5 h. The reaction
was cooled to 25 °C, diluted with sat. aq. NH4Cl (30 mL) and extracted with Et2O (2 x 50
mL). The combined extracts were dried (Na2SO4), filtered, and concentrated under

176
vacuum. The resulting residue was purified by flash column chromatography (SiO2,
10-40% EtOAc-hexane) to afford starting material 234 (0.524 g, 41% yield), coupled
acetylene 239 (0.240, 18% yield) and product 240 (0.440, 33% yield). 239: Rf = 0.45
(40% EtOAc in hexanes); IR (thin film): 2937, 2852, 1695, 1456, 1395, 1371, 1332,
1281, 1100, 1075, 1011 cm-1; 1H NMR (300 MHz, CDCl3): δ 10.45 (1 H, s), 7.53-7.32
(10 H, m), 5.07 (2 H, s), 4.79 (2 H, s), 4.53 (2 H, s), 3.91 (6 H, s), 2.30 (3 H, s) ppm; 13C
NMR (75 MHz, CDCl3): δ 189.3, 157.4, 152.9, 149.8, 137.4, 136.2, 132.9, 129.0, 128.6,
128.4, 128.3, 128.2, 128.0, 127.7, 112.9, 94.7, 79.5, 76.4, 71.3, 60.7, 60.6, 57.8, 10.0
ppm; HRMS calcd. for C27H27O5 (MH+) 431.1858 Da. Found 431.1846 Da. 240: Rf =
0.43 (40% EtOAc in hexanes); IR (thin film) 2936, 2857, 1589, 1454, 1394, 1359, 1317,
1079, 1004, 735, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ 9.45 (1 H, s), 7.91 (1 H, s),
7.54 (2 H, d, J = 8.01 Hz), 7.46-7.31 (8 H, m), 5.18 (2 H, s), 4.84 (2 H, s), 4.73 (2 H, s),
4.01 (3 H, s), 3.90 (3 H, s), 2.43 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3) δ 150.8,
149.2, 148.9, 147.0, 143.0, 138.0, 136.7, 128.8, 128.4, 128.3, 128.2, 127.7, 127.3, 126.7,
122.3, 111.6, 75.8, 73.0, 72.6, 61.4, 60.6, 10.0 ppm; HRMS calcd. for C27H28NO4 (MH+)
430.2018 Da. Found 430.2027 Da.

177
Optimized formation of 8-Benzyloxy-3-benzyloxymethyl-5,7-dimethoxy-6-methyl
isoquinoline (240)
NOBn
OMeMe
MeOOBn
To a solution of 234 (0.490 g, 1.19 mmol) in toluene (12.5 mL) was added 4 Å molecular
sieves (ca. 2 g) and t-butyl amine (0.375 mL, 3.57 mmole) at 25 °C. The mixture was
stirred for 3 h, filtered through Celite®, washed with toluene, and concentrated under
vacuum to afford imine 238, which was used without further purification.
To a degassed solution of 238 (0.58 g, 1.19 mmol) and progargyl benzyl ether4 (0.183 g,
1.25 mmol) in triethylamine (6.0 mL) at 25 °C under argon was added CuI (0.283 g, 1.49
mmol) in small portions. The mixture was stirred for 24 h and a yellow precipitate
formed. The mixture was diluted with triethylamine (8.0 mL), degassed, warmed to 85
°C, and stirred for 1 h. The mixture turned dark brown and the precipitate dissolved. The
solution was cooled to 25 °C, a black precipitate formed, and the mixture was filtered,
washed with triethylamine, and the solvent was removed under reduced pressure to give a
black oil, which was purified by flash column chromatography (SiO2, 10% to 20%
EtOAc in hexanes with 1% triethylamine) to afford 240 (0.465 g, 91% yield) as a yellow
oil: characterized as above.

178
8-Benzyloxy-3-benzyloxymethyl-5,7-dimethoxy-6-methyl-1-phenylsulfanylmethyl-
1H-isoquinoline-2-carboxylic acid methyl ester (241)
NOBn
OMeMe
MeOOBn
SPh
CO2Me
To a stirred, degassed solution of thioanisole (0.136 mL, 1.16 mmol) and (−)-sparteine
(0.266 mL, 1.16 mmol) in anhydrous THF (1.45 mL) at 0 °C was added n-BuLi (2.10 M
in hexanes, 0.551 mL, 1.16 mmol) dropwise within 10 min. The yellow solution was
allowed to warm to 25 °C and was stirred for 1 h. The solution of
phenylthiomethyllithium and (−)-sparteine was added dropwise within 10 min. to a
degassed solution of 240 (0.166 g, 0.386 mmol) in toluene (3.10 mL) at –78 °C under an
atmosphere of argon. The resulting orange solution was allowed to warm to 25 °C over
15 min. at which point the solution became black in color. To the reaction was added
methylchloroformate (0.149 mL, 1.93 mmol) at 25 °C and the solution became pale
orange. The solution was cooled to 0 °C, quenched with sat. aq. NaHCO3 (10 mL), and
then extracted with EtOAc (2 x 20 mL). The organic extracts were combined, washed
with sat. aq. NaCl (10 mL), dried (Na2SO4), filtered, and the solvent was removed under
vacuum. The crude yellow oil was purified by flash column chromatography (SiO2, 10%-
20% EtOAc in hexanes) to provide 241 (0.195 g, 83% yield) as a colorless oil: Rf = 0.26
(20% EtOAc in hexanes); IR (thin film) 2951, 2936, 2857, 1715, 1463, 1454, 1439, 1414,
1327, 1295, 1122, 1070, 1027, 1005 737, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.41-
7.29 (12 H, m), 7.10-7.06 (3 H, m), 6.67 (1 H, s), 5.91 (1 H, brs), 5.15 (1 H, d, J = 10.8
Hz), 4.90 (1 H, d, J = 10.8 Hz), 4.63-4.48 (4 H, m), 3.86 (3 H, s), 3.71 (3 H, s), 3.58 (3 H,

179
brs), 3.12-2.98 (2 H, m), 2.22 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3) δ 150.1, 143.4,
138.1, 137.0, 136.4, 128.6, 128.4, 128.2, 128.0, 127.9, 127.5, 127.4, 125.3, 125.1, 119.7,
119.7, 75.3, 72.3, 70.0, 61.2, 60.3, 52.6, 50.1, 34.9, 9.1 ppm; HRMS calcd. for
C36H38NO6S (MH+) 612.2120 Da. Found 612.2413 Da. HPLC conditions for separation
of enantiomers using ananlytical column Chiracel OD: loaded 12 μL of 0.53 mg/mL
sample in 1% isopropanol/hexanes, gradient 2-8% over 15 min., enantiomers elute at 12.4
min. and 13.1 min. in a ratio of 1:1.
8-Benzyloxy-5,7-dimethoxy-3,6-dimethyl-1-phenylsulfanylmethyl-3,4-dihydro-1H-
isoquinoline-2-carboxylic acid methyl ester (242)
8-Benzyloxy-3-benzyloxymethyl-5,7-dimethoxy-6-methyl-1-phenylsulfanylmethyl-
3,4-dihydro-1H-isoquinoline-2-carboxylic acid methyl ester (243)
N
OMeMe
MeOOBn
SPh
CO2Me
NOBn
OMeMe
MeOOBn
SPh
CO2Me
To a stirred solution of 241 (53 mg, 0.087 mmol) and triethylsilane (0.138 mL, 0.867
mmol) in CH2Cl2 (0.50 mL) at –10 °C was added TFA (pre-cooled to –20 °C, 34 µL,
0.435 mmol) in one portion. The solution was allowed to warm to 25 °C and was stirred
20 min. The reaction was then quenched with sat. aq. NaHCO3 (5 mL) and extracted with
CH2Cl2 (2 x 10 mL). The combined extracts were dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting crude oil was purified by flash column
chromatography (SiO2, 5% to 7% to 10% EtOAc in hexanes) to afford 242 (27 mg, 61%

180
yield) as a colorless oil and 243 (17 mg, 31% yield) as a colorless oil. 242: Rf = 0.44
(40% EtOAc in hexanes); IR (thin film) 2988, 2952, 2935, 1697, 1452, 1412, 1313, 1254,
1116, 1083, 1061, 738, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.47-7.28 (7 H, m),
7.13-7.08 (3 H, m), 6.07 (0.4 H, brs), 5.80 (0.6 H, brs), 5.07 (1 H, brs), 4.86 (1 H, brs),
4.13-4.01 (1 H, m), 3.85 (3 H, s), 3.69 (3 H, s), 3.67-3.43 (3 H, m), 3.31 (1 H, dd, J =
16.1, 7.3 Hz), 3.26-3.06 (2 H, m), 2.51 (1 H, dd, J = 16.1, 10.4 Hz), 2.23 (3 H, s), 1.47 (3
H, d, J = 6.2 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 151.6, 150.0, 144.0, 137.2, 128.8,
128.7, 128.4, 128.2, 128.1, 127.9, 127.7, 127.6, 125.5, 125.4, 124.6, 122.5, 95.1, 75.2,
60.6, 60.3, 52.2, 48.0, 28.1, 9.2 ppm; HRMS calcd. for C29H34NO5S (MH+) 508.2158 Da.
Found 508.2165 Da. 243: Rf = 0.21 (20% EtOAc in hexanes); IR (thin film), 2950, 2935,
2856, 1697, 1453, 1442, 1412, 1314, 1256, 1115, 1082, 1061, 1008, 738, 697 cm-1; 1H
NMR (300 MHz, CDCl3) δ 7.47-7.24 (12 H, m), 7.04 (3 H, brs), 6.08 (0.4 H, brs), 5.78
(0.6 H, brs), 5.12-4.98 (1 H, s), 4.91-4.78 (1 H, m), 4.66 (1 H, d, J = 12.1 Hz), 4.57 (1 H,
d, J = 12.0 Hz), 4.23-4.08 (1 H, m), 3.85 (4 H, brs), 3.77 (1 H, dd, J = 9.3, 3.0 Hz), 3.69
(3 H, s), 3.55 (3 H, brs), 3.35-3.11 (3 H, m), 2.91-2.85 (1 H, m), 2.23 (3 H, s) ppm;
HRMS calcd. for C36H40NO6S (MH+) 614.2576 Da. Found 614.2565 Da.

181
Improved Synthesis of 8-Benzyloxy-3-benzyloxymethyl-5,7-dimethoxy-6-methyl-1-
phenylsulfanylmethyl-3,4-dihydro-1H-isoquinoline-2-carboxylic acid methyl ester
(243)
NOBn
OMeMe
MeOOBn
SPh
CO2Me
To a solution of 241 (107 mg, 0.175 mmol), triethylsilane (0.280 mL, 1.75 mmol), and
benzyl alcohol (0.271 mL, 2.63 mmol) at –20 °C was added TFA (pre-cooled to –20 °C,
0.338 mL, 4.37 mmol) in one portion. The solution was allowed to warm to 0 °C and was
stirred for 16 h. The reaction was then quenched with sat. aq. NaHCO3 (10 mL) and
extracted with CH2Cl2 (2 x 10 mL). The combined extracts were dried (Na2SO4), filtered,
and concentrated under vacuum. The resulting crude oil was purified by flash column
chromatography (SiO2, 5% to 7% to 10% EtOAc in hexanes) to afford 242 (20 mg, 22%
yield) as a colorless oil and 243 (76 mg, 71% yield) as a colorless oil: characterized as
above.

182
8-Benzyloxy-3-hydroxymethyl-5,7-dimethoxy-6-methyl-1-phenylsulfanylmethyl-3,4-
dihydro-1H-isoquinoline-2-carboxylic acid methyl ester (248)
NOH
OMeMe
MeOOBn
SPh
CO2Me
To a solution of 243 (0.133 g, 0.217 mmol) in CH2Cl2 (1.0 mL) at –40 °C was added
BCl3 (1.0 M in CH2Cl2, 0.434 mL, 0.434 mmol) dropwise. Upon complete addition the
solution was stirred for 2 h and treated with methanol (0.5 mL), diluted with sat. aq.
NaHCO3 (5 mL), and extracted with CH2Cl2 (2 x 10 mL). The combined extracts were
dried (Na2SO4), filtered, and concentrated under vacuum. The crude residue was purified
by flash column chromatography (SiO2, 20%-50% EtOAc in hexanes) to afford 248
(0.100 g, 88% yield) as a colorless oil: Rf = 0.27 (40% EtOAc in hexanes); IR (thin film)
3448, 2936, 1697, 1412, 1396, 1349, 1315, 1257, 1115, 1056, 1006, 738, 697 cm-1; 1H
NMR (300 MHz, CDCl3) δ 7.52-7.28 (7 H, m), 7.12 (3 H, brs), 6.14 (0.4 H, brs), 5.86
(0.6 H, brs), 5.15-5.08 (1 H, m), 4.86-4.79 (1 H, m), 4.20 (1 H, dd, J = 11.8, 3.0 Hz),
4.06-3.98 (1 H, m), 3.86 (4 H, s), 3.75-3.70 (4 H, m), 3.68-3.46 (3 H, m), 3.27-2.82 (4 H,
m), 2.24 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3) δ 157.4, 151.9, 150.0, 143.7, 137.1,
135.0, 128.8, 128.6, 128.4, 128.1, 128.0, 125.7, 125.0, 124.9, 122.6, 95.0, 75.3, 65.3,
60.7, 60.3, 55.4, 52.3, 48.5, 38.3, 22.2, 9.2 ppm; HRMS calcd. for C29H34NO6S (MH+)
524.2107 Da. Found 524.2109 Da.

183
3-Azidomethyl-8-benzyloxy-5,7-dimethoxy-6-methyl-1-phenylsulfanylmethyl-3,4-
dihydro-1H-isoquinoline-2-carboxylic acid methyl ester (250)
NN3
OMeMe
MeOOBn
SPh
CO2Me
To a solution of 248 (0.10 g, 0.191 mmol) in THF (0.640 mL) at 0 °C was added
triphenylphosphine (0.11 g, 0.420 mmol) and diisopropyl azodicarboxylate (83 μL, 0.420
mmol). After 2 min. a white precipitate formed and diphenylphosphoryl azide (91 μL,
0.42 mmol) was added. The mixture was allowed to warm to 25 °C and was stirred for
1.5 h. The mixture was treated with H2O (0.4 mL) and concentrated to a crude residue
that was purified by flash column chromatography (SiO2, 5% to 10% EtOAc in hexanes)
to afford 250 (89 mg, 85% yield) as a colorless oil: Rf = 0.46 (20% EtOAc in hexanes);
IR (thin film) 3060, 2951, 2104, 1698, 1443, 1412, 1392, 1340, 1314, 1256, 1192, 1116,
1062, 1006, 737, 698 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.52-7.28 (7 H, m), 7.09 (3 H,
brs), 6.10 (0.4 H, brs), 5.82 (0.6 H, brs), 5.15-4.98 (1 H, m), 4.78-4.91 (1 H, m), 4.04 (1
H, m), 3.86 (4 H, s), 3.82-3.70 (4 H, m), 3.50 (3 H, brs), 3.38-3.09 (3 H, m), 2.76-2.67 (1
H, m), 2.24 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3) δ 156.4, 151.8, 150.1, 144.0,
137.1, 136.3, 129.2, 129.1, 129.7, 128.9, 128.7, 128.5, 128.3, 128.0, 127.9, 125.2, 124.9,
121.6, 75.2, 60.8, 60.4, 54.2, 52.5, 52.2, 48.8, 38.2, 23.3, 9.3 ppm; HRMS calcd. for
C29H33N4O5S (MH+) 549.2172 Da. Found 549.2478 Da.

184
3-Benzyloxy-4,6-dimethoxy-5-methyl-12-oxo-11,13-diaza-tricyclo[7.3.1.02,7]trideca-
2(7),3,5-triene-13-carboxylic acid methyl ester (251)
N
H
H CO2Me
NH
OBnO
MeO
MeOMe
To a solution of 250 (33 mg, 0.060 mmol) in chlorobenzene (0.300 mL) at 0 °C was
added N-chlorosuccinimide (8 mg, 0.060 mmol). The mixture was allowed to warm to 25
°C and was stirred for 2 h, then filtered through a pad of glass wool into a dry flask under
argon and washed with chlorobenzene (0.700 mL). The solution was cooled to 0 °C and
SnCl4 (3.0 M in CH2Cl2, 71 μL, 0.060 mmole) was added dropwise. The solution
immediately became orange and a precipitate formed. Upon complete addition no starting
material could be detected by TLC. The mixture was diluted with sat. aq. NaHCO3 until
basic (1.0 mL) and extracted with EtOAc (2 x 10 mL). The combined extracts were dried
(Na2SO4), filtered, and the solvent was removed under vacuum. The crude residue was
purified by flash column chromatography (SiO2, 20%-80% EtOAc in hexanes) to afford
251 (13 mg, 51% yield) as a colorless oil: Rf = 0.38 (100% EtOAc); IR (thin film) 2955,
1716, 1674, 1455, 1413, 1378, 1291, 1250, 1110, 1065, 1042, 973, 699 cm-1; 1H NMR
(500 MHz, CDCl3) δ 8.18 (1 H, s), 7.47 (2 H, d, J = 7.0 Hz), 7.43-7.39 (2 H, m), 7.37-
7.34 (1 H, m), 6.60 (1 H, brs), 5.15 (1 H, 11.0 Hz), 4.97 (1 H, 11.0 Hz), 4.87-4.85 (1 H,
m), 3.83-3.80 (4 H, m), 3.70 (3 H, s), 3.66 (3 H, s), 3.26 (1 H, ddd, J = 17.3, 5.3, 1.4 Hz),
3.17 (1 H, d, J = 11.5 Hz), 2.69 (1 H, dd, J = 7.3, 1.0 Hz), 2.18 (3 H, s) ppm; 13C NMR
(125 MHz, CDCl3) δ 158.6, 153.7, 143.8, 137.25, 128.7, 128.3, 127.9, 126.3, 120.0,

185
75.64, 64.4, 60.5, 59.9, 53.1, 51.6, 30.1, 29.6, 9.46 ppm; HRMS calcd. for C23H27N2O6
(MH+) 427.1869 Da. Found 427.1867 Da.
1-Benzyloxymethyl-1H-isoquinoline-2-carboxylic acid methyl ester (261)
NCO2Me
OBn
To a stirred solution of BnOCH2SnBu36 (0.35 g, 0.85 mmol) and (−)-sparteine (0.32 mL,
1.41 mmol) in toluene (6.0 mL) at −78 °C was added n-butyllithium (2.1 M in hexanes,
0.40 mL, 0.85 mmol) dropwise within 5 min. The brown solution was stirred for 10 min.
at −78 °C, and isoquinoline (0.10 mL, 0.71 mmol) was added as a solution in THF (0.10
mL). The brown solution was allowed to warm to 25 °C and stirred for 15 min. The
reaction was quenched with ClCO2Me (0.16 mL, 2.12 mmol) and the solution became
colorless. The solution was cooled to 0 °C, diluted with sat. aq. NaHCO3 (5 mL), and
extracted with EtOAc (3 x 15 mL). The extracts were combined and washed with sat. aq.
NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The resulting
residue was purified by flash column chromatography (SiO2, 10-20% EtOAc in hexanes)
to provide 261 (144 mg, 66% yield) as a colorless oil: Rf = 0.30 (20% EtOAc in
hexanes); IR (thin film): 3063, 3028, 2953, 2856, 1716, 1632, 1456, 1441, 1416, 1354,
1330, 1292, 1235, 1196, 1125 cm-1; 1H NMR (300 MHz, C6D6): δ 7.21-6.68 (10 H, m),
5.79 (0.6 H, t, J = 6.6 Hz), 5.54 (0.4 H, d, J = 7.8 Hz), 5.49 (0.6 H, d, J = 7.8 Hz), 5.40
(0.4 H, t, J = 6.8 Hz), 4.34 (0.6 H, d, J = 12.1 Hz), 4.19 (0.6 H, d, 12.2 Hz), 4.15 (0.4 H,
d, J = 12.4 Hz), 4.07 (0.4 H, d, J = 12.3 Hz), 3.54 (0.6 H, dd, J = 9.9, 7.5 Hz), 3.45-3.29

186
(4 H, m), 3.14 (0.4 H, dd, J = 9.7, 5.9 Hz) ppm; 13C NMR (75 MHz, C6D6): δ 153.6,
152.8, 138.4, 138.2, 130.9, 130.6, 129.8, 129.5, 127.9, 127.0, 126.9, 126.8, 126.7, 126.4,
126.0, 125.4, 124.4, 124.2, 107.8, 72.2, 70.2, 55.2, 54.3, 52.3 ppm; 1H NMR (500 MHz,
toluene-d8, 100 °C): δ 7.07-6.85 (9 H, m), 6.79 (1 H, d, J = 7.5 Hz), 5.55 (1 H, d, J = 7.7
Hz), 5.54 (1 H, brs), 4.29 (1 H, d, J = 12.2 Hz), 4.22 (1 H, d, J = 12.1 Hz), 4.30-4.20 (2
H, brs), 3.53 (1 H, dd, J = 9.9, 7.1 Hz), 3.49 (3 H, s), 3.36 (1 H, dd, J = 9.7, 6.0 Hz) ppm; 13C NMR (125 MHz, toluene-d8, 100 °C): δ 154.0, 139.3, 131.7, 130.9, 129.2, 129.0,
128.8, 128.3, 128.1, 127.9, 127.6, 126.9, 125.8, 125.5, 125.3, 108.4, 73.5, 71.1, 56.1, 52.8
ppm; HRMS calcd. for C19H20NO3 (MH+) 310.1443 Da. Found 310.1444 Da. HPLC
conditions: loaded 15 µL of 0.50 mg/mL sample solution in 1% isopropanol in hexanes,
gradient = 2% to 5% isopropanol in hexanes, enantiomers elute at 12.85 min. and 16.47
min. in a ratio of 2:1, 33% ee.
Benzyloxy-5,7-dimethoxy-6-methyl-3-triisopropylsilanyloxymethyl-isoquinoline
(262)
OMe
Me
MeOOBn
NOTIPS
To a stirred solution of o-iodoimine 238 (12.1 g, 26.0 mmol) and acetylene 200 (5.61 g,
28.6 mmol) in triethylamine (130 mL) was added CuI (6.09 g, 31.2 mmol) in small
portions over 20 min. The reaction was stirred under argon at 25 °C for 24 h, and treated
with sat. aq. NaHCO3 (200 mL). The solution was extracted with EtOAc (3 x 200 mL).

187
The combined extracts were washed with sat. aq. NaHCO3 (75 mL) until aqueous layer
was no longer blue, washed with sat. aq. NaCl (100 mL), dried (Na2SO4), filtered, and
concentrated under vacuum to a pale orange solid. The coupled product was dissolved in
dry DMF (260 mL) and degassed (argon bubbling for 3 h). CuI (0.989 g, 5.19 mmol) was
added. The reaction flask was sealed and heated to 100 °C, and the clear solution was
stirred for 2 h. The reaction was cooled to 25 °C, treated with sat. aq. NH4Cl (300 mL),
and extracted with EtOAc (3 x 300 mL). The combined extracts were washed with sat.
aq. NH4Cl (200 mL), sat. aq. NaCl (200 mL), dried (Na2SO4), filtered, and concentrated
under vacuum. The resulting brown oil was purified by flash column chromatography
(SiO2, 5-10% EtOAc-hexane) to afford isoquinoline 262 (10.8 g, 76% yield) as a yellow
oil: Rf = 0.23 (10% EtOAc in hexanes); IR (thin film): 2942, 2865, 1623, 1591, 1456,
1394, 1363, 1315, 1121, 1106, 1082, 1014, 883 cm-1; 1H NMR (300 MHz, CDCl3): δ
9.38 (1 H, s), 8.05 (1 H, s), 7.54 (2 H, d, J = 7.0 Hz), 7.41-7.37 (3 H, m), 5.18 (2 H, s),
5.10 (2 H, s), 4.0 (3 H, s), 3.90 (3 H, s), 2.43 (3 H, s), 1.29-1.20 (3 H, m), 1.15 (18 H, d, J
= 5.5 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 154.4, 149.3, 148.6, 146.6, 146.4, 143.2,
137.0, 128.8, 128.6, 128.4, 128.3, 122.2, 109.5, 75.9, 66.1, 61.3, 60.7, 18.0, 12.0, 10.1
ppm; HRMS calcd. for C29H42NO4Si (MH+) 496.2883 Da. Found 496.2887 Da.

188
8-Benzyloxy-1-benzyloxymethyl-5,7-dimethoxy-6-methyl-3-triisopropylsilanyloxy-
methyl-1H-isoquinoline-2,4-dicarboxylic acid dimethyl ester (264)
OMe
Me
MeOOBn
NOTIPS
OBn
CO2Me
CO2Me
To a stirred solution of BnOCH2SnBu36 (0.193 g, 0.47 mmol) in THF (0.71 mL) at −78
°C was added n-BuLi (1.90 M in hexanes, 83 µL, 0.157 mmol) dropwise. The solution
became yellow in color and was stirred for 15 min. at −78 °C, then a solution of
isoquinoline 262 (78 mg, 0.157 mmol) in THF (0.20 mL) was added dropwise. The
yellow solution was allowed to warm to 25 °C and stirred 15 min. The brown/orange
solution was quenched with ClCO2Me (61 µL, 0.785 mmol). The pale yellow solution
was diluted with sat. aq. NaHCO3 (5 mL) and extracted with EtOAc (2 x 10 mL). The
combined extracts were washed with sat. aq. NaCl (10 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting yellow oil was purified by flash column
chromatography (SiO2, 5-20% EtOAc-hexane) to afford starting isoquinoline 262 (68 mg,
43% yield) and diacylated product 264 (37 mg, 32% yield) as a colorless oil: Rf = 0.27
(20% EtOAc in hexanes); IR (thin film): 2943, 2866, 1750, 1716, 1441, 1268, 1126, 1069
cm-1; 1H NMR (300 MHz, CDCl3): δ 7.41-7.25 (5 H, m), 7.17-7.03 (3 H, m), 6.85-6.81 (2
H, m), 5.74 (1 H, brs), 5.04-4.93 (4 H, m), 4.75 (1 H, d, J = 10.9 Hz), 4.53 (1 H, d, J =
14.7 Hz), 3.74 (6 H, s), 3.67 (3 H, s), 3.51 (3 H, brs), 2.93-2.87 (2 H, m), 2.22 (3 H, s),
1.29-1.01 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 155.3, 149.6, 143.5, 137.4,
135.7, 134.2, 131.0, 128.6, 128.1, 127.6, 126.4, 124.3, 119.9, 106.7, 74.7, 66.7, 63.3,

189
61.0, 60.1, 54.5, 52.5, 52.0, 35.3, 17.9, 11.9, 9.02 ppm; HRMS calcd. for C41H56NO9Si
(MH+) 734.3724 Da. Found 734.3732 Da.
8-Benzyloxy-1-benzyloxymethyl-5,7-dimethoxy-6-methyl-3-triisopropylsilanyloxy-
methyl-1H-isoquinoline-2-carboxylic acid methyl ester (263)
8-Benzyloxy-1-butyl-5,7-dimethoxy-6-methyl-3-triisopropylsilanyloxymethyl-1H-
isoquinoline-2-carboxylic acid methyl ester (265)
OMe
Me
MeOOBn
NOTIPS
OBn
CO2Me
Me
MeOOBn
OMe
NOTIPS
CO2Me
To a stirred solution of BnOCH2SnBu36 (0.348 g, 0.847 mmol) in THF (1.4 mL) at −78
°C was added n-BuLi (2.10 M in hexanes, 0.403 mL, 0.847 mmol) dropwise. The
solution became yellow in color and was stirred for 15 min. at −78 °C, then a solution of
isoquinoline 262 (140 mg, 0.282 mmol) in THF (0.5 mL) was added dropwise. The
red/brown solution was stirred at −78 °C for 1.5 h and quenched with ClCO2Me (109 µL,
1.41 mmol). The pale yellow solution was diluted with sat. aq. NaHCO3 (10 mL) and
extracted with EtOAc (2 x 15 mL). The combined extracts were washed with sat. aq.
NaCl (10 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The resulting
yellow oil was purified by flash column chromatography (SiO2, 5-10% EtOAc-hexane) to
afford n-butyl adduct 265 (57 mg, 33% yield) as a colorless oil and 1,2-
dihydroisoquinoline 263 (86 mg, 45% yield) as a colorless oil. 265: Rf = 0.26 (10%
EtOAc in hexanes); 1H NMR (300 MHz, CDCl3): δ 7.56-7.52 (2 H, m), 7.46-7.34 (3 H,

190
m), 6.68 (1 H, s), 5.70 (1 H, brs), 5.07 (1 H, d, J = 10.7 Hz), 4.97 (1 H, d, J = 10.7 Hz),
4.95 (1 H, brs), 4.57 (1 H, d, J = 14.0 Hz), 3.85 (3 H, s), 3.73 (3 H, s), 3.72 (3 H, s), 2.22
(3 H, s), 1.71-1.63 (2 H, m), 1.32-0.77 (28 H, m) ppm; 1H NMR (500 MHz, toluene-d8,
100 °C): δ 7.46-7.44 (2 H, m), 7.20-7.17 (3 H, m), 6.88 (1 H, t, J = 1.5 Hz), 5.92 (1 H,
dd, J = 9.4, 4.7 Hz), 5.05 (1 H, dd, J = 14.0, 1.3 Hz), 5.05 (1 H, d, J = 11.3 Hz), 4.93 (1
H, d, J = 11.2 Hz), 4.79 (1 H, dd, J = 13.9, 1.5 Hz), 3.64 (3 H, s), 3.55 (3 H, s), 3.46 (3 H,
s), 2.20 (3 H, s), 1.85-1.80 (1 H, m), 1.55-1.49 (1 H, m), 1.47-1.40 (2 H, m), 1.32-1.23 (2
H, m), 1.18-1.14 (21 H, m), 0.81 (3 H, t, J = 3.9 Hz) ppm; 13C NMR (125 MHz, toluene-
d8, 100 °C): δ 155.1, 152.0, 151.5, 144.4, 138.9, 138.1, 128.8, 128.7, 128.2, 124.3, 121.0,
109.1, 76.0, 65.0, 60.9, 60.2, 52.9, 52.4, 33.3, 28.4, 22.9, 18.5, 14.1, 13.1, 9.5 ppm. 263:
Rf = 0.21 (10% EtOAc in hexanes); IR (thin film): 2943, 2865, 1714, 1456, 1321, 1128,
1090, 1067 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.54-7.51 (2 H, m), 7.45-7.29 (6 H, m),
7.21-7.17 (2 H, m), 6.74 (1 H, s), 6.05 (1 H, brs), 5.10 (1 H, brs), 5.09 (1 H, d, J = 10.8
Hz), 4.98 (1 H, d, J = 10.8 Hz), 4.50 (2 H, m), 4.38 (1 H, d, J = 12.2 Hz), 3.82 (3 H, s),
3.73 (3 H, s), 3.71 (3 H, s), 3.56 (1 H, t, J = 10.3 Hz), 3.27 (1 H, dd, J = 10.6, 4.3 Hz),
2.21 (3 H, s), 1.21-1.08 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 150.0, 144.2,
138.6, 137.7, 128.6, 128.2, 127.4, 125.0, 121.2, 107.1, 95.1, 75.4, 73.5, 72.4, 68.4, 67.2,
63.4, 61.3, 60.5, 53.1, 51.0, 18.2, 12.2, 9.4 ppm; 1H NMR (500 MHz, toluene-d8, 100
°C): 7.46-7.44 (2 H, m), 7.25-6.96 (8 H, m), 6.91 (1 H, t, J = 1.5 Hz), 6.28 (1 H, dd, J =
8.8, 4.7 Hz), 5.20 (1 H, dd, J = 14.5, 1.5 Hz), 5.05 (1 H, d, J = 11.1 Hz), 4.95 (1 H, d, J =
11.2 Hz), 4.76 (1 H, dd, J = 14.5, 1.6 Hz), 4.42 (1 H, d, J = 12.2 Hz), 4.31 (1 H, d, J =
12.2 Hz), 3.71 (1 H, dd, J = 10.6, 8.8 Hz), 3.61 (3 H, s), 3.56 (3 H, s), 3.45 (3 H, s), 3.41
(1 H, dd, J = 10.6, 4.8 Hz), 2.20 (3 H, s), 1.16-1.14 (21 H, m) ppm; 13C NMR (125 MHz,
toluene-d8, 100 °C): 155.0, 151.9, 151.4, 145.0, 138.8, 137.9, 137.5, 129.3, 128.7, 128.4,

191
128.3, 127.8, 127.5, 122.0, 108.1, 76.0, 73.7, 73.1, 69.9, 67.9, 64.6, 61.0, 60.2, 52.3, 18.5,
13.1, 9.5 ppm; HRMS calcd. for C39H54NO7Si (MH+) 676.3591 Da. Found 676.3601 Da.
Optimized formation of 8-Benzyloxy-1-benzyloxymethyl-5,7-dimethoxy-6-methyl-3-
triisopropylsilanyloxy-methyl-1H-isoquinoline-2-carboxylic acid methyl ester (263)
OMe
Me
MeOOBn
NOTIPS
OBn
CO2Me
To a degassed (argon bubbling) and stirred solution of BnOCH2SnBu36 (9.0 g, 21.9
mmol) in THF (109 mL) at −78 °C was added n-BuLi (2.10 M in hexanes, 7.81 mL, 16.4
mmol) within 8 min. The solution became yellow in color and was stirred for 5 min, then
a degassed (argon bubbling) solution of isoquinoline 262 (2.71 g, 5.47 mmol) in THF
(5.0 mL) was added dropwise within 10 min. The reaction solution became deep
red/brown in color. The solution was stirred for 2 h at −78 °C, then quenched rapidly with
ClCO2Me (2.1 mL, 27.4 mmol). The dark color of the solution faded to pale yellow. The
reaction was diluted with sat. aq. NaHCO3 (150 mL) and extracted with EtOAc (2 x 250
mL). The combined extracts were washed with sat. aq. NaCl (100 mL), dried (Na2SO4),
filtered, and concentrated under vacuum. The resulting yellow oil was purified by flash
column chromatography (SiO2, 5-10% EtOAc-hexane) to afford 1,2-dihydroisoquinoline
263 (3.22 g, 79% yield) as a colorless oil: characterized as above.

192
8-Hydroxy-1-hydroxymethyl-5,7-dimethoxy-6-methyl-3-triisopropylsilanyloxy-
methyl-1H-isoquinoline-2-carboxylic acid methyl ester (273)
NOTIPS
CO2Me
OMeMe
MeOOH
OH
To a solution of 1,2-dihydroisoquinoline 263 (38 mg, 0.056 mmol) in EtOH (1.0 mL) was
added PtO2 (48 mg, 0.211 mmol). The solution was charged with balloon pressure of H2
and stirred for 2 h. The solution was filtered through Celite® and concentrated under
vacuum. The residue was purified by flash column chromatography (SiO2, 20-40%
EtOAc in hexanes) to provide 273 (24 mg, 87% yield) as a colorless oil: Rf = 0.27 (40%
EtOAc in hexanes); IR (thin film): 3387, 2943, 2866, 1713, 1693, 1465, 1444, 1414,
1337, 1310, 1258, 1128, 1102, 1063, 1012, 882 cm-1; 1H NMR (300 MHz, CDCl3): δ
6.51 (1 H, s), 5.80 (1 H, t, J = 7.6 Hz), 5.59 (1 H, brs), 5.07 (1 H, brs), 4.46 (1 H, d, J =
11.9 Hz), 3.78 (6 H, s), 3.68 (3 H, s), 3.63-3.50 (2 H, m), 2.21 (3 H, s), 1.21-1.09 (21 H,
m) ppm; 13C NMR (75 MHz, CDCl3): δ 162.5, 147.5, 145.0, 140.8, 135.6, 123.0, 120.4,
117.4, 110.9, 95.1, 64.2, 61.4, 60.7, 52.9, 52.4, 17.7, 11.8, 9.2 ppm; HRMS calcd. for
C25H41NO7Si (M+) 495.2652 Da. Found 495.2652 Da.

193
8-Benzyloxy-1-benzyloxymethyl-3-hydroxymethyl-5,7-dimethoxy-6-methyl-1H-
isoquinoline-2-carboxylic acid methyl ester (274)
OMe
Me
MeOOBn
NOH
OBn
CO2Me
To a stirred solution of 263 (22 mg, 0.033 mmol) in CH2Cl2 (0.33 mL) at 0 °C was added
TFA dropwise with reaction monitoring until all starting material was consumed. The
reaction was diluted with sat. aq. NaHCO3 (15 mL) and extracted with EtOAc (2 x 20
mL). The combined extracts were washed with sat. aq. NaCl (10 mL), dried (Na2SO4),
filtered, and concentrated under vacuum. The residue was purified by flash column
chromatography (SiO2, 25-40% EtOAc-hexane) to afford 274 (15 mg, 88% yield) as a
colorless oil: Rf = 0.28 (50% EtOAc in hexanes); IR (thin film): 3462, 2938, 2862, 1699,
1456, 1414, 1395, 1346, 1331, 1290, 1251, 1114, 1064, 1008 cm-1; 1H NMR (300 MHz,
CDCl3): δ 7.51-7.19 (10 H, m), 6.44 (1 H, s), 5.98 (1 H, s), 5.10 (1 H, d, J = 11.1 Hz),
4.99 (1 H, d, J = 11.1 Hz), 4.54 (2 H, m), 4.31 (2 H, m), 3.83 (3 H, s), 3.76 (3 H, s), 3.71
(3 H, s), 3.47 (1 H, t , J = 10.2 Hz), 3.20 (1 H, m), 2.21 (3 H, s) ppm; 13C NMR (75 MHz,
CDCl3): δ 154.1, 143.8, 137.2, 128.2, 128.0, 127.3, 127.2, 125.1, 120.3, 111.2, 95.1, 75.2,
72.1, 67.6, 63.8, 61.3, 60.2, 53.1, 20.6, 14.0, 9.1 ppm; HRMS calcd. for C30H33NO7
(MH+) 519.2257 Da. Found 519.2253 Da.

194
1-Benzyloxymethyl-8-hydroxy-3-hydroxymethyl-5,7-dimethoxy-6-methyl-1H-
isoquinoline-2-carboxylic acid methyl ester (275)
NOH
CO2Me
OMeMe
MeOOH
OBn
To a solution of 1,2-dihydroisoquinoline 274 (13 mg, 0.025 mmol) in EtOH (0.30 mL)
was added 5% platinum on carbon (3 mg). The solution was charged with balloon
pressure of H2 and stirred for 16 h. The solution was filtered through Celite® and
concentrated under vacuum. The residue was purified by flash column chromatography
(SiO2, 10-30% EtOAc in hexanes) to provide 275 (10 mg, 94% yield) as a colorless oil:
Rf = 0.10 (40% EtOAc in hexanes); IR (thin film): 3392, 2959, 2861, 1686, 1445, 1445,
1414, 1335, 1261, 1101, 1061, 1011 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.35-7.27 (5 H,
m), 6.43 (1 H, s), 6.01 (1 H, brs), 5.74 (1 H, s), 4.63 (1 H, d, J = 12.0 Hz), 4.52 (1 H, brs),
4.50 (1 H, d, 12.0 Hz), 4.29 (1 H, dd, J = 12.8, 4.5 Hz), 3.83 (3 H, s), 3.80 (3 H, s), 3.70
(3 H, s), 3.56 (1 H, dd, J = 10.3, 8.8 Hz), 3.43 (1 H, dd, J = 10.1, 5.0 Hz), 2.22 (3 H, s)
ppm; 13C NMR (75 MHz, CDCl3): δ 155.1, 147.6, 145.6, 141.1, 137.9, 136.5, 128.4,
127.7, 127.5, 123.7, 120.4, 111.1, 72.8, 68.4, 64.1, 61.6, 60.9, 53.4, 50.6, 9.5 ppm; m/z
(C.I.) 412 (100%, MH+−H2O).

195
6-Benzyloxy-5-benzyloxymethyl-7,9-dimethoxy-8-methyl-1,5-dihydro-oxazolo[3,4-
b]isoquinolin-3-one (276)
OMe
Me
MeOOBn
N
OBn
O
O
To a stirred solution of 275 (15 mg, 0.029 mmol) in MeOH (0.33 mL) at 25 °C was
added NaBH4 (31 mg, 0.813 mmol). The reaction was stirred for 15 h at 25 °C, quenched
with H2O (0.5 mL), diluted with sat. aq. NaHCO3 (5 mL), and extracted with EtOAc (2 x
10 mL). The combined extracts were washed with sat. aq. NaCl (10 mL), dried (Na2SO4),
filtered, and concentrated under vacuum. The residue was purified by flash column
chromatography (SiO2, 10-30% EtOAc-hexane) to afford 276 (12 mg, 86% yield) as a
colorless oil: Rf = 0.23 (30% EtOAc in hexanes); IR (thin film): 2937, 2862, 1773, 1675,
1457, 1364, 1228, 1119, 1049, 1008 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.45-7.35 (5 H,
m), 7.24-7.21 (3 H, m), 7.18-7.15 (2 H, m), 5.77 (1 H, t, J = 2.0 Hz), 5.57 (1 H, t, J = 3.9
Hz), 5.03-4.99 (4 H, m), 4.60 (1 H, d, J = 12.0 Hz), 4.24 (1 H, d, J = 11.9 Hz), 3.79 (3 H,
s), 3.62 (3 H, s), 3.62-3.58 (2 H, m), 2.23 (3 H, s) ppm; HRMS calcd. for C29H30NO6
(MH+) 488.2073 Da. Found 488.2051 Da.

196
Optimized formation of 6-Benzyloxy-5-benzyloxymethyl-7,9-dimethoxy-8-methyl-
1,5-dihydro-oxazolo[3,4-b]isoquinolin-3-one (276)
OMe
Me
MeOOBn
N
OBn
O
O
To a stirred solution of 1,2-dihydroisoquinoline 275 (11.2 g, 16.6 mmol) in THF (110
mL) at −10 °C was added TBAF (1.0 M in THF, 16.6 mL, 16.6 mmol) dropwise within 5
min. The yellow solution was stirred for 15 min. at −10 °C, warmed to 0 °C and stirred
30 min., warmed further to 25 °C and stirred for 10 min. The reaction was quenched with
sat. aq. NaHCO3 (200 mL) and extracted with EtOAc (3 x 200 mL). The combined
extracts were washed with sat. aq. NaCl (200 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting yellow oil was purified by flash column
chromatography (SiO2, 20-30% EtOAc-hexane) to afford oxazolidinone 276 (7.3 g, 91%
yield) as a yellow oil: characterized as above.

197
6-Benzyloxy-5-benzyloxymethyl-7,9-dimethoxy-8-methyl-1,5,10,10a-tetrahydro-
oxazolo[3,4-b]isoquinolin-3-one (279)
MeO
Me
MeOO
N
OPhPh
1
11
7
5
3
9
10
15
1416
Ha Hb
O
O
HaHb
H
To a stirred solution of oxazolidinone 276 (1.09 g, 2.24 mmol) and Et3SiH (5.40 mL,
33.6 mmol) in CH2Cl2 (11 mL) at −10 °C was added TFA (pre-cooled to −10 °C, 2.60
mL, 33.6 mmol). The yellow solution was stirred for 15 min. at −10 °C to 0 °C, then
warmed to 25 °C and stirred for 3 h. The reaction was cooled to 0 °C and slowly
quenched with 10% aq. Na2CO3 until the aqueous layer was basic, and extracted with
CH2Cl2 (3 x 30 mL). The combined extracts were washed with sat. aq. NaCl (30 mL),
dried (Na2SO4), filtered, and concentrated under vacuum to a yellow oil. The yellow oil
was purified by flash column chromatography (SiO2, 20-40% EtOAc-hexane) to afford
the tetrahydroisoquinoline 279 (1.04 g, 95% yield) as a yellow oil: Rf = 0.16 (40%
EtOAc in hexanes); IR (thin film): 2936, 1751, 1456, 1411, 1117, 1030 cm-1; 1H NMR
(300 MHz, CDCl3): δ 7.36-7.28 (5 H, m), 7.19-7.16 (3 H, m), 7.00-6.96 (2 H, m), 5.01
(C1, 1 H, t, J = 2.2 Hz), 4.96 (C16, 1 H, d, J = 11.3 Hz), 4.92 (C16, 1 H, d, J = 11.3 Hz),
4.37 (C11-Hb, 1 H, t, J = 7.6 Hz), 4.40 (C15, 1 H, d, J = 12.2 Hz), 4.29 (C14, 1 H, dd, J =
9.8, 2.8 Hz), 4.22 (C15, 1 H, d, J = 12.2 Hz), 3.94 (C11-Ha, 1 H, dd, J = 10.8, 8.2 Hz),
3.78 (C7-OMe, 3 H, s), 3.77-3.71 (C3, 1 H, m), 3.64 (C5-OMe, 3 H, s), 3.52 (C14, 1 H,
dd, J = 9.8, 2.0 Hz), 3.14 (C4-Hb, 1 H, dd, J = 14.1, 3.0 Hz), 2.61 (C4-Ha, 1 H, dd, J =
14.0, 1.6 Hz), 2.24 (C6-Me, 3 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 156.5, 151.6,

198
150.5, 144.8, 138.7, 137.1, 128.5, 128.1, 128.0, 127.3, 127.1, 126.8, 124.6, 124.3, 74.4
(C16), 72.9 (C15), 70.0 (C14), 68.6 (C11), 60.7 (C5), 60.3 (C7), 53.9 (C3), 50.9 (C1),
27.7 (C4), 9.5 (C6) ppm; HRMS calcd. for C29H32NO6 (MH+) 490.2230 Da. Found
490.2216 Da.
(8-Benzyloxy-1-benzyloxymethyl-5,7-dimethoxy-6-methyl-1,2,3,4-tetrahydro-
isoquinolin-3-yl)-methanol (283)
MeO
Me
MeOO
NH
OPhPh
1
11
7
5
3
9
10
141315
Ha Hb
OH
To a stirred solution of oxazolidinone 276 (7.0 g, 14.4 mmol) and Et3SiH (34 mL, 215
mmol) in CH2Cl2 (72 mL) at −10 °C was added TFA (pre-cooled to −10 °C, 16.7 mL,
215 mmol). The yellow solution was stirred for 45 min. at −10 °C to 0 °C, then warmed
to 25 °C and stirred for 2 h. The reaction was cooled to 0 °C and slowly quenched with
10% aq. Na2CO3 until the aqueous layer was basic, and extracted with CH2Cl2 (3 x 150
mL). The combined extracts were washed with sat. aq. NaCl (100 mL), dried (Na2SO4),
filtered, and concentrated under vacuum to a yellow oil. The crude tetrahydroisoquinoline
279 was dissolved in ethylene glycol (144 mL) then KOH (21 g, 373 mmol) and
hydrazine monohydrate (3.48 mL, 72.0 mmol) were added at 25 °C. The reaction mixture
was warmed to 150 °C and became homogeneous, then stirred for 3 h. The heat was
removed and solution cooled to 25 °C. The mixture was made homogeneous by the

199
addition of CH2Cl2 and minimal amounts of MeOH, then purified by flash column
chromatography (SiO2 pre-treated with TEA, 100% CH2Cl2) to afford amino alcohol 283
(5.7 g, 86% yield) as a brown solid: recrystallized from EtOAc and hexanes; X-ray (see
Appendix D); mp = 84-89 °C; Rf = 0.19 (100% EtOAc); IR (thin film): 3333, 2935, 2864,
1455, 1410, 1336, 1113, 1060, 1026 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.38-7.29 (5 H,
m), 7.27-7.19 (5 H, m), 5.02 (C15, 1 H, d, J = 11.3 Hz), 4.85 (C15, 1 H, d, J = 11.2 Hz),
4.43 (C14, 1 H, d, J = 12.1 Hz), 4.38 (C14, 1 H, d, J = 12.0 Hz), 4.35 (C1, 1 H, dd, J =
8.0, 2.6 Hz), 4.03 (C12, 1 H, dd, J = 9.3, 3.2 Hz), 3.80 (C7-OMe, 3 H, s), 3.72 (C11, 1 H,
dd, J = 10.9, 3.4 Hz), 3.67 (C5-OMe, 3 H, s), 3.61 (C12, 1 H, dd, J = 9.2, 7.4 Hz), 3.52
(C11, 1 H, dd, J = 11.0, 6.6 Hz), 2.83-2.78 (C3, C4-Hb, 2 H, m), 2.75 (NH, OH, 2 H, brs),
2.32 (C4-Ha, 1 H, dd, J = 15.9, 11.6 Hz), 2.21 (C6-Me, 3 H, s) ppm; 13C NMR (75 MHz,
CDCl3): δ 152.3 (C5), 149.9 (C7), 145.4 (C8), 138.5 (C14Ph-ipso, 137.7 (C15Ph-ipso),
128.4, 128.2, 128.0, 127.9, 127.6 (C9), 127.5, 127.4, 125.8 (C10), 123.5 (C6), 74.5
(C15), 74.1 (C12), 72.9 (C14), 65.9 (C11), 60.3 (C7-OMe), 60.2 (C5-OMe), 53.6 (C3),
53.5 (C1), 26.5 (C4), 9.3 (C6) ppm; HRMS calcd. for C28H34NO5 (MH+) 464.2437 Da.
Found 464.2423 Da.

200
tert-Butoxycarbonylamino-acetic acid 8-benzyloxy-1-benzyloxymethyl-5,7-
dimethoxy-6-methyl-1,2,3,4-tetrahydro-isoquinolin-3-ylmethyl ester (286)
MeOMe
MeOO
NH
O
O
O
HN
PhPh
1
11
7
5
3
9
10
13 14
212022
O
O16
Ha Hb
18
19
To a stirred solution of 283 (100 mg, 0.216 mmol) and Boc-Gly-OH (40 mg, 0.227
mmol) in CH2Cl2 (1.1 mL) at 0 °C was added Py-BOP® (118 mg, 0.227 mmol), and after
5 min. Hünig’s base (0.12 mL, 0.681 mmol) was added. After stirring at 0 °C for 1 h the
solution was warmed to 25 °C and stirred 3 h. The reaction was treated with H2O (5 mL)
and extracted with EtOAc (2 x 15 mL). The combined extracts were washed with 5% aq.
NaHSO4 (15 mL), sat. aq. NaHCO3 (15 mL), sat. aq. NaCl (15 mL), dried (Na2SO4),
filtered, and concentrated under vacuum. The residue was purified by flash column
chromatography (SiO2, 20-40% EtOAc-hexane) to afford 286 (110 mg, 82% yield) as a
pale yellow oil: Rf = 0.26 (30% EtOAc in hexanes); IR (thin film): 3421, 3357, 1754,
1716, 1455, 1366, 1165, 1058 cm-1; 1H NMR (500 MHz, CDCl3): δ 7.38-7.30 (5 H, m),
7.29-7.19 (5 H, m), 5.03 (C22, 1 H, d, J = 11.1 Hz), 5.00 (BocNH, 1 H, brs), 4.82 (C22, 1
H, d, J = 11.1 Hz), 4.43 (C21, 1 H, d, J = 12.3 Hz), 4.40 (C21, 1 H, d, J = 12.1), 4.38
(C1, 1 H, m), 4.30 (C11, 1 H, dd, J = 10.9, 4.4 Hz), 4.14 (C11, 1 H, dd, J = 11.0, 7.2 Hz),
4.12 (C20, 1 H, dd, J = 9.2, 3.0 Hz), 3.93 (C14, 2 H, brs), 3.79 (C7-OMe, 3 H, s), 3.67
(C5-OMe, 3 H, s), 3.54 (C20, 1 H, dd, J = 9.0, 7.4 Hz), 2.98-2.93 (C3, 1 H, m), 2.85 (C4-
Hb, dd, J = 15.4, 2.2 Hz), 2.30 (C4-Ha, dd, J = 15.1, 11.0 Hz), 2.20 (C6-Me, 3 H, s), 1.43

201
(Boc, 9 H, s) ppm; 13C NMR (125 MHz, CDCl3): δ 170.4 (C13), 155.7 (C16), 152.3 (C5),
150.2 (C7), 145.6 (C8), 138.6 (C21Ph-ipso), 137.7 (C22Ph-ipso), 128.4, 128.3, 128.0,
127.9, 127.5, 127.4, 127.2 (C9), 125.1 (C10), 123.6 (C6), 80.0 (C18), 74.6 (C22), 74.4
(C20), 73.0 (C21), 68.9 (C11), 60.4 (C7-OMe), 60.2 (C5-OMe), 53.9 (C1), 51.0 (C3),
42.4 (C14), 28.3 (C19), 27.4 (C4), 9.3 (C6-Me) ppm; HRMS calcd. for C35H45N2O8
(MH+) 621.3176 Da. Found 621.3186 Da.
tert-Butoxycarbonylamino-acetic acid 8-benzyloxy-1-benzyloxymethyl-2-(2-tert-
butoxycarbonylamino-acetyl)-5,7-dimethoxy-6-methyl-1,2,3,4-tetrahydro-
isoquinolin-3-ylmethyl ester (288)
OMeMe
MeOOBn
NNHBoc
OOBn
O
ONHBoc
To a stirred solution of amino alcohol 283 (75 mg, 0.162 mmol) and Boc-Gly-OH (85
mg, 0.485 mmol) in CH2Cl2 (0.81 mL) at 0 °C was added Py-BOP® (252 mg, 0.485
mmol), and after 5 min. Hünig’s base (0.260 mL, 1.45 mmol) was added. After 1 hour
stirring at 0 °C the solution was warmed to 25 °C and stirred 15 h. The reaction was
treated with H2O (5 mL) and extracted with EtOAc (2 x 15 mL) The combined extracts
were washed with aq. 1 N HCl (15 mL), sat. aq. NaHCO3 (15 mL), sat. aq. NaCl (15
mL), dried (Na2SO4), filtered, and concentrated under vacuum. The residue was purified
by flash column chromatography (SiO2, 20-40% EtOAc-hexane) to afford 288 (107 mg,

202
85% yield) as a pale yellow oil: Rf = 0.33 (50% EtOAc in hexanes); IR (thin film): 3419,
3362, 2977, 2936, 1716, 1652, 1498, 155, 1417, 1366, 1249, 1166, 1055 cm-1; 1H NMR
(300 MHz, CDCl3): δ 7.51-7.12 (10 H, m), 5.36 (1 H, brs), 5.23 (1 H, dd, J = 9.1, 4.1
Hz), 5.12-5.04 (3 H, m), 4.56 (1 H, brs), 4.45-4.34 (3 H, m), 4.29-4.13 (3 H, m), 3.90-
3.82 (2 H, m), 3.81 (3 H, s), 3.67 (3 H, s), 3.46 (1 H, t, J = 9.6 Hz), 3.32 (1 H, brs), 3.21
(1 H, dd, J = 16.4, 7.5 Hz), 2.61 (1 H, dd, J = 16.2, 9.7 Hz), 2.23 (3 H, s), 1.46 (9 H, s),
1.44 (9 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 169.8, 169.5, 155.5, 151.7, 149.8,
144.0, 137.3, 137.1, 128.5, 128.3, 128.2, 128.1, 128.0, 127.6, 127.5, 127.3, 125.5, 125.3,
121.6, 79.7, 79.0, 74.8, 72.8, 70.8, 65.6, 60.6, 60.2, 60.0, 50.5, 49.1, 42.7, 42.2, 28.1,
22.6, 9.3 ppm; HRMS calcd. for C42H56N3O11 (MH+) 778.3915 Da. Found 778.3924 Da.
[2-(8-Benzyloxy-1-benzyloxymethyl-3-hydroxymethyl-5,7-dimethoxy-6-methyl-3,4-
dihydro-1H-isoquinolin-2-yl)-2-oxo-ethyl]-carbamic acid tert-butyl ester (287)
OMe
Me
MeOOBn
NNHBoc
OOBn
OH
To a stirred solution of dicoupled product 288 (75 mg, 0.096 mmol) in DMSO (1.0 mL)
was added 1 N aq. NaOH (1.0 mL). The solution was stirred for 5 min., diluted with H2O
(10 mL), and extracted with EtOAc (3 x 20 mL). The combined extracts were dried
(Na2SO4), filtered, and concentrated under vacuum. The yellow residue was purified by
flash column chromatography (SiO2, 20-50% EtOAc-hexane) to afford amide 287 (36
mg, 60% yield) as a colorless oil: Rf = 0.42 (75% EtOAc in hexanes); IR (thin film):

203
3420, 2975, 2935, 1714, 1656, 1455, 1415, 1366, 1251, 1168, 1054 cm-1; 1H NMR (300
MHz, CDCl3, signals from minor, 20%, carbamate resonance isomer not reported): δ
7.53-7.17 (10 H, m), 6.38-6.34 (0.3 H, m), 5.56 (0.3 H, brs), 5.37 (0.7 H, brs), 5.24 (0.7
H, dd, J = 10.3, 4.6 Hz), 5.17-5.04 (1.7 H, m), 4.88 (0.3 H, d, J = 10.4 Hz), 4.66 (0.3 H,
d, J = 11.8 Hz), 4.42 (0.7 H, d, J = 11.6 Hz), 4.37-3.91 (5 H, m), 3.82 (3 H, s), 3.68 (3 H,
s), 3.61-3.39 (2 H, m), 3.21-2.97 (3 H, m), 2.83-2.73 (1 H, m), 2.06 (3 H, s), 1.47 (9 H, s)
ppm; 13C NMR (75 MHz, CDCl3): δ 170.0, 155.5, 151.8, 149.6, 143.8, 137.1, 136.8,
128.6, 128.4, 128.3, 128.2, 128.0, 127.8, 127.6, 125.6, 125.0, 124.8, 122.8, 79.1, 74.9,
73.0, 70.1, 64.4, 60.6, 60.0, 54.0, 53.0, 42.8, 28.2, 22.2, 9.2 ppm; 1H NMR (500 MHz,
toluene-d8, 100 °C): δ 7.40 (2 H, d, J = 7.6 Hz), 7.25 (2 H, d, J = 7.6 Hz), 7.13-7.96 (6 H,
m), 5.60 (1 H, brs), 5.43 (1 H, brs), 4.98 (1 H, d, J = 11.1 Hz), 4.91 (1 H, d, J = 11.2 Hz),
4.28-4.20 (4 H, m), 3.99 (1 H, d, J = 14.4 Hz), 3.80 (1 H, brs), 3.59 (3 H, s), 3.53-3.43 (3
H, m), 3.39 (3 H, s), 2.98 (1 H, brs), 2.66-2.61 (2 H, m), 2.19 (3 H, s), 1.41 (9 H, s) ppm;
HRMS calcd. for C35H45N2O8 (MH+) 621.3176 Da. Found 621.3170 Da.

204
Optimized Formation of [2-(8-Benzyloxy-1-benzyloxymethyl-3-hydroxymethyl-5,7-
dimethoxy-6-methyl-3,4-dihydro-1H-isoquinolin-2-yl)-2-oxo-ethyl]-carbamic acid
tert-butyl ester (287)
OMe
Me
MeOOBn
NNHBoc
OOBn
OH
To a stirred solution of amino alcohol 283 (6.6 g, 14.2 mmol) in THF (71 mL) at 0 °C
was added triethylamine (5.95 mL, 42.7 mmol) then freshly distilled TMS-Cl (3.70 mL,
29.2 mmol) dropwise within 10 min. A white precipitate formed. The reaction was
warmed to 25 °C and stirred for 3 h.
To a stirred solution of Boc-Gly-OH (2.74 g, 15.7 mmol) and triethylamine (6.0 mL, 42.7
mmol) in THF (78 mL) at −20 °C was added pivalyl chloride (1.93 mL, 15.7 mmol)
dropwise within 10 min. The reaction was stirred for 2.5 h, then cooled to −65 °C. The
above solution of disilylated 290 was added with filtration (cotton plug) within 30 min.
The reaction was stirred at −65 °C to −10 °C for 3 h, warmed to 25 °C and stirred for 10
h. The reaction was treated with aq. 1 N HCl (100 mL), stirred vigorously for 1 min. and
extracted with EtOAc (3x 150 mL). The combined extracts were washed with aq. 1 N
HCl (100 mL), H2O (150 mL), aq. sat. NaHCO3 (150 mL), sat. aq. NaCl (150 mL), dried
(Na2SO4), filtered, and concentrated under vacuum. The resulting pale yellow oil was
purified by flash column chromatography (SiO2, 30-60% EtOAc-hexane) to afford amide
287 (7.2 g, 82% yield) as a colorless oil: characterized as above.

205
1-(8-Benzyloxy-1-benzyloxymethyl-3-hydroxymethyl-5,7-dimethoxy-6-methyl-3,4-
dihydro-1H-isoquinolin-2-yl)-2,2-dimethyl-propan-1-one (292)
Me
MeOOBn
OMe
N
OBn
H
H
O
OH
The reaction was conducted as above but scaled for 277 mg of amino alcohol 283 and the
addition to mixed anhydride 289 was done at −20 °C, which afforded 287 (212 mg, 57%
yield, characterized above) and tert-butyl amide 292 (69 mg, 21% yield) as a colorless
oil: 292: Rf = 0.19 (50% EtOAc in hexanes); IR (thin film): 3434, 2935, 2860, 1625,
1455, 1412, 1366, 1115, 1080 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.46-7.36 (5 H, m),
7.28-7.24 (3 H, m), 7.19-7.15 (2 H, m), 6.03 (1 H, t, J = 7.2 Hz), 5.22 (1 H, d, J = 11.1
Hz), 4.92 (1 H, d, J = 11.1 Hz), 4.43 (1 H, d, J = 11.9 Hz), 4.37 (1 H, d, J = 11.8 Hz),
4.35 (1 H, brs), 3.87 (1 H, dd, J = 11.6, 3.3 Hz), 3.79 (3 H, s), 3.69 (3 H, s), 3.67-3.57 (3
H, m), 3.49 (1 H, brs), 3.23 (1 H, dd, J = 16.2, 7.6 Hz), 2.65 (1 H, dd, J = 16.4, 9.6 Hz),
2.25 (3 H, s), 1.30 (9 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 179.4, 151.9, 149.8,
144.1, 137.8, 137.1, 128.2, 128.1, 127.6, 127.5, 126.9, 124.9, 122.6, 74.6, 73.0, 70.7,
66.7, 60.4, 60.1, 54.2, 50.2, 39.5, 28.5, 25.8, 9.2 ppm; HRMS calcd. for C33H42NO6
(MH+) 548.3012 Da. Found 548.3019 Da.

206
Benzyloxy-6-benzyloxymethyl-1-hydroxy-8,10-dimethoxy-9-methyl-4-oxo-
1,3,4,6,11,11a-hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic acid tert-butyl
ester (293)
OMe
Me
MeOOBn
NNBoc
OOBn
OHH
H
To a stirred solution of oxalyl chloride (73 μL, 0.834 mmol) in CH2Cl2 (3.6 mL) at −78
°C was added DMSO (119 μL, 1.67 mmol). The reaction was stirred for 10 min. and
amide 287 (0.42 g, 0.642 mmol) was added as a solution in CH2Cl2 (0.5 mL) within 10
min. After 8 min. triethylamine (0.447 mL, 3.21 mmol) was added dropwise. The
reaction was stirred at −78 °C for 10 min. and the solution allowed to warm for 8 min.
(when the reaction mixture was allowed to completely warm to 25 °C and stir for more
than 15 to 30 minutes, degradation was observed). The reaction mixture was diluted with
H2O (15 mL) and extracted with CH2Cl2 (3 x 25 mL). The combined extracts were
washed with sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The crude oil was purified by flash column chromatography (SiO2, 20-40%
EtOAc-hexane) to afford hemi-aminal 293 (0.364 g, 87% yield) as a pale yellow oil and a
3:2 mixture of diastereomers: Rf = 0.23/0.18 (40% EtOAc in hexanes); IR (thin film):
3400, 2976, 2936, 2869, 1700, 1653. 1455, 1413, 1393, 1367, 1339, 1164, 1115, 1061
cm-1; 1H NMR (300 MHz, CDCl3, only listed signals for dominant isomer): δ 7.44-7.28
(5 H, m), 7.25-7.19 (3 H, m), 7.15-7.05 (2 H, m), 5.94 (1 H, t, J = 4.9 Hz), 5.59 (0.6 H,
brs), 5.47 (0.4 H, brs), 4.98 (2 H, s), 4.52-4.38 (2 H, m), 4.23-4.18 (1 H, m), 3.95 (1 H, d,
J = 6.6 Hz), 3.88 (1 H, d, J = 17.5 Hz), 3.81 (3 H, s), 3.80- 3.70 (1 H, m), 3.69 (3 H, s),

207
3.59-3.38 (2 H, m), 3.19-3.01 (2 H, m), 2.26 (3 H, s), 1.50 (9 H, s) ppm; 13C NMR (75
MHz, CDCl3): δ 171.0, 165.9, 153.8, 151.3, 150.3, 144.5, 137.6, 137.0, 128.4, 128.3,
128.2, 128.0, 127.8, 127.5, 127.3, 127.2, 126.4, 124.8, 122.9, 81.2, 75.2, 74.6, 72.6, 71.7,
64.2, 60.7, 60.1, 55.5, 47.7, 46.0, 28.1, 23.7, 9.2 ppm; HRMS calcd. for C35H43N2O8
(MH+) 619.3019 Da. Found 619.3016 Da.
7-Benzyloxy-6-benzyloxymethyl-1,8,10-trimethoxy-9-methyl-4-oxo-1,3,4,6,11,11a-
hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic acid tert-butyl ester (297)
OMe
Me
MeOOBn
NNBoc
OOBn
OMeH
H
To a stirred solution of 293 (73 mg, 0.118 mmole) and trimethylorthoformate (0.30 mL)
in MeOH (1.2 mL) at 0 °C was added (1S)-(+)-10-camphorsulfonic acid (55 mg, 0.236
mmole). The solution was stirred 30 min at 0 °C, then quenched with sat. aq. NaHCO3
(10 mL) and extracted with EtOAc (2 x 15 mL). The combined extracts were washed
with sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under vacuum to
afford 297 (71 mg, 95% yield) as a pale yellow oil: Rf = 0.20 (30% EtOAc in hexanes);
IR (thin film): 2971, 2936, 2862, 1704, 1653, 1455, 1393, 1367, 1328, 1165, 1118, 1069,
907 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.39-7.27 (5 H, m), 7.21-7.12 (5 H, m), 5.77-
5.71 (1 H, m), 5.39 (0.6 H, s), 5.19 (0.4 H, s), 5.06 (1 H, d, J = 11.0 Hz), 5.00-4.97 (1 H,
m), 4.52-4.46 (1 H, m), 4.41 (1 H, d, J = 12.1 Hz), 4.30-4.17 (1 H, m), 3.91-3.80 (4 H,
m), 3.71-3.66 (5 H, m), 3.37-3.24 (4 H, m), 3.02-2.85 (2 H, m), 2.25 (3 H, s), 1.51 (9 H,

208
s) ppm; 13C NMR (75 MHz, CDCl3): δ 164.5, 153.9, 153.0, 151.1, 150.9, 150.2, 144.8,
138.6, 138.5, 136.9, 128.4, 128.2, 128.1, 128.0, 127.8, 127.6, 127.5, 127.1, 124.3, 124.1,
81.8, 81.1, 80.5, 75.1, 74.5, 72.5, 71.6, 69.4, 60.9, 60.2, 56.4, 55.3, 54.9, 49.2, 45.0, 44.1,
28.1, 25.0, 9.2 ppm; HRMS calcd. for C36H45N2O8 (MH+) 633.3176 Da. Found 633.3173
Da.
7-Benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-
1,3,4,6,11,11a-hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic acid tert-butyl
ester (300)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
H
To a stirred solution of oxalyl chloride (1.32 mL, 15.1 mmol) in CH2Cl2 (66 mL) at −78
°C was added DMSO (2.14 mL, 30.2 mmol). The reaction was stirred for 10 min. and
amide 187 (7.2 g, 11.6 mmol) was added as a solution in CH2Cl2 (15 mL) within 10 min.
After 8 min. triethylamine (4.85 mL, 34.8 mmol) was added dropwise. The reaction was
stirred at −78 °C for 10 min. and the solution was allowed the solution to warm for 8 min.
(when the reaction mixture was allowed to completely warm to 25 °C and stir for more
than 15 to 30 minutes, degradation was observed). The reaction mixture was diluted with
H2O (100 mL) and extracted with CH2Cl2 (3 x 150 mL). The combined extracts were
washed with sat. aq. NaCl (100 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The product was used directly for thioaminal formation.

209
To a stirred solution of the crude product mixture of hemi-aminal 293 (11.6 mmol) in
CH2Cl2 (60 mL) at 0 °C was added thiophenol (6.0 mL, 58.0 mmol) and
p-toluenesulfonic acid monohydrate (2.65 g, 13.9 mmol). The reaction was stirred for 1.5
h at 0 °C to 5 °C, quenched with sat. aq. NaHCO3 (100 mL), and extracted with CH2Cl2
(3 x 150 mL). The combined extracts were washed with sat. aq. NaCl (150 mL), dried
(Na2SO4), filtered, and concentrated under vacuum. The resulting yellow oil was purified
by flash column chromatography (SiO2, 10-30% EtOAc-hexane) to afford thioaminal 300
(7.1 g, 86% yield over two steps) as a white solid: recrystallized from EtOAc and
hexanes; X-ray (see Appendix E); mp = 162-164 °C; Rf = 0.26 (30% EtOAc in hexanes);
IR (thin film): 2977, 2936, 2864, 1702, 1658, 1455, 1415, 1391, 1368, 1305, 1163, 1118,
cm-1; 1H NMR (300 MHz, CDCl3): δ 7.57-7.52 (2 H, m), 7.42-7.24 (8 H, m), 7.20-7.12 (5
H, m), 5.98 (0.5 H, d, J = 2.5 Hz), 5.81-5.75 (1 H, m), 5.65 (0.5 H, d, J = 2.5 Hz), 5.06-
4.96 (2 H, m), 4.53-4.40 (2 H, m), 4.20-4.05 (2 H, m), 3.84 (1.5 H, s), 3.83 (1.5 H, s),
3.82-3.77 (1 H, m), 3.72 (1.5 H, s), 3.67 (1.5 H, s), 3.70-3.55 (2 H, m), 3.14-3.06 (2 H,
m), 2.28 (1.5 H, s), 2.26 (1.5 H, s), 1.26 (4.5 H, s), 1.15 (4.5 H, s) ppm; 1H NMR (500
MHz, DMSO-d6, 130 °C, signals were still significantly broad at 90 °C): δ 7.56-7.54 (2
H, m), 7.39-7.27 (8 H, m), 7.22-7.18 (3 H, m), 7.13-7.12 (2 H, m), 5.87 (1 H, brs), 5.76
(1 H, t, J = 4.4 Hz), 5.02 (1 H, d, J = 11.4 Hz), 4.95 (1 H, d, J = 11.2 Hz), 4.41 (1 H, d, J
= 12.0 Hz), 4.36 (1 H, d, J = 12.2 Hz), 4.13 (1 H, d, J = 17.8 Hz), 3.91 (1 H, d, J = 17.8
Hz), 3.77 (3 H, s), 3.73 (1 H, d, J = 12.2 Hz), 3.69-3.63 (2 H, m), 3.65 (3 H, s), 3.15 (1 H,
dd, J = 14.8, 3.3 Hz), 2.99 (1 H, t, J = 14.5 Hz), 2.20 (3 H, s), 1.25 (9 H, s) ppm; 13C
NMR (125 MHz, DMSO-d6, 130 °C): δ 163.7, 150.7, 149.5, 143.8, 137.9, 136.8, 133.2,
128.3, 127.5, 127.3, 127.2, 127.0, 126.4, 126.3, 125.8, 123.2, 122.9, 79.9, 73.8, 71.7,
59.9, 59.3, 56.3, 48.2, 27.1, 25.4, 8.5 ppm; HRMS calcd. For C41H47N2O7S (MH+)
711.3104 Da. Found 711.3113 Da.

210
3-Allyl-7-benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-1-
phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic
acid tert-butyl ester (301)
MeO
Me
MeOO
N
O
SPh
PhPh
17
5
3
9
10
1820
Ha Hb
NH
O
O
O11
1314
15
17
16
19
24
23
Hb
Ha
21
H
To a stirred solution of thioaminal 300 (30 mg, 0.042 mmol) in THF (0.33 mL) at −78 °C
was added LiHMDS (0.57 M in THF, 86 µL, 0.049 mmol) dropwise within 5 min. The
yellow solution was stirred for 15 min. at −78 °C, and allyl bromide (4.1 μL, 0.049
mmol) was added. After 20 min. the reaction was quenched with sat. aq. NH4Cl (5 mL)
and extracted with EtOAc (2 x 10 mL). The combined extracts were washed with sat. aq.
NaCl (5 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The resulting
pale yellow oil was purified by flash column chromatography (SiO2, 10-30% EtOAc-
hexane) to afford product 301 (22 mg, 70% yield) as a white solid: recrystallized from
benzene and hexanes; X-ray (see Appendix F); mp = 119-122 °C; Rf = 0.38 (30% EtOAc
in hexanes); IR (thin film): 2976, 2935, 2865, 1703, 1661, 1455, 1414, 1368, 1338, 1289,
1163, 1115 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.57-7.15 (15 H, m), 5.92 (1 H, t, J =
5.2 Hz), 5.73-5.54 (2 H, m), 5.06-4.92 (4 H, m), 4.53-4.36 (3 H, m), 3.85-3.77 (6 H, m),
3.71-3.52 (4 H, m), 3.26-3.10 (1 H, m), 3.05-2.85 (1 H, m), 2.66 (1 H, brs), 2.25 (3 H, s),
1.41 (4.5 H, s), 1.14 (4.5 H, s) ppm; 1H NMR (500 MHz, toluene-d8, 100 °C): δ 7.55
(PhS-ortho, 2 H, d, J = 6.7 Hz), 7.10-6.94 (13 H, m), 6.23 (C1, 1 H, t, J = 5.2 Hz), 5.79-
5.71 (C11, C16, 2 H, m), 5.07 (C20, 1 H, d, J = 11.0 Hz), 4.99 (C20, 1 H, d, J = 11.0 Hz),

211
4.98-4.95 (C17-Ha, 1 H, m), 4.83-4.80 (C17-Hb, 1 H, m), 4.59 (C13, 1 H, brs), 4.41 (C19,
1 H, d, J = 12.0 Hz), 4.34 (C19, 1 H, d, J = 12.1 Hz), 4.03 (C18, 2 H, d, J = 5.0 Hz),
3.85-3.80 (C3, 1 H, m), 3.61 (C7-OMe, 3 H, s), 3.40-3.34 (C4-Ha, 1 H, m), 3.36 (C5-
OMe, 3 H, s), 2.98 (C4-Hb, 1 H, dd, J = 14.7, 3.4 Hz), 2.80-2.73 (C15, 2 H, m), 2.23 (C6-
Me, 3 H, s), 1.24 (C24, 9 H, s) ppm; 13C NMR (125 MHz, toluene-d8, 100 °C): δ 168.2,
153.5, 152.5, 151.5, 146.1, 139.8, 138.5, 137.9, 137.5, 135.2, 134.8, 134.4 (C16), 129.3,
128.9, 128.6, 128.4, 128.0, 127.7, 127.3, 125.5, 124.8, 124.1, 118.1 (C17), 81.2 (C23),
75.8 (C20), 73.7 (C18), 73.4 (C19), 69.5 (C11), 60.5 (C5-OMe), 60.1 (C7-OMe), 59.1
(C13), 56.9 (C3), 50.3 (C1), 38.9 (C15), 28.4 (C24), 27.0 (C4), 9.6 (C6-Me) ppm; HRMS
calcd. for C44H51N2O7S (MH+) 751.3417 Da. Found 751.3410 Da.
3-Allyl-7-benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-1-
phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic
acid tert-butyl ester (302)
MeO
Me
MeOO
N
O
SPh
PhPh
17
5
3
9
10
1820
Ha Hb
NH
O
O
O11
1314
15
17
16
19
24
23
Hb
Ha
21
H
To a solution of 301 (75 mg, 0.10 mmol) in THF (1.2 mL) at −78 °C was added t-BuLi
(0.9 M in pentane, 0.28 mL, 0.25 mmol). Upon addition of t-BuLi the solution became
orange in color. Within 10 seconds of complete t-BuLi addition the reaction was
quenched with a solution of butylated hydroxyltoluene (0.110 g, 0.50 mmol) in THF (0.5

212
mL) and the solution became pale yellow in color. The solution was diluted with H2O (10
mL) and extracted with EtOAc (3 x 10 mL). The extracts were combined, washed with
sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The
residue was purified by flash column chromatography (SiO2, 10-20% EtOAc-hexane) to
yield 302 (0.145 g, 73% yield) as a colorless oil: Rf = 0.41 (30% EtOAc in hexanes); IR
(thin film): 2977, 2935, 2863, 1696, 1653, 1455, 1413, 1382, 1316, 1162, 1117 cm-1; 1H
NMR (300 MHz, CDCl3): δ 7.47-7.45 (2 H, m), 7.35-7.23 (8 H, m), 7.22-7.15 (5 H, m),
6.17 (0.5 H, s), 6.08-5.94 (0.5 H, m), 5.93-5.73 (2 H, m), 5.01-4.93 (3 H, m), 4.91-4.79 (1
H, m), 4.56-4.51 (0.5 H, m), 4.47 (2 H, s), 4.44-4.40 (0.5 H, m), 3.81 (3 H, s), 3.76 (1.0
H, d, J = 5.2 Hz), 3.68 (1.0 H, d, J = 3.9 Hz), 3.65-3.45 (4 H, m), 3.20-2.91 (3 H, m),
2.80-2.61 (1 H, m), 2.22 (3 H, s), 1.40 (6 H, s), 1.27 (3 H, s) ppm; 1H NMR (500 MHz,
toluene-d8, 100 °C): δ 7.52 (PhS-ortho, 2 H, d, J = 7.7 Hz), 7.09-6.95 (13 H, m), 6.26
(C16, 1 H, brs), 6.12 (C1, 1 H, t, J = 4.8 Hz), 6.08 (C11, 1 H, brs), 5.08 (C17-Ha, 1 H, d,
J = 16.5 Hz), 5.03 (C20, 2 H, s), 4.99 (C17-Hb, 1 H, d, J = 9.4 Hz), 4.50 (C13, 1 H, brs),
4.40 (C19, 1 H, d, J = 12.0 Hz), 4.30 (C19, 1 H, d, J = 12.1 Hz), 3.98 (C18, 1 H, dd, J =
9.9, 5.3 Hz), 3.83 (C18, 1 H, dd, J = 9.8, 4.0 Hz), 3.66 (C7-OMe, 3 H, s), 3.36 (C5-OMe,
3 H, s), 3.35-3.29 (C15, C3, C4-Ha, 3 H, m), 2.97-2.87 (C15, C4-Hb, 2 H, m), 2.25 (C6-
Me, 3 H, s), 1.32 (C24, 9 H, s) ppm; 13C NMR (125 MHz, toluene-d8, 100 °C): δ 167.8,
153.6, 152.4, 151.5, 145.7, 139.7, 138.3, 137.9, 137.5, 135.3, 132.7 (C16), 129.3, 128.9,
128.6, 128.4, 128.0, 127.7, 127.4, 125.5, 124.8, 124.7, 116.1 (C17), 81.3 (C23), 75.5
(C20), 73.9 (C18), 73.6 (C19), 60.6 (C5-OMe), 60.2 (C7-OMe), 58.6 (C3), 57.8 (C13),
50.6 (C1), 41.2 (C15), 28.5 (C24), 28.2 (C4), 9.6 (C6-Me) ppm; HRMS calcd. for
C44H51N2O7S (MH+) 751.3417 Da. Found 751.3419 Da.

213
(3-Bromo-propoxy)-triisopropyl-silane (304)
Br OTIPS
To a stirred solution of 3-bromopropanol (2.6 mL, 28.8 mmol), triethylamine (8.0 mL,
57.7 mmol), and dimethylaminopyridine (0.70 g, 5.76 mmol) in CH2Cl2 (144 mL) at 0 °C
was added TIPS-Cl (7.3 mL, 34.5 mmol) dropwise. The reaction was warmed to 25 °C
and stirred for 16 h, then treated with aq. 1 N HCl (100 mL), and extracted with CH2Cl2
(3 x 50 mL). The combined extracts were washed with washed with sat. aq. NaCl (50
mL), dried (Na2SO4), filtered, and concentrated under vacuum. The resulting pale yellow
oil was purified by flash column chromatography (SiO2, 1-2% EtOAc-hexane) then by
distillation (96 °C, 1 mmHg) to afford the TIPS ether 304 as a colorless liquid (4.1 g,
48% yield): Rf = 0.34 (2% EtOAc in hexanes); IR (neat): 2943, 1463, 1382, 1261, 1148,
1106, 1062, 952, 882, 733, 681 cm-1; 1H NMR (300 MHz, CDCl3): δ 3.82 (2 H, t, J = 5.5
Hz), 3.56 (2 H, t, J = 6.5 Hz), 2.06 (2 H, quintet, J = 6.1 Hz), 1.09-1.03 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 60.5, 35.6, 30.6, 17.8, 11.7 ppm; HRMS calcd. for
C12H28BrOSi (MH+) 297.1072 Da. Found 297.1075 Da.
(3-Iodo-propoxy)-triisopropyl-silane (306)
I OTIPS
To a stirred solution of TIPS ether 304 (4.1 g, 13.9 mmol) in acetone (35 mL) was added
NaI (20.9 g, 139.4 mmol). The reaction was heated at reflux for 3 h and cooled to 25 °C

214
then stirred for another 12 h. The solution was diluted with H2O (75 mL) and extracted
with Et2O (3 x 100 mL). The combined extracts were washed with sat. aq. NaCl (75 mL),
dried (Na2SO4), filtered, and concentrated under vacuum to afford pure iodide 3067 (4.0
g, 84% yield) as a pale yellow oil: Rf = 0.40 (2% EtOAc in hexanes); IR (neat): 2938,
1462, 1385, 1261, 1150, 1108, 955, 884, 736, 678 cm-1; 1H NMR (300 MHz, CDCl3): δ
3.75 (2 H, t, J = 5.4 Hz), 3.32 (2 H, t, J = 6.6 Hz), 2.01 (2 H, quintet, J = 6.2 Hz), 1.10-
1.03 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 62.4, 36.3, 17.8, 11.7 ppm; HRMS
calcd. for C12H28IOSi (MH+) 342.0876 Da. Found 342.0892 Da.
7-Benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-3-
(3-triisopropylsilanyloxy-propyl)-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-
b]isoquinoline-2-carboxylic acid tert-butyl ester (305)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
HOTIPS
To a stirred solution of thioaminal 300 (52 mg, 0.075 mmol) in THF (0.40 mL) at −78 °C
was added KHMDS (0.5 M in toluene, 180 µL, 0.090 mmol) dropwise within 5 min. The
yellow, clear solution was stirred for 5 min. and bromide 304 (20 µL, 0.090 mmol) was
added dropwise. The reaction was allowed to warm to 25 °C and stirred 30 min. The
reaction was quenched with sat. aq. NaHCO3 (5 mL) and extracted with EtOAc (3 x 10
mL). The combined extracts were washed with sat. aq. NaCl (10 mL), dried (Na2SO4),
filtered, and concentrated under vacuum. The resulting residue was purified by flash

215
column chromatography (SiO2, 5-10% EtOAc-hexane) to afford alkylated product 305
(33 mg, 48% yield) as a colorless oil and starting material 300 (16 mg, 30% yield). 305:
Rf = 0.34 (20% EtOAc in hexanes); IR (thin film): 2940, 2865, 1701, 1663, 1457, 1414,
1367, 1337, 1304, 1164, 1106 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.60-7.15 (15 H, m),
5.98 (1 H, t, J = 5.3 Hz), 5.71 (1 H, brs), 5.03 (2 H, s), 4.51 (2.5 H, brs), 4.30 (0.5 H, brs),
3.95-3.84 (2 H, m), 3.82 (3 H, s), 3.64-3.54 (6 H, m), 3.23-3.14 (1.5 H, m), 3.02-2.90 (0.5
H, m), 2.27 (3 H, s), 2.05-1.95 (1 H, m), 1.80-1.48 (3 H, m), 1.42 (6 H, brs), 1.16 (3 H,
brs), 1.06-0.99 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 168.3, 151.1, 150.2, 145.3,
138.3, 137.4, 134.6, 133.1, 128.9, 128.8, 128.4, 128.2, 128.1, 128.0, 127.8, 127.7, 127.3,
127.2, 126.9, 124.6, 122.8, 81.1, 74.7, 72.4, 62.9, 60.7, 60.0, 55.7, 48.4, 28.1, 27.9, 17.8,
17.5, 13.5, 11.7, 9.2 ppm; 1H NMR (500 MHz, DMSO-d6, 90 °C): δ 7.52 (2 H, d, J = 7.6
Hz), 7.43 (2 H, d, J = 7.2 Hz), 7.39-7.27 (6 H, m), 7.19-7.16 (3 H, m), 7.11-7.09 (2 H,
m), 5.83 (1 H, t, J = 5.4 Hz), 5.79 (1 H, s), 4.99 (1 H, d, J = 10.9 Hz), 4.91 (1 H, d, J =
10.9 Hz), 4.42 (1 H, d, J = 12.3 Hz), 4.39 (1 H, d, J = 12.1 Hz), 4.14 (1 H, dd, J = 7.5, 4.2
Hz), 3.95 (1 H, ddd, J = 11.9, 4.0, 1.6 Hz), 3.78-3.74 (1 H, m), 3.76 (3 H, s), 3.64 (1 H,
dd, J = 9.4, 6.9 Hz), 3.61-3.59 (2 H, m), 3.60 (3 H, s), 3.19 (1 H, brs), 3.05-2.99 (1 H, m),
2.18 (3 H, s), 1.90-1.78 (2 H, m), 1.61-1.54 (1 H, m), 1.48-1.39 (1 H, m), 1.27 (9 H, s),
1.01-0.91 (21 H, m) ppm; HRMS calcd. for C53H73N2O8SiS (MH+) 925.4857 Da. Found
925.4833 Da.

216
Optimized formation of 7-Benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-
4-oxo-1-phenylsulfanyl-3-(3-triisopropylsilanyloxy-propyl)-1,3,4,6,11,11a-
hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic acid tert-butyl ester (305)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
HOTIPS
To a stirred solution of thioaminal 300 (3.04 g, 4.28 mmol) in THF (21 mL) at −78 °C
was added KHMDS (0.5 M in toluene, 10.3 mL, 5.14 mmol) dropwise within 5 min. The
pale orange, clear solution was stirred for 5 min. and iodide 306 (1.9 g, 5.56 mmol) was
added dropwise. No color change was observed. The reaction appeared complete by TLC
analysis after 10 min. The reaction was quenched with water (10 mL), diluted with sat.
aq. NH4Cl (75 mL), and extracted with EtOAc (3 x 150 mL). The combined extracts were
washed with sat. aq. NaCl (100 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The resulting pale yellow oil was purified by flash column chromatography
(SiO2, 5-10% EtOAc-hexane) to afford alkylated product 305 (3.10 g, 78% yield) as a
colorless oil and starting material 300 (0.31 g, 10% yield).

217
7-Benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-3-
(3-triisopropylsilanyloxy-propyl)-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-
b]isoquinoline-2-carboxylic acid tert-butyl ester (307)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
HOTIPS
To a stirred, degassed solution of 305 (0.20 g, 0.22 mmol) in THF (2.2 mL) at −78 °C
was added t-BuLi (1.2 M in pentane, 0.40 mL, 0.48 mmol). Upon addition of t-BuLi the
solution became brown/orange in color. Within 10 seconds of complete t-BuLi addition
the reaction was quenched with a solution of butylated hydroxyltoluene (0.19 g, 0.86
mmol) in THF (0.5 mL) and the solution became pale yellow in color. The solution was
diluted with H2O (15 mL) and extracted with EtOAc (3 x 15 mL). The extracts were
combined, washed with sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and concentrated
under vacuum. The residue was purified by flash column chromatography (SiO2, very
slow gradient of 5-10-20% EtOAc-hexane) to yield 307 (0.145 g, 73% yield) as a
colorless oil: Rf = 0.38 (20% EtOAc in hexanes); IR (thin film): 2941, 2865, 1698, 1653,
1456, 1414, 1384, 1367, 1310, 1163, 1116 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.52-
7.47 (2 H, m), 7.37-7.19 (13 H, m), 6.17 (0.5 H, s), 5.85-5.79 (1.5 H, m), 5.06-4.96 (2 H,
m), 4.50-4.45 (2.5 H, m), 4.34-4.30 (0.5 H, m), 3.83 (3 H, s), 3.79-3.48 (8 H, m), 3.20-
2.97 (2 H, m), 2.38-2.10 (2 H, m), 2.25 (3 H, s), 2.05-1.84 (2 H, m), 1.43 (6 H, brs), 1.27
(3 H, brs), 1.07-0.94 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 168.3, 153.3, 151.0,
150.2, 144.6, 138.5, 137.1, 133.3, 131.2, 128.8, 128.7, 128.5, 128.3, 128.1, 127.8, 127.7,
127.2, 126.9, 126.8, 124.4, 123.8, 81.3, 74.4, 72.7, 72.3, 63.3, 62.2, 60.8, 60.2, 57.7, 56.7,

218
49.4, 33.1, 31.8, 31.4, 27.9, 27.7, 26.8, 17.9, 11.8, 9.2 ppm; 1H NMR (500 MHz, DMSO-
d6, 120 °C): δ 7.53-7.51 (2 H, m), 7.38-7.27 (8 H, m), 7.23-7.18 (3 H, m), 7.16-7.14 (2 H,
m), 6.01 (1 H, s), 5.78 (1 H, t, J = 5.2 Hz), 4.99 (1 H, d, J = 11.1 Hz), 4.95 (1 H, d, J =
11.0 Hz), 4.42 (1 H, d, J = 12.2 Hz), 4.37 (1 H, d, J = 12.3 Hz), 4.27 (1 H, t, J = 5.4 Hz),
3.77 (3 H, s), 3.69-3.59 (5 H, m), 3.61 (3 H, s), 3.03 (2 H, d, J = 7.5 Hz), 2.28-2.22 (1 H,
m), 2.19 (3 H, s), 1.98-1.88 (2 H, m), 1.67-1.62 (1 H, m), 1.34 (9 H, s), 1.11-1.01 (21 H,
m) ppm; 13C NMR (125 MHz, DMSO-d6, 120 °C): δ 167.2, 150.7, 149.5, 143.8, 138.0,
136.8, 131.3, 128.3, 127.6, 127.4, 127.2, 127.1, 126.6, 126.5, 126.4, 125.9, 123.2, 123.1,
80.1, 73.8, 72.0, 71.8, 62.7, 60.0, 59.4, 56.8, 48.3, 32.0, 30.7, 27.2, 26.1, 17.2, 17.1, 11.1,
8.6 ppm; HRMS calcd. for C53H73N2O8SiS (MH+) 925.4857 Da. Found 925.4824 Da.
7-Benzyloxy-6-benzyloxymethyl-3-(3-hydroxy-propyl)-8,10-dimethoxy-9-methyl-4-
oxo-1-phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-b]isoquinoline-2-
carboxylic acid tert-butyl ester (308)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
HOH
To a stirred, degassed solution of 305 (1.37 g, 1.48 mmol) in THF (15 mL) at −78 °C was
added t-BuLi (1.2 M in pentane, 2.71 mL, 3.26 mmol) within 30 sec. After 10 seconds
the orange solution was quenched with a solution of butylated hydroxyltoluene (1.31 g,
5.92 mmole) in THF (3.0 mL). The pale yellow solution was diluted with H2O (75 mL)
and extracted with EtOAc (3 x 100 mL). The combined extracts were washed with sat.

219
aq. NaCl (75 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The residue
was dissolved in CH3CN (15 mL), cooled to 0 °C, and 48% aq. HF (1.5 mL) added
dropwise. The solution became dark orange in color. After stirring for 30 min. at 0 °C the
reaction was quenched with sat. aq. NaHCO3 (100 mL) and extracted with EtOAc (3 x
100 mL). The combined extracts were washed with sat. aq. NaCl (75 mL), dried
(Na2SO4), filtered, and concentrated under vacuum. The resulting yellow residue was
purified by flash column chromatography (SiO2, 30-50% EtOAc-hexane) to afford
alcohol 308 (0.829 g, 83% yield) as a colorless oil: Rf = 0.28 (50% EtOAc in hexanes);
IR (thin film): 3472, 2936, 2866, 1696, 1652, 1454, 1414, 1388, 1368, 1336, 1262, 1162,
1117, 1059 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.53-7.42 (2 H, m), 7.33-7.17 (13 H,
m), 6.19 (0.5 H, s), 5.76-5.73 (1.5 H, s), 5.05-4.98 (2 H, m), 4.49 (2 H, s), 4.43-4.35 (1 H,
s), 3.84 (3 H, s), 3.81-3.45 (8 H, m), 3.25-2.97 (2 H, m), 2.61-2.57 (0.5 H, m), 2.41-2.37
(0.5 H, m), 2.25 (3 H, s), 1.95-1.85 (2.5 H, m), 1.64-1.61 (0.5 H, m), 1.43 (3 H, brs), 1.24
(6 H, brs) ppm; 13C NMR (75 MHz, CDCl3): δ 168.9, 152.3, 150.8, 150.3, 144.6, 144.5,
138.4, 136.8, 133.7, 132.4, 130.9, 129.6, 128.9, 128.7, 128.5, 128.1, 127.8, 127.7, 127.6,
128.2, 126.8, 126.7, 124.4, 123.4, 81.5, 74.5, 72.6, 72.1, 65.6, 62.1, 61.0, 60.7, 60.2, 57.5,
56.2, 54.1, 49.6, 33.3, 32.6, 31.4, 30.6, 28.1, 27.7, 26.8, 26.5, 9.2 ppm; 1H NMR (500
MHz, DMSO-d6, 120 °C): δ 7.54-7.53 (2 H, m), 7.39-7.27 (8 H, m), 7.27-7.18 (3 H, m),
7.16-7.14 (2 H, m), 6.01 (1 H, s), 5.78 (1 H, t, J = 5.2 Hz), 5.01 (1 H, d, J = 11.2 Hz),
4.95 (1 H, d, J = 11.1 Hz), 4.42 (1 H, d, J = 12.3 Hz), 4.38 (1 H, d, J = 12.3 Hz), 4.27 (1
H, t, J = 6.2 Hz), 3.90 (1 H, brs), 3.77 (3 H, s), 3.67-3.60 (3 H, m), 3.61 (3 H, s), 3.43-
3.40 (2 H, m), 3.04-3.00 (2 H, m), 2.28-2.20 (1 H, m), 2.19 (3 H, s), 1.92-1.75 (2 H, m),
1.59-1.54 (1 H, m), 1.35 (9 H, s) ppm; HRMS calcd. for C44H53N2O8S (MH+) 769.3523
Da. Found 769.3525 Da.

220
7-Benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-3-(3-oxo-propyl)-1-
phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-b]isoquinoline-2-carboxylic
acid tert-butyl ester (309)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
H O
To a solution of oxalyl chloride (0.139 mL, 1.60 mmol) in CH2Cl2 (7.0 mL) at −78 °C
was added DMSO (0.227 mL, 3.20 mmol). The reaction was stirred 10 min. and added
alcohol 308 (0.95 g, 1.23 mmol) as a solution in CH2Cl2 (1.5 mL) within 5 min. The
solution became dark orange and a precipitate formed. After 10 min. triethylamine (0.514
mL, 3.69 mmol) was added dropwise. The reaction was stirred at −78 °C for 5 min. and
allowed to warm for 9 min. The reaction was quenched with H2O (25 mL) and extracted
with CH2Cl2 (3 x 40 mL). The combined extracts were washed with sat. aq. NaCl (50
mL), dried (Na2SO4), filtered, and concentrated under vacuum. A small portion of
product was purified, for the purposes of characterization, by flash column
chromatography (SiO2, 20-30% EtOAc-hexane) to afford 309 as a colorless oil: Rf = 0.19
(30% EtOAc in hexanes); IR (thin film): 2976, 2937, 2863, 2721, 1722, 1694, 1651,
1454, 1415, 1385, 1368, 1336, 1291, 1162, 1117, 1078 cm-1; 1H NMR (300 MHz, CDCl3,
pronounced carbamate resonance led to two aldehyde signals for CHO): δ 9.74 (0.5 H, s),
9.65 (0.5 H, s), 7.50-7.42 (2 H, m), 7.34-7.10 (13 H, m), 6.21 (0.5 H, s), 5.82-5.75 (1.5 H,
m), 5.01 (2 H, s), 4.48 (2 H, s), 4.40-4.25 (1 H, m), 3.83 (3 H, s), 3.78-3.45 (6 H, m),
3.18-2.98 (2 H, m), 2.96-2.64 (2 H, m), 2.60-2.21 (2 H, m), 2.26 (3 H, s), 1.28 (4 H, s),
1.09 (5 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 201.9, 201.4, 167.8, 167.5, 153.1,

221
152.3, 150.8, 150.3, 144.6, 138.4, 137.0, 136.8, 133.1, 132.6, 132.5, 131.1, 129.6, 128.9,
128.5, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.6, 127.2, 127.0, 126.6, 126.2, 124.4,
123.6, 81.8, 81.4, 74.4, 72.2, 65.0, 61.7, 61.0, 60.8, 60.2, 57.5, 55.1, 53.2, 49.5, 42.3,
28.0, 27.8, 26.6, 14.0, 9.2 ppm; 1H NMR (500 MHz, DMSO-d6, 100 °C): δ 9.64 (1 H, s),
7.53 (2 H, d, J = 7.0 Hz), 7.38-7.28 (8 H, m), 7.23-7.19 (3 H, m), 7.16-7.14 (2 H, m),
6.04 (1 H, s), 5.76 (1 H, t, J = 5.1 Hz), 5.00 (1 H, d, J = 11.1 Hz), 4.95 (1 H, d, J = 11.0
Hz), 4.43 (1 H, d, J = 12.2 Hz), 4.38 (1 H, d, J = 12.2 Hz), 4.29 (1 H, t, J = 6.8 Hz), 3.77
(3 H, s), 3.70-3.59 (3 H, m), 3.63 (3 H, s), 3.12-3.08 (1 H, m), 3.05-3.01 (1 H, m), 2.72-
2.66 (1 H, m), 2.55-2.40 (2 H, m), 2.19 (3 H, s), 2.14-2.08 (1 H, m), 1.32 (9 H, s) ppm;
13C NMR (125 MHz, DMSO-d6, 100 °C): δ 201.6, 166.9, 150.7, 150.0, 143.9, 138.0,
136.8, 132.5, 131.6, 128.5, 127.7, 127.4, 127.2, 126.9, 126.6, 126.5, 125.9, 123.4, 123.1,
80.5, 73.8, 71.9, 71.8, 60.1, 59.5, 56.7, 54.9, 48.4, 41.0, 27.4, 27.3, 25.9, 8.7 ppm; HRMS
calcd. for C44H51N2O8S (MH+) 767.3366 Da. Found 767.3366 Da.

222
7-Benzyloxy-6-benzyloxymethyl-8,10-dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-3-
((E)-3-triisopropylsilanyloxy-allyl)-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-
b]isoquinoline-2-carboxylic acid tert-butyl ester (310)
OMe
Me
MeOOBn
NNBoc
OOBn
SPhH
HOTIPS
The crude aldehyde product 309 was dissolved in Et2O (12.3 mL) and triethylamine
(0.343 mL, 2.46 mmol), cooled to 0 °C, and freshly distilled TIPSOTf (0.495 mL, 1.85
mmol) added dropwise. The solution was warmed to 25 °C and stirred for 16 h. The
reaction was quenched with sat. aq. NaHCO3 (50 mL) and extracted with EtOAc (3 x 75
mL). The combined extracts were washed with sat. aq. NaCl (75 mL), dried (Na2SO4),
filtered, and concentrated under vacuum. The resulting yellow residue was purified by
flash column chromatography (SiO2, 5-10-20% EtOAc-hexane) to afford silyl ether 310
(1.01 g, 88% yield) as a colorless foam: Rf = 0.33 (20% EtOAc in hexanes); IR (thin
film): 2943, 2866, 1695, 1653, 1456, 1414, 1384, 1330, 1166, 1118 cm-1; 1H NMR (300
MHz, CDCl3): δ 7.51-7.48 (2 H, m), 7.39-7.16 (13 H, m), 6.39-6.11 (2 H, m), 5.85-5.76
(1 H, m), 5.06-4.84 (3 H, m), 4.50 (2 H, s), 4.46-4.42 (1 H, m), 3.83 (3 H, s), 3.77-3.48 (6
H, m), 3.21-2.82 (3 H, m), 2.76-2.58 (1 H, m), 2.23 (3 H, s), 1.46 (6 H, s), 1.39-1.26 (3
H), 1.05-0.85 (21 H, m) ppm; 13C NMR (75 MHz, CDCl3): δ 167.6, 153.4, 151.1, 150.3,
144.5, 141.6, 138.5, 137.0, 134.0, 132.6, 131.9, 130.5, 128.9, 128.7, 128.5, 128.3, 128.1,
127.8, 127.6, 127.2, 126.9, 126.5, 124.3, 123.8, 107.9, 81.3, 74.5, 72.7, 72.3, 62.2, 60.7,
60.1, 58.1, 57.5, 49.5, 33.5, 27.9, 26.8, 17.6, 11.7, 9.2 ppm; HRMS calcd. for
C53H71N2O8SiS (MH+) 923.4700 Da. Found 923.4702 Da.

223
aldehyde 311
OMeMe
MeOOBn
N
OOBn
N
HO
BocH
H
To a stirred solution of silyl ether 310 (23 mg, 0.025 mmol) in THF (1.0 mL) at 15 °C
was added AgBF4 as a solution in THF (0.3 mL from 44 mg AgBF4 in 1.0 mL THF). A
precipitate immediately formed. After 15 min. at 15 °C the reaction was warmed to 25 °C
and stirred for another 4 h. The reaction was quenched with sat. aq. NaHCO3 (2 mL) and
extracted with EtOAc (3 x 5 mL). The combined extracts were washed with sat. aq. NaCl
(5 mL), dried (Na2SO4), and concentrated under vacuum. The residue could be purified
by flash column chromatography (SiO2, 20-60% EtOAc-hexane) to afford aldehyde 311
(13 mg, 78% yield) as a colorless oil: Rf = 0.46 (60% EtOAc in hexanes); IR (thin film):
2975, 2936, 2868, 1726, 1700, 1663, 1456, 1412, 1368, 1317, 1161, 1114 cm-1; 1H NMR
(300 MHz, CDCl3): δ 9.56 (1 H, s), 7.38-7.27 (5 H, m), 7.22-7.19 (3 H, m), 7.05-7.02 (2
H, m), 5.43 (1 H, s), 5.02-4.95 (2 H, m), 4.63 (1 H, brs), 4.57 (1 H, brs), 4.29 (1 H, d, J =
11.8 Hz), 4.24 (1 H, d, J = 11.7 Hz), 4.15-4.09 (1 H, m), 3.81 (3 H, s), 3.69 (3 H, s), 3.66-
3.57 (1 H, m), 3.34 (1 H, dd, J = 9.4, 1.9 Hz), 3.08 (1 H, t, J = 7.9 Hz), 2.92 (1 H, dd, J =
14.3, 2.5 Hz), 2.71 (1 H, t, J = 12.9 Hz), 2.42-2.32 (1 H, m), 2.27 (3 H, s), 2.07-1.99 (1
H, m), 1.43 (9 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 198.9, 167.9, 150.9, 150.3,
144.2, 138.1, 136.9, 128.4, 128.2, 128.1, 127.8, 127.4, 126.4, 124.6, 124.3, 81.0, 74.4,
72.9, 71.4, 60.7, 60.1, 57.5, 56.8, 49.9, 48.5, 32.2, 28.0, 25.2, 9.3 ppm; HRMS calcd. for
C38H45N2O8 (MH+) 657.3176 Da. Found 657.3201 Da.

224
oxime 314
OMeMe
MeOOBn
N
OOBn
N
HN
BocH
H
HO
To a solution of hydroxylamine hydrochloride (12 mg, 0.165 mmol) in H2O (0.10 mL) at
0 °C was added KOAc (40 mg, 0.411 mmol) then aldehyde 311 (90 mg, 0.137 mmol) as
a solution in EtOH (1.0 mL). The reaction was warmed to 25 °C and stirred for 2 h. The
reaction mixture was extracted with Et2O (3 x 15 mL). The combined extracts were
washed with sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The resulting residue was purified by flash column chromatography (SiO2, 20-
40% EtOAc-hexane) to afford two fractions, each with predominantly one isomer: the
trans-oxime (50 mg, 45% yield) with Rf = 0.32 (50% EtOAc in hexanes) as a 1:4 mixture
of cis:trans oxime and cis-oxime (44 mg, 38% yield) as a white solid in 4:1 mixture of
cis:trans oxime: recrystallized from benzene and hexanes; X-ray (see Appendix G); mp =
168-169 °C; Rf = 0.30 (50% EtOAc in hexanes); IR (thin film): 3334, 2930, 2857, 1702,
1666, 1455, 1412, 1368, 1318, 1290, 1162, 1114 cm-1; 1H NMR (300 MHz, CDCl3): δ
7.37-7.31 (5 H, m), 7.27-7.18 (3 H, m), 7.06-7.01 (2 H, m), 6.78 (1 H, brs), 5.45 (1 H,
brs), 5.00-4.94 (2 H, m), 4.57-4.44 (2 H, m), 4.34-4.26 (2 H, m), 4.08-4.03 (1 H, m), 3.80
(3 H, s), 3.67 (3 H, s), 3.57 (1 H, brs), 3.38-3.25 (2 H, m), 2.94-2.78 (2 H, m), 2.38-2.25
(1 H, m), 2.26 (3 H, s), 2.19-1.95 (1 H, m), 1.46 (9 H, s) ppm; 13C NMR (75 MHz,
CDCl3): δ 168.0, 152.2, 150.9, 150.1, 144.3, 138.2, 128.2, 128.0, 127.9, 127.8, 127.3,

225
127.1, 125.3, 125.0, 124.2, 124.1, 80.9, 74.4, 72.7, 71.2, 60.7, 60.2, 49.9, 29.5, 28.1, 25.4,
9.2 ppm; HRMS calcd. for C38H46N3O8 (MH+) 672.3285 Da. Found 672.3298 Da.
diol 315
OMeMe
MeOOH
N
OOH
N
HO
BocH
H
To a stirred solution of silyl ether 310 (0.22 g, 0.238 mmol) in THF (7.0 mL) at 15 °C
was added AgBF4 as a solution in THF (1.7 mL from 1.0 g AgBF4 in 20 mL THF). A
precipitate immediately formed. After 15 min. at 15 °C the reaction was warmed to 25 °C
and stirred for another 4 h. The reaction was quenched with sat. aq. NaHCO3 (7 mL) and
extracted with EtOAc (3 x 15 mL). The combined extracts were washed with sat. aq.
NaCl (15 mL), dried (Na2SO4), filtered through a plug of silica gel, and concentrated
under vacuum. The resulting residue was carried to hydrogenolysis without further
purification. To a stirred solution of crude aldehyde 311 (156 mg, 0.238 mmol) in MeOH
(5 mL) at 25 °C was added Pd(OH)2 (moist, Pd content 20%, 64 mg) and the reaction
stirred under an atmosphere of H2 (balloon pressure) for 4 h. The reaction mixture was
filtered through a plug of Celite®, washed with MeOH, and concentrated under vacuum.
The residue was purified by flash column chromatography (SiO2, 50-100% EtOAc-
hexane, then 5% MeOH in EtOAc) to afford diol 315 (64 mg, 79% yield from
cyclization) as a colorless foam: Rf = 0.20 (100% EtOAc in hexanes); IR (thin film):

226
3384, 2937, 1700, 1684, 1653, 1472, 1417, 1368, 1162, 1115, 1055 cm-1; 1H NMR (300
MHz, CDCl3): δ 9.74 (1 H, s), 5.93 (1 H, brs), 5.63 (1 H, t, J = 4.3 Hz), 4.74 (1 H, s),
4.67 (1 H, brs), 3.97 (1 H, dd, J = 11.1, 3.6 Hz), 3.78 (3 H, s), 3.68 (3 H, s), 3.66-3.60 (1
H, m), 3.33 (1 H, t, J = 8.0 Hz), 3.07 (1 H, dd, J = 14.7, 2.2 Hz), 2.61 (1 H, dd, J = 14.4,
6.8 Hz), 2.57-2.47 (1 H, m), 2.41-2.37 (1 H, m), 2.24 (3 H, s), 2.21 (1 H, m), 1.43 (9 H, s)
ppm; HRMS calcd. for C24H33N2O8 (MH+) 477.2237 Da. Found 477.2219 Da.
aldehyde hydrate 316
MeOMe
MeOOH
N
OOH
NH
OHHO
H
HTFA
1
1511
16
137
5
3
18
9
Ha Hb
10
Ha
Hb17
To a stirred solution of diol 315 (40 mg, 0.084 mmol) in MeOH (1.0 mL) at 0 °C was
added 6 M HCl (1:1 H2O:MeOH, 1.0 mL). The solution was warmed to 25 °C, stirred for
3 h, and the solvents removed under vacuum. The white residue was used directly for
oxidation to quinone. Some material was purified by reverse phase HPLC (preparatory
column, rate = 8.0 mL/min, gradient = 100% to 70% H2O with 0.1% TFA in CH3CN
with 0.1% TFA over 20 min., injection volume = 1.0 mL, product elution from 17-19
min.) and lyophilized to afford 316 as white powder: IR (thin film): 3350, 2944, 1668,
1466, 1417, 1286, 1258, 1201, 1136, 1056 cm-1; 1H NMR (500 MHz, D2O): δ 5.31 (C1, 1
H, t, J = 3.0 Hz), 5.12 (C16, 1 H, d, J = 4.6 Hz), 4.37 (C11, 1 H, s), 4.28 (C13, 1 H, d, J =
6.4 Hz), 3.90 (C18, 1 H, dd, J = 11.8, 4.0 Hz), 3.79 (C3, 1 H, dt, J = 12.2, 2.4 Hz), 3.66

227
(C7-OMe, 3 H, s), 3.61 (C5-OMe, 3 H, s), 3.58 (C18, 1 H, dd, J = 11.7, 2.8 Hz), 2.98 (C4
Hb, 1 H, dd, J = 15.3, 2.6 Hz), 2.86-2.82 (C15, 1 H, m), 2.76 (C4 Ha, 1 H, dd, J = 15.1,
12.3 Hz), 2.44 (C14 Ha, 1 H, dd, J = 14.1, 9.1 Hz), 2.25 (C14 Hb , 1 H, dt, J = 14.2, 6.6
Hz), 2.13 (C6-Me, 3 H, s) ppm; 13C NMR (125 MHz, D2O, carbon signals for C6-10 are
inferred by chemical shift and analogous compounds): δ 167.4 (C17), 147.6 (C5), 144.8
(C7), 142.3 (C8), 124.9 (C9/10), 124.6 (C9/10), 118.6 (C6), 90.0 (C16), 62.1 (C18), 61.3
(C5-OMe), 60.8 (C7-OMe), 59.5 (C13), 59.1 (C11), 56.4 (C3), 52.3 (C1), 41.4 (C15),
31.6 (C14), 24.5 (C4), 9.0 (C6-Me) ppm; HRMS calcd. for C19H27N2O (MH+) 395.1818
Da. Found 395.1825 Da.
(±)-lemonomycinone amide (317)
OMe
MeOO
N
OOH
NH
OHHO
H
HTFA
1
1511
16
137
5
3
18
9
Ha Hb
10
Ha
Hb17
To a stirred solution of amine 316 (0.084 mmol) in H2O (1.0 mL) at 25 °C was added
ammonium cerium(IV) nitrate (0.138 g, 0.252 mmol) in one portion. The reaction was
monitored by reverse phase HPLC and after 4 hours no starting material remained. The
reaction solution was then purified without concentration on reverse phase HPLC (semi-
preparatory column, rate = 1.5 mL/min, gradient = 98% to 60% H2O with 0.1% TFA in
CH3CN with 0.1% TFA over 50 min., injection volume = 0.3 mL, product elution from
25-26 min.) and lyophilized to afford (±)-lemonomycinone amide (317) (14 mg, 35%

228
yield) as an orange residue: 1H NMR (500 MHz, D2O ): δ 5.13 (C16, 1 H, d, J = 4.7 Hz),
5.04 (C1, 1 H, brs), 4.38 (C11, 1 H, s), 4.31 (C13, 1 H, d, J = 6.3 Hz), 4.09 (C18, 1 H, dd,
J = 12.1, 3.4 Hz), 3.84 (C3, 1 H, dt, J = 11.5, 2.9 Hz), 3.84 (C7-OMe, 3 H, s), 3.51 (C18,
1 H, dd, J = 12.2, 2.1 Hz), 2.93 (C4-Hb, 1 H, dd, J = 16.5, 2.8 Hz), 2.81-2.77 (C15, 1 H,
m), 2.48-2.43 (C4-Ha, C14-Ha, 2 H, m), 2.26 (C14-Hb, 1 H, dt, J = 14.4, 6.5 Hz), 1.92
(C6-Me, 3 H, s) ppm; 13C NMR (125 MHz, D2O, carbon signals for C6-10 are inferred by
chemical shift and comparison with analogous compounds, no long-range carbon-
hydrogen correlation was conducted): δ 187.0 (C5), 181.5 (C8), 167.3 (C17), 155.8 (C7),
142.7 (C10), 136.5 (C9), 131.2 (C6), 89.9 (C16), 61.5 (C7-OMe), 61.0 (C18), 59.6 (C13),
58.7 (C11), 55.1 (C3), 52.2 (C1), 41.6 (C15), 31.6 (C14), 23.5 (C4), 8.7 (C6-Me) ppm;
HRMS calcd. for C18H23N2O7 (MH+) 379.1505 Da. Found 379.1535 Da.
5-Chloromethyl-2,4-dimethoxy-3-methyl benzaldehyde (322)
OMe
Me
MeO
HO
Cl
To a refluxing suspension of commercially available 2,4-dimethoxy-3-
methylbenzaldehyde (11.6 g, 64.5 mmol) and zinc chloride (9.3 g, 68.2 mmol) in 37%
aqueous formaldehyde (26 mL) was bubbled HCl gas for 90 min. The reaction solution
became homogeneous and dark orange in color. The reaction was cooled to 25 °C, poured
over ice, and extracted with Et2O (3 x 100 mL). The organic extracts were combined,
washed with aq. NaHCO3 (3 x 50 mL), sat. aq. NaCl (100 mL), dried (Na2SO4), filtered,

229
and concentrated under vacuum. The resulting brown oil was purified by flash column
chromatography (SiO2, 5-15% EtOAc-hexane) to afford benzyl chloride 322 (10.8 g,
73% yield) as a white solid: Rf = 0.41 (20% EtOAc in hexanes); mp = 67-68 °C; IR (thin
film): 2946, 2854, 1682, 1595, 1472, 1472, 1417, 1390, 1307, 1268, 1244, 1187, 1112,
1001 cm-1; 1H NMR (300 MHz, CDCl3): δ 10.29 (1 H, s), 7.79 (1 H, s), 4.63 (2 H, s),
3.92 (3 H, s), 3.90 (3 H, s), 2.28 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 188.7,
163.1, 128.4, 127.7, 125.9, 125.5, 63.1, 61.3, 40.6, 9.26 ppm; HRMS calcd. for
C11H14O3Cl (MH+) 229.0631 Da. Found 229.0637 Da.
Formic acid 5-chloromethyl-2,4-dimethoxy-3-methyl-phenyl ester (323)
OMe
Me
MeOO
Cl
O
H
To a stirred solution of benzyl chloride 322 (4.0 g, 17.5 mmol) in CH2Cl2 (35 mL) at 0 °C
was added m-CPBA (77% max., 6.6 g, 29.7 mmol max.) in small portions. The reaction
was warmed to 25 °C and stirred for 3 h. The reaction was treated with sat. aq. NaHCO3,
extracted with CH2Cl2 (3 x 40 mL). The combined organic extracts were dried (Na2SO4),
filtered, and concentrated under vacuum to afford the ester 323 (4.1 g, 95% yield) as a
yellow oil: Rf = 0.44 (20% EtOAc in hexanes); IR (thin film): 2944, 2831, 1747, 1484,
1457, 1417, 1319, 1243, 1218, 1100, 1005 cm-1; 1H NMR (300 MHz, CDCl3): δ 8.27 (1
H, s), 7.04 (1 H, s), 4.60 (2 H, s), 3.82 (3 H, s), 3.75 (3 H, s), 2.25 (3 H, s) ppm; 13C NMR

230
(75 MHz, CDCl3): δ 159.0, 155.4, 139.1, 126.8, 126.6, 121.3, 61.3, 60.5, 40.4, 9.7 ppm;
HRMS calcd. for C11H14O4Cl (MH+) 245.0581 Da. Found 245.0575 Da.
5-Chloromethyl-2,4-dimethoxy-3-methyl phenol (324)
OMe
Me
MeOOH
Cl
To a stirred solution of the ester 324 (4.1 g, 16.7 mmol) in THF (94 mL) at 0 °C was
added LiBH4 (0.36 g, 16.7 mmol) in small portions. After 10 min. analysis by TLC
showed that starting material was consumed. The reaction was treated with aq. 1 N HCl
(50 mL) and extracted with EtOAc (1 x 150 mL, 2 x 50 mL). The combined extracts were
washed with sat. aq. NaCl (50 mL), dried (Na2SO4), filtered, and concentrated under
vacuum to afford the phenol as an unstable orange oil. Rf = 0.22 (20% EtOAc in
hexanes); IR (thin film): 3405, 2942, 2834, 1596, 1485, 1457, 1418, 1346, 1307, 1248,
1197, 1114, 1049, 1005 cm-1; 1H NMR (300 MHz, CDCl3): δ 6.85 (1 H, s), 5.58 (1 H,
bs), 4.60 (2 H, s), 3.80 (3 H, s), 3.79 (3 H, s), 2.26 (3 H, s) ppm; 13C NMR (75 MHz,
CDCl3): δ 150.2, 146.1, 145.2, 129.5, 127.7, 126.7, 113.9, 61.4, 60.6, 41.1, 9.6 ppm;
HRMS calcd. for C10H14O3Cl (MH+) 217.0631 Da. Found 217.0629 Da.

231
1-Chloromethyl-2,4-dimethoxy-3-methyl-5-trityloxy benzene (321)
OMe
Me
MeOOTrt
Cl
To solution of the phenol 324 (3.8 g, 16.7 mmol) and triethylamine (7.0 mL, 50.1 mmol)
in CH2Cl2 (90 mL) at 25 °C was added trityl chloride (4.7 g, 16.7 mmol). The reaction
was stirred for 2 h then quenched with sat. aq. NaHCO3 (100 mL) and extracted with
EtOAc (1 x 150 mL, 2 x 50 mL). The combined extracts were washed with sat. aq. NaCl
(50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The brown residue
was purified by flash column chromatography (SiO2, slow gradient 0-10% EtOAc-
hexane) to afford benzyl chloride 321 (3.89 g, 51% yield over 3 steps) as a white solid.
Approximately 20% decomposition was observed when stored under argon in the dark at
room temperature for two weeks. Rf = 0.46 (20% EtOAc in hexanes); recrystallized from
EtOAc/hexanes, mp = 136-140 °C; IR (thin film): 3059, 3024, 2932, 2853, 1596, 1483,
1448, 1418, 1230, 1116, 1064, 1010, 905, 701 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.48
(6 H, d, J = 6.9 Hz), 7.31-7.20 (9 H, m), 6.35 (1 H, s), 4.26 (2 H, s), 3.87 (3 H, s), 3.68 (3
H, s), 2.13 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 150.8, 145.3, 143.7, 128.6,
127.5, 127.0, 125.0, 124.0, 120.1, 90.9, 61.2, 60.3, 41.2, 9.4 ppm; HRMS calcd. for
C29H26O3Cl (MH+) 457.1570 Da. Found 457.1569 Da.

232
7-Benzyloxy-6-benzyloxymethyl-3-(2,4-dimethoxy-3-methyl-5-trityloxy-benzyl)-8,10-
dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-
b]isoquinoline-2-carboxylic acid tert-butyl ester (325)
N
MeOMe
MeOOBn
OBnO
SPh
NH
OTrt
OMeMe
OMeBoc
H
To a degassed solution (freeze/pump/thaw) of thioaminal 300 (0.38 g, 0.54 mmol),
benzyl chloride 321 (0.30 g, 0.65 mmol), and 18-crown-6 (0.16 g, 0.59 mmol) in THF
(4.15 mL) at −78 °C was added KHMDS (0.5 M in toluene, 1.3 mL, 0.65 mmol)
dropwise within 2 min. The reaction became orange and remained clear. After 20 min. no
starting material was observed by TLC analysis. The reaction was quenched with sat. aq.
NaHCO3 (25 mL) and extracted with EtOAc (3 x 25 mL). The combined extracts were
washed with sat. aq. NaCl (25 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The yellow oil was purified by flash column chromatography (SiO2, 15-25%
EtOAc-hexane) to afford alkylated product 325 (0.595 g, 98% yield) as a colorless foam:
Rf = 0.35 (30% EtOAc in hexanes); IR (thin film): 2936, 1701, 1668, 1479, 1449, 1368,
1306, 1162, 1064, 1011, 907, 733 cm-1; 1H NMR (300 MHz, CDCl3), pronounced
carbamate resonance was observed giving sharp signals in ratio of 1:2, (high temperature
NMR [100 °C] led to decomposition): δ 7.66-7.53 (4 H, m), 7.44-7.06 (26 H, m), 6.20
(0.3 H, s), 6.11 (0.7 H, s), 6.02 (0.3 H, t, J = 6.2 Hz), 5.97 (0.7 H, t, J = 6.2 Hz) 5.26 (0.3
H, s), 5.17 (0.3 H, s), 5.13 (1.4 H, s), 5.06 (1 H, t, J = 5.2 Hz), 4.53-4.46 (2.3 H, m), 4.37
(0.7 H, m), 3.86-3.65 (2.0 H, m), 3.80 (2.3 H, s), 3.76 (0.7 H, s), 3.59 (2.3 H, s), 3.54 (0.7
H, s), 3.48 (0.7 H, s), 3.42 (2.3 H, s), 3.36 (3 H, s), 3.06-2.76 (3 H, m), 2.55-2.25 (2 H,

233
m), 2.22 (0.7 H, s), 2.20 (2.3 H, s), 1.53 (0.7 H, s), 1.49 (2.3 H, s), 1.31 (6.3 H, s), 1.17
(2.7 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 167.9, 153.2, 152.5, 152.2, 150.9, 150.6,
150.0, 145.1, 144.1, 138.6, 137.7, 134.5, 131.4, 129.5, 129.0, 128.8, 128.6, 128.3, 128.1,
128.0, 127.7, 127.4, 127.3, 127.1, 126.8, 126.7, 123.9, 123.2, 122.7, 122.3, 91.3, 81.0,
80.7, 74.3, 72.3, 72.2, 72.0, 69.0, 68.7, 60.5, 60.4, 60.3, 60.2, 60.1, 60.0, 59.0, 58.6, 54.8,
54.7, 48.3, 33.3, 32.3, 28.1, 27.6, 24.9, 24.2, 9.1, 8.9 ppm; HRMS calcd. for
C70H73N2O10S (MH+) 1133.4986 Da. Found 1133.4980 Da.
7-Benzyloxy-6-benzyloxymethyl-3-(2,4-dimethoxy-3-methyl-5-trityloxy-benzyl)-8,10-
dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-
b]isoquinoline-2-carboxylic acid tert-butyl ester (326)
N
MeOMe
MeOOBn
OBnO
SPh
NH
OTrt
OMeMe
OMeBoc
H
To a degassed solution (freeze/pump/thaw) of 325 (0.132 g, 0.117 mmol) in THF (1.2
mL) at −78 °C was added t-BuLi (1.3 M in pentane, 0.224 mL, 0.291 mmole). Upon
complete addition the solution became deep orange/brown in color. Within 10 seconds of
complete t-BuLi addition a degassed solution (freeze/pump/thaw) of butylated
hydroxytoluene (0.103 g, 0.466 mmol) in THF (0.25 mL) was added in one portion. The
brown color faded to pale yellow. The solution was diluted with sat. aq. NaHCO3 (10
mL) and extracted with EtOAc (3 x 15 mL). The combined extracts were washed with
sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under vacuum. 1H

234
NMR of the crude product revealed a 6:1 ratio of 326 to 325. The mixture was purified
by flash column chromatography (SiO2, very slow gradient of 10-20% EtOAc-hexane) to
yield 326 (0.064 g, 48% yield) as a colorless foam and a 3:2 mixture of 326:325 (0.033 g,
25% yield). 326: Rf = 0.39 (30% EtOAc in hexanes); IR (thin film): 3061, 3032, 2936,
2863, 1695, 1653, 1479, 1456, 1417, 1386, 1330, 1289, 1250, 1160, 1115, 1064, 1011,
910, 699 cm-1; 1H NMR (300 MHz, CDCl3) carbamate resonance was observed giving
broad signals rather than sharp splitting: δ 7.57-7.20 (30 H, m), 6.50 (1 H, s), 6.18 (1 H,
s), 5.85 (1 H, brs), 5.15 (2 H, brs), 4.56 (2 H, s), 4.45 (1 H, brs), 3.87 (3 H, s), 3.84 (3 H,
s), 3.86-3.77 (2 H, m), 3.70-3.60 (1 H, m), 3.63 (3 H, s), 3.54 (3 H, s), 3.50-3.43 (1 H,
m), 3.06 (1 H, m), 2.80 (1 H, m), 2.63 (1 H, m), 2.28 (3 H, brs), 2.18 (3 H, brs), 1.32 (2
H, brs), 1.18 (7 H, brs) ppm; 13C NMR (75 MHz, CDCl3): δ 167.4, 153.0, 151.4, 151.1,
150.2, 148.7, 145.0, 144.7, 144.2, 138.6, 137.2, 134.5, 133.3, 131.2, 129.1, 128.9, 128.2,
127.9, 127.8, 127.6, 127.5, 127.4, 127.2, 127.0, 126.8, 126.7, 126.4, 126.3, 124.4, 124.0,
120.7, 90.5, 80.9, 74.5, 72.7, 72.3, 63.0, 60.7, 60.2, 60.1, 60.0, 58.8, 57.3, 49.7, 35.8,
27.8, 27.5, 26.9, 9.6, 9.3 ppm; HRMS calcd. for C70H73N2O10S (MH+) 1133.4986 Da.
Found 1133.5018 Da.

235
7-Benzyloxy-6-benzyloxymethyl-3-(5-hydroxy-2,4-dimethoxy-3-methyl-benzyl)-8,10-
dimethoxy-9-methyl-4-oxo-1-phenylsulfanyl-1,3,4,6,11,11a-hexahydro-pyrazino[1,2-
b]isoquinoline-2-carboxylic acid tert-butyl ester (327)
N
MeOMe
MeOOBn
OBnO
SPh
NH
OH
OMeMe
OMeBoc
H
To a degassed solution (freeze/pump/thaw) of 325 (90 mg, 0.080 mmol) in THF (1.2 mL)
at −78 °C was added t-BuLi (1.1 M in pentane, 0.18 mL, 0.20 mmol). Upon complete
addition the solution became deep orange/brown in color. After 10 seconds a degassed
solution (freeze/pump/thaw) of butylated hydroxyltoluene (0.103 g, 0.466 mmol) in THF
(0.25 mL) was added in one portion. The brown color faded to pale yellow. To the
reaction mixture was added anhydrous HCl (1.0 M in Et2O, 1.0 mL). The solution was
warmed to 0 °C and added HCl in small portions over 1 h until the trityl ether could not
be detected by TLC. The solution was diluted with sat. aq. NaHCO3 (10 mL) and
extracted with EtOAc (2 x 15 mL). The combined extracts were washed with sat. aq.
NaCl (15 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The resulting
yellow residue was purified by flash column chromatography (SiO2, slow gradient, 10-
25% EtOAc-hexane) to afford phenol 327 (0.052 g, 73% yield) as a colorless oil: Rf =
0.26 (30% EtOAc in hexanes); IR (thin film): 3395, 2936, 2863, 1686, 1653, 1456, 1417,
1385, 1334, 1255, 1162, 1115, 1009, 735 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.56 (2 H,
d, J = 7.6 Hz), 7.40-7.17 (13 H, m), 6.64 (0.2 H, s), 6.34 (0.8 H, s), 6.04 (0.8, s), 5.86 (0.2
H, s), 5.86-5.81 (1 H, m), 5.03 (2 H, s), 5.01-4.93 (1 H, m), 4.80 (1 H, dd, J = 8.8, 3.4
Hz), 4.54 (2 H, s), 3.90-3.45 (4 H, m), 3.84 (3 H, s), 3.74 (6 H, s), 3.63 (3 H, s), 3.21-3.00

236
(3 H, m), 2.26 (3 H, s), 2.22 (3 H, s), 1.28 (2.3 H, s), 1.12 (6.7 H, s) ppm; 13C NMR (75
MHz, CDCl3): δ 167.4, 153.5, 151.0, 150.5, 150.3, 144.5, 144.4, 143.9, 138.5, 137.0,
134.0, 131.9, 129.6, 129.1, 128.9, 128.6, 128.4, 128.3, 128.1, 127.8, 127.7, 127.5, 127.2,
127.0, 126.6, 124.4, 123.8, 123.7, 123.5, 114.7, 114.2, 81.1, 74.4, 72.3, 65.2, 61.3, 60.8,
60.3, 60.2, 58.0, 57.5, 56.3, 49.9, 49.5, 35.3, 34.7, 27.7, 27.4, 26.8, 9.6, 9.2 ppm; HRMS
calcd. for C51H59N2O10S (MH+) 891.3890 Da. Found 891.3887 Da
phenol 332
N
MeOMe
MeOOBn
OBnO
NHH
H
HOOMe
Me
OMeH
H
To a solution of 327 (0.043 g, 0.048 mmol) in dry chlorobenzene (1.6 mL) at 25 °C was
added AgBF4 (0.20 mL of a solution of 0.18 g AgBF4 in 0.50 PhCl). A precipitate formed
and the solution remained pale yellow in color. The reaction was complete upon first
TLC analysis (within 5 minutes). After 10 minutes the reaction mixture was diluted with
20% aq. Rochelle’s salt (15 mL) and extracted with EtOAc (3 x 15 mL). The combined
extracts were washed with sat. aq. NaCl (15 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The resulting yellow residue was purified by flash column
chromatography (SiO2 treated with triethylamine, 100% EtOAc to 5% MeOH in EtOAc)
to afford 332 (0.019 g, 56% yield) as a pale yellow cloudy oil: Rf = 0.17 (5% MeOH in
EtOAc); IR (thin film): 3304, 3030, 2937, 2862, 1635, 1457, 1412, 1361, 1302, 1254,

237
1193, 1112, 1060, 1007, 911, 733 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.42-7.24 (5 H,
m), 7.12-7.10 (3 H, m), 6.81 (2 H, d, J = 3.5 Hz), 5.74 (1 H, t, J = 3.8 Hz), 5.04 (1 H, d, J
= 11.0 Hz), 4.96 (1 H, d, J = 11.0 Hz), 4.60 (1 H, d, J = 3.3 Hz), 4.06-3.95 (3 H, m), 3.80
(3 H, s), 3.78-3.67 (2 H, m), 3.65 (3 H, s), 3.62 (3 H, s), 3.59 (3 H, s), 3.54-3.37 (2 H, m),
3.23-3.03 (2 H, m), 2.33 (1 H, dd, J = 15.0, 12.9 Hz), 2.26-2.22 (NH, ArOH, 2 H, m),
2.23 (3 H, s), 2.16 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 169.5, 151.0, 149.9,
149.6, 144.6, 143.3, 142.0, 138.5, 137.2, 128.2, 128.1, 127.9, 127.9, 127.8, 127.7, 127.6,
127.5, 126.6, 126.5, 126.2, 123.9, 123.3, 122.9, 117.8, 74.5, 72.5, 72.0, 61.1, 60.5, 60.1,
59.8, 53.2, 49.6, 48.8, 29.5, 26.2, 9.4, 9.2 ppm; HRMS calcd. for C40H45N2O8 (MH+)
681.3176 Da. Found 681.3173 Da.
N-methylamine 333
N
MeOMe
MeOOBn
OBnO
NH
H
HOOMe
Me
OMeH
H
Me
To a stirred solution of phenol 327 (0.20 g, 0.224 mmol) in dry chlorobenzene (7.5 mL)
at 25 °C was added AgBF4 (0.93 mL of a solution of 1.0 g AgBF4 in 2.8 PhCl). A
precipitate formed and the solution remained pale yellow in color. The reaction was
complete upon first TLC analysis (within 5 minutes). After 10 min. the reaction mixture
was diluted with 20% aq. Rochelle’s salt (25 mL) and extracted with EtOAc (3 x 20 mL).
The combined extracts were washed with sat. aq. NaCl (20 mL), dried (Na2SO4), filtered,

238
and concentrated under vacuum. The yellow residue was dissolved in MeOH (16 mL).
37% Aq. formaldehyde (1.2 mL), sodium cyanoborohydride (0.282 g, 4.48 mmol), and
acetic acid (4.0 mL) were added. The yellow solution became colorless. After 40 min. the
solution was concentrated under vacuum, the resultant residue treated with sat. aq.
NaHCO3 (25 mL) and extracted with EtOAc (3 x 25 mL). The combined extracts were
washed with sat. aq. NaCl (20 mL), dried (Na2SO4), filtered, and concentrated under
vacuum. The pale yellow residue was purified by flash column chromatography (SiO2
treated with triethylamine, 50-100% EtOAc in hexanes) to afford methylamine 333
(0.101 g, 65% yield over two steps) as a pale yellow oil: Rf = 0.27 (100% EtOAc in
hexanes); IR (thin film): 3319, 2986, 2937, 2869, 1635, 1455, 1412, 1361, 1302, 1257,
1109, 1058, 1008, 753 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.44-7.27 (5 H, m), 7.11-
7.08 (3 H, m), 6.80-6.77 (2 H, m), 5.92 (1 H, ArOH, s), 5.75 (1 H, t, J = 3.1 Hz), 5.06 (1
H, d, J = 11.0 Hz), 4.97 (1 H, d, J = 11.0 Hz), 4.31 (1 H, d, J = 3.0 Hz), 4.04 (1 H, d, J =
12.1 Hz), 3.98 (1 H, d, J = 12.2 Hz), 3.91-3.83 (1 H, m), 3.80 (3 H, s), 3.72-3.67 (2 H,
m), 3.64 (3 H, s), 3.59 (3 H, s), 3.58 (3 H, s), 3.43-3.38 (2 H, m), 3.14 (1 H, dd, J = 18.1,
6.7 Hz), 2.98 (1 H, d, J = 18.0 Hz), 2.46 (3 H, s), 2.33 (1 H, dd, J = 15.0, 12.9 Hz), 2.23
(3 H, s), 2.15 (3 H, s) ppm; 13C NMR (75 MHz, CDCl3): δ 170.4, 151.0, 149.9, 149.3,
144.6, 143.4, 142.9, 138.4, 137.2, 128.2, 128.0, 127.8, 127.7, 127.6, 126.6, 126.5, 126.2,
126.1, 123.8, 122.7, 116.4, 74.4, 72.5, 71.9, 60.5, 60.1, 59.8, 59.2, 57.0, 55.0, 49.6, 40.1,
25.8, 24.6, 9.4, 9.2 ppm; HRMS calcd. for C41H47N2O8 (MH+) 695.3332 Da. Found
695.3337 Da.

239
diol 334
N
MeOMe
MeOOH
OBnO
NH
H
HOOMe
Me
OMeH
H
Me
To a solution of methylamine 333 (0.31 g, 0.446 mmol) in MeOH (9.0 mL) at 25 °C was
added Pd(OH)2 (moist, Pd content 20%, 62 mg) and the reaction was stirred under an
atmosphere of H2 (balloon pressure) for 1 h. The reaction mixture was filtered through a
plug of Celite®, washed with MeOH, and concentrated under vacuum. The pale yellow
residue was purified by flash column chromatography (SiO2 treated with triethylamine,
75-100% EtOAc in hexanes then 2% MeOH in EtOAc) to afford diol 334 (0.229 g, 85%
yield) as a fine white solid: recrystallized from MeOH with slow diffusion of EtOAc;
X-ray (see Appendix H); mp = 209-211°C (dec.); Rf = 0.45 (6% MeOH in EtOAc); IR
(thin film): 3326, 2938, 2870, 1635, 1464, 1414, 1362, 1302, 1273, 1193, 1109, 1059,
1006, 910 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.19-7.12 (3 H, m), 6.91-6.88 (2 H, m),
6.04 (1 H, s), 5.77 (2 H, brs), 4.33 (1 H, s), 4.20 (1 H, d, J = 12.1 Hz), 4.11 (1 H, d, J =
12.1 Hz), 4.01-3.96 (1 H, m), 3.76 (3 H, s), 3.74-3.68 (1 H, m), 3.64-3.58 (1 H, m), 3.63
(3 H, s), 3.62 (3 H, s), 3.59 (3 H, s), 3.49-3.40 (1 H, m), 3.14 (1 H, dd, J = 18.1, 6.8 Hz),
2.99 (1 H, d, J = 18.0 Hz), 2.48 (3 H, s), 2.27-2.20 (1 H, m), 2.22 (3 H, s), 2.17 (3 H, s)
ppm; 13C NMR (75 MHz, CDCl3): δ 149.3, 148.1, 144.4, 143.4, 142.9, 142.1, 138.0,
127.8, 126.9, 126.6, 125.3, 122.6, 119.6, 77.0, 72.8, 71.7, 60.6, 60.5, 59.8, 59.3, 57.1,
55.0, 49.4, 40.0, 25.6, 24.5, 9.4 ppm; HRMS calcd. for C34H41N2O8 (MH+) 605.2863 Da.
Found 605.2864 Da.

240
triol 335
N
MeOMe
MeOOH
OHO
NH
H
HOOMe
Me
OMeH
H
Me
To a solution of methylamine 333 (0.10 g, 0.144 mmol) in MeOH (2.9 mL) at 25 °C was
added Pd(OH)2 (moist, Pd content 20%, 45 mg), and the reaction was stirred under an
atmosphere of H2 (balloon pressure) for 6 h. The reaction mixture was filtered through a
plug of Celite®, washed with MeOH, and concentrated under vacuum. The pale yellow
residue was purified by flash column chromatography (SiO2 treated with triethylamine,
75-100% EtOAc in hexanes then 5% MeOH in EtOAc) to afford triol 335 (0.073 g, 94%
yield) as a fine white solid: Rf = 0.36 (6% MeOH in EtOAc); IR (thin film): 3333, 2937,
1634, 1463, 1413, 1362, 1302, 1272, 1192, 1110, 1060, 1005 cm-1; 1H NMR (300 MHz,
DMSO-d6): δ 8.74 (1 H, s), 8.48 (1 H, s), 5.45 (1 H, t, J = 4.9 Hz), 4.26 (1 H, brs), 4.19 (1
H, d, J = 3.2 Hz), 3.71 (1 H, dt, J = 12.7, 2.7 Hz), 3.58 (6 H, s), 3.54 (3 H, s), 3.53 (3 H,
s), 3.53-3.51 (1 H, m), 3.28 (1 H, d, J = 15.3 Hz), 3.17 (1 H, m), 2.96 (2 H, m), 2.63 (1 H,
d, J = 17.6 Hz), 2.28 (3 H, s), 2.09 (3 H, s), 2.08 (3 H, s), 2.01 (1 H, dd, J = 15.0, 13.0
Hz) ppm; 13C NMR (75 MHz, DMSO-d6): δ 170.7, 148.3, 147.4, 144.7, 144.1, 144.0,
143.2, 125.7, 122.5, 122.3, 122.1, 120.4, 117.6, 63.7, 60.8, 60.2, 59.7, 59.0, 57.8, 54.8,
50.5, 39.0, 25.6, 24.2, 9.6 ppm; HRMS calcd. for C27H35N2O8 (MH+) 515.2393 Da.
Found 515.2394 Da.

241
bis-isoquinolinequinone 337
N
MeO
MeOO
OHO
NH
OOMe
Me
OMe
1 137
5
3
22
9
10
21
11H16
1518
17
19Ha Hb
HaHb
To a solution of the triol 335 (73 mg, 0.135 mmol) in dry acetonitrile (5.0 mL) at −15 °C
was added ammonium cerium(IV) nitrate (0.369 g, 0.674 mmol) as a solution in water
(0.2 mL), pre-cooled to 0 °C. The orange/brown solution was stirred for 15 minutes as
the temperature rose to −5 °C. The solution was diluted with CHCl3 (15 mL) and 5% aq.
NaHCO3 (5.0 mL), aqueous layer extracted with CHCl3 (2 x 10 mL). The combined
organic extracts were washed with sat. aq. NaCl (5.0 mL), dried (Na2SO4), filtered, and
concentrated under vacuum. The brown residue was purified by flash column
chromatography (SiO2 treated with triethylamine, 50-100% EtOAc in hexanes) to afford
bis-isquinolinequinone 337 (0.036 g, 55% yield) as a burnt orange residue; Rf = 0.35 (5%
MeOH in EtOAc); IR (thin film): 3365, 2925, 2853, 1653, 1617, 1429, 1307, 1232, 1150
cm-1; The numbering scheme used in the isolation paper8 is not consistent with other
renieramycins. Using the numbering scheme consistent with other renieramycins: 1H NMR (500 MHz, CD2Cl2): δ 5.23 (C1, 1 H, dd, J = 4.0, 1.4 Hz), 4.14 (C11, 1 H, brs),
3.98 (C7/17-OMe, 3 H, s), 3.97 (C7/17-OMe, 3 H, s), 3.89 (C3, 1 H, brd, J = 11.4 Hz),
3.73 (C22, 1 H, dd, J = 11.0, 3.5 Hz), 3.68 (C13, 1 H, d, J = 6.7 Hz), 3.43 (C22, 1 H, dd,
J = 11.2, 4.1 Hz), 3.05 (C4-Hb, 1 H, dd, J = 16.4, 2.6 Hz), 2.88 (C14-Hb, 1 H, dd, J =
20.7, 6.8 Hz), 2.66 (C14-Ha, 1 H, brd, J = 20.6 Hz), 2.65 (OH, 1 H, brs), 2.00 (NMe, 3 H,
s), 1.95 (C6/16-Me, 3 H, s), 1.94 (C6/16-Me, 3 H, s), 1.68 (C4-Ha, 1 H, ddd, J = 16.3,

242
12.3, 1.3 Hz) ppm; 13C NMR (125 MHz, CD2Cl2): δ 186.8 (C15), 185.7 (C5), 182.7
(C18), 181.2 (C8), 171.2 (C21), 156.0 (C17), 155.9 (C7), 142.3 (C20/10), 142.2
(C20/10), 137.1 (C9), 135.2 (C19), 129.7 (C6/C16), 129.2 (C6/C16), 65.6 (C22), 61.3
(C7 and C17-OMe), 59.2 (C13), 57.0 (C3), 53.4 (C11), 52.4 (C1), 39.9 (NMe), 25.8 (C4),
24.3 (C14), 8.9 (C6 and C16-Me) ppm; HRMS calcd. for C25H27N2O8 (MH+) 483.1767
Da. Found 483.1786 Da.
(±)-renieramycin G (2g)
N
MeO
MeOO
OO
NH
OOMe
Me
OMe
OMe
Me
H
1 137
5
3
22
9
10
21
11H16
24
15
25
18
26
17
19Ha Hb
HaHb
To a solution of alcohol 337 (6 mg, 0.012 mmol) in CH2Cl2 (0.30 mL) at 25 °C was
added angeloyl chloride9 (approx. 15 μL, 0.120 mmol) and the solution was allowed to
stand for 24 h at 25 °C in the dark. The solution was concentrated under vacuum then
purified by reverse phase HPLC (preparatory column, rate = 8.0 mL/min, gradient = 65%
to 30% H2O without TFA in CH3CN without TFA over 20 min., then to 0% H2O over
next 10 min., injection volume = 1.0 mL, product elution from 23-24 min.) and
lyophilized to afford (±)-renieramycin G (2g) (5 mg, 74% yield) as an orange residue: Rf

243
= 0.25 (100% EtOAc); IR (thin film): 2929, 2854, 1717, 1655, 1616, 1420, 1373, 1351,
1307, 1263, 1229, 1149, 1120 cm-1;
Renieramcyin G was originally characterized in CD2Cl2, however trace amounts of
CH2Cl2 and water made NMR interpretation difficult. C6D6 proved to be a better solvent
for analysis of all signals without overlap or distortion.
The numbering scheme used in the isolation paper8 is not consistent with other
renieramycins. Using the numbering scheme consistent with other renieramycins: 1H NMR (500 MHz, C6D6): δ 5.56 (C1, 1 H, d, J = 2.2 Hz), 5.34 (C26, 1 H, q of q, J =
7.3, 1.4 Hz), 4.92 (C22, 1 H, dd, J = 11.6, 2.4 Hz), 4.52 (C22, 1 H, dd, J = 11.7, 2.3 Hz),
3.82 (C11, 1 H, d, J = 3.5 Hz), 3.79 (C7-OMe, 3 H, s), 3.62 (C17-OMe, 3 H, s), 3.47
(C13, 1 H, d, J = 7.2 Hz), 3.36 (C3, ddd, J = 12.3, 3.2, 3.0 Hz), 3.10 (C4-Hb, 1 H, dd, J =
16.5, 2.7 Hz), 2.87 (C14-Ha, 1 H, d, J = 20.9 Hz), 2.59 (C14-Hb, 1 H, dd, J = 20.5, 7.0
Hz), 1.90 (C6-Me, 3 H, s), 1.87 (C16-Me, 3 H, s), 1.80 (NMe, 3 H, s), 1.55 (C26-Me, 3
H, dq, J = 7.3, 1.5 Hz), 1.51 (C4-Ha, 1 H, ddd, J = 16.7, 12.2, 2.0 Hz), 1.37 (C25-Me, 3
H, dq, J = 1.6, 1.4 Hz) ppm; 13C NMR (125 MHz, C6D6): δ 185.9 (C15), 185.0 (C5),
182.7 (C18), 180.4 (C8), 169.9 (21), 166.8 (C24), 156.4 (C7), 155.5 (C17), 142.2 (C20),
141.1 (C10), 139.1 (C26), 136.3 (C9), 135.1 (C19), 128.8 (C16), 127.4 (C6), 126.9
(C25), 62.8 (C22), 60.6 (C7-OMe), 60.5 (C17-OMe), 59.3 (C13), 56.0 (C3), 53.3 (C11),
50.8 (C1), 39.3 (NMe), 25.9 (C4), 23.6 (C14), 20.3 (C25-Me), 15.3 (C26-Me), 8.6 (C16-
Me and C6-Me) ppm; HRMS calcd. for C30H33N2O9 (MH+) 565.2186 Da. Found
565.2203 Da.

244

245

246

247

248

249

250

251
5.2. REFERENCES
1) Perrin, D. D.; Armarego, W. L. F. Purification of Laboratory Chemicals; Third ed.; Pergamon Press: New York, 1993. 2) Larock, R. C.; Doty, M. J.; Cacchi, S. J. J. Org. Chem. 1993, 58, 4579. 3) Ireland, R. E.; Anderson, R. C.; Badoud, R.; Fitzsimmons, B. J.; McGarvey, G. J.; Thaisrivongs, S.; Wilcox, C. S. J. Am. Chem. Soc. 1983, 105, 1988. 4) Turos, E.; Ren, X. F.; Lake, C. H.; Churchill, M. R. J. Org. Chem. 1995, 60, 6468. 5) D. Saito, T.; Morimoto, M.; Akiyama, C.; Matsumoto, T.; Suzuki, K. J. Am. Chem. Soc. 1995, 117, 10757. 6) Kaufman, T. S. Synlett 1997, 1377. 7) Clayden, J.; Knowles, F. E.; Baldwin, I. R. J. Am. Chem. Soc. 2005, 127, 2412. 8) Davidson, B. S. Tetrahedron Lett. 1992, 33, 3721. 9) Beeby, P.J. Tetrahedron Lett. 1977, 38, 3379.

252
Appendix A: X-Ray Data for the Chloride 198
NCO2Me
SPh
ClH
H
198

253
Table 1. Crystal data and structure refinement for chloride 198
Empirical formula C19 H20 Cl N O2 S
Formula weight 361.87
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Monoclinic
Space group P21/c
Unit cell dimensions a = 12.1743(2) Å α = 90°
b = 13.8873(2) Å β = 105.929(1)°
c = 10.3218(1) Å γ = 90°
Volume 1678.08(4) Å3
Z 4
Density (calculated) 1.432 Mg/m3
Absorption coefficient 0.364 mm-1
F(000) 760
Crystal size 0.40 x 0.22 x 0.06 mm
Theta range for data collection 2.93 to 27.48°
Index ranges -15<=h<=15, -13<=k<=18, -13<=l<=13
Reflections collected 6556
Independent reflections 3841 [R(int) = 0.0182]
Completeness to theta = 27.48° 99.9 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 3841 / 0 / 298

254
Goodness-of-fit on F2 1.055
Final R indices [I>2sigma(I)] R1 = 0.0341, wR2 = 0.0796
R indices (all data) R1 = 0.0442, wR2 = 0.0841
Largest diff. peak and hole 0.329 and -0.246 e.Å-3

255
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for chloride 198. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________
N1 2227(1) 4245(1) 1789(1) 17(1) C2 1713(1) 3812(1) 452(1) 17(1) C3 2483(1) 3972(1) -453(1) 17(1) C4 2038(1) 4207(1) -1806(2) 21(1) C5 2768(1) 4333(1) -2623(2) 25(1) C6 3936(1) 4224(1) -2087(2) 26(1) C7 4383(1) 4007(1) -732(2) 24(1) C8 3660(1) 3883(1) 97(1) 19(1) C9 4091(1) 3631(1) 1564(2) 21(1) C10 3460(1) 4153(1) 2468(1) 18(1) C11 1514(1) 4777(1) 2321(1) 18(1) O12 485(1) 4834(1) 1843(1) 23(1) O13 2097(1) 5236(1) 3449(1) 22(1) C14 1413(2) 5837(1) 4064(2) 29(1) C15 1456(1) 2747(1) 659(1) 19(1) S16 763(1) 2072(1) -836(1) 23(1) C17 -701(1) 2385(1) -1181(1) 20(1) C18 -1129(1) 3120(1) -538(2) 22(1) C19 -2296(1) 3304(1) -904(2) 26(1) C20 -3037(2) 2768(1) -1900(2) 31(1) C21 -2604(2) 2036(1) -2532(2) 32(1) C22 -1447(1) 1837(1) -2180(2) 25(1) C23 3627(1) 3601(1) 3794(2) 25(1) Cl24 5093(1) 3600(1) 4749(1) 34(1)
________________________________________________________________________

256
Appendix B: X-Ray Data for the Bicyclo[3.3.1] 213
NO
CO2Me
SPh213

257
Table 1. Crystal data and structure refinement for Bicyclo[3.3.1] 213
Empirical formula C19 H19 N O3 S
Formula weight 341.41
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Monoclinic
Space group P21/c
Unit cell dimensions a = 11.0472(2) Å α = 90°
b = 7.6308(1) Å β = 92.423(1)°
c = 19.6869(3) Å γ = 90°
Volume 1658.10(4) Å3
Z 4
Density (calculated) 1.368 Mg/m3
Absorption coefficient 0.212 mm-1
F(000) 720
Crystal size 0.29 x 0.25 x 0.04 mm
Theta range for data collection 3.25 to 27.50°
Index ranges -14<=h<=14, -9<=k<=9, -25<=l<=25
Reflections collected 6798
Independent reflections 3797 [R(int) = 0.0218]
Completeness to theta = 27.50° 99.7 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 3797 / 0 / 294

258
Goodness-of-fit on F2 1.025
Final R indices [I>2sigma(I)] R1 = 0.0391, wR2 = 0.0753
R indices (all data) R1 = 0.0566, wR2 = 0.0811
Extinction coefficient 5.2(10)x10-6
Largest diff. peak and hole 0.27 and -0.26 e.Å-3

259
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for Bicyclo[3.3.1] 213. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________
O1 2818(1) 5353(1) 2863(1) 23(1) C2 2587(2) 7130(2) 2654(1) 28(1) C3 3362(1) 8469(2) 3045(1) 25(1) C4 4713(2) 8445(2) 2906(1) 28(1) C5 5424(1) 7073(2) 3310(1) 21(1) C6 6657(1) 6836(2) 3206(1) 25(1) C7 7325(2) 5578(2) 3564(1) 27(1) C8 6766(1) 4511(2) 4029(1) 26(1) C9 5546(1) 4744(2) 4144(1) 22(1) C10 4879(1) 6052(2) 3801(1) 19(1) C11 3565(1) 6370(2) 3967(1) 19(1) C12 2723(1) 5110(2) 3572(1) 20(1) N13 3204(1) 8163(2) 3775(1) 22(1) S14 1176(1) 5428(1) 3869(1) 28(1) C15 591(1) 3277(2) 3768(1) 25(1) C16 734(1) 2315(2) 3178(1) 29(1) C17 251(2) 647(2) 3116(1) 41(1) C18 -394(2) -61(3) 3635(1) 51(1) C19 -548(2) 901(3) 4219(1) 54(1) C20 -55(2) 2573(3) 4291(1) 40(1) C21 3331(1) 9559(2) 4203(1) 26(1) O22 3395(1) 11071(1) 4022(1) 37(1) O23 3347(1) 9053(1) 4861(1) 30(1) C24 3490(2) 10477(3) 5345(1) 42(1) ________________________________________________________________________

260
Appendix C: X-Ray Data for the Lactam 229
NNH
CO2Me
O229

261
Table 1. Crystal data and structure refinement for lactam 229
Empirical formula C13 H14 N2 O3
Formula weight 246.26
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Orthorhombic
Space group P212121
Unit cell dimensions a = 7.5720(3) Å α = 90°
b = 10.0310(5) Å β= 90°
c = 15.7540(8) Å γ = 90°
Volume 1196.59(10) Å3
Z 4
Density (calculated) 1.367 Mg/m3
Absorption coefficient 0.099 mm-1
F(000) 520
Crystal size 0.36 x 0.35 x 0.06 mm
Theta range for data collection 2.98 to 27.49°
Index ranges -9<=h<=9, -12<=k<=13, -20<=l<=20
Reflections collected 2615
Independent reflections 2615 [R(int) = 0.0000]
Completeness to theta = 27.49° 99.5 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 2615 / 0 / 220

262
Goodness-of-fit on F2 1.018
Final R indices [I>2sigma(I)] R1 = 0.0410, wR2 = 0.0784
R indices (all data) R1 = 0.0696, wR2 = 0.0878
Absolute structure parameter -1.4(12)
Extinction coefficient 3.2(3)x10-5
Largest diff. peak and hole 0.159 and -0.166 e.Å-3

263
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for lactam 229. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________
N1 882(2) 3998(2) 5360(1) 30(1) C2 2512(2) 3639(2) 5140(1) 26(1) C3 3977(2) 4687(2) 5212(1) 25(1) C4 4293(2) 5291(2) 4348(1) 25(1) C5 5648(3) 4855(2) 3819(1) 30(1) C6 5902(3) 5440(2) 3032(1) 35(1) C7 4802(3) 6463(2) 2776(1) 36(1) C8 3468(3) 6912(2) 3299(1) 31(1) C9 3199(2) 6336(2) 4096(1) 27(1) C10 1778(3) 6874(2) 4676(1) 33(1) C11 1754(3) 6331(2) 5590(1) 27(1) C12 362(3) 5278(2) 5739(1) 32(1) N13 3471(2) 5740(2) 5803(1) 28(1) O14 2844(2) 2522(1) 4847(1) 32(1) C15 4251(3) 5800(2) 6577(1) 30(1) O16 5564(2) 5177(1) 6773(1) 41(1) O17 3424(2) 6672(1) 7090(1) 40(1) C18 4122(4) 6759(3) 7947(1) 48(1) ________________________________________________________________________

264
Appendix D: X-Ray Data for the Amino Alcohol 283
Me
MeOOBn
OMe
NHOH
OBn
H
H
283

265
Table 1. Crystal data and structure refinement for amino alcohol 283
Empirical formula C28 H33 N O5
Formula weight 463.55
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Triclinic
Space group P-1
Unit cell dimensions a = 9.3397(2) Å α = 79.242(2)°
b = 11.4953(3) Å β = 83.470(2)°
c = 12.0017(3) Å γ = 80.402(2)°
Volume 1243.62(5) Å3
Z 2
Density (calculated) 1.238 Mg/m3
Absorption coefficient 0.084 mm-1
F(000) 496
Crystal size 0.30 x 0.30 x 0.10 mm
Theta range for data collection 2.93 to 27.46°
Index ranges -12<=h<=12, -14<=k<=14, -15<=l<=15
Reflections collected 9792
Independent reflections 5646 [R(int) = 0.0454]
Completeness to theta = 27.46° 99.0 %
Max. and min. transmission 0.9916 and 0.9751
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 5646 / 0 / 316

266
Goodness-of-fit on F2 0.995
Final R indices [I>2sigma(I)] R1 = 0.0512, wR2 = 0.1074
R indices (all data) R1 = 0.1546, wR2 = 0.1376
Extinction coefficient 1.9(2)x10-5
Largest diff. peak and hole 0.261 and -0.253 e.Å-3

267
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for amino alcohol 283. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________ N1 4257(2) 1779(2) 4475(1) 31(1) C2 4021(2) 2924(2) 3678(2) 30(1) C3 2419(2) 3466(2) 3740(2) 28(1) C4 1954(2) 4521(2) 3008(2) 31(1) C5 485(2) 5003(2) 3003(2) 33(1) C6 -564(2) 4448(2) 3724(2) 32(1) C7 -72(2) 3435(2) 4492(2) 32(1) C8 1393(2) 2953(2) 4535(2) 30(1) C9 1865(2) 1935(2) 5468(2) 37(1) C10 3479(2) 1814(2) 5604(2) 32(1) C11 4584(2) 2656(2) 2501(2) 32(1) O12 6110(1) 2243(1) 2525(1) 35(1) C13 6766(2) 1930(2) 1469(2) 43(1) C14 8343(2) 1470(2) 1604(2) 35(1) C15 8934(3) 308(2) 1505(2) 57(1) C16 10380(3) -122(3) 1718(2) 69(1) C17 11219(3) 613(3) 2031(2) 58(1) C18 10645(3) 1775(3) 2103(2) 58(1) C19 9227(2) 2192(2) 1895(2) 49(1) O20 2971(1) 5106(1) 2271(1) 34(1) C21 3355(2) 6103(2) 2671(2) 44(1) C22 4429(2) 6681(2) 1817(2) 34(1) C23 5426(2) 6035(2) 1133(2) 46(1) C24 6431(3) 6591(3) 380(2) 58(1) C25 6466(3) 7789(3) 309(2) 65(1) C26 5478(3) 8435(3) 976(2) 62(1) C27 4460(3) 7888(2) 1730(2) 47(1) O28 46(2) 6084(1) 2303(1) 44(1) C29 241(3) 6069(2) 1113(2) 66(1) C30 -2167(2) 4939(2) 3690(2) 43(1) O31 -1086(2) 2924(1) 5287(1) 40(1) C32 -1589(2) 1926(2) 4983(2) 53(1) C33 3979(2) 671(2) 6404(2) 36(1) O34 5532(2) 397(1) 6374(1) 38(1) ________________________________________________________________________

268
Appendix E: X-Ray Data for Thioaminal 300
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
300

269
Table 1. Crystal data and structure refinement for thioaminal 300
Empirical formula C41 H46 N2 O7 S
Formula weight 710.86
Temperature 153(2) K
Wavelength 0.71070 Å
Crystal system Monoclinic
Space group P21/c
Unit cell dimensions a = 17.3357(3) Å α = 90°
b = 18.3236(4) Å β = 92.4980(13)°
c = 11.5161(5) Å γ = 90°
Volume 3654.64(19) Å3
Z 4
Density (calculated) 1.292 Mg/m3
Absorption coefficient 0.142 mm-1
F(000) 1512
Crystal size 0.30 x 0.28 x 0.10 mm
Theta range for data collection 3.05 to 27.43°
Index ranges -18<=h<=21, -21<=k<=23, -13<=l<=14
Reflections collected 18879
Independent reflections 7594 [R(int) = 0.0804]
Completeness to theta = 27.43° 91.0 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 7594 / 0 / 460

270
Goodness-of-fit on F2 1.143
Final R indices [I>2sigma(I)] R1 = 0.0819, wR2 = 0.1831
R indices (all data) R1 = 0.1809, wR2 = 0.2095
Largest diff. peak and hole 0.660 and -0.299 e.Å-3

271
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for thioaminal 300. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________ N1 2863(2) 6022(2) 2742(3) 24(1) C2 2909(3) 5958(2) 1577(4) 28(1) C3 3562(3) 6321(3) 974(4) 31(1) N4 4164(2) 6641(2) 1738(3) 23(1) C5 4214(2) 6405(2) 2930(4) 23(1) C6 3430(2) 6448(2) 3448(4) 24(1) C7 3405(2) 6236(2) 4724(4) 25(1) C8 2600(3) 6374(2) 5097(4) 26(1) C9 2440(3) 6779(2) 6078(4) 27(1) C10 1694(3) 7013(2) 6298(4) 30(1) C11 1109(3) 6832(2) 5493(4) 29(1) C12 1243(3) 6397(2) 4536(4) 27(1) C13 1989(3) 6166(2) 4329(4) 25(1) C14 2178(3) 5723(2) 3280(4) 26(1) O15 2422(2) 5621(2) 968(3) 42(1) C16 4637(3) 7149(2) 1279(4) 26(1) O17 4625(2) 7288(2) 237(3) 32(1) O18 5084(2) 7467(2) 2107(3) 36(1) C19 5723(3) 7970(2) 1850(4) 31(1) C20 5428(3) 8599(3) 1119(6) 58(2) C21 6339(3) 7551(3) 1282(6) 61(2) C22 5979(4) 8226(4) 3047(5) 69(2) S23 4552(1) 5442(1) 3045(1) 36(1) C24 5488(3) 5519(2) 2490(4) 32(1) C25 6133(3) 5571(3) 3239(5) 43(1) C26 6866(3) 5594(3) 2792(6) 53(2) C27 6946(4) 5573(3) 1613(6) 56(2) C28 6305(4) 5525(3) 874(6) 57(2) C29 5574(3) 5495(3) 1293(5) 43(1) O30 3054(2) 7014(2) 6821(3) 30(1) C31 3316(3) 6473(3) 7645(4) 40(1) C32 1532(3) 7461(3) 7351(4) 42(1) O33 367(2) 7100(2) 5667(3) 35(1) C34 179(3) 7714(3) 4934(5) 45(1) O35 669(2) 6242(2) 3694(3) 31(1) C36 -42(3) 5932(3) 4053(4) 44(1)

272
C37 -415(3) 5567(2) 3020(4) 30(1) C38 -254(3) 5752(3) 1891(4) 35(1) C39 -606(3 ) 5407(3) 955(5) 45(1) C40 -1128(3) 4854(3) 1113(6) 52(2) C41 -1299(3) 4666(3) 2232(6) 56(2) C42 -962(3 ) 5022(3) 3182(5) 46(1) C43 2304(3) 4905(2) 3549(4) 31(1) O44 1615(2) 4551(2) 3845(3) 41(1) C45 1151(3) 4325(3) 2843(4) 39(1) C46 1448(3) 3651(2) 2269(4) 30(1) C47 1348(3) 2963(3) 2754(4) 40(1) C48 1597(3) 2347(3) 2208(5) 46(1) C49 1937(3) 2400(3) 1151(5) 48(2) C50 2052(3) 3070(3) 664(5) 45(1) C51 1801(3) 3695(3) 1218(4) 39(1) ________________________________________________________________________

273
Appendix F: X-Ray Data for the Allylated Product 301
Me
MeOOBn
OMe
N
OBn
H
HNBoc
O
SPh
301

274
Table 1. Crystal data and structure refinement for allylated product 301
Empirical formula C47 H53 N2 O7 S
Formula weight 789.97
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Triclinic
Space group P-1
Unit cell dimensions a = 11.1857(2) Å α = 87.112(1)°
b = 12.2691(3) Å β = 74.288(1)°
c = 16.4143(4) Å γ = 76.143(1)°
Volume 2105.14(8) Å3
Z 2
Density (calculated) 1.246 Mg/m3
Absorption coefficient 0.130 mm-1
F(000) 842
Crystal size 0.30 x 0.30 x 0.10 mm
Theta range for data collection 2.92 to 27.47°
Index ranges -14<=h<=14, -15<=k<=15, -21<=l<=18
Reflections collected 15723
Independent reflections 9454 [R(int) = 0.0895]
Completeness to theta = 27.47° 98.1 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 9454 / 0 / 515

275
Goodness-of-fit on F2 0.972
Final R indices [I>2sigma(I)] R1 = 0.0577, wR2 = 0.1094
R indices (all data) R1 = 0.2078, wR2 = 0.1489
Extinction coefficient 1.49(11)x10-5
Largest diff. peak and hole 0.308 and -0.348 e.Å-3

276
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for allylated product 301. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________ N1 4072(2) 2290(2) 3859(2) 24(1) C2 5273(3) 2124(3) 3182(2) 28(1) C3 5120(3) 1980(3) 2305(2) 28(1) N4 3980(2) 2418(2) 2149(1) 24(1) C5 3864(3) 2376(3) 1275(2) 28(1) C6 2864(3) 3364(3) 1126(2) 25(1) C7 3037(3) 3982(3) 387(2) 27(1) C8 2079(3) 4902(3) 287(2) 29(1) C9 917(3) 5201(3) 900(2) 28(1) C10 764(3) 4564(3) 1632(2) 26(1) C11 1725(3) 3675(3) 1759(2) 24(1) C12 1637(3) 3061(3) 2578(2) 25(1) C13 2870(3) 2944(3) 2838(2) 24(1) C14 2882(3) 2308(2) 3653(2) 24(1) C15 4198(3) 2103(3) 4670(2) 28(1) O16 5227(2) 1907(2) 4827(1) 38(1) O17 3065(2) 2187(2) 5239(1) 29(1) C18 3024(3) 2029(3) 6148(2) 29(1) C19 3504(3) 2939(3) 6458(2) 51(1) C20 1609(3) 2152(3) 6559(2) 41(1) C21 3766(3) 863(3) 6278(2) 45(1) C22 5969(3) 3069(3) 3154(2) 35(1) C23 5242(3) 4200(3) 2974(2) 38(1) C24 4761(3) 5035(3) 3518(2) 56(1) O25 6071(2) 1514(2) 1746(1) 37(1) C26 3603(3) 1239(3) 1135(2) 33(1) O27 3577(2) 1168(2) 277(1) 37(1) C28 3202(3) 174(3) 118(2) 40(1) C29 1846(3) 174(3) 575(2) 30(1) C30 1539(3) -756(3) 997(2) 41(1) C31 276(4) -770(4) 1386(2) 55(1) C32 -689(4) 156(5) 1367(3) 63(1) C33 -378(4) 1088(4) 960(3) 67(1) C34 868(4) 1108(3) 564(2) 53(1) O35 4200(2) 3720(2) -225(1) 31(1) C36 4150(3) 3237(3) -1002(2) 37(1)

277
C37 5453(3) 3021(3) -1598(2) 26(1) C38 6284(3) 3701(3) -1611(2) 32(1) C39 7465(3) 3504(3) -2185(2) 37(1) C40 7841(3) 2631(3) -2772(2) 36(1) C41 7019(3) 1953(3) -2773(2) 39(1) C42 5846(3) 2142(3) -2185(2) 33(1) O43 2263(2) 5514(2) -451(1) 36(1) C44 3038(3) 6302(3) -475(2) 43(1) C45 -107(3 ) 6192(3) 776(2) 36(1) O46 -375(2) 4885(2) 2276(1) 30(1) C47 -1253(3) 4199(3) 2298(2) 39(1) S48 2718(1) 852(1) 3595(1) 33(1) C49 1028(3) 1035(3) 3885(2) 27(1) C50 287(3) 1513(3) 4659(2) 41(1) C51 -1031(3) 1647(3) 4881(2) 48(1) C52 -1603(3) 1283(3) 4341(2) 44(1) C53 -871(3) 801(3) 3571(2) 36(1) C54 442(3) 678(3) 3338(2) 31(1) C1A -759(5) 4897(4) 4501(3) 74(1) C2A 369(5) 5220(3) 4161(3) 67(1) C3A 1128(4) 5324(3) 4664(3) 68(1) ________________________________________________________________________

278
Appendix G: X-Ray Data for Oxime 314
NN
H
O
MeOMe
OBn
H
H
OBn
Boc
MeO
NHOH
H
314

279
Table 1. Crystal data and structure refinement for oxime 314
Empirical formula C38 H45 N3 O8
Formula weight 671.77
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Triclinic
Space group P-1
Unit cell dimensions a = 8.8203(5) Å α = 102.634(1)°
b = 12.4682(3) Å β = 90.790(1)°
c = 16.9791(2) Å γ = 98.378(1)°
Volume 1800.57(11) Å3
Z 2
Density (calculated) 1.239 Mg/m3
Absorption coefficient 0.087 mm-1
F(000) 716
Crystal size 0.30 x 0.13 x 0.12 mm
Theta range for data collection 3.05 to 27.49°
Index ranges -11<=h<=11, -15<=k<=16, -21<=l<=21
Reflections collected 13638
Independent reflections 13638
Completeness to theta = 27.49° 98.6 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 13638 / 14 / 459

280
Goodness-of-fit on F2 1.443
Final R indices [I>2sigma(I)] R1 = 0.0905, wR2 = 0.1693
R indices (all data) R1 = 0.2415, wR2 = 0.1939
Extinction coefficient 1.15(12)x10-5
Largest diff. peak and hole 0.331 and -0.309 e.Å-3

281
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for oxime 314. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________ N1 2139(3) 5707(2) 2757(2) 29(1) C2 992(5) 5067(3) 3052(3) 37(1) C3 1126(4) 3882(3) 3018(2) 34(1) C4 2281(4) 3829(3) 3689(2) 38(1) C5 3848(4) 4178(3) 3350(2) 31(1) C6 3485(4) 4076(3) 2433(2) 27(1) C7 3307(4) 5177(3) 2249(2) 25(1) C8 4784(4) 6009(3) 2308(2) 28(1) C9 4394(4) 7007(3) 2035(2) 24(1) C10 5278(4) 7502(3) 1494(2) 29(1) C11 4933(4) 8455(3) 1263(2) 27(1) C12 3690(4) 8875(3) 1572(2) 31(1) C13 2750(4) 8381(3) 2102(2) 29(1) C14 3122(4) 7440(3) 2316(2) 25(1) C15 2106(4) 6907(3) 2880(2) 30(1) O16 -82(3) 5495(2) 3402(2) 43(1) N17 1935(3) 3400(2) 2305(2) 30(1) C18 1277(5) 3084(3) 1530(3) 36(1) O19 2021(3) 2888(2) 935(2) 41(1) O20 -260(3) 3005(2) 1559(2) 43(1) C21 -1235(5) 2635(4) 794(3) 59(1) C22 -1037(6) 1465(4) 396(3) 80(2) C23 -869(5) 3448(4) 257(3) 78(2) C24 -2811(5) 2687(5) 1121(3) 94(2) C25 5049(5) 3488(4) 3477(3) 45(1) N26 6495(5) 3641(4) 3449(3) 44(1) O27 6859(4) 4710(3) 3297(2) 45(1) N26' 6281(12) 4208(12) 3701(12) 57(6) O27' 7691(13) 3875(9) 3854(7) 44(3) O28 6604(3) 7077(2) 1218(2) 33(1) C29 6290(4) 6202(3) 498(2) 47(1) C30 5960(4) 9007(3) 715(2) 42(1) O31 3272(3) 9818(2) 1343(2) 41(1) C32 3645(5) 10833(3) 1937(3) 52(1) O33 1525(3) 8866(2) 2452(1) 32(1) C34 178(4) 8757(3) 1930(2) 46(1)

282
C35 -925(4) 9407(3) 2387(2) 31(1) C36 -858(4) 10517(3) 2400(2) 36(1) C37 -1898(4) 11156(3) 2825(3) 41(1) C38 -2996(4) 10636(4) 3248(2) 39(1) C39 -3076(4) 9531(3) 3261(2) 38(1) C40 -2037(5) 8930(3) 2834(3) 43(1) C41 2468(4) 7492(3) 3769(2) 34(1) O42 4065(3) 7565(2) 3953(2) 32(1) C43 4431(4) 8002(3) 4802(2) 37(1) C44 6111(4) 8167(3) 4955(2) 27(1) C45 6829(4) 9079(3) 5522(2) 36(1) C46 8409(5) 9235(3) 5676(2) 41(1) C47 9289(5) 8497(4) 5253(2) 40(1) C48 8577(5) 7578(3) 4702(2) 40(1) C49 7007(4) 7428(3) 4562(2) 34(1) ________________________________________________________________________

283
Appendix H: X-Ray Data for the Diol 334
OMeMe
MeOOH
NN
HOOMe
Me
OMeMe
H
H
H
H
OBnO
334

284
Table 1. Crystal data and structure refinement for diol 334
Empirical formula C34.50 H42 N2 O8.50
Formula weight 620.70
Temperature 153(2) K
Wavelength 0.71073 Å
Crystal system Triclinic
Space group P-1
Unit cell dimensions a = 9.0359(4) Å α = 87.720(2)°
b = 12.6771(6) Å β = 78.039(2)°
c = 14.4654(7) Å γ = 79.337(2)°
Volume 1593.03(13) Å3
Z 2
Density (calculated) 1.294 Mg/m3
Absorption coefficient 0.093 mm-1
F(000) 662
Crystal size 0.50 x 0.06 x 0.05 mm
Theta range for data collection 2.99 to 27.41°
Index ranges -11<=h<=8, -16<=k<=13, -18<=l<=18
Reflections collected 9982
Independent reflections 6911 [R(int) = 0.0950]
Completeness to theta = 27.41° 95.3 %
Absorption correction None
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 6911 / 0 / 424

285
Goodness-of-fit on F2 0.956
Final R indices [I>2sigma(I)] R1 = 0.0781, wR2 = 0.1116
R indices (all data) R1 = 0.3121, wR2 = 0.1601
Extinction coefficient 5.9(10)x10-6
Largest diff. peak and hole 0.260 and -0.236 e.Å-3

286
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2 x 103) for diol 334. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________
x y z U(eq) ________________________________________________________________________ N1 5746(4) 3576(3) 6039(2) 27(1) C2 4944(6) 2807(3) 5948(3) 27(1) C3 5811(5) 1723(3) 5589(3) 29(1) C4 6107(5) 1001(3) 6429(3) 32(1) C5 7517(5) 1162(3) 6776(3) 28(1) C6 7862(6) 659(3) 7599(3) 35(1) C7 9238(6) 673(4) 7896(3) 35(1) C8 10268(6) 1225(3) 7325(3) 34(1) C9 9953(6) 1733(3) 6506(3) 30(1) C10 8569(6) 1733(3) 6237(3) 27(1) C11 8185(5) 2346(3) 5375(3) 27(1) C12 7381(5) 3515(3) 5569(3) 27(1) C13 8185(5) 4131(3) 6135(3) 29(1) C14 7377(5) 5269(3) 6281(3) 26(1) C15 8162(6) 6120(3) 6297(3) 30(1) C16 7397(6) 7175(3) 6406(3) 29(1) C17 5818(5) 7376(3) 6461(3) 27(1) C18 5022(6) 6553(3) 6424(3) 29(1) C19 5796(5) 5486(3) 6357(3) 24(1) C20 4853(5) 4606(3) 6465(3) 30(1) N21 7238(4) 1821(3) 4888(2) 30(1) O22 3533(4) 2936(2) 6234(2) 36(1) C23 8048(5) 769(3) 4473(3) 36(1) O24 6787(4) 93(2) 8141(2) 44(1) C25 5721(6) 754(4) 8852(3) 63(2) C26 9590(6) 103(4) 8774(3) 55(2) O27 11689(4) 1243(2) 7556(2) 41(1) C28 11604(6) 2102(4) 8210(3) 59(2) O29 11004(4) 2260(2) 5933(2) 35(1) O30 9765(4) 5912(2) 6170(2) 36(1) C31 10350(6) 5475(4) 6975(3) 54(2) C32 8237(5) 8087(3) 6481(3) 34(1) O33 5059(3) 8446(2) 6503(2) 36(1) C34 4155(5) 8752(3) 7417(3) 47(1) O35 3446(4) 6775(2) 6501(2) 32(1) C36 4214(6) 4447(4) 7513(3) 42(1)

287
O37 5445(4) 4127(2) 7981(2) 50(1) C38 4971(7) 3914(4) 8950(3) 66(2) C39 6364(9) 3775(4) 9411(4) 64(2) C40 6378(11) 3215(5) 10233(5) 126(3) C41 7588(13) 3145(6) 10704(6) 124(4) C42 8800(11) 3615(6) 10320(6) 108(3) C43 8823(8) 4190(6) 9492(5) 95(2) C44 7586(8) 4272(5) 9035(4) 68(2) O1A 6645(10) -1764(6) 9328(5) 89(3) C2A 7563(16) -2716(9) 8954(9) 121(6) ________________________________________________________________________

288
Abbreviations
Ac acetyl AIBN 2,2’-azobisisobutyronitrile Ar aryl atm atmosphere BHT butylated hydroxytoluene Bn benzyl Boc tert-butyloxycarbonyl BOM benzyloxymethyl BOPCl bis(2-oxo-3-oxazolidinyl)phosphinic chloride br broad Bu butyl CAN ammonium cerium(IV) nitrate cat. catalytic Cbz benzyloxycarbonyl CI chemical ionization m-CPBA m-chloroperoxybenzoic acid CSA camphorsulfonic acid d doublet DABCO 1,3-diazabicyclo[2.2.2]octane DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DCC 1,3-dicyclohexylcarbodiimide DCE 1,2-dichloroethane DCM dichloromethane dd double doublet DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone DEAD diethyl azodicarboxylate DIAD diisopropyl azodicarboxylate DIBAL diisobutylaluminium hydride DMAP 4-N,N-dimethylaminopyridine DMDO dimethyldioxirane DMF N,N-dimethylformamide DMP Dess-Martin periodinane DMSO dimethylsulfoxide DMTS dimethylthexylsilyl equiv. equivalents Et ethyl Fmoc 9-fluorenylmethoxycarbonyl HRMS high resolution mass spectrometry imid. imidazole IR infrared KHMDS potassium bis(trimethylsilyl)amide LAH lithium aluminum hydride

289
m multiplet m meta M molar Me methyl mol mole MOM methoxymethyl mp melting point Ms methanesulfonyl MS mass spectrometry NCS N-chlorosuccinimide NIS N-iodosuccinimide NMR nuclear magnetic tesonance o ortho p para PCC pyridinium chlorochromate Ph phenyl Pht phthalimidyl PIFA [bis(trifluoroacetoxy)iodo]benzene PMB para-methoxybenzyl ppm parts per million Pr propyl pyr. pyridine PyBOP® benzotriazolyloxy-tris[pyrrolidino]-phosphonium hexafluorophosphate ORTEP Oak Ridge thermal ellipsoid program oxid. oxidation R alkyl q quartet s singlet t triplet TBAF tetrabutylammonium fluoride TBDPS tert-butyldiphenylsilyl TBDMS or TBS tert-butyldimethylsilyl TEA triethylamine TFA trifluoroacetic acid TFAA trifluoroacetic anhydride THF tetrahydrofuran TIPS triisopropylsilyl TLC thin layer chromatography TMEDA N,N,N',N'-tetramethylethylenediamine TMS trimethylsilyl TMSOTf trimethylsilyl trifluoromethanesulfonate Trt trityl Ts toluenesulfonyl TsOH or pTSA toluenesulfonic acid

290
Vita
Kenneth Stanley Matthews was born in Stockton, CA on December 16th, 1976,
the son of Mr. Stanley and Gay Matthews. He attended Lincoln High School in Stockton,
CA and graduated in May of 1995. From September 1995 to June 1999 he attended The
University of California at San Diego and graduated with a Bachelor of Science degree in
chemistry. After graduation he began work at Corvas International, Inc. as a Research
Associate until leaving for graduate school in August 2000. He entered the graduate
program of The University of Texas at Austin to obtain a Ph.D. in organic chemistry
under the supervision of Prof. Philip D. Magnus.
Permanent address: 3717 Moultrie Dr. Stockton CA 95129
This dissertation was typed by the author.