colloid transport in natural porous media: influence of surface chemistry and flow velocity
TRANSCRIPT

Pergamon Phys. Chem. Earrh, Vol. 23, No. 2. pp. 133-139, 1998
0 1998 Elsevier Science Ltd. All rights reserved Printed in Great Britain
0079-1946/98 $19.00 + 0.00
PII: SOO79-1946(98)00003-2
Colloid Transport in Natural Porous Media: Influence of Surface Chemistry and Flow Velocity
R. Kretzschmar and H. Sticher
Institute of Terrestrial Ecology, Swiss Federal Institute of Technology, Grabenstrasse 3, CH-8952 Schlieren, Switzerland
Received 25 April 1997; accepted 15 December 1997
Abstract. Mobile colloids in soils and groundwater aquifers can act as carriers for strongly sorbing chemicals and may enhance contaminant transport. Field studies suggest that mobile colloids can include clay minerals, oxides or hydroxides of Fe and Al, colloidal silica, organic matter, and biocolloids such as viruses and bacteria. The transport of colloids through porous media strongly depends on the kinetics of colloid deposition and release. Important chemical factors controlling colloid deposition and release kinetics in natural porous media are the charge of matrix and colloid surfaces and electrolyte type and concentration in the solution phase. The surface charge is strongly influenced by solution pH and the presence of specifically adsorbing ions, including organic polyelectrolytes such as humic substances. Surface charge heterogeneities on colloids and matrix surfaces also play an important role. Colloid transport experiments conducted under well-controlled conditions were conducted to investigate the influence of surface chemistry, solution chemistry, and flow velocity on the kinetics of colloid deposition in natural porous media.
Q 1998 Elsevier Science Ltd.
1 Introduction
Transport of some contaminants through groundwater aquifers and the vadose zone may be enhanced in the presence of mobile colloids (McDowell-Boyer et al., 1986; McCarthy and Zachara, 1989; Ryan and Elimelech, 1996). Colloids are usually defined as suspended particles having a dimension between 1 and 1000 nm in at least one direction (Stumm, 1993). Because of their small size, colloids have a large specific surface area and a large number of reactive surface functional groups per unit mass. Therefore, they are efficient sorbents for contaminants such as heavy metals, nonpolar organic compounds, and radionuclides, and can potentially enhance contaminant mobility. Field investigations have shown that mobile subsurface colloids are mostly composed of clay minerals,
oxyhydroxides of Fe and Al, silica, carbonates, and/or natural organic matter (NOM) (McCarthy and Degueldre, 1993). Colloidal particles can be mobilized in-situ by changes in groundwater or soil water composition, for example, decreases of ionic strength, increases of solution pH, or replacement of divalent cations by monovalent cations such as Na+ (Grolimund et al., 1996; Kaplan et al., 1996; Roy and Dzombak, 1996). Colloids composed of Fe(m) oxyhydroxides can also be generated by infiltration of oxygen-rich water into anoxic aquifers containing dissolved Fe(B) (Liang et al., 1993). Other factors that can result in colloid mobilization include dissolution of cementing agents and infiltration of specifically adsorbing anions or surfactants (Ryan and Gschwend, 1990; Ryan and Elimelech, 1996). Reported concentrations of mobile colloids in subsurface waters vary considerably. For example, Waber et al. (1990) detected colloid concentrations less than 0.2 mg/L in groundwater, Ryan and Gschwend (1990) reported colloid concentrations between 1 and 60 mg/L in groundwater, and Kaplan et al. (1993) found mobile colloid concentrations of 100 to 1700 mg/L in a field lysimeter study. Once mobile colloids have been generated, they can be transported through natural porous media at a greater velocity than conservative solute tracers due to size exclusion effects (Kretzschmar et al., 1995). For evaluating the risk of contaminant transport by mobile colloids it is essential to understand how colloids and associated pollutants will behave when they are transported into uncontaminated soil or aquifer zones. The mobilization, transport, and deposition of colloidal particles in natural subsurface porous media are still poorly understood (McCarthy and Zachara, 1989), making it difficult to predict the possible importance of colloid- facilitated contaminant transport at a contaminated site. The aim of this paper is to discuss the influence of surface chemistry, solution chemistry, and flow velocity on colloid transport and deposition in natural porous media. Examples of colloid transport experiments conducted under carefully controlled conditions are presented.
Correspondence to: R. Kretzschmar
133

134 R. Kretzschmar and H. Sticher
2 Materials and Methods
2.1 Colloids Submicron sized hematite (a-FezOx) particles were prepared using a condensed ferric Fe hydroxide gel method (Sugimoto and Sakata, 1992). The resulting particles were. characterized by transmission electron microscopy (TEM), X-ray diffraction (XRD), multi-point BET surface area analysis, potentiometric titrations, and electrophoretic mobility measurements (Kretzschmar and Sticher, 1997; Schudel et al., 1997). The colloids were 122 ~t29 nm in diameter and had a point of zero charge (PZC) at pH 9.2.
Soil humic was extracted from the surface horizon of an acidic soil (sandy skeletal, mixed, thermic Typic Umbraquult) from North Carolina and purified using standard procedures (Kretzschmar et al., 1997).
2.2 Soil columns Soil material was collected from the EB horizon of an acidic, sandy alluvial soil (Winzlerboden, Psammentic Hapludalf) in northern Switzerland. Some properties of the soil are listed in Table 1. For column experiments, a dry- sieved fraction (mostly aggregates of primary particles) between 0.20 and 0.63 mm was packed into glass chromatography columns (Omnifit, Cambridge, England) with 1.0 or 2.5 cm inner diameter and 6 to 45 cm length. The short-pulse method for determining colloid deposition rates and collision efficiencies in porous media was described in detail by Kretzschmar et al. (1997). Briefly, short-pulse breakthrough experiments were conducted with a conservative tracer (NOs) and colloids by injecting small pulses (0.002 to 0.03 pore volumes) into chromatographic columns packed with soil material under steady-state, saturated flow conditions. The Nos. concentrations in the effluent were monitored on-line by absorbance at 220 nm and colloid concentrations by absorbance at 430 nm using a flow-through UV-VIS detector. The total amounts of colloids injected (No) were determined from separate bypass experiments using the identical set-up but short- circuiting the soil column. The column influents and effluents had pH values between 5.6 and 5.8 in all breakthrough experiments.
2.3 Data analysis Under steady-state, saturated flow conditions the transport of colloids through porous media can be described by a convective-dispersive transport equation including a term for first-order colloid deposition:
ac at- - Llg-“,,F-kc
X (1)
where C is the colloid concentration in solution, t is the elapsed time, x is the travel distance, D is the dispersion coefficient for colloidal particles, v,, is the average travel
Table 1. Selected properties of the soil material used for columns
experiments (Winzlerbden. EB horizon).
f+opetiY
sieved panicle diameter (mm) +
sad C&g)
silt &/kg)
clay (g/kg)
Nz-BET surface area (m*/g)
CEC (mmol&g)
% (g/kg)
total carbon (g/kg)
’ single particles or aggregates of smaller particles
Value
0.20 - 0.63
905
42
53
3.4
23.1
2.14
I .38
velocity of colloidal particles, and k is the colloid deposition rate coefficient. In this equation, colloid release is neglected, which is justified if colloid release is slow on the time scale of a breakthrough experiment. For columns with high Peclet number, the first-order colloid deposition rate coefficient k can be calculated by numerical integration of the breakthrough pulse as (Kretzschmar et al., 1997):
k = -‘In $-]C(r)dr P [ 1 0 0
where t, = L/V p is the average travel time of the colloidal
particles through the column, q is the flux, No is the total amount of colloids injected, t, is the time after which the colloid pulse has completely moved through the column, and L is the column length.
3 Results and Discussion
3.1 Influence of humic substances The influence of humic acid on the electrophoretic mobility and colloidal stability of the hematite suspended in 10.’ M CaClz at pH 5.7 is shown in Figure 1. Since the pH was well below the PZC, pure hematite exhibited positive electrophoretic mobility. Addition of 0.1 mg/L TOC as humic acid to the suspensions caused reversal of surface charge from positive to negative. The stability of the suspensions was evaluated by optical density measurements after a 20 h time period. Pure hematite suspensions remained stable, but addition of small amounts of humic acid (0.1 or 0.2 mg/L TOC) resulted in aggregation and settling of the particles within 20 h. Addition of larger amounts of humic acid (>0.2 mg/L TOC) again increased colloidal stability due to increased negative surface charge (Figure 1B).

Colloid Transport in Natural Porous Media
4 0 lh A
F 0 20h
~I”““““.““““““” 0 1 2 3 4 5
1 IB
p 0.8 I % - 0.6 8
f 0.4
P ( 0.2 t
l
I \ eYeye
1
Obrr,.““““““.‘.“‘.‘.’ 0 1 2 3 4 5
HUMICACID (mg/L OC)
Fig.1. Influence of humic acid on the electrophotetic mobility and colloidal stability of hematite colloids (122 nm) in lOa M CaCl2 solution at pH 5.7. (A) Electrophoretic mobility measurements conducted by Laser Doppler Velocimetty - Photon Correlation Spectmscopy. (8) Colloidal stability of the 30-mg/L hematite suspensions evaluated by optical density measurements (430 nm) after 20 h flocculation and settling period.
Similar effects of humic acid, fulvic acid, and polyacryhc acid on the surface charge and stability of iron oxide colloids have been reported previously (Tipping, 1981; Amal et al., 1992; Amirbahman and Olson, 1993; Stumm, 1993; Zhang and Buffle, 1995). Humic and fulvic acids adsorb to mineral surfaces by a variety of mechanisms (Murphy and Zachara, 1995). Electrostatic attraction and specific adsorption via ligand exchange are the dominant mechanism for humic matter binding to oxide surfaces and edges of clay minerals, especially at low pH (Parfitt et al., 1977; Gu et al., 1994; Murphy and Zachara, 1995). Small amounts of adsorbed anionic polyelectrolytes result in charge neutralization and destabilization of oxide suspensions (Amal et al., 1992; Zhang and Buffle, 1995). Aggregation may also occur according to the so-called electrostatic patch model in which small amounts of adsorbed polyelectrolyte create negatively charged patches on the positively charged oxide surface, thus creating local electrostatic attraction between the colloids (Elimelech et al., 1995). Larger amounts of specifically adsorbed polyelectrolytes cause complete charge reversal and strongly increase’ colloidal stability
- NO, Tracer n O.OmgILDC
0 0.2 mg/LDC l 0.5 mg/L DC
A 1.0 mg/LDC
l 2.0 mg/L DC
1 ,
PORE VOLUMES
8 u 0.8 - 6 9 1 P s 0.6 -
2 = Q 0.4
-P 1
5 s 0.2 - l v = 153 cm/h (L-45 cm; d=l.o cm) II. -g 0 v - 25 cm/h (L-10 cm; d=2.5 cm)
oe”“‘“’ I’, se”‘t ht”a’n
135
A
B
0 1 2 3 4 5
HUMIC ACID (mg TOC L”)
10’ RapId 153 cmflt 0 v9153cmh C
‘; 5 0 v=25cm/h
I~““““““““““““‘1 0 1 2 3 4 5
HUMIC ACID (mg TOC L”)
Fig. 2. Influence of humic acid on the tmuspott and deposition kinetics of hematite colloids in a sandy soil. (A) Breakthrough curves resulting liutn short-pulse inputs of a cooserwttive tracer (N01’) and hematite colloids (30 mg/L) with different humic acid concentrations (O-4 mg/L OC; lOA M Cat&; pH 5.8). (B) Integtnted peak aceas correqondiig to the tiactions of colloids recovered in the column effluents. (C) Colloid deposition rate coefficient determined at two different flow velocities as a function of the humic acid concentmtion in the injected suspension.
(Tipping, 1981; Tipping and Higgins, 1982; Kretzschmar et al., 1997).
The influence of humic acid on the transport of hematite colloids through Ca” saturated soil columns is presented in Figure 2A. No colloids were detected in the column effluent when pure hematite was injected into 45- cm long soil columns. However, colloid mobility was strongly enhanced by small additions of humic acid to the injected suspensions. Generally, colloid breakthrough

136 R. Kretzschmar and H. Sticher
occurred well ahead of the conservative solute tracer. This can be explained by a size exclusion chromatographic effect, in which colloids are excluded from smaller pores. In our experiments this exclusion volume was on the order of 15% of the total pore space. Similar size exclusion effects were reported previously for the transport of colloids and macromolecules through natural porous media (Enfield et al., 1989; Higgo et al., 1993; Kretzschmar et al., 1995).
Colloid deposition rate coefficients were calculated from the integrated peak areas using equation (2). A summary of all breakthrough experiments on the influence of humic acid is given in Figures 2B and 2C. Experiments were conducted at two different pore water velocities using columns of different dimensions. With increasing humic acid additions the fraction of colloids recovered in the column effluents increased up to approximately 80% in the presence of 21 mg/L TOC (Figure 2B). Correspondingly, the colloid deposition rate coefficients decreased by approximately two orders of magnitude (Figure 2C). Flow velocity did not have a pronounced effect on the colloid deposition rate coefficients, however, a slight increase in deposition rate with increasing flow velocity was observed. The influence of flow velocity will be discussed later in greater detail. The dashed line in Figure 2C represents the fast deposition rate (k’““‘G3.3 he’) measured for humic- coated hematite colloids in the presence of 22~10-~ M CaCls (at v=153 cm/h). Using this experimental fast deposition rate, colloid-matrix collision efficiencies can be calculated as
(3).
As evident in Figure 2C, pure hematite was deposited almost at the fast rate (~1) even at low electrolyte concentration ( 10e4 M Car&). This can be. explained by the predominance of attractive double layer interactions between positively charged hematite colloids and negatively charged matrix surfaces in the soil column, which results in favorable deposition conditions (Elimelech, 1991). At humic acid concentrations above 1 mg/L TOC the collision efficiency a was reduced to approximately 0.01.
The results presented demonstrate that pure Fe oxide colloids, which have a PZC around pH 9, are very immobile in soils or aquifers that are composed mainly of quartz, feldspars, layer silicates, or other negatively charged minerals. In such systems, colloid-bound natural organic matter is likely to play a key role in the transport and deposition of oxide colloids. Positively charged colloids could only be mobile in strongly acidic, oxide dominated aquifers that are very low in organic matter content. The matrix surfaces in such porous media are predominantly positively charged at low pH, so that deposition of positively charged colloids would not be favorable. Indeed, Seaman et al. (1995) observed
mobilization and transport of positively charged Fe oxide colloids as a result of pH decreases in an acidic, strongly weathered aquifer material from the coastal plain of South Carolina.
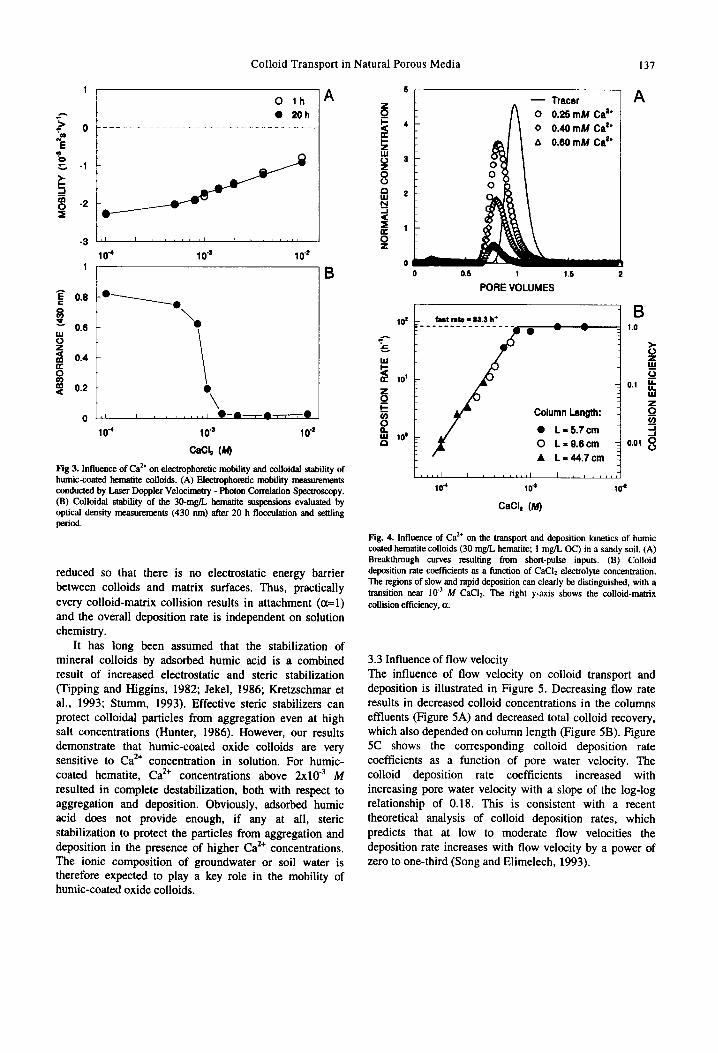
3.2 Influence of Ca” The solution chemistry in many natural soils and groundwaters is dominated by Ca’+. Therefore, it seems of great importance to understand the influence of Ca** on colloid stability and transport. The influence of solution CaCls concentration on the electrophoretic mobility and stability of humic-coated hematite colloids (1 mg/L TOC; pH 5.7) is displayed in Figure 3. Increasing Cat& concentration resulted in a decrease in negative electrophoretic mobility. This can be explained by a combination of two factors. Firstly, increasing ionic strength results in compression of diffuse double layers around charged surfaces, thus, in decreased electrical potential at the plane of shear (Hunter, 1986). Secondly, increasing Ca2+ concentration in solution results in increased complexation of functional groups of colloid- bound humic acid with Ca*+ ions (Amirbahman and Olson, 1995). This also reduces the negative surface charge and electrophoretic mobility of humic-coated oxide particles.
Increasing CaCls electrolyte concentration also resulted in reduced colloidal stability, as demonstrated by optical density measurements after a 20 h time period (Figure 3B). At CaCls concentrations above 10e3 M the suspensions aggregated and settled almost completely within 20 h. Aggregation rate measurements by dynamic light scattering confirmed that the critical coagulation concentration for this system was approximately 2x10” M CaC12.
The influence of Ca*+ on colloid breakthrough curves and deposition rate coefficients is illustrated in Figure 4. Increasing solution Ca2+ concentration from 10e4 to 10e3 M drastically reduced colloid breakthrough (Figure 4A). Figure 4B summarizes the deposition rate coefficients and collision efficiencies determined from breakthrough experiments using columns with lengths between 6 and 45 cm. It is evident that the length of the columns had no influence on the colloid deposition rates. Figure 4B represents a typical stabiliry plot for colloid deposition, in which two deposition regimes can be distinguished. The first is the regime of slow or reaction limited deposition at electrolyte concentrations below approximately 10e3 M Ca’+. In this concentration range repulsive double layer interactions are important and significantly reduce the colloid-matrix collision efficiency (a<l). With increasing CaCla concentration, the charge of soil matrix and humic- coated colloid surfaces is reduced due to specific interaction of Ca” and double layer compression, resulting in increased deposition rates. The second regime is that of fast or transport limited deposition at electrolyte concentrations greater than approximately 1O-3 M Ca*+. In this deposition regime, surface charges are significantly
Colloid Transport in Natural Porous Media
1
s- 5 b 0
“E 0 s _,
g
p -2
-3
1
z 0.8
8 z 0.6
zo gi 0.4
8
3 0.2
0
0 lh A 0 20h
Fig 3. Influence of Ca” on electmphoretic mobility and colloidal stability of humic-coated hematite colloids. (A) Electrophoretic mobility measurements conducted by Laser Doppler Velwimetry - Photon Correlation Speumswpy. (B) Colloidal stability of the 30-mg/L hematite suspensions evaluated by optical density measurements (430 nm) after 20 h flocculation and settling period.
reduced so that there is no electrostatic energy barrier between colloids and matrix surfaces. Thus, practically every colloid-matrix collision results in attachment (ml) and the overall deposition rate is independent on solution chemistry.
It has long been assumed that the stabilization of mineral colloids by adsorbed humic acid is a combined result of increased electrostatic and steric stabilization (Tipping and Higgins, 1982; Jekel, 1986; Kretzschmar et al., 1993; Stumm, 1993). Effective steric stabilizers can protect colloidal particles from aggregation even at high salt concentrations (Hunter, 1986). However, our results demonstrate that humic-coated oxide colloids are very sensitive to Ca’+ concentration in solution. For humic- coated hematite, Ca*+ concentrations above 2x10” M resulted in complete destabilization, both with respect to aggregation and deposition. Obviously, adsorbed humic acid does not provide enough, if any at all, steric stabilization to protect the particles from aggregation and deposition in the presence of higher Ca*+ concentrations, The ionic composition of groundwater or soil water is therefore expected to play a key role in the mobility of humic-coated oxide colloids.
0 0.6 1 1.6 2
PORE VOLUMES
10’
7 5
ii 10’
E F B P ; 10’
fast rat. = 83.2 h* B ________________ 90 - - 1.0
Column LenQth:
0 L-5.7cm
0 L=Q&cm
A L=44.7cm
Fig. 4. Influence of Ca*’ on the transport and deposition kinetics of humic coated hematite colloids (30 mgL hematite; 1 mg/L OC) in a sandy soil. (A) Breakthrough curves resulting from short-pulse inputs. (B) Colloid deposition rate coefficients as a function of CaC12 electrolyte concentration. The regions of slow and rapid deposition can clearly be distinguished, with a transition near Iv1 M Cat& The tight y-axis shows the colloid-matrix collision efficiency, n.
3.3 Influence of flow velocity The influence of flow velocity on colloid transport and deposition is illustrated in Figure 5. Decreasing flow rate results in decreased colloid concentrations in the columns effluents (Figure 5A) and decreased total colloid recovery, which also depended on column length (Figure 5B). Figure 5C shows the corresponding colloid deposition rate coefficients as a function of pore water velocity. The colloid deposition rate coefficients increased with increasing pore water velocity with a slope of the log-log relationship of 0.18. This is consistent with a recent theoretical analysis of colloid deposition rates, which predicts that at low to moderate flow velocities the deposition rate increases with flow velocity by a power of zero to one-third (Song and Elimelech, 1993).

R. Kretzschmar and H. Sticher
6 A
5
4
3 0 0.1 mUmin
2
1
0 0 0.5 1 1.5 2
PORE VOLUMES
‘IB
1 2 10 20 100 200 PORE WATER VELOCITY (cm h”)
1 2 10 20 100 200
PORE WATER VELOCITY (cm h”)
Fig. 5. Influence of flow velocity on colloid transport and deposition klnetics of htmtic coated hematite colloids in a sandy soil. (A) Breakthrough curves resulting from short-pulse inputs at different flow rates. (B) Colloid recovery normalized to the amount of colloids injected as a function of flow velocity and column length. (C) Colloid deposition rate coefficients as a function of flow velocity. Note, that column dimension has no influence on the first-order kinetic rate coefficients determined horn the breakthmugh curves.
4 Conclusions
The present study demonstrates that adsorbed NOM plays a key role in the transport of Fe oxide colloids through natural soils and groundwater aquifers. Humic- coated Fe oxide colloids are negatively charged at most relevant pH conditions and are therefore much more mobile than positively charged, pure Fe oxide colloids. In natural field situations, mobilized colloids and associated pollutants may be transported into uncontaminated zones
of a soil or groundwater aquifer in which the solution chemistry is dominated by Ca’+. Our results show that colloidal stability and mobility of humic-coated Fe oxide colloids are very sensitive to solution Ca*+ concentration. The data suggest that humic-coated oxide colloids would not be expected to be very mobile when the solution Ca*+ concentration exceeds about 10e3 M, provided that the overail geochemical conditions are comparable. Flow velocity was shown to have only a minor effect on the first- order kinetic colloid deposition rate coefficients. For predicting colloid transport in subsurface porous media, the kinetics of colloid release and deposition must known.
Acknowledgements. We thank Kurt Bamrettler for his technical suppon.
This research was funded by ETH Zurich.
References
be.
Amal, R.. Raper, 1. A., and Waite, T. D., Effect of fulvic acid adsorption on the aggregation kinetics and structure of hematite patticles, J. Colloid Inrerface Sci.. 151,244-257. 1992.
Amirbahmatt. A., and Olson, T. M., Deposition kinetics of humic matter- coated hematite in porous media in the presence of Car+, Colloids und SwfacesA, 99, I-10, 1995.
Amirbahman, A., and Olson, T. M., Transport of humic-matter coated hematite in packed beds, Environ. Sci. Tech&., 27, 2807-2813, 1993.
Elimelech, M.. Kinetics of capture of colloidal patticles in packed beds under attractive double layer interactions, J. Colloid Inrer@ce Sci., 146, 337- 352, 1991.
Elimelech. M.. Gregory. J., Jia, X., and Williams, R. A., Parri& Deposirion and Aggregation. Measurement. Model&, and Simulation, Butterworth-Heinemann, 1995.
Enfield. C. G., Bengtsson, G., and Lindqvist. R., Influence of macromolecules on chemical transport, Environ. Sci. Technol.. 23. 1528- 1535. 1989.
Gmlimund, D.. Borkovec, M., Barmettler, K., and Sticher, H., Colloid- facilitated transport of strongly sorbing contaminants in natural porous media: A laboratory column study, Environ. Sci. Z'e~hd.. 30, 3 1113~ 3123. 1996.
Gu, B.. Schmitt, J., Chen, Z., Liang. L.. and McCarthy, J. F., Adsorption and desorption of natural organic matter on iron oxide: Mechanisms and models, Environ. Sci. Technol.. 28, 38-46. 1994.
Higgo, J. J. W.. Williams, G. M.. Harrison, I., Warwick, P.. Gardiner. M. P.. and Longwotth, 0.. Colloid transport in a glacial sand aquifer. Laboratory and field studies. Colloids and Sutfaces A, , 179-200, 1993.
Hunter, R. J.. Foundarionr of CoNoid Science, Oxford University Press, Oxford, 1986.
Jekel. M. R., The stabilization of dispersed mineral particles by adsorption of humic substances, Water Res., 20, 1543-1554. 1986.
Kaplan. D. 1.. Bertsch, P. M.. Adriano, D. C., and Miller. W. P.. Soil-borne mobile colloids as influenced by water flow and organic carbon, Environ. Sci. Technol., 27, 1193-1200, 1993.
Kaplan. D. I., Sumner, M. E.. Bertsch. P. M.. and Adriano. D. C., Chemical conditions conducive to the release of mobile colloids from Ultisol profiles, Soil Sci. Sot. Am. .I.. 60, 269-274, 1996.
Kretzschmar, R.. Barmettler, K.. Grolimund, D., Yan, Y. D., Borkovec. M.. and Sticher, H., Experimental detetmination of colloid deposition rates and collision efficiencies in natural porous media, W&r Resow. Res., 33, 1129-1137, 1997.
Kretzschmar. R., Hesterberg, D.. and Sticher. H.. Influence of adsorbed htmtic acid on surface charge and flocculation of kaolinite. Soil Sci. Sot. Am. J., 61. 101-108. 1997.
Ktutrachmar. R., Robarge, W. P., and Amoozegar. A., Influence of natural organic matter on transport of soil colloids through saprolite, Water Resow. Res., 31.435-445. 1995.

Colloid Transport in Natural Porous Media 139
Kretzschmar, R., Robarge. W. P., and Weed, S. B., Flocculation of kaolinitic Ryan, J. N., and Gschwend, P. M., Colloid mobilization in two atlantic soil clays: Effects of humic substances and iron oxides, Soil Sci. Sot. Am. coastal plain aquifers: Field studies, Water Resow. Res., 26, 307-322. J.. 57, 1277-1283. 1993. 1990.
Kretzschmar, R., and Sticher, H.. Transport of humic-coated iron oxide colloids in a sandy soil: influence of Ca*’ and trace metals, Environ. Sci. Technol.. 1997 (in oress).
Schudel. M., Behrens. H., Holthoff, H.. Kretzschmar, R.. and Borkovec. M.. Absolute aggregation rate constants of hematite particles in aqueous suspensions: A comparison of two different surface morphologies. J, Colloid Interjbce Sci. (accepted). Liang, L.. McCarthy, j. F.. Volley, L. W., McNabb, J. A., and Mehlhom, T.
L., Iron dynamics: Transformation of Fe(lI)/Fe(III) during injection of natural organic matter in a sandy aquifer, Geochim. Cosmochim. Actu, 57, 1987-1999. 1993.
McCarthy, J. F., and Degueldre, C. Sampling and characterization of colloids and particles in groundwater for studying their role in contaminant transport, pp. 247-315. In J. Buftle and H. P. van Leeuwen @is.) Envimnmenrcrl Panicles, Vol. 2. Lewis Publishers, Boca Raton, 1993.
McCarthy, 1. F.. and Zachara, J. M., Subsurface transport of contaminants, Environ. Sci. Technol., 23.496-502, 1989.
McDowell-Boyer, L. M.. Hunt, J. R., and Sitar, N.. Particle transpott through porous media, Wafer Resow. Rex.. 22. 1901-1921, 1986.
Murphy, E. M.. and Zachara. I. M.. The role of sorbed humic substances on the distribution of organic and inorganic contaminants in groundwater. Geodermu, 67, 103-124. 1995.
Parfitt, R. L.. Fraser, A. R.. and Farmer, V. C.. Adsorption on hydrous oxides. III Fulvic acid and humic acid on goethite. gibbsite and imogolite. J. Soil Sci., 28, 289-296, 1977.
Roy, S. B., and Dzombak. D. A.. Colloid release and transport processes in natural and model porous media, Colloidc and Sutfaces A, 107.245-262. 1996.
Ryan, I. N., and Elimelech. M., Colloid mobilization and tmnspott in groundwater, Col1oid.v and Surfaces A, 107, l-56, 19%.
Seaman. J. C.. Bcrtsch. P. M.. and Miller. W. P.. Chemical controls on colloid generation and transpott in a sandy aquifer. Environ. Sci. Technol.. 29, 1808-1815, 1995.
Stumm, W.. Aquatic colloids as chemical reactants: Surface structure and reactivity, Colloids andSutface.r A, 73, I-18, 1993.
Sugimoto. T.. and Sakata, K., Preparation of monodisperse pseudocubic a- Fez03 particles horn condensed ferric hydroxide. J. Colloid Inteflace Sci., 152.587-590, 1992.
Tipping, E.. ‘IIe adsorption of aquatic humic substances by imn oxides, Geochim. Cosmochim. Acta, 45, 191-199. 1981.
Tipping, E.. and Higgins. D. C.. The effect of adsorbed humic substances on the colloid stability of haematite particles, CoNoids and Surfaces, 5, 85- 92, 1982.
Waber, U. E.. Lienert. C., and van Gunten, H. R., Colloid-related infiltration of trace metals from a river to shallow groundwater, J. Conram. IIydroL. 6.251-265, 1990.
Zhang, J., and BuBle. J., Kinetics of hematite aggregation by polyacrylic acid: Importance of charge neutralization. 1. Colloid Interfke Sci.. 174, 500-509. 1995.