cloning the cystic fibrosis gene: implications ... · a serum protein, cf antigen, consistently...
TRANSCRIPT

Thorax 1990;45:46-55
Cysticfibrosis * 3 Series editor: D J Shale
Cloning the cystic fibrosis gene: implications fordiagnosis and treatment
David J Porteous, Julia R Dorin
In this article we trace the steps that led to themolecular cloning of the cystic fibrosis gene
and identification of the major mutation, dis-cuss detection assays that have been designedand applied to prenatal diagnosis and theproblems associated with extending genemutation assays to population carrier detec-tion, and finally speculate on the prospects forimproved treatment in the light of the likelybiochemical function of the normal gene
product.
The genetics of cystic fibrosisCystic fibrosis is the most common autosomalrecessive genetic disease in white populations;with a carrier frequency of about 1 in 25; itaffects around 1 in 2500 neonates. The mostcharacteristic physiological defect is inchloride ion transport, which causes a raisedchloride concentration in sweat, a long recog-
nised diagnostic marker of the disease.Exocrine pancreatic insufficiency is a frequentcomplication, leading to meconium ileus andmalabsorption. Several fetal intestinal andmicrovillar enzymes are characteristicallypresent at reduced concentrations in affectedprenancies as a consequence of the disease.These enzymes provided a basis for rapidprenatal diagnosis in the second trimester in"at risk" families, without the possibleproblems of "crossovers" and "non-infor-mativeness" associated with the early DNAprobes.'
Medical ResearchCouncilHuman Genetics Unit,Western GeneralHospital,Edinburgh EH4 2XUD J PorteousJ R DorinReprint requests to:Dr Porteous
Candidate genesTwo complementary genetic approaches havebeen applied to cystic fibrosis.2 The "clas-sical" approach is to study candidate genes forthe proteins concerned either in terms of theirinferred or proved biochemical function or
through consistent association of their muta-tions (alteration or deletion) witlh the disease.A serum protein, CF antigen, consistentlyoccurred in affected individuals, was absentfrom normal serum, and was found at inter-mediate concentrations in heterozygotes.Monoclonal antibodies to CF antigen madepossible its purification and tissue localisation.Peptide sequencing facilitated cloning viacomplementary DNA (cDNA) librariesderived from tissues that expressed the pep-tide.45 These studies showed that CF antigen
comprises two peptides, both of which havebeen mapped on the same region of chromo-some 1 and show homology to the S-100family of calcium binding proteins.6 Althoughthis is not the primary defect, the intermediateconcentrations of CF antigen seen inheterozygotes, and the expression profile innormal individuals (high concentrations ingranulocytes and moderate concentrations inepithelial cells of the tongue, oesophagus, andbuccal cavities and in cultured nasal epithe-lium in these carriers) may indicate a role inthe functional expression of the cystic fibrosisgene.
Electrophysiological studies on intactsecretory epithelial cells from patients withcystic fibrosis show defective chloride iontransport78 (see next article in this series). Anobvious, if difficult, route to the basic defectwould be the isolation of chloride ion channelproteins.9 The "reverse" genetic approach,however, succeeded in identifying the cysticfibrosis gene, which is most likely a mutant ofthe gene regulating chloride ion transportrather than that coding for the ion channelitself.
From genetic linkage to the disease gene"Reverse" genetics does not presuppose thenature of the abnormal gene but dependsessentially on establishing where the generesides on the chromosome. Once a gene hasbeen linked to a particular chromosomal regionthen somatic cell and molecular geneticmethods* can be applied to determine preciselywhere the gene lies.Solomon and Bodmer'0 first suggested that,
just as protein variants can be used in pedigreeanalysis to track disease susceptibility, so theinherent variability in DNA sequence betweendifferent individuals could be exploited. DNAsegments not genetically linked to the diseasesegregate randomly through the pedigreewhereas linked fragments associate positivelywith the disease. The frequency with which theassocation breaks down, or a "crossover" isobserved, provides an estimate of the geneticdistance, in terms of recombination events,between theDNA marker and the disease gene.This method of genotyping relies on thespecificity of restriction endonucleases asmolecular scissors for revealingDNA sequencevariation between individuals through the*For explantions of molecular biology methods see Thorax1990;45:52-6 (January) and 147-53 (February).
46
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from
Cloning the cystic fibrosis gene: implications-for diagnosis and treatment
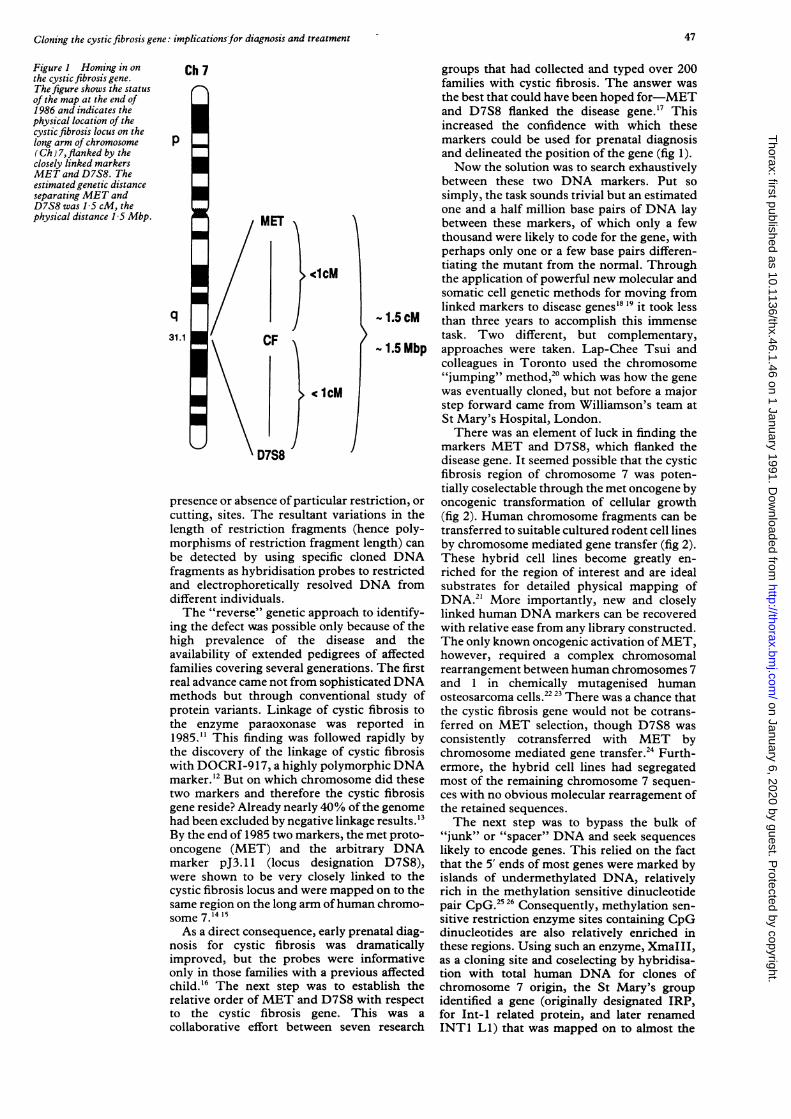
Figure I Homing in onthe cystic fibrosis gene.The figure shows the statusof the map at the end of1986 and indicates thephysical location of thecystic fibrosis locus on thelong arm of chromosome(Ch) 7,flanked by theclosely linked markersMET and D7S8. Theestimated genetic distanceseparating MET andD7S8 was 1-5 cM, thephysical distance 1 5 Mbp.
Ch 7
p
q31.1
MET
CF
D7S8
<lcM
ccM
presence or absence of particular resticutting, sites. The resultant variatiolength of restriction fragments (heimorphisms of restriction fragment lebe detected by using specific clonfragments as hybridisation probes toand electrophoretically resolved DIdifferent individuals.The "reverse" genetic approach to
ing the defect was possible only becahigh prevalence of the diseaseavailability of extended pedigrees o
families covering several generations.real advance came not from sophisticamethods but through conventionalprotein variants. Linkage of cystic fthe enzyme paraoxonase was rep1985." This finding was followed r
the discovery of the linkage of cystiwith DOCRI-9 17, a highly polymorpmarker.'2 But on which chromosometwo markers and therefore the cystigene reside? Already nearly 40% of thhad been excluded by negative linkageBy the end of 1985 two markers, the noncogene (MET) and the arbitramarker pJ3. 11 (locus designationwere shown to be very closely linkicystic fibrosis locus and were mappedsame region on the long arm ofhumansome 7.14 '5As a direct consequence, early pren
nosis for cystic fibrosis was draimproved, but the probes were in:only in those families with a previouchild. 6 The next step was to estarelative order ofMET and D7S8 wilto the cystic fibrosis gene. Thicollaborative effort between seven
groups that had collected and typed over 200families with cystic fibrosis. The answer wasthe best that could have been hoped for-METand D7S8 flanked the disease gene. 17 Thisincreased the confidence with which thesemarkers could be used for prenatal diagnosisand delineated the position of the gene (fig 1).Now the solution was to search exhaustively
between these two DNA markers. Put sosimply, the task sounds trivial but an estimatedone and a half million base pairs of DNA laybetween these markers, of which only a fewthousand were likely to code for the gene, withperhaps only one or a few base pairs differen-tiating the mutant from the normal. Throughthe application of powerful new molecular andsomatic cell genetic methods for moving fromlinked markers to disease genes'8 19 it took less
-1.5 cM than three years to accomplish this immensetask. Two different, but complementary,
- 1.5 Mbp approaches were taken. Lap-Chee Tsui andcolleagues in Toronto used the chromosome"jumping" method,20 which was how the genewas eventually cloned, but not before a majorstep forward came from Williamson's team atSt Mary's Hospital, London.There was an element of luck in finding the
markers MET and D7S8, which flanked thedisease gene. It seemed possible that the cysticfibrosis region of chromosome 7 was poten-tially coselectable through the met oncogene by
riction, or oncogenic transformation of cellular growthins in the (fig 2). Human chromosome fragments can bence poly- transferred to suitable cultured rodent cell linesngth) can by chromosome mediated gene transfer (fig 2).ed DNA These hybrid cell lines become greatly en-restricted riched for the region of interest and are idealNA from substrates for detailed physical mapping of
DNA.2' More importantly, new and closelyidentify- linked human DNA markers can be recovereduse of the with relative ease from any library constructed.and the The only known oncogenic activation ofMET,
If affected however, required a complex chromosomalThe first rearrangement between human chromosomes 7LtedDNA and 1 in chemically mutagenised humanstudy of osteosarcoma cells.22 23 There was a chance thatibrosis to the cystic fibrosis gene would not be cotrans-ported in ferred on MET selection, though D7S8 wasapidly by consistently cotransferred with MET byic fibrosis chromosome mediated gene transfer.24 Furth-hic DNA ermore, the hybrid cell lines had segregateddid these most of the remaining chromosome 7 sequen-ic fibrosis ces with no obvious molecular rearragement ofLe genome the retained sequences.results.'3 The next step was to bypass the bulk of
net proto- "junk" or "spacer" DNA and seek sequencestry DNA likely to encode genes. This relied on the factD7S8), that the 5' ends of most genes were marked by
ed to the islands of undermethylated DNA, relativelyI on to the rich in the methylation sensitive dinucleotidei chromo- pair CpG.2526 Consequently, methylation sen-
sitive restriction enzyme sites containing CpGLatal diag- dinucleotides are also relatively enriched inLmatically these regions. Using such an enzyme, XmaIII,formative as a cloning site and coselecting by hybridisa-Is affected tion with total human DNA for clones ofblish the chromosome 7 origin, the St Mary's groupth respect identified a gene (originally designated IRP,s was a for Int-1 related protein, and later renamedresearch INTl Ll) that was mapped on to almost the
47
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Porteous, Dorn
(a) MET SELECTED CHROMOSOME MEDIATED GENE TRANSFER
MNNG-HOS cell line carrying activatedMET oncogene on Ch 7 Mitotic arrest Chromosome isolation
Ca PO4co-precipitate
_... 6
Foci weeks NIH3T3 monolayer
human MET selected CMGTmouse hybrid retaining - 3Mbpof Ch 7 DNA including METand D7S8
(b) CpG ISLAND CLONING OF INTI LI
- 3Mbp human Ch 7, MET and D7S8 + veI
prepare DNA
Xmalll - HindlIl library screenedfor human sequences to selectfor candidate CF gene
CpGdeave with Xmalli+ Hindll, cloneinto Loist 6b
Alu
X HHybridisation select for - 0.1%
1. map to MET-D7S8 interval human recombinants2. isolate overlapping clones from standard DNA library CpA3. identify RFLPs Alu4. map closer to CF5. sequence to identify INT1 Ll Probable location of the CF gene
linka Disequilibrium Gradi'net D met H XV2c CS.7 KM19 pJ3.11 RFLPs
a a CpG islandINT1 Li
ient
D7S8 LociTranscription
100 kbp Scale
Figure 2 Met selected chromosome mediated gene transfer and CpG island cloning of INTI Ll. (a) Method ofchromosome mediated gene transfer was used by the St Mary's team to derive a somatic cell hybrid whose humancomponent was reduced to about 3 Mbp ofDNA subtending the cysticfibrosis (CF) locusfrom the MNNG-HOSchromosome. (b) Restricting the DNA with XmaIII and screeningfor human sequences enrichedfor CpG islandassociatedfragments of chromosome (Ch) 7. One suchfragment identified a gene, INTI LI, that was mapped betweenMET and D7S8 and was in strong linkage disequilibrium with cysticfibrosis.
exact site where the cystic fibrosis gene was
predicted to lie.27 Polymorphic fragments ofthis gene showed strong linkage disequilibrium(that is, a bias towards an association of one
haplotype with the disease by comparison withits frequency in the normal population), com-
pelling evidence that they were either in or veryclose to the gene.The finding of informative polymorphisms
and strong linkage disequilibrium with theXV2C, KM19, and CS7 probes derived from
around this CpG island improved prenataldiagnosis still further, but neither the nature ofthe gene as an inferred secreted growth factornor its pattern of expression fitted with thatpredicted for the cystic fibrosis gene28 and no
differences were detected by sequencing bet-ween normal and affected individuals. Finallyand conclusively, crossovers between the CpGisland associated with INT1 LI and cysticfibrosis were discovered.2930 The hunt for thereal CF gene was not over.
n
MET_F-0
48
h y -Y :.OcNo )t j k 4-1%-1
r- 0
,1
0No 00
0 0
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Cloning the cystic fibrosis gene: implications for diagnosis and treatment
:ACS enrichment for Ch 7 markers (b) Saturation mapping of Ch 7 (c) "Walking" (d) "Jumping"
man Ch 7 only CHO hybrid
cSP14I Flumscenty
stainedftomosomes
| |FACS
O1 0 a
0 00 0
CHO Human DNA librayChs Ch 7
,.-,258
45!f
i llI I I
I III I
I
Il B-ElB-E7
High Mwt DNAMET
, Partal EcoRldest
E1 E2 E3E4Es E6 E7
B B
Add tag marker, (NM)4, religate to form circles
R;ec with second enzyrselect for tag molecule,recover both ends of IonDNA fragment
plasmid
DS78 prepare B-E7 and B-E1 fragmentsas separate pbes
Figure 3 Saturation cloning, "jumping", and "walking" to cystic fibrosis transmembrane regulator gene. The figure illustrates the strategy initiatedby Lap-Chee T'Sui and coworkers, laterjoined by Collins and colleagues, for cloning the gene. (a) A FACS sorted library of human chromosome 7fragments was searched exhaustively for markers mapped between MET and D7S8. (b) Each new marker was located precisely to a subregion ofchromosome 7 by mapping it on an extensive panel of somatic cell hybrids carrying deleted or translocated chromosome 7s. (d) Bidirectional "jumps"were madefrom MET,from D7S8, andfrom the two new start points, the gaps between one "jumping" clone and the next beingfilled in by "walking"along the chromosome in small steps, until (c) the INT1 Li gene was reached and passed. Each new segment ofDNA was checkedfor polymorphismsto follow the gradient towards maximum linkage disequilibrium andfor species conservation as an indicator of coding sequence. The isolation of thecystic fibrosis transmembrane regulator gene and the identification of the AS508 mutation was announced in September 1989. Of 258 clones mapping onto chromosome 7, 45 were shown to map close to the cystic fibrosis region and two to map between MET and D7S8.
Saturation cloning and chromosome"jumping" and "walking" identify thecystic fibrosis geneThe St Mary's team continued to "walk" fromthe INT LI gene towards the cystic fibrosisgene while the teams in Toronto and Michigancollaborated to saturation clone the regionbetween MET and D7S8. As a first step, Tsuiand colleagues searched through 258 differenthuman chromosome 7 probes from a FACSsorted library to find two that were mappedbetween MET and D7S8.3' These became thestarting points for "jumps" along thechromosome in either direction20 (fig 3).Chromosome jumping libraries are con-
structed by cleaving genomic DNA to producea representative set of long, linear molecules-upwards of 100 000 base pairs (100 Kbp).These are ligated with a "tag" molecule underconditions favouring the formation of circularmonomers rather than linear concatemers. The
DNA is then cleaved to produce many smallfragments and the selectable tag molecule isused to isolate only those derived from eitherend of the original molecule, originallyseparated on the chromosome by 100 Kbp ormore (fig 3). Two consecutive jumps in thesame direction took them to the CpG islandassociated with INT LI. This indicated thatthey were moving towards D7S8 and the regionof maximum linkage disequlibrium.32 Furtherjumps, complemented by extensive isolation ofoverlapping, intervening clones ("walking"),enabled them to bypass the unclonable regionthat had stalled the progress of the St Mary'steam. In the region with maximum linkagedisequilibrium with cystic fibrosis they sear-ched for sequences conserved across speciesboundaries (that is, homologous DNA seg-ments present in mouse, hamster, bovine, orchicken DNA, and sometimes all of these-referred to as zoo blots) as an indication of
49
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Porteous, Dorin
coding rather than junk or spacer DNA.32Unfortunately, the new candidate gene seg-
ments failed to detect RNA transcripts intissues predicted to express the cystic fibrosisgene. DNA sequence analysis did show,however, a short length of open reading framedistal to a CpG rich, undermethylated stretchof DNA. Thus the region had some of thehallmarks of a bona fide gene. For verification,they searched for homologous sequences incomplementary DNA (cDNA) libraries con-structed from cultured epithelial sweat gland, atissue predicted to express the gene.33 Even-tually, a single cDNA clone was isolated. Itrecognised a 6-5 Kbp transcript, the expressionof which was essentially restricted to exocrinetissues. Sequentially isolated cDNAs wereclearly related one to another, but contained ahigh level of sequence rearragement, plusextraneous sequences of uncertain origin. Nofull length cDNA clones could be isolated, buteventually the entire coding sequence wasabstracted from overlapping clones. Much ofthe difficulty of first identifying genic sequen-ces in the cystic fibrosis region and thenisolating large cDNAs is explained by the factthat the mRNA is transcribed from 27 shortexons stretched over about 250 Kbp ofgenomicDNA. Nevertheless, DNA sequence homologysearches and translation of the cDNA sequenceinto a predicted protein structure have alreadytold us much about the likely function of thecystic fibrosis gene product (to be discussed indetail in the next article). Furthermore, iden-tification of disease associated mutations in thecystic fibrosis transmembrane regulator havenot only aided prenatal diagnosis but have alsocontributed to a growing understanding ofdemographic and clinical anomalies.
Disease associated mutations in cysticfibrosis transmembrane regulatorComparison of the cDNA sequences of thecystic fibrosis transmembrane regulatorisolated from normal and affected individualsrevealed a major mutation comprising a threebase pair deletion, causing the loss of a singlephenylalanine at residue 508 in the first ATPbinding domain. This mutation accounts forabout 70% of cystic fibrosis mutations inNorth American34 and slightly more in north-ern European populations,35 46% in southernEuropean populations,36 30% in Ashkenazifamilies,37 and 35% in the closed community ofHutterites.38 Initially, there appeared to be agood correlation between homozygosity for theA508 mutation and pancreatic insufficiency,39 40but this is not absolute.4' Similarly, althoughmild lung disease is often associated withpancreatic sufficiency, individuals with onlymoderate lung disease in a severely affectedfamily, all carrying the A508 mutation, havebeen described.42The search for mutations accounting for the
remaining 30%, or more, of affected chromo-somes continues. This relies on the polymerasechain reaction to amplify regions of the gene fordetailed analysis (fig 4). Analysis may be byvarious methods, including high resolution
polyacrylamide gel electrophoresis, to look forlength polymorphisms (micro deletions orinsertions)43 chemical cleavage methods todetect and position single base mismatches (forexample, the hydroxylamine osmium tetroxidemethod44), neutral gel analysis of singlestranded DNA mobility to look for sequencecomposition or length variations that lead toconformational polymorphisms,45 or simply bydirect DNA sequence analysis. The first fewdocumented cases used the last two methods todetect mutations on non-A508 chromo-somes.4647 In one case neither parent of anaffected child carried the A508 mutation, but atwo nucleotide insertion in exon 13 was foundin the mother that would result in a severelytruncated and non-functional protein.' Thenature of the paternal mutation was not dis-covered. In a study searching for mutations inthe transmembrane region three different basechange mutations, each causing an amino acidchange from positively charged to less polar,were detected in exons 4 and 7 of non-A508chromosomes in 46 patients.47 One of thesemutations was common to two of the A508compound heterozygote families where thepenetrance was quite variable. Most of thepatients had a mild form of the disease, but oneindividual was severely affected. Additionalenvironmental, epigenetic, or genetic factorsclearly contribute to the clinical picture withthese atypical mutations.
It has been assumed that chloride ion trans-port is an essential function and thatindividuals homozygous for null mutations ofthe cystic fibrosis transmembrane regulator,such as frameshift terminations, will notsurvive. Such mutations will be found only incompound heterozygotes with dysfunctionalmutations, such as A508. It will be of con-siderable interest to identify further mutationsof the dysfunctional class (amino acid substitu-tions or, possibly, aberrantly spliced products)as these will indicate functionally importantregions of the gene. Possibly the common A508mutation may represent a partial "gain offunction" or "over expression" defect. Anadvantage for A508 heterozygotes-forexample, increased resistance to chloridesecreting diarrhoea, might explain the highprevalence of cystic fibrosis in whitepopulations,48 although this is hard to reconcilewith the data for the southern Mediteraneanand Ashkenazi populations, where the A508mutation accounts for less than half the muta-tions yet all are associated with the samehaplotype (defined by the closely linked mark-ers and dubbed B), which in turn accounts forover three quarters of the mutated cysticfibrosis transmembrane regulator chromo-somes.3637 Certainly, a second mutation (per-haps three or more) must have arisen on themajor haplotype. The peak of linkage dis-equilibrium in these atypical populationspoints clearly to the region between INTl LIand the cystic fibrosis gene. Additionalintervening sequences, coding or controlling,may play a part in the aetiology of cystic fibrosisand contribute to the heterozygous advantageof the B haplotype and the high prevalence ofcystic fibrosis.
50
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Cloning the cystic fibrosis gene: implications for diagnosis and treatment
Figure 4 Polymerasechain reaction baseddetection of the A508mutation. The PolymeraseChain Reaction is used togenerate 1lo or more copiesof exon 10, site of the A508mutation. The sequence isthen compared with thecorresponding DNAsegment from a normali.ndividual either by directsequence analysis, byaccurate sizing on apolyacrylamide gel, or byhybridisation with allelespecific oligonucleotides.CFTR-cysticfibrosistransmembrane regulatorgene; T-thymine; G-guanine; C-cytosine;A-adenine.
CFTR E 1 OOKbp I L
i ~~~~~~~~~~~~~p
Exon 10 40bp I
codon 465 5281. Denature template DNA (1 min, 92°C)2. Anneal exon 10 oligonucleotides specific to each DNA strand (1 min, 53TC)3. Synthesise new strand with Taq polymerase (2 min, 72T)4. Repeat cycle x 25, or more
25 cycles
225 products pertemplate molecule
x105
(a) Direct sequence (b) Size difference/allele specific oligonucleotide
T
GG
T
T
A
C
NORMALG AT C
TTGTGGTTAC
A508G ATC
+/-3bp
Prenatal diagnosisEach mutation discovered sheds light on thefunctional constraints of the cystic fibrosistransmembrane regulator protein, but com-
plicates unequivocal prenatal diagnosis as a
specific detection assay must be devised foreach mutation. For this reason, an immuno-logical assay to distinguish functional and dys-functional protein in accessible tissue would beideal. Efforts are focusing on the putative cellsurface domain, with peptides as immunogensor fusion protein constructs. The polymerasechain reaction49 makes detection of the A508mutation and screening for new mutations atthe DNA level much easier (fig 4). The poly-merase chain reaction allows any segment ofDNA lying between two sequence specific sitesdefined by two chemically synthesised oligo-nucleotide primers to be repeatedly amplified,
-- - - N oligo-A508
oligo
generating unlimited copies of the interveningDNA corresponding to the genotype of theoriginal template. The source ofDNA templatecan be a minute quantity derived from a bloodspot, Guthrie card, mouth wash, microvillarsample, or even a single cell removed from the 8cell stage of a human embryo fertilised invitro."The A508 mutation can be detected in
several ways-for instance, on the basis of theintrinsic size difference between the ampli-fication products derived from normalchromosomes and those with the deletion ofthree base pairs, and by using allele specificoligonucleotides that encompass the mutation.Unaffected, carrier, and affected individualsare typed according to the presence or absenceof hybridisation with the polymerase chainreaction products (fig 4). Substituting fluores-
51
......... rrrTTlrrTTn
ffTTTTT77TT17TTT71rm
rrr,rn,rTTrrrrrrrrrn
rrrrrrrrm
rrrrrrrrrrrrrrrrrm"" 4 a t ttyrTTTrrrm
t 2 I I I rrrrrrrrm
rrrrTTrrrrrrrrTTTTn
rrrrrrTrrTrrrrrTIrm
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Porteous, Dorin
Figure 5 Gene targettingiin miouse stem cells tocreate atn animal modelforcysticfibrosis (CF).Outline of theexperimental scheme forderivitng chimeric micefroyn steml cells mutated atthe uioiuse cystic fibrosistransniembrane regulator(CFTR} locus by genetargettinig.
_~~x
i ~~~CFTR targetingBlastocyst / 0i
Al
MicroinjectElectroporateLipofect
Pluripotent ES cells E
Select and/or PCRscreenfor homologous recombination|
BlastocystInjection
A508o+ germline chimera
A508/+: +1+
x
A5081+ As08/+
Ao508+ A508/+CF heterozygotes
carriers
x 2
+1+
+1+ +1+
+1+
Normal
cently labelled primers and probes offers excit-ing opportunities for simultaneous screeningfor several different mutations in a single testtube, the result being indicated simply by theabsorption spectrum of the product.
Despite the power of the polymerase chainreaction and an increasingly elegant set ofmutation screening and detection assays, scan-ning all 27 exons, totalling 6 5 Kbp of codingsequence, for new mutations is an enormoustask. If, as seems likely, some defects are due tonon-coding mutations affecting transcriptionthe task will be greater as the cystic fibrosislocus encompasses about 250 Kbp of genomicsequence. It may be of practical value tosubdivide cystic fibrosis into A508 and non-A508 disease for the purpose of prenataldiagnosis and population screening. Recognis-ing that clinical variation among the haemo-globinopathies may be related to mutationaldifferences within the globin locus has been agreat help in population screening, prenataldiagnosis, genetic counselling, and clinicaltreatment. With cystic fibrosis, the freq-uency
of A508 approaches 75%, in most populationswhere the mutation is prevalent, and prenataldiagnosis for this one mutation is available nowfor almost half the families with cysticfibrosis.35
Population screeningThe arguments for and against populationscreening are complex and strongly felt.5' 52There is much to learn from the failure of thescreening programme for sickle cell anaemiaand the success of the Tay-Sachs and ,B thalas-samia screening programmes. The scientificknowledge and technology required for screen-ing for the major cystic fibrosis mutationalready exists. The major outstanding re-quirement is a comprehensive programme ofinformation, education, counselling, and sup-port. The American Society of Human Gene-tics considers it premature to offer carrierdetection as part of antenatal care. Meanwhile,commercial companies are eager to tap theperceived public demand for testing, but are
constructligonucleotideactedal vectorAC recombinant
A508/A508CF homozygote
affected?
52
1-ft.0
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Cloning the cystic fibrosis gene: implications for diagnosis and treatment
unable or unwilling to accept the responsibi-lities for genetic counselling that must accom-pany the test results. In Britain pilot schemesdesigned to ascertain the acceptability andeffectiveness of screening through schools,general practitioners, or antenatal clinics areunder way. Whatever programmes arefavoured, carrier screening gives prospectiveparents informed choice and may reduce theincidence of cystic fibrosis, freeing valuableresources for the care and treatment of existingpatients. Screening for A508 alone in theUnited Kingdom would identify over 100fetuses with cystic fibrosis annually. In NorthAmerica the figure would exceed 1000. Such aprogramme would reduce the a priori riskestimate to 1 in 10 000 if one parent is shownnot to carry the A508 mutation, and 1 in 40 000if both parents are found not to carry it. Ofcourse, the opposite is also true, with theattendant risks proportionally increased if oneor both parents are found to be carriers. Theneed for informed consent to testing and acomprehensive genetic counselling and followup programme cannot be overemphasised.
Prospects for treatmentThe prospects for survival with cystic fibrosisto early adulthood have increased over recentyears. The most aggressive population screen-ing programme imaginable would still take ageneration to have full effect. Until then, mostnew cases of cystic fibrosis will continue to arisein families with no previous history of thedisease. As a direct result of the molecularcloning of the cystic fibrosis gene, we canconfidently anticipate progress by several com-plementary approaches to improved treatment.The major mutations, and perhaps most
mutations, cause dysfunction rather than func-tional replacement and the prospects for drugor antibody treatment are good. Elucidation ofthe complete ion transport pathway in vivo andan understanding of the specific role of thecystic fibrosis transmembrane regulatorprotein in that complex process may provide arational approach to compensatory interven-tion.For this purpose we and others are establish-
ing an animal model for the disease. This willcomplement studies with cultured humancystic fibrosis cells and provide a basis forphysiological and pharmacological testing. Theexperimental approach is outlined in figure 5and hinges on two recent developments. Thefirst is the ability to derive pluripotentembryonal stem cells from mouse blastocysts,which can be maintained and manipulated inculture.53 On being reintroduced to mouseblastocysts, genetically manipulated stem cellscan contribute to both the somatic and the germline tissues of the resultant chimeric mouse.The second requirement is to mutate, in the
stem cells, the mouse equivalent of the cysticfibrosis transmembrane regulator gene byhomologous recombination, or gene target-ing.54 Cloned fragments of the cystic fibrosisgene are constructed in such a way that ontransfection of cultured stem cells and
homologous recombination with the corres-ponding chromosomal sequences they mutatethe endogenous transmembrane regulatorgene. This process is inherently inefficient, butvarious selection or screening strategies havebeen devised to identify the rare homologousrecombination events. Such is the promise ofthis approach for the analysis of development,cancer, and genetic disease that despite itstechnical difficulty the approach has alreadybeen successful with genes as diverse as thosecoding for hypoxanthine phosphoribosyl-transferase,55 the cellular homologue of theAbelson murine leukaemia virus (c-abl).56
In the simplest gene targeting experimentthe homologous recombination event com-pletely disrupts gene function, though mimick-ing the natural mutations will be important.We have designed vectors that may be used tointroduce precise chain termination events andthese will be valuable for recreating clinicallyinteresting natural mutations.4657 Indeed, thisstrategy has been used successfully to create atruncation mutation of c-abl in mouse stemcells.56
Ideally, we wish to mimic the dysfunctionalA508 mutation. This calls for a more preciseapproach to gene targeting, which we will testin various ways. One option is to microinjectA508 specific oligonucleotides into stem cellsand screen exhaustively by a stringentpolymerase chain reaction protocol for DNAstrand displacement events. We will also isolatethe entire mouse cystic fibrosis gene in the formof a yeast artificial chromosome recombinant58and then be able to introduce any chosenmutation by standard yeast genetic proceduresinto the recombinant.A final and direct approach to management
of cystic fibrosis is somatic gene treatment.Developments in three directions encourageoptimism and deserve consideration. Ethicaldilemmas impinge on the use of fetal tissuetransplants. Safety considerations temper theuse of differentiated epithelial cells from nor-mal patients transformed in vitro. One solutionto the consequences of pacreatic insufficiencymight be to enclose secretory exocrine cells inGor-Tex fibres to form a contained, implant-able organoid.59The second approach would be to use retro-
viral vectors to transfer a functional trans-membrane regulator cystic fibrosis gene, orcompensatory regulator gene, to secretory epi-thelial cells. Cell specificity of infection wouldbe achieved by using ecotropic murine retro-viruses, which can infect only the chosen targetcell type through antibody mediated binding ofa cell surface receptor.60 A third, relatedapproach uses polycations such as poly-L-lysine to encapsulate and deliver the thera-peutic DNA construct to the cell surface. Cellspecific transfer is achieved by covalent linkageof an appropriate ligand to the polycation. Thisreceptor mediated endocytosis gene transferstrategy has successfully directed intra-venously injected DNA constructs to rat hepa-tocytes and achieved efficient transfection ofcultured haemopoietic cells.6' Possibly abenign procedure, such as the regular inhala-
53
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

Porteous, Dorin
tion of DNA aerosols, could be used to restrictor relieve the pulmonary complications ofcystic fibrosis.
ConclusionsThe cloning of the cystic fibrosis gene stands as
a dual testament-to the power of moleculargenetic approaches to human disease and to thededication and ingenuity of the community ofresearchers whose immediate triumph this was.There has been talk of "winners" and "losers",but without the spirit of competition andcollaboration the race would still be inprogress. The identification of the gene and themajor mutation associated with the disease hashad an immediate impact in improved prenataland clinical diagnosis. This has spawned awealth of studies to characterise new muta-tions, to deduce protein structure and function,and to construct valid disease models in cultureand in whole animals from which to devise newimmunological, drug, and gene replacementtreatments. The challenge to the scientists,clinicians and counsellors are great, but thepromised reward for the cystic fibrosis com-
munity is unquestionable and paramount.
We gratefully acknowledge the advice and criticism of our
colleagues in the University of Edinburgh Human GeneticsUnit and the MRC Human Genetics Unit and the support of theMedical Research Council and Cystic Fibrosis Research Trust.
I Brock DJH. Prenatal diagnosis. In: Goodfellow P, ed. Cysticfibrosis. Oxford: Oxford University Press, 1989:66-91.
2 van Heyningen V, Porteous DJ. Molecular genetics.In: Goodfellow P, ed. Cystic fibrosis. Oxford: OxfordUniversity Press, 1989:41-65.
3 van Heyningen V, Hayward C, Fletcher J, McAuley C.Tissue localization and chromosomal assignment of aserum protein that tracks the cystic fibrosis gene. Nature1985;315:513-5.
4 Dorin JR, Novak M, Hill RE, Brock DJ, Secher DS, van
Heyningen V. A clue to the basic defect from cloning theCF antigen gene. Nature 1987;326:614-7.
5 Odink K, Cerletti N, Bruggen J, et al. Two calcium bindingproteins in infiltrate macrophages of rheumatoid arthritis.Nature 1987;330:80-2.
6 Dorin JR, Emslie E, van Heyningen V. Related calcium-binding proteins map to the same sub-region of chromo-some 1q and to an extended region of synteny on murinechromosome 3. Genomics (in press).
7 Quinton PM. Chloride impermeability in cystic fibrosis.Nature 1983;301:421-2.
8 Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT,Boucher RC. Abnormal ion permeation through cysticfibrosis respiratory epithelium. Science 1983;221:1067-70.
9 Levitan IB. The basic defect in cystic fibrosis. Science1989;224: 1423.
10 Solomon E, Bodmer W. Evolution of the sickle variant gene.Lancet 1979;i:923.
11 Eiberg H, Mohr J, Schmiegelow K, Neilson LS, WilliamsonR. Linkage relationships of paraoxonase (PON) with othermarkers: identification of PON-cystic fibrosis synteny.Clin Genet 1985;28:265-71.
12 Tsui L-C, Buchwald M, Barker D, et al. Cystic fibrosis locusdefined by a genetically linked polymorphic DNA marker.Science 1985;230:1054-7.
13 Eighth International Workshop on Human Gene Mapping.Human gene mapping 8. Cytogenet Cell Genet 1985;40:Nos 1-4.
14 White R, Woodward S, Zeppert M, et al. A closely linkedgenetic marker for cystic fibrosis. Nature 1985,318:382-4.
15 Wainright BJ, Scambler PJ, Schmidtke J, et al. Localizationof cystic fibrosis locus to human chromosome 7cen-q22.Nature 1985;318:384-5.
16 Farrall M, Law HY, Rodek CH et al. First trimester prenataldiagnosis of cystic fibrosis with linked DNA probes.Lancet 1 986;i: 1402-5.
17 Beaudet A, Bocock A, Buchwald M, et al. Linkage of cysticfibrosis to two tightly linked DNA markers: joint report
from a collaborative study. Am J Hum Genet 1986;39:681-93.
18 Dean M. Molecular and genetic anslysis of cystic fibrosis.Genomics 1988;3:93-9.
19 Porteous DJ, van Heyningen V. Cystic fibrosis from linkedmarkers to the gene. Trends in Genetics 1989;2:149-52.
20 Collins FS, Drumm ML, Cole JL, et al. Construction of ageneral human chromosome jumping library with applica-tion to cystic fibrosis. Science 1987;235:1046-9.
21 Porteous DJ. Chromosome mediated gene transfer: a func-tional assay for complex loci and an aid to human genomemapping. Trends in Genetics 1987;3:177-82.
22 Cooper CS, Park M, Blair DG, et al. Mollecular cloning of anew transforming gene from a chemically transformedhuman cell line. Nature 1984;311:29-33.
23 Park M, Dean M, Cooper CS, et al. Mechanism of metoncogene activation. Cell 1986;45:895-904.
24 Scambler PJ, Law HY, Williamson R, Cooper CS.Chromosome mediated gene transfer of sic DNA markerslinked to the cystic fibrosis locus on human chromosome 7.Nuc Acids Res 1986;14:7159-74.
25 Bird A. CpG-rich islands and the function ofDNA methyla-tion. Nature 1986;321:209-13.
26 Lindsay S, Bird A. Use of restriction enzymes to detectpotential gene sequences in mammalian DNA. Nature1987;327:336.
27 Estivill X, Farrell M, Scambler PJ, et al. A candidate for thecystic fibrosis locus isolated by selection for methylation-free islands. Nature 1987;326:840-5.
28 Wainright BJ, Scambler PJ, Scannier P, et al. Isolation of ahuman gene with protein sequence similarity to humanand murine int-1 and the Drosophila segment polaritymutant wingless. EMBO J 1988;7:1743-8.
29 Berger W, Hein J, Gedschold J, et al. Crossovers in twoGerman cystic fibrosis families determine probe order forMET 7C22 and XV-2c/CS.7. Hum Genet 1987;77:197-9.
30 Farrall M, Wainright BJ, Feldman GL, et al. Recombina-tions between IRP and cystic fibrosis. Am J Hum Genet1988;43:741-75.
31 Rommens JM, Zengalin S, Burns J, et al. Identification andregional localization of DNA markers on chromosome 7(for the cloning of the cystic fibrosis gene). Am J HumGenet 1988;43:645-63.
32 Rommens JM, Lannuzzi MC, Kerem B, et al. Identificationof the cystic fibrosis gene chromosome walking andjumping. Science 1989;245:1059-65.
33 Riordan JR, Rommens JM, Kerem B, et al. Identification ofthe cystic fibrosis gene cloning and characterisations ofcDNA. Science 1989;245:1066-72.
34 Kerem B, Rommens JM, Buchanan JA, et al. Identificationof the cystic fibrosis gene: genetic analysis. Science1989;245:1073-80.
35 McIntosh L, Lorenzo M-L, Brock D. Frequency of deltaF508 mutation on cystic fibrosis chromosomes in UK.Lancet 1989;ii: 1404-5.
36 Estivill X, Chillon M, Casals T, et al. Delta F508 gene deletionin cystic fibrosis in Southern Europe. Lancet 1989;ii: 1404.
37 Lemna WK, Feldman GL, Kerem BS, et al. Mutationanalysis for heterozygote detection and the prenatal diag-nosis of cystic fibrosis. N Engl JMed 1990;322:291-6.
38 Klinger K, Vaughn GT, Stanislovitis E, Schwartz RH,Fujiwara TM, Morgan K. Cystic fibrosis mutations in theHuttierite brethren. Am J Hum Genet 1990;46:983-7.
39 Kerem BS, Buchannan JA, Durie P, et al. DNA markerhaplotype association with pancreatic sufficiency in cysticfibrosis. Am J Hum Genet 1989;44:827-34.
40 Ferrari M, Antonelli M, Bellini F, et al. Genetic differencesin cystic fibrosis patients with and without pancreaticinsufficiency. Hum Genet 1990;84:435-8.
41 Struhrmann M, Macek M, Reis A, et al. Genotype analysisof cystic fibrosis patients in relation to pancreatic suffi-ciency. Lancet 1990;i:738-9.
42 Santis G, Osbourne L, Knight RA, Hodson ME. Linkedmarker haplotypes and the delta F,50 mutation in adultswith mild pulmonary disease and cystic fibrosis. Lancet1990;i: 1426-9.
43 Rommens J, Kerem B, Greer W, Chang P, Tsui L-C, Ray P.Rapid non radioactive detection of the major cysticfibrosis mutation. Am J Hum Genet 1990;16:395-6.
44 Montandon AJ, Green PM, Giannelli F, Bentley DR. Directdetection of point mutations by mismatch analysis: appli-cation to haemophilia B. Nucl Acids Res 1989;17:3347-58.
45 Orita M, Suzuki Y, Sekiya T, Hayashi K. Rapid andsensitive detection of point mutations and DNA poly-morphisms using the polymerase chain reaction. Genomics1989;5:874-9.
46 White MB, Amos J, Hsu JMC, Gerrard B, Finn P, Dean M.A frame-shift mutation in the cystic fibrosis gene. Nature1990;344:665-7.
47 Dean M, White M, Amos J. Multiple mutations in highlyconserved residues are found in mildly affected cysticfibrosis patients. Cell 1990;81:863-70.
48 Romeo G, Devoto M, Galietta L. Why is the cystic fibrosisgene so frequent? Hum Genet 1989;84:1-5.
49 Mullis K, Faloona F. Specific synthesis ofDNA in vitro via apolymerase-catalysed chain reaction. Methods Enzymol1987;155(part F):335-50.
50 Handyside AH, Kontogianni EH, Hardy K, Winton RML.
54
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from

55Cloning the cystic fibrosis gene: implications for diagnosis and treatment
Pregnancies from biopsied human preimplantationembryos sexed by Y-specific DNA amplification. Nature1990;344:768-70.
51 Gilbert F. Is population screening for cystic fibrosisappropriate now? Am J Hum Genet 1990;46:394-5.
52 Brock D. Population screening for cystic fibrosis. Am JHumGenet 1990;47:164-5.
53 Robertson EJ. Teratocarcinomas and embryonic stem cells: a
practical approach. Oxford: IRL Press, 1987.54 Capecchi M. The new mouse genetics; altering the genome
by gene targeting. Trends in Genetics 1989;5:70-6.55 Thompson, et al. S Germ line transmission and expression of
a corrected HPRT gene produced by gene targeting in EScells. Cell 1989;56:313-21.
56 Schwartzberg P, Goff S, Robertson E. Germ-line trans-mission of a c-abl mutation produced by targeted genedisruption in ES cells. Science 1989;246:799-803.
57 Dorin J, Inglis J, Porteous D. Selection for precise
chromosomal targeting of a dominant marker by homo-logous recombination. Science 1989;243:1357-60.
58 Burke DT, Carle G, Olson M. Cloning of large segments ofexogenous DNA into yeast by means of artificial chromo-some vectors. Science 1987;236:806-12.
59 Culliton BJ. Gore-Tex organoids and genetic drugs. Science1989;246:747-9.
60 Roux P, Jeanteur P, Prechaczyk M. A versatile and poten-tially general approach to the targeting of specific celltypes by retroviruses: application to the infection ofhuman cells by means of major histocompatibility com-plex class I and class II antigens by mouse ecotropicmurine leukemia virus-derived viruses. Proc Natl AcadSci USA 1989;86:9079-83.
61 Zenke M, Steinlein P, Wagner E, Cotten M, Beug H,Birnstiel ML. Receptor mediated endocytosis of trans-ferrin-polycation conjugates: an efficient way to introduceDNA into haematopoietic cells. Proc Natl Acad Sci USA1 990;87:3655-9.
on January 6, 2020 by guest. Protected by copyright.
http://thorax.bmj.com
/T
horax: first published as 10.1136/thx.46.1.46 on 1 January 1991. Dow
nloaded from