classification of amyloidosis
TRANSCRIPT

CLASSIFICATION OF AMYLOIDOSISABHINEET DEY
Department of PathologyGauhati Medical College & Hospital

According to a combined biochemical-clinical classification, Amyloid may be
Systemic (generalized), involving several organ systems,
Localized, when deposits are limited to a single organ, such as the heart.
As should become evident, several different biochemical forms of amyloid are encompassed by such segregation.
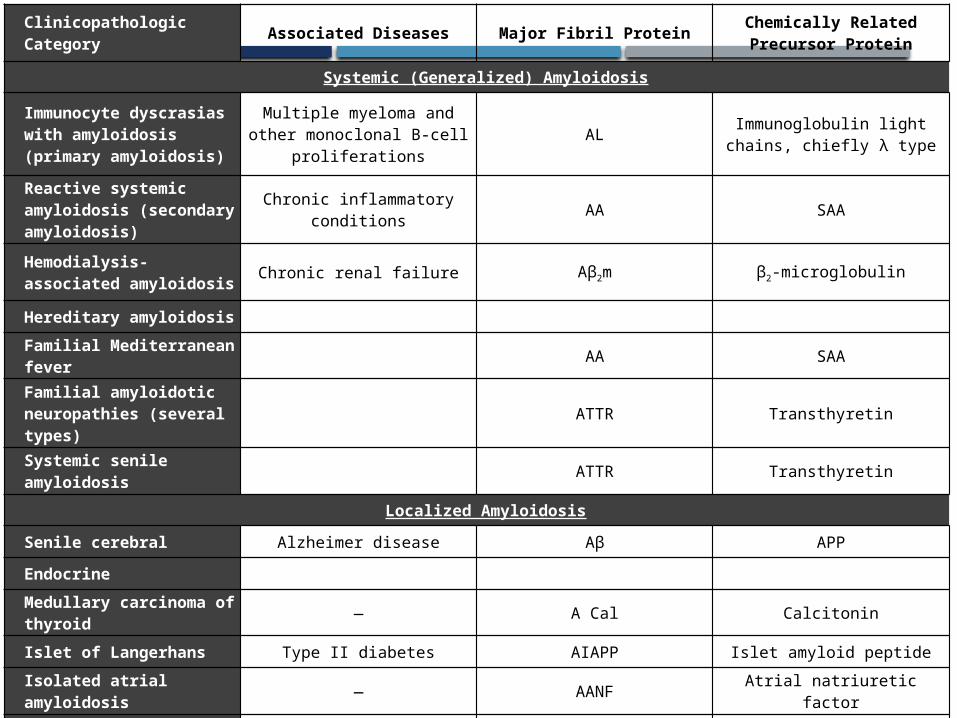
Clinicopathologic Category
Associated Diseases Major Fibril ProteinChemically Related Precursor Protein
Systemic (Generalized) Amyloidosis
Immunocyte dyscrasias with amyloidosis (primary amyloidosis)
Multiple myeloma and other monoclonal B-cell
proliferationsAL
Immunoglobulin light chains, chiefly λ type
Reactive systemic amyloidosis (secondary amyloidosis)
Chronic inflammatory conditions
AA SAA
Hemodialysis-associated amyloidosis
Chronic renal failure Aβ2m β2-microglobulin
Hereditary amyloidosis
Familial Mediterranean fever
AA SAA
Familial amyloidotic neuropathies (several types)
ATTR Transthyretin
Systemic senile amyloidosis
ATTR Transthyretin
Localized Amyloidosis
Senile cerebral Alzheimer disease Aβ APP
Endocrine
Medullary carcinoma of thyroid
— A Cal Calcitonin
Islet of Langerhans Type II diabetes AIAPP Islet amyloid peptide
Isolated atrial amyloidosis
— AANF Atrial natriuretic factor
Prion diseasesVarious prion diseases of the
CNSMisfolded prion protein
(PrPSC)Normal prion protein PrP

SYSTEMIC (GENERALISED)On clinical grounds, the systemic, or generalized, pattern is again sub classified into some groups. These are as follows:

PRIMARY AMYLOIDOSIS
Plasma Cell Disorder associated with Amyloidosis.
Amyloid in this category is usually systemic in distribution and is of the AL type. This is the most common form of amyloidosis.
This disorder is caused by clonal proliferation of plasma cells that synthesise Ig.
About 30% cases of AL Amyloid have some form of plasma cell dyscrasias, most commonly multiple myeloma.
The remaining 70% cases do not have evident B Cell proliferative disorder.

REACTIVE SYSTEMIC AMYLOIDOSISSECONDARY AMYLOIDOSIS
The fibril protein contains AA Amyloids.
It occurs typically as a complication chronic infection, non infectious chronic inflammatory conditions.

HEREDOFAMILIAL AMYLOIDOSIS
This constitutes a separate, albeit heterogeneous group, with several distinctive patterns of organ involvement.
Most of them are rare and occur in limited geographical area.
These are as under:
1. Hereditary Poly-Neuropathy Amyloidosis
2. Amyloid in Familial Mediterranean Fever
3. Rare Hereditary Form
LOCALISED AMYLOIDOSIS
Amyloid deposition are limited to a single organ or tissue without involvement of any other site in the body.
The deposits may produce detectable nodular masses, most often encounter in lung, larynx, skin, tongue and urinary bladder.

LOCALISED AMYLOIDOSIS may be described under the following headings:
1. Endocrine Amyloidosis
Microscopic deposits may be found in certain endocrine tumours such as:
• Medullary Carcinoma of the thyroid gland
• Islet Tumour of the pancreas
• Undifferentiated Carcinoma of the stomach.
2. Senile Cardiac Amyloidosis
3. Senile Cerebral Amyloidosis
4. Localised Tumour forming Amyloid

Morphological features of amyloidosis of organs

AMYLOIDOSIS OF KIDNEY
It is most common and most serious because of its ill effect on renal functions.
The deposits in the kidney are found in most cases of secondary amyloidosis and in about one third of cases of primary amyloidosis.

AMYLOIDOSIS OF KIDNEY (contd.)
GROSSLY
The kidney may be of normal size and colour.
In advanced cases they may be shrunken because of ischemia caused by vascular narrowing induced by deposition of amyloid.
HISTOLOGICALLY
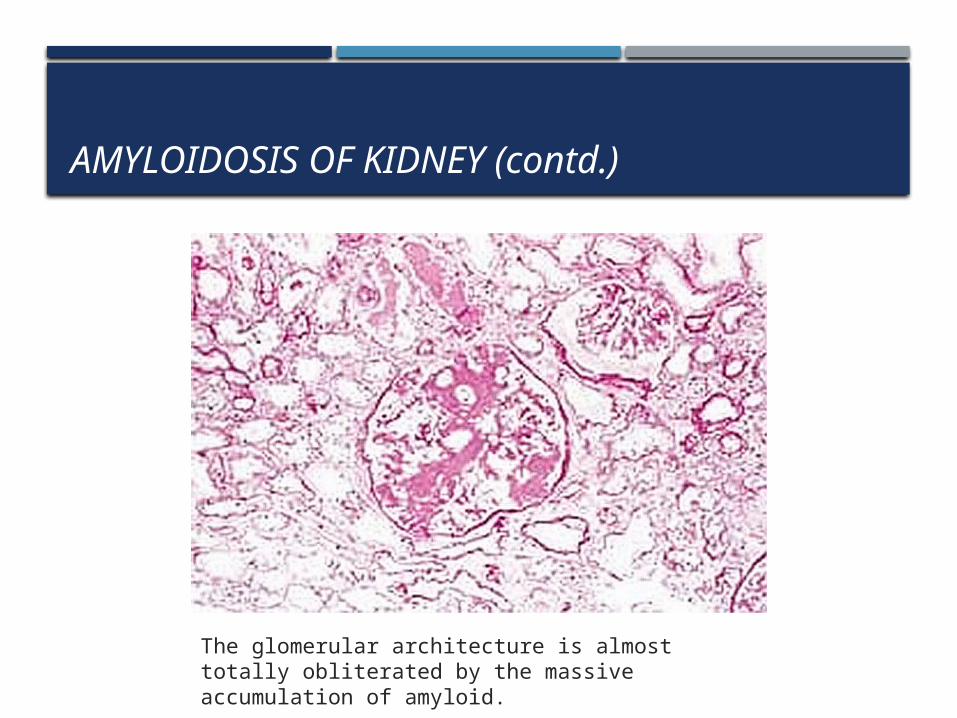
The amyloid is deposited primarily in the glomeruli but the interstitial peritubular tissues, arteries and arterioles are also affected.
The glomerular architecture is almost totally obliterated by the massive accumulation of amyloid.
AMYLOIDOSIS OF KIDNEY (contd.)
The glomerular architecture is almost totally obliterated by the massive accumulation of amyloid.

AMYLOIDOSIS OF SPLEEN
Amyloidosis of Spleen may be not apparent grossly or may cause moderate to marked splenomegaly.
Amyloid deposition in the spleen may have on of the following two pattern:
1. Sago Spleen: The deposits are largely limited to splenic follicle producing tapioca-like granules.
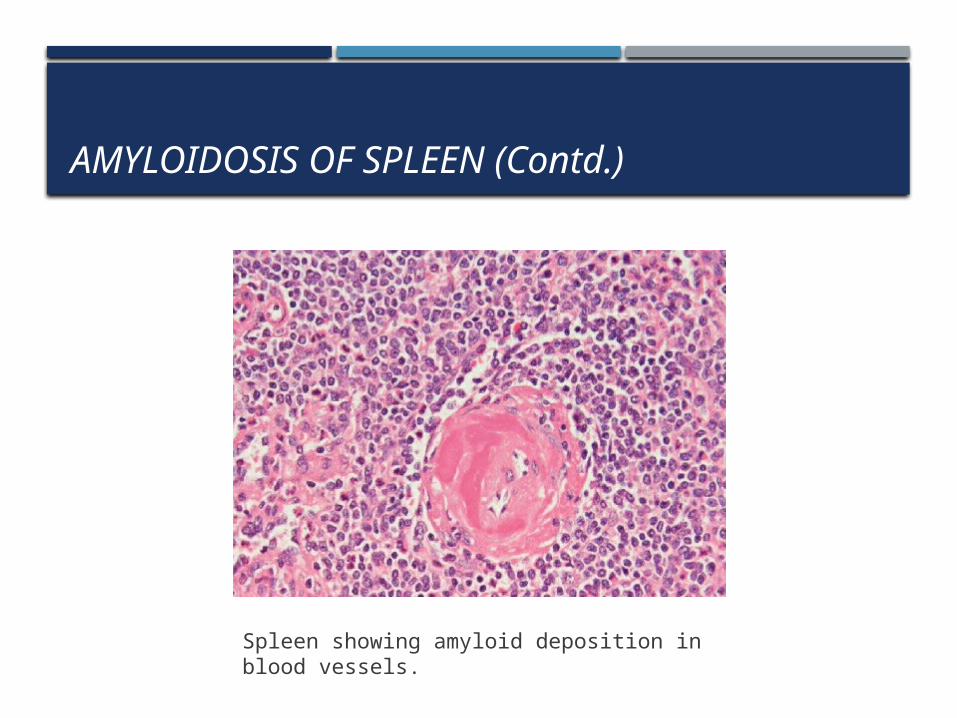
2. Lardaceous Spleen: Amyloid involves the wall of splenic sinuses and connective tissue in red pulp. Fusion of the early deposits gives rise to large, map like areas of amyloidosis.
AMYLOIDOSIS OF SPLEEN (Contd.)
Spleen showing amyloid deposition in blood vessels.

AMYLOIDOSIS OF LIVER
GROSSLY
The liver is often enlarged, pale, waxy and firm.
HISTOLOGICALLY
The amyloid initially appears in the Space of Disse.
Later as the deposit increases, they compress the chords of hepatocytes.

PROGNOSIS
The prognosis of patient with generalised amyloidosis is generally poor.
Primary amyloidosis, if left untreated, is rapidly progressive and fatal.
For secondary amyloidosis, control of inflammation is the main step of treatment.

THANK YOU
d!