cellular epigenetic stability and cancer
TRANSCRIPT

Cellular epigenetic stability and cancerPeter Sarkies and Julian E. Sale
Medical Research Council Laboratory of Molecular Biology, Division of Protein and Nucleic Acid Chemistry, Hills Road, Cambridge,
CB2 0QH, UK
Review
Glossary
Bromodomain: a protein domain that can interact with acetylated lysine, such
as those found on histone tails.
Chromatin immunoprecipitation (ChIP): a method for identifying the proteins,
including histones and their modifications, associated with a particular frag-
ment of DNA. Proteins are crosslinked to the DNA, which is then fragmented.
Fragments are immunoprecipitated with antibodies against the histone modifi-
cation of interest. The amount of a particular fragment of DNA can be assessed
with quantitative PCR or levels across the entire genome monitored by array
hybridisation (ChIP on chip) or by high-throughput sequencing (ChIP-seq).
Chromodomain: a protein domain that promotes binding to methylated lysines
within the tail of histone H3.
Dicer: a ribonuclease that cleaves double-stranded RNA into short interfering
RNAs that can mediate gene silencing.
G4 DNA motif: a G-rich DNA sequence of the general form L1–7-G3-5-L1–7-G3–5-L1–7-
G3–5 (where L can be any base) that is capable of forming a G-quadruplex structure.
G quadruplex: a DNA secondary structure that can form in single-stranded DNA
conforming to a G4 DNA motif. It comprises stacks of planar rings of four
Hoogsteen-bonded dG bases. The structures are thermodynamically very stable
and yet can form under physiological conditions and pose a potent barrier to
transcription and replication [81].
Heterochromatin: chromatin with a condensed, tightly packed structure asso-
ciated with repression of gene expression. The prefix ‘pericentric’ denotes
heterochromatin formed around the centromere.
Histone marks: a commonly used term for any post-translational modification
of histone proteins (Box 2).
Hox genes: an evolutionarily conserved cluster of genes that specify the body
pattern of multicellular organisms from insects to vertebrates.
Position effect variegation: the stochastic inactivation of a gene caused by its
placement adjacent to a region of heterochromatin. The classic example is that
of the Drosophila white gene brought close to pericentric heterochromatin by a
translocation. In the translocated position, white is stochastically silenced by
spreading of the heterochromatic state from the centromere [49].
Polycomb group proteins: a set of proteins, first identified in Drosophila
melanogaster, that are involved in epigenetic gene silencing of targets, includ-
ing the Hox genes, through the introduction of H3K27 trimethylation.
Polycomb repressive complex 2 (PRC2): a histone methyltransferase complex
whose core comprises the histone methyltransferase EZH2, along with embry-
onic ectoderm development (EED), suppressor of zeste 12 (SUZ12) and retino-
blastoma-binding protein 4 (RbAp48). It is responsible for the introduction of the
repressive H3K27me3 mark [82].
Polycomb response element (PRE): a specific DNA sequence responsible for
recruitment of the PRCs. Although well defined in organisms such as Drosophi-
la, they are less clearly conserved in vertebrates.
Proliferating cell nuclear antigen (PCNA): the DNA sliding clamp that tethers
polymerases and other factors to DNA.
Ribonucleotide reductase: an enzyme responsible for the conversion of ribo-
nucleotides to deoxyribonucleotides. Its inhibition results in depletion of deoxy-
ribonucleotide pools and stalling of DNA replication.
SET domain: a protein domain found in lysine methyltransferases. Most histone
lysine methyltransferases contain a SET domain, a notable exception being the
H3K79 methyltransferase Disruptor of telomeric silencing 1 (Dot1) [83].
Translesion synthesis: a mechanism for bypassing DNA base lesions during
replication that relies on specialised DNA polymerases whose active sites are
able to accommodate bulky or distorted DNA templates.
X chromosome inactivation: mammalian female cells contain two X chromo-
somes, one of which is inactivated to maintain the same gene dosage as the
male. The mechanism of inactivation is complex but involves the packaging of
When a cell divides, it must not only accurately duplicateits genome, but also restore its previous levels of geneexpression. The information determining gene expres-sion is often not directly encoded in the DNA and ishence termed ‘epigenetic’. The molecular basis of epige-netic memory remains a subject of intense debate, but islikely to arise from the collaboration of several mecha-nisms, including histone post-translational modifica-tions, transcription factors, DNA methylation andnoncoding RNAs. In this article, we look at how thesemechanisms interact to generate robust epigeneticstates. We then consider recent observations that mi-totic inheritance of stable gene expression can be com-promised by interruption of DNA replication. We discusshow these data may provide direct evidence for a centralrole for histone modifications in transcriptional memoryand how they could potentially provide an explanationfor the some of the widespread alterations in transcrip-tion seen in cancer cells.
The importance of epigenetic informationAlthough all cells in an organism carry the same genome,cells at different developmental stages and with differentfunctions must express only a subset of genes. For differentcell types to maintain their identity when they divide, theyhave to retain a ‘memory’ of which proteins must beproduced and, hence, which genes are to be transcribed.The different transcriptional potentials of any particulargene in different cell types are not determined by changesin DNA sequence; therefore, the information specifyingthese differences is termed ‘epigenetic’ (Box 1). Failureto propagate epigenetic information accurately results indeviations from the normal pattern of gene expression,which in turn can lead to failure of developmental pro-grammes and, potentially, to cancer.
Despite the clear importance of epigenetic memory, itsmolecular basis remains a subject of much experiment anddebate. Post-translational modifications of histone pro-teins have occupied the centre stage in most thinking aboutepigenetic memory (Box 2) and have been proposed to actas a ‘code’ that determines the transcriptional state ofchromatin [1,2]. However, although the association be-tween histone modifications and transcriptional states isclear, there is still doubt over their ability to actuallyspecify such states, rather than simply being effectors ofother modes of transcriptional memory [3,4]. Thus, manycurrent models consider how histone modifications interactwith other potential sources of epigenetic information toproduce robust epigenetic states. In the first part of this
Corresponding author: Sale, J.E. ([email protected]).
118 0168-9525/$ – see front matter � 2011 Elsevier Ltd. All righ
review, we examine this idea, paying particular attentionto the interaction of histone modifications with transcrip-tion factors, DNA methylation and noncoding RNAs. Wethen go on to look at some recent studies that expose the
the entire chromosome into heterochromatin, a process driven by a long
noncoding RNA called Xist [84].
ts reserved. doi:10.1016/j.tig.2011.11.005 Trends in Genetics, March 2012, Vol. 28, No. 3

Box 1. Epigenetic processes
Originally coined by Waddington in 1942 [71], the term ‘epigenetic’
was initially taken to mean processes generating biological diversity
that cannot be attributed directly to differences in DNA sequence.
This article, however, focuses on epigenetics at the cellular level, for
which this definition has been refined to include an additional
criterion, namely the heritability of the state through cell division
[29]. In this view, a cellular epigenetic process is one that meets the
following criteria: (i) it constitutes an observable difference between
two cells with the same genotype; (ii) this difference must persist in
the absence of the initial signal; and (iii) the difference must be
heritable through mitotic division. Some epigenetic marks might
also be inherited through meiosis.
This definition might be considered to be overly restrictive, and
may therefore not be appropriate in all contexts. However, in this
article, we follow it because it provides a useful framework for
understanding the molecular mechanism whereby information
additional to the DNA sequence might be perpetuated through cell
division.
Review Trends in Genetics March 2012, Vol. 28, No. 3
impact of interruptions in DNA replication on epigeneticstability. We consider how these experiments provide sup-port for an important role for parental histone recycling inmaintaining transcriptional states at many loci and, fur-thermore, how replication-associated epigenetic instabilitymight play an important role in the development of cancer.
Histone post-translational modifications andtranscriptional memorySeveral recent genome-wide studies have confirmed thecorrelation between certain post-translational modifica-tions of histone proteins and defined transcriptional states
Box 2. Histone post-translational modifications and gene
expression states
Both locus-specific and genome-wide experiments carried out in a
variety of different model systems have demonstrated an impress-
ive correlation between the presence of certain histone post-
translational modifications and the expression state of the under-
lying gene (e.g. [5,72,73]). Indeed, some histone modifications have
actually been used to uncover hitherto unknown transcripts [74].
These correlations could reflect a direct influence of histone
modifications on gene expression; alternatively, different histone
post-translational modifications might be a consequence of differ-
ent transcriptional mechanisms. These possibilities are a matter of
on-going debate [1–4]. Importantly, however, these possibilities are
not necessarily mutually exclusive. The connections between
chromatin modifications, differing gene expression states and their
potential roles in transcription have been extensively reviewed (e.g.
[75,76]). Among a plethora of histone modifications that have been
reported, it is generally accepted that acetylation of histone H3 on
lysines 9 and 14, and acetylation of the N terminus of H4, are
associated with transcriptional activation. Another marker of active
genes is H3K4 trimethylation, whose presence is maximal within a
fairly tight region around the promoter of genes. The body of active
genes tends to be enriched for H3K36 methylation, which is
deposited directly by the transcription machinery. By contrast,
regions of the genome that are not producing functional transcripts,
known generally as heterochromatin, tend to be hypoacetylated.
These regions can be further subdivided into developmentally silent
genes, known also as facultative heterochromatin, such as the Hox
loci, which accumulate H3K27 trimethylation, and constitutively
silenced regions, such as centromeres and repetitive regions, which
are enriched for H3K9 trimethylation. However, there is clearly
overlap between these two types of chromatin, with H3K9
dimethylation in particular being found at high levels at facultative
as well as constitutive heterochromatin (reviewed in [77]).
(Box 2). Furthermore, locus-specific [5] and genome-wide [6]maps of histone modifications, derived by chromatin immu-noprecipitation, show that different patterns of histonemarks (see Glossary) can distinguish different cell types.This has led to the suggestion that the pattern of histonemodifications could directly influence transcription of aparticular locus [1,2]. Potentially, this would also allowhistone modifications to carry epigenetic information, be-cause their pattern appears to be preserved along with geneexpression when cells divide. However, this possibility relieson the existence of a mechanism to copy histone modifica-tions directly when cells divide. How might this work?
During DNA replication, parental nucleosomes arerecycled and rapidly segregated evenly between daughterstrands [7,8]. To maintain nucleosome density on the DNA,newly synthesised histones must also be incorporated inidentical, or very similar, amounts to the parental his-tones. A weight of biochemical evidence has converged on amodel whereby the more stable [H3/H4]2 tetramer at theheart of the nucleosome stays together during parentalhistone recycling [9,10], whereas two newly synthesisedhistone 3/histone 4 (H3/H4) dimers form new nucleosomes.H2A/H2B dimers then appear to assort randomly betweenold and new nucleosomes. Given that histones are depos-ited on newly synthesised DNA within 200 bp or so of thereplication fork [8,11], this assembly pathway would beexpected to give rise to an intermediate state where old andnew [H3/H4]2 tetramers lie adjacent to one another. Po-tentially, this could allow the modifications present onparental histones to be copied onto newly synthesised ones,restoring the pattern of modifications (Figure 1a).This mechanism requires enzymes that can recognise amodification and then introduce the same modification onan adjacent nucleosome. A prime example of such a systemis seen in the formation and spreading of di/trimethylationof histone H3 at lysine 9 (H3K9me2/3)-dependent hetero-chromatin. Heterochromatin protein 1 (HP1) can bind tothe H3K9me2 mark and simultaneously recruit the H3K9methyltransferase, suppressor of variegation 3-9 (SUV39),suggesting how marks could be copied from a parental tonewly synthesised nucleosome [12,13]. A similar model hasbeen proposed for maintaining the Polycomb-dependentrepressive mark H3K27me3 involving recognition ofH3K27me3 by Polycomb repressive complex 2 (PRC2)itself, thus bringing the histone methyltransferase, en-hancer of zeste homologue 2 (EZH2), in proximity to anadjacent, unmodified nucleosome to copy the mark [14].
Evidence for the importance of histone mark propaga-tion in the inheritance of active transcriptional states ismore limited. Increased gene expression and histone acet-ylation induced by treatment with histone deacetylaseinhibitors can be mitotically heritable in both Schizosac-charomyces pombe [15] and early mouse embryos [16] afterremoval of the drug. It is possible that this mechanismworks analogously to that proposed for H3K9 methylation,given that some histone acetyltransferase (HAT) com-plexes, such as the bromodomain-containing Gcn5, canrecognise the histone modification that they introduce[17]. Additionally, a model for the maintenance of histonetail acetylation has been proposed based on crosstalkbetween H3 tail acetylation and the H3K4me3 mark
119

ESC subunitE(Z) subunit
E(Z)
PHO ko ESC ko
Replication
Incomplete restoration of H3K27me3and progressive loss of silencing
Incomplete restorationand progressive loss of silencing
Complete restoration of H3K27me3
(d)
PRE
PRC2
PHO-RC
SUV39
HP1
Gene X
Initiation
Maintenance
EFG1
WOR1
WOR2
White Opaque
Signal
X
EFG1
WOR1
WOR2
X
(a)
(c)
Replication
(b)
Gene X
Gene Y
TRENDS in Genetics
Figure 1. Histone post-translational modifications, transcription factors and their interplay in maintaining transcriptional memory through cell division. (a) Restoration of
parental histone modifications following DNA replication. A tract of nucleosomes carrying the H3K9me2 mark (small red circles), characteristic of heterochromatin, is
replicated. The new [H3/H4]2 tetramers (magenta centres) do not carry the H3K9me2 mark but can have it restored by the histone methyltransferase suppressor of
variegation 3-9 (SUV39), which is recruited in complex with heterochromatin protein 1 (HP1), which binds through its chromodomain to an adjacent H3K9me2-modified
nucleosome [12,13]. (b) Principle of epigenetic signal carriage by a transcription factor network. Transcription of Gene X (a transcription factor) is activated by an external
signal. The product of Gene X, Protein X (orange box), can activate transcription of its own gene, providing positive feedback. In addition, Protein X activates transcription of
Gene Y. Thus, as long as sufficient Protein X is carried through mitosis, expression of Gene Y will be maintained even in the absence of the initial signal. (c) A simplified
diagram of the genetic circuit that determines the epigenetically stable white and opaque phenotypes of Candida albicans. Green indicates an actively transcribed gene, and
red a repressed gene. The default state of the circuit is proposed to be the inactive, white state. Activation of white-opaque regulator 1 (WOR1) results in a series of positive
feedback loops that can maintain the opaque state for many generations. For more details, see [78]. (d) Cooperation between trans-factor binding and histone modification
in establishment of the H3K27me3 mark in Drosophila melanogaster. The polycomb repressive complex 2 (PRC2; purple), including at its core the methyl-binding protein
extra sex combs (ESC) and histone methyltransferase Enhancer of zeste [E(Z)], is recruited to specific DNA sequences termed ‘Polycomb response elements’ (PRE) through
the pleiohomeotic–recessive complex (PHO–RC) (light-green oval). This promotes trimethylation of H3K27. PRC2 is also able to bind H3K27me3 through its ESC subunit,
providing a second mechanism for the recruitment of the enzymatic activity of E(Z) and the propagation of the H3K27me3 mark [28]. Cooperation between these two
mechanisms is illustrated by the loss of silencing seen if either PHO or ESC is knocked out (ko) [26,28].
Review Trends in Genetics March 2012, Vol. 28, No. 3
120

Review Trends in Genetics March 2012, Vol. 28, No. 3
[16]. Supporting an important role for the H3K4me3 mark,transcriptional memory of myogenic differentiation(MyoD) expression in Xenopus laevis is impaired followingdepletion of the histone variant H3.3, which is often foundat active promoters. Replacing depleted H3.3 with a pointmutant (H3.3K4A) that cannot be methylated failed torescue this phenotype, whereas overexpression of wild typeH3.3 appeared to enhance transcriptional memory [18].More recently, live imaging of the transcription of a report-er locus in the social amoeba Dictyostelium discoideumrevealed that the correlation between the transcriptionalactivity in parent and daughter cells was much reduced inthe absence of H3K4 methylation [19]. Together, thesereports suggest the inheritance of an active histone modi-fication, but further work will be needed to confirm wheth-er H3K4 trimethylation is a true epigenetic mark.
Although the histone mark-copying model has someexperimental and theoretical support, there remain someserious objections as to whether histone marks alone aretrue carriers of epigenetic information. One of the mostimportant issues is that of nucleosome turnover, which israpid even in regions of apparently stable repressive chro-matin [20,21]. Thus, the lifetime of individual modifica-tions on chromatin is likely to be short relative to the cellcycle, challenging their ability to act as templates to intro-duce modifications onto newly synthesised nucleosomesduring cell division [4,21]. One possible way to resolve thisis through the interaction of histone modification withother forms of epigenetic information, which we considerin the next section.
Interplay between histone post-translationalmodifications and other sources of epigeneticinformationTranscription factor loops
Concerns about the role of histone modifications in epige-netic memory has led to the argument that, even in com-plex developmental scenarios, transcription factors alonemay be enough to maintain distinct gene expression states[3]. Here, epigenetic inheritance is dependent on a positivefeedback loop whereby, once activated by a transient sig-nal, a transcription factor is able to maintain its ownexpression in the absence of the signal. If sufficient quan-tity of the transcriptional activator can be carried throughmitosis to restore expression in the daughter cells, then thetranscription factor itself will carry epigenetic information(Figure 1b). A well-characterised example of such a net-work is white-opaque switching in Candida albicans(Figure 1c). The white form of C. albicans is the mostcommon, but occasionally cells can switch to a stable,morphologically distinct, opaque form, which is geneticallyidentical. The transcriptional programme of the opaquestate is maintained by a simple positive feedback loopinvolving the transcription factor white-opaque regulator1 (Wor1). Wor1 stimulates its own transcription whilerepressing transcription of enhanced filamentous growth1 (EFG1), the key gene responsible for maintaining thewhite state. Thus, the two states are mutually exclusiveand stable through cell division [22].
However, it is unlikely that transcription factor net-works alone account for all instances of transcriptional
memory. Even in the white–opaque system, histone-modi-fying complexes have been shown to be important modu-lators of switching frequency, although the effects ofremoving histone-modifying complexes are smaller thanthe effects of deleting core members of the transcriptionloop shown in Figure 1c [23]. Interestingly, histone modi-fication appears to act upstream of Wor1 in the pathway,leading to the suggestion that histone modifications bufferfluctuations in Wor1 levels [24], thereby increasing therobustness of a primarily transcription factor-specifiedepigenetic state. Such an interaction may therefore beparticularly important for transcription factor loops whosecomponents are expressed at very low copy number, suchthat the stochastic partitioning of the proteins into thedaughter cells at mitosis could have a significant impact onthe ability to re-establish the transcriptional potential of agene.
Further evidence for crosstalk between transcriptionfactors and histone modifications can also be drawn fromsystems classically regarded as dependent on histone mod-ifications. At loci in Drosophila melanogaster whose silenc-ing depends on the Polycomb group proteins, H3K27trimethylation is essential for maintaining the silent state[25], but so to are cis-acting DNA elements called Polycombresponse elements (PREs) [26]. PREs are bound by Poly-comb proteins to form a Polycomb repressive complex.Given that Polycomb repressive complexes remain boundto PREs during replication [27], this could allow them topromote duplication of histone modifications, facilitated bytheir interaction with parental H3K27 trimethylatednucleosomes [14,28] (Figure 1d).
DNA methylation
In mammals, CpG methylation at promoters is generallyassociated with transcriptionally silent genes and, onceintroduced, can remain stable for many generations [29].When DNA is replicated, the newly synthesised strand isnot methylated, meaning that the daughter strands willonly be methylated on one strand (‘hemi-methylated’).However, specific ‘maintenance’ DNA methyltransferasesare able to introduce DNA methylation onto the otherstrand. Importantly, maintenance methyltransferases,such as DNA (cytosine-5-)-methyltransferase 1 (DNMT1),are restricted to acting on hemi-methylated DNA due to anallosteric inhibition of the catalytic activity of the proteinspecifically by unmethylated DNA [30] (Figure 2a). Addi-tionally, maintenance methyltransferases are recruited tothe sites of hemi-methylation by the protein ubiquitin-likewith PHD and ring finger domains 1 (UHRF1), enablingthem to introduce the modification onto the other strand[31–33]. This elegant mechanism to ensure CpG methyla-tion is preserved through cell division, making it an at-tractive candidate as a source of epigenetic information.However, there is no clear evidence for a direct influence ofDNA methylation on transcription; thus, DNA methyla-tion would need to act through effector mechanisms, suchas histone modifications. Furthermore, CpG DNA methyl-ation is not universally conserved in eukaryotes. Althoughthere is some suggestion that it is an evolutionary ancientmodification, it is not present in yeast and worms, despiteboth these lineages having epigenetic processes [34]. Thus,
121

CG
GC
DNMT1
CG
GC
G9aUHRF1
GC
DNA methylation directinghistone methylation
Histone methylation directingDNA methylation
(b)
Replication
CG
(a)
Replication
CG
GC
CG
DNMT1
DNMT1 catalytic domain
C
GC
CG
GC
CG
GC
C
G
TRENDS in Genetics
Figure 2. Propagation of DNA methylation through replication and its interplay with histone modifications. (a) Restoration of parental DNA methylation patterns after DNA
replication. Unmethylated DNA is excluded from the active site of DNA (cytosine-5-)-methyltransferase 1 (DNMT1), the major maintenance mammalian DNA
methyltransferase (DNMT1 in purple; the active site in pink). This inhibition is relieved by hemi-methylated DNA, thereby providing a mechanism for restriction of the
activity of DNMT1 to maintenance of DNA methylation after replication but preventing it from introducing de novo methylation at new sites [30]. (b) Cooperation between
DNA methylation and histone methylation. Key to crosstalk between DNA methylation and H3K9me3 is the multi-domain protein ubiquitin-like with PHD and ring finger
domains 1 (UHRF1; also known as Np95 or ICBP90). UHRF1 is able to recognise hemi-methylated DNA [31–33,79] and is also able to bind to the histone methyltransferase
G9a [80], suggesting a mechanism by which DNA methylation reinforces histone methylation. Conversely, UHRF1 can bind H3K9me3 through its tandem Tudor domains
[37], and it also interacts with DNMT1 [79], suggesting a mechanism by which histone methylation reinforces maintenance DNA methylation.
Review Trends in Genetics March 2012, Vol. 28, No. 3
DNA methylation needs to be combined with other sourcesof epigenetic information. In particular there is abundantevidence of significant crosstalk between DNA methylationand histone modifications.
Genome-wide H3K9 methylation is extremely well cor-related with DNA methylation [35]. Experimental datahave provided support both for stimulation of histonemodification by DNA methylation [36] and the recruitmentof DNA methyltransferases by histone modifications [37].Key to this process is the protein UHRF1. As mentionedabove, UHRF1 recruits DNA methyltransferases to hemi-methylated DNA. However, it can also recognise histoneH3K9 methylation [38] and interacts with the histonemethyltransferase G9a [39]. This strongly suggests thatH3K9 methylation and DNA methylation are self-reinfor-cing, providing additional robustness to the epigeneticstate (Figure 2b). A powerful example of this synergycomes from comparison of X chromosome inactivation inmarsupials and placental mammals. In marsupials, Xchromosome inactivation is not accompanied by the samedegree of DNA methylation that coats mammalian Xchromosomes [40,41]. The marsupial X chromosome is
122
nevertheless still transcriptionally silent, suggesting thatDNA methylation is not essential to repress transcription.However, in the absence of extensive DNA methylation, Xchromosome inactivation appears to be less robust becausea much higher frequency of reporter gene reactivation isobserved on the marsupial X chromosome than is seen inplacental mammals [42,43].
Noncoding RNAs
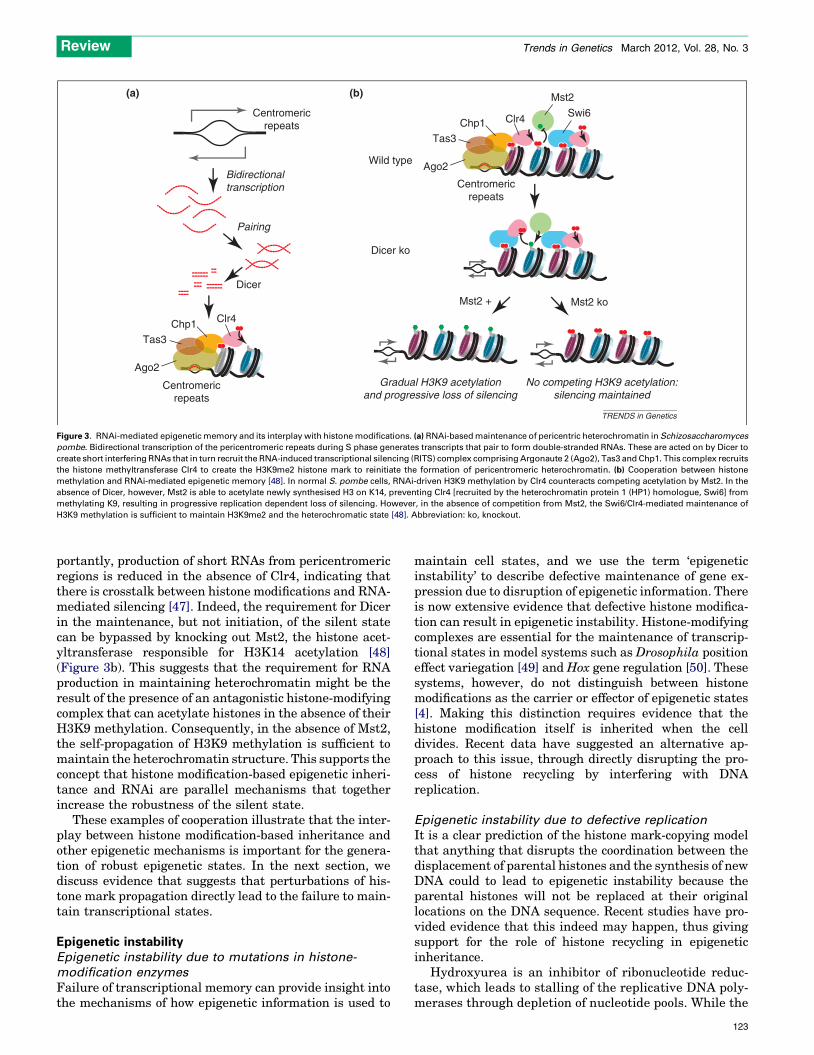
The best-understood mechanism for how noncoding RNAcontributes to the maintenance of an epigenetic state is theS. pombe centromere. Short double-stranded RNAs tran-scribed from pericentromeric tandem repeats and cleavedby the exonuclease Dicer recruit the RNA-induced tran-scriptional silencing complex (RITS), which includes the S.pombe H3K9 methyltransferase Clr4, resulting in thesilencing of the centromere [44] (Figure 3a). Dicer is re-quired for both the initiation and maintenance of the silentstate, implicating RNA as being central to the inheritanceprocess [45]. The process requires the production of newRNAs during each S phase to drive the re-establishment ofthe pericentromeric heterochromatin [46]. However, im-
Mst2
Swi6
Dicer ko
Wild type
Mst2 + Mst2 ko
(b)
Centromericrepeats
Gradual H3K9 acetylationand progressive loss of silencing
No competing H3K9 acetylation:silencing maintained
Tas3
Ago2
Chp1 Clr4
Tas3
Ago2
Chp1 Clr4
Dicer
Pairing
Bidirectionaltranscription
Centromericrepeats
Centromericrepeats
(a)
TRENDS in Genetics
Figure 3. RNAi-mediated epigenetic memory and its interplay with histone modifications. (a) RNAi-based maintenance of pericentric heterochromatin in Schizosaccharomyces
pombe. Bidirectional transcription of the pericentromeric repeats during S phase generates transcripts that pair to form double-stranded RNAs. These are acted on by Dicer to
create short interfering RNAs that in turn recruit the RNA-induced transcriptional silencing (RITS) complex comprising Argonaute 2 (Ago2), Tas3 and Chp1. This complex recruits
the histone methyltransferase Clr4 to create the H3K9me2 histone mark to reinitiate the formation of pericentromeric heterochromatin. (b) Cooperation between histone
methylation and RNAi-mediated epigenetic memory [48]. In normal S. pombe cells, RNAi-driven H3K9 methylation by Clr4 counteracts competing acetylation by Mst2. In the
absence of Dicer, however, Mst2 is able to acetylate newly synthesised H3 on K14, preventing Clr4 [recruited by the heterochromatin protein 1 (HP1) homologue, Swi6] from
methylating K9, resulting in progressive replication dependent loss of silencing. However, in the absence of competition from Mst2, the Swi6/Clr4-mediated maintenance of
H3K9 methylation is sufficient to maintain H3K9me2 and the heterochromatic state [48]. Abbreviation: ko, knockout.
Review Trends in Genetics March 2012, Vol. 28, No. 3
portantly, production of short RNAs from pericentromericregions is reduced in the absence of Clr4, indicating thatthere is crosstalk between histone modifications and RNA-mediated silencing [47]. Indeed, the requirement for Dicerin the maintenance, but not initiation, of the silent statecan be bypassed by knocking out Mst2, the histone acet-yltransferase responsible for H3K14 acetylation [48](Figure 3b). This suggests that the requirement for RNAproduction in maintaining heterochromatin might be theresult of the presence of an antagonistic histone-modifyingcomplex that can acetylate histones in the absence of theirH3K9 methylation. Consequently, in the absence of Mst2,the self-propagation of H3K9 methylation is sufficient tomaintain the heterochromatin structure. This supports theconcept that histone modification-based epigenetic inheri-tance and RNAi are parallel mechanisms that togetherincrease the robustness of the silent state.
These examples of cooperation illustrate that the inter-play between histone modification-based inheritance andother epigenetic mechanisms is important for the genera-tion of robust epigenetic states. In the next section, wediscuss evidence that suggests that perturbations of his-tone mark propagation directly lead to the failure to main-tain transcriptional states.
Epigenetic instabilityEpigenetic instability due to mutations in histone-
modification enzymes
Failure of transcriptional memory can provide insight intothe mechanisms of how epigenetic information is used to
maintain cell states, and we use the term ‘epigeneticinstability’ to describe defective maintenance of gene ex-pression due to disruption of epigenetic information. Thereis now extensive evidence that defective histone modifica-tion can result in epigenetic instability. Histone-modifyingcomplexes are essential for the maintenance of transcrip-tional states in model systems such as Drosophila positioneffect variegation [49] and Hox gene regulation [50]. Thesesystems, however, do not distinguish between histonemodifications as the carrier or effector of epigenetic states[4]. Making this distinction requires evidence that thehistone modification itself is inherited when the celldivides. Recent data have suggested an alternative ap-proach to this issue, through directly disrupting the pro-cess of histone recycling by interfering with DNAreplication.
Epigenetic instability due to defective replication
It is a clear prediction of the histone mark-copying modelthat anything that disrupts the coordination between thedisplacement of parental histones and the synthesis of newDNA could to lead to epigenetic instability because theparental histones will not be replaced at their originallocations on the DNA sequence. Recent studies have pro-vided evidence that this indeed may happen, thus givingsupport for the role of histone recycling in epigeneticinheritance.
Hydroxyurea is an inhibitor of ribonucleotide reduc-tase, which leads to stalling of the replicative DNA poly-merases through depletion of nucleotide pools. While the
123

Review Trends in Genetics March 2012, Vol. 28, No. 3
polymerases are stalled, the replicative helicase can con-tinue to unwind DNA for some distance, extruding a tractof single-stranded DNA. Under such conditions, massspectrometry analysis of the histones associated withthe chaperone anti-silencing function 1 (ASF1) at thereplication fork in human cells showed an increase inhistones carrying post-translational modifications typicalof chromatin, such as higher methylation states of H3K9[51]. This supports the idea that, when new DNA synthesisis interrupted, parental histones are displaced and buff-ered by ASF1. However, it is not yet clear whether theseparental ASF1-bound histones are returned to the generalpool with their marks, a situation that could give rise to
Parental his
N
Reinitiation ofDNA synthesis
(a)
(b)
(c) Quadruplex resolution an with only new, unm
ASF1
RecyclingBuffering
ASF1
Figure 4. A model for replication arrest-induced epigenetic instability. (a) Histone manag
example, H3K9me2 marks (red circles) are displaced ahead of the replicative helicase (gr
are deposited behind the fork in an approximately 50:50 distribution between the two
nucleosomes by heterochromatin protein 1 (HP1; light blue), which in turn recruits sup
tetramers. (b) Stalling of replication by a G-quadruplex structure in REV1 or Fanconi ane
shown having formed between the helicase and polymerase [orange oval attached to
FANCJ, the fork remains paused at the structure, allowing the helicase to run ahead. D
unreplicated gap. The advancing replicative helicase continues to displace parental nuc
[H3/H4]2 tetramers are split and buffered by anti-silencing function 1 (ASF1) [51]. (c) Re
presumably by specialised helicases, and the gap is filled in. However, because this DNA
parental histones. The gap is therefore chromatinised only with newly synthesised his
124
epigenetic instability through misincorporation of markedhistones at inappropriate locations.
Replication forks can also be stalled by problems on theDNA template, such as base damage or secondary struc-tures in DNA, such as the G-quadruplex. In the chicken cellline DT40, sequences that can form G-quadruplexes re-quire the translesion polymerase REV1, and the helicasesFanconi anemia, complementation group J (FANCJ), Wer-ner syndrome, RecQ helicase-like (WRN) and Bloom syn-drome, RecQ helicase-like (BLM) for their efficientreplication [52,53]. In the absence of REV1, impedimentsto the replication fork lead to the formation of gaps in theDNA duplex, which are filled in after bulk replication
tone recycling
ew histone deposition
SUV39HP1
Polymerase stalledat G-quadruplex
d gap filling remote from forkarked histones available
Leading
Lagging
Leading
Lagging
Leading
Lagging
TRENDS in Genetics
ement during replication. Parental [H3/H4]2 tetramers (cyan centre) carrying, in this
ey star). The parental and unmarked newly synthesised tetramers (magenta centre)
strands. Mark copying takes place by recognition of the H3K9me2 on the parental
pressor of variegation 3-9 (SUV39; pink) to methylate H3K9 in newly synthesised
mia, complementation group J (FANCJ)-deficient cells. A G-quadruplex structure is
the proliferating cell nuclear antigen (PCNA) in green]. In the absence of REV1 or
NA synthesis can reinitiate downstream of the stalled polymerase, but leaves an
leosomes but because only the lagging strand is available for redeposition, excess
solution and chromatinisation of the gap. The G-quadruplex structure is resolved,
synthesis takes place remote from the replication fork, there is no supply of marked
tones lacking the marks present at the locus prior to replication [52,53].

Review Trends in Genetics March 2012, Vol. 28, No. 3
[54,55]. This uncouples DNA synthesis from the displace-ment of histones by the replicative helicase. In turn, thisleads to a failure to maintain the expression state of someloci that contain G-quadruplex-forming DNA as the depo-sition of chromatin during gap filling has only naı̈ve, newlysynthesised histones available, which lack the histonemarks present on the parental nucleosomes (Figure 4).This failure of histone recycling gives rise to widespreadepigenetic changes that are significantly associated withthe presence of potential G-quadruplex-forming sequences[52,53]. Interestingly, both active and repressed genes areaffected. Importantly, loss of silencing in this context is notassociated with accumulation of modifications typical ofactive chromatin. Equally, loss of activation is not associ-ated with accumulation of marks typical of repressivechromatin. Instead, affected genes appear to be left inan apparently ‘neutral’ state, in which they are neitherfully expressed nor fully active. These observations suggestthat passive loss of the epigenetic information on parentalhistones is responsible for the observed epigenetic changes,rather than active conversion of the chromatin state. Theyalso suggest that the maintenance of both transcriptionallyactive and transcriptionally silent chromatin states isdependent on histone recycling.
Replication-associated epigenetic instability and disease
Epigenetic instability due to replication stress may be partof the explanation for the human syndrome caused by lossof the helicase a thalassaemia/mental retardation syn-drome X-linked (ATRX). The key features of this syndromeare thalassaemia, due to reduced expression of the geneencoding a-globin, and mental retardation (reviewed in[56]). The ATRX protein can bind to G-quadruplex DNA invitro, and is found at G4 DNA motifs in vivo, leading to thesuggestion that ATRX controls expression of genes nearG4 DNA motifs [57], for example through its interactionwith the H3.3 histone chaperone death-domain associatedprotein (DAXX) [58]. However, deletion of ATRX does notalter H3.3 distribution except at telomeres [59]. An alter-native possibility, yet to be tested, is that ATRX is requiredfor the processive replication of G4 DNA sequences in amanner similar to that described above for REV1. Giventhat mutations in the ATRX helicase domain cause thedisease [60], it is possible that ATRX could contribute tounwinding G-quadruplex sequences, thus acting to main-tain replication fork progression around G4 DNA motifsand preventing uncoupling of histone recycling from DNAsynthesis in a similar manner to REV1 and FANCJ[52,53].
Recent genomic surveys have identified histone-modify-ing genes as frequent mutational targets in a range ofdisparate cancers [61–64], and this provides a direct mech-anism by which a genetic change could lead to alterationsin the epigenome. Indeed, cancer cells show widespreadepigenetic changes, many of which are important in theprogression of the disease. A particularly striking exampleis the loss in cancer cell lines, such as HeLa, of the largeblocks of H3K9 methylation normally found in differenti-ated cells [65]. However, many tumours also exhibit con-stitutive replication stress that is induced by oncogeneexpression, leading to proliferative drive that exceeds
the resources available to the cell. This stress manifestsas increased replication fork stalling and collapse [66–68]and appears to be focussed particularly on difficult-to-replicate sequences, such as common fragile sites [69].Therefore, at least some examples of epigenetic instabilityin cancer may be directly linked to defective parentalhistone recycling. If this is so, one might expect regionswhere replication forks are predicted to stall more fre-quently to be particularly liable to epigenetic as well asgenetic changes.
Recent evidence suggests this may well be the case. Inhuman cancers, there is an excellent correlation betweenG4 DNA motifs and common DNA breakpoints at whichcopy number changes had occurred in human cancers,suggesting that replication fork perturbation and collapseis more probable at these regions [70]. However, theauthors also observed changes in DNA methylation aroundthese sites. A fascinating hypothesis is that this mighthave arisen in association with changes in histone mod-ifications caused by replication fork stalling at G-quadru-plex structures, as observed at the gene encoding r-globinin DT40 cells lacking REV1 [52].
Concluding remarksHistone modifications have many appealing features toexplain the maintenance of cellular phenotypes essentialfor development and for the prevention of disease, such ascancer. The ability of these modifications to copy them-selves is likely to prove key to this role, but it is alsoprobable that histone modifications must cooperate withother modes of epigenetic inheritance to ensure theircorrect propagation in the face of the upheaval in his-tone–DNA contacts caused by DNA replication. The intri-cate connections between transcription factors, RNAi,DNA methylation and histone modifications could explainthe apparent deficiencies in each as an individual mode ofepigenetic inheritance. By separating processes that copyhistone modifications from the recycling of parental his-tones, experiments perturbing DNA replication may provea useful tool in evaluating the contribution of histonemodifications to this complex interplay.
AcknowledgementsWe thank Daniela Rhodes and members of the Sale lab for helpfuldiscussions. Work in the lab is supported by the Medical ResearchCouncil, Association for International Cancer Research and the FanconiAnemia Research Fund.
References1 Turner, B.M. (1993) Decoding the nucleosome. Cell 75, 5–82 Jenuwein, T. and Allis, C. (2001) Translating the histone code. Science
293, 1074–10803 Ptashne, M. (2007) On the use of the word ‘epigenetic’. Curr. Biol. 17,
R233–R2364 Henikoff, S. and Shilatifard, A. (2011) Histone modification: cause or
cog? Trends Genet. 27, 389–3965 Litt, M.D. et al. (2001) Correlation between histone lysine methylation
and developmental changes at the chicken beta-globin locus. Science293, 2453–2455
6 Filion, G.J. et al. (2010) Systematic protein location mapping revealsfive principal chromatin types in Drosophila cells. Cell 143, 212–
2247 Jackson, V. and Chalkley, R. (1985) Histone segregation on replicating
chromatin. Biochemistry 24, 6930–6938
125

Review Trends in Genetics March 2012, Vol. 28, No. 3
8 Sogo, J.M. et al. (1986) Structure of replicating simian virus 40minichromosomes. The replication fork, core histone segregation andterminal structures. J. Mol. Biol. 189, 189–204
9 Jackson, V. (1988) Deposition of newly synthesized histones: hybridnucleosomes are not tandemly arranged on daughter DNA strands.Biochemistry 27, 2109–2120
10 Xu, M. et al. (2010) Partitioning of histone H3-H4 tetramers duringDNA replication-dependent chromatin assembly. Science 328, 94–98
11 Radman-Livaja, M. et al. (2011) Patterns and mechanisms of ancestralhistone protein inheritance in budding yeast. PLoS Biol. 9, e1001075
12 Bannister, A.J. et al. (2001) Selective recognition of methylated lysine 9on histone H3 by the HP1 chromo domain. Nature 410, 120–124
13 Lachner, M. et al. (2001) Methylation of histone H3 lysine 9 creates abinding site for HP1 proteins. Nature 410, 116–120
14 Hansen, K. et al. (2008) A model for transmission of the H3K27me3epigenetic mark. Nat. Cell Biol. 10, 1291–1300
15 Ekwall, K. et al. (1997) Transient inhibition of histone deacetylationalters the structural and functional imprint at fission yeastcentromeres. Cell 91, 1021–1032
16 VerMilyea, M.D. et al. (2009) Transcription-independent heritability ofinduced histone modifications in the mouse preimplantation embryo.PLoS ONE 4, e6086
17 Li, S. and Shogren-Knaak, M.A. (2009) The Gcn5 bromodomain of theSAGA complex facilitates cooperative and cross-tail acetylation ofnucleosomes. J. Biol. Chem. 284, 9411–9417
18 Ng, R.K. and Gurdon, J.B. (2008) Epigenetic memory of an active genestate depends on histone H3.3 incorporation into chromatin in theabsence of transcription. Nat. Cell Biol. 10, 102–109
19 Muramoto, T. et al. (2010) Methylation of H3K4 Is required forinheritance of active transcriptional states. Curr. Biol. 20, 397–406
20 Mellor, J. (2006) Dynamic nucleosomes and gene transcription. TrendsGenet. 22, 320–329
21 Deal, R.B. et al. (2010) Genome-wide kinetics of nucleosome turnoverdetermined by metabolic labeling of histones. Science 328, 1161–1164
22 Zordan, R.E. et al. (2006) Epigenetic properties of white-opaqueswitching in Candida albicans are based on a self-sustainingtranscriptional feedback loop. Proc. Natl. Acad. Sci. U. S. A. 103,12807–12812
23 Klar, A.J. et al. (2001) A histone deacetylation inhibitor and mutantpromote colony-type switching of the human pathogen Candidaalbicans. Genetics 158, 919–924
24 Hnisz, D. et al. (2009) Transcriptional loops meet chromatin: a dual-layer network controls white-opaque switching in Candida albicans.Mol. Microbiol. 74, 1–15
25 Papp, B. and Muller, J. (2006) Histone trimethylation and themaintenance of transcriptional ON and OFF states by trxG and PcGproteins. Genes Dev. 20, 2041–2054
26 Sengupta, A.K. et al. (2004) General transcriptional silencing by aPolycomb response element in Drosophila. Development 131, 1959–
196527 Francis, N.J. et al. (2009) Polycomb proteins remain bound to
chromatin and DNA during DNA replication in vitro. Cell 137, 110–12228 Margueron, R. et al. (2009) Role of the polycomb protein EED in the
propagation of repressive histone marks. Nature 461, 762–76729 Holliday, R. (1987) The inheritance of epigenetic defects. Science 238,
163–17030 Song, J. et al. (2011) Structure of DNMT1–DNA complex reveals a role
for autoinhibition in maintenance DNA methylation. Science 331,1036–1040
31 Hashimoto, H. et al. (2008) The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455, 826–829
32 Avvakumov, G.V. et al. (2008) Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455,822–825
33 Arita, K. et al. (2008) Recognition of hemi-methylated DNA by the SRAprotein UHRF1 by a base-flipping mechanism. Nature 455, 818–821
34 Krauss, V. (2008) Glimpses of evolution: heterochromatic histoneH3K9 methyltransferases left its marks behind. Genetica 133, 93–106
35 Meissner, A. et al. (2008) Genome-scale DNA methylation maps ofpluripotent and differentiated cells. Nature 454, 766–770
36 Sarraf, S. and Stancheva, I. (2004) Methyl-CpG binding protein MBD1couples histone H3 methylation at lysine 9 by SETDB1 to DNAreplication and chromatin assembly. Mol. Cell 15, 595–605
126
37 Rottach, A. et al. (2010) The multi-domain protein Np95 connects DNAmethylation and histone modification. Nucleic Acids Res. 38, 1796–
180438 Karagianni, P. et al. (2008) ICBP90, a novel methyl K9 H3 binding
protein linking protein ubiquitination with heterochromatinformation. Mol. Cell. Biol. 28, 705–717
39 Zhang, J. et al. (2011) S phase-dependent interaction with DNMT1dictates the role of UHRF1 but not UHRF2 in DNA methylationmaintenance. Cell Res. 21, 1723–1739
40 Kaslow, D.C. and Migeon, B.R. (1987) DNA methylation stabilizes Xchromosome inactivation in eutherians but not in marsupials: evidencefor multistep maintenance of mammalian X dosage compensation.Proc. Natl. Acad. Sci. U. S. A. 84, 6210–6214
41 Rens, W. et al. (2010) Epigenetic modifications on X chromosomes inmarsupial and monotreme mammals and implications for evolution ofdosage compensation. Proc. Natl. Acad. Sci. U. S. A. 107, 17657–17662
42 Migeon, B.R. et al. (1989) Frequent derepression of G6PD and HPRT onthe marsupial inactive X chromosome associated with cell proliferationin vitro. Exp. Cell Res. 182, 597–609
43 Al Nadaf, S. et al. (2010) Activity map of the tammar X chromosomeshows that marsupial X inactivation is incomplete and escape isstochastic. Genome Biol. 11, R122
44 Verdel, A. et al. (2004) RNAi-mediated targeting of heterochromatin bythe RITS complex. Science 303, 672–676
45 Sadaie, M. et al. (2004) A chromodomain protein, Chp1, is required forthe establishment of heterochromatin in fission yeast. EMBO J. 23,3825–3835
46 Chen, E.S. et al. (2008) Cell cycle control of centromeric repeattranscription and heterochromatin assembly. Nature 451, 734–737
47 Motamedi, M.R. et al. (2004) Two RNAi complexes, RITS and RDRC,physically interact and localize to noncoding centromeric RNAs. Cell119, 789–802
48 Reddy, B.D. et al. (2011) Elimination of a specific histone H3K14acetyltransferase complex bypasses the RNAi pathway to regulatepericentric heterochromatin functions. Genes Dev. 25, 214–219
49 Ebert, A. et al. (2006) Histone modification and the control ofheterochromatic gene silencing in Drosophila. Chromosome Res. 14,377–392
50 Ringrose, L. and Paro, R. (2004) Epigenetic regulation of cellularmemory by the Polycomb and Trithorax group proteins. Annu. Rev.Genet. 38, 413–443
51 Jasencakova, Z. et al. (2010) Replication stress interferes with histonerecycling and predeposition marking of new histones. Mol. Cell 37, 736–
74352 Sarkies, P. et al. (2010) Epigenetic instability due to defective
replication of structured DNA. Mol. Cell 40, 703–71353 Sarkies, P. et al. (2011) FANCJ coordinates two pathways that
maintain epigenetic stability at G-quadruplex DNA. Nucleic AcidsRes. DOI: 10.1093/nar/gkr868
54 Edmunds, C.E. et al. (2008) PCNA ubiquitination and REV1 definetemporally distinct mechanisms for controlling translesion synthesisin the avian cell line DT40. Mol. Cell 30, 519–529
55 Jansen, J. et al. (2009) Separate domains of Rev1 mediate two modes ofDNA damage bypass in mammalian cells. Mol. Cell Biol. 29, 3113–3123
56 Gibbons, R. (2006) Alpha thalassaemia-mental retardation, X linked.Orphanet J. Rare Dis. 1, 15
57 Law, M.J. et al. (2010) ATR-X syndrome protein targets tandemrepeats and influences allele-specific expression in a size-dependentmanner. Cell 143, 367–378
58 Lewis, P.W. et al. (2010) Daxx is an H3.3-specific histone chaperone andcooperates with ATRX in replication-independent chromatin assemblyat telomeres. Proc. Natl. Acad. Sci. U. S. A. 107, 14075–14080
59 Goldberg, A.D. et al. (2010) Distinct factors control histone variantH3.3 localization at specific genomic regions. Cell 140, 678–691
60 Mitson, M. et al. (2011) Functional significance of mutations in the Snf2domain of ATRX. Hum. Mol. Genet. 20, 2603–2610
61 Morin, R.D. et al. (2011) Frequent mutation of histone-modifying genesin non-Hodgkin lymphoma. Nature 476, 298–303
62 Morin, R.D. et al. (2010) Somatic mutations altering EZH2 (Tyr641) infollicular and diffuse large B-cell lymphomas of germinal-center origin.Nat. Genet. 42, 181–185
63 Dalgliesh, G.L. et al. (2010) Systematic sequencing of renal carcinomareveals inactivation of histone modifying genes. Nature 463, 360–363

Review Trends in Genetics March 2012, Vol. 28, No. 3
64 van Haaften, G. et al. (2009) Somatic mutations of the histoneH3K27 demethylase gene UTX in human cancer. Nat. Genet. 41,521–523
65 Wen, B. et al. (2009) Large histone H3 lysine 9 dimethylated chromatinblocks distinguish differentiated from embryonic stem cells. Nat.Genet. 41, 246–250
66 Bartkova, J. et al. (2005) DNA damage response as a candidateanti-cancer barrier in early human tumorigenesis. Nature 434, 864–
87067 Gorgoulis, V.G. et al. (2005) Activation of the DNA damage checkpoint
and genomic instability in human precancerous lesions. Nature 434,907–913
68 Halazonetis, T.D. et al. (2008) An oncogene-induced DNA damagemodel for cancer development. Science 319, 1352–1355
69 Tsantoulis, P.K. et al. (2008) Oncogene-induced replication stresspreferentially targets common fragile sites in preneoplastic lesions.A genome-wide study. Oncogene 27, 3256–3264
70 De, S. and Michor, F. (2011) DNA secondary structures and epigeneticdeterminants of cancer genome evolution. Nat. Struct. Mol. Biol. 18,950–955
71 Waddington, C.H. (1942) The epigenotype. Endeavour 1, 18–2072 Ernst, J. et al. (2011) Mapping and analysis of chromatin state
dynamics in nine human cell types. Nature 473, 43–4973 Gardner, K.E. et al. (2011) Operating on chromatin, a colorful language
where context matters. J. Mol. Biol. 409, 36–46
74 Guttman, M. et al. (2009) Chromatin signature reveals over a thousandhighly conserved large non-coding RNAs in mammals. Nature 458,223–227
75 Kouzarides, T. (2007) Chromatin modifications and their function. Cell128, 693–705
76 Li, B. et al. (2007) The role of chromatin during transcription. Cell 128,707–719
77 Trojer, P. and Reinberg, D. (2007) Facultative heterochromatin: isthere a distinctive molecular signature? Mol. Cell 28, 1–13
78 Lohse, M.B. and Johnson, A.D. (2009) White-opaque switching inCandida albicans. Curr. Opin. Microbiol. 12, 650–654
79 Bostick, M. et al. (2007) UHRF1 plays a role in maintaining DNAmethylation in mammalian cells. Science 317, 1760–1764
80 Kim, J.K. et al. (2009) UHRF1 binds G9a and participates in p21transcriptional regulation in mammalian cells. Nucleic Acids Res. 37,493–505
81 Lipps, H.J. and Rhodes, D. (2009) G-quadruplex structures: in vivoevidence and function. Trends Cell Biol. 19, 414–422
82 Margueron, R. and Reinberg, D. (2011) The Polycomb complex PRC2and its mark in life. Nature 469, 343–349
83 Nguyen, A.T. and Zhang, Y. (2011) The diverse functions of Dot1 andH3K79 methylation. Genes Dev. 25, 1345–1358
84 Wutz, A. (2011) Gene silencing in X-chromosome inactivation:advances in understanding facultative heterochromatin formation.Nat. Rev. Genet. 12, 542–553
127