ca2+/calmodulin-dependent kinase ii signaling causes ... · ca2+/calmodulin-dependent kinase ii...
TRANSCRIPT

Available online at www.sciencedirect.com
17 (2008) 132–146www.elsevier.com/developmentalbiology
Developmental Biology 3
Ca2+/Calmodulin-dependent kinase II signaling causes skeletal overgrowthand premature chondrocyte maturation
Michael J. Taschner 1, Mehran Rafigh, Fabienne Lampert, Simon Schnaiter 2, Christine Hartmann⁎
Institute of Molecular Pathology, Dr. Bohr-Gasse 7, A-1030 Vienna, Austria
Received for publication 27 August 2007; revised 1 February 2008; accepted 5 February 2008Available online 15 February 2008
Abstract
The long bones of vertebrate limbs originate from cartilage templates and are formed by the process of endochondral ossification. This processrequires that chondrocytes undergo a progressive maturation from proliferating to postmitotic prehypertrophic to mature, hypertrophicchondrocytes. Coordinated control of proliferation and maturation regulates growth of the skeletal elements. Various signals and pathways havebeen implicated in orchestrating these processes, but the underlying intracellular molecular mechanisms are often not entirely known. Here wedemonstrated in the chick using replication-competent retroviruses that constitutive activation of Calcium/Calmodulin-dependent kinase II(CaMKII) in the developing wing resulted in elongation of skeletal elements associated with premature differentiation of chondrocytes. Thepremature maturation of chondrocytes was a cell-autonomous effect of constitutive CaMKII signaling associated with down-regulation of cell-cycle regulators and up-regulation of chondrocyte maturation markers. In contrast, the elongation of the skeletal elements resulted from a non-cellautonomous up-regulation of the Indian hedgehog responsive gene encoding Parathyroid-hormone-related peptide. Reduction of endogenousCaMKII activity by overexpressing an inhibitory peptide resulted in shortening of the skeletal elements associated with a delay in chondrocytematuration. Thus, CaMKII is an essential component of intracellular signaling pathways regulating chondrocyte maturation.© 2008 Elsevier Inc. All rights reserved.
Keywords: Chicken; Chondrocyte maturation; Cyclin; AP1; PTHrP-signaling
Introduction
The vertebrate skeleton is a complex structure with over 200skeletal elements, which differ in shape and size. The majorityof skeletal elements are formed by endochondral ossification,whereby a cartilaginous scaffold that prefigures the futureskeletal element is eventually replaced by bone. Chondrocyteswithin these cartilaginous scaffolds have to undergo a highlyregulated maturation process that controls the size of the futureelement and is absolutely required for the remodeling into bone.During maturation chondrocytes cease to proliferate, becomepostmitotic prehypertrophic chondrocytes, which mature
⁎ Corresponding author. Fax: +43 17987153.E-mail address: [email protected] (C. Hartmann).
1 Present address: Cancer Research UK, Clare Hall, South Mimms,Hertfordshire, EN6 3LD, UK.2 Present address: Department of Histology and Molecular Cell Biology,
Medical University of Innsbruck, A-6020 Innsbruck, Austria.
0012-1606/$ - see front matter © 2008 Elsevier Inc. All rights reserved.doi:10.1016/j.ydbio.2008.02.007
further into hypertrophic chondrocytes. Various growth factorsand hormones are involved in the regulation of chondrocytematuration (Church et al., 2002; Karsenty and Wagner, 2002;Yasoda et al., 2004), yet little is known about the intracellularsignaling pathways involved.
Many of the so far known pathways seem to feed into acentral regulatory negative feedback loop between the secretedmolecules Indian hedgehog (Ihh) and Parathyroid-relatedpeptide (PTHrP) coordinating chondrocyte proliferation andmaturation (Kronenberg, 2003). This feedback loop is con-served in different species such as chicken, mouse and humans.Ihh is synthesized by prehypertrophic chondrocytes andregulates the expression of Pthrp by periarticular chondrocytes(Lanske et al., 1996; St-Jacques et al., 1999; Vortkamp et al.,1996). Ihh has PTHrP-dependent and independent effects onchondrocyte maturation; it acts through PTHrP to keepchondrocytes in the proliferative pool and controls chondrocyteproliferation independent of PTHrP (Chung et al., 1998; Karp etal., 2000; Long et al., 2001). PTHrP as well as the calcium

133M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
regulating hormone, parathyroid hormone (PTH), bind to the G-protein coupled PTH/PTHrP-receptor (Ppr). Ppr is expressed atlow levels by proliferating chondrocytes and at high levels byprehypertrophic and early hypertrophic chondrocytes. PTHrP/Ppr signaling is important in controlling chondrocyte prolifera-tion and maturation; high levels of PTHrP, or constitutive PPRsignaling, delay the appearance of maturated, hypertrophicchondrocytes, while loss of PPR signaling results in shorteningof long bones and accelerated chondrocyte maturation (Lanskeet al., 1999; Schipani et al., 1997; Weir et al., 1996). PPR signalsvia heterotrimeric G-proteins such as Gs, which stimulatesadenylyl cyclase or Gq/11, which activates phospholipase C(PLC), thereby generating multiple second messengers (Abou-Samra et al., 1992; Guo et al., 2002; Juppner et al., 1991). Gssignaling leads to activation of cAMP-dependent protein kinaseA (PKA) and is required to keep chondrocytes in theproliferative pool (Bastepe et al., 2004). In contrast, PPRsignaling via Gq/11 and PLC stimulates chondrocyte maturation(Guo et al., 2002). Thus, activation of different intracellularpathways downstream of PPR plays a critical role indetermining the rate of chondrocyte maturation during endo-chondral ossification.
PLC-signaling leads to the activation of different proteinkinases such as protein kinase C (PKC) and Calcium/Calmodulin (CaM) dependent kinases (CaMKs) includingCaMKIIs. Four different CaMKII genes have been reported invertebrates, encoding the four isoforms, CaMKII α, β, γ, δ,with numerous splice-variants. CaMKII acts as a multimericholoenzyme composed out of 4–14 subunits of either homo- orheteromers (Lantsman and Tombes, 2005; Rosenberg et al.,2006). Ca2+/CaM binding leads to Thr286-autophosphorylationand releases the enzyme from autoinhibition, rendering it in aCa2+/CaM independent active form (Colbran, 2004; Hudmonand Schulman, 2002). CaMKII has been shown to playimportant roles in synaptic plasticity and memory (Cammarotaet al., 2002; Fox, 2003). In addition, CaMKII signaling has beenimplicated in mediating mechanical transduction in human andbovine articular cartilage (Shimazaki et al., 2006; Valhmu andRaia, 2002) and in osteoblast and osteoclast differentiation(Quinn et al., 2000; Seales et al., 2006; Zayzafoon, 2006; Zhanget al., 2005).
Since CaMKII is activated downstream of PLC and all fourisoforms are expressed in chicken chondrocytes, its activitycould potentially be involved in various signaling pathways,such as the PTHrP/PPR, FGF or the non-canonical Wnts, whichare known to regulate chondrogenesis. In order to gain insightinto a potential role of CaMKII in skeletogenesis we used aretroviral misexpression approach in chicken. Constitutiveactivation of CaMKII signaling in the developing limb usinga retrovirus expressing the mutated CaMKII-T286D form(daCaMKII) resulted in skeletal overgrowth of infected longbones and premature maturation of chondrocytes. In contrastinhibition of endogenous CaMKIIs using retroviral-mediatedoverexpression of a peptide-inhibitor resulted in shortening ofthe infected long bones. Thus, our results suggest that intra-cellular CaMKII activity is involved in the control of chon-drocyte maturation.
Materials and methods
Construction of retroviruses and retroviral misexpression
The retroviral constructs RCASBP(A) carrying the ORF of the ratCamkIIα gene with the point mutation T286D, rendering the CaMKIIconstitutively active, or K42M, converting it to a kinase-dead molecule (Kuhlet al., 2000), as well as the viruses carrying the corresponding eGFP fusionproteins (eGFP was fused in frame with the ORF of CaMKII-T286D andCaMKII-K24M via an BamHI site located in a short linker at the 5′end ofthe eGFP gene) were engineered as outlined in (Logan and Tabin, 1998). TheRCASBP(A) expressing the a fusion of eGFP and the rat CaM-KIIN fusion-protein (gift from T. Soderling) was generated by PCR and cloning of theGFP-CaM-K2N (NcoI–HindIII) into the Slax13 shuttle vector. Transfectionand growth of the RCAS viruses were performed in principle according to(Morgan and Fekete, 1996) with slight modifications. Concentrated viruseswith a titer of 0.8–1.0×109 pfu/ml were injected as described previously(Hartmann and Tabin, 2000).
Skeletal preparations, histology, in situ hybridization andimmunohistochemistry
Whole mount alcian blue stainings of days 8.5 and 9.5 chick embryos wereperformed as previously described (Goff and Tabin, 1997). Weigert–Safranin Oand alcian blue stainings of paraffin sections were done according to standardprocedures. Non-radioactive in situ hybridizations on alternating paraffinsections of blocks containing both the contra-lateral wing and the infected wingwere done using DIG-labeled anti-sense RNA probes as previously described(Murtaugh et al., 2001). Double fluorescent in situ-hybridizations were doneusing DIG- and Biotin-labeled RNA probes modified after (Tylzanowski et al.,2003). Briefly after hybridization, slides were washed, quenched and blocked.Probes were then detected sequentially by incubation of slides withstreptavidine-HRP (Perkin Elmer, dilution 1 in 100) developed with Cy3 orCy5 tyramide labelled fluorescent dyes, followed by a quenching step withstreptavidine solution (Roche, dilution 1 in 75) and subsequent detection of theDIG labeled probe using anti-DIG-HRP (Roche, dilution 1 in 50), followed byCy5 or Cy3 tyramide labelled fluorescent dyes (see also instructions of the TSAplus fluorescent systems kit, Perkin Elmer, NEL760). Tissue was mounted inusing SlowFade Light Antifade (Molecular Probes). Chicken probes for Ihh,ColX, Pthrp, and Ppr have been published previously. Probes for c-Fos (ChEST517o21) and c-Jun (ChEST 947b5) were generated from UMIST clones. Probesfor cyclinA (for: CAGCTCCGACGATCAACC; rev: TCAGACTTGGTCTT-CATGG) and cyclinD1 (for: GACCCGACGAGTTACTGC; rev:GATTCCTTCTCAGATGCCC) were generated by RT-PCR using the indicatedprimer pairs and cloned into pGEMTeasy (Promega).
Cell culture and cell sorting
Primary chondrocytes were isolated from E18.5 chicken as previouslydescribed (Koyama et al., 1999), using the caudal 1/3 of the sternum to enrichfor proliferating chondrocytes. Chondrocytes were cultures at 5×105 cells perwell in a 6-well plate. Chondrocytes were infected at 50–60% confluence with2×106 viral particles in 500 μl medium. Infection of chondrocytes wasdetermined by immunohistochemical staining using 1:3 diluted 3C2 hybridomacell supernatant (Developmental Studies Hybridoma Bank). Gfp-virus infectedcells were FACS sorted for GFP 2 days post infection and plated for one extraday prior to RNA or protein isolation. For the in vitro studies at least threeindependent experiments were carried out.
Northern blot and Real-time PCR analyses
RNA from cultured primary chondrocytes or freshly isolated sterna wasisolated using TRIZOL according to manufacturers instructions. 15 μg totalRNA/lane were used for Northern blot analysis following standard protocols.[P32]-labeled probes for CamkIIα, β, γ, and δ were generated using therediprime kit (Amersham) and inserts of the appropriate UMIST EST clones(ChEST 141m12; ChEST 712p21; ChEST 213l23) or from a PCR product (for:

134 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
GCAGACCCTAATGAAGATGG; rev: ACAGTGTGGATGAAAGGTGG)cloned into pGEMT in the case of CamkIIβ. For Real time PCR analysis1 μg of DNase treated, total RNAwas used to produce first strand cDNA. Realtime PCR was performed using SYBR green 1 nucleic acid gel stain (MolecularProbes), TAKARA Taq, and the DNA-engine Opticon 2 (MJ Research). Realtime PCR analyses were performed in duplicates and results were reproduced inthree independent experiments. Values were calculated using the comparative C(t) method, and normalized to cGapdh expression. Primer sequences areavailable by request. P-values were determined by performing a paired Studentt-test.
Results
Expression of CaMKII isoforms in chick chondrocytes
CamkIIs are more or less ubiquitously expressed, however,some isoforms are more prevalent in certain tissues, for exampleCamkIIα and β are very abundant in the brain, while the γ andδ-isoforms are more broadly expressed (Rongo, 2002; Soder-ling et al., 2001). In the chicken, we detected all four isoforms,α, β, γ, δ, by RT-PCR and Northern blot in primarychondrocytes (Fig. 1A; data not shown). We confirmed thepresence of the γ, δ isoforms byWestern blots (data not shown).However, we were unable to detect any specific expressionpatterns for the different isoforms within the cartilage elementsby in situ hybridization.
Constitutive activation of CaMKII results in skeletalovergrowth
We performed retroviral misexpression experiments usingreplication competent avian retroviruses (RCAS) to analyze apotential role of CaMKII signaling in chondrogenesis. SinceCaMKII acts as multimeric holoenzyme, which can becomposed of different isoforms, we used a retrovirusexpressing a constitutively active form of one CaMKII isoform(CaMKIIα-T286D; Hanson et al., 1994), referred to in thefollowing as daCaMKII, to transform the entire holoenzymeinto a dominant active complex through incorporation of oneto two dominant-active subunits (Hudmon and Schulman,2002). Misexpression of daCaMKII in the posterior half ofchicken wing buds at day 3.5 (HH22–23) resulted in a
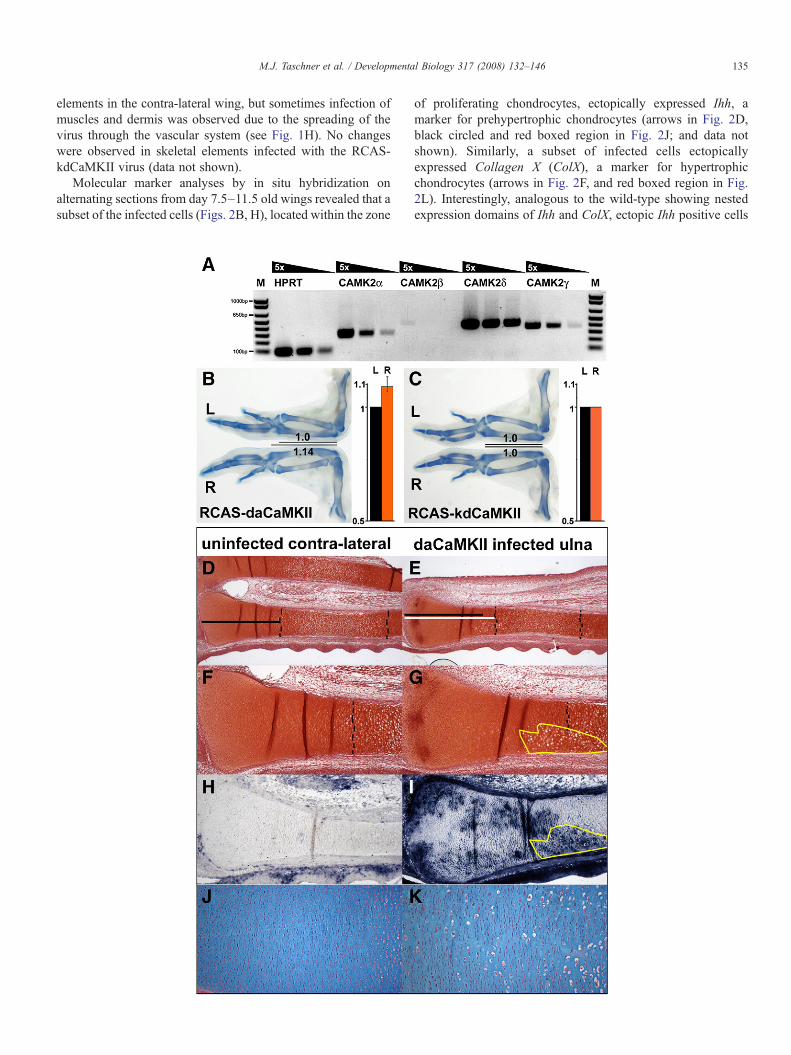
Fig. 1. Phenotypic changes in response to retroviral daCaMKII overexpression. (A) R(B–C) Alcian blue stained whole skeletal preparations of wing pairs at d9.5; the lowserved as the internal control. The black bars and the numbers indicate the length of thinfected with RCAS-daCaMKII shows elongation of the skeletal elements, such asrelative increase in length of infected R-ulnae (orange bar) in comparison to the contrrepresents the standard deviation of the mean of the results. (C) RCAS-kdCaMKIcomparison to contra-lateral wing. Bar diagram showing no difference in the relative l(black bar), n=10. (D–K) Histological and in situ analyses on sections of uninfected(D–E) Low magnification of a contra-lateral uninfected wing, showing the radius (up(D) indicates the length of the proliferating zone in the uninfected ulna in comparisonshown in (E), the broken lines demarcate the zone of hypertrophic chondrocytes.magnification of the infected ulna showing a localized expansion of hypertrophic ceshowing no infection in chondrocytes in the contra-lateral ulna (H), but high levels oectopic differentiation into hypertrophic chondrocytes correlates in part with the infeshowing the orderly array of flattened proliferating chondrocytes in the contra-lateravacuolized (appear white) cells (K).
noticeable lengthening of the infected skeletal elements at days8.5 and 9.5 by 6–14% compared to corresponding elements inthe contra-lateral wing (n=20, Fig. 1B, and data not shown).The lengthening of the infected elements upon misexpressionof daCaMKII is a very peculiar phenotype, since misexpres-sion of various factors commonly leads to shortening of theaffected elements.
In addition, we misexpressed a RCAS virus encoding aCaMKII subunit with a K24M mutation in the ATP-bindingpocket (kdCaMKII), which did not affect the growth of theinfected skeletal elements (n=9; Fig. 1C). This result was notentirely unexpected since retroviral mediated expression levelsare probably insufficient to replace all subunits and replacementof only some of the subunits of the holoenzyme with a kinase-dead molecule will not render the enzyme inactive (Rich andSchulman, 1998).
CaMKII signaling causes premature differentiation ofchondrocytes
We next examined why the daCaMKII infected skeletalelements were elongated, performing histological and markergene analyses at various stages of development. The histolo-gical analyses on sections of infected and uninfected (contra-lateral control limbs) using Safranin O/Weigert revealed anectopic expansion of enlarged, vacuolized, hypertrophicchondrocytes into the prehypertrophic region (Figs. 1E, G)compared to wild-type skeletal elements (Figs. 1D, F). Theexpansion of hypertrophic chondrocytes overlapped in part withthe infected regions, detected by in situ hybridization using ananti-sense probe for the exogenous rat CamkII transcript(compare yellow encircled regions Figs. 1G, I). In addition, aslight elongation of the zone of round and flattened proliferativechondrocytes (from the articular region to the endogenous zoneof hypertrophic chondrocytes) was observed in the infectedskeletal elements (white line in Fig. 1E, compared to black linein Fig. 1D). Alcian-blue staining on infected and control limbsrevealed the presence of abnormal looking, enlarged cellswithin the zone of flattened proliferative chondrocytes ininfected elements (compare Figs. 1K, J). In all samples analyzedno retroviral infection was detected in chondrocytes of skeletal
T-PCR for CaMKII-isoforms, α, β, γ, δ, and HPRT as control on dilution series.er, right (R) wing was virally infected and the upper, contra-lateral left (L) winge ulnae; note length of the uninfected L-wing is always set to one. (B) Right wingthe ulna, in comparison to the contra-lateral element. Bar diagram showing thea-lateral L-ulnae (black bar), n=20, pb0.00005 (paired Student t-test), error barI infected wing shows no size difference in the length of skeletal elements inength of infected R-ulnae (orange bar) in comparison to the contra-lateral L-ulnaeand infected ulnae at day 9.5 (D–I) and day 12.5 (J–K), showing the distal ends.per) and ulna (lower) elements stained with Safranin O-Weigert. The black bar into the white bar indicating the length of the proliferation zone in the infected ulna(F) Higher magnification of the distal end of the uninfected ulna. (G) Higherlls (area demarcated by yellow line). (H–I) In situ hybridization for rat CamkIIf patchy infection in chondrocytes in the ulna of the infected limb (I). Note thected regions (area demarcated in yellow). (J–K) Alcian blue staining on sectionsl ulna (J), which is disturbed in the infected ulna by the appearance of enlarged,
135M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
elements in the contra-lateral wing, but sometimes infection ofmuscles and dermis was observed due to the spreading of thevirus through the vascular system (see Fig. 1H). No changeswere observed in skeletal elements infected with the RCAS-kdCaMKII virus (data not shown).
Molecular marker analyses by in situ hybridization onalternating sections from day 7.5–11.5 old wings revealed that asubset of the infected cells (Figs. 2B, H), located within the zone
of proliferating chondrocytes, ectopically expressed Ihh, amarker for prehypertrophic chondrocytes (arrows in Fig. 2D,black circled and red boxed region in Fig. 2J; and data notshown). Similarly, a subset of infected cells ectopicallyexpressed Collagen X (ColX), a marker for hypertrophicchondrocytes (arrows in Fig. 2F, and red boxed region in Fig.2L). Interestingly, analogous to the wild-type showing nestedexpression domains of Ihh and ColX, ectopic Ihh positive cells

Fig. 2. DaCaMKII misexpression causes premature chondrocyte maturation. (A–N) Non-radioactive in situ hybridizations on alternating sections of contra-lateral andRCAS-daCaMKII infected wings at day 7.5 (A–F) and day 9.5 (G–N). (A) Absence of ectopic CamkIIα expression in chondrocytes of the contra-lateral, uninfectedulna. (B) Ectopic CamkII expression in the infected ulna. (C) Ihh expression in contra-lateral, uninfected ulna. (D) Presence of ectopic Ihh-positive cells closer to thearticular region (arrows) in the infected ulna. (E) ColX expression domain in contra-lateral, uninfected ulna (black bar). (F) Presence of ectopic ColX-positive cells inthe infected ulna (arrows). The red bar in (E) and (F) indicates the size of the ColX expression domain, not including the region in which ectopic differentiation isobserved. (G) Absence of exogenous CamkII in the contra-lateral ulna. (H) Ectopic CamkII expression in the infected ulna. (I) Wild-type expression of Ihh. (J)Presence of ectopic Ihh positive cells in the zone of proliferating chondrocytes (red square and black circle). (K) Wild-type ColX expression in hypertrophicchondrocytes. (L) Ectopic ColX expression within the zone of proliferative chondrocytes (red square), however, cells closer to the articular region do not show ectopicColX expression (black circle). (M, N) Pthrp expression in the articular region of the contra-lateral, uninfected ulnae and of the infected ulnae, showing increasedexpression levels of Pthrp in the articular region of the infected ulnae (asterisk).
136 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
were always found closer to the articular region (black circle inFig. 2J), while ectopic ColX positive cells were detected inregions closer to the diaphysis (red box in Fig. 2L).Furthermore, analysis of ColX at day 7.5 revealed a ratherunexpected reduction in the size of the ColX domain in theinfected skeletal elements in comparison to the control elements(see red brackets in Figs. 2E, F compared to black bracket inFig. 2E). This size reduction is indicative for a delay in the
maturation of hypertrophic chondrocyte. Thus activation ofCaMKII signaling leads to an overall elongation of the infectedskeletal elements associated with ectopic maturation of infectedchondrocytes within the proliferative zone, while at the sametime the size of the endogenous zone of hypertropic, ColXexpressing chondrocytes was reduced in the infected elementssuggesting an overall delay in chondrocyte maturation. Thesepartially contradictory observations suggest that CaMKII

137M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
signaling must lead to the production of secondary signalsinvolved in proliferation and negative regulation of chondrocytematuration, one of them being Ihh.
We hypothesized that the presence of cells ectopicallyexpressing Ihh closer to the articular region should lead to anup-regulation of Pthrp expression in articular chondrocytes.Therefore, we performed in situ hybridizations using a Pthrpriboprobe and found that Pthrp was indeed up-regulated inarticular chondrocytes of the elbow region in the infected (Fig.2N), compared to the contra-lateral limbs (Fig. 2M). The markeranalyses on alternating sections suggested that the ectopicexpression of maturation markers always coincided withdaCaMKII infected areas, but we could not rule out thepossibility that CaMKII signaling up-regulated a secretedmolecule stimulating maturation of surrounding cells. Tounambiguously show in vivo that the ectopic maturation ofchondrocytes was a cell-autonomous event down-stream ofCaMKII signaling, we performed double-fluorescence in situhybridizations for CamkII/Ihh (Figs. 3A–C) and CamkII/ColX(Figs. 3D–F). Cells ectopically expressing Ihh always coloca-lized with daCaMKII expressing cells (see arrows in Figs. 3A,B) and cells ectopically expressing ColX also colocalize withdaCaMKII positive cells (see arrows in Figs. 3D, E). However,
Fig. 3. Cell autonomous maturation of daCaMKII-infected cells. (A–F) Double fluordetected with Cy3 using a digoxygenin-labeled anti-sense riboprobe. (B, E) Ectopic(C) Merged image of Ihh in red and CamkII in green. (D) Cy3 channel showing ColXin red and CamkII in green. Note: yellow arrows mark cells expressing ectopically
like in the non-fluorescent in situ hybridizations, not everyCaMKII positive cell expressed Ihh or ColX. These resultsdemonstrate that the ectopic maturation is a cell-autonomousevent, since it occurred only in chondrocytes where the CaMKIIsignaling pathway had been activated. Nevertheless, theseobservations do not rule out that the ectopic up-regulation ofmaturation markers is still under the influence of extrinsicsignals, which would explain why not all cells expressingdaCaMKII ectopically maturated, especially those closest to thearticular region.
Down-regulation of cyclins and AP1 family members precedesectopic maturation of chondrocytes
The finding that mature chondrocytes, which express Ihhand ColX, are found closer to the articular region, resembles thephenotypes of Ppr−/− and Gnas−/− (encoding Gαs) chimericgrowth plates, where chondrocytes lacking either PPR or theGαs subunit of heterotrimeric G-proteins ectopically matureinto hypertrophic chondrocytes (Bastepe et al., 2004; Chung etal., 1998). Since CyclinA and CyclinD1, as well as c-Fos and c-Jun have been shown to be regulated by PPR signaling (Beierand LuValle, 2002; Ionescu et al., 2001) we analyzed the
escent hybridizations on daCaMKII infected ulnae at day 10. (A) Ihh expressionCamkII expression detected with Cy5 using a biotinylated anti-sense riboprobe.expression using a DIG-labeled anti-sense riboprobe. (F) Merged image of ColXIhh (A) or ColX (D) and are infected with daCaMKII (B, E).

Fig. 4. CaMKII signaling causes down-regulation of the cell-cycle genes cyclinA andD1. (A–R)Double fluorescent hybridizations on sections of the contra-lateral ulna(A, D, G, J,M, P) of daCaMKII-infected specimens (B, E, H, K, N, Q), and of kdCaMKII-infectedwings at day 9.5, showing the distal end of the ulnae. (A–C) Black andwhite images of Cy5 channel showing absence of infection in chondrocytes of the contra-lateral element (A) and ectopicCamkII expression in infected elements (B, C).(D–F) Black and white images of the Cy3 channel showing ubiquitous CyclinA expression in proliferating chondrocytes in wild-type (D) and kdCaMKII-infectedelements (F), and patchy down-regulation in daCaMKII-infected elements (yellow encircled areas in panel E), using a digoxygenin-labeled anti-sense riboprobe. (G–I)Merged images showing CamkII in red and CyclinA in green. (J–L) Cy5 channel showing absence of infection in the contra-lateral element (J) and detecting ectopicCamkII expression in infected elements (K, L) using a biotinylated anti-sense riboprobe. (M–O) Cy3 channel showing CyclinD1 expression in the wild-type (J) beinghighest in a subpopulation of proliferating chondrocytes (zone II, see panels M, P) using a digoxygenin-labeled anti-sense riboprobe. (N) Patchy down-regulation ofCyclinD1 in daCaMKII-infected elements (yellow encircled areas in panel N) and increase in CyclinD1 in uninfected chondrocytes of zone I (see asterisk in panels N,Q). (O) No alterations were detected kdCaMKII-infected elements. (P–R) Merged images showing CamkII in red and CyclinD1 expression in green.
138 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146

139M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
expression of these four genes in daCaMKII and kdCaMKII-infected skeletal elements using double-fluorescence in situhybridizations. The kdCaMKII infected elements served as acontrol for possible quenching effect of the probe detecting theinfected areas on the second fluorescent signal. CyclinA in thecontra-lateral wing, which showed no exogenous CaMKII-signal (Fig. 4A), was more or less ubiquitously expressed inproliferating chondrocytes (Fig. 4D). In contrast its expressionwas down-regulated (Fig. 4E) in regions that were infected withRCAS-daCaMKII (Fig. 4B). No apparent expression changeswere detected in kdCaMKII infected chondrocytes (Figs. 4C, F),showing that the down-regulation was not due to a quenching ofthe second signal. CyclinD1 was expressed in wild-type skeletalelements at low levels in chondrocytes close to the articularregion (region I), while its expression was then up-regulated(region II) and went down again as chondrocytes matured(region III) (Figs. 4M, P). A similar expression profile has beenreported for the CyclinD1 protein (Yang et al., 2003). In contrastto the controls, we observed in the infected elements that Cy-clinD1 expression in region I was elevated in cells that were notinfected with the daCaMKII virus (see asterisks in Figs. 4N, Q),while its expression was down-regulated (Fig. 4N) in daCaMKIIinfected proliferating chondrocytes (Fig. 4K). No such changeswere observed in kdCaMKII infected chondrocytes (Figs. 4L,O). Expression of c-Fos and c-Jun, which are ubiquitously ex-pressed in proliferating chondrocytes of wild-type skeletal ele-ments (Figs. 5C, I), was also down-regulated (Figs. 5D, J) indaCaMKII infected cells (Figs. 5B, H), while their expressionwas not affected by kdCaMKII misexpression (data not shown).Thus, misexpression of daCaMKII led to down-regulation of thePPR-responsive genes Cyclin A and D1, and AP1 family mem-bers, c-Fos and c-Jun in the majority of daCaMKII infectedcells. In contrast, ectopic expression of Ihh and ColX was onlydetected in a subset of the infected chondrocytes and interest-ingly never in the articular regions.
DaCaMKII accelerates chondrocyte maturation in vitro
To independently confirm the cell-autonomous effects ofCaMKII activation in vitro we used sternal primary chondrocyte
Fig. 5. CaMKII signaling causes down-regulation of the AP1 genes, c-Fos andc-Jun. (A–L) Double fluorescent hybridizations on sections of the contra-lateralulna (A, C, E, G, I, K) of daCaMKII-infected specimens (B, D, F, H, J, K) at day9.5, showing the distal end of the ulnae. (A, B) Cy5 channel detecting absence ofectopic CamkII expression in the contra-lateral element (E) and ectopicexpression in infected elements (F) using a biotin-labeled anti-sense riboprobe.(C, D) Cy3 channel showing ubiquitous c-Fos expression in proliferatingchondrocytes in wild-type (C) and patchy down-regulation in daCaMKII-infected elements (yellow encircled areas in panel D) using a digoxygenin-labeled anti-sense riboprobe. (E, F) Merged images showing CamkII in red andc-Fos expression in green. (G, H) Cy5 channel detecting absence of ectopicCamkII expression in the contra-lateral element (G) and ectopic expression inthe infected elements (L) using a biotin-labeled anti-sense riboprobe. (I, J) Cy3channel showing ubiquitous c-Jun expression in proliferating chondrocytes inthe wild-type (I) and patchy down-regulation of c-Jun in daCaMKII-infectedelements (yellow encircled areas in panel J) using a digoxygenin-labeled anti-sense riboprobe. (K, L) Merged images showing CamkII in red and c-Junexpression in green.
cultures, which were infected either with RCAS-Gfp, or RCAS-daCaMKII-Gfp, or RCAS-kdCaMKII-Gfp virus. Chick caudal(lower) sternal chondrocytes are a population of poliferative,undifferentiated, immature chondrocytes that do not sponta-neously proceed toward a terminal differentiation stage(Gerstenfeld et al., 1990; Szuts et al., 1998). Retrovirusescarrying Gfp-fusion proteins of the mutated forms of CaMKIIwere used in order to enrich for the infected chondrocytes bysorting, since infections with untagged virus constructs resultedonly in 25–35% infected chondrocytes (based on immunohis-tochemical stainings using an anti-gag-AB; data not shown).
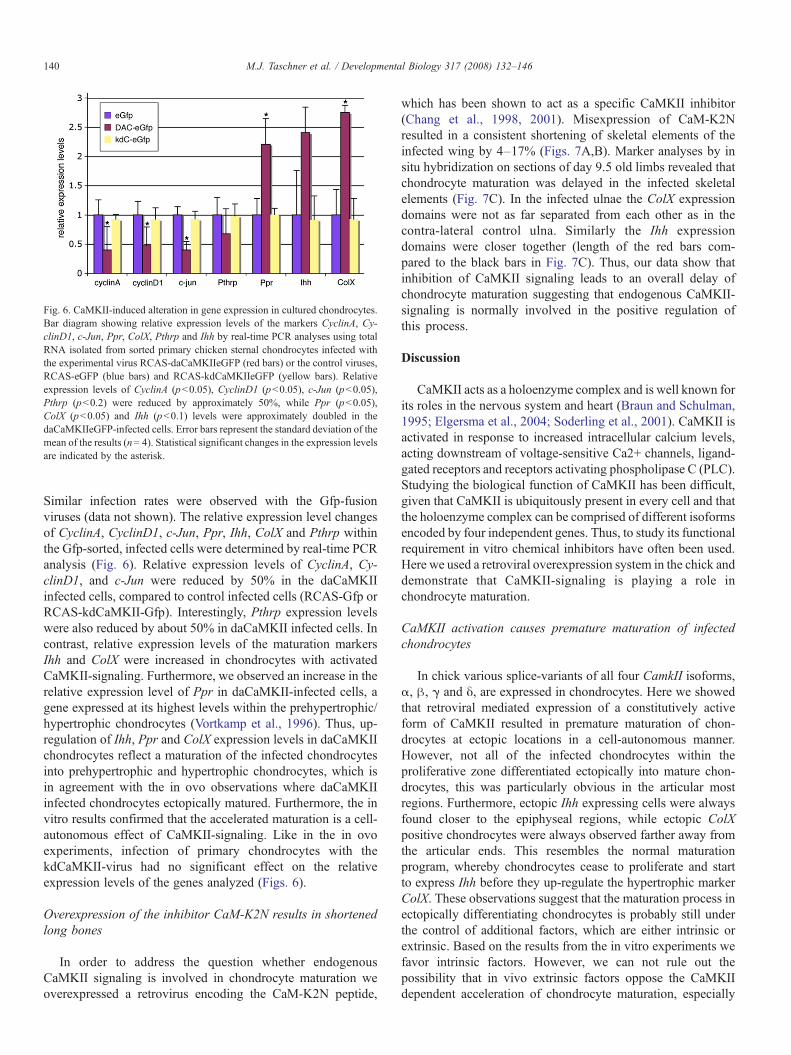
Fig. 6. CaMKII-induced alteration in gene expression in cultured chondrocytes.Bar diagram showing relative expression levels of the markers CyclinA, Cy-clinD1, c-Jun, Ppr, ColX, Pthrp and Ihh by real-time PCR analyses using totalRNA isolated from sorted primary chicken sternal chondrocytes infected withthe experimental virus RCAS-daCaMKIIeGFP (red bars) or the control viruses,RCAS-eGFP (blue bars) and RCAS-kdCaMKIIeGFP (yellow bars). Relativeexpression levels of CyclinA (pb0.05), CyclinD1 (pb0.05), c-Jun (pb0.05),Pthrp (pb0.2) were reduced by approximately 50%, while Ppr (pb0.05),ColX (pb0.05) and Ihh (pb0.1) levels were approximately doubled in thedaCaMKIIeGFP-infected cells. Error bars represent the standard deviation of themean of the results (n= 4). Statistical significant changes in the expression levelsare indicated by the asterisk.
140 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
Similar infection rates were observed with the Gfp-fusionviruses (data not shown). The relative expression level changesof CyclinA, CyclinD1, c-Jun, Ppr, Ihh, ColX and Pthrp withinthe Gfp-sorted, infected cells were determined by real-time PCRanalysis (Fig. 6). Relative expression levels of CyclinA, Cy-clinD1, and c-Jun were reduced by 50% in the daCaMKIIinfected cells, compared to control infected cells (RCAS-Gfp orRCAS-kdCaMKII-Gfp). Interestingly, Pthrp expression levelswere also reduced by about 50% in daCaMKII infected cells. Incontrast, relative expression levels of the maturation markersIhh and ColX were increased in chondrocytes with activatedCaMKII-signaling. Furthermore, we observed an increase in therelative expression level of Ppr in daCaMKII-infected cells, agene expressed at its highest levels within the prehypertrophic/hypertrophic chondrocytes (Vortkamp et al., 1996). Thus, up-regulation of Ihh, Ppr and ColX expression levels in daCaMKIIchondrocytes reflect a maturation of the infected chondrocytesinto prehypertrophic and hypertrophic chondrocytes, which isin agreement with the in ovo observations where daCaMKIIinfected chondrocytes ectopically matured. Furthermore, the invitro results confirmed that the accelerated maturation is a cell-autonomous effect of CaMKII-signaling. Like in the in ovoexperiments, infection of primary chondrocytes with thekdCaMKII-virus had no significant effect on the relativeexpression levels of the genes analyzed (Figs. 6).
Overexpression of the inhibitor CaM-K2N results in shortenedlong bones
In order to address the question whether endogenousCaMKII signaling is involved in chondrocyte maturation weoverexpressed a retrovirus encoding the CaM-K2N peptide,
which has been shown to act as a specific CaMKII inhibitor(Chang et al., 1998, 2001). Misexpression of CaM-K2Nresulted in a consistent shortening of skeletal elements of theinfected wing by 4–17% (Figs. 7A,B). Marker analyses by insitu hybridization on sections of day 9.5 old limbs revealed thatchondrocyte maturation was delayed in the infected skeletalelements (Fig. 7C). In the infected ulnae the ColX expressiondomains were not as far separated from each other as in thecontra-lateral control ulna. Similarly the Ihh expressiondomains were closer together (length of the red bars com-pared to the black bars in Fig. 7C). Thus, our data show thatinhibition of CaMKII signaling leads to an overall delay ofchondrocyte maturation suggesting that endogenous CaMKII-signaling is normally involved in the positive regulation ofthis process.
Discussion
CaMKII acts as a holoenzyme complex and is well known forits roles in the nervous system and heart (Braun and Schulman,1995; Elgersma et al., 2004; Soderling et al., 2001). CaMKII isactivated in response to increased intracellular calcium levels,acting downstream of voltage-sensitive Ca2+ channels, ligand-gated receptors and receptors activating phospholipase C (PLC).Studying the biological function of CaMKII has been difficult,given that CaMKII is ubiquitously present in every cell and thatthe holoenzyme complex can be comprised of different isoformsencoded by four independent genes. Thus, to study its functionalrequirement in vitro chemical inhibitors have often been used.Here we used a retroviral overexpression system in the chick anddemonstrate that CaMKII-signaling is playing a role inchondrocyte maturation.
CaMKII activation causes premature maturation of infectedchondrocytes
In chick various splice-variants of all four CamkII isoforms,α, β, γ and δ, are expressed in chondrocytes. Here we showedthat retroviral mediated expression of a constitutively activeform of CaMKII resulted in premature maturation of chon-drocytes at ectopic locations in a cell-autonomous manner.However, not all of the infected chondrocytes within theproliferative zone differentiated ectopically into mature chon-drocytes, this was particularly obvious in the articular mostregions. Furthermore, ectopic Ihh expressing cells were alwaysfound closer to the epiphyseal regions, while ectopic ColXpositive chondrocytes were always observed farther away fromthe articular ends. This resembles the normal maturationprogram, whereby chondrocytes cease to proliferate and startto express Ihh before they up-regulate the hypertrophic markerColX. These observations suggest that the maturation process inectopically differentiating chondrocytes is probably still underthe control of additional factors, which are either intrinsic orextrinsic. Based on the results from the in vitro experiments wefavor intrinsic factors. However, we can not rule out thepossibility that in vivo extrinsic factors oppose the CaMKIIdependent acceleration of chondrocyte maturation, especially

Fig. 7. Inhibition of endogenous CaMKII activity results in shortening of skeletal elements and delayed chondrocyte maturation. (A) Alcian blue stained whole skeletalpreparations of a wing pair at d9.5; showing a shortening of the lower right (R) RCAS-CaM-K2N infected wing in comparison to the upper contra-lateral left (L)uninfected wing. The black bars and the numbers indicate the length of the ulna; note length of the uninfected L-wing is set to one. (B) Bar diagram showing therelative length of the CaM-K2N infected ulnae (black bar) in comparison to the length of the ulnae of the contra-lateral wing (orange bar). Error bar represents thestandard deviation of the mean of the results, pb0.0003 (paired Student t-test). (C) Non-radioactive in situ hybridizations on alternating sections of a day 9.5uninfected contra-lateral and the CaM-K2N-infected ulna using digoxygenin-labeled anti-sense riboprobes for CaM-K2N, ColX and Ihh. CaM-K2N staining showsthe absence of infection in the contra-lateral ulna, the rate of infection in the infected ulna. ColX staining shows that the expression domains are closer together in theinfected ulna (red bar compared to black bar). The Ihh staining shows that the Ihh expression domains are closer together in the infected ulna (red bar compared to black bar).
141M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
since in vivo only a subpopulation of the infected chondrocytesup-regulates maturation markers. The phenotype we observedin the mosaic growth plates of chicken long bones, where cellsexpressing the constitutively active form of CaMKII areintermingled with non-infected wild type cells, remarkablyresembles the phenotype observed in Ppr−/− chimeric growthplates where Ppr deficient chondrocytes located within theproliferative zone of otherwise wild-type chondrocytes ectopi-cally mature into prehypertrophic/hypertrophic chondrocytes(Chung et al., 1998, 2001).
Our attempt to inhibit the endogenous CaMKII activity inskeletal elements by overexpression of a specific inhibitorypeptide resulted a slight, but statistically significant, shorteningof the infected skeletal elements and was associated withdelayed chondrocyte maturation. The delay is reflected in theobservations that the Ihh positive zones of prehypertrophic andthe ColX positive zones of hypertrophic chondrocytes are closertogether than in the contra-lateral control element. Thisphenotype is similar to the one observed in mouse expressinga mutant form of PPR, which cannot activate phospholipase C(Guo et al., 2002).
DaCaMKII retroviral expression causes non-cell autonomousphenotypes
Retroviral misexpression of growth factors or signalingmodifiers very often leads to shortening of infected skeletalelements, while lengthening of the long bones has only beenobserved in a few cases. In particular, mice lacking the Fgfr3, ormice with too high levels of Insulin-like growth factor 1 (IGF1),or increased C-type natriuretic peptide (CNP) signaling activityhave elongated long bones (Colvin et al., 1996; Jaubert et al.,1999; Mathews et al., 1988). Interestingly, overexpression ofdaCaMKII also resulted in an overall elongation in length of theinfected skeletal elements. This elongation is the complex resultof a combination of cell-autonomous and non-cell-autonomouseffects. The acceleration of chondrocyte maturation is a cell-autonomous effect of activated CaMKII signaling and isassociated with two non-cell autonomous effects, elongationof the proliferative zone and delay of hypertrophic chondrocytematuration. The latter two effects are due to the cell-autonomous acceleration of chondrocyte maturation resultingin ectopic differentiation of Ihh expressing chondrocytes closer

142 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
to the articular region. Ectopic Ihh expression contributes to thelengthening of skeletal elements by two mechanisms. First, itserves as a direct proliferative stimulus for the surroundingchondrocytes due to the proliferation promoting activity ofIHH, acting independently of PTHrP (Karp et al., 2000). Thisactivity is probably reflected in the observed up-regulation ofCyclinD1 in cells outside of the infected regions, since Cy-clinD1 has recently been shown to be a direct target of IHHsignaling (Long et al., 2001). Second, ectopic expression of Ihhalso led to an up-regulation of PTHrP, an IHH responsive factor,in articular chondrocytes thereby keeping chondrocytes in theproliferative pool. In addition to IHH, the increase in PTHrPsignaling could be responsible for the up-regulation of Cy-clinD1. Since we did not observe a distinct up-regulation of theexpression of CyclinA, c-Fos or c-Jun, which presumably arealso regulated by PTHrP-signaling, we favor the hypothesis thatthe up-regulation of CyclinD1 is an effect of IHH signaling. Asa consequence the proliferative zone of chondrocytes isenlarged in the infected skeletal elements in a non-cellautonomous manner due to the combination of PTHrP-independent and -dependent activities of IHH, acting on thenon-infected pool of chondrocytes. The two effects, increasedproliferation and higher PTHrP levels, which keep cells withinthe proliferative pool, have been previously shown to delaychondrocyte maturation (Vortkamp et al., 1996; Weir et al.,1996). Therefore, those activities are eventually responsible forthe observed non-cell autonomous delay in the maturation ofhypertrophic, ColX expressing chondrocytes in the early (day7.5) daCaMKII infected skeletal elements. In contrast, down-regulation of Cyclin A and D1, c-Jun, and c-Fos, as well asthe up-regulation of ColX, Ihh and Ppr observed in vivo andin vitro are cell-autonomous effects of activated CaMKIIsignaling.
Surprisingly, in vitro Pthrp expression was down-regulatedin the infected chondrocytes unlike in ovo where Pthrpexpression in articular chondrocytes was increased. This canbe explained by the differences in chondrocyte populations invitro versus in ovo. In the sternum, but also the long bonesPthrp is expressed in proliferating chondrocytes as well andbecomes down-regulated in maturated chondrocytes (Pateder etal., 2001, 2000). Thus, the down-regulation of Pthrp expressionin the cultures most likely reflects that these cells haveundergone maturation.
CaMKII signaling acts down-stream of PPR controllingchondrocyte maturation
As mentioned earlier CaMKII can be activated in response todifferent events, including signaling pathways mediatedthrough heterotrimeric G-protein coupled receptors or recep-tor-tyrosine-kinases, which activate different PLC isoforms(Schlessinger, 2004). Among the signaling pathways that leadto PLC activation and possibly CaMKII are the non-canonicalWnt-, the PTHrP/PPR- and Fgf-signaling pathway, all of whichhave been implicated in regulating chondrocyte maturation(Kronenberg, 2006; Ornitz and Marie, 2002; Yang et al., 2003).Activation of non-canonical Wnts, such as Wnt5a and Wnt5b,
delayed chondrocyte maturation in chick and mouse and did notresult in a down-regulation of CyclinD1 expression (Church etal., 2002; Hartmann and Tabin, 2000; Yang et al., 2003). Fgf-signaling has been shown to induce CyclinD1 expression and toinhibit chondrocyte proliferation (Krejci et al., 2004; Zhang etal., 2004). Furthermore, it has been suggested that Fgf-signalingpromotes chondrocyte maturation in part by down-regulation ofIhh expression (Dailey et al., 2003; Minina et al., 2002).However, this is controversial, since there is evidence that Fgf-signaling positively regulates Ihh expression during earlyskeletal development (Hung et al., 2007; Spater et al., 2006).Based on our results we would rule out that CaMKII is actingdownstream of non-canonical Ca2+/Wnt-signaling that delayschondrocyte maturation. However, CaMKII might still beacting downstream of Fgfs mediating their positive effect onchondrocyte maturation, but it is generally thought that Fgfsexert their activities on chondrocytes via the MAPK-pathwayand Stat1 activation (Murakami et al., 2004; Raucci et al., 2004;Sahni et al., 2001). This clearly needs to be investigated furtherin the future, particularly, because Stat1 has been reported beinga CaMKII substrate (Nair et al., 2002).
Ectopic CaMKII activation led to an up-regulation of Pprexpression in vivo and in vitro (Fig. 6, and data not shown).Based on this observation we exclude the possibility thatectopic maturation is due to transcriptional down-regulation ofPpr levels in the infected chondrocytes, like it has beenobserved in some PTH-responsive cell-lines (Fukayama et al.,1994). Instead we favor the idea that CaMKII activity isintracellularly interfering with the proliferation promotingactivity of PPR signaling and that it is acting downstream ofthe PPR-Gq/11 signaling branch. This branch is thought tostimulate chondrocytes maturation and to oppose the prolif-erative branch acting via PKA (Guo et al., 2002). PPR hasbeen shown to activate via PKA and the cAMP-responseelement binding protein (CREB) a number of genes, includingCyclinA and CyclinD1 (Beier et al., 2001; Beier and LuValle,2002; Ionescu et al., 2001). Mice mutant for CyclinD1 aregrowth retarded (Fantl et al., 1995; Sicinski et al., 1995). PPRsignaling also leads to an increase in c-Jun and c-Fosexpression (Kameda et al., 1997; McCauley et al., 1997),which have been shown to inhibit chondrocyte maturation(Kameda et al., 1997; Thomas et al., 2000; Watanabe et al.,1997). Therefore, it is likely that the down-regulation of thecyclins and AP1 family members is a necessary prerequisitefor chondrocyte maturation.
Active CaMKII can exert its effect on chondrocyte maturationthrough a variety of mechanisms
There are numerous possibilities how CaMKII activity couldinterfere with PPR/PKAmediated gene regulation. Downstreamof PPR, CREB transcriptional activity is supposedly stimulatedby phosphorylation of Ser133 mediated by PKA (Gonzalez andMontminy, 1989). Interestingly, it has been shown that CaMKIIcan also phosphorylate CREB in vitro (Sheng et al., 1991). Thephenotype observed in chondrocytes, however, suggests an in-activation of CREB activity rather than stimulation. Peculiarly,

143M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
CaMKIIα can attenuate gene transcription by phosphorylatingCREB at Ser133 and Ser142, thereby preventing its dimeriza-tion with the CREB binding protein (CBP) and subsequent geneactivation (Wu and McMurray, 2001). However, CREB levelsand CREB-pSer133 levels were not altered in extracts fromdaCaMKII-infected chondrocytes compared to controls and wecould not detect CREB-Ser142 phosphorylation by Westernblot or immunohistochemistry using a phospho-Ser142 specificantibody (data not shown).
Not only CREB, but also the activating transcription factor 2(ATF2) and the retinoblastoma susceptibility gene product/Sp1signaling pathway control the expression of Cyclins (A andD1), and c-Fos and CyclinD1, respectively (Beier et al., 1999,2000; Sohm et al., 1999). ATF2, like ATF1 or CREB, could be apotential target for phosphorylation by CaMKII, leading to theinhibition of its transcriptional activity (Sun et al., 1994, 1996).Disruption of ATF2 activity, but also overexpression of adominant-negative form of CREB (A-CREB) in mice result inchondrodysplasia, showing that members of this transcriptionfactor family are involved in chondrocyte differentiation (Beieret al., 2000, 2001; Long et al., 2001; Reimold et al., 1996).However, unlike the loss of PPR (Chung et al., 1998), neitherloss of ATF2 nor overexpression of A-CREB resulted inpremature differentiation of chondrocytes, suggesting thatCaMKII activation and PPR-signaling must have additionaleffects.
Furthermore, CaMKII has been shown to inhibit Rb/Sp1transcriptional activity through direct phosphorylation of Rband Sp1, leading to increased degradation and inhibiting DNAbinding activity, respectively, thereby affecting the transcriptionof c-Fos and CyclinD1 (Sohm et al., 1999).
The cAMP-phosphodiesterase (PDE) has also been shown tobe a substrate of CaMKII in the central nervous system(Yoshimura et al., 2000). Phosphorylation of cAMP-phospho-diesterase can increase the activity of PDE (Macphee et al.,1988; Shaulsky et al., 1998) and this could lead eventually to adown-regulation of PKA activity. Inhibition of PKA activity hasbeen shown to lead to up-regulation of chondrocyte maturationmarkers such as Runx2 and subsequent activation of Runx2target genes such as Ihh and ColX (Li et al., 2004; Takeda et al.,2001; Yoshida et al., 2004; Zheng et al., 2003). Active CaMKIIsignaling could further stimulate hypertrophy by increasingeither Runx2 or Mef2c activity through inhibition of their co-repressor HDAC4 by phosphorylation (Arnold et al., 2007;Linseman et al., 2003; Little et al., 2007; Vega et al., 2004).Inhibition of HDAC4 could be directly through HDAC4phosphorylation or indirectly through 14-3-3 phosphorylationby CaMKII (Ellis et al., 2003; Little et al., 2007). The exactmechanism how activated CaMKII signaling promotes chon-drocyte maturation will be subject of future investigations.
Unfortunately, our study did not allow us to determine whichCaMKII-isoform(s) are the relevant ones. Although our datasuggest that activation of CaMKII signaling has multiplepotential targets and differential phenotypic effects in chon-drocytes, the overexpression of the activated CaMKIIα-isoformcould potentially not only lead to a change in total CaMKIIactivity, but might affect the subcellular activity of different
CaMKII holoenzyme complexes within the cell, which mighthave different targets (Hudmon and Schulman, 2002). Despitethese potential limitations, our gain- and loss of functionoverexpression data suggest that CaMKII signaling is involvedin chondrocyte differentiation.
Acknowledgments
We thank Michael Kühl and Randy Moon for the mutatedCaMKII clone, Jenna Galloway for help with cloning, AnnetteNeubüser for the RCAS-GFP virus, Günther Schütz for anti-p-S142-CREB and Tom Soderling for the Gfp-CaM-K2N con-struct. ESTs were received from the UK chicken EST depositoryat HGMP-RC. We thank Latifa Bakiri and Erwin Wagner forcritical comments. The IMP is supported by BoehringerIngelheim. This work was supported in part by the EU networkof Excellence: Cells into Organs (LSHM-CT-2003-504468).
References
Abou-Samra, A.B., Juppner, H., Force, T., Freeman, M.W., Kong, X.F.,Schipani, E., Urena, P., Richards, J., Bonventre, J.V., Potts Jr., J.T., et al.,1992. Expression cloning of a common receptor for parathyroid hormoneand parathyroid hormone-related peptide from rat osteoblast-like cells: asingle receptor stimulates intracellular accumulation of both cAMP andinositol trisphosphates and increases intracellular free calcium. Proc. Natl.Acad. Sci. U. S. A. 89, 2732–2736.
Arnold, M.A., Kim, Y., Czubryt, M.P., Phan, D., McAnally, J., Qi, X., Shelton,J.M., Richardson, J.A., Bassel-Duby, R., Olson, E.N., 2007. MEF2Ctranscription factor controls chondrocyte hypertrophy and bone develop-ment. Dev. Cell. 12, 377–389.
Bastepe, M., Weinstein, L.S., Ogata, N., Kawaguchi, H., Juppner, H.,Kronenberg, H.M., Chung, U.I., 2004. Stimulatory G protein directlyregulates hypertrophic differentiation of growth plate cartilage in vivo. Proc.Natl. Acad. Sci. U. S. A. 101, 14794–14799.
Beier, F., LuValle, P., 2002. The cyclin D1 and cyclin A genes are targets ofactivated PTH/PTHrP receptors in Jansen's metaphyseal chondrodysplasia.Mol. Endocrinol. 16, 2163–2173.
Beier, F., Lee, R.J., Taylor, A.C., Pestell, R.G., LuValle, P., 1999. Identificationof the cyclin D1 gene as a target of activating transcription factor 2 inchondrocytes. Proc. Natl. Acad. Sci. U. S. A. 96, 1433–1438.
Beier, F., Taylor, A.C., LuValle, P., 2000. Activating transcription factor 2 isnecessary for maximal activity and serum induction of the cyclin A promoterin chondrocytes. J. Biol. Chem. 275, 12948–12953.
Beier, F., Ali, Z., Mok, D., Taylor, A.C., Leask, T., Albanese, C., Pestell, R.G.,LuValle, P., 2001. TGFbeta and PTHrP control chondrocyte proliferation byactivating cyclin D1 expression. Mol. Biol. Cell 12, 3852–3863.
Braun, A.P., Schulman, H., 1995. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol. 57,417–445.
Cammarota, M., Bevilaqua, L.R., Viola, H., Kerr, D.S., Reichmann, B.,Teixeira, V., Bulla, M., Izquierdo, I., Medina, J.H., 2002. Participation ofCaMKII in neuronal plasticity and memory formation. Cell. Mol. Neurobiol.22, 259–267.
Chang, B.H., Mukherji, S., Soderling, T.R., 1998. Characterization of a calmodulinkinase II inhibitor protein in brain. Proc. Natl. Acad. Sci. U. S. A. 95,10890–10895.
Chang, B.H., Mukherji, S., Soderling, T.R., 2001. Calcium/calmodulin-dependent protein kinase II inhibitor protein: localization of isoforms inrat brain. Neuroscience 102, 767–777.
Chung, U.I., Lanske, B., Lee, K., Li, E., Kronenberg, H., 1998. The parathyroidhormone/parathyroid hormone-related peptide receptor coordinates endo-chondral bone development by directly controlling chondrocyte differentia-tion. Proc. Natl. Acad. Sci. U. S. A. 95, 13030–13035.

144 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
Chung, U.I., Schipani, E., McMahon, A.P., Kronenberg, H.M., 2001. Indianhedgehog couples chondrogenesis to osteogenesis in endochondral bonedevelopment. J. Clin. Invest. 107, 295–304.
Church, V., Nohno, T., Linker, C., Marcelle, C., Francis-West, P., 2002. Wntregulation of chondrocyte differentiation. J. Cell. Sci. 115, 4809–4818.
Colbran, R.J., 2004. Targeting of calcium/calmodulin-dependent protein kinaseII. Biochem. J. 378, 1–16.
Colvin, J.S., Bohne, B.A., Harding, G.W., McEwen, D.G., Ornitz, D.M., 1996.Skeletal overgrowth and deafness in mice lacking fibroblast growth factorreceptor 3. Nat. Genet. 12, 390–397.
Dailey, L., Laplantine, E., Priore, R., Basilico, C., 2003. A network oftranscriptional and signaling events is activated by FGF to inducechondrocyte growth arrest and differentiation. J. Cell. Biol. 161, 1053–1066.
Elgersma, Y., Sweatt, J.D., Giese, K.P., 2004. Mouse genetic approaches toinvestigating calcium/calmodulin-dependent protein kinase II function inplasticity and cognition. J. Neurosci. 24, 8410–8415.
Ellis, J.J., Valencia, T.G., Zeng, H., Roberts, L.D., Deaton, R.A., Grant, S.R.,2003. CaMkinase IIdeltaC phosphorylation of 14-3-3beta in vascular smoothmuscle cells: activation of class II HDAC repression. Mol. Cell. Biochem.242, 153–161.
Fantl, V., Stamp, G., Andrews, A., Rosewell, I., Dickson, C., 1995. Mice lackingcyclin D1 are small and show defects in eye and mammary glanddevelopment. Genes Dev. 9, 2364–2372.
Fox, K., 2003. Synaptic plasticity: the subcellular location of CaMKII controlsplasticity. Curr. Biol. 13, R143–R145.
Fukayama, S., Schipani, E., Juppner, H., Lanske, B., Kronenberg, H.M., Abou-Samra, A.B., Bringhurst, F.R., 1994. Role of protein kinase-A inhomologous down-regulation of parathyroid hormone (PTH)/PTH-relatedpeptide receptor messenger ribonucleic acid in human osteoblast-like SaOS-2 cells. Endocrinology 134, 1851–1858.
Gerstenfeld, L.C., Kelly, C.M., Von Deck, M., Lian, J.B., 1990. Comparativemorphological and biochemical analysis of hypertrophic, non-hypertrophicand 1,25(OH)2D3 treated non-hypertrophic chondrocytes. Connect. TissueRes. 24, 29–39.
Goff, D.J., Tabin, C.J., 1997. Analysis of Hoxd-13 and Hoxd-11 misexpressionin chick limb buds reveals that Hox genes affect both bone condensation andgrowth. Development 124, 627–636.
Gonzalez, G.A., Montminy, M.R., 1989. Cyclic AMP stimulates somatostatingene transcription by phosphorylation of CREB at serine 133. Cell 59,675–680.
Guo, J., Chung, U.I., Kondo, H., Bringhurst, F.R., Kronenberg, H.M., 2002. ThePTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo withoutactivating phospholipase C. Dev. Cell. 3, 183–194.
Hanson, P.I., Meyer, T., Stryer, L., Schulman, H., 1994. Dual role of calmodulinin autophosphorylation of multifunctional CaM kinase may underliedecoding of calcium signals. Neuron 12, 943–956.
Hartmann, C., Tabin, C.J., 2000. Dual roles of Wnt signaling duringchondrogenesis in the chicken limb. Development 127, 3141–3159.
Hudmon, A., Schulman, H., 2002. Structure–function of the multifunctionalCa2+/calmodulin-dependent protein kinase II. Biochem. J. 364, 593–611.
Hung, I.H., Yu, K., Lavine, K.J., Ornitz, D.M., 2007. FGF9 regulates earlyhypertrophic chondrocyte differentiation and skeletal vascularization in thedeveloping stylopod. Dev. Biol. 307, 300–313.
Ionescu, A.M., Schwarz, E.M., Vinson, C., Puzas, J.E., Rosier, R., Reynolds,P.R., O'Keefe, R.J., 2001. PTHrP modulates chondrocyte differentiationthrough AP-1 and CREB signaling. J. Biol. Chem. 276, 11639–11647.
Jaubert, J., Jaubert, F., Martin, N., Washburn, L.L., Lee, B.K., Eicher, E.M.,Guenet, J.L., 1999. Three new allelic mouse mutations that cause skeletalovergrowth involve the natriuretic peptide receptor C gene (Npr3). Proc.Natl. Acad. Sci. U. S. A. 96, 10278–10283.
Juppner, H., Abou-Samra, A.B., Freeman, M., Kong, X.F., Schipani, E.,Richards, J., Kolakowski Jr., L.F., Hock, J., Potts Jr., J.T., Kronenberg, H.M.,et al., 1991. A G protein-linked receptor for parathyroid hormone andparathyroid hormone-related peptide. Science 254, 1024–1026.
Kameda, T., Watanabe, H., Iba, H., 1997. C-Jun and JunD suppress maturationof chondrocytes. Cell Growth Differ. 8, 495–503.
Karp, S.J., Schipani, E., St-Jacques, B., Hunzelman, J., Kronenberg, H.,McMahon, A.P., 2000. Indian hedgehog coordinates endochondral bone
growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development 127, 543–548.
Karsenty, G., Wagner, E.F., 2002. Reaching a genetic and molecularunderstanding of skeletal development. Dev. Cell. 2, 389–406.
Koyama, E., Golden, E.B., Kirsch, T., Adams, S.L., Chandraratna, R.A.,Michaille, J.J., Pacifici, M., 1999. Retinoid signaling is required forchondrocyte maturation and endochondral bone formation during limbskeletogenesis. Dev. Biol. 208, 375–391.
Krejci, P., Bryja, V., Pachernik, J., Hampl, A., Pogue, R., Mekikian, P., Wilcox,W.R., 2004. FGF2 inhibits proliferation and alters the cartilage-likephenotype of RCS cells. Exp. Cell Res. 297, 152–164.
Kronenberg, H.M., 2003. Developmental regulation of the growth plate. Nature423, 332–336.
Kronenberg, H.M., 2006. PTHrP and skeletal development. Ann. N.Y. Acad.Sci. 1068, 1–13.
Kuhl, M., Sheldahl, L.C., Park, M., Miller, J.R., Moon, R.T., 2000. The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape. TrendsGenet. 16, 279–283.
Lanske, B., Karaplis, A.C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., Karperien,M., Defize, L.H., Ho, C., Mulligan, R.C., Abou-Samra, A.B., Juppner, H.,Segre, G.V., Kronenberg, H.M., 1996. PTH/PTHrP receptor in earlydevelopment and Indian hedgehog-regulated bone growth. Science 273,663–666.
Lanske, B., Amling, M., Neff, L., Guiducci, J., Baron, R., Kronenberg, H.M.,1999. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leadsto distinct abnormalities in bone development. J. Clin. Invest. 104,399–407.
Lantsman, K., Tombes, R.M., 2005. CaMK-II oligomerization potentialdetermined using CFP/YFP FRET. Biochim. Biophys. Acta 1746, 45–54.
Li, T.F., Dong, Y., Ionescu, A.M., Rosier, R.N., Zuscik, M.J., Schwarz, E.M.,O'Keefe, R.J., Drissi, H., 2004. Parathyroid hormone-related peptide(PTHrP) inhibits Runx2 expression through the PKA signaling pathway.Exp. Cell Res. 299, 128–136.
Linseman, D.A., Bartley, C.M., Le, S.S., Laessig, T.A., Bouchard, R.J.,Meintzer, M.K., Li, M., Heidenreich, K.A., 2003. Inactivation of themyocyte enhancer factor-2 repressor histone deacetylase-5 by endo-genous Ca(2+)//calmodulin-dependent kinase II promotes depolariza-tion-mediated cerebellar granule neuron survival. J. Biol. Chem. 278,41472–41481.
Little, G.H., Bai, Y., Williams, T., Poizat, C., 2007. Nuclear calcium/calmodulin-dependent protein kinase II delta preferentially transmits signals to histonedeacetylase 4 in cardiac cells. J. Biol. Chem. 282, 7219–7231.
Logan, M., Tabin, C., 1998. Targeted gene misexpression in chick limb budsusing avian replication-competent retroviruses. Methods 14, 407–420.
Long, F., Zhang, X.M., Karp, S., Yang, Y., McMahon, A.P., 2001. Geneticmanipulation of hedgehog signaling in the endochondral skeleton reveals adirect role in the regulation of chondrocyte proliferation. Development 128,5099–5108.
Macphee, C.H., Reifsnyder, D.H., Moore, T.A., Lerea, K.M., Beavo, J.A., 1988.Phosphorylation results in activation of a cAMP phosphodiesterase inhuman platelets. J. Biol. Chem. 263, 10353–10358.
Mathews, L.S., Hammer, R.E., Behringer, R.R., D'Ercole, A.J., Bell, G.I.,Brinster, R.L., Palmiter, R.D., 1988. Growth enhancement of transgenicmice expressing human insulin-like growth factor I. Endocrinology 123,2827–2833.
McCauley, L.K., Koh, A.J., Beecher, C.A., Rosol, T.J., 1997. Proto-oncogene c-fos is transcriptionally regulated by parathyroid hormone (PTH) and PTH-related protein in a cyclic adenosine monophosphate-dependent manner inosteoblastic cells. Endocrinology 138, 5427–5433.
Minina, E., Kreschel, C., Naski, M.C., Ornitz, D.M., Vortkamp, A., 2002.Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyteproliferation and hypertrophic differentiation. Dev. Cell. 3, 439–449.
Morgan, B.A., Fekete, D.M., 1996. Manipulating gene expression withreplication-competent retroviruses. Methods Cell Biol. 51, 185–218.
Murakami, S., Balmes, G., McKinney, S., Zhang, Z., Givol, D., deCrombrugghe, B., 2004. Constitutive activation of MEK1 in chondrocytescauses Stat1-independent achondroplasia-like dwarfism and rescues theFgfr3-deficient mouse phenotype. Genes Dev. 18, 290–305.

145M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
Murtaugh, L.C., Zeng, L., Chyung, J.H., Lassar, A.B., 2001. The chicktranscriptional repressor Nkx3.2 acts downstream of Shh to promote BMP-dependent axial chondrogenesis. Dev. Cell. 1, 411–422.
Nair, J.S., DaFonseca, C.J., Tjernberg, A., Sun, W., Darnell Jr., J.E., Chait, B.T.,Zhang, J.J., 2002. Requirement of Ca2+ and CaMKII for Stat1 Ser-727phosphorylation in response to IFN-gamma. Proc. Natl. Acad. Sci. U. S. A.99, 5971–5976.
Ornitz, D.M., Marie, P.J., 2002. FGF signaling pathways in endochondral andintramembranous bone development and human genetic disease. Genes Dev.16, 1446–1465.
Pateder, D.B., Rosier, R.N., Schwarz, E.M., Reynolds, P.R., Puzas, J.E.,D'Souza, M., O'Keefe, R.J., 2000. PTHrP expression in chondrocytes,regulation by TGF-beta, and interactions between epiphyseal and growthplate chondrocytes. Exp. Cell Res. 256, 555–562.
Pateder, D.B., Ferguson, C.M., Ionescu, A.M., Schwarz, E.M., Rosier, R.N.,Puzas, J.E., O'Keefe, R.J., 2001. PTHrP expression in chick sternalchondrocytes is regulated by TGF-beta through Smad-mediated signaling.J. Cell. Physiol. 188, 343–351.
Quinn, C.O., Rajakumar, R.A., Agapova, O.A., 2000. Parathyroid hormoneinduces rat interstitial collagenase mRNA through Ets-1 facilitated bycyclic AMP response element-binding protein and Ca(2+)/calmodulin-dependent protein kinase II in osteoblastic cells. J. Mol. Endocrinol. 25,73–84.
Raucci, A., Laplantine, E., Mansukhani, A., Basilico, C., 2004. Activation of theERK1/2 and p38 mitogen-activated protein kinase pathways mediatesfibroblast growth factor-induced growth arrest of chondrocytes. J. Biol.Chem. 279, 1747–1756.
Reimold, A.M., Grusby, M.J., Kosaras, B., Fries, J.W., Mori, R., Maniwa, S.,Clauss, I.M., Collins, T., Sidman, R.L., Glimcher, M.J., Glimcher, L.H.,1996. Chondrodysplasia and neurological abnormalities in ATF-2-deficientmice. Nature 379, 262–265.
Rich, R.C., Schulman, H., 1998. Substrate-directed function of calmodulinin autophosphorylation of Ca2+/calmodulin-dependent protein kinase II.J. Biol. Chem. 273, 28424–28429.
Rongo, C., 2002. A fresh look at the role of CaMKII in hippocampal synapticplasticity and memory. Bioessays 24, 223–233.
Rosenberg, O.S., Deindl, S., Comolli, L.R., Hoelz, A., Downing, K.H., Nairn,A.C., Kuriyan, J., 2006. Oligomerization states of the association domainand the holoenyzme of Ca2+/CaM kinase II. FEBS J. 273, 682–694.
Sahni, M., Raz, R., Coffin, J.D., Levy, D., Basilico, C., 2001. STAT1 mediatesthe increased apoptosis and reduced chondrocyte proliferation in miceoverexpressing FGF2. Development 128, 2119–2129.
Schipani, E., Lanske, B., Hunzelman, J., Luz, A., Kovacs, C.S., Lee, K., Pirro,A., Kronenberg, H.M., Juppner, H., 1997. Targeted expression ofconstitutively active receptors for parathyroid hormone and parathyroidhormone-related peptide delays endochondral bone formation and rescuesmice that lack parathyroid hormone-related peptide. Proc. Natl. Acad. Sci.U. S. A. 94, 13689–13694.
Schlessinger, J., 2004. Common and distinct elements in cellular signaling viaEGF and FGF receptors. Science 306, 1506–1507.
Seales, E.C., Micoli, K.J., McDonald, J.M., 2006. Calmodulin is a criticalregulator of osteoclastic differentiation, function, and survival. J. Cell.Biochem. 97, 45–55.
Shaulsky, G., Fuller, D., Loomis, W.F., 1998. A cAMP-phosphodiesterasecontrols PKA-dependent differentiation. Development 125, 691–699.
Sheng, M., Thompson, M.A., Greenberg, M.E., 1991. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependentkinases. Science 252, 1427–1430.
Shimazaki, A., Wright, M.O., Elliot, K., Salter, D.M., Millward-Sadler, S.J.,2006. Calcium/calmodulin-dependent protein kinase II in human articularchondrocytes. Biorheology 43, 223–233.
Sicinski, P., Donaher, J.L., Parker, S.B., Li, T., Fazeli, A., Gardner, H., Haslam,S.Z., Bronson, R.T., Elledge, S.J., Weinberg, R.A., 1995. Cyclin D1provides a link between development and oncogenesis in the retina andbreast. Cell 82, 621–630.
Soderling, T.R., Chang, B., Brickey, D., 2001. Cellular signaling throughmultifunctional Ca2+/calmodulin-dependent protein kinase II. J. Biol.Chem. 276, 3719–3722.
Sohm, F., Gaiddon, C., Antoine, M., Boutillier, A.L., Loeffler, J.P., 1999. Theretinoblastoma susceptibility gene product/Sp1 signalling pathway ismodulated by Ca2+/calmodulin kinases II and IV activity. Oncogene 18,2762–2769.
Spater, D., Hill, T.P., O'Sullivan, R.J., Gruber, M., Conner, D.A., Hartmann, C.,2006. Wnt9a signaling is required for joint integrity and regulation of Ihhduring chondrogenesis. Development 133, 3039–3049.
St-Jacques, B., Hammerschmidt, M., McMahon, A.P., 1999. Indian hedgehogsignaling regulates proliferation and differentiation of chondrocytes and isessential for bone formation. Genes Dev. 13, 2072–2086.
Sun, P., Enslen, H., Myung, P.S., Maurer, R.A., 1994. Differential activation ofCREB by Ca2+/calmodulin-dependent protein kinases type II and type IVinvolves phosphorylation of a site that negatively regulates activity. GenesDev. 8, 2527–2539.
Sun, P., Lou, L., Maurer, R.A., 1996. Regulation of activating transcriptionfactor-1 and the cAMP response element-binding protein by Ca2+/calmo-dulin-dependent protein kinases type I, II, and IV. J. Biol. Chem. 271,3066–3073.
Szuts, V., Mollers, U., Bittner, K., Schurmann, G., Muratoglu, S., Deak, F., Kiss,I., Bruckner, P., 1998. Terminal differentiation of chondrocytes is arrested atdistinct stages identified by their expression repertoire of marker genes.Matrix Biol. 17, 435–448.
Takeda, S., Bonnamy, J.P., Owen, M.J., Ducy, P., Karsenty, G., 2001.Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncoversits ability to induce hypertrophic chondrocyte differentiation and partiallyrescues Cbfa1-deficient mice. Genes Dev. 15, 467–481.
Thomas, D.P., Sunters, A., Gentry, A., Grigoriadis, A.E., 2000. Inhibition ofchondrocyte differentiation in vitro by constitutive and inducible over-expression of the c-fos proto-oncogene. J. Cell. Sci. 113 (Pt. 3), 439–450.
Tylzanowski, P., De Valck, D., Maes, V., Peeters, J., Luyten, F.P., 2003. Zfhx1aand Zfhx1b mRNAs have non-overlapping expression domains during chickand mouse midgestation limb development. Gene Expr. Patterns 3, 39–42.
Valhmu, W.B., Raia, F.J., 2002. myo-Inositol 1,4,5-trisphosphate and Ca2+/calmodulin-dependent factors mediate transduction of compression-inducedsignals in bovine articular chondrocytes. Biochem. J. 361, 689–696.
Vega, R.B., Matsuda, K., Oh, J., Barbosa, A.C., Yang, X., Meadows, E.,McAnally, J., Pomajzl, C., Shelton, J.M., Richardson, J.A., Karsenty, G.,Olson, E.N., 2004. Histone deacetylase 4 controls chondrocyte hypertrophyduring skeletogenesis. Cell 119, 555–566.
Vortkamp, A., Lee, K., Lanske, B., Segre, G.V., Kronenberg, H.M., Tabin, C.J.,1996. Regulation of rate of cartilage differentiation by Indian hedgehog andPTH-related protein. Science 273, 613–622.
Watanabe, H., Saitoh, K., Kameda, T., Murakami, M., Niikura, Y., Okazaki, S.,Morishita, Y., Mori, S., Yokouchi, Y., Kuroiwa, A., Iba, H., 1997.Chondrocytes as a specific target of ectopic Fos expression in earlydevelopment. Proc. Natl. Acad. Sci. U. S. A. 94, 3994–3999.
Weir, E.C., Philbrick, W.M., Amling, M., Neff, L.A., Baron, R., Broadus, A.E.,1996. Targeted overexpression of parathyroid hormone-related peptide inchondrocytes causes chondrodysplasia and delayed endochondral boneformation. Proc. Natl. Acad. Sci. U. S. A. 93, 10240–10245.
Wu, X., McMurray, C.T., 2001. Calmodulin kinase II attenuation of genetranscription by preventing cAMP response element-binding protein(CREB) dimerization and binding of the CREB-binding protein. J. Biol.Chem. 276, 1735–1741.
Yang, Y., Topol, L., Lee, H., Wu, J., 2003. Wnt5a and Wnt5b exhibit distinctactivities in coordinating chondrocyte proliferation and differentiation.Development 130, 1003–1015.
Yasoda, A., Komatsu, Y., Chusho, H., Miyazawa, T., Ozasa, A., Miura, M.,Kurihara, T., Rogi, T., Tanaka, S., Suda, M., Tamura, N., Ogawa, Y., Nakao,K., 2004. Overexpression of CNP in chondrocytes rescues achondroplasiathrough a MAPK-dependent pathway. Nat. Med. 10, 80–86.
Yoshida, C.A., Yamamoto, H., Fujita, T., Furuichi, T., Ito, K., Inoue, K.,Yamana, K., Zanma, A., Takada, K., Ito, Y., Komori, T., 2004. Runx2 andRunx3 are essential for chondrocyte maturation, and Runx2 regulates limbgrowth through induction of Indian hedgehog. Genes Dev. 18, 952–963.
Yoshimura, Y., Aoi, C., Yamauchi, T., 2000. Investigation of protein substratesof Ca(2+)/calmodulin-dependent protein kinase II translocated to thepostsynaptic density. Brain Res. Mol. Brain Res. 81, 118–128.

146 M.J. Taschner et al. / Developmental Biology 317 (2008) 132–146
Zayzafoon, M., 2006. Calcium/calmodulin signaling controls osteoblast growthand differentiation. J. Cell. Biochem. 97, 56–70.
Zhang, Y., Lin, Y., Bowles, C., Wang, F., 2004. Direct cell cycle regulation bythe fibroblast growth factor receptor (FGFR) kinase through phosphoryla-tion-dependent release of Cks1 from FGFR substrate 2. J. Biol. Chem. 279,55348–55354.
Zhang, L., McKenna, M.A., Said-Al-Naief, N., Wu, X., Feng, X., McDonald,J.M., 2005. Osteoclastogenesis: the role of calcium and calmodulin. Crit.Rev. Eukaryot. Gene Expr. 15, 1–13.
Zheng, Q., Zhou, G., Morello, R., Chen, Y., Garcia-Rojas, X., Lee, B., 2003. TypeX collagen gene regulation by Runx2 contributes directly to its hypertrophicchondrocyte-specific expression in vivo. J. Cell. Biol. 162, 833–842.