april 2006 xosé mª fernández european bioinformatics institute ensembl sep 2006
Post on 20-Dec-2015
215 views
TRANSCRIPT

April 2006
XosXosé Mª Fernándezé Mª FernándezEuropean Bioinformatics InstituteEuropean Bioinformatics Institute
EnsemblEnsembl
Sep 2006Sep 2006

2 of 68
• Overview of Ensembl• Making genomes useful• Beyond Ensembl
Outline of talkOutline of talk

3 of 68
• Overview of Ensembl– The era of sequencing genomes– Exploring genomes– Gene annotation
• Making genomes useful• Beyond Ensembl
Outline of talkOutline of talk

4 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases
and APIs)

5 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases and
APIs)

6 of 68
Beyond classical Beyond classical ab initioab initio gene predictiongene prediction
• Ensembl automatic gene prediction relies on homology ‘supporting evidence’ to avoid overprediction.
• Classical ab initio gene prediction (eg GENSCAN) relies partly on global statistics of protein coding potentials, not used in the cell
• Genes are just a series of short signals– Transcription start site– Translation start site– 5’ & 3’ Intron splicing signals– Termination signals
• Short signal sequences difficult to recognise over background noise in large genomes

7 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases and
APIs)

8 of 68
Species in Ensembl v40Species in Ensembl v40

9 of 68
The era of sequencing genomesThe era of sequencing genomes
360
450
990 25
70
140
?
550
25070?
1002003004005001000
Million years
340
1500?
?
Chordata
Vertebrata
AmniotaTetrapoda
Teleostei
Urochordata
Arthropoda
NematodaFungi
Red : whole genome assembly availableGreen : whole genome assembly due within the next year in Ensembl
* 19 species currently in Ensembl* 19 species currently in Ensembl+ 10 + 10 Pre! Pre! EnsemblEnsembl
S. cerevisiae (baker’s yeast) *
C. elegans (nematode) *
A. mellifera (honey bee) *
D. rerio (zebrafish) *
D. melanogaster (fruitfly) *A. gambiae (African malaria mosquito) *A. aegypti (yellow fever mosquito) +
C. intestinalis (transparent sea squirt) * C. savignyi (sea squirt) +
T. rubripes (torafugu) *T. nigroviridis (spotted green pufferfish) *
O. latipes (Japanese medaka)
G. aculeatus (Stickleback) +
23
O. aries (sheep)
G. gallus (chicken) *
X. laevis (African clawed frog)
M. musculus (house mouse) *R. norvegicus (Norway rat) *
M. mulatta (rhesus macaque) *P. troglodytes (chimpanzee) *
C. familiaris (dog) *F. catus (cat)E. caballus (horse)S. scrofa (pig)B. taurus (cow) *
310
197
92
M. domestica (opossum) *
170
L. africana (elephant) +
105
41
91
4574
83
65
20
H. sapiens (human) * +
X. tropicalis (western clawed frog) *Amphibia
AvesMetatheria
Mammalia
Eutheria

10 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases and
APIs)

11 of 68
http://das.sanger.ac.uk/registryhttp://das.sanger.ac.uk/registry
DAS DAS RegistryRegistry

12 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.orghttp://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases and
APIs)

13 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases and
APIs)

14 of 68
• Object model– standard interface makes it easy for others to build
custom applications on top of Ensembl data
• Open discussion of design ([email protected])• Most major pharma and many academics represented
on mailing list and code is being actively developed externally
• Ensembl locally– Both industry & academia
Open source open Open source open standardsstandards
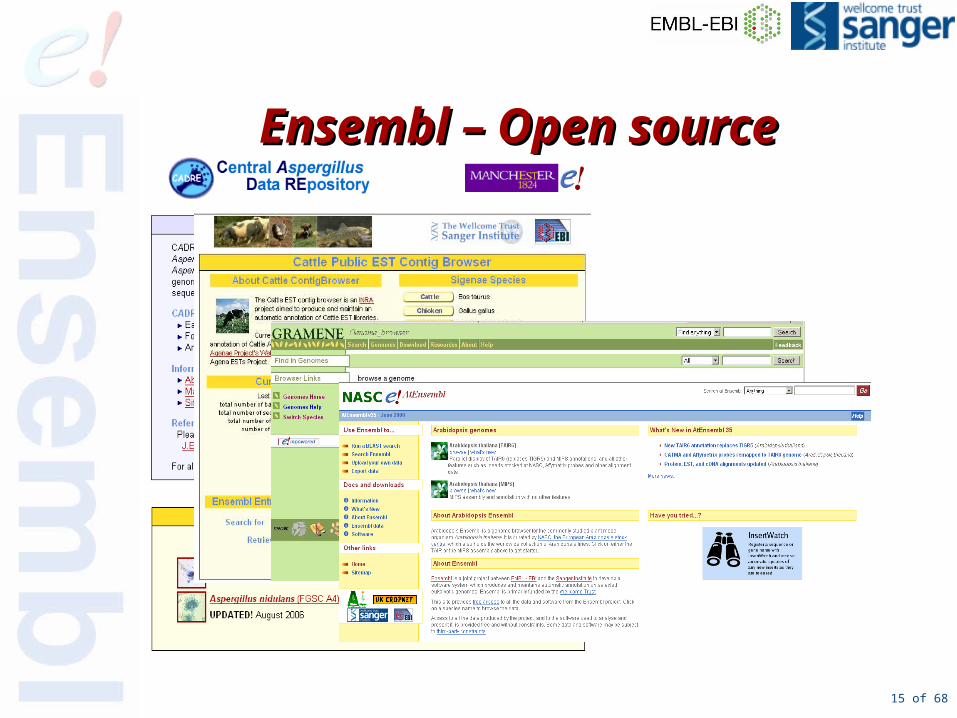
15 of 68
Ensembl – Open sourceEnsembl – Open source

16 of 68
Genome ReviewsGenome Reviews

17 of 68
Ensembl - ProjectEnsembl - Project
• Joint project– EMBL – European Bioinformatics Institute (EBI) – Wellcome Trust Sanger Institute
• Produce accurate, automatic genome annotation• Focused on selected eukaryotic genomes • Integrate external (distributed) biological data• Presentation of the analysis to all via the Web at
http://www.ensembl.org • Open distribution of the analysis the community• Development of open, collaborative software (databases
and APIs)

18 of 68
APIsAPIs• Used to retrieve data from and to store data in
Ensembl databases.• Ensembl Perl API;
– Written in Object-Oriented Perl,
– Foundation for the Ensembl Pipeline and Ensembl Web interface.
• Ensembl Java API;– Written in Java, but similar in layout to the
Perl API,– Foundation for Apollo,– Development will stop in December 2006.

19 of 68
• Overview of Ensembl– The Era of Sequencing Genomes– Exploring genomes– Gene annotation
• Making genomes useful• Beyond Ensembl

20 of 68
ACCCAATAGCAGAACAGCTACTGGAACTAAAATCCTCTGATTTCAAATAACAGCCCCGCCCACTACCACTAAGTGAAGTCATCCACAACCACACACCGACCACTCTAAGCTTTTGTAAGATCGGCTCGCTTTGGGGAACAGGTCTTGAGAGAACATCCCTTTTAAGGTCAGAACAAAGGTATTTCATAGGTCCCAGGTCGTGTCCCGAGGGCGCCCACCCAAACATGAGCTGGAGCAAAAAGAAAGGGATGGGGGACTTGGAGTAGGCATAGGGGCGGCCCCTCCAAGCAGGGTGGCCTGGGACTCTTAAGGGTCAGCGAGAAGAGAACACACACTCCAGCTCCCGCTTTATTCGGTCAGATACTGACGGTTGGGATGCCTGACAAGGAATTTCCTTTCGCCACACTGAGAAATACCCGCAGCGGCCCACCCAGGCCTGACTTCCGGGTGGTGCGTGTGCTGCGTGTCGCGTCACGGCGTCACGTGGCCAGCGCGGGCTTGTGGCGCGAGCTTCTGAAACTAGGCGGCAGAGGCGGAGCCGCTGTGGCACTGCTGCGCCTCTGCTGCGCCTCGGGTGTCTTTTGCGGCGGTGGGTCGCCGCCGGGAGAAGCGTGAGGGGACAGATTTGTGACCGGCGCGGTTTTTGTCAGCTTACTCCGGCCAAAAAAGAACTGCACCTCTGGAGCGGGTTAGTGGTGGTGGTAGTGGGTTGGGACGAGCGCGTCTTCCGCAGTCCCAGTCCAGCGTGGCGGGGGAGCGCCTCACGCCCCGGGTCGCTGCCGCGGCTTCTTGCCCTTTTGTCTCTGCCAACCCCCACCCATGCCTGAGAGAAAGGTCCTTGCCCGAAGGCAGATTTTCGCCAAGCAAATTCGAGCCCCGCCCCTTCCCTGGGTCTCCATTTCCCGCCTCCGGCCCGGCCTTTGGGCTCCGCCTTCAGCTCAAGACTTAACTTCCCTCCCAGCTGTCCCAGATGACGCCATCTGAAATTTCTTGGAAACACGATCACTTTAACGGAATATTGCTGTTTTGGGGAAGTGTTTTACAGCTGCTGGGCACGCTGTATTTGCCTTACTTAAGCCCCTGGTAATTGCTGTATTCCGAAGACATGCTGATGGGAATTACCAGGCGGCGTTGGTCTCTAACTGGAGCCCTCTGTCCCCACTAGCCACGCGTCACTGGTTAGCGTGATTGAAACTAAATCGTATGAAAATCCTCTTCTCTAGTCGCACTAGCCACGTTTCGAGTGCTTAATGTGGCTAGTGGCACCGGTTTGGACAGCACAGCTGTAAAATGTTCCCATCCTCACAGTAAGCTGTTACCGTTCCAGGAGATGGGACTGAATTAGAATTCAAACAAATTTTCCAGCGCTTCTGAGTTTTACCTCAGTCACATAATAAGGAATGCATCCCTGTGTAAGTGCATTTTGGTCTTCTGTTTTGCAGACTTATTTACCAAGCATTGGAGGAATATCGTAGGTAAAAATGCCTATTGGATCCAAAGAGAGGCCAACATTTTTTGAAATTTTTAAGACACGCTGCAACAAAGCAGGTATTGACAAATTTTATATAACTTTATAAATTACACCGAGAAAGTGTTTTCTAAAAAATGCTTGCTAAAAACCCAGTACGTCACAGTGTTGCTTAGAACCATAAACTGTTCCTTATGTGTGTATAAATCCAGTTAACAACATAATCATCGTTTGCAGGTTAACCACATGATAAATATAGAACGTCTAGTGGATAAAGAGGAAACTGGCCCCTTGACTAGCAGTAGGAACAATTACTAACAAATCAGAAGCATTAATGTTACTTTATGGCAGAAGTTGTCCAACTTTTTGGTTTCAGTACTCCTTATACTCTTAAAAATGATCTAGGACCCCCGGAGTGCTTTTGTTTATGTAGCTTACCATATTAGAAATTTAAAACTAAGAATTTAAGGCTGGGCGTGGTGGCTCACGCCTGTAATCCCAGCACTTTGGGAGGCCGAGGTGGGCGGATCACTTGAGGCCAGAAGTTTGAGACCAGCCTGGCCAACATGGTGAAACCCTATCTCTACTAAAAATACAAAAAATGTGCTGCGTGTGGTGGTGCGTGCCTGTAATCCCAGCTACACGGGAGGTGGAGGCAGGAGAATCGCTTGAACCCTGGAGGCAGAGGTTGCAGTGAGCCAAGATCATGCCACTGCACTCTAGCCTGGGCCACATAGCATGACTCTGTCTCAAAACAAACAAACAAACAAAAAACTAAGAATTTAAAGTTAATTTACTTAAAAATAATGAAAGCTAACCCATTGCATATTATCACAACATTCTTAGGAAAAATAACTTTTTGAAAACAAGTGAGTGGAATAGTTTTTACATTTTTGCAGTTCTCTTTAATGTCTGGCTAAATAGAGATAGCTGGATTCACTTATCTGTGTCTAATCTGTTATTTTGGTAGAAGTATGTGAAAAAAAATTAACCTCACGTTGAAAAAAGGAATATTTTAATAGTTTTCAGTTACTTTTTGGTATTTTTCCTTGTACTTTGCATAGATTTTTCAAAGATCTAATAGATATACCATAGGTCTTTCCCATGTCGCAACATCATGCAGTGATTATTTGGAAGATAGTGGTGTTCTGAATTATACAAAGTTTCCAAATATTGATAAATTGCATTAAACTATTTTAAAAATCTCATTCATTAATACCACCATGGATGTCAGAAAAGTCTTTTAAGATTGGGTAGAAATGAGCCACTGGAAATTCTAATTTTCATTTGAAAGTTCACATTTTGTCATTGACAACAAACTGTTTTCCTTGCAGCAACAAGATCACTTCATTGATTTGTGAGAAAATGTCTACCAAATTATTTAAGTTGAAATAACTTTGTCAGCTGTTCTTTCAAGTAAAAATGACTTTTCATTGAAAAAATTGCTTGTTCAGATCACAGCTCAACATGAGTGCTTTTCTAGGCAGTATTGTACTTCAGTATGCAGAAGTGCTTTATGTATGCTTCCTATTTTGTCAGAGATTATTAAAAGAAGTGCTAAAGCATTGAGCTTCGAAATTAATTTTTACTGCTTCATTAGGACATTCTTACATTAAACTGGCATTATTATTACTATTATTTTTAACAAGGACACTCAGTGGTAAGGAATATAATGGCTACTAGTATTAGTTTGGTGCCACTGCCATAACTCATGCAAATGTGCCAGCAGTTTTACCCAGCATCATCTTTGCACTGTTGATACAAATGTCAACATCATGAAAAAGGGAAATGATTCCATAGCGTTATTATGAAAGTAGTTTTGAACTGTAATGGTAGAGGATGAATAGCTCACAATACAAATTTGTCATTTCCCTTTAAGAGAGAATTCCCATTTTATGTGAGAGTCCACATGTTCCTCATACCCATAGTTTGCCACATCTTGAGTACTCTTCAGAATTATTTGAATTTTTTGAATTTTATCTGTGGAATGTATTTTTTTTTTTTTCTTTTTTGAGACACAGTCTTGCT
3500 bps in chr 13…3500 bps in chr 13…
TT
A
T
G
T
C
C
C
C

21 of 68
Exploring genomesExploring genomes
• Browse genes in genomic context• Display features in and around a particular gene• Explore larger chromosome regions• Search and retrieve information on a gene- and
genome-scale• Investigate genome organisation• Compare genomes

22 of 68
We make genomes usefulWe make genomes useful

23 of 68
Making genomes usefulMaking genomes useful
• Interpretation– Where are the interesting parts of the
genome?– What do they do?– How are they related to elements in other
genomes?

24 of 68
EnsemblEnsemblContigView

26 of 68
ContigViewContigView - Close-up
Manualannotationvia Vega
Ensembl predictions
Ensembl EST-based predictions
Chromosomes with manual annotation (http://vega.sanger.ac.uk): 1, 6, 7, 9, 10, 13, 14, 16, 18, 19, 20, 22, X and Y

27 of 68
ContigViewContigView - Navigation
Click and drag mouse to select region

28 of 68
CytoViewCytoView

30 of 68
BioMart - a distributed BioMart - a distributed architecturearchitecture
XML XML XML
MySQL ORACLE PostgreSQL
ANSI SQL
XML
XML
XML
XML
XML
XML

31 of 68
SNPVega
Retrieval
BioMart API
JAVA Perl
MartExplorer MartShell MartView
Schema transformation
MartBuilder MartEditor
Configuration
Databases
Public data (local or remote)
BioMart architectureBioMart architecture
MSD UniProt Ensembl
XML
myMartmyDatabase

32 of 68
Comparing genomesComparing genomes

33 of 68
MultiContigMultiContigViewView

34 of 68
ChallengesChallenges
• What is the right way to calculate evolutionary relationships between these genomes?– How different is the gene build for each
new genome?• Is there novel information to be deduced
from the set of related genomes?• How do we integrate “close” genomes and
genome variation?

35 of 68
DNA/DNA matches web DNA/DNA matches web displaydisplay
ContigView human BRCA2

36 of 68
DotterViewDotterView

37 of 68
Multiple alignmentsMultiple alignments
• Currently 3 sets:– MLAGAN-amniota vertebrates:
– MLAGAN-eutherian mammals:
– MLAGAN primates:
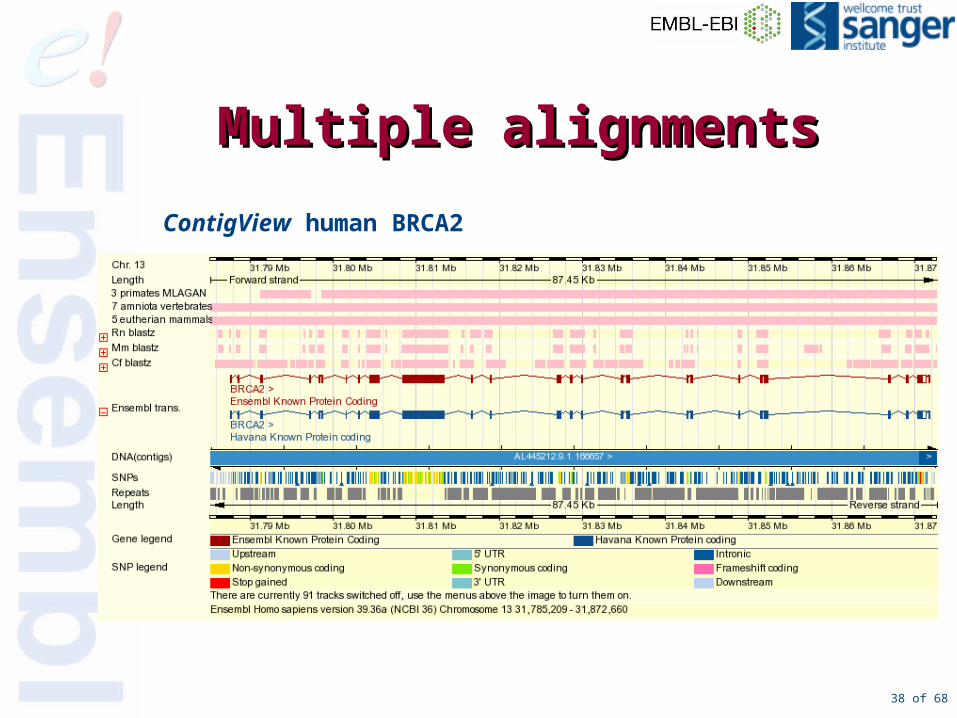
38 of 68
MultipleMultiple alignmentsalignments
ContigView human BRCA2

39 of 68
Multiple alignmentsMultiple alignments GeneSeqalignView human BRCA2 v other mammals
...

40 of 68
GeneGene EvolutionEvolution
• Divergence
• Speciation / Duplication
• Change within allelic population
• Point Mutations / Selection / Drift
• Exon/domain shuffling
• Transposition / Translocation
• Retroposition (reverse transcription)
Orthologues and ParaloguesOrthologues and Paralogues
Reconstruct the Molecular Evolutionary history from the evidence visible within the known extant genes

41 of 68
Para/Orthologue Para/Orthologue predictionspredictions
• Homology system (BRH/RHS)
• Pairwise species analysis
• Conservative / missed predictions
• Family system
• MCL Clustering all species (+UniProt)
• No ranking of relationships within cluster
• Liberal / over predict
∆ Tree based analysis to get best of both worlds

42 of 68
Species treeSpecies tree

43 of 68
Sequence variation in Sequence variation in EnsemblEnsembl

44 of 68
Two types of variation dataTwo types of variation data
Natural• Limitless• Dense markers
required• Need for optimal
experimental design (HapMap)
• Human and Anopheles
Managed• Limited strain number• Light density adequate
for some uses• (dense for complete
dataset)• Mouse, Rat

45 of 68
SNPs in EnsemblMapView: SNP density on chromosome

46 of 68
TransView &ProtView: SNPs in transcript/ protein
SNPs in Ensembl

48 of 68
GeneSNPView
• Gene Variation Report• Variations in region of gene• Variations and
consequences
SNPs in Ensembl

49 of 68
GeneSNPGeneSNPViewView

50 of 68
Variation dataVariation data
• Recombination variability and population history of a species– “HapMap”
• Includes individual, cohort, population and genotype concepts
• Population substructure, close species and individual variation – Understanding positive and negative selection
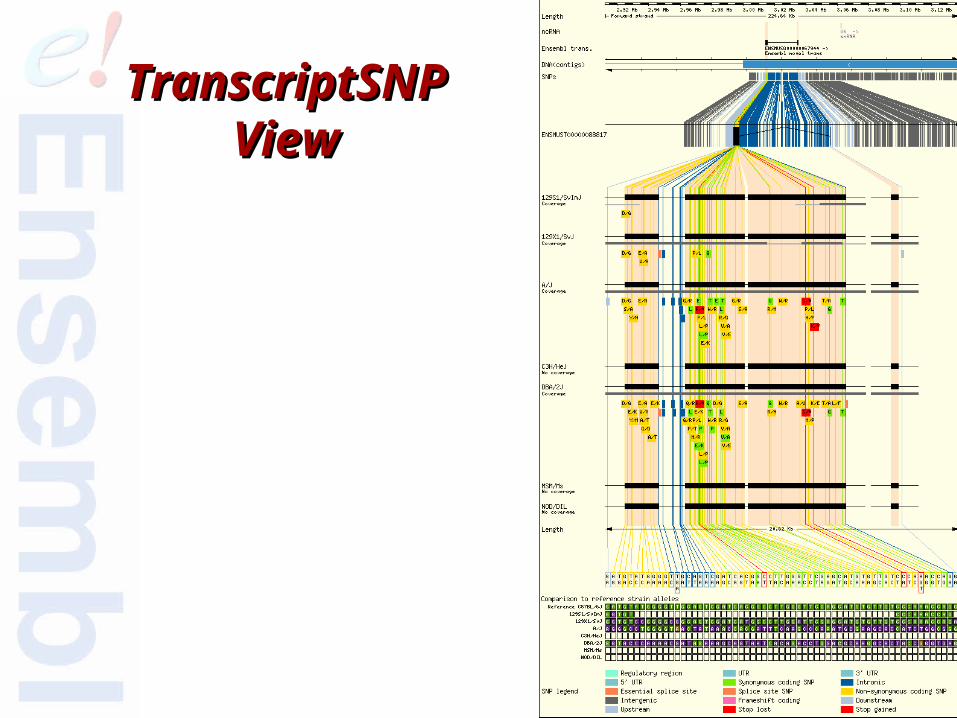
51 of 68
TranscriptSNPTranscriptSNPViewView

52 of 68
Linkage DisequilibriumLinkage Disequilibrium
LD values between two variants are displayed by means of inverted coloured triangles going from white (low LD) to red (high LD).
LDView

53 of 68
Manual CurationManual Curation• People are the best at
– Resolving conflicting heterogeneous information– Recognising “out of the ordinary” biology
• For high investment genomes an automated pipeline with human intervention is the endgame– Human and Mouse

54 of 68
Automatic vs ManualAutomatic vs Manual
AutomaticAutomatic AnnotationAnnotation• Quick whole genome
analysis ~ weeks• Consistent annotation• Use unfinished
sequence or shotgun assembly
• No polyA sites or signals, pseudogene
• Predicts ~ 70% loci
ManualManual AnnotationAnnotation• Extremely slow
~ 3 months for Chr 6• Need finished sequences• Flexible, can deal with
inconsistencies• Most rules have exception• Consult publications as
well as databases

55 of 68
Manual CurationManual Curation
– 7 (Washington University)– 14 (Genoscope)– 18 (Broad Institute)– 16, 19 (DOE Joint Genome Institute)
• Other groups will also contribute to Vega
Manual annotation of finished clonesVega Genome Browser http://vega.sanger.ac.uk/Currently only chromosomes
– 1, 6, 9, 10, 13, 20, 22, X and Y (Sanger Institute)

56 of 68
• Several human gene sets– Ensembl, Vega, NCBI, UCSC, UniProt
• Aim: Converge on, and maintain, a set of fixed CDS structures we are confident in– Unique identifier number and version number (e.g.,
CCDS1.1, CCDS234.1). • Version number will update if either the CDS structure or
the underlying genome sequence at that location changes
• The CCDS set will be mapped forward, maintaining identifiers. All changes to existing CCDS genes will be done by collaboration agreement.
Human gene set Human gene set convergenceconvergence

57 of 68
Conserved CoDing Conserved CoDing SequenceSequence
CCDS quality requirements:
1. Coding region annotated as full-length (with an initiating ATG and valid stop-codon).
2. Protein can be translated from the genome without frameshifts.
3. Consensus splice-sites are used.
14,795 CDS 16,085 transcripts 13,031 genes

58 of 68
There are more than genes!There are more than genes!
• RNA genes– “well known” structural RNA genes– Newer miRNA genes– Pseudogenes/duplications a massive headache
• Cis-regulatory motifs– Transcriptional motifs– RNA processing motifs
• Yet unknown other stuff

59 of 68
PseudogenesPseudogenesFilter retro-transposed (processed) pseudogenes
Query: 3 SRLLLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEK 62 S ++LNNG K +LGLGTWKSPPGQV EAVKVAI+ YRHIDC+HV+QN++ QE+Sbjct: 2 SHIMLNNGTKTDMLGLGTWKSPPGQVAEAVKVAINTVYRHIDCSHVHQNKD------QEQ 55
Query: 63 LREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEF 122 L+EQVV+RE LFI+SK W H K LV+G+C+K LS L+LDYLDL+LIHWPTG PGKEFSbjct: 56 LKEQVVRREWLFIISKPWGICHRKCLVRGSCRKVLSGLELDYLDLHLIHWPTGCHPGKEF 115
Query: 123 FPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAV 182 LDESG + +GLVKA GISNF HLQ E LNK GLK Sbjct: 116 SFLDESGLI-------------------QGLVKAAGISNF-HLQAERTLNKSGLKLSATG 155
Query: 183 NQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHN 242 LTQE LIQY QSK VTAYSPLGSPDRP AKPEDPSLLEDPRIK IAAKHNSbjct: 156 RS------LTQENLIQYYQSKA-AVTAYSPLGSPDRPRAKPEDPSLLEDPRIKVIAAKHN 208
Query: 243 KTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRN 295 + T+QVL+ QRNLVV P SVT +RIAENFKVFDFELSSQDMT+LLS NRNSbjct: 209 E-TSQVLMWLLTQRNLVVTPTSVTLDRIAENFKVFDFELSSQDMTSLLSCNRN 260
Chromosome 18:8,535,635-8,536,757
Chromosome 7:133,584,367-133,601,097
Mostly dead-on-arrival
Intronless, poly-A tail, short direct repeats

60 of 68
ncRNAsncRNAs• Functional RNAs (tRNAs, miRNA)• Families share conserved secondary
structure• Low sequence identity• Ribosome• Spliceosome

61 of 68
miRNAmiRNA• Highly conserved across species• Identified using BLAST genomic vs miRBase
precursors• RNAfold used to test for stem loop• Precursor stem loop sequence ~ 70nt• Mature miRNA ~ 21nt(only 2 nt changes tolerated)
Start with ~ 290,000 BLAST hits
End with 222 miRNA

62 of 68
• Overview of Ensembl– The Era of Sequencing Genomes– Exploring genomes– Gene annotation
• Making genomes useful– Website– BioMart
• Beyond Ensembl

63 of 68
Display Display 3D structures3D structures

64 of 68
Display Display 3D structures3D structures

65 of 68
GeneDASGeneDAS

66 of 68

67 of 68
Example: Epigenomic Example: Epigenomic ChIP-chip dataChIP-chip data

68 of 68
Help!Help!
• context sensitive help pages - click
• access other documentation via generic home page
• email the helpdesk

Ensembl TeamEnsembl TeamJuly 2006July 2006

70 of 68
Leaders Ewan Birney (EBI), Tim Hubbard (Sanger Institute)
Database Schema and Core API Glenn Proctor, Ian Longden, Patrick Meidl
BioMart Arek Kasprzyk, Damian Smedley, Richard Holland, Syed Haldar
Distributed Annotation System (DAS) Andreas Kähäri, Eugene Kulesha
Outreach Xosé M Fernández, Bert Overduin, Giulietta Spudich, Michael Schuster
Web Team James Smith, Fiona Cunningham, Anne Parker, Steve Trevanion (VEGA), Matt Wood
Comparative GenomicsAbel Ureta-Vidal, Kathryn Beal, Benoît Ballester, Stephen Fitzgerald, Javier Herrero Sánchez, Albert Vilella
Analysis and Annotation PipelineVal Curwen, Steve Searle, Browen Aken, Juilo Banet, Laura Clarke, Sarah Dyer, Jan-Hinnerck Vogel, Kevin Howe, Felix Kokocinski, Stephen Rice, Simon White
Functional Genomics Paul Flicek, Yuan Chen, Stefan Gräf, Nathan Johnson, Daniel Rios
Zebrafish Annotation Kerstin Howe, Mario Caccamo, Ian Sealy
VectorBase Annotation Martin Hammond, Dan Lawson, Karyn Megy
Systems & Support Guy Coates, Tim Cutts, Shelley Goddard
Research Damian Keefe, Guy Slater, Michael Hoffman, Alison Meynert, Benedict Paten, Daniel Zerbino
Ensembl TeamEnsembl Team
Sep 2006Sep 2006

71 of 68
Training...Training... Somewhere near you Somewhere near you

72 of 68
Ensembl annotation pipelineEnsembl annotation pipeline

73 of 68
SNPViewSNPView

74 of 68
Measures of LD
• D = P(AB) – P(A)P(B)– D ranges from – 0.25 to + 0.25
– D = 0 indicates linkage equilibrium
– dependent on allele frequencies, therefore of little use
• D’ = D / maximum possible value– D’ = 1 indicates perfect LD
– estimates of D’ strongly inflated in small samples
• r2 = D2 / P(A)P(B)P(a)P(b)– r2 = 1 indicates perfect LD
– measure of choice

75 of 68
BioMart - a distributed BioMart - a distributed architecturearchitecture
XML XML XML
MySQL ORACLE PostgreSQL
ANSI SQL
XML
XML
XML
XML
XML
XML

76 of 68
SNPVega
Retrieval
BioMart API
JAVA Perl
MartExplorer MartShell MartView
Schema transformation
MartBuilder MartEditor
Configuration
Databases
Public data (local or remote)
BioMart architectureBioMart architecture
MSD UniProt Ensembl
XML
myMartmyDatabase

77 of 68
SNPs in Ensembl
ContigView: SNPs in genomic context

78 of 68
Linkage DisequilibriumLinkage DisequilibriumLDTableView

79 of 68
Atime
Duplication
M 2’
Speciation
Duplication
M 2
A 1 A 2
M 1 H 1
H 2
Inparalogues
OutparaloguesOrthologues
Inparalogues
Inparalogues
Orthologous genes have originated from a single ancestor (often have equivalent functions).Paralogous are genes related via duplication:
•Inparalogues (ortholog_one2one, ortholog_one2many, etc.) duplication follows speciation and •Between_species_paralog (outparalogues). Duplication precedes speciation
Homologue RelationshipsHomologue Relationships

80 of 68
……in Ensembl…in Ensembl…

81 of 68
Genome ReviewsGenome Reviews

82 of 68
• Several human gene sets– Ensembl, Vega, NCBI, UCSC, UniProt
• Aim: Converge on, and maintain, a set of fixed CDS structures we are confident in– Unique unique identifier number and version number
(e.g., CCDS1.1, CCDS234.1). • Version number will update if either the CDS structure or
the underlying genome sequence at that location changes
• The CCDS set will be mapped forward, maintaining identifiers. All changes to existing CCDS genes will be done by collaboration agreement.
Human gene set Human gene set convergenceconvergence

83 of 68
PseudogenesPseudogenesFilter retro-transposed (processed) pseudogenes
Query: 3 SRLLLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEK 62 S ++LNNG K +LGLGTWKSPPGQV EAVKVAI+ YRHIDC+HV+QN++ QE+Sbjct: 2 SHIMLNNGTKTDMLGLGTWKSPPGQVAEAVKVAINTVYRHIDCSHVHQNKD------QEQ 55
Query: 63 LREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEF 122 L+EQVV+RE LFI+SK W H K LV+G+C+K LS L+LDYLDL+LIHWPTG PGKEFSbjct: 56 LKEQVVRREWLFIISKPWGICHRKCLVRGSCRKVLSGLELDYLDLHLIHWPTGCHPGKEF 115
Query: 123 FPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAV 182 LDESG + +GLVKA GISNF HLQ E LNK GLK Sbjct: 116 SFLDESGLI-------------------QGLVKAAGISNF-HLQAERTLNKSGLKLSATG 155
Query: 183 NQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHN 242 LTQE LIQY QSK VTAYSPLGSPDRP AKPEDPSLLEDPRIK IAAKHNSbjct: 156 RS------LTQENLIQYYQSKA-AVTAYSPLGSPDRPRAKPEDPSLLEDPRIKVIAAKHN 208
Query: 243 KTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRN 295 + T+QVL+ QRNLVV P SVT +RIAENFKVFDFELSSQDMT+LLS NRNSbjct: 209 E-TSQVLMWLLTQRNLVVTPTSVTLDRIAENFKVFDFELSSQDMTSLLSCNRN 260
Chromosome 18:8,535,635-8,536,757
Chromosome 7:133,584,367-133,601,097
Mostly dead-on-arrival
Intronless, poly-A tail, short direct repeats

84 of 68
miRNAmiRNA• Highly conserved across species• Identified using BLAST genomic vs miRBase
precursors• RNAfold used to test for stem loop• Precursor stem loop sequence ~ 70nt• Mature miRNA ~ 21nt(only 2 nt changes tolerated)
Start with ~ 290,000 BLAST hits
End with 222 miRNA

89 of 68
Distributed Annotation SystemDistributed Annotation SystemDASDAS

90 of 68
Server
Distributed AnnotationDistributed AnnotationExternal Contributors
Server
Database providers
UsersViewer
xml
html
SequencePrograms
Annotation
Server
CoordinateSynchronisation
Server
xml
Dowell, R.D. et al. (2001) “The Distributed Annotation System” BMC Bioinformatics. 2, 7

91 of 68
Example: Epigenomic Example: Epigenomic ChIP-chip dataChIP-chip data

92 of 68
Example: Epigenomic Example: Epigenomic ChIP-chip dataChIP-chip data
Histone-3-Lysine-4-Trimethylation data of Mus musculus chr 17 (GEN-AU)
• DAS serverhttp://www.ebi.ac.uk/das-srv/test3
• dsn requesthttp://www.ebi.ac.uk/das-srv/test3/dsn
• Example feature requesthttp://www.ebi.ac.uk/das-srv/test3/das/
GEN-AU_MEFB1_H3K4me3_vs_MEFB1_NCBIm35/
features?segment=17:19558400,19658400

93 of 68
Display of uploaded dataDisplay of uploaded data

94 of 68
browser position chr2:1-10000track name=Ensembl_test description="Ensembl workshop (BED)" color=000000 url=http://www.ebi.ac.uk/~xose/ensembl_test.html2 1000 1100 bed_feature_1 1000 + 2 2000 2100 bed_feature_2 500 + 2 3000 3100 bed_feature_2 100 +
URL-basedURL-based

95 of 68
Display of data via URLDisplay of data via URL
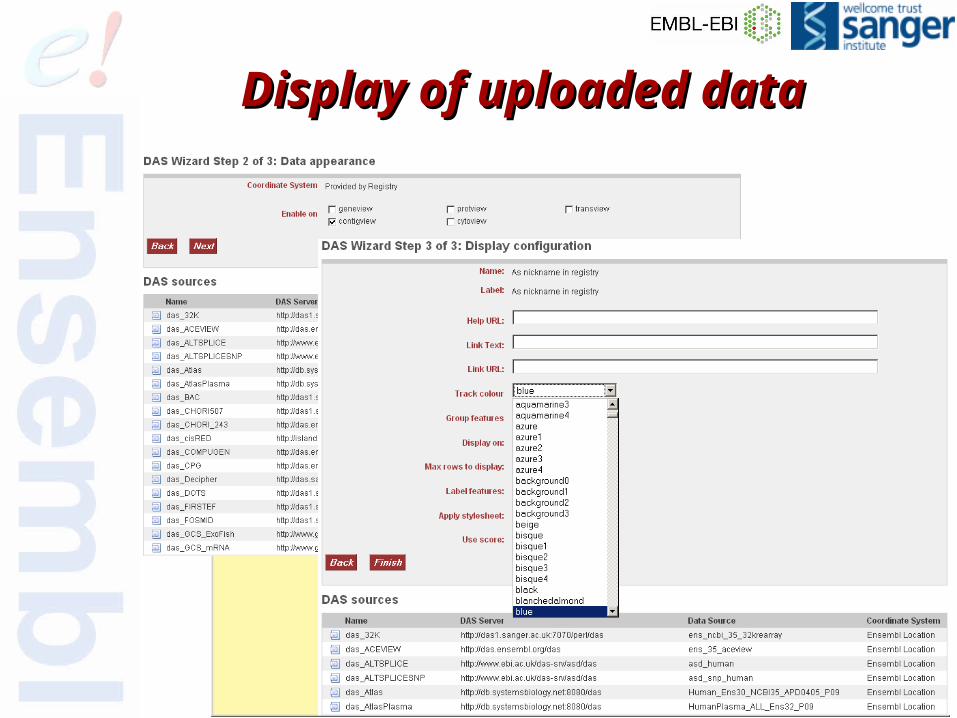
96 of 68
Display of uploaded dataDisplay of uploaded data

97 of 68
#<col1> <col2> <col3> <col4>#<group> <name> <type> <subtype>
Similarity Fake_match_1 homology wublastn Transcription Fake_tscr_1 transcript exonTranscription Fake_tscr_1 transcript exon
#<col5> <col6> <col7> <col8> <col9> <col10>#<chr> <start> <end> <strand><phase> <score>
2 4000 4050 + . 100 2 4200 4300 + . 1002 4400 4500 + . 100
File upload-basedFile upload-based

98 of 68
ExonerateExonerate

99 of 68
Display Display 3rd party data3rd party data
100 of 68
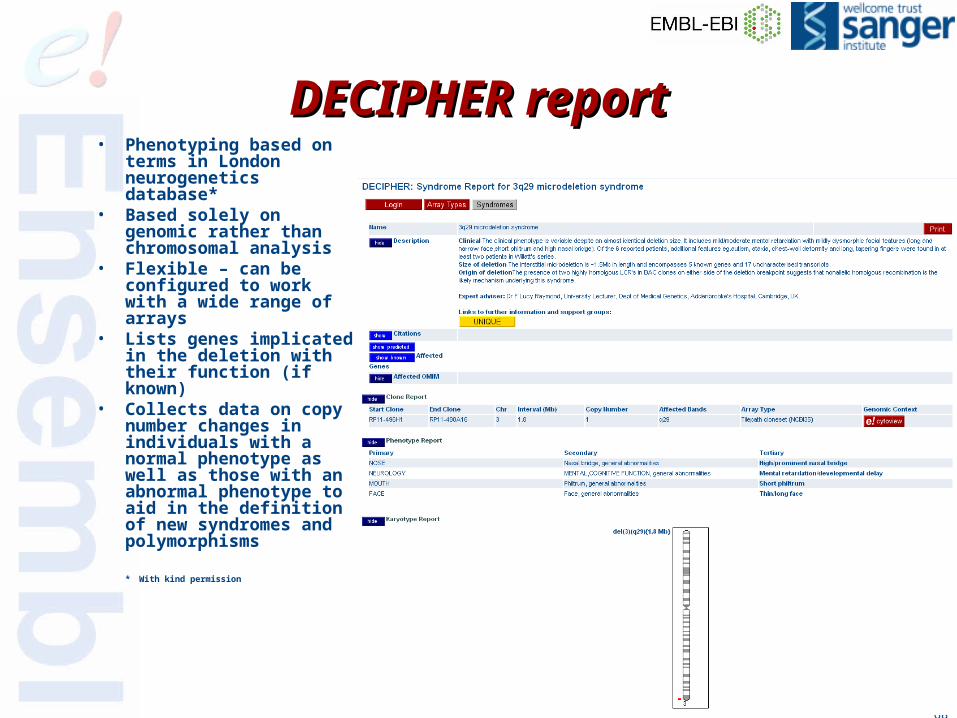
• Phenotyping based on terms in London neurogenetics database*
• Based solely on genomic rather than chromosomal analysis
• Flexible – can be configured to work with a wide range of arrays
• Lists genes implicated in the deletion with their function (if known)
• Collects data on copy number changes in individuals with a normal phenotype as well as those with an abnormal phenotype to aid in the definition of new syndromes and polymorphisms
* With kind permission
DECIPHER reportDECIPHER report

101 of 68
• Displays the microdeletion/duplication in its genomic location
• Shows whether a similar change has been reported to the database before
• Phenotype visible by ‘mouse-over’, enabling individual records to be compared
DECIPHER – Ensembl DECIPHER – Ensembl interfaceinterface

102 of 68
GeneDASGeneDAS

103 of 68
• New data– More species– Variation data– Comparative data– Phylogenetic info
• More comparative views– OrthoView
• LDView add export data in Haploview formathttp://www.broad.mit.edu/mpg/haploview
• Greater integration of user data– New developments in DAS
Future plansFuture plans

104 of 68
Insulin clusterInsulin cluster
Duplication nodeSpeciation node or leaf

105 of 68
HaplotypesHaplotypes

106 of 68
PARPAR
There are 2 pseudoautosomal regions in the human chrX/chrY chromosomes.
•PAR1 region (chrX:1-2709520 2.7 Mb), comprises the beginning tip of the short arm for both. •PAR2 (chrX:154,494,747-154,824,264 about 330kb), comprises the ending tip of the long arm for both.