apoptotic cell death - icksh.org · intracellular atp concentration: a switch in the decision...
TRANSCRIPT

Session SS05 - Cell Death Mechanism
Soo-Youl Kim, Ph.D. Division of Cancer Biology
National Cancer Center, Korea
Apoptotic Cell Death : Opportunity of Oncological Application

What is Apoptosis? A Greek word meaning
“falling off” as leaves from a tree

The term apoptosis was first used in a paper by Kerr, Wyllie, and Currie to
describe a morphologically distinct form of cell death.
Br J Cancer 26, 239–57, 1972
Dr. Horvitz found that programmed cell death occurs during the development
of the nematode Caenorhabditis elegans (Horvitz, Cancer Res 59, 1701s–
1706s,1999)
In this organism 1090 somatic cells are generated in the formation of the
adult worm, of which 131 of these cells undergo apoptosis or “programmed
cell death.”
The Nobel Prize in Physiology or Medicine
2002
Prof. H. Robert Horvitz
Apoptosis

(A) Large nuclei and scant cytoplasm / (B) Early during the chromatin condensation phase, arrow is a
fragmented section of nucleus and the arrowhead indicates an apoptotic body/ (C) Extensive plasma
membrane blebbing occurs followed by karyorrhexis and separation of cell fragments into apoptotic
bodies during a process called “budding”/ (D) These bodies are subsequently phagocytosed by
macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes.
Lymphocytes TEM
B
A C
D
Morphology of Apoptosis
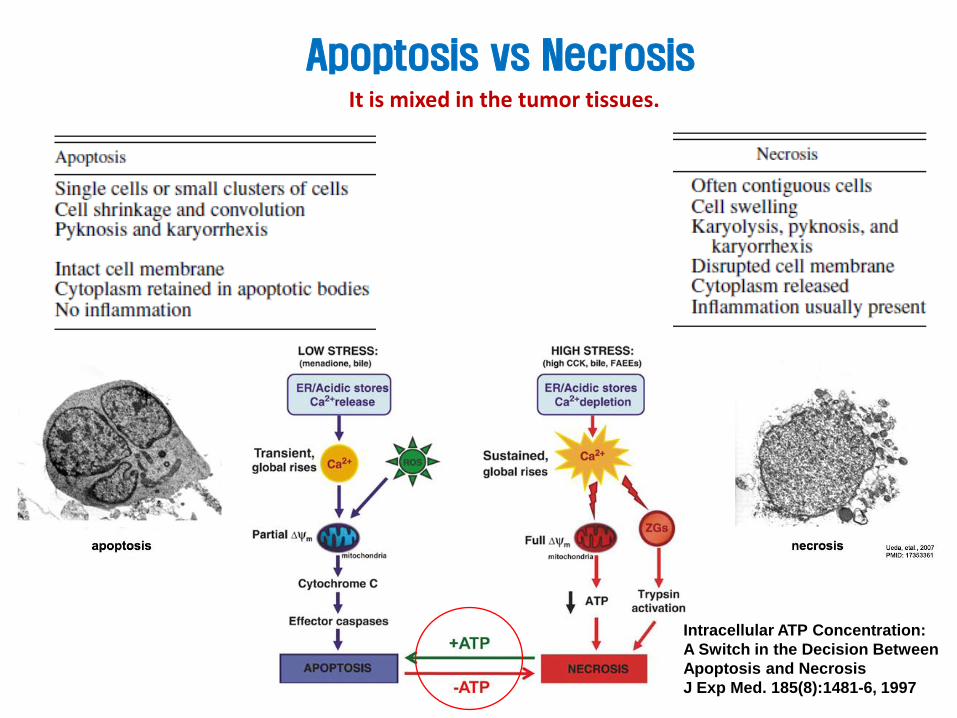
Apoptosis vs Necrosis It is mixed in the tumor tissues.
Intracellular ATP Concentration:
A Switch in the Decision Between
Apoptosis and Necrosis
J Exp Med. 185(8):1481-6, 1997

Apoptosis: Point of No Return to Life
(1) apoptotic cells do not release their cellular
constituents into the surrounding interstitial
tissue; (2) they are quickly phagocytosed by
surrounding cells thus likely preventing
secondary necrosis; and, (3) the engulfing cells
do not produce anti-inflammatory cytokines.
Most Confused Idea
Mitochondria outer membrane permeabilization

Toxicologic Pathology, 35:495–516, 2007
Major Pathways of Apoptosis
DISK: death inducing signaling complex MPT: mitochondrial perfusion transformation SET: nucleosome assembly protein

Intrinsic Apoptosis

Intrinsic Apoptosis
The intrinsic signaling pathways
that initiate apoptosis involve a
diverse array of non receptor
mediated stimuli that initiates
mitochondrial events.
Negative signals: the absence of certain
growth factors, hormones and cytokines that
can lead to failure of suppression of death
programs, thereby triggering apoptosis.
Positive signals: radiation, toxins, hypoxia,
hyperthermia, viral infections, and free
radicals.

Intrinsic Apoptosis
mitochondrial permeability transition
All of these stimuli cause
changes in the inner
mitochondrial membrane that
results in an opening of the
mitochondrial permeability
transition pore, loss of the
mitochondrial
transmembrane potential and
release of two main pro-
apoptotic proteins including
cytochrome c and Smac
from the intermembrane
space into the cytosol.

Mitochondria outer
membrane
permeabilization (MOMP)
is considered the “point of
no return” for apoptosis.
The steps leading up to
MOMP can be stopped in
their tracks by inhibitor
molecules, but once
MOMP has been achieved,
the cell will complete the
death process.
Intrinsic Apoptosis
BAX (Green) forms ring like structure On the fragmented mitochondria
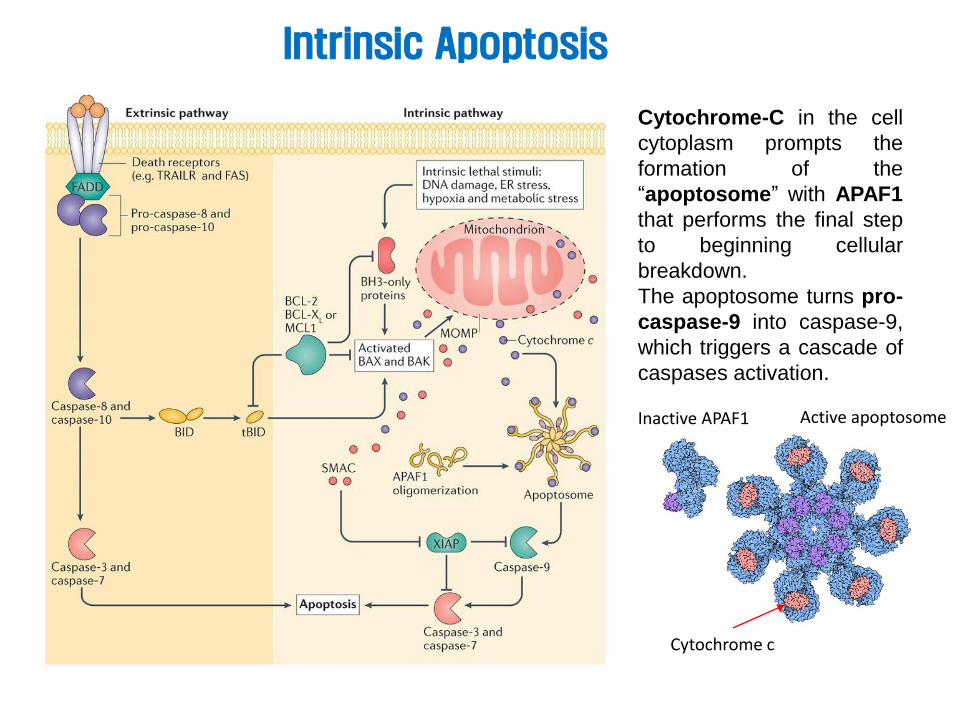
Cytochrome-C in the cell
cytoplasm prompts the
formation of the
“apoptosome” with APAF1
that performs the final step
to beginning cellular
breakdown.
The apoptosome turns pro-
caspase-9 into caspase-9,
which triggers a cascade of
caspases activation.
Intrinsic Apoptosis
Cytochrome c
Inactive APAF1 Active apoptosome

Intrinsic Pathway Proteins
Cancer cells employ this molecules

Extrinsic Apoptosis

Extrinsic Apoptosis
Extrinsic Pathways initiate apoptosis involved with transmembrane receptor-mediated
Interactions such as FasL/FasR, TNF-α/TNFR1, Apo3L/DR3, Apo2L/DR4 and
Apo2L/DR5. After signaling, a death-inducing signaling complex (DISC) is formed,
resulting in the autocatalytic activation of procaspase-8. Once caspase-8 is activated, the
execution phase of apoptosis is triggered.

cFLIP is an Inhibitor of Apoptosis
Death receptor-mediated apoptosis can be inhibited
by a protein called c-FLIP which will bind to FADD
and caspase-8, rendering them ineffective.
Extrinsic Pathway Proteins

TNF-Induced Apoptosis
TNFα, cachexin, or cachectin: discovered in 1975
Discovery of tumor necrosis
factor (TNF), a key immune
signaling molecule (cytokine)
that, in addition to its promise
for the treatment of cancer
and other diseases, has
provided a powerful research
tool in biomedicine.
Dr. Lloyd J. Old, M.D., c. 1995
Lloyd John Old

TNF inhibitor
• autoimmune and immune-mediated disorders such as rheumatoid
arthritis, ankylosing spondylitis, inflammatory bowel disease,
psoriasis, hidradenitis suppurativa and refractory asthma.
• The drugs inhibiting TNF include Remicade (infliximab), Enbrel
(etanercept), Humira (adalimumab), Cimzia (certolizumab pegol) and
Simponi (golimumab).

TRAIL and FAS Ligand Induced Apoptosis
TNF-related apoptosis-inducing ligand
(TRAIL)
The FAS receptor (FasR), also known as
apoptosis antigen 1 (APO-1 or APT), cluster
of differentiation 95 (CD95) or tumor necrosis
factor receptor superfamily member 6
(TNFRSF6)

Perforin/Granzyme Pathway

Cytotoxic T lymphocytes
(CTLs) are able to kill
target cells via the
extrinsic pathway and the
FasL/FasR interaction is
the predominant method.
CTL Induced Apoptosis

Perforin/Granzyme Pathway
A novel pathway involves secretion
of the transmembrane pore-forming
molecule perforin with a
subsequent exophytic release of
cytoplasmic granules including
granzymes A and B through the
pore and into the target cell.
Granzyme B can directly activate
Caspase 3, BID, and ICAD which
triggers apoptosis.
Granzyme A can activates
DNAse NM23-H1, which blocks
the maintenance of chromatin
structure integrity.
ICAD: Inhibitor of Caspase Activated DNAse

Cancer PD-L1 Expression Avoids TCR Activated FasL/TRAIL by SHP2
T cell death
Activation of cytotoxic T cells (Tc) is an antigen-specific process requiring the interaction of the TCR–CD3
complex with a processed tumor antigen–derived peptide bound to a MHC class I molecule. Tc activation
Induces FASL and TRAIL to kill cancer cells. However, PD-1 activation by cancer PD-L1 stops Tc activation through SHP2 and triggers T cell death.

How to Cure Cancer with Apoptosis Inducers ?

Treatment with Coley’s toxins
A patient with round cell sarcoma of the jaw and
abdominal metastases seen by coley in 1899.
Photograph after 63 injections with coley’s toxins;
tumour had diminished to about half its original
size.
Mouse bearing subcutaneous
human tumour xenograft
Wild-type mouse treated with the
carcinogen DMBA and the
tumour promoter TPA
Tnf–/– mouse
TNF treat
TNF-a Between Promotion and Suppression in Cancer
Coley’s toxins : a mixture consisting
of killed bacteria of species
Streptococcus pyogenes and Serratia
marcescens

Apoptosis Inducers

Oncogenic Apoptosis

Oncogenic Apoptosis
Apoptosis can also cause unwanted effects that may even promote cancer.
Understanding this may help maximizing anti-cancer apoptosis while
minimizing pro-tumorigenic effects.

Enhancing Apoptosis
pink boxes: Possible enhancers of apoptosis
blue boxes: inhibitors of unwanted effects
Nat Rev Cancer. 2016 Aug;16(8):539-48.
MOMP: mitochondria outer membrane permeabilization
SMAC: second mitochondria-derived activator of
Caspases.
XIAP: X-linked inhibitor of apoptosis protein
(caspase inhibitor)
CAD: caspase-activated Dnase
TAM: tumour associated macrophage
celecoxib

Novel Approach to Apoptosis Induction Via p53 Stabilization

p53 is a Key Regulator in Cancer

Nature Reviews Cancer 9, 749-758 (2009)
Regulator of p53

- + -
IB: p53
170 130
95 70
55 43 34 26
17
+ + + + P53
TGase2 p53
TGase2
BSA
Coomassie
p53
monomer
+ +
p53
polymer
IP: p53, IB: TGase2
INPUT: TGase2
p53
1-3
93
1-3
56
1-3
23
Δ3
23
-356
Δ1
02
-292
55
43
34 28
IP: IgG, IB: TGase2
INPUT: p53
Caki-1/p53-/-
1-50 63-97 102-292 323-356 363-393
Trans-
activation SH3 DNA binding domain Tetramer-
ization
Regulatory
domain
K Q K Q
p53 is the Major Target of Transglutaminase 2
132 144 164 192
FASEB J. 2013 Sep;27(9):3487-95.

By Microarray of TGase 2 using NCC 72 Cell Lines
Transglutaminase 2 is Universally Increased in RCC
RCC
TGase 2
b-actin
Oncogene. 30, 4780-4790, 2011
FASEB J. 2013 Sep;27(9):3487-95
A New Regulator of p53

p53 Mutation is Only 4% in ccRCC TGase 2 is highly increased in ccRCC
p62/SQSTM1 (p-value=0.00618) TGM2 (p-value=0.00579)
TGM2 (p-value=3.5708×10-15)
mR
NA
Expre
ssio
n
(RN
A S
eq v
2 R
SE
M)
Normal Tumor
Frequent mutations in clear cell renal cell carcinoma In COSMIC database
Cell Death and Disease (2016) 7, e2163
Instability of p53 in RCC is not associated with mutations because the COSMIC database showed only 4% mutation of p53 in clear cell RCC.
+ high + low
+ high + low

National Cancer Center, KOREA
p53 MDM 2
Proteasome degradation
p53 TGase 2
Autophagy degradation
1) Normal cells
2) RCC MDM 2
TGase 2
Mechanism of TGase 2-p53 Regulation in RCC
Nutlin-3 KN383
Cell Death and Disease (2016) 7, e2163
FASEB J. 2013 Sep;27(9):3487-95
Autophagosome
p53-polymer
Ca++
Autolysosome
Lysosome
Phagophore
LC3II
LC3II
N’
C’
p62 p53
C’ C’
N’
N’
LC3 binding
N’
C’ p62 p53
TG2
C’
C’
N’
N’
PB1 UBA
Ca++
LC3
N’
C’
p53 TG2
C’
N’
CD DBD
Ca++
C’
CD DBD
C’
CD DBD C’
CD DBD
C’
CD DBD
TG2

National Cancer Center, KOREA
p53 H&E BrdU
CT
L
GK
92
1
TGase 2 Inhibitor Reverses RCC via p53 Stabilization
J Cancer Res Clin Oncol. 140(5):757-67, 2014
By collaboration with Prof. Gong Y.D

β-actin
p53
p-p53(S15)
TG2
Doxo (hr)
siTG2
0 8 0 8
- - + +
CAKI-1
p21
Bad
Bax
Puma
Control GK921
Doxorubicin GK921+Doxorubicin
3.57 23.52
9.52 53.64
DNA intercalation of Doxo
Inhibits topo II
ATM/ATR
P-p53 apoptosis
p53
HDM2
TG2 GK921
DNA damage + TGase 2 Inhibition = Synergy
Cell Death and Disease (2016) 7, e2163
Control Doxorubicin 1mg/kg
GK921 2mg/kg Combination
32
12
0
500
1000
1500
2000
0 1 2 3 4 5 week
Tu
mo
r v
olu
me(m
m3)
(1 mM)
(1 mM)

Novel Approach to Apoptosis Induction Via ATP Depletion

Cancer
receptors Signaling
mTOR
mTOR produces
survival signal molecules
& nutrient uptake/catabolic
Enzymes for proliferation
receptor tyrosine
kinase EGFR, VEGFR, IGFR, PDGFR
RAS
RAF
ME
K
ERK
PI3
K
AKT
mTO
R
Gefitinib, Erlotinib, Imatinib, Sutent
Rapamycin, Temsirolimus
Sorafenib
transcription
Translation
Survival Proliferation
Conventional Anti-Cancer Drug Development
Transcription
regulation
Translation
regulation
Kinase
inhibition
Rapamycin
Ribavirin
Pateamine A
Targeting Anabolism

41
ALDH Is Required for ATP Production in Cancer
Oncotarget. 7:49397-49410, 2016
Experimental & Molecular Medicine (2016) 48, e272
ALDH1L1
low
ALDH1L1
high
(P-value = 0.01135)
Cancer Normal
ALDH1L1
KN817

ALDH Inhibitor + Mito Complex I Inhibitor
A549
(P-value = 0.01135)
ALDH1L1
ALDH1L1 low
ALDH1L1 high
Cancer Normal
ALDH1L1
Xenograft mice model
Oncotarget. 7:49397-49410, 2016
-300
200
700
1200
1700
0 5 9 12 15 19 22 25 29 32 35
★ ★
★
★ ★
★
★ ★
KN817

Cancer
cNADH ATP
mTOR
Cytosolic NADH
production increase
via eg. ALDH
ATP
Production
via ETC
ATP suppresses
AMPK & activates
mTOR
mTOR produces
survival signal molecules
& nutrient uptake/catabolic
Enzymes for proliferation
Opportunity for anti-cancer drug development via regulation of oxidative cancer energy metabolism
receptor tyrosine kinase
EGFR, VEGFR, IGFR, PDGFR
RAS
RAF
MEK
ERK
PI3K
AKT
mTOR
Gefitinib, Erlotinib, Imatinib, Sutent
Rapamycin, Temsirolimus
Sorafenib
transcription
Translation
Survival Proliferation
NADH
transport
Conventional anti-cancer drug development
ATP
Summary : New Approach for Drug Dev
Cell death
Cancer energy metabolism