api assessment: approaches and considerations, 19 january 2011 1 |1 | api assessment: approaches and...
TRANSCRIPT

API assessment: Approaches and considerations, 19 January 20111 |
API assessment:Approaches and considerations
Antony Fake
WHO Medicines Prequalification Programme

API assessment: Approaches and considerations, 19 January 20112 |
OverviewOverview
API information is assessed within the PQ programme in two ways:
as part of the FPP assessment, or
during the APIMF assessment
The new guidance for generic medicines emphasises that the data requirements in either situation are the same.
There is only one standard for the assessment of APIs.

API assessment: Approaches and considerations, 19 January 20113 |
Overview (2)Overview (2)
This is a point of interest review of API assessment, it is not a line by line review of technical requirements.
There is a focus on the API starting material (API-SM) and the API starting material for synthesis.
Some requirements are scientific in nature others are regulatory in nature.

API assessment: Approaches and considerations, 19 January 20114 |
3.2.S.1 – General properties3.2.S.1 – General properties
This section summarises information that is already known about the API.
Despite this applicants often do not provide complete information.
Particularly, information on: partition coefficients and solubility.
Optical rotation is sometimes performed using a different solvent or temperature .

API assessment: Approaches and considerations, 19 January 20115 |
3.2.S.1 – General properties3.2.S.1 – General properties
Different manufacturers make the same API in different polymorphs, particle sizes, solvates, hydration states.
It is important to understanding the molecule in question.
Unless this information is accurately recorded it is difficult to know in the future whether another API manufacturer is making the exact same molecule. This could potentially lead to bioinequivalence of the FPP.
An exact chemical and physical description of the API is desired.

API assessment: Approaches and considerations, 19 January 20116 |
3.2.S.2 – Manufacture3.2.S.2 – Manufacture
Ensure the unit and block is specified and recorded.
If it is not stated ask.
This affects future variations for changes to the manufacturing facility.
It is needed for GMP inspection purposes.

API assessment: Approaches and considerations, 19 January 20117 |
3.2.S.2 – Manufacture (2)3.2.S.2 – Manufacture (2)
A detailed description of manufacture is required.
Detailed flow diagram of synthesis indicating chemical structures, molecular weights, solvents, reagents.
Detailed narrative of each synthetic step. Types and quantities of reagents and solvents. Reaction conditions, critical steps, in-process controls, yields.
Alternative processes, reprocessing steps or recovery steps should be equally detailed.

API assessment: Approaches and considerations, 19 January 20118 |
3.2.S.2 – Manufacture (3)3.2.S.2 – Manufacture (3)
API starting material
API intermediate(s)
Final API intermediate
Final API
Choosing the API starting material (APl-SM)
The practise of buying reaction intermediates is common. Increasingly intermediates late in the synthesis are being purchased.

API assessment: Approaches and considerations, 19 January 20119 |
3.2.S.2 – Manufacture (4)3.2.S.2 – Manufacture (4)
Choice of API-SM
Simplermolecules
Final API
INDUSTRY
ASSESSORS

API assessment: Approaches and considerations, 19 January 201110 |
3.2.S.2 – Manufacture (5)3.2.S.2 – Manufacture (5)
The API-SM is the point at which GMP applies to manufacture.
This can be advantageous when API manufacturers buy reaction intermediates from secondary manufacturers with limited GMP experience.
API manufacturers prefer to have the API-starting material (API-SM) defined as late in the synthesis as possible because:

API assessment: Approaches and considerations, 19 January 201111 |
3.2.S.2 – Manufacture (6)3.2.S.2 – Manufacture (6)
The problem for assessors is:
Information on the preparation of a complex API from one or two steps makes determination of impurities in the API very difficult.
If the SM is complex it is hard to judge the acceptability of the SM specifications.
The guarantee of quality API is the result of good manufacture and control throughout all the steps. Comprehensive testing of the final API does not replace this. Therefore knowledge of all steps is important.

API assessment: Approaches and considerations, 19 January 201112 |
3.2.S.2 – Manufacture (7)3.2.S.2 – Manufacture (7)
There is a clever compromise,
The API starting-material-for-synthesis.
This defines the point in manufacture from which detailed manufacturing information should be provided.
This allows assessors to judge whether the controls on the API-SM are sufficient, without obliging the preparation of the API-SM to be in compliance with GMP regulations.

API assessment: Approaches and considerations, 19 January 201113 |
3.2.S.2 – Manufacture (8)3.2.S.2 – Manufacture (8)
API starting material
Starting materialfor synthesis
API intermediate(s)
Final API intermediate
Final API
Reaction intermediate(s)
Detailed information
provided dossier
GMP compliance
API starting material
Starting materialfor synthesis
API intermediate(s)
Final API intermediate
Final API
Reaction intermediate(s)

API assessment: Approaches and considerations, 19 January 201114 |
3.2.S.2 – Manufacture (9)3.2.S.2 – Manufacture (9)
Information on the preparation of the API commencing from the API-SM-for-synthesis must be provided in section 3.2.S.2.
However, information on the steps from the API-SM-for-synthesis to the API-SM is usually supplied as an annex.
There is often more than one supplier of API-SM. Each API-SM supplier must provide detailed information.

API assessment: Approaches and considerations, 19 January 201115 |
3.2.S.2 – Manufacture (10)3.2.S.2 – Manufacture (10)
The assessor has to:
1. Judge whether the choice of API-SM is appropriate.
2. Judge whether the choice of API-SM-for-synthesis is appropriate.
It is subjective, but there is some additional guidance.

API assessment: Approaches and considerations, 19 January 201116 |
3.2.S.2 – Manufacture (11)3.2.S.2 – Manufacture (11)
Simplermolecules
Final API
Poorer information provided on the preparation of the API-SM.
Detailed information provided on the preparation of the API-SM.
Choice of API-SM

API assessment: Approaches and considerations, 19 January 201117 |
3.2.S.2 – Manufacture (12)3.2.S.2 – Manufacture (12)
The API-SM for synthesis is a synthetic precursor of one or more synthesis steps prior to the final API intermediate. Acids, bases, salts, esters and similar derivatives of the API, as well as the racemate of a single enantiomer API, are not considered final intermediates…Be incorporated as a significant structural fragment into the structure of the API.
GUIDELINE ON SUBMISSION OF DOCUMENTATION FOR A MULTISOURCE (GENERIC) FINISHED PHARMACEUTICAL PRODUCT (FPP): QUALITY PART

API assessment: Approaches and considerations, 19 January 201118 |
3.2.S.2 – Manufacture (13)3.2.S.2 – Manufacture (13)
N
NH
N N
OCH3
N
NH
N N
OCH3
N
NH
N
O CH3
ClNH
A company submission proposes molecule 3 as the AP-SM. Can this be accepted?
Nevirapine
Crude Nevirapine
Proposed API SM
(1)
(2)
(3)

API assessment: Approaches and considerations, 19 January 201119 |
3.2.S.2 – Manufacture (14)3.2.S.2 – Manufacture (14)
First identify the final intermediate, as logically the API-SM and API-SM-for-synthesis must occur prior to this molecule.
N
NH
N N
OCH3
N
NH
N N
OCH3
N
NH
N
O CH3
ClNH
Nevirapine
Crude Nevirapine
Proposed API-SM
(1)
(2)
(3)
API assessment: Approaches and considerations, 19 January 201120 |
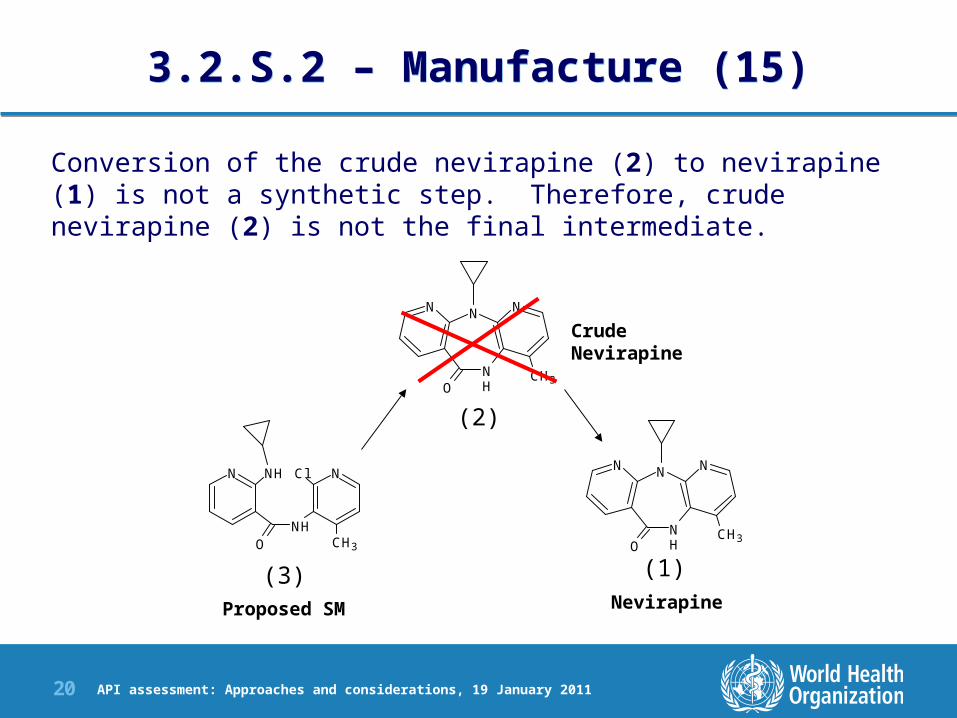
3.2.S.2 – Manufacture (15)3.2.S.2 – Manufacture (15)
Conversion of the crude nevirapine (2) to nevirapine (1) is not a synthetic step. Therefore, crude nevirapine (2) is not the final intermediate.
N
NH
N N
OCH3
N
NH
N N
OCH3
N
NH
N
O CH3
ClNH
Nevirapine
Crude Nevirapine
Proposed SM
(1)
(2)
(3)

API assessment: Approaches and considerations, 19 January 201121 |
3.2.S.2 – Manufacture (16)3.2.S.2 – Manufacture (16)
N
NH
N N
OCH3
N
NH
N N
OCH3
N
NH
N
O CH3
ClNH
The conversion of precursor 3 to crude nevirapine (2) is a synthetic step. Therefore, precursor 3 is the final intermediate. Further information on how molecule 3 is prepared is required.
Final intermediate(3)
(2)
(1)Proposed SM

API assessment: Approaches and considerations, 19 January 201122 |
3.2.S.2 – Manufacture (17)3.2.S.2 – Manufacture (17)
N
NH
N N
OCH3
N
NH
N N
OCH3
N
NH
N
O CH3
ClNH
The applicant states that molecule 3 is prepared from molecule 4. Molecule 4 can be assigned as the API-SM, but as it is a complex molecule further information on its preparation is required.
N
NH
N
O CH3
ClCl
Final intermediate
API Starting material
(4)
(3)
(2)
(1)

API assessment: Approaches and considerations, 19 January 201123 |
3.2.S.2 – Manufacture (18)3.2.S.2 – Manufacture (18)
N
NH
N N
OCH3
N
NH
N N
OCH3
N
NH
N
O CH3
ClNH
Molecules 5 and 6 can be accepted as the API-SM-for-synthesis. Each are significant structural fragments of the final API and commercially available.
N
NH
N
O CH3
ClCl
N Cl
Cl
O
N
CH3
NH2
Cl
+
NevirapineFinal intermediate
API Starting material
Starting materials for synthesis
(4)
(6)(5)(3)
(2)

API assessment: Approaches and considerations, 19 January 201124 |
3.2.S.2 – Manufacture (19)3.2.S.2 – Manufacture (19)
Controls on the API-SM should typically include identity, assay and impurity content.
The controls should take into account the API-SM preparation.
Controls on reaction intermediates should typically include identity, assay and impurity content, but will vary depending on the length of synthesis and controls placed on subsequent intermediates.

API assessment: Approaches and considerations, 19 January 201125 |
3.2.S.3 Characterisation3.2.S.3 Characterisation
It would be rare that the manufacturer has synthesised a completely different molecule. Even the wrong enantiomer or diastereomer.
It’s more a question of verification than being truly worried, but you never know! And, the consequences could be complete therapeutic failure, or worse.

API assessment: Approaches and considerations, 19 January 201126 |
3.2.S.3 Characterisation (2)3.2.S.3 Characterisation (2)
For non-pharmacopoeial substances a range of different techniques can and should be used. UV, IR, 1H and 13C 19F, 31P NMR, elemental analysis, HRMS, MS.
For pharmacopoeial substances comparison to the pharmacopoeial reference standard is desirable. Comparative IR is suggested, however, proton NMR wouldn’t hurt either.
Comparative IR will not address the question of enantiomers (maybe not even diastereomers), or particle size. It may address polymorphism.

API assessment: Approaches and considerations, 19 January 201127 |
3.2.S.3 Characterisation (3)3.2.S.3 Characterisation (3)
If the molecule can exhibit stereoisomerism
Ensure the techniques used can distinguish these.
Optical rotation comparisons to a reference standard is simplest.
Through-space proton NMR (NOE, NOESY, ROESY) experiments are increasingly used in larger organisations, but its really just showing off.

API assessment: Approaches and considerations, 19 January 201128 |
3.2.S.3 Characterisation (4)3.2.S.3 Characterisation (4)
Polymorphism
pXRD and DSC can be used to distinguish polymorphs.
Comparative IR is often used, but IR may not always be able to distinguish all polymorphs, or mixtures of polymorphs.
The use of IR must be validated by suitable experiments. Literature can be used for this purpose.

API assessment: Approaches and considerations, 19 January 201129 |
3.2.S.3.2 Impurities3.2.S.3.2 Impurities
This is covered in another talk, but:
Always look at the method of preparation.
Pharmacopoeial monographs are not magic, they may not control all potential impurities.
If the test method can’t detect the impurity, you will never see it!

API assessment: Approaches and considerations, 19 January 201130 |
3.2.S.4.1 Specifications3.2.S.4.1 Specifications
Do the tests cover all the applicable physical and chemical tests?
Compliance with a pharmacopoeial monograph may not be sufficient control, especially for impurities.
If a pharmacopoeial standard is claimed then the API specifications must meet the monograph requirements.
A one sentence statement in the report about compliance, or not, with a specific monograph is useful.
Microbial quality is not an issue unless the API is sterile.

API assessment: Approaches and considerations, 19 January 201131 |
3.2.S.4.1 Specifications (2)3.2.S.4.1 Specifications (2)
It is important that the manufacturer provides a signed, dated and version-numbered specification. This is to ensure traceability of testing requirements in case of GMP audits and future variations.
Skip testing
Often manufacturers will propose the elimination of certain tests based upon batch manufacturing data.
It is best to retain the test in specifications as a non-routine test, with a comment that the test is to be performed if there is a change to to manufacture, material suppliers etc.

API assessment: Approaches and considerations, 19 January 201132 |
3.2.S.4.1 Specifications (3)3.2.S.4.1 Specifications (3)
API specifications should be provided from the FPP manufacturer and from all of the API manufacturers.
The FPP manufacturer should have one API specification, even if there are multiple API suppliers.
The FPP manufacturer’s specifications may be a compilation of all the API manufacturers‘ specifications.

API assessment: Approaches and considerations, 19 January 201133 |
3.2.S.4.1 Specifications (4)3.2.S.4.1 Specifications (4)
if one API supplier controls the API to the EP monograph, plus an additional impurity,
the FPP must also control for the additional impurity. Adoption of the EP monograph only is not acceptable.
The FPP manufacturer's specifications should take into account those of the API manufacturers. Therefore,

API assessment: Approaches and considerations, 19 January 201134 |
3.2.S.4.2 Analytical Procedures3.2.S.4.2 Analytical Procedures
Non-pharmacopoeial test methods should be explicitly described, and numbered.
In an APIMF it is the API manufacturer’s procedures that are described.
In an FPP dossier it is the FPP manufacturer’s API test methods that are described.

API assessment: Approaches and considerations, 19 January 201135 |
3.2.S.4.3 Validation of Analytical Procedures3.2.S.4.3 Validation of Analytical Procedures
The provision of validation data for pharmacopoeial methods is not usually required.
If an in-house method is used when claiming a pharmacopoeial standard, equivalence of the in-house and pharmacopoeial methods must be demonstrated.

API assessment: Approaches and considerations, 19 January 201136 |
3.2.S.4.4 Batch Analyses3.2.S.4.4 Batch Analyses
Batch analysis should be provided for at least two pilot scale batches from each API supplier.
This should include the API batch used in the manufacture of the biobatch.
The size, manufacturing site and manufacturing date for each batch should be identified.
If testing was not to proposed specifications, a justification should be provided.
Certificates of Analysis from the API and FPP manufacturer should be provided for each batch.

API assessment: Approaches and considerations, 19 January 201137 |
3.2.S.5 Reference Standards3.2.S.5 Reference Standards
The primary and secondary reference standards used in the tests for assay, impurities and identification should be stated.
Primary reference standards obtained from the Ph.Int., BP, EP or USP should be used if possible.
If a pharmacopoeial standard is claimed then the primary reference standard of that pharmacopoeia should be used.

API assessment: Approaches and considerations, 19 January 201138 |
3.2.S.6 Container Closure System3.2.S.6 Container Closure System
Copies of the specifications for the primary packaging should be provided.
The materials used in the construction of the primary packaging must be suitable for use with pharmaceuticals
It should be verified that the proposed packaging is that used in the stability studies.

API assessment: Approaches and considerations, 19 January 201139 |
3.S.7.1 Stability data3.S.7.1 Stability data
This will be covered in detail in a further talk, but…
If the drug substance assessment is occurring as part of the FPP dossier assessment the drug substance stability data must be assessed for each API manufacturer.
Long-term storage conditions.

API assessment: Approaches and considerations, 19 January 201140 |
3.S.7.1 Stability data (3)3.S.7.1 Stability data (3)
They should be unless it has been demonstrated that the API is inherently unstable at 30ºC/65%RH or 30ºC/75%RH.
Instability during accelerated stability trials is not acceptable evidence of instability.
Are the primary stability studies conducted at 30ºC/65%RH or 30ºC/75%RH?

API assessment: Approaches and considerations, 19 January 201141 |
3.S.7.1 Stability data (4)3.S.7.1 Stability data (4)
If the API is stable at 30ºC/65%RH or 30ºC/75%RH to 24 months then a lower storage condition is not really justified.
If the API is unstable at 30ºC/65%RH. studies at 25ºC/60%RH should be attempted. 2ºC - 8ºC is not the next logical storage condition.

API assessment: Approaches and considerations, 19 January 201142 |
3.S.7.1 Stability data (5)3.S.7.1 Stability data (5)
commit to initiating long-term stability trials on the next two batches (pilot scale or greater) manufactured at either 30ºC/65%RH or 30ºC/75%RH.
provide a signed, authorised stability protocol for the new stability studies.
specify a date when they will submit an APIMF amendment or FPP variation to change the recommended storage conditions to stored below 30ºC.
If there is no data available at 30ºC/65%RH or 30ºC/75%RH then the API manufacturer should:

API assessment: Approaches and considerations, 19 January 201143 |
QuestionsQuestions
1. Does the APIMF need to be in the CTD format?
Yes, it should be. The question is why wouldn't it be?
2. If the API manufacturers have DMF in the US-DMF format, will it be acceptable? Do they need to convert to the CTD format, or can the applicant submit it as it is, as annex to 3.2.S and fill the sections S.1 – S.7 in the PD accordingly?
This course of action is not encouraged! But I would rather have a DMF than not

API assessment: Approaches and considerations, 19 January 201144 |
QuestionsQuestions
3. If we use US-DMF or EDMF that already got reviewed by the USFDA or European authority, will the WHO review it again?
Yes, unless the evaluation report can be provided. In which case a peer review of the report will be conducted.
4. If the DMF is old (e.g. >5 years ago) but can still meet the current requirements, will it be acceptable? Does the WHO have restriction on that, or require additional data e.g. annual product review, ongoing stability data?
No there are no specific requirements on the age of the APIMF. But be honest, will a 5 year old APIMF really meet current requirements? Or, accurately reflect current manufacture and control?

API assessment: Approaches and considerations, 19 January 201145 |
QuestionsQuestions
5. Can the WHO share the names of API manufacturers or APIMF holders that were already passed the review by the WHO as part of the prequalified FPP? This can help the FPP manufacturer knows the good source of API that can meet the WHO requirements and in turn reduce the WHO workload in reviewing the DMF.
No. Not at this time. But, the names and details of Prequalified APIs will be published for this very purpose.

API assessment: Approaches and considerations, 19 January 201146 |
QuestionsQuestions
Variations:
1. When the manufacture of an API is scaled up, a variation needs to be introduced to the WHO. This is the responsibility of the API manufacturer and not the FPP manufacturer. May be the FPP manufacturer could have a “quality agreement” with the API manufacturer asking for this commitment. Nevertheless when an API has a CEP is this required?

API assessment: Approaches and considerations, 19 January 201147 |
QuestionsQuestions
When there is an API related change and there is an APIMF then an APIMF amendment should first be submitted by the APIMF holder. Once the amendment is accepted then a variation from the FPP manufacturer maybe required, depending on the change involved.
If there is a CEP involved, no notification to the WHO is required unless the CEP is reissued.
If there is no APIMF or CEP supporting the FPP then the FPP manufacturer should submit a variation for all API related changes.

API assessment: Approaches and considerations, 19 January 201148 |
QuestionsQuestions
2. Will the reduction in number of batches for stability study affect the variations? For example, if some changes require stability of 3 pilot lots, will it be reduced to 2 pilot lots as per the new guideline? It should, yes, but not for requirements that concern production lots.

API assessment: Approaches and considerations, 19 January 201149 |
QuestionsQuestions
Revised stability requirements for generic FPPs were introduced to the PQ programme as part of the new guidance document for generic FPPs
Therefore, when stability data is required to support a variation, the following requirements apply:
For uncomplicated FPPs (e.g. immediate release solid oral dose forms) the minimum number of FPP batches required to establish the shelf-life is now two, of which one batch must be at least pilot scale.
For complicated FPPs the minimum number of FPP batches required to establish the shelf life is now two, both of which must be pilot scale or larger.