anöploi̇di̇
TRANSCRIPT

Otozomal Anöploidi
Mesut Akpunar

KROMOZOM BOZUKLUKLARI
SAYISAL
BOZUKLUKLAR
YAPISAL
BOZUKLUKLAR
Poliploidi
Anöploidi
Cinsiyet kromozomları anöploidisi
Otozomal kromozomların anöploidisi
Delesyon
Duplikasyon
İnversiyon
İnsersiyon
Ring kromozom
İzokromozom
Mozaisizm
Translokasyonlar
Robertsonian
Resiprokal

ÖPLOİDİ
İnsan eşey hücrelerinde 23 adet kromozom bulunur ve bu sayı haploid(n) dir.
Haploid kromozom sayısının tam katlarına “öploid denir.
Hücrelerdeki kromozom sayısı o organizma türü için temel gametik sayının tam katı kadar
artmıştır.
Haploidi
Diploidi
Triploidi
Tetraploidi
( n=23)
(n= 46)
(n=69)
( n=92)

ANÖPLOİDİ
Temel kromozom sayısının tam katı kadar olamayan artma yada azalmadır. (2n +1 veya 2n-1)
-Trizomi 21, monozomi X
İnsan kromozom bozuklukları içerisinde en sık görülen ve klinik olarak en önemli tipidir.
Eğer bir kromozom parçasının artması veya azalması şeklinde ise parsiyel anöploidi adını alır.
Mekanızması :
Kromozom ayrılmaması (non-disjunction)Anafazda geri kalma (anaphase lagging)

Otozomal anöploidi: otozomal kromozom çiftinin veya çiftlerinin
ikiden eksik veya fazla sayıda olmasını tanımlamaktadır. Bu olay
sayısal olabileceği gibi yapısalda olabilir. Trizomilerin büyük
çoğunluğu sadece bazı hücrelerde (mosaic aneuploidy)
görülürken, diğer kısmı tüm hücrelerde (nonmosaic)
görülmektedir. Yeni doğanlardaki ozomal anöploidinin insidansı
%20. Birçok otozomal anöploidi yaşamla bağdaşmamakta ve
insidansı sppontan düşüklerde %27-30. Sitogenetik araştırmalar
tüm frekans içerisindeki anormalliklerin %20’sinin yumurta
kaynaklı olduğunu %10 kısmının ise sperm kaynaklı olduğunu
söylemekte. Sayısal olarak %90 dan daha fazla anormallik
yumurta hücresinde gözlemlenirken %50 den daha azı sperm
hücresinde görülmekte.
Otozomal Anöploidi

Bu çalışmalarda fluorescent in situ hybridization (FISH) veya
primed in situ labelling (PRINS) kullanılmakta ve otozomal
anöploidi sperm hücrelerindeki kromozomlarda (kromozom
3,7,8,9,10,11,13,16,17 ve 21) uniform olmakla beraber oranı
%26-34 arasında. Ayrıca FISH ile tanımlanan en yüksek
frekansa sahip anöploidi 21. kromozom ve görülme sıklığı
%29. Diğer kromozomlarda (kromozom 1,2,4,9,12,15,16,18
ve 20) görülme sıklığı ise %8-19 arasında. Genelde mayoz
bölmedeki rastgele ayrılmamaya (nondisjunction) bağlı
olarak gelişir dolayısıyla düşüklere sebep olur. Aynı zamanda
canlı doğumda olabilir.

Otozomlardaki tüm trizomilerde (1. kromozomda dahil olmak üzere)hamileliğin 8-9 haftasında düşük rapor edilmiş. Klinik tanı IVF ilehamileliğin 6 haftasında yapılabilir.
Her bir trizominin frekansı farklılık göstemekle birlikte ÖR. trizomi 16 herbir trizomi içerisindeki düşüklerde %30’u teşkil etmekte.
Canlı doğumlarda, ‘’kromozom 1 ve 11’’ ile alakalı mosaiklik ve nonmosaiklik rapor edilmemiş çünkü 13,18 ve 21 kromozom dışındakileroldukça nadir.
Trizomi 21 dışındaki, diğer trizomilerin frekensı gebelik sırasında benzerfakat düşük ile canlı doğum arasında oransal olarak büyük farklar var. Buolay kromozomlardaki yıkıcı dengesizlikle alakalı.
Pre-embriyonik safhada birçok otozomal anöploidi çok zararlı ve öldürücü,bundan dolayı spontan düşüklerin nedenleri tanımlanmamış ve çalışılmamış.
Parsiyel otozomal anöploidideki ölüm kromozomun içerdiği gen içeriğiylebağlantılı.


Not: Gen bakımından zengin
kromozomlardaki trizomilerin yaşamla
bağdaşlaşması daha az. 13,18 ve 21.
kromozomlar gen içeriği bakımından
daha fakir olduklarından bu
kromozomlardaki trizomiler de hayatla
bağdaşma olur ve etkide hafiftir.

Mekanizma ve etiyolojisi
Mayozdaki hatalar (non-disjunction) sonucunda, gametleranormal sayılarda kromozomlar içerirler buda döllenme sırasındaanöploidiye sebep olur.
DNA markırlar kulllanılarak ebeveyn orijinli ekstra kromozomlarotozomal anöploidi içerisinde çalışılmış (trizomi2,7,13,14,15,16,18,21 ve 22).
Tüm bu çalışmalar gösterdi ki birçok trizomi maternal kaynaklıama bu oran farklı kromozomlar arasında farklı oranlardagerçekleşmekte.
Kromozom 7 ve 18 maternal ayrılmama daha çok mayoz IIaşamasında görülüyor.

Table1: meiotic/mitotic origin of autosomal trisomies determined by molecular
studies (number of cases)

Otozomal anöploidi ile anne yaşı arasındaki ilişki uzun zamanönce tanımlandı.
1933 te Penrose anne yaşının Down sendromlu doğmada kilitfaktör olduğunu belirtti.
Mekanizmasıyla alakalı mayoz I ve mayoz II dekiayrılmamanın temel faktör olduğunu belirtti.
Bu ayrılmama iki şekilde olabilir; birincisi ayrılmamış
bivalent kromozomlar ile homolog kromozomların aynı kutba
gitmesi ve bu mekanizma Angell tarafından gösterilmiştir.
İkincisi ise, kardeş kromatitlerde ki erken ayrılmayla alakalı.
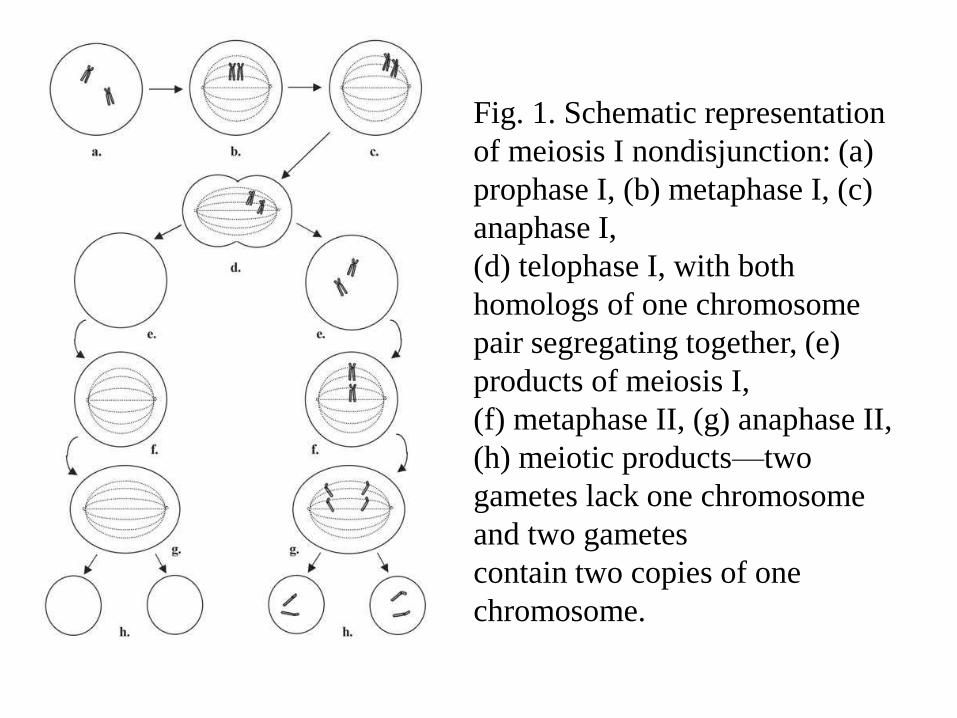
Fig. 1. Schematic representation
of meiosis I nondisjunction: (a)
prophase I, (b) metaphase I, (c)
anaphase I,
(d) telophase I, with both
homologs of one chromosome
pair segregating together, (e)
products of meiosis I,
(f) metaphase II, (g) anaphase II,
(h) meiotic products—two
gametes lack one chromosome
and two gametes
contain two copies of one
chromosome.

Fig. 2. Schematic representation of meiosis
I error resulting from premature sister
chromatid separation:
(a) prophase I, (b) metaphase I, (c)
anaphase I, with premature separation of
centromere of one chromosome,
(d) telophase I, with one prematurely
separated chromatid segregating with its
homologous chromosome, (e)
products of meiosis I, (f) metaphase II, (g)
anaphase II, (h) meiotic products—two
gametes with a normal
chromosome complement, one gamete
lacking one chromosome, and one gamete
containing two copies of one
chromosome.

Mayoz II de kardeş kromatitlerde ayrılma görülür.
Ayrılmama olayında ise bu kromatitler aynı kutba giderler
Fig. 3. Schematic representation
of meiosis II nondisjunction: (a)
prophase I, (b) metaphase I, (c)
anaphase I, (d) telophase I, (e)
products of meiosis I, (f)
metaphase II, (g) anaphase II,
with both sister chromatids
segregating together, (h) meiotic
products—two gametes with a
normal chromosome
complement, one gamete lacking
one chromosome, and one
gamete containing two copies of
one chromosome.

Eğer maternal mayoz I deki ayrılmama olayı ileri
yaşlarda artıyorsa buna ne sebep oluyordur?
Farklı mekanizmalardan bahsediliyor.
Bir tanesi production line hipotezi;
Buna göre oositlerin olgunlaşması yetişkinlerde benzer basamaklara göre ilerliyor
ve oogonia mayoza giriyor daha anne karnındayken (fetal life). Eğer oogonia
mayoza daha geç girerse kiazma yapılardaki gelişim bozuk oluyor böylece
ayrılmama gerçekleşiyor. Bu hipotezi destekleyen ilk sitogentik çalışma
ayrılmamış homolog kromozomların frekensının moyoz I deki pakiten ve
diploten kromozomlarına sahip oositlerin düşük materyalinden (13 ile 24.
haftalar arsındaki) alınması ve altı kromozomun (X, 7, 13, 16, 18 ve 21)
çalışılmasıyla belirlendi. Ayrılma hatalarının erken dönemdeki oogonialarda (0-
1.2 %) daha geç dönemde mayoza giren oogonyalarda ise (1.3-5.5 %) olduğu
görülmüş ve budaki önemli farkda gösterilmiş.

Yinede daha fazla araştırmaya ihtiyaç var bu hipotezi desteklemek için. Bir
diğer örnek sınırlı oosit havuzu (limited oocyte pool) modeli menstrual
döngünün antral basamağında, birçok folikül gelişiminin farklı aşamasında
bulunuyor.
Eğer yüksek miktarda bunlar FSH ile uyarılırlarsa, sadece bir folikül
muhtemelen optimal stage geçecek ve M I bitirip ovulasyon aşamasına
geçecek.
Maternal yaş arttıkça antral basamaktaki folikül sayısı azalacak, folikül sayısı
azaldıkça, tam tersi optimal stage erişmemiş oosit ovulasyon için
seçilemeyecek.
Eğer daha az optimal (less optimal) basamaktaki oosit mayoz I’e girerse
böylece ayrılmama gerçekleşir. Sonuç olarak yaş arttıkça ovulayondaki
oositler anöploidi için yüksek risk taşımış olacaklar.
Ama son araştırmalardaki datalar bu modeli desteklememekte!.. Bu olaydaki
muhtemel bir faktörde, ayrılmamaya uygun hale gelen gametlerin (predisposes
gametes) anormal rekombinasyonu.

Otozomal trizomiler
Trizomi 21
Trizomi 21 [47,XX veya XY, +21] insanda tanımlanan ilk
kromozomal anormalite.
Bu fenotip Jon Langdon Down tarafından 1866 da tanımlandı ve
Down sendromu ismini aldı.
Populasyon içerisindeki frekansı 1/700.
Bu sendromun erkeklerdeki frekansı daha fazla (male to female
ratio 1.2:1).
Multicolor FISH tekniğiyle yapılan çalışmalarda önemli miktarda
Y (X kromozomuna göre) taşındığı görülmüş, sperm disomicler
arasında kromozom 21 için.
Bu sonuçlar trizomi 21’in erkeklerde fazla olması parental
mayozdaki hatalardan kaynaklandığı sonucunu göstermekte.

Trisomy 21 Down syndrome female kayotype [47,X,+21]

95% Down sendromunda trizomi 21 görülmüş.
Mozaisizm ve Robertsonian translokasyon ise 5%’lik kısmını kaplamakta.
Down sendromunun fenotipiDown sendromunun klinik fenotipi iyi tanımlanmıştır.
Bunlar;
Baş nispeten ufaktır, ense kısa ve geniştir. Burun kökü yassılaşmıştır, kulaklar kafada
normalden düşük bir seviyede durur ve gözler birbirinden ayrık ve çekik görünür. Dil
ağıza göre genellikle çok büyük olduğundan dışarı taşmış gözükür.
Ense cildi oldukça gevşek olduğundan ensede genellikle boğumlar vardır. Bu bebeklerin
tonusları (vücut gerginliği) düşüktür. Parmaklar kısa ve tombuldur ve sıklıkla
avuçiçlerin den birinde ya da ikisinde simian çizgisi adı verilen tek bir çizgi vardır.
Ellerin serçe parmakları genellikle içe doğru kıvrımlıdır. Bunun nedeni bu parmağın
orta falanksının az gelişmiş olmasıdır. Down sendromlu bebeklerde en sık kalp
hastalıkları (40-50% her hastada) ve sindirim sistemi hastalıkları görülür. Down
sendromlu bebeklerde yenidoğan ya da çocukluk çağında lösemi (kan kanseri) daha sık
gözlenir (10 ile 20 kat daha fazla).

Fig. 5. The hand of a Down
syndrome child showing
small hand, clinodactyly,
only one crease in the fifth
finger, and single palmar
crease.
Feet of a boy with Down syndrome
Loose Nuchal Skin

Down sendromunun birçok aile için en üzücü özelliği bebek büyüdükçe barizleşen
zeka geriliğidir. Bunun şiddeti bebekler arasında önemli farklılıklar gösterir.
Mozaik down sendromlu hastalarda klinik fenotip daha hafif ama trizomi 21’li
hücreler ile klinik anlatımının ağırlığı arasında bir korralasyon yoktur. Fakat
nonmozaik trizomi 21’li bireyler kadar mozaik trizomi 21’li hastalarda da fenotip
ağır olabilir.
Çeşitli moleküler metodlar kullanarak bu sendromun çeşitli özellikleri açıklana
bilir.
Bu çalışmalar; CuZn-superoxide dismutase (SOD1) ve amiloid prekürsör
proteinlerin (APP) 21q22.1 proximal kısmına yerleşirler ve belirleyici olurlar
yinede 21q 22.2 ve 21q22.3 bantlar (D21S55 lokusunu içerirler) minimal bölge ve
çok önemli bir bölgedir Down sendromunun gelişiminde.
Korenberg ve ark, göre tek kritik bölge yerine birçok bölge, kromozom 21 de ki,
Down sendromun gelişimine neden olabilir.

Tekrarlama sıklığı (recurrence)
30 yaş altında deneysel tekrarlama riski trizomilerde (trizomi 21 hariç)
yaklaşık %1.
30 yaş üzerinde tekrarlama riski önemli değil (not significant), yaşa bağlı
spesifik riskten.
İki trizomi 21 çocuğu olan 13 ailede yapılan çalışmada, üç ebeveyn de
mosaik trizomi 21 olduğu belirlenmiş, iki ailede potansiyel olarak mozaiklik
belirlenmiş.
Üç trizomi 21’li çocuğu olan bir aile, Harris ve ark, tarfından çalışılmış
annenin lemfosit ve deri fibroblastları trizomi 21 için mozaik olduğu
belirlenmiş.
Bir diğer durumda dört trizomi 21’li çocuğu olan bir ailede, anneye
ovaryum biopsisi yapıldığında alınan hücre hatlarının trizomi 21’li olduğu
tespit edilmiş.
Sonuç olarak gonadal mozaiklik trizomi 21’in tekrarlama sıklığında çok
önemli olduğu anlaşılmış. Böylece tekrarlama riskinin gonadlarda trizomi
21’li hücre bulunmasına bağlı olarak daha yüksek risk taşıdığı açıklanmış.


Trizomi 18
Trizomi 18 ilk kez [47,XX or XY, +18] Edwards ve ark,
tarafından tanımlanmıştır 1960 yılında.
Ayrıca Edward sendromu olarak da bilinir.
Görülme sıklığı 1/6000-8000.
Bayanlarda daha sık görülüyor 1:3-4 (male to female ratio).
Maternal yaş arttıkça trizomi 18 riski de artar. Trizomi 21 den
sonra en sık görülen trizomi çeşididir.

Trizomi 18’in fenotipi
Doğan bebeklerin çoğunun ilk bakışta en dikkati çeken özellikleri normaldoğum kilolarından belirgin bir şekilde daha düşük kilolu olmaları,mikrosefali, mikrognati, kulaklarda yapısal anomaliler ve kulaklarınnormalden daha aşağı konumda yer almaları gibi özelliklerdir.
Yapılan ayrıntılı incelemelerde bu bebeklerin %90’ından fazlasında kalpanomalileri (ventriküler septal defekt (VSD)) ve yine önemli bir kısmındaböbrek ve sindirim sistemi anomalileri bulunduğu saptanmış. Yinekriptorşidi sendromu taşıyan erkek bebeklerde sıklıkla gözlenen diğer birdurumdur.
Trizomi 18’li vakaların en azından %87’sinde gelişme geriliği vardır.Trizomi 18’de gözlenen el ve ayak anomalileri tipiktir (“clenched hand”,yumru ayak ya da “rocker-bottom feet”), ve yapılan çalışmalarda en sıkdikkati çeken bulgu fetal parmakların anormal pozisyonudur.

Tekrarlama sıklığı
(recurrence)
Bu sendrom için yeterli
data yoktur. Tek durum
rapor edilmiş trizomi 18
için kardeşlerde ve abort
materyalde. Genetik
danışmanlık amaçlı, risk
durumu %1 den daha az.
Fig. 6. Profile of a trisomy 18 child showing
prominent occiput, low-set malformed ear, and
micrognathia

Bu anomalinin rastlandığı gebeliklerin % 95'i daha doğum aşamasınagelmeden bebeğin ölümü ile sonuçlanırken, bir haftalık hayatta kalmayüzdeleri %35-45 ve ortalama ömürleri 5 gün bir hafta içerisinde.
Doğan çocukların %10'un dan daha azı bir yılın üzerindeyaşayabilmektedir ve çok azı on yaşını geçebiliyor.
Bazı istisnalarda var, yapılan çalışmalarda en az altı trizomi 18 hastasınınnormal zekaya sahip olduğu ve uzun yaşadıkları saptanmış.
Son iki moleküler çalışmada toplamda on parsiyel trizomi 18’li hastada18q12 kromozomunun proximal bandının bu semptom üzerinde etkisininolmadığı yinede iki kritik bölgenin, biri proximal (18q12.1 q21.2) vedistal (18q22.3 qter) bölgelerinin birlikte çalışmasıyla trizomi 18fenotipinin oluştuğu belirtilmiş.
Daha fazlası bu hastalardaki ağır mental retardasyonun(18q112.3 q21.1)ile ilgili olduğu saptanmış.

Trizomi 13
Trizomi 13 [47,XX or XY,+13] ilk kez Patau ve ark, 1960’ta tanımlandı. Tahminedilen insidansı 1/12,000. bayanlarda erkeklere göre daha az görülüyor. Maternal yaşarttıkça trizomi 13 riskide artıyor.
Fenotipi:
Normal gününde bile doğsa bu bebekler daha düşük kiloda doğarlar. Küçük başçevresi ve eğimli alın yapısı gözlenir. Genellikle doğumdan hemen sonra görülenyapısal beyin problemleri vardır. Çoğunlukla beynin ön kısmı düzgün bir şekildeayrılmamıştır. Bu gözlerin yakınlığı, burun ve burun deliği gelişimi gibi bebeğin yüzgelişiminde değişikliklere neden olur. Yarık dudak ve çene bu bebeklerde sıradandır.
Göz problemleri ve kulak pozisyonu ile şekli farklı olabilir. Deriye yakın ince kandamarlarından dolayı hastalığa özel bir bir cilt rengi görülür.
Trizomi 13’lü bebeklerin bir çoğunda fazladan bir parmak görülebilir. Kalp (%80hastada gözlenir) ve böbrek problemleri , erkeklerde inmemiş testis, kızlarda rahimşekil bozukluğu diğer görülen anomalilerdir.
Ortalama hayatta kalmaları 2.5 gün ve 6 ay yaşama olasılığı %5. Ağır zeka geriliği,gelişim bozukluğu ve nöbetler görülür yaşamları boyu.
Mozaik trizomi 13’lü bireyler daha az etkilenmişlerdir ama bunun derecesi deçeşitlidir.

Yapılan çalışmalarda proximal segmenteki
(13pter q14) trizomi 13 üzerinde az etkisinin olduğubulunmuş bunun yerine distal segmentteki (tüm veyabir parça 13q14 qter) trizomi 13’te ki ana nedenneden olduğu belirtilmiştir.
Tekrarlama sıklığı (recurrence)
Güvenilir bir data yok ama risk çok düşük (%1 dendaha az) ama genetik danışmanlıkta tırnak içinealınmış.

ypical features of an infant with Trisomy 13. A. Midline defect withcleft lip & palate. B. Clenched hand with overlapping fingers. C. Postaxial polydactyly. D. Equinovarus deformity. E. Punched outaplasia cutis scalp lesions. From http://health-7.com

Trizomi 8Trizomi 8 [47,XX veya XY,+8] ilk kez Grouchy ve ark, tarafından 1971yılında tanımlandı.
•Çok nadir ve insidansı da bilinmiyor.
•Literatürde rapor edilen 100 vaka var.
•Bunların büyük çoğunluğu mozaik [47,+8/46].
•Erkek ve bayandaki oran ise 2-3:1 (male to female ratio).
Fenotipi;
Mental retardasyon, kısa boy, kilo azlığı, vertebral anomaliler. Dismorfik kafa,alın çıkıklığı, displazik kulaklar, strabismus, alt dudak sarkıklığı, yüksekdamak, yarık yumuşak damak, mikrognati. Konjenital kalp hastalığı. Ürineryol anomalisi, dar pelvis. patellar displazi, eklem hareketlerinde kısıtlılık, elayası ve ayak tabanlarında derin fleksiyon katlantıları. Birkaç tane dehematolojik malingnansi de rapor edilmiş.
Tekrarlama sıklığı (recurrence)
Bilinmiyor.

Fig. 1: Facial appearance of the patient: (A) the neonatal period, (B) at the age of six weeks,(C) age of 14 months. Fig. 2 (A) Bifid tip of tongue and grooved uvula, deep palmar creases,single flexion crease on the fifth finger. (B) Foot image - “hallmark “ sign of the syndrome.
Fig. 1
Fig. 2

Trizomi 9 İlk vakalar 1973 yılında rapor edilmiş trizomi 9 için hem nonmosaic [47,XX veya
XY, +9] hem de mozaik [47,+9/46].
•40’tan fazla trizomi 9 için canlı ve ölü doğum rapor edilmiş.
•Büyük çoğunluğu mozaik.
•Erkek bayan oranı 1:1
Fenotipi
Ağır mental retardasyon, mikrosefali, alın çıkıklığı, çıkıntılı kulaklar, sivri burun,
balık ağzı, mikrognati, Konjenital kalp hastalığı (%60 hastada görülüyor), böbrek
yapısındaki bozukluklar (%40 hastada görülüyor), Üriner yol anomalisi, konjenital
kalça/diz çıkıklığı, klinodaktili, dijital hipoplazi, tırnak hipoplazisi, sindaktili.
Not: Mozaik hastalar daha fazla yaşama eğilimindedirler fakat trizomi 9 hücrelerin
oranı yaşam uzunluğu ve semptomların ağırlığıyla ilgili tahmin vermez.
Ayrıca son çalışmalara göre maternal yaş arttıkça trizomi 9’lu bireylerin sayısında
genel populasyonda önemli bir artış var.
Ama tekrarlama sıklığı (recurrance)bilinmiyor.

Trizomi 16
Trizomi 16 spontan abortuslarda görülen en sık otozomal anöploidi. Hamileliğin 35.haftasında bir ölü doğum vakası vardır. Nonmozaik trizomi 16 her zaman lethalolmuştur erken embriyonik veya fötal gelişim boyunca.
Mozaik trizomi 16’lı fetüsler bazen hayatta kalırlar. 10’dan az rapor edilmiş böyle vakavar.
Fenotipi;
Uterus içindeki büyüme gecikmeleri sabit. %50’den fazla vakada maternal serum hCG(human chorionic gonadotropin) veya a-fetoprotein seviyesi hamilelik boyunca yüksek.
Konjenital kalp bozuklukları %60 hastada gözleniyor. Doğum sonrası büyüme geriliği,hafif gelişimsel konuşma gecikmesi, yüzde asimetri, ptosis (organlarda sarkma), burunkemiğinde yassılaşma, kulakların normalden aşşağıda konumlanması, skolyoz, tırnakhipoplazisi….vb. yaklaşık olarak %50 hasta hayatlarının ilk bir yılında ölmekte.
Not: Uzun süreli bir takip bu hastalıkla alakalı yok ama Hajianpour ve Wang’in kişiselgözlemlerinde 5 yıldan fazla yaşayan vakalarda görülmüş.
Tekrarlama riski muhtemelen önemsiz!...

Trizomi 20
•Sık görülen otozomal bir anöploidi olasına rağmen, canlı doğumlarda oldukça nadir.
•Mozaik trizomi 20 ile ilgili çok az canlı doğum rapor edilmiş ve bunların hepsi fenotipolarak normallermiş.
•Vakalar uzun süre takip edildiğinde üç vakada hipopigmentasyon görülmüş ama önemlibir deformite ve zeka gerililiği görülmemiş.
•Nonmozaik trizomi 20 kaydedilmemiş.
Abort materyaldeki anormal fenotip ise;
Mikrosefali, yüz bozuklukları, kalp hasarları, üriner sistem anomalileri.
Trizomi 20 hücreleri çeşitli fötal dokuda görülmekte örneğin; böbrek, akciğer, özafagus,ince bağırsak ve deride.
Deri fibroblastlarının ve idrar çöküntülerinin (urine sediment) kültüründen postanalolarak belirlenmekte.
Göbek kordonundan alınan kanla da bir vaka tespit edilmiş ama daha sonraki çalışmalarperiferal kandan 4 aylıkken alınarak incelenmiş.
Bunlar dışında başka rapor yok !..
Terarlama riski önemsiz…

Trizomi 22
Trizomi 22 ilk 1971’de rapor edilmiş.
O günden sonra literatürde 20 canlı doğum var.
Fenotipi;
Mental-motor gerilik, mikrosefali, dış kulak kanalının olmayışı, lakrimal kanal
stenozu, ptozis, strabismus, kulak malformasyonu, yarık dudak, hipertelorizm,
mikrognata, yele boyun, konjenital kalp hastalığı (%80 hastada).
Deride hipopigmentasyon genellikle mozaik vakalarda görülmekte.
Birçok non mozaik hasta hayatının ilk yılında ölüyor. Raporlara geçen en
uzun yaşam süresi 3 yıl.
Mozaik hastalarda 20 yaş üzeri vaka var. Trizomi 22’li hücreler hem kan
lemfositlerinden hemde deri fibroblastların dan belirlenebilir.
Recurransı bilinmiyor.

Diğer nadir otozomal trizomiler
Başlangıçta da söylendiği gibi mozaik ve nonmozaik otozomal trizomiler(1 ve 11 den başka) canlı doğumlarda rapor edilmiş.
Trizomilerin tespiti sıklıkla spontan abortlarda ve prenatal testlerden eldeedilmekte.
Sonuç olarak canlı doğumlarda trizomilerin görülmesi oldukça nadirsadece izole vakalarda tespit edilmiş.
Mozaik trizomi 2 ile ilgili rapor edilen tek vaka var. 16 aylık bu çocuktamikrosefali, kaslarda zayıflık, büyüme ve gelişme geriliği tespit edilmiş.
Üç vaka var trizomi 3 ile alakalı, bunlardan bir tanesi ağır mentalretardasyonu olan 32 yaşında bir bayan. Klinik özelliği üç vaka için defarklı fakat benzer olan yüz anomalileri (göz ve kulak).

Mozaik trizomi 4 ve 5 için birer durum rapor edilmiş, her iki durumdada prenatal
amniyosentez yöntemi ile, trizomili hücreler kan lemfositlerinden ve deri
hücrelerinden belirlenmiş. Her iki vakanında konjenital anomalileri olduğu
belirlenmiş.
Bir durumda trizomi 6 ile ilgili, bu çocuk hamileliğin 25. haftasında doğuyor, klinik
özelliği kalp hasarları, büyük kulaklar, ayrık sağ el, sindaktili, düzensiz şekilde ve
uzunluktaki ayaklar, derideki kalınlaşma. Bu vakada büyüme yavaşlamasına rağmen
gelişme normal olarak ilerliyor 2 yaşına kadar. Trizomi 6 da deri fibrobllastlarından
tespit ediliyor.
En az altı tane trizomi 7 ile ilgili vaka var, trizomi 7’li hücreler deri fibroblastlarından
elde edilmiş. Tüm hastalar fenotipiksel olarak anormaller. Hepsinin ortak özelliği
büyüme ve gelişme geriliği, deri pigmentasyonunda kusur (hipopigmentasyon veya
hiperpigmentasyon), yüz ve vücutta asimetri. Bu vakayla ilgili bir örnek 18 yaşındaki
zeka geriliği olan bir erkek.

Birkaç vaka da mozaik trizomi 10 ile ilgili, bir vaka mozaik trizomi 10
idi. Deri fibroblastlarıda monozom X içeriyordu ve yine kan hücreleri
monozomi X içeriyordu. Bu vaka 7 haftalıkken kalp krizinden ölmüş.
Ortak klinik fenotip; büyüme hataları, yüz anormallikleri,
hipertelorizm, kalp hasarları, kısa ömür …vb.
En az 6 vaka da trizomi 12 için var, hepsi mozaik bir vaka kısır erkek,
bir diğeri birçok anormal kusur ve pigment bozukluğu ile doğuyor ve
2 aylıkken ölüyor. Fenotipik özelliği; yüz anormallikleri, skolyoz, kalp
hasarları, boy kısalığı ve zeka geriliği.
En az 15 vaka mozaik trizomi 15 ile ilgili, fenotipik abnormaliteler;
büyüme ve zeka geriliği, geniş burun, düşük kulak, küçük çene, kalp
hasarları. Yaşam şansları değişiyor 1 günden 29 yıla kadar.

Trizomi 14’lü hücreler hem lenfositlerden hemde fibroblastlardan belirlene bilir.Vucut asimetrisi olan hastalarda trizomik hücreler atrofik bölgelerde sınırlıdır.
En az on vakada trizomi 15 için kaydedilmiştir, bunlardan iki tanesi nonmozaik.Bazı vakalarda sadece trizomi 15 li hücreler deri hücrelerinde görünmüş fakatkan lenfositlerinde görülmemiş. İki vakada ise maternal uniparental dizomi 15,normal hücre hatlarının içerisinde bulunmuş ve bu vakalar çok ağır fenotipleresahiplerdi. Bu fenotipik anormallikler ise; hipotoni, çeşitli yüz anormalliği,minör iskelet anomalisi, kalp hasarları ve kısa ömür.
Mozaik trizomi 17 ile alakalı iki vaka var. Trizomi hücreler lemfositlerdegörülmedi ama yüksek oranda deri fibroblastlarında görüldü. Bir hasta 8yaşındaydı, zeka ve büyüme geriliği, mikrosefali, nöbet, duyma kaybı, dikkateksikliği ve hiperaktivite ve otistik davranışlar gösteriyordu.
Mozaik trizomi 19 ile ilgili literatürde iki vaka var bunlardan biri ölü doğmuşerkek, diğeri ise 13. günde ölmüş. Klinik özellikleri çok çeşitli ama anakusurlarla ilgili bir rapor yok.

Otozomal monozomiler
Otozomal monozomiler oldukça nadirler hem canlıdoğumlarda
hemde düşüklerde.
Semptomlarının ağırlığı genetik dengesizlik den dolayı bir
kromozomun tamamen ortadan kalmasıyla bağlantılı.
Monosomi 20, 21 ve 22 rapor edilen monosomiler. 3,5 yaşında
mozaik monozomi 20 hastası bir erkek çocuğundaki fenotipik
özllikler; atipik konuşma, dil öğreniminin gecikmesi, davranış
problemleri, mikrosefali ve derideki pigmentasyon sorunları.

Monosomi 21
Mosaik monozomi 21 için literatürde dört canlı doğumda rapor edilmiş.
En sık görülen özellikler; uterustaki büyüme geriliği, postnatal büyüme ve zeka geriliği,
hipertoni, göz kapaklarının normalden aşşağı olaması, kulakların daha aşşağı konuma
yerleşmesi ve küçük çene. En son rapor edilen vaka 20 haftalık bir dişi fetüs, yukarıdaki
anormalliklere ek olarak kompleks kalp hasarları, bağırsaklardaki yanlış rotasyon, çift rahim,
küçük olgunlaşmamış ovaryumlar ve akciğer hasarları rapor edilmiş.
Canlı doğumlarda yaklaşık on vaka rapor edilmiş mozaik monozomi 21 için. Bu vakalardan
bazılarının sonradan parsiyel monosomi 21 olduğu anlaşılmış çok ince bir translokasyondan
dolayı.
Gözlemler mozaik monozomi 21in nonmozaik monozomi 21’e göre daha az yaygın olduğunu
göstemiş, tam monozomi 21’in ise hayatla bağdaşmadığı açıklanmış.
Bir erkek çocuğu 11 yaşına kadar yaşamasına rağmen bir çok hasta 2 yaşından önce ölmüş.
Fenotipik özellikler;
Diğer mozaikliklere benziyor. Ör, büyüme ve zeka geriliği, mikrosefali, çıkık burun, ayrık
dudak, küçük çene..vb. ayrıca anormal kas tonusu ve çoğunlukla hipertoni oldukça yaygın. Kalp
anormallikleride birkaç vakada görülmüş.

Monozomi 22
Mozaik monozomi 22 ile ilgili dört vaka rapor edilmiş canlı doğumlarda.
Bir tanesi 34 haftalık premeture bebek, gastroşizis ile birlikte doğmuş doğumdan kısa bir süre sonra beyin kanamasından ölmüş.
Dismorfik özellikleri kaydedilmemiş ve otopsi de yapılmamış.
İki hastada büyüme ve gelişme geriliği, mikrosefali ve hafif yüz dismofizimi görülmüş.
Dördüncü hasta ise 30 haftalık premeture, yüz özellikleri DiGeorge sendromuna benziyor, yüksek tansiyon, ana eklemlerde sınırlı gerilme ve tüm parmakların bükülmesi gözlenen problemler.

Poliploidi
Poliploidiler toplam kromozom setindeki sayısal anormallikler. Genellikle hayatlabağdaşmamasına rağmen nadiren de canlı doğumlar görülebilir.
TriploidiKromozom sayısının 3n=69 olması durumudur. Tahmin edilen görülme oranı %1.Abortlarda ise %17-18 olduğu tespit edilmiş.
Hayatla bağdaşmaları çok nadir. İki farklı fenotip tanımlanmış. Bunlardan birisi iyibüyümüş fetüs, diğer özellikleri; mikrosefali, anormal büyüklükte plesanta (ayrıcaplesantadaki kistler) ve molar gebelik.
Gebelik ürününün genetik yapısı hem anne hem baba orjinlidir. Bunun sonucu triploidyapıdadır (en sık 69,XXY). Ebeveynlerden birinden 2n (diploid) sayıda kromozomgelirken diğerinden n (haploid) sayıda kromozom gelmektedir.
Triploidi ile ilgili çalışmalar sitogenetik heteromorfizim ve DNA polimorfizim analizi ileyapılmakta.
En son yapılan çalışmalarda, 87 triploidi vakasının hamileliğin 5-18 haftaları arası düşüklesonuçlandığı tespit edilmiş.

Fig. 9. Karyotype of a triploid fetus (69,XXX).

•Sex kromozomlarında ki triploidi hem XXX hemde XXY çok nadir olarak
da XYY görülür. Yapılan iki çalışmada spontan abortus sayısı (XXX; XXY;
XYY- 82:92:2 (3)) & (XXX; XXY; XYY- 26:36:1).
• Bir çalışmada amniyon sıvısındaki hücrelerle yapılmış ve oran 6:8:0
olarak rapor edilmiş. Fenotipik özelliklerin ise parental orijine bağlı olduğu
belirtilmiş.
•Elliden fazla nonmozaik triploidi rapor edilmiş hem 69,XXX hem de 69,
XXY. Bunların büyük çoğunluğu doğumdan kısa süre sonra ölmüş. Sekiz
hasta 2 aydan daha fazla yaşamış ve en uzun yaşayan ise ancak 10. aya
erişebilmiş.
•Ekstra set kromozomun kaynağı sitogenetik polimorfizim veya insan
lökosit antijeni (HLA) ile maternal 3 vakada paternal 1 vakada tespit
edilmiş. Bir çalışma DNA polimorfizm ile yapılmış bu vaka 46 gün
yaşamış maternal mayoz II deki hata sonucu triploidi görülmüş.
•Bu bulgular (digynic) maternal kaynaklı triploidilerin, (diandric) paternal
kaynaklı triploidilere göre daha uzun yaşadıklarını göstermiş.
•En sık görülen anormallikler; büyüme geriliği, hipotoni, baş, yüz
anormallikleri, ektrimitelerdeki bozukluklar, kalp hasarları ve beyin
anomalileri.

Mozaik triploidi (diploid/triploid mixoploidy) yaklaşık 20 hastada tespitedilmiş. Triploid hücreler hem lenfositler hemde fibroblastlardabulunmuş (fibroblastlarda nispeten daha az).
Bu hastalar nonmozaiklere göre daha az etkilenmişler ve 10 yılyaşadıkları gözlemlenmiş.
Genel klinik özellikleri büyüme geriliği, fizikomotor geriliği, asimetrikbüyüme, geniş burun, sindaktili, genital anomaliler, düzensiz deripigmentasyonu. Bazı hastalarda Truncal obezite görülmüş. Bu bireylerdeayrıca mitotik ayrılmama görülmemiş.
Mekanizması;
Muhtemel mekanizamsı çift döllenme (bir ovumun iki ayrı spermledöllenmesi). Bir sperm nukleusu ovum nukleusu ile birleştikten sonraerken blastomer safhasında başka bir spermin daha döllemesiyle triploidhücre oluşuyor. En az bir vaka için bu mekanizma kanıtlanmış.
Bir diğer mekanizmada moleküler olarak kanıtlanmıştır buna göre; erkenblastomer safhalarından birindeki ikinci polar body nin birleşmesiningecikmesi ile triploid hücre hatları oluşmaktadır (digynic).


Tetraploidi
Kromozom sayısı 4n=92. Triploidiye göre daha nadir. Spontanabortuslardaki görülme sıklığı %6-7. Tetraploidinin muhtemel orijini,zigotta kromozomların duplikasyonundan sonra sitoplazmabölünmesinin olmaması.
En az sekiz nonmozaik canlı doğum rapor edilmiş. Sex kromozomlarıbir birinin komplementi (XXXX veya XXYY). 92, XYYY veya XYYYhiç görülmemiş.

En sık görülen anormalite büyüme ve gelişme geriliği, hipotoni, baş-yüz
anormallikleri, ektremite anormallikleri ve böbrek anormallikleri. Bir çok hasta 1
yaşını doldurmadan öldü. Raporlanan sadece bir kız 22 aya ulaşıyor.
Raporlanan mozaik tetraploidi (diploid/tetraploid mixoploid) vaka sayısı 12.
teetraploid hücreler periferal kan lemfositlerinde, deri fibroblastlarında ve kemik
iliğinde görülmüş (tetraploid hücreler kemik iliği hücrelerinin %95’de görülüyor).
Yaşları 11 ve 21 olan iki kız çocuğunda ise ağır zeka özürü ve deri
pigmentasyonu displazisi görülmüş. Tetraploid hücreler ise sadece deri
fibroblastlarında bulunmuş. Lemfositlerde tetraploidi görülmesi yaşa bağlı olarak
azalıyor.
Sonuç olarak klinik özellikleri aynı ama nonmozaik tetraploidiye göre daha hafif
ve tahmini yaşam süreleri daha uzun. Rapor edilen en uzun yaş, 6 olarak kayda
geçmiş.

Parsiyel otozomal anöploidiler
• Tetrazomi 5p
Tetrazomi 5p [47,XX veya XY, +i(5) (p10)] ekstra isokromozomunolması 5. kromozomun kısa kolunda.
Oldukça nadir sadece üç canlı doğum var ve bunların hepsi mozaik.Bu anormal hücre hatları lemfositler de, deri fibroblastlarında vekondrositlerde bulunmuş.
Fenotip trizomi 5p’ye çok benziyor. Hipotoni, nöbetler ve anormalEEG, fizikomotor retardasyon, makrosefali, yüz anormallikleri vesolunum zorlukları gözlemlenen bulgular. Bir hastada deridehiperpigmentasyon görülmüş. Yaşam süreleri değişken bir hasta 6aylıkken ölmüş, bir diğeri ise 5 yaşında ölmüş.

Tetrazomi 8p
Tetrazomi 8p [47,XX veya XY, +(8)(p10)] genellikle eksta
isokromozomun 8. kromozomun kısa kolundan köken alır.
Tüm vakaların mozaik olduğu rapor edilmiş. Bu anormal hücre
hatları lemfositlerde hemde deri fibroblastlarında bulunmuş.
11 vaka tespit edilmiş bunlardan bazıları hayatlarının 5. yılında
ölmüş ama 5 yılın üzeri yaygın değil.
Doğumda kilo ve baş circumference normaldi. Sıklıkla gözlenen
fenotipik özlliklerik ise; mental gerilik, konuşma ve motor
haretlerdeki yavaşlık, serebral ventriküllerin dilasyonu hafif yüz
anomalileri ve omurga anomalileri.

Tetrazomi 9p
• Tetrazomi 9p [47,XX veya XY, +i(9)(p10)] ekstra isokromozomdurumudur. 20 canlıdoğumda rapor edilmiştir.
• İzokromozom bazı durumlarda hem tüm kısa kolu içerdiği gibi, bazı durumlardada kısa kolun hepsini ve uzun kolunda heterokromatinbölgesini içere bilir.
• Bazı durumlardada yine uzun kolun ökromatin bölgesini içere bilir. Gözlemlenen fenotipik özellikler üç çeşit.
• Hem mozaik hemde nonmozaik hastalar rapor edilmiş. Yine hücre hatları deri fibroblastlarından b-ve lemfositlerden alına bilir.
• Yaşam şansları çeşitli birkaç saat ile 10 yıl arasında değişmekte. En sık görülen fenotipik anormalite; düşük doğum kilosu, büyüme ve gelişme gecikmesi, baş-yüz anomalileri, kısa boyun, iskelet anomalileri, eklem kontraktürü, tırnak hipopalazisi, ürogenital anomaliler. Hastalrınyarısında kalp hasarları vardı.

Tetrazomi 12p
• Tetrazomi 12p (Pallister-Killian syndrome) ekstra izokromozom sonucunda görülür. 12. kromozomun tüm kısa kolunda [i(12)(p10) veya i(12p).
• İlk olarak 1977 yılında Pallister ve ark, tarafından 37 yaşında bir erkekte ve 19 yaşında bir bayanda tanımladı. 60’dan fazla vaka var ve bunalrın hepsi mozaik.
• Maternal yaş arttıkça vakaların görülme sıklığı artmış. Bu gözlemler mayozdaki hatalar sonucu izokromozomun arttığını göstermiş. i(12p) yüksek oranda deri fibroblastlarında, amniositlerde ve nadirende lemfositlerdebulunmuş.
• Ayrıca bu izokromozomun bulunma oranı yaş arttıkça azalmış. Bir çok hasta doğumdan hemen sonra ölmüş.
• Genellikle büyüme parametreleri doğumda normal. Yenidoğanlarda şiddetli hipotoni ve kontraktürlerin geç oluşması. Belirgin alın, büyük kulaklar, hipertelorizm, epikantal katlanmalar, geniş, kısa ve yassılaşmış burun, kalkık burun delikleri, ince üst dudak, yüksek damak. Birçok hastada hiper- ve hipopigmentasyon. Diğer anormaliteler; kısa boyun, uzun dil, küçük çene ve ürogenital anormaliteler. Ağır zeka geriliği ve nöbetler yaşam boyu görülen durumlar. Tüm vakalar sporadik ve tekrarlama sıklığı önemsiz.

Tetrazomi 8p
Tetrazomi 18p [47,XX veya XY, +i(18)(p10)] kromozom 18’in kısa kolunun hepsinin izokromozom olmasıyla alakalı.
Bu sendrom ilk kez Froland ve ark, tarafından 1963 yılında tanımlandı. En az 50 vaka rapor edilmiş ve bunların çoğunluğu nonmozaik. Tetrazomi 18 lemfositlerden kolaylıklar tespit edşle bilir.
En sık gözlenen klinik bulgular; düşük kilo, küçük kafa, beslenme problemleri, çeşitli fizikomotor retardasyon, nöbetler, oval yüz, hilal kaşlar, displazik kulaklar, üçgen ağız, yüksek damak, küçük çene, dar omuzlar ve dar toraks, kalp hasarları, ürogenital anomaliler. Tetrazomi 18’in anne yaşıyla olan ilişkisi bilinmemekte.

Diğer parsiyel anöploidiEkstra marker kromozom
Tetrazomilere ek olarak parsiyel otozomal anöploidiler, küçük ekstra marker kromozomların varlığıyla alakalı.
Frekansı 1000 yenidoğumda yaklaşık 0.7. Sitogenetik orijinleri bilinmemekte.
FISH tekniğiyle bu marker kromozomlar belirlenmekte. Mozaik formlar içerisinde bu ekstra markır kromozomlar stallit ve non-stallit diye sınıflandırılırlar.
Markır kromozomlar sadece heterokromatin bölgeleri içerirler. Diğeryandan bu kromozomlar üzerindeki ökromatinler genellikle iyi huylu değil ve fenotipik abnormalitelere sebep olur.
NOT: Markırlar tüm otozomal kromozomlarda rapor edilmiş. En yaygın markır 15. kromozomdaki inversiyon duplikasyonu [invdup(15)].
Se ddistribuie ggratuit - Pucioasa 2017. 5. 8.آ ploi. أژn Liturghier, la slujba care se oficiazأ£ la
Lucian Vasile - obiectiv- Ploi n SPORT n POLITIC£’ n POLITIC£’ n ACTUALITATE Verde pentru promovare!