analysis of the decarboxylation step in mammalian
TRANSCRIPT

ANALYSIS OF THE DECARBOXYLATION STEP IN MAMMALIAN HISTIDINE DECARBOXYLASE: A COMPUTATIONAL STUDY*
Aurelio A. Moya-García1, Javier Ruiz-Pernía2, Sergio Martí3, Francisca Sánchez-Jiménez1 and Iñaki Tuñón2 .
From 1Procel Lab. Departamento de Biología Molecular y Bioquímica. Facultad de Ciencias, Universidad de Málaga, CIBER-ER. Campus Teatinos, 29071 Málaga. Spain.
2Departamento de Química Física, Universitat de València, 46100 Burjassot, València, Spain 3Departament de Química-Física i Analítica, Universitat Jaume I, 12071 Castelló, Spain
E-mail to: Aurelio A. Moya-García [email protected] and Iñaki Tuñón [email protected]
We report a hybrid quantum mechanics/molecular mechanics theoretical study on the reaction mechanism of mammalian histidine decarboxylase, that allows us to obtain valuable insights on the structure of the cofactor-substrate adduct (external aldimine) in the active site of rat histidine decarboxylase. By means of molecular dynamics simulations, we t r a c e d t h e p o t e n t i a l o f m e a n f o rc e corresponding to the decarboxylation reaction of the adduct in both the active site of the enzyme and in aqueous solution. By comparing this process in both media, we have identified the key electrostatic interactions that explain the lowering of the free energy barrier in the enzyme. Our analysis also offers a validation of Dunathan’s hypothesis [Proc. Natl. Acad. Sci. 1966, 55 , 712] regarding the role of stereoelectronic effects in the enzymatic decarboxylation process.
Introduction
Computational approaches to investigation of enzymatic catalysis rely on the ability to obtain a proper initial structure for the system under study. Three-dimensional structures are best determined by experimental methods such as X-ray diffraction, nuclear magnetic resonance (NMR) spectroscopy, or even electron microscopy (1). However, experimental methods cannot always be applied. Many proteins are too large for NMR analysis and cannot be expressed or purified in an amount substantial enough to be crystallized for X-ray diffraction; consequently, there are no corresponding 3D structures for the majority of protein sequences. In such cases, prediction of the protein structure by computational methods can frequently result in a useful model. However, the application of a comparative model depends on its accuracy. A strong predictor is the sequence identity between the target and the known protein structure used as a template; there is a broad application spectrum based on this identity. The high end of the accuracy spectrum corresponds to models based on 50% identity or more; the average accuracy of these models is about 1 Å root
mean square (RMS) for the main-chain atoms, which is comparable to that of low-resolution X-ray structures (2).
Mammalian histidine decarboxylase (HDC. EC 4.1.1.22) is one of the enzymes for which there is no experimentally solved structure due to its instability in vivo, crude extracts and purified solutions (3,4). This is a Pyridoxal 5’-Phosphate (PLP) dependent enzyme that synthesizes histamine by decarboxylation of histidine. The importance of this enzyme is obvious considering that the biogenic amine histamine (2-(4-imidazolyl)ethylamine) is a multifunctional intercellullar mediator involved in many different physiological responses in animals (hematopoiesis, gastric secretion, neurotransmission) (5-8). It is also related to many pathological processes (gas t r ic u lcer, a l lergy and many other inflammatory diseases, bone lost, tumor progression, learning deficiency and epilepsy, etc) (9-13). Thus, the control of its synthesis should be interesting for therapy against these pathologies that affect a high population rate at any stage of their lives. Nevertheless, nowadays, anti-histamine pharmacology is based on agonists and antagonists of the histamine reception in target cells, but not on the development of modulators of its synthesis. This is mainly due to the lack of information about the proper targets for intervention at the level of its synthesis in the different histamine-producing cell types (3).
Gram positive bacteria express a non-homologous pyruvoyl-dependent HDC, but Enterobacteria express a PLP-dependent HDC that is evolutionary related to the mammalian enzyme (14). Besides, several mammalian cell types (mainly neurons) also express a highly homologous PLP-dependent enzyme which is responsible for the synthesis of several neurotransmitters, named dopa decarboxylase or L-aromatic amino acid decarboxylase (DDC/AADC) (15-17). According to these facts, it has been difficult to develop specific modulators of mammalian histidine decarboxylase activity. In actuality, synthetic substrate analogs such as fluoromethylhistidine and histidine methyl ester
1
http://www.jbc.org/cgi/doi/10.1074/jbc.M707434200The latest version is at JBC Papers in Press. Published on February 29, 2008 as Manuscript M707434200
Copyright 2008 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

can inhibit both the Gram-negative and the mammalian HDC (18); other inhibitors, such as epigalocathequine-3-gallate, have been described as inhibitors of both mammalian HDC and DDC/AADC (19). As a consequence, a deeper structural characterization of the mammalian enzyme is needed to locate the best targets for selective intervention.
The first 3D model of a mammalian enzyme (rat HDC or rHDC) was obtained in 2003 based on a pig dopa decarboxylase (pDDC) structure as the template (sequence identity of 52%) (18). This structure was then validated with experimental results which were obtained from more than 25 direct mutants (20,21) so that the functional data on the enzyme fit well with the proposed model of the enzyme (3). The agreement between our proposed rHDC structure and the experimental data available has led us to perform a detailed computational study aimed to unravel special topics of the catalytic mechanism, namely the location of the cofactor-substrate adduct in the active site, as well as its correct conformation for decarboxylation. Such an effort offers additional and definitive validation of our model as a reliable representation of the structure of mammalian histidine decarboxylase, and contributes to extending the scope of computational focus on enzyme catalysis.
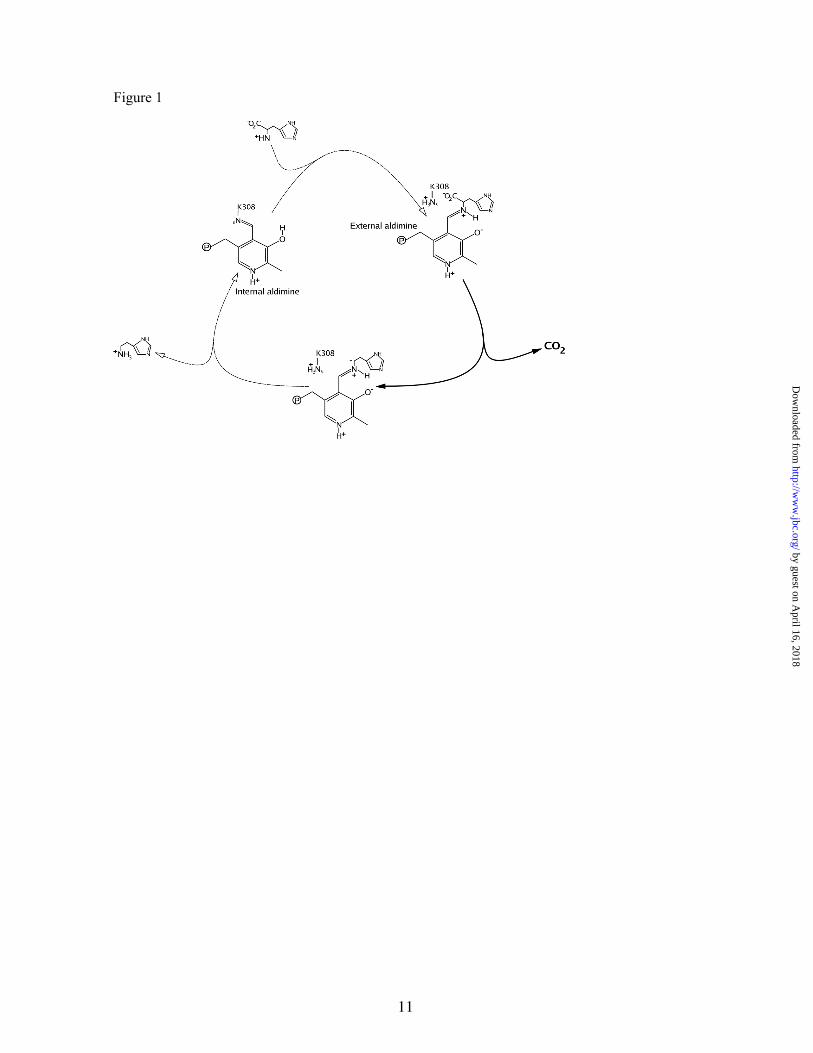
Catalysis by pyridoxal 5’-phosphate (PLP)-dependent enzymes has been extensively reviewed (23,24). The PLP-dependent decarboxylase catalytic cycle begins with the cofactor covalently bound to the ε -amino group of a specific and conserved Lys residue (K308 in rHDC) in a Schiff base known as internal aldimine. After substrate binding, the external aldimine intermediate (PLP-His) is formed via a reversible transaldimination process; then, decarboxylation occurs, histamine is released from the catalytic site, and the enzyme recovers the internal aldimine state (Figure 1). Any of these processes may be the rate-limiting step of the entire reaction rate. In fact, kcat of the mammalian enzyme is extremely low (lower than 0.1 s-1), in contrast to those higher values (2-3 orders of magnitude) reported for both homologous enzymes, mammalian DDC/AADCs, and HDC from Enterobacteria (25,26).
As revealed by structure/function relationship studies carried out in ours and other labs, both mammalian HDCs and DDC/AADCs are described as homodimeric enzymes (18-21). Their active sites must be similar and involve residues of both monomers. PLP-binding residues are mainly located in one of them, but both monomers
contribute to the substrate binding site (16,18,21). By means of different biophysical techniques (circular dichroism, electrophoresis under semi-denaturing conditions and gel fi l tration chromatography), it was detected that local conformational changes occur after histidine binding and then are transmitted to the global conformation of the mammalian histidine decarboxylase (18). These facts have not been observed in other homologous enzymes, at least in such extent (21,26-28). Transaldimination leads to formation of the external aldimine while the conserved lysine residue is released. This external aldimine can exist in different prototropic forms, although the predominant form seems to be that which presents the pyridine and the amide nitrogen atoms as protonated while the 3’-hydroxyl group is deprotonated (29) (see Figure 2).
There are two possible ways of histidine binding to PLP which lead to two different conformations for the external aldimine. We have named them as conformation 1 , for the conformation in which the side chain of the substrate points towards the re face of the imino sp2 center (i.e. opposite to the PLP-binding lysine), and conformation 2, for that in which the substrate points towards the si face of the cofactor. Kern et al. (30) asserted that the PLP-His position in the group II amino acid decarboxylase folding family corresponds to the conformation 1. When the structure of pDDC was solved, a conformation similar to conformation 2 was observed for the PLP-carbiDOPA adduct in the catalytic site (16).
In this work, we carry out a theoretical analysis of external aldimine decarboxylation in the active site of rHDC. The structure of the enzyme, obtained from homology models, is used to explore the catalytic properties of the enzyme by means of Quantum Mechanics/Molecular Mechanics (QM/MM) simulations. The first part of the study is devoted to investigation of an appropriate model of the external aldimine in the active site. We investigate different possible structures for this external aldimine, concluding that conformation 1 seems to be more appropriate for the external aldimine considered in this case. In the second part, we simulate the decarboxylation reaction in the active site and compare it to the uncatalyzed process in water.
2
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Methodology
A homology model for the structure of rat histidine decarboxylase reported by the authors (18) and validated by directed mutagenesis (3,21) was used to manually fit the Schiff base formed by the histidine substrate and the cofactor PLP into the active cleft in two different ways; this was done in order to obtain the two starting conformations of the external aldimine.
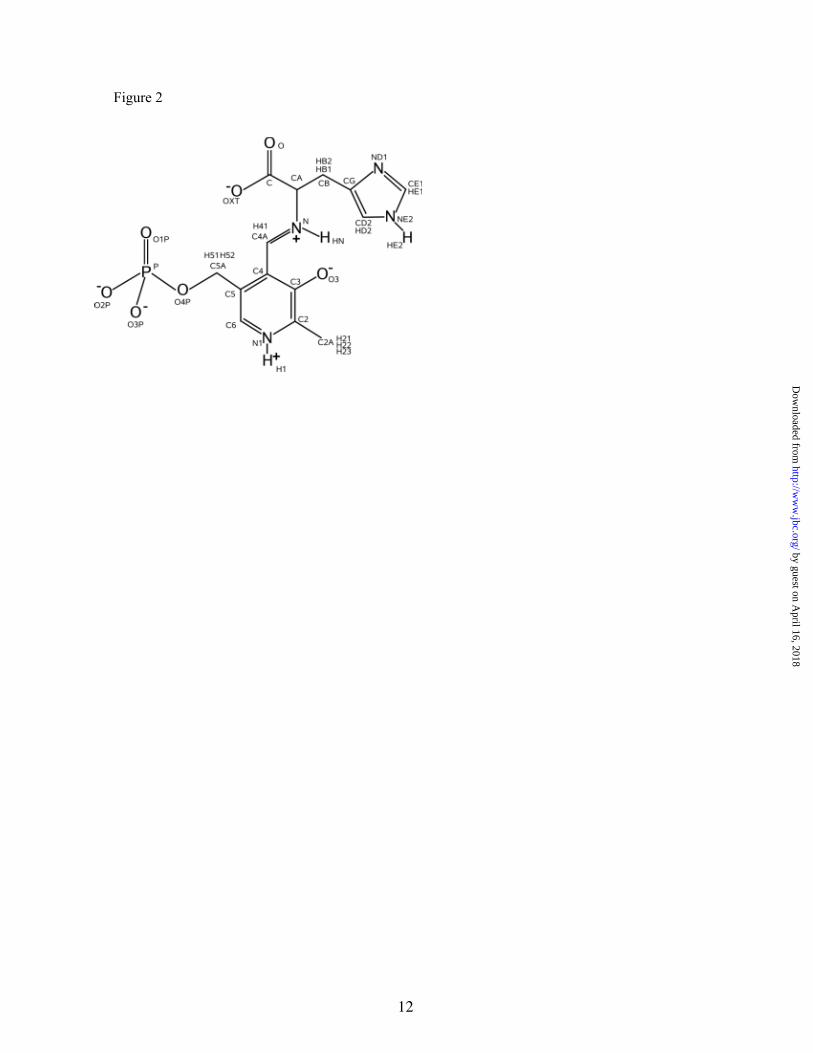
Calculations were performed by means of a QM/MM computational scheme (31-33) using the CHARMM 27 package (34). The external aldimine was chosen as the QM subsystem (described using the AM1 hamiltonian) (35) while the MM subsystem was composed of both protein residues and water molecules. To achieve a reasonable agreement between performance and calculation time for this large protein (15054 atoms), a simulation protocol was followed (36,37). First, the coordinates of the hydrogen atoms of the protein were determined using the HBUILD program of the CHARMM package, where all the ionizable groups were set to their normal ionization state at pH 7. Then, the protein was relaxed using a potential restraining scheme with a low force constant (25 kcal·mol-1Å-2) applied only on the peptidic backbone. Finally, the optimized protein was solvated with a 24 Å radius sphere of TIP3P water molecules, using atom C4 of the substrate (see Figure 2 for atom numbering) as the geometrical center of the system. Water molecules that were within a distance of 2.5 Å of any non-hydrogen atom were removed. The resulting system was resolvated four more times using different relative orientations between the protein and the water sphere to ensure good solvation of the system. Then, water positions were optimized using the Adopted Basis Newton-Raphson (ABNR) algorithm, followed by optimization of that part of the system included in the sphere of 24 Å around C4. Finally, a molecular dynamics simulation of the water molecules (5 ps) was carried out to relax energetically unfavorable contacts, and the 3-fold cycle of superposition, deletion, and rotation was then repeated to fill in additional cavities generated during the dynamic simulation. Thus, the final model has 16352 atoms; 15054 of them are atoms from the protein, 41 from the cofactor-substrate adduct, and 1257 atoms are from the water molecules. The residues lying outside of this 24 Å sphere were kept fixed for subsequent calculations, while the residues inside the 20 Å sphere from C4 were totally free. A buffer zone was described for the residues and water molecules in the zone between 20 and 24 Å. The
movement of the atoms in this buffer region was restrained following a protocol detailed in ref. 36. In addition, a switched cut-off radius from 12 to 13.5 Å was used for all interactions. The final structure was first heated to 310 K during 30 ps and then equilibrated using hybrid QM/MM Langevin-Verlet molecular dynamics (NVT) over 500 ps. We also studied the decarboxylation step in solution, using the same QM/MM methodology. In this case, the aldimine (QM subsystem) was placed in a cavity deleted from a 55.8 Å cubic box of water molecules, described using the classical TIP3P potential (a total of 5804 water molecules). For all calculations performed in water, periodic boundary conditions were applied, making use of the same swi tched cut -off radius . The conformational changes and decarboxylation reactions were studied through computation of the corresponding Potentials of Mean Force (PMFs) using the canonical ensemble (NVT). A PMF was traced using the χ dihedral angle (see Figure 2) as the distinguished coordinate in order to investigate the relative orientation of the carboxylate group and the pyridine ring. A total of 12 simulation windows were run to cover coordinate values from -80 to -180 degrees for this χ transition. Each window was started from the final conformation of the previous window and consisted of 10 ps of equilibration and 80 ps of production, using a time step of 1 fs. Changes in the selected reaction coordinate were restrained using an umbrella potent ia l wi th a force constant of 1 .2 kcal·mol-1degree-2 and an additional biasing potential (39). A Potential of Mean Force was also computed using the distance between CA and C (see Figure 2) as the reaction coordinate. In this case, we ran 17 windows to cover a reaction coordinate range from 1.4 Å to 3.0 Å. Each consecutive window consisted of 10 ps of production at 310 K, with a time step of 1 fs and a force constant of 500 kcal·mol-1Å-2. In both cases, the reference temperature was 310 K. The different values of the variable sampled during the simulations were then pieced together by means of the weighted histogram analysis method (WHAM) (38) to construct the distribution functions from which the PMFs were derived.
Finally, the same chemical step was studied in water solution. The reaction coordinate, defined also as the distance between the CA and C atoms, was sampled along a total of 65 windows. Langevin-Verlet NVT molecular dynamics were applied to each window, running for a total of 10 ps for relaxation, followed by 15 ps for production. The umbrella constant applied on each particular value of the reaction coordinate was 600
3
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

kcal·mol-1·Å-2, and the temperature of the bath was 310 K. As before, WHAM was used to render the PMF.
Results and Discussion
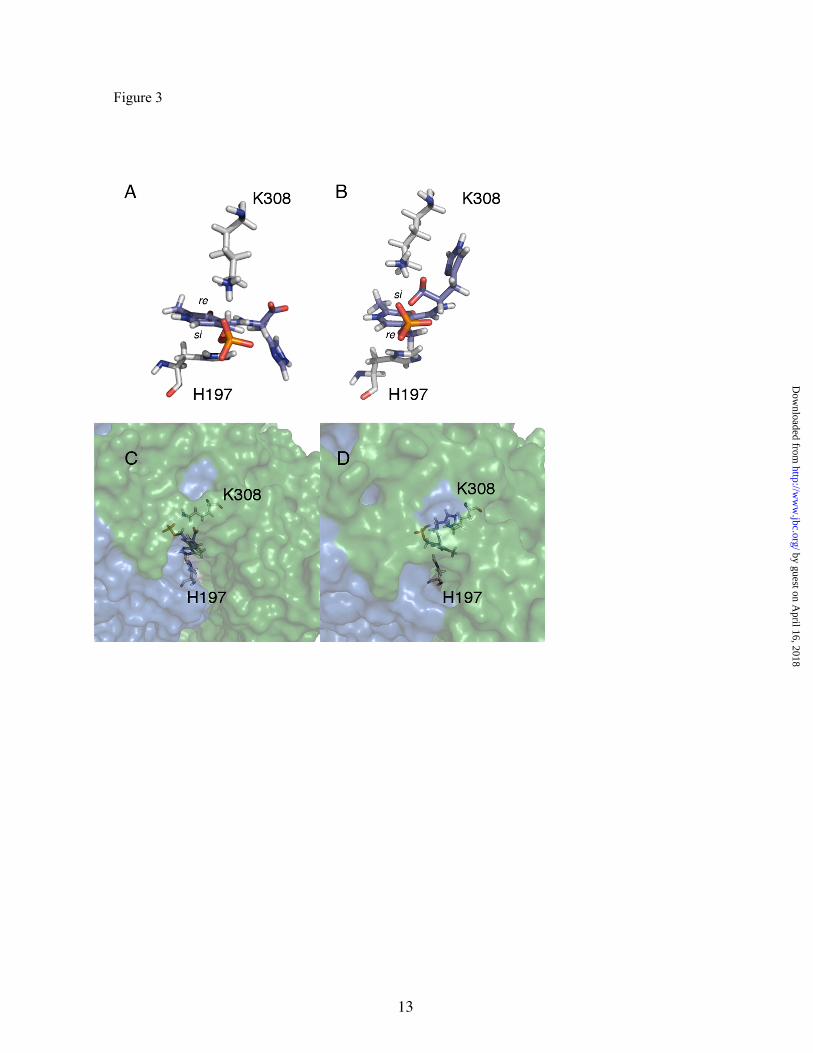
External aldimine conformation- The external aldimine is formed in the pDDC active site from PLP and the inhibitor carbiDOPA, the substrate analog pointed towards the si face of the cofactor in a conformation similar to conformation 2 in Figure 3 (16). An interesting discussion has been reported on the suitability of binding the substrate to PLP towards the re or si face (40). Matsuda and coworkers suggest that because the inhibitor carbiDOPA has an α-hydrazino group with an extra nitrogen atom (compared to the α-amino group of DOPA and His), the PLP-carbiDOPA complex is expected to adopt a significantly different conformation from that of PLP-DOPA, and consequently the PLP-His in the HDC active site. This issue has important consequences for the relative orientation of the substrate with respect to the enzyme. In the conformation 2, the sidechain of the substrate points towards the interior of the protein, while in the conformation 1 points outwards the enzyme (see figure 3, panels C and D).
To solve this question, we built both conformations for the external aldimine complex. The two conformations of the external aldimine were fully minimized in the active site using the QM/MM hybrid potential described in the previous section. After geometry optimization, we obtained the conformations presented in Figure 3.
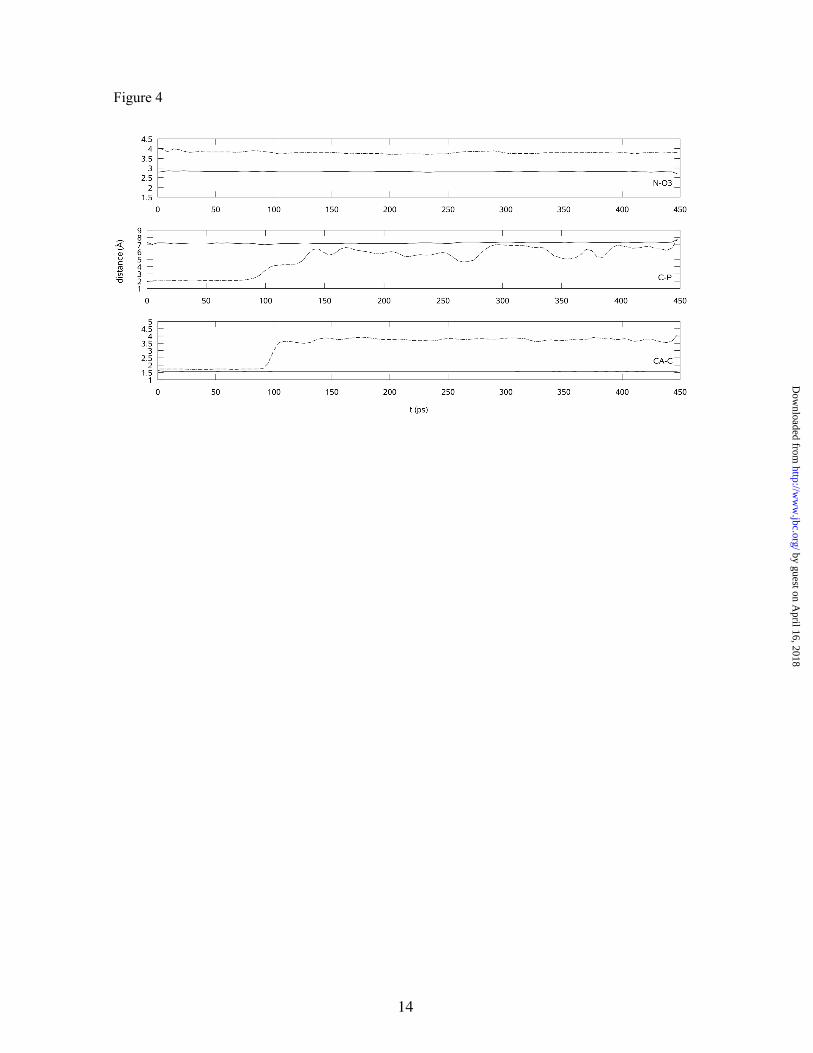
It is important to note that conformation 2 presents a too-short distance between two negatively charged groups of the molecule: the phosphate and the carboxylate groups. This high-energy conformation of the external aldimine is then very unlike to be appropriate for describing the product of the transaldimination. To confirm this point, the full system (cofactor-substrate complex, protein, and water molecules) was heated to 310 K and then 450 ps of NVT/MD simulation were run for each of the two conformations. Figure 4 displays the time evolution of some key distances for both cases: the distance between the carbon atom of the carboxylate group and the phosphorous atom of the phosphate group (C-P), the distance between the imino nitrogen atom and the negatively charged O3 atom (N-O3), and the CA-C bond length. As can be observed, the conformation 2 evolves through spontaneous decarboxylation after 100 ps. The only way found in this conformation for diminishing the
electrostatic repulsion between the phosphate and carboxylate groups is to lengthen the CA-C distance, leading to decarboxylation of the transaldimination product. On the other hand, the conformation 1 remains stable during the simulation. Another noticeable difference between the two conformations is that the first form retains a strong and biologically relevant intramolecular hydrogen bond between the imino nitrogen and the O3 atom (23) (the O3-N distance fluctuates around 2.2Å), while in the other form, the N atom forms a hydrogen bond with a water molecule. We can thus consider that the conformation 2 does not properly represent the external aldimine, while the first conformation may be considered as a realistic model in agreement with Matsuda’s proposal; therefore, subsequent studies were performed on this conformation.
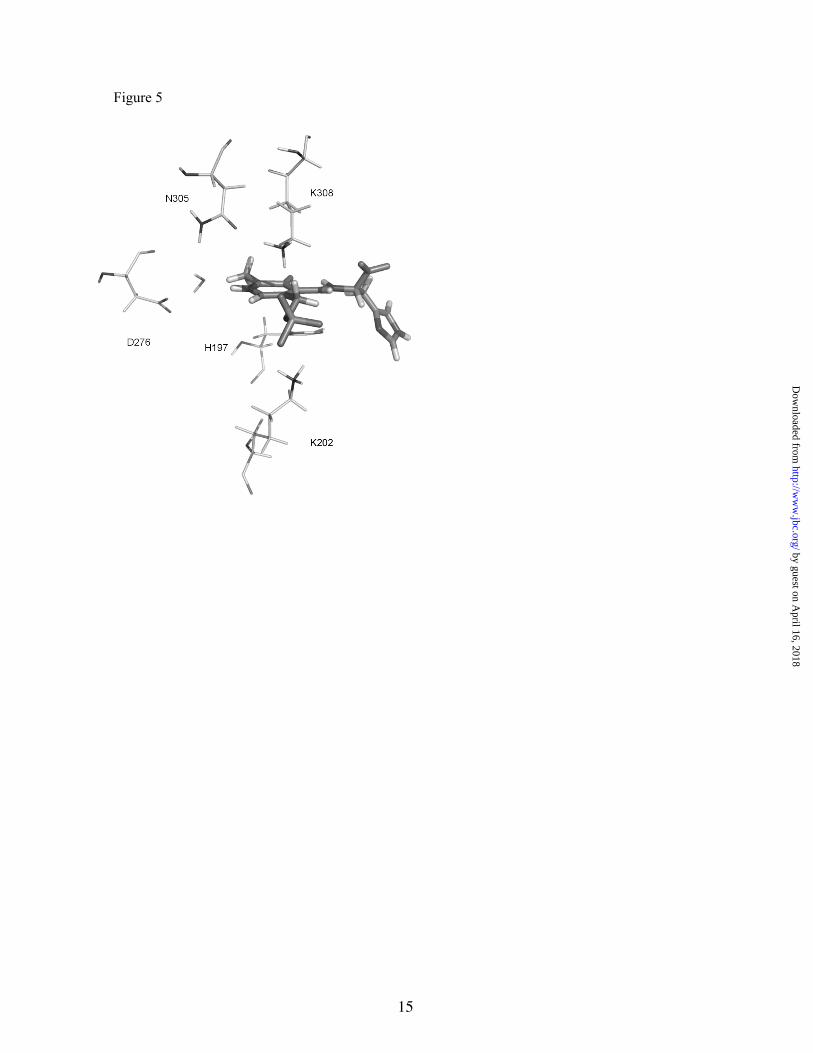
Active site description- Once the conformation of the external aldimine in the active site seemed to be well-established, we could concentrate on the most important interactions observed with the active site residues. We monitored distances between heteroatoms of PLP-His and those of the closest-neighbor amino acids to localize important hydrogen bonds. Figure 5 shows the position of some important residues of the active site interacting with the external aldimine in the conformation 1.
Residues located less than 5Å from the PLP moiety are (A and B are used to distinguish both monomers): V100A, T152A, V153A, S154A, H197A, S199A, K202A, T247A, T250A, T251A, D276A, K308A, F314A, P355B, and L356B; those closer to the substrate moiety are: Y83A, Y84A, I439A, F331B, M350B, H351B, and I354B. There is experimental evidence for an essential role in mammalian HDC activity for those residues highlighted in italics (21).
The active site is located in the monomer-monomer interface but is mainly composed of residues from one monomer; as in pDDC (16), residues of the opposite monomer participating in the active site are mainly hydrophobic and noteworthy, there are a number of water molecules participating in direct interactions with both the cofactor and substrate. There are several non-polar residues that are meant to increase the hydrophobicity of the active site in order to facilitate the introduction of an aromatic substrate such as histidine. This is also a common feature of aromatic amino acid decarboxylases (23). It is also noteworthy that there are a number of water molecules (only one is shown in the figure)
4
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

participating in direct interactions with both the cofactor and the substrate.
K308 is the residue that binds PLP covalently to form the internal aldimine. This residue is shifted during transaldimination and is responsible for product release (23). Interestingly, in the external aldimine state, this lysine, together with K202, interacts with the phosphate group of PLP, providing extra binding energy to the adduct.
K202 can establish this interaction after a conformational change of its sidechain. The charge-charge interactions established between these two lysine residues and the phosphate group of PLP play a very important role in providing a favorable environment for the external aldimine. The averaged distances from the Nε atoms of lysine residues to the closest oxygen atoms of the phosphate groups are 2.73 and 3.27 Å for K308 and K202, respectively.
H197, another highly conserved residue, is stacked parallel to the cofactor pyridine ring. Its analogue position in pDDC (H192) is predicted to form a hydrogen bond with the carboxylate group of carbiDOPA (16). The authors assume that the natural substrate, L-DOPA, should be located even closer to the imidazole ring of H192, so they suggested a direct catalytic role for this residue. In our model, taking into account the different location of the histidine substrate in the conformation 1, H197 cannot interact with the carboxylate group of the substrate, so an active catalytic role is not expected for this residue. This finding is supported by the previous observation that a H197G direct mutant still retains 10% of the wild-type HDC activity (21).
D276 and its counterpart in pDDC D271 were predicted to contact the pyridine ring of PLP (16,18). In the case of the pDDC crystal structure, D271 can form a hydrogen bond with the protonated nitrogen of the pyridine ring (N1). This direct interaction is not observed in rHDC, although D276 can still stabilize the protonated state of the pyridine ring by a water-mediated hydrogen bonding interaction (see Figure 5). The external aldimine is free to move in the active site cavity and prior to the molecular dynamics simulations, it is allowed to relax and adopt a suitable position in the active site. The permanence of this water molecule, acting as a bridge between D276 and the external aldimine, over the equilibration and production molecular dynamics trajectory, indicates that this water molecule has a structural (and functional) role. Moreover, a translation of the substrate cofactor adduct deeply in the active site cavity towards D276, would mean
the loss of important stabilizing interactions such as those established by K308 and K202 with the phosphate group of PLP, and clashes between the sidechain of the substrate and other active site residues.
According to a previously reported model of the structure of rat aromatic amino acid decarboxylase (rAADC) and the subsequent directed mutagenesis analysis (40), D271 corresponding in rAADC to D276 in rHDC is also essential for rAADC activity. Interestingly, a D271E mutant conserves 2% of the wild-type activity while noting that lengthening the distance between the PLP and the negatively charged group leads to an important reduction of the enzymatic activity. The assumed role for this interaction is to enhance the electron delocalization along the conjugated π system, which is commonly accepted to stabilize PLP-dependent enzyme intermediates (32,41). The distance between the carboxylate group of D271 and the N1 atom in the pDDC crystal structure is 2.50 Å, much lower than the corresponding distance between D276 and the N1 in our model (4.38 Å) where a water molecule is placed in between the carboxylate group of aspartate and PLP. As said, this larger separation could partly explain the lower catalytic efficiency of rHDC when compared to pDDC and rAADC.
The equivalent residue to N305, N300 in pDDC, was predicted to play a relevant role in catalysis due to its close interaction to the phosphate group (16). This residue has an identical counterpart in other mammalian group II decarboxylases , such as g lu tamic ac id decarboxylase, in addition to pDDC (21). In our system, N305 is not predicted to have such importance in the external aldimine state. This residue is found 5 Å away from the oxygens of the phosphate group of PLP. As far as we know, there is no experimental support for a role of this residue, neither in pDDC nor in rHDC.
Dunathan’s hypothesis- Another important question related to the structure of the external aldimine in the rHDC active site is the orientation of the bond to be broken during decarboxylation (CA-C bond) with respect to the conjugated π system. Dunathan (42) suggested that PLP-dependent enzymes can favor the reaction by means of stereoelectronic effects, orienting the nascent p orbital parallel to the p orbitals of the π electronic system. Crystal structures of several PLP-dependent enzymes (43,44) support this observation. In the gas phase, there are three main contributions which determine the relative positioning of the CA-C bond relative to the plane
5
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

of the conjugated system: the intramolecular hydrogen bond between the carboxylate and the protonated imino groups (which should favor a dihedral angle of 180 degrees), the electrostatic repulsion between the carboxylate group and the O3 atom (favoring a dihedral angle of 0 degrees), and the stereoelectronic effect (which stabilizes a dihedral angle of ±90 degrees). Using a reduced model for the external aldimine (where the histidine moiety is substituted by a hydrogen atom), we determined that the gas phase AM1 energy minima appeared at the χ (C-CA-N-C4A) dihedral angle of approximately ±50 degrees. It seems that the value of this dihedral angle in the gas phase minimum is dominated by electrostatic repulsion and stereoelectronic effects, although the minimum energy orientation is far from the preferred value for decarboxylation (±90 degrees).
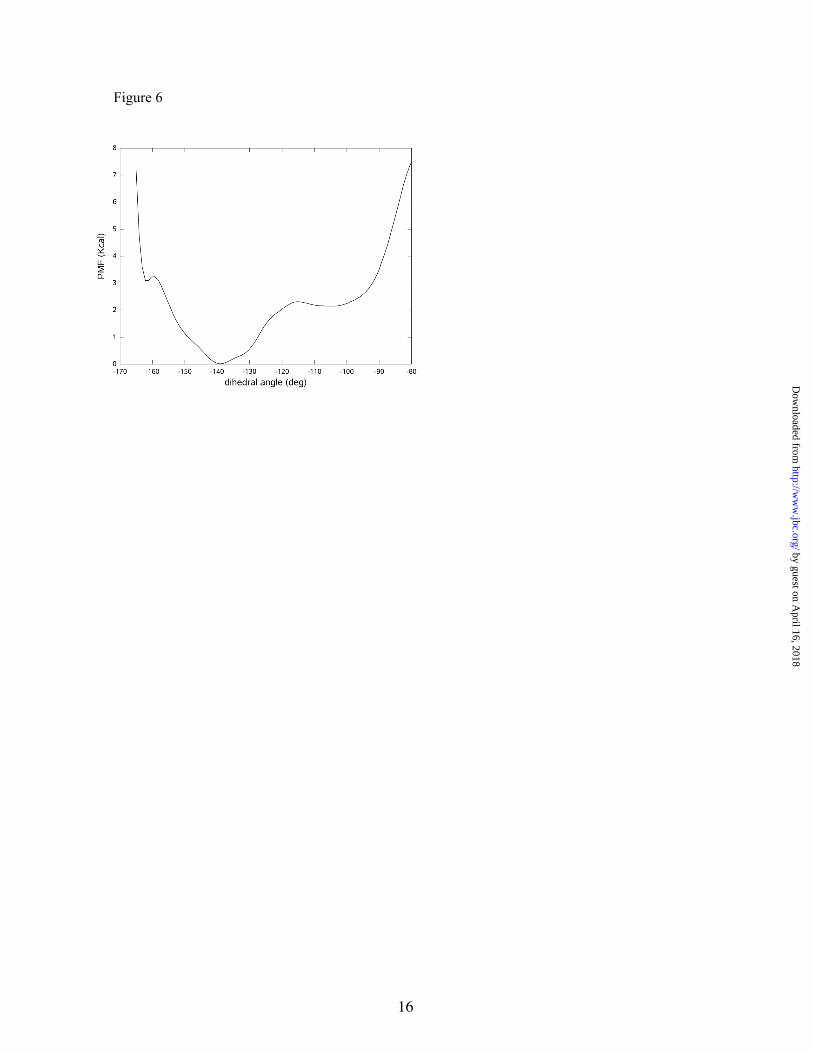
To investigate Dunathan’s hypothesis, we traced the free energy profile associated with the χ dihedral angle in the enzyme. This PMF, presented in Figure 6, shows two free energy minima placed at approximately -105 and -140 degrees, with the latter being the more stable by about 2.2 kcal/mol.
This result means that the enzymatic environment induces considerable changes in the conformation of the external a ldimine. Intermolecular interactions in the protein residues and water molecules very efficiently shield the electrostatic repulsion between the negatively charged carboxylate group and the O3 atom. In this way, both the formation of an intramolecular hydrogen bond between the carboxylate and the protonated imino groups and the stereoelectronic effect dominate the energy profile associated with the χ dihedral angle in the enzyme. In the free energy minimum appearing at -140 degrees, the position of the carboxylate group is favored by means of intramolecular hydrogen bonds with the protonated imino nitrogen (average O-N distance of 2.85 Å), with the hydroxyl group of T251 (average O-Oγ distance of 3.17 Å), and with a water molecule (average O-OW distance of 3.03 Å). In the case of the Dunathan’s minimum appearing at a dihedral angle value of -105 degrees, the orientation of the carboxylate group is favored by means of hydrogen bond interactions with two water molecules (with average O-OW distances of 3.43 Å) and with the backbone amino group of Y84 (average O-N distance of 3.58 Å). This secondary minimum found in the PMF presents the CA-C bond nearly perpendicular to the conjugated π system, and thus, according to Dunathan’s hypothesis, the decarboxylation process should be favored in this case. Although this is not the most stable conformation, the free
energy difference with respect to the absolute minimum is not too high, and a significant proportion of external aldimine complexes should present a value of the χ dihedral angle optimum for the decarboxylation. Nonetheless, we decided to investigate the reaction paths that began with both conformers (see below).
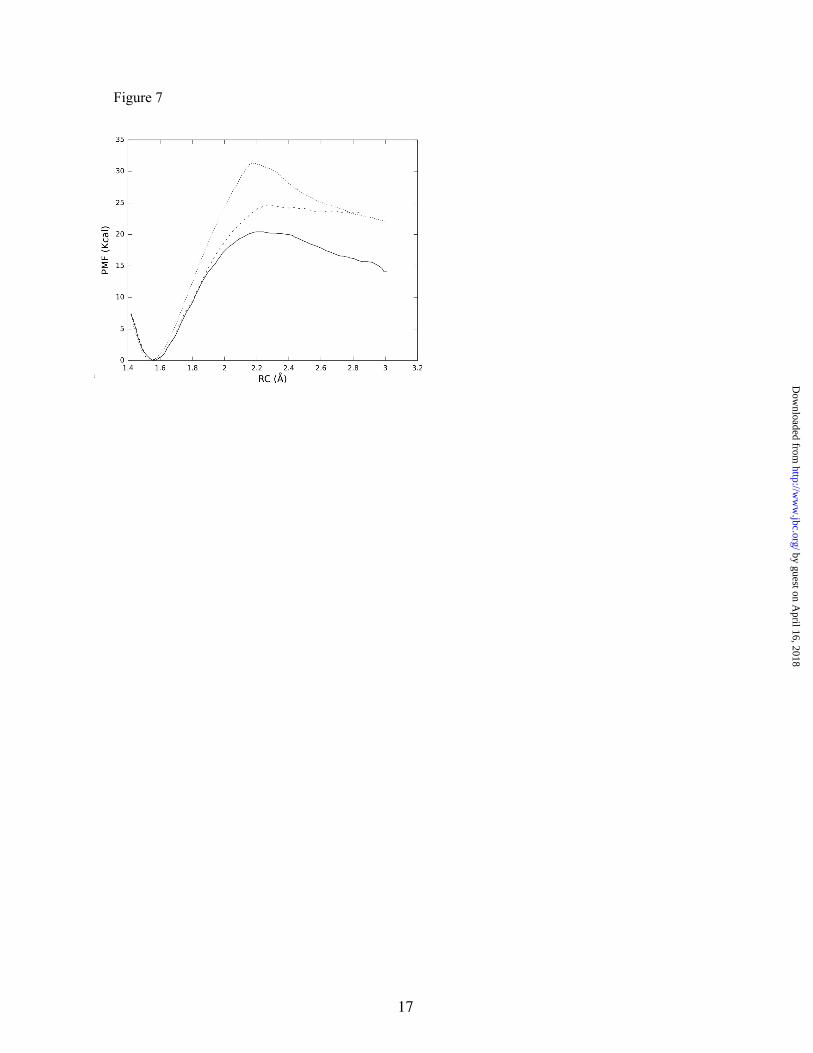
Decarboxylation reaction and origin of the catalytic effect- Experimental data on the reaction mechanism and its kinetics have been obtained on a fully active recombinant version of the rat enzyme by using different spectroscopic approaches (UV-Vis, fluorescence and circular dichroism). A kcat of 0.077 s-1 was determined at 310K (25), from which we can estimate an activation free energy of 19.7 kcal/mol by direct application of the transition state theory (assuming a transmission coefficient equal to unity). These studies pointed out that the rate limiting step in the catalytic cycle is one of the steps occurring after transaldimination (26). Decarboxylation is therefore an excellent candidate for the rate-limiting step of the global catalytic process. Simulations of this step also offer an excellent chance to validate Dunathan’s hypothesis, an experimentally unaffordable task. To this end, we have traced the free energy profile associated with this process in terms of a PMF obtained as a function of the CA-C distance. We considered the reaction from the two different conformations obtained for the external aldimine. This is (with two different orientations of the leaving carboxylate group with respect to the plane of the conjugated system) the absolute energy minimum complex (with a χ dihedral angle of -140 degrees) and the Dunathan’s complex (with a χ dihedral angle value of -105 degrees). The results are shown in Figure 7. The calculated barriers for the decarboxylation processes are 20.4 kcal/mol for the Dunathan’s complex and 24.7 kcal/mol for the other. Thus, the relative reactivities of these two conformers are in agreement with Dunathan’s hypothesis that the conformer with a perpendicular orientation of the CA-C bond relative to the conjugated system is the most reactive.
However, it must be taken into account that in order to estimate the total activation free energy associated to the decarboxylation process through the Dunathan’s conformer, one must add the free energy difference with respect to the absolute minimum energy complex (2.2 kcal/mol, see Figure 6), thus resulting in a total free energy barrier of 22.6 kcal/mol. This result compares satisfactorily with the experimental estimation (19.7 kcal/mol). Although we are using a low-level semiempirical Hamiltonian to describe the process,
6
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

it has been previously shown that the AM1 Hamiltonian gives energy barriers in very good agreement with those obtained using much higher theoretical treatments (such as post-Hartree-Fock or density functional based methods) for this particular type of reaction (45). Thus, the agreement of the reported barrier with the experimental activation free energy gives us confidence in our model for the rHDC active site.
The presence of two external aldimine conformations could justify a mechanism selected by evolution to restrict the efficiency of the mammalian enzyme in order to maintain homeostasis of the tissues. Intracellular concentrations of the substrate can be over 10-fold that of the HDC KM value (1-2 mM compared to 0.2-0.4 mM) (26,46), which means that the enzyme could work at its maximum activity. The product, histamine, can elicit its physiological roles at very low concentrations (in the nano- and micromolar order) in target cells (47). To ensure a minimum of histamine efflux from the major histamine-producing cells, the amine needs to be stored in secretion granules until the proper e x t e r n a l s t i m u l u s ( p r o - i n f l a m m a t o r y, neuroendocrine, etc) is received (47,48). Since there is no need of a high enzymatic activity, mammalian HDC is under a low evolutionary pressure to achieve optimal catalysis. The cell can coordinate both histamine synthesis and storage, minimizing histamine efflux to biological fluids which could have lethal effects on the whole organism.
To quantify the catalytic power of the rHDC enzyme in the decarboxylation process, we also t r a c e d t h e P M F c o r r e s p o n d i n g t o t h e decarboxylation of the external aldimine in aqueous solution. In this case, we verified that in the initial conformation of the substrate, the CA-C bond was perpendicular to the conjugated system, similar to the Dunathan’s conformer in the enzyme active site, and that this conformation remained stable during simulation. As shown in Figure 7, the free energy barrier obtained was 31.2 kcal/mol. This result is in good agreement with experimental and theoretical determinations of the activation free energy barriers for the decarboxylation of similar compounds (44,45). It must be stressed that the uncatalyzed reaction presents a free energy barrier 10.8 kcal/mol above the best of the enzymatic pathways, which translates to a rate acceleration of 4.38x107 at 310 K caused by the enzyme.
There is a growing consensus on the fact that enzymes catalyzes chemical reactions by selective
transition state stabilization by means of specific electrostatic interactions (46,47). In the reaction we are considering the carboxylate group, which formally supports a charge of -1, leaves the substrate as a neutral entity, an electron being transferred to the rest of the molecule. Thus, there is a great charge reorganization when passing from the reactant state to the transition state that should be favored in the enzymatic environment with respect to the aqueous solution. To analyse this point we have computed the averaged atomic Mulliken charges for the reactant and transition states in aqueous solution and in the enzyme (in this last case for the most reactive pathway taking place through the Dunathan’s conformer). Although these charges cannot be taken as a quantitative measure of the electronic distribution, they are very informative when employed for comparative purposes. For the sake of simplicity, the charges have been gathered to represent different groups of the external aldimine and are represented in Figure 8.
According to the results presented in Figure 8, we can observe that the PLP acts as an electron sink, which is the role usually assumed for this cofactor (23,34). It must be noted that this effect is substantially larger in aqueous solution than in the enzyme. Effectively, the electron charge on the pyridine ring becomes more negative when going from the reactant to the transition state, by -0.68 a.u. in solution and -0.21 a.u. in the enzyme. While the largest portion of the charge transferred from the carboxylate group goes to the pyridine ring in solution, in the enzyme active site, the electron density increases noticeably on the CA atom (B group in Figure 8), the charge of this atom becomes more negative by -0.35 a.u.
Afeter decarboxylation occurs, a quinoid structure seems to be favored in solution, resulting from the addition of an electron to the pyridine ring while the enzyme seems to preferentially stabilize a form that localizes the negative charge on the B group. According to Figure 8, the electron flow that occurs during the decarboxylation in aqueous solution diminishes the partial positive charge on the pyridine ring. Consequently, the hydrogen bonds established between the protonated nitrogen atom of the pyridine ring and the water solvent molecules are weakened in the transition state relative to the reactant state. This effect obviously contributes to increase the activation energy in aqueous solution because a stabilizing interaction is partially lost when passing from reactants to the transition state.
7
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

In the enzyme active site, the electrostatic environment is quite different from that found in solution. The quite rigid water-mediated interaction between D276 and the protonated nitrogen atom of the pyridine ring (see Figure 5) should favor the presence of a formal positive charge on this atom and then, the quinoid structure is not especially favored The averaged distance between the pyridine nitrogen atom and the oxygen atom of the water molecule is nearly the same in the reactant and the transition states (2.86 and 2.85 Å respectively). The net effect, comparing the reaction in solution and in the enzyme, is a relative transition state stabilization in this latter environment through the interactions established with the protonated nitrogen atom of the pyridine ring . It is also interesting to notice that the different polarization induced by the enzymatic environment, favoring the localization of negative charge on the B group, is expected to also favor proton transfer to this atom, which must take place in the next step of the catalytic cycle (see Figure 1).
The enzyme effect on the carboxylate group can be interpreted as the consequence of an active site electrostatically designed to favor the transition state charge distribution, where the charge is transferred towards the B group and to
the pyridine ring. Positioning of K308, K202, and D276 are the origin of this electrostatic complementarity with the transition state.
The changes observed in the reactant state are not an exclusive feature of this reaction. In fact, it has been often observed that the enzyme can induce changes on the substrate to drive it (geometrically or electronically) to the transition state (48). This effect has been recently recognized a s t h e c o n s e q u e n c e o f a n a c t i v e s i t e electrostatically designed to accommodate the transition state (49-52).
Summarizing, in the present work, QM/MM based molecular simulations on a previously validated mammalian HDC model made possible to solve some questions that are hard or impossible to tackle by classical experimentation. We have analyzed in detail the decarboxylation reaction step in HDC and we have been able to validate the Dunathan’s hypothesis about stereoelectronic effects contributing to the reactivity of the external aldimine. The presence of two positively charged lysine residues (K308 and K202) and a water-mediated hydrogen bond interaction with D276 seem to be the key electrostatic factors contributing to the reduction of the activation free energy in the active site.
REFERENCES
1. Chiu, W.; Baker, M.; Jiang, W.; Dougherty, M.; Schmid, M. Structure (2005) 13, 363–72. 2. Baker, D.; Sali, A. Science (2001) 294, 93–6. 3. Moya-García, A.; Medina, M.; Sánchez-Jiménez, F. Bioessays (2005) 27, 57–63.4. Furuta, K.; Nakayama, K.; Sugimoto, Y.; Ichikawa, A.; Tanaka, S. J. Biol. Chem. (2007) 282,
13438-46.5. Haas, H.; Panula, P. Nat Rev. Neurosci. (2003) 4, 121–30. 6. Ohtsu, H. et al. FEBS Lett. (2001) 502, 53–6. 7. Schneider, E.; Rolli-Derkinderen, M.; Arock, M.; Dy, M. Trends Immunol. (2002) 23, 255–63. 8. Schubert, M. Curr. Opin. Gastroenterol. (2005) 21, 636–43. 9. Fitzpatrick, L.; Buzas, E.; Gagne, T.; Nagy, A.; Horvath, C.; Ferencz, V.; Mester, A.; Kari, B.;
Ruan, M.; Falus, A.; Barsony, J. Proc. Natl. Acad. Sci. U. S. A. (2003) 100, 6027–32. 10. Medina, M.; Urdiales, J.; Rodríguez-Caso, C.; Ramírez, F.; Sánchez-Jiménez, F. Crit. Rev. Biochem.
Mo.l Bio.l (2003) 38, 23–59. 11. Panula, P.; Lintunen, M.; Karlstedt, K. Semin. Cancer. Biol. (2000) 10, 11–4. 12. Parsons, M.; Ganellin, C. Br. J. Pharmacol. (2006) 147 Suppl 1, S127–35. 13. Pos, Z.; Safrany, G.; Muller, K.; Toth, S.; Falus, A.; Hegyesi, H. Cancer Res. (2005) 65, 4458–66. 14. Kamath, A.; Vaaler, G.; Snell, E. J. Biol. Chem. (1991) 266, 9432–7. 15. Sandmeier, E.; Hale, T.; Christen, P. Eur. J. Biochem. (1994) 221, 997–1002. 16. Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.; Malashkevich, V. Nat. Struct. Biol.
(2001) 8, 963–7. 17. Siaterli, M.; Vassilacopoulou, D.; Fragoulis, E. Neurochem. Res. (2003) 28, 797–803. 18. Rodríguez-Caso, C.; Rodríguez-Agudo, D.; Moya-García, A.; Fajardo, I.; Medina, M.;
Subramaniam, V.; Sánchez-Jiménez, F. Eur. J. Biochem. (2003) 270, 4376–87.
8
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

19. Rodríguez-Caso, C.; Rodríguez-Agudo, D.; Sánchez-Jiménez, F.; Medina, M. Cell. Mol. Life Sci. (2003) 60, 1760–3.
20. Fleming, J.; Fajardo, I.; Langlois, M.; Sánchez-Jiménez, F.; Wang, T. Biochem. J. (2004) 381, 769–78.
21. Fleming, J.; Sánchez-Jiménez, F.; Moya-García, A.; Langlois, M.; Wang, T. Biochem. J. (2004) 379, 253–61.
22. Madhusudhan, M.; Martí-Renom, M. A.; Eswar, N.; John, B.; Pieper, U.; Karchin, R.; Min-yi Shen,; Sali, A. In The Proteomics Protocols Handbook; Walker, J., Ed.; Humana Press Inc., Totowa, NJ, 2005; Chapter Comparative Protein structure Modeling, pp 831–860.
23. Hayashi, H. J. Biochem. Tokyo. (1995) 118, 463–73. 24. Eliot, A.; Kirsch, J. Annu. Rev. Biochem. (2004) 73, 383–415. 25. Tanase, S.; Guirard, B.; Snell, E. J. Biol. Chem. (1985) 260, 6738–46. 26. Olmo, M.; Sánchez-Jiménez, F.; Medina, M.; Hayashi, H. J. Biochem. Tokyo. (2002) 132, 433–9. 27. Hayashi, H.; Mizuguchi, H.; Kagamiyama, H. Biochemistry (1993) 32, 812–8. 28. Ishii, S.; Hayashi, H.; Okamoto, A.; Kagamiyama, H. Protein Sci. (1998) 7, 1802–10. 29. Auld, D.S.; Bruice, T.C. J. Am. Chem. Soc. (1967) 89, 2098-210630. Kern, A. D.; Oliveira, M. A.; Coffino, P.; Hackert, M. L. Structure. (1999) 7, 567-8131. Warshel, A.; Levitt, M. J. Mol. Biol. (1976) 103, 227–49. 32. Roca, M.; Moliner, V.; Ruiz-Pernía, J.; Silla, E.; Tuñón, I. J. Phys. Chem. A Mol. Spectrosc. Kinet.
Environ. Gen. Theory (2006) 110, 503–9. 33. Gao, J.; Truhlar, D. Annu. Rev. Phys. Chem. (2002) 53, 467–505. 34. MacKerell, Jr., A. D. et al. J. Phys. Chem. B (1998) 102, 3586–3616. 35. Dewar, M.; Zoebisch, E.; Healy, E.; Stewart, J. J. Am. Chem. Soc. (1985) 107, 3902–3909. 36. Poulsen, T.; García-Viloca, M.; Truhlar, D. J. Phys. Chem. B (2003) 107, 9567–9578. 37. Ferrer, S.; Ruiz-Pernía, J.; Tuñón, I.; Moliner, V.; García-Viloca, M.; González-Lafont, A.;
Lluch, J. M. J. Chem. Theor. Comput. (2005) 1, 750–761. 38. Gao, J. J. Am. Chem. Soc. (1991) 113, 7796. 39. Torrie, G. M. Valleau, J. P. J. Comput. Phys. (1977) 23, 187–199.40. Matsuda, N.; Hayashi, H.; Miyatake, S.; Kuroiwa, T.; Kagamiyama, H. J. Biochem. Tokyo. (2004)
135, 33–42. 41. Poupon, A.; Jebai, F.; Labesse, G.; Gros, F.; Thibault, J.; Mornon, J.; Krieger, M. Proteins (1999)
37, 191–203. 42. Dunathan, H. Proc Natl Acad Sci U S A, (1966) 55, 712-643. Hyde, C.; Ahmed, S.; Padlan, E.; Miles, E.; Davies, D. J. Biol. Chem. (1988) 263, 17857–71. 44. Jansonius, J.; Eichele, G.; Ford, G.; Kirsch, J.; Picot, D.; Thaller, C.; Vincent, M.; Gehring, H.;
Christen, P. Biochem. Soc. Trans. (1984) 12, 424–7. 45. Wu, N.; Mo, Y.; Gao, J.; Pai, E. Proc. Natl. Acad. Sci. U. S. A. (2000) 97, 2017–22. 46. Olsson, M. H. M.; Parson, W. W.; Warshel, A. Chem. Rev. 2006, 106, 1737-5647. Roca, M.; Andrés, J.; Moliner, V.; Tuñón, I.; Bertrán, J.; J. Am. Chem. Soc. (2005) 127, 10648–55.
48. Warshel, A.; Strajbl, M.; Villa, J.; Florian, J. Biochemistry (2000) 39, 14728–38. 49. Bruice, T. Acc. Chem. Res. (2002) 35, 139–48. 50. Warshel, A. J. Biol. Chem. (1998) 273, 27035–8. 51. Martí, S.; Roca, M.; Andrés, J.; Moliner, V.; Silla, E.; Tuñón, I.; Bertrán, J. Chem. Soc. Rev. (2004)
33, 98–107. 52. García-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D. Science (2004) 303, 186–95.
FOOTNOTES
*This work has been granted by SAF2005-01812 and CTQ2006-15447-CO2-02 (MEC, Spain), CVI-267 (Junta de Andalucía), GV06-021 (Generalitat Valenciana) and Fundación Ramón Areces. J.J.R-P and A. A. M-G thank Ministerio de Educacion y Ciencia and Junta de Andalucía, respectively, for doctoral fellowships. Authors also thanks Silvia Ferrer Castillo and Alejandro Soriano for valuable advice and help during this research.
9
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE LEGENDS
Figure 1. Catalytic cycle of mammalian HDC. Only main steps are depicted. Decarboxylation reaction is highlighted.
Figure 2. Representation of the external aldimine with atom labelling. χ is the C-CA-N-C4A dihedral angle that describes the relative orientation between substrate and cofactor.
Figure 3. Proposed external aldimine conformations. Panels A and C, conformation 1; panels B and D, conformation 2. For clarity, panels A and B only show the external aldimine conformations with respect to two essential residues of the catalytic center (K308 and H197) and the rest of the protein has been omitted (in these upper views, the entrance of the catalytic site would be located on the right side of the pictures). In panels C and D, the surface of both monomers have also been represented, and the views has been clock-wise rotated by 90 degrees approximately on the longitudinal axes with respect to the upper panels, so that the catalytic hole entrance is now in front of the viewer.
Figure 4. Time evolution of key distances in conformations 1 (solid lines) and 2 (dashed lines) for the production MD trajectory.
Figure 5. Snapshot of the external aldimine with selected residues of the rHDC active site.
Figure 6. Potential of Mean Force associated to the change in the χ (C-CA-N-C4A) dihedral angle.
Figure 7. Computed free energy activation profiles for the decarboxylation reactions of the PLP-His adduct in aqueous solution (dotted line) and in rat HDC. Absolute energy minimum conformer (dashed line) and Dunathan’s conformer (solid line). RC, reaction coordinate, distance between CA and carboxylate group.
Figure 8. Schematic representation of Mulliken charge distribution of the external aldimine in the enzyme (left) and aqueous solution (right). Black, reactive state. White, transition state.
10
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from


Iñaki TuñónAurelio A. Moya-García, Javier Ruiz-Pernía, Sergio Martí, Francisca Sánchez-Jiménez and
computational studyAnalysis of the decarboxylation step in mammalian histidine decarboxylase: A
published online February 29, 2008J. Biol. Chem.
10.1074/jbc.M707434200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from