an introduction to bioinformatics 2. comparing biological sequences: sequence alignment (cont’d)
TRANSCRIPT

An Introduction to Bioinformatics
2. Comparing biological sequences:
sequence alignment (cont’d)

An Introduction to Bioinformatics
Outline
• Global Alignment • Scoring Matrices• Local Alignment• Alignment with Affine Gap Penalties

An Introduction to Bioinformatics
From LCS to Alignment: Change up the Scoring
• The Longest Common Subsequence (LCS) problem—the simplest form of sequence alignment – allows only insertions and deletions (no mismatches).
• In the LCS Problem, we scored 1 for matches and 0 for indels• Consider penalizing indels and mismatches with negative
scores• Simplest scoring schema: +1 : match premium -μ : mismatch penalty -σ : indel penalty

An Introduction to Bioinformatics
Simple Scoring
• When mismatches are penalized by –μ, indels are penalized by –σ,
and matches are rewarded with +1,
the resulting score is:
#matches – μ(#mismatches) – σ (#indels)

An Introduction to Bioinformatics
The Global Alignment ProblemFind the best alignment between two strings under a given
scoring schema
Input : Strings v and w and a scoring schemaOutput : Alignment of maximum score
↑→ = -б = 1 if match = -µ if mismatch
si-1,j-1 +1 if vi = wj
si,j = max s i-1,j-1 -µ if vi ≠ wj
s i-1,j - σ s i,j-1 - σ
: mismatch penaltyσ : indel penalty

An Introduction to Bioinformatics
Scoring Matrices
To generalize scoring, consider a (4+1) x(4+1) scoring matrix δ.
In the case of an amino acid sequence alignment, the scoring matrix would be a (20+1)x(20+1) size. The addition of 1 is to include the score for comparison of a gap character “-”.
This will simplify the algorithm as follows:
si-1,j-1 + δ (vi, wj)
si,j = max s i-1,j + δ (vi, -)
s i,j-1 + δ (-, wj)

An Introduction to Bioinformatics
Measuring Similarity
• Measuring the extent of similarity between two sequences• Based on percent sequence identity• Based on conservation

An Introduction to Bioinformatics
Percent Sequence Identity
• The extent to which two nucleotide or amino acid sequences are invariant
A C C T G A G – A G A C G T G – G C A G
70% identical
mismatchindel

An Introduction to Bioinformatics
Making a Scoring Matrix
• Scoring matrices are created based on biological evidence.
• Alignments can be thought of as two sequences that differ due to mutations.
• Some of these mutations have little effect on the protein’s function, therefore some penalties, δ(vi , wj), will be less harsh than others.
An Introduction to Bioinformatics
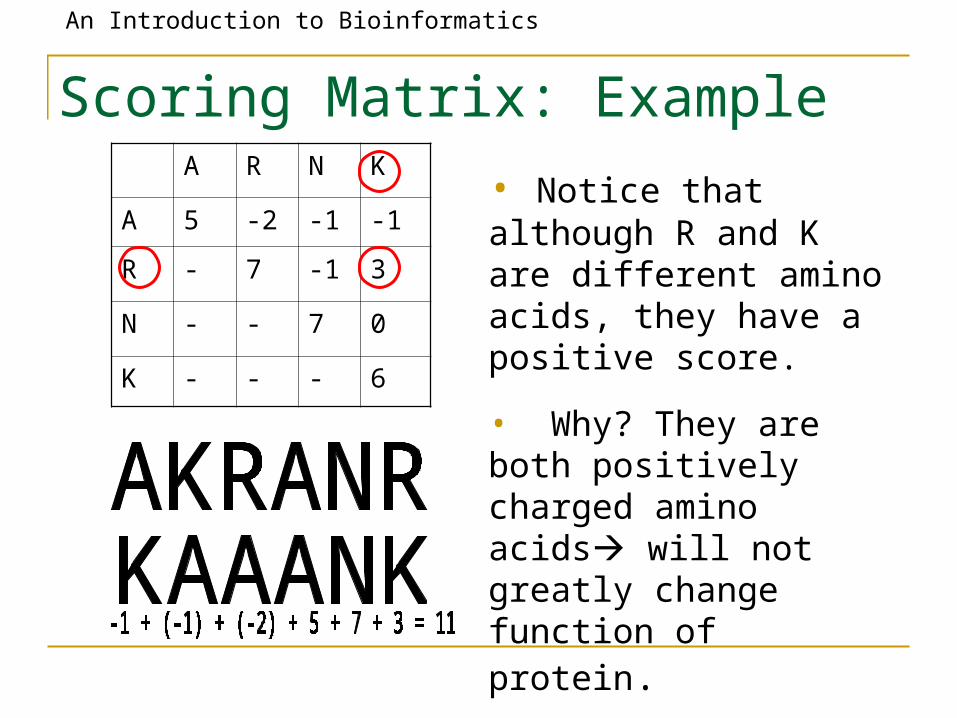
Scoring Matrix: ExampleA R N K
A 5 -2 -1 -1
R - 7 -1 3
N - - 7 0
K - - - 6
• Notice that although R and K are different amino acids, they have a positive score.
• Why? They are both positively charged amino acids will not greatly change function of protein.

An Introduction to Bioinformatics
Conservation
• Amino acid changes that tend to preserve the physico-chemical properties of the original residue• Polar to polar
• aspartate glutamate• Nonpolar to nonpolar
• alanine valine• Similarly behaving residues
• leucine to isoleucine

An Introduction to Bioinformatics
Scoring matrices
• Amino acid substitution matrices• PAM• BLOSUM
• DNA substitution matrices• DNA is less conserved than protein
sequences• Less effective to compare coding regions at
nucleotide level

An Introduction to Bioinformatics
PAM• Point Accepted Mutation (Dayhoff et al.)• 1 PAM = PAM1 = 1% average change of all amino
acid positions• After 100 PAMs of evolution, not every residue will
have changed• some residues may have mutated several
times• some residues may have returned to their
original state• some residues may not changed at all

An Introduction to Bioinformatics
PAMX
• PAMx = PAM1x
• PAM250 = PAM1250
• PAM250 is a widely used scoring matrix:
Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys ... A R N D C Q E G H I L K ...Ala A 13 6 9 9 5 8 9 12 6 8 6 7 ...Arg R 3 17 4 3 2 5 3 2 6 3 2 9Asn N 4 4 6 7 2 5 6 4 6 3 2 5Asp D 5 4 8 11 1 7 10 5 6 3 2 5Cys C 2 1 1 1 52 1 1 2 2 2 1 1Gln Q 3 5 5 6 1 10 7 3 7 2 3 5...Trp W 0 2 0 0 0 0 0 0 1 0 1 0Tyr Y 1 1 2 1 3 1 1 1 3 2 2 1Val V 7 4 4 4 4 4 4 4 5 4 15 10

An Introduction to Bioinformatics
BLOSUM
• Blocks Substitution Matrix • Scores derived from observations of the
frequencies of substitutions in blocks of local alignments in related proteins
• Matrix name indicates evolutionary distance• BLOSUM62 was created using sequences
sharing no more than 62% identity

An Introduction to Bioinformatics
The Blosum50 Scoring Matrix

An Introduction to Bioinformatics
Local vs. Global Alignment
• The Global Alignment Problem tries to find the longest path between vertices (0,0) and (n,m) in the edit graph.
• The Local Alignment Problem tries to find the longest path among paths between arbitrary vertices (i,j) and (i’, j’) in the edit graph.

An Introduction to Bioinformatics
Local vs. Global Alignment• The Global Alignment Problem tries to find the
longest path between vertices (0,0) and (n,m) in the edit graph.
• The Local Alignment Problem tries to find the longest path among paths between arbitrary vertices (i,j) and (i’, j’) in the edit graph.
• In the edit graph with negatively-scored edges, Local Alignmet may score higher than Global Alignment
An Introduction to Bioinformatics
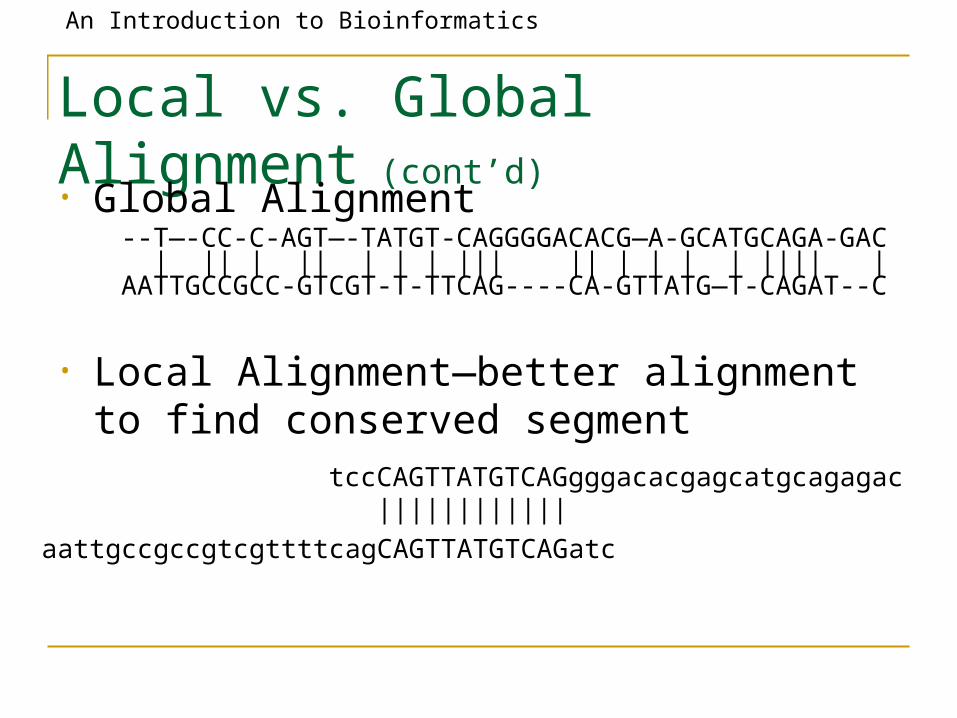
Local vs. Global Alignment (cont’d)• Global Alignment
• Local Alignment—better alignment to find conserved segment
--T—-CC-C-AGT—-TATGT-CAGGGGACACG—A-GCATGCAGA-GAC | || | || | | | ||| || | | | | |||| | AATTGCCGCC-GTCGT-T-TTCAG----CA-GTTATG—T-CAGAT--C
tccCAGTTATGTCAGgggacacgagcatgcagagac ||||||||||||
aattgccgccgtcgttttcagCAGTTATGTCAGatc

An Introduction to Bioinformatics
Local Alignment: Example
Global alignment
Local alignment
Compute a “mini” Global Alignment to get Local

An Introduction to Bioinformatics
Local Alignments: Why?• Two genes in different species may be similar over
short conserved regions and dissimilar over remaining regions.
• Example:• Homeobox genes have a short region called the
homeodomain that is highly conserved between species.
• A global alignment would not find the homeodomain because it would try to align the ENTIRE sequence

An Introduction to Bioinformatics
The Local Alignment Problem
• Goal: Find the best local alignment between two strings
• Input : Strings v, w and scoring matrix δ• Output : Alignment of substrings of v and w
whose alignment score is maximum among all possible alignment of all possible substrings

An Introduction to Bioinformatics
The Problem with this Problem• Long run time O(n4):
- In the grid of size n x n there are ~n2 vertices (i,j) that may serve as a source.
- For each such vertex computing alignments from (i,j) to (i’,j’) takes O(n2) time.
• This can be remedied by giving free rides

An Introduction to Bioinformatics
Local Alignment: Example
Global alignment
Local alignment
Compute a “mini” Global Alignment to get Local

An Introduction to Bioinformatics
Local Alignment: Example

An Introduction to Bioinformatics
Local Alignment: Example

An Introduction to Bioinformatics
Local Alignment: Example
An Introduction to Bioinformatics
Local Alignment: Example

An Introduction to Bioinformatics
Local Alignment: Example

An Introduction to Bioinformatics
Local Alignment: Running Time • Long run time O(n4):
- In the grid of size n x n there are ~n2 vertices (i,j) that may serve as a source.
- For each such vertex computing alignments from (i,j) to (i’,j’) takes O(n2) time.
• This can be remedied by giving free rides

An Introduction to Bioinformatics
Local Alignment: Free Rides
Vertex (0,0)
The dashed edges represent the free rides from (0,0) to every other node.
Yeah, a free ride!

An Introduction to Bioinformatics
The Local Alignment Recurrence
• The largest value of si,j over the whole edit graph is the score of the best local alignment.
• The recurrence:
0 si,j = max si-1,j-1 + δ (vi, wj)
s i-1,j + δ (vi, -)
s i,j-1 + δ (-, wj)
Notice there is only this change from the original recurrence of a Global Alignment

An Introduction to Bioinformatics
The Local Alignment Recurrence
• The largest value of si,j over the whole edit graph is the score of the best local alignment.
• The recurrence:
0 si,j = max si-1,j-1 + δ (vi, wj)
s i-1,j + δ (vi, -)
s i,j-1 + δ (-, wj)
Power of ZERO: there is only this change from the original recurrence of a Global Alignment - since there is only one “free ride” edge entering into every vertex

An Introduction to Bioinformatics
The Smith-Waterman algorithm
Idea: Ignore badly aligning regions
Modifications to Needleman-Wunsch:
Initialization: F(0, j) = F(i, 0) = 0
0
Iteration: F(i, j) = max F(i – 1, j) – d
F(i, j – 1) – d
F(i – 1, j – 1) + s(xi, yj)

An Introduction to Bioinformatics
Scoring Indels: Naive Approach
• A fixed penalty σ is given to every indel:• -σ for 1 indel, • -2σ for 2 consecutive indels• -3σ for 3 consecutive indels, etc.
Can be too severe penalty for a series of 100 consecutive indels

An Introduction to Bioinformatics
Affine Gap Penalties
• In nature, a series of k indels often come as a single event rather than a series of k single nucleotide events:
Normal scoring would give the same score for both alignments
This is more likely.
This is less likely.

An Introduction to Bioinformatics
Accounting for Gaps• Gaps- contiguous sequence of spaces in one of the
rows
• Score for a gap of length x is: -(ρ + σx) where ρ >0 is the penalty for introducing a gap: gap opening penalty ρ will be large relative to σ: gap extension penalty because you do not want to add too much of a
penalty for extending the gap.

An Introduction to Bioinformatics
Affine Gap Penalties
• Gap penalties:• -ρ-σ when there is 1 indel• -ρ-2σ when there are 2 indels• -ρ-3σ when there are 3 indels, etc. • -ρ- x·σ (-gap opening - x gap extensions)
• Somehow reduced penalties (as compared to naïve scoring) are given to runs of horizontal and vertical edges

An Introduction to Bioinformatics
Compromise: affine gaps
ρσ
Gap cost

An Introduction to Bioinformatics
Affine Gap Penalties and Edit Graph
To reflect affine gap penalties we have to add “long” horizontal and vertical edges to the edit graph. Each such edge of length x should have weight
- - x *

An Introduction to Bioinformatics
Adding “Affine Penalty” Edges to the Edit Graph
There are many such edges!
Adding them to the graph increases the running time of the alignment algorithm by a factor of n (where n is the number of vertices)
So the complexity increases from O(n2) to O(n3)
An Introduction to Bioinformatics
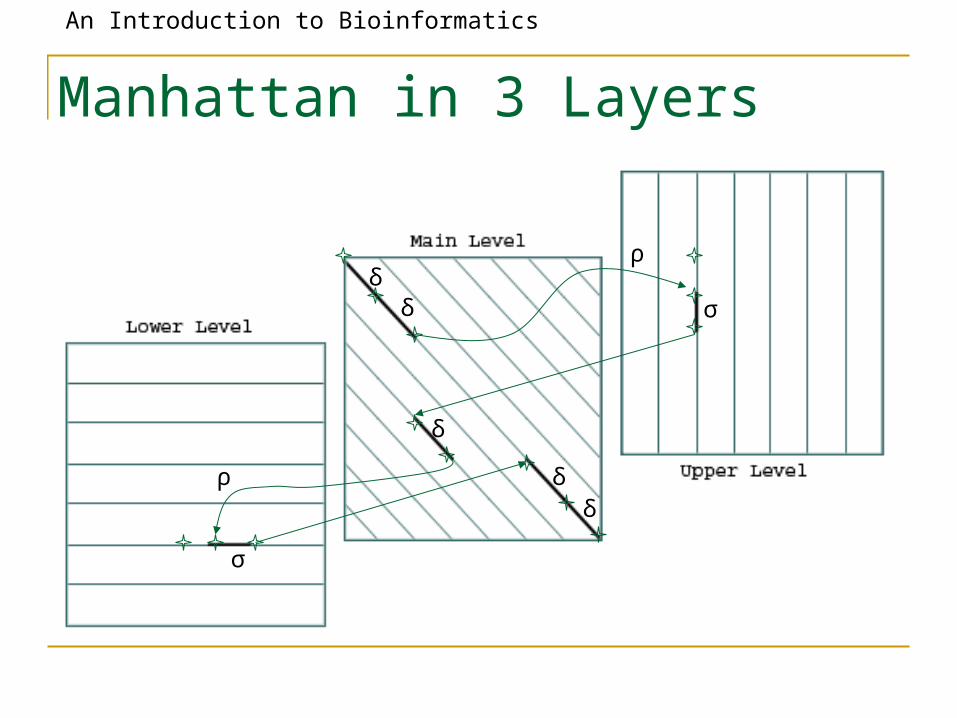
Manhattan in 3 Layers
ρ
ρ
σ
σδ
δ
δ
δδ

An Introduction to Bioinformatics
Affine Gap Penalties and 3 Layer Manhattan Grid
• The three recurrences for the scoring algorithm creates a 3-layered graph.
• The top level creates/extends gaps in the sequence w.
• The bottom level creates/extends gaps in sequence v.
• The middle level extends matches and mismatches.

An Introduction to Bioinformatics
Switching between 3 Layers
• Levels:• The main level is for diagonal edges • The lower level is for horizontal edges• The upper level is for vertical edges
• A jumping penalty is assigned to moving from the main level to either the upper level or the lower level (-)
• There is a gap extension penalty for each continuation on a level other than the main level (-)

An Introduction to Bioinformatics
The 3-leveled Manhattan Grid

An Introduction to Bioinformatics
Affine Gap Penalty Recurrencessi,j = s i-1,j - σ
max s i-1,j –(ρ+σ)
si,j = s i,j-1 - σ
max s i,j-1 –(ρ+σ)
si,j = si-1,j-1 + δ (vi, wj)
max s i,j
s i,j
Continue Gap in w (deletion)
Start Gap in w (deletion): from middle
Continue Gap in v (insertion)
Start Gap in v (insertion):from middle
Match or Mismatch
End deletion: from top
End insertion: from bottom

An Introduction to Bioinformatics
Bounded Dynamic ProgrammingInitialization:
F(i,0), F(0,j) undefined for i, j > k
Iteration:
For i = 1…M
For j = max(1, i – k)…min(N, i+k)
F(i – 1, j – 1)+ s(xi, yj)
F(i, j) = max F(i, j – 1) – d, if j > i – k(N)
F(i – 1, j) – d, if j < i + k(N)
Termination: same
Easy to extend to the affine gap case
x1 ………………………… xM
y1 …
……
……
……
……
…
yN
k(N)

An Introduction to Bioinformatics
Computing Alignment Path Requires Quadratic Memory
Alignment Path• Space complexity for
computing alignment path for sequences of length n and m is O(nm)
• We need to keep all backtracking references in memory to reconstruct the path (backtracking)
n
m

An Introduction to Bioinformatics
Computing Alignment Score with Linear MemoryAlignment Score• Space complexity of
computing just the score itself is O(n)
• We only need the previous column to calculate the current column, and we can then throw away that previous column once we’re done using it
2
nn

An Introduction to Bioinformatics
Computing Alignment Score: Recycling Columns
memory for column 1 is used to calculate column 3
memory for column 2 is used to calculate column 4
Only two columns of scores are saved at any given time

An Introduction to Bioinformatics
Crossing the Middle Line
m/2 m
n
Prefix(i)
Suffix(i)
We want to calculate the longest path from (0,0) to (n,m) that passes through (i,m/2) where i ranges from 0 to n and represents the i-th row
Define
length(i)
as the length of the longest path from (0,0) to (n,m) that passes through vertex (i, m/2)

An Introduction to Bioinformatics
m/2 m
n
Prefix(i)
Suffix(i)
Define (mid,m/2) as the vertex where the longest path crosses the middle column.
length(mid) = optimal length = max0i n length(i)
Crossing the Middle Line

An Introduction to Bioinformatics
Computing Prefix(i)• prefix(i) is the length of the longest path from
(0,0) to (i,m/2)• Compute prefix(i) by dynamic programming in
the left half of the matrix
0 m/2 m
store prefix(i) column

An Introduction to Bioinformatics
Computing Suffix(i)• suffix(i) is the length of the longest path from (i,m/2) to (n,m)• suffix(i) is the length of the longest path from (n,m) to (i,m/2)
with all edges reversed• Compute suffix(i) by dynamic programming in the right half
of the “reversed” matrix
0 m/2 m
store suffix(i) column

An Introduction to Bioinformatics
Length(i) = Prefix(i) + Suffix(i)
• Add prefix(i) and suffix(i) to compute length(i):• length(i)=prefix(i) + suffix(i)
• You now have a middle vertex of the maximum path (i,m/2) as maximum of length(i)
middle point found
0 m/2 m
0
i

An Introduction to Bioinformatics
Finding the Middle Point0 m/4 m/2 3m/4 m

An Introduction to Bioinformatics
Finding the Middle Point again0 m/4 m/2 3m/4 m

An Introduction to Bioinformatics
And Again0 m/8 m/4 3m/8 m/2 5m/8 3m/4 7m/8 m

An Introduction to Bioinformatics
Time = Area: First Pass• On first pass, the algorithm covers the entire
area
Area = nm

An Introduction to Bioinformatics
Time = Area: First Pass• On first pass, the algorithm covers the entire
area
Area = nm
Computing prefix(i)
Computing suffix(i)

An Introduction to Bioinformatics
Time = Area: Second Pass• On second pass, the algorithm covers only
1/2 of the area
Area/2
An Introduction to Bioinformatics
Time = Area: Third Pass• On third pass, only 1/4th is covered.
Area/4

An Introduction to Bioinformatics
Geometric Reduction At Each Iteration1 + ½ + ¼ + ... + (½)k ≤ 2• Runtime: O(Area) = O(nm)
first pass: 1
2nd pass: 1/2
3rd pass: 1/4
5th pass: 1/16
4th pass: 1/8

An Introduction to Bioinformatics
Indexing-based Local Aligners
BLAST, WU-BLAST, BlastZ, MegaBLAST, BLAT, PatternHunter, ……

An Introduction to Bioinformatics
Some useful applications of alignments• Given a newly discovered gene,
• Does it occur in other species?• How fast does it evolve?
• Assume we try Smith-Waterman:
The entire genomic database
Our new gene
104
1010 - 1011

An Introduction to Bioinformatics
Some useful applications of alignments• Given a newly sequenced organism,• Which subregions align with other organisms?
• Potential genes• Other biological characteristics
• Assume we try Smith-Waterman:
The entire genomic database
Our newly sequenced mammal
3109
1010 - 1011

An Introduction to Bioinformatics
Indexing-based local alignment
(BLAST- Basic Local Alignment Search Tool)
Main idea:
1. Construct a dictionary of all the words in the query
2. Initiate a local alignment for each word match between query and DB
Running Time: O(MN)
However, orders of magnitude faster than Smith-Waterman
query
DB

An Introduction to Bioinformatics
Indexing-based local alignmentDictionary:
All words of length k (~10)
Alignment initiated between words of alignment score T
(typically T = k)
Alignment:
Ungapped extensions until score
below statistical threshold
Output:
All local alignments with score
> statistical threshold
……
……
query
DB
query
scan

An Introduction to Bioinformatics
Indexing-based local alignment—Extensions A C G A A G T A A G G T C C A G T
C
C
C
T
T
C C
T
G
G
A T
T
G
C
G
A
Example:
k = 4
The matching word GGTC initiates an alignment
Extension to the left and right with no gaps until alignment falls < T below best so far
Output:
GTAAGGTCC
GTTAGGTCC

An Introduction to Bioinformatics
Indexing-based local alignment—Extensions A C G A A G T A A G G T C C A G T
C
T
G
A
T
C C
T
G
G
A
T
T
G C
G
A
Gapped extensions
• Extensions with gaps in a band around anchor
Output:
GTAAGGTCCAGTGTTAGGTC-AGT

An Introduction to Bioinformatics
Indexing-based local alignment—Extensions A C G A A G T A A G G T C C A G T
C
T
G
A
T
C C
T
G
G
A
T
T
G C
G
A
Gapped extensions until threshold
• Extensions with gaps until score < T below best score so far
Output:
GTAAGGTCCAGTGTTAGGTC-AGT

An Introduction to Bioinformatics
Indexing-based local alignment—The index• Sensitivity/speed
tradeoff
long words
(k = 15)
short words
(k = 7)
Sensitivity
Speed
Kent WJ, Genome Research 2002
Sens.
Speed

An Introduction to Bioinformatics
Indexing-based local alignment—The indexMethods to improve sensitivity/speed
1. Using pairs of words
2. Using inexact words
3. Patterns—non consecutive positions
……ATAACGGACGACTGATTACACTGATTCTTAC……
……GGCACGGACCAGTGACTACTCTGATTCCCAG……
……ATAACGGACGACTGATTACACTGATTCTTAC……
……GGCGCCGACGAGTGATTACACAGATTGCCAG……
TTTGATTACACAGAT T G TT CAC G

An Introduction to Bioinformatics
Measured improvement
Kent WJ, Genome Research 2002

An Introduction to Bioinformatics
Non-consecutive words—Patterns Patterns increase the likelihood of at least one
match within a long conserved region
3 common
5 common
7 common
Consecutive Positions Non-Consecutive Positions
6 common
On a 100-long 70% conserved region: Consecutive Non-consecutive
Expected # hits: 1.07 0.97Prob[at least one hit]: 0.30 0.47

An Introduction to Bioinformatics
Advantage of Patterns
11 positions
11 positions
10 positions

An Introduction to Bioinformatics
Multiple patterns
• K patterns• Takes K times longer to scan• Patterns can complement one another
• Computational problem:• Given: a model (prob distribution) for homology between two regions• Find: best set of K patterns that maximizes Prob(at least one match)
TTTGATTACACAGAT T G TT CAC G T G T C CAG TTGATT A G
Buhler et al. RECOMB 2003Sun & Buhler RECOMB 2004
How long does it take to search the query?

An Introduction to Bioinformatics
Variants of BLAST• NCBI BLAST: search the universe http://www.ncbi.nlm.nih.gov/BLAST/• MEGABLAST:
• Optimized to align very similar sequences• Works best when k = 4i 16• Linear gap penalty
• WU-BLAST: (Wash U BLAST)• Very good optimizations• Good set of features & command line arguments
• BLAT• Faster, less sensitive than BLAST• Good for aligning huge numbers of queries
• CHAOS• Uses inexact k-mers, sensitive
• PatternHunter• Uses patterns instead of k-mers
• BlastZ• Uses patterns, good for finding genes

An Introduction to Bioinformatics
ExampleQuery: gattacaccccgattacaccccgattaca (29 letters) [2 mins]
Database: All GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or phase 0, 1 or 2 HTGS sequences) 1,726,556 sequences; 8,074,398,388 total letters
>gi|28570323|gb|AC108906.9| Oryza sativa chromosome 3 BAC OSJNBa0087C10 genomic sequence, complete sequence Length = 144487 Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus
Query: 4 tacaccccgattacaccccga 24 ||||||| |||||||||||||
Sbjct: 125138 tacacccagattacaccccga 125158
Score = 34.2 bits (17),
Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus
Query: 4 tacaccccgattacaccccga 24 ||||||| |||||||||||||
Sbjct: 125104 tacacccagattacaccccga 125124
>gi|28173089|gb|AC104321.7| Oryza sativa chromosome 3 BAC OSJNBa0052F07 genomic sequence, complete sequence Length = 139823 Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus
Query: 4 tacaccccgattacaccccga 24 ||||||| |||||||||||||
Sbjct: 3891 tacacccagattacaccccga 3911

An Introduction to Bioinformatics
ExampleQuery: Human atoh enhancer, 179 letters [1.5 min]
Result: 57 blast hits1. gi|7677270|gb|AF218259.1|AF218259 Homo sapiens ATOH1 enhanc... 355 1e-95 2. gi|22779500|gb|AC091158.11| Mus musculus Strain C57BL6/J ch... 264 4e-68 3. gi|7677269|gb|AF218258.1|AF218258 Mus musculus Atoh1 enhanc... 256 9e-66 4. gi|28875397|gb|AF467292.1| Gallus gallus CATH1 (CATH1) gene... 78 5e-12 5. gi|27550980|emb|AL807792.6| Zebrafish DNA sequence from clo... 54 7e-05 6. gi|22002129|gb|AC092389.4| Oryza sativa chromosome 10 BAC O... 44 0.068 7. gi|22094122|ref|NM_013676.1| Mus musculus suppressor of Ty ... 42 0.27 8. gi|13938031|gb|BC007132.1| Mus musculus, Similar to suppres... 42 0.27
gi|7677269|gb|AF218258.1|AF218258 Mus musculus Atoh1 enhancer sequence Length = 1517 Score = 256 bits (129), Expect = 9e-66 Identities = 167/177 (94%),
Gaps = 2/177 (1%) Strand = Plus / Plus Query: 3 tgacaatagagggtctggcagaggctcctggccgcggtgcggagcgtctggagcggagca 62 ||||||||||||| ||||||||||||||||||| |||||||||||||||||||||||||| Sbjct: 1144 tgacaatagaggggctggcagaggctcctggccccggtgcggagcgtctggagcggagca 1203
Query: 63 cgcgctgtcagctggtgagcgcactctcctttcaggcagctccccggggagctgtgcggc 122 |||||||||||||||||||||||||| ||||||||| |||||||||||||||| ||||| Sbjct: 1204 cgcgctgtcagctggtgagcgcactc-gctttcaggccgctccccggggagctgagcggc 1262
Query: 123 cacatttaacaccatcatcacccctccccggcctcctcaacctcggcctcctcctcg 179 ||||||||||||| || ||| |||||||||||||||||||| |||||||||||||||
Sbjct: 1263 cacatttaacaccgtcgtca-ccctccccggcctcctcaacatcggcctcctcctcg 1318