advanced microarray analysis courselausanne.isb-sib.ch/~pwirapat/amac2009/day4/day4.pdf · 7....
TRANSCRIPT

Advanced Microarray Analysis Course
Day 4: Integrative Analysis
PJ “Asa” Wirapati
BioinformaticsCore Facility
Swiss Instituteof Bioinformatics
Lausanne, 3 November 2009
1

Today’s Topics
• Genome-wide scans and summary statistics
• Comparing genome-wide summary profiles
• Combined analysis
2

Overview
We’ve seen different types of regression analysis and unsupervisedmethod such as normal mixture models.
They can be applied to:
• Each gene separately (typically correlating to a certain variable,e.g. phenotype)
• Pairs of genes (such as in hierarchical clustering, or trying tofind pairs than predict a response)
• Multiple variables in the problem of building classifiers.
• Dependency network reconstruction (where a respone variablein one model is a predictor in some other models)
We’ll focus on the first one.
3

Genome/Transcriptome Scan
Basic idea:
• Formulate a question in terms of the appropriate statistical model(specific regression formula, mixture model, etc.), involving oneunknown variable (usually a gene) and some fixed ones. Theunknown can be response or explanatory variable in a regression(whichever is appropriate).
• Fit the model to each gene, separately (“scan” the genome)
• Choose some summary statistics to select/rank interesting genes(e.g. test of significance, fold-change, etc.)
• Often a cutoff (e.g. corrected p-value, FDR) is chosen to binarizethe summaries, to produce “significant” gene list. This may not beoptimal in the context of integrative analysis.
4

Types of summary statistics
Effect size : regression coefficients, model parameters
Measure strength of physical/biological relationship
⇒ The scale (unit of measurement) has to be meaningful
Dependency : Pearson’s correlation, R2, mutual information, etc.
How much we can predict; “proportion of explained variation”
⇒ The scale doesn’t matter, but the design (sampling criteria) does
Significance : p-value, t-stat, Z-test, deviance, etc.
Strength of evidence
⇒ reflects the strength of the study design (sample size, methodology,
etc.) and strength of relationship, but the role of each isn’t clear
Decision : accept or reject, gene lists, “signatures”
Which hypothesis (null or alternative) generates the observations
⇒ depends on the choice of cut-off (acceptable trade-off between “risk”
and “reward”)
5

An example
1. We are interested in metastasis-free survival in breast cancer
2. Use simple formulation:coxph(Surv(t.mfs,d.mfs) ∼ gex[,i]) ,
applied to every single gene i
3. Extract relevant summary statistics:
• Trivial stats: mean, SD of expression values
• Effect size: the estimate of coefficient and its SE
• Dependence/correlation: standardized coeff. and its SE[In this case (with no other covariates): coeff times SD]
• Log likelihood (alternate measure of significance)
• Sample size and number of events per gene (not necessarilythe same for all genes due to missing values)
Significance, p-value, rank, FDR, etc. are derived from the above.
6

Demo of an example summary table
Load data and scripts
Examine table headers
Plot (histogram, scatter, etc.) some (pairs of) summaries
Learn nplot (or others) and identify function
Derive Z-scores, p-value, deviance
Derive pseudo R2
(Near) equivalence of some of the summaries
Comparing significance, effect size, correlation (volcano plots)
7

Selecting interesting genes
Ranking is typically based on significance, not effect size (because ofgene-specific standard error), nor correlation/R2 (because it’s nearlyidentical to significance since each variable has the same sample size).
Effect size criteria are used informally, in conjuction with significance(Volcano plot).
Cutoff is decided by various criteria.
• The traditional one: p-value with multiple testing correction (e.g.Bonferroni)
• False discovery rate (e.g. Benjamini-Hochberg): treat the summarydistribution as a kind of mixture, with most genes generated by thenull, and interesting ones are outliers
[Demo: histogram Z-score, p-value, scatter plots; use p.adjust()]
8

Comparing summary profiles
Note: If the summary profiles have been turned in to gene lists, ouronly tool is contingency tables (or Venn diagram). There will be lostof information due to premature decision making.
⇒ Don’t make decision if you don’t have to!
We will use a summary, the Z-test (a.k.a. Z-score) to comparegenome-wide results of different questions on the same data.
[Demo: Load already precomputed table]
[Examine the different questions]
[Compare overlap/Venn diagram with scatter plot]
9

SwissBrodSwiss Breast Oncogenomic Database
SusanneKunkel
Dataset No. of Institution Reference Platform Data source No. ofsymbol arrays GeneIDs
Genomic platformsNKI 337 Nederlands Kanker Instituut van’t Veer 2002, van de Vijver 2002 Agilent author’s website 13120EMC 286 Erasmus Medical Center Wang 2005 Aff. U133A GEO:GSE2034 11837UPP 249 Karolinksa Institute (Uppsala) Miller 2005, Calza 2006 Aff. U133A,B GEO:GSE4922 15684STOCK 159 Karolinska Institute (Stockholm) Pawitan 2005, Calza 2006 Aff. U133A,B GEO:GSE1456 15684DUKE 171 Duke University Huang 2005, Bild 2006 Aff. U95Av2 author’s website 8149UCSF 161+8 UC San Francisco Korkola 2003 cDNA author’s website 6178UNC 143+10 University of Carolina Hu 2006 Agilent HuA1 author’s website 13784NCH 135 Nottingham City Hospital Naderi 2006 Agilent HuA1 AE:E-UCON-1 13784STNO 115+7 Stanford Univ./Norwegian Radium Hosp. Sorlie 2003 cDNA author’s website 5614JRH1 99 John Radcliffe Hospital Sotiriou 2003 cDNA journal’s website 4112JRH2 61 John Radcliffe Hospital Sotiriou 2006 Aff. U133A GEO:GSE2990 11837MGH 60 Massachusetts General Hospital Ma 2004 Agilent GEO:GSE1379 11421
expO 239 International Genomic Consortium http://www.intgen.org Aff. U133v2 GEO:GSE2109 16634TGIF1 49 EORTC trial 10994 Farmer 2005 Aff. U133A GEO:GSE1561 11837BWH 40+7 Brigham and Women’s Hospital Richardson 2006 Aff. U133v2 GEO:GSE3744 16634
Small diagnostic platformsTRANSBIG 253 TRANSBIG Consortium Buyse 2006 Agilent AE:E-TABM-77 1052EMC2 180 Erasmus Medical Center Foekens 2006 Aff. (custom) GSE3453 86HPAZ 96 Hospital La Paz, Madrid Espinosa 2005 RT-PCR paper’s appendix 61
Total 2865 = 2833 carcinomas No. of the union of all GeneIDs: 17198+ 32 non-malignant breast tissues No. of GeneIDs common to genomic platforms: 1963
• Abbreviations: No. = number, GEO: = Gene Expression Omnibus accession, AE: = ArrayExpress accession, Aff. = Affymetrix
• Curation: (re)group datasets into independent, non-redundant cohorts• Few genes are common to all platforms ⇒ use the union, not the intersection
10
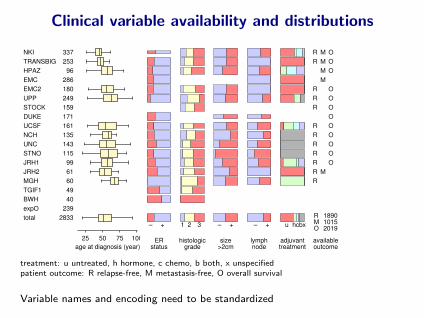
Clinical variable availability and distributions
NKI 337TRANSBIG 253HPAZ 96EMC 286EMC2 180UPP 249STOCK 159DUKE 171UCSF 161NCH 135UNC 143STNO 115JRH1 99JRH2 61MGH 60TGIF1 49BWH 40expO 239total 2833
25 50 75 100age at diagnosis (year)
– +
ERstatus
1 2 3
histologicgrade
– +
size>2cm
– +
lymphnode
u hcbx
adjuvanttreatment
R M OR M O
M OM
R OR OR O
OR OR OR OR OR OR MR
R 1890M 1015O 2019
availableoutcome
treatment: u untreated, h hormone, c chemo, b both, x unspecifiedpatient outcome: R relapse-free, M metastasis-free, O overall survival
Variable names and encoding need to be standardized
11

Heterogeneity in survival data
total 6311890 1415 1147 799 469
STNO 60115 39 10 1UNC 32128 33 10 4JRH1 4599 75 59 30 1MGH 2560 50 42 28 18NCH 47135 110 97 81 66NKI 121319 260 216 131 75TRANSBIG 101253 207 170 147 118UPP 88249 185 158 140 107JRH2 1561 55 44 38 33STOCK 40159 140 124 68EMC2 37180 164 149 94 40UCSF 20132 97 68 37 11
STNOUNCJRH1MGH
NCH
NKITRANSBIGUPP
JRH2STOCK
EMC2UCSF
40
60
80
100
rela
pse-
free
surv
ival (
%)
group: number at risk: eventsfollow-up: 0 2.5 5 7.5 10 (year)
total 4842019 1628 1313 867 512
STNO 46115 54 14 3 1DUKE 43170 86 46 12 3UNC 22129 39 14 5JRH1 4599 85 70 32 1UCSF 37132 104 74 43 14NKI 74319 290 248 147 89STOCK 40159 148 130 64NCH 34135 122 111 96 81TRANSBIG 57253 240 212 190 154UPP 51232 198 173 152 122EMC2 23180 175 166 103 44HPAZ 1296 87 55 20 3
STNODUKE
UNC
JRH1
UCSFNKI
STOCKNCH
TRANSBIGUPPEMC2HPAZ
40
60
80
100
over
all s
urviv
al (%
)
group: number at risk: eventsfollow-up: 0 2.5 5 7.5 10 (year)
Variability between studies greater than that due to natural risk factors ortreatments ⇒ typical in retrospective studies
12

Quirks in the original normalized data
Plot SD-vs-mean of each probe in a dataset⇒ A characteristic trend for each (platform,normalization) combo
NKI: Agilent (Rosetta) + ? EMC: Affy U133 + MAS 5.0 TGIF1: Affy U133A + RMA
Raw data for renormalization from scratch not always available⇒ post-hoc correction:
13

Emerging Statistical/Computational Challenges
Adapt to multi-dataset contexts:
• Gene-by-gene association or differential expression
• Clustering of genes (to find coexpression modules)
• Clustering of patients (to find disease subtypes)
• Prediction/classification
• Others, e.g. mixture models
The first one has been addressed, to some extent, by borrowingtechniques from meta analysis
14

A spectrum of approaches to combined estimates
1. Pool (treat as a single cohort)
assume the data can be “normalized” somehow beforehand
2. Fixed-effect models
study-specific nuisance parameters (e.g. as covariates)
3. Hierarchical/multilevel/mixed/empirical Bayes models
stages of sampling, with between study variance; shrinkage
4. Combined significance measures
e.g., Z-test (inverse normal), p-value (Fisher’s or inverse χ2)
5. Combined ranks or decisions
e.g., Venn diagram, overlaps, vote counting
The extremes are more easily applicable and, hence, popular
15

Illustration: Berkeley grad school admission 1973
A
B
C
D
E
F
0.25 0.5 1 2
women menodds ratio
Pooled
FE
REΣ0
2
Q = 19.9, p = 0.003
combined Z-test = -1.46, p = 0.14vote count: 1�6 significant women
Problem with pooling:“The whole can be outside the range of its parts”
• Simpson’s (reversal) paradox
(e.g. this example)
• attenuation of effect size
(e.g. when pooling centered variables)
FE: total under fixed-effect model
β =P
i βi/σ2i
. Pi 1/σ2
i
RE: total under random-effect model
β0 =P
i βi/(σ20 + σ2
i ). P
i 1/(σ20 + σ2
i )
σ20 : random effect variance (between strata)
Q: Cochran’s heterogeneityP
i(yi− µ)2/σ2i ∼ χ
2k−1
Combined Z-test:P
i Zi/√k
16

Hierarchical Modelsβ0
ww��
((βi ∼ N(β0, σ
20) β1
��
· · · βi
��
· · · βk
��
β
Yij ∼ f(βi, σ2i ) Y 1 · · · Y i · · · Y k
?>=<89:;?
BC
EDREoo
//
FE
OO
Single study:
• Inference about βi (β0 + study bias: technical, design,. . . )
Fixed-effect models
• Inference about β =P
i βi/k (mean of the specific set in hand)
• C.I. not affected by between study variability σ20
Random-effect/hierarchical models
• Inference about β0 (the “truth”; expectation of future studies)
• When Q is small behaves like FE models (weighted average);when Q is large, behaves like the “average-of-average” (insensitive toimbalance in study precisions)
17

Genome-wide scan of summary statistics
Venn diagram
160 27 411
overlap fixed-effect random-effect
18

Which quantity should be linked across studies?
Example: breast cancer survival vs expression (Cox proportional hazard).For an example gene:
MELK
-1 0 1 2beta, coefficient
DUKE 43/170EMC 107/286EMC2 0/0HPAZ 25/96JRH1 45/98JRH2 15/61MGH 0/0NKI 109/319STNO 0/0STOCK 40/159UCSF 0/0UNC 22/128UPP 53/232NCH 35/135TRANSBIG 67/253
FE mean, CI p=1e-12heterogeneity Q=42.3RE sigma0^2RE mean, CI p=6e-05
0 0.5 1Z/sqrt(Ne), correlation
p=4e-19 Q=7.6
p=4e-19
0 2.5 5 7.5 10Z-test, significance
p=2e-18 Q=11.2
p=2e-16
-log10(p)0 1 2 3 4
p = 1e-14 chi^2 = 115.1 df = 2 x 11
-log10(rank/m)0 1 2
p = 8e-08 chi^2 = 75.8 df = 2 x 11
FWER
––
–––
–
–
––––
FDR
––
–––
+
–
–+––
19

AURKA
-0.5 0 0.5 1 1.5beta, coefficient
DUKE 43/170EMC 107/286EMC2 0/0HPAZ 0/0JRH1 44/98JRH2 15/61MGH 28/60NKI 109/319STNO 46/115STOCK 40/159UCSF 38/132UNC 22/129UPP 53/232NCH 35/135TRANSBIG 67/253
FE mean, CI p=4e-19heterogeneity Q=21.1RE sigma0^2RE mean, CI p=6e-13
0 0.5 1 1.5Z/sqrt(Ne), correlation
p=3e-20 Q=13.3
p=6e-18
0 5 10Z-test, significance
p=1e-17 Q=24.9
p=3e-09
-log10(p)0 2.5 5
p = 2e-15 chi^2 = 128.0 df = 2 x 13
-log10(rank/m)0 1 2 3
p = 2e-07 chi^2 = 80.5 df = 2 x 13
FWER
––
–––+–––––––
FDR
–+
–––+–––––––
20
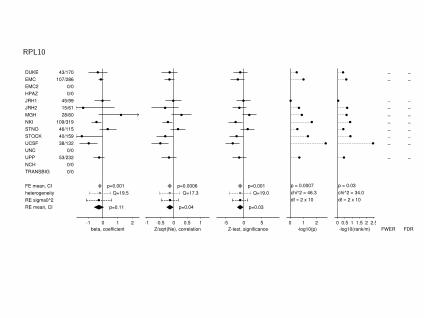
RPL10
-1 0 1 2beta, coefficient
DUKE 43/170EMC 107/286EMC2 0/0HPAZ 0/0JRH1 45/99JRH2 15/61MGH 28/60NKI 109/319STNO 46/115STOCK 40/159UCSF 38/132UNC 0/0UPP 53/232NCH 0/0TRANSBIG 0/0
FE mean, CI p=0.001heterogeneity Q=19.5RE sigma0^2RE mean, CI p=0.11
-1 -0.5 0 0.5 1Z/sqrt(Ne), correlation
p=0.0006 Q=17.3
p=0.04
-5 0 5Z-test, significance
p=0.001 Q=19.0
p=0.03
-log10(p)0 1 2
p = 0.0007 chi^2 = 46.3 df = 2 x 10
-log10(rank/m)0 0.5 1 1.5 2 2.5
p = 0.03 chi^2 = 34.0 df = 2 x 10
FWER
––
–––––––
–
FDR
––
–––––––
–
21
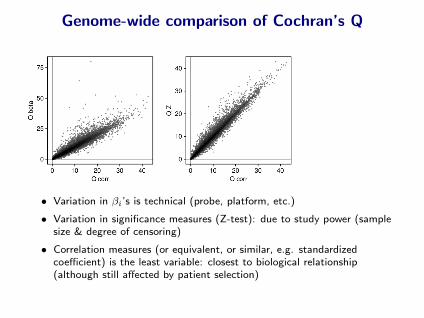
Genome-wide comparison of Cochran’s Q
• Variation in βi’s is technical (probe, platform, etc.)
• Variation in significance measures (Z-test): due to study power (samplesize & degree of censoring)
• Correlation measures (or equivalent, or similar, e.g. standardizedcoefficient) is the least variable: closest to biological relationship(although still affected by patient selection)
22

Genome-Wide Applications
23

Hierarchical Agglomerative Clustering of Genes
(Clustering arrays is a different problem)
• Use hierarchical model for pairwise Pearson correlations rijk(i studies, j, k genes)
• Transform to approximately normal quantities:
zijk = tanh−1(rijk), Var(zijk) = 1/(n− 3)ortijk = rijk√
1−r2ijk
, Var(tijk) = 1/(n− 2)
• Treat the combined correlations as similarity measures inhierarchical agglomerative clustering. No need to backtransform.
• Display the heatmaps in stratified manner
24
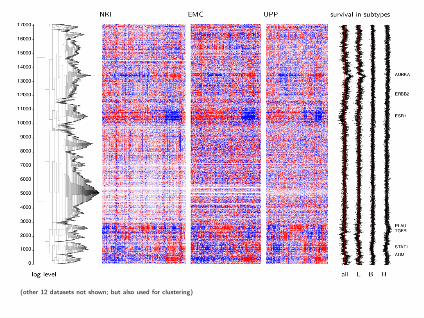
(other 12 datasets not shown; but also used for clustering)
25
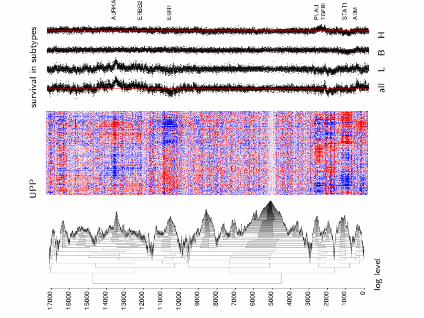
26

Glioblastoma (courtesy of Eugenia Miggliavacca)
27

Colon cancer (courtesy of Mauro Delorenzi)
28

Between array correlationsUse pairwise correlations (using variable genes)Each pair of studies (platforms) has its own common genesPool all correlation, make a single tree, use the ordering
glioblastoma colon
So far, performed separately per disease (cancer type)
Additional sampling hierarchy: disease → studies → patients
29
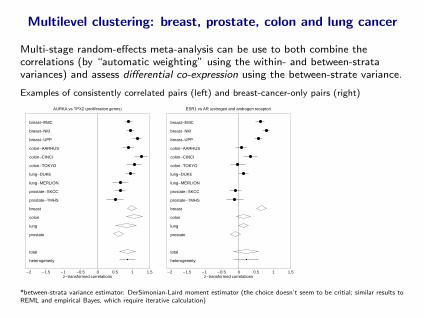
Multilevel clustering: breast, prostate, colon and lung cancer
Multi-stage random-effects meta-analysis can be use to both combine thecorrelations (by “automatic weighting” using the within- and between-stratavariances) and assess differential co-expression using the between-strate variance.
Examples of consistently correlated pairs (left) and breast-cancer-only pairs (right)
-2 -1.5 -1 -0.5 0 0.5 1 1.5z-transformed correlations
AURKA vs TPX2 Hproliferation genesL
breast-EMC
breast-NKI
breast-UPP
colon-AARHUS
colon-CINCI
colon-TOKYO
lung-DUKE
lung-MERLION
prostate-SKCC
prostate-TMHS
breast
colon
lung
prostate
total
heterogeneity
-2 -1.5 -1 -0.5 0 0.5 1 1.5z-transformed correlations
ESR1 vs AR Hestrogen and androgen receptorL
breast-EMC
breast-NKI
breast-UPP
colon-AARHUS
colon-CINCI
colon-TOKYO
lung-DUKE
lung-MERLION
prostate-SKCC
prostate-TMHS
breast
colon
lung
prostate
total
heterogeneity
*between-strata variance estimator: DerSimonian-Laird moment estimator (the choice doesn’t seem to be critial; similar results toREML and empirical Bayes, which require iterative calculation)
30

breast cancer
Dendogram of 16742 genes NKI EMC UPP
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
proliferation
estrogen receptor
androgen receptor
stroma
ERBB2
EGFR
immuneresponse
"junk"
branch depth: log(level) n = 337 n = 286 n = 249
31

prostate cancer colon cancer lung cancer
SKCC TMHS AARHUS CINCI TOKYO DUKE MERLION
n = 148 n = 65 n = 155 n = 105 n = 84 n = 198 n = 72
32

Prediction
Naıve/idiot Bayes predictors: assume conditional independenceamongst predictor variables (conditioned on the response).
DLDA, Tukey’s compound covariate, etc. are based on this.Penalized regression is similar, if the penalty is large.
• Fit gene-by-gene models
• Select top genes
• Use the gene-by-gene coefficients or significance (t-stat orZ-score, or simply the ± signs) as weights in linear predictor:∑wixi; the cutoff is to be calibrated from the training set
Still one of the best for microarray data.
⇒ Most amenable to cross-platform applications, because it’sinsensitive to the exact weights or missing genes.
33

Cross validation schemes
1. Within dataset
• Split each dataset into learning and test parts
• Select top genes (ranking based on total significance)
• In each dataset, apply the model with dataset-specificparameters to the test part
• Combine performance
2. Cross-dataset
• Split datasets into learning and test datasets
• Fit model in the test datasets
• Apply to test datasets: global weights, local cutoff (needits own CV)
⇒ “Leave-one-dataset out CV” is particularly simple
34

Example of LODOCV: Breast cancer datasets
Use Z-test as weights in linear predictor
Cutoff is 30% low-risk
total 6752426 1913 1533 1029 592
hi-risk 5561633 1216 941 617 363lo-risk 119793 697 592 412 229
hi-risk
lo-risk
0
20
40
60
80
100
DMFS
(%)
group: number at risk: eventsfollow-up: 0 2.5 5 7.5 10 (year)
DUKEEMCEMC2HPAZJRH1JRH2MGHNKISTNOSTOCKUCSFUNCUPPNCHTRANSBIGTotal
0 25 50 75 1005-year DMFS (%)
hi-risk lo-risk
hazard ratio1 3 10 30
35

Summary
Hierarchical models can be used to co-analyze datasets from multiplestudies
Random-effect meta-analysis is a decent approximation to analyzingall raw data in a stratified/hierarchical manner.
Heterogeneity/consistency is an important property that should beintegral part of the analysis and presentation
More works are needed to embed hierarchical models into othermodes of analysis: finite mixture models, conditional independentgraphs (dependency networks), . . .
36