activation of cytosolic phospholipase a2 by transforming growth
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL C H E M I S ~ ~ Y Q 1993 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 268, No. 22, Issue of August 5, pp. 16795-16802,1993 Printed in U.S.A.
Activation of Cytosolic Phospholipase Az by Transforming Growth Factor-ar in HEL-30 Keratinocytes*
(Received for publication, September 25, 1992, and in revised form, February 11,1993)
Raimund Kast, Gerhard Furstenbergert, and Friedrich Marks From the Research Program Tumor Cell Regulation, German Cancer Research Center, 0-6900 Heidelberg, Germany
In the mouse keratinocyte line HEL-30 the epidermal mitogen transforming growth factor-a (TGF-a) stimu- lated the rapid release of arachidonic acid in a dose- and time-dependent manner. The liberation of arachi- donic acid was due to the activation of a Caz+-depend- ent cytosolic phospholipase Az (cPLAz). The activation mechanism critically depended on a functionally active epidermal growth factor receptor tyrosine kinase and occurred independently of phospholipase C-mediated increases in cellular diacylglycerol and inositol 1,4,5- trisphosphate concentrations and protein kinase C ac- tivation. The activation included an increase in cyto- solic PLAz (cPLAz) activity and an association of the enzyme with the membrane fraction. Both activation steps apparently occurred in the presence of basal cy- toplasmic Caz+ concentrations. Moreover, cPLAz or a closely associated protein was found to be phosphoryl- ated on tyrosine upon TGF-a challenge of the cells. The data suggest that tyrosine phosphorylation is involved in the TGF-a-induced activation of cPLAz.
Phospholipases A2 (PLA2)’ are a family of enzymes catalyz- ing the hydrolysis of the sn-2-acyl ester bond in membrane phospholipids. The mammalian enzymes are subdivided into two major families: the low molecular mass (10-14 kDa) enzymes which are either cell-associated or extracellular, and the structurally unrelated high molecular mass (85 kDa) en- zymes localized in the cytosol (cPLA2). The former comprise type I or pancreatic PLA2s originally found in pancreatic secretions but are also present in other mammalian tissues, and type I1 or nonpancreatic PLA2s found at high levels in synovial fluids from patients with inflammatory diseases, but also in macrophages and thrombocytes (for reviews, see Refs. 1 and 2). cPLA2s have been purified to homogeneity from different cell types and tissues such as human platelets (3) and monocytes (4, 5), rat kidney (6), and mouse spleen (7). cDNAs of the corresponding human and mouse genes have
*This investigation was supported in part by a grant from the Mildred-Scheel-Stiftung/Deutsche Krebshilfe. The costs of publica- tion of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertise- ment” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
‘# To whom correspondence should be addressed Research Program Tumor Cell Regulation, German Cancer Research Center, Im Neuen- heimer Feld 280, 6900 Heidelberg, Germany.
The abbreviations used are: PLAz, phospholipase AP; PLC, phos- pholipase C; BK, bradykinin; PBS, phosphate-buffered saline; TPA, 12-O-tetradecanoylphorbol-13-acetak PKC, protein kinase C; TGF- a, transforming growth factor a; Ins-1,4,5-P3, inositol trisphosphate, DAG, diacylglycerol; DTT, dithiothreitol; EGF, epidermal growth factor; LPC, lysophosphatidylcholine; MEM, modified Eagle’s me- dium; PAGE, polyacrylamide gel electrophoresis; HBSS, Hanks’ bal- anced salt solution.
been cloned from cDNA libraries of human U 937 and mouse RAW 264.7 monocytes, and the recombinant proteins ex- pressed in bacteria and eukaryotic cells (8,9).
Both PLA2 families share some common properties such as neutral to alkaline pH optimum and Ca” dependence. How- ever, in contrast to the low molecular weight PLAzs, the cPLA2s are already activated in the presence of submicro- molar Ca2+ concentrations (2, 4, 5) and cannot be inhibited by reducing agents which split the intramolecular disulfide bonds essential for the activity of the former (2,4). Moreover, a Ca2+-dependent shift of cPLA2 activity and protein from the cytosol to the membrane has been observed (6,8,10).
PLA2s are involved in a variety of physiological and path- ophysiological processes (11-13). Since cPLA2s preferentially hydrolyze the sn-2-acyl ester bond carrying an unsaturated fatty acid residue such as arachidonic acid (4, 5), they seem to play an important role in the generation of lipid mediators such as the eicosanoids (14) which exert regulatory functions both as first and second messengers (15, 16).
In skin keratinocytes various physiological stimuli such as bradykinin (17, 18), thrombin, histamine (19), and epidermal growth factor (EGF; Ref. 20), but also irritant and mitogenic agents such as the phorbol esters (21-23) or the ionophore A23187 (24, 25), stimulate the release of arachidonic acid, which in some studies has been attributed to an activation of a PLA2 acting predominantly on phosphatidylcholine (18,25, 26). Subsequent formation of eicosanoids has been shown to be critically involved in keratinocyte mitogenesis in vivo and in vitro (for a review, see Ref. 27).
Transforming growth factor-a (TGF-a) is thought to be an important endogenous stimulator of keratinocyte prolifera- tion in uiuo and in vitro (28, 29). In epidermis, TGF-a rather than EGF seems to be the cognate EGF receptor ligand (30). TGF-a binds to the EGF receptor and activates its intrinsic tyrosine kinase activity (31, 32). The signalling cascade thus initiated and finally leading to keratinocyte proliferation is not known in detail. Here, we report that the release of arachidonic acid via a stimulation of a Ca2+-dependent cPLA, is an early response of keratinocytes to TGF-a. The TGF-a- activated cPLAz preferentially hydrolyzes arachidonoyl phos- pholipids indicating an important role of this type of PLA, in the initiation of eicosanoid biosynthesis and mitogenic sig- nalling in keratinocytes.
EXPERIMENTAL PROCEDURES
Materials-Recombinant human TGF-a was provided by Bissen- dorf-Bachem, Hannover, Germany. Bradykinin (BK) triacetate, ara- chidonic acid, 1,2-diacylglycerol (DAG), 1,3-diacylglycerol, lysophos- phatidylcholine (LPC), dithiothreitol (DTT), phenylmethanesulfonyl fluoride, and other reagents were obtained from Sigma, Munchen, Germany; ionomycin and indo-l-acetoxymethylester were from Cal- biochem, Frankfurt, Germany. Tyrphostin was purchased from GIBCO-BRL, Karlsruhe, Germany. Monoclonal anti-EGF receptor antibodies clone 29.1.1 and Ab-3 were obtained from Sigma and
16795

16796 Regulation of Cytosolic Phospholipase A2
Dianova, Hamburg, Germany, respectively, monoclonal antiphospho- tyrosine antibodies (IG2 and clone 3-365-10) from Boehringer Mann- heim, a monoclonal anti-a-protein kinase C (PKC) antibody from Amersham, Braunschweig, Germany. The polyclonal anti-6-PKC an- tibody was kindly provided by Drs. Leibersperger and Gschwendt, and the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) was a generous gift from Dr. Hecker, all of the German Cancer Research Center, Heidelberg, Germany. Methyl-[3H]choline (specific activity 1.5-2-2 TBq/mmol), my0-[2-~H]inositol (659 GBq/mmol), [l-14C]arachidonic acid (2.04 GBq/mmol), and l-stearoyI-2-[l-"C] arachidonoyl phosphatidylcholine (2.07 GBq/mmol) and 1-palmitoyl- 2-1 1-"C]palmitoyl phosphatidylcholine (2.14 GBq/mmol) were pur- chased from Amersham Buchler. [-y-32P]ATP (111 TBq/mmol) were obtained from Du Pont-New England Nuclear, Dreieich, Germany. Silica Gel 60 TLC plates were purchased from Merck, Darmstadt, Germany.
Cell Culture-The murine epidermal cell line HEL-30 (18) was grown in 100-mm plastic dishes in a modified Eagle's minimal essen- tial medium with a 4-fold concentration of amino acids and vitamins (4 X MEM) supplemented with 10% fetal calf serum and antibiotics (penicillin, 100 IU/ml, streptomycin 100 pg/ml). The cells were kept at 30 "C in a humidified atmosphere of 95% air and 5% COz. For experiments, confluent cultures were harvested by trypsinization and diluted in 35-mm plastic dishes (2.5 X lo6 cells/cm2) with 4 X MEM supplemented with 5% fetal calf serum (growth medium). The cells were incubated with growth medium at 34 "C and used after 3-4 days, when they had reached 80-90% confluency (1-1.5 X lo6 cells/dish). The human carcinoma cell line A-431 was maintained in Dulbecco's modified Eagle's medium plus 10% fetal calf serum in the presence of glucose (4.5 g/liter) at 37 "C in an atmosphere of 95% air and 5% COz, and subcultured for experiments by plating lo4 cells/cm' in a 100-mm dish.
Preparation, Treatment, and Analysis of (l-14C]Arachidonic Acid- labeled Cells-Keratinocytes grown in 35-mm dishes to near con- fluency were labeled by incubation with 0.3 pCi of [l-'4C]arachidonic acid in growth medium for 16 h. For measurements of arachidonic acid liberation the cells were rinsed free of unincorporated [1-"C] arachidonic acid with PBS, and fresh growth medium was added containing either acetone or PBS (final concentrations 0.1%; con- trols), the phorbol ester TPA or TGF-a. After the appropriate incu- bation times 1 ml of medium was removed, acidified to pH 3 with 1 N HCI, and extracted twice with ethyl acetate as described in Ref. 22. Eicosanoids were analyzed by thin layer chromatography on Silica Gel 60 plates with the solvent system obtained from the upper phase of isooctane/ethyl acetate/acetic acid/water (50:110:20100; by vol- ume). Reference spots were visualized with iodine vapor. The radio- activity was determined using a TLC Linear Analyzer (Berthold B 2722-2). The impulses/min measured with the TLC analyzer corre- spond to 2.2% disintegrations/min. Linearity was checked using [ l - "C]arachidonic acid.
For experiments showing the effect of protein kinase C down- regulation on ["C]arachidonic acid liberation, keratinocytes were treated with 1 p M TPA for 18 h. To label the cellular phospholipids the TPA-treated cells were washed with PBS and then refed with new growth medium containing 1.5 pCi of [I-"Clarachidonic acid. After a 1-h incubation the cells were washed with PBS once again and new growth medium was added containing TGF-a and/or TPA. After appropriate time intervals the growth medium was removed and processed as described before.
LPC Analysis-For LPC analysis, methyl-[3H]choline (0.3 pCi) was introduced into keratinocytes grown to near confluency for a period of 16 h through a growth medium change. After rinsing the cells with PBS, TGF-a, or TPA were added to the cells together with new growth medium for the times indicated. LPC was extracted from the cells with 1-butanol (33) and assayed by two-dimensional thin layer chromatography on Silica Gel 60 plates using the solvent chloroform, methanol, 25% ammonia (65:35:4; by volume) in the first and chloroform/acetone/methanol/acetic acid/water (5020:10:105; by volume) in the second direction. Finally, the plates were dried and the phospholipids were visualized with comigrated standards using iodine vapor. The spots were scraped from the plates and the radio- activity was measured by liquid scintillation counting.
Determination of DAGS-Confluent keratinocytes were labeled with [l-14C]arachidonic acid (0.3 pCi/35-mm dish, see above) and stimulated with BK or TGF-a without growth medium change. To stop the reaction the growth medium was aspirated and 1-ml of ice- cold methanol was added. The cells were scraped from the dishes and
the methanol/cell suspension was extracted and analyzed according to Ref. 18.
Determination of Inositol Phosphates-Confluent cells were labeled for 24 h with my0-[2-~H]inositol (7.5 pCi/35-mm dish) in growth medium. Prior to BK or TGF-a activation, the cells were incubated with 10 mM LiCl for 1 h to inhibit inositol phosphatase activity. BK or TGF-a were then added directly to the growth medium and the keratinocytes were incubated for the times indicated. Metabolic ac- tivity was terminated after aspiration of growth medium followed immediately by the addition of 1 ml of ice-cold 10% (w/v) trichloro- acetic acid. The inositol phosphates were extracted according to Ref. 18 and quantitated by liquid scintillation counting.
Preparation of Cell-free Extracts-HEL-30 cells grown in 100-mm dishes to confluency were incubated in the presence of 50 p~ Na3V04 for 2 h and then treated with PBS or acetone (0.1% final concentra- tion; controls) or TGF-a, TPA, or BK for various time periods. The cells were washed once with PBS and subsequently incubated with homogenization buffer containing 50 mM TrislHCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 1 mM PMSF, 1 mM Na3V0,, 1 mM DTT, 150 mM NaCl for 15 min at 4 "C. All subsequent steps were performed at 4 "C. The cells were scraped from the culture dish and homogenized by sonication for 60 s. The lysate was centrifuged at 1000 x g for 5 min. The supernatant was recentrifuged at 100,000 X g for 60 min. The high-speed supernatant was recovered as cytosolic fraction and the pellet as particulate fraction. The latter was resuspended in 0.5 ml of homogenization buffer at a protein concentration of 1 mg/ml. cPLA, activity and protein were determined as described below. To study agonist-induced cPLAz translocation, cells were lysed in ho- mogenization buffer in the absence of EDTA and EGTA. PLA, distribution in untreated cells was determined in the absence of NaaV04.
Assay of cPLAz Actiuity-The PLAZ assay measured the hydrolysis of [l-14C]arachidonic acid from the substrate l-stearoyl-2-[ l-"C] arachidonoyl phosphatidylcholine. The assay was performed accord- ing to Bonventre et al. (34) using homogenization buffer containing 4 mM CaCI,, 1 mM DTT, and 15 pM radiolabeled substrate in a final volume of 40 pl. Substrate was prepared by drying down the solvent under vacuum evaporation and resuspending the residue in dimethyl sulfoxide by vigorous vortexing for 2 min. The assay was initiated by addition of the keratinocyte fractions adjusted to a protein concen- tration of 1 mg/ml. After 30 min at 37 "C the reaction was stopped by the addition of ice-cold ethanol containing 2% of 1 N HCl and 20 pgjml arachidonic acid as carrier. 50 pl of the mixture was then spotted onto silica gel thin layer chromatography plates which were developed and analyzed for [I-"Clarachidonic acid as described above.
Down-regulation of PKC-In order to down-regulate the keratino- cyte PKC isoenzymes a and 6 (351, confluent HEL-30 cells were treated with M TPA for 18 h. Total PKC activity in the homog- enate was measured as described previously (18). To analyze down- regulation of the PKC protein the homogenate was applied to a protamine-agarose column pre-equilibrated with 0.5 M NaCl in col- umn buffer containing 50 mM Tris/HCl, pH 8.0, 1 mM EGTA, and 20 mM 2-mercaptoethanol. After washing, PKC isoenzymes were eluted with column buffer containing 1.5 M NaCI. The proteins were separated by SDS-polyacrylamide gel electrophoresis on a 7.5% ac- rylamide gel and transferred to an Immobilon P membrane. PKC isoenzymes a and 6 were visualized by a monoclonal anti-PKC-a antibody or a polyclonal affinity purified anti-PKC-6 antibody and immunostained with alkaline phosphatase-conjugated goat anti- mouse IgG or goat anti-rabbit IgG, respectively.
Preparation of Anti-cPLAZ-Peptide Antiserum and Immunoblotting Procedures-Immunization was performed by injection of 100 pg of the C-terminal cPLAz-peptide (amino acids 709-726) emulsified in Freund's complete adjuvant in New Zealand rabbits. Booster injec- tions with 100 pg of antigen emulsified in Freund's incomplete adju- vant were given at a 4-week interval. Blood was collected 2 weeks after booster injections. cPLA2 was detected by Western blotting using an purified anti-cPLA2-peptide antiserum (dilution 1:lOOO) and anti-rabbit IgG-alkaline phosphatase conjugates (36).
Immunoprecipitation of Tyrosine-phosphorylated Proteins-HEL- 30 cells treated with PBS (0.1%; controls) or TGF-a (20 nM) for 5 min were washed with PBS and then incubated with homogenization buffer containing 10 mM NaF, 10 pg/ml leupeptin for 15 min at 4 "c. Preparation of cytosol was done as described above. Immunoprecipi- tation was performed by incubation of 0.2 ml of cytosol (1 mg of protein/ml) with 5 pg of a monoclonal antiphosphotyrosine antibody (clone 3-365-10, Boehringer) in the presence or absence of varying
Regulation of Cytosolic Phospholipase A2 16797
concentrations of phenyl phosphate overnight at 4 "C. The immune complexes were precipitated by incubation with 20 pl of a 10% solution of Pansorbin for 1 h. The immunoprecipitates were subjected to SDS-PAGE and immunoblot analysis using the purified anti- cPLAz antiserum as primary antibody (dilution 1:1000).
EGF Receptor !Prosine Kinase Assay-A-431 cells grown to con- fluency were solubilized with lysis buffer containing 25 mM Hepes, pH 7.8,5 mM EGTA, 50 mM NaF, 50 PM Na3V04, 10 pg/ml leupeptin, 1% Triton X-100. After removing insoluble material by centrifugation at 4 'C for 30 min at 40,000 X g, the supernatants were assayed for EGF receptor tyrosine kinase activity. The assays were performed in 25 mM Hepes, pH 7.8, 1 mM EGTA, 5 mM Mg% 50 mM NaF, 100 p~ Na3V04, 10 pg/ml leupeptin, 0.2% Triton X-100 in the presence or absence of TGF-CY or monoclonal EGF receptor antibodies (clone 29.1.1; Sigma) in a final volume of 50 ~ 1 . After incubation of the samples for 15 min at 25 'C, the reaction was terminated by adding 12.5 pl of 5 X Laemmli sample buffer. The samples were boiled at 100 'C for 5 min and subjected to SDS-polyacrylamide gel electro- phoresis. The state of tyrosine phosphorylation of the EGF receptor was investigated by a Western blot procedure using a monoclonal anti-phosphotyrosine antibody (1G2, Boehringer Mannheim) and anti-mouse IgG-alkaline phosphatase conjugates.
Ca2+ Measurements-To determine intracellular Caz+ concentra- tions, HEL-30 keratinocytes (0.5 X lo6 cells) were seeded on plastic coverslips. The cells were cultured in growth medium and reached confluency after 3 days of incubation. After rinsing once in PBS, cells were loaded with indo-1/AM by incubation at 34 "C for 30 min in 4 X MEM, 0.5% fetal calf serum to which 2 PM indo-1/AM was added. Subsequently, cells were washed three times in HBSS containing 137 mM NaCl, 5.4 mM KCl, 0.8 mM MgS04. 7Hz0, 0.9 mM NazHP04, 1.0 mM CaC12.2Hz0, 0.4 mM KH2P04, 5.6 mM D-glUCOSe, 0.1 mg/ml bovine serum albumin, 10 mM Hepes, pH 7.4, and then incubated in HBSS at 25 'C for 15 min (37).
For the experiments, the coverslips were placed in quartz cuvettes containing HBSS. Indo-1 fluorescence was measured using a Perkin- Elmer LS-58 luminescence spectrometer. The excitation wavelength was 330 nm, the emission wavelength was recorded at 500 nm. Calibrations were accomplished by the ionomycin/Mn2+ method as described by Moolenaar et al. (38).
RESULTS
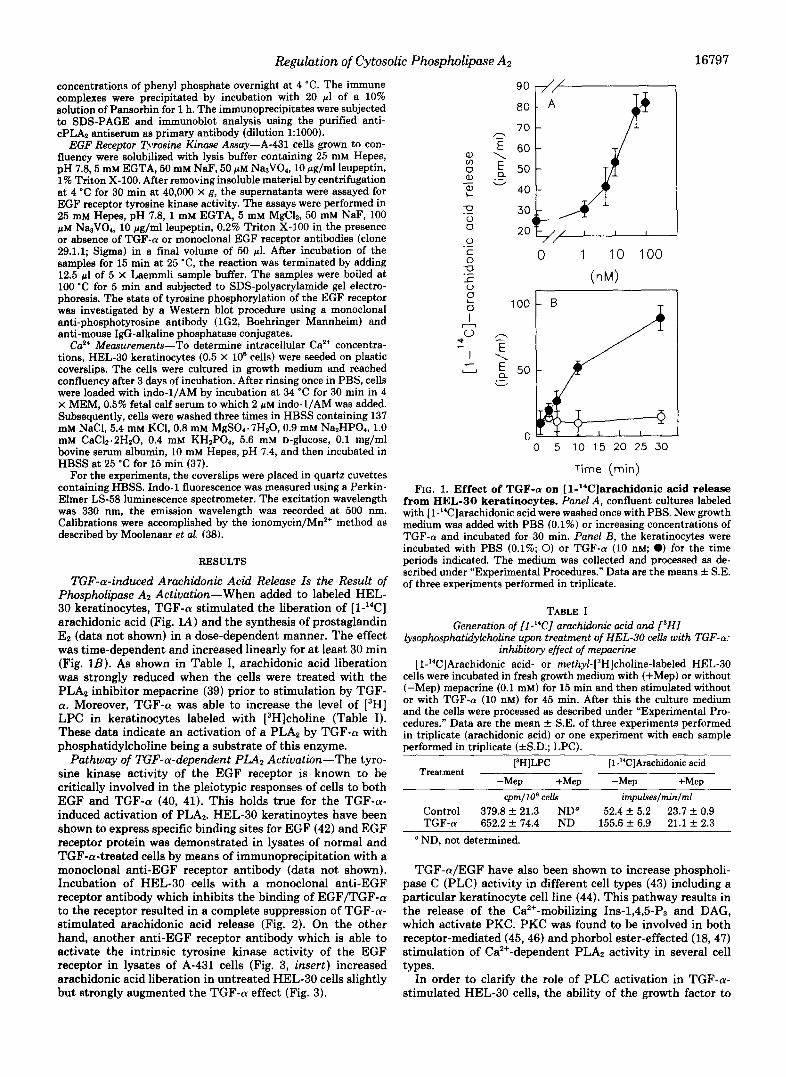
TGF-a-induced Arachidonic Acid Release Is the Result of Phospholipase A2 Activation-When added to labeled HEL- 30 keratinocytes, TGF-a stimulated the liberation of [l-"CC] arachidonic acid (Fig. 1A) and the synthesis of prostaglandin E2 (data not shown) in a dose-dependent manner. The effect was time-dependent and increased linearly for at least 30 min (Fig. 1B). As shown in Table I, arachidonic acid liberation was strongly reduced when the cells were treated with the PLA, inhibitor mepacrine (39) prior to stimulation by TGF- a. Moreover, TGF-a was able to increase the level of [3H] LPC in keratinocytes labeled with [3H]choline (Table I). These data indicate an activation of a PLA2 by TGF-a with phosphatidylcholine being a substrate of this enzyme.
Pathway of TGF-a-dependent PLA2 Actiuatwn-The tyro- sine kinase activity of the EGF receptor is known to be critically involved in the pleiotypic responses of cells to both EGF and TGF-(Y (40, 41). This holds true for the TGF-a- induced activation of PLA2. HEL-30 keratinoytes have been shown to express specific binding sites for EGF (42) and EGF receptor protein was demonstrated in lysates of normal and TGF-a-treated cells by means of immunoprecipitation with a monoclonal anti-EGF receptor antibody (data not shown). Incubation of HEL-30 cells with a monoclonal anti-EGF receptor antibody which inhibits the binding of EGF/TGF-a to the receptor resulted in a complete suppression of TGF-a- stimulated arachidonic acid release (Fig. 2). On the other hand, another anti-EGF receptor antibody which is able to activate the intrinsic tyrosine kinase activity of the EGF receptor in lysates of A-431 cells (Fig. 3, insert) increased arachidonic acid liberation in untreated HEL-30 cells slightly but strongly augmented the TGF-a effect (Fig. 3).
90
50
40 W .-
100
50
0 5 10 15 20 25 30
Time (min)
FIG. 1. Effect of TGF-(r on [1-"C]arachidonic acid release from HEL-30 keratinocytes. Panel A, confluent cultures labeled with [l-14C]arachidonic acid were washed once with PBS. New growth medium was added with PBS (0.1%) or increasing concentrations of TGF-a and incubated for 30 min. Panel B, the keratinocytes were incubated with PBS (0.1%; 0) or TGF-a (10 nM; 0) for the time periods indicated. The medium was collected and processed as de- scribed under "Experimental Procedures." Data are the means f S.E. of three experiments performed in triplicate.
TABLE I Generation of [l-"C] arachidonic acid and P H I
lysophosphatidykholine upon treatment of HEL-30 cells with TGF-a: inhibitory effect of mepacrine
[1-"CIArachidonic acid- or rnethyl-[3H]choline-labeled HEL-30 cells were incubated in fresh growth medium with (+Mep) or without (-Mep) mepacrine (0.1 mM) for 15 min and then stimulated without or with TGF-a (10 nM) for 45 min. After this the culture medium and the cells were processed as described under "Experimental Pro- cedures." Data are the mean f S.E. of three experiments performed in triplicate (arachidonic acid) or one experiment with each sample performed in triplicate (fS.D.; LPC).
13H]LPC Treatment
[1-"CJArachidonic acid
-Mep +Mep -Mep +Mep cpm/lO' celb impw!ses/minlml
Control 379.8 f 21.3 ND" 52.4 f 5.2 23.7 f 0.9 TGF-a 652.2 f 74.4 ND 155.6 f 6.9 21.1 k 2.3 ' ND, not determined.
TGF-a/EGF have also been shown to increase phospholi- pase C (PLC) activity in different cell types (43) including a particular keratinocyte cell line (44). This pathway results in the release of the Ca'+-mobilizing Ins-1,4,5-P3 and DAG, which activate PKC. PKC was found to be involved in both receptor-mediated (45,46) and phorbol ester-effected (18,47) stimulation of Ca2+-dependent PLA2 activity in several cell types.
In order to clarify the role of PLC activation in TGF-a- stimulated HEL-30 cells, the ability of the growth factor to

Cytosolic Phospholipase A:! 16798 Regulation
L 0 I
PBS TGFa
FIG. 2. Inhibition of TGF-a-induced [1-“Clarachidonic acid release from HEL-30 keratinocytes by a neutralizing anti-EGF receptor antibody. [1-“CIArachidonic acid-labeled HEL-30 cells were incubated with IgG (20 pg/ml; -ab; control) or the monoclonal anti-EGF receptor antibody Ab-3 (Dianova; 20 pg/ml; +ab) together with new growth medium for 30 min and then stimu- lated with PBS (0.5%) or TGF-a (10 nM) for 45 min. After that, the growth medium was removed and processed as described under “Ex- perimental Procedures.’’ The data represent the means & S.D. from one experiment with each sample performed in triplicate.
e, (0 0 e, e, L
PBS TGFa
FIG. 3. Effect of a tyrosine kinase-activating anti-EGF receptor antibody on normal and TGF-a-stimulated release of [ 1-“Clarachidonic acid from HEL-30 keratinocytes. Insert, effect of this antibody on EGF receptor autophosphorylation in lysates of A-431 cells in the absence and presence of TGF-a. [l-“C] Arachidonic acid-labeled HEL-30 cells were incubated with new growth medium containing mouse IgG (20 pg/ml; -ab; control) or the EGF receptor tyrosine kinase-activating antibody (clone 29.1.1, Sigma; 20 pg/ml; +ab) and PBS (0.1%) or TGF-a (10 nM) for 30 min. After that, the growth medium was removed and processed as de- scribed under “Experimental Procedures.” The data are the results from one experiment with each sample performed in triplicate (& S.D.). Autophosphorylation of the EGF receptor was analyzed by immunoblotting using a monoclonal antiphosphotyrosine antibody as described under “Experimental Procedures.” Arrow indicates the position of the EGF receptor. A molecular mass marker (in kilodal- tons) is indicated on the left. Inset: lane I , A-431 lysate alone (control); lane 2, control plus TGF-a (10 nM); lane 3, control plus antibody (4 pg/ml); lane 4, control plus TGF-a (10 nM) plus antibody (4 pg/ml).
activate the DAG/Ins-1,4,5-P3 cascade was examined. In con- trast to BK, a known activator of the epidermal PLC (18), TGF-a was found to be completely inactive in stimulating the release of these second messengers (Fig. 4, A and B ) and in increasing the concentration of cytoplasmic free Ca2+ in cells preloaded with indo-1/AM (Fig. 4C). Moreover, the PKC inhibitor K-252a (48) only slightly inhibited the TGF-a- induced arachidonic acid release, whereas it completely pre-
0)
Time (min)
- I 8000
5 -0 7000
Q E
O ) m
s
3000 .- 2\ 2 k 1000 m 2000
I - 0 .- .- - I
m 0 5 10 40 45 U c. Time (min)
1000 ,
.- +” c.\ 600 N - r 800h 0 - 400 V I
U
0 1 2 3 4 5
Time (min) FIG. 4. Effects of TGF-a and BR on the release of [l-“C]
diacylglycerol ( A ) and [*H]inositol phosphates ( B ) from and on the cytoplasmic Ca2+ concentration (C) in HEL-30 keratin- ocytes. A, [1-”Clarachidonic acid-labeled HEL-30 cells were stimu- lated with PBS (0.1%; 0), TGF-a (10 nM; O), or BK (1 p ~ ; m) for different time periods. Cellular lipids were extracted and analyzed as described under “Experimental Procedures.” Data are the mean f S.E. of two experiments performed in triplicate. B, my~-[~H]inositol- labeled cells were incubated with PBS (0.5%; 01, TGF-a (10 nM; O), or BK (1 p ~ ; W) for the time periods indicated. Total inositol phosphates were isolated as described under “Experimental Proce- dures.” The data represent the means & S.E. from two independent experiments performed in triplicate. C, HEL-30 keratinocytes were plated on plastic coverslips in Leighton tubes and used after reaching confluency. The cells were loaded with indo-1/AM and then stimu- lated with TGF-a (10 nM; 0) or BK (1 p ~ ; W) for various periods of time. Ca2+ measurements were carried out in HBSS test medium as described under “Experimental Procedures.’’ Each point is the mean & S.E. of four independent determinations.

Regulation of Cytosolic Phospholipase A2 16799
vented the effect of the PKC agonist TPA (Ref. 18; Table 11). Likewise, TPA-induced down-regulation of phorbol ester- sensitive PKC isozymes (data not shown) in HEL-30 cells did not affect the analogous TGF-a-induced arachidonic acid release but completely prevented the TPA effect (Ref. 18; Fig. 5). Vice versa, the tyrosine kinase inhibitor tyrphostin was able to prevent TGF-a-induced arachidonic acid liberation only when applied in concentrations known to inhibit the kinase activity in keratinocytes (Ref. 49; Fig. 6). Taken to- gether, these results indicate the effect of TGF-a on keratin-
TABLE I1 Effect of the PKC inhibitor K252a on TGF-a-induced [l--“Cl
arachidonic acid release from HEL-30 keratinocytes [1-”ClArachidonic acid-labeled HEL-30 cells were incubated with
TGF-a (10 nM) in the presence (fK252a) or absence (-K252a) of K252a (0.1 pM) for 30 min. After this the culture medium was removed and processed as described under “Experimental Procedures.” Data are the means f S.E. of three experiments performed in triplicate.
Treatment [1-“CIArachidonic acid release
-K525a +K252a impulses/min/ml
Control 33.8 * 3.5 28.0 f 2.9 TGF-a 90.8 f 4.2 75.0 f 2.3
o n a, ;5- 200
150 U 100
0 50 0 0
0 acetone
.- 0
.- C
-0
1 A 1
I
TPA .- L 0
F ? I - 0
400 - z 350
c‘ - ’ E 300 -
E 250 - ;; 200 -
150 - 100 - “-LI
Q
50 - 0
acetone TGFa FIG. 5. Effects of TPA-induced down-regulation of PKC on
TPA- and TGF-a-induced [l-“C]arachidonic acid release from HEL-30 keratinocytes. Cells were incubated with acetone (0.5%; +PKC) and TPA (1 p ~ ; -PKC) for 18 h, washed once with PBS, and radiolabeled with [l-’‘C]arachidonic acid (1 pCi/ml) for 1 h. Subsequently, the cells were stimulated with acetone (0.5%; con- trol), TPA (1 JLM: panel A), or TGF-a (10 nM; panel B ) , dissolved in new growth medium for 30 min. The growth medium was then removed and processed as described under “Experimental Proce- dures.” Data are the means k S.E. of two experiments performed in triplicate.
r
I U -
0
n
acetone TGFa
L TPA
FIG. 6. Effect of tyrphostin on TPA- and TGF-a-induced release of [1-“C]arachidonic acid from HEL-30 keratino- cytes. Cells were labeled with [I-“Clarachidonic acid for 16 h in the absence (-tyr) or the presence of the tyrosine kinase inhibitor tyr- phostin (20 PM; +tyr). The cells were then washed once with PBS and new growth medium was added containing acetone (0.5%; con- trols), TPA (1 p ~ ) , or TGF-a (10 nM). After 30 min of incubation the growth medium was removed and processed as described under “Experimental Procedures.” The data represent the means from two independent experiments performed in triplicate. Maximal deviation was less than 15%.
ocyte PLA2 to be more directly induced by the EGF receptor tyrosine kinase rather than to be mediated via the DAG/Ins- 1,4,5-P3 cascade.
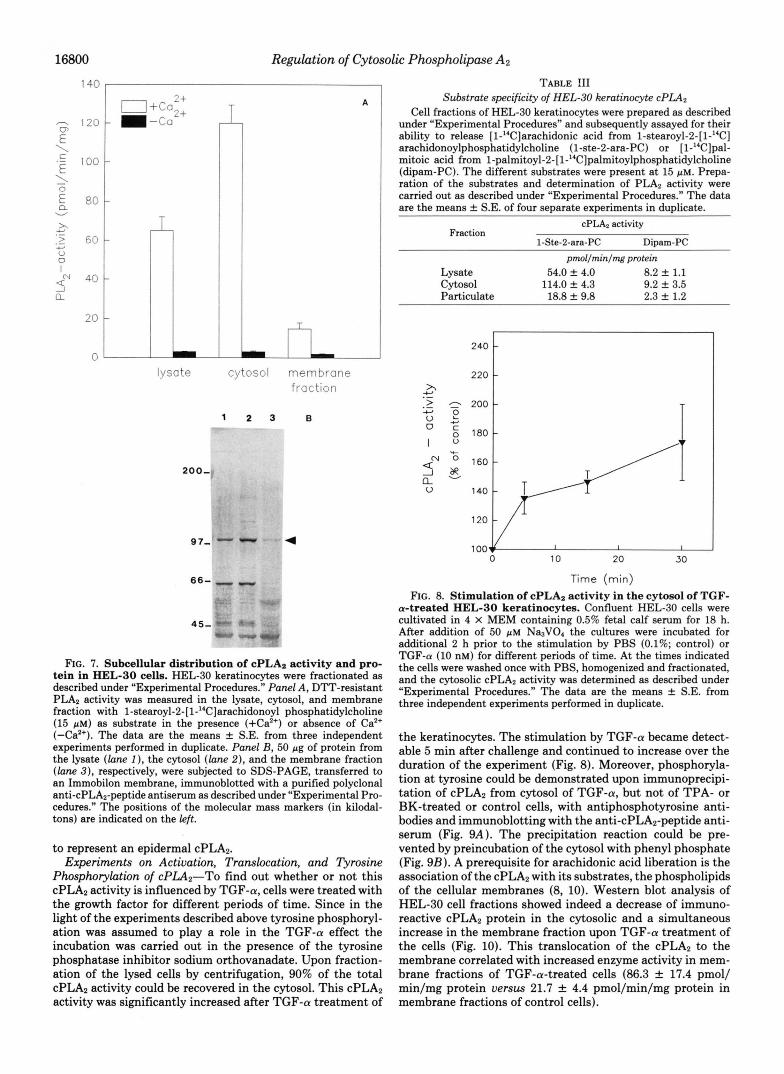
TGF-(U Activates a cPLAz-In the following experiments an attempt was made to characterize the PLA, type responsible for TGF-a-induced arachidonic acid liberation in HEL-30 keratinocytes. Upon lysis of the cells, homogenization and subsequent fractionation under reducing conditions, and in the presence of EGTA and EDTA, to prevent membrane adsorption of the enzyme, the Ca2+-dependent PLA, activity of the lysate as determined by incubation with the substrate l-stearoyl-2-[l-’4C]arachidonoyl phosphatidylcholine was re- covered predominantly in the cytosol, while only 10% of the activity determined in the cytosol was found in the particulate fraction (Fig. 7A). Neither the intracellular distribution nor the total PLA2 activity were altered in the absence of the reducing agent DTT, excluding the presence of low molecular weight PLA2 species (2, 4). The cytosolic PLA2 activity showed a remarkable substrate selectivity in that it catalyzed the release of arachidonic acid 7-15 times more efficiently than that of palmitoic acid (Table 111). Such a selectivity has also been observed for other cPLA2s (4-7, 10). Thus, the keratinocyte PLA, exhibits several properties characteristic for the high molecular mass cPLA2 (85 kDa) which was shown to exhibit an apparent molecular mass of 100-110 kDa on
In fact, such a 100-kDa protein species was detected in immunoblots of cell lysates derived from HEL-30 keratino- cytes using a cPLA2-specific antiserum that we had raised against a C-terminal oligopeptide (36) derived from the mouse cPLA2 sequence which has recently been published (8). More- over, when equal amounts of cell lysates, cytosol, and partic- ulate fraction obtained in the presence of EDTA and EGTA were immunoblotted, the subcellular distribution of this 100- kDa protein (Fig. 7 B ) was found to be similar to that of PLA, activity (Fig. 7A). These results indicate the 100-kDa protein
SDS-PAGE (4, 5).
16800 Regulation of Cytosolic Phospholipase AP
140 I
lysate
200.
97
66
45
Cytosol membrane fraction
1 2 3 B
4
FIG. 7. Subcellular distribution of cPLA2 activity and pro- tein in HEL-30 cells. HEL-30 keratinocytes were fractionated as described under "Experimental Procedures." Panel A, Dm-resistant PLA2 activity was measured in the lysate, cytosol, and membrane fraction with l-stearoyl-2-[l-14C]arachidonoyl phosphatidylcholine (15 pM) as substrate in the presence (+Ca2') or absence of Ca2+ (-Ca2'). The data are the means f S.E. from three independent experiments performed in duplicate. Panel B, 50 pg of protein from the lysate (lone I ) , the cytosol (lane 2), and the membrane fraction (lane 3), respectively, were subjected to SDS-PAGE, transferred to an Immobilon membrane, immunoblotted with a purified polyclonal anti-cPLA2-peptide antiserum as described under "Experimental Pro- cedures." The positions of the molecular mass markers (in kilodal- tons) are indicated on the left.
to represent an epidermal cPLA2. Experiments on Activation, Translocation, and Tyrosine
Phosphorylation of cPLA2"To find out whether or not this cPLA2 activity is influenced by TGF-a, cells were treated with the growth factor for different periods of time. Since in the light of the experiments described above tyrosine phosphoryl- ation was assumed to play a role in the TGF-a effect the incubation was carried out in the presence of the tyrosine phosphatase inhibitor sodium orthovanadate. Upon fraction- ation of the lysed cells by centrifugation, 90% of the total cPLA2 activity could be recovered in the cytosol. This cPLA2 activity was significantly increased after TGF-a treatment of
TABLE !I1 Substrate specificity of HEL-30 keratinocyte cPLA2
Cell fractions of HEL-30 keratinocytes were prepared as described under "Experimental Procedures" and subsequently assayed for their ability to release [ 1-"Clarachidonic acid from l-stearoyl-2-[l-"C] arachidonoylphosphatidylcholine (1-ste-2-ara-PC) or [1-"Clpal- mitoic acid from l-palmitoyl-2-[l-14C]palmitoylphosphatidylcho~ine (dipam-PC). The different substrates were present at 15 p ~ . Prepa- ration of the substrates and determination of PLA, activity were carried out as described under "Experimental Procedures." The data are the means f S.E. of four separate experiments in duplicate.
Fraction cPLA2 activity
1-Ste-2-ara-PC Dipam-PC pmollrninlmg protein
Lysate 54.0 f 4.0 Cytosol
8.2 f 1.1 114.0 f 4.3 9.2 f 3.5
Particulate 18.8 f 9.8 2.3 f 1.2
220 x 2401
10 20 30
Time (rnin)
FIG. 8. Stimulation of cPLAz activity in the cytosol of TGF- a-treated HEL-30 keratinocytes. Confluent HEL-30 cells were cultivated in 4 X MEM containing 0.5% fetal calf serum for 18 h. After addition of 50 p~ Na3V04 the cultures were incubated for additional 2 h prior to the stimulation by PBS (0.1%; control) or TGF-a (10 nM) for different periods of time. A t the times indicated the cells were washed once with PBS, homogenized and fractionated, and the cytosolic cPLA2 activity was determined as described under "Experimental Procedures." The data are the means f S.E. from three independent experiments performed in duplicate.
the keratinocytes. The stimulation by TGF-a became detect- able 5 min after challenge and continued to increase over the duration of the experiment (Fig. 8). Moreover, phosphoryla- tion at tyrosine could be demonstrated upon immunoprecipi- tation of cPLA2 from cytosol of TGF-a, but not of TPA- or BK-treated or control cells, with antiphosphotyrosine anti- bodies and immunoblotting with the anti-cPLA2-peptide anti- serum (Fig. 9A). The precipitation reaction could be pre- vented by preincubation of the cytosol with phenyl phosphate (Fig. 9B). A prerequisite for arachidonic acid liberation is the association of the cPLA2 with its substrates, the phospholipids of the cellular membranes (8, 10). Western blot analysis of HEL-30 cell fractions showed indeed a decrease of immuno- reactive cPLA2 protein in the cytosolic and a simultaneous increase in the membrane fraction upon TGF-a treatment of the cells (Fig. 10). This translocation of the cPLA2 to the membrane correlated with increased enzyme activity in mem- brane fractions of TGF-a-treated cells (86.3 f 17.4 pmol/ min/mg protein uersus 21.7 f 4.4 pmol/min/mg protein in membrane fractions of control cells).

A 1
2 0 0 -
97-
66-
B 1
97 kDa -
Regulation of Cytosolic Phospholipase AP
2 3 4 1 2 3 4
97 -
16801
c
1
2 3 4 5
4
FIG. 9. Immunoprecipitation of cPLA2 phosphorylated on tyrosine from cytosol of TGF-a- (A and B ) , but not of BK- and TPA (A)-treated HEL-30 keratinocytes. Cells were treated with Na3V04 as described in the legend to Fig. 8 and incubated with PBS (0.1%; controls), TGF-a (20 nM), TPA (1 pM), or BK (1 p M ) for 5 min. Fractionation and immunoprecipitation in the absence or presence of increasing concentrations of phenyl phosphate and im- munoblotting were performed as described under “Experimental Pro- cedures.” A: lane 1, control; lane 2, BK; lane 3, TGF-a; lane 4, TPA. B: lane 1, control; lane 2, TGF-a; lane 3, TGF-cY/~O-~ M phenyl phosphate, lane 4, TGF-cY/~O-~ M phenyl phosphate; lane 5, cytosol. The positions of the molecular mass markers (in kilodaltons) are indicated at the left. The arrow indicates the position of the cPLAz.
DISCUSSION
TGF-a is a major autocrine growth factor for epidermal keratinocytes in uiuo and in vitro (28-30). Increased TGF-a expression is observed in hyperplastic states of the skin such as epidermal wound healing or chronic hyperproliferative skin diseases (50), and in epidermal tumorigenesis (51, 52), i.e. processes which have been found to be critically dependent on endogenous eicosanoid generation. The experiments pre- sented here show an activation of high molecular weight cPLA2 to be responsible for the growth factor TGF-a-induced arachidonic acid liberation in keratinocytes, which is a pre- requisite for subsequent eicosanoid biosynthesis. As shown here, TGF-a indeed provokes a rapid release of arachidonic acid from HEL-30 keratinocytes. The conclusion that this response is due to PLA, activation is first of all based on the inhibitory effect of the PLA, inhibitor mepacrine (39) and the generation of LPC as a second reaction product of a PLA,- catalyzed phosphatidylcholine hydrolysis.
FIG. 10. Association of cPLA2 protein with the particulate fraction of HEL-30 keratinocytes upon stimulation with TGF- a. Confluent HEL-30 cells were cultivated in 4 X MEM containing 0.5% fetal calf serum for 18 h. After addition of 50 p~ Na3V04 the cultures were incubated for an additional 2 h prior to the stimulation by PBS (0.1%; control) or TGF-a (10 nM) for 30 min. The lysis and fractionation of the cells was carried out in the absence of EDTA and EGTA as described under “Experimental Procedures.” 100 pg of protein from the cytosol and particulate fractions of untreated and TGF-a-treated HEL-30 cells were subjected to SDS-PAGE as de- scribed under “Experimental Procedures” and immunoblotted with a purified polyclonal anti-cPLAz-peptide antiserum. Lane 1, control cytosol; lane 2, cytosol from TGF-a-treated cells; lane 3, control particulate fraction; lane 4, particulate fraction from TGF-a-treated cells. Arrow indicates the position of the cPLAz. A molecular mass marker (in kilodaltons) is indicated on the left.
The rapid activation kinetics of the keratinocyte PLA2 point to a constitutively expressed enzyme. The predominant cytosolic localization of the enzyme activity, its Ca2+ depend- ence and insensitivity to reducing agents, as well as the preferential hydrolysis of the sn-2-arachidonoyl ester bond of phosphatidylcholine, as compared with the corresponding pal- mitoyl ester, suggest a cPLA2 to be responsible for the TGF- a effect. Moreover, the PLA, protein in HEL-30 keratinocytes shows the expected (3, 5-7) molecular mass of 100-110 kDa in SDS-PAGE. In addition, this band can be detected by an oligopeptide-directed anti-cPLAp antibody and a strict corre- lation exists between the subcellular distribution of this pro- tein and PLA, activity. On the basis of these results we conclude the TGF-a-stimulated PLAz in HEL-30 keratino- cytes to belong to the cPLA2 family.
A functionally active EGF receptor appears to be a prereq- uisite for the activation of the epidermal cPLA2 by TGF-a confirming results obtained in cells expressing nonfunctional or overexpressing functional EGF receptors (34, 53-55). A major pathway for the transduction of TGF-a-induced signals is provided by the EGF receptor-dependent tyrosine phos- phorylation and activation of PLC-7 (40,43). However, such an activation could not be demonstrated in HEL-30 cells as well as in other cellular systems (34,53,54) including primary human keratinocytes (30). A critical parameter for PLC--y activation by TGF-a/EGF seems to be the number of EGF receptors/cell (53). Thus, in cell types expressing lo6 recep- tors/cell, EGF/TGF-a has been found to be unable to stimu- late the generation of DAG and Ins-1,4,5-P3 (30), whereas EGFITGF-a-induced PLC activation has been observed in EGF receptor-overexpressing cells including the A-431 carci- noma cell line (41, 56) and, to a minor extent, in Balb/MK epidermal cells which have been selected for EGF-induced mitogenesis (44). This may also explain the insensitivity of HEL-30 cells which express only about lo4 EGF receptors/ cell (data not shown). Thus, the inability of HEL-30 to respond to TGF-a with activation of the PLC/DAG/Ins-1,4,5- P3 cascade indicates that a PLC/PKC-independent pathway of cPLA2 activation requires the intrinsic tyrosine kinase activity of the receptor. In line with these results is the observation that a PLC- and PKC-independent increase of PLA, activity was also found to occur in mesangial cells in response to EGF stimulation (34) and in the cytosol of EGF- treated HER-14 cells expressing the normal human EGF

16802 Regulation of Cytosolic Phospholipase Az
receptor but not K-721A cells transfected with an EGF recep- tor defective in tyrosine kinase activity (53).
The TGF-a-induced EGF receptor-mediated stimulation of arachidonic acid release in HEL-30 cells is not only due to an increase in the enzymatic activity and but also to a translo- cation of cPLAZ from the cytosol to the membrane. The demonstration of a so-called CaLB (Ca2+-dependent lipid- binding) domain in the structure of the cPLAz (9) and other proteins which are translocated to the membrane, such as the cytosolic PKCs (57), support the importance of Ca" in the translocation process. Accordingly, a transient increase in the intracellular Ca2+ concentration as induced by the cPLA, agonist BK (via Ins-1,4,5-P3 release) may allow the translo- cation observed in BK-treated HEL-30 cells.2 Such an in- crease in cytosolic Ca2+ has not been detectable, however, upon TGF-a treatment. Therefore, alternative mechanisms of activation have to be considered. These may include a sensitization of the cPLAz to Caz+, for instance, by EGF receptor tyrosine kinase-catalyzed phosphorylation, which would allow the translocation process under normal cellular Ca2+ concentrations. The Caz+-dependent translocation of lipocortin I from the cytosol to the membrane has indeed been found to be influenced by EGF receptor-catalyzed tyro- sine phosphorylation in that the sensitivity of this process towards Caz+ was markedly increased (58).
Taken together, these data provoke the suggestion of TGF- a-induced tyrosine phosphorylation to be involved in the activation process of cPLAz. Support for this view has been provided in a very recent paper (59) presenting evidence that EGF-induced activation of cPLAz may be due to a stable modification of the enzyme. In addition, we could specifically immunoprecipitate cPLAZ from the cytosol of TGF-a, but not of BK- or TPA-treated or control cells, using antiphospho- tyrosine antibodies. This result indicates that the enzyme or a strongly associated protein is phosphorylated on tyrosine. However, neither the role of tyrosine phosphorylation in the activation of cPLAz nor the kinase responsible for this effect are presently known. A prime candidate would be the EGF receptor tyrosine kinase provided that cPLAZ is able to phys- ically associate with this kinase. If so, this is most likely not due to a direct interaction which is known to be mediated by SH-2 and/or SH-3 domains (43) being absent in the structure
In summary, the results presented here provide evidence for a cPLAz to be responsible for the growth factor TGF-a- stimulated arachidonic acid release and eicosanoid production in keratinocytes. Activation of cPLAz involves both an in- crease in the activity of the cytosolic enzyme and the trans- location to the membrane. On the basis of these results it may be postulated that EGF receptor-catalyzed tyrosine phos- phorylation plays a crucial role in both translocation and activation of the enzyme.
cPLAz (9).
REFERENCES 1. Mukhejee, A. B., Cordella-Miele, E., and Miele, L. (1992) DNA CeU Biol.
2. Dennis, E. A., Rhee, S. G., Billah, M. M., and Hannun Y . A. (1991) FASEB 11,233-243
J. R. m " 2 m 3. Kramer, R. M., Jakubowski, J. A,, and Deykin, D. (1988) Biochim. Biophys.
4. Clark, J. D., Milona, N., and Knopf, J. L. (1990) Proc. Natl. Acad. Sci.
5. Kramer, R. M., Roberta, E. F., Manetta, J., and Putnam, J. E. (1991) J.
6. Gronich, J. H., Bonventre, J. V., and Nemenoff, R. A. (1990) Bwchem. J.
. . - , - - - - -. . . Acta 959,269-279
U. S. A. 87,7708-7712
Bwl. Chem. 2 6 6 , 5 2 6 5 2 7 2
271,37-43
* R. Kast, G. Fiirstenberger, and F. Marks, unpublished results.
7. Wijkander, J., and Sunder, R. (1991) Eur. J. Biochem. 202,873-880 8. Clark, J. D., Lin, L. L., Kriz, R. W., Ramesha, C. S., Sultzman, L. A., Lln
9. Sharp, J. D. White, D. L., Ehiou, X. G., Goodson, T., Gamboa, G. C., A. Y., Milona, N., and Kno f, J. L. (1991) Cell 65,1043-1051
McClure, D., Burgett, S., Hoskins, J., Skatrud, P. L. Sportaman J. R.
Biol. C h m . 2 6 6 , 14&0-14853 Becker G. W., Kang L. H., Roberta, E., and KramLr, R. M. (1691) JI
10. Channon, J. Y., and Leslie, C. C. (1990) J. Bid. Chem. 266,5409-5413 11. Dennis, E. A. (1990) Adu. Exp. Med. Biol. 275, l -25 12. Chang, J., Musser, J. H., and McGregor, H. (1987) Biochem. Phurmucol.
13. Pruzanski, W., and Vadas, P. (1991) ZmmunoL Today 12,143-146 14. Needleman, P., Turk, J., Jaschik, B. A., Morrison, A. R., and Lefkowith, J.
B. (1986) Annu. Reu. Biochem. 56,69-102 15. Shimizu, T., and Wolfe, L. S. (1990) J. Neurochem. 55,1-15
17. Talwar, H. S., Fisher, G. J., and Voorhees, J. J. (1990) J. huest. Dermutol. 16. Axelrod, J. (1990) Biochem. Sot. Trans. 18,503-507
18. Kast, R., Fiirstenberger, G., and Marks, F. (1991) Eur. J. Biochem. 2 0 2 ,
19. Fisher, G. J., Talwar, H. S., Ryder, N. S., and Voorhees, J. J. (1989)
20. Kvedar, J., and Levine, L. (1987) J. Inuest. Dermutol. 88, 124-129 21. Hammarstrom, S., Lindgren, J. A., Marcello, C. Duell, E. A., Anderson, T.
22. Fiirstenberger, G., Richter, K. H., Fusenig, N. E., and Marks, F. (1980)
23. Fiacher, S. M., Patrick, K. E., Lee, M. L., and Cameron, G. S. (1991) Cancer
24. Ganss, M., Seemannn, D., Fiirstenberger, G., and Marks, F. (1982) FEBS
36,2429-2436
95,705-710
941-950
Bioehem. Biophys. Res. Commun. 163 , 1344-1350
F., and Voorhees, J. J. (1979) J. Inuest. Dermkol. 73.180-183
Cancer Lett. 11, 191-198
Res. 61,850-856
Lett. 142,54-58
Inuest. Dermatol. 86. 319-323 25. Galey, C. I., Ziboh, V. A., Marcello, C. L., and Voorhees, J. J. (1985) J.
31.
33. 32.
34.
35.
37. 36.
38.
39.
40.
41.
42. 43.
44.
45.
47. 46.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58. 59.
26. Fiirstenberger, G., Rogers, M., Ganss, M. Richter, K. H., and Marks, F. 27. Fiirstenberger, G., and Marks, F. (1990) in Eicosanoids and the Skin
28. Coffey, R. J., Derynck R. Wilcox, J. N. Bringman T Goustin, A., Moses,
30. Klein, S. B,. Fisher, G. J., Jensen, T. C., Mendelsohn, J., Voorhees, J . J., 29. Barrandon, Y., and Green, H. (1987) Cell 50, 1131-1137
11987) J. Cancer Res. Clin. Oncot. 113,310-318
(Ruzicka, T., ed) pp. 107-124, CRC Press, Boca Raton, FL
H., and Pittelkow, M. (1987) Nature 328,817d20"
, Derynck, R.'(1988) CeU 64,593-595 , Velu, T. J. (1990) Mol. Cell. Endocrinol. 7 0 , 205-216
and Elder J. T. (1992) J. Cell. Physwl. 151,326-336
Bjiere, K. S., Dee, L. N. W., and Bremer, J. (1974) Anal. Ewchem. 5 8 , 9 R P " A K
Bonventre, J. V., Gronich, J., and Nemenoff, R. A. (1990) J. Bwl. Chem.
hikrsperger, H., Gschwendt, M., Gernold, M., and Marks, F. (1991) J .
1" "- 265.4934-4938
Bwl. Chem. 266.14778-14784 Kast, R., Fiirsienberger, G.,and Marks, F. (1993) FEBS Lett., in press Poenie, M., Alderton, J., Tsien, R. Y., and Steinhardt R. A. (1985) Nature R I K 1~7-1.m
Moolenaar, W. H., Aerts, R. J., Tertoolen, L. G . J., and De Laat, S. W. "V, ". .X"
(1986) J. Bioi. Chem. 262.279-284 Blackwell, G. J., Duncombe,.W. G., Flower, R. J., Parsons, M., and Vane
J. R. (1977) Br. J. Phnrmocol. 59,353-366 Moolenaar, W. H., Bierman, A. J., Tilly, B. C., Verlaan, I., Defize L. K. H.,
Honegger, A. M., Ullrich, A., and Schlessinger, J. (1988) EMBO J. 7 ,
Mar olis, B., Rbee, S . G.! Felder, S., Mervic, M., Lyall, R., Levitzki, A., 707-710
Murray, A. W., and F'usenig, N. E. (1979) Cancer Lett. 7 , 71-77 Ufirich, A., and Schlessmger, J. (1989) Cell 67,1101-1107
Rhee, S. G., and Choi, K. D. (1992) Adu. Second Messenger Phosphoprotein
Moscat, J , Molloy, C. J., Fleming, T. P., and Aaronson, S. A. (1988) Mol.
Takuwa, N., Kumada, M., Yashamita, K., and Takuwa Y . (1991) J. Biol.
Weiss, B. A., and Insel, P. A. (1991) J. Biol. Chem. 266,2126-2133 Parker, J., Daniel, L. W., and Waite, M. (1987) J. Biol. Chem. 262,5385-
Tamoki, T., Nomoto, H., Takahashi, Y., Kato, M., and Tomitn, F. (1986) 5393
Dvir, A., Milner, Y., Chomsky, O., Gilom, C., Gazit, A., and Levitzki, A. Biochem. Biophys. Res. Commun. 135,5036-5041
Elder, J. T., Fisher, G . J., Lind uist, P. B., Bennet, G. L., Pittelkow, M. (1991) J. Cell Biol. 113,857-865
R., Coffey R. J., Ellingsworth,%., Derynck, R., and Voorhees, J. J. (1989)
Imamoto, A,, Beltran, I. M., and DiGiovanni, J. (1991) Mol. Carcinog. 4 ,
Gottlieb, A. B., Chang C. K Posnett, D. N., Fanelli, B., and Tam, J. P.
Goldberg H. J., Viegas, M. M., Mar olis B L Schlessinger, J., and
Margolis, B. L., Holub, B. J., Troyer, D. A., and Skorecki, K. L. (1988)
Margolis, B. L., Bonventre, J. V., Kremer, S. G., Kudlow, J. E., and
Wahl, M. J., Nishibe, S., Suh, P. G., Rhee, S. G., and Carpenter, G . (1989)
Res. 2 6 , 35-60
Endocrinol. 2,799-805
Chem. 266,14237-14243
Title 243,811-814
52-60
(1988) J. Exp. Med. i67.670-675
Skorecki, K. L. (1990) Biochem. J. 2 8 7 , h - 4 6 ;
Bwehem J . 266.469-474
Skorecki, K. L. (1988) Biochem J. 249,587-592
Proc. Natl. Acad. Sci. U. S. A. 86. 1568-1572 Ono, Y., FujiiT., Igarashi K., Kuho, T., Tanaka, C., Kikkawa, U., and
Schlaepfer, D. D., and Hai ler, H. T. (1987) J. BWL Chem. 262,6931-6937 Spaargaren, M., Wissink, l., Defize, L. H. K., De Laat, S. W., and Boonstra,
Nishizuka, Y. (1989) Prdc. Nati. Acad. Sci. U. S. A. 86,48694871
J. (1992) Biochem. J. 287,37-43