a transcriptional repressor - university of toronto t-space · charactereation of c-myc as a...
TRANSCRIPT

CHARACTEREATION OF C-MYC AS A TRANSCRIPTIONAL REPRESSOR Wilson Marhin 1998, Doctor of Philosophy Graduate Department of Molecular and Medical Genetics University of Toronto, Canada
ABSTRACT
The c-myc proto-oncogene encodes a nuclear phosphoprotein, whose expression
has been tightly linked to cellular transformation in vivo and in vitro. The strong
tumorigenic potential of Myc is reflected in its biological activities. Myc can promote cell
cycle progression, inhibit growth arrest in the form of quiescence and senescence, and
abrogate cellular differentiation in a diverse array of cell types. In essence, the net effect of
Myc is to enhance cell growth by promoting cell proliferation and inhibiting growth arrest.
Myc is believed to mediate these diverse biological activities by acting as a transcription
factor, inducing or repressing the expression of specific subsets of genes. Analysis of the
function of the Myc-induced genes that have been identified, reveals that some of these
genes are required for progression through the cell cycle, particularly the transition from
G1 to S phase. This observation suggests that Myc may enhance cell growth through the
induction of genes which are essential to cell cycle progression. In comparison, less is
known about the importance or mechanism of Myc repression activities, due to the small
number of genes that have been reported to be repressed by Myc. Identification of the
subset of genes which are repressed by Myc will help us to understand the mechanism of
Myc-mediated gene repression, and how the repression of genes by Myc can promote cell
proliferation. The crux of my research has been to identify and characterize novel gene
targets for Myc repression, with the intention of revealing novel Myc-dependent pathways
to drive cells out of growth arrest and promote cell proliferation. We have focused
primarily on three genes: the cyclin Dl gene; the Platelet-derived growth factor P receptor
@d@-pr) gene; and the growth arrest gene, gadd45; analyzing their expression in response

to Myc-activation. In contrast to previous resuits obtained in selected immortalized cell
lines, we clearly demonstrate that in primary mouse embryonic fibroblasts, cyclin Dl
expression is not repressed by Myc. Indeed, loss of cyclin Dl expression is only evident
in cells which exhibit Myc-activation and lack retinoblastoma tumour suppressor protein
expression. Our data hrther suggest that the decrease in cyclin Dl expression is an indirect
effect of cellular transformation rather than a direct effect of Myc. In addition, we have
identified and characterized the repression of gadd45 and thepdgf-pr genes by Myc.
Analysis of these Myc-mediated repression mechanisms revealed that unlike Myc-mediated
transactivation, Myc-mediated repression is comparatively more complicated and can be
mediated by multiple pathways. The precise nature of these repression mechanisms is
currently under study. The identification of these genes as targets for Myc repression has
revealed that Myc repression pathways serve an important role in the ability of Myc to
rebwlate the cellularresponse to changes in the extracellular environment, to drive cells out
of growth arrest, and aid in tumour progression. Thus, it would appear that we are at the
dawn of a new era of Myc research. Deciphering the significance of Myc repression will
help us to understand how both Myc transactivation and Myc repression cooperate together
to regulate normal cell proliferation, through the induction of cell cycle progression and the
inhibition of growth arrest.

ACKNOWLEDGMENTS
As I sit here thinking about my acknowledgments, I realize that this is one of the
most difficult things that I have ever had to write. A number of individuals have helped
and guided me to this moment. How do you adequately thank everyone for their support
and friendship over the years? My appreciation and gratitude to these individuals cannot be
solely expressed in mere words, but I will try. I will start with thanking the one person
who has been and still is the biggest influence in my life, Dr. Linda J. Z. Penn. I am not
exaggerating when I say that she has helped me become who I am today. I reminisce about
the old days when I started my research career, Linda was in the lab helping me to do my
first Northern. That was typical of Linda, she was always available for help and
discussion. She was also very understanding, allowing me the freedom to pursue
additional experiments during my training, even though they were completely unrelated to
my main project. I have benefited greatly ftom her guidance, and so I would like to
express my gratitude to her for her support, and more importantly her friendship over the
years.
The next person on my list is Mr. Gerald Yates, my grade 10 science teacher. He
made science more than just another high school subject. In my distant past, when I was
unsure about my future, he steered me onto the path of science. As such, I owe my life in
research to him. I thank him for his mentorship, and stimulating my interest in science. I
also thank the members of my committee, Drs. Eleanor Fish and Andrew Bognar, whose
knowledge and advice supported me through my training. I would also like to thank the
members of the lab, past and present, for their friendship during my studies. The days just
fly past with guys like that in the lab. Thanks to Linda Facchini, it is upon her shoulders
that I stand. The other members of the triangle of knowledgeable sarcasm, Patrick
"Captain Quebecois" Dion and Justin "Kahuna" Lear. Gihane for being my sympatico.
The current gang Mary, Cynthia, Sara, Gina and Jim for making me laugh. Mary for the

caring, supportive person that she is. Cynthia and Sara for teaching me more than I ever
wanted to know about life. Jim for being just Jim. I reserve special thanks to my parents
and my siblings for their continual love, and support. I acknowledge the financial support
of the Ontario Graduate Scholarship Program and the University of Toronto.
Hey .... it is true, once you write that first sentence the rest is easy.

ATTRIBUTION OF DATA
I am responsible for all of the work described in this thesis with the following
exceptions. The in vitro kinase assays in Chapter 2 were performed by a collaborator, Dr.
Yong-J. Hei. The somatic cell hybrids Rat-1 x NIH3T3v3 and Rat-1 v-myc x NIH3T3v3
v-myc cells in Chapter 3 were generated by Dr. Linda M. Facchini. The H015.19 gfp and
H015.19 gfpmyc cells in Chapters 3 and 4 were generated by Sara K. Oster.

TABLE OF CONTENTS
Page
ABSTRACT
ACKNOWLEDGMENTS
AlTRIBUTION OF DATA
TABLE OF CONTENTS
LIST OF FIGURES
LIST OF ABBREVIATIONS
CHAPTER 1 INTRODUCTION
The molecular basis of cancer.
1.1.1 Normal cell signalling cascades in growth control.
1.1.2 The cell cycle.
1.1.3 Cancer: genetic aberration of growth control genes.
Myc activation and cancer.
The c-myc proto-oncogene.
1.3.1 The myc family of genes.
1.3.2 Structure of the myc gene.
1.3.3 Regulation of Myc expression.
1.3.4 Structure of the Myc protein.
1.3.5 Post-translational modification of the Myc protein.
The biological activities of Myc.
1.4.1 Myc in normal cell proliferation.
1.4.2 Myc: inhibitor of differentiation.
1.4.3 Myc and apoptosis.
vii
ii
iv
vi
vii
xiii
xvi

1.4.4 Myc and cdkIs.
1.4.5 Myc transformation in vitro and in vivo.
1.4.6 Myc cooperativity in transformation.
1.5 Mechanism of action.
1.5.1 Myc interacting proteins.
1.5.2 Transcriptional activation.
1.5.3 Transcriptional repression.
1.6 The emerging significance of Myc repression.
1.6.1 Cotrelation of repression with transformation.
1.6.2 Activation and repression co-operate to promote
cell proliferation.
1.7 Objectives.
CHAPTER 2 LOSS OF RB AND MYC ACTIVATION CO-OPERATE TO
SUPPRESS CYCLIN Dl AND CONTRIBUTE TO
TRANSFORMATION
2.1 Abshct
2.2 Introduction
2.3 Materials and Methods
2.3.1 Cell culture.
2.3.2 Genomic DNA extraction and mouse embryonic
fibroblast genotyping.
2.3.3 Retroviral infection.
2.3.4 RNase protection.
2.3.5 Immunoblotting and immunoprecipitation.
2.3.6 In vitro kinase assay.

2.4 Results
2.4.1 Primary mouse embryonic fibroblasts lacking pRb
protein expres normal levels of cyclin Dl protein.
2.4.2 Constitutive Myc expression suppresses cyclin Dl
protein in pRb-deficient but not wild-type MEFs.
2.4.3 The suppression of cyclin Dl protein in MEFs can
occur by regulatory mechanisms controlling either
RNA or protein expression.
2.4.4 pRb-deficient MEFs exhibit a delay in growth
compared to wildtype MEFs.
2.5 Discussion
2.6 Acknowledgments
CHAPTER 3 MYC IS AN ESSENTIAL NEGATIVE REGULATOR OF
PLATELET DERIVED GROWTH FACTOR BETA
RECEPTOR EXPRESSION
3.1 Abstract
3.2 Introduction
3.3 Materials and Methods
3.3.1 Cell culture, and somatic cell hybridizations.
3.3.2 Rekoviral vectors.
3.3.3 Retroviral infection.
3.3.4 RNase protection.
3.3.5 Northern blot analysis.
3.3.6 PDGF BB binding studies.
3.3.7 Luciferase assays.
3.4 Results

PDGF P receptor mRNA expression is suppressed
following serum or PDGF BB stimulation of Rat-1
cells.
3.4.2 PDGF P receptor RNA is down-regulated in Myc
-activated non-transfomed cells.
3.4.3 The suppression of PDGFP receptor mRNA levels
following Myc-activation occurs with rapid kinetics.
3.4.4 Myc-induced repression of celi surface PDGF P receptors.
3.4.5 The mechanism of PDGF P receptor suppression
differs from Myc autosuppression.
3.4.6 Myc represses transcription from thepdgf-Pr promoter.
3.4.7 Characterization of the mousepdgf-jr promoter.
3.4.8 Myc is required for the suppression inpdgf-fir mRNA
levels following mitogen stimulation.
3.5 Discussion
3.6 Acknowledgments
CHAPTER 4 MYC REPRESSES THE GROWTH ARREST GENE GADD45
4.1 Abstract
4.2 Introduction
4.3 Materials and Methods
4.3.1 Cell 4ture.
4.3.2 Retroviral infection.
4.3.3 RNase protection and luciferase assays.
4.3.4 Immunoblotting and irnmunoprecipitation.
4.4 Results

Sequential induction of c-myc and suppression of
gadd45 mRNA levels following serum stimulation
of quiescent Rat-1 fibroblasts.
Ectopic Myc expression in primary cell culhxes and
immortalized cell l i e s results in a suppression of
endogenous gadd45 mRNA levels.
4.4.3 The suppression ofgadd45 in response to Myc
-activation occurs with rapid kinetics.
4.4.4 The suppression ofgadd45 in response to Myc does
not require wildtype p53 activity.
4.4.5 Myc and p53 co-regulategadd45 transcription.
4.4.6 Myc regulates transcription fiom the gadd45 promoter.
4.4.7 Deletion analysis of the hamstergadd45 promoter.
4.4.8 Myc is an essential negative regulator ofgadd45
expression.
4.5 Discussion
4.6 Acknowledgments
CHAPTER 5 FUTURE DIRECTIONS
Summary of research.
Analysis of the relationship between Myc, pRB, and cyclin Dl.
Characterization of the repression of PDGF-PR expression by Myc.
5.3.1 Biological role of PDGF-PR suppression by Myc
following ligand stimulation.
5.3.2 Analysis of the mechanism ofpdgf-pr mRNA
suppression by Myc.
Characterization of the mechanism of gadd45 repression by Myc.
xi

5.4.1 13iological role ofgadd45 suppression by Myc 179
following rnitogen stimulation.
5.4.2 Myc abrogates the stress-induced upregulation in 181
gadd45 expression.
5.4.3 Analysis of the mechanism ofgadd45 mRNA 182
suppression by Myc.
5.5 Models of Myc repression. 183
5.6 Does Myc transactivation or Myc repression lead to cell transformation? 186
5.7 Perspectives 188
CHAPTER 6 REFERENCES 190

LIST OF FIGURES
Page
Chanter 1
Figure 1.1 Simplified schematic of a mitogenic signal transduction
pathway.
1.2 The cell cycle.
1.3 C-myc gene organization.
1.4 Myc mRNA and protein expression, in non-transformed and
transformed cells, in response to external stimuli.
1.5 Functional and structural features of the human cMyc protein.
1.6 Myc-induced Apoptosis.
1.7 Possible mechanisms for the role of Myc in driving cell cycle
progression, based on the induction of the indicated target
genes by Myc.
Chaoter 2
Figure 2.1 Determination of RB-1 gene status and pRb protein expression
in primary mouse embryonic fibroblast (MEF) cultures derived
from 3 litters of embryos produced *om the mating of RB-1
heterozygotes.
2.2 MEFs lacking pRb protein express cyclin Dl protein and
cyclin Dl associated kinase activity at levels comparable to
their wildtype littermates.
2.3 Cyclin Dl protein expression is induced in quiescent wildtype
and pRb-deficient MEFs in response to serum-stimulation.
xiii

2.4 Constitutive ectopic Myc expression in primary MEFs lacking 77
pRb protein suppresses cyclin Dl protein expression and activity.
2.5 Cyclin Dl expression in Myc-activated, pRb-deficient MEFs is 80
suppressed either at Ihe RNA or protein level.
2.6 Proliferation profiles of wildtype (MEF 4) and pRb-deficient 82
MEFs (MEF I), infected with control retrovirus (CONTROL)
or with retrovirus canying a v-myc gene (MYC).
2.7 pRb-deficient MEFs constitutively expressing ectopic Myc
protein exhibit an increased proliferative capacity.
Chapter 3
Figure 3.1 Expression of c-myc andpdgf-pr mRNAs in quiescent Rat-1
cells stimulated with serum or PDGF BB.
3.2 Suppression of endogenous c-nryc andpdgf-Dr mRNA levels
in rodent cells constitutively expressing exogenous Myc.
3.3 Pdgf-pr mRNA expression is suppressed in response to Myc
induction.
3.4 Myc suppresses PDGF-PR expression in Rat-1 cells.
3.5 The suppression of endogenouspdgf-pr and c-myc mRNA
levels by Myc appear to be mediated by different pathways.
3.6 Pdgf--P, mRNA transcription is suppressed by Myc.
3.7 Deletion analysis of the mousepdgf-fir promoter.
3.8 Myc is required for the repression ofpdgf-fir mRNA levels in
both serum stimulated quiescent cells and in proliferating cultures.
Chapter 4
Figure 4.1 Serum stimulation of confluent, quiescent Rat-1 fibroblasts

results in the induction of c-n~yc mRNA expression followed
by a suppression ofgadd45 mRNA levels.
Ectopic Myc expression in primary and immortalized fibroblasts
results in the suppression ofgadd45 mRNA and protein levels.
gadd45 mRNA expression is suppressed with rapid kinetics in
response to Myc induction.
p53 mRNA and protein expression is not responsive to ectopic
Myc expression in Rat-1 cells.
The suppression ofgadd45 by Myc does not require wildtype
p53 activity.
Myc and p53 co-regulategadd45 transcription.
gadd45 transcription is suppressed in response to Myc.
Deletion analysis of the hamstergadd45 promoter.
Myc is an essential negative regulator of gadd45 expression.

LIST OF ABBREVIATIONS
AdMLP
AKT
aMEM
B
bFGF
Pgal
BSA
cad
cak
CAT
cdk
cdkl
C-fms
CKII
CSF-1 R
CTS
dhfr
D m 1
EGF
eIF-2a
eIF-4E
ERTAD
FBS
gadd
Adenovims major late promoter
Protein kinase B
Alpha modified Eagle's medium
Myc basic region (specific DNA binding domain)
Basic fibroblast growth factor
P-galactosidase gene
Bovine serum albumin
Carbamoyl-phosphate synthaselaspartate carbamoyl transferasel
dihydroorotase
Cyclin-dependent kinase activating kinase
Chloramphenicol acetyltransferase
Cyclin-dependent kinase
Cyclin-dependent kinase inhibitor
Colony stimulating factor receptor
Casein kinase I1
Colony stimulating factor receptor
Charcoal-treated serum
Dihydrofolate reductase
Cyclii D-interacting myb-like protein
Epidermal growth factor
Eukaryotic initiation factor-2a
Eukaryotic initiation factor-4E
Estrogen receptor transcriptional activation domain
Fetal bovine serum
Growth arrest and DNA damage-inducible gene
xvi

GAP
GAPDH
gas
GFP
GSK3
h
HLH
IGF-I
Inr
LDH-A
LTag
LZ
MAP Kinase
MBI
MBII
MEF
MIDb
NBD
NLS
NLS M1
NLS M2
odc
OH-T
PBS
PCNA
PDGF
PDGF BB
The GTPase-activating protein of Ras
Glyceraldehyde-3-phosphate dehydrogenase
Growth arrest specific gene
Green fluorescent protein
Glycogen synthase kinase 3
Hour(s)
Helix-loop-helix
Insulin growth factor-1
Initiator element
Lactate dehydrogenase gene-A
SV40 Large T antigen
Leucine zipper
Mitogen-activated protein kinase
Myc box I
Myc box I1
Mouse embryonic fibroblast
Myc-regulated DEAD box-related gene
Myc non-specific DNA binding domain
Nuclear localization signal
Myc nuclear localization signal 1
Myc nuclear localization signal 2
Omithine decarboxylase
4-hydroxy (Z) tamoxifen
Phosphate buffered saline
Proliferating cell nuclear antigen
Platelet-derived growth factor BB
Platelet-derived growth factor BB
xvii

pdgf-cu
pdgf-Pr
PI3K
PKA
PKB
PKC
PLcy
P R ~
REF
RB-1
sdr
TAD
TBP
TCR
TdT
TGF-P
wtMycER
Ah4ycER
W 1
Platelet-derived growth factor alpha receptor
Platelet-derived growth factor beta receptor
Phosphoinositol3' kinase
Protein kinase A
Protein kinase B
Protein kinase C
Phospholipase C-yl
Retinoblastoma susceptibility protein
Rat embryo fibroblast
The retinoblastoma susceptibility gene
Serum deprivation response gene
Transactivation domain
TATA-binding protein
T cell receptor
Terminal deoxy-transferase
Transforming growth factor P Human c-myc(wt)/ER
Human c-myc(A106-143)IER
Yin-Yang-1

Chapter 1
INTRODUCTION

1.1 The molecular basis of cancer
1.1.1 Normal cell signalling cascades in growth control
Cells are sensitive to both positive and negative growth stimuli in their
environment. Stimuli may take a number of forms including: growth factors, inhibitory
cytokines, cell-cell interactions, and cellular contacts with the extracellular matrix. These
extracellular changes are detected by specific receptors on the cell surface, which initiate
pathways that transmit signals into the cell, eliciting a cellular response to these stimuli. In
general, different signal transduction pathways are tasked with the transmission of specific
signals. Yet, several of these signalling pathways are organizationally related, insofar as
interaction of a ligand to its receptor elicits receptor phosphorylation; and the sequential
activation of a series of intermediate components of the signal transduction mechanism.
The nature of these intermediate components is diverse and includes: soluble factors, such
as calcium ions released from intracellular stores, membrane restricted proteins, such as
small GTPases, and serine/threonine protein kinases. The overall effect is the amplification
and transmission of a signal by a cascade of intermediate proteins to the nucleus (Figure
1.1).
At the nucleus, the endpoint of these pathways, the transduced signal elicits the
induction andlor post-translational modification of nuclear protein effectors. A variety of
nuclear effectors may be induced in response to the activation of a given signal transduction
pathway. As an example, the activation of a growth promoting signal transduction
pathway, following the exposure of cells to mitogenic growth factors, elicits the induction
of a class of genes called the immediate early response genes which include the proto-
oncogenes c-myc, c-fos, and c-jzm (Cochran et al., 1983; Greenberg et al., 1985; Kelly et
al., 1983; Lau and Nathans, 1987). The expression of these genes is up-regulated within 1
to 2 h following mitogen stimulation and occurs in the absence of protein synthesis. The

MITOGEN
MITOGEN RECEPTOR CASCADE OF
SECOND MESSENGERS
UIESCENCE (GO)
IMMEDIATE EARLY
RESPONSE GENES
Figure 1.1. Simplified schematic of r mitogenic signal transduction pathway. The interaction of a mitogenic growth factor with its receptor, elicits receptor phosphorylation and activation. The activated receptor initiates a signalling cascade, delivering a signal from the cell surface to the nucleus. The expression of immediate early genes such as c-myc, c-fos, and c-jtrr~ are induced, and they function to inhibit the expression or activity of growth arrest genes such as thegadd group of genes, as well as drive entry and progression through the cell cycle.

products of the immediate early genes function to drive cells out of a growth arrested
condition known as quiescence, into an actively proliferating state (Pledger et al., 1977;
Pledger et al., 1978). Conversely, the absence of growth factors results in the repression
of these growth promoting genes in conjunction with the induction of genes which serve to
either affect or maintain a variety of growth arrested states such as that associated with
quiescence and differentiation. These are the growth arrest genes, and include genes such
as: gax; sd,; as well as the gas and gadd group of genes. The activity and interplay
between the proto-oncogenes and the growth arrest genes evoke the appropriate
modifications in gene expression, allowing the cell to adapt to changes in its environment.
1.1.2 The cell cycle
Positive or negative growth stimuli activate distinct signaling pathways which
impact on the proliferation of cells. Therefore at a molecular level, both pathways must
feed into and modulate the cell cycle. The cell cycle is tasked with orchestrating the
accurate replication and segregation of the genome of a cell into the emerging daughter
cells, within the constraints of a strict temporal program. Duplication of the genome of a
cell occurs in S phase, and the subsequent segregation of the chromosome pairs into each
daughter cell occurs during mitosis or M phase. Each of these critical phases is separated
by a gap, the GI and G2 phases respectively. During these gaps, the cell synthesizes the
requisite machinery to effect the successful completion of the forthcoming phase (reviewed
in Hatakeyama et al., 1994; Heichman and Roberts, 1994).
The cell is sensitive to the inputs of signal transduction pathways initiated by
external stimuli predominantly in the G1 phase of the cell cycle. It was observed that
during the early part of this phase, cells receive information about the state of the extcrnal
milieu, and on the basis of these signals the cell will either commit to proceeding through
the cell cycle or enter quiescence. Transit through this phase and hence cell cycle

progression is dependent upon the presence of growth factors. The position at which cells
are mitogen independent, and are committed to progressing through the cell cycle, is called
the restriction point (Pardee, 1974; Temin, 1971). Beyond this point, cells are refractory
to the absence of growth promoting signals until their entry into the subsequent G1 phase.
Progression through the cell cycle is driven by the activity of cyclin-dependent
kinases (cdks). As such the activities of these proteins are common targets of nuclear
transcription factors, which are the endpoints of several signal transduction pathways. cdk
activity is regulated via a combination of three mechanisms: binding to a cyclin which is the
regulatory subunit, the phosphorylation state of the cdk, and the interaction of the cdk with
a cdk inhibitor (cdkI) (reviewed in Fisher, 1997; Harper, 1997; Nigg, 1995). The
expression of each cyclin and hence its associated kinase activity is generally restricted to
specific phases of the cell cycle (Figure 1.2). Cyclin abundance is a balance of
transcriptional induction versus rapid degradation via post-translational mechanisms, such
as the ubiquitination-proteosome pathway (reviewed in Hershko, 1997; Pagano, 1997).
Therefore each cyclidcdk complex is responsible for promoting the progression of cells
through that associated phase of the cell cycle. There are a number of cyclins which have
been shown to drive cell cycle progression, and they can be divided on the basis of when
their activity is maximal. The GI cyclins include the D-type cyclins, Dl, D2, D3, and
cyclin E. Cyclin A expression and likewise its associated kinase activity predominates in S
and early G2 phases, while cyclin B expression is restricted to the G2 phase (reviewed in
Nigg, 1995; Sherr and Roberts, 1995).
There are essentially four cdks whose phosphorylation capabilities are required for
cell cycle progression: cdk4, cdk6, cdk2, and cdc2 (cdkl). cdk4 and cdk6 interact with
and are r.sgulated by the D-type cyclins. cdk2 is regulated by cyclin E, as well as cyclin A,
while cdc2 (cdkl) is regulated by both cyclins A and B. Cyclin-associated kinase activity
is also regulated through phosphorylation. cdk activity is activated upon phosphorylation
by the cdk-activating kinase (cak), on a single threonine residue in both cdk2 and cdk4.


Cak itself is comprised of a cyclin, cyclin H, associated with a kinase subunit, cdk7
(reviewed in Sclafani, 1996). Conversely, the phosphorylation of cdks on specific
threonine and tyrosine residues by kinases such as weel, will inhibit kinase activity
(reviewed in Lew and Kombluth, 1996). These inhibitory phosphates are removed by the
cdc25 family of phosphatases (reviewed in Draetta and Eckstein, 1997; H o f i a n n and
Karsenti, 1994). One member of this family, cdc25A is active in G1 phase and it has been
suggested that the activity of this phosphatase is required for progression through the
restriction point ( H o f i a m et al., 1994; Jim0 et al., 1994).
The final mechmism through which cdk activity is regulated is via its interaction
with cdk inhibitors (cdkI). Based on sequence and functional similarities, there are two
families of cdkI: first, the INK4 family consisting of p16, p15, p18, and p19, which
specifically inhibit the activity of the D-type cyclin-associated cdks 4 and 6; second, the
cipikip family comprising p21, p27, and p57. These inhibitors interact with and inhibit all
cdks. In addition to the differences in binding specificities, these two groups of inhibitors
also interact with their respective cyclidcdk complexes via different mechanisms. Binding
of the INK4 family to cdk4 or cdk6 inhibits the interaction of the D cyclins with the cdk.
In contrast p21, p27, and p57 are found in ccmplexes with both the cyclin and cdk
subunits (Harper, 1997; Lees, 1995).
Activation of the cyclin-associated kinase cascade initiates a number of events
which stimulate the timely progression of cells through each phase of the cell cycle.
Among these the inactivation of the retinoblastoma tumour suppressor protein (pRb) in late
G1, appears to be the critical step for the transition through the G1 phase of the cell cycle
(reviewed in Bartek et al., 1997). The pRb protein is active in its hypophosphorylated
state. As the cell progresses through the cell cycle, pRb is sequentially phosphorylated by
successive cyclidcdk complexes, resulting in the inactivation of pRb activity. In late M
phase, pRb is dephosphorylated, and hence reactivated prior to entry into the subsequent
cell cycle (Figure 1.2). Hypophosphorylated pRb functions as a cell cycle arrestor by

binding to and inhibiting the activity of members of the E2F family of transcription factors.
The pRblE2F complex is an active repressor of E2F responsive genes whose activity is
required for progression through S phase. In GI phase, both cyclins D and E with their
associated cdks are required for the phosphorylation and inhibition of pRb function.
Therefore the inactivation of pRb permits the release of E2F ftom pRb/EZF complexes,
facilitating the transcription of S phase genes and S phase transition (reviewed in Nevins et
al., 1997; Reed, 1997).
1.1.3 Cancer: genetic aberration of growth control genes
Both growth stimulatory and inhibitory signals elicit modifications to gene
expression to drive either proliferation or growth arrest, in part through the modulation of
components of the cell cycle. Changes to the cell cycle are manifested in a number of
ways, through the regulation of either one or all of the following: cyclin expression, the
activity of cak and cdc25A, or the activity of cdkIs. Several players in signal transduction
pathways function either as proto-oncogenes conferring a strong proliferative impetus,
such as the c-myc proto-oncogene, or as growth inhibitors which guard against
uncontrolled cell proliferation; for example, the pRb tumour suppressor protein. Normal
cell proliferation entails a balance between proto-oncogenes and growth inhibitors, and
perturbations in this fine balance commonly lead to tumour formation. For example,
mutations which result in the deregulated expression of a component of a growth
promoting signal transduction pathway, such as a proto-oncogene, will enhance normal
cell proliferation and elicit unrestrained growth. A similar fate may arise if there is a
mutation in the expression or activity of growth inhibitors. In either case, there is the
potential that the increase in the number of cell divisions, with the associated elevation in
the number of DNA replication events, will facilitate the genesis of additional oncogenic
mutations which promote tumorigenesis. This model introduces one of the tenets of

turnorigenesis. A single genetic aberration may enhance the proliferative capacity of a cell,
yet will not induce turnour formation; additional cooperating oncogenic events are required
to elicit full cellular transformation. The reasoning for this cooperative relationship is still
unclear. On the basis of our knowledge of the functions of individual oncogenes which
have been reported to synergize to elicit tumour growth, we can hypothesize that each
oncogene within the partnership serves a complementary function. Each mutation is
required for full transformation. This may reflect the number of extracellular signals which
are needed by a normal cell to proliferate. The c-myc proto-oncogene is one well-described
example of an oncogene which is a component of many proliferative signal transduction
pathways and is also frequently mutated in a variety of tumours (reviewed in Field and
Spandidos, 1990; Garte, 1993; Schwab and Amler, 1990).
1.2 Myc activation and cancer
The v-gag-myc gene was the first member of the myc gene family to be isolated.
V-gag-myc is a potent transforming oncogene found in aggressive avian retroviruses, such
as MC29,OK10, CMII, and MH2. These viruses all induce acute lymphoid malignancies
in chickens with high frequency (reviewed in Cole, 1986; Graf and Beug, 1978). The
identification and analysis of the cellular homologues of this oncogene, revealed the strong
oncogenic potential of members of this gene family. C-myc, the first cellular homologue to
be identified, is frequently activated in human tumours originating from, but not restricted
to, cells of hematopoietic origin. Indeed, in humans the array of tumour types which
frequently exhibit alterations in Myc expression is extensive: hematopoietic malignancies
such as lymphoma and leukemia, connective tissue cancers such as osteosarcoma,
carcinomas derived from the epithelial layers of the breast, lung, cervix, ovary, stomach,
prostate, and colon, as well as squamous cell carcinomas of head and neck (reviewed in
Field and Spandidos, 1990; Garte, 1993; Schwab and Amler, 1990). Structurally and

functionally related family members, N-myc and L-myc also have a significant impact in
carcinogenesis. The N-myc gene is amplified in human neuroblastoma, retinoblastoma,
and small cell lung carcinoma (Brodeur et al., 1984; Brodeur et al., 1985; Kohl et al.,
1984; Schwab et al., 1983). L-myc expression is commonly elevated in small cell lung
carcinoma (Barren et al., 1992; Bernard et al., 1992; Kawashima et al., 1992; Makela et
al., 1992; Nau et al., 1985; Schreiber-Agus et al., 1993). Thus, the members of the myc
oncogene family possess a strong oncogenic potential and play a significant role in a large
number of human cancers arising from a wide variety of tissues.
In many turnours, Myc activation can be attributed to structural alterations affecting
the myc gene, including chromosomal translocations, retroviral promoter or enhancer
insertions, and gene amplification (reviewed in Cole, 1986; Kelly and Siebenlist, 1986;
Marcu et al., 1992). For example, B-cell neoplasias, including human Burkitt's
lymphoma, rat immunocytoma, and mouse plasmacytomas, consistently display a
reciprocal chromosomal translocation involving the c-myc locus with one of three
immunoglobulin loci (Kelly and Siebenlist, 1986). The chimeric gene consists of c-myc
coding sequences juxtaposed to transcriptional regulatory elements of one of the
immunoglobulin genes, resulting in a B cell-specific transcription unit driving the
constitutive expression of the c-myc oncogene. Although deregulated expression can be
associated with an elevation of Myc expression, many Burkitt's lymphoma cells exhibit c-
myc mRNA expression at levels comparable to physiological levels seen in proliferating B
cells (Keath et al., 1984; Taub et al., 1984). It has been shown that the translocated c-myc
allele frequently harbors point mutations in exon 11-coding sequences. However, the
individual mutations are not consistent among Burkitt's lymphoma cells, and they are not
correlated with specific phenotypes of the tumour (reviewed in Zimrnerman and Alt, 1990).
Thus, the primary event converting the proto-oncogene to its oncogenic form is
deregulation of the intact c-myc coding region, leading to continuous Myc protein
expression (Figure 1.4). Elevated expression or alterations in the protein product through

point mutations likely further potentiates the activity of the activated allele. It remains to be
determined whether deregulated, elevated Myc levels promote tumorigenesis by
constitutively activating the physiologically normal functions of Myc or through the
initiation of novel mechanisms which are specific to the turnongenic pathway.
Another c-myc activating mutation which commonly leads to deregulation of c-myc
expression is retroviral promoter or enhancer insertion (reviewed in Kelly and Siebenlist,
1986). The c-myc gene is a common site for integration of the avian leukosis virus in pre-
B cells of the bursa, resulting in the genesis of bursal lymphomas (Hayward et al., 1981;
Payne et al., 1982). Integrations occur predominantly 5' to e-myc coding sequences and in
the same transcriptional orientation as the c-myc gene. As a result of the juxtaposition of
the retroviral LTR and c-myc sequences, c-myc mRNA expression is enhanced due to
transcription from the retroviral LTR (Hayward et al., 1981; Payne et al., 1982). On
occasion, the proviral DNA integrates in the opposite transcriptional orientation to or
downstream of the c-myc gene (Corcoran et al., 1984; Li et al., 1984). In these instances,
elevated c-myc mRNA transcription appears to be due to the activity of retroviral enhancer
elements (Payne et al., 1982). T cell lymphomas resulting from the infection of newborn
mice with murine leukemia virus (Selten et al., 1984; Steffen, 1984) also exhibit proviral
integrations at the c-myc gene. In both T and B cell lymphomas, c-myc expression is
elevated in comparison to normal cells, yet it is not clear whether the increase in c-myc
expression is due to an elevation in the rate of transcription or due to the disruption of
transcriptional regulatory elements in the c-myc gene (Kelly and Siebenlist, 1986). The
consistent observation in these tumours of proviral integrations at the c-myc gene, suggests
that deregulated Myc expression may play an important role in the genesis of these
malignancies.
The third somatic cell aberration which leads to Myc activation is gene
amplification. Amplification does not alter the structure of the myc gene, it simply
increases the number of normal myc genes within the cell, resulting in an elevation in the

number of Myc protein molecules per cell (reviewed in Cole, 1986; Zirnmerman and Alt,
1990). The amplified myc genes are not necessarily deregulated, and retain their sensitivity
to growth regulatory agents in the external environment. For example, the promyelocytic
cell line HL60 exhibits enhanced c-Myc expression due to a 10 to 50 fold amplification of
the c-myc gene (Dalla-Favera et al., 1982), yet exposure to differentiation agents will
trigger the usual reduction in endogenous c-myc mRNA and protein levels associated with
myeloid cell differentiation (Reitsma et al., 1983). Myc amplification plays an important
role in the promotion of tumorigenesis, demonstrated by an analysis of a large number of
primary malignant tumours derived from a wide variety of different tissues, which revealed
that 10% contained an amplification of a c-myc allele (Yokota et al., 1986). Interestingly,
ntyc amplification appears to correlate with more aggressive stages of malignancies. In
neuroblastomas, N-my amplification is only observed in the more tumorigenic forms,
stages 111 and IV, of this disease (Brodeur et al., 1984; Brodeur et al., 1985; Seeger et al.,
1985). In addition, in small cell lung carcinoma, c-myc amplification is found only in the
more malignant variants of this cancer (Little et al., 1983). These observations suggest
Myc activation by amplification confers a growth advantage to a wide variety of cells, but
this mechanism of activation may be restricted to more genetically unstable transformed
cells. Thus, activation of Myc expression can arise from gross somatic mutations which
elicit either deregulated andlor constitutive Myc expression. Taken together the evidence
strongly suggests that temporally inappropriate Myc expression is the critical component of
Myc activation which promotes the tumorigenic program.
Gross genetic alterations may not be the only mechanism leading to Myc activation.
Myc activities may be enhanced through mutations which inactivate any one of the many
mechanisms controlling normal Myc expression, or the Myc protein may engage in novel
protein-protein interactions initiating transformation specific functions. These mechanisms
may potentially confer a growth advantage to the affected cell, and contribute to the
stepwise progression of tumorigenesis. If so, then the estimated frequency of Myc

activation in human cancer is presently grossly underestimated, as it is based on detection
of gross genetic abnormalities of the myc family members. The more subtle Myc activation
events which likely occur in cells from malignant transformations should be readily
ascertainable in the near future, primarily through the identification and characterization of
Myc-interacting proteins and the recent advances in diagnostic methods to measure changes
in gene expression, such as RNA-FISH and quantitative RT-PCR. By these approaches,
the absolute frequency of Myc activation, the specificity of tumour types affected and the
nature of collaborating mutations involved in human cancer will be understood. Moreover,
with these advances, the molecular mechanisms and the underlying cause of the more
subtle activation mechanisms can be better understood and key regulators of Myc
expression uncovered. Thus, quantitating subtle as well as gross Myc activation events in
human tumours will significantly advance both clinical and basic aspects of the field.
Whether gross or subtle mutations are involved, the net result of these somatic aberrations
is the activation of the myc proto-oncogene and the promotion of unrestrained cell
proliferation, leading ultimately to cellular transformation.
1.3 The c-myc proto-oncogene
1.3.1 The myc family of genes
The importance of the myc proto-oncogene is emphasized by its evolutionary
conservation. Homologues of c-myc have been identified in the genomes of a variety of
organisms including trout, zebrafish, frog, sea star, chicken, rat, mouse, and more
recently, the fruitfly, drosophila (Dalla-Favera et al., 1982; Dalla-Favera et al., 1982;
Eladari et al., 1992; Gallant et al., 1996; Hayashi et al., 1987; King et al., 1986; Schreiber-
A y s et al., 1993; Stanton et al., 1984; Van Beneden et al., 1986; Walker et al., 1992).

However, as yet no c-n~yc homologues have been detected in yeast or C. elegans, two
well-characterized organisms. In eukaryotic cells, the myc family members c-myc, N-
nyc, and L-myc are functionally related, executing similar biochemical activities. Yet they
do exhibit distinct temporal and spatial expression patterns in the developing embryo and in
adult tissues, suggesting that each member plays distinct roles during embryogenesis.
Indeed, transgenic and gene disruption studies in the developing mouse embryo have
revealed that each Myc family member plays a non-complementary role during
development. The loss of function of one Myc family member can only be partially
compensated through the expression of the other Myc proteins (Davis and Bradley, 1993;
Davis et al., 1993).
B-myc (Asker et al., 1995; Ingvarsson et al., 1988), and S-myc (Sugiyama et al.,
1989), are additional closely related genes which were both isolated From rat genomic
libraries. The importance of B-tyc and S-tnyc gene expression in normal cell growth is
not fully understood as their protein products remain to be detected in vivo. B-myc mRNA
is expressed in a variety of tissues with its highest expression in the brain (Ingvarsson et
al., 1988). The B-myc mRNA is homologous to exon 2 of the c-myc gene, and encodes a
putative 168 amino acid protein. The S-myc gene consists of sequences homologous to
exons 2 and 3 of N-myc, but lacks the intervening intron (Asai et al., 1994; Sugiyama et
al., 1989). The S-myc mRNA has only been detected in rat embryo chrondocytes.
Although the precise roles of the putative S-Myc and B-Myc proteins in the cell are not
known, some studies indicate that these proteins can antagonize the activity of Myc (Resar
et al., 1993). This may serve to fine-tune the activities of Myc and determine how Myc
responds to changes in the external environment.

1.3.2 Structure of the myc gene
The c-nzyc gene is located on chromosome 8 in humans and chromosome 15 in
mice (Dalla-Favera et al., 1982; Nee1 et al., 1982). The structure of the c-myc gene is
highly conserved between species, and among family members (reviewed in Lemaitre et
al., 1996). The c-myc gene consists of three exons; the first exon being largely
untranslated, with the remaining two highly conserved exons encoding the Myc protein
(Bernard et al., 1983; Colby et al., 1983; Wan et al., 1983). C-myc transcription is driven
from distinct promoters. The major promoters are PI and P2, P1 being located at the start
of exon I and P2, 150 bp downstream of P1. P1 accounts for only 10 % of total c-myc
mRNA, while P2 is responsible for over 85 % of the c-myc transcripts. There are also two
additional promoters, PO and P3; PO being found 550 bp upstream of PI, and P3 in intron
1. Less than 5 % of total c-myc mRNA originates from both of these promoters.
Transcription is also affected by the presence of two transcriptional attenuation sites, each
downstream of either P1 or P2, which are responsible for the pausing of RNA polymerase
11. Antisense c-myc transcripts originating from exon I1 and intron I have also been
reported; however, the importance of these transcripts to Myc expression or function is
unknown. Transcripts terminate at one of two polyadenylation signals in the untranslated
portion of exon 111; the 3' signal being the predominant site of transcriptional termination
(reviewed in Lemaitre et al., 1996). A schematic diagram of the genomic structure of the
human c-mjc gene is illustrated in Figure 1.3.

Figure 1.3. C-myc gene organization. The c-myc gene consists of three exons. The promoters (PO, PI, P2, P3), translation initiation sites (AUG, CUG), and the polyadenylation sites (AAAI, AAA2) are indicated. The translated sequences in each exon are represented by open rectangles.

1.3.3 Regulation of Myc expression
Expression of the myc proto-oncogene is induced as a consequence of signaling
pathways downstream of a variety of mitogens such as platelet-derived growth factor
(PDGF), epidermal growth factor (EGF), basic fibroblast growth factor @FGF), and 12-
0-tetradecanoylphorbol-13-acetate (Cutry et al., 1989; Dean et al., 1986; Kelly et al.,
1983). The components of the signal transduction pathways which drive myc transcription
have not been fully elucidated, yet studies have identified a few of the players such as
protein kinase C (PKC), protein kinase A (PKA), src, E2F, ets, and abl (Barone and
Courineidge, 1995; Cleveland et al., 1989; Coughlin et al., 1985; Hamel et al., 1992;
Kelly et al., 1983; Mudryj et al., 1990; Ran et al., 1986; Roussel et al., 1991; Roussel et
al., 1994; Wong et al., 1995).
A role for src in the induction of Myc expression, following mitogen stimulation,
was suggested in studies utilizing a mutant CSF-1 receptor (Roussel et al., 1991).
Activation of the mutant receptor elicited the induction of the immediate early genes. c-fos
and c-jlm, but c-myc induction was abrogated. In addition, activation of the src tyrosine
kinase was reduced, suggesting a possible link between src activation and Myc expression.
A definitive role for src in the signaling pathway responsible for Myc induction was
revealed upon the analysis of the PDGF-induced signaling mechanisms which drive
fibroblasts out of quiescence. Ectopic expression of dominant negative mutants of src, or
inhibitory antibodies to src inhibited c-Myc transcription and progression of cells into the
replicative phase (Barone and Courtneidge, 1995).
Evidence suggests that activated src induces the transcription of the c-myc gene by
regulating the activity of the ets family of transcription factors (Roussel et al., 1994).
There is a conserved ets binding site in the c-myc promoter between P1 and P2. Mutation
of this site will abrogate the src-dependent induction in c-myc expression. This
observation may suggest that inhibiting the binding of ets proteins will abrogate c-myc

transcription, yet the answer may not be so simple. The ets binding site is a part of a
highly conserved binding site for the E2F family of transcriptional regulators. The binding
of E2F to this site has also been shown to be required for the induction of c-myc
transcription in response to stimulation with mitogens such as serum (Mudryj et al., 1990).
In addition, the nonreceptor tyrosine kinase c-abl, another component of common signal
transduction pathways, also induces c-myc transcription through the E2F site (Wong et al.,
1995). Hence, mutation of the ets and consequently the E2F binding site, potentially
negates three separable signaling pathways. Thus, recent studies have identified a few
important regulators of myc transcription; however, further characterization is required to
determine the contribution of each of these mechanisms to the regulation of Myc expression
following exposure to external stimuli.
Given the strong proliferative capacity of an activated myc allele it is not surprising
that, in non-transformed cells, endogenous c-myc expression is tightly regulated at both the
mRNA and protein level (reviewed in Spencer and Groudine, 1991). Indeed, Myc
expression is tightly linked to the growth state of the cell, and is immediately responsive to
changes in the external environment (reviewed in Lemaitre et al., 1996). Myc mRNA and
protein expression are low in quiescent, growth arrested cells. Upon mitogen stimulation,
Myc mRNA and protein expression levels are rapidly elevated approximately 40 fold
within 2 h, concomitant with the entrance of cells into the cell cycle (Campisi et al., 1984;
Greenberg et al., 1986; Kelly et al., 1983). The induction in Myc expression is transient: 6
to 12 h following mitogen stimulation Myc expression decreases to levels approximately 10
fold above that observed in quiescent cells. Myc expression is maintained at this level
through the remainder of, and in subsequent cell cycles (Rabbitts et al., 1985; Rabbitts et
al., 1985). Removal of mitogens or induction to differentiate results in a rapid loss of Myc
expression and exit from the cell cycle (Figure 1.4) (Dean et al., 1986; Kelly and
Siebenlist, 1986; Rabbitts et al., 1985; Waters et al., 1991). As Myc expression is so
closely associated with the growth state of the cell, it is possible that Myc functions as an

internal barometer of conditions in the external mi!ieu, deciding when it is possible to
proliferate and maintaining the cell in this state until conditions are no longer favourable for
growth. The half-lives of the c-Myc protein and mRNA are short (Dani et al., 1984; Ham
and Eisenman, 1984), and are well suited to the role of Myc as a sensor and responder to
external changes. c-Myc protein and mRNA expression is controlled via a host of
processes which act at multiple stages: at the levels of mRNA transcription initiation,
elongation, and stability, as well as at the level of protein degradation (reviewed in Cole,
1986). The existence of multiple mechanisms to govern Myc expression emphasizes the
need to control the strong proliferative potential of Myc.

MYC mRNA and protein
EXPRESSION
t GROWTHARRESTED QUIESCENT PROLIFERATING
DIFFERENTIATED
TRANSFORMED CELLS
NON-TRANSFORMED
I CELLS
Mitogen Stimulation
Growth inhibitors or
Differentiation Agents
Figure 1.4. Myc mRNA and protein expression, in non-transformed and transformed cells, in response to external stimuli. In non-transformed primary and immortalized cells, Myc expression is highly responsive to changes in the external environment, with Myc expression being tightly linked to actively proliferating cells (solid line). In transformed cells, Myc expression is frequently deregulated, and is refractory to the nature of the extracellular milieau (hatched line). Levels of Myc expression in transformed cells vary, depending upon the nature of the activating mutation of the nlyc gene.

1.3.4 Structure of the Myc protein
The c-myc gene encodes two polypeptides with molecular weights of
approximately 64 and 67 kd (Hann et al., 1988). The half-life of the Myc proteins are very
short, approximately 20 to 30 mins, mimicking the half-life of the c-myc mRNA (Luscher
and Eisenman, 1990). Translation of the 64 kd (Myc-2) protein initiates from an AUG
start codon in exon 11, while the larger Myc-1 protein, containing 14 additional amino acids
at the amino-terminus, originates from a cryptic CUG at the end of exon I (Figure 1.3)
(Ham et al., 1988). Myc-2 consists of 439 amino acids and is more highly expressed in
most cell types, compared to Myc-1. At a functional level there is no apparent difference in
the activities of these two species (Blackwood et al., 1994). Yet it was observed that
expression of the p67Myc protein increases in quiescent cells, suggesting a potential
distinct role for this isoform of Myc in growth arrest (Hann, 1995; Ham et al., 1992).
Indeed, in support of this hypothesis, ectopic expression of p67Myc by Cos cells appeared
to induce growth arrest (Hann et al., 1994). Recently, it was recognized that smaller
proteins were also translated from the c-myc mRNA transcript, arising from conserved
internal translational initiation codons within the coding region of c-myc. C-Myc S
proteins lack the first 100 amino acids of the amino terminus of the full-length Myc-1
protein (Spotts et al., 1997). C-Myc S proteins are ubiquitously expressed, and are
evident in human, avian, and murine cells. Additional studies are required to fully
comprehend the relationship among the different Myc isoforms, and the role of these
species in the regulation of normal cell proliferation.
Computer analysis of the amino acid sequence of human c-Myc predicts that the
structure of the amino-terminus (amino acids 1 to 203) consists of a-helical and P-sheet
domains and the carboxyl end (amino acids 238 to 439) is composed primarily of a-
helices, while the intervening region appears to be an unstructured hinge (reviewed in Penn
et al., 1990). There is evidence for a number of functional domains, such as a

transcriptional regulatory domain, nuclear localization motifs, as well as sequences which
effect specific DNA binding, and protein dimerization (Figure 1 SA).
The transcriptional activation domains of Myc comprise the amino-terminal 143
amino acids and were identified through studies by Kato et al. (Kato et al., 1990). They
assayed the ability of different portions of Myc, fused to the DNA binding domain of the
yeast transcription factor Ga14, to transcriptionally activate a heterologous promoter
containing Gal4 DNA binding sites. This transactivation domain can be further divided
into three autonomous portions. The region spanning amino acids 1 to 41 is glutamine-
rich, and shares sequence homology with the transcriptional activation domains of the well-
characterized transcription factors Spl and herpesvirus VP16 proteins (Mitchell and Tjian,
1989). The second region, amino acids 42 to 103, is rich in prolines, similar to the
transcriptional regulatory domain of the CTFMFl family of transcriptional regulators
(Mitchell and Tjian, 1989). The final region, amino acids 104 to 143, is not homologous
to the transactivation domain of any known transcription factor (Kato et al., 1990). Myc
repression of gene expression has also been mapped to this region; however, the precise
sequences required for specific repression events vary. Amino acids 106 to 143 are
required for c-myc negative autosuppression; amino acids 92 to 106 are involved in the
suppression of cyclin Dl; while residues 122 to 143 effect repression of the adenovims
major late promoter (Li et al., 1994; Penn et al., 1990; Philipp et al., 1994). The existence
of this discrepancy suggests that there are multiple mechanisms for Myc repression.
Sequence analysis of the members of the Myc protein fami!? have highlighted regions
which are highly conserved among the members of the Myc protein family. Conservation
of these regions suggests that they may be critical for the functions of Myc. Two Myc
boxes, Myc Box 1 and 2, are found in the transcriptional activation domain; Myc Box 1
comprises amino acids 45 to 63 in humans, and Myc box 2 consists of amino acids 129 to
141 (reviewed in Ingvarsson, 1990). Myc box 2 is essential for all of the biological
activities of Myc such as cell cycle progression, inhibition of differentiation, exit

Figure 1.5. Functional and structural features of the human c-Myc protein.
A. Myc 1 (p67) initiates at a CUG in exon I, and contains 14 additional amino acids
compared to Myc 2 which initiates at an AUG in exon 11. Functionally important domains
are indicated: c-Myc harbours three independent transcriptional activation domains (I, 11,
111); a non-specific DNA binding domain (NDB); a basic region (B); a helix-loop-helix
motif (HLH); and a leucine zipper domain (LZ). Filled rectangles in the transcriptional
activation domains represent Myc Box I (MBI) and Myc Box I1 (MBII), two highly
conserved regions found in all Myc family members. At the carboxy terminus there are
two nuclear localization signals (NLS MI, NLS M2). Phosphorylation sites (P) and their
putative kinases are indicated at the top. B. The domains of Myc which are required for
Myc functions and involved in the interaction with other proteins are shown.


from quiescence, induction of apoptosis, and tumorigenesis (Eilers et al., 1989; Evan et
al., 1992; Freytag et al., 1990; Stone et al., 1987). The transcription regulatory domains
are also required for interactions with other proteins such as the transcription factor AP-2
(Gaubatz et al., 1995).
The c-Myc protein contains two nuclear localization domains, which facilitate the
transport and translocation of c-Myc into the nucleus (Dang and Lee, 1988; Stone et al.,
1987). The predominant nuclear localization signal (NLS) called NLS MI, comprises
amino acids 320 to 328 in human, and is homologous with the well characterized NLS of
the SV40 and polyoma virus T antigens (Kalderon et al., 1984; Richardson et al., 1986).
These sequences are able to direct the normally cytoplasmic protein pyruvate kinase into the
nucleus (Dang and Lee, 1988). In the absence of NLS MI, the weaker NLS M2 can
substitute partially for NLS Ml. NLS M2 consists of amino acids 364 to 374 in humans,
and coincides with the DNA binding domain (Dang and Lee, 1988).
c-Myc can bind nonspecifically to single-stranded and double-stranded DNA, in
vitro (Beimling et a]., 1985; Persson and Leder, 1984; Watt et al., 1985). The region
responsible for this activity is contained within amino acids 265 to 3 18 in human c-Myc
(NDB) (Dang et a]., 1989). It is believed that this activity allows c-Myc to b i d loosely to
and move along DNA, until it encounters a high affinity binding site. As yet, a role for this
domain in vivo has not been established; however, deletion of this region will abolish the
ability of c-Myc to transform cells (Stone et al., 1987).
Structure-function analysis of the carboxyl terminus of the c-Myc protein revealed
the presence of a number of conserved motifs which are common among well-characterized
transcription factors. There is a basic region (B) (amino acids 355 to 367 in humans),
which serves as a specific DNA binding domain. In addition, there are two protein-protein
interaction motifs, a helix-loop-helix domain (HLH) (amino acids 368 to 410 in humans),
and a leucine zipper motif (LZ) (amino acids 41 1 to 439) (Kerkhoff and Bister, 1991).
Deletion of this region will inhibit all of the biological activities of Myc, suggesting that

protein-protein interaction and DNA binding is required for Myc function. The
combination and contiguous arrangement of these domains is common within a family of
B-HLH-LZ-containing transcriptional regulators including USF, TFEB, AP-4, and TFE3
(Beckmann et al., 1990; Carr and Sharp, 1990; Gregor et al., 1990; Hu et al., 1990).
Both USF and TFE3 bind specifically to a consensus DNA element, CANNTG, called an
E-box, suggesting that Myc may also bind DNA through this site. This hypothesis was
substantiated in a variety of studies utilizing either the bacterially expressed c-Myc
carboxyl-terminus, chimeric proteins consisting of the c-Myc basic region linked to the
helix-loop-helix domain of either the drosophila El2 or the yeast pH04 proteins, or the full
length Myc protein (Blackwell et al., 1990; Fisher et al., 1991; Halazonetis and Kandil,
1991; Kerkhoff et al., 1991). All of these studies demonstrated that c-Myc binds
specifically to the sequence CACGTG in vitrv. Myc is also able to bind to specific
noncanonical sites related in sequence to the E-box, with high affinity (Blackwell et al.,
1993; Grandori et al., 1996). The identification of the specific DNA binding property of
Myc was a significant observation, as it suggested that Myc may effect its biological
activities through the regulation of gene expression. Myc homodimers are not observed in
vivo, due to steric interference between the protein-protein interaction domains of the c-
Myc proteins (Amati et al., 1993). Specific DNA binding by Myc is dependent upon
interaction with a ubiquitously expressed B-HLH-LZ protein, Max (Blackwood and
Eisenman, 1991; Prendergast et al., 1991). The formation of MycIMax heterodimers is
favoured over MadMax homodimers; and as a consequence Myc is predominantly found
complexed with Max (Blackwood and Eisenman, 1992; Kato et al., 1992; Littlewood et
al., 1992). Heterodimerization of Myc and Max is mediated through the protein
dimerization motifs, the helix-loop-helix and the leucine zipper (Blackwood and Eisenman,
1991).

1.3.5 Post-translational modification of the Myc protein
The c-Myc protein is phosphorylated at multiple serine and threonine sites in vivo,
suggesting that phosphorylation may play some role in the regulation of Myc activity. The
sites of phosphorylation include: Thr58, Ser62, Ser71, Ser82, Ser164, five sites between
residues 240 and 262, as well as Ser293, Thr343, Ser344, Ser347, and Ser348. In vitro,
the amino-terminus of Myc is phospholylated on three sites, threonine 58, serine 62, and
serine 71 by a variety of kinases including mitogen-activated protein kinase (MAP kinase),
glycogen synthase kiiase 3 (GSK3), cyclin-dependent kinase 1 (cdkl), and a pl07/cyclin
AIcdk2 complex (Alvarez et al., 1991; Gu et al., 1994; Henriksson et al., 1993; Hoang et
al., 1995; Lutterbach and Ham, 1994; Pulverer et al., 1994; Seth et al., 1991). The
importance of these phosphorylation events in Myc function is at present unclear, yet the
high incidence of mutations at these sites in tumours such as Burkitt's lymphoma suggests
that they may play a role in the capacity of Myc to promote hunour formation (Gupta et al.,
1993). Threonine 58 is also reported to be the target of glycosylation; however, whether
phosphorylation or glycosylation of this residue has any effect on Myc-mediated cellular
transformation is not known (Chou et al., 1995; Chou et al., 1995). Residues in the
carboxyl-terminus are phosphorylated by the ubiquitous kinase, casein kinase I1 (CKII), in
vitro (Luscher et al., 1989). CKII phosphorylates c-Myc at sites located behveen amino
acids 240 to 262, and amino acids 342 to 357 in the human c-Myc protein. The proximity
of the latter phosphorylation events to the basic region suggested that they may influence
Myc function by affecting DNA binding. However, studies have failed to attribute a
hnction to any of these phosphorylations (Street et al., 1990).

1.4 The biological activities of Myc
1.4.1 Myc in normal cell proliferation
Several studies have supported the model that c-Myc protein expression is essential
for the promotion of cell cycle progression and the maintenance of cell proliferation.
Constitutive ectopic Myc expression in fibroblast cells will abrogate the requirement for
growth factors to progress from the G1 to S phase of the cell cycle (Armelin et al., 1984;
Kam et al., 1989; Mougneau et al., 1984; Sorrentino et al., 1986; Stem et al., 1986).
Elevated expression will increase the growth rate, predominantly through the shortening of
the GI phase (Kam et al., 1989). Using MycER, an estrogen-inducible conditional allele
of Myc, it has been demonstrated that induction of Myc activity alone can induce cells to
exit growth arrest and progress through the cell cycle (Daksis et al., 1994; Eilers et al.,
1989). Indeed, microinjection of Myc alone into quiescent fibroblasts will induce cell cycle
entry (Kaczmarek et al., 1985). Moreover, an inhibition of Myc expression through the
use of antisense RNA will result in growth arrest (Heikkila et al., 1987; Holt et al., 1988).
Interestingly, a subclone of the immortalized cell line Rat-I, in which both alleles of the c-
myc gene are deleted through homologous recombination, is viable but exhibits a slower
growth rate and reduced ability to enter the cell cycle after serum stimulation (Mateyak et
al., 1997). The importance of Myc expression in effecting cell cycle entry and hence cell
proliferation in serum-stimulated quiescent cells, was illustrated via elegant studies by
Roussel eta[. (Roussel et al., 1991). Ectopic expression of the colony stimulating factor
receptor (CSF-I R) by the immortalized mouse NIH3T3 cell line renders these cells
responsive to stimulation with the cytokine, CSF-1. A Y809F substitution in the CSF-1
R, specifically abrogates the induction of c-myc expression inhibiting the growth
promoting affects of CSF-1 treatment. Ectopic expression of Myc in these cells will rescue

the CSF-1-mediated entry into cell cycle. Taken together, these observations suggest that
Myc activity is crucial for cells to exit growth arrest and enter the cell cycle.
As Myc protein is continuously expressed in asynchronously proliferating cells,
Myc may have additional functions at other points of the cell cycle. Indeed, a potential role
for Myc in the G2 phase has recently been suggested through the studies of c-myc null rat
fibroblasts. Disruption of Myc expression does not inhibit growth, but both the G1 and
G2 phases are noticeably lengthened while the duration of S phase is unaltered (Mateyak et
al., 1997). This was the first study to directly demonstrate a role for Myc in progression
through the G2 phase of the cell cycle, although its hnction in this period is unclear and
studies are currently underway which will shed light on this question.
Thus, considerable evidence exists to support a role for Myc in driving cells out of
quiescence and into the cell cycle. Yet the continued expression of Myc in asynchronously
proliferating cells suggests that additional roles for Myc in the promotion of cell cycle
progression remain to be defined. Studies in Myc-null cells hint at a role for Myc in the
progression of cells through the G2 phase; however, the function and the importance of
Myc in this phase has not been fully explored.
1.4.2 Myc: inhibitor of differentiation
Consistent with the role of Myc as a strong proliferative stimulus, Myc expression
can inhibit the differentiation program in a number of cellular systems. Indeed, in a variety
of cultured cells exposure to inducers of differentiation results in the repression of Myc
expression and a withdrawal from the cell cycle (Bianchi Scarra et al., 1986; Gonda and
Metcalf, 1984; Gowda et al., 1986; Griep and DeLuca, 1986; Lachman and Skoultchi,
1984; Reitsma et al., 1983). Myc downregulation is evident upon the differentiation of a
number of cell lines: 3T3L1 pre-adipocytes (Freytag, 1988; Freytag et al., 1990), F9
tetracarcinoma cells (Dony et al., 1985), U-937 monoblastic cells (Larsson et al., 1988),

murine myeloid M1 cells (Einat et al., 1985), and proerythroid K562 cells (Bianchi Scarra
et al., 1986). In other differentiating systems, Myc repression is associated with the
terminal stages of the differentiation process. In these cells, the repression of myc mRNA
is biphasic, exhibiting a transient reduction in myc mRNA expression before returning to
levels close to that observed in proliferating cultures prior to the loss of Myc expression
upon terminal differentiation. This biphasic pattern has been reported in the differentiation
of the human promyelocytic HL60 (Siebenlist et a]., 1988; Simpson et al., 1987), mouse
erythroleukemia MEL cells (Mechti et al., 1986; Nepveu et al., 1987), the mouse P19
embryonic carcinoma cell line, and L6E9-B rat muscle cells (Endo and Nadal-Ginard,
1986).
A direct role for Myc in inhibiting cellular differentiation was demonstrated in
several studies. Constitutive ectopic Myc expression will inhibit the differentiation of
MEL, U937, F9, and 3T3L1 cells (Coppola and Cole, 1986; Dmitrovsky et al., 1986;
Freytag, 1988; Kaneko-Ishino et al., 1988; Larsson et al., 1988; Onclercq et al., 1989;
Prochownik and Kukowska, 1986). The introduction of antisense c-myc RNA in HL60
cells will induce growth arrest (Griep and Westphal, 1988; Holt et al., 1988; Prochownik
et al., 1988). Myc can antagonize myogenic differentiation induced by myogenin and
MyoD, a process which is independent of Id, an inhibitor of both myogenin and MyoD
(Miner and Wold, 1991). Further evidence supporting a role for Myc in t!~e inhibition of
differentiation can be found in the analysis of B lymphoid cell development in mice
expressing the c-myc transgene governed by the immunoglobulin heavy chain enhancer
(Adams et al., 1985). These mice exhibit a B cell population consisting of a large
proportion of undifferentiated pre B cells. Thus, studies in a variety of cell types indicate
that Myc is a potent inhibitor of differentiation, inhibiting growth arrest and promoting the
expansion of an undifferentiated cell population.
Yet there are a few contradictory observations which seem to suggest that the role
of Myc in the inhibition of differentiation is dependent upon the cell type. Reexpression of

Myc in differentiated myoblasts can not reverse the differentiated phenotype (Endo and
Nadal-Ginard, 1986). Elevated ectopic Myc expression cannot inhibit the y-interferon
induced differentiation of U937 cells (Oberg et al., 1991). Myc expression promotes the
differentiation of human epidermal stem cells (Gandarillas and Watt, 1997). Similarly,
targeted expression of ectopic c-myc in the developing mouse lens did not inhibit the
elaboration of differentiation-specific markers, yet it did inhibit proliferative arrest
(Morgenbesser et al., 1995).
The molecular role of Myc in the inhibition of differentiation is not well
understood. It is still not clear whether Myc can directly inhibit both the elaboration of
differentiation-specific markers such as the mim-1 gene, andlor inhibit the growth arrest
program associated with cellular differentiation. Yet, in the pre-adipocyte cell line, 3T3-
L1, Myc appears to effect both pathways. Differentiation in these cells is accompanied at
the molecular level with a gradual decrease in Myc expression, and the induction of the
differentiation-specific transcriptional activator clebpa (Cao et al., 1991; Christy et al.,
1991; Christy et al., 1989). The upregulation in clebpa expression results in the
subsequent transcriptional induction of the growth arrest gene, gadd45 (Constance et al.,
1996), and differentiation-specific genes such as: senim albumin, lysozyme, and mim-1
(Burk et al., 1993; Christy et al., 1989; Friedman et al., 1989; Mink et al., 1996). The
upregulation and activity of clebpa is crucial and sufficient to induce the differentiation
process (reviewed in Lane et al., 1996; MacDougald and Lane, 1995, Lin and Lane, 1992;
Lin and Lane, 1994; Ron et al., 1992). Myc can inhibit the activity of clebpa, thereby
inhibiting the induction of differentiation-specific genes including gadd45. Myc effects this
activity in two ways: Myc has been reported to repress the transcription of clebpa;
alternatively, it has been shown in transcription promoter-reporter assays, that Myc can
inhibit the activity of debpa via an as yet uncharacterized mechanism (Antonson et al.,
1995; Constance et al., 1996; Li et al., 1994; Mink et al., 1996). Additional studies
support the view that clebpa is the critical target for Myc in the inhibition of adipocyte cell

differentiation, as introduction of ectopic clebpa into 3T3-L1 cells expressing ectopic Myc
protein will rescue the differentiation program. Therefore in differentiating 3T3-L1
adipocytes, Myc inhibits cellular differentiation by inhibiting both the elaboration of
differentiation-specific genes and the expression of growth arrest genes. As such, Myc is a
potent inhibitor of the differentiation program, serving a pivotal role in inhibiting growth
arrest and forcing cells into a proliferative state.

1.4.3 Myc and apoptosis
It is clear from both in vivo and in vitro studies that Myc confers a strong
proliferative stimulus, inhibiting growth arrest states such as quiescence, senescence, and
differentiation. Indeed, it can be universally stated that Myc expression and growth arrest
are mutually exclusive. The consequence of opposing this restrictive relationship is
illustrated in the Myc-dependent apoptotic mechanism. Ectopic Myc expression imparts
such a strong proliferative impulse upon cells that they are unable to arrest upon exposure
to growth arrest agents: instead they undergo apoptosis.
Expression of ectopic Myc in the IL3-dependent myeloid cell line 32D results in an
acceleration of apoptosis in cells cultured in the absence of IL3 (Askew et al., 1991).
Activation of a conditional Myc allele in serum starved rodent fibroblasts rapidly induces
apoptosis (Evan et al., 1992), and sensitizes rodent fibroblasts to apoptosis induced by
treatment with TNFa (Klefstrom et al., 1994). Identical results were observed upon Myc
activation in NIH3T3 cells and primary MEFS (Hermeking and Eick, 1994). A direct role
for Myc in promoting apoptosis was observed in studies of T cell receptor (TCR)-activated
T cell hybridomas (Shi et al., 1992). Activation of the TCR induces an apoptotic program
which requires Myc expression. The regions of Myc which are required to effect apoptosis
include MBII, DNA binding, and protein dimerization domains (Amati et al., 1993; Stone
et al., 1987). In addition, Myc-induced apoptosis is dependent upon the interaction of Myc
with its protein partner, Max (Amati et al., 1993). These results suggest that the ability of
Myc to transactivate gene expression is required for the induction of apoptosis. In support
of a role for transcriptional regulation by Myc in apoptosis, two Myc-regulated genesp53
(Hermeking and Eick, 1994), and odc (Packham and Cleveland, 1994; Packham and
Cleveland, 1995), appear to be required for Myc-induced apoptosis. Both these genes
contain Myc DNA binding sites, and are reported to be transcriptionally regulated by Myc . The importance of p53 expression in Myc-induced death, can be seen in studies performed

in cells lacking p53. Fibroblasts lacking p53 expression due to targeted gene disruption are
resistant to Myc-induced apoptosis (Hermeking and Eick, 1994). Similar studies
employing inhibitors of odc function have also implicated a role for odc in this mechanism.
For example, inhibiting odc expression via an antisense approach, abrogates the apoptotic
program in 32D cells (Packham and Cleveland, 1994). Ongoing dissection of the Myc-
induced apoptotic mechanism has revealed additional players in this pathway. Studies by
Evan et al, have elucidated that the interaction of Fas with its ligand, and the resultant Fas-
mediated signaling pathway is required for apoptosis (Hueber et al., 1997). In addition,
caspase 3, a cysteine aspartase, was shown to be required for Myc-induced apoptosis,
through the use of specific protease inhibitors (Kagaya et al., 1997; Kuchino et al., 1996).
Apoptosis can be inhibited by ectopic expression of bcl-2, or treatment with the growth
factors, insulin growth factor-1 (IGF-I), or platelet-derived growth factor BB (PDGF)
(Hamngton et al., 1994). IGF-1 or PDGF elicits a signaling pathway which involves
phosphoinositol 3' kinase (PI3K) and its downstream target AKT (Protein Kinase B)
(Figure 1.6) (Datta et al., 1997; Kauffinann-Zeh et al., 1997; Kennedy et al., 1997).
It is a paradox that Myc is a strong promoter of cell proliferation and apoptosis
(reviewed in Henriksson and Luscher, 1996; Packham and Cleveland, 1995). Yet, such
an arrangement may serve as an internal control responsible for inhibiting cell
transformation by the elimination of cells which have suffered c-rnyc activation as a
primary or early oncogenic hit in the step-wise progression of hunorigenesis.


1.4.4 Myc and cdkIs
The expression of the cdkIs is commonly elevated in a variety of growth arrest
states including quiescence, senescence, contact inhibition, exposure to growth inhibitory
agents. and in response to DNA damage. Thus, the cdkls could potentially be involved in
initiating or maintaining cells in a growth arrested state. A number of studies have clearly
shown that Myc can overcome many of these growth arrest states, although the precise
mechanism through which this is achieved is not known. Recent evidence has
demonstrated that Myc can overcome many of these growth inhibitory stimuli.
One of the first hints that Myc could overcome a cdkI-mediated block to cell
proliferation arose from the studies by Alexandrow et al. They demonstrated that Myc
circumvented a transforming growth factor P (TGF-P)-mediated growth arrest, which is
mediated by the activities of the cdkIs p27, p15, and p21 (Alexzndrow et al., 1995;
Alexandrow and Moses, 1995). More compelling evidence emerged from the studies of
Steiner et al. as well as Rudolph et al. It was demonstrated that Myc aciivation in quiescent
cells leads to the rapid induction in cyclin EIcdk2 activity and exit from growth arrest
(Rudolph et al., 1996; Steiner et al., 1995). The activation of cyclin EIcdk2 is due to the
absence of p27 activity in these Myc-activated cells. The mechanism effecting the loss of
p27 activity is controversial: two mechanisms have been proposed. First, the p27 within
cyclin EIcdk2lp27 complexes is phosphorylated by cyclin EIcdk2 followed by the
degradation of p27 possibly via a ubiquitin-dependent pathway (Clurman et al., 1996;
Muller et al., 1997; Sheaff et al., 1997; Steiner et al., 1995). Second, an alternate
mechanism was proposed by Vlach et a/., suggesting that p27 was converted into a
functionally inactive form which is incapable of biidiig to the cyclinlcdk complex (Vlach et
al., 1996). The reason for the dissimilar observations is puzzling, and cannot be explained
at this time. Rescue from p27-mediated growth arrest by Myc requires the amino-terminal
transcriptional regulatory domain (amino acids 7 to 145), the DNA binding domain, and

interaction with Max (Vlach et al., 1996). These domains are required for the
transactivztion function of Myc, suggesting that Myc may overcome the activity of p2i,
through the induction of gene expression. As p27 is bound predominantly to cyclin EIcdk2
complexes during quiescence, it was not surprising that the inhibition of p27 function by
Myc induces cyclin EIcdk2 kinase activity. Yet it is apparent that the induction in cyclin E-
associated cdk2 activity is not the sole function of Myc activation, as constitutive ectopic
expression of cyclin E alone in rat fibroblasts could not substitute for Myc. In addition,
elevated expression of the other G1 cyclins, cyclin A or the D-type cyclins, in rat
fibroblasts, could not circumvent the growth inhibito~y effects of p27. Neither can the p27
block be inhibited by overexpressing the cdk phosphatase, cdc25A. Interestingly, ectopic
expression of E2Fs alone cannot abrogate the p27 block. This supports an alternate role
for cyclin EIcdk2 activity distinct fiom pRb phosphorylation, and liberation of active E2F,
in the promotion of GUS transit (Vlach et al., 1996).
Thus, Myc induces exit from a G1 cell cycle block, potentially by antagonizing the
activity of the cdkI p27, resulting in the activation of cyclin EIcdk2 in late G1 phase
promoting entry into the cell cycle. Yet, while the induction in cyclin Elcdk activity is
important for the abrogation of the p27 cell cycle arrest by Myc, it clearly is not sufficient.
Cyclin EIcdk2 induction must be complemented by additional Myc-regulated activities
which remain to be identified and characterized (Vlach et al., 1996). It must be noted that
the story is not so simple. Ectopic expression of cyclin E in NIH3T3 cells can overcome a
p27-induced G1 block to cell growth (Sheaff et al., 1997). In addition, ectopic expression
of E2F-1 alone will overcome a p27-induced growth arrest in the immortalized rat cell line
REF52 (DeGregori et al., 1995). Both of these results contradict the observations made in
Rat-1 cells, hinting that the observed mechanisms which abolish the p27-dependent growth
arrest are influenced by unknown factors specific to individual immortalized cell lines.
Analyses in primary cell cultures may be the ideal system within which these discrepancies
may be resolved.

Myc activation can similarly bypass the inhibitory effects of either p21 or p16 in
fibroblasts. Myc can overcome a p2lcdkI-dependent G1 arrest by inducing the expression
or activity of an as yet uncharacterized heat labile inhibitor of this cdkI, abrogating its
inhibitory effect on cyclin associated kinase activity (Hermeking et al., 1995). An alternate
mechanism through which Myc may relieve a p21-induced growth arrest was proposed by
the observations of Saha et al. (Saha et al., 1997). Myc can transcriptionally induce the
expression of the phosphatase cdc25A. Both cdc25A and the p2lcdkI share the same
binding site on the cyclin/cdk dimer, hence binding by either of the two proteins is
mutually exclusive. Hence a Myc-dependent increase in the expression of cdc25A may
hinder the interaction and inhibition of cyclin E-dependent kinases by p21, allowing cell
proliferation.
In conjunction with the inhibition of p21 activity by Myc, Myc can also circumvent
a pl6-dependent growth arrest (Alevizopoulos et al., 1997). The precise mechanism
through which Myc abrogates the growth inhibitory effect of pl6 is unknown. However,
it is clear that this mechanism exhibits some similarities to the pathway via which Myc
circumvents the activity of the p27 cdkI. The end result ofboth of these mechanisms is a
loss of p27 activity concomitant with the activation of cyclin Wcdk2. Yet, there are notable
differences between these two mechanisms. In contrast to the p27-mediated growth arrest,
the pl6-dependent block to proliferation is also rescued by ectopic expression of cyclin E,
or E2Fs 1,2, and 3, indicating that these cell cycle effectors may function within the same
pathway as Myc. In addition, while Myc overcomes a p27-induced cell cycle arrest via a
pathway which involves the phosphorylation of the tumour suppressor pRb, the latter
pathway is dispensable for the Myc circumvention of the pl6-dependent growth arrest.
This discrepancy suggests that separable pathways are responsible for the Myc-induced
abrogation cf a p16- versus p27-mediated block to cell proliferation. Although our
understanding of these mechanisms is minimal, these pathways may represent the
mechanism through which Myc overcomes a variety of growth arrest states such as

senescence and quiescence and promotes cell proliferation. Hence, the elucidation of the
mechanisms through which Myc overcomes the growth inhibitory effects of the cdkIs will
aid in our understanding of how Myc confers a strong growth stimulus, and contributes to
tumour formation.
1.4.5 Myc Transformation in vitro and in vivo
Ectopic Myc expression in primary fibroblasts will abrogate the senescence
program and elicit cellular immortalization. Yet, immortalization does not render the cell
autonomous, as proliferation is still dependent upon culture in the presence of the
appropriate mitogens (reviewed in Henriksson and Luscher, 1996). Indeed in the absence
of mitogen, cells expressing ectopic Myc undergo apoptosis (Evan et al., 1992),
suggesting that the abrogation of the apoptotic program by mitogenic signals is critical to
the immortalization activity of Myc and permits immortalization to proceed unrestrained. In
combination with an additional cooperating oncogenic lesion such as an activated Ras, or
Raf allele, Myc will induce transformation in primaxy cells derived from a variety of tissue
types (Adams and Cory, 1992).
Transgenic mouse studies provide the strongest evidence for the role of Myc in the
genesis of tumours. Transgenic mice were generated which ectopically express human c-
Myc under the control of the immunoglobulin heavy chain enhancer, mimicking
translocation events observed in Burkitt's lymphoma. These transgenic mice exhibit
elevated levels of immature undifferentiated pre-B cells, suggesting that elevated ectopic
Myc expression can inhibit the normal differentiation program and enhance the proliferative
potential of the pre-differentiation lymphoid population. These mice develop monoclonal
B-lymphoid malignancies which are phenotypically characteristic of Bwkitt's lymphoma
(Adams et al., 1985). The monoclonal nature of these tumours indicate that secondary co-
operating mutations are required to elaborate the full transformed phenotype. In accord

with the functional similarity among the Myc family members, expression of an L-myc
transgene driven by an immunoglobulin enhancer induced T-lymphoid tumours in Ep-L-
niyc transgenic mice (Moroy et al., 1990). Ep-N-myc transgenic animals succumb to
clonal lymphoid malignancies, primarily B-cell lymphomas (Dildrop et al., 1989;
Rosenbaum et al., 1989). Importantly, targeted expression of the N-Myc protein in the
neuroectodermal cells of the developing mouse embryo results in the development of
neuroblastomas (Weiss et al., 1997). Therefore, it is clear from both the transgenic animal
and tissue culture studies that Myc can play a role in the initiation of cellular transformation
in a wide variety of cell types, and additional secondary genetic alterations are needed to
produce full transformation.
What is the contribution of Myc to the tumorigenic process? The work of
Tikhonenko et al., illustrates the conventional thinking regarding the characteristics of the
transformed phenotype which are thought to be directly controlled by Myc (Tikhonenko et
al., 1995). Avian embryo fibroblasts expressing a glucocorticoid-inducible v-gag-myc
allele, were transformed upon activation of Myc activity following treatment with the
glucocorticoid dexamethasone. Transformed cells exhibited an increased growth rate,
anchorage-independent growth and increased refractivity. However, removal of the
glucocorticoid, and the resulting loss in ectopic Myc activity, resulted either in growth
arrest or a reduced growth rate, while refractivity as well as anchorage-independent growth
were unaffected. Therefore it is apparent, at least in this Myc-dependent tumour model,
that escape from growth arrest and enhanced proliferation may be the primary effects of
Myc, and secondary mutations are responsible for the transformed morphology. The
ability of Myc to enhance the cellular proliferative potential, concomitant with the increased
number of DNA replications, may facilitate the emergence of additional genetic aberrations
which will permit full cellular transformation. Indeed, Myc has been reported to induce the
non-random amplification of the dihydrofolate reductase (dhfr) locus in a variety of cell

types originating from different species. Yet, it is not clear whether this activity is a direct
or indirect effect of Myc (Denis et al., 1991; Mai, 1994; Mai et al., 1996; Mai et al., 1996).
In essence Myc may promote cell transformation via two complementary
mechanisms: first, Myc inhibits growth arrest programs such as senescence and
differentiation; and second, Myc drives progression through the cell cycle. The provision
of both activities would confer an increased growth advantage to cells, facilitating the
emergence of additional mutations which permit the evolution of the fully transformed
phenotype.
1 .4 .6 Myc cooperativity in transformation
This past year has provided a wealth of information regarding how Myc interacts
with different components of the cell cycle machinery, particularly in the GI phase of the
cell cycle. These reports have provided new insights into the requirement of oncogene
cooperation for full cellular transformation by Myc. Myc is a component in two types of
cooperative relationships, these two categories are divided on the basis of the contribution
of the cooperating oncogene to the partnership. First, the cooperating oncogene within the
partnership may supply an independent and complementary function. The synergy of both
activities is sufficient to permit unrestrained cell growth. The most commonly studied of
these relationships is the synergy of activated Myc with Ras. Myc and Ras exist on
separable signal transduction pathways originating either i?om a single or multiple mitogen
activated receptors, depending upon the cell (Barone and Courtneidge, 1995). Hence the
ectopic constitutive expression of activated alleles of both c-myc and ras may circumvent
the necessity for mitogen stimulation in the promotion of continual cell proliferation.
Although it is well known that at a phenotypic level, Myc in conjunction with Ras
can elicit cellular transformation, the biochemical basis of this synergy is unclear. Studies
by Leone et al. sought to answer this question, by determining the contribution or the

impact each of these two oncogenes has on components of the cell cycle machinery. The
expression of both Myc and Ras is necessary for serum-stimulated quiescent cells to exit
growth arrest and enter the cell cycle. However, ectopic expression of activated Myc or
Ras in the immortalized cell line REF52 were ineffective in elaborating progression into S
phase. Moreover, ectopic Ras expression elicited growth arrest in G1 phase of the cell
cycle, likely the consequence of the absence of Gl cdk activity. Indeed, both cyclin E and
cyclin Dl-associated kinase activity was inhibited due to the activities of the cdkI p27.
Interestingly, coexpression of Myc with Ras permitted DNA replication, preceded by a
reduction in p27 levels and a concomitant induction in cyclin EIcdk2 activity. The
reduction in p27 levels is due to the synergistic effect of both Myc and Ras coexpression,
as expression of ectopic Myc or Ras did not affect p27 levels. The reduction in p27 was
not an indirect consequence of cell cycle progression as inhibition of cell proliferation
through the expression of the cdkI p21 did not abrogate the Myc, Ras-induced loss of p27
expression (Leone et al., 1997). Thus, the synergy of Myc with Ras in the induction of
cyclin EIcdk2 activity is a pathway, which may explain how Myc and Ras can cooperate to
elicit full cellular transformation in primary and immortalized fibroblasts.
The studies of Alevizopoulos et al. suggested another possible explanation for this
cooperative relationship (.4levizopoulos et al., 1997). Activated Myc can overcome a p16-
mediated cell cycle block, suggesting that the function of Myc within the Myc, Ras
partnership may involve inhibiting the activity ofp16. This hypothesis is supported by the
observation that while Myc and Ras are required for the transformation of wildtype MEF
cultures, Ras alone can transform pl6-null MEFs. Therefore, the loss of pl6 expression
precludes the requirement for Myc in cellular transformation. Irrespective of the identity of
the cdkI whose activity must be silenced to effect full transformation, the primary function
of Myc appears to be the inhibition of the activity of growth inhibitors which hinder cell
proliferation, facilitating cell transformation.

The second category is based on the observation that expression of activated Myc
promotes both cell proliferation as well as apoptosis. The ability of Myc to successfully
promote tumour formation is dependent upon the abrogation of the Myc-induced apoptotk
pathway. This can be achieved in two ways: first, the activation of inhibitors of apoptosis
such as bcl-2, and secondly, mutations in genes downstream of Myc, such as p53, which
effect the apoptotic mechanism. Hence, it was not surprising to discover that mutations in
bcl-2 or p53 can cooperate with Myc to promote tumour progression (reviewed in Adams
and Cory, 1992). A third of primary Burkitt's lymphoma isolates and most of the derived
cell lines exhibit point mutations inp53 (Farrell et al., 1991; Gaidano et al., 1991; Wiman
et al., 1991). In addition,p53 mutations were identified in some pre B and B-lymphomas
in Ep-myc mice (reviewed in Adams and Cory, 1992). The evidence for an interaction
between myc and bcl-2 originated primarily from transgenic mouse studies. Many of the
spontaneous tumours which arose in bcl-2 transgenic mice contained a aberrant myc allele
(McDonnell and Korsmeyer, 1991). In addition, crosses of Ep-myc and Ep-bcl-2 mice
produced progeny exhibiting an expanded population of pre B cells and rapidly succumbed
to B lymphoid malignancies (Strasser et al., 1990).
Both of these models are readily supported in experimental studies, indicating that
either pathway is equally likely and that the pathways are probably not mutually exclusive.
In addition, these observations support the view that multiple mechanisms can cooperate
with Myc to promote cell transformation. Yet what can we learn of the contribution of Myc
to transformation in both of these categories? In the first category, Myc activation will
overcome a G1 arrest resulting from Ras activation, allowing continued cell proliferation
and tumorigenesis. In the second category, Myc both enhances cell proliferation as well as
promotes apoptosis, thus there is an inherent suicide program within cells to inhibit Myc
activation from promoting the turnorigenic pathway. Activation of bcl-2 inhibits the Myc-
induced apoptotic program and permits Myc to drive cells through the cell cycle. Thus, in
both instances, we can hypothesize that the role of Myc is to confer enhanced proliferation,

which in conjunction with additional mutations facilitates tumorigenesis potentially via the
induction of genomic instability.
1.5 Mechanism of action
At a phenotypic level the biological effects of Myc expression are clear. Myc exerts
a strong proliferative impulse, inhibiting diverse growth arrest states such as quiescence,
senescence, or differentiation, and promoting cell proliferation. Yet at a molecular level, it
is not known how Myc executes these biological effects. It is known that Myc can bind to,
and influence the activity of components of the transcription machinery, as well as
companents of the cell cycle. In addition, Myc can activate as well as repress gene
transcription. Hence, Myc may utilize one or all of these activities to exert its strong
proliferative capacity. Further characterization of each of these Myc-dependent
mechanisms will help us to determine the importance of these pathways for Myc to drive
cells out of growth arrest and progess through the cell cycle.
1.5.1 Myc-interacting proteins
The identification of Max as a dimerization partner of Myc was a significant
breakthrough in the field of Myc research. It has contributed to our understanding of the
molecular mechanisms through which Myc effects its activities. Max was identified by
screening a human expression library with a radiolabeled carboxyl-terminal fragment of c-
Myc (Blackwood and Eisenman, 1991). This discovery was rapidly followed by the
identification of a murine homologue, nyn (Prendergast et al., 1991). The Max protein is
highly stable with a half-life of greater than 14 hours, and its expression is invariant
throughout all phases of the cell cycle (Berberich et al., 1992; Blackwood and Eisenman,
1992). There are two predominant isoforms of Max, resulting from alternative splicing,

p2 1 Max (1 51 amino acids) and p22 Max (160 amino acids) (Blackwood and Eisenman,
1991). They differ only in the presence of a 9 amino acid insertion amino-terminal to the
basic region. The reason for the two isoforms is unknown, as both proteins are
equivalently and ubiquitously expressed. Both isoforms bind to Myc with equal affinity
and facilitate the binding of Myc to DNA. The structure of Max is highly analogous to the
carboxyl-terminus of Myc. Max possesses a basic region, followed by HLH and LZ
domains; however unlike Myc, the NLS is located downstream of the LZ (reviewed in
Henriksson and Luscher, 1996). Alternate splicing is also responsible for additional
transcripts. These isoforms include AMax proteins p16 and p17 which lack the carboxyl
terminal NLS. The importance of these proteins is unclear as the respective endogenous
proteins remain to be isolated (Makela et al., 1992; Vastrik et al., 1995). In addition, there
is also a dMax variant, which has been identified in cell extracts. dMax lacks the basic
region and helix 1 and the loop of the helix-loop-helix domain (Arsura et al., 1995).
Ectopic expression of the AMax and dMax variants revealed that they are capable of
binding with high affinity to Myc, but MycIdMax heterodimers do not bind to DNA.
Thus, the dMax isoform may serve as an inhibitor of Myc function. Both Myc/Max and
MadMax dimers bind the same consensus DNA element, CACGTG. Some studies have
reported that binding is influenced by the nature of the sequences flanking the core.
MycIMax complexes appear to bind DNA with higher affinity compared to MadMax
homodimers, possibly due to the phosphorylation state of the MadMax homodimer
(Berberich and Cole, 1992).
Co-immunoprecipitation studies have demonstrated that Max is the predominant
binding partner of Myc. However, Myc has been reported to bind to a host of additional
proteins in vivo, including the transcription factors Yin-Yang-1 (YYl), AP-2, and Miz-1,
components of the basal transcription machinery, such as the TATA-binding protein
(TBP), regulators of the cell cycle such as p107, the putative tumour suppressor Bin-1, the
transformation-associated protein TR-AP, as well as cell structural elements such as a-

tubulin. Myc is also reported to bind in virro to TFII-I, a transcriptional initiator (Figure
1.5B) (reviewed in Henriksson and Luscher, 1996; Lemaitre et al., 1996).
YY-1, is a zinc-finger containing transcriptional regulator which regulates a number
of genes, inducing, repressing or initiating gene expression depending upon the context. A
search for proteins which interact with YY-1 using the yeast two hybrid approach yielded
c-Myc (Shrivastava et al., 1993; Shrivastava et al., 1996). YY-1 interacts with the
carboxyl terminus of cMyc, amino acids 250 to 439. As this region is also required for
binding to Max, the interaction of YY-1 or Max to c-Myc is mutually exclusive. Although
this interaction does not prevent YY-1 from binding to DNA, it inhibits the transcriptional
regulatory activity of YY-1, suggesting a DNA-independent mode of Myc action
(Shrivastava et al., 1993).
The carboxyl terminus of the c-Myc protein is also required for binding to the
transcription factor AP-2 (Gaubatz et al., 1995). Unlike YY-1, the interaction of AP-2
with Myc does not preclude Max binding; however, the AP-UMyc complex is incapable of
binding to DNA. Moreover, in some promoters, the Myc E box and the AP-2 binding site
overlap such as in the a-pr.othyrnosirz gene, and ectopic AP-2 expression can inhibit
MycIMax binding to DNA and consequently Myc-dependent transactivation. Hence the
interaction of Myc with AP2 results in either a direct or indirect inhibition of Myc
transcriptional activation function.
Miz-1, is a novel PO2 domain-containing protein which binds to the initiator in the
cycliit Dl and adenovims major late promoters (AdMLP), inducing transcription. Miz-1
was identified via a yeast two-hybrid approach, as a protein which can bind to the HLH
domain of c-Myc and not to Max or USF (Schneider et al., 1997). The association of Myc
with Miz-1 inhibits the transcription initiating activity of Miz-1, suggesting another
mechanism through which Myc may negatively regulate gene transcription. Interestingly,
ectopic expression of Miz-1 results in growth arrest and; as such, its activities are logical
candidates for Myc repression.

TFII-I is another example of an initiator-binding protein whose activity is inhibited
by Myc (Roy et al., 1993). TFII-I fulfills a unique role in transcription regulation; as a
component of the basal transcription machinery: TFII-I initiates transcription of TATA-less
promoters which possess initiator sites. In vitro, Myc HLH and LZ domains mediate the
interaction with TFII-I, and are responsible for the inhibition of TFII-I-dependent
transcriptional initiation. The recent cloning and characterization of TFII-I will allow an in-
depth characterization of the activities of this transcriptional regulator and permit an
analysis of the importance of the MycmFII-I interaction within the context of Myc function
and regulation. Myc has also been shown to interact with another component of the basal
transcription machinery. The amino-terminal transactivation domain of Myc can interact
with the TATA-binding protein (TBP), a subunit of the basal transcription factor, TFII-D,
in vivo and in vifro (Hateboer et al., 1993; Maheswaran et al., 1994). This interaction
substantiates a role for Myc in the regulation of transcription at core regulatory elements.
Myc can interact with p107, a member of the retinoblastoma susceptibility family of
tumour suppressors, in vivo and in vitro (Beijersbergen et al., 1994; Gu et al., 1994;
Hoang et al., 1995). p107 is a strong growth inhibitor and when ectopically expressed in
cultured cells, will induce growth arrest. The interaction between these two proteins is
mediated through the pocket region of p107 and amino acids 45 to 103 of Myc. The
binding of plO7 to Myc facilitates the formation of a quaternary protein complex consisting
of Myc, p107, and cyclin Ncdk2 (Gu et al., 1994; Hoang et al., 1995). The recruitment
of cyclin Ncdk2 to Myc facilitates the phosphorylation of Myc, hindering the ability of
Myc to transactivate gene expression. In support of this model, while p107 can bind to
both wildtype and mutant c-Myc proteins from Burkitt's lymphoma, the mutant c-Myc
proteins are not phosphorylated by cyclin Ncdk2, and p107 does not inhibit the
transactivation activity of these mutant Myc proteins. Thus, this correlative evidence
suggests that transcriptional activation by Myc and Myc-mediated transformation may be
linked, and that the phosphorylation of Myc by cyclin Ncdk2 appears to be a mechanism

through which Myc transcriptional activation function, and hence the tumorigenic potential
of Myc, is negatively regulated.
Bin-1, a novel Myc-interacting protein was isolated by screening a murine
embryonic cDNA library using the conserved Myc box I of Myc (Sakamuro et al., 1996).
Both MBI and MBII are required for binding to Myc. Bin-1 is a putative tumour
suppressor, its expression is absent in a variety of tumour cells, and ectopic Bin-1
expression antagonizes MycRas-dependent transformation (Sakamuro et al., 1996).
Consistent with its apparent role as a negative regulator of cell growth, the expression of
Bin-1 is induced upon the differentiation of C2C12 myoblasts (Wechsler-Reya et al.,
1998). Depending on the context, Bin-1 can inhibit Myc transactivation. Bin-1 can
abrogate the Myc-dependent induction of the omithine decarboxylase promoter, but is
ineffective against a promoter construct consisting of four CACGTGs upstream of a
chloramphenicol acetyltransferase (CAT) reporter gene. Thus, Bin-l may antagonize Myc
function by modulating the ability of Myc to regulate gene expression.
TR-AP, was identified as a Myc-binding protein via biochemical means (Brough et
al., 1995). TR-AP binds to the MBII region of Myc. As this region is also required for
Myc-mediated cellular transformation, TR-AP may play a role in effecting or enhancing the
tumorigenic activities of Myc. Indeed, this hypothesis was substantiated by studies which
demonstrate that dominant negative mutants of TR-AP will inhibit Myc-induced
transformation. It is not known how TR-AP functions. The carboxyl-terminus contains a
domain found in the phosphoinositol3' kinase (PI3K) family; however, TR-AP lacks the
catalytic kinase domain, hence it may not exhibit kinase activity. The model currently
under investigation proposes that TR-AP may function as a molecular scaffold recmiting
additional proteins which will execute the biochemical directives of Myc.
Thus, in addition to Max there are a host of Myc interacting proteins, which either
facilitate or impair Myc function. Miz-1, TBP, TR-AP, and TFII-I serve to enhance or
effect the activities of Myc; while AP2, YY-1, p107, and Bin-1 may antagonize Myc

function. The importance of each of these interactions within the context of the execution
of the biological activities of Myc is unknown, as the characterization of these novel
regulatory pathways is still in its infancy. Yet, these pathways may be the key to
unraveling the complexities that surround Myc hnction and regulation.
1.5.2 Transcriptional activation
One avenue of research which has been the primary focus for a number of
researchers in this field, involves determining how Myc can execute its biological functions
by regulating gene transcription. The evidence to support a role for Myc as a regulator of
gene transcription is considerable as well as convincing. This wealth of substantiation can
be attributed primarily to the simple fact that more effort has been devoted to this venture,
compared to any other, rather than a testament to the validity or importance of this
mechanism.
Structural analysis of the c-Myc protein revealed motifs commonly associated with
well characterized transcription factors, suggesting that Myc may function as a
transcriptional regulator (reviewed in Lemaitre et al., 1996). A number of studies support
this notion. First, Myc together with Max (Blackwood and Eisenman, 1991; Blackwood
and Eisenman, 1992; Blackwood et al., 1992; Blackwood et al., 1992; Prendergast et al.,
1991), can bind specifically to DNA at a consensus E-box element, CACGTG, as well as
to other related noncanonical sites (Blackwell et al., 1993; Fisher et al., 1993; Grandori et
al., 1996; Prendergast et al., 1991; Solomon et al., 1993). Second, in vitro transcription
assays using the Myc binding site linked in tandem to a reporter gene, have demonstrated
that Myc can activate transcription of the reporter gene upon binding to the Myc binding
site (Amati et al., 1992; Amin et al., 1993; Gu et al., 1993; Ham et al., 1994; Kretzner et
al., 1992; Kretzner et al., 1992). Third, a relatively small number of genes have been
identified as being transcriptionally induced in response to Myc activation (Bello-Fernandez

and Cleveland, 1992; Bello-Femandez et al., 1993; Benvenisty et al., 1992; Galaktionov et
al., 1996; Miltenberger et al., 1995; Rosenwald et al., 1993; Sears et al., 1997; Wagner et
al., 1993). Therefore, it is clearly evident that Myc can activate gene expression at a
transcriptional level. This led to the hypothesis that Myc promotes cell proliferation by
inducing transcription of genes which either promote or are required for progression
through the cell cycle.
The list of reported Myc-induced genes is small but contains a number of genes
whose cellular function may explain how Myc promotes cell growth (Figure 1.7). Myc-
regulated genes can be divided into distinct categories on the basis of their physiological
function. There are a subset of genes which are required for progression into S phase
consisting of ornithine decarboxylase (odc), the rate-limiting enzyme for polyamine
biosynthesis (Bello-Femandez and Cleveland, 1992; Bello-Femandez et al., 1993; Wagner
et al., 1993), and carbamoyl-phosphate syntltase/aspartate carbamoyl
transferase/dihydroorotase (cad), necessary for pyrimidine biosynthesis (Miltenberger et
al., 1995). E2F-2 (Sears et al., 1997), is required for the expression of genes such as
cyclin E, thymidim kinase, as well as the additional members of the E2F family of
proteins. A G1-specific phosphatase, cdc25A, activates cdk kinase activity (Galaktionov et
al., 1996). Other genes are involved in protein translation, such as eIF-4E and eIF-Za,
which encode enzymes that are involved in translation initiation (Rosenwald et al., 1993).
A DEAD box-related gene (MrDb), is a potential RNA helicase and mediator of RNA
stability (Grandori et al., 1996). Lactate dehydrogenase gened (LDH-A), which
functions in anaerobic glycolysis, is also a target of Myc (Shim et al., 1997). Lastly, Myc
has been reported to regulate genes of uncertain function: a-protl~ymosin (Desbarats et
al., 1996; Eilers et al., 1991; Gaubatz et al., 1994), and ECA39 (Benvenisty et al., 1992).
Both odc and c a d catalyze essential biosynthetic reactions producing
macromolecules which are required for DNA replication. The transcription factor E2F-2,
drives the expression of a host of genes whose expression is required for progression

odc + Polyamine biosynthesis \ /
DNA Replication cad -+ Pyrimidine biosynthesis
E2F-2 -- Gl and S phase genes A / Cell cycle progression ---8-
cdc25A + cdk activation
eIF-2a \ Initiation of protein translation =-
eIF-4E
MrDb -+ mRNA stability and transport m
LDH-A - Anaerobic glycolysis - Gmwth in hyporic conditions*
Figure 1.7. Possible mechanisms for the role of Myc in driving cell cycle progression, based on the induction of the indicated target genes bv Mvc. Abbreviations used: odc, omithine decarbox&G; cad, & r b ~ m ~ ~ l - ~ h o s ~ h a t c synthaselaspartate carbamoyl transferaseldihydroorotase; cdk, cyclin-dependent kinase; LDH-A, lactate
Cell Growth

through late G1 and S phase. Cdc25A is an intriguing target for Myc regulation, since it is
active in late G1 phase of the cell cycle and is required for transit from G1 to S phase.
Cdc25A may activate cdk activity via either of two mechanisms: first, it is a phosphatase
which can activate cdks by removing inhibitory phosphate groups (Jinno et al., 1994);
second, cdc25A can compete with the cdkI p21 for binding to cyclin E and A in vitro,
thereby abrogating the inhibitory effects of p21, permitting cell cycle progression (Saha et
al., 1997). EIF-4E, a translation initiation factor which recognizes the cap structure at the
5' end of rnRNAs, has been demonstrated to enhance specifically the translation of cyclin
Dl. Hence, this mechanism represents one mode via which Myc promotes cyclin Dl
expression. Thus, Myc induces a diverse array of genes whose finctions impact on a
number of proliferative systems. Myc promotes cyclin/cdk activation by enhancing the
expression or activation of cyclins, and Myc also induces genes which supply essential
reagents for the process of DNA replication. In addi.tion, Myc can elevate the expression
of growth promoting genes through the induction of eIF-4E and MrDb. All of these Myc-
initiated activities make a significant contribution to the progression of cells from GOIGl to
S phase of the cell cycle, enhancing cell proliferation.
Analysis of the promoters of these Myc target genes has provided further evidence
that Myc can induce the transcription of these genes. It was generally assumed that Myc
served a critical role in the induction of these genes upon serum stimulation, because of the
overwhelming evidence supporting a role for Myc in the exit from quiescence upon
mitogen stimulation. This is true for most of the induced genes; however, analysis of the
regulation of odc by Myc has refined and expanded this perception of its role in the
promotion of cell cycle progression. Packham et al. analyzed the role of Myc in the
induction of odc in quiescent murine myeloid progenitor cells, following stimulation with
the mitogen IL-3 (Packham and Cleveland, 1997). They reported that the transcriptional
regulation of ode occurs in two sequential stages. The first stage encompasses the initial
induction in odc transcription following IL-3 stimulation; it is effected via a pathway which

is independent of Myc and de novo protein synthesis. The second stage entails the
maintenance of odc expression throughout the cell cycle and is dependent upon Myc
activity and protein synthesis. In support of this model, inducible expression of a
dominant negative mutant of c-Myc inhibited the delayed second stage in odc expression
but did not affect the induction of odc following exposwe to IL-3. Thus, while Myc is
required for the transcriptional induction of odc, Myc appears to be responsible for the
maintenance of gene expression rather than the mitogen-stimulated transient activation of
gene transcription (Packham and Cleveland, 1997). This result proposes a novel role for
Myc, and prompts the question of whether the other Myc-induced genes are similarly
regulated.
1.5.3 Transcriptional repression
Recent studies have demonstrated that Myc can repress gene transcription, in
addition to activating gene transcription (Antonson et al., 1995; Facchini et al., 1997;
Inghirami et al., 1990; Jansen-Dun et al., 1993; Lee et al., 1997; Li et al., 1994; Marhin et
al., 1997; Penn et al., 1990; Philipp et al., 1994; Prendergast et al., 1990; Tikhonenko et
al., 1996; Versteeg et al., 1988). Characterization of the functions of the genes which are
repressed by Myc suggests that these Myc-mediated repression activities may play an
important role in the ability of Myc to control normal cell proliferation and consequently
tumorigenesis. In comparison with Myc-mediated transcriptional activation, Myc
repression is less well understood. Repression appears to be mediated through core
regulatory elements, and unlike Myc transactivation, repres~ion is not dependent upon the
presence of the conserved CACGTG within the gene. For some genes, repression appears
to be effected through the initiator element (Inr), where Myc is believed to inhibit the
binding and transcriptional activity of proteins which interact with the Inr. Two candidate
Inr-binding proteins whose activity has been reported to inhibited by Myc are TFII-I and

Miz-1. More than one mechanism of suppression may be orchestrated by Myc as
heterodimerization with its protein partner Max is dispensable for Myc repression of cyclin
Dl, yet is required for Myc autosuppression (Facchini et al., 1997; Philipp et al., 1994).
Indeed, structure/function analysis of the transcriptional regulatory domain of the Myc
protein has suggested that the regions which are important for Myc activation and
repression are separable. A few targets for Myc repression have been identified. Myc can
repress debpa (Antonson et al., 1995; Constance et al., 1996; Li et al., 1994; Mink et al.,
1996), cyclitl DI (Philipp et al., 1994), the adenovims 5 major-late promoter (AdMLP) (Li
et al., 1994), and c-myc (Facchini et al., 1997) promoter activities.
The importance of d e b p a to the differentiation program in some cell systems
makes it a logical candidate for Myc repression. Although it is clear that Myc can repress
the activity of this differentiation-specific transcriptional regulator, the precise mechanism
through which repression is effected is controversial. Using a transient transfection
approach in the immortalized cell line NIH3T3, it was demonstrated that Myc represses
transcription via a mechanism which is dependent upon the initiator element in the basal
promoter (Li et al., 1994). Repression requires both the MBII region and the B-HLH-LZ
of c-Myc. These domains coincide with the domains of Myc which are crucial to Myc-
dependent cell transformation, suggesting that Myc repression and Myc-mediated
transformation may be linked. In HIB-lB, a murine brown adipocyte cell line, ectopic
Myc expression did repress the transcription of debpa promoter reporter constructs;
however, mutation of the initiator element did not abolish the repression. Myc-dependent
repression appeared to be mediated through the core promoter, potentially through
interactions with the basal transcription machinely (Antonson et al., 1995). These
conflicting observations may be due to differences in cell type, or methodology, and need
to be further explored.
The relationship between Myc and cyclin Dl is complex, as in primary MEF
cultures, ectopic Myc expression does not regulate cyclin Dl expression. Yet, in some

immortalized fibroblasts, Myc can repress cyclin Dl expression, depending on the cellular
context. The precise nature of the discrepancy between primary cell cultures and
immortalized fibroblasts which facilitate the repression of cyclin Dl by Myc is unknown.
In sensitive cell lines, the repression of transcription of the cyclin Dl promoter by Myc
occurs via a mechanism which is dependent on the initiator site. Myc is suggested to effect
repression via its interaction with initiator-binding proteins such as Miz-1, and TFII-I,
thereby inhibiting the transactivation activities of these transcriptional initiators. The
domains in the human c-Myc protein which are required for the repression of cyclin Dl
mapped to amino acids 92 to 106, the carboxyl terminal DNA binding and protein
dimerization motifs. These regions do not coincide with the domains of Myc which are
crucial to cellular transformation. Hence, the relevance of cyclin Dl repression by Myc, to
the Myc turnorigenic program is questionable.
The regulation of the AdMLP by Myc is complex. The AdMLP possesses two
CACGTG motifs as well as an initiator site. In transient promoter reporter assays uing a
AdMLP-driven luciferase reporter construct, expression of low concentrations of ectopic
Myc protein results in the activation of transcription of the luciferase reporter. Myc
transactivation is dependent upon the consensus E boxes. Expression at high Myc
concentrations repressed transcription via a mechanism dependent upon the initiator.
Repression may be mediated through the interaction of Myc with the initiator-binding
protein, Miz-1, although this has yet to be demonstrated (Schneider et al., 1997).
The c-myc gene is one of the better characterized Myc-repressed target genes. C-
myc auto-suppression is evident in all primary and some immortalized cell lines. It is a
homeostatic regulatory mechanism which results in the repression of endogenous c-myc
expression at the level of transcription initiation. This feedback mechanism functions in a
Myc dose-dependent manner, and requires the interaction of Myc with Max (Facchini et
al., 1997; Grignani et al., 1990; Pem et al., 1990; Penn et al., 1990). Although there is an
initiator in the c-myc basal promoter, mutational analysis clearly demonstrated that it is not

required for Myc repression. Evidence to date suggests Myc represses gene transcription
through core regulatory elements via a mechanism which does not require the presence of
previously identified c-myc transcription regulatory sites (Antonson et al., 1995; Facchini
et al., 1997; Li et al., 1994; Lucas et al., 1993; Philipp et al., 1994; Roy et al., 1993).
The analysis of these few Myc-repressed genes has hinted at the potential
importance of Myc repression in ow understanding of Myc function. The identification
and analysis of additional Myc-repressed genes is essential to aid our understanding of the
mechanisms of Myc repression, and the significance of Myc repression in the context of
the biological activities of Myc.
1.6 The emerging significance of Myc repression
1.6.1 Correlation of repression with transformation
What is the significance of Myc repression of gene expression? R i s is a difficult
question given the small number of genes that have been identified as hlyc-repressed
targets. Characterization of the mechanisms through which these genes are regulated by
Myc has revealed that Myc repression may be as important as Myc transactivation in
effecting Myc function. Analysis of the Myc-mediated repression of the AdMLP
demonstrated that deletion of the MBII region of c-Myc will abrogate both transcription
repression, and the ability of Myc to cooperate with activated Ras, in a rat embryo
fibroblast transformation assay (Li et al., 1994; Stone et al., 1987). Importantly, this
mutant Myc protein can still transactivate, suggesting that Myc-mediated activation and
repression may be executed via distinct pathways (Li et al., 1994). This view was
substantiated through the characterization of mutant c-Myc proteins derived from Burkitt's
lymphoma cells (Lee et al., 1996). These mutants contained specific point mutations in the
Myc TAD, permitting a detailed analysis of this region to ascertain which residues in the c-

Myc protein were required for Myc-dependent transformation versus Myc activation or
repression. The results of this study suggested that Myc transactivation and Myc-mediated
repression are separable. Moreover, Myc repression seems to correlate with Myc-mediated
transformation (Lee et al., 1996; Li et al., 1994), suggesting that Myc repression of gene
transcription may serve an important role in Myc-dependent transformation. Further
evidence supporting the view that Myc transactivation was not sufficient to effect Myc
transformation arose in studies where the weak Myc TAD was replaced with the strong
TAD of the herpesvirus VP16 transcriptional activator (Brough et al., 1995). Although
this chimeric protein could transactivate, it was unable to repress gene transcription, nor
could it transform cells. Lastly, the domains required for Myc repression coincide with
those that are crucial to Myc transformation, supporting a role for Myc repression in
effecting cell transformation (Li et al., 1994; Penn et al., 1990; Philipp et al., 1994; Stone
et al., 1987).
1.6.2 Activation and Repression cooperate to promote cell proliferation
Given the potential importance of the mechanism of Myc repression in the
execution of the tumorigenic program, the conventional model depicting the role of Myc in
normal cell proliferation and tumorigenesis may need to be broadened. The ability of Myc
to promote cell proliferation may require the cooperation of Myc repression as well as
activation. For example, Myc may suppress the expression or activity of the products of
growth arrest genes, driving cells out of growth arrest. Myc transactivation will serve to
promote the progression through the GI phase into the DNA replicative phase, through the
induction of genes which are required for the passage through the cell cycle. Thus, both
pathways, Myc activation and Myc repression, are most probably essential for Myc-
mediated cell cycle progression, and tumorigenesis.

1.7 Objectives
Our appreciation of the potential role of Myc repression in facilitating enhanced cell
proliferation and tumorigenesis is hampered by the small number of Myc-repressed genes
that have been identified. The identification and characterization of additional genes which
are repressed by Myc will help us to understand both the mechanism of Myc-mediated gene
repression and how the repression of genes by Myc can stimulate cell proliferation and
ultimately promote tumour progression. To approach this objective, we attempted to
resolve the controversy surrounding the relationship between Myc, pRb, and cyclin Dl, by
analyzing the expression of cyclin Dl in response to Myc activation andlor pRb loss in
primary cell cultures. In addition, we have identified and characterized two genes which
are repressed in response to Myc activation: the PDGF-jR gene, and the growth arrest
gene, gadd45.
In a variety of tumour cell lines and selected immortalized cell lines, studies have
revealed a number of novel regulatory relationships among three key cell cycle regulators:
Myc, pRb, and cyclin Dl. For example, pRb and cyclin Dl are reported to exist in a
negative feedback loop, a mechanism proposed to regulate normal cell proliferation. In
addition, Myc activation has been reported to either repress, promote, or exert no effect on
the expression of cyclin Dl (Daksis et al., 1994; Marhin et al., 1996; Philipp et al., 1994;
Solomon et al., 1995). Clearly, the immortalized nature of these cell lines has influenced
the analysis of these putative normal regulatory mechanisms. Therefore, to assay the
validity of these reported pathways, primary MEF cultures were analyzed. Neither Myc
activation nor pRb loss in primary MEF cultures exerted an effect on cyclin Dl expression.
Interestingly, ectopic Myc expression in MEFs which lack expression of the retinoblastoma
tumour suppressor protein (pRb) exhibit a loss of cyclin Dl protein expression,
concomitant with an increase in cellular growth rate and a tumorigenic phenotype. Cyclin
Dl loss is mediated at both the mRNA and protein level, arguing that the suppression of

cyclin Dl is an indirect consequence of the transformed nature of these cells. Ow results in
nontransformed cells clearly contradict earlier studies conducted in turnow cell lines, and
support a novel cross-regulatory mechanism between Myc, cyclin Dl, and pRb.
Moreover, our results suggest a novel role for cyclin Dl in tumorigenesis (Marhin et al.,
1996).
The PDGF-PR is an important mitogenic receptor which initiates signal
transduction pathways required for cells to exit quiescence and enter the cell cycle. The
interaction of PDGF-PR with its ligand PDGF induces c-Myc expression and cell
proliferation. Following transmission of the signal, the ligand receptor complex is
internalized, and receptor expression is repressed in a putative negative feedback
mechanism. Recent studies have revealed some candidates for this inhibitory pathway, the
GTPase Ras, and the nonreceptor tyrosine kinase src (Vaziri and Faller, 1995). The
components downstream of these signal transducars responsible for the repression ofpdgf-
pr mRNA expression were unknown. We demonstrated that Myc is a potential candidate
in this pathway. Indeed, the expression of the pdgf-pr gene is repressed at the
transcriptional level in response to Myc. The Myc-mediated suppression in PDGF-PRs on
the cell surface represents a putative pathway through which Myc is able to regulate the
proliferative response of a cell to mitogens in the extracellular environment (WM,
submitted).
The growth arrest gene gadd45, functions as a negative regulator of cell growth
keeping cells in a non-proliferative, growth-arrested state. Gadd45 expression is elevated
in response to a variety of growth arrest signals, including contact inhibition, mitogen
withdrawal, and exposure to growth inhibitory agents. We demonstrate that the
transcription of gadd45 is repressed by Myc and as such this repression mechanism defines
a novel pathway in which Myc promotes cell growth by inhibiting the expression of a
growth inhibitor, driving cells out of growth arrest (Marhin et al., 1997).

Identification of these genetic targets of Myc repression will allow us to understand
the mechanism(s) by which Myc represses gene expression, and has elaborated as well as
defined novel roles for Myc in the control of normal cell proliferation md the promotion of
tumorigenesis.

Chapter 2
LOSS OF RB AND MYC ACTIVATION CO-OPERATE TO
SUPPRESS CYCLIN Dl AND CONTRIBUTE TO
TRANSFORMATION
This chapter is a modified version of the following manuscript: Loss of Rb and Myc activation co-operate to suppress cyclin Dl and contribute to transformation. 1996. Marhin, W.W., Hei, Y.-J., Chen, S., Rang, Z., Gallie, B., Philips, R.A. and Pem,
L.Z. Oncogene, 12,43-52.

2.1 Abstract
Cyclin Dl can bind and phosphorylate the product @Rb) of the retinoblastoma gene
(RE-I) and recent evidence suggests pRb, in turn, may regulate cyclin Dl protein
expression. In transformed cell lines, loss of pRb activity strongly correlates with a
decrease in cyclin Dl protein expression, and conversely, introduction of pRb can induce
cyclin D l promoter activity. Yet we show pRb does not regulate cyclin Dl directly as
basal and serum-stimulated levels of cyclin Dl protein and kinase activity are similar in
wildtype and pRb-deficient primary mouse embryonic fibroblasts (MEFs). These
observations suggest that the suppression of cyclin Dl in pRb-minus turnour cell lines
requires loss of pRb in conjunction with at least one additional genetic event. We have
determined that constitutive, ectopic Myc expression in pRb-deficient, but not wildtype,
MEFs suppresses cyclin Dl protein expression and kinase activity. The suppression of
cyclin Dl protein expression is mediated by a post-transcriptional mechanism.
Phenotypically, pRb-deficient MEFs consistently exhibited a delayed growth response in
comparison to wildtype MEFs. This growth delay is abrogated in pRb-deficient MEFs
which are expressing ectopic Myc protein, coincident with the loss of cyclin Dl protein
expression. Moreover, these cells exhibit an increased proliferative capacity, and they no
longer show contact inhibition. Our results support a cross-regulatory mechanism between
Myc, pRb and cyclin Dl and suggest a novel role for cyclin Dl in tumorigenesis.

2 . 2 Introduction
The retinoblastoma susceptibility gene is mutated in a variety of tumors including,
retinoblastomas, small cell lung carcinomas and osteosarwmas, implicating the loss of pRb
function as a key contributor to hunorigenesis (reviewed in Goodrich and Lee, 1993; Wang
et al., 1994; Weinberg, 1995; Zacksenhaus et al., 1992). This view was fhrther
substantiated by studies which showed that introduction of pRb into pRb-deficient tumour
cell lines, arrested cells in G1 phase of the cell cycle (Ewen et al., 1993; Fung et al., 1993;
Goodrich and Lee, 1993; Hinds et al., 1992; Huang et al., 1988; Templeton et al., 1991).
pRb is thought to play a critical role in the control of normal cell proliferation by negatively
regulating the progression of cells into the S phase of the cell cycle (DeCaprio et al., 1989;
Ewen et al., 1993; Goodrich and Lee, 1993; Hinds et al., 1992). The growth suppressive
activity of pRb is controlled at the level of phosphorylation as pRb protein levels are
invariant throughout the cell cycle. The hypophosphorylated form of pRb in GO and early
GI phase of the cell cycle is functionally active. Phosphorylation on serine and threonine
residues, in mid-GI and then throughout S and G2 phases, leads to the inactivation of pRb
which is then dephosphorylated following mitosis (Buchkovich et al., 1989; Chen et al.,
1989; DeCaprio et al., 1992; Lees et al., 1991; Lin et al., 1991; Ludlow et al., 1993;
Ludlow et al., 1990; Mihara et al., 1989; Mittnacht et al., 1994). pRb may exert its growth
suppressive properties by interacting with and inacrivating at least some members of the
E2F family of transcriptional transactivators (Chellappan et al., 1991; Helin and Harlow,
1992; Kaelin et al., 1992; Nevins, 1992; Shirodkar et al., 1992). It is believed that
phosphorylation of pRb in late GI disrupts complex formation with E2F allowing
expression of a subset of EZF-regulated genes required for progression into S phase
(Hamel et al., 1992; Hiebert et al., 1992; Weintraub et al., 1992). Thus, phosphorylation
of pRb is key to pRb regulation and cell cycle progression.

Thct phosphorylation sites on the pRb protein are consensus recognition sites for
cyclins and their catalytic partners, cyclin dependent kinases (cdk). The kinetics of pRb
phosphorylation suggest that the G1 cyclins, particularly the D-type cyclins (Baldin et al.,
1993; Motokura et al., 1990; Sherr, 1993; Xiong et al., 1991) together with their cdk
partners (Bates et al., 1994; Kato et al., 1993; Meyerson and Harlow, 1994; Tam et al.,
1994), function as pRb kinases. Indeed, the D-type cyclins are able to interact with and
phosphorylate pRb in vitro at sites identical to those phosphorylated in vivo (Dowdy et al.,
1993; Ewen et al., 1993; Horton et al., 1995; Kato et al., 1993; Matsushime et al., 1994;
Meyerson and Harlow, 1994). Moreover, ectopic expression of D-cyclins can overcome
the proliferation block imposed by pRb and stimulate the cells to enter S phase (Dowdy et
al., 1993; Ewen et al., 1993; Hinds et al., 1992).
Cyclin Dl is a delayed early response gene which is transiently induced following
mitogen stimulation, peaking late in G1 phase of the cell cycle. In human and rodent
fibroblasts, cyclin Dl is essential for the G1 to S phase transition (Baldin et al., 1993;
Lukas et al., 1995; Lukas et al., 1994; Roussel et al., 1995; Tam et al., 1994), and it is
believed that cyclin Dl regulates this step through phosphorylation and inactivation of pRb
(Lukas et al., 1995). In addition, it was demonstrated that pRb can induce transcription of
the cyclin Dl gene (Muller et al., 1994). Therefore, a cross-regulatory pathway involving
cyclin D l and pRb can be envisaged. Hypophosphorylated pRb induces cyclin Dl
expression, and in turn cyclin Dl together with its associated cdk can phosphorylate and
thereby inactivate pRb. A correlation between loss of pRb function and absence of cyclin
Dl protein in human tumour cells further supports such a cross-regulatory mechanism
(Lukas et al., 1994; Muller et al., 1994; Tam et al., 1994), yet recent results show the
suppression of cyclin D l is not a direct effect of pRb (Lukas et al., 1995). Therefore, the
molecular mechanism involved in the regulatory interaction of pRb and cyclin Dl remains
unknown.

Cyclin Dl expression is highly influenced by the concentration of cellular Myc
protein; however, the pattern of cyclin Dl expression in response to Myc is complex
(Daksis et al., 1994; Jansen et al., 1985; Philipp et al., 1994; Rosenwald et al., 1993).
Myc is a key regulator of normal cell proliferation whose expression is required for cell
cycle progression (reviewed in Dang, 1991; Penn et al., 1990). In its activated form,
deregulated Myc expression confers a strong cellular self-renewal potential and is a
common feature of a diverse array of tumors (reviewed in Spencer and Groudine, 1991).
Daksis et al., showed that induction of Myc activity in quiescent non-transformed Rat-1
cells transiently induced cyclin Dl mRNA and protein expression, and promoted the
transition from GI to S phase of the cell cycle. This induction is rapid and occurs at the
transcriptional level in the absence of de novo protein synthesis, suggesting Myc can
directly induce cyclin Dl gene transcription (Daksis et al., 1994). The work of Rosenwald
et al. also shows Myc can up-regulate cyclin Dl protein expression in NIH 3T3 cells at the
translational level by inducing the rate-limiting eukaryotic initiation factor-4E (eIF-4E)
(Rosenwald et al., 1993). Experimentally, constitutive Myc expression can lead to cyclin
Dl suppression in some (Jansen et al., 1985; Philipp et al., 1994), but not other fibroblast
cell lines (Rosenwald et al., 1993); Daksis and Penn, unpublished results). Thus, Myc
induction results in the elevation of cyclin Dl expression, whereas the effect of
constitutively expressed, activated Myc on cyclin Dl expression remains unclear.
Myc, cyclin Dl and pRb are highly regulated proteins which function as effectors
of cell proliferation in a wide variety of cells. To ascertain whether regulatory pathways
linking these molecules exist in non-transformed cells, we analyzed cyclin Dl protein
expression in mouse embryonic fibroblasts (MEFs) where pRb protein expression has
been abrogated through targeted gene disruption (Jacks et al., 1992). Neither basal nor
mitogen-induced cyclin Dl protein was suppressed in the pRb-deficient MEFs, and cyclin
Dl protein showed kinase activity. Expression of ectopic Myc in pRb-deficient, but not in
wildtype, cells suppresses cyclin Dl protein expression as well as its associated kinase

activity. The suppression of cyclin Dl protein in MEFs can occur by regulatory
mechanisms involving either RNA or protein expression.

2.3 Materials and Methods
2.3.1 Cell culture
129 mice heterozygous for the RB-I gene were obtained fiom Jacks et al. (Jacks et
al., 1992). Primary mouse embryonic fibroblasts (MEF) were prepared fiom embryos at
day 12.5 of gestation and derived fibroblast cell lines were cultured in alpha modified
Eagle's medium (&EM) supplemented with 10% fetal bovine serum (FBS, Gibco/BRL),
100 pg/ml kanamycin (Sigma) and 2 pg/ml gentamicin (Roussel). Early passage cell
cultures were used for all assays. Unless otherwise stipulated all analyses were performed
on asynchronous, subconfluent, exponentially growing cells. Growth kinetics were
conducted by trypsinizing near-confluent cells, seeding 1 x 103 cells per well of a 24-well
dish (Nunc) in 10% FBSIaMEM, then trypsinizing and counting the number of cells per
well at the time points indicated. Each time point represents an average of three wells. All
cell cultures were assayed in at least two independent experiments.
2.3.2 Genomic DNA extraction and mouse embryonic fibroblast
genotyping
Genomic DNA was extracted fiom primary MEFs using the QIAamp Blood Kit
(Qiagen). Genotype of the MEFs was detected by PCR analysis of 1 ng of genomic DNA
for the presence of the wildtype and mutant RB-1 alleles with Amplitaq (Perkin-Elmer-
Cetus). Primer sequences: 5' primer RX3 [AAlTGCGGCCGCATCTGCATCmATCG
C] (within R E - I exon 3); 3' primers, for the wildtype R E - I allele R13
[CCCATGTTCGGTCCCTAG] (within RB-1 intron 3), and for the mutant allele PGK3'
[GAAGAACGAGATCAGCAG] (within the 3' pgk promoter in the pgk neomycin
cassette). The PCR was performed for thirty cycles, each cycle consisting of 94"C, 1 min,

5 8 T , 1 min, and 72"C, 1 min, and the PCR products were resolved on a 1.5% agarose
gel.
2.3.3 Retroviral infection
The pBabeIpuro retroviral vector has been described previously (Morgenstem and
Land, 1990). The pBabelv-mycpuro vector was constructed by subcloning the 3 Kb Bgl I1
v-gag-myc fragment from the pRasImyc 9 retroviral vector (kind gift of Tim Thompson)
(Thompson et al., 1989) into the Barn HI site of the pBabe1puro plasmid. To produce
infectious replication-defective ecotropic retroviral particles, recombinant retroviral
constructs were trnnsfected, using calcium phosphate precipitation (Graham and Van der
Eb, 1973), into GP+E packaging cell lines (Markowitz et al., 1988), and selected in 2
y d m l puromycin (Sigma) or 150 ydml hygromycin B (Sigma). Drug resistant clones
were pooled and expanded for virus production. Primary MEFs were subsequently
infected with retrovirus and selected in either puromycin or hygromycin B. Individual
drug-resistant colonies were isolated and the remaining colonies combined, generating
pooled populations.
2.3 .4 RNase protection
RNA was prepared by the guanidinium isothiocyanate method (Chirgwin et al.,
1979) and assayed by RNase protection (Penn et al., 1990) essentially as described. The
probes were generated using T3 RNA polymerase (Stratagene) from linearized Bluescript
KS and SK cloning vectors (Stratagene) containing the following DNA fragments: rat c-
myc exon 1 (Penn et al., 1990); the gag gene derived from the avian myelocytomatosis
virus MC29 (Penn et al., 1990); and the murine glyceraldehyde-3-phosphate
dehydrogenase (gavdh) cDNA (Facchini et al., 1994). The probe for the mouse cyclin Dl

gene was transcribed using Sp6 RNA polymerase (Stratagene) from a linearized
pGEM7zf+ vector (Promega) containing the full-length murine cyclin Dl cDNA (kind gift
of M. Kiess and P. Hamel). The protected probes were resolved by electrophoresis on
6% denaturing polyacrylamide gels and visualized by autoradiography on X-OMAT film
(Kodak).
2.3.5 Immunoblotting and immunoprecipitation
Antibodies used in this study include the monoclonal antibodies: anti-cyclin Dl
HDI (kind gift of E. Lees and E. Harlow), anti-Rb 14001A (Pharmingen), and a
polyclonal pan-myc antibody (kind gift of G. Evan). Total cell lysates (5 x 105 cells) were
immunoblotted as previously described (Daksis et al., 1994). Cyclin Dl, v-Myc, and pRb
protein were detected by incubating the membrane with their respective antibody for 1 h in
20 mM Tris pH 7.6, 137 mM NaCI, 0.05 % Tween 20 (TBS-T) containing 1% non-fat
milk. The membrane was washed, and incubated with a 112000 dilution of either
horseradish peroxidase-conjugated goat anti-mouse IgG (Biorad) or swine anti-rabbit IgG
antibody (Dako) in TBS-T containing 1% non-fat milk for 0.5 h. After washing, signals
were detected using the ECL (Amersham) detection system. As a positive control for
cyclin Dl protein, we used whole cell extracts of confluent, quiescent BALBlc 3T3 cells
stimulated with serum for 10 h. (Lanahan et al., 1992). Immunoprecipitations using the
anti-pRb monoclonal antibody (14001A) were performed as described (Hamel et al., 1990)
and immune complexes were resolved on a 7.5% SDSlacrylamide gel.
2.3.6 In vitro kinase assay
Total cell lysates were prepared and in vitro kinase assays were performed as
previously described (Matsushime et al., 1994), except lysates were sonicated at 4 "C (full

microtip power 3 times for 15 secs each). Lysates were clarified by centrifugation at
13,000 rpm for 13 min., and cyclin Dl complexes were immunoprecipitated for 1 h at 4 'C
with 2 pg of Dl-72-13G, a monoclonal antibody specific for murine cyclin Dl (Santa
Cruz). Full-length pRb protein (2 ug; QED Adv. Res. Tech.) was used as substrate in the
kinase assays. Samples were boiled in Laemmli sample buffer and resolved on a 7.5%
SDSlacrylamide gel. Phosphorylated proteins were detected by autoradiogaphy.

2 . 4 Results
2.4 .1 Primary mouse embryonic fibroblasts lacking pRb protein express
normal levels of cyclin Dl protein
In a number of transformed cell lines, loss of pRb function correlates with an
absence of cyclin Dl protein expression (Lukas et al., 1994; Tam et al., 1994). In
addition, it was previously demonstrated that pRb can induce the transcription from the
cyclit~ Dl promoter (Mullet et al., 1994). Taken together these results suggest the
existence of a pRbIcyclin Dl cross-regulatory mechanism. To ascertain whether this
regulatory mechanism is present in non-transformed cell lines, we analyzed cyclin Dl
protein expression in primary MEFs lacking pRb protein. MEF cultures were prepared
using embryos derived from mating mice in which one RE-1 allele was mutated through
targeted gene disruption (Jacks et al., 1992). We analyzed three independent litters and
from each litter, cultures were derived from one wildtype embryo (MEF 4, MEF 40, MEF
53), and its pRb-deficient littermate (MEF 1, MEF 37, and MEF 47). The genotype of
each culture was verified by PCR (Figure 2.1A) and confirmed by assaying the level of
endogenous pRb protein expressed (Figure 2.1B). Analysis of cyclin Dl protein
expression revealed the level of cyclin Dl protein expressed in pRb-deficient cells was not
suppressed, and was similar to that seen in wildtype MEFs (Figure 2.2A). To determine if
the cyclin Dl protein expressed in the pRb-deficient cultures was functionally active, we
performed in vitro kinase assays. Our results indicate that, irrespective of the RE-1 status,
all six cultures exhibited readily detectable levels of cyclin Dl associated kinase activity
(Figure 2.2B). As pRb has been shown to induce cyclin Dl promoter activity [Miiller,
1994 #925], we determined whether pRb protein was required for the induction of cyclin
Dl protein following mitogen-stimulation of confluent, quiescent cells (Baldin et al., 1993;
Daksis et al., 1994; Lanahan et al., 1992; Lukas et al., 1994; Won et al., 1992). Serum-

nnnnnn PRIMERSET wt A wt A wt A wt A wt A wt A
mutant - wildtype -
LITTER 1 2 3 b i
MEF 4 1 40 37 53 47
Figure 2.1. Determination of RB-I gene status and pRb protein expression in primary mouse embryonic fibroblast (MEF) cultures derived from 3 Litters of embryos produced from the mating of RB-I heterozygotes. (A) Screening of genomic DNA extracted from 6 mouse embryonic fibroblast cultures was performed by PCR analysis of the disruption of the RB-I gene through the insertion of the neomycin cassette in intron 3. The wildtype allele was distinguished from the mutant allele through the use of specific primer sets. RX3 and R13 for the wildtype allele generated a 380 bp amplicon, RX3 and PGK3' for the mutant allele produced a 400 bp amplicon. MEF 4, MEF 40, MEF 53 are homozygous for the wildtype RB-1 allele and MEF 1, MEF 37, MEF 47 are homozygous for the mutant RB-I allele. (B) Total cell lysates of each of the MEF cultures in (A) were immunoprecipitated with an anti-Rb monoclonal antibody (14001A), resolved on a 7.5 % SDSlpolyactylamide gel, transferred to PVDF membrane and immunoblotted with the pRb antibody. Equivalent amounts of protein were loaded in each lane as determined by Coomassie blue staining.

L I r n R 1 2 3 -nn
MEF C 4 1 40 37 53 47
LITTER 1 2 3
MEF 4 1 40 37 53 47
Figure 2.2. MEFs lacking pRb protein express cyelin D l protein and cyclin Dl associated kinase activity at levels comparable to their wildtype littermates. (A) Immunoblot analysis of total cell lysates prepared from primary MEFs using a monoclonal antibody specific to mouse cyclin Dl. Equivalent amounts of total cell extracts were resolved on a 7.5% SDSIpolyacrylamide gel, transferred to PVDF membrane, and cyclin Dl protein expression detected with the monoclonal antibody HD1. MEFs lacking pRb protein expression (MEF 1, MEF 37, MEF 47) exhibit significant levels of cyclin Dl protein compared to that of their wildtype littermates (MEF 4, MEF 40, MEF 53). (C= Positive control) (B) Whole cell extracts of asynchronously growing MEFs were immunoprecipitated with a monoclonal antibody specific for mouse cyclin Dl and tested for kinase activity. Significant phosphorylation of the pRb protein substrate by cyclin Dl associated kinase activity was evident in lysates from both wildtype MEFs and MEFs lacking pRb protein expression.

stimulation of confluent, quiescent wild-type and pRb-deficient MEFs induces cyclin D 1
protein (Figure 2.3) suggesting that mitogen-induced cyclin Dl expression does not require
pRb protein. Therefore, in contrast to studies performed in transformed cell lines and in
agreement with recent studies in non-transformed cells (Lukas et al., 1995), loss of pRb
function in primary MEFs does not affect cyclii Dl protein expression or kinase activity.
2.4 .2 Constitutive Myc expression suppresses cyclin D l protein in pRb-
deficient but not wild-type MEFs
A second genetic event resulting from the transformation process may act in
conjunction with the lack of pRb expression to cause the observed suppression of cyclin
Dl in tumour-derived cell lines (Lukas et al., 1994; Tam et al., 1994). As Myc can
suppress cyclin D l expression in BALBlc 3T3 cells, Myc was a strong candidate for this
second event. To investigate this hypothesis, we constitutively expressed ectopic Myc
protein in both wildtype MEF 4 cells and in cells of the corresponding mutant sibling MEF
1. MEF cultures were infected with either a control retrovirus carrying only the puromycin
resistance gene (pBabepuro) or with a retrovirus canying the ectopic v-myc gene @Babe/v-
myc puro). Following selection, individual clones were isolated and the remaining clones
combined to generate pooled populations. Two forms of cyclin Dl were consistently
detected in immunoblots, as previously reported (Matsushime et al., 1991). Both forms
are phosphoproteins, and the biochemical difference between them has not been
characterized. Cyclin Dl protein expression in wildtype MEF 4 cells was not affected by
expression of ectopic ivIyc protein (Figure 2.4A, compare lanes 5-8 with lanes 1-4). In
contrast, elevated expression of ectopic Myc in pRb-deficient MEF 1 cells (Figure 2.4A,
lanes 13-16) resulted in a significant suppression in cyclin Dl protein compared to MEF 1
cells not expressing ectopic Myc (Figure 2.4A, lanes 9-12). Results obtained with clonal
populations were consistent with those obtained with pools. Indeed, the dose dependent

Cydin Dl--) -0
1 2 3 4 5 6 7 8
Figure 2.3. Cycl i D l protein expression is induced in quiescent wildtype and pRb-deficient MEFs in response to serum-stimulation. Imrnunoblot analysis of cyclin Dl protein levels was performed using whole cell extracts prepared from confluent MEF cultures rendered quiescent in 0.1% FBSIuMEM for 3 days and then stimulated with 10% FBSIuMEM at the times indicated. Cyclin Dl protein was detected using a monoclonal antibody (HDI).

effect of ectopic Myc protein expression on the degree of cyclin Dl suppression in the
pRb-deficient MEFs is particularly striking (Figure 2.4A, lanes 13 - 16). Introduction of
activated Myc in two additional wild-type and pRb-deficient MEFs resulted in an identical
pattern of cyclin Dl expression (data not shown). The abundant number of colonies
observed following selection of control or Myc-containing retrovirus indicated that Myc
suppression of cyclin Dl protein in a pRb-minus cell is a directed molecular event, and not
a selected rare event. As a further control, we assayed the protein lysates of the pooled
populations for in vifro cyclin Dl kinase activity and found the level of kinase activity
directly rtilects the level of cyclin Dl protein expressed (Figure 2.4B). Thus, suppression
of cyclin Dl occurs in response to Myc-activation in pRb-deficient MEFs.
To determine whether the lack of cyclin Dl suppression in the wild-type MEFs was
due to insufficient ectopic Myc activity, the expression and activity level of exogenous Myc
was measured. Ectopic Myc expression was clearly evident in wildtype and pRb-deficient
MEFs as assayed at the RNA level by RNase protection (Figure 2.4C) and at the protein
level by western blot analysis (Figure 2.4A). To determine whether this ectopic v-Myc
protein was functionally active in the MEF cultures, we assayed for Myc negative auto-
regulation. Through this homeostatic feedback mechanism, elevated expression of ectopic
Myc will suppress the transcription of the endogenous c-myc gene (Cleveland et al., 1988;
Grignani et al., 1990; Pem et al., 1990). RNase protection analysis of endogenous c-myc
mRNA expression showed that expression of ectopic Myc protein in both wildtype (MEF
4) and pRb-deficient (MEF 1) MEFs (Figure 2.4C) suppressed endogenous c-myc mRNA
levels. These results suggest that the lack of suppression of cyclin Dl protein in wildtype
cells expressing ectopic Myc is likely not due to a lack of Myc activity in these cells.

Figure 2.4. Constitutive ectopic Myc expression in primary MEFs lacking
pRb protein suppresses cyclin D l protein expression and activity. (A) Clonal
and pooled (P) populations of wildtype (MEF 4) and pRb-deficient (MEF 1) MEFs
infected with control retrovirus (lanes 1-4 and 9-12) or with retrovirus carrying the v-ntyc
gene (lanes 5-8 and 13-16), were assayed by immunoblot for cyclin Dl and v-Myc protein
expression. The weak signal in the control lanes of the v-Myc blot represents a cross-
reactive band evident with this batch of pan myc antibody. Equivalent amounts of protein
were loaded in each lane as determined by Coomassie blue staining. (B) Whole cell
extracts of asynchronously growing MEFs were immunoprecipitated with a monoclonal
antibody specific for mouse cyclin Dl and tested for kinase activity. Phospholylation of
the pRb protein substrate by cyclin Dl associated kinase activity was significantly reduced
in lysates from pRb-deficient MEFs which are expressing ectopic Myc protein, consistent
with the reduction in cyclin Dl protein expression. (C) The ectopic v-Myc protein is
functionally active in both wildtype and mutant fibroblasts, as Myc negative autoregulation
is evident in both cell types. RNase protection analysis of endogenous c-myc, ectopic v-
myc, and gapdh mRNA expression in wildtype (MEF 4) and pRb-deficient (MEF 1)
MEFs, showed endogenous c-niyc mRNA was clearly evident in control cells (lanes 1-4
and 9-12) and suppressed in cells expressing ectopic Myc protein (lanes 5-8 and 13-16).
(P= Pooled populations).

MEF 4 (+I+) MEF 1 (-I-) 1 U
CONTROL VMYC CONTROL VMYC I
CLONES CLONES CLONES CLONES I---
1 4 6 P l 4 6 P l 5 6 P 2 4 5 P
Cyclin Dl-+ *
a.ry*l).
. .
MEF4 MEFl (+I+) (-I-)
eRb - substrate
MEF 4 (+I+) MEF 1 (-I-) I a
CONTROL VMYC CONTROL VMYC I I
CLONES CLONES CLONES CLONES I---
1 4 6 P l 4 6 P 1 5 6 P 2 4 5 P
v-myc +
Endogenous- c-myc

2.4 .3 The suppression of cyclin D l protein in MEFs can occur by
regulatory mechanisms controlling either RNA or protein
expression
The suppression of cyclin Dl protein levels in pRb-deficient cells expressing
ectopic Myc is highly consistent as an identical pattern of regulation was observed among
MEFs derived from independent litters (Figure 2.5A). To determine whether the Myc-
induced suppression of cyclin Dl protein in pRb-deficient cells is the result of a repression
of cyclin Dl mRNA levels, we assayed endogenous cyclin Dl mRNA expression using
RNase protection. For both wildtype MEF 4 and pRb-deficient MEF 1 cells, pooled
cultures expressing ectopic Myc protein were compared to controls lacking ectopic Myc
expression. Expression of ectopic Myc protein had no effect on cyclin Dl mRNA levels in
either wildtype MEF 4 (Figure 2.5B, compare lanes 1 and 2) or pRb-deficient MEF 1
(Figure 2SB, compare lanes 3 and 4). Similar results were observed in an additional Rb-
deficient MEF culture (data not shown). However, analysis of another pair of wildtype
and Rb-deficient MEF cells gave a different result. Introduction of ectopic Myc did not
affect cyclin Dl mRNA expression in wildtype MEF 40 cells (Figure 2SB, compare lanes
5 and 6), yet resulted in suppression of cyclin Dl mRNA levels in pRb-minus MEF37 cells
(Figure 2SB, compare lanes 7 and 8). Thus, in wildtype MEFs, ectopic expression of
Myc does not suppress cyclin Dl protein (Figure 2.4A and 2.5A), kinase activity (Figure
2.4B) or mRNA (Figure 2.5B). By contrast, in pRb-deficient MEFs, introduction of
activated Myc consistently suppressed cyclin Dl protein (Figure 2.4A and 2.5A) and
kinase activity (Figure 2.4B). This regulation is at the level of RNA in MEF 37 cells
(Figure 2.5B, lanes 7 and 8) and at the protein level in MEF 1 cells (Figure 2.5B, lanes 3
and 4).

MEF 4 MEF 1 MEF 40 MEF 37 (+I+) (-1-1 (+I+) (-1-1 --
v-myc - + - $ - + - +
MEF 4 MEF I MEF 40 MEF 37 (+I+) (-I-) (+I+) (-I-) n-
v-myc - + - + - + - +
Cgclin 111 w 1
v-myc + ?
Figure 2.5. Cycl i D l expression in Myc-activated, pRb-deficient MEFs is suppressed either at the RNA or protein level. (A) Pooled populations of wildtype (MEF4, MEF40) and pRb-deficient (MEF 1, MEF 37) MEFs infected with control retrovirus (lanes 1,3,5 and 7) or with retrovirus carrying the v-myc gene (lanes 2,4,6 and 8), were assayed by immunoblot for cyclin Dl and v-Myc protein expression. The weak signal in the control lanes of the v-Myc blot represents a cross-reactive band evident with this batch of pan myc antibody. Equivalent amounts of protein were loaded in each lane as determined by Coomassie blue staining. A longer exposure of lanes 1 and 2 are shown for clarity. (B) Total RNA was extracted from control (lanes 1,3,5 and 7) and Myc-expressing (lanes 2,4,6 and 8) asynchronous pooled populations of both wildtype (MEF 4: lanes 1 and 2), (MEF40: lanes 5 and 6) and pRb-deficient (MEF 1: lanes 3 and 4), (MEF 37: lanes 7 and 8) cells. RNA was analysed by RNase protection for the expression of the endogenous cyclin Dl and gapdh, as well as ectopic v-myc mRNAs.

2.4.4 pRb-deficient MEFs exhibit a delay in growth compared to
wildtype MEFs
To examine the effect of pRb expression on the growth of primary MEF cultures,
we compared the growth of subconfluent cultures of pRb-deficient MEFs with that of
wildtype MEFs. It was immediately evident that subconfluent MEF cultures lacking pRb
protein exhibited a delayed growth response of approximately 1 to 2 days compared to
wildtype cultures (Figure 2.6, compare Ato E). This result was verified with two
additional pairs of wildtype and pRb-deficient MEF cultures, as well as with their
respective earliest-passage parental cultures derived prior to retroviral infection and
selection (data not shown). The delay can be attributed primarily to an initial lag, as
growth rate and saturation density were not affected (data not shown). Expression of
ectopic Myc in the pRb-deficient cell culture (0) significantly alleviated the delay in their
growth response and resulted in proliferation comparable to both control (A) and Myc-
expressing (A) wildtype cultures (Figure 2.6). This Myc-abrogated growth delay also
correlated with a loss of contact inhibition (Figure 2.7). The lack of contact inhibition was
not evident in control (panel A and E) or Myc-expressing (panel B and F) wildtype MEFs,
nor in control pRb-deficient MEFs (panel C and G), but was clearly visible in pRb-
deficient, Myc-activated MEFS (panel D and H). In summary, these results show that
cyclin Dl expression in pRb-deficient primary cells correlates with a reduced proliferative
capacity compared to wildtype MEFs. In pRb-deficient cells ectopic Myc expression can
lead to down-regulation of cyclin Dl and an increased proliferative capacity. The nature of
the additional factors which co-operate with a loss of pRb and Myc-activation in the
transformation process is the subject of further investigation.

J
: + MEF 4 (+I+) CONTROL - MEF4(+I+)MYC
& MEF I (-/-) CONTROL
-4- MEF 1 (-1-1 MYC
I I I a I I 1 I
0 1 2 3 4 5 6 7 8 9
TIME (days)
Figure 2.6. Proliferation profiles of wildtype (MEF 4) and pRb-deficient MEFs (MEF I), infected with control retrovirus (CONTROL) or with retrovirus carrying a v-riyc gene (MYC). Triplicate cultures of MEF 4 CONTROL cells (A), MEF 4 MYC cells (A), MEF 1 CONTROL cells (P), and MEF 1 MYC cells (0) were plated sub con fluent!^ at 1000 cells per well in 10% FBSlcLMEM and daily cell counts were performed. Mean values of the triplicates, together with standard errors, are shown plotted against time.

Figure 2.7. pRb-deficient MEFs constitutively expressing ectopic Myc protein exhibit an increased proliferative capacity. Photomicromaphs of confluent MEF cultures maintained in 10% FBS/&EM for 3 dais. MEF 4 C O N T R ~ L @ ~ ~ ~ I A and E), MEF 4 MYC (panel B and F), MEF 1 CONTROL (panel C and G), and MEF 1 MYC (panel D and H). Photomicrographs are shown at 40X (A - D), and 100X (E - H) magnification. The arrowheads point to examples of cells which have lost contact-inhibition, forming dense, refkctile foci.

2.5 Discussion
Our results in primary MEFs contrast with those conducted in established cell lines
and clearly show that loss ofpRb or Myc-activation alone is insufficient to suppress cyclin
Dl expression; yet in collaboration, loss of pRb and Myc activation can suppress cyclin Dl
and contribute to the transformation process. Previous analyses of human tumow cell lines
originating from a variety of tissues, showed a strong positive correlation between loss of
pRb function and a decrease in cyclin Dl protein expression (Lukas et al., 1994; Muller et
al., 1994; Tam et al., 1994). Under these conditions the residual cyclin Dl protein could
not associate with cdk4, its predominant catalytic partner in fibroblasts, resulting in loss of
cyclin Dl-associated kinase activity (Parry et al., 1995). In contrast, we have shown that
pRb-deficient MEFs express cyclin Dl protein and kinase activity further supporting Lukas
et al. (Lukas et al., 1995). The results presented here clearly show the down-regulation of
cyclin Dl expression and activity in transformed cells is complex and not dependent solely
on pRb. Downregulation of cyclin Dl activity likely depends upon other genetic changes
in addition to the loss of pRb function.
We reasoned that the additional transforming event may involve expression of the c-
myc proto-oncogene, as it is commonly activated in tumorigenesis and has recently been
shown to play a role in cyclin Dl regulation. Induction of Myc results in the rapid
elevation of cyclin Dl protein through transcriptional (Daksis et al., 1994) or post-
transcriptional (Rosenwald et al., 1993) mechanisms. Constitutive expression of Myc can
suppress cyclin Dl mRNA and protein through a transcriptional repression mechanism in
some (Jansen-Diirr et al., 1993; Philipp et al., 1994) but not other cell lines (Rosenwald et
al., 1993; Daksis and Penn, unpublished results). Ectopic expression of Myc in primary
MEFs, as shown in this thesis, does not result in a concomitant decrease in cyclin Dl
expression. Insufficient exogenous Myc activity cannot account for the apparent lack of
cyclin Dl suppression as exogenous Myc can trigger Myc negative autoregulation and

suppress endogenous MEF c-myc expression. Thus, in primary MEFs, constitutive
expression of Myc alone does not lead to the suppression of cyclin Dl expression or
activity.
To test whether the suppression in cyclin Dl observed in pRb-negative tumour cells
results from cooperative interaction between Myc-activation and pRb loss, we measured
their combined effect in early passage pr imw MEFs. Indeed, constitutive expression of
ectopic Myc protein in pRb-deficient, but not in wildtype MEFs, led to a suppression of
both cyclin Dl protein and its associated kinase activity. Thus, a second genetic event is
required in conjunction with the loss of pRb function to suppress cyclin Dl expression in
MEFs. It remains unclear whether Myc-activation is the only second transforming event
which can co-operate with pRb-inactivation to suppress cyclin Dl expression and whether
Myc-activation per se is evident in all or a limited subset of pRb-minus tumour cells.
Interestingly, Squire er a/. showed amplification of N-myc in retinoblastoma tumors,
demonstrating that this cooperativity can exist iiz vivo (Squire et al., 1986).
Cyclin Dl is a highly regulated molecule whose expression can be affected at both the
transcriptional and post-transcriptional levels. The complexity with which the activity of
this integral cell cycle regulator is modulated, is supported by our results showing the
suppression of cyclin Dl in response to pRb-loss and Myc-activation in MEFs can be
regulated at both the RNA and protein levels. Analysis of primary MEFs derived from
independent litters is key to hlly elucidating the multiple mechanisms which govern cyclin
Dl expression and activity. At the post-transcriptional level Myc may suppress cyclin Dl
protein expression by suppressing the translation of cyclin Dl or reducing the stability of
the cyclin Dl protein. e1F-4E, a translation initiation factor can be induced in response to
Myc and, in turn, specifically up-regulate cyclin Dl protein expression (Rosenwald et al.,
1993). It is tempting to speculate that e1F-4E may also serve as a control point for the
translational suppression of cyclin Dl in response to Myc in pRb-deficient MEFs, as
reported here. It is interesting that cyclin Dl mRNA was suppressed in the pRb-deficient

MEF 37 culture which expressed ectopic Myc protein. It will be important to ascertain
whether the suppression of mRNA expression in MEF37 is mediated at the transcriptional
level and whether pRb-dysfunction may be involved in Myc-repression of cyclin Dl
transcription as reported in (Philipp et al., 1994). Interestingly, the Myc negative
autoregulation mechanism remains intact in the pRb-deficient MEFs, and serves as a
positive control to show loss of pRb does not result in a general, non-specific disruption of
homeostatic regulatory mechanisms. Activation of c-myc expression in pRb-minus cells
reproducibly leads to downregulation of cyclin Dl activity. The mechanisms of this down-
regulation are complex involving RNA with some cells and protein in others: the different
mechanisms probably reflect the different secondary genetic changes in the cultured cell
lines.
Analysis of the growth of MEFs derived kom three independent litters of mouse
embryos revealed that pRb-deficient MEFs consistently exhibited a delay in growth
compared to wildtype MEFs. In contrast, Lukas el al. recently reported that pRb-deficient
MEFs have a shortened G1 phase compared to their wildtype counterparts (Lukas et al.,
1995). This difference in results may be attributed to our distinct experimental conditions.
The delay in growth, reported here, reflects an initial lag in proliferation following seeding
as subconfluent cultures. It remains unclear whether the lag in growth is a result of
decreased plating efficiency, an increased rate of apoptosis or a direct lengthening of the
cell cycle. Apoptosis is associated with pRb-inactivation (Clarke et al., 1992; Haas-Kogan
et al., 1995; Haupt et al., 1995; Jacks et al., 1992; Lee et al., 1992; Morgenbesser et al.,
1994), but is not likely contributing to the lag in population growth, as we did not observe
an increase in programmed cell death in pRb-deficient cells grown as subconfluent (Figure
2.6) or confluent (Figure 2.7) cultures (GW, WWM, Y-JH, LJZP, unpublished results).
Expression of Myc in pRb-deficient MEFs can suppress cyclin Dl, abrogate the
characteristic delay in cell growth and push the cells to a more transformed state. The
suppression of cyclin Dl in a pRb-deficient cell is not the only molecular event

downstream of Myc potentially involved in increasing cellular proliferative potential, but it
likely contributes and may be required in a pRb-deficient cell. These results are consistent
with observations reported in the literahue. Cellular transformation by other nuclear
oncogenes, adenovirus ElA or SV40 large T antigen, results in the suppression of cyclin
Dl expression as well as the inactivation of the product of the retinoblastoma gene (Bates et
al., 1994; Buchou et al., 1993; Lukas et al., 1994; Muller et al., 1994; Tam et al., 1994;
Xiong et al., 1993). Moreover, human cell lines harboring a loss of pRb uniformly show
a suppression in cyclin Dl protein expression (Bates et al., 1994; Tam et al., 1994; Xiong
et al., 1993). These human tumour cells also demonstrate an increase in the product of the
INK4IMTS1 gene, p16, which forms inhibitory complexes with cdk 4 or cdk 6 (Parry et
al., 1995), &her demonstrating that suppression of cyclin Dl kinase activity appears
crucial in pRb-deficient tumors.
Although cyclin Dl expression has been strongly implicated in tumour formation, the
precise role of cyclin Dl in tumorigenesis has not been elucidated. Cyclin Dl is
overexpressed in several types of tumors such as parathyroid adenomas (Motokura et al.,
1991), and centrocytic B-cell lymphomas (Motokura and Arnold, 1993), and is amplified
in breast cancers: (Motokura and Arnold, 1993). These observations suggested that cyclin
Dl may function as a promoter of cell proliferation. This view was supported by studies
demonstrating that cyclin Dl activity was required for progression through the cell cycle.
Microinjection of anti-cyclin Dl antibodies into wildtype MEFs (Lukas et al., 1995) or
human diploid fibroblasts (Baldin et al., 1993) blocks cells in the G1 phase. Furthermore,
expression of elevated levels of ectopic cyclin Dl shortens the G1 phase of the cell cycle
(Quelle et al., 1993; Resnitzky et al., 1994). However, in vivo its role as an oncogene is
not definitive. When constitutively overexpressed in transgenic mice, cyclin Dl provides
only a weak oncogenic stimulus (Bodrug et a]., 1994; Wang et al., 1994). Indeed, recent
evidence establishes a role for cyclin Dl as a negative regulator of cell proliferation
(Freeman et al., 1994; Lucibello et al., 1993; Quelle et al., 1993; Resnitzky et al., 1994).

Cyclin D l is induced in post-mitotic neurons (Freeman et al., 1994) and in senescent
h u m a cells (Lucibello et al., 1993). Constitutive expression of high levels of cyclin Dl in
a variety of mammalian cell lines inhibits cell growth (Quelle et al., 1993; Resnitzky et al.,
1994). Interestingly, microinjection of neutralizing antibodies to cyclin Dl in pRb-
deficient MEFs can accelerate S phase entry (Lukas et al., 1995), which agrees with our
results and further suggests suppression of cyclin Dl in an pRb-deficient cell can
contribute to cellular growth potential.
Clearly the regulation of cyclin Dl is highly dependent on the genetic context of the
cell. The tissue of origin, stage of differentiation and acquired genetic lesions will play a
significant role in determining the net effect of cyclin Dl expression on cell proliferation
and tumorigenesis.

2.6 Acknowledgments
We extend special thanks to R. Weinberg and T. Jacks for RB-I heterozygous
mice. We also thank S. Egan, P. Hamel, M. Kiess, L. Facchini and J. Lear for helpful
discussions and critical reading of the manuscript; E. Harlow and E. Lees for cyclin Dl
antibody; G. Evan for anti-myc antibody; M. Kiess and P. Hamel for murine cyclin Dl
cDNA; and T. Thompson for the pRasImyc 9 retroviral vector. This work was supported
by grants from the Medical Research Council of Canada (LJZP)( BLG), the National
Cancer Institute of Canada with funds from the Canadian Cancer Society (LJZP) and from
the Terry Fox Run (RAP & BLG), and by an Ontario Graduate Studentship Award
(WWM).

Chapter 3
M c
PLATELET-DERIVED GROWTH FACTOR
BETA RECEPTOR EXPRESSION
This chapter is a modified version of the following manuscript: Myc is an essential
negative regulator of platelet-derived growth factor beta receptor expression. 1998.
Wilson W. Marhii, Charlotte Asker, Shaojun Chen, Sara K. Oster, Linda M. Facchini, Patrick A. Dion, Martin Post, Akira Ishisaki, Keiko Funa, John M. Sedivy and Linda Z. Penn. Molecular and Cellular Biologv, submitted.

3.1 Abstract
Platelet derived growth factor BB (PDGF BB) is a potent mitogen for fibroblasts as
well as many other cell types. Interaction of PDGF BB with the PDGF P receptor (PDGF-
PR) activates numerous signaling pathways and leads to a decrease in receptor expression
on the cell surface. PDGF-PR downregulation is effected at two levels, the immediate
internalization of ligandlreceptor complexes and the reduction in pdgf-pr mRNA
expression. Our studies show thatpdgf-pr mRNA suppression is regulated by the c-myc
proto-oncogene. Both constitutive and inducible ectopic Myc protein can suppresspdgf-pr
mRNA and protein. Suppression of pdgf-pr mRNA in response to Myc is specific, as
expression of the related receptor, pdgf u receptor is not affected. We further show that
Myc suppresses pdgf-pr mRNA expression at the transcriptional level by a mechanism
which is distinguishable from Myc autosuppression. Analysis of c-Myc-null fibroblasts
demonstrates that Myc is required for the repression of pdgf-pr mRNA expression in
quiescent fibroblasts following mitogen stimulation. In addition, it is evident that the Myc-
mediated repression ofpdgf-jr mRMA levels plays an important role in the regulation of
basal pd&r expression in proliferating cells. Thus, our studies suggest an essential role
for Myc in a negative feedback loop regulaiing the expression of the PDGF-PR.

3 . 2 Introduction
The PDGF P receptor (PDGF-PR) has been extensively studied in the context of
wound healing and carcinogenesis where many of the biological activities of the PDGF-PR
clearly play a role (reviewed in Bennett and Schultz, 1993; Bomfeldt et al., 1995;
Claesson-Welsh, 1996; Claesson-Wdsh, 1994). The activated PDGF-PR can elicit
diverse and seemingly paradoxical biological activities, including cell growth, cell survival,
differentiation, and cellular transformation. Activation of the PDGF-PR occurs rapidly
following PDGF ligand-receptor interaction. PDGF A and B polypeptides can form
homodimers (AA and BB), or a heterodimer (AB), and bind two distinct PDGF receptors
(PDGF-aR, PDGF-PR) with differing affinity. In fibroblasts, the PDGF-aR is expressed
at low abundance and binds with high affinity to all three PDGF isofonns, whereas the
PDGF-PR is highly abundant and exhibits restricted binding to two isoforms; PDGF BB
with high affinity, and PDGF AB with lower affinity (reviewed in Westermark and Heldin,
1993). Ligand binding results in receptor dimerization which induces autophosphorylation
at multiple tyrosine residues and activates the receptor (Ullrich and Schlessenger, 1990).
The phosphorylated tyrosine residues serve as binding sites for a host of SH2-domain-
containing proteins which include: phospholipase C-yl (PLCy), the GTPase-activating
protein of Ras (GAP), the regulatory subunit of phophatidylinositol-3-kinase (PI3K), the
phosphotyrosine phosphatase Syp, Src-family members (Src, Yes and F p ) , Nck, Shc, as
well as additional uncharacterized proteins (see Claesson-Welsh, 1996). Following
receptor-binding, these proteins are activated through phosphorylation and subsequently
stimulate a number of signal transduction cascades. These signaling pathways include the
activation of MAP kinase and the induction of immediate early growth response genes such
as the c-myc proto-oncogene. The net outcome of receptor signaling is dependent upon the
combination of receptor-associated proteins recruited. For example, PI3K and PLCy are
required for transmission of the mitogenic signal of the PDGF-PR (Valius and Kazlauskas,

1993), but recruitment of GAP to the receptor can inhibit PLCy-mediated mitogenesis
(Valius et al., 1995). The mechanisms used to fine tune receptor-induced signaling and the
full diversity of the signaling potential of the receptor remain unclear.
The induction of Myc protein expression, following mitogen stimulation, is an
essential prerequisite for G1 to S phase progression of the cell cycle in non-transformed
cells (Campisi et al., 1984; Heikkila et al., 1987; Kaczmarek et al., 1985; Kelly et al.,
1983; Prochownik et al., 1988; Roussel et al., 1995; Shichiri et al., 1993). Consistent
with the role of Myc as a strong stimulator of cell proliferation, deregulated Myc
expression is a potentiator of tumorigenesis, and is a common hallnxk of a diverse array
of tumors (reviewed in Garte, 1993; Prins et al., 1993; Spencer and Groudine, 1991). In
addition to the well-established role of Myc as a positive growth regulator, Myc can also
induce the apoptotic death program (Askew et al., 1991; Evan et al., 1992; Shi et al.,
1992). Myc is thought to function as a central regulator of such diverse cellular activities
by regulating gene expression. Myc can function as a transcriptional activator when
heterodiierized with its partner Max, a basic helix-loop-helix leucine zipper protein (Amati
et al., 1992; Blackwell et al., 1993; Blackwell et al., 1990; Blackwood and Eisenman,
1991; Halazonetis and Kandil, 1991; Kretzner et al., 1992; Prendergast et al., 1991).
Transcription activation is dependent upon the binding of the Myc-Max heterodimer to a
canonical DNA sequence CACGTG as well as to specific non-canonical elements. In
addition to its role as a transcriptional activator, studies suggest Myc can also repress gene
transcription. The precise mechanism through which Myc represses gene transcription is
not clear, as only a relatively small number of Myc-repressed target genes have been
identified. Myc can repress d e b p a (Antonson et al., 1995; Li et al., 1994), cyclitt Dl
(Philipp et al., 1994), the adenovincs 5 major-late promoter (AdMLP) (Li et al., 1994), and
c-ntyc (Facchini et al., 1997) promoter activities. The c-myc gene is one of the better
characterized Myc repressed target genes. Myc autosuppression is a homeostatic
regulatory mechanism which results in the repression of endogenous c-myc expression at

the level of transcription initiation. This feedback mechanism functions in a Myc dose-
dependent manner, and requires the interaction of Myc with Max (Facchini et al., 1997;
Grignani et al., 1990; Penn et al., 1990). Evidence to date suggests Myc represses gene
transcription through core regulatory elements and does not require Myc-Max binding sites
(Antonson et al., 1995; Facchini et al., 1997; Li et al., 1994; Lucas et al., 1993; Philipp et
al., 1994; Roy et al., 1993). Identification of additional Myc-regulated target genes is
required to further delineate the molecular mechanisms through which Myc regulates gene
transcription, and help us to better understand how Myc modulates normal cell proliferation
in response to the external environment.
Given the potent mitogenic stimulus initiated by the ligand-activated PDGF-PR, it
is not surprising that the cell possesses negative feedback mechanisms to regulate receptoi.
expression. Proliferation of non-transformed cells following exposure to mitogen is
controlled through the down-regulation of growth factor receptors. Receptor down-
regulation involves the internalization and degradation of receptor-ligand complexes
(Nilsson et al., 1983; Rosenfeld et al., 1985; Sorkin et al., 1991). Additional mechanisms
also play a role in this process, as ligandheceptor interaction also leads to receptor mRNA
suppression, as shown for the CSF-1 (c-fms) receptor (Gliniak and Rohrschneider, 1990),
the c-kit receptor (Gliniak and Rohrschneider, 1990), and the PDGF-PR (Vaziri and Faller,
1995), all of which are members of the PDGF receptor family (Yarden et al., 1987). The
mechanism of receptor mRNA suppression is unclear; however recent studies have
provided clues as to the constituents of this pathway. Ectopic expression of activated Ras
or Src in fibroblasts results inpdgf-/3r mRNA down-regulation (Vaziri and Faller, 1995;
Zhang et al., 1995). As activated Src can target Myc (Barone and Courtneidge, 1995), the
latter may serve as an effector of~dgf-pr mRNA suppression. Further support for this
hypothesis comes from studies of small cell lung carcinoma by Plummer et al. (1993),
which showed an inverse correlation between the expression of Myc and the c-kit receptor.
In this thesis, we show that PDGF-PR expression is down-regulated in non-

transformed fibroblast cells following exposure to either serum, PDGF BB ligand or Myc-
activation. The suppression of PDGF-PR levels is specific as Myc activation has no effect
on pdgf-ffr expression. Myc suppression of pdgf-jr mRNA is effected at the
transcriptional level by a mechanism which appears to be uncoupled from Myc
autosuppression. We demonstrate that Myc is required for the repression of pdgf-pr
expression following serum stimulation. Moreover, we show that Myc repression ofpdgf-
j r mRNA levels plays an important role in regulating basal pdgf-fir expression in
proliferating cells. Our results support a role for Myc in the homeostatic regulation of
pdgf-pr mRNA levels in proliferating fibroblasts.

3 . 3 Materials and Methods
3 .3 .1 Cell culture, and somatic cell hybridizations
Primary rat embryo fibroblasts (REF) were prepared as described previously (Land
et al., 1983). Rat-1; Rat-1 pMV7hc-myc(wt)/ER (wtMycER), and Rat-1 pMV7hc-
myc(Al06-143)lER (AMycER) were previously described (Daksis et al., 1994; Evan et
al., 1992; Pem et al., 1990). The KPREF cell line is a population of spontaneously
immortalized rat embryo fibroblasts (Pem et al., 1990). Rat-1 wtMycERTM, and Rat-1
AMycERRVI cell lines were generated by infecting Rat-1 cells with replication incompetent
retroviruses either Babepuro c-mycERm or Babepuro AI06-143c-mycERm. Cells were
selected in medium supplemented with puromycin, and individual drug resistant colonies
were cloned. CIones were characterized for MycERTM expression, to ascertain that
expression was within physiological levels and that wtMycERm activation will induce
cells to progress from GOIG1 to S phase of the cell cycle. Clones of our variant NIH3T3
cell line, NIH3T3v3 and NIH3T3v10, were derived from and possess identical properties
to the parental population first described by Pem et al. (Pem et al., 1990). Wildtype
TGR-1 (+I+) and c-Myc null H015.19 (-I-) cell lines were previously described (Mateyak
et al., 1997). c-Myc null H015.19 gfp (-1- gfp) and c-Myc null H015.19 gfpmyc (-1-
gfpmyc) cells were generated by infecting c-Myc null H015.19 (-I-) cells with either the
retrovirus BabeMNIRESgfp or BabeMNIRESgfpmyc. Rat-1 wtMycER and Rat-1
AMycER cells were maintained in phenol red minus alpha modified Eagle's medium
((rMEM) supplemented with 10% charcoal treated serum (CTS)(Hyclone). Wildtype Rat-1
clone TGR-1 (+I+), c-Myc null H015.19 (-I-), H015.19 gfp (-1- gfp) and H015.19
gfpmyc (-1- gfpmyc) cells were grown in DMEM H21 supplemented with 10% calf serum,
as described (Prouty et al., 1993). A11 other fibroblast cell lines were cultured in aMEM
supplemented with 10% fetal bovine serum (FBS)(Gibco). Culture media was

supplemented with 100 pg/ml kanamycin and 2 pghl gentamycin or 100 pglml penicillin
and 100 pglml streptomycin sulfate. Somatic cell hybridizations were conducted as
previously described (Facchini et al., 1994; Penn et al., 1990). To analyze the response to
PDGF BB stimulation, subconfluent cells were cultured for three days in 0.3%
FBSIaMEM prior to stimulation with 40 nglml PDGF BB (Upstate Biotechnology
Incorporated). To activate ectopic Myc activity, Rat-1 wtMycER cells were exposed to 100
nM P-estradiol (Sigma) in ethanol, Rat-1 wtMycERTM cells were exposed to 100 nM 4-
Hydroxy (2) Tamoxifen (OH-T) in ethanol (Research Biochemical International, Natick,
MA), while controls were exposed to ethanol alone. To analyze the response to serum
stimulation, subconfluent wildtype TGR-1 (+I+), and c-Myc null H015.19 (-I-) cells were
maintained in 0.25% calf semm1DMEM HZ1 for 2 days, then stimulated with 10% calf
serum/DMEM H21. Unless otherwise stated, all cells were analyzed as subconfluent
proliferating cultures.
3.3.2 Retroviral vectors
The pDORheo, pDoklv-mycneo , pBabehygro , pBabelv-mychygro, pBabeIpuro,
pBabelv-mycpuro, retroviral vectors were constructed as previously described (Facchini et
al., 1994; Morgenstem and Land, 1990; Penn et al., 1990; Ridley et al., 1988). The
pBabepuro c-mycERTM, and pBabepuro A106-143c-mycERTM retroviral vectors were the
kind gift of G. Evan and T. Littlewood and were described previously (Littlewood et al.,
1995). To generate the pBabeMNIRESgfpmyc retroviral vector, human c-myc exon 11, I11
sequences excised from pBluescript KS+ Hc-myc were cloned into EcoRI, MtoI digested
pBabeMNIRESgfp (a kind gift of G. Nolan).

3.3 .3 Retroviral infection
To produce infectious replication-defective ecotropic retroviral particles,
recombinant retrovital constructs were transfected, using calcium phosphate precipitation
(Graham and Van der Eb, 1973), into either the GP+E packaging cell line (Markowitz et
al., 1988), or the Phoenix-Eco cell line (ATCC), and selected in 0.5 mg/ml G418 sulphate
(Sigma), 2 mglml puromycin (Sigma) or 300 pg/ml hygromycin B (Sigma). After drug
selection, drug resistant clones were pooled and expanded for virus production. Primary
rodent embryo fibroblasts and cell lines were infected with retrovirus and selected in either
G418, hygromycin B or puromycin. Individual drug-resistant colonies were either isolated
to produce clonal populations or combined to generate pooled populations. Green
fluorescent protein (gfp) positive c-Myc null H015.19 gfp (-1- gfp) and H015.19 gfpmyc
(-I- gfpmyc) cells were isolated with a Coulter 753 cell sorter using an absorption
wavelength of 488 nrn, and pooled.
3.3.4 RNase protection
RNA was prepared by the guanidinium isothiocyanate method of Chirgwin et al.
(Chirgwin eta]., 1979). RNaseprotection was conducted essentially as described (Penn et
al., 1990). The probes were generated using T3 RNA polymerase (Stratagene) from
linearized Bluescript KS and SK cloning vectors (Stratagene) containing the following
DNA fragments: rat c-myc exon I (Pem et al., 1990); thegag gene derived from the avian
myelocytomatosis virus MC29 (Penn et al., 1990); rat (Penn et al., 1990), and the murine
glyceraldehyde-3-phosphate dehydrogenase (gapdh) gene (Facchini et al., 1994). The
mousepdgf-pr probe comprised nucleotides 1 to 227 of the mouse pdgf--pv cDNA. This
cDNA fragment was excised from h-N16 (kind gift of Y. Yarden) with EcoRI, and SmaI,
then cloned into a Bluescript KS vector (Stratagene). The Bluescript KS mousepdgf-pr

cDNA plasmid was linearized with BamHI, and thepdgf-fir RNase protection probe was
transcribed using T3 RNA polymerase (Stratagene). The aminoglycoside
phosphotransferasefileo) probe comprised nucleotides 1 to 245 of the neomycin resistance
gene. These sequences were excised from pBabeneo (Morgenstern and Land, 1990) with
Hind11 and ClaI, and cloned into a EcoRV and CIaI digested pBluescript KS+ vector
(Stratagene). The Bluescript KS+ aminoglycoside phosphotransferase plasmid was
linearized with NcoI, and the RNase protection probe transcribed with T3 RNA
polymerase (Stratagene). The protected probes were resolved by electrophoresis on 6%
denaturing polyacrylamide gels and visualized using a phosphoimager or by
autoradiography on X-OMAT film (Kodak). Densitometry was performed by
phosphoimager analysis.
3.3 .5 Northern blot Analysis
RNA was prepared by the yanidinium isothiocyanate method of Clrirgwin et al.
(Chirgwin et al., 1979). Northern Blot analysis was conducted as described previously
(Daksis et al., 1994). The ratpdgf-ar probe consisted of a 510 bp EcoRV fragment
derived from pCRlIPDGFaR (Souza et al., 1995). The probe for 36B4, a gene encoding
an acidic ribosomal protein, consisted of a 800 bp Pstl fragment derived from p36B4
(Masiakowski et al., 1982). Densitometry was performed by phosphoimager analysis.
3.3.6 PDGF BB binding studies
Subconfluent rodent fibroblasts were washed twice with ice-cold Phosphate
Buffered Saline containing 1% (w:v) bovine serum albumin (l%BSA/PBS). To determine
high-affinity saturable [ ~ ~ ~ I I P D G F BB binding, cells were incubated at 4OC in 500 yl of
l%BSAIPBS with increasing concentrations of [ ~ ~ ~ I I P D G F BB in the absence (total

binding) or presence (non-specific binding) of an 100-fold excess unlabelled PDGF BB.
After 2 hrs of incubation, the medium was aspirated and the cells were washed 5 times
with 1 ml ice-cold l%BSA/PBS. The remaining cells were extracted for 1 hour by adding
lml/well lysing buffer (1% (v:v) Triton X-100, 10 mM glycine and 20mM HEPES, pH
7.4), and total radioactivity determined by gamma spectrophotometry.
3.3.7 Luciferase assays
The mousepdgf-pr promoter luciferase constructs have been previously described
(Ballagi et al., 1995). Each plate was transfected with 10 pg of each of the pdgf-pr
promoter luciferase constructs, 0.1 pg of a vector carrying the P-galactosidase gene
(pCMVpgal), and 10 pg of pBluescript KS+ plasmid (Stratagene), using the calcium
phosphate method. Luciferase and P-galactosidase assays were performed as previously
described (Facchini et al., 1997).

3.4 RESULTS
3.4.1 PDGF P receptor mRNA expression is suppressed following
serum or PDGF BB stimulation of Rat-1 cells
PDGF BB is one of the primary growth factors for fibroblasts. Yet little is known
of the pattern or regulatory mechanisms which negatively govern the mRNA expression of
its key receptor, PDGF-PR, following ligandlreceptor interaction in normal and
transformed cells. Earlier reports have implicated Src and Ras as negative regulators of
pdgf-fir expression (Vaziri and Faller, 1995). As Myc has been placed downstream of src
in the PDGF BB signaling pathway (Barone and Courtneidge, 1995), we explored the
relationship between c-myc and pdgf-br expression following serum-stimulation of
quiescent cells. Subconfluent Rat-1 cells were incubated in low fetal bovine serum (0.3%
FBSlaMEM) for 3 days to reduce the levels of PDGF in the medium, and subsequently
exposed to either fresh 10% FBSIcdvlEM or PDGF BB growth factor. RNA was extracted
at the times indicated and analyzed by RNase protection to detect endogenous c-myc,pdgf-
pr and gapdh-specific transcripts. As expected (Campisi et al., 1984; Kelly et al., 1983),
exposure of serum-deprived Rat-1 cells to 10% serum induced a rapid and transient
increase of endogenous c-myc mRNA expression (Figure 3.1A). C-myc mRNA
expression peaked at approximately 2 h, then gradually decreased reaching steady-state
levels 12 to 18 h post stimulation. Exposure to PDGF BB alone also induced c-myc
mRNA expression, however, compared to serum stimulation, c-myc mRNA expression
was not sustained, as levels returned to basal approximately 4 h post-stimulation (Figure
3.1B). Basal pdgf-pr RNA is readily detectable in cells cultured under low serum
conditions, and is down-regulated approximately 3 to 4 h after exposure of cells to serum
or PDGF BB alone (Figure 3.1A, 3.1B). The sequential induction of Myc expression

Serum I I 0 1 2 3 6 121824 h
c-myc
pdgf-Pr
PDGF BB
Figure 3.1. Expression of c-myc andpdgf-pr mRNAs in quiescent Rat-1 cells stimulated with serum o r PDGF BB. Rat-1 cells were maintained in low serum 0.3% FBSIaMEM for 3 days then treated with either (A) 10% FBSIaMEM or (B) 40ndml PDGF BB. At the indicated intervals RNA was extracted and analyzed by RNase protection using single stranded probes complementary to endogenous c-nzyc, pdgf- p,; and gapdlz-specific sequences.

followed by the suppression ofpdgflpr RNA suggested Myc may play a role in initiating
receptor repression following growth factor stimulation.
3.4.2 PDGF P receptor RNA is down-regulated in Myc-activated non-
transformed cells
To determine whether constitutive ectopic Myc expression can regulate the
expression ofpdgf-br mRNA, primary rat embryo fibroblasts (REFS) and the immortal,
non-transformed cell lines, KPREF and Rat-1, were infected with control replication-
incompetent retrovims (DORIneo) carrying only the neomycin resistance gene, or with
retrovirus (Doklv-mycneo) canying the v-myc gene in addition to the neomycin resistance
gene. RNA from drug resistant, subconfluent proliferating cultures was analyzed by
RNase protection assay (Figure 3.2A). Pdgf-pr mRNA expression was readily visible in
control cells and clearly suppressed in the cells expressing ectopic Myc (Figure 3.2A;
compare lanes 1,3,5 with 2,4,6, respectively). As a positive control for exogenous Myc
activity, ectopic v-myc and endogenous c-myc mRNA expression were assayed. Our
results demonstrate ectopic v-myc protein was also expressed and functionally active as
endogenous c-myc mRNA levels are suppressed in v-Myc expressing cells, due to the Myc
negative feedback mechanism as previously reported (Figure 3.2B; compare lanes 1 ,3 ,5
with 2, 4, 6, respectively) (Cleveland et al., 1988; Grignani et al., 1990; Penn et al.,
1990). These results show that enforced Myc expression down-regulates pdgf-pr
expression in primary and non-transformed cells.

REF KPREF Rat-1 I nn
v-mnyc - + - + - +
REF JXPREF Rat-1 n
v-mnyc - + - +I I - +(
Figure 3.2. Suppression of endogenous c-myc and pdgf-pr mRNA levels in rodent cells constitutively expressing exogenous Myc. RNA (10 pg) fiom subconfluent rodent fibroblasts infected with a control retrovirus (-), or retrovirus carrying the v-myc gene (+) was analyzed by RNase protection assay to detect (A) endogenous pdgf-pr and control gapdh, (B) endogenous c- myc, ectopic v-myc, and control gapdh expression.

3.4.3 The suppression of PDGF P receptor mRNA levels following
Myc-activation occurs with rapid kinetics
To examine the kinetics ofpdgf-pr mRNA suppression we first employed an
estrogen-inducible MycER system in Rat-1 cells (Rat-1 wtMycER). These Rat-1 cells
constitutively express an inactive fusion protein consisting of the human c-Myc protein
fbsed to the estrogen binding domain of the estrogen receptor. This MycER fusion protein
is rapidly activated in response to P-estradiol or the P-estradiol antagonist 4-hydroxy-
tamoxifen (OH-T), allowing Myc activity to develop within minutes of ligand exposure
(Daksis et al., 1994; Eilers et al., 1989; Evan et al., 1992; Selvakumaran et al., 1993). As
a control, we analyzed the Rat-1 AMycER cell line, which expresses a AMycER fusion
protein containing a deletion of amino acids 106 to 143 within the Myc protein, rendering
the fusion protein inactive for all known biological activities of Myc (reviewed in Penn et
al., 1990). To determine the kinetics ofpdgf-jr suppression Rat-1 wtMycER and Rat-1
AMycER cell lines were analyzed during different states of cellular proliferation (Figure
3.3A). Subconfluent, proliferating Rat-1 wtMycER and Rat AMycER cells were exposed
to P-estradiol for the times indicated. In addition, confluent quiescent serum-deprived Rat-
1 wtMycER cells were either exposed to ethanol as a control or treated with P-estradiol
dissolved in ethanol, to induce Myc activity. Suppression of endogenouspdgf-jr mRNA
is clearly detectable in both subconfluent and confluent Rat-1 wtMycER cells at 6 h
following induction of Myc activity (Figure 3.3A. compare lanes 5 and 6; lanes 17 and
18). Thepdgf-jr mRNA remained suppressed in the P-estradiol-treated Rat-1 wtMycER
cells over the course of the experiment. Pdgf-pr expression is elevated in confluent
quiescent cells in comparison to subconfluent proliferating cells. Yet, despite the
difference in their basal level of RNA expression, the kinetics and degree of suppression
were similar in both subconfluent and confluent cultures. Suppression of the receptor was
not evident in control ethanol treated Rat-1 wtMycER cells (Figure 3.3A. lanes 13, 15,17,

19 & 21), and in Rat-1 AMycER cells which were or were not exposed to J3-estradiol
(Figure 3.3A. lanes 23 and 24). Receptor expression also remained invariant in
subconfluent Rat-1 AMycER cells in response to P-estradiol (Figure 3.3A. lanes 1,3, 5,
7, 9, 1 I) as apparent differences are due to lane loading variability. Thus our results
demonstrate that pdgf-pr mRNA expression is suppressed upon Myc induction in both
quiescent and proliferating cells with similar kinetics. To eliminate the possibility that the
intrinsic transactivation domain present in the estrogen receptor portion of the wtMycER
chimaera (ERTAD) could contribute to the suppression in pdgf-pr mRNA, we next
employed Rat-1 cells expressing a novel wtMycERm construct in which the ERTAD has
been deleted (Rat-1 wtMycERTM) (Littlewood et al., 1995). Confluent, quiescent Rat-1
wtMycERTM cells stably expressing the inactive wtMycERm protein were treated with
OH-T for the times indicated to induce Myc activity. The kinetics and overall level of
suppression observed with the wtMycERm construct are comparable to that observed in
Rat-1 wtMycER cells (compare Figure 3.3A, lanes 13 to 22 and Figure 3.3B). Hence, the
ERTAD in the wtMycER construct did not contribute significantly to Myc suppression of
endogenous pdgf-pr mRNA levels. As a positive control for exogenous wtMycERTM
activity, endogenous c-myc mRNA expression was assayed and showed the expected
suppression due to the Myc negative feedback mechanism as was previously described
(data not shown) (Facchini et al., 1997). Myc suppression ofpdgf-prmRNA levels is
specific, as induction of Myc activity has no effect on the mRNA expression of the related
growth factor receptor, Platelet-Derived Growth Factor a receptor (pdgf-ar) (Figure
3.3C). The suppression of endogenouspdgf-pr mRNA levels is not the result of a Myc-
induced autocrine negative feedback pathway, as MycER activation had no effect on the
expression of pdgf-B mRNA (data not shown). Therefore, the suppression ofpdgf-pr
mRNA by Myc represents a specific regulatory mechanism.

Figure 3.3. Pdgf-pr mRNA expression is suppressed in response to Myc
induction. (A)pdgf-jr mRNA expression in Rat-1 wtMycER (wt) and Rat-1 AMycER
(A) cells was analyzed under both subconfluent and confluent conditions. Cell lines were
grown either as asynchronous subconfluent populations and treated with P-estradiol (lanes
1-12) or as confluent quiescent cultures which were treated with either ethanol as control (-
) or with P-estradiol (+) (lanes 13-24). At the indicated intervals after P-estradiol
treatment RNA was extracted and total RNA (10 yg) was analyzed by RNase Protection to
detect the expression of endogenous pdgf-pr, and control gapdh genes. (B) Confluent
Rat-1 wtMycERm cells were maintained in low serum 0.1% FBSIaMEM for 2 days and
then exposed to 100 nM OH-T to induce exogenous MycERm activity. At the indicated
intervals after OH-T treatment RNA was extracted and 10 pg total RNA analyzed by
RNase Protection assay using single stranded probes complementary to endogenouspdgf-
pr, and gapdh-specific sequences. The plotted data indicates thepdgf-jr signal normalized
to gapdh, as determined by densitometry. (C) Northern blot analysis ofpdd-ar , and
36B4 expression in OH-T stimulated Rat-1 wtMycERTM cells. The constitutively
expressed 3684 mRNA codes for an acidic ribosomal protein, and serves as a measure of
rhe amount of RNA present in each lane. Confluent quiescent Rat-1 wtMycERTM cells
were exposed to 100 nM OH-T to induce exogenous MycERTM activity. At the indicated
intervals after OH-T treatment RNA was extracted and 10 yg total RNA analyzed by
Northern blot using gene-specific probes for endogenous pdgf-cr,, and 3684 specific
sequences. The plotted data is thepdgf-m- signal as normalized to 3684, as determined by
densitometry.

Subconfluent cells Confluent cells I Rat-1 Rat-l Rat-l Rat-l 1 I
AMycER Or wtMycER wtMycER AMycER I i I i n
0 3 6 9 1 2 2 4 1 3 6 9 2 4 2 4 h
m n n n n n n n n n n n A w t A w t A w t A w t A w t A w t - + - + - + - + - + - + - . - -
pdgf-Pr g b* wxl u c 4 &she 9 s. @ .Z . - ' . A -

. . . .
gffpdh

3.4.4 Myc-induced repression of cell surface PDGF P receptors
To determine whether Myc suppression ofpdd-pr mFWA affected receptor protein
expression, the number of PDGF-PRs expressed at the cell surface before and after ectopic
Myc-induction was measured in direct ligand binding assays. Specifically, subconfluent
Rat-1 wtMycER cells were exposed to ethanol as a control or to P-cstradiol in ethanol for
24 h and then incubated with increasing concentrations of [1251]-PDGF BB. Saturable
binding was observed for both control and P-estradiol treated cells at 12 nglml of [125~]-
PDGF BB. The binding capacity of control Rat-1 wtMycER cells was 5.0 h o l of [1251]-
PDGF BB per 3x105 cells (Figre 3.4, closed circles), whereas 24 h following MycER
activation Rat-1 wtMycER cells bound only 2.4 h o l of [1251]-PDGF BB per 3 x 1 0 ~ cells
at saturation (Figure 3.4, open triangles). Scatchard analysis revealed that the estimated
KD of binding for the PDGF ligand was 1.2 X 10-12M, and the estimated number of
binding sites was 12 X 103 for control cells. The specific binding affinity was not
significantly altered in response to Myc-activation, suggesting that our results reflect a
decrease in the number of receptors at the cell surface. Indeed, while the estimated KD of
binding for ligand in P-estradiol treated cells was 0.9 X 10-'ZM, the number of binding
sites was reduced approximately two-fold to 5 X 103. The results obtained from the
PDGF BB binding assays correlates with the decrease in pdgf-pr mRNA expression as
shown by RNase protection analysis. Thus, induced Myc expression leads to
approximately a 50% reduction in cell surface PDGF-PRs compared to control cells.


3.4.5 The mechanism of PDGF fi receptor suppression differs from Myc
autosuppression
A number of similarities exist between the mechanisms employed by Myc to
repress both the endogenous pdgf-pr as well as c-myc mRNA expression. First,
constitutive Myc expression resulted in the suppression of both endogenous pdgf-fir as
well as c-ntyc mRNA levels (Figure 3.2A and 3.2B) (Penn et al., 1990; Pem et al., 1990).
Second, the suppression of both endogenous c-myc (Facchini et al., 1997) andpdgf-pr
(Figure 3.3A and 3.3B) mRNA levels in response to Myc activation occurs with similar
kinetics, given that the half-life of c-myc mRNA is approximately 20 mins while the half-
life of pdgf-fir mRNA is 4 to 6 h. Taken together, these results suggested that the
repression of c-myc andpdgf-pr mRNA by Myc may be functioning through a common
pathway. To test this hypothesis, we employed a variant mouse NIH3T3v3 cell line. In
contrast to other NIH3T3 cells (K.F., unpublished data) and Rat-1 cells (Figure 3.2B),
expression of ectopic Myc in this variant NIH3T3v3 cell line, did not result in the
suppression of either c-myc orpdgf-fir mRNAs (Figure 3.5, lanes 1 and 2). Stable
intraspecies somatic cell hybrids were generated by fusion of the Rat-1 cell line with the
mouse NIH3T3v3 cell line. By this approach we can deterrn'w whether the trans-acting
factors in Rat-1 cells can complement the dysfunction in the mouse NIH3T3v3 cells for
Myc suppression of pdgf-fir mRNA, as we have previously shown for Myc
autosuppression (Penn et al., 1990).
To establish basal levels of endogenous mousepdgf-fir and c-nzyc mRNAs in the
control somatic cell hybrids, Rat-1 cells were fused with NIH3T3v3 cells in the absence of
ectopic Myc expression. Following drug selection, stable somatic cell hybrids were cloned
and assayed by RNase protection for the expression of mousepdgf-fir and c-ntyc mRNAs
(Figure 3.5, lanes 3-5). To distinguish whether the Myc suppression of endogenous c-
myc andpdgf-pr mRNAs is mediated by the same or different pathways, Rat-1 cells

expressing ectopic Myc were fused to Myc-activated mouse NIH3T3v3 cells. The
resultant stable hybrids were similarly analyzed (Figure 3.5, lanes 6-8). As previously
shown, endogenous mouse c-myc mRNA is readily detectable in the control hybrids, and
is suppressed in somatic cell hybrids expressing ectopic Myc (Figure 3.5; compare lanes 3-
5 to 6-8). In contrast, expression of pdgfpr mRNA in control and Myc-expressing
somatic cell hybrids is identical (Figure 3.5; compare lanes 3-5 with 6-8). Thus, trans-
acting components of Rat-1 cells were able to complement the Myc autosuppression, but
not thepdgf-pr repression mechanism in NIH3T3v3 cells. Identical results were obtained
in similar somatic cell hyhrids derived from another clone, NIH3T3v10, as well as the
original parental variant NIH3T3 cell line (Penn et al., 1990) (data not shown). Therefore,
the repression of mouse c-myc andpdgf-pr mRNAs in response to Myc appear to be
mediated by different pathways.

NIH3T3v3 Rat-lNH3T3v3 HYBRIDS - I I v-mvc - + - - - + + +
Figure 3.5. The suppression of endogenous pdgf$r and c-myc mRNA levels by Myc appear to be mediated by different pathways. Total RNA (10 pg) was analyzed by RNase protection to detect the expression of endogenous mouse c-myc, pdgf-pr and gapdh genes. The cells analyzed include: control NIH3T3v3 (lane l), NIH3T3v3 v-myc (lane 2), as well as three random, independent clones of the stable somatic cell hybrids, generated from the intraspecific fusion of Rat-1 x NIH3T3v3 cells (lanes 3-5), and Rat- 1 v-myc x NIH3T3v3 v-myc (lanes 6-8).

3.4.6 Myc represses transcription from the pdgf-fir promoter
To determine whether the pdgf-Pr promoter is responsive to Myc activity, we
performed in vitro transient-transfection-transcription assays. Subconfluent Rat-l
wtMycERTM and control Rat-l AMycERm cells were transfected with a 1.6 Kb, Sac1
reporter plasmid containing sequences between nucleotides -1994 and -396, relative to the
translation start site in the mousepdgf-pr gene (Figure 3.6A). To control for transfection
efficiency a vector canying the P-galactosidase gene, was co-transfected with the promoter
luciferase construct. Cells were treated with OH-T to induce ectopic Myc activity, and at
the indicated time intervals, whole cell lysates were prepared and then assayed for both
luciferase and P-galactosidase activity. Induction of Myc activity, in Rat-l wtMycERTM
cells, resulted in a clear and reproducible suppression of luciferase activity (Figure 3.6B),
reflecting the suppression of transcription from the mousepdgf-pr promoter by Myc. The
loss in luciferase activity is first evident approximately 6 h following Myc-activation. This
suppression is due solely to the activity of Myc as OH-T treated Rat-1 AMycERTM cells
exhibited no change in luciferase activity (Figure 3.6B). As the half-lives ofpdgj'+
mRNA and luciferase are both approximately 4 to 6 hours (Thompson et al., 1991; Vaziri
and Faller, 1995), the level and kinetics of repression as measured in the pdgf-pr
promoter-luciferase assay (Figure 3.68) closely mimicked the suppression of endogenous
pdgf-Pr mRNA in response to MycERm activation (Figure 3.3B). The repression of the
pdgf-jr promoter by Myc is specific, as induction of Myc activity had no significant effect
on transcription from the CMV promoter, as P-galactosidase activity remained invariant
(Figure 3.6C). Thus, Myc expression results in a suppression of transcription from the
pdgf-Pr promoter, and this suppression activity requires the sequences between nucleotides
-1994 and -396, relative to the translation start site of the mousepdgf-jr promoter.

Figure 3.6. Pdgf-fir mRNA transcription is suppressed by Myc. (A)
Schematic diagram of the genomic structure of the mousepdgf-pr promoter, showing
putative transcription factor binding sites. Numbers indicate position relative to the
translational start site (+l). Rat-1 AMycERTM and Rat-1 wtMycERTM cells were
transfected with a 1.6 Kb mousepdgfpr promoter luciferase construct, containingpdgf-pr
sequences fiom nucleotides -1994 to -396. To assay transfection efficiency, cells were co-
transfected with a plasmid carrying the p-galactosidase gene under the control of the CMV
promoter. Cell lines were subsequently treated with 100 nM OH-T for the times indicated
to induce ectopic MycERm activity. Whole cell lysates were prepared and analyzed for
luciferase and p-galactosidase activity. (B) Histograms represent the measured luciferase
activity. Data was normalized to 0-galactosidase activity to adjust for minor differences in
transfection efficiency amongst the samples. (C) Histograms represent the p-galactosidase
activity. Data are a representative out of 3 repeated experiments. Error bars denote
standard deviations.

Mouse pdg@r promoter
NFI SRF NFI
NF-Y APZ
GATA-I AP2 -1
0 1 3 6 9 1 2 1 8 2 4 TIME (h)
TIME (h)
0 1 3 6 9 1 2 1 8 2 4 TIME (h)

3.4.7 Characterization of the mouse pdgf-pr promoter.
The 1.6 Kb Sac I fragment of the mousepdgf-pr promoter contains consensus
sequences for a variety of known transcription factors including: GATA-1, SRF, NFl,
AP2, APl, and NF-Y. To determine which sequences within this fragment are responsible
for the repression of pdgf-pr mRNA transcription by Myc, we constructed a series of
deletion mutants within the Sac I mousepdgf-jr promoter luciferase construct (Figure
3.7). Thesepdgf-pr promoter constructs were transfected into Rat-1 wtMycERTM cells
and assayed for the responsiveness of the promoter to activation of Myc activity. Upon
activation of ectopic Myc activity in Rat-1 wtMycERTM cells, all of thepdgf-pr promoter
constructs, displayed a 2 to 3 fold reduction in luciferase activity, relative to the basal
luciferase activity exhibited by each promoter construct. Deletion of 558 bp at the 5' end of
the Sac I fragment, reduced basal luciferase activity to approximately 30% of that observed
with the full length Sac I fragment, suggesting the presence of a positive regulatory element
in this region. An additional deletion of 361 bp, further reduces basal luciferase activity to
about 17% of the full length Sac I fragment. Subsequent deletions between -975 and -745
do not impact upon basal transcription, however, deletion of sequences between -745 and
-430 results in a loss of basal transcription. Moreover, this region of the mouse pdgf-jr
promoter contains sequences which are responsive to Myc suppression.
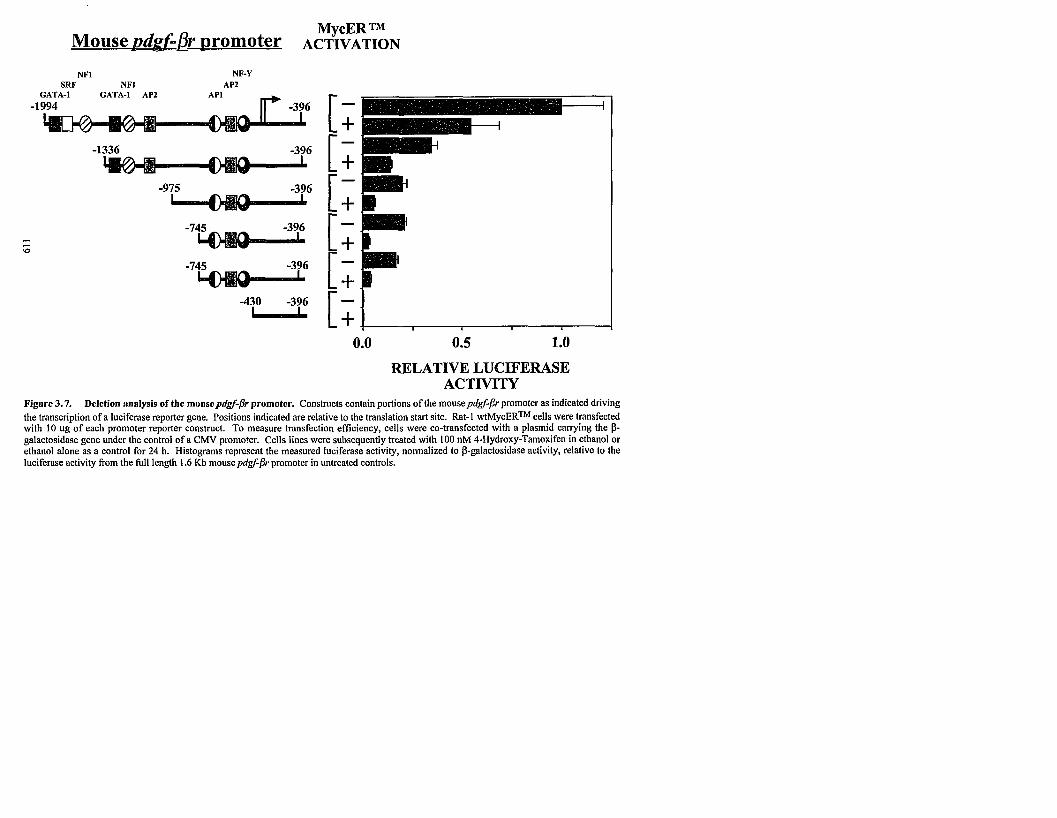
NFI SRF NFI
NF-Y AP2
RELATIVE LUCIFERASE ACTIVITY
Figure 3.7. Deletion analysis of the mousepdsf-pr promoter. Constructs contain portions of the mousepdgf-j?rpromoter as indicated driving the transcription of a luciferase reporter gene. Positions indicated are relative to the translation start site. Rat-l w t ~ y c ~ ~ ~ cells were transfected with 10 ug of each promoter reporter construct. To measure transfection efficiency, cells were co-transfected with a plasmid carrying the P- galactosidase gene under the control of a CMV promoter. Cells lines were subsequently treated with 100 nM 4-Hydroxy-Tamoxifen in ethanol or ethanol alone as a control for 24 h. Histograms represent the measured luciferase activity, normalized to P-galactosidase activity, relative to the luciferase activity from the full length 1.6 Kb mousepdgf--P,promoter in untreated controls.

3.4.8 Myc is required for the suppression in pdgf-jr mRNA levels
following mitogen stimulation.
To examine the role of Myc in the repression ofpdgf-jr mRNA levels following
mitogen stimulation, we analyzedpdgf-jr mRNA expression in serum-deprived c-Myc-
null H015.19 (-I-) rodent fibroblasts upon stimulation with 10% serum. H015.19 (-I-)
cells, derivatives of parental Rat-1 fibroblasts, lack c-Myc expression due to targeted
disruption of both alleles of the c-myc gene with aminoglycoside transferase(neo) and
histidine-marked targeting vectors (Mateyak et al., 1997). Transcription of the disrupted c-
myc genes produces hybrid truncated transcripts consisting of c-myc exon I sequences
fused to the coding sequences of either the neo or histidine genes. c-Myc null (-I-) rodent
cells were cultured in 0.25% calf senun/DMEM H21 for two days then stimulated with
10% serum. RNA was extracted at the indicated time intervals and analyzed for
endogenous pdgf-jr and gapdh expression via RNase protection analysis. As shown in
Figure 3.1A, wildtype Rat-l (+I+) fibroblasts exhibit a gradual reduction in pdgf-fir
mRNA levels upon mitogen stimulation of serum-deprived cultures. In contrast, serum
stimulation of c-Myc null (-I-) fibroblasts failed to elicit a reduction in endogenouspdgf-jr
mRNA levels (Figure 3.8A). Indeed, pdgf-fir mRNA expression was invariant despite
prolonged exposure to serum for 48 h. Thus, Myc is essential for the repression ofpdgf-
j r mRNA levels in serum-deprived cells following serum stimulation.
Myc also plays a role in the regdation of basal pdgf-jr mRNA levels in
subconfluent, proliferating cells. Analysis of endogenous pdgf-jr mRNA levels in
asynchronously proliferating cultures of wildtype (+I+) and c-Myc null (-I-) fibroblasts
revealed that pdgf-pr mRNA levels in c-Myc null cells are elevated in comparison to
wildtype fibroblasts (Figure 3.8B, compare lane 2 to lane 1). Thus, in subconfluent,
proliferating cultures, Myc represses basal pdgf-jr mRNA expression. In addition to the
loss ofpdgf-jr mRNA repression due to the absence of endogenous Myc activity, there is

an abrogation of the c-myc autosuppression mechanism, resulting in an elevation in
expression of the disrupted c-myc allele as detected with a c-myc exon I specific probe
(Figure 3.8B, compare lane 2 to lane 1). The endogenous pdgf-pr and c-myc genes
remain responsive to Myc as repression of both genes can be rescued through the
reconstitution of c-Myc expression in H015.19 (-I-) cells. c-Myc null H015.19 gfpmyc (-
I- gfpmyc) cells constitutively expressing both ectopic human c-Myc, and green fluorescent
protein as a selectable marker, elicits a clear repression ofpd&3r mRNA levels compared
to the parental c-Myc null (4) cells as well as the control cell line H015.19 gfp (-1- gfp),
which constitutively expresses only the green fluorescent protein (Figure 3.8B, compare
lane 4 to lanes 2 and 3). In addition, there is also a rescue of the Myc autosuppression
mechanism, as detected by probes to either c-myc exon I or the neo sequences in the hybrid
transcripts from one of the disrupted c-myc alleles (Figure 3.8B, compare lane 4 to lanes 2
and 3). Analysis of human c-myc mRNA expression clearly demonstrated that ectopic
human c-myc is expressed in the appropriate cell populations (Figure 3.8C). Thus, studies
in c-Myc null fibroblasts have demonstrated that Myc is required for the suppression of
pdgf-/3r mRNA levels in serum-starved rodent fibroblasts following serum stimulation.
Moreover, c-Myc plays a role in maintaining basalpdgf-/3r mRNA levels by repressing
pdgf-/3r mRNA expression in asynchronously, proliferating cells.

Figure 3.8. Myc is required for the repression of pdgJ-jr mRNA levels in
both serum stimulated quiescent cells and in proliferating cultures. (A) Rat-1
cells, lacking c-Myc expression (-I-), were maintained in low serum 0.25% calf
serumJDMEM H21 for 2 days then treated with 10% calf semm/DMEM H21. At the
indicated intervals, RNA was extracted and analyzed by RNase protection using single
stranded probes complementary to endogenous pdgf-jr, and gapdh-specific sequences.
RNA(I0 yg), extracted from subconfluent cultures of wildtype rodent fibroblasts (+I+),
rodent fibroblasts lacking endogenous Myc expression (-I-), and c-Myc null fibroblasts
which have been stably infected with either a control retrovirus expressing gfp (-1- gfp) or a
retrovirus containing human c-myc exon 11,111 cDNA as well as gfp (-1- gfpmyc) was
analyzed by RNase protection for (B) endogenous pdgf-pr, c-myc, aminoglycoside
phosphotransferase (rteo) and gapdh (C) ectopic human c-myc, and endogenous gapdlt-
specific sequences. Similar results were obtained in three separate experiments.

3.5 Discussion
We show that the physiological down-regulation of cell surface PDGF-PRs
following mitogen stimulation involves the product of the c-myc proto-oncogene and
occurs at the RNA level. Constitutive or induced expression of Myc results inpdgf-pr
mRNA down-regulation in non-transformed cells by a mechanism which appears to be
distinct from that of the Myc negative feedback mechanism. The repression ofpdgf-Pr
levels by Myc occurs at the transcriptional level. This Myc-mediated repression ofpdgf-Pr
mRNA levels affects basal expression in subconfluent, proliferating rodent fibroblasts, as
well as mitogen-regulated expression. Taken together, our results show Myc is integral to
the regulation ofpdgf-pr expression.
The molecular mechanism ofpdgf-Pr RNA suppression in response to Myc protein
was explored using the inducible MycER system in Rat-1 cells. We show Myc induction
specifically down-regulates pdgf-pr RNA and does not affect RNA expression of the
related growth factor receptor, thepdgfa receptor. This effect is evident in both growth
arrested confluent, and asynchronous proliferating cells, suggesting receptor down-
regulation occurs in response to Myc per se and is not a consequence of the cellular
transition from GOlGl to S phase of the cell cycle. Ectopic Myc expression does not
induce the expression of PDGF B mRNA thus it is unlikely that Myc is suppressing
receptor expression via a ligand-induced autocrine feedback mechanism. Receptor
suppression is detectable within 3 to 6 h of either growth-factor stimulation or Myc-
induction. As the half-life ofpdgf-Pr RNA is 4 to 6 h, this is the earliest point at which
down-regulation of receptor RNA can be visualized (Vaziri and Faller, 1995). Indeed,
given the long half-life of the~dgf-Pr RNA, the repression of thepdgf-pr RNA following
induction of ectopic Myc activity is rapid, suggesting that Myc may exert a direct effect on
thepdgf-jr promoter.

The decrease in pdgf-pr mRNA expression in response to Myc activation is
mediated at the level of gene transcription. We demonstrate that Myc suppresses
transcription from a Sac I fragment of the mouse pdgf-pr promoter, consisting of
sequences between -1994 and -396, relative to the translational start site. Analysis of the
few Myc-repressed genes identified to date, suggests that Myc can repress gene
transcription through core regulatory elements such as initiator elements or TATA-boxes
(Desbarats et al., 1996; Facchini et al., 1997; Li et al., 1994; Philipp et al., 1994; Roy et
al., 1993). Myc can directly bind to components of the basal transcription complex such as
TFII-I and TBP, inhibiting the activity of these transcriptional initiators. However, the
upstream regulatoly region of the mousepdgf-pr gene is relatively simple, lacking both of
these elements. Yet, there are several putative binding sites for transcription factors such
as GATA-I, SRF, AP1, NF1, AP2, and NF-Y. Deletion analysis of the 1.6 Kb Sac I
fragment of the mousepdgf-pr promoter revealed that multiple elements found between
-1994 and -975 contribute to full transcriptional activity. Moreover, there exist sequences
within -745 to -396 which are required for the maintainence of basal transcription. This
region also contains elements which are responsive to Myc repression. Interestingly, this
fragment contains sequences which are binding sites for three well characterized
transcription factors: AP1, AP2, and NF-Y. It is possible that one or all of these sites are
required to mediate the repression ofpdgf-jr transcription by Myc. The NF-Y site is of
particular interest as mutational analysis of the mousepdgf-pr promoter by Ishisaki el al.,
has demonstrated that the NF-Y binding site is singularly crucial to maintaining basal
mRNA transcription (Ishisaki et al., 1997). 111 vivo studies have also demonstrated that
Myc can regulate the activity of the members of this CTF/NF-1 family of transcriptional
activators. Myc represses transcription of thepro-alpha 2 (1) collagen gene via an indirect
mechanism involving phosphorylation of NFI which inhibits its potential to activate
transcription (Yang et al., 1991; Yang et al., 1993). Hence this site is a likely candidate
through which Myc represses basal transcription of thepdgf-pr gene. The nature of this

Myc-mediated suppression of transcription of thepdgf-br gene, the DNA binding proteins
involved, and the DNA sequences required are currently under investigation.
Many characteristics of Myc-suppression of pdgf-pr are similar to features
associated with the Myc negative autoregulation mechanism (Cleveland et al., 1988;
Facchini et al., 1997; Grignani et al., 1990; Penn et al., 1990; Penn et al., 1990). Myc-
suppression ofpdgf-br and c-myc RNA expression are both evident in primary REFS and
established non-transformed cell lines. The critical Myc-box I1 domain of Myc which is
required for negative autoregulation and for all known biological activities of Myc is
mandatory forpdgf-Dr suppression. Both Myc autosuppression and Myc-suppression of
thepdgf-br occurs at the level of gene transcription. Interestingly, constitutive expression
of Myc in a variant NIH3T3v3 cell line does not lead to the suppression of either c-myc or
pdgf-pr RNA expression. Using a somatic cell hybridization approach with mouse
NIH3T3v3 and Rat-1 cell lines showed that rat cell factors restored suppression of the
mouse c-myc gene (Penn et al., 1990), however, the mousepdgf-br gene was unaffected
in the hybrid cellular background. These results suggest that the regulatory mechanisms
through which Myc suppresses the endogenous c-myc and thepdgf-br genes are distinct,
and further suggests that Myc repression of gene transcription may occur by multiple
mechanisms.
Interestingly, introduction of transforming ras or src oncogenes can lead to reduced
pdgf-pr RNA expression in rodent fibroblasts (Vaziri and Faller, 1995; Zhang et al.,
1995). Indeed, the characteristics of this suppression are similar to those seen in response
to Myc and suggests that Myc may lie in the same pathway, downstream of Ras and Src in
the repression of PDGF-PR. Studies by Barone et al., lend support to this model, as it
was demonstrated that Myc is the target of the Src signaling pathway following PDGF-PR
activation (Barone and Courtneidge, 1995). Thus pathway(s) downstream of Src and
possibly Ras likely signal through Myc to suppresspdgf--& RNA and protein expression,

suggesting Myc is a component of a homeostatic regulatoly mechanism controllingpdgf-jr
RNA expression.
Indeed, through the use of c-Myc null rodent fibroblasts, we are able to
demonstrate that Myc is an essential component of a regulatory pathway effecting
repression of pdgf-jr mRNA levels. While repression of pdgf-jr mRNA levels in
wildtype Rat-1 cells is maximal 12 h post mitogen exposure, c-Myc null fibroblasts did not
exhibit a reduction inpdgf-jr mRNA levels up to 48h after serum stimulation. Transit
through one full cell cycle was monitored in both wildtype and null Rat-1 cells, as the
doubling time is 17 to 22h and 45 to 60h, respectively. Thus the abrogation of the serum
induced repression ofpdgf-fir nJWA expression is due to the absence of Myc expression,
and not the indirect result of a reduced rate of transit through the cell cycle. This is further
demonstrated through our analysis of endogenous pdgf-jr mRNA expression in
proliferating c-Myc null fibroblasts. Basal expression ofpdgf-jr mRNA in asynchronous,
subconfluent, growing cells is elevated in null cells compared to wildtype Rat-1 cells.
Moreover, reconstitution of the null cells with ectopic human c-Myc protein elicited a
reduction inpdgf-jr mRNA levels, showing that thepdgf-jr gene is indeed responsive to
Myc.
The repression of PDGF-PR levels by Myc represents a negative feedback
pathway, which is functionally analogous to the Myc negative auto-regulation mechanism,
in that through both pathways Myc functions to curtail a proliferative stimulus. This
mechanism will serve to temper the growth impetus, guarding against deregulated cell
proliferation. Indeed, experimental evidence suggests that the inhibition of this negative
feedback loop can contribute to the formation of tumors. Glioblastomas and astrocytomas
constitutively express high levels of both the PDGF-PR and its ligand, which creates an
autocrine feedback pathway that contributes to the deregulated growth of these tumors
(Guha et al., 1995; Silver, 1992). The critical nature of receptor down-regulation is
supported by the many mechanisms which have evolved to ensure its successful execution;

internalization, intracellular degradation, and RNA suppression. We show that Myc can
suppress pdgf-pr expression at a transcriptional level, and propose that Myc is a
component of the pathway responsible for receptor suppression following ligand
stimulation.

3.6 Acknowledgments
We thank the members of the lab for helpful discussions and critical reading of the
manuscript; Y. Yarden, G. Evan, T. Littlewood, and G. Nolan for valuable reagents. In
addition, we extend our thanks to E. Fish, and C. Lingwood for their assistance with the
PDGF BB ligand binding assays.
Studentship support was kindly provided through the Ontario Graduate Scholarship
Program (W.W.M., S.K.O.) and the Medical Research Council of Canada (L.M.F.).
C.A. was supported by the Swedish Medical Research Council, the Karolinska-University
of Toronto Exchange Program, and a grant from the Swedish Cancer Society. K.F. was
supported by grants from the Swedish Medical Research Council, the Swedish Cancer
Society and the Barncancerfonden. 1% work was supported by a grant from the National
Cancer Institute of Canada with funds from the Canadian Cancer Society (L.Z.P.).

Chapter 4
MYC REPRESSES THE GROWTH ARREST GENE GADD45
This chapter is a modified version of the following manuscript: Myc represses the growth arrest gene gadd45. 1997. Marhin, W., Chen, S., Facchini, L., Fornace Jr., A., and Penn, L. Oncogene, 14 (23), 2825-2834.

4.1 Abstract
The c-Myc protein strongly stimulates cellular proliferation, inducing cells to exit
GOIGI and enter the cell cycle. At a molecular level, Myc prevents growth arrest and
drives cell cycle progression through the transcriptional regulation of Myc-target genes.
Expression of the growth arrest and DNA damage inducible gene 45 (gadd45) is elevated
in response to DNA damaging agents, such as ionizing radiation via a p53-dependent
mechanism, upon nutrient deprivation, or during differentiation. Gadd45 holds a vital role
in growth arrest as ectopic expression confers a strong block to proliferation. Exposure of
quiescent cells to mitogen stimulates a rapid increase in c-Myc expression which is
followed by the subsequent reduction in gadd45 expression. The kinetics of these two
regulatory events suggest that Myc suppresses the expression of gadd45, contributing to
entry into the cell cycle. Indeed, ectopic Myc expression in primary and immortalized
fibroblasts results in the suppression ofgadd45 mRNA levels, by a mechanism which is
independent of cell cycle progression. Using an inducible MycERTM system, rapid
suppression of gadd45 mRNA is first evident approximately 0.5h following Myc
activation. The reduction in gadd45 mRNA expression occurs at the transcriptional level
and is mediated by a p53-independent pathway. Notably, the repression in gadd45 mRNA
levels following serum-stimulation is absent in c-Myc null cells, demonstrating that Myc is
required for this activity. Moreover, we show that the Myc-mediated repression ofgadd45
is an essential regulator of basal gadd45 expression in proliferating fibroblasts. Thus, Myc
plays an essential role in the regulation of gadd45 expression; defining a novel pathway
through which Myc promotes cell cycle entry and prevents growth arrest of transformed
cells.

4 .2 Introduction
The ability of Myc to inhibit cellular growth arrest and promote GOIG1 to S phase
transition likely contributes to its strong oncogenic potential. Physiologically, c-myc
expression is tightly linked to the growth state of the cell (Dean et al., 1986; Kelly and
Siebenlist, 1986; Waters et al., 1991). Expression is nearly undetectable in growth-
arrested cells and is transiently elevated in response to mitogens, yet remains evident
throughout the entire cell cycle (Eilers et al., 1989; Eilers et al., 1991; Ham et al., 1985;
Kaczmarek et al., 1985). Exposure of non-transformed cells to growth arrest signals such
as se~m-withdrawal or inducers of differentiation, triggers a rapid drop in Myc expression
which results in cellular growth arrest. This responsiveness to extracellular signals is lost
with c-myc deregulation, such that a Myc-activated cell which encounters a growth-arrest
signal can no longer exit the cell cycle, and will undergo programmed cell death (apoptosis)
(Askew et al., 1991; Evan et al., 1992). Thus, Myc elicits its effects in a dose-dependent
manner; elevated levels stimulate cell proliferation and apoptosis, whereas the absence of
Myc may be required for growth arrest and differentiation. How Myc can so effectively
and universally control the balance between cell growth arrest and proliferation remains
unclear.
Evidence to date suggests c-Myc mediates such diverse activities by bctioning as
a transcriptional activator when complexed with Max, another basic region helix-loop-helix
leucine zipper protein (Amati et al., 1992; Blackwell et al., 1990; Blackwood and
Eisenman, 1991; Blackwood et al., 1992; Prendergast et al., 1991; Prendergast and Ziff,
1991). MycIMax heterodiiers bind to the canonical DNA hexamer CACGTG as well as to
specific non-canonical elements (Blackwell et a]., 1993; Blackwood and Eisenman, 1991;
Fisher et al., 1993; Grandori et al., 1996). Despite Myc being well-defined as an activator
of gene transcription, only a few direct genetic targets of Myc transactivation have been
identified in vivo, such as ornithine decarboxylase (odc) (Bello-Fernandez et al., 1993;

Wagner et al., 1993), carbamoyl-phosphate syt~tl~ase/aspartarecarbamoyltransferase/
dil~ydroorotase (cad) (Miltenberger et al., 1995), cdc25A (Galaktionov et al., 1996); eIF-
4E (Rosenwald et al., 1993), a DEAD box-related gene (MrDb) (Grandori et al., 1996);
a-prothymosin (Desbarats et al., 1996; Eilers et a]., 1991; Gaubatz et a]., 1995) and
ECA39 (Benvenisty et al., 1992).
As well as a transcriptional activator, Myc plays an important role as a repressor of
gene expression (Desbarats et al., 1996; Inghirami et al., 1990; Lee et al., 1996; Li et al.,
1994; Penn et al., 1990; Philipp et al., 1994; Versteeg et al., 1988; Yang et a]., 1993).
Recent structure-function analysis of the amino-terminus of the c-Myc protein shows Myc
repression and Myc kansactivation are separable, with Myc repression being more closely
associated with neoplastic transformation (Brough et al., 1995; Lee et al., 1996; Li et al.,
1994; Penn et al., 1990). Myc can repress c/ebpa(Antonson et al., 1995; Li et al., 1994),
c y c h D l (Philipp et al., 1994), the adenovims 5 major late promoter (AdMLP) (Li et al.,
1994), and c-myc (Facchini et al., 1997) promoter activities. Myc suppression appears to
be mediated through core regulatoly elements and does not require the consensus Myc/Max
binding sites within the target promoter sequence (Antonson et al., 1995; Facchini et al.,
1997; Li et al., 1994; Lucas et al., 1993; Philipp et al., 1994). Interestingly, Max
heterodimerization is not required for cyclin D l suppression (Philipp et al., 1994), yet it is
obligatory for Myc autosuppression (Facchini et a]., 1997) and remains unexplored for
d e b p a and AdMLP suppression. It remains unclear whether Myc repression of these
target genes occurs by direct or indirect pathways; however, Myc can clearly repress gene
expression by more than one mechanism (Marhin et al., 1996).
By identifying cellular Myc-regulated gene targets, the molecular mechanism(s) of
Myc as a regulator of gene transcription can be delineated. Moreover, the link between
Myc repression and transformation suggests the identification of genes whose expression
is suppressed in response to Myc will also prove crucial to understanding the role of Myc
in neoplastic transformation. Intuitively, candidate Myc-repressed genes could include

those whose pattern of expression contrasts that of c-Myc and are highly expressed during
cellular growth arrest or quiescence. Known genes belonging to this category include the
growth arrest-specific (gas) genes (Schneider et al., 1988), the serum deprivation response
gene (sdr) (Gustincich and Schneider, 1993), the homeobox gar gene (Weir et a]., 1995),
and the growth-arrest and DNA damage inducible (gadd) genes (reviewed in Fomace,
1992; Papathanasiou and Fomace, 1991). With the exception of gas1 andgadd45, the
hctional role of these genes in effecting growth arrest is not clear.
The gadd genes are coordinately expressed in response to growth arrest conditions
as well as to a variety of DNA damaging agents (Fomace et al., 1988; Fomace et al.,
1989). Elevated gadd45 expression has also been linked to growth arrest triggered either
by developmental signals (Constance et al., 1996) or to nutrient-deprivation associated
with GO phase of the cell cycle (Fomace et al., 1989; Smith and Fomace, 1996; Zhan et
al., 1994). Upon exposure to ionizing radiationgadd45 is induced (Hollander et al., 1993;
Papathanasiou et al., 1991), and this induction is dependent upon the p53 transcription
factor (Zhan et al., 1993). Functionally, Gadd45 con?ers a strong block to proliferation as
shown through ectopic expression in tumorigenic carcinoma cell lines such as HeLa and
RKO as well as non-transformed NIH3T3 cells (Vairapandi et al., 1996; Zhan et al.,
1994). Gadd45 is thought to mediate growth arrest through its interaction with two cell
cycle components: the cyclin dependent kinase inhibitor p21, and proliferating cell nuclear
antigen (PCNA) (Chen et al., 1995; Hall et al., 1995; Kearsey et al., 1995). The
mechanism to overcome the growth-inhibitory affects of genes such as gadd45 and exit
GO, in response to mitogen-stimulation or Myc-activation, remains unclear.
In this thesis we show gadd45 gene transcription is repressed in response to Myc
activation. Ectopic Myc expression in both primary and immortalized fibroblasts results in
a reduction in gadd45 mRNA expression. Myc suppression of gadd45 mRNA
corresponds to a similar reduction in Gadd45 protein levels in Rat-1 cells expressing
ectopic Myc protein. Although basal levels of gadd45 are low in subconfluent

asynchronous cultures, expression of ectopic Myc can further down-regulate gadd45
expression, demonstrating that Myc suppression ofgadd45 is not a simple consequence of
cellular transition fiom quiescence into the cell cycle. The suppression ofgadd45 by Myc
occurs with rapid kinetics and occurs at the level of gene transcription in a p53-independent
manner. Myc suppression affects the overall level of Gadd45 expression, as cells
expressing an activated c-myc allele do not show maximal induction of gadd45 expression
in response to gamma-radiation, We demonstrate that Myc plays an essential role in the
regulation of gadd45 expression in mitogen-stimulated and asynchronous proliferating
cells. The suppression ofgadd45 in response to Myc represents a novel and biologically
critical pathway, elucidating one of the mechanisms via which Myc stimulates cells to exit
growth arrest, enter the cell cycle and contribute to transformation.

4.3 Materials and Methods
4.3.1 Cell culture
Primary rat embryonic fibroblasts (REF) and primary mouse embryonic fibroblasts
(MEF) were prepared as previously described (Jacks et al., 1992; Land et al., 1983). Rat-
1 fibroblasts are a subclone of the Fischer rat embryo fibroblast line F2408 (Lania et al.,
1980). Wildtype TGR-1 (+I+) and c-Myc null H015.19 (-I-) cell lines were previously
described (Mateyak et al., 1997). c-Myc null H015.19 gfp (-1- gfp) and c-Myc null
H015.19 gfpmyc (-1- gfpmyc) cells were generated by infecting c-Myc null H015.19 (-I-)
cells with either the retrovirus BabeMNIRESgfp or BabeMNIRESgfpmyc.
Wildtype Rat-1 clone TGR-1 (+I+), c-Myc null H015.19 (-I-), H015.19 gfp (-1-
gfp) and H015.19 gfpmyc (-1- gfpmyc) cells were grown in DMEM HZ1 supplemented
with 10% calf serum, as described (Prouty et al., 1993). All other fibroblast cell lines
were cultured in alpha modified Eagle's medium (aMEM) supplemented with 10% fetal
bovine serum (FBS)(Gibco). Culture media was supplemented with 100 pdml kanamycin
(Sigma) and 2 pdml gentamycin (Roussel) or 100 pglml penicillin (Sigma) and 100 pglml
streptomycin sulfate (Sigma). To activate ectopic Myc activity, Rat-1 wtMycERTM and
AMycERm cells were exposed to 100 nM 4-hydroxy (2) Tamoxifen (OH-T) (Research
Biochemical International, Natick, MA). Rat-1 fibroblasts were exposed to 10 Gy of
ionizing radiation using a source at 1.13 Gylmin. To analyze the response to
serum stimulation, subconfluent wildtype TGR-1 (+I+), and c-Myc null H015.19 (-I-)
cells were maintained in 0.25% calf serudDMEM HZ1 for 2 days, then stimulated with
10% calf serudDMEM H21. Unless otherwise stated, all cells were analyzed as
subconfluent proliferating cultures. Early passage cell cultures were used for all assays.

4.3.2 Retroviral infection
The pDORheo, pBabePnygro, pBabeIpuro, pDoklv-mycneo, pBabelv-mychygro,
pBabelv-mycpuro, pSVX.1, pSVX.6, pBabepuro c-mycERTM, or pBabepuro 4106-143c-
mycERTM retroviral vectors were constructed as previously described (Facchini et al.,
1997; Littlewood et al., 1995; Morgenstem and Land, 1990; Ridley et al., 1988). To
generate the pBabeMNIRESgfpmyc retroviral vector, human c-myc exon 11,111 sequences
excised from pBluescript KS+ Hc-myc were cloned into EcoRI, XhoI digested
pBabeMNIRESgfp (a kind gift of G. Nolan).
To produce infectious replication-defective ecotropic retroviral particles,
recombinant retroviral constructs were transfected, using calcium phosphate precipitation
(Graham and Van der Eb, 1973), into either the GP+E packaging cell line (Markowitz et
al., 1988), or the Phoenix-Eco cell line (ATCC), and selected in 0.5 m g h l G418 sulphate
(Sigma), 2 mglml puromycin (Sigma) or 300 pglml hygromycin B (Sigma). Drug
resistant clones were pooled and expanded for virus production. Fibroblast cultures were
subsequently infected with retrovirus and stable drug-resistant colonies were combined
generating pooled populations. To generate clonal populations of Rat-1 wtMycERTM and
Rat-1 AMycERTM cells, individual drug resistant colonies were isolated. Green
fluorescent protein (gfp) positive c-Myc null H015.19 gfp (-1- gfp) and H015.19 gfpmyc
(-1- gfpmyc) cells were isolated with a Coulter 753 cell sorter using an absorption
wavelength of 488 nm, and pooled.
4.3.3 RNase Protection and luciferase assays
RNA was prepared by the guanidinium isothiocyanate method (Chirgwin et al.,
1979) and assayed by RNase protection assay as described (Penn et al., 1990). The
probes were generated using T3 RNA polymerase (Stratagene) from linearized Bluescript

KS and SK cloning vectors (Stratagene) containing: rat c-myc exon 1, thegag gene derived
from the avian myelocytomatosis virus MC29, murine and rat gapdh cDNA (Facchini et
al., 1994; Penn et al., 1990). The probe for the rat gadd45 gene was transcribed using
Sp6 RNA polymerase (Stratagene) from a linearized pGEM3ZF+ vector (Promega)
containing a 450 bp PstI fiagment of the rat gadd45 cDNA (kind gift of T. Yoshida). The
aminoglycoside phosphotransferase(neo) probe comprised nucleotides 1 to 245 of the
neomycin resistance gene. These sequences were excised from pBabeneo (Morgenstern
and Land, 1990) with Hind111 and ClaI, and cloned into a EcoRV and ClaI digested
pBluescript KS+ vector (Stratagene). The Bluescript KS+ aminoglycoside
phosphotransferase plasmid was linearized with NcoI, and the RNase protection probe
transcribed with T3 RNA polymerase (Stratagene). The protected probes were resolved by
electrophoresis on 6% denaturing polyacrylarnide gels and visualized by autoradiography
on X-OMAT film (Kodak). Luciferase and P-galactosidase assays were performed as
previously described (Facchini et al., 1997).
4.3.4 Immunoblotting and Immunoprecipitation
Antibodies used in this study include the monoclonal antibodies, anti-p53 Pab421
and anti-SV40 Large T antigen Pab419 (kind gifts of S. Benchiiol), as well as polyclonal
anti-gadd45 antibody Sc797 (Santa Cruz), and a polyclonal pan-myc antibody (kind gift of
G. Evan). Whole cell extracts were prepared as recommended (Santa Cruz), and total cell
lysates (5 x 10s cells) were immunoblotted as previously described (Daksis et al., 1994).
Proteins were detected by incubating the membrane with their respective antibody for 1 h in
TBS-T containing 1% non-fat milk. The membrane was washed, and incubated with a
112000 dilution of either horseradish peroxidase-conjugated goat anti-mouse IgG (Biorad)
or swine anti-rabbit IgG antibody (Dako) in TBS-T containing 1% non-fat milk for 0.5 h.
After washing, signals were detected using the ECL (Amersham) detection system and

visualized by autoradiography (Kodak). Imrnunoprecipitations using the anti-gadd45
polyclonal antibody (Sc797) were performed as recommended by the manufacturer (Santa
Cruz). Immune complexes were resolved on a 10% SDSlpolyacrylamide gel, transferred
to PVDF membrane (Millipore), developed with the same antibody, and then visualized by
autoradiography (Kodak).

4.4 Results
4.4.1 Sequential induction of c-myc and suppression of gadd45 mRNA
levels following serum stimulation of quiescent Rat-1 fibroblasts
To determine the relative kinetics of c-myc and gadd45 expression during cell cycle
progression, we assayed the endogenous mRNA expression of these two genes in
quiescent Rat-1 fibroblasts following serum stimulation. Confluent Rat-1 fibroblasts were
cultured in low serum (0.1% FBSIaMEM) for 2 days to induce quiescence and then
mitogen stimulated by changing the medium to 10% FBSIaMEM. RNA was harvested at
0, 0.5, 1, 2, 3, 6 , 12, 18, 24 hours post serum-stimulation and assayed by RNase
protection using single-stranded probes complementary to mRNA of endogenous rat c-
myc, rat gadd45 and rat glyceraldehyde-3-phosphate dehydrogenase (gapdh). Mitogen-
stimulation of quiescent Rat-1 fibroblasts resulted in a transient increase in c-myc mRNA
expression first evident at approximately 0.5 hours after serum-stimulation (Figure 4.1,
compare lane 2 with 1). This was followed by a gradual suppression in endogenous
gadd45 mRNA levels which was clearly evident approximately 1 hour post-serum (Figure
4.1, compare lane 3 with lane 1). To ascertain whether the suppression ofgadd45 mRNA
levels was dependent upon de novo protein synthesis, confluent quiescent Rat-1 cells were
treated simultaneously with 10% FBSIaMEM and 50 bg/ml cycloheximide for 3 hours.
Expression of the endogenous gadd45 gene was not suppressed in the presence of
cycloheximide, showing gadd45 suppression was dependent upon de novo protein
synthesis following serum stimulation. The timing of these two mechanistic events
suggested a role for Myc in effecting the suppression of gadd45 mRNA expression.

Figure 4.1. Serum stimulation of confluent, quiescent Rat-1 fibroblasts results in the induction of c-nryc mRNA expression followed by a suppression of gadd45 mRNA levels. Rat-1 cells were maintained in low serum 0.1% FBSIaMEM for 2 days then left untreated (lane 1) or exposed to 10% FBS (lanes 2 to 9) or 10% FBS and 50 pg/ml cycloheximide (lane 10). At the indicated intervals RNA was extracted and analyzed by RNase protection using single stranded probes complementary to endogenous c-myc, gadd45, and gapdh- specific sequences.

4.4.2 Ectopic Myc expression in primary cell cultures and immortalized
cell lines results in a suppression of endogenous gadd45 mRNA
levels
To determine whether ectopic Myc expression can suppress gadd45 mRNA levels,
primary rat embryo fibroblasts (REF) and the immortal non-tumorigenic Rat-1 cell line,
were infected with control retrovirus (DORheo) canying the neomycin resistance gene or
retrovirus (DoWv-mycneo) also carrying the v-myc gene. Primary mouse embryo
fibroblast cultures (MEF) were similarly infected with either a control retrovirus
(Babelpuro) or a retrovirus (Babelv-mycpuro) carrying the v-myc gene. Drug resistant
colonies were pooled and analyzed as asynchronous proliferating cultures. Subconfluent-
growing cells were analyzed to ensure changes in endogenous gadd45 gene expression
were not due to an indirect effect of Myc on cell cycle (Evan et al., 1992). Under the
conditions of these experiments, populations of both control cells and cells expressing
ectopic Myc protein showed similar rates of growth and a similar distribution of cells
throughout all phases of the cell cycle (data not shown). RNA was extracted and analyzed
by RNase protection assay for the expression of the endogenous gadd45 and gapdh
mRNAs. Expression of ectopic Myc protein in primary cultures as well as in Rat-1 cells
resulted in a 4 to 5 fold repression in endogenous gadd45 mRNA levels compared to
controls as determined by densitometry (Figure 4.2A, compare lanes 2,4,6 to lanes 1,3,5,
respectively). Suppression of gadd45 was also clearly evident in Myc-expressing cell
cultures which were grown to confluence (data not shown). The ectopic v-myc gene was
expressed as demonstrated by RNase protection assay (Figure 4.2B). Thus, constitutive
ectopic Myc expression leads to a suppression in gadd45 mRNA levels.
To determine if the suppression in gadd45 mRNA levels results in a corresponding
decrease in Gadd45 protein expression, whole cell lysates were prepared from
subconfluent asynchronous cultures of both control Rat-1 and Rat-1 cells expressing

Figure 4.2. Ectopic Myc expression in primary and immortalized
fibroblasts results in the suppression of gadd45 mRNA and protein levels.
(A) 10 pg of RNA from subconfluent proliferating rat embryo fibroblasts (REF), Rat-1
fibroblasts (Rat-I), and mouse embryo fibroblasts (MEF) infected with control retrovirus
(-) (lanes 1,3,5), or retrovirus carrying the v-myc gene (+) (lanes 2,4,6) was analyzed by
RNase protection assay to detect the presence of endogenous gadd45 and gapdh-specific
sequences; (B) 10 yg of RNA from subconfluent proliferating rodent fibroblasts infected
with control retrovirus (-) (lanes 1,3,5), or retroviruses canying the v-myc gene (+) (lanes
2,4,6) was analyzed by RNase protection assay to detect the presence of ectopic v-myc
and gapdh-specific sequences (C) 2 mg of total cell lysate from subconfluent Rat-1
fibroblasts infected with a control retrovirus (-), or retroviruses canying the v-myc gene
(+) was immunoprecipitated using a polyclonal anti-Gadd45 antibody (Sc797).
Immunoprecipitated proteins were resolved on a 10% polyacrylamide gel and Gadd45
protein detected by western blot analysis using antibody Sc797. The weak signal in the
control lanes of the western blot probed for exogenous Myc expression represents across-
reactive band evident with this batch of pan-myc antibody.

A. REF Rat-1 MEF
B. REF Rat-1 MEF + "--I - + '
I + I v-myc -

ectopic v-Myc protein and analyzed by immunoprecipitation followed by immunoblot
analysis. Rat-1 cells constitutively expressing ectopic Myc protein exhibited a 5 fold
decrease in Gadd45 protein expression compared to control cells (Figure 4.2C). This level
of suppression correlated with the observed 4 to 5 fold decrease in gadd45 mRNA
expression. Western blot analysis clearly demonstrated that the ectopic v-Myc protein is
expressed in Rat-1 Myc cells compared to the control Rat-1 cells. The weak signal in the
control lanes of the western blot probed for v-Myc expression represents a non-specific
cross-reactive band. Thus, constitutive ectopic Myc expression results in a suppression in
gadd45 mRNA levels with a corresponding decrease in Gadd45 protein expression.
4.4.3 The suppression of gadd45 in response to Myc-activation occurs
with rapid kinetics
To determine the kinetics of suppression of gadd45 in response to Myc we
employed an estrogen-inducible M ~ C E R ~ ~ system in Rat-1 cells (Littlewood et al., 1995).
Rat-1 cells were infected with a retrovirus carrying a chimeric gene consisting of the human
c-myc cDNA linked in frame to the estrogen binding regulatory domain of the estrogen
receptor, and individual drug-resistant clones were isolated. These Rat-1 wtMycERTM
cells constitutively express an inactive MycERTM fusion protein, which is rapidly activated
following exposure of cells to hydroxy-tamoxifen (OH-T). By this approach, Myc can be
induced in the absence of mitogen-stimulation. Confluent Rat-1 wtMycERTM cells were
cultured in low serum (0.1% FBSIaMEM) for two days to induce quiescence and then
treated with OH-T to induce ectopic Myc activity. RNA was extracted at 0,0.5, 1,2,3,6,
9, 12, 18, 24 hours following exposure of cells to OH-T and expression of gadd45 and
gapdh mRNA determined by RNase Protection assay (Figure 4.3). Induction of ectopic
Myc activity triggered a reduction ingadd45 mRNk levels, with a 2 fold reduction at 3 h
and a maximal 5 fold suppression after approximately 12 hours, as determined by

Figure 4.3. gadd45 mRNA expression is suppressed with rapid kinetics in response to Myc induction. Confluent Rat-1 wtMycERm cells were maintained in low serum 0.1% FBSIaMEM for 2 days and then exposed to 100 nM OH-T to induce exogenous MycERm activity. At the indicated intervals after OH-T treatment RNA was extracted and 10 pg total RNA analyzed by RNase protection assay using single stranded probes complementary to endogenous gadd45, and gapdh-specific sequences.

densitometry. Similar results were observed in multiple independent Rat-1 wtMycERm
cell clones. To determine whether Myc directly or indirectly suppresses gadd45, Rat-1
wtMycERm cells were exposed to cycloheximide at the time of MycER activation;
however, the short 20-30 minute half-life of MycER quickly depleted the cell of effector
protein making results uninterpretable (data not shown). As the half-life of gadd45 mRNA
is approximately 20 to 80 mins (Jackman et al., 1994), Myc repression of gadd45 RNA
was rapid and first evident as early as 0.5 h following OH-T treatment. gadd45 repression
was not evident in OH-T treated control Rat-1 AMycERTM cells expressing a non-
functional MycERTM fusion protein harbouring a deletion in amino acids 106-143 of the
Myc portion of the fusion protein (data not shown). This region of the c-Myc protein is
required for all known biological activities of Myc (Evan et al., 1992; Freytag et al.,'1990;
Garte, 1993; Penn et al., 1990; Stone et al., 1987). This cell line serves as a control for
the expression and hormone-activation of the M ~ C E R ~ ~ fusion protein. Thus, our results
show gadd45 mRNA expression is suppressed in response to Myc with rapid kinetics.
4.4.4 The suppression of gadd45 in response to Myc does not require
wildtype p53 activity
It is well established that induction of the gadd45 gene following exposure to
ionizing radiation, is mediated by the transcription factor p53 (Kastan et al., 1992; Zhan et
al., 1994; Zhan et al., 1993). To determine whether Myc may indirectly repress gadd45
expression by affecting wildtype p53 expression, RNA isolated from subconfluent control
Rat-l and Rat-l cells expressing ectopic v-Myc protein was assayed for endogenous p53
mRNA and protein expression. There was no change in endogenous p53 mRNA
expression in Rat-1 Myc cells compared to control Rat-1 cells (Figure 4.4A).
Furthermore, comparable levels of endogenous wildtype p53 protein were expressed in

Figure 4.4. p53 mRNA and proteh expression is not responsive to ectopic Myc expression in Rat-1 cells. (A) 10 pg of RNA from subconlluent Rat-1 cells infected with a control retrovirus (-), or retrovirus canying the v-myc gene (+) was analyzed by RNase protection assay to detect the presence of endogenous p53 and gapdh-specific sequences. (B) Immunoblot analysis of 200 pg of total cell lysates from subconfluent Rat-1 cells infected with a control retrovirus (-), or retroviruses canying the v-myc gene (+) to detect endogenous p53 protein expression.

control and Myc-expressing Rat-1 cells (Figure 4.4B). Thus, constitutive ectopic Myc
expression does not affect the expression of endogenous p53 mRNA or protein in Rat-1
cells.
To determine whether wildtype p53 activity is required for the suppression of
gadd45 by Myc, control Rat-1 and Rat-1 Myc cells were infected with either a control
retrovirus (SVX.1) carrying only the neomycin resistance gene, or a retrovirus (SVX.6)
also canying the SV40 LTag gene. Following selection, drug-resistant colonies were
pooled and analyzed as subconfluent proliferating cultures. The expression of SV4O LTag
results in the inactivation of wildtype p53 activity (Jiang et al., 1993; Lenahan and Ozer,
1996; Mietz et al., 1992). Constitutive expression of ectopic SV40 LTag had no
significant effect on gadd45 expression in control Rat-l cells or in Rat-1 Myc cells (Figure
4.5A). To ensure that the products of the ectopic genes were expressed, whole cell lysates
from each of the Rat-1 cultures were assayed by Western blot analysis for the expression
of ectopic v-Myc and LTag proteins. Ectopic v-Myc protein was clearly expressed in the
appropriate cell lines (Figure 4.5B, compare lanes 3 and 4 with 1 and 2). Ectopic SV40
LTag protein expression was also highly expressed in Rat-1 cells which were infected with
the SVX.6 retrovirus canying the LTag gene (Figure 4.5B, compare lanes 2 and 4 with 1
and 3). Thus, wildtype p53 activity was not required for either basal level gadd45
expression nor Myc suppression of gadd45.

Figure 4.5. The suppression of gadd45 by Myc does not require wildtype
p53 activity. (A) 10 pg of RNA from subconfluent Rat-1 cells infected with a control
retrovirus (-), retrovirus carrying the v-myc gene andlor retrovirus carrying the SV40 large
T antigen (+) was analyzed by RNase protection assay to detect the presence of
endogenous gadd45 and gapdh-specific sequences. Histograms represent gadd45 mRNA
abundance relative to the control as quantitated by densitometry. All data was normalized
to the level ofgapdh mRNA expressron. (B) Immunoblot analysis of total cell lysates from
subconfluent Rat-1 fibroblasts as described in (A), using a pan-myc polyclonal antibody to
detect ectopic v-Myc protein, and the monoclonal antibody Pab419 to detect ectopic SV40
LTag expression.

1 .o
0.8 RELATIVE
gadd45 0.6 infWA
ABUNDANCE 0.4
0.2
0.0
v-myc 6 I + + SV40LTag - + - +
Rat-1 I
v-myc I - + + I SV40LTag - + I. +

4.4.5 Myc and p53 co-regulate gadd45 transcription
gadd45 mRNA expression is suppressed in response to Myc activation and induced
by p53 following exposure to ionizing radiation (Kastan et al., 1992; Zhan et al., 1994;
Zhan et al., 1993). To determine whether these two pathways interact, subconfluent
proliferating control Rat-1 and Rat-1 Myc cells were exposed to 10 Gy of ionizing
radiation. Following treatment of cells, total RNA was extracted at 0, 1, 3, 8, 12 and 18
hours and expression of endogenous gadd45 and gapdh mRNA was analyzed by RNase
protection assay. gadd45 mRNA expression in unirradiated Rat-1 Myc cells was
suppressed compared to unirradiated control Rat-1 cells (Figure 4.6, compared lane 7 to
1 j. Furthermore, exposure of either of the two cell lines to ionizing radiation led to the
gradual induction of gadd45 mRNA expression (Figure 4.6, compare lanes 1 to 6 with
lanes 7 to 12). Importantly, maximal achievable levels ofgadd45 expression evident in the
control cells were not reached in the Myc-activated cells after 18 hours (Figure 4.6).
Extended time courses show similar results (data not shown). Hence both mechanisms;
the suppression of gadd45 by Myc and the induction of gadd45 by p53, are independent
regulatory mechanisms which combine to co-regulate gadd45 expression.
4.4.6 Myc regulates transcription from the gadd45 Promoter
To ascertain whether Myc represses gadd45 expression at the level of gene
transcription, we conducted transient-transfection reporter assays in both Rat-1
W ~ M ~ C E R T M and control Rat-1 AM~CERTM cells. Thegadd45 promoter lacks both
TATA and Inr elements and contains few putative transcription factor binding sites. There
are two potential OCT and two CAT sites located within 100 bp upstream of the
transcription start site, and an APl site as well as a p53 binding site in intron 3 which are


conserved in the human, mouse and hamster genes (Figure 4.7A) (Hollander et al., 1993).
As our results show that Myc suppression is p53 independent, we focused ow analysis on
upstream regulatory sequences using a hamstergadd45 promoter luciferase construct
consisting of the sequences from -1675 to +I49 relative to the transcription start site. This
construct was transiently transfected into subconfluent proliferating Rat-1 wtMycERTM
and control Rat-1 AM~CERTM cells. To control for transfection efficiency a vector
carrying the lacZ gene, was co-transfected with the promoter luciferase construct. Cells
were treated with OH-T and at 0,3,6, 9, 12, lg, 24 hours thereafter, whole cell lysates
were prepared and then assayed for both luciferase and P-galactosidase activity. Treatment
of Rat-1 AMycERm cells with OH-T had no effect on luciferase activity (Figure 4.7B).
However, OH-T induction of ectopic Myc activity in Rat-1 wtMycERm cells resulted in
the down-regulation of luciferase activity. As the half-life of luciferase is 4 to 6 hours
(Thompson et al., 1991), the level and kinetics of repression as measured in the gadd45
promoter-luciferase assay (Figure 4.7B) closely mimicked the suppression of endogenous
gadd45 mRNA in response to MycERTM activation (Figure 4.3). The repression of the
gadd45 promoter by Myc is specific, as induction of Myc aztivity had no significant effect
on P-galactosidase activity (Figure 4.7C). Thus, Myc can suppress transcription from the
gadd45 promoter, and this suppression activity can be localized to nucleotide sequences
found between -1675 and +149, relative to the transcription start site.
4.4.7 Deletion analysis of the hamster gadd45 promoter
The 1.8 Kb Xba I fragment of the hamster gadd45 promoter is responsive to Myc
activation. To determine which sequences in this promoter fragment are required for the
repression of transcription by Myc, consecutive deletions were made at the 5' end of the
Xba I fragment of the hamster gadd45 promoter, generating a series ofgadd45 promoter
luciferase constructs (Figure 4.8). These gadd45 promoter luciferase constructs were

transfected into Rat-1 wtMycERTM cells and assayed for the responsiveness of each
promoter to Myc activation. For each promoter construct, induction of Myc activity elicited
a 3 to 4 fold repression in luciferase activity relative to basal luciferase activity. Deletion of
419 bp at the 5' end of the Xba I hgment of the hamstergadd45 promoter elicited a 3 fold
elevation in basal luciferase activity suggesting that negative regulatory sequences are
present at the 5' end of the promoter. The subsequent deletion of 613 bp, truncating the
promoter to approximately 1 Kb, eliminates the elevation in basal transcription from this
promoter fragment, hinting at a positive regulator of transcription between nucleotides
-1256 and -643. Further deletions to position -222, relative to the transcription start site,
do not significantly impact on basal transcription. However deletion of a region of the
promoter consisting of nucleotides -222 to -72, which contains the putative transcription
factor binding sites, an Oct and a Cat site, results in a loss of basal luciferase activity.
Thus, sequences found between -222 and +I49 are required to maintain basal transcription
of the hamster gadd45 promoter, and contained within this region are sequences which
mediate the repression ofgadd45 transcription by Myc.

Figure 4.7. gadd45 transcription is suppressed in response to Myc. (A)
Schematic diagram of the genomic structure of the hamstergadd45 gene, showing putative
transcription factor binding sites. Numbers indicate position relative to the transcriptional
start site (+I). Rat-1 AMycERm and Rat-1 wtMycERW cells were transfected with a 1.8
Kb hamster gadd45 promoter luciferase construct, containing gadd45 sequences from
nucleotides -1675 to +149. To assay transfection efficiency, cells were co-transfected with
a plasmid carrying the Pgalacfosidase gene under the control of the CMV promoter. Cell
lines were subsequently treated with 100 nM OH-T for the times indicated to induce ectopic
MycERm activity. Whole cell lysates were prepared and analyzed for luciferase and P- galactosidase activity. (B) Histogmms represent the measured luciferase activity relative to
the untreated control. Data was normalized to P-galactosidase activity to adjust for minor
differences in transfection efficiency amongst the samples. (C) Histograms represent the
P-galactosidase activity. Data are a representative out of 3 repeated experiments. Error bars
denote standard deviations.

Hamster gadd45 promoter
TIME (h)
0 3 6 9 12 18 24 TIME (h)
0 3 6 9 12 18 24 TIME (h)
-- 0 3 6 9 12 18 24
TIME (h)
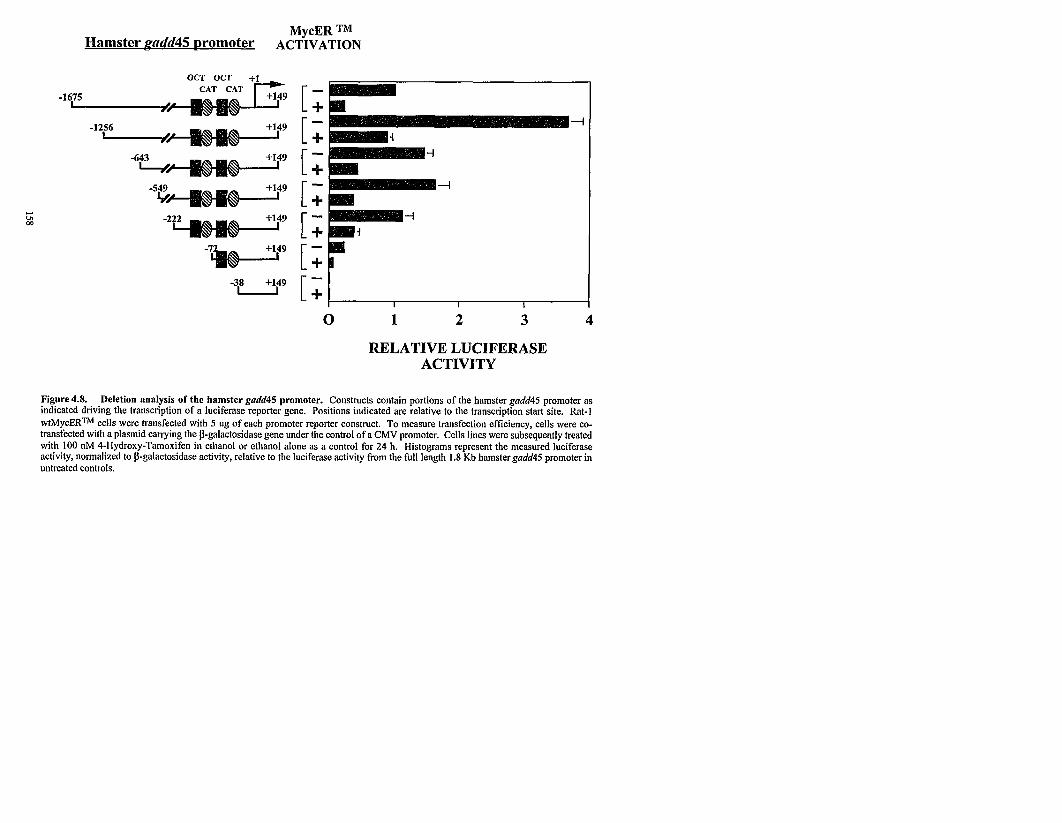
MycER TM Hamster gndd45 ~romoter ACTIVATION
OCT OCT
-1
RELATIVE LUCIFERASE ACTIVITY
Figure 4.8. Deletion analysis of the hamster gadd45 promoter. Constructs contain portions of the hamster gadd45 promoter as indicated driving the transcription of a luciferase reporter gene. Positions indicated are relative to the transcription start site. Rat-l w t ~ y c E R ~ M cells were transfected with 5 ug of each promoter reporter construct. To measure transfection efficiency, cells were co- transfected with a plasmid carrying the p-galactosidase gene under the control of a CMV promoter. Cells lines were subsequently treated with 100 nM 4-Hydroxy-Tamoxifen in ethanol or ethanol alone as a control for 24 h. Histograms represent the measured luciferase activity, normalized to p-galactosidase activity, relative to the luciferase activity from the full length 1.8 Kb hamstergadd45 promoter in untreated controls.

4.4.8 Myc is an essential negative regulator of gadd45 expression
Mitogen-stimulation of quiescent fibroblasts elicits a transient induction in
endogenous c-Myc protein expression; coincident with a reduction in gadd45 mRNA
expression. To determine if Myc is required for the repression in gadd45 mRNA levels
following serum-stimulation, we analyzed gadd45 mRNA expression in c-Myc null
H015.19 (-I-) cells, after stimulation with serum. H015.19 (-I-) cells, were cultured in
0.25% calf s e r u d M E M H21 for 2 days then treated with serum. At the indicated time
intervals, total RNA was extracted and analyzed for gadd45 and gapdh mRNA expression
by RNase protection assay. Serum-stimulation of quiescent wildtype Rat-1 (+I+)
fibroblasts elicits a clear repression in gadd45 mRNA levels (Figure 4.1). In contrast, in
serum-deprived c-Myc null H015.19 (-I-) rodent fibroblasts, gadd45 mRNA expression
was not repressed in response to serum-stimulation (Figure 4.9A). Moreover, gadd45
mRNA levels were invariant through the course of the experiment, up to 48 h post-
treatment. Thus, Myc is required for the suppression ofgadd45 mRNA levels following
mitogen-stimulation.
Analysis of exponentially growing cultures revealed that Myc also plays a role in
regulating basal gadd45 mRNA levels. In proliferating c-Myc null H015.19 (-I-) cells,
gadd45 mRNA levels are elevated in comparison to wildtype Rat-1 (+I+) fibroblasts
(Figure 4.9B). Hence, in addition to repressing gadd45 mRNA expression following
serum-stimulation, Myc also plays a key in regula!ing basal expression of the gadd45 gene
in proliferating cells. In conjunction with the elevated expression of the gadd45 gene, the
expression of the endogenous c-myc gene is also elevated (Figure 4.9B). Yet, both genes
remain responsive to Myc as the Myc-mediated repression of both genes can be rescued
upon expression of ectopic Myc in the c-Myc null (-I-) cells. Ectopic Myc expression in c-
Myc null H015.19 gfpmyc (-1- gfpmyc) cells results in a clear repression of endogenous
gadd45 as well as c-myc mRNA levels, in comparison with parental H015.19 (-I-) cells as

well as the control H015.19 gfp (-1- gfp) cells, which expresses only the green fluorescent
protein (Figure 4.9B). RNase protection analysis clearly demonstrates that exogenous
human c-myc mRNA is expressed in the appropriate cell cultures (Figure 4.9C). Thus,
Myc is required for the repression ofgadd45 expression in quiescent rodent fibroblasts,
following serum-stimulation. In addition, the repression ofgadd45 by Myc is an essential
regulatory mechanism maintaining basal gadd45 expression in proliferating fibroblasts.

Figure 4.9 Myc is an essential negative regulator of gadd45 expression (A)
Rat-1 cells, lacking c-Myc expression (-I-), were maintained in low serum 0.25% calf
serum/DMEM H21 for 2 days then treated with 10% calf serurn/DMEM H21. At the
indicated intervals, RNA was extracted and analyzed by RNase protection using single
stranded probes complementary to endogenous gadd45, and gapdh-specific sequences.
RNA(10 yg), extracted from subconfluent cultures of wildtype rodent fibroblasts (+I+),
rodent fibroblasts lacking endogenous Myc expression (-I-), and c-Myc null fibroblasts
which have been stably infected with either a control retrovirus containing the gfp cDNA (-
I- gfp) or a retrovirus containing human c-myc exon 11, 111 cDNA as well as gfp (-I-
gfpmyc) was analyzed by RNase protection for (B) endogenous gadd45, c-myc,
aminoglycoside phosphotransferase (neo) and gapdh (C) ectopic human c-myc, and
endogenous gapdlt-specific sequences. Similar results were ot:ained in three separate
experiments.

Human c-myc
gapdh
1 2 3 4

4.5 Discussion
Identifying the mechanisms by which Myc can overcome growth arrest and
stimulate cells to enter the cell cycle is crucial to our understanding of how Myc promotes
cell proliferation and contributes to cellular transformation. Our results show that the
suppression of the potent growth arrest gene gadd4S by Myc may constitute one such
mechanism. The reduction in gadd4S mRNA levels in response to mitogen stimulation of
quiescent cells coincided with the induction of endogenous c-Myc expression. Moreover,
simultaneous treatment of growth arrested cells with both cycloheximide and senun for 3h
abrogated the reduction in gadd4S mRNA expression, demonstrating that the down-
regulation of gadd4S mRNA levels was dependent upon de novo synthesis of a protein
which was expressed soon after serum stimulation. To investigate the role of Myc in
gadd4S suppression, we ectopically expressed Myc protein in primary cultures as well as
in immortalized non-tumorigenic cell lines. Our results showed a reduction of gadd4S
expression in Myc-activated cells. The 4 to 5 fold suppression of endogenous gadd4S
mRNA levels in Rat-1 cells expressing ectopic Myc protein, correlated with a similar
reduction in Gadd4S protein expression. Importantly, this suppression was evident in
subconfluent proliferating cells where the cell population was asynchronous, showing that
Myc suppression ofgadd45 was not an indirect result of the GOIGl to S phase transition of
the cell cycle.
To determine the kinetics ofgadd45 suppression in response to Myc induction we
employed Rat-1 MycERm cells. The MycERm fusion protein is composed of the human
c-Myc protein (Myc) fused in frame at its carboxyl terminus to the hormone regulatory
domain of the estrogen receptor (ERm). Rat-1 cells stably and constitutively express the
inactive MycERm hsion protein and exposure of cells to 4-Hydroxy-Taznoxifen (OH-T)
rapidly activates MycERm in the absence of the broader response to mitogen. Activation
of the MycERm protein resulted in the suppression of gadd4S mRNA levels; suppression

was first evident as early as 0.5 h following OH-T stimulation and half-maximal
suppression occurred after 3 hours. The kinetics of suppression were rapid, as the half-life
ofgadd45 mRNA is 20 to 80 mins (Jaclanan et al., 1994), showing gadd45 expression is
a downstream target of Myc regulation.
Transcription of thegadd45 gene is directly induced by the p53 transcription factor.
Exposure to ionizing radiation results in an increase in p53 protein levels which in turn
activates transcription through a p53 responsive element in intron 3 of the gadd45 gene
(Hollander et al., 1993; Kastan et al., 1992; Zhan et al., 1994; Zhan et al., 1993). Rat-1
cells express wildtype p53 protein as determined by immunoblot analysis, hence it is
plausible that Myc suppression of gadd45 is either mediated through p53 or requires
wildtype p53 activity. Our results demonstrated that constitutive expression of ectopic Myc
protein in Rat-1 cells did not affect the expression of the endogenous p53 mRNA or
protein. To determine if wildtype p53 activity is rcquired for the suppression ofgadd45 by
Myc, SV40 LTag was ectopically expressed in both control and Rat-1 Myc cells to inhibit
wildtype p53 activity (Jiang et al., 1993; Lenahan and Ozer, 1996; Mietz et al., 1992). We
observed that gadd45 mRNA expression was suppressed in Rat-1 cells which are
expressing ectopic Myc protein, irrespective of the p53 status of the cell. We further show
the suppression ofgadd45 by Myc and the induction ofgadd45 by p53 following exposure
to ionizing radiation were non-competitive regulatory mechanisms which co-regulated
gadd45 expression. Thus, the suppression ofgadd45 by Myc is p53 independent.
The results of the transient transfection reporter assays rlemonstrate that gadd45
transcription was suppressed in response to Myc activation. This response was specific to
Myc as it was not evident in the control Rat-1 AMycERm cells. The region of the gadd45
gene which was sufficient to confer suppression in response to Myc encompassed 371 bp
of the hamster gadd45 promoter containing nucleotides -222 to +149, relative to the
transcription start site. This region of the promoter also lacks the p53 binding site
(Hollander et al., 1993), further supporting our observation that the effects of Myc and p53

on gadd45 regulation are separable. Sequence analysis of the gadd45 promoter revealed
that this regulatory region lacks canonical Myc binding sites; a characteristic which is
common among genes which are repressed by Myc (Antonson et al., 1995; Facchini et al.,
1997). Recent studies suggest that Myc may repress gene expression either by directly
interacting with components of the basal transcription machinery or by inhibiting
transcription through an initiator element (Li et al., 1994; Philipp et al., 1994; Roy et al.,
1993). Interestingly, the gadd45 gene lacks both an initiator site and a TATA box. The
suppression ofgadd45 transcription by Myc appears to be universal, as this mechanism is
evident in both rat and mouse fibroblasts. This suggests that the suppression mechanism
may be effected through shared enhancer elements found in the promoters of the gadd45
gene from different species. Indeed this region of the gadd45 gene possesses only two
putative enhancers which are conserved among human, hamster, and mouse (Hollander et
al., 1993). These include two OCT sites and two CAT sites located within 100 bp
upstream of the transcription start site. The CAT sites are of particular interest as earlier
studies suggest Myc can repress gene transcription by inhibiting the activities of CAT-
binding transcription factors belonging to the CTF/NFl family and the clebp family of
trans-acting factors (Martin, 1991; Wedel and Ziegler-Heitbrock, 1995). Freytag and
colleagues suggest that Myc is able to mediate a change in the phosphorylation state of
CTF/NFl, modifying its potential to activate transcription of the pro alpha 2(I) collagen
promoter (Yang et al., 1991; Yang et al., 1993). Recently, transient transfection reporter
assays demonstrate that in 3T3-L1 cells, ectopic Myc expression can abrogate the clebpa
mediated activation of gadd45 transcription associated with adipocyte differentiation
(Constance et al., 1996). In addition, Mink et al. show ectopic v-Myc can suppress the
expression of myelomonocytic specific genes by inhibiting both the expression and the
fhction of clebpa transcription factors (Mink et al., 1996) Clebpa is predominantly
expressed in differentiated cells (Birkenmeier et al., 1989), hence the role of clebpa in the
GOIGl-associated up-regulation, or Myc-mediated repression of gadd45 in cycling cells,

remains unclear. Indeed, Myc may repress gadd45 directly by interacting with components
of the core transcription complex, indirectly in an enhancer-dependent manner or by a yet
unknown mechanism of action. With the identification of gadd45 as a Myc-suppressed
gene target, the molecular mechanism of Myc-repression of this uncomplicated promoter
can now be delineated.
Although the precise mechanism via which Myc represses transcription of the
gadd45 gene is not known, it is clear that Myc plays an important role in regulating the
expression of the gadd45 gene. Myc is required for the repression of gadd45 expression
following serum-stimulation of serum-deprived fibroblasts. The duration of the cell cycle
is prolonged in c-Myc null fibroblasts in comparison to their wildtype counterparts, 45 to
60 h, and 17 to 22 h, respectively. As such, in our serum-stimulation experiments, we
intentionally monitored gadd45 rnRNA expression through one full cell cycle, in both
wildtype and c-Myc null cultures. In wildtype Rat-1 fibroblasts, gadd45 mRNA levels are
maximally repressed approximately 9 h following serum-stimulation; approximately 4 h
prior to the GI to S transition (data not shown). Yet, in the c-Myc null cells, gadd45
mRNA expression is invariant despite prolonged exposure to serum for 48 h. Hence, the
abrogation of the serum-induced repression in gadd45 mRNA levels, in c-Myc null cells, is
due solely to the absence of c-Myc expression, and not the indirect consequence of a
reduced rate of transit through the cell cycle. The importance of the Myc-mediated gadd45
repression mechanism was further substantiated by the analysis of gadd45 mRNA
expression in subconfluent, proliferating cultures. Basal expression of endogenous
gadd45 mRNA is elevated in comparison with wildtype Rat-1 cells. Yet, the gadd45 gene
is responsive to Myc, as reconstitution of c-Myc null cells with ectopic human c-Myc will
result in the repression in gadd45 mRNA levels.
The identification of Myc-suppressed target genes is of fundamental importance, as
recent studies have linked Myc repression of gene transcription with Myc-induced
transformation (Brough et al., 1995; Lee et al., 1996; Li et al., 1994). Indeed, we show

that the region of the human c-Myc protein, amino acids 106 to 143, required for Myc-
suppression of gadd45, is also required for Myc-induced cell cycle progression, and
cellular transformation (Evan et al., 1992; Freytag et al., 1990; Garte, 1993; Pem et al.,
1990; Stone et al., 1987). Interestingly this region of the c-Myc protein was dispensable
for Myc suppression of cyclin Dl (Philipp et al., 1994), suggesting the molecular
mechanisms of Myc suppression of gadd45 and cyclin Dl are likely non-overlapping and
Myc may repress gene transcription by a number of pathways.
gadd45 expression is strongly associated with growth arrest, and identifying
gadd45 as a Myc-suppressed gene target contributes to our understanding of how Myc can
stimulate cells to exit growth arrest and enter the cell cycle. Myc suppression of gadd45
likely contributes to its turnongenic potential, precluding transformed cells from entering a
growth arrested state. In this report we propose thatgadd45 is a target of Myc repression,
but may not be the sole target of Myc repression to permit the GO to GUS transition.
Indeed we have observed that the growth arrest genes gasl , and gadd153 are also
suppressed in response to Myc, while the gas genes 3 and 6 are not regulated by Myc
(WWM, LZP, unpublished data). We propose that mitogen-stimulation induces Myc
which in turn plays an im,ortant role in co-ordinating the suppression of specific growth
arrest genes, thereby permitting cells to exit GOIGl and enter the cell cycle.

4 .6 Acknowledgments
We thank J. Lear, M. Shago, C. Ho, J. Dimitroulakos, and S. Lee for helpful
discussions and critical reading of the manuscript; S. Benchimol for SV40 large T antigen
and p53 antibodies; T. Yoshida for the rat gadd45 cDNA, and G . Nolan for the
pBabeMNIRESgfp retroviral vector.
Studentship support was kindly provided through the Ontario Graduate Scholarship
Program (W.M.) and the Medical Research Council of Canada (L.F.). This work was
supported by a grant from the National Cancer Institute of Canada with funds from the
Canadian Cancer Society (L.P.).

Chapter 5
FUTURE DIRECTIONS

5 . 1 Summary of research
Accumulating evidence suggests a role for Myc repression in Myc-mediated
tumorigenesis (Lee et al., 1996; Li et al., 1994; Stone et al., 1987). Yet the scarcity of
Myc-repressed cellular target genes has hampered the assessment of the significance and
contribution of Myc repression to enhancing cellular proliferation and tumorigenesis. This
concern is presently being addressed as a search of the literature illustrates the recent
interest in this avenue of Myc research. Realizing the potential of this subset of genes as
regulatory targets for Myc, we sought to identify and characterize genes whose expression
is repressed by Myc. We have pursued three avenues of investigation: we analyzed the
regulatory relationships between three key cell cycle regulators: Myc, cyclin Dl, and pRb
in primary embryonic cells. In addition, we have idxtified two genes pdgf-pr; and
gadd45 whose expression are repressed by Myc at the hanscriptional level. The analysis
of Myc-mediated repression events has contributed to our understanding of the
mechanism(s) of Myc repression, and the role these repression activities play in the
biological functions of Myc.
5 . 2 Analysis of the relationship between Myc, pRb, and cyclin D l
A variety of studies conducted in tumorigenic and immortalized cell lines have led
to the emergence of a number of conflicting regulatory pathways involving three key cell
cycle regulators: Myc, pRb and cyclin Dl. For example, in transformed cell lines, Myc
has been reported to suppress cyclin Dl expression. This repression activity is also
evident in a few selected immortalized cell lines but not others. Similarly, in a variety of
tumour cell lines, it has been demonstrated that hypo-phosphorylated pRb can transactivate
the expression of cyclin Dl. This observation is corroborated by the observation that in
many tumour lines pRb loss is associated with a loss in cyclin Dl expression. This led to

the hypothesis that a feedback mechanism exists between pRb and cyclin Dl .
Hypophosphorylated pRb transcriptionally induces the cyclin Dl gene, cyclin Dl with its
respective cdk, phosphorylates pRb, inactivating its activity. The universality of these
regulatory mechanisms and hence the importance of these findings to our understanding of
cell cycle regulation in non-transformed cells, was unclear. Conceivably, the transfonned
nature of these systems may taint many of these obsetvations. My goal was to assay the
validity of these reported mechanisms in a relatively normal cellular model, primary mouse
embryo fibroblasts.
We demonstrate that these three critical cell cycle regulators: Myc, pRb, and cyclin
Dl exist within a complex interdependent relationship (Marhin et al., 1996). Contrary to
the results obtained in transformed cell lines, in primary cells, pRb is not required for the
expression of cyclin Dl. In addition, Myc does not repress cyclin Dl transcription in
primary cells. Interestingly, cyclin Dl expression is lost in MEFs lacking pRb and
expressing ectopic Myc protein. The repression of cyclin Dl is comprehensive, as it is
evident in Myc-activated pRb-null MEFs derived fiom independent litters. The repression
of cyclin Dl expression can be effected at multiple levels, as we have described earlier.
Thus, irrespective of the mechanism of repression, the ultimate goal in these Myc-activated
pRb-null MEFs is the elimination of cyclin Dl expression. In the context of Myc-
activation and loss of pRb, what is the function of cyclin Dl? A survey of the literature
reveals a strong bias towards a singular function for cyclin Dl, namely the phosphorylation
and inactivation of pRb, facilitating cell cycle progression (reviewed in Sherr, 1996; Sherr
et al., 1994). This model was supported by studies which demonstrate that cyclin Dl
activity was dispensable for cell cycle progression in pRb-null MEFs (Baldin et al., 1993;
Lukas et al., 1995; Quelle et al., 1993). Yet, in pRb-null MEFs, loss of cyclin Dl
expression did affect cell growth, as there was a consistent acceleration into S phase
(Lukas et al., 1995). This observation suggested that cyclin D l expression may exert a
growth inhibitory function which is separable from its role as a pRb kinase in a pRb-null

MEF. Indeed, recent studies support the view that cyclin Dl exerts an activity independent
of the phosphorylation of pRb, and hint at additional novel functions for cyclin Dl. For
example, cyclin Dl can bind to and stimulate the estrogen receptor, activating the
transcriptional activity of the receptor. Importantly, this activity is independent of cdk4
activation (Neuman et al., 1997).
Thus, one can hypothesize that the repression of cyclin Dl in a pRb-null cell will
confer a selective proliferative advantage. This hypothesis is consistent with our
observation that the loss of cyclin D 1 expression in a pRb-null MEF, expressing ectopic
Myc, correlates with an increased proliferative potential (Marhin et al., 1996). Thus, the
cooperation of these two genetic events, Myc-activation and loss of pRb, may constitute a
novel mechanism for tumorigenesis, a mechanism which appears to require the repression
of cyclin Dl. This model is consistent with observations made in surveys of human
tumour cell lines, where there exists a strong correlation between the loss of pRb and the
absence of cyclin Dl expression. This hypothesis poses three questions: first, what is the
mechanism of cyclin Dl repression? Second, the cooperation of Myc-activation and pRb
loss is a novel pathway to enhance tumorigenesis. Is there evidence to support this
relationship in known tumors? Third, is cyclin Dl expression in apRb-null Myc-activated
cell growth inhibitory?
The question as to the mechanism of cyclin Dl repression is a difficult one to
answer. Repression is mediated via mechanisms which affect either mRNA or protein
expression, suggesting that secondaly genetic aberrations within each clone may influence
which pathway is adopted. As the ultimate goal appears to be the loss of cyclin Dl
activi~y, cyclin Dl repression may be effected by one of a multiple array of mechanisms,
depending upon the history of the cell. As such, it is not immediately apparent what can be
gained from characterizing one out of potentially multiple repression mechanisms in one or
two MEF cultures.

To determine if the association of Myc-activation and loss of pRb expression, with
the concomitant loss of cyclin Dl expression, is evident in vivo in known tumour samples
or hunorigenic cell lines, these cell sources will be surveyed to support the existence of this
relationship as a potentially new mechanism for tumorigenesis. In vivo evidence for the
existence of this regulatory association can be found in the cervical carcinoma cell line
HeLa, which possesses deregulated Myc expression, lacks pRb activity, and does not
exhibit cycli D 1 expression.
Our studies in cell culture have revealed a potentially novel mechanism for
tumorigenesis. What is the contribution of each player: Myc-activation and pRb loss, to
this mechanism? Ectopic expression of Myc did not impart an enhanced proliferative
potential to subconfluent asynchronously growing cultures of wildtype MEFs, yet ectopic
Myc expression in pRb-null cells resulted in a dramatic reproducible increase in the
proliferative potential of these cells. What is the reason for this phenomenon? This
question could be addressed by assaying the expression patterns of players in the cell cycle
machinery in control and Myc-activated pRb-null MEFs. In particular, the expression
patterns of cyclins, cdks, cdkls, the temporal activation of the cdks, and the activity of
activating and inhibitory kinases such as the cdc25 and wee1 group of kinases, throughout
the cell cycle will be ascertained. Earlier studies have demonstrated that the specific loss of
cyclin Dl expression will accelerate the transition of pRb null cells into S phase (Lukas et
al., 1995). Is this the reason for the increased proliferative and tumorigenic potential of
Myc-activated pRb-null MEFs? This question can be directly addressed by inhibiting the
expression of cyclin Dl in pRb-null MEFs, through the constitutive expression of
antisense cyclin Dl (Arber et al., 1997; Driscoll et al., 1997; Kornmann et a]., 1998;
Skotzko et al., 1995). These cells can then be assayed for their proliferative and
tumorigenic potential through live cell counts, and their ability to form foci and grow in
soft agar. This analysis will help to determine the molecular basis for the significant
difference in growth potential exhibited in these cells.

The increased expression of cyclin Dl in both senescent fibroblasts and upon
cellular differentiation in a variety of cell types suggests that cyclin Dl may exert a
heretofore uncharacterized function in growth arrest states, associated with senescence and
differentiation (Freeman et al., 1994; Fukami et al., 1995; Lucibello et al., 1993; Xiong et
al., 1997). Cyclin Dl expression is induced upon differentiation in the promyelocytic cell
line HL60, in C2C12 muscle cells, in the rat hippocampal cell line H19-7, and in PC12
pheochromocytoma cells (Horiguchi-Yamada et al., 1994; Jahn et al., 1994; Tamaru et al.,
1994; van Grunsven et al., 1996; Xiong et al., 1997; Yan and Ziff, 1995). This
mechanism appears to be distinct from its function as a regulator of pRb phosphorylation
(Skapek et al., 1996). These observations provide correlative evidence to suggest a role
for cyclin Dl in regulating growth arrest associated with cellular differentiation. Indeed,
cyclin Dl has been implicated in the inhibition of cell proliferation, as ectopic expression of
cyclin Dl in immortalized fibroblasts or mammary epithelial cells inhibits growth (Han et
al., 1995; Quelle et al., 1993; Resnitzky et al., 1994). What is the role of cyclin Dl in
instances of growth arrest such as differentiation and senescence? The answer is not clear
as recent reports have indicated that cyclin Dl can participate in an array of activities
independent of cdk activation. For example, cyclin Dl can interact with the estrogen
receptor inducing estrogen receptor transcriptional activity via a mechanism which is
independent of cdk activation (Neuman et al., 1997). Cyclin Dl can also bind to the
proliferating cell nuclear antigen (PCNA), and the novel transcription factor, cyclin D-
interacting myb-like protein (DMPI) (Hirai and Sherr, 1996; Inoue and Shen; 1998). In
addition, in differentiating neurons, cyclin Dl binds to cdk5, a cdk which is required for
neuronal differentiation, yet does not activate its intrinsic kinase activity (Xiong et al.,
1997). Thus, emerging evidence has expanded the biological activities of cyclin Dl.
Hence one of these activities, or an as yet unidentified cyclii Dl function, may explain the
role of cyclin Dl in growth arrest staks. Analysis of the role of cyclin Dl in differentiating
systems will potentially expose and delineate novel cyclin-dependent mechanisms.

Consequently, we may be able to translate these findings to aid our understanding of the
role of cyclin Dl not only in cell differentiation but also in the control of cell cycle
progression.
5.3 Characterization of the repression of PDGF-PR expression by
MY c
The activated PDGF-PR confers a strong proliferative response in a variety of cell
types. Moreover, deregulated activation of the PDGF-PR has been implicated in the
promotion of tumorigenesis (Guha et al., 1995; Silver, 1992). Hence, the suppression of
this gene by Myc represents a novel example of how Myc is tasked by the cell with
regulating a proliferative response through the inhibition of the proliferative stimulus at its
source, the cell surface. This is the second example of the involvement of Myc in a
negative regulatory pathway. The function of Myc in negatively regulating its own
transcription has been well described (Facchini et al., 1997; Penn et al., 1990; Penn et al.,
1990). However, in contrast to Myc negative auto-regulation, little is known of either the
mechanism or function of the Myc-mediated repression ofpdgf-PR mRNA expression.
The answers to these questions will be pursued in future.
5.3.1 Biological role of PDGF-PR suppression by Myc following ligand
stimulation
The repression of pdgf-pr mRNA by Myc potentially represents a negative
feedback mechanism through which the proliferative potential of PDGF-PR signalling can
be controlled. In addition, this mechanism will also serve to restrict PDGF-PR expression
to cellular growth states during which PDGF-PR expression is required to provide a
proliferative stimulus. Indeed, PDGF-PR expression is elevated in confluent, quiescent

fibroblasts compared to exponentially proliferating cells (Vaziri and Faller, 1995). The
elevated expression levels in growth arrested cells pennit the cell to respond quickly to the
presence of mitogens in the external environment, ensuring that the cell will be able to
quickly re-enter the cell cycle.
Is the mechanism of PDGF-PR repression by Myc necessary? Indeed, what are the
consequences of enforced PDGF-PR expression in a cell which is induced to enter the cell
cycle following mitogen-stimulation? To address this question, PDGF-PRs would be
ectopically expressed in Rat-1 cells, and the effect of deregulated PDGF-PR expression on
cell proliferation and turnongenesis in exponentially growing cells ascertained. Analysis of
tumors which exhibit deregulated PDGF-PR expression demonstrated that constitutive
PDGF-PR signaling constitutes a potent tumorigenic mechanism. This observation
provides correlative evidence to support the hypothesis that the repression of PDGF-PR by
Myc may serve to curtail a putative tumorigenic program.
5.3.2 Analysis of the mechanism of pdd-fir mRNA suppression by Myc
The repression of the pdgf-br by Myc occurs at the transcriptional level, and
requires sequences found between nucleotides -1994 and -396, relative to the translation
start site. The transcriptional start site as yet has not been precisely determined and lies
between nucleotides -421 to -41 1. The mousepdgf-pr promoter contains a variety of
putative transcription factor binding sites (Ishisaki et al., 1997). To determine which
sequences within this region are required for the repression of pdgf-pr by Myc, we
undertook deletion analysis of the mousepdgf-br promoter. The transcriptional repression
of thepdgf-pr gene by Myc appears to be mediated through sequences contained within
nucleotides -745 and -396 of the mouse pdgf-pr promoter. This region also contains
sequences necessary to maintain basal transcription. Repression is potentially effected
through one of the three putative transcription factor binding sites in this region. One of

these sites, the NF-Y site is particularly intriguing because of findings from two studies.
First, the NF-Y site is reported to be the sole element required for the maintenance of basal
transcription of thepdgf-jr gene in NIH3T3 cells (Ishisaki et al., 1997). Second, Myc can
repress the activity of the CTFNFI family of transcription factors, of which NF-Y is a
member (Yang et al., 1991; Yang et al., 1993). Myc can effect the phosphorylation of
NFl, another member of the CTFINFI family, inhibiting the ability of this transcription
factor to transactivate the expression of the pro-alpha 2(I) collagen gene. Hence, NF-Y is a
highly probable candidate through which Myc may repress transcription of thepdgf-Dr
gene.
Myc has also been reported to inhibit the activity of components of the basal
transcription machinery such as the Initiator binding proteins, Miz-1 and TFII-I, as well as
the TATA-binding protein (TBP), via direct association (Hateboer et al., 1993;
Maheswaran et al., 1994; Roy et al., 1993; Schneider et al., 1997). However, the absence
of both an initator element or a TATA-box in the mousepdgf--P,. promoter excludes these
pathways. To ascertain which sequences are cmcial for the Myc-mediated repression
mechanism, each of the putative transcription factor binding sites APl, AP2, and NF-Y in
this 31 5 bp region could be specifically altered via site directed mutagenesis. The effect of
these mutations on transcription from thepdgf-pr promoter will be assayed via promoter
luciferase assays in Rat-1 cells expressing an inducible Myc construct. Ascertaining
whether the NF-Y site is the site required for the repression of thepdgf-pr gene by Myc
will be difficult, as deletions in this site may abolish basal transcription in Rat-1 cells as
was reported in the NIH3T3 cell line.
An alternative approach, DNA-footprinting, will also be employed to analyze the
fragment of the mousepdgf-pr promoter comprising nucleotides -745 and -396. Using
nuclear lysates extracted from either control or Myc-activated rodent fibroblasts, the sites
on this promoter fragment which are protected with each nuclear extract will be compared
and consequently elements which are responsive to Myc identified. The responsiveness of

these elements to Myc can be assayed in promoter reporter assays, by fusing these
elements to heterologous promoters driving a reporter gene and performing transient
transfection promoter reporter assays. To identify sites which are specifically bound by
Myc or Myc containing complexes, electrophoretic mobility shift assays with nuclear
lysates from control and Myc-activated rodent fibroblasts will be performed. The presence
of Myc in DNA bound complexes can be assayed through the use of specific antibodies to
Myc; these will either produce a supershift in Myc containing complexes or will eliminate
these DNA bound complexes. Identification of the elements in thepdgf-P. promoter which
are responsive to Myc repression will allow us to better comprehend the mechanism via
which Myc represses gene transcription and consequently how Myc repression
contribute to cellular transformation.
5.4 Characterization of the mechanism of gadd45 repression by Myc
The repression of the gadd45 gene by Myc signals a change in direction in the M
field, as it reveals a novel function for Myc: the suppression of a growth arrest gene
(Marhin et al., 1997). Thus, Myc may promote cell proliferation by two pathways: first,
Myc induces genes which are required for progression through the cell cycle, and second
Myc represses the expression of genes which effect or maintain growth arrest, keeping
cells in cycle. Logically, both pathways must be equally essential, as a Myc enforced
initiation of a proliferative program in the presence of a strong block to growth, provided
by growth arrest genes, will induce apoptosis (Evan et al., 1992). Therefore, Myc-
mediated transcriptional repression and transactivation may both play an essential role not
only in enhancing cell proliferation, but at a more fundamental level, permitting normal cell
proliferation. Another member of the gadd group of genes, gadd153, and the fhctionally
related gene gasl, have also been shown to be repressed in response to Myc-activation
(Chen et al., 1996; Lee et al., 1997). However, whether the gaddgenes or gasl are direct

targets for Myc repression has to be ascertained. Not all of the known growth arrest genes
are suppressed by Myc. Preliminary studies indicate that the effect of Myc on the
expression of gas3 and gas6 is negligible (WM, unpublished results). Thus, Myc can
suppress the expression of specific growth arrest genes, potentially enhancing cell
proliferation.
5.4.1 Biological role of gadd45 suppression by Myc following mitogen
stimulation
Gadd45 is a physiologically important target for Myc repression. Gadd45
expression is associated with a variety of growth arrest states, suggesting that gadd45 may
play a role in the initiation or maintenance of growth arrest (reviewed in Fornace, 1992;
Papathanasiou and Fornace, 1991). Indeed, ectopic expression of gadd45 is growth
inhibitory in both immortalized fibroblasts and in tumorigenic cell lines, emphasizing its
strong growth inhibitory impetus (Vairapandi et al., 1996; Zhan et al., 1994). Yet, gadd45
is only one member of a group of growth arrest specific genes, which are functionally
highly related. Given that the growth arrest genes gadd153, and gas1 are also repressed by
Myc (Chen et al., 1996; Lee et al., 1997), what is the biological importance of the
repression of gadd45 by Myc? Is the repression of gadd45 by Myc required for cell cycle
progression?
To address this question, gadd45 will be ectopically expressed in control and Myc-
activated Rat-1 cells to ascertain the consequences of co-expression of a strong proliferative
with a strong growth inhibitory stimulus. I anticipate that, similar to the observations made
in NIH3T3 cells (Vairapandi et al., 1996), ectopic gadd45 expression in Rat-1 cells will be
growth inhibitory. Indeed, ectopic expression of the functionally related gene gadd153 in
rodent fibroblasts inhibited the propagation of these cells, hinting that ectopic gadd45
expression may elicit an analogous phenotype (Chen et al., 1996). Co-expression of

ectopic Myc and gadd45 in Rat-1 cells may elicit one of three outcomes: first, the growth
inhibitory effect of gadd45 expression will dominate, driving the cells into growth arrest.
Evidence to support this outcome arose from studies which have shown that ectopic
gadd45 expression in the hunorigenic Myc-activated cell line HeLa is growth inhibitory
(Zhan et al., 1994). Alternatively, the proliferative impetus of Myc may predominate,
overcoming a gadd45-induced block to cell proliferation. Again we could make some
predictions based on studies of the ectopic expression of Myc and gadd153 in rodent
fibroblasts (Chen et al., 1996). The gaddl53-induced block to cell proliferation was
rescued by ectopic Myc expression. Cells co-expressing Myc and gadd153 were able to
progress through the cell cycle and proliferate, yet the rescue was incomplete as these cells
exhibited prolonged GI and G2 phases. Another alternative outcome would be that the
conflict in growth signals may elaborate an apoptotic mechanism, a program distinct from
that dictated by gadd45 or Myc alone. A conflict between growth promoting and inhibitoly
stimuli is a well characterized trigger of a Myc-dependent apoptotic pathway (Evan et al.,
1992). Thus, there exist parallel studies which can support each of these hypothetical
outcomes, and as such each is equally possible.
Two of the three possible outcomes involve a gradual loss in cell viability, therefore
the growth of cells expressing ectopic gadd45 must be assayed quickly following
transduction of the gadd45 gene into Rat-1 cells. For this reason, control and Myc-
activated Rat-l cells will be infected with recombinant retroviruses which will deliver two
cDNAs into the cell, the gadd45 cDNA together with a cDNA encoding the green
fluorescent protein (GFP). Following infection, gadd45 expressing cells will be isolated
on the basis of GFP expression using a fluorescence based cell sorter. Pooled populations
will be derived, their growth assayed, and the growth response characterized.

5.4.2 Myc abrogates the stress induced upregulation in gadd45
expression
We have demonstrated that ectopic Myc expression can retard the induction of
gadd45 in Rat-1 cells exposed to ionizing radiation (Marhin et al., 1997). The biological
significance of this mechanism is unclear. Exposure of both control and Myc-activated
Rat-1 cells to ionizing radiation, resulted in apoptosis in both cell populations. However,
Rat-1 cells expressing ectopic Myc are more sensitive to exposure to ionizing radiation. At
a dose of 10 Gy, the Dl0 value (the dose at which only 10 % of the population survives)
for control Rat-1 cells is approximately 2 Gy greater than that of Myc-activated cells (WM,
unpublished data). The reason for the increase in sensitivity is not known. It is interesting
to hypothesize that the decreased expression of gadd45 following exposure to ionizing
radiation in Myc-activated cells may play a role in the initiation of the apoptotic mechanism.
One can envisage a model where the induction of gadd45 following exposure to DNA-
damaging agents such as ionizing radiation, serves to arrest cell growth, permitting the cell
to repair damaged DNA prior to progression into S phase. However, in a Myc-activated
cell, ectopic Myc expression confers a strong proliferativc stimulus, tempers gadd45
induction, inhibiting growth arrest, and DNA repair, driving the cells into the cell cycle.
The Myc-driven inability to arrest growth to repair damaged DNA prior to cell division may
trigger an apoptotic program.
To determine if the reduced expression of gadd45 in Myc-activated cells is
responsible for the increased sensitivity of these cells to ionizing radiation, control Rat-1,
Myc-activated Rat-1, Rat-1 cells expressing ectopic gadd45, and Rat-1 cells co-expressing
Myc and gadd45 will be exposed to 10 Gy of ionizing radiation. The growth of each of
these three cell culhlres will then be monitored to determine if the deregulated expression of
gadd45 could inhibit the apoptotic response. The caveat of this study is that one will be

capable of co-expressing Myc and gadd45 in Rat-1 fibroblasts without inducing apoptosis.
This concern will be addressed in the earlier described studies.
5.4.3 Analysis of the mechanism of gadd45 mRNA suppression by Myc
The suppression of gadd45 transcription by Myc is dependent upon sequences
located within a 1.8 Kb Xba I fragment of the hamster gadd45 gene, consisting of
nucleotides - 1675 to +149, relative to the transcription start site. Thegadd45 promoter is
relatively simple, possessing neither a TATA box, nor an initiator element. In addition,
there are only a few putative transcription factor binding sites. An analysis of thegadd45
promoters derived from human, hamster, and mouse species revealed the presence of a
conserved OCT, CAT, OCT, CAT motif located within 100 bp of the transcription start
site. To determine which sequences within the hamstergadd45 promoter are required for
the repression ofgadd45 expression by Myc, we employed deletion analysis of the 1.8 Kb
Xba I fragment of the hamstar gadd45 promoter. The sequences which are responsive to
Myc repression are located within a region of the promoter, consisting of nucleotides -222
to +149, which contains elements required for basal transcription.
Further analysis is required to reveal the sequences which are required for Myc
repression of gadd45 transcription. The relatively large region of the gadd45 promoter
consisting of nucleotides -222 to +I49 requires additional characterization via progressive
deletion analysis, and site-directed mutagenesis of the only conserved sequences within
this region, the OCT and CAT sites. Deletion or mutational analysis of this region is
potentially difficult, as the elements required for Myc repression may overlap with elements
which are required for mediating basal transcription. Thus, alternative approaches such as
DNA-footprinting, and electrophoretic mobility shift assays can be employed, via an
identical approach as proposed earlier for the analysis of thepdgf-B. promoter.

5.5 Models of Myc repression
The mechanism(s) through which Myc represses gene transcription have not been
fully determined. It is clear that contrary to Myc transactivation of gene expression, Myc
repression is not dependent upon the presence of a consensus E box, CACGTG, in the
target gene. Furthermore, analysis of the few Myc-repressed genes that have been
identified, has revealed that Myc-mediated repression may be effected through multiple
pathways. For example, repression of cyclin Dl by Myc is mediated through the initiator
element (Inr), and does not require heterodimerization with Max (Philipp et al., 1994).
Conversely c-myc negative autoregulation is not mediated through the Inr, yet binding to
Max is obligatory for this repression activity to occur (Facchini et al., 1997). What are the
possible mechanisms through which Myc represses gene transcription? Myc may repress
gene transcription via direct or indirect mechanisms.
Myc may repress gene expression by inhibiting the activity of enhancers or
initiators of gene transcription. Indeed, a variety of reports have demonstrated that Myc is
capable of fulfilling this role. Myc can interact with and inhibit the activity of the
transcriptional regulators AP2, YY1, Miz-1, TFII-I, and TBP (Gaubatz et al., 1995;
Hateboer et al., 1993; Maheswaran et al., 1994; Roy et al., 1993; Schneider et al., 1997;
Shrivastava et al., 1993; Shrivastava et al., 1996). Myc can inhibit the activity of the
enhancer AP-2 via two mechanisms (Gaubatz et al., 1995). First, as we have described
earlier, AP-2 is a Myc interacting protein. The interaction of Myc with AP-2 renders AP-2
incapable of binding to DNA and activating transcription. Alternatively, in the a-
prothymosin promoter there are overlapping AP-2 and Myc binding sites, hence both
factors compete for binding to this promoter. Thus, the interaction of Myc with AP-2 can
repress gene expression via both DNA-dependent as well as independent mechanisms.
Myc can also mediate the repression of gene transcription through Inr elements.
The list of promoters which belong to this category include: adenovirus 5 major late

promoter (AdMLP); cyclin Dl; clebpcr, serum albumin; terminal deoxy-transferme (Tdl);
and A5 (Antonson et al., 1995; Li et al., 1994; Mai and Martensson, 1995; Philipp et al.,
1994). Myc is reported to repress initiat0r-mfXbted transcription through the interaction
with Inr binding proteins including W 1 , Miz-1, and TFII-I (Roy et al., 1993; Schneider et
al., 1997; Shrivastava et al., 1993; Shrivastava et al., 1996). Both W1 and Miz-1 bind
specifically to Inr elements, initiating gene transcription. The interaction of Myc with either
of these factors directly curtails the transactivation role of these proteins, potentially
through the inhibition of contacts between YY1 or hypothetically Miz-1 with components
of the basal transcription machinery such as TBP or TFIIB (Shrivastava et a]., 1993;
Shrivastava et al., 1996). On TATA-less promoters, the transcription initiation factor
TFII-I, binds to the Inr and initiates formation of the RNA polymerase I1 pre-initiation
complex at the transcription start site. Myc complexes with and sequesters TFII-I,
preventing the initiation of transcription (Roy et al., 1993). In an analogous fashion, with
TATA-containing promoters, Myc can also directly bind to and sequester TBP, inhibiting
formation of the RNA polymerase I1 pre-initiation complex (Hateboer et al., 1993;
Maheswaran et al., 1994). These studies illustrate that Myc can interact with a variety of
proteins influencing the formation or activity of the basal transcription complex. Thus, this
may constitute the mechanism through which Myc effects transcriptional repression on a
variety of genes, yet it must be noted that all of these studies were conducted using isolated
promoters rather than the full genes. Hence, the significance of these mechanisms in Myc
repression and ultimately the biological activities of Myc need to be further characterized.
Myc can also indirectly inhibit the expression of genes by either repressing an
enhancer of transcription or inducing a repressor of transcription. Myc has been reported
to repress the transcriptional induction of specific genes through the inhibition of the
expression of transcriptional enhancers. During the differentiation of the pre-adipocyte cell
line, 3T3-L1, the transcription factor clebpa is induced (Cao et al., 1991; Christy et al.,
1991; Christy et al., 1989). Clebpa is responsible for the transcriptional induction of both

growth arrest genes such as gadd45 and differentiation specific genes such as mim-I
(Christy et al., 1989; Constance et al., 1996). Myc indirectly represses the induction of the
gadd45 gene by repressing the expression of clebpa (Constance et al., 1996). The Myc-
mediated repression of clebpa is mediated through either the Inr element or interactions
with components of the basal transcription machinery (Antonson et al., 1995; Li et al.,
1994). Myc is also capable of directly inhibiting the ability of clebpa to activate the
transcription of these genes via an unknown mechanism (Mink et al., 1996). Myc has also
been reported to modulate the phosphorylation state of the enhancer CTFMFI, inhibiting
the ability of this transcription factor to drive the expression of the pro alpha 2(I) collagen
gene (Yang et al., 1991; Yang et al., 1993). The most revealing approach to determining
the mechanism through which Myc represses the transcription of the gadd45 andpdgf-br
genes would be EMSAs as I have described earlier. EMSAs will demonstrate firstly, if
Myc represses gene expression through the regulation of complex formation on the
promoters of these genes and secondly, whether Myc effects these modulations via direct
or indirect means.
To date the analysis of the few genes which have been reported to be repressed by
Myc has exposed only a few components of Myc-dependent repression mechanisms. As
such, the delineation and understanding of how Myc represses gene transcription is still in
its infancy, primarily due to the limited number of genes that have been identified as targets
of Myc repression. It will be through the identification and characterization of additional
targets for Myc repression such as gadd45 and thcpdgf-br that we will be able to reveal
common themes from among seemingly different repression mechanisms to fully elaborate
the pathways of Myc repression.

5.6 Does Myc transactivation or Myc repression lead to cell
transformation?
The transforming domains of the c-Myc protein were initially identified via deletion
analysis in rat embryonic fibroblasts (Stone et al., 1987). The regions which were
required for cellular transformation include the amino ternlinal amino acids 106 to 143
(MBII), and the C-terminal89 amino acids. The amino terminal domain was subsequently
discovered to function as a transcriptional activation domain in transient transfection
reporter assays. When fused to the DNA binding domain of the yeast Gal4 transcription
factor, this domain can transactivate a reporter gene containing Gal4 DNA-binding sites
(Kato et al., 1990). Yet the link between Myc-mediated transformation and transactivation
is not concrete, as there are Myc mutants which can disassociate these two functions.
Ectopic expression of an N-terminal262 amino acid fragment of Myc will competitively
inhibit MycIRas cotransformation (Brough et al., 1995). Moreover, this inhibition is
dependent upon the presence of Myc box I1 amino acids 129 to 145. Yet, this Myc mutant
when fused to the DNA-binding domain of the yeast transcription factor GAL4, is able to
transactivate a heterologous reporter gene driven by GAL4 DNA binding sites. Thus,
MBII is required for Myc-dependent cell transformation, but is dispensable for the
transactivation of a synthetic promoter.
In the amino-terminus of Myc can also be found regions which are necessary for
Myc repression of gene expression. The domains required for repression are different for
each gene, mapping to amino acids 92 to 106 for cyclin Dl, but amino acids 122 to 143 for
the AdMLP (Li et al., 1994; Philipp et al., 1994). Further analysis of specific amino acid
mutations in Myc revealed a stronger correlation between Myc transformation and Myc
repression in comparison to Myc transactivation. For example, a Phe-115 mutation
enhanced both the transcriptional repression and transformation capabilities of Myc (Lee et
al., 1996).

Does Myc-mediated transcriptional repression or transactivation drive neoplastic
transfomatio-9 This question may be addressed in two ways. First, the identification of
additional g~netic targets for Myc repression may reveal novel functions of Myc which
directly contribute to cellular transformation. Second, preliminary mutational analysis of a
few mutants of the Myc protein has suggested that a structure-function analysis of the Myc
protein may be an ideal approach to dissect the regions of the Myc protein which are
required for Myc repression, transactivation, and transformation. Undertaking a structure
function analysis of the Myc protein with greater resolution, utilizing smaller deletions and
site directed mutagenesis may be the key to determining whether the domains required for
neoplastic transformation overlap with the regions required for Myc repression or Myc
transactivation.

5.7 Perspectives
The product of the c-myc proto-oncogene is a highly regulated protein which is
essential for cell proliferation, and has been implicated in the genesis and progression of
tumors arising from a wide variety of cell types. Delineating the mechanisms through
which Myc elicits cell proliferation in normal cells will aid in our understanding of the role
of Myc in tumorigenesis. In the last 18 years of Myc research, our progress in realizing
this goal has not been linear, but punctuated by bursts of discovery which have provided
us with novel views on the role of Myc.
More recently, the Myc field has embarked in a new direction centered around the
ability of Myc to abrogate molecular blocks to cell proliferation. Myc exerts this function
in three ways: directly associating with and inhibiting the activity of transcriptional
activators, bypassing the growth inhibitory effects of the cdkI proteins, and the repression
of gene transcription. Our understanding of these mechanisms is still in its infancy, the
role of the reported Myc binding proteins in the context of the biological functions of Myc
is uncertain. Likewise, the Myc-mediated circumvention of cell cycle blocks induced by
assorted cdkls has not been fully characterized. Yet, correlative evidence does suggest that
the ability of Myc to overcome a p27-induced arrest is indirectly dependent upon the
transcription regulatory activities of Myc. Myc repression is an intriguing puzzle which
holds a great deal of promise, due to the correlative evidence linking Myc-mediated
repression to tumorigenesis. Through my research I have expanded our knowledge of the
contribution of Myc repression to the activities of Myc in enhancing normal cell
proliferation. The repression ofpdgf-Pr expression by Myc represents a putative feedback
mechanism. Myc, as the endpoint of a potent proliferative signal transduction pathway,
can tightly control this stimulus by repressing the expression of the receptor which initiated
the signal, hence maintaining normal cell proliferation. In addition, Myc can also
contribute to an enhanced proliferative and tumorigenic potential. Myc-activation and pRb

loss appear to cooperate to repress cyclin Dl activity, and enhance turnongenic potential as
evidenced by loss of contact inhibition. Lastly, Myc represses the expression of growth
arrest specific genes, such as gadd45, gadd153, as well as gasl ; which are potent
inhibitors of cell proliferation and viability. The growth arrest genes have physiologically
important functions, antagonizing the activity of genes which drive cell proliferation.
Normal cell proliferation is achieved through the fine balance between these two opposing
stimuli, hence the repression of growth arrest genes by Myc helps to explain how Myc
promotes enhanced cell proliferation and consequently tumorigenesis. Yet it remains to be
determined if these genes are directly repressed by Myc and whether they are all repressed
via the same or different mechanisms.
The abrogation of growth arrest mechanisms by Myc expands our understanding of
the proliferative potential of Myc, that Myc may promote cell growth through both the
repression and activation of the functions of specific proteins. The circumvention of cell
cycle inhibitors by Myc will keep cells out of growth arrest, and the induction of growth
promoters such as: E2Fs; cdc25A; cad and ode promotes the transition of cells through G1
and into S phase of the cell cycle. These new developments arrive at a time when concerns
were emerging as to the part Myc transcriptional regulation plays in effecting the biological
activities of Myc. The chronic absence of biologically important targets for transcriptional
regulation by Myc, and the futility of current means of identification of Myc regulated
genes should prompt us to rethink conventional approaches and our views of how Myc
may function. Indeed the c-Myc deficient fibroblasts may represent a powerful tool to
screen and identify the biologically important genes from among the existing pool of
reported Myc regulated targets. In addition, this system may result in the identification of
novel targets for Myc, which wil: ultimately allow us to understand Myc function.
Eventually, with the adoption of novel -views and approaches we will develop the tools to
unravel the enigma that is Myc.

Chapter 6
REFERENCES

Adarns, J. M., and Cory, S. (1992). Oncogene co-operation in leukaemogenesis. Cancer
Sum. 15, 119-41.
Adams, J. M., Harris, A. W., Pinkert, C. A., Corcoran, L. M., Alexander, W. S., Cory, S., Palmiter, R. D., and Srinster, R. L. (1985). The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318,
533-8.
Alevizopoulos, K., Vlach, J., Hemecke, S., and Amati, B. (1997). Cyclin E and c-Myc promote cell proliferation in the presence of p16(INK4a) and of hypophosphorylated retinoblastoma family proteins. EMBO J. 16,5322-33.
Alexandrow, M. G., Kawabata, M., Aakre, M., and Moses, H. L. (1995). Overexpression of the c-Myc oncoprotein blocks the growth-inhibitory response but is required for the mitogenic effects of transforming growth factor beta 1. Proc. Natl. Acad.
Sci. USA 92. 3239-43.
Alexandrow, M. G., and Moses, H. L. (1995). Transforming growth factor beta 1 inhibits mouse keratinocytes late in G1 independent of effects on gene transcription. Cancer Res.
55, 3928-32.
Alvarez, E., Northwood, I. C., Gonzalez, F. A., Latour, D. A., Seth, A., Abate, C., Curran, T., and Davis, R. J. (1991). Pro-Leu-SerIThr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein
kinase. J. Biol. Chem. 266, 15277-85.
Amati, B., Brooks, M. W., Levy, N., Littlewood, T. D., Evan, G. I., and Land, H.
(1993). Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 72,
233-45.
Amati, B., Dalton, S., Brooks, M. W., Littlewood, T. D., Evan, G. I., and Land, H.
(1992). Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 359,423-6.

Amati, B., Littlewood, T. D., Evan, G. I., and Land, H. (1993). The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 12, 5083-7.
Amin, C., Wagner, A. J., and Hay, N. (1993). Sequence-specific transcriptional activation by Myc and repression by Max. Mol. Cell. Biol. 13,383-90.
Antonson, P., Pray, M. G., Jacobsson, A., and Xanthopoulos, K. G. (1995). Myc inhibits CCAATIehancer-binding protein alpha-gene expression in HIB-IB hibemoma
cells through interactions with the core promoter region. Eur. J. Biochem. 232,397-403.
Arber, N., Doki, Y., Han, E. K., Sgambato, A., Zhou, P., Kim, N. H., Delohery, T., Klein, M. G., Holt, P. R., and Weinstein, I. B. (1997). Antisense to cyclin Dl inhibits the growth and tumorigenicity of human colon cancer cells. Cancer Res. 57, 1569-74.
Armelin, H. A., Armelin, M. C., Kelly, K., Stewart, T., Leder, P., Cochran, B. H., and Stiles, C. D. (1984). Functional role for c-myc in mitogenic response to platelet-derived growth factor. Nature 310,655-60.
Arsura, M., Deshpande, A., Ham, S. R., and Sonenshein, G. E. (1995). Variant Max protein, derived by alternative splicing, associates with c-Myc in vivo and inhibits
transactivation. Mol. Cell. Biol. 15, 6702-9.
Asai, A., Miyagi, Y., Sugiyama, A., Nagashima, Y., Kanemitsu, H., Obinata, M., Mishima, K., and Kuchino, Y. (1994). The s-Myc protein having the ability to induce
apoptosis is selectively expressed in rat embryo chondrocytes. Oncogene 9,2345-52.
Asker, C. E., Magnusson, K. P., Piccoli, S. P., Anderson, K., Klein, G., Cole, M. D., and Wiman, K. G. (1995). Mouse and rat B-myc share amino acid sequence homology with the c-myc transcriptional activator domain and contain a B-myc specific carboxy terminal region. Oncogene 11,1963-9.
Askew, D. S., Ashmun, R. A., Simmons, B. C., and Cleveland, J. L. (1991). Constitutive c-myc expression in an ILJ-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6,1915-22.

Baldin, V., Lukas, J., Marcote, M. J., Pagano, M., and Draetta, G. (1993). Cyclin Dl is a nuclear protein required for cell cycle progression in Gl. Genes Dev. 7,812-821.
Ballagi, A. E., Ishizaki, A., Nehlin, J. O., and Funa, K. (1995). Isolation and
characterization of the mouse PDGF beta-receptor promoter. Biochem. Biophys. Res. Comm. 210. 165-173.
Barone, M. V., and Courtneidge, S. A. (1995). Myc but not Fos rescue of PDGF signalling block caused by kiiase-inactive Src. Nature 378,509-12.
Barrett, J., Birrer, M. J., Kato, G. J., Dosaka-Akita, H., and Dang, C. V. (1992). Activation domains of L-Myc and c-Myc determine their transforming potencies in rat embryo cells. Mol. Cell. Biol. 12, 3 130-7.
Bartek, J., Bartkova, J., and Lukas, J. (1997). The retinoblastoma protein pathway in cell
cycle control and cancer. Exp. Cell. Res. 237, 1-6.
Bates, S., Bonetta, L., MacAllan, D., Pany, D., Holder, A., Dickson, C., and Peters, G.
(1994). CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin- dependent kinases that associate with cyclin Dl. Oncogene 9,71-79.
Bates, S., Parry, D., Bonetta, L., Vousden, K., Dickson, C., and Peters, G. (1994). Absence of cyclin Dlcdk complexes in cells lacking functional retinoblastorna protein. Oncogene 9,1633-1640.
Bechann, H., Su, L. K., and Kadesch, T. (1990). TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer muE3 motif. Genes Dev. 4,
167-79.
Beijersbergen, R. L., Hijmans, E. M., Zhu, L., and Bemards, R. (1994). Interaction of c- Myc with the pRb-related protein p107 results in inhibition of c-Myc-mediated
transactivation. EMBO J. 13,4080-6.
Beimling, P., Benter, T., Sander, T., and Moelling, K. (1985). Isolation and characterization of the human cellular myc gene product. Biochemistry 24,6349-55.

Bello-Fernandez, C., and Cleveland, J. L. (1992). c-myc transactivates the ornithine decarboxylase gene. Cum. Top. Microbiol. Immunol. 182,445-52.
Bello-Fernandez, C., Packham, G., and Cleveland, J. L. (1993). The ornithine
decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 90, 7804-8.
Bennett, N. T., and Schultz, G. S. (1993). Growth factors and wound healing: biochemical properties of growth factors and their receptors. Am. J. Surg. 165,728-37.
Benvenisty, N., Leder, A., Kuo, A., and Leder, P. (1992). An embryonically expressed gene is a target for c-Myc regulation via the c-Myc-binding sequence. Genes Dev. 6,2513-
2523.
Berberich, S., Hyde-DeRuyscher, N., Espenshade, P., and Cole, M. (1992). max encodes a sequence-specific DNA-binding protein and is not regulated by serum growth
factors. Oncogene 7, 775-9.
Berberich, S. J., and Cole, M. D. (1992). Casein kinase I1 inhibits the DNA-binding activity of Max homodimers but not MyJMax heterodimers. Genes Dev. 6,166-76.
Bernard, O., Cory, S., Gerondakis, S., Webb, E., and Adams, J. M. (1983). Sequence of the murine and human cellular myc oncogenes and two modes of myc transcription resulting from chromosome translocation in B lymphoid tumours. EMBO J. 2,2375-83.
Bernard, O., Drago, J., and Sheng, H. (1992). L-myc and N-myc influence lineage determination in the central nervous system. Neuron 9,1217-24.
Bianchi Scarra, G. L., Romani, M., Coviello, D. A,, Garre, C., Ravazzolo, R., Vidali, G., and Ajmar, F. (1986). Terminal erythroid differentiation in the K-562 cell line by 1- beta-D-arabinofuranosylcytosine: accompaniment by c-myc messenger RNA decrease.
Cancer Res. 46.6327-32.
Birkenmeier, E. H., Gwynn, B., Howard, S., Jeny, J., Gordon, J. I., Landschulz, W. H., and McKnight, S. L. (1989). Tissue-specific expression, developmental regulation,

and genetic mapping of the gene encoding CCAATIenhancer binding protein. Genes Dev.
3, 1146-56.
Blackwell, T. K., Huang, J., Ma, A., Kretnzer, L., Alt, F. W., Eisenman, R. N., and Weintraub, H. (1993). Binding of Myc proteins to canonical and noncanonical DNA sequences. Mol. Cell. Biol. 13,5216-24.
Blackwell, T. K., Kretner, L., Blackwood, E. M., Eisenman, R. N., and Weintraub, H. (1990). Sequence specific DNA binding by the c-Myc protein. Science 250, 1149-52.
Blackwood, E. M., and Eisenman, R. N. (1991). Max: a helix-loop-helix zipper protein
that forms a sequence-specific DNA-binding complex with Myc. Science 251,121 1-7.
Blackwood, E. M., and Eisenman, R. N. (1992). Regulation of Myc: Max complex
formation and its potential role in cell proliferation. Tohoku J. Exp. Med. 168, 195-202.
Blackwood, E. M., Kretzner, L., and Eisenman, R. N. (1992). Myc and Max function as a nucleoprotein complex. Curr. Opin. Genet. Dev. 2,227-35.
Blackwood, E. M., Lugo, T. G., Kretzner, L., King, M. W., Street, A. J., Wide, 0. N., and Eisenman, R. N. (1994). Functional analysis of the AUG- and CUG-initiated forms of
the c-Myc protein. Mol. Biol. Cell. 5, 597-609.
Blackwood, E. M., Luscher, B., and Eisenman, R. N. (1992). Myc and Max associate in
vivo. Genes Dev. 6, 71-80.
Bodrug, S. E., Warner, B. J., Bath, M. L., Lindeman, G. J., Hanis, A. W., and Adams, J. M. (1994). Cyclin Dl transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 13,2124-30.
Bornfeldt, K. E., Raines, E. W., Graves, L. M., Skinner, M. P., Krebs, E. G., and Ross, R. (1995). Platelet-derived growth factor. Distinct signal transduction pathways associated with migration versus proliferation. Ann. N. Y. Acad. Sci. 766,416-30.

Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E., and Bishop, J. M. (1984). Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121-4.
Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E., and Bishop, J. M. (1985). Amplification of N-myc sequences in primary human neuroblastomas: correlation with advanced disease stage. Prog. Clin. Biol. Res. 175, 105-13.
Brough, D. E., Hofinann, T. J., Ellwood, K. B., Townley, R. A., and Cole, M. D. (1995). An essential domain of the c-myc protein interacts with a nuclear factor that is also required for El A-mediated transfornation. Mol. Cell. Biol. 15, 1536-44.
Buchkovich, K., Duffy, L. A., and Harlow, E. (1989). The retinoblastoma protein in
phosphorylated during specific phases of the cell cycle. Cell 58, 1097-1 105.
Buchou, T., Kranenburg, O., van Dam, H., Roelen, D., Zamtema, A., Hall, F. L., and van der Eb, A. (1993). Increased cyclin A and decreased cyclin D levels in adenovirus 5
El A-transformed rodent cell lines. Oncogene 8,1765-1773.
Burk, O., Mink, S., Ringwald, M., and Klempnauer, K. H. (1993). Synergistic activation of the chicken mim-1 gene by v-myb and clebp transcription factors. EMBO J. 12,2027- 38.
Campisi, J., Gray, H. E., Pardee, A. B., Dean, M., and Sonenshein, G. (1984). Cell- cycle contrd of c-myc but not c-ras expression is lost following chemical transformation. Cell 36, 241-7.
Cao, Z., Umek, R. M., and McKnight, S. L. (1991). Regulated expression of three clebp isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 5, 1538-52.
Cam, C. S., and Sharp, P. A. (1990). A helix-loop-helix protein related to the immunoglobulin E box-binding proteins. Mol. Cell. Biol. 10,4384-8.
Chellappan, S. P., Hiebert, S., Mudryj, M., Horowitz, J. M., and Nevins, J. R. (1991). The E2F transcription factor is a cellular target for the RB protein. Cell 65, 1053-1061.

Chen, C., Nussenzweig, A., Guo, M., Kim, D., Li, G. C., and Ling, C. C. (1996). Down-regulation of gadd153 by c-myc in rat fibroblasts and its effect on cell growth and radiation-induced apoptosis. Oncogene 13,1659-65.
Chen, I. T., Smith, M. L., O'Connor, P. M., and Fomace, A. J., Jr. (1995). Direct interaction of Gadd45 with PCNA and evidence for competitive interaction of Gadd45 and p21 WaflICipl with PCNA. Oncogene 11,193 1-7.
Chen, P.-L., Scully, P., Shew, J.-Y., Wang, J. Y. J., and Lee, W.-H. (1989).
Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 58, 1 193-1 198.
Chirgwin, J. M., Przybyla, A. E., MacDonald, R. J., and Rutter, W. J. (1979). Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18, 5294-5299.
Chou, T. Y., Dang, C. V., and Hart, G. W. (1995). Glycosylation of the c-Myc
transactivation domain. Proc. Natl. Acad. Sci. USA 92,4417-21.
Chou, T. Y., Hart, G. W., and Dang, C. V. (1995). c-Myc is glycosylated at threonine
58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 270, 18961-5.
Christy, R. J., Kaestner, K. H., Geiman, D. E., and Lane, M. D. (1991). CCAATIenhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc. Natl. Acad. Sci. USA 88,2593-7.
Christy, R. J., Yang, V. W., Ntambi, J. M., Geiman, D. E., Landschulz, W. H.,
Friedman, A. D., Nakabeppu, Y., Kelly, T. J., and Lane, M. D. (1989). Differentiation-
induced gene expression in 3T3-L1 preadipocytes: CCAATIenhancer binding protein interacts with and activates the promoters of two adipocyte-specific genes. Genes Dev. 3,
1323-35.
Claesson-Welsh, L. (1996). Mechanism of action of platelet-derived growth factor. Int. J. Biochem. Cell. Biol. 28. 373-85.

Claesson-Welsh, L. (1994). Signal transduction by the PDGF receptors. Prog. Growth Factor Res. 5, 37-54.
Clarke, A. R., Maandag, E. R., van Roon, M., van der Lugt, N. M. T., van der Valk, M., Hooper, M. L., Bems, A., and te Riele, H. (1992). Requirement for a functional RB- 1 gene in murine development. Nature 359,328-330.
Cleveland, J. L., Dean, M., Rosenberg, N., Wang, J. Y., and Rapp, U. R. (1989).
Tyrosine kinase oncogenes abrogate interleukin-3 dependence of murine myeloid cells through signaling pathways involving c-myc: conditional regulation of c-myc transcription
by temperature-sensitive v-abl. Mol. Cell. Biol. 9,5685-95.
Cleveland, J. L., Huleihel, M., Bressler, P., Siebenlist, U., Akiyama, L., Eisenrnan, R. N., and Rapp, U. R. (1988). Negative regulation of c-myc transcription involves myc family proteins. Oncogene Res. 3,357-375.
Clurman, B. E., Sheaff, R. J., Thress, K., Groudine, M., and Roberts, J. M. (1996). Turnover of cyclin E by ihe ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phospho~ylation. Genes Dev. 10, 1979-90.
Cochran, B. H., Reffel, A. C., and Stiles, C. D. (1983). Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell 33,939-47.
Colby, W. W., Chen, E. Y., Smith, D. H., and Levinson, A. D. (1983). Identification
and nucleotide sequence of a human locus homologous to the v-myc oncogene of avian myelocytomatosis virus MC29. Nature 301,722-5.
Cole, M. D. (1986). The myc oncogene: its role in transformation and differentiation. Annu. Rev. Genet. 20,361-84.
Constance, C. M., Morgan, J. I. t., and Umek, R. M. (1996). Clebp alpha regulation of
the growth-mest-associated gene gadd45. Mol. Cell. Biol. 16,3878-83.
Coppola, J. A., and Cole, M. D. (1986). Constitutive c-myc oncogene expression blocks mouse erythroleukaemia cell differentiation but not commitment. Nature 320,760-3.

Corcoran, L. M., Adams, J. M., Dunn, A. R., and Cory, S. (1984). Murine T lymphomas in which the cellular myc oncogene has been activated by retroviral insertion. Cell 37, 113-22.
Coughlin, S. R., Lee, W. M., Williams, P. W., Giels, G. M., and Williams, L. T. (1985). c-myc gene expression is stimulated by agents that activate protein kinase C and does not account f ~ r the mitogenic effect of PDGF. Cell 43,243-51.
Cutry, A. F., Kinniburgh, A. J., Krabak, M. J., Hui, S. W., and Werner, C. E. (1989). Induction of c-fos and c-myc proto-oncogene expression by epidermal growth factor and transforming growth factor alpha is calcium- independent. J. Biol. Chem. 264,19700-5.
Daksis, J. I., Lu, R. Y., Facchini, L. M., Marhin, W. W., and Penn, L. J. 2. (1994). Myc induces cyclin Dl expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene 9 ,36354.
Dalla-Favera, R., Bregni, M., Erikson, J., Patterson, D., Gallo, R. C., and Croce, C. M.
(1982). Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 79,7824-7.
Dalla-Favera, R., Gelmann, E. P., Martinotti, S., Franchini, G., Papas, T. S., Gallo, R. C., and Wong-Staal, F. (1982). Cloning and characterization of different human sequences related to the onc gene (v-myc) of avian myelocytomatosis virus (MC29). Proc. Natl.
Acad. Sci. USA 79,6497-501.
Dalla-Favera, R., Wong-Staal, F., and Gallo, R. C. (1982). Onc gene amplification in promyelocytic leukaemia cell line HL-60 and primary leukaemic cells of the same patient.
Nature 299. 61-3.
Dang, C. V. (1991). c-Myc oncoprotein function. Biochim. Biophys. Acta 1072, 103- 113.
Dang, C. V., and Lee, W. M. (1988). Identification of the human c-myc protein nuclear
translocation signal. Mol. Cell. Biol. 8,4048-54.

Dang, C. V., van Dam, H., Buckmire, M., and Lee, W. M. (1989). DNA-binding domain of human c-Myc produced in Escherichia coli. Mol. Cell. Biol. 9,2477-86.
Dani, C., Blanchard, J. M., Piechaczyk, M., El Sabouty, S., Marty, L., and Jeanteur, P. (1984). Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. USA 81, 7046-50.
Datta, S. R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., and Greenberg, M. E. (1997). Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell 91,231-41.
Davis, A., and Bradley, A. (1993). Mutation of N-myc in mice: what does the phenotype tell us? Bioessays 15,273-5.
Davis, A. C., Wims, M., Spotts, G. D., Ham, S. R., and Bradley, A. (1993). A null c-
myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7,671-82.
Dean, M., Levine, R. A., and Campisi, J. (1986). c-myc regulation during retinoic acid-
induced differentiation of F9 cells is posttranscriptional and associated with growth arrest. Mol. Cell. Biol. 6, 518-24.
Dean, M., Levine, R. A., Ran, W., Kindy, M. S., Sonenshein, G. E., and Campisi, J. (1986). Regulation of c-myc transcription and mRNA abundance by serum growth factors and cell contact. J. Biol. Chem. 261,9161-6.
DeCaprio, J. A., Furukawa, Y., Ajchenbaum, F., Griffin, J. D., and Livingston, D. M. (1992). The retinoblastoma susceptibility gene product becomes phosphorylated in multiple stages during cell cycle entry and progression. Proc. Natl. Acad. Sci., USA 89, 1795- 1798.
DeCaprio, J. A., Ludlow, J. W., Lynch, D., Furukawa, Y., Griffin, J., Piwnica-Jones, H., Huang, C.-H., and Livingston, D. M. (1989). The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 58, 1085-1095.

DeGregori, J., Leone, G., Ohtani, K., Miron, A., and Nevins, J. R. (1995). E2F-1 accumulation bypasses a G1 arrest resulting from the inhibition of G1 cyclin-dependent kinase activity. Genes Dev. 9,2873-87.
Denis, N., Kitzis, A., Kruh, J., Dautry, F., and Corcos, D. (1991). Stimulation of methotrexate resistance and dihydrofolate reductase gene amplification by c-myc. Oncogene 6, 1453-7.
Desbarats, L., Gaubatz, S., and Eilers, M. (1996). Discrimination between different E- box-binding proteins at an endogenous target gene of c-myc. Genes Dev. 10,447-60.
Dildrop, R., Ma, A., Zimmerman, K., Hsu, E., Tesfaye, A., DePinho, R., and Alt, F. W. (1989). IgH enhancer-mediated deregulation of N-myc gene expression in transgenic
mice: generation of lymphoid neoplasias that lack c-myc expression. EMBO J. 8, 1 121-8.
Dmitrovsky, E., Kuehl, W. M., Hollis, G. F., Kirsch, I. R., Bender, T. P., and Segal, S. (1986). Expression of a transfected human c-myc oncogene inhibits differentiation of a mouse erythroleukaemia cell line. Nature 322,748-50.
Dmitrovsky, E., Kuehl, W. M., Hollis, G. F., Kirsch, I. R., Bender, T. P., and Segal, S. (1986). A transfected c-myc oncogene inhibits mouse erythroleukemic differentiation. Cum. Top. Microbiol. Irnrnunol. 132,327-30.
Dony, C., Kessel, M., and Gruss, P. (1985). Post-transcriptional control of myc and p53 expression during differentiation of the embryonal carcinoma cell line F9. Nature 31 7, 636-9.
Dowdy, S. F., Hinds, P. W., Louie, K., Reed, S. I., Arnold, A., and Weinberg, R. A.
(1993). Physical interaction of the retinoblastoma protein with human D cyclins. Cell 73, 499-5 1 1.
Draetta, G., and Eckstein, J. (1997). Cdc25 protein phosphatases in cell proliferation.
Biochim. Biophys. Acta 1332, 53-63.

Driscoll, B., Wu, L., Buckley, S., Hall, F. L., Anderson, K. D., and Warburton, D. (1997). Cyclin Dl antisense RNA destabilizes pRb and retards lung cancer cell growth. Am. J. Physiol. 273, 941-9.
Eilers, M., Picard, D., Yamamoto, K. R., and Bishop, J. M. (1989). Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature
340, 66-8.
Eilers, M., Schirm, S., and Bishop, J. M. (1991). The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 10, 133-41.
Einat, M., Resnitzky, D., and Kirnchi, A. (1985). Close link between reduction of c-myc expression by interferon and, GOIGl arrest. Nature 313,597-600.
Eladari, M. E., Mohammad-Ali, K., Argaut, C., and Galibert, F. (1992). Gibbon and marmoset c-myc nucleotide sequences. Gene 116,23 1-43.
Endo, T., and Nadal-Ginard, B. (1986). Transcriptional and posttranscriptional control of c-myc during myogenesis: its rnRNA remains inducible in differentiated cells and does not
suppress the differentiated phenotype. Mol. Cell. Biol. 6, 1412-21.
Evan, G. I., Wyllie, A. H., Gilbert, C. S., Littlewood, T. D., Land, H., Brooks, M.,
Waters, C. M., Penn, L. Z., and Hancock, D. C. (1992). Induction of apoptosis in
fibroblasts by c-myc protein. Cell 69, 119-128.
Ewen, M. E., Sluss, H. K., Sherr, C. J., Matsushime, H., Kato, J.-Y., and Livingston, D. M. (1993). Functional interactions of the retinoblastorna protein with mammalian D-
type cyclins. Cell 73,487-497.
Facchini, L. M., Chen, S., Marhin, W. W., Lear, J. N., and Penn, L. Z. (1997). The Myc negative autoregulation mechanism requires Myc-Max association and involves the c- myc P2 minimal promoter. Mol. Cell. Biol. 17, 100-14.
Facchini, L. M., Chen, S., and Penn, L. J. Z. (1994). Dysfunction of the Myc-induced apoptosis mechanism accompanies c-myc activation in the tumorigenic L929 cell line. Cell Growth Differ. 5, 637-46.

Farrell, P. J., Allan, G. J., Shanahan, F., Vousden, K. H., and Crook, T. (1991). p53 is frequently mutated in Burkitt's lymphoma cell lines. EMBO J. 10,2879-87.
Field, J. K., and Spandidos, D. A. (1990). The role of ras and myc oncogenes in human solid tumours and their relevance in diagnosis and prognosis. Anticancer Res. 10, 1-22.
Fisher, F., Crouch, D. H., Jayaraman, P. S., Clark, W., Gillespie, D. A., and Goding, C. R. (1993). Transcription activation by Myc and Max: flanking sequences target activation to a subset of CACGTG motifs in vivo. EMBO J. 12,5075-82.
Fisher, F., Jayaraman, P. S., and Goding, C. R. (1991). C-myc and the yeast
transcription factor pH04 share a common CACGTG- binding motif. Oncogene 6, 1099- 104.
Fisher, R. P. (1997). CDKs and cyclins in transition(s). Curr. Opin. Genet. Dev. 7,32-8.
Fomace, A. J., Jr. (1992). Mammalian genes induced by radiation; activation of genes
associated with growth control. Annu. Rev. Genet. 26,507-26.
Fomace, A. J., Jr., Alamo, I., Jr., and Hollander, M. C. (1988). DNA damage-inducible transcripts in mammalian cells. Proc. Natl. Acad. Sci. USA 85,8800-4.
Fomace, A. J., Jr., Nebert, D. W., Hollander, M. C., Luethy, J. D., Papathanasiou, M., Fargnoli, J., and Holbrook, N. J. (1989). Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol. Cell. Biol. 9,4196-203.
Freeman, R. S., Estus, S., and Johnson, E. M., Jr. (1994). Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of Cyclin Dl during programmed cell death. Neuron 12,343-55.
Freytag, S. 0. (1988). Enforced expression of the c-myc oncogene inhibits cell differentiation by precluding entry into a distinct predifferentiation state in GOIGI. Mol. Cell. Biol. 8, 1614-24.

Freytag, S. O., Dang, C. V., and Lee, W. M. (1990). Definition of the activities and properties of c-myc required to inhibit cell differentiation. Cell Growth Differ. 1,339-43.
Friedman, A. D., Landochulz, W. H., and McKnight, S. L. (1989). CCAATIenhancer binding protein activates the promoter of the serum albumin gene in cultured hepatoma cells. Genes Dev. 3, 13 14-22.
Fukami, J., Anno, K., Ueda, K., Takahashi, T., and Ide, T. (1995). Enhanced expression of cyclin Dl in senescent human fibroblasts. Mech. Ageing Dev. 81, 139-57.
Fung, Y. T., T'Ang, A., Murphree, A. L., Zhang, F., Qiu, W., Wang, S., Shi, X., Lee, L., Driscoll, B., and Wu, K. (1993). The Rb gene suppresses the growth of normal cells.
Oncogene 8,2659-2672.
Gaidano, G., Ballerini, P., Gong, J. Z., Inghirami, G., Neri, A,, Newcomb, E. W.,
Magrath, I. T., Knowles, D. M., and Dalla-Favera, R. (1991). p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 88, 5413-7.
Galaktionov, K., Chen, X., and Beach, D. (1996). Cdc25 cell-cycle phosphatase as a target of c-myc. Nature 382,511-7.
Gallant, P., Shiio, Y., Cheng, P. F., Parkhurst, S. M., and Eisenman, R. N. (1996). Myc and Max homologs in Drosophila. Science 274, 1523-7.
Gandarillas, A., and Watt, F. M. (1997). c-Myc promotes differentiation of human
epidermal stem cells. Genes Dev. 11,2869-82.
Garte, S. J. (1993). The c-myc oncogene in tumor progression. Crit Rev Oncogenesis 4,
435-49.
Gaubatz, S., Imhof, A., Dosch, R., Werner, O., Mitchell, P., Buettner, R., and Eilers, M. (1995). Transcriptional activation by Myc is under negative control by the transcription factor AP-2. EMBO J. 14, 1508-19.

Gauoatz, S., Meichle, A., and Eilers, M. (1994). An E-box element localized in the first intron mediates regulation of the prothyrnosin alpha gene by c-myc. Mol. Cell. Biol. 14,
3853-62.
Gliniak, B. C., and Rohrschneider, L. R. (1990). Expression of the M-CSF receptor is
controlled by the dominant actions of GM-SF or multi-CSF. Cell 63,1073-83.
Gonda, T. J., and Metcalf, D. (1984). Expression of myb, myc and fos proto-oncogenes during the differentiation of a murine myeloid leukaernia. Nature 310,249-51.
Goodrich, D. W., and Lee, W.-H. (1993). Molecular characterization of the
retinoblastoma susceptibility gene. Biochim. Biophys. Acta 1155,43-61.
Gowda, S. D., Koler, R. D., and Bagby, G. C., Jr. (1986). Regulation of c-myc expression during growth and differentiation of normal and leukemic human myeloid
progenitor cells. J. Clin. Invest. 77,271-8.
Graf, T., and Beug, H. (1978). Avian leukemia vimses: interaction with their target cells in vivo and in vitro. Biochim. Biophys. Acta 516,269-99.
Graham, F. L., and Van der Eb, A. J. (1973). A new technique for the assay of infectivity
of human adenovims 5 DNA. Virology 52,456-467.
Grandori, C., Mac, J., Siebelt, F., Ayer, D. E., and Eisenman, R. N. (1996). Myc-Max
heterodimers activate a DEAD box gene and interact with multiple E box-related sites in vivo. EMBO J. 15,4344-57.
Greenberg, M. E., Greene, L. A., and Ziff, E. B. (1985). Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J. Biol. Chem. 260, 14101-10.
Greenberg, M. E., Hermanowski, A. L., and Ziff, E. B. (1986). Effect of protein synthesis inhibitors on growth factor activation of c-fos, c-myc, and actin gene transcription. Mol. Cell. Biol. 6, 1050-7.

Gregor, P. D., Sawadogo, M., and Roeder, R. G. (1990). The adenovims major late
transcription factor USF is a member of the helix-loop-helix group of regulatory proteins and binds to DNA as a dimer. Genes Dev. 4,1730-40.
Griep, A. E., and DeLuca, H. F. (1986). Decreased c-myc expression is an early event in retinoic acid-induced differentiation of F9 teratocarcinoma cells. Proc. Natl. Acad. Sci.
USA 83,5539-43.
Griep, A. E., and Westphal, H. (1988). Antisense Myc sequences induce differentiation of F9 cells. Proc. Natl. Acad. Sci. USA 85, 6806-10.
Grignani, F., Lombardi, L., Giorgio, I., Stemas, L., Cechova, K., and Dalla-Favera, R. (1990). Negative autoregulation of c-myc gene expression is inactivated in transformed cells. EMBO J. 9,3913-22.
Gu, W., Bhatia, K., Magrath, I. T., Dang, C. V., and Dalla-Favera, R. (1994). Binding and suppression of the Myc transcriptional activation domain by p107. Science 264,251- 4.
Gu, W., Cechova, K., Tassi, V., and Dalla-Favera, R. (1993). Opposite regulation of gene transcription and cell proliferation by c-Myc and Max. Proc. Natl. Acad. Sci. USA
90, 2935-9.
Guha, A., Dashner, K., Black, P. M., Wagner, J. A., and Stiles, C. D. (1995). Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int. J. Cancer. 60, 168-73.
Gupta, S., Seth, A., and Davis, R. J. (1993). Transactivation of gene expression by Myc is inhibited by mutation at the phosphorylation sites Thr-58 and Ser-62. Proc. Natl. Acad. Sci. USA 90, 3216-20.
Gustincich, S., and Schneider, C. (1993). Senun deprivation response gene is induced by serum starvation but not by contact inhibition. Cell Growth Differ. 4,753-60.

Haas-Kogan, D. A., Kogan, S. C., Levi, D., Dazin, P., T'Ang, A., Fung, Y.-K. T., and Israel, M. A. (1995). Inhibition of apoptosis by the retinoblastoma gene product. EMBO J. 14, 461 -472.
Halazonetis, T. D., and Kandil, A. N. (1991). Determination of the c-MYC DNA-binding site. Proc. Natl. Acad. Sci. USA 88, 6162-6.
Hall, P. A., Kearsey, J. M., Coates, P. J., Norman, D. G., Warbrick, E., and Cox, L. S.
(1995). Characterisation of the interaction between PCNA and Gadd45. Oncogene 10, 2427-35.
Hamel, P. A., Cohen, B. L., Sorce, L. M., Gallie, B. L., and Phillips, R. A. (1990). Hyperphosphorylation of the retinoblastoma gene product is determined by domains
outside the simian virus 40 large-t-antigen-binding regions. Mol. Cell. Biol. 10, 6586- 6595.
Hamel, P. A., Gill, R. M., Phillips, R. A., and Gallie, B. L. (1992). Transcriptional
repression of the E2-containing promoters EIIaE, c-myc, and RB1 by the product of the RBI gene. Mol. Cell. Biol. 12, 3431-8.
Han, E. K., Sgambato, A., Jiang, W., Zhang, Y. J., Santella, R. M., Doki, Y., Cacace,
A. M., Schieren, I., and Weinstein, I. B. (1995). Stable overexpression of cyclin Dl in a human mammary epithelial cell line proiongs the S-phase and inhibits growth. Oncogene 10, 953-61.
Hann, S. R. (1995). Methionine deprivation regulates the translation of hnctionally- distinct c-Myc proteins. Adv. Exp. Med. Biol. 375, 107-16.
Ham, S. R., Dixit, M., Sears, R. C., and Sealy, L. (1994). The alternatively initiated c-
Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes Dev. 8,2441-52.
Hann, S. R., and Eisenman, R. N. (1984). Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol. Cell. Biol. 4,2486-97.

Hann, S. R., King, M. W., Bentley, D. L., Anderson, C. W., and Eisenman, R. N. (1988). A non-AUG translational initiation in c-myc exon 1 generates an N- terminally
distinct protein whose synthesis is disrupted in Burkitt's lymphomas. Cell 52, 185-95.
Ham, S. R., Sloan-Brown, K., and Spotts, G. D. (1992). Translational activation of the non-AUG-initiated c-myc 1 protein at high cell densities due to methionine deprivation. Genes Dev. 6, 1229-40.
Ham, S. R., Thompson, C. B., and Eisenman, R. N. (1985). C-myc oncogene protein synthesis is independent of the cell cycle in human and avian cells. Nature 314,366-369.
Harper, J. W. (1997). Cyclin dependent kinase inhibitors. Cancer Surv. 29,91-107.
Harrington, E. A., Bennett, M. R., Fanidi, A., and Evan, G. I. (1994). cMyc-induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO J. 13,3286-9s.
Hatakeyama, M., Herrera, R. A., Makela, T., Dowdy, S. F., Jacks, T., and Weinberg, R. A. (1994). The cancer cell and the cell cycle clock. Cold Spring Harb. Symp. Quant. Biol. 59, 1-10.
Hateboer, G., Timmers, H. T., Rustgi, A. K., Billaud, M., van't Veer, L. J., and Bemards, R. (1993). TATA-binding protein and the retinoblastoma gene product bind to
overlapping epitopes on c-Myc and adenovirus E1A protein. Proc. Natl. Acad. Sci. USA 90, 8489-93.
Haupt, Y., Rowan, S., and Oren, M. (1995). PS3-mediated apoptosis in HeLa cells can be overcome by excess pRB. Oncogene 10, 1563-1571.
Hayashi, K., Makino, R., Kawamura, H., Arisawa, A., and Yoneda, K. (1987). Characterization of rat c-myc and adjacent regions. Nucleic Acids Res. 15,6419-36.
Hayward, W. S., Neel, B. G., and Ashin, S. M. (1981). Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 290,475-80.
Heichman, K. A., and Roberts, J. M. (1994). Rules to replicate by. Cell 79, 557-62.

Heikkila, R., Schwab, G., Wickstrom, E., Loke, S. L., Plumik, D. H., Watt, R., and Neckers, L. M. (1987). A c-myc antisense oligodeoxynucleotide inhibits entry into S phase but not progress from GO to G1. Nature 328,445-9.
Helin, K., and Harlow, E. (1992). The retinoblastoma protein as a transcriptional repressor. Trends Cell Biol. 3,43-46.
Henriksson, M., Bakardjiev, A., Klein, G., and Luscher, B. (1993). Phosphorylation
sites mapping in the N-terminal domain of c-myc modulate its transforming potential. Oncogene 8,3 199-209.
Henriksson, M., and Luscher, B. (1996). Proteins of the Myc network: essential regulators of cell gowth and differentiation. Adv. Cancer Res. 68, 109-82.
Hermeking, H., and Eick, D. (1994). Mediation of c-Myc-induced apoptosis by p53. Science 265,2091-3.
Hermeking, H., Funk, J. O., Reichert, M., Ellwart, J. W., and Eick, D. (1995).
Abrogation of p53-induced cell cycle arrest by c-Myc: evidence for an inhibitor of p2lWAFl/CIPl/SDII. Oncogene 11,1409-15.
Hershko, A. (1997). Roles of ubiquitin-mediated proteolysis in cell cycle control. Cum. Opin. Cell Biol. 9, 788-99.
Hiebert, S. W., Chellappan, S. P., Horowitz, J. M., and Nevins, J. R. (1992). The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of
E2F. Genes Dev. 6, 177-85.
Hinds, P. W., Mittnacht, S., Dulic, V., Arnold, A., Reed, S. I., and Weinberg, R. A. (1992). Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70, 993-1006.
Hirai, H., and Shen; C. J. (1996). Interaction of D-type cyclins with a novel myb-like transcription factor, DMPI. Mol. Cell. Biol. 16,6457-67.

Hoang, A. T., Lutterbach, B., Lewis, B. C., Yano, T., Chou, T. Y., Barrett, J. F., Raffeld, M., Hann, S. R., and Dang, C. V. (1995). A link between increased transforming activity of lymphoma-derived MYC mutant alleles, their defective regulation by p107, and altered phosphorylation of the c-Myc transactivation domain. Mol. Cell.
Biol. IS, 4031-42.
Hoffinann, I., Draetta, G., and Karsenti, E. (1994). Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the GUS transition. EMBO J. 13,4302-10.
Hoffinann, I., and Karsenti, E. (1994). The role of cdc25 in checkpoints and feedback controls in the eukqotic cell cycle. J. Cell Sci. Suppl. 18,75-9.
Hollander, M. C., Alamo, I., Jackman, J., Wang, M. G., McBride, 0. W., and Fomace, A. J., Jr. (1993). Analysis of the mammalian gadd45 gene and its response to DNA
damage. J. Biol. Chem. 268,24385-93.
Holt, J. T., Redner, R. L., and Nienhuis, A. W. (1988). An oligomer complementary to
c-myc mRNA inhibits proliferation of HL-60 promyelocytic cells and induces
differentiation. Mol. Cell. Biol. 8, 963-73.
Horiychi-Yamada, J., Yamada, H., Nakada, S., Ochi, K., and Nemoto, T. (1994). Changes of G1 cyclins, cdk2, and cyclin A during the differentiation of HL60 cells induced by TPA. Mol. Cell Biochem. 132,31-7.
Horton, L. E., Qian, Y., and Templeton, D. J. (1995). G1 Cyclins control the Retinoblastoma Gene Product Growth Regulation Activity via Upstream Mechanisms. Cell Growth Differ. 6, 395-407.
Hu, Y. F., Luscher, B., Admon, A., Mermod, N., and Tjian, R. (1990). Transcription
factor AP-4 contains multiple dimerization domains that regulate dimer specificity. Genes Dev. 4, 1741-52.
Huang, H. J., Yee, J. K., Shew, J. Y., Chen, P. L., Bookstein, R., Friedmann, T., Lee,
E. Y., and Lee, W.-H. (1988). Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 242,1563-1566.

Hueber, A. O., Zomig, M., Lyon, D., Suda, T., Nagata, S., and Evan, G. I. (1997). Requirement for the CD95 receptor-ligand pathway in c-myc-induced apoptosis. Science
278, 1305-9.
Inghirami, G., Grignani, F., Sternas, L., Lombardi, L., Knowles, D. M., and Dalla- Favera, R. (1990). Down-regulation of LFA-1 adhesion receptors by C-myc oncogene in human B lymphoblastoid cells. Science 250,682-6.
Ingvarsson, S. (1990). The myc gene family proteins and their role in transformation and differentiation. Semin. Cancer Biol. 1,359-69.
Ingvarsson, S., Asker, C., Axelson, H., Klein, G., and Surnegi, J. (1988). Structure and expression of B-myc, a new member of the myc gene family. Mol. Cell. Biol. 8,3168-74.
Inoue, K., and Sherr, C. J. (1998). Gene expression and cell cycle arrest mediated by transcription factor DMPl is antagonized by D-type cyclins through a cyclin-dependent- kinase- independent mechanism. Mol. Cell. Biol. 18,1590-600.
Ishisaki, A., Murayama, T., Ballagi, A. E., and Funa, K. (1997). Nuclear factor Y
controls the basal transcription activity of the mouse platelet-derived-growth-factor beta- receptor gene. Eur. J. Biochem. 246, 142-6.
Jackman, J., Alamo, I., Jr., and Fomace, A. J., Jr. (1994). Genotoxic stress confers
preferential and coordinate messenger RNA stability on the five gadd genes. Cancer Res. 54, 5656-62.
Jacks, T., Fazelli, A., Schmitt, E. M., Bronson, R. T., Goodell, M. A., and Weinberg, R. A. (1992). Effects of an Rb mutation in the mouse. Nature 359,295-300.
Jahn, L., Sadoshima, J., and Izumo, S. (1994). Cyclins and cyclin-dependent kinases are
differentially regulated during terminal differentiation of C2C12 muscle cells. Exp. Cell Res. 212, 297-307.

Jansen, H. W., Patschinsky, T., Walther, N., Lurz, R., and Bister, K. (1985). Molecular and biological properties of MH2D12, a spontaneous mil deletion mutant of avian oncovirus MH2. Virology 142, 248-62.
Jansen-Dun; P., Meichle, A., Steiner, P., Pagano, M., Finke, K., Botz, J., Wessbecher, J., Draetta, G., and Eilers, M. (1993). Differential modulation of cyclin gene expression
by MYC. Proc. Natl. Acad. Sci. USA 90, 3685-9.
Jiang, D., Srinivasan, A., Lozano, G., and Robbins, P. D. (1993). SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene 8,2805-12.
Jinno, S., Suto, K., Nagata, A., Igarashi, M., Kanaoka, Y., Nojima, H., and Okayama, H. (1994). Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 13, 1549-56.
Kaczmarek, L., Hyland, J. K., Watt, R., Rosenberg, M., and Baserga, R. (1985). Microinjected c-myc as a competence factor. Science 228, 1313-5.
Kaelin, W. G. J., Krek, W., Sellers, W. R., DeCaprio, J. A., Ajchenbaum, F., Fuchs,
C. S., Chettenden, T., Li, Y., Farnham, P. J., Blanar, M. A., Livingston, D. M., and Flemington, E. K. (1992). Expression cloning of a cDNA encoding a retinoblastoma-
binding protein with E2F-like properties. Cell 70,351-364.
Kagaya, S., Kitanaka, C., Noguchi, K., Mochizuki, T., Sugiyama, A., Asai, A., Yasuhara, N., Eguchi, Y., Tsujimoto, Y., and Kuchino, Y. (1997). A functional role for death proteases in s-Myc- and c-Myc-mediated apoptosis. Mol. Cell. Biol. 17,6736-45.
Kalderon, D., Richardson, W. D., Markham, A. F., and Smith, A. E. (1984). Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature 311,33-8.
Kaneko-Ishino, T., Kume, T. U., Sasaki, H., Obinata, M., and Oishi, M. (1988). Effect of c-myc gene expression on early inducible reactions required for erythroid differentiation in vitro. Mol. Cell. Biol. 8, 5545-8.
Karn, J., Watson, J. V., Lowe, A. D., Green, S. M., and Vedeckis, W. (1989). Regulation of cell cycle duration by c-myc levels. Oncogene 4,773-87.

Kastan, M. B., Zhan, Q., el-Deiry, W. S., Canier, F., Jacks, T., Walsh, W. V.,
Plunken, B. S., Vogelstein, B., and Fomace, A. J., Jr. (1992). A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71, 587-97.
Kato, G. J., Barren, J., Villa-Garcia, M., and Dang, C. V. (1990). An amino-terminal c- myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 10, 5914-20.
Kato, G. J., Lee, W. M., Chen, L. L., and Dang, C. V. (1992). Max: functional domains and interaction with c-Myc. Genes Dev. 6,81-92.
Kato, J.-Y., Matsushime, H., Hiebert, S. W., Ewen, M. E., and Sherr, C. J. (1993). Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 7,331-342.
Kauffmann-Zeh, A., Rodriguez-Viciana, P., Ulrich, E., Gilbert, C., Coffer, P., Downward, J., and Evan, G. (1997). Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 385,544-8.
Kawashima, K., Nomura, S., Hirai, H., Fukushi, S., Karube, T., Takeuchi, K., Naruke, T., and Nishimwa, S. (1992). Correlation of L-myc RFLP with metastasis, prognosis and multiple cancer in lung-cancer patients. Int. J. Cancer 50,557-61.
Kearsey, J. M., Coates, P. J., Prescott, A. R., Warbrick, E., and Hall, P. A. (1995). Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cipl. Oncogene 11, 1675-83.
Keath, E. J., Kelekar, A., and Cole, M. D. (1984). Transcriptional activation of the translocated c-myc oncogene in mouse plasmacytomas: similar RNA levels in tumor and proliferating normal cells. Cell 37, 521-8.
Kelly, K., Cochran, B., Stiles, C., and Leder, P. (1983). Cell specific regulation of the c-
myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 35,603-10.

Kelly, K., and Siebenlist, U. (1986). The regulation and expression of c-myc in normal and malignant cells. Annu. Rev. Imrnunol. 4, 317-338.
Kennedy, S. G., Wagner, A. J., Conzen, S. D., Jordan, J., Bellacosa, A., Tsichlis, P.
N., and Hay, N. (1997). The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11,701-13.
Kerkhoff, E., and Bister, K. (1991). Myc protein structure: localization of DNA-binding and protein dimerization domains. Oncogene 6,93-102.
Kerkhoff, E., Bister, K., and Klempnauer, K. H. (1991). Sequence-specific DNA
binding by Myc proteins. Proc. Natl. Acad. Sci. USA 88,4323-7.
King, M. W., Roberts, J. M., and Eisenman, R. N. (1986). Expression of the c-myc proto-oncogene during development of Xenopus laevis. Mol. Cell. Biol. 6,4499-508.
Klefstrom, J., Vastrik, I., Saksela, E., Valle, J., Eilers, M., and Alitalo, K. (1994). c- Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J. 13, 5442-50.
Kohl, N. E., Gee, C. E., and Alt, F. W. (1984). Activated expression of the N-myc gene
in human neuroblastomas and related tumors. Science 226, 1335-7.
Kommann, M., Arber, N., and Korc, M. (1998). Inhibition of basal and mitogen- stimulated pancreatic cancer cell growth by cyclin Dl antisense is associated with loss of tumorigenicity and potentiation of cytotoxicity to cisplatinum. J. Clin. Invest. 101,344-52.
Kretzner, L., Blackwood, E. M., and Eisenman, R. N. (19923. Myc and Max proteins possess distinct transcriptional activities. Nature 339,426-9.
Kretzner, L., Blackwood, E. M., and Eisenman, R. N. (1992). Transcriptional activities
of the Myc and Max proteins in mammalian cells. Curr. Top. Microbial. Immunol. 182, 435-43.
Kuchino, Y., Asai, A., and Kitanaka, C. (1996). Myc-mediated apoptosis. Prog Mol
Subcell. Biol. 16, 104-29.

Lachman, H. M., and Skoultchi, A. I. (1984). Expression of c-myc changes during differentiation of mouse erythroleukaemia cells. Nature 310,5924
Lanahan, A., Williams, J. B., Sanders, L. I<., and Nathans, D. (1992). Growth factor- induced delayed early response genes. Mol. Cell Biol. 12,3919-3929.
Land, H., Parada, L. F., and Weinberg, R. A. (1983). Tumorigenic conversion of
primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304,596- 602.
Lania, L., Gandini-Attardi, D., Griffiths, M., Cooke, B., DeCicco, D., and Fried, M. (1980). The polyoma vims lOOK large T-antigen is not required for the maintenance of transformation. Virology 101,217-232.
Larsson, L. G., Ivhed, I., Gidlund, M., Pettersson, U., Vennstrom, B., and Nilsson, K. (1988). Phorbol ester-induced terminal differentiation is inhibited in human U-937 monoblastic cells expressing a v-myc oncogene. Proc. Natl. Acad. Sci. USA 85,2638-42.
Lau, L. F., and Nathans, D. (1987). Expression of a set of growth-related immediate early genes in BALBIc 3T3 cells: coordinate regulation with c-fos or c-myc. Proc. Natl. Acad. Sci. USA 84. 1182-6.
Lee, E. Y.-H., Chang, C.-Y., Hu, N., Wang, Y.-C. J., Lai, c.-c., Hemp, K., Lee, W.- H., and Bradley, A. (1992). Mice deficient for Rb are nonviable and show defects in
neurogenesis and haematopoiesis. Nature 359,288-294.
Lee, L. A,, Dolde, C., Barren, J., Wu, C. S., and Dang, C. V. (1996). A link between c- Myc-mediated transcriptional repression and neoplastic transformation. J. Clin. Invest. 97, 1687-95.
Lee, T. C., Li, L., Philipson, L., and Ziff, E. B. (1997). Myc represses transcription of the growth arrest gene gasl. Proc. Natl. Acad. Sci. USA 94, 12886-91.
Lees, E. (1995). Cyclin dependent kinase regulation. Cum. Opin. Cell Biol. 7,773-80.

Lees, J. A., Buchkovich, K. J., Marshak, D. R., Anderson, C. W., and Harlow, E. (1991). The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J. 10,4279-4290.
Lemaitre, J. M., Buckle, R. S., and Mechali, M. (1996). c-Myc in the control of cell proliferation and embryonic development Adv. Cancer Res. 70,95-144.
Lenahan, M. K., and Ozer, H. L. (1996). Induction of c-myc mediated apoptosis in SV40-
transformed rat fibroblasts. Oncogene 12,1847-54.
Leone, G., DeGregori, J., Sears, R., Jakoi, L., and Nevins, J. R. (1997). Myc and Ras collaborate in inducing accumulation of active cyclin EICdk2 and E2F. Nature 387,422-6.
Lew, D. J., and Kombluth, S. (1996). Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Cum. Opin. Cell Biol. 8,795-804.
Li, L. H., Nerlov, C., Prendergast, G., MacGregor, D., and Ziff, E. B. (1994). c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and
Myc box 11. EMBO J. 13,4070-9.
Li, Y., Holland, C. A., Hartley, J. W., and Hopkins, N. (1984). Viral integration near c- myc in 10-20% of mcf 247-induced AKR lymphomas. Proc. Natl. Acad. Sci. USA 81,
6808-1 1.
Lin, B. T.-Y., Gmenwald, S., Mona, A. O., Lee, W.-H., and Wang, J. Y. J. (1991). Retinoblastoma cancer suppressor gene product is a substrate of the cell cycle regulator cdc2 kinase. EMBO J. 10,857-864.
Little, C. D., Nau, M. M., Carney, D. N., Gazdar, A. F., and Minna, J. D. (1983). Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306, 194-6.
Littlewood, T. D., Amati, B., Land, H., and Evan, G. I. (1992). Max and c-MycIMax
DNA-binding activities in cell extracts. Oncogene 7,1783-92.

Littlewood, T. D., Hancock, D. C., Danielian, P. S., Parker, M. G., and Evan, G. I.
(1995). A modified oestrogen receptor ligand-binding domain as an improved switch for
the regulation of heterologous proteins. Nucleic Acids Res. 23, 1686-90.
Lucas, J. M., Wilkie, N. M., and Lang, J. C. (1993). c-MYC repression of promoter activity through core promoter elements. Biocbem. Biophys. Res. Comrnun. 194, 1446-
52.
Lucibello, F. C., Sewing, A., Brusselbach, S., Burger, C., and Muller, R. (1993). Induction of cyclin Dl and cyclin-E and suppression of cdk2 and cdk4 in senescent human cells. J. Cell Sci. 105, 123-133.
Ludlow, J. W., C.L., G., D.M., L., and J.A., D. (1993). Specific enzymatic
dephosphorylation of the retinoblastoma protein. ~Mol. Cell. Biol. 13,367-372.
Ludlow, J. W., Shon, J., Pipas, J. M., Livingston, D. M., and DeCaprio, J. A. (1990). The retinoblastoma susceptibility gene product undergoes cell cycle dependent dephosphorylation and binding to and release from SV40 large T. Cell 60,387-396.
Lukas, J., Bartkova, J., Rohde, M., Strauss, M., and Bartek, J. (1995). Cyclin Dl is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol. Cell. Biol. 15, 2600-261 1.
Lukas, J., Pagano, M., Staskova, Z., Draetta, G., and Bartek, J. (1994). Cyclin Dl protein oscillates and is essential for cell cycle progression in human tumour celi lines.
Oncogene 9,707-71 8.
Luscher, B., and Eisenman, R. N. (1990). New light on Myc and Myb. Part I. Myc.
Genes Dev. 4, 2025-35.
Luscher, B., Kuenzel, E. A,, Krebs, E. G., and Eisenman, R. N. (1989). Myc oncoproteins are phosphorylated by casein kinase 11. EMBO J. 8, 11 11-9.
Lutterbach, B., and Hann, S. R. (1994). Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis.
Mol. Cell. Biol. 14, 5510-22.

Maheswarm, S., Lee, H., and Sonenshein, G. E. (1994). Intracellular association of the protein product of the c-myc oncogene with the TATA-binding protein. Mol. Cell. Biol.
14. 1147-52.
Mai, S. (1994). Overexpression of c-myc precedes amplification of the gene encoding
dihydrofolate reductase. Gene 148,253-60.
Mai, S., Fluri, M., Siwarski, D., and Huppi, K. (1996). Genomic instability in MycER- activated RatlA-MycER cells. Chromosome Res. 4,365-71.
Mai, S., Hanley-Hyde, J., and Fluri, M. (1996). c-Myc overexpression associated DHFR
gene amplification in hamster, rat, mouse and human cell lines. Oncogene 12,277-88.
Mai, S., and Martensson, I. L. (1995). The c-myc protein represses the lambda 5 and TdT initiators. Nucleic Acids Res. 23, 1-9.
Makela, T. P., Koskinen, P. J., Vastrik, I., and Alitalo, K. (1992). Alternative forms of Max as enhancers or suppressors of Myc-ras cotransformation. Science 256,373-7.
Makela, T. P., Saksela, K., and Alitalo, K. (1992). Amplification and rearrangement of L-
myc in human small-cell lung cancer. Mutat. Res. 276,307-15.
Marcu, K. B., Bossone, S. A,, and Patel, A. J. (1992). Myc function and regulation. Annu. Rev. Biochem. 61,809-60.
Marhin, W. W., Chen, S., Facchini, L. M., Fomace Jr, A. J., and Pem, L. Z. (1997). Myc represses the growth arrest gene gadd45. Oncogene 14,2825-34.
Marhin, W. W., Hei, Y. J., Chen, S., Jiang, Z., Gallie, B. L., Phillips, R. A., and Penn,
L. Z. (1996). Loss of Rb and Myc activation co-operate to suppress cyclin Dl and contribute to transformation. Oncogene 12,43-52.
Markowitz, D., Goff, S., and Bank, A. (1988). A safe packaging line for gene transfer: separating viral genes on two different plasmids. J. Virol. 62, 1120-1 124.

Martin, K. J. (1991). The interactions of transcription factors and their adaptors, coactivators and accessory proteins. Bioessays 13,499-503.
Masiakowski, P., Breathnach, R., Bloch, J., Gannon, F., Krust, A., and Chambon, P.
(1982). Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res. 10,7895-903.
Mateyak, M. K., Obaya, A. J., Adachi, S., and Sedivy, J. M. (1997). Phenotypes of c-
Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 8, 1039-48.
Matsushime, H., Quelle, D. E., Shurtleff, S. A., Shibuya, M., Shen; C. J., and Kato, J.- Y . (1994). D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 14, 2066-2076.
Matsushime, H., Roussel, M. F., Ashmun, R. A., and Sherr, C. J. (1991). Colony- stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 65, 701-713.
McDonnell, T. J., and Korsmeyer, S. J. (1991). Progression from lymphoid hyperplasia
to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 349,254-6.
Mechti, N., Piechaczyk, M., Blanchard, J. M., Marly, L., Bonnieu, A., Jeanteur, P., and Lebleu, B. (1986). Transcriptional and post-transcriptional regulation of c-myc expression during the differentiation of murine erythroleukemia Friend cells. Nucleic Acids Res. 14, 9653-66.
Meyerson, M., and Harlow, E. (1994). Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 14,2077-2086.
Mietz, J. A., Unger, T., Huibregtse, J. M., and Howley, P. M. (1992). The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T- antigen and by HPV-16 E6 oncoprotein. EMBO J. 11,5013-20.

Mihara, K., Cao, X.-R., Yen, A., Chandler, S., Driscoll, B., Murphee, A. L., T'Ang,
A., and Fung, Y.-K. T. (1989). Cell cycle-dependent regulation of phosphorylation of the human retinoblastoma gene product. Science 246, 1300-1303.
Miltenberger, R. J., Sukow, K. A., and Famham, P. J. (1995). An E-box-mediated increase in cad transcription at the GlIS-phase boundary is suppressed by inhibitory c-myc mutants. Mol. Cell. Biol. 15, 2527-2535.
Miner, J. H., and Wold, B. J. (1991). c-myc inhibition of MyoD and myogenin-initiated myogenic differentiation. Mol. Cell. Biol. 11,2842-51.
Mink, S., Mutschler, B., Weiskirchen, R., Bister, K., and Klempnauer, K. H. (1996). A novel function for Myc: inhibition of clebp-dependent gene activation. Proc. Natl. Acad. Sci. USA 93, 6635-40.
Mitchell, P. J., and Tjian, R. (1989). Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 245,371-8.
Mittnacht, S., Lees, J. A., Desai, D., Harlow, E., Morgan, D. O., and Weinberg, R. A. (1994). Distinct sub-populations of the retinoblastoma protein show a distinct pattern of phosphorylation. EMBO J. 13, 118-127.
Morgenbesser, S. D., Schreiber-Agus, N., Bidder, M., Mahon, K. A., Overbeek, P. A., Homer, J., and DePinho, R. A. (1995). Contrasting roles for c-Myc and L-Myc in the regulation of cellular gowth and differentiation in vivo. EMBO J. 14,743-56.
Morgenbesser, S. D., Williams, B. O., Jacks, T., and DePinho, R. A. (1994). p53- dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature 371,
72-74.
Morgenstem, J. P., and Land, H. (1990). Advanced mammalian gene transfer: high titre
retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587-3596.

Moroy, T., Fisher, P., Guidos, C., Ma, A,, Zimmerman, K., Tesfaye, A., DePinho, R., Weissman, I., and Alt, F. W. (1990). IgH eilhancer deregulated expression of L-myc:
abnormal T lymphocyte development and T cell lymphomagenesis. EMBO J. 9,3659-66.
Motokura, T., and Amold, A. (1993). PRADllCyclin Dl proto-oncogene: Genomic organization 5' DNA sequence, and sequence of a tumor-specific rearrangement breakpoint. Gene Chrom. Canc. 7, 89-95.
Motokura, T., Bloom, T., Kim, H. G., Jiippner, H., Ruderman, J. V., Kronenberg, H. M., and Arnold, A. (1991). A novel cyclin encoded by a bcll- linked candidate oncogene. Nature 350, 512-5 15.
Motokura, T., Loom, T., Kim, H. G., Jiippner, H., and Ruderman, J. (1990). A novel cyclin encoded by bcl-1 linked candidate oncogene. Nature 350,512-515.
Mougneau, E., Lemieux, L., Rassoulzadegan, M., and Cuzin, F. (1984). Biological activities of v-myc and rearranged c-myc oncogenes in rat fibroblast cells in culture. Proc. Natl. Acad. Sci. USA 81, 5758-62.
Mudryj, M., Hiebert, S. W., and Nevins, J. R. (1990). A role for the adenovims inducible E2F transcription factor in a proliferation dependent signal transduction pathway. EMBO J. 9, 2179-84.
Muller, D., Bouchard, C., Rudolph, B., Steiner, P., Stuckmann, I., Saffrich, R.,
Ansorge, W., Huttner, W., and Eilers, M. (1997). Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin EIcdk2 complexes. Oncogene 15,2561-
76.
Muller, H., Lukas, J., Schneider, A., Warthoe, P., Bartek, J., Eilers, M., and Strauss,
M. (1994). Cyclin Dl expression is regulated by the retinoblastoma protein. Proc. Natl. Acad. Sci. USA 91,2945 - 2949.
Nau, M. M., Brooks, B. J., Battey, J., Sausville, E., Gazdar, A. F., Kirsch, I. R., McBride, 0. W., Bertness, V., Hollis, G. F., and Minna, J. D. (1985). L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 318,69-
73.

Neel, B. G., Jhanwar, S. C., Chaganti, R. S., and Hayward, W. S. (1982). Two human c-onc genes are located on the long arm of chromosome 8. Proc. Natl. Acad. Sci. USA 79, 7842-6.
Nepveu, A., Marcu, K. B., Skoultchi, A. I., and Lachman, H. M. (1987). Contributions
of transcriptional and post-transcriptional mechanisms to the regulation of c-myc expression in mouse erythroleukemia cells. Genes Dev. 1,938-45.
Neuman, E., Ladha, M. H., Lin, N., Upton, T. M., Miller, S. J., DiRenzo, J., Pestell,
R. G., Hinds, P. W., Dowdy, S. F., Brown, M., and Ewen, M. E. (1997). Cyclin Dl stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol. Cell. Biol. 17, 5338-47.
Nevins, J. R. (1992). E2F: A link between the Rb tumor suppressor protein and viral
oncoproteins. Science 258,424-429.
Nevins, J. R., Leone, G., DeGregori, J., and Jakoi, L. (1997). Role of the RbIE2F pathway in cell growth control. J. Cell Physiol. 173,233-6.
Nigg, E. A. (1995). Cyclin-dependent protein kinases: key regulators of the eukaryotic cell
cycle. Bioessays 17,471-80.
Nilsson, J., Thyberg, C. H., Heldin, B., Westermark, A., and Wasteson, A. (1983). Surface binding and internalization of platelet-derived growth factor in human fibroblasts. Proc. Natl. Acad. Sci. USA 80, 5592-6.
Oberg, F., Larsson, L. G., Anton, R., and Nilsson, K. (1991). Interferon gamma abrogates the differentiation block in v-myc- expressing U-937 monoblasts. Proc. Natl.
Acad. Sci. USA 88,5567-71.
Onclercq, R., Babinet, C., and Cremisi, C. (1989). Exogenous c-myc gene overexpression interferes with early events in F9 cell differentiation. Oncogene Res. 4,
293-302.

Packham, G., and Cleveland, J. L. (1995). c-Myc and apoptosis. Biochim. Biophys. Acta 1242, 11-28.
Packham, G., and Cleveland, J. L. (1997). Induction of omithine decarboxylase by IL-3 is mediated by sequential c-Myc-independent and c-Myc-dependent pathways. Oncogene 15, 1219-32.
Packham, G., and Cleveland, J. L. (1994). Ornithine decarboxylase is a mediator of c- Myc-induced apoptosis. Mol. Cell. Biol. 14,5741-7.
Packham, G., and Cleveland, J. L. (1995). The role of omithine decarboxylase in c-Myc-
induced apoptosis. Cum Top. Microbial. Imrnunol. 194,283-90.
Pagano, M. (1997). Cell cycle regulation by the ubiquitin pathway. Faseb J. 11, 1067-75.
Papathanasiou, M. A., and Fomace, A. J., Jr. (1991). DNA-damage inducible genes. Cancer Treat Res. 57, 13-36.
Papathanasiou, M. A., Kerr, N. C., Robbins, J. H., McBride, 0. W., Alamo, I., Jr., Barrett, S . F., Hickson, I. D., and Fornace, A. J., Jr. (1991). Induction by ionizing
radiation of the gadd45 gene in cultured human cells: lack of mediation by protein kinase C. Mol. Cell. Biol. 11, 1009-16.
Pardee, A. B. (1974). A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 71, 1286-90.
Parry, D., Bates, S., Mann, D. J., and Peters, G. (1995). Lack of cyclin D-Cdk in Rb- negative cells correlates with high levels of p161NK4'MTS1 tumour suppressor gene product. EMBO J. 14, 503-51 1.
Payne, G. S., Bishop, J. M., and Varmus, H. E. (1982). Multiple arrangements of viral
DNA and an activated host oncogene in bursa1 lymphomas. Nature 295,209-14.
Penn, L. J., Brooks, M. W., Laufer, E. M., and Land, H. (1990). Negative
autoregulation of c-myc transcription. EMBO J. 9,1113-21.

Penn, L. J., Brooks, M. W., Laufer, E. M., Littlewood, T. D., Morgenstem, J. P., Evan, G. I., Lee, W. M., and Land, H. (1990). Domains of human c-myc protein required for autosuppression and cooperation with ras oncogenes are overlapping. Mol. Cell. Biol. 10, 4961-6.
Penn, L. J., Laufer, E. M., and Land, H. (1990). C-MYC: evidence for multiple regulatory functions. Semin. Cancer Biol. 1,69-80.
Penn, L. J. Z., Brooks, M. W., Laufer, E. M., and Land, H. (1990). Negative autoregulation of c-myc transcription. EMBO J. 9, 11 13-1 121.
Penn, L. J. Z., Brooks, M. W., Laufer, E. M., Littlewood, T. D., Morgenstem, J. P.,
Evan, G. I., Lee, W. M. F., and Land, H. (1990). Domains of human c-myc protein required for autosuppression and cooperation with ras oncogenes are overlapping. Mol. Cell. Biol. 10, 4961-4966.
Persson, H., and Leder, P. (1984). Nuclear localization and DNA binding properties of a protein expressed by human c-myc oncogene. Science 225,718-21.
Philipp, A., Schneider, A., Vasrik, I., Finke, K., Xiong, Y., Beach, D., Alitalo, K., and Eilers, M. (1994). Repression of cyclin Dl: a novel function of MYC. Mol. Cell. Biol. 14, 4032-43.
Pledger, W. J., Stiles, C. D., Antoniades, H. N., and Scher, C. D. (1977). Induction of DNA synthesis in BALB/c 3T3 cells by serum components: reevaluation of the commitment process. Proc. Natl. Acad. Sci. USA 74,4481-5.
Pledger, W. J., Stiles, C. D., Antoniades, H. N., and Scher, C. D. (1978). An ordered
sequence of events is required before BALBIc-3T3 cells become committed to DNA synthesis. Proc. Natl. Acad. Sci. USA 75, 2839-43.
Plummer 111, H., Catlett, J., Leflwich, J., Armstrong, B., Carlson, P., Huff, T., and Krystal, G. (1993). c-myc expression correlates with suppression of c-kit protooncogene
expression in small cell lung cancer cell lines. Cancer Res. 53,4337-42.

Prendergast, G. C., Diamond, L. E., Dahl, D., and Cole, M. D. (1990). The c-myc- regulated gene mrl encodes plasrninogen activator inhibitor 1. Mol. Cell. Biol. 10, 1265-9.
Prendergast, G. C., Lawe, D., and Ziff, E. B. (1991). Association of Myn, the murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and Ras cotransformation. Cell 65,395-407.
Prendergast, G. C., and Ziff, E. B. (1991). Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 251, 186-189.
Prins, J., Devries, E., and Mulder, N. (1993). The myc family of oncogenes and their
presence and importance in small-cell lung carcinoma and other tumor types. Anticancer Res. 13, 1373-85.
Prochownik, E. V., and Kukowska, J. (1986). Deregulated expression of c-myc by murine erythroleukaemia cells prevents differentiation. Nature 322,848-50.
Prochownik, E. V., Kukowska, J., and Rodgers, C. (1988). c-myc antisense transcripts accelerate differentiation and inhibit G1 progression in murine erythroleukernia cells. Mol.
Cell. Biol. 8, 3683-95.
Prouty, S. M., Hanson, K. D., Boyle, A. L., Brown, J. R., Shichiri, M., Follansbee, M. R., Kang, W., and Sedivy, J. M. (1993). A cell culture model system for genetic analyses of the cell cycle by targeted homologous recombination. Oncogene 8,899-907.
Pulverer, B. J., Fisher, C., Vousden, K., Littlewood, T., Evan, G., and Woodgett, J. R. (1994). Site-specific modulation of c-Myc cotransformation by residues phosphorylated in
vivo. Oncogene 9,59-70.
Quelle, D. E., Ashmun, R. A., Shurtleff, S. A., Kato, J. Y., Bar-Sagi, D., Roussel, M. F., and Shen; C. J. (1993). Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 7, 1559-71.
Rabbitts, P. H., Watson, J. V., Lamond, A., Forster, A., Stinson, M. A., Evan, G., Fischer, W., Atherton, E., Sheppard, R., and Rabbitts, T. H. (1985). Metabolism of c-

myc gene products: c-myc mRNA and protein expression in the cell cycle. EMBO J. 4, 2009-15.
Rabbins, T. H., van Straaten, P., Rabbins, P. H., and Watson, J. (1985). Discussion on the metabolism of c-myc mRNA and protein. Proc. R. Soc. Lond. B. Biol. Sci. 226,79- 82.
Ran, W., Dean, M., Levine, R. A., Henkle, C., and Campisi, J. (1986). Induction of c- fos and c-myc mRNA by epidermal growth factor or calcium ionophore is CAMP
dependent. Proc. Natl. Acad. Sci. USA 83, 8216-20.
Reed, S. I. (1997). Control of the GUS transition. Cancer S w . 29,7-23.
Reitsma, P. H., Rothberg, P. G., Astrin, S. M., Trial, J., Bar-Shavit, Z., Hall, A., Teitelbaum, S. L., and Kahn, A. J. (1983). Regulation of myc gene expression in HL-60 leukaemia cells by a vitamin D metabolite. Nature 306,492-4.
Resar, L. M., Dolde, C., Barrett, J. F., and Dang, C. V. (1993). B-myc inhibits neoplastic transformation and transcriptional activation by c-myc. Mol. Cell. Biol. 13,
1130-6.
Resnitzky, D., Gossen, M., Bujard, H., and Reed, S. I. (1994). Acceleration of the Gl/S
phase transition by expression of cyclins Dl and E with an inducible system. Mol. Cell. Biol. 14, 1669-1679.
Richardson, W. D., Roberts, B. L., and Smith, A. E. (1986). Nuclear location signals in polyoma virus large-T. Cell 44,7745.
Ridley, A. J., Paterson, H. F., Noble, M., and Land, H. (1988). Ras-mediated cell cycle
arrest is altered by nuclear oncogenes to induce Schwann cell transformation. EMBO J. 7, 1635-45.
Rosenbaum, H., Webb, E., Adams, J. M., Cory, S., and Hanis, A. W. (1989). N-myc transgene promotes B lymphoid proliferation, elicits lymphomas and reveals cross- regulation with c-myc. EMBO J. 8,749-55.

Rosenfeld, M. E., Bowen-Pope, D., and Ross, R. (1985). Platelet-derived growth factoc morphologic and biochemical studies of binding, internalization, and degradation. J. Cell Physiol. 122, 263-74.
Rosenwald, I. B., Lazaris-Karatzas, A., Sonenbeg, N., and Schmidt, E. V. (1993). Elevated levels of cyclin Dl protein in response to increased expression of eukaryotic
initiation factor 4E. Mol. Cell. Biol. 13,7358-7363.
Rosenwald, I. B., Rhoads, D. B., Callanan, L. D., Isselbacher, K. J., and Schmidt, E. V. (1993). Increased expression of eukaryotic translation initiation factors eIF-4E and eIF- 2 alpha in response to growth induction by c-myc. Proc. Natl. Acad. Sci. USA 90, 6175- 8.
Roussel, M. F., Cleveland, J. L., Shurtleff, S. A., and Sherr, C. J. (1991). Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature 353,361-3.
Roussel, M. F., Davis, J. N., Cleveland, J. L., Ghysdael, J., and Hiebert, S. W. (1994). Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene 9,405-15.
Roussel, M. F., Theodoras, A. M., Pagano, M., and Sherr, C. J. (1995). Rescue of defective mitogenic signaling by D-type cyclins. Proc. Natl. Acad. Sci. USA 92, 6837-41.
Roy, A. L., Carruthers, C., Gutjahr, T., and Roeder, R. G. (1993). Direct role for Myc in transcription initiation mediated by interactions with TFII-I. Nature 365,359-61.
Rudolph, B., Saffrich, R., Zwicker, J., Henglein, B., Muller, R., Ansorge, W., and Eilers, M. (1996). Activation of cyclin-dependent kinases by Myc mediates induction of cyclin A, but not apoptosis. EMBO J. 15,3065-76.
Saha, P., Eichbaurn, Q., Silbennan, E. D., Mayer, B. J., and Dutta, A. (1997). p21CIPl and Cdc25A: competition between an inhibitor and an activator of cyclin-dependent kinases. Mol. Cell. Biol. 17,4338-45.

Sakarnuro, D., Elliott, K. J., Wechsler-Reya, R., and Prendergast, G. C. (1996). BIN1 is a novel MYC-interacting protein with features of a turnour suppressor. Nat. Genet. 14,69- 77.
Schneider, A., Peukert, K., Eilers, M., and Hanel, F. (1997). Association of Myc with the zinc-finger protein Miz-1 defines a novel pathway for gene regulation by Myc. Cum. Top. Microbiol. Imrnunol. 224, 137-46.
Schneider, C., King, R. M., and Philipson, L. (1988). Genes specifically expressed at growth arrest ofmammalian cells. Cell 54,787-793.
Schreiber-Agus, N., Homer, J., Torres, R., Chiu, F. C., and DePinho, R. A. (1993). Zebra fish myc family and max genes: differential expression and oncogenic activity throughout vertebrate evolution. Mol. Cell. Biol. 13,2765-75.
Schreiber-Agus, N., Torres, R., Homer, J., Lau, A., Jamrich, M., and DePinho, R. A. (1993). Comparative analysis of the expression and oncogenic activities of Xenopus c-, N- , and L-myc homologs. Mol. Cell. Biol. 13,2456-68.
Schwab, M., Alitalo, K., Klempnauer, K. H., Varmus, H. E., Bishop, J. M., Gilbert, F., Brodeur, G., Goldstein, M., and Trent, J. (1983). Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 305,245-8.
Schwab, M., and Amler, L. C. (1990). Amplification of cellular oncogenes: a predictor of
clinical outcome in human cancer. Genes Chromosomes Cancer 1,181-93.
Sclafani, R. A. (1996). Cyclin dependent kinase activating kiases. Curr. Opin. Cell. Biol. . 8, 788-94.
Sears, R., Ohtani, K., and Nevins, J. R. (1997). Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol. 17, 5227-35.

Seeger, R. C., Brodeur, G. M., Sather, H., Dalton, A., Siegel, S. E., Wong, K. Y., and Hammond, D. (1985). Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 313, 11 11-6.
Selten, G., Cuypers, H. T., Zijlstra, M., Melief, C., and Berns, A. (1984). Involvement of c-myc in MuLV-induced T cell lymphomas in mice: frequency and mechanisms of
activation. EMBO J. 3,3215-22.
Selvakumaran, M., Liebermann, D., and Hoffman-Liebermann, B. (1993). Myeloblastic leukemia cells conditionally blocked by myc-estrogen receptor chimeric transgenes for terminal differentiation coupled to growth arrest and apoptosis. Blood 81,2257-62.
Seth, A., Alvarez, E., Gupta, S., and Davis, R. J. (1991). A phospholylation site located
in the NH2-terminal domain of c-Myc increases transactivation of gene expression. J. Biol. Chem. 266,23521-4.
Sheaff, R. J., Groudine, M., Gordon, M., Roberts, J. M., and Clurman, B. E. (1997). Cyclin E-CDK2 is a regulator of p27Kipl. Genes Dev. 11, 1464-78.
Sherr, C. J. (1996). Cancer cell cycles. Science 274, 1672-7.
Shen; C. J. (1993). Mammalian G1 cyclins. Cell 73, 1059-1065.
Sherr, C. J., Kato, J., Quelle, D. E., Matsuoka, M., and Roussel, M. F. (1994). D-type
cyclins and their cyclin-dependent kinases: G1 phase integrators of the mitogenic response. Cold Spring Harb Symp Quant Biol59,ll-9.
Sherr, C. J., and Roberts, J. M. (1995). Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149-63.
Shi, Y., Glynn, J. M., Guilbert, L. J., Cotter, T. G., Bissonnette, R. P., and Green, D. R. (1992). Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 257,2 12-4.

Shichiri, M., Hanson, K. D., and Sedivy, J. M. (1993). Effects of c-myc expression on
proliferation, quiescence, and the GO to G1 transition in nontransformed cells. Cell Growth Differ. 4, 93-104.
Shim, H., Dolde, C., Lewis, B. C., Wu, C. S., Dang, G., Jungmann, R. A., Dalla- Favera, R., and Dang, C. V. (1997). c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 94,6658-63.
Shirodkar, S., Ewen, M., DeCaprio, M. A., Morgan, J., Livingston, D. M., and Chittenden, T. (1992). The transcriptional factor E2F interacts with the retinoblastoma product and a pl07-cyclin A complex in a cell cycle-regulated manner. Cell 68, 157-166.
Shrivastava, A., Saleque, S., Kalpana, G. V., Artandi, S., Goff, S. P., and Calame, K. (1993). Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science 262, 1889-92.
Shrivastava, A., Yu, J., Artandi, S., and Calame, K. (1996). YY1 and c-Myc associate in vivo in a manner that depends on c-Myc levels. Proc. Natl. Acad. Sci. USA 93, 10638-41.
Siebenlist, U., Bressler, P., and Kelly, K. (1988). Two distinct mechanisms of transcriptional control operate on c-myc during differentiation of HL6O cells. Mol. Cell. Biol. 8, 867-74.
Silver, B. J. (1992). Platelet-derived growth factor in human malignancy. Biofactors 3,
217-27.
Simpson, R. U., Hsu, T., Begley, D. A., Mitchell, B. S., and Alizadeh, B. N. (1987). Transcriptional regulation of the c-myc protooncogene by 1,25- dihydroxyvitamin D3 in HL-60 promyelocytic leukemia cells. J. Biol. Chem. 262,4104-8.
Skapek, S. X., Rhee, J., Kim, P. S., Novitch, B. G., and Lassar, A. B. (1996). Cyclin-
mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol. Cell. Biol. 16, 7043-53.

Skotzko, M., Wu, L., Anderson, W. F., Gordon, E. M., and Hall, F. L. (1995). Retroviral vector-mediated gene transfer of antisense cyclin G1 (CYCG1) inhibits proliferation of human osteogenic sarcoma cells. Cancer Res. 55,5493-8.
Smith, M. L., and Fomace, A. J., Jr. (1996). Mammalian DNA damage-inducible genes associated with growth arrest and apoptosis. Mutat. Res. 340, 109-24.
Solomon, D. L., Amati, B., and Land, H. (1993). Distinct DNA binding preferences for the c-MydMax and MaxIMax diiers. Nucleic Acids Res. 21,5372-6.
Solomon, D. L., Philipp, A., Land, H., and Eilers, M. (1995). Expression of cyclin Dl
mRNA is not upregulated by Myc in rat fibroblasts. Oncogene 11,1893-7.
Sorkin, A., Westermark, B., Heldin, C. H., and Claesson-Welsh, L. (1991). Effect of receptor kinase inactivation on the rate of internalization and degradation of PDGF and the
PDGF beta-receptor. J. Cell. Biol. 112,469-78.
Somentino, V., Drozdoff, V., McKinney, M. D., Zeitz, L., and Fleissner, E. (1986). Potentiation of growth factor activity by exogenous c-myc expression. Proc. Natl. Acad. Sci. USA 83, 8167-71.
Souza, P., Kuliszewski, M., Wang, J., Tseu, I., Tanswell, A. K., and Post, M. (1995). PDGF-AA and its receptor influence early lung branching via an epithelial-mesenchymal interaction. Development 121,2559-67.
Spencer, C. A., and Groudine, M. (1991). Control of c-myc regulation in normal and neoplastic cells. Adv Cancer Res. 56, 1-48.
Spotts, G. D., Patel, S. V., Xiao, Q., and Hann, S. R. (1997). Idc~tification of downstream-initiated c-Myc proteins which are dominant-negative inhibitors of transactivation by full-length c-Myc proteins. Mol. Cell. Biol. 17, 1459-68.
Squire, J., Goddard, A,, Canton, M., Becker, A,, Phillips, R., and Gallie, B. (1986).
Tumour induction by the retinoblastoma mutation is independent of N-myc expression. Nature 322, 555-7.

Stanton, L. W., Fahrlander, P. D., Tesser, P. M., and Marcu, K. B. (1984). Nucleotide sequence comparison of normal and translocated murine c-myc genes. Nature 31 0,423-5.
Steffen, D. (1984). Proviruses are adjacent to c-myc in some murine leukemia virus-
induced lymphomas. Proc. Natl. Acad. Sci. USA 81,2097-101.
Steiner, P., Philipp, A., Lukas, J., Godden-Kent, D., Pagano, M., Mittnacht, S., Bartek,
J., and Eilers, M. (1995). Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 14,4814-26.
Stem, D. F., Roberts, A. B., Roche, N. S., Sporn, M. B., and Weinberg, R. A. (1986). Differential responsiveness of myc- and ras-transfected cells to growth factors: selective stimulation of myc-transfected cells by epidermal growth factor. Mol. Cell. Biol. 6,870-7.
Stone, J., de Lange, T., Ramsay, G., Jakobovits, E., Bishop, J. M., Varmus, H., and Lee, W. (1987). Definition of regions in human c-myc that are involved in transformation and nuclear localization. Mol. Cell. Biol. 7, 1697-709.
Strasser, A., Barria, A. W., Bath, M. L., and Cory, S. (1990). Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348, 33 1-3.
Street, A. J., Blackwood, E., Luscher, B., and Eisenman, R. N. (1990). Mutational analysis of the carboxy-terminal casein kinase I1 phosphorylation site in human c-myc. Cur. Top. Microbiol. Immunol. 166,251-8.
Sugiyama, A., Kume, A., Nemoto, K., Lee, S. Y., Asami, Y., Nemoto, F., Nishirnura, S., and Kuchino, Y. (1989). Isolation and characterization of s-myc, a member of the rat myc gene family. Proc. Natl. Acad. Sci. USA 86,9144-8.
Tam, S. W., Theodoras, A. M., Shay, J. W., Draetta, G. F., and Pagano, M. (1994). Differential expression and regulation of cyclin Dl protein in normal and tumor human cells: association with Cdk4 is required for Cyclin Dl function in G1 progression.
Oncogene 9,2663-2674.

Tamaru, T., Okada, M., and Nakagawa, H. (1994). Differential expression of D type cyclins during neuronal maturation. Neurosci. Lett. 168,229-32.
Taub, R., Kelly, K., Battey, J., Latt, S., Lenoir, G. M., Tantravahi, U., Tu, Z., and Leder, P. (1984). A novel alteration in the structure of an activated c-myc gene in a variant
t(2;8) Bwkitt lymphoma. Cell 37,511-20.
Temin, H. M. (1971). Stimulation by serum of multiplication of stationary chicken cells. J. Cell Physiol. 78, 161-70.
Templeton, D. J., Park, S. H., Lanier, L., and Weinberg, R. A. (1991). Nonfunctional mutants of the retinoblastoma protein are characterized by defects in phosphorylation, viral
oncoprotein association, and nuclear tethering. Proc. Natl. Acad. Sci. USA 88, 3033-
3037.
Thompson, J. F., Hayes, L. S., and Lloyd, D. B. (1991). Modulation of firefly luciferase
stability and impact on studies of gene regulation. Gene 103, 171-7.
Thompson, T. C., Southgate, J., Kitchener, G., and Land, H. (1989). Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell 56,917-
930.
Tikhonenko, A. T., Black, D. J., and Linial, M. L. (1995). v-Myc is invariably required to sustain rapid proliferation of infected cells but in stable cell lines becomes dispensable
for other traits of the transformed phenotype. Oncogene 11,1499-508.
Tikhonenko, A. T., Black, D. J., and Linial, M. L. (1996). Viral Myc oncoproteins in infected fibroblasts down-regulate thrombospondin-1, a possible tumor suppressor gene. J. Biol. Chem. 271, 30741-47.
Ullrich, A., and Schlessenger, J. (1990). Signal transduction by receptors with tyrosine kinase activity. Cell 61,203-12.
Vairapandi, M., Balliet, A. G., Fomace, A. J., Jr., Hoffman, B., and Liebermann, D. A. (1996). The differentiation primary response gne MyD118, related to GADD45, encodes

for a nuclear protein which interacts with PCNA and p21 WAFIICIPI. Oncogene 12,
2579-94.
Valius, M., and Kazlauskas, A. (1993). Phospholipase C-yl and phosphatidylinositol 3
kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell 73,
321-34.
Valius, M., Serist, J. P., and Kazlauskas, A. (1995). The GTPase-activating protein of ras suppresses platelet-derived growth factor I3 receptor signaling by silencing phospholipase C-yl. Mol. Cell. Biol. 15, 3058-71.
Van Beneden, R. J., Watson, D. K., Chen, T. T., Lautenberger, J. A., and Papas, T. S. (1986). Cellular myc (c-myc) in fish (rainbow trout): its relationship to other vertebrate myc genes and to the transforming genes of the MC29 family of viruses. Proc. Natl. Acad.
Sci. USA 83. 3698-702.
Van Grunsven, L. A., Thomas, A., Urdiales, J. L., Machenaud, S., Choler, P., Durand,
I., and Rudkin, B. B. (1996). Nerve growth factor-induced accumulation of PC12 cells expressing cyclin Dl: evidence for a G1 phase block. Oncogene 12,855-62.
Vastrik, I., Makela, T. P., Koskinen, P. J., and Alitalo, K. (1995). Determination of sequences responsible for the differential regulation of Myc function by delta Max and
Max. Oncogene 11,553-60.
Vaziri, C., and Faller, D. V. (1995). Repression of platelet-derived growth factor beta- receptor expression by mitogenic growth factors and transforming oncogenes in murine 3T3 fibroblasts. Mol. Cell. Biol. 15, 1244-53.
Versteeg, R., Noordermeer, I. A., Kruse-Wolters, M., Ruiter, D. J., and Schrier, P. I.
(1988). c-myc down-regulates class I HLA expression in human melanomas. EMBO J. 7, 1023-9.
Vlach, J., Hennecke, S., Alevizopoulos, K., Conti, D., and Amati, B. (1996). Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J.
15, 6595-604.

Wagner, A. J., Meyers, C., Laimins, L. A., and Hay, N. (1993). C-myc induces the expression and activity of ornithime decarboxylase. Cell Growth Differ. 4, 879-883.
Walker, C. W., Boom, J. D., and Marsh, A. G. (1992). First non-vertebrate member of
the myc gene family is seasonally expressed in an invertebrate testis. Oncogene 7,2007- 12.
Wang, J. Y. J., Knudsen, E. S., and Welch, P. J. (1994). The retinoblastoma tumor
suppressor protein. Adv. Can. Res. 64, 25-85.
Wang, T. C., Cardiff, R. D., Zukerberg, L., Lees, E., Arnold, A., and Schmidt, E. V.
(1994). Mammary hyperplasia and carcinoma in MMTV-cyclin Dl transgenic mice. Nature 369, 669-671.
Waters, C. M., Littlewood, T. D., Hancock, D. C., Moore, J. P., and Evan, G. I. (1991). c-myc protein expression in untransfonned fibroblasts. Oncogene 6,797-805.
Watt, R., Stanton, L. W., Marcu, K. B., Gallo, R. C., Croce, C. M., and Rovera, G.
(1983). Nucleotide sequence of cloned cDNA of human c-myc oncogene. Nature 303,
725-8.
Watt, R. A., Shatzman, A. R., and Rosenberg, M. (1985). Expression and characterization of the human c-myc DNA-binding protein. Mol. Cell. Biol. 5,448-56.
Wechsler-Reya, R. J., Elliott, K. J., and Prendergast, G. C. (1998). A role for the putative tumor suppressor Bin1 in muscle cell differentiation. Mol. Cell. Biol. 18,566-75.
Wedel, A., and Ziegler-Heitbrock, H. W. (1995). The clebp family of transcription
factors. Imrnunobiology 193, 171-85.
Weinberg, R. A. (1995). The Retinoblastoma Protein and Cell Cycle Control. Cell 81, 323-330.
Weintraub, S. J., Prater, C. A., and Dean, D. C. (1992). Retinoblastoma protein switches the E2F site from positive to negative element. Nature 358,259-261.

Weir, L., Chen, D., Pastore, C., Isner, J. M., and Walsh, K. (1995). Expression of gax, a growth arrest homeobox gene, is rapidly down-regulated in the rat carotid artery during the proliferative response to balloon injury. J. Biol. Chem. 270,5457-61.
Weiss, W. A., Aldape, K., Mohapatra, G., Feuerstein, B. G., and Bishop, J. M. (1997). Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16,
2985-95.
Westermark, B., and Heldin, C. H. (1993). Platelet-derived growth factor. Structure, function and implications in normal and malignant cell growth. Acta Oncol. 32, 101-5.
Wiman, K. G., Magnusson, K. P., Ramqvist, T., and Klein, G. (1991). Mutant p53 detected in a majority of Burkitt lymphoma cell lines by monoclonal antibody PAb240. Oncogene 6, 1633-9.
Won, K.-A., Xiong, Y., Beach, D., and Gilman, M. Z. (1992). Growth-regulated expression of D-type cyclin genes in human diploid fibroblasts. Proc. Natl. Acad. Sci.,
USA 89, 9910-9914.
Wong, K. K., Zou, X., Merrell, K. T., Patel, A. J., Marcu, K. B., Chellappan, S., and
Calame, K. (1995). v-Abl activates c-myc transcription through the E2F site. Mol. Cell. Biol. 15, 6535-44.
Xiong, W., Pestell, R., and Rosner, M. R. (1997). Role of cyclins in neuronal differentiation of immortalized hippocampal cells. Mol. Cell. Biol. 17,6585-97.
Xiong, Y., Connolly, T., Futcher, B., and Beach, D. (1991). Human D-type cyclin. Cell 65, 691 -699.
Xiong, Y., Zhang, H., and Beach, D. (1993). Subunit rearrangement of the cyclin- dependent kinases is associated with cellular transformation. Genes Dev. 7, 1572-1583.
Yan, G. Z., and Ziff, E. B. (1995). NGF regulates the PC12 cell cycle machinery through specific inhibition of the Cdk kinases and induction of cyclin Dl. J. Neurosci. 15,6200-
12.

Yang, B. S., Geddes, T. J., Pogulis, R. J., de Crombrugghe, B., and Freytag, S. 0.
(1991). Transcriptional suppression of cellular gene expression by c-Myc. Mol. Cell. Biol. 11, 2291-5.
Yang, B. S., Gilbert, J. D., and Freytag, S. 0. (1993). Overexpression of Myc suppresses CCAAT transcription factor/nuclear factor 1-dependent promoters in vivo. Mol. Cell. Biol. 13, 3093-102.
Yarden, Y., Kuang, W. J., Yang-feng, T., Coussens, L., Munemitsu, S., Dull, T. J., Chen, E., Schlessinger, J., Francke, and Ulrich, A. (1987). Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 6, 3341- 51.
Yokota, J., Tsunetsugu-Yokota, Y., Battifora, H., Le Fevre, C., and Cline, M. J. (1986). Alterations of myc, myb, and rasHa proto-oncogenes in cancers are frequent and show clinical correlation. Science 231,261-5.
Zacksenhaus, E., Bremner, R., Jiang, Z., Gill, R. M., Muncaster, M., Sopta, M., Phillips, R. A., and Gallie, B. L. (1992). Unraveling the function of the retinoblastoma gene. Adv. Cancer Res. 61, 1 15-41.
Zhan, Q., Bae, I., Kastan, M. B., and Fomace, A. J., Jr. (1994). The p53-dependent gamma-ray response of GADD45. Cancer Res. 54,2755-60.
Zhan, Q., Canier, F., and Fomace, A. J., Jr. (1993). Induction of cellular p53 activity by
DNA-damaging agents and growth arrest. Mol. Cell. Biol. 13,4242-50.
Zhan, Q., Lord, K. A., Alamo, I., Jr., Hollander, M. C., Carrier, F., Ron, D., Kohn, K.
W., Hoffman, B., Liebermann, D. A., and Fomace, A. J., Jr. (1994). The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that synergistically suppress cell growth. Mol. Cell. Biol. 14, 2361-71.
Zhang, Q. X., Walker, F., Burgess, A. W., and Baldwin, G. S. (1995). Reduction in
platelet-derived growth factor receptor mRNA in v-src-transformed fibroblasts. Biochim. Biophys. Acta 1266,9-15.

Zimmennan, K., and Ah, F. W. (1990). Expression and function of myc family genes. Crit. Rev. Oncog. 2, 75-95.