应用化学专业实验
DESCRIPTION
应用化学专业实验. 实验目录. 实验一 十二烷基硫酸钠的合成及性能测试 实验二 环氧树脂中间体双酚 A 的合成 实验三 聚醋酸乙烯乳液的制备和性能测试 实验四 农药福美双的合成 实验五 植物生长调节剂 2 , 4— 二氯苯氧乙酸的合成 实验六 从番茄中提取番茄红素和 β— 胡萝卜素 实验七 超声辐射辅助壳聚糖的制备及性质分析 实验 八 短碳链甜菜碱的制备及检测 实验九 药物安妥明的制备 实验十 从猫豆粉中提取抗震颤药左旋多巴 实验十一 微波辅助合成松香甘油酯 附 录 几种常用仪器的使用说明. 实验注意事项 - PowerPoint PPT PresentationTRANSCRIPT
应用化学专业实验

实验目录实验一 十二烷基硫酸钠的合成及性能测试实验二 环氧树脂中间体双酚 A 的合成实验三 聚醋酸乙烯乳液的制备和性能测试 实验四 农药福美双的合成 实验五 植物生长调节剂 2 , 4— 二氯苯氧乙酸的合成实验六 从番茄中提取番茄红素和 β— 胡萝卜素实验七 超声辐射辅助壳聚糖的制备及性质分析实验 八 短碳链甜菜碱的制备及检测实验九 药物安妥明的制备实验十 从猫豆粉中提取抗震颤药左旋多巴实验十一 微波辅助合成松香甘油酯附 录 几种常用仪器的使用说明

实验注意事项参看有机化学实验安全教育相关内容

实验一 十二烷基硫酸钠的合成及性能测试
一、目的要求1 、掌握高级醇硫酸酯盐型阴离子表面活性剂的主要性质和
用途;2 、了解高级醇硫酸酯盐型阴离子表面活性剂的合成原理和
方法;3 、掌握固含量、表面张力和泡沫性能的测定方法。
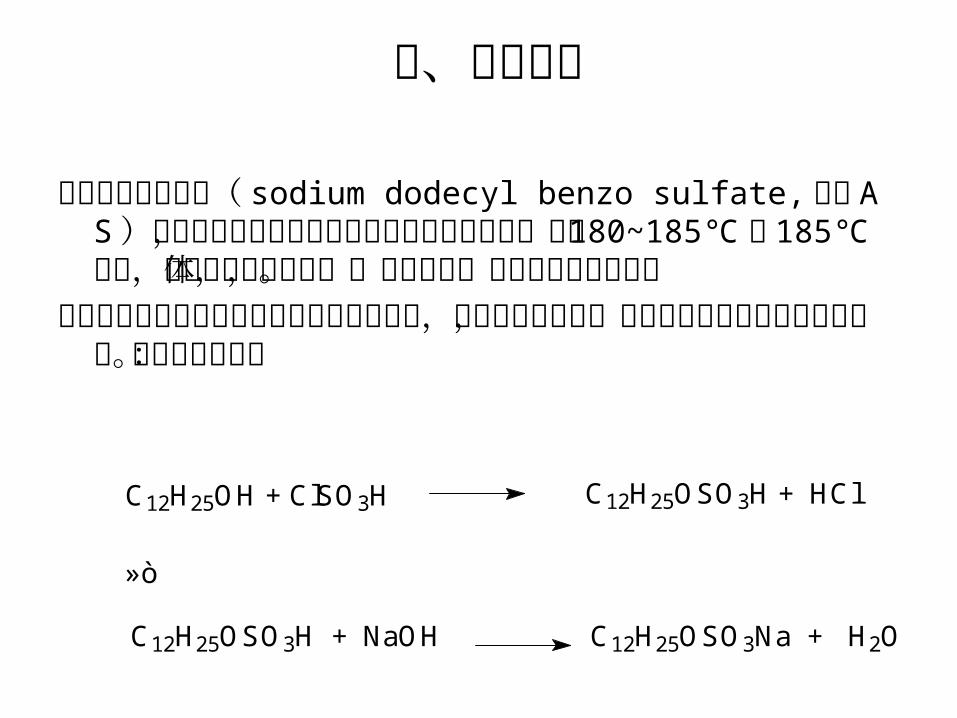
二、实验原理
十二烷基硫酸酯盐( sodium dodecyl benzo sulfate, 代号 AS )是硫酸酯盐类阴离子表面活性剂的典型代表,熔点 180~185℃ , 185℃ 分解,是白色至淡黄色固体,易溶于水,水溶液呈半透明状。
十二烷基硫酸酯钠是由月桂醇与氯磺酸反应,再加碱中和而成,或由月桂醇与氨基磺酸作用得到。其反应式如下:
C12H25OH + ClSO3H C12H25OSO3H + HCl
C12H25OSO3H + NaOH C12H25OSO3Na + H2O
»ò

三、主要试剂及仪器
月桂醇 氯磺酸 氢氧化钠 双氧水 三口烧瓶 搅拌器 温度计 气体吸收装置( 5% 氢氧化钠溶
液) 泡沫测定仪等

四、实验步骤合成方法一1. 氯磺酸法 --在装有搅拌器、温度计和氯化氢气体吸收装
置的 250ml 三口烧瓶中加入 47g 月桂醇,室温下( 25℃ )慢慢滴入 32g 氯磺酸,滴加时温度不要超过 30℃ ,注意反应体系起泡沫,控制勿使物料溢出。加完后在 40~45℃ 反应 2 小时。冷却至 25℃ ,充分搅拌下慢慢滴入 30%氢氧化钠溶液直至反应物的 PH 为 7.0~8.5 。将反应物倒入500ml 烧杯中,烧瓶中少量的反应物用少量水全部洗出,搅拌下滴入 25ml30% 双氧水,继续搅拌 30 分钟,得十二烷基硫酸钠粘稠液体。(也可按以下操作:硫酸化后,将反应物缓慢倒入盛有 100g2 1﹕ 冰水混合物的 250mL 烧杯中,稀释均匀,充分搅拌下滴加 30% 氢氧化钠溶液至反应物的 PH 为 7.0~8.5 ,烧杯外面用冰水浴冷却)。用 50 毫升烧杯取 2 克样品测固形物含量和泡沫性能。
2. 产物性能测试泡沫测定:称取约 5g 粘稠产品,加入 300ml 去离子水,轻轻
搅拌均匀,测定其泡沫性能。

• 合成方法二1. 氨基磺酸法 在装有搅拌器、温度计的三颈瓶中加入 74g
月桂醇。称取 40g 氨基磺酸、 8g 尿素在研钵中研细,混合均匀。控制反应体系温度为 30~40℃ ,开动搅拌下慢慢将研细的固体混合物分多次加入三颈瓶中,使混合物分散均匀,加完后升温至 105~110℃ 反应 1.5~2h 。反应结束后,加入 150ml 水,搅拌均匀,趁热倒出,在搅拌下用30% 氢氧化钠溶液中和至 PH 为 7.0~8.5 。测固形物含量和泡沫性能。

固含量测定在已称好的铝箔(或表面皿)中加入 0.5g 左右样品(精确
至 0.0001g ),放在平面电炉(或烘箱)上烘烤至恒重。按下式计算固含量:
固含量 = 其中 m1 、、 m2 、分别为干燥前、后样品与铝箔质量之和, m0 为铝箔的质量。
泡沫性能一般有稳定性和起泡能力,本实验只测量泡沫的稳定性(罗氏泡沫测定仪)。
产品的表面张力使用 JZHY—180 型界面张力仪测定。仪器使用见后附。

五、注意事项
1 、 因氯磺酸遇水会分解,故所用玻璃仪器必须干燥。2 、氯磺酸为腐蚀性很强的酸,使用时必须戴好橡皮手套,
在通风橱内量取。3 、 氯化氢吸收装置要密封好。

六、思考题1 、阴离子型表面活性剂有哪几种?试写出其结构式。
2 、高级醇硫酸酯盐有哪些特性和用途?3 、产品的 PH 为什么控制在 7.0~8.5?
4 、使用 30% 双氧水的主要目的是什么?参 考 文 献
[1] 赵何为 朱承炎,精细化工实验,华东化工学院出版社:48~50 ;
[2] 强亮生等,精细化工综合实验,哈尔滨工业大学出版社: 47~49 。
返回

实验二 环氧树脂中间体双酚 A 的合成 一、目的要求1 、掌握双酚 A 的合成原理和合成方法。2 、学习和掌握离心机的操作方法。3 、熟悉有机物熔点的测定方法及重结晶的操作方法。

二、基本原理1 、主要性质和用途• 双酚 A(bisphenol A) 又称二酚基丙烷,化学名称为 2,2,— 二对羟基苯基丙烷 [2,2—bis(p-hydroxyphenyl)propane] ,结构如下: 本品为无色结晶粉末,熔点 155~C-158℃ ,密度 1.95(20 )℃ 。溶于甲醇、乙醇、异丙醇、丁醇、乙酸、丙酮及二乙醚,微溶于水。易被硝化、卤化、硫化、烃化等。
• 双 A酚是用途很广的化工原料,可作为双酚 A 型环氧树脂、聚碳酸酯、聚砜、聚苯醚等树脂的合成原料和中间体,也可作为塑料和油漆用抗氧剂,是聚氯乙烯的热稳定剂,电线防老剂,塑料油漆、油墨等的抗氧剂和增塑剂。
COH
CH3
CH3
OH
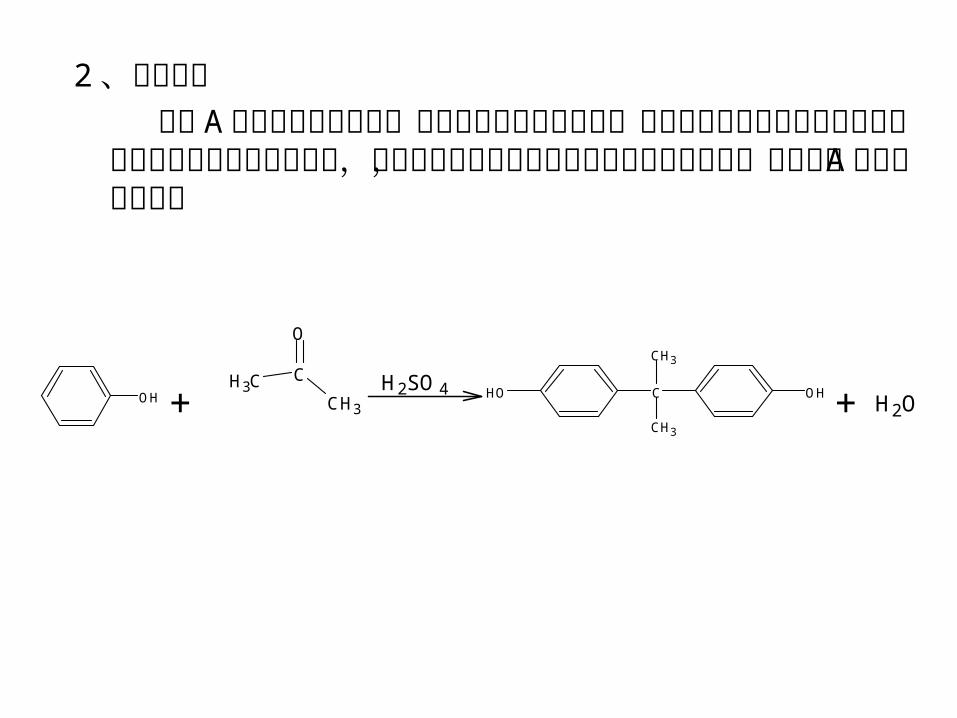
2 、合成原理 双酚 A 的合成方法有多种,大都由苯酚与丙酮合成,不同
之处是采用不同的催化剂。本实验采用的是硫酸催化法,即苯酚与过量丙酮在硫酸的催化下缩合脱水,生成双酚 A ,其反应式为:
OH +CH3
C
O
CH3
H2SO4 + H2OCOH
CH3
CH3
OH

三、主要仪器和试剂
分液漏斗 (500m1) 、布氏漏斗 (φ8) 、吸滤瓶 (500m1) 、电动搅拌器、水浴锅、电热干燥箱、电动离心机、三口烧瓶(250m1) 、球形冷凝管、玻璃水泵、温度计 (0-100 )℃ 、烧杯 (500m1) 、量筒 (100m1) 、滴液漏斗 (60m1) 、托盘天平。
苯酚、丙酮、甲苯、 79%硫酸、二甲苯、巯基乙酸。

四、实验步骤1 、双酚 A 的合成 在三口烧瓶中加入 30g 熔融的苯酚、 60g甲苯, 40g 79%硫酸,将三口烧瓶放入冷水浴冷却物料至 28℃ 以下。在搅拌下加入 0.2g催化剂巯基乙酸,然后一边搅拌一边用滴液漏斗滴加 10g丙酮,滴加期间,瓶内物料温度控制在 32℃~ 35℃ ,不得超过 40℃ ,同时开启回流冷凝管的上水。约在 30min 内滴加完丙酮,在 36 ~40℃ ℃ 搅拌 2h 以上(出现淡黄色致棕红色沉淀)。移入分液漏斗用热水洗涤三次,第一次水洗量为 50ml ,第二、三次水洗量均为 80ml( 水温为 82 )℃ 。每次水洗时,一边搅拌一边滴加热水,加完水后,振荡使之混合均匀,再静止分层。放出下层液,将上层的物料移至烧杯中,一边搅拌一边用冷水冷却、结晶。当冷至 25℃ 以下,吸滤,用水洗涤滤饼,吸滤至干,得粗双酚 A 。滤液可回收。

2 、双酚 A 的精制 称取 5g 双酚 A粗品于 100ml圆底烧瓶,加入 30ml 二甲苯,水浴回流 10min ,冷却结晶,烘干,测其熔点,计算收率。
双酚 A 的精制采用重结晶法,按粗双酚 A∶水∶二甲苯 =1 l 6 (∶ ∶ 质量比 ) 的配料投入三口烧瓶中,搅拌下加热升
温至 92 ~95℃ ℃ 。加热回流 15~30min 。停止搅拌,将物料移入分液漏斗中静置分层,放出下层水液后,冷却结晶,当冷至 35℃ 以下后,离心脱出二甲苯 (回收 ) ,将双酚 A烘干后称重,计算收率。并测定熔点。
产品质量标准如下:外观 白色结晶粉末 酚含量(%) <0.02熔点(℃) 155~158 水分(%) <0.4铁含量( l0-6mo1/l ) <15 色相(铂 -钴比色) <30

五、注意事项洗涤反应液时切勿剧烈振荡,否则反应液发生乳化现象。六、思考题1 、滴加丙酮时为什么控制温度 ?2 、水洗时水温的控制依据是什么 ?3 、本实验使用的硫酸催化剂为什么不能太浓( 98% )?参 考 文 献[1] 强亮生等,精细化工综合实验,哈尔滨工业大学出版社,
1996 : 69~70 。[2] 章思规主编,精细有机化学品技术手册,科学出版社,
1991 。
返回

实验三 聚醋酸乙烯乳液的制备和性能测试
一、目的要求1 、掌握实验室制备聚醋酸乙烯酯乳液的方法;2 、了解自由基引发的聚合反应原理和乳液聚合的方法及各组分的作用。

• 二、基本原理• 烯类单体的自由基型加聚反应可按本体、溶液、悬浮和乳
液等方法进行,采用何种方法主要决定于产物的用途。悬浮和乳液聚合都是在水介质中聚合成聚醋酸乙烯分散体,悬浮聚合一般用来生产相对分子质量较高的聚醋酸乙烯,用少量聚乙烯醇为分散剂,用过氧苯甲酰等能溶于单体的引发剂,聚合反应在分散的单体液滴中进行,可制得颗粒 0.2~1.0mm 的聚合物珠体;乳液聚合就是烯类单体,在乳化剂的作用下,分散在水相中呈乳状液,并在水溶性引发剂的作用下进行聚合反应,得到以微胶粒 (0.1~ 1.0微米 ) 状态分散在水中的聚合物乳液。乳化剂以阴离子型和非离子型表面活性剂为主,阴离子型表面活性剂有 SDS 、 LAS 等,用量为单体的 0.5~2% ,制得的乳液粘度较低,与盐混合时稳定性差;非离子型表面活性剂有环氧乙烷的各种烷基醚或缩醛,用量较多,为单体的 1~5% ,制得的乳液粘度大。广泛应用于涂料、粘合剂、纺织印染和纸张助剂等的制造。乳液聚合也可在保护胶体的作用下进行,一般以聚乙烯醇作保护胶体,有利于提高乳液稳定性和调节乳液粘度。

• 本实验醋酸乙烯酯通过乳液聚合的主要反应式如下:
O-S-O-O-S-O
O
O O
O O
O
O-S-O
R CH2=CH
OCOCH3
RCH2CH
OCOCH3
CH2=CH
OCOCH3
CH2CH
OCOCH3
CH2CH
OCOCH3
CH2CH2 CH=CH
OCOCH3OCOCH3
- - -2 .
+ +. . .
2 . +

三、主要试剂及仪器醋酸乙烯酯、 乳化剂聚乙烯醇和 OP-10 、去离子水、过硫
酸铵、碳酸氢钠、邻苯二甲酸二丁酯。( OP-10 :烷基苯酚聚氧乙烯醚)
三口烧瓶、搅拌器、温度计、球形冷凝管、滴液漏斗等。
R O(CH2CH2O)nH

四、实验步骤
1 、聚乙烯醇的溶解 在装有搅拌器、温度计的球形冷凝管的 250 毫升三口烧瓶中加入 44 毫升去离子水和 0.5 克乳化剂 OP-10 ,开动搅拌,逐渐加入 3 克聚乙烯醇。加热升温,在 80~ 90℃保持 0.5 小时左右,直至聚乙烯醇全部溶解,冷却备用。
2 、将 0.3 克过硫酸铵溶于水中,配成 5%的溶液。

3 、聚合 把 10g蒸馏过的醋酸乙烯酯、 2 毫升 5%过硫酸铵水溶液
加至上述三口烧瓶中。开动搅拌器,水浴加热,保持温度在 65-75℃ 。当回流基本消失时,用滴液漏斗在 1.5—2
小时内缓慢地、按比例地滴加 34g醋酸乙烯酯和余量的过硫酸铵水溶液,加料完毕后升温至 90~ 95℃ ,至无回流为止,冷却至 50℃ 。加入 2—4 毫升 5%碳酸氢钠水溶液,调整 PH 至 5~ 6 。然后慢慢加 5 克邻苯二甲酸二丁酯。搅拌冷却 1 小时,即得白色稠厚的乳液。

测定乳液的固含量、转化率和粘度: ( 1 )、 固含量按前述方法测定;
( 2 )、 转化率 =
其中 mc 为取样干燥后样品的固含量; S 为实验中加入的乳化剂、引发剂、增塑剂总质量; ma 为三颈瓶内乳液体系总质量; mb 为取样湿质量; G 为实验中醋酸乙烯酯单体加入总质量。
( 3 )、用 NDJ-79 型旋转式粘度计测定样品在 25±0.5℃时的粘度。
粘度计使用见后附。
%100/
/
ab
abc
mmG
mmSm

五、注意事项1. 醋酸乙烯酯为无色液体,性易变,不溶于水,沸点 71~7
3℃ 。高度易燃,应远离火种存放,使用时应避免吸入蒸气。
2. 聚乙烯醇是聚醋酸乙烯酯乳液聚合最常用的乳化剂,在水中溶解速度较慢,必须溶解完全,并保持原来的体积。如使用工业品聚乙烯醇,可能会有少量皮屑状不溶物悬浮于溶液中,可用粗孔铜丝网过滤除去。
3. 滴加单体的速度要均匀,防止加料太快发生爆聚冲料等事故。过硫酸铵水溶液数量少,注意均匀,按比例地与单体同时加完。
4. 搅拌速度要适当,升温不能过快。5. 在台秤上称量 0.5 克、 0.3 克物质时可能较难,实验中几组同学可以一起称量配成溶液后再进行称量平分。滴加 34g醋酸乙烯酯和余量的过硫酸铵水溶液时须从两个滴液漏斗分别进行。

六、思考题1 、聚乙烯醇在反应中起什么作用 ? 为什么要与乳化剂 OP-1
0 混合使用 ?2 、为什么大部分的单体和过硫酸铵用逐步滴加的方式加入 ?3 、过硫酸铵反应中起什么作用 ? 其用量过多或过少对反应
有何影响 ?4 、为什么反应过程中酸度会增加?反应结束后要用碳酸氢
钠调整 pH=5~ 6 ?5 、实验中加入邻苯二甲酸二丁酯的目的是什么?参 考 文 献[1] 鲍慈光等,应用化学实验,科学出版社: 58~62 ;[2] 张兴英等,高分子科学实验,化学工业出版社: 96~99 ;[3] 强亮生等,精细化工综合实验,哈尔滨工业大学出版社:
114~116 。[4] 赵何为,朱承炎,精细化工实验,华东化工学院出版社:
70~75 。[5] Brandrup J.and Immergut E.H.,eds.,Polymer Handbook,
John Wielry & Sons,Somerset,NJ,1989 。返回

实验四 农药福美双的合成 一、目的要求1 、了解福美双的性能及用途;2 、掌握福美双的制备方法。

性质与用途• 福美双又名四甲基秋兰姆 (thiram) ,化学名称是二硫化双
(硫羧基二甲胺) [bis(dimethylthiocarbamyl)disulfide] ,结构式如下:
CNCH3
CH3S
S S C NCH3
CH3
S

• 产品为白色结晶状粉末,由氯仿乙醇混合溶剂重结晶所得产品熔点 155~ 156℃ ,电解法含量 99.14%.熔点 148.8℃ 。相对密度 1.29 。微溶于乙醇、乙醚。溶于苯、丙酮、氯仿、二硫化碳,不溶于水、稀碱和汽油。与水共热时,生成二甲胺和二硫化碳,有毒,对呼吸道和皮肤有刺激作用。有特臭的气味。
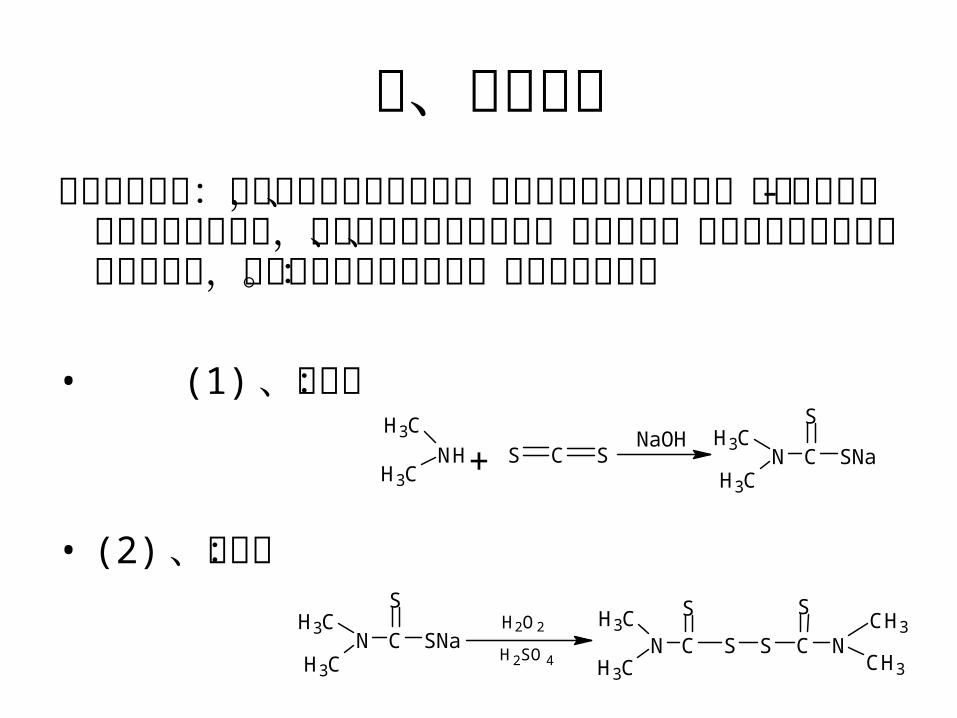
二、基本原理福美双的制备:福美双的合成方法很多,主要有亚硝酸钠氧化法、氯气 -空气氧化法和电解氧化法等,本实验采用经由二甲胺、二硫化碳、氢氧化钠进行加成反应得福美钠,再经双氧水氧化为产品。反应过程如下:
• (1) 、加成:
• (2) 、氧化:
NH
CH3
CH3+ CS S
NaOHCN
CH3
CH3SNa
S
CNCH3
CH3S
S S C NCH3
CH3
S
CNCH3
CH3SNa
SH2O2
H2SO4

三、实验试剂与仪器
40% 二甲胺(工业品)、 98% 二硫化碳(工业品)、 30%氢氧化钠(工业品)、 30% 双氧水 ( 试剂 ) 、 98% 硫酸( 试剂 ) 。
电动搅拌器、真空泵、三口烧瓶等。

四、实验步骤
• 1 、二甲基二硫代氨基甲酸钠 (福美钠 ) 的制备• 在装有回流冷凝管的 250ml 三口圆底烧瓶中,加入 36g 3
3%二甲胺水溶液, 35.2g30% NaOH 溶液,然后再加入约 10g碎冰,搅拌均匀,在 30 分钟内逐渐滴加 25g 二硫化碳,控制温度在 25℃ 以下,加完后继续搅拌 30 分钟,直到反应液的 PH值为 8~ 9 时为反应终点。之后再冷凝管上口接上真空装置,抽真空 10 分钟,即可得到淡黄色或草绿色的二甲基二硫代氨基甲酸钠透明溶液。(若要制取二甲基二硫代氨基甲酸钠,把反应装置改为蒸馏装置,继续减压蒸馏,蒸出溶液里的水,冷却后就可得到二甲基二硫代氨基甲酸钠结晶)。

• 福美双的制备• 在上述二甲基二硫代氨基甲酸钠溶液中,逐渐滴加氧化
剂(水∶硫酸∶双氧水= 120ml 10g 10g∶ ∶ ),反应温度控制在 25℃ 以下,加完后,反应 1 小时,之后检测 PH值,当溶液的 PH值达到 3~ 4 时为终点。把溶液倒入 500ml 烧杯,用水洗至中性,抽滤,在 80℃ 烘干即可得到产品。测熔点。
• 3 、福美双含量测定:主要方法有黄原酸盐法、极谱法和紫外分光光度法等。

五、思考题
1. 为什么在加成反应滴加二硫化碳时要控制温度?反应结束抽真空目的是什么?
2. 为什么氧化剂中加入较大量的水?3. 本反应也可用氨水代替氢氧化钠,请写出化学反应式。参 考 文 献[1] 朱建军等,福美双合成方法的改进实验,河北师范大学
学报 (自然科学版 ) , Vol.22,No.1 : 97-99 ;[2] 周艺峰等,福美双的新合成工艺, pesticides Vol.34,N
o.9(1995):8-10 。
返回

实验五 植物生长调节剂 2 , 4— 二氯苯氧乙酸的合成
一、目的要求: 1 、掌握多步反应制备 2 , 4- 二氯苯氧乙酸的原理及方法; 2 、了解植物生长调节剂 2 , 4- 二氯苯氧乙酸的性质和用
途。

二、基本原理
• 植物生长调节剂是在任何浓度条件下能影响植物生长和发育的一类化合物,包括肌体内产生的天然化合物和来自外界的一些天然产物。人类已经合成出一些与生长调节剂功能相似的化合物,如本实验的 2,4- 二氯苯氧乙酸( 2,4-dichloro-phenoxyacetic acid )就是一种用于除草的植物生长调节剂。它是由苯酚钠和氯乙酸通过 Williamson 合成法先制备苯氧乙酸,然后通过苯氧乙酸的氯化,得到对氯苯氧乙酸,进一步氯化就得到 2,4- 二氯苯氧乙酸(简称 2,4D )。苯氧乙酸作为防霉剂又称防落剂,可以减少农作物落花落果。 2,4- 二氯苯氧乙酸又名除绣剂,可选择性地除掉杂草,二者都可作为植物生长调节剂。总的化学反应式如下:

ClCH2CO2HNa2CO3
ClCH2CO2Na NaOH
OH
ClCH2CO2Na
ClH
OCH2CO2H
OCH2CO2H
+ ClH + H2O2
FeCl3OCH2CO2H
Cl
H+
2NaOCl
OCH2CO2H
Cl Cl
O

三、实验试剂
• 3.8g(0.04mol) 氯乙酸, 2.5g(0.027mol)苯酚,饱和碳酸钠溶液, 35% 氢氧化钠溶液,冰醋酸,浓盐酸,过氧化氢( 33% ),次氯酸钠,乙醇,乙醚,四氯化碳

四、实验步骤
1 、 苯氧乙酸的制备• 在装有搅拌器、回流冷凝管和滴液漏斗的 100ml 三颈瓶中,
加入 3.8g 氯乙酸和 5ml 水。开动搅拌,慢慢滴加饱和碳酸钠溶液(约需 7ml ),至溶液 pH 为 7~8 。然后加入 2.5g苯酚,在慢慢滴加 35% 的氢氧化钠溶液至反应混合物 pH为 12 。将反应物在沸水浴中加热约半小时。反应过程中 pH值会下降,应补加氢氧化钠溶液,保持 pH值为 12 ,在沸水浴上在继续加热 15 分钟。反应完毕后,将三颈瓶移出水浴,趁热转入锥形瓶中,在搅拌下用浓盐酸酸化至 pH为 3~4 。在冰浴中冷却,析出固体,待结晶完全后,抽滤,粗产物用冷水洗涤 2~3 次,在 60~65℃ 下干燥,产量约 3.5~4g ,测熔点。粗产物可直接用于对氯苯氧乙酸的制备。
• 纯粹的苯氧乙酸熔点为: 98~99℃ 。

对氯苯氧乙酸的制备 在装有搅拌器、回流冷凝管和滴液漏斗的 100ml 的三颈瓶
中加入 3g ( 0.02mol )上述制备的苯氧乙酸和 10ml 冰醋酸。将锥形瓶置于水浴加热,同时开动搅拌。待水浴温度上升至 55℃ 时,加入少许(约 20mg )三氯化铁和 10ml浓盐酸。当水浴温度升至 60~70℃ 时,在 10分钟内慢慢滴加 3ml 过氧化氢( 33% ),滴加完毕后保持此温度再反应 20 分钟。升高温度使瓶内固体全溶,慢慢冷却,析出结晶。抽滤,粗产物用水洗涤 3 次。粗品用 1 3∶ 乙醇—水重结晶,干燥后产量约 3 克。纯粹对氯苯氧乙酸的熔点: 158~159℃ 。

2,4— 二氯苯乙酸( 2,4-D )的制备 在 100ml锥形瓶中,加入 1g (0.0066mol) 干燥得对氯苯
氧乙酸和 12ml 冰醋酸,搅拌使固体溶解。将锥形瓶置于冰浴中冷却,在摇荡下分批加入 19ml5% 的次氯酸钠溶液。然后将锥形瓶从冰浴中取出,待反应物温度升至室温后再保持 5 分钟。此时反应液颜色变深。向锥形瓶中加入 50ml 水,并用 6mol/L 的盐酸酸化至刚果红试纸变蓝。反应物每次用 25ml乙醚萃取 2 次。合并乙醚萃取液,在分液漏斗中用 15ml 水洗涤后,再用 15ml10%的碳酸钠溶液萃取产物(小心!有二氧化碳气体逸出)。将碱性萃取液移至烧杯中,加入 25ml 水,用浓盐酸酸化至刚果红试纸变蓝。抽滤析出的晶体,并用冷水洗涤2~3 次,干燥后产量约为 0.7g ,粗品用四氯化碳重结晶,熔点: 134~136℃ 。纯粹 2,4— 二氯苯乙酸的熔点138℃ 。

注意事项• 五、注意事项• 1 、为防止氯乙酸水解,先用饱和碳酸钠溶液使之成盐,并且
加碱的速度要慢;• 2 、步骤 2 中开始滴加浓盐酸时,可能有沉淀产生,不断搅拌
后又会溶解,盐酸不能过量太多,否则会生成烊盐而溶于水。若未见沉淀生成,可再补加 2~3ml 盐酸;
• 3 、若次氯酸钠过量,会使产量降低。也可直接用市售洗涤漂白剂,不过由于次氯酸钠含量不稳定,所以常会影响反应。
• 六、思考题• 1 、说明本实验中各步反应 pH值的目的和意义。• 2 、以苯氧乙酸为原料,如何制备对溴苯氧乙酸?能用本法制备对碘苯氧乙酸吗?
• 参 考 文 献:• 兰州大学,复旦大学,有机化学实验,高等教育出版社: 279-
280 。
返回

实验六 从番茄中提取番茄红素和 β— 胡萝卜素
一、实验目的:1 、熟悉使用柱层析从天然物中提取分离色素的方法和操作技能;
2 、掌握薄层层析鉴别番茄红素和 β -胡萝卜素。
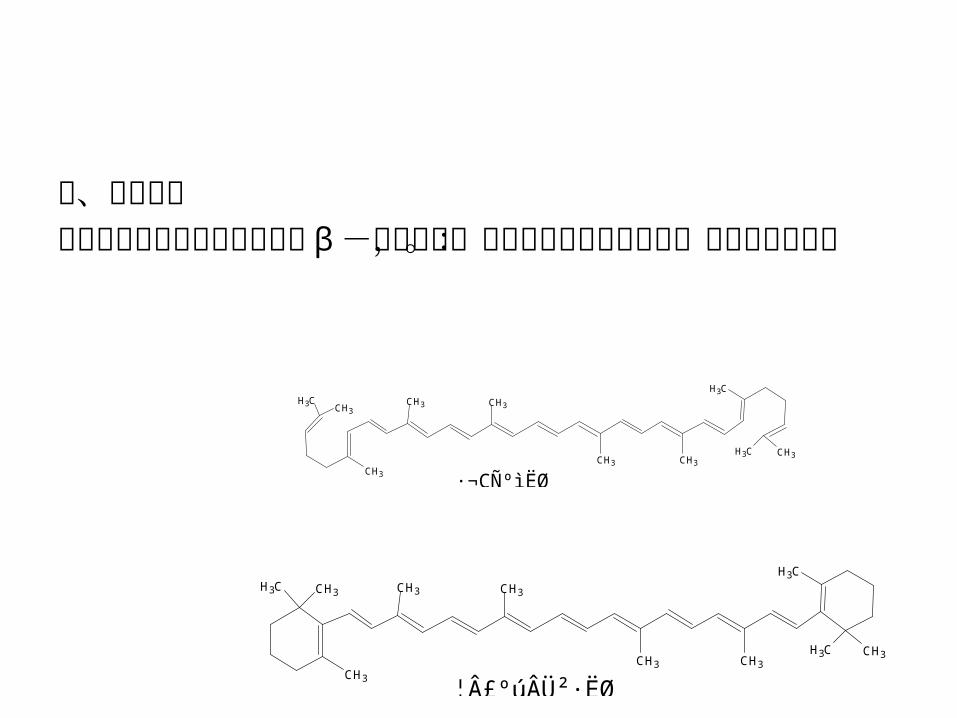
二、基本原理番茄中含有番茄红素和少量的 β -胡萝卜素,二者均属于类胡萝卜素。其结构式如下:
CH3
CH3CH3
CH3 CH3
CH3
CH3 CH3
CH3 CH3
·¬ÇѺìËØ
CH3
CH3CH3
CH3 CH3
CH3
CH3 CH3
CH3 CH3
¦Â£ºúÂܲ·ËØ

类胡萝卜素为多烯类色素,不溶于水而溶于酯溶性有机溶剂。本实验先用 95%乙醇将番茄中的水脱去,再用二氯甲烷萃取类胡萝卜素。因为二氯甲烷不与水混溶,故只有除去水分后才能有效地从组织中萃取出类胡萝卜素。根据番茄红素与 β -胡萝卜素极性的差别,用柱层析可以将它们分离。分离效果可以用薄层层析进行检验。

实验材料和试剂
• 新鲜番茄(或番茄酱)。• 95%乙醇,二氯甲烷,石油醚( 60~ 90℃ ),氯仿,
中性或酸性氧化铝,环己烷,硅胶 G ,饱和氯化钠溶液,无水硫酸钠。

实验步骤
原料处理与色素提取• 称取新鲜番茄酱 20g 于 100ml圆底烧瓶中,加 95%
乙醇 40ml ,摇匀,装上回流冷凝管,在水浴上加热回流 5 分钟,趁热抽滤,只将溶液倒出,残渣留在瓶内,再加入 30ml 二氯甲烷,水浴上加热回流 5 分钟,冷却,将上层溶液倾出抽滤,固体仍保留在烧瓶内,再加 10ml 二氯甲烷重复萃取一次。合并乙醇和两次二氯甲烷提取液,倒入分液漏斗中,加 5ml饱和氯化钠溶液(有利分层),振摇,静置分层。分出橙黄色有机相,使其流经一个在颈部塞有疏松棉花且在棉花上铺一层1cm厚的无水硫酸钠的三角漏斗(或直接在锥形瓶中用无水硫酸钠干燥),以除去微量水分。将此溶液储存于干燥的有塞子的锥形瓶中。层析之前,将此溶液在通风橱中用热水浴蒸发至干。

• 2 、柱层析分离• 取一支长 15cm 左右内径为 1~ 1.2cm 的层析柱,柱内装
有用石油醚调制的氧化铝。将粗制的类胡萝卜素溶解于 4ml苯中,用滴管在氧化铝表面附近沿柱壁缓缓加入柱中(留 1~ 2 滴供以后的薄层层析用 ) ,打开活塞,至有色物料在柱顶刚流干时即关闭活塞。用滴管取几毫升石油醚,沿柱壁洗下色素,并通过放出溶剂至柱顶刚流干,从而使色素吸附在柱上。然后加大量的石油醚洗脱(也可用 1 :1 的环己烷:石油醚洗脱)。黄色的 β 一胡萝卜素在柱中移动较快,红色的番茄红素移动较慢。收集洗脱液至黄色的 β—胡萝卜素从柱上完全除去,然后用极性较大的氯仿作洗脱剂洗脱番茄红素 ( 注意更换接收瓶 ) 。将收集到的两个部分在通风橱内用热水浴蒸发至干,加塞,留待薄层色谱用。(也可将样品分别溶于尽可能少的二氯甲烷中,尽快进行薄层层析)

• 3 、薄层层析检验• 在用硅胶 G铺成的薄板上距离底边约 lcm处,分别用毛
细管点上三个样品,中间点未分离的混合物,两边分别点上分离得到的 β—胡萝卜素和番茄红素。可以多次点样,即点完一次,待溶剂挥发后再在原来的位置上点样。但要注意,必须在同一位置上点,而且样品斑点尽量小。点样时毛细管只要轻轻接触板面即可,切不可划破硅胶层。样品之间的距离为 1~ 1.5cm 。将此板放入装有环已烷作展开剂(也可用 1 : 9 的苯 /环己烷)的层析缸中,盖上盖子。切勿让展开剂浸没样品斑点。待溶剂展开至 10cm 左右时,取出层析板。因斑点会氧化而迅速消失,故要用铅笔立即圈出。计算不同样品的 Rf值,比较不同样品 Rf值大小的原因以及分离效果。
离溶剂前沿至原点中心距点中心的距离溶质最高浓度中心至原fR

五、注意事项1 、 新鲜番茄浆的制备:将新鲜番茄洗净,用捣碎机捣碎或用市售的番茄酱。
2 、 氧化铝层析柱的装填方法:将层折柱垂直固定于铁架上,铺上一层薄薄的石英砂,关闭活塞。称取 15g 氧化铝置于 50ml锥形瓶中,加入 15ml石油醚 (顺序不能反 )边加边搅,且不断旋摇直至成均匀浆液 ( 稠厚但能流动 ) ,向柱内加入溶剂 (石油醚 ) 至半满,然后开启活塞让溶剂以每秒一滴的速度流入小锥形瓶中,摇动浆液,不断地逐渐倾入正在流出溶剂的柱子中,不断用木棒或带橡皮管的玻璃棒轻轻敲击柱身,使顶部成水平面,将收集到的溶剂在柱内反复循环几次,以保证沉降完全和装紧柱。整个过程不能让柱流干。待溶剂刚好放至柱顶刚变干时即可上样。
3 、 硅胶G薄层板的制备。将 4g硅胶G 置于一小烧杯中,加入 11ml蒸馏水不断搅拌至浆糊状,倾倒在洗净的玻璃板上 (18×6cm) ,流平,或用涂布器铺板,并轻轻敲打均匀,在室温放置半小时凉干,然后移入烘箱,缓慢升温至 105℃~ 110℃ 恒温活化半小时,取出放入干燥器中备用。
4 、 β—胡萝卜素对光及氧极其敏感,对酸、碱也较敏感,在弱碱条件下比酸性条件下稳定。重金属离子特别铁离子可使其颜色消失。番茄红素的耐光、耐氧性能也很差,与铁离子接触会变褐。所以提取、分离及使用时要特别注意。

六、思考题
1. 一般每千克新鲜成熟的西红柿含 0.02 克番茄红素,试分析可能导致提取率降低的原因。
2. 柱色谱分离 β—胡萝卜素和番茄红素时若效果不好的可能原因有哪些?
参 考 文 献[1] 樊开明等,食品添加剂,四川科学技术出版社, 1987 。[2] Goodwin ed.,Chemistry and Biochemistry of Plant Pigm
ents,Academic Press,1965.[3] Bernfeld ed.,Rodd¹s Chemistry of Carbon Compounds,E
lsevier(2 nd ed.):Vol.Iib,Ch.7,1968.
返回

实验七 超声辐射辅助壳聚糖的制备及性质分析
一、目的要求:了解并掌握壳聚糖的制备原理和操作方法;
掌握微波辐射辅助合成有机物的新技术。

二、基本原理
壳聚糖( chitosan,简称 CTS )学名聚氨基葡萄糖,又名可溶性甲壳质,是由虾、蟹壳脱钙、蛋白质和脂肪后经脱乙酰基得到的天然高分子聚合物,为白色或灰白色、略有珍珠光泽、半透明片状固体,不溶于水和碱溶液,可溶于大多数稀酸,与盐酸、醋酸、苯甲酸、环烷酸等可成盐,这是壳聚糖最重要、有用的性质之一,但其溶液应随配随用,不宜久置,因为壳聚糖会缓慢水解,溶液的粘度逐步降低。壳聚糖不同的脱乙酰度和相对分子量有不同的物理性能和药理性能,也是制备过程中较难控制的因素,传统的方法需高温加热 1h 以上,非常容易得到副产物。本实验使用超声波辐射法处理甲壳素,具有脱乙酰度高,反应速度快提高等优点,是近来使用的一项有机合成新技术。

三、仪器与试剂
• 数控超声细胞粉碎机;电热恒温真空干燥箱;乌氏粘度计(内径 0.8-0.9 , a-b间球容积为 4.2ml );片状甲壳素(预备好的); 0.5g/ml ( 34% ) NaOH 溶液; 3%乙酸; 10% NaOH 溶液; 0.2mol/L乙酸和 0.1mol/L乙酸钠体系;无水乙醇。

四、实验步骤
1 、甲壳素的准备 虾壳用 30g/LHCl浸泡约 6h 至无 CO2 放出,过滤,清水
洗至中性,然后用 10% NaOH 溶液浸泡,搅拌,加热至沸腾 1 小时脱蛋白质,过滤,清水洗至中性,太阳晒干脱色素得到片状甲壳素。脱色素可按下列方法操作:将已经脱钙、脱蛋白质处理的甲壳素在烧杯中用 0.2M KMnO4溶液浸泡 1 小时,取出过滤,清水洗净后再加入 1%NaHSO3 溶液浸泡 10 分钟,还原掉多余的 KMnO4 ,加入少量稀盐酸使溶液呈酸性,继续浸泡 20 分钟,以除去 MnO2 和过量的 NaHSO3 ,过滤,清水洗净。

2 、超声波辐射制备壳聚糖 在 250mL 烧杯中加入 25mL0.5g/mL ( 34% ) NaOH 溶
液,和 1g 已剪成 1cm×1cm 的片状甲壳素浸泡,在数控超声细胞粉碎机中进行超声波辐射处理 2 分钟后,搅拌并加热至 110℃ ,反应 1 小时,停止反应,冷却,过滤,滤渣用清水洗涤到中性,红外灯下干燥、称量,得粗产品。
3 、壳聚糖的纯化 粗品壳聚糖溶解于一定量 3%乙酸溶液中,抽滤去除不溶杂质,滤液用 10% NaOH 溶液调 PH 至 10 左右,即有纯净壳聚糖析出。抽滤,清水洗涤到中性,无水乙醇脱水,室温真空干燥。

4 、脱乙酰度的测定:壳聚糖重要的质量指标之一,是指甲壳素分子中脱除乙酰基
的链节数占总链节数的百分数,一般脱乙酰度在 70% 以上的产品工业上即为合格品。测定方法的原理如下:
A 、定量法:当一定量的壳聚糖与适当浓度的硫酸煮沸后会水解生成一定量的醋酸, 蒸馏出 醋酸,以酚酞作指示剂,用 0.01MNaOH 溶液进行滴定,由消耗的标准碱溶液计算出脱乙酰度。
O
NHCOCH3
CH2OHO
OHO
NH2
CH2OHO
OHH2SO4
CH3COOH[ ] n [ ] nH2
O £¬+

测定步骤:称取 0.1000 克样品,放入 250 毫升三口烧瓶,用移液管加
入 4ml W氏硫酸,并在烧瓶上作好液位标记,加入少许沸石,用温火回流 30 分钟,期间应不断摇动烧瓶,以免水解产物粘在器壁上。然后迅速冷却,并用少量蒸馏水冲洗冷凝管(约 5-6 毫升),再假如几粒沸石,改为蒸馏装置,加热蒸馏,待烧瓶中液面降至 4ml标记时,开始从分液漏斗加入 2ml 水,再蒸馏至标记,如此反复交错进行,直到收集得 30ml馏液(不包括最初蒸出的冲洗水),加入 3 滴 1%酚酞乙醇溶液,用 0.01M NaOH标准溶液滴定至粉红色( 3 分钟不褪色),记下所消耗的碱体积,按以下公式计算脱乙酰度。
%1001000
1000
AVN
A
2
1
M
MVNmA
脱乙酰度 (%)=
;其
中

• 其中: m 为样品重量 (g) ;• N 为 NaOH标准溶液的摩尔浓度;• V 为消耗的 NaOH标准溶液体积 (ml) ;• M1 为甲壳素的分子量;• M2 为壳聚糖的分子量。• 注 1 :本方法有时因回流和蒸馏时火力过猛,溶液剧烈沸腾而导致失败。因此回流时使用温火控制冷凝管顶部试纸不变色;蒸馏时控制温度 99℃ (不能超过 100℃ 以上)。
• 注 2 : wenzel,s 硫酸(即W氏硫酸):将 100 毫升浓硫酸搅拌加入装有 200 毫升蒸馏水的敞口瓶中,在通风橱中煮沸 30 分钟以消除任何挥发性酸气,期间要自始至终加入蒸馏水保持原有体积。

B 、红外光谱定性法:在红外光谱中各官能团的吸收位置如下:
酰胺的羰基为 1660cm-1
酰胺的 -NH- 为 1558cm-1
酰胺的 -CN- 为 1315cm-1
氨基 (-NH2) 为 1590cm-1

通过上述波数上吸收峰强弱的对比,可大致地看出甲壳素的脱乙酰度。步骤如下:
a 、制备 1%壳聚糖醋酸溶液:用台秤称取 0.1 克壳聚糖,放入烧杯中加入 10 克 1%醋酸溶液(约 10ml ),搅拌使之溶解。
b 、制备壳聚糖薄膜:在光滑干净的玻璃片上滴加几滴 1%壳聚糖醋酸溶液,然后用加热器慢慢升温烤干,放入 5%NaOH 溶液中浸泡,直至壳聚糖膜片脱落,小心取出,用蒸馏水洗涤到中性,再慢慢升温烤干,即得壳聚糖膜片。
c 、红外光谱测定:把制好的壳聚糖膜片放到已调好的红外光谱仪的样品池上,打开测量开关进行扫描,记录壳聚糖的红外光谱图。

5 、相对分子质量的测定 用乌氏粘度计按一点法测定样品的特性粘度 [η] ,按下式计
算粘均相对分子质量。溶剂为 0.2mol/L乙酸和 0.1mol/L乙酸钠体系,温度为 25℃ 。
[η]=1.81×10-3M0.93

6 、粘度的测定粘度也是壳聚糖的主要质量指标之一,不同粘度的产品有不同的用途,目前国内外产品因粘度的不同分为三大类:
高粘度壳聚糖: 1%壳聚糖溶液, 1%醋酸溶液,粘度大于1000厘泊;
中粘度壳聚糖: 1%壳聚糖溶液, 1%醋酸溶液,粘度在 100-200厘泊;
低粘度壳聚糖: 2%壳聚糖溶液, 2%醋酸溶液,粘度在 25-50厘泊之间。

测定步骤:仪器: NDJ-79 型旋转式粘度计测定:在台秤上称取 2.8 克壳聚糖样品,加入 277.2 克 1%醋酸溶液,搅拌溶
解得到 1%壳聚糖溶液。将此溶液放入直径不小于 70mm 的烧杯中,把选择好合适的转子放入壳聚糖溶液中央,使溶液刚好浸在转子上的刻度,调好粘度计的水平,打开粘度计的测量开关,等到转子旋转平衡(约需 30秒钟),读数,记下粘度值。重复三次,取其平均值。测定在室温下进行。
五、思考题1 、写出由甲壳素制备壳聚糖的化学反应式。2 、微波辐射时间过长,强度过大,对脱乙酰度、相对分子量有何影响?参考文献[1] 丁盈红等,微波辐射制备壳聚糖,化学试剂, 2003 , 25 ( 1 ), 41-42 。[2] 姜俊艳,壳聚糖的制备及应用,化学工程师, 2003 , 98 ( 5 ), 57-58 。[3] Jan Knapczyk 等,中国海洋药物, 1993 , 12 ( 4 ): 53 。
返回

实验 八 短碳链甜菜碱的制备及检测目的要求:
了解甜菜碱的性质、用途和合成方法;掌握离子交换树脂纯化甜菜碱的操作技巧;掌握凯氏定氮法测定化合物的含氮量。

• 甜菜碱 (betaine)又名三甲铵乙内酯 (1-carboxy-N,N,N,-trimethylmethanaminium hydroxide inner salt) ,是普遍存在于动植物体内水溶性的一种季铵碱类化合物,在生物的代谢中起着重要的作用,它可以调节渗透压、增进食欲、稳定维生素、减少脂肪、缓和应激,还可作为甲基供体,代替部分蛋氨酸和氯化胆碱。甜菜碱作为饲料添加剂应用广泛,其盐酸盐还可用于人类的胃酸缺乏、动脉粥样硬化、肝脏疾病、风湿病、糖尿病等的治疗。甜菜碱原来的主要来源是从甜菜的糖蜜中提取,但由于产量少、工艺复杂,现在主要从化学合成得到。

基本原理
• 本实验甜菜碱的合成反应式如下:
• 反应生成的甜菜碱和氯化钠通过强酸性阳离子交换树脂进行分离。
ClCH2COOH NaOH ClCH2COONa
ClCH2COONa (CH3)3N (CH3)3NCH2COO NaCl
+
++ -
+

氮含量测定1. 提纯后的甜菜碱再使用凯氏定氮装置测定含氮量。2. 将含氮有机化合物在凯氏定氮烧瓶中用浓硫酸与催化剂
加热分解 (消化 ) ,使样品中所含的氮转变成为硫酸氢铵;
3. 反应液在蒸馏器中用氢氧化钠碱化后,析出的氨用水蒸汽蒸馏出来,并用饱和的硼酸溶液吸收;
4. 以甲基红—溴甲酚绿混合液作指示剂,用标准盐酸溶液滴定,从而计算样品中的含氮量;
5. ( 理论含 N值 11.97%) ,反应原理可分为以下几个步骤表示:
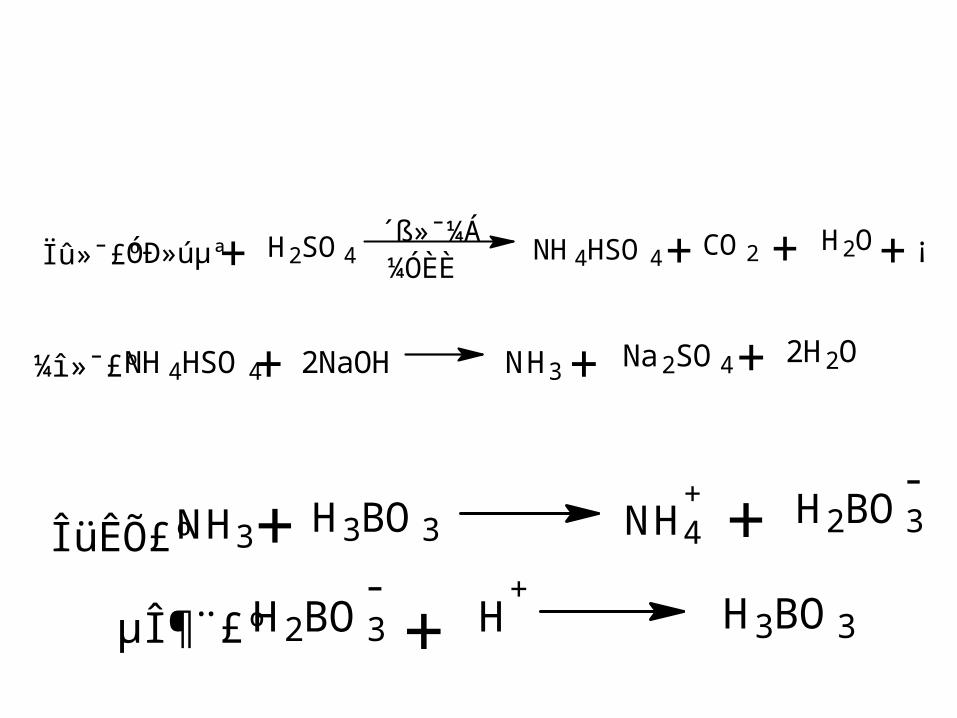
Óлúµª H2SO4´ß»¯¼Á¼ÓÈÈ
NH4HSO4+ CO2 H2O+ + ¡ Ïû»¯£º +
NH4HSO4 2NaOH NH3Na2SO4 2H2O+¼î»¯£º + +
NH3+ H3BO3 NH4+ + H2BO3
-
ÎüÊÕ£º
H3BO3+H2BO3-
H+
µÎ¶¨£º

• 计算公式如下:
• 式中:• Vl—— 样品消耗 HCl 的毫升数• V2――空白试验消耗 HCl 的毫升数• M——HCl标准溶液的摩尔浓度• W 一-样品重 ( 克 )
1000100
10
101.14)(
% 21
W
MVVN

三、实验试剂及仪器:• 30% 三甲胺、氯乙酸、 4mol/L 氢氧化钠、强酸性阳离子交换树脂( Na 型)、 2mol/L 的盐酸、 5% 的氨水、浓硫酸 (比重 1.84 ,分析纯 ) 、催化剂( CuSO4 K2SO4∶ = 1 3∶ )、 30% NaOH 溶液、混合指示剂( 5 体积 0.1%溴甲酚绿乙醇溶液加 1 体积 0.1%甲基红乙醇溶液)、4% H3BO3 溶液、 0.02N标准盐酸;
• Ф1×20cm层析柱,凯氏烧瓶,重迭式水蒸汽馏装置。

四、实验步骤:
1 、甜菜碱的合成将 19 克氯乙酸在 100 毫升烧杯中用 20 毫升蒸馏水溶解后,
加入 4mol/L 氢氧化钠中和至中性,再将所得溶液加到安装搅拌器、回流冷凝管的三口烧瓶中,搅拌下缓缓滴加 40 毫升 30% 三甲胺(滴液漏斗应插入液面下)。室温下反应 30 分钟后,在 50~55℃ 的水浴中加热 1 小时,升温 80~85℃ 下再反应 1 小时,抽真空至无氨味,冷却,反应液用蒸馏水稀释(一般稀释 5~6倍)到约 600 毫升待用。

2 、甜菜碱的纯化在甜菜碱合成反应的间隙,事先将 300 毫升强酸性阳离子交换树脂( Na 型)装入层析柱中,加入蒸馏水至水面刚超过树脂层,除去树脂中的空气泡,用 2mol/L 的盐酸使树脂由 Na 型充分转变为 H 型(如何判断?用多少量?)。将得到的反应液以 2.5L/h 的速度通过树脂柱,然后用蒸馏水以 2L/h 的速度淋洗至中性,收集废水约 200~300 毫升,再用 5% 的氨水以 0.6L/h 的速度洗脱,收集含有甜菜碱的洗脱液。将洗脱液真空浓缩、结晶、烘干。纯净甜菜碱的m.p.293℃ (分解)。

3 、凯氏定氮法测定甜菜碱的含氮量 ( 1 )、消化 准确称取 0.1~ 0.15 克样品于凯氏烧瓶中,
加 1 克催化剂,然后加 6 毫升浓硫酸将附在瓶颈上的样品及催化剂冲至瓶底,呈 45 度角斜置(见下图 A ),瓶口放一小漏斗,放在通风橱中,加热至微沸,并保持微沸至溶液从黑色变为透明的绿色止, ( 要避免激烈的沸腾,以免 NH4HSO4被 H2S04蒸汽带走而影响结果,如发现有发泡现象,应赶紧改用小火 ) ,放冷后,将反应溶液定量地移到 100 毫升容量瓶中, ( 容量瓶先放 10~ 15 毫升的蒸馏水 ) ,此时溶液发热,待溶液冷却至温后,才加蒸馏水至刻度。
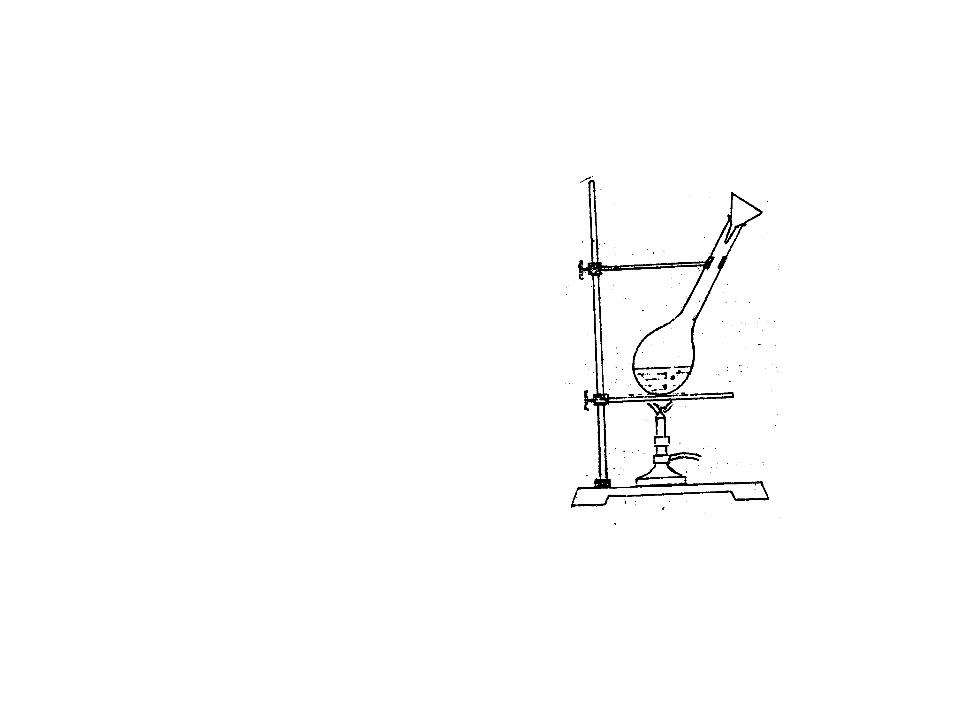
消化装置

( 2 )、蒸馏及滴定1. 在下图的吸收瓶 I 和瓶Ⅱ中分别加入 15 毫升 4% H3BO
3 ,指示剂 8 滴。注意要使管 H 的一端尽可能接近Ⅰ瓶内的液面,而另一端插入Ⅱ瓶的底部。
2. 拨开接在管G 的胶管,用移液管从管G向蒸馏瓶 B 内加入 10 毫升的消化液,接上胶管和漏斗,并从漏斗加入 12 毫升 30%的 NaOH ,立即夹紧螺旋夹 K ,再在漏斗中加进一些水封口,进行蒸镏,从沸腾开始计算 8~ 10 分钟,即可停止加热,此时由于 A 瓶停止沸腾,逐渐冷却,引起压力降低。因此吸收瓶口的溶液被吸至 I 瓶,当Ⅱ瓶的溶液全部被吸进Ⅰ瓶后,用少量蒸馏水冲洗 H管和Ⅱ瓶的内壁,并利用回吸作用将洗涤掖合并于 I 瓶中,如此将Ⅱ瓶冲洗二、三次,移去Ⅱ瓶,取下 I 瓶,再以少量蒸馏水冲洗管 F 及 H ,将洗液接入Ⅰ瓶,然后进行滴定。

将Ⅰ瓶中的吸收液,用 0.02N标准盐酸滴定,终点时指示剂由兰色变为粉红色。 空白实验:用完全相同的手续与试剂用量,进行两次空白实验,从每次含有样的滴定值减去两次空白实验的滴定平均值,即为样品所消耗的酸液数值。实验结束后,要将仪器彻底洗干净。

五、注意事项
• 树脂的预处理:新出厂的树脂要用水浸泡,使之充分吸水膨胀,然后用酸碱处理,以除去不溶于水的杂质。一般步骤为:先用水浸泡 24h ,倾出水后洗至澄清,加入 2-3倍量 2mol/L 盐酸搅拌 2h (或在柱中流洗),除酸后水洗至中性,再加入 4-5倍量 2mol/L 氢氧化钠搅拌 2h (或流洗),除碱液后水洗至中性。
• 强酸性阳离子交换树脂 (H 型 ) 的制备:预处理后装柱的阳离子交换树脂加入 2-3倍量 2mol/L 盐酸在柱中流洗即可。不用时加水保存,一般阳离子交换树脂均要转化为钠型,而阴离子交换树脂转化为氯型保存。
• 3 、树脂的再生:阳离子交换树脂按酸、碱、酸的预处理法;而阴离子交换树脂则采用碱、酸、碱的步骤处理,最后加水保存即可。

六、思考题
1 、说明本实验甜菜碱的合成中采用了分阶段加热法的目的。 2 、为什么实验中使用了过量的三甲胺原料?参 考 文 献 [1] 柳恒等,三甲胺乙内酯(甜菜碱)的合成,陕西化工,
Vol.28,No.1:21-22 。[2] 赵树凯等,有机分析实验讲义,广西大学化学化工学院:
7-8 。
返回

实验九 药物安妥明的制备目的要求:
了解相转移催化反应和卡宾反应的基本原理;掌握相转移催化法合成安妥明的操作及检测方法;熟悉油水分离操作。

1. 安妥明( Clofibratun )化学名为 α -对-氯苯氧异丁酸乙酯,结构式如下:
2. 常用于降低血中胆固醇,其合成分两步进行:首先制备对氯苯氧异丁酸,然后进行酯化反应即得安妥明。
3. 相转移催化( PTC )反应是上个世纪 60年代发展起来的合成新技术,已在烃基化、亲核取代、加成、氧化-还原和 WITTIG 反应等方面得到应用。 PTC 反应一般条件温和,无需太高温度,常用溶剂有二氯甲烷、苯、乙腈等。 PTC具有反应快、条件温和、收率高和产品纯的特点。
Cl O C
CH3
COOC2H5
CH3

基本原理 • 相转移催化( PTC )反应是上个世纪 60年代发展起来的
合成新技术,已在烃基化、亲核取代、加成、氧化-还原和 WITTIG 反应等方面得到应用。 PTC 反应一般条件温和,无需太高温度,常用溶剂有二氯甲烷、苯、乙腈等。PTC具有反应快、条件温和、收率高和产品纯的特点。
• 制备对-氯苯氧异丁酸乙酸通常按下述反应进行:
Cl OH + CHCl3 + CH3COCH3¹ÌÌåNaOH
Cl O C COOH
CH3
CH3

• 由于 NaOH 在 CH3COCH3 中溶解度小,需要长时间搅拌,若用浓 NaOH ,则催化能力降低,并引起 CH3COCH3 水解。
• 当用苄基三乙基氯化铵( TEBA )作相转移催化剂催化上述反应,则可使上述反应易于进行,并提高收率。
• 合成路线如下:
Cl OH + CHCl3 CH3COCH3
TEBA
50%NaOHCl O C
CH3
CH3
COOH
Cl O C
CH3
CH3
COOC 2H5
C2H5OH
H2SO4
+

三、主要试剂及用量
• 对氯苯酚( 13.5g )、丙酮 ( 120ml )、 50% 和 0.5%的氢氧化钠溶液、氯仿( 13mL )、甲苯、盐酸、浓硫酸、无水乙醇、苄基三乙基氯化铵 (TEBA) ( 1.0 克)、苯( 20mL )。

四、操作步骤
对氯苯氧异丁酸的制备 250ml 三口烧瓶中放入对氯苯酚 13.5g ( 0.105mol )、丙酮 120ml 、 50%氢氧化钠溶液 50ml 和 TEBA1.0g ( 0.005mol ),在搅拌滴加氯仿 13ml ( 0.15mol ),保持 30~ 40℃ ,加完待反应平衡后,加热回流 0.5 小时,于 60℃ 左右用盐酸调节 PH值为 2 ,析出晶体,冷却,过滤,依次用水和甲苯洗涤,干燥后测定其熔点(文献值为 117~ 122℃ )和红外光谱,计算收率。若纯度不够,可用水重结晶。

安妥明的制备• 100ml圆底烧瓶中放入对氯苯氧异丁酸 15g ( 0.07mo
l )、 25ml无水乙醇、 20ml苯及 4ml浓硫酸,摇匀后加入沸石,其上装一水分离器,水分离器上放一冷凝管,在水分离器中放入水至支管处,然后放去约 8ml 。
• 将烧瓶在水浴加热回流,开始时回流速度要慢,随着回流的进行,水分离器中出现了三层液体,回流至中层液体达 7~ 8ml 左右,即停止加热,放出中、下呈液体并记下体积。继续用水浴加热,使多余的苯和乙醇蒸发至水分离器中(当充满时可由活塞放出)。

• 瓶中残留液倒入盛有 100ml 冷水的烧杯中,有机层先用 0.5%氢氧化钠洗涤,再用水洗至中性,尽量分去水分,然后减压蒸馏,收集 148~ 150 /20mmHg℃ 的馏份。用收集到的馏份测定红外光谱。
• 把两次的红外光谱图进行比较,注意比较吸收基团波数的变化。
• 注: TEBA 可按以下方法制备:• ( 1 )、 1moL 氯苄 +1moL 三乙胺 +1moL丙酮(或乙腈)混合室温下放置过夜;
• ( 2 )、将氯苄和三乙胺在甲苯中回流 4~5天,即可得到接近理论产率的 TEBA 。

思考题
1. 对氯苯氧异丁酸制备时为什么使用相转移催化法进行?能用其他方法吗?
2. 合成安妥明时使用油水分离法的目的是什么?油水分离器中的三层液体各是什么?
参 考 文 献:[1]A.T.Babayan,N.Gambaryan,andN.P.Gambaryan,Zh.Obs
hch.Khim.,24,1887(1954);Chem.Abstr.,49,10879(1955);Chem.Zentralbl.Sonderb.,1950/54,4532.
[2] G.Maerker,J.F.Carmichael,and W.Port, J.Org.Chem.,26,2681(1961).
返回

实验十 从猫豆粉中提取抗震颤药左旋多巴
目的要求1 、了解左旋多巴的性质和用途; 2 、掌握阴离子交换法提取植物有效成分的条件及操作。

• 左旋多巴( Levodopa ; 3,4-dihydroxyphenyl-L-alanine )是目前治疗震颤麻痹症的主要药物,白色晶体,分子式为 C9H11NO4 ,是苯丙氨酸类化合物,按干燥品计算,含 C9H11NO4 不得少于 98.0% 。其等电点为 PH3.5 左右,易溶于水,不溶于乙醇等有机溶剂,分子中的邻二酚羟基在酸性下稳定,在中性或碱性条件下易缩合。其结构式如下:
OH
OH
OH
O
H NH2

• 左旋多巴一般是从油麻藤属植物种子中获得,其中猫豆的左旋多巴含量大约为 6-7% ,含量高,易栽培,一年生,适合作为工业化生产的原料。但由于植物成分复杂,溶剂法工艺的提取液的多种杂质对产品收率影响较大,如果用离子交换法处理水提取液,去除大量杂质,可以提高产品的收率。

实验仪器和试剂
猫豆粉 (40 目 ) ,去离子水,蒸馏水, D296 阴离子交换树脂(按说明活化), 5%醋酸溶液,三氯化铁溶液, Ф1×20cm层析拄。

实验步骤1. 左旋多巴提取液的制备:取猫豆种子粉碎过 40 目 100g ,
以 50℃ 去离子水 50倍量分三次浸提,每次浸提时搅拌15min ,静置 15min ,然后吸取上清液离心去除悬浮物,5℃贮藏备用。
2. 离子交换提取左旋多巴:在 Ф1×20cm层析拄中装入 10ml D296 阴离子交换树脂(事先按有关说明书进行活化),洗涤平衡后,加入定量的提取液,使其以 1.5BV/h(or1.0ml/min) 的流速下行通过树脂拄,溶液流至树脂床上表面后,用蒸馏水快速冲洗拄床,然后加入 5%醋酸溶液下行通过树脂拄进行解吸,收集流出液直到三氯化铁试剂检验至无蓝色。

1. 左旋多巴产品的提取:将洗脱液合并, 40-50℃真空浓缩至原体积的 1/3-1/4 , 5℃ 放置 24h 结晶,减压过滤得到粗晶。
2. 精制:粗晶加 30-40倍蒸馏水, 3% 冰醋酸, 1%Vc 搅拌溶解后加热至沸,加入 5% 活性炭并需煮沸 2-3min进行脱色,趁热过滤, 5℃ 放置 24h ,离心过滤得到白色晶体,用少量蒸馏水及无水乙醇洗涤后, 40℃鼓风干燥,收率为 4.2-4.5% 。

检验:精制后的晶体按 2005版《中国药典》要求方法测定其比旋光度及含量等项目。
A 、定性鉴别:(1) 、取本品约 5mg 加盐酸溶液 (9→1000)5ml 使溶解,加
三氯化铁试液 2 滴,即显绿色。分取溶液 2.5m1, 加过量的稀氨溶液 ,即显紫色;剩余的溶液中加过量的氢氧化钠试液,即显红色。
(2) 、取本品约 5mg ,加水 5m1 使溶解,加 1%茚三酮溶液1ml ,置水浴中加热,溶液渐显紫色。
( 3 )本品的红外光吸收图谱与文献图谱(光谱图集)对照。

B 、定量测定:取本品约 0.1g ,精密称定,加无水甲酸 2ml 使溶解,加冰醋酸 20m1摇匀,加结晶紫指示液 2 滴,用高氯酸滴定液(0.1mol/L) 滴定,至溶液显绿色,并将滴定的结果用空白试验校正。每 1m1 高氯酸滴定液( 0.1mol/L )相当于 19.72mg 的 C9H11NO4 。
C 、比旋度:取本品约 0.2g, 精密称定,置 25ml棕色量瓶中,加乌洛托品 5g, 再加盐酸溶液( 9→100 )溶解并稀释至刻度,摇匀,避光放置 3 小时,依法测定,比旋度为 -159o 至 -168o 。

思考题1. 左旋多巴提取时如果使用温度过高的去离子水有何不好?2. 用酸性或碱性过强的溶液提取左旋多巴对收率均有影响,为什么?
3. 使用真空浓缩的目的是缩短时间,为什么浓缩时间过长不好?4. 能用阳离子交换树脂进行实验吗?需要什么条件?在此条件
下对收率有何影响?参 考 文 献[1] 周荣汉,杨星昊,唐正平,中药材, 1988 , 11 ( 3 ), 2
2[2] 中国药典 2005版 P85[3] 杨星昊,张晓彤,周荣汉,基层中药杂志, 1997 , 11
( 4 ), 45-46[4] 杨星昊,顾觉奋,周荣汉,离子交换与吸附, 1998 , 14
( 3 ), 210-214
返回

实验十一 微波辅助合成松香甘油酯目的要求:1. 了解松香及松香甘油酯的性质及用途;2. 掌握微波辐射法制备松香酯类化合物的原理和操作。

• 松香 (Rosin) 是一种可再生天然树脂。我国是松香生产大国,脂松香年产 40万吨以上,居世界第一,广西则是脂松香生产大省,可以说脂松香是广西的特产之一。随着石油等一次性资源的渐渐枯竭,如何利用松香这种可再生的天然资源来替代部分一次性资源已成为日益重要的研究课题。
• 松香是分子式为 C19H29COOH 的各种脂酸混合物,微黄色至黄红色透明、硬脆、有松脂气味的玻璃状固体。软化点≥ 72-74℃ ,沸点约 300 (0.67℃ 千帕 ) 。易溶于乙醇、乙醚、丙酮、苯、二氯乙烷、二硫化碳、松节油、石油醚、汽油;溶于油类和碱溶液,不溶于水。

• 松香是各种松香酸异构体的混合物,主要由下列几种松香酸组成:
CH3 COOH
CH3
CH(CH3)2
CH3 COOH
CH3CH3
CH3
CH3
CH3 COOH
CH3
CH2
松香酸 (枞酸 ) 新松香酸 异右旋海松酸

• 松香按来源分为脂松香、木松香、浮油松香等,各种松香的组成大致如下表:
组成 脂松香 木松香 浮油松香松香酸
脱氢松香酸新松香酸异海松酸异松香酸海松酸其他
204
1818182
20
352047
103
21
4587
11103
16

• 松香甘油酯 (glycerine rosinate ; ester gum)又名酯胶,松香酸甘油酯。为淡黄色不规则透明玻璃块状固体,微有特殊香气,溶于植物油,不溶于水。工业级产品为油漆的重要原料,也可用作胶粘剂 ( 热用胶、压敏胶、薄膜胶等 ) ,本品对提高 EVA 热用胶的热初粘性及改善难粘物质的粘着强度,效果显著,同时对胶的耐寒性的提高有独特作用。食品级常用作乳化剂,是口香糖的基础剂和乳化香精。

1. 松香甘油酯的传统制法大致有:松香与脂肪醇催化酯化法、松香树脂酸盐与卤代烃催化酯化法以及酯交换法等。
2. 松香与甘油催化酯化法目前仍是工业上生产松香甘油酯的主流方法,它是由松香和甘油在催化剂存在下经加热反应制得,反应式如下:
C19H29COOH CH2OHCHOHCH2OH
CH2OOCC19H29
CHOH
CH2OH
+ + H2O´ß»¯¼Á

与一般的羧酸不同,松香的羧基位于叔碳原子上,空间位阻很大,反应时活化能较低,所以它的酯化条件比较苛刻,除了要求高温 (200-300 )℃ 、时间长 (6-11h) 外,还必须借助于高活性催化剂才能使反应顺利进行。
多年来在寻找新的方法和手段以缩短松香酯化反应时间方面做了大量的研究工作,但进展不大。
微波辐射对许多有机化学反应有显著的加速作用,许多反应的反应速率能提高数百倍乃至上千倍,大大缩短了化学反应时间。本实验以氧化锌为催化剂,将松香与甘油在微波辐射下进行酯化反应。

主要原料、试剂及仪器 • 松香 ( 酸值 165mgKOH/ g ,软化点 72.0℃ ,加纳色泽
6) ;甘油 (AR) ; ZnO 、乙醇、苯和甲苯 (AR) ;常压微波辅助合成/萃取反应仪。

实验步骤• 松香甘油酯的制备 在 100mL 三口瓶中加入 20.0g颗粒状松香、 0.068g 氧
化锌及 2.4g甘油,置于外部与搅拌器、冷凝管相连接的微波炉中。用 600W功率 (10档 )微波进行辐射,当反应物熔化后开始搅拌,常压下反应 30min 。在连续抽真空的条件下,使反应体系的压力在小于 5kPa 下继续反应 30min ,以尽可能蒸出反应生成的水及多余的甘油,反应结束后趁热将产品倒入一干净的烧杯中,冷却待用。

松香甘油酯各项指标的测定按照国家标准 GBl0287—88 测定松香甘油酯的酸值、软化
点等指标。1 、酸值的测定 测定松香酸值的方法有内指示剂法和电位滴定法。 A 、内指示剂法 本法适用于浅色和中等色度的松香甘油
酯。 (1) 、试剂:中性乙醇,酚酞指示剂 ,0.5N 氢氧化钾乙醇标准溶液(根据试验,用氢氧化钾水溶液完全可以代替氢氧化钾乙醇溶液)。

(2) 、操作:将除去外表层并粉碎的松香甘油酯立即称取试样约 2克(准确至 0.001 克)于 250 毫升锥形瓶中,加中性乙醇 50毫开溶解(必要时在电热板或水浴上加热,使试样全部溶解后放冷),加酚酞指示剂 0.5 毫升,然后用 0.5N 氢氧化钾乙醇标准溶液滴定至微红色 30秒不褪为止。
( 3 )、计算:酸值按下式计算:酸值 =V×N×56.11/W( 毫克氢氧化钾 / 克 )
式中: V- 滴定时消耗氢氧化钾乙醇标准溶液的体积(毫升) N— 氢氧化钾乙醇标准溶液的当量浓度W— 试样重(克)56.11- 氢氧化钾的毫克当量 ( 毫克/毫克当量 )
两次平行试验允许相差 0.5 ,以算术平均值为结果(精确至小数点后第一位)。

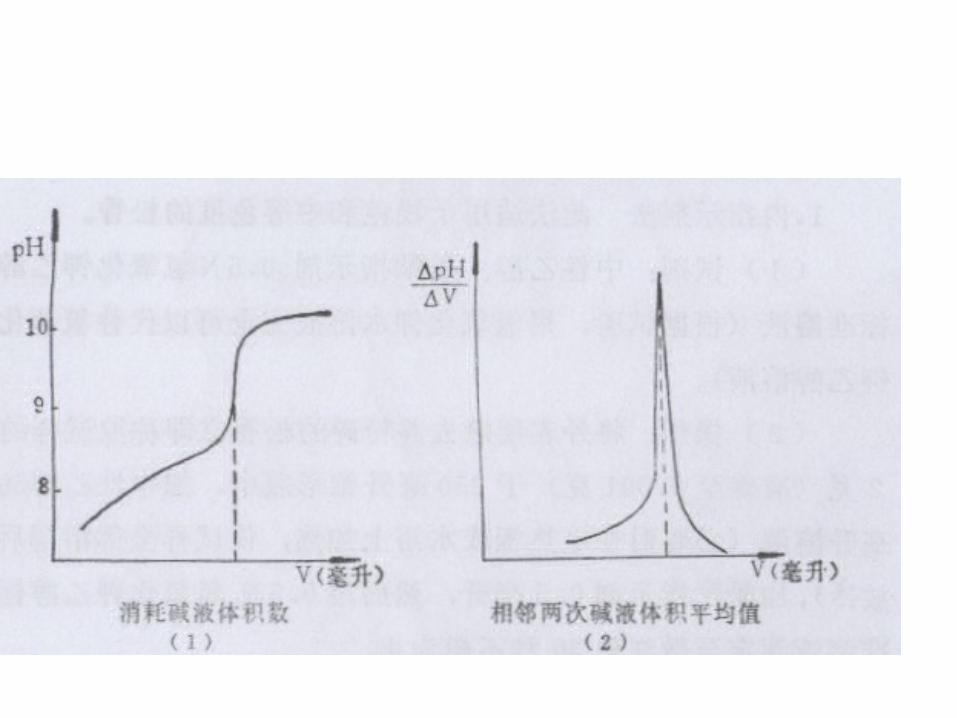
• B 、电位滴定法 (附) • 本法同时适用于浅色和深色的松香甘油酯。松香中的树脂酸
在溶液中能发生电离作用,形成的氢离子能和碱溶液中的氢氧离子起中和反应生成难电离的水。碱液不断加入,松香甘油酯溶液的 pH值也不断增加, pH值的这种变化,在中和反应达到等当点附近时极为明显。碱液的加入量和松香溶液 pH值的关系见下图 (1) 和图 (2) 。

• 软化点的测定:操作见附录部分。• 质量规格
软化点 (环球法 )/℃色泽 (加纳色号 )酸值 /(mgKOH/g)杂质 /(mg/L)
≥85℃≤7≤7≤30

• 五、注意事项:本实验在微波辐射下进行,操作过程中应尽量远离微波炉,切不可在打开炉盖状态下启动微波辐射。
• 六、思考题:• 1 、试简述松香甘油酯的酸值及软化点的大小对其应用有何影响。
• 2 、反应结束前为什么必须减压除去生成的水和多余的甘油?
• 参考文献• [1] 林中祥 ,陈日清,微波辐射下松香与多元醇的酯化反
应研究,林产化学与工业 ,2003,23(3) : 37 - 40• [2] 林产化工手册: 411-427• [3] 精细化学品手册大全: 2084-2085
返回

附 录 几种常用仪器的使用说明一、 NDJ-79 型旋转式粘度计及使用方法二、 JZHY-180 界面张力仪的使用方法三、罗氏泡沫测定仪的使用方法四、乌氏 (Ubbelchde) 粘度计的使用五 福美双含量测定的黄原酸盐法介绍返回

一、 NDJ-79 型旋转式粘度计及使用方法
1 、原理 仪器的驱动是靠一个微型的同步电动机,它以 750r/min 的
恒速旋转,几乎不受荷载和电源电压变化的影响。电动机的壳体采用悬挂式来安装,它通过转轴和挂钩带动转筒旋转。当转筒在被测液体中旋转受到粘滞阻力作用时,产生反作用而使电动机壳体偏转,电动机壳体与两根具有正反力矩的金属游丝相连,壳体的转动使游丝产生扭矩,当游丝的力矩与粘滞阻力矩达到平衡时,与电动机壳体相连接的指针便在刻度盘上指示出某一数值,此数值与转筒所受粘滞阻力成正比,于是刻度读数乘上转筒因子就表示动力粘度的量值。

2 、旋转式粘度计示意图旋转式粘度计如下图所示:1-柱座; 2- 电源插座; 3- 电源开关; 4- 安放测定器的托架;
5-悬吊转筒的挂钩; 6-读数指针; 7-同步电动机; 8-指针调零螺丝; 9-具有反射镜的刻度盘; 10- 测定器; 11-温度计; 12 、 13 、 14-因子分别为 1 、 10 、 100 的转筒

3 、操作步骤 (1) 、通过粘度计的电源插座连接 220V 50Hz 的交流电源; (2) 、调整零点。开启电源开关,使电动机在空载时旋转,待稳定后用调零螺丝将指针调到刻度的零点,关闭开关。
(3) 、将被测液体小心地注人测定器,直至液面达到锥形面下部边缘为止,约需液体 15 m1 左右,将转筒浸人液体直到完全浸没为止,连上专用温度计,接通恒温水源,将测定器放在粘度计托架上,并将转筒悬挂于挂钩上。
(4) 、开启电源开关,起动电动机,转筒从开始晃动到对准中心。为加速对准中心,可将测定器在托架上向前后左右微量移动。
(5) 、当指针稳定后即可读数,将所用转筒的因子乘以刻度读数即得以 Pa·s 表示的粘度。如果读数小于 10格,应当调换直径大一号的转筒。记下读数后,关闭电源开关。将测定器内孔和转筒洗净擦干。

4 、注意事项 (1) 、本粘度计为精密测量仪器,必须严格按规定的步骤操
作。 (2) 、开启电源开关后,电动机就应起动旋转,如负荷过大
或其他原因迟迟不能起动,就应关闭电源开关,查找原因后再开,以免烧毁电动机和变压器。
(3) 、电动机不得长时间连续运转,以免损坏。 (4) 、使用前和使用后都应将转筒及测定器内孔洗净擦干,
以保证测量精度。(5) 、以上所述粘度的测量范围为 10-104cP 。对于更小或更
高粘度的测量,请详见仪器说明书。
返回

二、 JZHY-180 界面张力仪的使用方法
1 、原理 JZHY-180 型界面张力仪主要由扭力丝、铂环、支架、拉杆架、蜗轮副等组成。使用时通过蜗轮副的旋转对钢丝施加扭力,并使该扭力与液体表面接触的铂环对液体的表面张力相平衡。当扭力继续增加,液面被拉破时,钢丝扭转的角度,通过刻度盘上的游标指示出来,此值就是界面张力(P)值 ,单位mN/m 。

2 、张力仪示意图附图 JZHY-180 型表面张力仪样品座; B- 样品座螺丝; J-臂的制止器; L-指针; M-涡轮把手;
P-微调涡轮把手; Q- 固定钢丝手母; E- 水平螺旋; K-臂的制止器 (2) ; C-游码

3 、操作步骤(1) 、准备工作A 、将仪器放在平稳的地方,通过调节螺母 E 将仪器调到水
平状态,使横梁上的水准泡位于中央位置。B 、将铂环放在吊杆端的下末端,小纸片放在铂环的圆环上,打开臂之制止器 J 和 K ,调好放大镜,使臂上的指针 L 与反射镜上的红线重合,如果刻度盘上游标正好指示为零,则可进行下一步。如果不指零的话,可以旋转微调蜗轮把手 P进行调整。

• C 、用质量法校正。在铂圆环的小纸片上放一定质量的砝码,当指针与红线重合时游标指示正好与计算值一致。若不一致可调整臂 F 和 G 的长度,臂的长度可以用两臂上的两个手母来调整。调整时这两个手母必须是等值旋转,以便使臂保持相同的比例,保证铂环在试验中垂直地上下移动,再通过游码 C 的前后移动达到调整结果。具体方法是将 500-800mg砝码放在铂环的小纸片上;旋转蜗轮把手,直到指针 L 与反射镜上红线精确地重合。记下刻度盘的读数(精确到 0.1 分度)。如果用 0.8g 的砝码,刻度盘上的读数为
• 如记录的读数比计算值大,应调节杠杆臂的两个手母,使两臂的长度等值缩短;如过小,则应使臂的长度伸长。如此重复几次,直到刻度盘上的读数与计算值一致为止。
6530.062
17.9808.0
2/ 1
L
mgmNP

D 、在测量以前,应把铂环和玻杯用洗涤剂清洗。 (2) 、表面张力的测量 A 、将铂环插在吊杆臂上,将被测溶液倒在玻杯中,高约 2
0~ 25mm ,将玻杯放在样品座的中间位置上,旋转螺母B,铂环上升到溶液表面,且使臂上的指针与反射镜上的红线重合。
B 、旋转螺母 B 和蜗轮把手M来增加钢丝的扭力,保持指针 L始终与红线相重合,直至薄膜破裂时,刻度盘上的读数指出了溶液的表面张力值。测三次取其平均值。
仪器使用完毕,铂环取下清洗后放好,扭力应处于不受力的状态。杠杆臂应用偏心轴和夹板固定好。
返回

三、罗氏泡沫测定仪的使用方法 1 、原理 泡沫稳定性是泡沫最主要的性能。此外表面活性剂(或其他起泡剂)的起泡能力亦属与泡沫有关的重要性质。因而一般泡沫性能的测量,主要是对稳定性及起泡性进行研究。
泡沫稳定性的测量方法很多。根据成泡的方式主要分为两类:气流法及搅动法。在生产及实验室中比较方便而又准确地测量泡沫性能的方法是“倾注法”,它也属于搅动法一类。
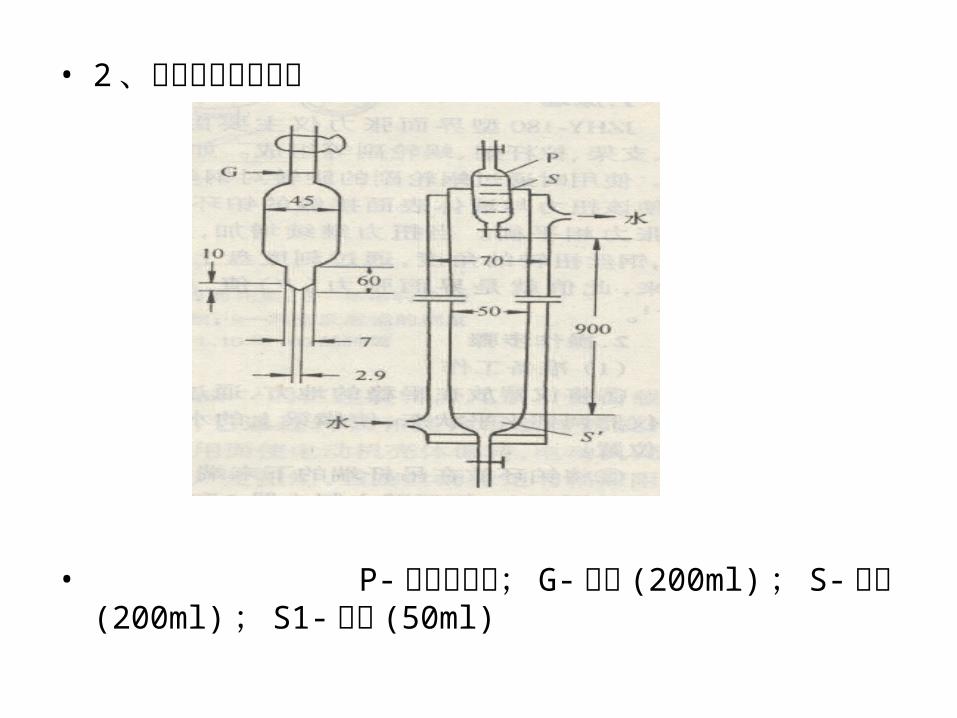
• 2 、罗氏泡沫仪示意图
• P- 泡沫移液管; G-刻度 (200ml) ; S- 试液 (200ml) ; S1- 试液 (50ml)

3 、操作步骤 ①、用蒸馏水将柱刷洗两次。 ②、控制恒温槽的温度在 (50±0.1)℃ 。然后将循环恒温水通过恒温槽打入仪器的外套管中,使其在恒温条件下工作。
③、将盛有待测溶液的容量瓶放入恒温槽内,以保持一定的温度。
④、恒温后,沿柱内壁缓慢地加入待测溶液至 50 ml刻度处,并将吸满待测溶液的泡沫移液管垂直夹牢,使其下端与柱上的刻度线对齐。
⑤、打开泡沫移液管的旋塞使溶液全部流下,待溶液流至250ml刻度处,记录一次泡沫高度, 5min 后再记录一次泡沫高度。测量三次取其平均值。
返回

四、乌氏 (Ubbelchde) 粘度计的使用
1 、基本原理高分子稀溶液的粘度主要反映了液体分子之间因流动或相对运动所产生的内摩擦阻力。内摩擦阻力越大,表现出来的粘度就越大,且与高分子的结构、溶液浓度、溶剂的性质、温度以及压力等因素有关。实验证明,对于给定聚合物在给定的溶剂和温度下,其特性粘度 [η] 的数值仅由样品的粘均相对分子质量所决定,其关系如下:
K 、 α 与温度、聚合物种类和溶剂性质有关, K值受温度的影响较明显,而 α值主要取决于高分子线团在溶剂中舒展的程度,一般介于 0.5-1.0 之间。对给定的聚合物 - 溶剂体系,一定的相对分子质量范围内 K 、 α值可从有关手册中查到,或采用几个标准样品由进行确定,标准样品的相对分子质量由绝对方法(如渗透压和光散射法等)确定。
MK

在一定温度下,聚合物溶液的粘度对浓度有一定的依赖关系。描述溶液猫度的浓度依-赖的方程式很多,而应用较多的有:
哈斯金 (Huggins) 方程
克拉默 (Kraemer) 方程
ckcsp 2'
ccr 2ln
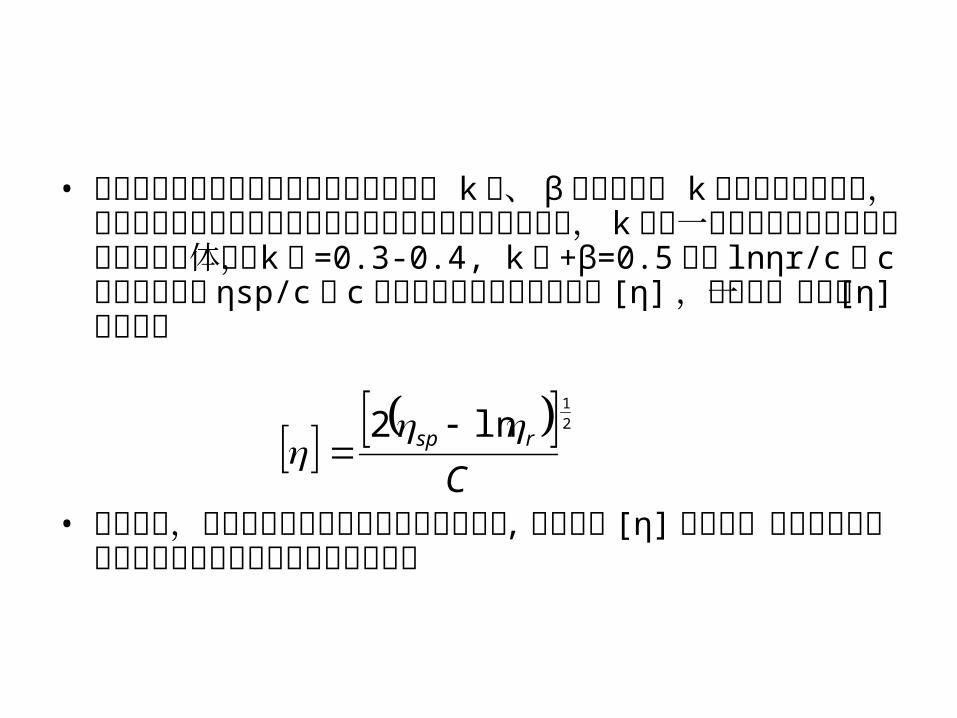
• 对于给定的聚合物在给定温度和溶液时, k︱、 β 应是常数。 k︱称为哈金斯常数,它表示溶液中高分子间和高分子与溶剂分子间的相互作用, k︱值一般说来对线形柔性链高分子良溶剂体系, k︱ =0.3-0.4, k︱ +β=0.5 。用 lnηr/c对 c 作图外推和用 ηsp/c对 c 作图外推可得到共同的截距 [η] ,由此得一点法求 [η] 的方程:
• 由上可见,用粘度法测定高分子溶液相对分子量 , 关键在于 [η] 的求得,最为方便的是用毛细管粘度计测定溶液的相对粘度。
21
ln2
Crsp

• 常用的粘度计为乌氏 (Ubbelchde) 粘度计,其特点是溶液的体积对测量没有影响,所以可以在粘度计内采取逐步稀释方法得到不同浓度的溶液。
• 式中, ρ 、 ρo 分别为溶液和溶剂的密度,因溶液很稀, ρ=ρo ; A 、 B 为粘度计常数; t 、 to 分别为溶液和溶剂在毛细管中的流出时间,即液面经过刻线 a 、 b所需时间 (见下图a) 。在恒温条件下,用同一支粘度计测定溶液和溶剂的流出时间,如果溶剂在该粘度计中的流出时间大于 100s,则动能较正项 B/At2值远小于 1 ,可忽略不计,因此溶液的相对粘度为: ηr=t/to
• 样品溶液浓度一般在 0.01g/ml 以下,使 ηr 在 1.05-2.5 之间较为适宜, ηr最大不应大于 3.0 。
2
2
1
1
ooo
r
At
Bt
At
Bt
o

• 2 、仪器:
– 乌氏粘度计– b 、装置图: 1. 水槽 2.毛细管 3. 加热棒 4.探温头 5. 温控仪 6. 搅拌器

3 、操作步骤:A 、溶液配制取洁净干燥的样品,在分析天平上准确称取 2. OOOg±0.001
g ,溶于 500ml 烧杯内(加纯溶剂 200m1 左右),微微加热,使其完全溶解,但温度不宜高于 60℃ ,待完全溶解后用砂芯漏斗滤至 2000ml 容量瓶内(用纯溶剂将烧杯洗 2-3 次滤入容量瓶内),稀释至刻度,反复摇匀后待用。
B 、安装粘度计 将干净干燥的粘度计,用过滤后的纯溶剂洗 2-3 次,然后
将过滤好的纯溶剂从 A管加入至 F球的 2/3-3/4 ,再固定在恒温 30.0士 0.1℃ 的水槽中,使其保持垂直,并尽量使 E球全部浸泡在水中,最好使 a 、 b两刻度线均没入水面以下(如图 b所示)。安装时除注意垂直外,还应注意固定的是否牢固,在测量的过程中不至引起数据的误差。

C 、纯溶剂流出时间 to 的测定恒温 10-15min 后,开始测定。紧闭 C管上的乳胶管,用吸耳球从 B管口将纯溶剂吸至 G球的一半,拿下吸耳球打开 C管,记下纯溶剂流经 a, b刻度线之间的时间 to ,重复几次测定,直到出现三个数据,两两误差小于 0.2s, 取这三次时间的平均值。

D 、溶液流经时间 t 的侧定
将毛细管内的纯溶剂倒掉,用溶液润洗 1-2 次,加入溶液至 F球的 2/3-3/4, 固定在水槽中,恒温 15min 左右,开始测定。紧闭 C管上的乳胶管,用吸耳球从 B管口将溶液吸至 G球的一半(注意 B管中溶液表面不能有汽泡,若有汽泡可从 B管上方将其吸出),拿下吸耳球打开 C管,记下溶液流经 a , b刻度线之间的时间 t 。重复几次测定,直到出现三个数据,两两误差小于 0.2s ,取这三次时间的平均值。测定结束后倒出粘度计中的溶液,倒入纯溶剂,将其吸至 a线上方小球的一半清洗毛细管,反复几次,倒挂毛细管粘度计以待后用。

• E 、数据整理• 1 、测量数据记入下表
• 记 录 次 数 to t ηr ηsp [η]
• 平 均
• 2 、根据特定温度和溶剂,从文献中查找到 K 和 α值,求出粘均分子质量。
返回

五、 福美双含量测定的黄原酸盐法介绍 原理:当采用氯化亚锡为还原剂,以醋酸为裂解剂与福美双作用时,
会释放出定量的 CS2 ,用甲醇氢氧化钾吸收后生成的黄原酸钾,可以用碘进行定量滴定,从而计算出福美双含量。
(CH3)2N-C-S
(CH3)2N-C-S
S
S
[(CH3)2N-C-S]4Sn
S
[(CH3)2N-C-S]4Sn
S
(CH3COO)4Sn
CS2KOH CH3OH CH3OCSSK
CH3OCSSCOCH3
S S
2 + 2SnCl2 + SnCl4
+ 4CH3COOH 4(CH3)2NH + 4CS2 +
+ + + H2O
2CH3OCSSK + I2 + 2KI

2 、仪器:使用代森锌仪器装置 (见下图 ) ,增加一个吸收瓶。(1) 、反应瓶 (500ml);(2) 、分液漏斗 (50ml);(3) 、冷凝管 ;(4 、
5) 、第一、二吸收管 ;(6) 、下口瓶 (10L)

3 、测定步骤:称取 0.3-0.4g 样品 (准确至 0.2mg) ,置于反应瓶中,注入 0.1mol/L 氯化亚锡 50%乙酸溶液 50ml 。连接好三个吸收瓶,第一吸收瓶内盛 50ml10%乙酸铅溶液,第二、三吸收瓶内装入 2mol/L 氢氧化钾的甲醇溶液 25ml 。同时以 4-6 泡 /秒速度抽气,加热至微热,反应 30min 后用 200ml 水洗第二、三吸收瓶到碘瓶中,加二滴 10%酚酞溶液,用 30%醋酸溶液滴定至中性,加 5ml0.5%淀粉溶液作为指示剂,用 0.1mol/L碘标准溶液滴定至微蓝色,即为终点。同时做一空白测定。

4 、福美双含量 (X) 计算公式:
式中 c —碘标准溶液的摩尔浓度, mol/L ;V 1— 样品测定时消耗碘标准溶液的体积, ml ;V2—空白测定时消耗碘标准溶液的体积, ml ;m— 样品的重量, g ;0.1202—福美双的摩尔质量, kg/mol 。
%100
21202.021
m
VVcX
返回