2 risk management applicato ai dispositivi medici
TRANSCRIPT

INGEGNERIA CLINICA
Il Risk Management applicato ai dispositivi medici:
aspetti normativi di riferimento
Ing. P. Balestra – Servizio di Ingegneria Clinica

Risk Management: le apparecchiature biomediche
La rapida introduzione ed evoluzione delle dotazioni tecnologiche
all’interno delle strutture sanitarie, ha favorito la possibilità di
sviluppare metodiche medico-chirurgiche più sofisticate ed efficaci
a fini diagnostici e curativi, ma ha anche moltiplicato le fonti di
rischio legate sia alle anomalie accidentali di funzionamento, sia
all’uso improprio involontario.

Circa 600.000 prodotti:
Apparecchiature biomediche: hanno la caratteristica di essere
beni durevoli, di avere necessità di collaudi, manutenzione, controlli
periodici (TAC, elettrobisturi, laser chirurgici/terapeutici, …)
Strumentario: accessori (pinze, manipoli,…)
Materiale di consumo: pronto all’uso, monouso, pluriuso (lastre,
siringhe, piastre per elettrobisturi, aghi, cateteri, …)
Protesi: stent, protesi d’anca, …
Software: RIS-PACS, LIS, CIS, …
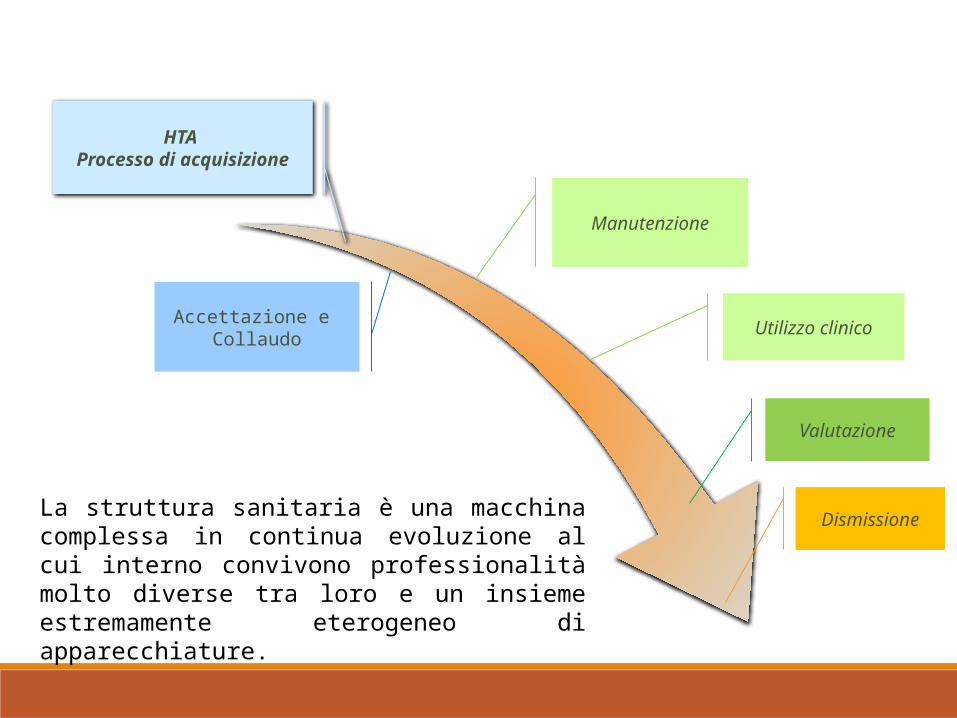
HTA Processo di acquisizione
Accettazione e Collaudo
Manutenzione
Utilizzo clinico
Valutazione
DismissioneLa struttura sanitaria è una macchina complessa in continua evoluzione al cui interno convivono professionalità molto diverse tra loro e un insieme estremamente eterogeneo di apparecchiature.
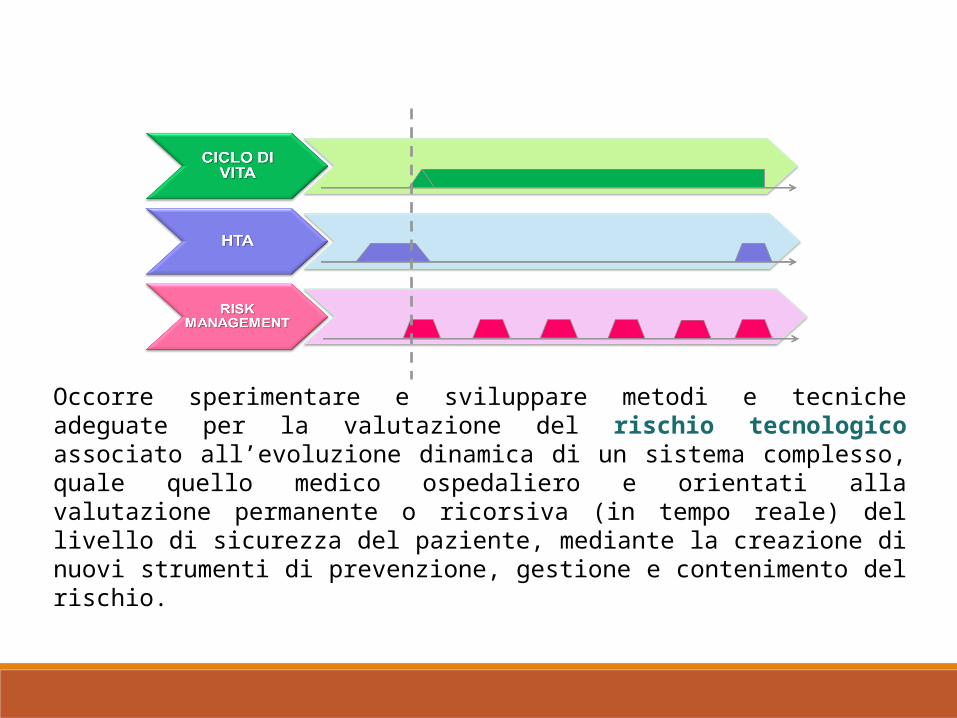
Occorre sperimentare e sviluppare metodi e tecniche adeguate per la valutazione del rischio tecnologico associato all’evoluzione dinamica di un sistema complesso, quale quello medico ospedaliero e orientati alla valutazione permanente o ricorsiva (in tempo reale) del livello di sicurezza del paziente, mediante la creazione di nuovi strumenti di prevenzione, gestione e contenimento del rischio.
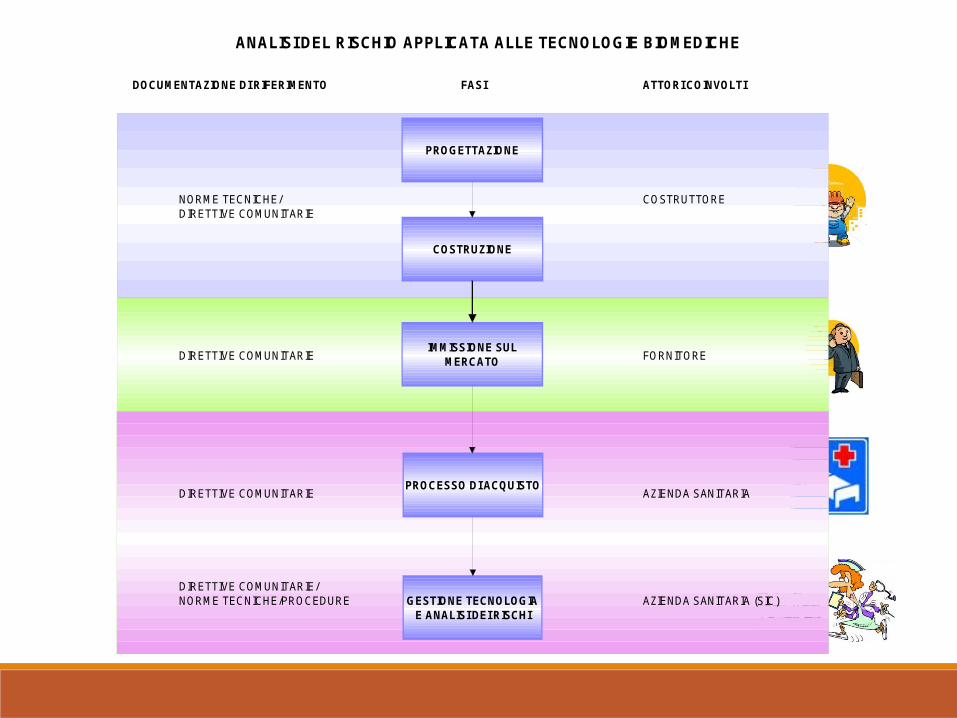
ANALISI DEL RISCHIO APPLICATA ALLE TECNOLOGIE BIOMEDICHE
COSTRUZIONE
IMMISSIONE SUL MERCATO
PROCESSO DI ACQUISTO
GESTIONE TECNOLOGIA E ANALISI DEI RISCHI
PROGETTAZIONE
DOCUMENTAZIONE DI RIFERIMENTO FASI ATTORI COINVOLTI
COSTRUTTORE
FORNITORE
AZIENDA SANITARIA
AZIENDA SANITARIA (SIC)DIRETTIVE COMUNITARIE/NORME TECNICHE/PROCEDURE
NORME TECNICHE/DIRETTIVE COMUNITARIE
DIRETTIVE COMUNITARIE
DIRETTIVE COMUNITARIE

Fino al 1968 Legislatura prevalentemente nazionale.
I dispositivi medici devono essere costruiti ‘a regola d’arte’.
Interpretazione SOGGETTIVA
1968 – 1990 Legge 186/1968Norme generali per la sicurezza degli apparecchi elettromedicali + Norme Particolari. Esse prescrivono modalità di progetto, costruttive e a volte d’uso corretto.
criterio OGGETTIVO :
un’apparecchiatura che soddisfa la norma è costruita ‘a regola d’arte’
Evoluzione degli aspetti normativi

1990 – 2000 Direttiva Dispositivi Medici 93/42. Le Norme generali e particolari per la sicurezza non vengono sostituite.
RESPONSABILIZZAZIONE del COSTRUTTORE
• seguire il rapido progresso della tecnologia• preferibile normare le procedure di progettazione, di costruzione, di distribuzione, di manutenzione e d’uso dei dispositivi medici
• sistematizzare l’attività produttiva e distributiva
Evoluzione degli aspetti normativi

Le direttive comunitarie hanno modificato l’approccio alla progettazione, produzione,
commercializzazione e gestione dei
DISPOSITIVI MEDICI
L’Obbiettivo principale delle direttive comunitarie è quello di garantire la libera circolazione nel mercato
di prodotti conformi e di fissare per tutti gli stati membri un elevato livello di sicurezza per gli
utenti/consumatori
Direttive comunitarie

L’ Unione Europea ha predisposto tre normative di riferimento che riguardano il settore sanitario:
DIRETTIVA 93/42/CEE
DIRETTIVA 90/385/CEE
DIRETTIVA 98/79/CEE
Direttive comunitarie

DIRETTIVA 93/42/CEE
Riguarda i dispositivi medici
Recepita in Italia con il Dlgs n. 46/97 modificato con i Dlgs 95/98, 332/00, 271/02 e 2007/47/CE
Direttive comunitarie

Qualsiasi strumento, apparecchio, impianto, sostanza o altro prodotto, utilizzato da solo
o in combinazione, compreso il software informatico impiegato per il corretto
funzionamento, e destinato dal fabbricante ad essere impiegato nell'uomo a scopo di:
diagnosi, prevenzione, controllo, terapia o attenuazione di una malattia;
diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un
handicap;
studio, sostituzione o modifica dell'anatomia o di un processo fisiologico;
intervento sul concepimento;
il quale prodotto non eserciti l'azione principale, nel o sul corpo umano, cui è destinato,
con mezzi farmacologici o immunologici né mediante processo metabolico ma la cui
funzione possa essere coadiuvata da tali mezzi
DISPOSITIVO MEDICO - [Direttiva 93/42/CEE aggiornata con la 2007/47/CE]

DIRETTIVA 90/385/CEE
Riguarda i dispositivi medici impiantabili attivi
Recepita in Italia con il Dlgs n. 507/92
Direttive comunitarie

DIRETTIVA 98/79/CEE
Riguarda i dispositivi medici diagnostici in vitro
Recepita in Italia con il Dlgs n. 46/97 modificato con i Dlgs 95/98 il 332/00 e 271/02
Direttive comunitarie

14/06/1993 Approvazione della Direttiva EuropeaEntro il 01/07/1994 Adozione e pubblicazione da parte degli stati membri delle
disposizioni legislative, regolamentari e amministrative01/01/1995 Applicazione da parte degli stati membri delle disposizioni
della direttiva ai nuovi dispositivi
24/02/1997 Recepimento della Direttiva in Italia con D.L. n°46 Fino al 14/06/1998 Gli stati membri ammettono l’immissione in commercio e
la messa i servizio dei dispositivi a condizione che essi siano conformi alla normativa in vigore sul loro territorio alla data del 31/12/1994
Dal 15/06/1998 Tutti i dispositivi medici sono soggetti a questa Direttiva
DIRETTIVA 93/42/CEE aggiornata con la 2007/47/CE

DIRETTIVA 93/42/CEE aggiornata con la 2007/47/CE
Definisce i dispositivi medici ed i loro fabbricanti
Definisce i requisiti essenziali cui devono soddisfare i Dispositivi Medici
Definisce quattro classi di appartenenza
Per ogni classe stabilisce i criteri per l’apposizione della marcatura CE
A chi è rivoltaFabbricanti
Gestori
Utilizzatori

Si applica a: dispositivi medici impiantabili non attivi dispositivi medici attivi non impiantabili dispositivi medici non attivi non impiantabili Accessori Software per il funzionamento dei dispositivi medici
Non si applica a: diagnostici in vitro (es: reagenti)
(si applica la direttiva diagnostici in vitro 98/79/CEE) dispositivi impiantabili attivi (es: cardiostimolatori)
(si applica la direttiva dispositivi medici impiantabili attivi 90/385/CEE)
Campo di applicazione
Direttiva 93/42/CEE
Dispositivi medici impiantabili non attivi
Campo di applicazione
Direttiva 93/42/CEE

Dispositivi medici attivi non impiantabili
Campo di applicazione
Direttiva 93/42/CEE

Dispositivi medici non attivi non impiantabili
Campo di applicazione
Direttiva 93/42/CEE

Accessori
Campo di applicazione
Direttiva 93/42/CEE

Dispositivi “di confine”
INCLUSIONI Autoclave Considerata come “accessorio” ad
esempio per sterilizzare i ferri chirurgici che sono DM
Lampada scialitica Considerata come “accessorio”, consente di assistere nell’uso altri DM in condizioni di maggiore sicurezza
Direttiva 93/42/CEE

• La sicurezza dei dispositivi
• L’affidabilità
• La minimizzazione del rischio
• La libera circolazione dei dispositivi all’interno della Comunità Europea
ObiettiviDirettiva 93/42/CEE

Per garantire questi obiettivi:
Fissa alcuni REQUISITI ESSENZIALI che ogni fabbricante deve dimostrare di avere rispettato durante le fasi di progettazione
e costruzione e commercializzazione dei singoli dispositivi
I REQUISITI ESSENZIALI
Definiscono i limiti di sicurezza e il livello di accettabilità di rischio di un dispositivo
(RISCHIO MINIMO ACCETTABILE)
ObiettiviDirettiva 93/42/CEE

Si limita a FISSARE i risultati che devono essere raggiunti senza imporre soluzioni tecniche per
ottenerli.
Questo per non limitare l’innovazione tecnologica
ObiettiviDirettiva 93/42/CEE

Il costruttore (oppure un suo mandatario nell’Unione Europea)
attesta che il prodotto soddisfa i requisiti essenziali delle direttive
applicabili a quel prodotto e che le procedure stabilite nelle
direttive sono state assolte, ad esempio è stata compilata la
dichiarazione di conformità, è stato predisposto il fascicolo
tecnico, ecc.
La marcatura CE è obbligatoria per un prodotto in uno Stato
membro che ha recepito almeno una direttiva applicabile a quel
prodotto, una volta terminato il periodo transitorio.
Marcatura CEDirettiva 93/42/CEE

Il marchio di qualità è volontario e attesta che il prodotto è conforme alle
norme relative, secondo le prove di tipo ed il controllo della produzione
condotte da un organismo indipendente (es. IMQ, TUV). Sullo stesso
prodotto possono coesistere la marcatura CE ed il marchio di qualità:
per distinguere anche per mezzo delle parole, oltre che nei significati, le
due modalità si parla di marcatura CE e di marchi di conformità alle
norme (o marchi di qualità).
Differenza fra marcatura CE e marchi di qualità
Direttiva 93/42/CEE
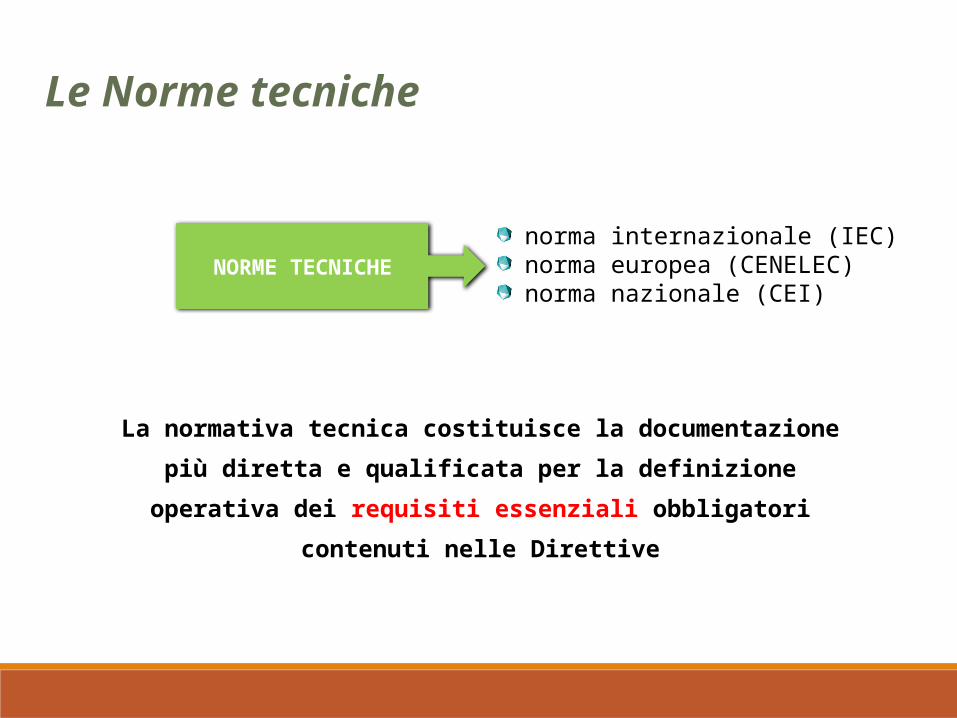
La normativa tecnica costituisce la documentazione più diretta e qualificata per la definizione operativa dei requisiti essenziali
obbligatori contenuti nelle Direttive
Le Norme tecniche
NORME TECNICHEnorma internazionale (IEC) norma europea (CENELEC) norma nazionale (CEI)

NORMA CEI 62-5
Generale per tutti gli apparecchi elettromedicali
NORME CEI PARTICOLARI 62-XX
Per apparecchi specifici
NORME CEI 64-8 sez. 710
Per gli impianti elettrici nei locali adibiti ad uso medico
Le Norme tecniche in ambito sanitario
LE NORME CEI (Comitato Elettrotecnico Italiano)
COMITATO TECNICO 62 (CT 62-) APPARECCHIATURE
ELETTROMEDICALI
COMITATO TECNICO 64 (CT 64-) IMPIANTI ELETTRICI

• Hanno carattere “Volontario”
• Rappresentano secondo la legge 186 del 1968 le “Regole di buona tecnica”
• “Regola d’arte”
Le Norme tecniche

La Legge 186 del 1968 dispone:
Art. 1
“Tutti i materiali, le apparecchiature, i macchinari, le
installazioni, gli impianti elettrici ed elettronici
devono essere realizzati e costruiti
a REGOLA D’ARTE”
Le Norme tecniche

Art. 2
“I materiali, le apparecchiature, i macchinari le
installazioni, gli impianti elettrici ed elettronici
realizzati secondo le norme CEI si considerano
a REGOLA D’ARTE”
Legge 186 del 1968

Specifica “una procedura mediante la quale il FABBRICANTE può identificare i pericoli associati a un dispositivo medico e ai suoi accessori, stimare e valutare i rischi associati a tali pericoli, controllare tali rischi e monitorare l’efficacia di tale controllo”
La Norma UNI CEI EN ISO 14971
“Dispositivi Medici – applicazione della gestione dei rischi ai Dispositivi Medici”

È basata su 4 principi fondamentali:
1. Analisi dei rischi
2. Valutazione dei rischi
3. Controllo dei rischi
4. Informazione post vendita
La Norma UNI CEI EN ISO 14971

Le direttive comunitarie e le norme tecniche tendono a garantire la sicurezza INTRINSECA del dispositivo medico. Permettono di ridurre al minimo il rischio ma non di eliminarlo completamente.
La valutazione del rischio eseguita dal fabbricante rappresenta un elemento importante ma non unico nella valutazione globale del rischio del dispositivo medico.
Pertanto…

La valutazione del rischio legata all’utilizzo del dispositivo medico (e non alla progettazione o costruzione) assume un’importanza strategica per la struttura ospedaliera in quanto permette la programmazione razionale degli interventi correttivi finalizzati alla minimizzazione dei rischi.
Ma…

E’ indiscutibile il coinvolgimento del DM negli incidenti, a causa
della sua sicura presenza e indubbio utilizzo durante l’erogazione
della prestazione sanitaria.
non è sempre facile stabilire• grado di coinvolgimento del DM• entità del contributo
all’interno del processo che ha condotto al verificarsi dell’incidente

Malfunzionamento
Errato utilizzo
Reazione allergica
Interazioni con l’ambiente
Alcune cause di coinvolgimento:
• difetti di fabbricazione• manutenzione preventiva e correttiva• manomissione• uso clinico inappropriato• cadute accidentali• utilizzo di accessori non consoni• pulizia non idonea
• connessione impianti• temperatura, umidità, luce• interferenza EMI o RFI
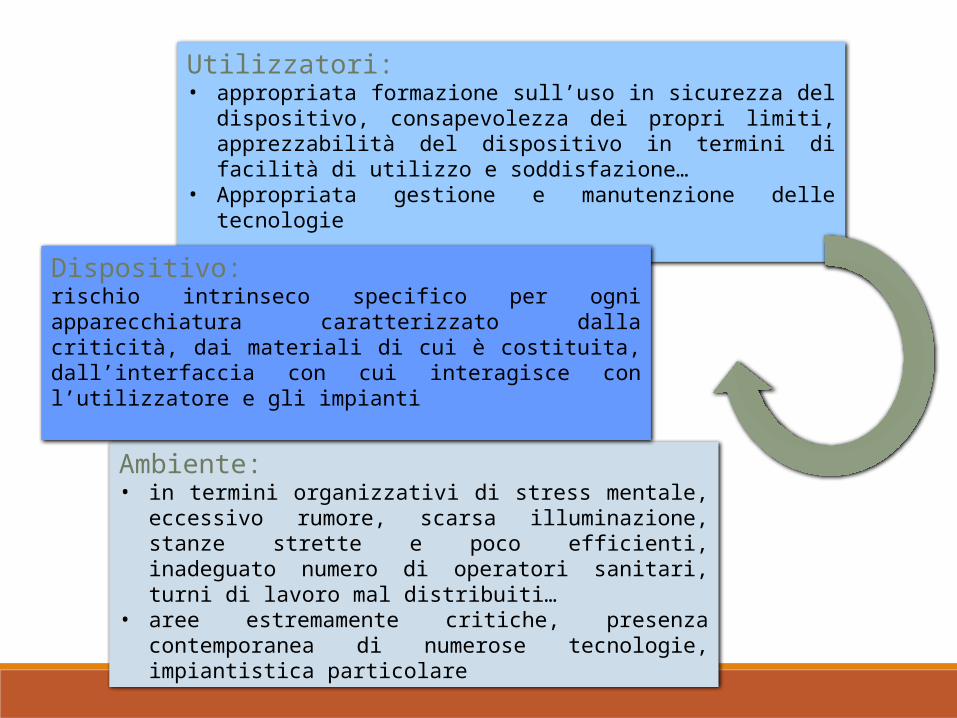
Utilizzatori: • appropriata formazione sull’uso in sicurezza del dispositivo,
consapevolezza dei propri limiti, apprezzabilità del dispositivo in termini di facilità di utilizzo e soddisfazione…
• Appropriata gestione e manutenzione delle tecnologie
Ambiente: • in termini organizzativi di stress mentale, eccessivo
rumore, scarsa illuminazione, stanze strette e poco efficienti, inadeguato numero di operatori sanitari, turni di lavoro mal distribuiti…
• aree estremamente critiche, presenza contemporanea di numerose tecnologie, impiantistica particolare
Dispositivo: rischio intrinseco specifico per ogni apparecchiatura caratterizzato dalla criticità, dai materiali di cui è costituita, dall’interfaccia con cui interagisce con l’utilizzatore e gli impianti
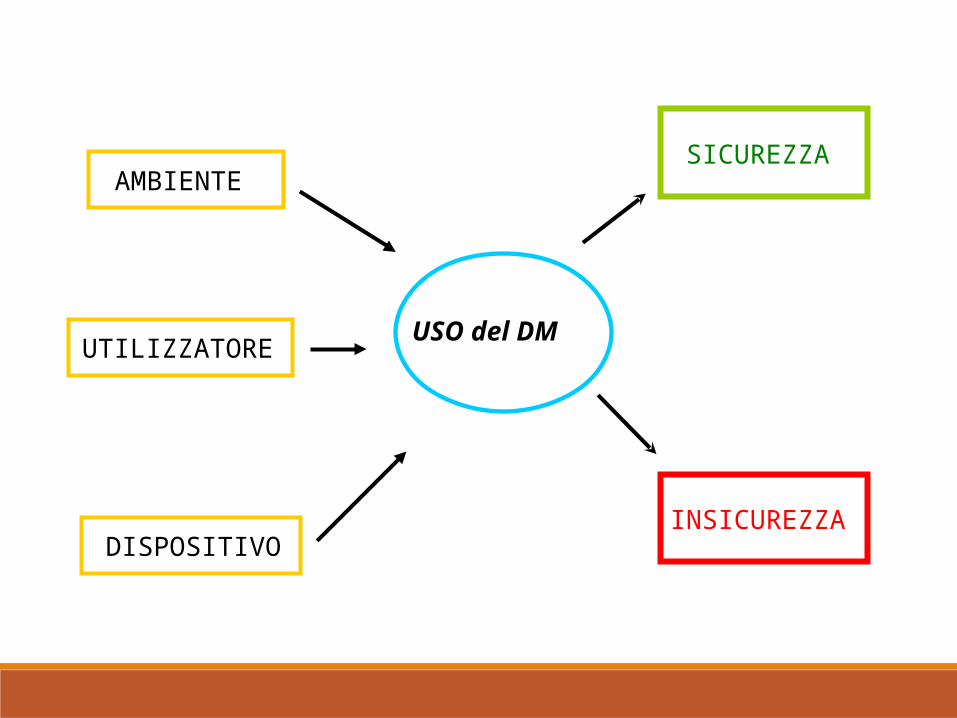
USO del DM
AMBIENTE
UTILIZZATORE
DISPOSITIVO
SICUREZZA
INSICUREZZA

Formazione
Training on the job
Mappatura dei rischi
Protocolli di sicurezza
Procedure manutenzione
preventiva
Azioni preventive e correttive da
implementare al fine di garantire
un uso sicuro, corretto ed
appropriato delle tecnologie
biomediche

…Cosa ci hanno insegnato SARS , Antrace, Mucca Pazza, Ebola, …
Evitare gli eccessi

Agire sui comportamenti
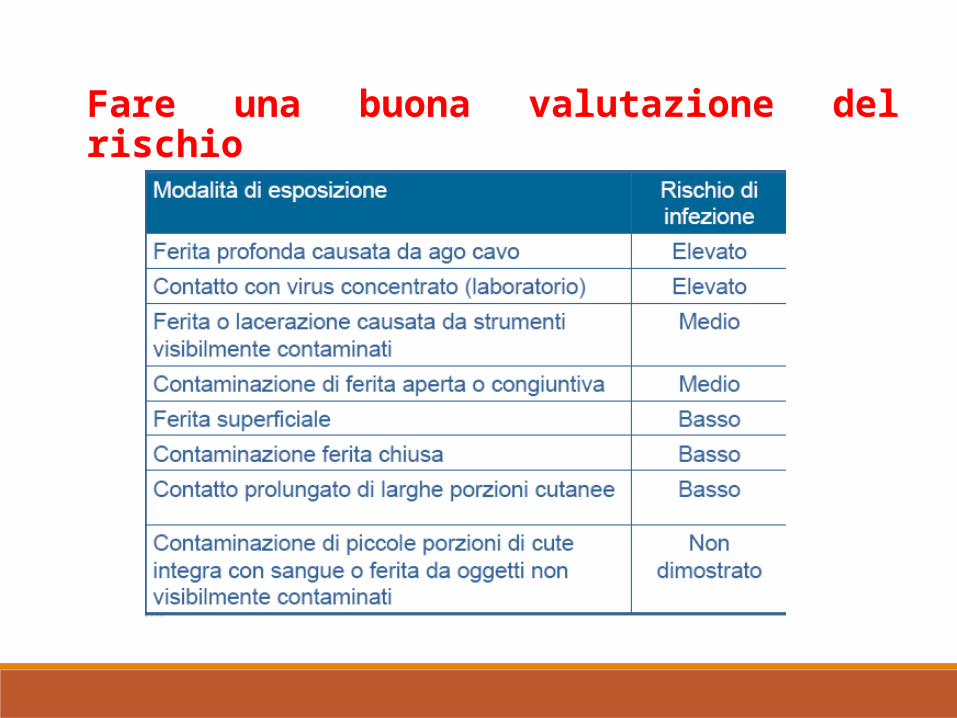
Fare una buona valutazione del rischio

Uso corretto dei Dispositivi di Protezione
