· coagulación y fibrinolítico y, por otro lado, el sangrado que se presenta en el contexto de...
TRANSCRIPT




Comité Editor
Comision DirectivaPresidente
Coordinadoras Asociadas
Secretaria
Tesorera
Vocales
Revisores de Cuenta
Diseño Gráfico
Imagen de Tapa
Diana AltunaAlicia N. BlancoCarlos FondevilaJuan Pablo FrontrothPaula HellerRoberto G. Pozner
Andrea Rossi
Paula HellerAna María Lauricella
Yolanda Adamczuk
Dolores Puente Mirta HepnerDavid VeronLaura Vrdoljak Roberto G. PoznerVerónica Cortés GuerrieriLourdes Herrera
Laura Sus
Mabel Nakkache

GRUPO COOPERATIVO ARGENTINO DE HEMOSTASIA Y TROMBOSIS
GUIA DE SANGRADO
2016DIAGNÓSTICO Y TRATAMIENTO


Estimados lectores,
Nos es grato presentar la tercer guía editada por el Grupo Cooperativo Argentino de Hemostasia y Trombosis (CAHT) titulada Guía de Sangrado: Diagnóstico y Tratamiento. En la misma se abarcan distintas patologías hemorrágicas, que involucran las plaquetas, el sistema de coagulación y fibrinolítico y, por otro lado, el sangrado que se presenta en el contexto de diversas condiciones clínicas y quirúrgicas en niños y adultos. El adecuado abordaje del paciente con hemorragia require del aporte conjunto del laboratorio y la clínica, aspectos que han sido abordados en forma integral en los distintos capítulos, los que reflejan la experta opinión de los autores responsables. Nuestro especial agradecimiento a los autores, todos ellos profesionales del área de la Hemostasia, quienes han colaborado con entusiasmo y dedicación para la confección de este manual. Esperamos que el contenido de esta guía sea de utilidad práctica para facilitar el enfoque y el manejo de los pacientes con sangrado en nuestro medio.
Comité Editor
PRÓLOGO


INDICE
1
2
3
4
5
6
7
8
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico.Carlos Fondevila y Juan Pablo Frontroth.
Valoración del riesgo de sangrado en el paciente anticoagulado.José M. Ceresetto.
Enfermedad de von Willebrand (VWD).Analía Sanchez Luceros, Adriana Woods y Gabriela Sciuccati.
Hemofilia.Daniela Neme, Ludmila Elhelou, Egle Honnorat, Miguel Tezanos Pinto y Alicia N. Blanco.
Desórdenes hemorrágicos raros del sistema de coagulación.Cristina Duboscq y Patricia Casais.
Inhibidores adquiridos de factores de la coagulación.Patricia Casais y Alicia N. Blanco.
Trombocitopenia inmune.Rosana Marta, Mónica Martinez y Daniel Fassi.
Desórdenes plaquetarios.Susana Meschengieser, Emilse Bermejo, Ana Glembotsky y María Esther Aris Cancela.
19
35
49
67
85
105
125
143
165
179
199
215
235
251
273
287
9
10
11
12
13
14
15
16
Trombocitopenia neonatal.Florencia Tisi Baña.
Coagulación intravascular diseminada.Alicia Grinspon y Dardo Riveros.
Coagulopatía en la enfermedad hepática y el trasplante hepático.Estela S. Viñuales y Marta E. Martinuzzo.
Sangrado en trauma.Carlos Fondevila y Sebastián Marún.
Sangrado quirúrgico.Hugo H. Ferro y Cecilia C. Colorio.
Menorragia.Beatriz E. Grand y Diana Altuna.
Hemorragia post-parto.Jorge D. Korin.
Complicaciones hemorrágicas del tratamiento antitrombótico.Alicia B. Vilaseca y Mirta Hepner.

GLOSARIO
A-RTPO: agonistas del receptor de trombopoyetina.a2-AP: a2-antiplasmina.AAS: ácido acetilsalicílico.ABC: asessment of blood consumption.AcTx: ácido tranexámico.ACCP: american college of chest physicians.ACOG: american college of obstetricians and gynecologist.ACV: accidente cerebrovascular.ADMP: altas dosis de metilprednisolona.ADAMTS-13: proteasa que degrada al factor von Willebrand.AICE: italian association of haemophilia centres.AINEs: antiinflamatorios no esteroides.AOD: anticoagulantes orales directos.AR: artritis reumatoidea.ARM: asistencia respiratoria mecánica.ARP: alta reactividad plaquetaria.AT: antitrombina.ATIII: antitrombina III.ATB: antibiótico.ATLS: advance trauma life support.AVK: antagonistas de vitamina K.aVWF: anti-factor von Willebrand.AVWS: síndrome de von Willebrand adquirido.
BAT: bleeding assessment tool.BRiSc: Papworth Bleeding Risk Score.BRP: baja reactividad plaquetaria.BSS: síndrome de Bernard-Soulier.
A
B
Guía de sangrado | 2016

2016 | Guía de sangrado
CAP: college of american pathologists.CAT: coagulopatía aguda del trauma.Cc: concentrado.CCP: concentrado de complejo protrombínico.CCPa: concentrado de complejo protrombínico activado.CCV: cirugía cardiovascular.CE: célula endotelial.CEC: circulación extracorpórea.CF: concentrado de factor.CFI: concentrado de fibrinógeno.CID: coagulación intravascular diseminada.Cl: clearence.CMV: citomegalovirus.COC: anticonceptivos orales combinados.Crio: crioprecipitados.CVWF: concentrado de factor von Willebrand.
DAMPs: patrones moleculares asociados a daño.DB: déficit de base.DBT: diabetes.DD: dímero D.DDAVP: desmopresina.DIU: dispositivo intrauterino.DRVVT: tiempo de veneno de víbora Russell diluido.
E: especificidad.EACH2: european acquired haemophilia registry.EPCR: receptor endotelial de la proteína C.ESA: european society of anaesthesiology.
C
D
E

Guía de sangrado | 2016
ETEV: enfermedad tromboembólica venosa.ETS: enfermedades de transmisión sexual.EV: endovenosa.
F: factor de coagulación.F1+2: fragmento 1+2 de la protrombina.FA: fibrilación auricular.FAST: focus assessment with sonography in trauma.FBG: fibrinógeno.FDA: food and drug administration.FHA: falla hepática aguda.FP AyB: fibrinopéptidos A y B.FR: factor(es) de riesgo.FT: factor tisular.
GCS: Glasgow coma scale.GGCX: g-glutamilcarboxilasa.GI: gastrointestinal.GP: glicoproteínas.GR: glóbulos rojos.GRDs: glóbulos rojos desplasmatizados.
HBPM: heparinas de bajo peso molecular.HES: hidroxietilamidón.HIC: hemorragia intracraneal.HMGB1: high mobility group box 1.HNF: heparina no fraccionada.HPA: antígenos plaquetarios humanos.HTA: hipertensión arterial.
F
G
H

2016 | Guía de sangrado
IAM: infarto agudo de miocardio.ICAM: molécula de adhesión celular intercelular.Ig: inmunoglobulina.IL: inhibidorlúpico.IL-: interleuquina.ISS: injury severity score.ISTH: international society on thrombosis and haemostasis.IT: inmunotolerancia.
LES: lupus eritematoso sistémico.LMAN1: lectin mannose-binding 1.LTA: agregación por transmisión de luz.
MBP: muy bajo peso.MCF: máxima firmeza del coágulo.MCFD2: multiple coagulation factor deficiency protein 2. ME: mepacrina.MFs: monómero de fibrina soluble.ML: lisis máxima.
NAO: nuevos anticoagulantes orales.NETs: trampas extracelulares de neutrófilos.NGS: next generation sequencing.NICE: national institute of clinical excellence.
OBRI: outpatient bleeding risk index.
P+N: mezcla de plasma del paciente y plasma normal.PAI-1: inhibidor del activador del plasminógeno tipo 1.
I
L
M
N
O
P

Guía de sangrado | 2016
PAMPs: patrones moleculares asociados a patógenos.PAP: plasmina-antiplasmina.PC: proteína C.PCa: proteína C activada.PCD: prueba de Coombs directa.PCR: proteína C reactiva.PDFs: productos de degradación de fibrinógeno/fibrina.PFC: plasma fresco congelado.POC: point of care.PS: proteína S.PTI: trombocitopenia inmune.
Qx: cirugía.
RC: remisión completa.RCDH; resucitación con control del daño hemostático.rFVIIa: factor VII activo recombinante.RIN: razón internacional normatizada.RNPT: recién nacidos pretérmino.RNT: recién nacidos de término.ROIs: intermediarios reactivos del oxígeno.ROTEM: tromboelastografía rotacional.RR: riesgo relativo.
S: sensibilidad.SAF: síndrome antifosfolípido.SIRS: síndrome de respuesta inflamatoria sistémica.SISET: società italiana per lo studiodell’ emostasi e della trombosi.
Q
R
S

2016 | Guía de sangrado
SNC: sistema nervioso central.SPD: enfermedad de pool de depósito.SPO: sangrado post-operatorio.
TAC: tomografía axial computarizada.TAS: tensión arterial sistólica.TASH: trauma-associated severe hemorrhage.TAT: trombina-antitrombina.TAFI: inhibidor de la fibrinolisis activado por trombina.TC: tiempo de coagulación.TCPH: trasplante de células progenitoras hematopoyéticas.TEG: tromboelastografía.TEP: tromboembolismo pulmonar.TER: tiempo en rango.TEV: tromboembolismo venoso.TFC: tiempo de formación del coáguloTFNA: trombocitopenia feto/neonatal alo-inmune.TFPI: inhibidor de la vía del factor tisular.TG: tromboastenia de Glanzmann.TH: trombocitopenias hereditarias.TLC: tiempo de lisis del coágulo.TLE: tiempo de lisis de euglobulinas.Tm: transfusión masiva.TM: trombomodulina.TNFa: Factor de necrosis tumoral a.TP: tiempo de protrombina.tPA: activador tisular del plasminógeno.TPH: trombocitopatías hereditarias.TR: tiempo de reptilase.
T

Guía de sangrado | 2016
TSP: trombospondina.TT: tiempo de trombina.TTPA: tiempo de tromboplastina parcial activada.Tx: transplante.
UB: unidad Bethesda.UI: unidades internacionales.UKHCDO: united kingdom haemophilia centre doctors’ organization.
VCAM: molécula de adhesión celular vascular.VEB: virus de Epstein Barr.VHB: virus de hepatitis B.VHC: virus de la hepatitis C.VIH: virus de inmunodeficiencia humana.VKCFD: déficit conjunto congénito de factores vitamina K dependientes.VKOR: vitamina K epóxido reductasa.VLDL: lipoproteína de muy baja densidad.VO: vía oral.VPM: volumen plaquetario medio.VPN: valor predictivo negativo.VPP: valor predictivo positivo.VWD: enfermedad de von Willebrand.VWF: factor von Willebrand.VWF:Ag: ensayo antigénico del VWF.VWF:CB: ensayo de unión del VWF al colágeno.VWF:RCo: ensayo de cofactor de ristocetina del VWF.VWFpp: propéptido del VWF.
U
V

18

19
Introducción
Hasta 25% de los sujetos con historia de sangrado no tendrá una coagulopatía. El costo monetario y humano de realizar estudios de coagulación innecesarios es enorme. Por otro lado, no diagnosticar un defecto puede tener consecuencias serias. La hemorragia es una complicación grave y aún fatal, y que afecta los resultados tanto de los procedimientos invasivos y cirugías cuanto de los tratamientos antitrombóticos. Disponer de una herramienta que permita predecir el riesgo de sangrado, anticipando cuál paciente va a sangrar, permitiría tomar medidas de prevención del sangrado procedimental así como buscar alternativas terapéuticas distintas de la anticoagulación. En otro capítulo se aborda el tema de la predicción del riesgo de sangrado asociado al uso de anticoagulantes. El resto del presente capítulo se referirá a las herramientas clínicas y de laboratorio que permiten confirmar la presencia de una falla de la hemostasia en una persona con síntomas sugestivos así como anticipar el riesgo de sangrado ante una cirugía o un procedimiento invasivo en sujetos sin defecto conocido.
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágicoCarlos Fondevila1 y Juan Pablo Frontroth2. 1 Médico Hematólogo. Clínica Bazterrica de Buenos Aires. Prof. Adjunto de M II- Hematología, Facultad de Medicina, Universidad del Salvador.2 Bioquímico Especialista en Hemostasia. Laboratorio de Hemostasia y Trombosis. Hospital de Pediatría Prof. Dr. Juan P. Garrahan.
1

20
Papel de la clínica
El sangrado es un motivo de consulta habitual. El 25% de los hombres y el 46% de las mujeres normales refieren, al menos, un síntoma de sangrado. El sangrado es un síntoma que conlleva una alta carga subjetiva por lo cual el hematólogo debe saber discernir cuándo un sangrado es esperable o anormal a fin de evitar estudios innecesarios. Muchos sangrados son triviales, esto es que no interfieren con la vida diaria o actividad social. Se consideran menores aquellos sangrados con poco impacto sobre la actividad cotidiana (por ej. la mujer con menstruación profusa que pierde algún día laboral; una epistaxis o un sangrado oral que duran más de 10 min; sangrado post-exodoncia que obliga a regresar al consultorio o a suturar). Se considera sangrado mayor aquél que pone en peligro el órgano o la función, o amenaza la vida o requiere transfusión. Algunos sangrados son poco orientadores como en caso de hematuria o hematoquezia. Sangrado de encías, equimosis o epistaxis fueron referidos por 13, 25 y 19%, de sujetos sanos, respectivamente. Excepto en casos extremos como epistaxis que requiere transfusión, sangrado post-mordedura de lengua que ocasiona un hematoma sofocante, estos síntomas generalmente no son útiles como indicadores de un trastorno de la hemostasia.Los trastornos leves de la coagulación se caracterizan por sangrado de tipo mucocutáneo. El patrón del sangrado no es característico ya que no hay síntomas de sangrado que sean sugestivos de enfermedad de von Willebrand (VWD), trombocitopatía o deficiencia leve de un factor de la cascada. El número y la severidad de los episodios de sangrado pueden resultar difíciles de diferenciar de aquellos que, ocasionalmente, refieren las personas sanas. En estos casos el laboratorio podrá ser determinante para identificar/excluir la presencia de un trastorno de la coagulación aunque las pruebas globales resultan inespecíficas y de baja sensibilidad. Finalmente, deberá tenerse presente que los trastornos leves de la coagulación pueden no mostrar síntomas de sangrado en la vida diaria, los que sólo aparecerán luego de un desafío hemostático.Una historia de sangrado mayor espontáneo es muy infrecuente en la población general en ausencia de organicidad (patología gastrointestinal o malformación anatómica o vascular cerebral) y debe tomarse como altamente específica de un trastorno grave de la coagulación, haciendo inexcusable la evaluación extensiva del laboratorio, que incluirá el dosaje de factores individuales VIII, IX, XI o VII (los más prevalentes).El sangrado post-trauma o cirugía (aparte del sangrado post-parto), sobre
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico

21
todo en presencia de una historia familiar de sangrado, es muy sugerente de una alteración de la hemostasia. También es un indicador de un trastorno de la cascada una historia de sangrado frecuente, severo o reiterado así como la presencia de hemartrosis, sangrado muscular o en cavidades y el antecedente de sangrado intracraneal o con la caída del cordón umbilical.La presencia de síntomas de sangrado frecuentes/severos a una edad temprana son sugestivos de un trastorno de la coagulación más severo.La Sociedad Internacional de Hemostasia y Trombosis (ISTH) ha establecido criterios sugestivos de una historia hemorrágica significativa: sangrado único que requirió transfusión, sangrado por un sitio único en 3 ocasiones diferentes o sangrado espontáneo o provocado por 2 o más sitios no relacionados. Probablemente nada resulte más tranquilizador para el cirujano que entrar al quirófano teniendo certeza de que su paciente no tiene una tendencia hemorrágica y que no va a presentar un sangrado anormal durante el procedimiento. Ni la anamnesis más exhaustiva ni ninguna prueba de laboratorio brindan esa certeza.El 25% de las cirugías sangran. El 5% sangra masivamente, en especial cirugía cardiovascular (CCV), cirugía de trasplante, cirugía del trauma y obstetricia. Existen factores de riesgo vinculados con la cirugía, tales como el tipo de procedimiento (urgencia, reoperación, cirugía oncológica), el sitio operatorio (tejidos con excesiva fibrinolisis, sitios incomprensibles) o la posibilidad de trauma vascular debido a la presencia de vasos anómalos o a un accidente. Finalmente, la habilidad del cirujano es un punto que no debe descuidarse y los casos a priori difíciles deben ser asignados al cirujano más capacitado del equipo.Algunas de estas causas de sangrado orgánico pueden anticiparse, permitiendo tomar recaudos previos a la operación. Como por ej, la colocación de un catéter balón que permita inflarlo en el momento quirúrgico y trabajar en un lecho exangüe.Existen causas no quirúrgicas que anticipan una mayor probabilidad de sangrado: la edad avanzada, la anemia o la presencia de infección o la sepsis son condiciones favorecedoras. El sangrado por falla hemostática, puede deberse a causas previas (como una diátesis hereditaria, presencia de hepatopatía, falla renal o consumo de antitrombóticos). En ocasiones el defecto no puede anticiparse como en caso de coagulación intravascular diseminada (CID), hiperfibrinolisis o trombocitopatía adquiridos intracirugía.Una publicación reciente, identificó en 24 estudios los factores de riesgo para
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1

22
sangrado quirúrgico. Los principales fueron: edad, presencia de enfermedad renal o cardiovascular e historia de sangrado. Las alteraciones del labo-ratorio ocuparon el último lugar. La guía ESA 2013 (European Society of Anaesthesiology) recomienda, antes de realizar un procedimiento invasivo, la utilización de un cuestionario estructurado en relación a la historia personal/familiar de sangrado y al consumo de fármacos en lugar del laboratorio convencional de coagulación (recomendación 1C).
El interrogatorio debe rescatar la historia personal de sangrado espontáneo o ante desafíos hemostáticos previos (cirugías, exodoncias, partos). La presencia de enfermedad hepática o renal, la historia de anemia o cáncer. El consumo de fármacos (incluyendo drogas con efecto potencial sobre la hemostasia como el ácido valproico o los inhibidores de la recaptación de serotonina -SSRI-) o productos de venta libre o herbáceos con efecto potencial sobre la coagulación (ajo, ginkgo, ginseng, cúrcuma). Un sangrado de tipo mucocutáneo caracteriza los defectos de la hemostasia primaria. Deberá prestarse atención a la semiología del sangrado. Un sangrado tardío, que aparece varios días después del desafío, sugiere defectos de la formación de fibrina o hiperfibrinolisis. Un sangrado reiterado pero limitado a un sitio (por ej. epistaxis) hace necesario descartar una causa local. El interrogatorio negativo no descarta la presencia de un defecto menor (VWD leve, defectos de secreción plaquetaria o deficiencia leve de algún factor), en especial si la persona no ha tenido desafíos hemostáticos. Finalmente, deberá interrogarse la historia de sangrado en familiares directos. Al menos en trastornos dominantes, la historia familiar suele ser positiva. La información obtenida tiene un valor limitado: hasta la mitad de los normales tendrá familiares con historia de sangrado.
Nada es más confiable que un buen interrogatorio para anticipar el riesgo hemorrágico.
La sensibilidad de los criterios clínicos para pesquisar un trastorno leve de la coagulación no supera el 40-50%.
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico

23
En ocasiones, el examen físico podrá ofrecer claves que faciliten el diagnóstico o, al menos, la sospecha de un trastorno de la hemostasia. La presencia de telangiectasias en labios, nariz o pulpejos caracteriza la telangiectasia hemorrágica hereditaria. La presencia de hiperlaxitud cutánea, hipermovilidad articular o cicatrices anormalmente amplias, de aspecto atrófico, así como alteraciones en el cristalino o válvulas cardíacas, orientan hacia patología hereditaria del colágeno. Anormalidades morfo-lógicas (faciales u otras) o la presencia de albinismo o eccema sugieren la coexistencia de una coagulopatía junto a un síndrome genético (Noonan, Hermansky-Pudlak, Wiskott-Aldrich).
Scores de sangrado
Casi la mitad de las mujeres refieren sangrado menstrual abundante al menos una vez durante su vida fértil. El 20% de la población refiere epistaxis (y hasta 1/3 de los niños). Equimosis fáciles y sangrado prolongado post-exodoncia son motivo frecuente de consulta. Al menos un síntoma de sangrado es referido por 40-50% de los hombres y 70% de las mujeres. El paciente sangrador es aquél que sangra con frecuencia y lo hace abundantemente. Por este motivo, desde los 2000´s se han confeccionado distintos scores o cuestionarios estructurados y guiados para recoger datos en relación a la existencia de patología asociada o manifestaciones previas de sangrado
Los síntomas de sangrado que mejor identifican a un sangrado genuino son: menorragia, sangrado luego de cirugía, extracción dentaria o trauma menor y sangrado post-parto. También el sangrado relacionado con el uso de aspirina.
El valor de un interrogatorio negativo es mayor en un paciente o familia con historia de múltiples desafíos hemostáticos.
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1

24
espontáneo o ante desafíos. La presencia y la severidad de los síntomas de sangrado son tabulados, dando como resultado un índice o probabilidad. El objetivo es determinar la probabilidad de sangrado o la posibilidad de que una alteración hemostática se encuentre presente o, por el contrario, establecer con cierto nivel de certeza que no lo está. En otro capítulo de esta guía se analizan diferentes scores que identifican los factores de riesgo de sangrado en anticoagulados, útiles para establecer el costo-eficacia del tratamiento con dicumarínicos. También se han diseñado scores para el uso preoperatorio en un intento de predecir el riesgo de sangrado quirúrgico. Con la probable excepción del Papworth bleeding risk score (BRiSc) para CCV, su utilidad está poco validada. Finalmente, se han publicado distintos scores para aplicar en el diagnóstico de enfermedades hemorrágicas específicas. En VWD se ha utilizado el score de Vicenza (2005) y el MCMDM-1 VWD (2006). En ocasiones, un score elevado >10 se utilizó como predictor de la necesidad del uso de desmopresina o de concentrados. A fin de abreviar el tiempo de administración del cuestionario, se ha publicado una forma condensada (2008), la que también fue utilizada para la evaluación de otros trastornos hemorrágicos. También se ha publicado una adaptación para uso pediátrico (2009). En 2010 el Scientific and Standardization Committee for von Willebrand Factor (VWF) desarrolló el score ISTH-BAT, el cual es el actualmente recomendado para el diagnóstico de los trastornos hemorrágicos. Sin embargo, y si bien un score anormal ha mostrado buena correlación con la presencia de VWD, la sensibilidad del score ISTH-BAT es baja para identificar trastornos plaquetarios y, en especial, las deficiencias leves de factores. Algunos trabajos detallan los diferentes scores y discuten su utilidad, ya sea aislada o en combinación con los resultados del laboratorio, para identificar la presencia de trastornos de la coagulación (en especial, los trastornos leves). En trombocitopenia inmune se han publicado dos scores. El ITP bleeding score (Khellaf M, 2005) categorizó la severidad del sangrado de manera que aquellos casos con sangrado más grave (score ≥8) fueron tratados inicialmente con esteroides más inmunoglobulina endovenosa. El score SMOG o ITP BAT (Rodeghiero F, 2013) sistematiza la descripción del fenotipo de sangrado así como la respuesta al tratamiento. Ambos requieren validación clínica adicional.
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico

25
La mayor utilidad de los scores radica en su alto valor predictivo negativo (VPN), que excluye la necesidad de estudios adicionales. Sin embargo, en pacientes jóvenes que no han tenido desafíos hemostáticos, deberá considerarse realizar una evaluación del laboratorio.
En pacientes con score de sangrado alto y laboratorio de rutina normal, deberá considerarse: 1. repetir las pruebas de rutina2. realizar pruebas adicionales que incluyan: dosaje individual de factores, VWF, FXIII y fibrinolisis (incluyendo a-2AP).
Se debe tener presente que, a pesar de todas nuestras precauciones, incluyendo un interroga- torio y score negativos, se producirán sangrados no-anticipables: ciertos pacientes, sometidos o no a desafíos hemostáticos, parecen empeñados en sangrar a pesar de todos los pronósticos: son los sangrados de causa desconocida.
En cirugía será inevitable y hasta tolerable un cierto grado de sangrado (hematuria transitoria siguiendo una resección prostática transuretral, sangrado de hasta 500 mL en un parto vaginal o en el drenaje post-cirugía de cadera).
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1

26
Lugar del laboratorio
Los trastornos graves de la coagulación, a pesar de ser infrecuentes, resultan sencillos de identificar con el laboratorio habitual. En cambio, los trastornos leves, que son mucho más prevalentes, suelen presentar dificultades para su identificación por el laboratorio, necesitando estudios reiterados.Del laboratorio de hemostasia se espera tanto el diagnóstico (¿por qué se produjo este sangrado?) cuanto el pronóstico (¿cuál es el riesgo de que vuelva a sangrar?). El laboratorio convencional fue utilizado con el objeto de confirmar que un sangrado tuvo una causa coagulopática, para identificar la deficiencia individual de un factor o plaquetas y para guiar el tratamiento sustitutivo. Tradicionalmente, los defectos de la función plaquetaria y de la fibrinolisis han quedado afuera de las pruebas de rutina. Del laboratorio también se espera que identifique a aquellos sujetos con riesgo aumentado de sangrado ante un procedimiento.Muchas publicaciones han analizado el lugar del laboratorio para estimar el riesgo de sangrado, sobre todo en procedimientos invasivos y cirugías mayores. Sin embargo, la evidencia publicada hace dudar de que las pruebas de laboratorio resulten útiles en este sentido. Esto se aplica tanto a los bien establecidos ensayos de coagulación como a las más nuevas pruebas de evaluación global de la hemostasia. La realidad muestra que un sangrado operatorio excesivo o exsanguinante se debe mucho más a menudo a una causa quirúrgica que a una falla hemostática. La guía de la ESA ya mencionada así como el British Committee for Standards in Haematology (BCSH) no recomiendan, en ausencia de una historia de hemorragia, el uso indiscriminado de los estudios de laboratorio, previo a cirugía o procedimientos invasivos, en la pesquisa del riesgo perioperatorio de sangrado (recomendaciones 1C y B III respectivamente). Si la historia de sangrado es negativa, no está indicada ninguna prueba de laboratorio (recomendación C IV). En presencia de historia de sangrado o de evidencia clínica (por ej. enfermedad hepática), deberá realizarse un estudio exhaustivo y guiado por los hallazgos (recomendación C IV).Por otro lado, hasta 1/3 de los sujetos con historia de sangrado negativa será portador de una coagulopatía. Esto es más evidente en sujetos que no tuvieron desafíos hemostáticos importantes previos. En estos casos, el laboratorio podría descubrir algún trastorno leve. Esta conducta resultará particularmente útil si el paciente será sometido a procedimientos de alta
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico

27
demanda hemostática (CCV con bomba, cirugía de aorta, endocarditis o a procedimientos re-do) o a cirugías de alto riesgo ante sangrado (sitios cerrados o incompresibles, SNC, ojo, médula espinal, biopsias cerradas). Tosetto y col. (2011) mostaron, en un estudio prospectivo, que la combinación de score de sangrado negativo y tiempo de tromboplastina parcial activada (TTPA) normal excluyó casi por completo la posibilidad de un trastorno de la coagulación.Las principales pruebas para la evaluación de la hemostasia son el TTPA y el tiempo de protrombina (TP). Estos miden el tiempo de coagulación en respuesta a reactivos exógenos, cada cual comprometiendo sólo a algunos de los constituyentes del complejo sistema de la coagulación. Por fuera quedan los mecanismos fibrinolíticos, de entrecruzamiento de fibrina como también los componentes sanguíneos y la influencia del sistema vascular. Además, las pruebas de rutina tienen poca sensibilidad para pesquisar trastornos leves. Así, no son el reflejo absoluto de la respuesta hemostática y, si la sospecha es alta, deberá recurrirse a un laboratorio más especializado (dosaje de factores individuales, medición de VWF, agregación y liberación plaquetarias, parámetros de fibrinolisis). La guía SISET (Società Italiana per lo Studio dell’ Emostasi e della Trombosi) sólo recomienda realizar la determinación de TP, TTPA y plaquetas. En cambio, desaconseja la medición de tiempo de sangría (TS), fibrinógeno, pruebas de agregación y tromboelastografía (TEG). En el caso de las exodoncias, sugiere no realizar prueba alguna.El ensayo de TTPA se lleva a cabo sobre plasma recalcificado con un reactivo que incluye fosfolípidos y un activador. Estos últimos pueden ser de diferentes orígenes dando una alta variabilidad en la sensibilidad del ensayo en respuesta a la deficiencia de factores e inhibidores. Idealmente, cada laboratorio debería determinar los rangos en el cual la prueba es sensible a la deficiencia de factores. Pero al mismo tiempo el sistema para detectar inhibidores no necesariamente es el mismo. Además de los inconvenientes en la fase pre-analítica y analítica, la situación se hace aún más compleja al tener en cuenta los factores epigenéticos como el estrés emocional o la respuesta física que pueden elevar, por ej, los niveles de FVIII y arrojar resultados normales.El ensayo de TP evalúa la integridad del sistema extrínseco y de la vía final común. Se realiza sobre un plasma recalcificado en presencia de tromboplastina. Aunque ha sido altamente estandarizado para el monitoreo de la terapéutica anticoagulante con antagonistas de la vitamina K
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1

28
(dicumarínicos), también es un componente común en los ensayos de pesquisa para predecir el sangrado previo a un procedimiento invasivo o cirugía mayor. Lamentablemente, los mismos inconvenientes mencionados para el TTPA se presentan en el TP. La principal causa de variabilidad de esta prueba es el origen de la tromboplastina utilizada.Los rangos de referencia definidos como normales para ambas pruebas se calculan habitualmente en base a dos desvíos estándar alrededor de una media calculada en un grupo de individuos normales. Al menos 2,5% de los plasmas analizados podrían reflejar falsos negativos o falsos positivos, pero sin significancia clínica. Un valor normal de TP o TTPA no excluye una deficiencia leve de algún factor. TP y TTPA son más sensibles a deficiencias múltiples que a deficiencias individuales de factores de las vías extrínseca, intrínseca o final.¿Por qué las diferentes guías no asignan un lugar relevante al laboratorio de hemostasia como forma de anticipar el riesgo de sangrado operatorio? Ya fue mencionado que el laboratorio convencional mide una porción pequeña de la hemostasia. En conjunto no más del 5% inicial de la trombina generada. Un aumento de algún factor (por ej. FVIII reactante de fase) puede enmascarar una deficiencia leve de otro factor. Esto es más evidente para el TTPA. En sujetos asintomáticos, la especificidad y el valor predictivo positivo (VPP) de un resultado anormal son bajas. Hasta 1/3 de la variabilidad de estas pruebas no se relaciona con el contenido de factores. Prolongaciones leves de TP o TTPA no correlacionan con la aparición de sangrado y frecuentemente desaparecen en un nuevo estudio, esta situación se da con mayor frecuencia en pediatría. En más de 1.600 niños sin antecedentes estudiados previo a amigdalectomía, se encontraron alteraciones en el coagulograma en el 2,3%. Sin embargo, en la repetición 7-10 días después, el 58% mostró resultados normales. Además de las variables pre-analíticas, es frecuente la presencia de inhibidores de tipo lúpico (en niños con patología amigdalina o adenoidea). En un estudio realizado en niños previo a una tonsilectomía, el 15-29% mostró prolongación del TP o TTPA: sin embargo, el sangrado operatorio inmediato sólo ocurrió en 3-8% de los casos. Una publicación que resumió datos de 25 estudios que analizaron pacientes con TP <70% / RIN >1,2 o >1,5 previo a un procedimiento, no encontró diferencias en la incidencia de sangrado procedimental. Esta conclusión no debe interpretarse como que un TP <70% o un RIN >1,5 sea seguro en todos ya que, lamentablemente, como se
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico

29
analizaron los datos en forma discreta (sí/no) el grupo de TP <70% contenía tanto pacientes con 69% como con <10%.Finalmente, todas las pruebas de coagulación que miden tanto la hemostasia primaria y secundaria cuanto la fibrinolisis, arrojan resultados normales en los trastornos de la hemostasia vascular, como en las enfermedades hereditarias del tejido conectivo, la enfermedad de Rendu Osler, la amiloidosis o el escorbuto. Ocasionalmente, en la enfermedad de Ehler Danlos el TS y los estudios de función plaquetaria pueden dar alterados.
¿Existe un valor de laboratorio lo suficientemente anormal como para predecir sangrado? La Tabla 1 muestra los valores que mejor correlacionan con la ocurrencia de sangrado clínico.
Las guías tradicionalmente han tomado un valor >1,5xN del TP o TTPA, o RIN <1,8 como indicadores de la necesidad de soporte hemostático. Este valor se corresponde con un nivel aproximado al 30% de factores individuales. Los valores de TP, TTPA, fibrinógeno y plaquetas que tradicionalmente se consideraron hemostáticos corresponden a deficiencias de un factor único, en condiciones de normalidad de otros factores, temperatura, pH y disponibilidad de calcio.La Tabla 2 muestra las correlaciones de un TP o TTPA prolongados. El TP tiene una sensibilidad (S) de 84% y una especificidad (E) de 88%. El TTPA tiene una S de 50% y una E de 100%. Un TTPA x1,5 se corresponde con ≈60% del nivel global de factores+fibrinógeno o ≈40% de factores vitamina K dependientes o ≈20% de factores individuales. Un RIN 1,5 se corresponde a un ≈50% de nivel global de factores+fibrinógeno o ≈40% de factores
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1
TP
RIN
TTPA
Fibrinógeno
Factores
Plaquetas
>1,6-1,8
>1,7
>1,5-2,0
<100 (<200 en CCV y obstetricia)
<20%
<50x109/L (<100x109/L en neurocirugía, trauma y obstetricia)
Tabla 1. Valores del laboratorio convencional que muestran mejor correlación con sangrado operatorio

30
vitamina K dependientes o ≈20% de factores individuales de vía extrínseca. Un RIN ≥1,5 tiene un VPP de 90% para deficiencia de un factor TP-sensible. Mientras que un RIN <1,5 tiene un VPN de 80% para no-deficiencia.
Otros métodos de evaluación global de la hemostasia
Tiempo de sangría. Utilizado desde hace muchos años, esta prueba se ha realizado en variadas formas; su reproducibilidad fue mejorada como resultado de la estandarización. No obstante, su sensibilidad para identificar VWD o trastornos plaquetarios es baja (está prolongado en sólo 40-50%). Sin embargo, un TS prolongado puede ser la única anomalía en hasta en 1/5 personas con sangrado sin causa aparente. Una extensa evaluación demostró que el TS es un pobre predictor de sangrado. Sólo resulta S si se encuentra prolongado 2 veces. Las guías actuales no incluyen al TS dentro de las pruebas a realizar. Se ha sugerido reemplazarlo por otros estudios de la función plaquetaria automatizados, como el PFA-100®.Dispositivos ex vivo para la evaluación de la función plaquetaria. Estos dispositivos han probado ser de utilidad en ciertas situaciones particulares. El PFA-100® fue diseñado para evaluar la hemostasia primaria en un entorno
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico
Tabla 2. Correlación de la prolongación de TP o TTPA con la deficiencia de factores de vía extrínseca (VE) o intrínseca (VI) así como el valor predictivo positivo (VPP) o negativo (VPN)
<1,5
≥1,5
Prolongación x N
40% K dep
20% factores individuales
de VE
40% K dep
20% factores individuales
de VI
Factores
TP
TTPA
TP
TTPA
Prueba
90%
100%
VPP (% CON deficiencia de algún factor)
80%(20% tendrá deficiencia)
60%(40% tendrá deficiencia)
VPN (% SIN deficiencia de algún factor)

31
de flujo de alto shear rate, en donde la concentración de VWF probó ser el determinante de su resultado, dejando baches en la S a ciertas disfunciones plaquetarias y dando resultados variables respecto a la acción de agentes antiagregantes. La S para identificar las formas más severas de VWD es de 60-70%, resultando aún menor (en el orden del 25-50%) para trastornos plaquetarios que involucran el almacenamiento o la secreción del contenido de los gránulos. Un resultado normal de PFA-100® no excluye aquellos casos más leves. Por otra parte, un resultado anormal deberá complementarse con estudios de agregación, liberación de ATP y la determinación de glicoproteínas (GP) de membrana. Tromboelastografía (TEG). Aunque es una técnica que se conoce hace 60 años, los desarrollos actuales de miniaturización y análisis por computadora han relanzado el interés por este método. Mide los cambios viscoelásticos en una muestra de sangre entera provocados por la polimerización de fibrina, con la participación de las plaquetas y la acción posterior de la fibrinolisis. La utilidad de este método para identificar trastornos de la coagulación no está clara aún. La evidencia actual muestra su utilidad en el manejo del sangrado crítico, dentro del quirófano o en el área de emergencia, así como para optimizar la transfusión de componentes sanguíneos en áreas específicas (transplante hepático o CCV). En contraposición al laboratorio convencional que se utiliza para gatillar la reposición de plasma fresco congelado o crioprecipitados, la información brindada por la TEG permite una política transfusional dirigida y el uso de concentrados específicos. La aplicación de TEG en la práctica rutinaria para la predicción del riesgo de sangrado parece promisoria aunque aguarda validación. El VPP de un resultado anormal es de apenas 40% (aunque muy superior al de las pruebas convencionales). El método resulta mucho más útil durante un sangrado para identificar la causa: el VPN de un TEG normal es del 80%. En el 97% de los sangrados operatorios con TEG normal se identificó una causa quirúrgica.
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1

32
Predicción del riesgo de sangrado operatorio. Conclusiones:
• No hay umbral de laboratorio que identifique todo paciente con riesgo de sangrar.
• Bajo VPP de alteraciones leves <1,5xN (la mayoría no sangra en exceso).• Posibilidad de sangrado a pesar de valores “suficientes”.• No se justifica evaluar la hemostasia: - En adultos o niños sin historia personal o familiar de sangrado a pesar de desafíos hemostáticos que serán sometidos a cirugía general u otorrinolaringológica.• Se debe evaluar la hemostasia: - En presencia de historia de sangrado personal o familiar. - En personas sin historia de desafíos hemostáticos. - En situación clínica de alteración potencial de la coagulación. - En cirugías de alta exigencia. • Beneficio aún incierto de TEG y pruebas que midan la generación global
de trombina.
Conflictos de interés
Los autores no presentan conflictos de interés.
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico

33
Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico | 1
Referencias
Boender J, Kruipp MJHA, Leebek FWG. A diagnostic approach to mild bleeding disorders. J Thromb Haemostas 2016. doi: 10.1111/jth.13368.
Rodeghiero F, Michel M, Gernsheimer Tet al. Standardization of bleeding assessment in immune thrombocytopenia: Report from the International Working Group. Blood 2013; 121: 2596-2606.
Hunt BJ, Allard S, Keeling D, Norfolk D, Stanworth SJ, Pendry K and on behalf of the British Committee for Standards in Haematology. A practical guideline for the haematological management of major haemorrhage. Br J Haematol 2015; 170: 788-803.
Levy JH, Szlam F, Wolberg A, Winkler A. Clinical Use of the Activated Partial Thromboplastin Time and Prothrombin Time for Screening.A Review of the Literature and Current Guidelines for Testing. Clin Lab Med 2014; 34: 453-477.
Spahn DR, Bouillon B, Cerny V et al. Management of bleeding and coagulopathyfollowing major trauma: an updated European guideline. Critical Care 2013; 17: R76.
Kozek-Langenecker SA, ArashAfshari A, Albaladejo P et al. Management of severe perioperative bleeding. Guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol 2013; 30: 270-382.
Tosetto A, Castaman G, Rodeghiero F. Bleeders, bleeding rates and bleeding score. J Thromb Haemostas 2013; 11: 142-150.
De Hert S, Imberger G, Carlisle J et al.; Task Force on Preoperative Evaluation of the Adult Noncardiac Surgery Patient of the European Society of Anaesthesiology. Preoperative evaluation of the adult patient undergoing non-cardiac surgery: guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol 2011; 28: 684-722.
Cosmi B, Alatri A, Cattaneo M et al. Assessment of the risk of bleeding in patients undergoing surgery or invasiveprocedures: Guidelines of the Italian Society for Haemostasisand Thrombosis (SISET). Thrombosis Research 2009; 124: e6-e12.

34
1 | Rol de la clínica y el laboratorio en la predicción del riesgo hemorrágico
Chee YL, Crawford CJ, Watson HG and Greaves M.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures. British Committee for Standards in Haematology. Br J Haematol 2008; 140: 496–504.
Segal JB and Dzik WH on behalf of the Transfusion Medicine/Hemostasis Clinical Trials Network. Paucity of studies to support that abnormal coagulation testresults predict bleeding in the setting of invasive procedures:an evidence-based review. Transfusion 2005; 45: 1413-1425.
Khellaf M, Michel M, Schaeffer A, Bierling P, Godeau B. Assessment of a therapeutic strategy for adults withsevere autoimmune thrombocytopenic purpura basedon a bleeding score rather than platelet count. Haematologica 2005; 90: 829-832.

35
La ventaja del tratamiento anticoagulante para la prevención del tromboembolismo arterial o venoso puede perderse fácilmente si el paciente tiene un episodio devastador de sangrado mayor. El sangrado es el principal efecto secundario de cualquier anticoagulante, principal motivo de consulta a emergencias y la causa más frecuente de abandono del tratamiento anticoagulante (1). La incidencia de sangrado mayor en la población anticoagulada difiere en los estudios clínicos vs. estudios poblacionales y según los criterios y definiciones de sangrado. En trabajos prospectivos fluctúa entre 1 y 3% por año, pero en series de casos, con pacientes más frágiles por edad o comorbilidades, llega a 2-15% por año con una mortalidad de 10-30% de los eventos. Un meta-análisis reciente (16 estudios randomizados, 31 estudios observacionales) confirmó un mínimo de 2% por año de sangrado mayor, pero la mayoría de los trabajos no incluía los 3 primeros meses de tratamiento anticoagulante (1). Por otro lado no será lo mismo un sangrado cerebral que la caída de hemoglobina de 2 g/dL (un evento con muy baja mortalidad o secuelas). El sangrado digestivo totaliza las dos
Valoración del riesgo de sangrado en el paciente anticoaguladoJosé M. Ceresetto.Jefe Sección Hemostasia del Hospital Británico de Buenos Aires. Docente UCA, UBA en Medicina Interna. Director de la Revista Hematología.
2

36
2 | Valoración del riesgo de sangrado en el paciente anticoagulado
terceras partes de los episodios de sangrado mayor pero con una mortalidad relativamente baja de hasta 18%. Una hemorragia cerebral, en cambio, en un paciente anticoagulado con dicumarínicos, tiene una mortalidad de 40-50% y totaliza cerca del 90% de todas las muertes asociadas a warfarina y 100% de las secuelas producto de sangrados. Sin embargo, su incidencia anual es muy baja, del orden de 0,2%, aunque en estudios poblacionales de pacientes añosos y frágiles la hemorragia intracraneal llega al 2,5% por año (1, 2). Los nuevos anticoagulantes orales directos (AOD) tienen menor riesgo de sangrado que los antagonistas de vitamina K (AVK, especialmente sangrado en sistema nervioso central (SNC) que se reduce 60-70%) pero de todas maneras conllevan un riesgo de hemorragia mayor de 2-3,5% por año (3). Sin embargo, en mayores de 75 años este beneficio se pierde. Debido a que los AOD se concentran en el tubo digestivo (dabigatran por menor absorción y los xabanes por la excreción activa del sistema de la glicoproteína p) el sangrado digestivo alto y bajo es mayor que con dicumarínicos y es la principal fuente de sangrado mayor con estas drogas.El riesgo de tener un sangrado grave debe balancearse con el riesgo trombótico. Existe el prejuicio en muchos médicos que consideran en menor medida el riesgo trombótico y sobrevaloran el riesgo de sangrado. En fibrilación auricular (FA) con indicación de anticoagulación, una cuarta parte de los pacientes suspende el tratamiento antes del año, porque se percibe al sangrado como muy riesgoso. La ecuación es simple, si el riesgo de trombosis es bajo estos pacientes con alto riesgo hemorrágico podrían eventualmente suspender el anticoagulante y pasar a ácido acetilsalicílico (AAS), tanto en FA como en enfermedad tromboembólica venosa (ETEV). Por el contrario, si el riesgo de trombosis es alto, la recomendación no será ineludiblemente suspender el tratamiento anticoagulante. El beneficio clínico neto favorece mantener la anticoagulación, aún en pacientes con máximo riesgo de sangrado. En ETEV el riesgo de nuevo evento trombótico en los primeros 3 meses de anticoagulación es 10 veces superior al riesgo de sangrado mayor. Tanto en FA como en ETEV tener el RIN por debajo de 2 resulta, o bien significativamente menos efectivo, o sin ventajas en reducir el riesgo de sangrado mayor, comparado con un RIN entre 2 y 3 (4). ¿Para qué sirve medir el riesgo de sangrado? Reconocer al paciente con mayor riesgo hemorrágico permite, eventualmente, modificar una recomendación médica. Se puede utilizar gastroprotección en el caso de AVK + anti-inflamatorios no esteroides (AINEs) o AAS, también se puede

37
Valoración del riesgo de sangrado en el paciente anticoagulado | 2
considerar una menor dosis con los AOD. Con los AVK se puede reducir el rango a RIN de 2-2,5, como se recomienda en pacientes con doble antiagregación por stent y portadores de FA. También se puede intentar, en los pacientes con mal tiempo en rango (TER), integrarlos a clínicas de anticoagulación y educarlos con su familia sobre los riesgos del tratamiento anticoagulante. Otra alternativa es hacer los controles más frecuentemente, especialmente en los primeros 3 meses del tratamiento. En otros casos se pueden corregir los factores de riesgo de sangrado reversibles, como evitar la ingesta de alcohol o el control estricto de hipertensión arterial (HTA) (1-4). Todas las guías internacionales recomiendan considerar el riesgo hemorrágico para ajustar el tratamiento anticoagulante. Por ej, para ETEV, las guías del American College of Chest Physicians (ACCP) consideran el riesgo de sangrado para evaluar la duración de la anticoagulación en pacientes con trombosis idiopática (nivel de evidencia 2B) al igual que las guías del Reino Unido de 2011. Para el manejo anticoagulante de FA la guía europea también utiliza un score de riesgo hemorrágico (5). Se han propuesto diversos scores para clasificar a los pacientes como de alto, moderado o bajo riesgo de sangrado. Sin embargo, ninguno de los evaluadores de riesgo desarrollados hasta hoy posee una adecuada capacidad para predecir sangrados frente al paciente individual. En las diferentes validaciones de pacientes anticoagulados sólo se obtuvo un valor predictivo leve de sangrado mayor (C entre 0,51 y 0,69) y en ningún caso el valor predictivo fue moderado (C ≥0,7) o alto (C ≥0,9). Para ponernos en contexto, un valor predictivo de 0,50 es lo mismo que tirar una moneda para ver si sale cara o cruz. El mejor de los scores con que contamos apenas tiene una capacidad predictiva de 0,66. Con lo que estamos lejos del predictor ideal y, por el momento, tal vez estos marcadores de riesgo no deberían utilizarse en la clínica para la toma de decisiones (3-7).
Factores de riesgo (FR) de sangrado
Algunos de estos factores de riesgo son reversibles como HTA no controlada, el RIN lábil, el uso de AAS o AINEs y el abuso de alcohol. Otros no los podemos modificar (4). Ver Tabla 1.

38
Antecedente de sangrado mayor. Es el factor más consistente, el sangrado digestivo triplica el riesgo de un nuevo evento, el antecedente de sangrado en SNC aumenta 10 veces el riesgo. Es una de las razones más frecuentes para no indicar o suspender AVK.
Edad. Es el segundo FR más consistente y actúa como un continuo, desde los 65 años (riesgo de sangrado 1,7 veces mayor) hasta los >80 años donde el riesgo se triplica. Intervienen múltiples factores: polifarmacia, uso de AINEs por patología osteomuscular, deterioro de la función renal, deterioro cognitivo, etc. Para sangrado en SNC: angiopatía amiloide, HTA, predisposición a las caídas. Para sangrado digestivo: divertículos, angiodisplasia, pólipos o cáncer.
RIN elevado. RIN >3,5 aumenta el riesgo de sangrado en SNC x3,5 (22% episodios de sangrado vs. 5% en controles). Un RIN >4,5 aumenta 12 veces el riesgo de sangrado mayor. En otro estudio, el riesgo de sangrado solo aumentó significativamente a partir de RIN >6 (8). En población frágil el RIN >4 aumenta hasta 20 veces el sangrado mayor. En el estudio TERRA (Tiempo En Rango en la República Argentina) el 18% de los controles fuera de rango en pacientes con FA tenían un RIN >3, pero en forma interesante, en determinados centros, tener el RIN elevado constituía un
Historia de sangrado mayor
Edad >65 años
RIN >3,5
Tiempo en rango (TER) inadecuado
Primeros 3 meses de anticoagulación
Manejo no especializado
Interferencia medicamentosa
Angiopatía amiloide
Comorbilidades
Farmacogenética
Aspectos psicosociales
Riesgo de caídas
Tabla 1. Principales marcadores del riesgo de sangrado
2 | Valoración del riesgo de sangrado en el paciente anticoagulado

39
patrón característico y posiblemente operador dependiente (RIN >3 en más de 2/3 de los valores fuera de rango) (9). Si bien globalmente el 40-50% de los sangrados ocurren con RIN >3, hasta 60% de las hemorragias en SNC ocurrieron con un rango adecuado de anticoagulación y el 20% con RIN <2. En estos casos, donde hay un sangrado con niveles adecuados de anticoagulación, se deberá descartar una causa orgánica local que favorezca la hemorragia.
Mal TER o RIN lábil. el riesgo de sangrado se triplica en pacientes con inadecuado control de anticoagulación. Se ha propuesto un TER >60%, medido por el método de Rosendaal, para que los AVK sean seguros. En pacientes con FA, la rama warfarina presentó 1,5% de sangrado mayor en pacientes con TER >70% pero 3,85% si el TER era inferior a 60%. Tiene la ventaja de que se puede modificar.
Inicio de tratamiento anticoagulante con AVK. Aumenta 3 veces el riesgo de sangrado. En los primeros 3 meses de tratamiento ocurren el 20-60% de los episodios de sangrado mayor. Factores relacionados con el sangrado en esta etapa inicial podrían ser la mayor sensibilidad a la dosis de carga de los AVK y la presencia de alguna patología orgánica predisponente, que se manifiesta clínicamente a partir de la anticoagulación (8).
Manejo del paciente por un médico general vs. clínicas de anticoagulación o algoritmos automáticos de cálculo de dosis. Relacionado al mal TER, menor frecuencia de controles o manejo diferente de pacientes irregulares o con menor educación.
Medicamentos que alteran la hemostasia o que interfieren con los AVK. Es uno de los factores de riesgo más frecuentes. Debemos considerar a los AINEs y antiagregantes plaquetarios (AAS y/o clopidogrel, prasugrel, ticagrelor) o drogas que potencian a los AVK (amiodarona, antibióticos, anti-inflamatorios, hipolipemiantes, etc.). En FA el 20-40% de los pacientes ingiere AAS y se duplica el riesgo de sangrado, en especial en el tubo digestivo, pero también se duplica el riesgo de sangrado en SNC. La doble antiagregación junto con un AVK tiene un riesgo de sangrado mayor de 10-20% por año, por lo que se sugiere bajar el RIN a 2-2,5, hacer controles más frecuentes y acortar todo lo posible la etapa de doble antiagregación.
Valoración del riesgo de sangrado en el paciente anticoagulado | 2

40
Angiopatía amiloide cerebral. De difícil diagnóstico, prevalece en ancianos. En muchas oportunidades es causa de sangrado cerebral en rango terapéutico y cuando se diagnostica, hace muy compleja la decisión de reiniciar el tratamiento con drogas anticoagulantes.
Enfermedades comórbidas. Por frecuencia debemos tener en cuenta a la insuficiencia cardíaca, accidente cerebrovascular (ACV) previo, infarto agudo de miocardio (IAM) o enfermedad coronaria aguda (uso de antiagregantes), HTA, especialmente si no está controlada (sangrado en SNC x2,7), y cáncer activo (aumenta el sangrado x3) (8). Otras enfermedades relacionadas con mayor riesgo hemorrágico son la falla hepática (riesgo relativo (RR) x2,3), anemia (hematocrito <30%, RR x2,2), abuso de alcohol y diabetes. La insuficiencia renal es un factor especialmente complejo que duplica el riesgo de sangrado. Particularmente debemos tener en cuenta la falla renal como factor de sangrado de aquellos agentes con mayor aclaramiento renal como algunos de los AOD y las heparinas de bajo peso molecular (HBPM)/pentasacáridos.
Factores genéticos. Las variables alélicas del citocromo hepático p450 (Polimorfismo 2 y 3 del CYP2C9) y los haplotipos AA de la enzima VKORC1 se han relacionado con un menor requerimiento de dosis de dicumarínicos por mayor sensibilidad a los AVK y, posiblemente, con mayor riesgo de sangrados. Pero el estudio de polimorfismos no es una práctica habitual y para cuando se obtiene el resultado, el paciente ya dejó atrás el período inicial de riesgo.
Características psicosociales de la población anticoagulada, como nivel de educación, capacidad de discernimiento, soporte familiar, edad juvenil, falta de adherencia al tratamiento, falta de recursos o de estructura de salud para un adecuado seguimiento del tratamiento.
Riesgo de caídas (>2 caídas que requieran internación). Es uno de los factores por los que se suspende o contraindica la anticoagulación más frecuentemente en pacientes añosos. Se suman factores como trastornos de la marcha, ataxia, debilidad muscular, hipoacusia, demencia o desorientación. En un estudio hasta 25% de episodios de sangrado en SNC se asoció a caídas. Sin embargo, en un modelo de toma de decisión en FA
2 | Valoración del riesgo de sangrado en el paciente anticoagulado

41
se requerían más de 200 caídas para que el riesgo de sangrado supere el riesgo de embolia en pacientes de riesgo, un dato difícil de sostener frente al paciente individual.
Predictores del riesgo de sangrado
Como vimos existen múltiples factores relacionados con mayor riesgo de sangrados. Para el manejo práctico se han agrupado en tablas o scores. Estos scores de riesgo deben tener ciertas características para ser útiles:
Sencillo de recordar. Generalmente para esto se usan acrónimos en inglés que menciona a cada uno de los factores.
Fácil de implementar. Si algún marcador requiere de tecnología compleja, como un estudio genético, seguramente será menos práctico que si solo usa parámetros clínicos o del interrogatorio.
Estar validado en la población enferma. Algunos scores se han validado solo en FA, como el denominado HAS-BLED, y no pueden simplemente trasladarse a ETEV.
Otro de los problemas con estos marcadores de riesgo es que, en FA los factores evaluados para sangrado se superponen con los marcadores de riesgo cardioembólico. Por ej, la edad >75 años, HTA y ACV previo están presentes en el score de riesgo trombótico CHADS2 y en el score de riesgo hemorrágico HAS-BLED (4-6).En ETEV el primer predictor de riesgo desarrollado en 1989 por Landefeld se denominó OBRI. Para ETEV el score del registro RIETE ha identificado a los pacientes con muy bajo riesgo de sangrado pero no ha sido validado adecuadamente en forma prospectiva. Para FA se han utilizado y validado 3 scores de riesgo de sangrado: HAS-BLED, HEMORR2HAGES y ATRIA.
Valoración del riesgo de sangrado en el paciente anticoagulado | 2

42
Predictor HAS-BLED (Tabla 2): Propuesto en 2010, utilizó pacientes del Euro Heart Survey con FA y ha sido validado en diversos estudios de cohorte. El valor predictivo en 7 trabajos fue de 0,65 (rango 0,60–0,69) y comparado con los otros evaluadores de riesgo resultó el mejor por su mayor sensibilidad. El máximo puntaje es 9. Cada categoría incluye aproximadamente a 1/3 de los pacientes anticoagulados. Se recomienda con puntaje ≥3 precaución con el tratamiento anticoagulante y revisión del paciente en forma frecuente. Se lo utiliza en las guías de la Sociedad Europea de Cardiología, la Sociedad Cardiovascular Canadiense, NICE 2014 y guías de la sociedad americana de cardiología AHA/ACC/HRS 2014. Debilidades: el punto de “RIN lábil” no se puede aplicar a los nuevos AOD y no podemos conocerlo hasta después de 3-6 meses de iniciado el tratamiento con AVK, por lo que su valor es relativo en la práctica. La inclusión de HTA, si está controlada, es controvertida (6).
Predictor HEMORR2HAGES (Tabla 3). Derivado del Registro Nacional Americano de 3.800 pacientes con FA (2006). Tiene un valor predictivo en 5 estudios de 0,63 (rango 0,61-0,66) menor que el HAS-BLED. Su máximo puntaje es 12. Debilidades: es el más extenso y más difícil de implementar por el estudio genético. Además, el riesgo alto de caídas es complejo de definir. Y su valor predictivo no es el más alto (4).
H
A
S
B
L
E
D
Hypertension
Abnormal
function
Stroke
Bleeding risk
Labile INR
Elderly
Drugs/alcohol
1 p
1 p
1 p
1 p
1 p
1 p
1 p
1 p
1 p
HTA sistólica no controlada (>160 mmHg)
falla renal (creatininemia >2,25 mg%)
falla hepática (Bilirrubinemia total >2 mg% o TGP x3)
ACV previo
Antecedente o predisposición al sangrado
RIN lábil es decir mal Tiempo en Rango (<60%)
Edad >65 años
Drogas afectan hemostasia como AAS o AINEs
Abuso de alcohol
Puntos
0
1-2
≥3
Riesgo
Bajo
Moderado
Alto
Sangrado Mayor
1-2,5% por año
3-5% por año
12,5% por año
Tabla 2. Predictor HAS-BLED
2 | Valoración del riesgo de sangrado en el paciente anticoagulado

43
Predictor ATRIA (Tabla 4). Surge a partir del registro ATRIA que evaluó 9.200 pacientes con FA (2011). Es el más sencillo de los predictores de riesgo pero su poder para discernir riesgo hemorrágico en 3 estudios fue el más bajo (0,63 rango 0,56-0,72). El máximo puntaje es 10. Debilidades: solo incluyó pacientes crónicamente anticoagulados por lo que hay selección del paciente que presenta sangrado inicial y no consideró el uso de AAS.
Anemia
Ins. Renal severa
Añoso
Sangrado
HTA
3 p
3 p
2 p
1 p
1 p
Hemoglobina <13 gr/dL en hombres, <12 gr/dL mujeres
Clearence de creatinina <30 mL/min o hemodiálisis
Edad >75 años
Antecedente de sangrado mayor
Puntos
0-3
4
≥5
Riesgo
Bajo
Moderado
Alto
Sangrado Mayor
0,8% por año
2,6% por año
5,8% por año
Tabla 4. Predictor ATRIA
H
E
M
O
R
R2
H
A
G
E
S
Hepatic or renal disease
Ethanol abuse
Malignancy
Older age
Reduced platelet count
Re-bleeding risk
Hypertension
Anemia
Genetic factors
Excesive fall risk
Stroke
1 p
1 p
1 p
1 p
1 p
2 p
1 p
1 p
1 p
1 p
1 p
Insuficiencia hepática o renal
Abuso de alcohol
Cáncer
Edad >75 años
Trombocitopenia, trombocitopatía
Antecedente de sangrado
HTA no controlada
Anemia
Polimorfismo CYP2C9
Riesgo alto de caídas
ACV previo
Tabla 3. Predictor HEMORR2HAGES
Puntos
0-1
2-3
≥4
Riesgo
Bajo
Intermedio
Alto
Sangrado Mayor
2% por año
5% por año
12% por año
Valoración del riesgo de sangrado en el paciente anticoagulado | 2
44
Predictor RIETE (Tabla 5). De diseño retrospectivo a partir de la base del registro RIETE sobre 19.000 pacientes con ETEV en los 3 primeros meses en que iniciaron anticoagulación (2008). El máximo puntaje es 8 pero el 74% de los pacientes eran de riesgo moderado y apenas 6% de alto riesgo. Debilidades: solo incluyó a pacientes que ya habían terminado los 3 meses de anticoagulación y en forma retrospectiva, no se evaluó TER o medicación concomitante y un estudio mostró que la capacidad predictiva fue 0,56 (7).
Predictor OBRI (outpatient bleeding risk index) (Tabla 6). Derivado de una serie de 550 pacientes con ETEV o patología arterial seguidos en una clínica de anticoagulación de Boston (1989). El máximo puntaje es 4. Debilidades: con un n tan pequeño es muy difícil sacar conclusiones. Además no se consideró el TER, se mezclaron pacientes con patología arterial y venosa, y es discutible considerar como factor de riesgo sólo las últimas 2 semanas desde un episodio de sangrado digestivo (10, 11).
Edad >75 años
Sangrado reciente
Cáncer activo
Falla renal (creatininemia >1,2 mg/dL)
Anemia
Tromboembolismo pulmonar (TEP) al diagnóstico
1 p
2 p
1 p
1,5 p
1,5 p
1p
Puntos
0
1-4
≥4
Riesgo
Bajo
Moderado
Alto
Sangrado Mayor
0,3% en 3 meses
2,5% en 3 meses
7,3 en 3 meses
Tabla 5. Predictor RIETE para ETEV
2 | Valoración del riesgo de sangrado en el paciente anticoagulado

45
Conclusión
Tener un predictor de riesgo de sangrado sería una herramienta deseable en la práctica clínica, especialmente para extremar cuidados y seguimiento de la anticoagulación. Pero ninguno de los predictores desarrollados hasta hoy es el ideal y todos ellos predicen apenas mejor que el azar al paciente que sangrará. A pesar de esto algunos predictores parecen comportarse en mejor medida que otros. El score HAS-BLED para pacientes con FA es práctico y sencillo de usar y se lo recomienda para los AVK y también para ajustar la dosis con los nuevos AOD. En el tratamiento inicial de ETEV, por el contrario, ninguno de los scores ha demostrado tener impacto clínico significativo y no se recomienda aplicarlos en la práctica habitual.
Conflictos de interés
El Dr. José M. Ceresetto ha recibido honorarios de los laboratorios Pfizer-BMS, Boehringer Ingelheim y Bayer. Participa del advisory board del laboratorio Boehringer Ingelheim.
Edad >65 años
Antecedentes de hemorragia digestiva (2 semanas)
Antecedente de ACV
Comorbilidades:
-IAM reciente
-Anemia (hematocrito <30%)
-Insuficiencia renal (creatininemia >1,5 mg/dL)
-Diabetes
1 p
1 p
1 p
1 p
Puntos
0
1-2
3-4
Riesgo
Bajo
Moderado
Alto
Sangrado Mayor
3% por año
10% por año
30% por año (18 pacientes evaluados)
Tabla 6. Predictor OBRI para ETEV y FA
Valoración del riesgo de sangrado en el paciente anticoagulado | 2

46
Referencias
1. Fang MC, Go AS, Chang Y et al. Death and disability from warfarin-associated intracranial and extracranial hemorrhages. Am J Med 2007; 120: 700-705.
2. Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed.: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141: e44S-88S.
3. Korin J. Bleeding during anti-vitamin K treatment. Incidence, risk factors and comparison with the new oral anticoagulants. Medicina (Buenos Aires) 2012; 72: 419-424.
4. Loewen P, Dahri K. Risk of bleeding with oral anticoagulants: an updated systematic review and performance analysis of clinical prediction rules. Ann Hematol 2011; 90: 1191-1200.
5. Heidbuchel H, Verhamme P, Alings M et al. European Heart Rhythm Association Practical Guide on the use of new oral anticoagulants in patients with non valvular atrial fibrillation. Europace 2013; 15: 625-651.
6. Zhu W, He W, Guo L, Wang X, Hong K. The HAS-BLED Score for Predicting Major Bleeding Risk in Anticoagulated Patients With Atrial Fibrillation: A Systematic Review and Meta-analysis. Clin Cardiol 2015; 38: 555-561.
7. Ruíz-Giménez N, Suárez C, González R et al. Predictive variables for major bleeding events in patients presenting with documented acute venous thromboembolism. Findings from the RIETE Registry. Thromb Haemost 2008; 100: 26-31.
8. Casais P, Sánchez Luceros A, Meschengieser S, Fondevila C, Santarelli MT,Lazzari MA. Bleeding risk factors in chronic oral anticoagulation with acenocoumarol. Am J Hematol 2000; 63: 192-196.
2 | Valoración del riesgo de sangrado en el paciente anticoagulado

47
9. Tajer C, Ceresetto J, Bottaro FJ, Martí A, Casey M; TERRA Trial investigators. Assessment of the quality of chronic anticoagulation control with time in therapeutic range in atrial fibrillation patients treated with vitamin K antagonists by hemostasis specialists: The TERRA registry: tiempo en rango en la República Argentina. Clin Appl Thromb Hemost 2016; Jan 5. pii: 1076029615623378.
10. Klok FA, Niemann C, Dellas C,Hasenfuß G, Konstantinides S, Lankeit M. Performance of five different bleeding-prediction scores in patients with acute pulmonary embolism. J Thromb Thrombolysis 2016; 41: 312-320.
11. Piovella C, Dalla Valle F, Trujillo-Santos J. Comparison of four scores to predict major bleeding in patients receiving anticoagulation for venous thromboembolism: findings from the RIETE registry. Intern Emerg Med 2014; 9: 847-852.
Valoración del riesgo de sangrado en el paciente anticoagulado | 2

48

49
Introducción
La VWD constituye la coagulopatía hemorrágica hereditaria más frecuente, con una prevalencia estimada en la población general del 1% y sólo 1/1.000 individuos sintomáticos. Se clasifica en tipo 1 y 3 a la deficiencia cuantitativa leve y severa de factor von Willebrand (VWF), respectivamente. La VWD tipo 2 (VWD2) agrupa a los defectos cualitativos del factor. La VWD1 y 2 se heredan en general, de forma autosómica dominante mientras que la VWD3 es heredada con un patrón autosómico recesivo. En esta revisión y guía que presentamos, hemos tratado de asignar un grado de recomendación, basado en el nivel de evidencia disponible, en algunos temas que hacen al diagnóstico y manejo de pacientes con VWD. Los niveles de recomendación y calidad de evidencia se resumen en la Tabla 1.
Enfermedad de von Willebrand (VWD)Analía Sanchez Luceros1, Adriana Woods2 y Gabriela Sciuccati3.1 Jefe de Depto. de Hemostasia y Trombosis, Instituto de Investigaciones Hematológicas “Mariano R. Castex”, Academia Nacional de Medicina. Inv. Adjunto. IMEX-CONICET-ANM.2 Bioquímica, PhD, Laboratorio de Hemostasia y Trombosis, IMEX- CONICET-ANM.3 Médica Hematóloga Pediatra, Servicio de Hematología y Oncología, Hospital de Pediatría “Prof. Dr. J. P. Garrahan”.
3
50
3 | Enfermedad de von Willebrand (VWD)
Manifestaciones clínicas
El diagnóstico se realiza mediante la evaluación de la historia personal y familiar de sangrado, el examen físico y los resultados de los ensayos específicos de laboratorio. El sangrado asociado a la VWD involucra la piel y las mucosas, y su severidad es muy variable. El sangrado muscular y la hemartrosis espontánea son manifestaciones hemorrágicas poco frecuentes que se presentan en la VWD3 y en la variante Normandy, una forma de tipo 2. Una historia clínica detallada constituye el primer paso en la evaluación de los pacientes con manifestaciones de sangrado. Muchos pacientes presentan hematomas y/o epistaxis como única manifestación hemorrágica, síntomas que son frecuentes en población normal, tanto en niños como adultos (hasta el 20% de la población general). La distinción entre un paciente sano y uno con una coagulopatía hemorrágica hereditaria puede ser dificultosa, y esta dificultad es mayor en niños de corta edad que no han enfrentado un desafío hemostático como cirugía, trauma o menarca.En la actualidad se recomienda la utilización de un sistema de puntaje estandarizado para valorar los síntomas de sangrado. Para ello se ha diseñado
Tabla 1. Grados y calidad de evidencia
Fuerte (1)
Débil (2)
A: Alta
B: Moderada
C: Baja
D: Muy baja
Grados de evidencia
Calidad de evidencia
Hay certeza en que los beneficios o no de una conducta superan a los riesgos.
Cuando se cree que los beneficios y riesgos están balanceados, o que hay apreciable incerteza acerca de la magnitud de beneficios y riesgos.Se tiene en cuenta la preferencia del paciente en la toma de decisión, y se sugiere o se pone en consideración la conducta.
Más investigaciones no cambiarán el grado de confianza en la estimación del efecto.
Investigaciones posteriores pueden tener impacto importante en la confianza del estimado, pudiendo cambiarlo.
Muy probable que investigaciones posteriores tengan impacto importante en la confianza del estimado, cambiándolo.
Cualquier estimado del efecto es incierto.
Adaptado de: www.gradeworkinggroup.org/index.htm

51
Enfermedad de von Willebrand (VWD) | 3
un cuestionario que evalúa la frecuencia y la severidad del sangrado (ISTH-BAT) (1) en niños y adultos (Tabla 2, nivel de recomendación 2C). Los niños, de ambos sexos, con un puntaje ≤2 tienen baja probabilidad de tener VWD, en cambio en adultos, se diferencia el punto de corte entre varones ≤3 y mujeres ≤5.
Epistaxis
Cutáneo
Heridas menores
Cavidad oral
Gastrointestinal
Exodoncia
Cirugía
Menorragia
Post-parto
Hematomas
musculares
No o trivial
(<5/año)
No o trivial (≤1cm)
No o trivial
(<5/año)
No
No
No realizada o
sin sangrado en 1
exodoncia
No realizada o sin
sangrado en 1 cirugía
No
No partos o
sin sangrado
en 1 parto
Nunca
>5/año o
duración >10 min
>1 cm y sin trauma
>5/año o
duración >5 min
Reportado,
no consultado
Causa
identificada
Reportada, no
consultada
Reportada, no
consultada
Sólo consulta
Sólo consulta
Post-trauma, sin
tratamiento
Sólo consulta
Sólo consulta
Sólo consulta
Sólo consulta
Espontáneo
Sólo consulta
Sólo consulta
Antifibrinolíticos
o anticonceptivos
orales
Curetaje,
suplemento con
hierro o terapia
antifibrinolìtica
Espontáneo sin
tratamiento
Compresión,
cauterización o
antifibrinolíticos
-
Hemostasia
quirúrgica
Hemostasia quirúrgica
o antifibrinolìticos
Hemostasia quirúrgica,
TCG, terapia de
reemplazo, DDAVP o
antifibrinolìticos
Sutura
Hemostasia quirúrgica
y antifibrinolíticos
Curetaje,
suplemento
con hierro
TCG, terapia de
reemplazo o DDAVP
Espontáneo o
traumático, requirió
DDAVP o terapia de
reemplazo
TCG, terapia de
reemplazo
o DDAVP
-
TCG, terapia de
reemplazo o DDAVP
TCG, terapia de
reemplazo o DDAVP
TCG, terapia de
reemplazo
o DDAVP
TCG, terapia de
reemplazo o DDAVP
TCG, terapia de
reemplazo, DDAVP o
histerectomía
Histerectomía
Espontáneo o
traumático, requirió
cirugía o TCG
Tabla 2. Cuestionario de síntomas de sangrado (ISTH-BAT)
Síntoma Puntaje0 1 2 3 4
52
Un examen físico minucioso, con atención especial al tamaño y la distribución de las hemorragias cutáneas (petequias, equimosis o hematomas) orienta el diagnóstico diferencial con otros desórdenes como síndromes de hiperlaxitud, vasculitis, trombocitopatías, etc.Las mujeres son más propensas a poner de manifiesto un trastorno hemo-rragíparo, incluso cuando éste es leve, ya que fisiológicamente tienen desafíos hemostáticos en su vida debido al ciclo natural de la menstruación, embarazo y parto.Menstruaciones abundantes - Menorragia. La VWD debe ser investigada en pacientes con menorragia, especialmente en presencia de síntomas de sangrado de otros sitios, para evitar procedimientos invasivos de riesgo o incluso innecesarios. Aunque la presencia de anemia acompañante puede claramente indicar la existencia de un sangrado excesivo, se sugiere el uso de registros que grafiquen (Figura 1) la cuantía de la pérdida, de modo de evitar subjetividades (2). En el 80% de las mujeres la menorragia está presente desde la menarca. El tratamiento sugerido se basa, de acuerdo a la preferencia
Hemartrosis
SNC
Otros:
Circuncisión
Cordón umbilical
Cefalohematoma
Hematuria
macroscópica
Post-venopuntura
Conjuntival
Nunca
Nunca
No
Post-trauma,
sin tratamiento
-
Reportado
Espontáneo
sin tratamiento
-
Sólo consulta
Espontáneo o
traumático, requirió
DDAVP o terapia de
reemplazo
Hemorragia subdural,
cualquier intervención
Hemostasia quirúrgica,
antifibrinolìticoso
suplemento con hierro
Espontáneo
o traumático,
requirió cirugía
o TCG
Parenquimatoso,
cualquier
intervención
TCG, terapia de
reemplazo o
DDAVP
Tabla 2. Cont.
Síntoma Puntaje0 1 2 3 4
DDAVP: desmopresina; TCG: transfusión de concentrado globular
3 | Enfermedad de von Willebrand (VWD)

53
individual de la mujer o los padres de adolescentes, en anticonceptivos orales, ácido tranexámico (AcTx) y/o desmopresina (DDAVP). Pacientes con defectos severos deberán recibir concentrados de FVIII/VWF.
Cambios en el embarazo. Normalmente, el embarazo se acompaña de una mayor concentración de fibrinógeno, FV, FVIII, FX y VWF. En conjunto los cambios hemostáticos y fibrinolíticos contribuyen al estado de hiper-coagulabilidad del embarazo. En mujeres con trastornos hemorragíparos, puede observarse cierta tendencia a la normalización del defecto, aunque la respuesta es variable aún en mujeres con el mismo defecto. Las mujeres con VWD1 muestran un aumento progresivo de los niveles de FVIII, antígeno (VWF:Ag) y actividad de VWF, alcanzando incluso niveles hemostáticos (50 UI/dL o 50%) en el tercer trimestre. Sin embargo, esta mejoría no se presenta
Figura 1.
Enfermedad de von Willebrand (VWD) | 3

54
en mujeres con formas severas de tipo 1 y en el tipo 3, ya que por definición la proteína está ausente y por ende tampoco aumentará el FVIII. En las variantes de tipo 2 el FVIII y el VWF:Ag en general aumentan, pero con incrementos mínimos de la actividad del VWF, ya que el defecto cualitativo no se corrige.Cuidados antenatales. Existen desafíos hemostáticos potenciales que se presentan en la primera mitad del embarazo y aumentan el riesgo de sangrado en mujeres con VWD, ya que en esta etapa no se han producido cambios relevantes en los niveles de factores. Estos son aborto espontáneo, hemorragia antenatal y procedimientos invasivos (para diagnóstico prenatal; cerclaje cervical). Debe tenerse presente el monitoreo de factores ante estas situaciones, para considerar la necesidad de profilaxis.El parto y la cesárea. El riesgo de hemorragia peri-parto es mayor en mujeres con VWD, especialmente si no alcanzan niveles hemostáticos de factores. Los datos sobre prevalencia de hemorragia post-parto (HPP) en mujeres con VWD son limitados. En un estudio reciente, se describió un mayor riesgo de HPP (odd-ratio (OR): 1,5; intervalo de confianza (IC)95%: 1,1-2,0) y cinco veces mayor riesgo de requerir transfusiones de sangre (OR: 4,7; IC95%: 3,2-7,0) en comparación con las mujeres sin VWD (3). En nuestro medio, la HPP en mujeres con VWD es también frecuente, con 40% de frecuencia entre las mujeres con fenotipo leve a moderado (4). El monitoreo en el tercer trimestre, entre el 7º y 8º mes, es fundamental para el manejo del parto. Las decisiones de cómo tratar o prevenir el sangrado dependerán de los controles de laboratorio durante el tercer trimestre, del tipo de VWD, la historia personal de sangrado, especialmente en partos previos, y del tipo de parto planeado. La profilaxis deberá ser indicada si los niveles de FVIII o la actividad de VWF se mantienen por debajo de 50 UI/dL. Las mujeres con VWD1 responderán generalmente a una dosis de DDAVP. En los otros tipos, salvo que una prueba de DDAVP con buena respuesta haya sido realizada con anterioridad al embarazo, será necesario el uso de concentrados de FVIII/VWF. Si se presenta HPP, a pesar de la profilaxis, deben excluirse siempre las causas locales y obstétricas de hemorragia. Opciones de analgesia obstétrica. El uso de bloqueo regional en mujeres con VWD y otros defectos ha sido controvertido debido al riesgo de hematoma espinal o epidural, con el daño neurológico consecuente. Sin embargo, si el defecto hemorrágico es corregido por el embarazo, por DDAVP o por concentrados de FVIII/VWF, el bloqueo regional ha sido empleado de manera segura (5). Cada mujer, sin embargo, debe evaluarse de forma individual y
3 | Enfermedad de von Willebrand (VWD)

55
la decisión sobre el uso de analgesia regional debe hacerse por adelantado conjuntamente por el anestesista, hematólogo, obstetra y la mujer.VWD neonatal. La necesidad de un diagnóstico de VWD durante el período neonatal es excepcional, y no es recomendado ya que los niveles de VWF se encuentran elevados hasta los 6 meses de vida. El sangrado relacionado a VWD en el neonato es poco frecuente, escasos reportes de complicaciones hemorrágicas asociadas a VWD3 han sido publicadas. Dado su alto peso molecular, el VWF no atraviesa la placenta por lo que el aumento de sus niveles plasmáticos durante el embarazo tiene escaso impacto en los niveles plasmáticos del VWF en el recién nacido. Las técnicas de monitoreo invasivo, tales como electrodos en el cuero cabelludo y/o muestreo de sangre fetal se deberían evitar. A diferencia de la hemofilia, no se recomienda el control de los niveles de factores en el recién nacido, salvo en caso de familias con VWD3.
Pruebas para el diagnóstico de VWD
Diagnóstico fenotípico. las formas leves son más difíciles de diagnosticar, por lo que se impone repetir los estudios en individuos que presenten clínica de VWD, en tres momentos diferentes si es necesario. Para llegar al diagnóstico se precisan varias pruebas (6). El laboratorio comienza con pruebas orientadoras, donde la alteración de una o varias de las mismas resultan sugestivas de VWD, se continúa con pruebas confirmatorias, y posteriormente pruebas para definir la variante de VWD.• Pruebas orientativas: Tiempo de sangría (TS) (Ivy o Template), es
inespecífica; se da por cohibida cuando persiste sangrado profuso luego del doble del rango superior normal; recuento de plaquetas en sangre (RP): cuando está disminuido es orientador del VWD2B; tiempo de tromboplastina parcial activada (TTPA): su prolongación indica probable disminución de FVIII:C; en este caso, realizar prueba de corrección con plasma normal para descartar presencia de inhibidores.
• Pruebas confirmatorias: FVIII:C, ensayo antigénico (VWF:Ag), ensayo de cofactor de ristocetina (VWF:RCo), (Nivel de recomendación 1A). Éste último mide actividad del VWF. Si los niveles de VWF están bajos y el cociente RCo/Ag es >0,6 el desorden es cuantitativo (Tabla 3). Si el cociente RCo/Ag es <0,6 el desorden es cualitativo (tipo 2) (Nivel de
Enfermedad de von Willebrand (VWD) | 3

56
recomendación 1B). Se debe continuar el estudio con pruebas más específicas para determinar la variante (Tabla 4).
Pruebas para definir la variante de la VWD
Análisis multimérico del VWF en gel de SDS-agarosa. En los de baja resolución (1%) se observa todo el perfil multimérico. A alta resolución (1,7%) se observan los multímeros intermedios y pequeños, y la estructura de los tripletes. Esto último es útil frente a la sospecha de proteólisis aumentada del VWF, dado por el aumento de las bandas satélites, siendo útil para diferenciar
TS
RP
TTPA
FVIII:C (UI/dL)
VWF:Ag (UI/dL)
VWF:RCo (UI/dL)
RCo/Ag
Composición
multimérica
Respuesta a la
DDAVP
VWF
intraplaquetario
RIPA
VWFpp/Ag
Prolongada
Normal
Prolongado
10-15
10-15
10-15
Normal
Normal
Respuesta
insuficiente
Disminuido
Disminuida o
ausente
Normal
Prolongada
Normal
Normal o
prolongado
30-49
30-49
30-49
Normal
Normal
Responde
Variable
Variable
Normal
Muy prolongada
o cohibida
Normal
Muy prolongado
<5
<5
<5
No aplicable
Ausente
No responde
Ausente
Ausente
No aplicable
Prolongada
Normal
Prolongado
15-30
15-30
15-30
Normal
Normal
Responde
Normal,
disminuido,
discordante
Disminuida
Normal
Prolongada
Normal
Prolongado
15-30
15-30
10-15
Normal
Aumento de
bandas satélites
ULMWM:
Vicenza
Respuesta
variable, con
sobrevida
acortada
Normal
Muy disminuida
Aumentado
≤4,5 min
150-400x109/L
Según reactivo
usado
50-150
50-150
50-150
>0,6
Todos los
multímeros
0,1-0,4
U/109 plaq
Positiva a
>0,8 mg/mL
0,9-2,14
Tabla 3. Diferencias en las pruebas de laboratorio entre variantes cuantitativas
Laboratorio Tipo 1 severo
Probable tipo 1
Tipo 3Tipo 1 Tipo 1C Valor normal
3 | Enfermedad de von Willebrand (VWD)
ULMWM: ultra-large molecular weight multimers o múltimeros de peso molecular ultra alto.
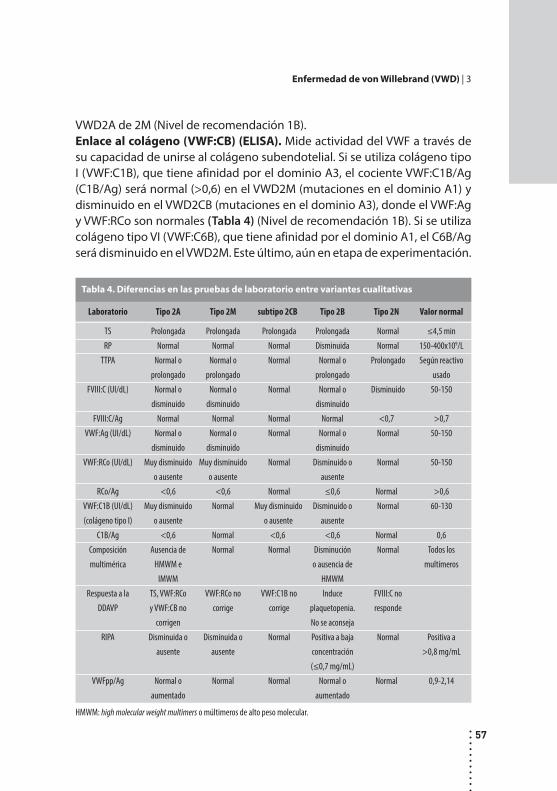
57
VWD2A de 2M (Nivel de recomendación 1B).Enlace al colágeno (VWF:CB) (ELISA). Mide actividad del VWF a través de su capacidad de unirse al colágeno subendotelial. Si se utiliza colágeno tipo I (VWF:C1B), que tiene afinidad por el dominio A3, el cociente VWF:C1B/Ag (C1B/Ag) será normal (>0,6) en el VWD2M (mutaciones en el dominio A1) y disminuido en el VWD2CB (mutaciones en el dominio A3), donde el VWF:Ag y VWF:RCo son normales (Tabla 4) (Nivel de recomendación 1B). Si se utiliza colágeno tipo VI (VWF:C6B), que tiene afinidad por el dominio A1, el C6B/Ag será disminuido en el VWD2M. Este último, aún en etapa de experimentación.
TS
RP
TTPA
FVIII:C (UI/dL)
FVIII:C/Ag
VWF:Ag (UI/dL)
VWF:RCo (UI/dL)
RCo/Ag
VWF:C1B (UI/dL)
(colágeno tipo I)
C1B/Ag
Composición
multimérica
Respuesta a la
DDAVP
RIPA
VWFpp/Ag
Prolongada
Normal
Normal o
prolongado
Normal o
disminuido
Normal
Normal o
disminuido
Muy disminuido
o ausente
<0,6
Muy disminuido
o ausente
<0,6
Ausencia de
HMWM e
IMWM
TS, VWF:RCo
y VWF:CB no
corrigen
Disminuida o
ausente
Normal o
aumentado
Prolongada
Normal
Normal
Normal
Normal
Normal
Normal
Normal
Muy disminuido
o ausente
<0,6
Normal
VWF:C1B no
corrige
Normal
Normal
Normal
Normal
Prolongado
Disminuido
<0,7
Normal
Normal
Normal
Normal
Normal
Normal
FVIII:C no
responde
Normal
Normal
Prolongada
Normal
Normal o
prolongado
Normal o
disminuido
Normal
Normal o
disminuido
Muy disminuido
o ausente
<0,6
Normal
Normal
Normal
VWF:RCo no
corrige
Disminuida o
ausente
Normal
Prolongada
Disminuida
Normal o
prolongado
Normal o
disminuido
Normal
Normal o
disminuido
Disminuido o
ausente
≤0,6
Disminuido o
ausente
<0,6
Disminución
o ausencia de
HMWM
Induce
plaquetopenia.
No se aconseja
Positiva a baja
concentración
(≤0,7 mg/mL)
Normal o
aumentado
≤4,5 min
150-400x109/L
Según reactivo
usado
50-150
>0,7
50-150
50-150
>0,6
60-130
0,6
Todos los
multímeros
Positiva a
>0,8 mg/mL
0,9-2,14
Tabla 4. Diferencias en las pruebas de laboratorio entre variantes cualitativas
Laboratorio Tipo 2A subtipo 2CB Tipo 2NTipo 2M Tipo 2B Valor normal
Enfermedad de von Willebrand (VWD) | 3
HMWM: high molecular weight multimers o múltimeros de alto peso molecular.

58
Enlace del FVIII:C al VWF (VWF:FVIIIB) (ELISA). se ensaya cuando el FVIII:C/Ag es <0,7, para diferenciar VWD2N de Hemofilia A (Tabla 3).Agregación plaquetaria con ristocetina (RIPA). La concentración de trabajo es 1,2 mg/mL, con hipo-respuesta en VWD1, 2A y 2M, o hiper-respuesta a bajas concentraciones de ristocetina cuando se sospecha VWD2B (Tabla 4). Algunos autores sugieren hacer la prueba con bajas concentraciones de ristocetina en todo paciente con cociente RCo/Ag<0,6 y/o trombocitopenia asociada (Nivel de recomendación 1B).Propéptido (VWFpp) (ELISA). nos informa de la síntesis y la depuración del VWF. El aumento en el cociente VWFpp/Ag nos indica vida media disminuida. Está normal en las variantes cuantitativas, excepto en el tipo VWD1C (incluido el Vicenza), en las formas adquiridas (AVWS) y en casos de VWD3 politransfundidos con desarrollo de alo-anticuerpos.Pruebas para diferenciar entre VWD2B y del tipo plaquetario (PT-VWD)• Análisis de mezclas cruzadas de PPP paciente/normal y plaquetas
lavadas paciente/normal.• Agregación del PRP/paciente con crioprecipitadoo concentrado de
VWF comercial.
Pruebas adicionales
Respuesta al DDAVP. Puede ser útil para discriminar entre VWD1 severo y VWD3, entre VWD2M y VWD1 severo cuando el VWF:RCo es muy bajo, en la pesquisa del VWD1C.VWF intraplaquetario. Ayuda a discriminar entre VWD1 severo y VWD3, en los últimos años ha perdido relevancia.PFA-100®. Más sensible que el TS; aún no se conoce su especificidad; se necesitan más datos para su recomendación a pesar de que su uso se inició en 1985.Presencia de anticuerpos (Acs). Se recomienda la detección de Acs anti-VWF por ELISA; éstos, aunque muy sensibles, han demostrado a veces presencia de falsos positivos. Los ensayos de mezclas sólo pueden demostrar la presencia de Acs inhibidores de la función del VWF con resultados positivos muy infrecuentes, por lo que resultados negativos no excluyen la presencia de inhibidor, ya que puede estar dirigido hacia epitopes no funcionales (7).Estudios genotípicos. La genética sólo influye entre el 60-70% de la
3 | Enfermedad de von Willebrand (VWD)

59
variación de los niveles del VWF, siendo el 30% restante producido por causas epigenéticas, como el grupo sanguíneo ABO, edad, factores raciales y hormonales. El diagnóstico genotípico es útil para diferenciar el VWD2N de la Hemofilia A leve o moderada, el VWD2B del PT-VWD, el VWD congénito del AVWS con fenotipo 2A o 2B, donde la búsqueda de mutaciones responsables dará negativa. También es útil en el momento de la toma de decisiones terapéuticas frente a casos severos, donde se pueden requerir estudios prenatales y donde también es de mucha ayuda estudiar el grupo familiar para establecer grados de penetrancia de las mutaciones responsables (Nivel de recomendación 1B).En la Figura 2 se muestra el algoritmo con la marcha diagnóstica.
Enfermedad de von Willebrand (VWD) | 3
Figura 2.
(*) hacer biología molecular para diferenciar y/o confirmar el diagnóstico; HA: Hemofilia A.

60
El laboratorio en cada subtipo
Variantes cuantitativas: tipo 1 (VWD 1) y tipo 3 (VWD 3) (Tabla 3).• VWD1: VWF plasmático: 10-40 UI/dL, niveles paralelamente bajos de
FVIII:C, VWF:Ag y VWF:RCo. Se han descripto subgrupos de acuerdo a los niveles de VWF, como se muestra en la Tabla 3.
• VWD3: VWF:Ag y VWF:RCo ausentes o <5 UI/dLy el FVIII:C <5-10 UI/dL.Variantes cualitativas: tipos 2 (Tabla 4); el VWF:Ag y FVIII:C suelen ser normales. La relación RCo/Ag es<0,6, excepto el subtipo 2N, donde es normal (>0,6). • VWD2A: caracterizada por mutaciones ubicadas en su mayoría en domi-
nio A2 (exón 28), que alteran la dimerización/multimerización del VWF.• VWD2M: se caracteriza por incapacidad del VWF de unirse a la GPIbα
plaquetaria, por mutaciones en su mayoría localizadas en el dominio A1 (exón 28). Incluye el VWD2CB, deficiencia aislada de VWF:CB, con mutaciones en dominio A3.
• VWD2B: causado por mutaciones en el dominio A1 (100% ubicadas en el exón 28) que producen ganancia de función, debido al aumento en la afinidad del VWF por la GPIbα plaquetaria.
• VWD2N: incapacidad del VWF de unir al FVIII:C por la presencia de mutaciones en la región comprendida entre los dominios D’-D3.
Controles de calidad relacionados con el laboratorio; condiciones pre-analíticas
El VWF es reactante de fase aguda, por lo tanto, es indispensable el reposo de 30 min previo a la toma de muestras. Como anticoagulante se utiliza citrato trisódico 0,11 M; para el análisis multimérico se agregan anti-proteasas (EDTA 50mM, n-etilmaleimida 60 mM, aprotinina 2.000 KUI/mL); para el análisis de ADN, se utiliza EDTA 2%. Evitar situaciones de estrés, en especial en niños. No se recomienda estudiar a menores de 6 meses de edad. Durante el ciclo menstrual, se sugiere realizar los estudios preferentemente durante los tres primeros días. El embarazo no es momento adecuado para el diagnóstico de pacientes con VWD (excepto ante la sospecha de variantes severas) y mucho menos, realizar la prueba de DDAVP.Es necesario aclarar que hasta ahora, no hay ninguna prueba de laboratorio,
3 | Enfermedad de von Willebrand (VWD)

61
fenotípica ni genotípica que se relacione como marcador de riesgo de sangrado. Se recomienda el uso de estándares domésticos para FVIII:C, VWF:Ag, VWF:RCo, VWF:CB y VWFpp, controlados frente al estándar internacional de WHO (World Health Organization-International Standards).
Tratamiento
Se indica para prevenir una complicación hemorrágica o controlar un sangrado excesivo. Los tratamientos disponibles son aquellos dirigidos a aumentar los niveles de VWF (endógeno: DDAVP o con VWF exógeno) y otros tratamientos adyuvantes.Terapias que aumentan los niveles del VWF y/o FVIII. El DDAVP es un análogo sintético de la vasopresina que aumenta los niveles plasmáticos del VWF y del FVIII. Se utiliza para la prevención del sangrado o el tratamiento de las complicaciones del mismo. Se puede administrar por vía endovenosa, como spray intranasal o subcutánea (Tabla 5). Se recomienda realizar la prueba de respuesta a la DDAVP a los pacientes con VWD1, 2A, 2M y 2N, al diagnóstico (Nivel de Recomendación 1C) y antes de iniciar su uso clínico, excepto en mujeres embarazadas o durante un episodio de sangrado (8). Si bien no hay acuerdo general, se recomienda determinar VWF:Ag, VWF:RCo y FVIII pre-infusión, a los 30-60 min y a las 2-4 hs después de administrada la DDAVP. Las definiciones de respuesta biológica a DDAVP varían, pero en general se requiere como mínimo un aumento de 2-3 veces del VWF:RCo y FVIII:C y niveles plasmáticos >30 UI/dL o preferentemente >50 UI/dL post-infusión. Estos niveles de respuesta podrían ser suficientes para procedimientos menores y mayores respectivamente. Si bien se observa buena respuesta en la mayoría de los pacientes con VWD1, la respuesta es menor en pacientes con niveles basales de VWF:RCo <10 UI/dL y FVIII <20 UI/dL (9). La respuesta es variable en VWD2A y 2M, mientras que en el VWD2N los niveles de FVIII aumentan en forma transitoria y solo en algunas variantes. No tiene beneficio en VWD3. La administración de DDAVP en los pacientes con VWD2B, puede causar un descenso transitorio de las plaquetas o agravar una trombocitopenia pre-existente, por lo que su uso no está recomendado en este grupo de pacientes y debe ser valorado individualmente. El enrojecimiento facial, hipo o hipertensión y la cefalea son las reacciones adversas más frecuentes. Por sus efectos antidiuréticos,
Enfermedad de von Willebrand (VWD) | 3

62
DDAVP puede favorecer la retención hídrica e hiponatremia secundaria. Se sugiere monitoreo del balance hidroelectrolítico y restricción del aporte de líquidos durante las 24 hs posteriores a su administración, especialmente en los niños más pequeños (10) (Nivel de recomendación 2C). No se recomienda su indicación en niños <2 años. Considerar otra alternativa terapéutica en niños con antecedentes de convulsiones.
Terapia de reemplazo. La administración de hemoderivados (concentrados comerciales) está indicada en (Nivel de recomendación 1A):• Sangrados moderados/severos o en cirugía mayor o procedimientos que
requieren tratamiento prolongado.• Pacientes sin respuesta a la DDAVP o en quienes esté contraindicada.Existen diferentes concentrados comerciales que se diferencian entre sí por su contenido en VWF (11). Las dosis y duración del tratamiento sugeridas se describen en las Tablas 6, 7 y 8.La indicación de tratamiento profiláctico a largo plazo es controvertida.Los pacientes con un fenotipo de sangrado que requieran infusión de concentrados con VWF en forma frecuente se pueden beneficiar con su utilización. Si bien todos los concentrados que contengan VWF con o sin FVIII son efectivos, no hay consenso sobre la dosis y la frecuencia de administración (12).Los crioprecipitados son derivados plasmáticos, sin procesos de atenuación viral. Su uso sólo está indicado en situaciones de riesgo de vida y sin disponibilidad de concentrados de VWF/FVIII.
Respuesta a la DDAVP
RIPA
Infusión EV
Vía subcutánea*
Spray intranasal*
Pico de VWF y FVIII
TS, VWF:RCo y VWF:CB no corrigen
Disminuida o ausente
0,3 µg/Kg diluidos en 50-100 mL de solución fisiológica durante 30 min
0,3 µg/Kg
300 µg intranasal (150µg en pacientes<30 kg) cada 12-24 hs
30-60 min post- infusión EV o 90-120 min post-inyección s.c. o spray intranasal
Tabla 5. DDAVP. Vías de administración y dosis
* Forma farmacéutica no disponible en Argentina
3 | Enfermedad de von Willebrand (VWD)

63
Terapias no específicas (adyuvantes)
Drogas antifibrinolíticas. Inhiben la lisis del coágulo. Se pueden adminis-trar como terapia única o asociadas a la terapia de reemplazo. Se eliminan por vía renal, se sugiere ajustar la dosis en casos de insuficiencia renal. • AcTx: dosis 10 a 30 mg/Kg, cada 8-12 hs, vía oral o EV.• Acido e-aminocaproico: 50-60 mg/Kg EV/oral cada 4-6 hs (dosis max 30
g/día).
Dosis de ataque*
Dosis de mantenimiento*
Monitoreo
Objetivo terapéutico
Parámetro de seguridadSe pueden alternar concentrados y DDAVP al final del tratamiento
40-60 UI/Kg
20-40 UI/Kg cada 8-24 hs
VWF:RCo y FVIII:C, valle y pico, diario
Valle VWF:RCo y FVIII: C>50 UI/dL durante 7-14 días
No superar VWF:RCo >200 UI/dL o FVIII:C 250-300 UI/dL
Tabla 6. Terapéutica de VWD con concentrados de VWF/FVIII
*Dosis en VWF:RCo (UI/dL). Modificado de Nichols WL y col.
Sangrado severo/cirugía mayor
Dosis de ataque*
Dosis de mantenimiento*
Monitoreo
Objetivo terapéutico
Parámetro de seguridadSe pueden alternar concentrados y DDAVP al final del tratamiento
30-60 UI/Kg
20-40 UI/Kg cada 12-48 hs
VWF:RCo y FVIII:C, valle y pico una vez
Valle VWF:RCo y FVIII:C >50 UI/dL durante 3-5 días
No superar VWF:RCo >200 UI/dL o FVIII:C 250-300 UI/dL
Tabla 7. Terapéutica de la VWD con concentrados de VWF/FVIII
* Dosis en VWF:RCo (UI/dL). Modificado de Nichols WL y col.
Sangrado leve/cirugía menor
Enfermedad de von Willebrand (VWD) | 3

64
Los niños pequeños con dificultad para tomar comprimidos, pueden recibir por vía oral la solución de uso EV.• Contraindicación: hematuria macroscópica, riesgo aumentado de
trombosis.• Sellantes de fibrina: son drogas de uso tópico se aplican localmente en
el sitio de sangrado, especialmente en ciertas situaciones quirúrgicas.
Manejo del paciente con inhibidor de VWF
Se describieron en pacientes con VWD3 con múltiples transfusiones de concentrados de VWF (13). Los pacientes que desarrollan inhibidores presentan pérdida de respuesta a la administración de concentrados de VWF y pueden asociarse con reacción alérgica. Como se describió en el apartado de laboratorio, los estudios de mezcla son métodos poco sensibles para su detección, en cuyo caso la pobre respuesta clínica a la infusión de concentrados junto a la baja recuperación y corta sobrevida del VWF post-infusión podrían ser el único indicador de su presencia (Nivel de recomendación 2A). Las opciones de tratamiento en estos casos son infusión de dosis altas de rFVIII, rFVIIa, transfusión de concentrado plaquetario y AcTx (Nivel de recomendación 2A).
Conflictos de interés
Las autoras no presentan conflictos de interés.
CardiotorácicaCesárea
CraneotomíaHisterectomía
Colecistectomía abiertaProstatectomía
Biopsia de mama y cervicalExodoncia complicada
Cirugía gingivalColocación de vía central
Procedimientos laparoscópicos
CateterismoCataratas
Endoscopia sin biopsiaBiopsia hepática
LaceracionesExodoncia simple
Tabla 8. Duración sugerida de la terapia de reemplazo con concentrados de VWF
*Casos individuales podrán requerir mayor o menor duración de acuerdo a la severidad de VWD y a las características del procedimiento. Modificado de Nichols WL y col.
Cirugía mayor 7-14 días
Cirugía menor 1-5 días
Otros procedimientosÚnica dosis
3 | Enfermedad de von Willebrand (VWD)

65
Referencias
1. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Hemophilia 2014, 20: 831-835.
2. Janssen CA, Scholten PC, Heintz AP. A simple visual assessment technique to discriminate between menorrhagia and normal menstrual blood loss. Obstet Gynecol 1995; 85: 977-982.
3. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007; 5: 1165-1169.
4. Sánchez-Luceros A, Meschengieser SS, Turdó K et al. Evaluation of the clinical safety of desmopressin during pregnancy in women with a low plasmatic VWF level and bleeding history. Thromb Res 2007; 120: 387-390.
5. Chi C, Lee CA, England A, et al. Obstetric analgesia and anaesthesia in women with inherited bleeding disorders. Thromb Haemost 2009; 101: 1104-1111.
6. Roberts JC, Flood VH. Laboratory diagnosis of von Willebrand disease. Int J Lab Hematol 2015; 37: 11-17.
7. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013; 122: 636-640. Review.
8. Sánchez-Luceros A, Meschengieser S, Woods AI, et al. Biological and clinical response to desmopressin (DDAVP) in a retrospective cohort study of children with low von Willebrand factor levels and bleeding history. Thromb Haemost 2010; 104: 984-989.
9. Mannucci PM, Franchini M, Castaman G,et al. Evidence-based recommendations on the treatment of von Willebrand disease in Italy. Blood Transfus 2009; 7: 117-126.
Enfermedad de von Willebrand (VWD) | 3

66
10. Bonduel M, Frontroth JP, Hepner M, et al. von Willebrand disease in children: Diagnosis and management of pediatric cohort in one single center in Argentina. Semin Thromb Hemost 2011; 37: 560-567.
11. Nichols WL, HultinMB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008; 14: 171-232.
12. Federici AB. Prophylaxis in patientswith von Willebrand disease: who, when, how? J Thromb Haemost 2015; 13: 1581-1584.
13. Woods AI, Sánchez-Luceros A, Meschengieser SS, Kempfer AC, Blanco AN, Lazzari MA. Diagnosis and management of von Willebrand disease in a single institution of Argentina. Semin Thromb Hemost 2011; 37: 568-575.
3 | Enfermedad de von Willebrand (VWD)

67
HemofiliaDaniela Neme1, Ludmila Elhelou2, Egle Honnorat3, Miguel Tezanos Pinto4 y Alicia N. Blanco5.1 Directora médica de la Fundación de la Hemofilia.2 Médica de la Fundación de la Hemofilia – Instituto de Investigaciones Hematológicas “Mariano R. Castex”, Academia Nacional de Medicina. 3 Médica de la Fundación de la Hemofilia.4 Asesor médico de la Fundación de la Hemofilia.5 Doctora en Bioquímica (UBA); Jefe División Hemostasia, Instituto de Investigaciones Hematológicas “Mariano R. Castex”, Academia Nacional de Medicina.
Manejo de pacientes con hemofilia congénita
Definición
La hemofilia es un desorden hemorrágico hereditario y congénito, originado por mutaciones en el cromosoma X, caracterizado por la disminución o ausencia de la actividad funcional de los FVIII o FIX. La hemofilia afecta a individuos varones de la rama materna; un tercio de los casos surge como consecuencia de mutaciones espontáneas. La frecuencia de la hemofilia A (FVIII) es de aproximadamente 1 cada 5.000 a 10.000 nacimientos de varones y la de la hemofilia B (FIX) es de 1 cada 30.000 a 50.000 nacimientos.
4

68
Manifestaciones clínicas
La expresión clínica es la hemorragia en diversas localizaciones del organismo, siendo las más características y frecuentes las de articulaciones (hemartrosis) y músculos. Las articulaciones más afectadas son los to-billos, rodillas y codos. Las hemartrosis repetidas originan una patología característica denominada artropatía hemofílica, que provoca una severa limitación de la función articular y dolor crónico. El objetivo primario del tratamiento es la prevención de su desarrollo.
Clasificación
Se resume en la Tabla 1; se basa en la severidad, que depende del nivel plasmático del FVIII/FIX.
Diagnóstico
Se sospecha por la prolongación del Tiempo de tromboplatina parcial activada (TTPA), que corrige con el agregado de plasma normal y es necesaria la determinación del nivel de FVIII/FIX para su diagnóstico.El diagnóstico de hemofilia se debe considerar en las siguientes situaciones clínicas:• Neonatos: Aquellos con presencia de hematomas musculares en los
sitios de administración de vitamina K o vacunas, hemorragia intracra-neal, cefalohematoma, hematomas en sitios de venopunción, etc.
4 | Hemofilia
Tabla 1. Clasificación en base al nivel plasmático de los FVIII/FIX, indicando las características clínicas
<1 U/dL de factor
Las hemorragias pueden ser espontáneas
Episodios hemorrágicos muy frecuentes; compromiso de varias articulaciones
1 a 5 U/dL
Pueden sangrar por traumatismos mínimos
Hemorragias menos frecuentes; pueden presentar compromiso articular
>5 U/dL de factor
Pueden sangrar por traumatismos severos, cirugías, etc.
Hemorragias muy infrecuentes; es raro el compromiso articular
Hemofilia Severa Hemofilia Moderada Hemofilia Leve

69
• Niños: El inicio de la deambulación y el gateo pueden originar hematomas glúteos, subcutáneos en piernas, hemartrosis en tobillos o rodillas en pacientes con hemofilia severa. El corte del frenillo del labio superior (traumático) suele ser otra localización habitual de hemorragia persistente en estos pacientes.
Diagnóstico Molecular. Constituye el método para identificar la mutación responsable de la hemofilia y es el método recomendado para detección de portadoras (Guías Británicas). Diagnóstico Prenatal. Se realiza por medio del estudio genético de una biopsia de vellosidades coriónicas, entre las semanas 9 y 11 de gestación, o de amniocentesis (alrededor de la semana 20 de gestación). Es posible realizar el estudio genético (preimplantatorio) en embriones.
Tratamiento
Agentes hemostáticos.• Concentrados de FVIII y FIX fraccionados del plasma o recombinantes.• Otras opciones terapéuticas: -Desmopresina (DDAVP). En hemofilia A leve, cuando el FVIII puede elevarse hasta un nivel terapéutico adecuado. Dosis: 0,3 μg/Kg/día. (Guías Británicas). -Antifibrinolíticos (Guías Británicas): ácido tranexámico 25 mg/Kg cada 8 horas por vía oral o 15 mg/Kg endovenoso (EV), ácido ε-amino- caproico: 50-100 mg/Kg cada 6 hs (dosis máxima 24 g/día). Modalidades de tratamiento.• A demanda: administración de los concentrados sólo ante la aparición
de un evento hemorrágico.• Profilaxis: administración de concentrados para disminuir o evitar la
presencia de hemorragias.Tratamiento de las hemorragias.Debe instaurarse lo más temprano posible (ver Tabla 2). Cálculo de dosis.• FVIII (UI): peso (Kg) x nivel deseado x 0,5• FIX (UI): peso (Kg) x nivel deseado (*)(*) si se utiliza FIX recombinante, la dosis deberá multiplicarse por 1,2 en adultos y 1,5 en niños.
Hemofilia | 4

70
La Tabla 2 resume las recomendaciones de tratamiento de hemorragias en pacientes con hemofilia sin inhibidor descriptas a continuación.Hemorragias menores. Hemartrosis, hematomas musculares, hema-turia, sangrados mucosos, epistaxis, etc. Elevar el nivel de factor a un mínimo de 30 U/dL con los primeros síntomas o inmediatamente después del trauma. Según la severidad de la hemorragia se continuará el tratamiento sustitutivo diario, así como las medidas adyuvantes (crioterapia, reposo temporal, analgésicos, kinesiología).Hematoma de músculo psoas-ilíaco. Esta hemorragia puede presentarse simulando un abdomen agudo con dolor en fosa ilíaca, ingle y/o región lumbar, dolor y limitación en la extensión del muslo. Ante la sospecha se recomienda internar al paciente, indicar reposo absoluto y elevar inmediatamente el nivel de factor por encima del 50 U/dL durante 48 a 96 horas. Mantener luego niveles superiores a 30 U/dL durante 5 a 7 días más. Si la evolución es adecuada, continuar con profilaxis durante 4 a 12 semanas.Hemorragias mayores. Hemorragias en sistema nervioso central (SNC) o traumatismo de cráneo, hemorragia en cuello, piso de la boca, digestiva, etc. Administrar factor inmediatamente, antes de la confirmación diagnóstica, con el objetivo de alcanzar un nivel de factor del 70 al 100 U/dL. En presencia de hemorragia en SNC continuar con factor diario durante al menos 15 días (nivel 50 U/dL). Luego indicar profilaxis por al menos 6 meses. La infusión continua de factor (2-4 UI/Kg/hora) permite mantener niveles constantes diarios en caso de hemorragias severas. Es necesario el monitoreo diario de los niveles plasmáticos alcanzados (Guías Británicas).Manejo odontológico y quirúrgico. Alcanzar un nivel plasmático cercano a 50 U/dL antes de procedimientos que requieran anestesia troncular. Ante exodoncias alcanzar un nivel mínimo de 50 U/dL previo a la misma y continuar tratamiento con una dosis diaria por 48 hs (nivel requerido ≥30 U/dL).Procedimientos invasivos menores. Infundir factor, 10 min antes del procedimiento, para alcanzar un nivel plasmático de 50 U/dL. Continuar con niveles ≥30 U/dL durante 48 hs.Procedimientos quirúrgicos. Evaluar la respuesta del paciente al factor correspondiente previo a la cirugía. Inmediatamente antes del procedimiento indicar una dosis de factor para elevar el nivel plasmático a 80-100 U/dL. Mantener niveles superiores a 50 U/dL en el post-operatorio inmediato. Mantener un nivel apropiado de factor durante 5 a 7 días para cirugías menores y de 10 a 14 días para cirugías mayores y/o traumatológicas. Indicar
4 | Hemofilia

71
profilaxis post-quirúrgica durante 1 a 3 meses.
Profilaxis
La profilaxis continua, a edad temprana (primaria), es el tratamiento ideal del paciente con hemofilia severa y es el único capaz de prevenir la artropatía hemofílica (Guía Argentina).Tipos de profilaxis. Se resumen en la Tabla 3.Dosis de profilaxis. El esquema deberá ajustarse en forma individualizada. Se puede iniciar un esquema de una dosis por semana e ir escalando hasta administrar el esquema óptimo:• FVIII: 20-40 UI/Kg tres veces por semana• FIX: 30-50 UI/Kg dos veces por semana. Duración de la profilaxis. La profilaxis continua debe realizarse hasta que los pacientes alcancen la madurez física (alrededor de los 18 años). Luego, se recomienda continuar la profilaxis ajustando el esquema (dosis y frecuencia) en base al fenotipo de sangrado.
Tabla 2. Recomendaciones para el tratamiento de hemorragias, en pacientes con hemofilia sin inhibidor
Articular, hematoma muscular superficial
Hematoma músculo psoas, muscular profundo/extenso
SNC, gastrointestinal, cuello y garganta
Cirugía menor
Cirugía mayor
>30
Inicial: >50Mantenimiento: >30
Inicial: 70-100Mantenimiento: >50
Pre-operatorio: >50Post-operatorio: >30
Pre-operatorio: 80-100Post-operatorio: >50
Post-operatorio: 30-50
1-3
2-35-7
(continuar con profilaxis)
1-78-15
(continuar con profilaxis)
2-7(según procedimiento)
5-710-15
(continuar con profilaxis)
Tipo de hemorragia Nivel de FVIII/FIX deseado (U/dL) Duración del tratamiento (días)
SNC: sistema nervioso central.
Hemofilia | 4

72
Inhibidores en hemofilia
Los inhibidores son anticuerpos dirigidos contra el FVIII o el FIX. Interfieren con la acción del concentrado de factor infundido, haciéndolos ineficaces e implican la necesidad de utilizar agentes hemostáticos alternativos como los agentes bypaseantes: FVII activado recombinante (rFVIIa) y concentrado de complejo protrombínico activado (CCPa). Es la complicación del tratamiento más significativa observada actualmente, en pacientes con hemofilia. La mayoría de los inhibidores en hemofilia son de tipo I que neutralizan completamente a los factores VIII o IX. El 20-30% de los pacientes con hemofilia A severa y menos del 5% de los pacientes con hemofilia B severa desarrollan inhibidores. La presencia de un inhibidor no modifica la frecuencia o severidad de las hemorragias, pero hace más dificultoso el manejo de las mismas.En un paciente con hemofilia, un TTPA prolongado que no corrige con el agregado de plasma normal, constituye un importante indicio de la presencia de inhibidor. El diagnóstico de los inhibidores se deberá realizar con el método de Bethesda modificado o Nijmegen (Guía Argentina). Se
Profilaxis primaria
Profilaxis secundaria
Profilaxis intermitente
Tratamiento regular continuo (mínimo de 45 semanas al año) que comienza a aplicarse:- luego de la primera/segunda hemartrosis, en ausencia de una enfermedad articular óseo-cartilaginosa- a partir de los 2 años, si no ha ocurrido aún una hemartrosis- posterior a una hemorragia en SNC (en ausencia de las 2 situaciones previas)
Tratamiento regular continuo, que comienza a aplicarse después de que se han producido más de 2 hemartrosis o en presencia de daño articular
Tratamiento que se aplica para prevenir hemorragias durante períodos cortos (entre 1 a 6 meses) en las siguientes situaciones:- posterior a hematomas musculares- pre y post-cirugías ortopédicas (junto a la kinesioterapia)- hemartrosis recurrentes en una misma articulación,post-hemorragia grave.
Tabla 3. Tipos de profilaxis
SNC: sistema nervioso central.
4 | Hemofilia

73
considera positivo un título >0,6 UB/mL. El mayor riesgo de desarrollo de inhibidor se presenta en las primeras 20-50 exposiciones al FVIII/IX.Se clasifican en:• Inhibidores de baja respuesta, cuando los títulos persisten por debajo
de 5 UB/mL, aún después de la administración de FVIII o FIX.• Inhibidores de alta respuesta, cuando el título de inhibidor es mayor a 5
UB/mL y aumenta, en general, con la administración de FVIII/IX (respuesta anamnésica). Estos pacientes pueden presentar en algún momento, títulos bajos (<5UB/mL) o negativos como consecuencia de la falta de exposición al factor.
Tratamiento de los episodios hemorrágicos
Inhibidor de baja respuesta.• Hemorragias menores: FVIII/IX 50-100 UI/Kg EV por día. La duración del
tratamiento es de 2-3 días o hasta resolución del cuadro clínico.• Hemorragias mayores: FVIII/IX 100 UI/Kg en bolo EV como dosis inicial,
seguida de 50-100 UI/Kg cada 8-12 hs en bolo EV o 10 UI/Kg/hora en infusión continua. Monitorear regularmente el nivel de FVIII/IX deseado (>50 U/dL). Si no hubiera respuesta, administrar el tratamiento indicado en sangrados severos en pacientes con inhibidor de alta respuesta.
Inhibidor de alta respuesta.• Hemorragias menores: Independiente del título de inhibidor al
momento del episodio se adminstra rFVIIa (270 µg/Kg/día en bolo EV, o 90 μg/Kg cada 2 horas de 1-3 dosis/día) o CCPa (50-100 UI/Kg en bolo EV por día). La duración del tratamiento es de 2-3 días o hasta resolución del cuadro clínico. Si no hubiera respuesta al tratamiento, implementar esquema de manejo de hemorragia mayor.
• Hemorragias mayores: Iniciar tratamiento en forma inmediata con rFVIIa 90-120 μg/Kg cada 2 hs o CCPa 100 UI/Kg y evaluar el título de inhibidor.
• Título bajo (<5 UB/mL). Seguir con el tratamiento indicado en hemorragia mayor, en pacientes con inhibidor de baja respuesta. Considerar que alrededor del 5° día de inicio de la administración de FVIII/IX el título de inhibidor aumenta; si fuese mayor a 5 UB/mL, se deberá utilizar agentes bypaseantes.
• Título alto (≥5 UB/mL). Administrar rFVIIa 90-120 µg/Kg en bolo EV
Hemofilia | 4

74
cada 2 hs durante las primeras 24-48 hs; luego prolongar el intervalo de aplicación cada 3-6 hs, según evolución clínica. En niños puede ser necesario usar una dosis mayor a 120 μg/Kg. Otra opción es administrar CCPa 50-100 UI/Kg cada 6-12 hs. Es posible que en estas circunstancias se exceda la dosis máxima diaria de 200 UI/Kg; en este caso debe evaluarse riesgo/beneficio.
Se recomienda incrementar la dosis o disminuir los intervalos de dosis si la respuesta clínica es parcial. Ante la falta de respuesta o deterioro del cuadro, se sugiere cambiar por el agente bypaseante alternativo. Si la respuesta al tratamiento con el agente alternativo no es adecuada debería considerarse el tratamiento secuencial con ambos productos.Terapia secuencial: CCPa 100 UI/Kg; a las 6 horas rFVIIa 90 µg/Kg cada 2 hs por 2-3 dosis; a las 4 hs nuevamente CCPa 100 UI/Kg.Hemofilia B e inhibidor: Si presentan antecedentes de reacciones alérgicas, se recomienda el uso de rFVIIa como tratamiento de elección para el manejo de los sangrados.
Profilaxis en pacientes con inhibidor
Se sugiere considerar profilaxis con agentes bypaseantes en los siguientes escenarios (Grado 2A):• Niños con diagnóstico de inhibidor reciente, candidatos a
inmunotolerancia (IT), antes y durante la IT, con el fin de disminuir los episodios hemorrágicos y prevenir artropatía. Se recomienda usar rFVIIa a fin de evitar el aumento del título de inhibidor (anamnesis).
• Pacientes con hemorragia en SNC, posterior al evento durante ≥6 meses.• Hemorragias graves con riesgo de vida, posterior al evento durante 1-3
meses.• Pacientes no candidatos a IT o con fracaso a la misma, durante un
lapso de 3 a 6 meses en caso de: hemorragias múltiples que afecten presentismo escolar/laboral, provoquen hospitalizaciones periódicas, etc; articulación blanco; posterior a hematoma muscular; previo/post cirugía ortopédica.
Esquemas de profilaxis:• rFVIIa 90 μg/Kg/día, o días alternos, o 90-270 μg/Kg trisemanal. • CCPa 85 UI/Kg (±15%) trisemanal.
4 | Hemofilia

75
Si el esquema inicial no es efectivo, incrementar la frecuencia de la infusión sería más efectivo que incrementar la dosis.
Cirugía en pacientes con inhibidor
Es siempre un procedimiento de alto riesgo, debido a que la hemorragia incontrolable es una amenaza grave. El inhibidor debe ser cuantificado previamente. En los pacientes con inhibidor de alta respuesta con bajo título (<2 UB/mL) previo a la cirugía se puede utilizar FVIII en los primeros días de la cirugía. Se deben mantener niveles de FVIII >50 U/dL y monitorear la respuesta anamnésica. El uso de altas dosis de FIX debe evitarse en pacientes con antecedentes de reacciones alérgicas.La duración del tratamiento es de 1-5 días para cirugías menores y de 7-14 días para las mayores. Luego, se deberá indicar profilaxis intermitente, según el caso.En pacientes con hemofilia A leve o moderada, inhibidor tipo II y prueba de DDAVP con respuesta adecuada, ésta puede utilizarse en procedimientos menores. Las dosis sugeridas de agentes bypaseantes, para cirugías en pacien-tes con hemofilia e inhibidor de alta respuesta, se resumen en la Tabla 4.
Tabla 4. Dosis sugeridas de agentes bypaseantes para cirugías en pacientes con hemofilia e inhibidor de alta respuesta
Cirugía menor
Cirugía mayor
rFVIIa
Adultos: 90-180 µg/KgNiños: 120-180 µg/Kg
CCPa
75-100 UI/Kg
rFVIIa
Adultos: 90-180 µg/KgNiños: 120-180 µg/Kg
CCPa
75-100 UI/Kg
rFVIIa
90-150 µg/Kg cada 2 hs por 4 dosis y luego cada 3-6 hs
CCPa
75-100 UI/Kg cada 12-24 hs
rFVIIa
90-120 µg/Kg cada 2 horas por 48 hs, luego cada 3 hs
CCPa
75-100 UI/Kg cada 8-12 hs
rFVIIa
90-120 µg/kg cada 4-6 hs
CCPa
75-100 UI/Kg cada 12 hs
Tipo de cirugía Pre-operatorio Días 1-5post-operatorio
Días 6-14post-operatorio
Hemofilia | 4

76
Tratamiento de inmunotolerancia
La erradicación del inhibidor es el objetivo fundamental del manejo de los pacientes con inhibidor. La IT es la única estrategia terapéutica exitosa demostrada, para erradicar el inhibidor dirigido contra el FVIII. Los pacientes con hemofilia A severa e inhibidor (positivo en más de una oportunidad), que interfiere en la profilaxis o en el tratamiento de las hemorragias con dosis estándar del FVIII, deberían iniciar IT para erradicar el inhibidor.Esquema inicial.• Pacientes con inhibidor de baja respuesta:-FVIII 50 UI/Kg 3 veces por semana o días alternos.• Pacientes con inhibidor de alta respuesta:-Título de inhibidor <10 UB/mL y pico histórico <200 UB/mL: FVIII 50-100 UI/Kg 3 veces por semana. También se podría utilizar esquema de altas dosis.-Título de inhibidor >10 UB/ml y/o pico histórico >200 UB/ml: esquema altas dosis (FVIII 200 UI/Kg/día).Resultado exitoso. Título del inhibidor indetectable y farmacocinética normal. El promedio de éxito de la IT en Hemofilia A severa es del 68-86%.Resultado parcial. Reducción del título del inhibidor (<5 UB/mL) sin normalización de la farmacocinética; ausencia de respuesta anamnésica en un período prolongado de tiempo.Fracaso. Ausencia de los criterios de éxito luego de 33 meses de tratamiento ininterrumpido o demostración de fracaso en la reducción progresiva de un 20% del título del inhibidor después de sucesivos controles cada 6 meses de IT.
Laboratorio
El laboratorio es importante en diferentes aspectos ligados al diagnóstico y seguimiento del paciente hemofílico, así como de las mujeres portadoras.
Diagnóstico y clasificación
La sospecha clínica requiere la confirmación de la deficiencia de FVIII o FIX, según se trate de hemofilia A o B. Sin embargo, igual nivel de factor no
4 | Hemofilia

77
corresponde en todos los casos a un fenotipo de sangrado similar; además, ciertos pacientes muestran discrepancias en la actividad medida por diferentes metodologías. La clasificación incorrecta, sea por error en el factor deficiente o en su nivel, llevará a un tratamiento inadecuado. En casos de defecto leve o moderado de FVIII, deberán incluirse estudios complementarios para diferenciar hemofilia A de enfermedad de von Willebrand (VWD).Control de la terapéutica. Permite estimar si se alcanzan los niveles deseados según el contexto clínico, o los niveles esperados según la cantidad de factor administrado. Si los valores post-infusión son inferiores a los esperados, se sospecha el desarrollo de inhibidores específicos, que bloquean la actividad del factor infundido; se refleja en una inadecuada respuesta clínica al tratamiento.Detección de inhibidores. Es fundamental la identificación y titulación de los inhibidores específicos de FVIII o FIX. Su presencia implica un manejo diferente, que varía según el título del inhibidor.Portadoras. Dada la herencia recesiva y ligada al sexo, las hijas de hemofílicos, las madres de un varón hemofílico con antecedentes por rama materna o de más de un hemofílico, son portadoras obligadas del gen anómalo. Las madres de un hemofílico sin antecedentes familiares, así como hermanas u otros grados de parentesco por vía materna, sin descendencia, son portadoras potenciales; la condición de portadora se determina, de ser factible, mediante análisis genético. La mayoría de las portadoras son asintomáticas (nivel promedio: 50 U/dL FVIII o FIX); algunas pueden presentar niveles semejantes a un hemofílico leve o moderado, con el consecuente riesgo de sangrado. Se recomienda (Grado 3, World Federation of Hemophilia) la evaluación del factor deficiente en familiares (madres, hermanas, hijas) de hemofílicos en especial previo a un procedimiento invasivo, parto o ante síntomas hemorrágicos.Embarazo. el FVIII aumenta, se recomienda (Grado 3 Guías WFH) medirlo en el 3er trimestre para decidir la conducta clínica al momento del parto; si bien el FIX no cambia significativamente, su nivel condiciona las decisiones clínicas.
Pruebas de coagulación
Se requiere conocimiento y experiencia en la realización e interpretación de las pruebas, uso correcto de equipos y reactivos, así como adecuados controles de calidad. El control de las variables pre-pre-analíticas, pre-
Hemofilia | 4

78
analíticas, analíticas y post-analíticas es crítico; abarca desde la adecuada evaluación clínica y pedido de estudios, hasta la correcta interpretación e informe de los resultados.Variables pre-analíticas. Las condiciones pre-analíticas son las habituales para los estudios de coagulación. El FVIII es un reactante de fase aguda, aumenta además por estrés o ejercicio previo a la toma de muestra y puede activarse por acción de trazas de trombina (producto de la contaminación del plasma con material tromboplástico); en todas estas circunstancias la actividad detectada será mayor a la real. En neonatos o en niños pequeños es posible obtener niveles más altos que los reales.Fase analítica. Se realizan las pruebas de screening para detectar posibles causas de sangrado; el diagnóstico se confirma determinando la actividad de los factores. La Figura 1 resume la sistemática del estudio; muestra el TTPA prolongado, la actividad funcional disminuida de FVIII o FIX (hemofilia A o B)
B: disminuido; A: prolongado; FC: fase de contacto; N: plasma normal; P: plasma del paciente; RP: recuento de plaquetas; TLC: tiempo de lisis del coágulo; TLE: tiempo de lisis de euglobulinas; TP: tiempo de protrombina; TS: tiempo de sangría; TTPA: tiempo de tromboplastina parcial activada.
Figura 1. Algoritmo del estudio de coagulación en el paciente hemofílico
4 | Hemofilia

79
y la corrección con plasma normal del TTPA y de la actividad del factor.TTPA. Utilizar un reactivo sensible al déficit de factores, en especial FVIII y FIX; preferentemente no sensible a inhibidor lúpico (IL).Determinación de la actividad del FVIII o FIX. Se utiliza comunmente la técnica en una-etapa; debe expresarse en unidades internacionales usando plasma de referencia calibrado contra el correspondiente estándar internacional de la World Health Organization (WHO). La determinación en una única dilución del plasma problema reduce la precisión del ensayo y puede llevar a resultados inexactos en presencia de inhibidores de interferencia. Al evaluar deficiencias severas o moderadas, se requieren diluciones adicionales del calibrador (curva ampliada o curva baja); no es correcto extender simplemente la curva por extrapolación porque la sensibilidad del sistema puede variar en un rango amplio de concentración (1-100 U/dL). Ante diferentes formas moleculares puede haber discrepancias según se aplique la técnica en una-etapa o la cromogénica (dos-etapas). Es conveniente contar con más de un tipo de ensayo para una mejor caracterización, en especial de las formas leves y para el control de los nuevos productos terapéuticos.Diagnóstico diferencial. La Tabla 5 señala los diagnósticos probables según los resultados de las pruebas de screening.
Normal
HemofiliaA o B*
VWD
Defecto plaquetario
Normal
Normal
Normal
Normal
Normal
Prolongado
Normal o prolongado
Normal
Normal
Normal
Normal o prolongado
Normal o prolongado
Normal
Normal
Normal o disminuido
Normal o disminuido
Tabla 5. Diagnóstico diferencial de hemofilia - Pruebas de screening
Diagnósticoprobable
TP TTPA Tiempo de sangría
Recuento de plaquetas
(*) o deficiencia de FXI, FXII o factores de la fase de contacto; TP: tiempo de protrombina; TTPA: tiempo de tromboplastina parcial activada; VWD: enfermedad de von Willebrand.
Hemofilia | 4

80
Detección de inhibidores. La Figura 2 muestra el algoritmo diagnóstico. Se sospecha inhibidor cuando el TTPA no corrige o corrige parcialmente por el agregado de normal. El resultado del índice de Rosner (ver capítulo 6 “Inhibidores adquiridos”), puede ser difícil de interpretar dado el posible defecto combinado (déficit+inhibidor). Además, el efecto progresivo tiempo y temperatura dependiente de los inhibidores de FVIII, puede no manifestarse si no se evalúa luego de la incubación (1-2 hs) a 37°C (ver capítulo 6 “Inhibidores adquiridos”). Siempre debe demostrarse la inhibición específica sobre la actividad del factor; la mezcla no corrige cuando la actividad
(-): negativo; (+): positivo; B: disminuido; A: prolongado; FC: fase de contacto; IL: inhibidor lúpico; N: plasma normal; P: plasma del paciente; RP: recuento de plaquetas; TLC: tiempo de lisis del coágulo; TLE: tiempo de lisis de euglobulinas; TP: tiempo de protrombina; TS: tiempo de sangría; TT: tiempo de trombina; TTPA: tiempo de tromboplastina parcial activada.
Figura 2. Algoritmo del estudio de coagulación en el paciente hemofílico con inhibidor
4 | Hemofilia

81
observada es menor a la teórica esperada. El análisis de paralelismo entre las curvas del paciente y del control aporta información adicional. Los títulos de inhibidor muy elevados pueden interferir en la determinación de otros factores medidos por el mismo sistema (ver cap. 6 “Inhibidores adquiridos”). Además debe descartarse IL, el más frecuente de la coagulación, cuyo efecto dependiente de fosfolípidos no inhibe específicamente un único factor sino que interfiere en la determinación de todos los factores de la vía.Titulación del inhibidor. Se aplica la modificación de Nijmegen, más sensi-ble y específica que el método Bethesda original (Grado 1 Guías WFH) (ver cap. 6 “Inhibidores adquiridos”). Los inhibidores aFIX actúan de modo inmediato, por lo cual la titulación no requiere incubación durante 2 hs a 37°C.
Control de calidad
Asegura confiabilidad en la evaluación y reporte de los resultados para sustentar las decisiones clínicas, monitorear la terapéutica y diagnosticar/clasificar adecuadamente la anormalidad.Controles internos (normales y anormales): son útiles para identificar el grado de precisión de los ensayos.Controles externos: permiten evidenciar la variabilidad inter-laboratorios; se recomienda la participación en esquemas como External Quality Assurance System (EQAS) de la WHO o el de la WFH, diseñado especialmente para centros de tratamiento de hemofílicos.
Conflictos de interés
Daniela Neme ha recibido honorarios de los laboratorios Novo Nordisk, Baxter, Pfizer en concepto de conferencias, asesorías y actividades educativas en las que ha participado.Ludmila Elhelou ha recibido honorarios de los laboratorios Novo Nordisk y Baxter, en concepto de conferencias y actividades educativas en las que ha participado.Miguel Tezanos Pinto ha recibido honorarios del laboratorio Novo Nordisk en concepto de conferencias, asesorías y actividades educativas en las que ha participado.Egle Honnorat y Alicia N. Blanco no presentan conflictos de interés.
Hemofilia | 4

82
Referencias
1. Blanchette VS, Key NS, Ljung LR, et al. Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost 2014; 12: 1935-1939.
2. Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol 2013; 160: 153-170.
3. Favaloro EJ, Meijer P, Jennings I, et al. Problems and solutions in laboratory testing for hemophilia. Semin Thromb Hemost 2013; 39: 816-833.
4. Favaloro EJ, Verbruggen B, Miller CH. Laboratory testing for factor inhibitors. Haemophilia 2014; 20: 94-98.
5. Guías para el Tratamiento de la Hemofilia, Federación Mundial de Hemofilia, 2012, Segunda edición, www.wfh.org.
6. Guía para el manejo de la hemofilia congénita 2015, Consenso de médicos especialistas en hemofilia de la República Argentina, Fundación de la Hemofilia, 2015, Segunda edición, Buenos Aires, Argentina.
7. MASAC Recommendation concerning prophylaxis, National Hemophilia Foundation, document #179, www.hemophilia.org.
8. MASAC Recommendation regarding prophylaxis with bypassing agents in patients with hemophilia and high titer inhibitors, National Hemophilia Foundation, document #220, www.hemophilia.org.
9. MASAC Recommendation regarding the use of bypassing agents in patients with hemophilia A or B and inhibitors, National Hemophilia Foundation, document #220, www.hemophilia.org.
4 | Hemofilia

83
10. Richards M, Williams M, Chalmers E et al. United Kingdom Haemophilia Centre Doctors’ Organization guideline approved by the British Committee for Standards in Haematology: guideline on the use of prophylactic factor VIII concentrate in children and adults with severe haemophilia A. Br J Haematol 2010; 149: 498-507.
11. Textbook of hemophilia, Christine A. Lee, Erik E. Berntorp, W. Keith Hoots. 2014, Third edition, Wiley-Blackwell, New Jersey, USA.
12. UKHCDO protocol for first line immune tolerance induction for children with severe hemophilia A: A protocol from the UKHCDO Inhibitor and Paediatric Working Parties, United Kingdom Haemophilia Centre Doctors’Organization, 2015, www.ukhcdo.org.
13. Young G, Auerswald G, Jimenez-Yuste V et al. When should prophylaxis therapy in inhibitor patients be considered? Haemophilia 2011; 17: 849-857.
Hemofilia | 4

84

85
Desórdenes hemorrágicos raros del sistema de coagulaciónCristina Duboscq1 y Patricia Casais2.1Directora Técnica del Laboratorio deHemostasia. Servicio de Hematología. Hospital Británico de Buenos Aires.2Doctora en Medicina (UBA); Instituto de Investigaciones Epidemiológicas, Academia Nacional de Medicina; Centro de Hematología Pavlovsky.
Introducción
Los déficits raros de factores de la coagulación representan el 3-5% de todos los trastornos hemorragíparos hereditarios. Comprenden las deficiencias congénitas de fibrinógeno (FBG), factor (F) II, FV, FVII, FX, FXI, FXIII y la deficiencia combinada de FV y FVIII; algunos autores incluyen también el déficit conjunto congénito de factores vitamina K dependientes que es extremadamente raro (VKCFD) (1). Habitualmente se trasmiten de modo autosómico recesivo, excepto para algunos déficits de FXI y alguna hipo o disfibrinogenemia.En la mayoría de los casos las mutaciones se encuentran en el gen que codifica para cada factor de coagulación con excepción de la deficiencia combinada de FV y FVIII y las alteraciones conjuntas de los factores vitamina K dependientes. La deficiencia combinada de FV y FVIII está causada por mutaciones en genes que codifican para proteínas de trasporte intracelular de estos factores (MCFD2 -multiple coagulation factor deficiency protein 2- y LMAN1 -lectin mannose-binding 1-). En el caso de VKCFD la alteración se encuentra en los genes que codifican para enzimas involucradas en la modificación post-traduccional de los factores vitamina K dependiente y/o
5
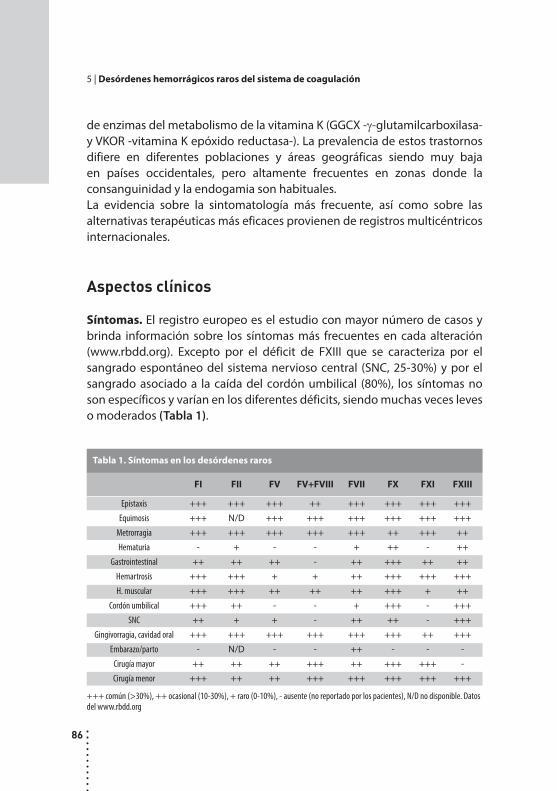
86
de enzimas del metabolismo de la vitamina K (GGCX -g-glutamilcarboxilasa- y VKOR -vitamina K epóxido reductasa-). La prevalencia de estos trastornos difiere en diferentes poblaciones y áreas geográficas siendo muy baja en países occidentales, pero altamente frecuentes en zonas donde la consanguinidad y la endogamia son habituales.La evidencia sobre la sintomatología más frecuente, así como sobre las alternativas terapéuticas más eficaces provienen de registros multicéntricos internacionales.
Aspectos clínicos
Síntomas. El registro europeo es el estudio con mayor número de casos y brinda información sobre los síntomas más frecuentes en cada alteración (www.rbdd.org). Excepto por el déficit de FXIII que se caracteriza por el sangrado espontáneo del sistema nervioso central (SNC, 25-30%) y por el sangrado asociado a la caída del cordón umbilical (80%), los síntomas no son específicos y varían en los diferentes déficits, siendo muchas veces leves o moderados (Tabla 1).
Epistaxis
Equimosis
Metrorragia
Hematuria
Gastrointestinal
Hemartrosis
H. muscular
Cordón umbilical
SNC
Gingivorragia, cavidad oral
Embarazo/parto
Cirugía mayor
Cirugía menor
FI
+++++++++
-++
+++++++++++
+++-
+++++
FVII
+++++++++
++++++++
+++++++++
+++
FV
+++++++++
-+++
++-+
+++-
++++
FXI
+++++++++
-++
++++--
++-
++++++
FII
+++N/D+++
+++
+++++++++
+++N/D++++
FX
++++++++++
++++++++++++++
+++-
++++++
FV+FVIII
++++++++
--+
++--
+++-
++++++
FXIII
++++++++++++
+++++
+++++++++
--
+++
+++ común (>30%), ++ ocasional (10-30%), + raro (0-10%), - ausente (no reportado por los pacientes), N/D no disponible. Datos del www.rbdd.org
Tabla 1. Síntomas en los desórdenes raros
5 | Desórdenes hemorrágicos raros del sistema de coagulación

87
El déficit de FBG plantea un caso especial, ya que incluye no sólo los defectos cuantitativos (afibrinogenemia e hipofibrinogenemia) sino también las alteraciones cualitativas (disfibrinogenemias) que pueden presentar síntomas hemorrágicos o trombóticos (20% de los casos). En pacientes asintomáticos o con sangrado leve, la sospecha clínica surge a partir de una alteración de laboratorio. Cuantificación del sangrado. se ha utilizado el Bleeding assessment tool (BAT), diseñado para la enfermedad de von Willebrand (VWD), aunque carece de especificidad y de poder de discriminación entre trastorno congénito y adquirido. A mayor puntaje del BAT, mayor probabilidad de alteración de la coagulación. Clasificación clínica del sangrado. teniendo en cuenta la localización, el potencial impacto clínico, el desencadenante del sangrado (espontáneo, asociado a trauma, o inducido por drogas) se ha propuesto la siguiente clasificación de la severidad del sangrado (1):• Grado I: sangrado asociado a trauma o drogas.• Grado II: sangrado espontáneo menor (equimosis, heridas menores,
cavidad oral, epistaxis, menorragia).• Grado III: sangrado espontáneo mayor (hematomas, hemartrosis, SNC,
tracto gastrointestinal, cordón umbilical).
Diagnóstico por el laboratorio de los deficits raros de los factores de coagulación
Este conjunto de desórdenes puede diagnosticarse por casualidad o porque se solicita el estudio de coagulación por manifestaciones de sangrado. En este último, si la clínica es importante es aconsejable realizar el dosaje de factores, aunque las pruebas de tiempo de protrombina (TP) y tiempo de tromboplastina parcial activada (TTPA) sean normales ya que son pruebas globales que, según el reactivo y el sistema de detección que se utilice, pueden tener baja sensibilidad para detectar un déficit leve de factores.Por otro lado, dada la baja prevalencia de estas alteraciones, cuando se identifica algún déficit siempre debe ser confirmado al menos en una nueva muestra y en familiares directos, en especial en niños.El siguientes es un algoritmo diagnostico clásico para evaluar los componen-tes plasmáticos del sistema de coagulación (Figura 1). El estudio comienza
Desórdenes hemorrágicos raros del sistema de coagulación | 5

88
por las pruebas globales TTPA, TP y tiempo de trombina (TT). Si alguna o ambas pruebas son anormales se realizan las pruebas de mezcla, es decir 1 volumen de plasma del paciente (PP) +1 volumen de plasma normal (PN), para excluir la presencia de un inhibidor. De acuerdo a cuál(es) prueba(s) de screening esté(n) alterada(s) se dirige la o las determinaciones de factores.
Tener en cuenta que en el caso de la deficiencia aislada de FXIII el TTPA y el TP son siempre normales. Otro punto para recordar es que solo niveles de FBG por debajo de 50 mg/dL modifican el TP y/o el TTPA (Tabla 2).
Def. de FII
Def. de FV
Def. de FVII
Def. de FX
Def. de FXI
Def. de FXIII
Def. de FI
Afibrinog.
Hipofibrinog.
Disfibrinog.
Def. combinada de FV/FVIII
VKCFD
Prol.
Prol.
Prol.
Prol.
Normal
Normal
Prol.
Normal/ Prol.
Normal/ Prol.
Prol.
Prol.
Prol.
Prol.
Normal
Prol.
Prol.
Normal
Prol.
Normal/ Prol.
Normal/ Prol.
Prol.
Prol.
Normal
Normal
Normal
Normal
Normal
Normal
<1 g/L
1-1,5 g/L
Normal/levemente reducido
Normal
Normal
Normal
Normal
Normal
Normal
Normal
Normal
Prol.
Poco prol.
Prol.
Normal
Normal
Def: deficiencia; Prol: prolongado; VKCFD: déficit conjunto congénito de factores vitamina K dependientes.
Tabla 2. Comportamiento de las pruebas básicas en los distintos déficits
VPP (% CON deficiencia de algún factor)
TP TTPA FI TT
5 | Desórdenes hemorrágicos raros del sistema de coagulación
Figura 1. Algoritmo de diagnóstico de laboratorio para el diagnóstico de los déficits raros
H/LMWK: quininógeno de bajo/alto peso molecular.

89
Variables pre-analíticas
Se deben cumplir estrictamente las recomendaciones para la extracción, conservación y procesamiento de la muestra. Las muestras deben conser-varse a temperatura ambiente y procesarse antes de las 4 hs, sobre todo si se estudian factores lábiles como el FV.
Dosaje de factores
Habitualmente se realizan por el método coagulable en una etapa, si es posible en más de una dilución para disminuir el coeficiente de variación (CV). Las distintas guías internacionales recomiendan para el dosaje de cualquier factor:• La curva de calibración debe realizarse contra un plasma calibrador
y contra los estándares internacionales. Las curvas de tres puntos en coagulómetros automáticos, en coagulómetros semi-automáticos o manuales deben realizarse por sesión. Actualmente en algunos coagulómetros automáticos existen curvas de varios puntos que son más estables, en este caso se debe realizar la misma por cambio de lotes de reactivo, o cuando los controles así lo indiquen.
• Los dosajes de factores deben realizarse al menos entres diluciones, en especial los que se determinan por la vía del TTPA, esto disminuye el CV y permite excluir la presencia de inhibidores.
• Los valores de coagulación deben estar dentro de la curva de calibración. De lo contrario, se debe hacer la prueba en una dilución diferente.
• Deben utilizarse controles internos al menos de dos niveles, uno normal y otro patológico, y el laboratorio debe participar en un programa externo de calidad para cada analito.
Para mayores detalles técnicos consultar Fundamentos para el Manejo Práctico del Laboratorio de Hemostasia, Grupo CAHT, Edición 2013.
Dosaje de factores por la vía del TP. Por esta vía se determinan el FII, FV, FVII y FX. El rango normal por lo general es 70 -120 UI/dL para los FV, FVII y FX. El límite inferior para la normalidad del FII es alrededor de 80 UI/dL.Los pacientes que presenten un nivel bajo de FV también deberían someterse a un ensayo de FVIII para descartar la deficiencia conjunta de estos factores.
Desórdenes hemorrágicos raros del sistema de coagulación | 5

90
Se han reportado algunas diferencias en los niveles de FVII coagulable de acuerdo al origen de las tromboplastinas utilizadas, por lo que es recomendable el uso de tromboplastina humana recombinante para el dosaje de FVII ya que este reactivo reflejaría mejor la actividad in vivo. En ese sentido, se han descripto niveles de FVII muy bajos con tromboplastina de conejo que presentaron valores normales realizando el ensayo con tromboplastina de origen humano. La tendencia al sangrado correlacionaría mejor con los ensayos que utilizan la tromboplastina de origen humano. En aproximadamente el 10% de los déficits de FVII el nivel antigénico es normal.Dosaje de factores por la vía del TTPA. Por esta vía se determinan el FVIII, FIX, FXI y FXII cuyo déficit no produce manifestaciones hemorrágicas. Los rangos de normalidad en general son entre 50 y 150 UI/dL para los FVIIII y FIX y el límite inferior normal para el FXI estaría entre 63 y 80 UI/dL, según distintos reportes.Pediatría. Para evaluar los desórdenes raros en niños hay que tener presente que los valores de referencia para neonatos a término y pre-término en los primeros 6 meses de vida difieren de los valores de adultos, en particular para todos los factores de coagulación excepto el FVIII. A causa de que los niveles de los factores aumentan durante los primeros meses de vida las deficiencias moderadas deben ser confirmadas cuando el niño sea mayor y/o estudiando a ambos padres.
Estudio de las alteraciones del FBG
El diagnóstico preciso de algunos desordenes del FBG es difícil aún en los laboratorios especializados. Esto es particularmente verdad en el caso de las disfibrinogenemia (discrepancia entre el nivel de FI coagulable e inmunológico) debido a que la sensibilidad de las distintas pruebas depende de las diferentes mutaciones, reactivos y técnicas utilizadas. La determinación de los niveles de FI debe realizarse por el método coagulable y no debe utilizarse el método derivado del TP.Afibrinogenemia. en el caso de la afibrinogenemia, hay una marcada prolongación del TP, TTPA y TT. El FI es indetectable tanto en el ensayo coagulable como en el inmunológico. Es importante excluir las causas adquiridas de hipofibrinogenemia principalmente la disfunción hepática. También son útiles los estudios familiares para confirmar el diagnóstico. El
5 | Desórdenes hemorrágicos raros del sistema de coagulación

91
tiempo de reptilase también es muy prolongado.Hipofibrinogenemia. Las pruebas de screening están prolongadas de acuerdo al nivel de la deficiencia de FI. El TT es la prueba más sensible. El FI coagulable e inmunológico están ambos reducidos en niveles similares. Es importante excluir las causas adquiridas de hipofibrinogenemia, princi-palmente la disfunción hepática. También son útiles los estudios familiares para confirmar el diagnóstico.Disfibrinogenemia. El TP y el TTPA pueden estar prolongados, pero el TT es generalmente la prueba más sensible en el screening para la disfibri-nogenemia (Tabla 3). El grado de prolongación depende de la mutación presente. Usualmente está prolongado, pero en algunos casos podría ser normal o incluso estar acortado. En estos casos es importante excluir la presencia de heparina y la interferencia con la polimerización de la fibrina por altos niveles de productos de degradación de la fibrina (PDF), para proteínas, o algún inhibidor de trombina. El tiempo de reptilase, en algunos casos más sensible que el TT, está usualmente prolongado, pero también puede ser normal o acortado. El FI coagulable medido por Clauss suele estar disminuido mientras que la concentración antigénica es normal o aumentada.
Prol.
Prol.Prol.Prol.
Prol.
Igualmente prol.
Marcadamente prol.
Normal
Levemente prol.
Igualmente
prol.
Hipofibrinogenemia o
afibrinodenemia
Disfibrinogenemia
Heparina
Heparina y algún tipo de
hipofibrinogenemia o
disfibrinodenemia
Coagulación intravascular
diseminada (CID)
Medir FBG
Congénita o adquirida
Un caso de disfibrinogenemia
reportado presenta este patrón
Medir dímeros-D
Prol.: prolongado.
Tabla 3. Evaluación de un TT prolongado
Tiempo de trombina Tiempo de reptilase Causa Comentarios
Desórdenes hemorrágicos raros del sistema de coagulación | 5

92
Diagnóstico de la deficiencia de FXIII
La investigación del FXIII debe realizarse siempre que un paciente presente signos hemorrágicos, con estudios de plaquetas y pruebas de coagulación (TP, TTPA y TT) normales, ya que la deficiencia aislada de FXIII no modifica el tiempo de formación del coágulo. La única prueba global que se altera (parcialmente) con el déficit de factor XIII es el tromboelastograma (TEG). En el caso de las deficiencias severas, también se ve afectada la solubilidad del coágulo. Para el diagnóstico y clasificación del déficit de FXIII (Tabla 1) el subcomité de estandarización de la Sociedad Internacional de Hemostasia y Trombosis (FXIII and Fibrinogen, SSC-ISTH) recomienda:• En primera línea utilizar un método funcional de FXIII, que detecte
todas las formas de deficiencia.• Si la actividad de FXIII está disminuida, se debe clasificar el subtipo de la
deficiencia utilizando las siguientes pruebas: -Medir la concentración antigénica de ambas subunidades en forma conjunta y si resulta baja, medir cada una por separado; también es válido determinar directamente la concentración antigénica de FXIII-A y FXIII-B por separado. -Medir la actividad de FXIII y la concentración de FXIII-A en el lisado plaquetario.• Evaluar la presencia de auto-anticuerpos contra FXIII de la siguiente
manera: -Realizar estudios de mezclas y medir la actividad de FXIII para detectar anticuerpos (Acs) neutralizantes contra FXIII-A. -Realizar ensayos de unión para la detección de anticuerpos no- neutralizantes contra FXIII-A y FXIII-B.• Evaluar el entrecruzamiento de la fibrina aplicando SDS-PAGE.• Identificar el defecto genético para confirmar la deficiencia hereditaria.La Tabla 4 muestra la clasificación recomendada por el FXIII and Fibrinogen, SSC-ISTH.
5 | Desórdenes hemorrágicos raros del sistema de coagulación
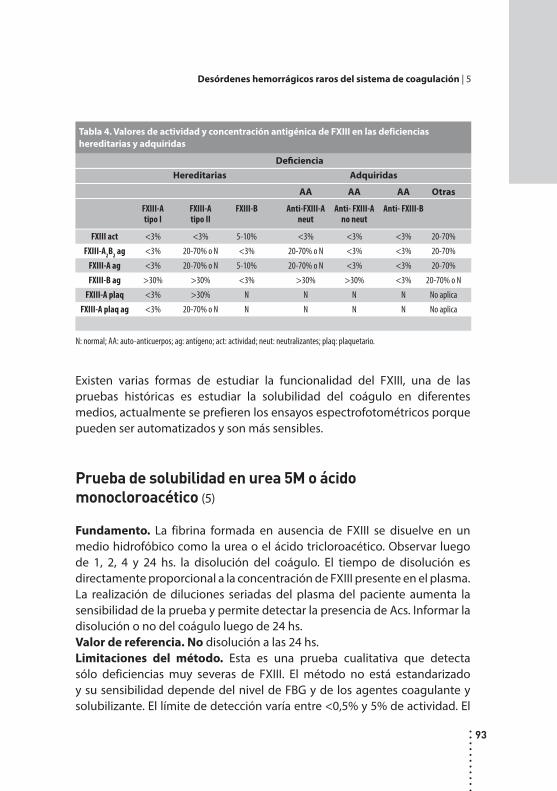
93
Existen varias formas de estudiar la funcionalidad del FXIII, una de las pruebas históricas es estudiar la solubilidad del coágulo en diferentes medios, actualmente se prefieren los ensayos espectrofotométricos porque pueden ser automatizados y son más sensibles.
Prueba de solubilidad en urea 5M o ácido monocloroacético (5)
Fundamento. La fibrina formada en ausencia de FXIII se disuelve en un medio hidrofóbico como la urea o el ácido tricloroacético. Observar luego de 1, 2, 4 y 24 hs. la disolución del coágulo. El tiempo de disolución es directamente proporcional a la concentración de FXIII presente en el plasma. La realización de diluciones seriadas del plasma del paciente aumenta la sensibilidad de la prueba y permite detectar la presencia de Acs. Informar la disolución o no del coágulo luego de 24 hs.Valor de referencia. No disolución a las 24 hs. Limitaciones del método. Esta es una prueba cualitativa que detecta sólo deficiencias muy severas de FXIII. El método no está estandarizado y su sensibilidad depende del nivel de FBG y de los agentes coagulante y solubilizante. El límite de detección varía entre <0,5% y 5% de actividad. El
FXIII actFXIII-A2B2 ag
FXIII-A agFXIII-B ag
FXIII-A plaqFXIII-A plaq ag
<3%
<3%
<3%
>30%
<3%
<3%
DeficienciaHereditarias Adquiridas
AA AA AA Otras
5-10%
<3%
5-10%
<3%
N
N
<3%
<3%
<3%
>30%
N
N
<3%
<3%
<3%
<3%
N
N
<3%
20-70% o N
20-70% o N
>30%
>30%
20-70% o N
<3%
20-70% o N
20-70% o N
>30%
N
N
20-70%
20-70%
20-70%
20-70% o N
No aplica
No aplica
N: normal; AA: auto-anticuerpos; ag: antígeno; act: actividad; neut: neutralizantes; plaq: plaquetario.
FXIII-Atipo I
FXIII-B Anti- FXIII-A no neut
Anti- FXIII-BFXIII-Atipo II
Anti-FXIII-A neut
Tabla 4. Valores de actividad y concentración antigénica de FXIII en las deficiencias hereditarias y adquiridas
Desórdenes hemorrágicos raros del sistema de coagulación | 5

94
FXIII and Fibrinogen, SSC-ISTH recomienda no utilizar esta prueba como screening de la deficiencia de FXIII.
Método espectrofotométrico
El FXIII es activado por la trombina en presencia de fibrina soluble. El FXIIIa libera amoníaco de un sustrato peptídico específico; luego, la concentración de amoníaco se determina midiendo la absorbancia a 340 nm, en una reacción acoplada. Los valores de referencia a modo orientativo son 70-140%, pero debe ser establecido por cada laboratorio. La ventaja de este método es que es rápido y al ser un ensayo cinético de un solo paso, puede ser fácilmente automatizado. El límite de detección está entre 3 y 5% de actividad. Para muestras con niveles de FXIII menores al 10% se debe realizar un blanco de muestra.
Ensayos de incorporación de aminas (4,6)
Estos ensayos están basados en la unión de una amina fluorescente o biotinilada a un residuo glutamina de una proteína, sustrato del FXIIIa. Luego de detener la reacción, las aminas libres, no unidas, son removidas y la fracción proteica es cuantificada por fluorescencia o espectrofotométricamente.La ventaja de los métodos de incorporación es su alta sensibilidad. Las desventajas son su laboriosidad y la falta de estandarización. Además, debido a los pasos de separación, no puede considerarse que sean un verdadero ensayo cinético. Los resultados de algunos ensayos de incorporación que miden la actividad transglutaminasa al inicio de la activación del FXIII son influenciados por el polimorfismo Val34Leu de este factor.
Método de incorporación con detección espectrofotométrica
El FXIIIa cataliza la incorporación de la amina biotinilada 5-(biotinamido)pentilamina (BAPA), en una proteína inmovilizada (fibrina). Luego, la amina
5 | Desórdenes hemorrágicos raros del sistema de coagulación

95
biotinilada reacciona con la estreptavidina en presencia de un sustrato específico, generando color. El incremento de la absorbancia es directamente proporcional a la concentración de FXIII presente en la muestra. Este método es capaz de detectar el cambio de actividad debido a la presencia del polimorfismo Val34Leu del FXIII. Los valores de referencia sólo a modo orientativo son 65-135%.
Método inmunoturbidimétrico para determinar la concentración de FXIII-A (2, 9)
Este método emplea partículas de poliestireno acopladas con un Ac policlonal dirigido contra la subunidad A del FXIII hecho en conejo. El grado de aglutinación es directamente proporcional a la concentración de antígeno de FXIII contenido en el plasma y se determina midiendo el descenso de la luz transmitida causado por los agregados. Hay reportes que muestran muy buena correlación entre este método y la actividad de FXIII medida espectrofotométricamente (2). Los CVs total están entre 3,5% para el control normal y 5,5% para el control patológico. Los valores de referencia solo orientativos son 79-150%.
Determinación inmunológica de FXIII
La concentración de FXIII se puede determinar por inmunoelectroforesis o ELISA utilizando anti-sueros específicos para cada subunidad (A o B). Se ha desarrollado recientemente un ELISA en un solo paso para la determinación de toda la molécula de FXIII. El valor de referencia para cada subunidad de 50-150%.
Diagnóstico diferencial
En todos los casos debe plantearse el diagnóstico diferencial con las patologías que se asocian a déficit adquirido, incluidos los inhibidores (Tabla 5).
Desórdenes hemorrágicos raros del sistema de coagulación | 5
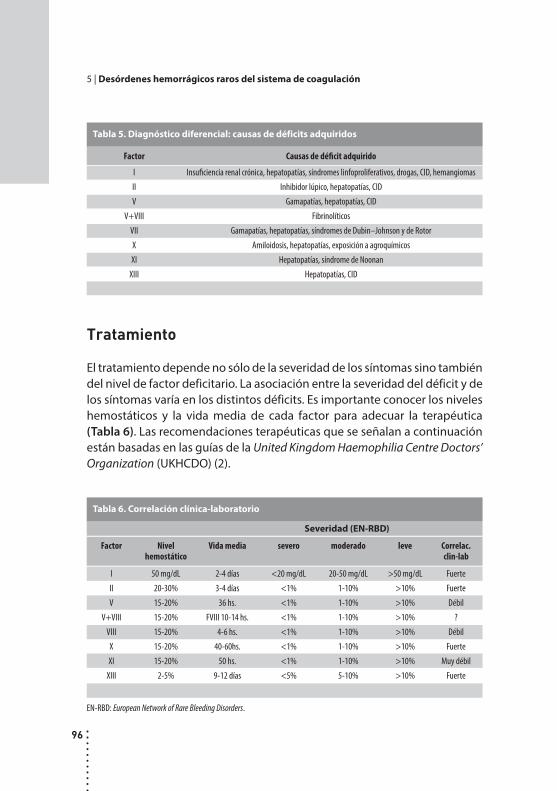
96
Tratamiento
El tratamiento depende no sólo de la severidad de los síntomas sino también del nivel de factor deficitario. La asociación entre la severidad del déficit y de los síntomas varía en los distintos déficits. Es importante conocer los niveles hemostáticos y la vida media de cada factor para adecuar la terapéutica (Tabla 6). Las recomendaciones terapéuticas que se señalan a continuación están basadas en las guías de la United Kingdom Haemophilia Centre Doctors’ Organization (UKHCDO) (2).
5 | Desórdenes hemorrágicos raros del sistema de coagulación
I
II
V
V+VIII
VII
X
XI
XIII
Insuficiencia renal crónica, hepatopatías, síndromes linfoproliferativos, drogas, CID, hemangiomas
Inhibidor lúpico, hepatopatías, CID
Gamapatías, hepatopatías, CID
Fibrinolíticos
Gamapatías, hepatopatías, síndromes de Dubin–Johnson y de Rotor
Amiloidosis, hepatopatías, exposición a agroquímicos
Hepatopatías, síndrome de Noonan
Hepatopatías, CID
Tabla 5. Diagnóstico diferencial: causas de déficits adquiridos
Factor Causas de déficit adquirido
I
II
V
V+VIII
VIII
X
XI
XIII
50 mg/dL
20-30%
15-20%
15-20%
15-20%
15-20%
15-20%
2-5%
2-4 días
3-4 días
36 hs.
FVIII 10-14 hs.
4-6 hs.
40-60hs.
50 hs.
9-12 días
<20 mg/dL
<1%
<1%
<1%
<1%
<1%
<1%
<5%
20-50 mg/dL
1-10%
1-10%
1-10%
1-10%
1-10%
1-10%
5-10%
>50 mg/dL
>10%
>10%
>10%
>10%
>10%
>10%
>10%
Fuerte
Fuerte
Débil
?
Débil
Fuerte
Muy débil
Fuerte
Severidad (EN-RBD)
Nivel hemostático
Vida media severo moderado leve Correlac.clin-lab
Factor
Tabla 6. Correlación clínica-laboratorio
EN-RBD: European Network of Rare Bleeding Disorders.

97
Recomendaciones generales. Las recomendaciones para el manejo de estos pacientes incluyen la consulta con un médico especialista y medidas generales tales como evitar las actividades de alto riesgo de sangrado, las drogas antiplaquetarias (aspirina y antiinflamatorios no esteroides) y evaluar los procedimientos invasivos según el riesgo de sangrado asociado y seleccionar, siempre que sea posible, aquel con el menor riesgo.• Sangrado espontáneo menor ó procedimientos invasivos menores.
En todos los déficits se recomienda: ácido tranexámico(AcTx) 15-20 mg/día ó 1g cada 6 hs. (Grado 2C); pero está contraindicado en casos de sangrado del tracto urinario o en pacientes con alto riesgo trombótico. En menorragia, las alternativas terapéuticas no quirúrgicas incluyen además el dispositivo intrauterino con levonorgestrel y los anticonceptivos combinados.
• Terapia de reemplazo del factor deficitario. El objetivo de la terapia de reemplazo está dirigido a alcanzar valores hemostáticos. La sustitución del factor deficitario puede realizarse con hemoderivados o, cuando está disponible, con concentrado de factor. Siempre que esté disponible se prefiere sustituir con concentrados ya que aportan sólo el factor faltante con menor volumen de infusión y mayor seguridad por los métodos de inactivación viral.
• Hemoderivados.- El plasma fresco congelado (PFC) contiene todos los factores y los inhibidores de la coagulación. Aproximadamente 1 mL de PFC contiene 1 U de Factor. Las principales desventajas están dadas por el gran volumen de infusión y el riesgo de infección (el PFC habitualmente no es sometido a métodos de inactivación viral).- Los crioprecipitados contienen 80 Ul de FVIII, 40-70% de Factor von Willebrand, 200 mg de FBG y 20-30 UI de FXIII, aproximadamente. Presentan las mismas desventajas que el PFC.- Los concentrados de complejo protrombínico (CCP) contienen FII, FVII, FIX, FX, Antitrombina III (ATIII), proteínas C y S (PC y PS, respectivamente) y heparina. Las cantidades de cada factor varían en los diferentes productos y deberán conocerse antes de administrar un concentrado determinado. La actividad del CCP se calcula según su contenido de FIX. Los CCP tienen mayor actividad de factor/mL por lo que el volumen y el tiempo de infusión son menores; todos son sometidos a procesos de inactivación viral.
Desórdenes hemorrágicos raros del sistema de coagulación | 5
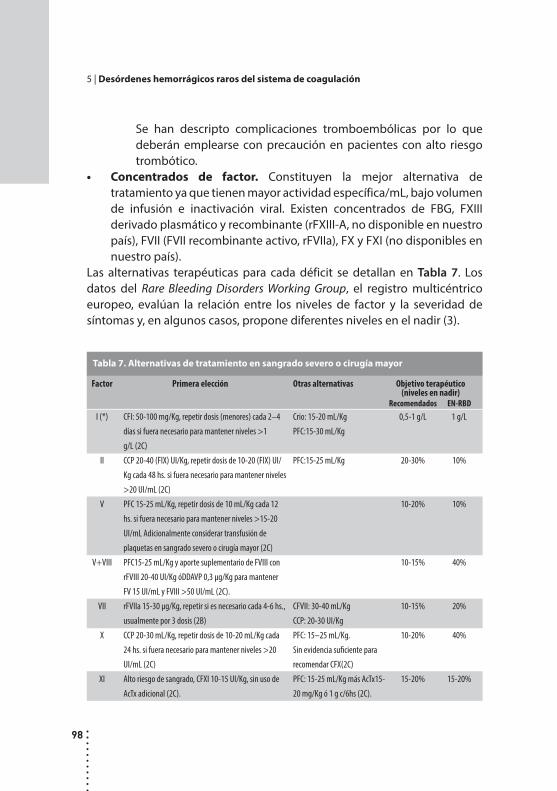
98
Se han descripto complicaciones tromboembólicas por lo que deberán emplearse con precaución en pacientes con alto riesgo trombótico.
• Concentrados de factor. Constituyen la mejor alternativa de tratamiento ya que tienen mayor actividad específica/mL, bajo volumen de infusión e inactivación viral. Existen concentrados de FBG, FXIII derivado plasmático y recombinante (rFXIII-A, no disponible en nuestro país), FVII (FVII recombinante activo, rFVIIa), FX y FXI (no disponibles en nuestro país).
Las alternativas terapéuticas para cada déficit se detallan en Tabla 7. Los datos del Rare Bleeding Disorders Working Group, el registro multicéntrico europeo, evalúan la relación entre los niveles de factor y la severidad de síntomas y, en algunos casos, propone diferentes niveles en el nadir (3).
I (*)
II
V
V+VIII
VII
X
XI
CFI: 50-100 mg/Kg, repetir dosis (menores) cada 2–4
días si fuera necesario para mantener niveles >1
g/L (2C)
CCP 20-40 (FIX) UI/Kg, repetir dosis de 10-20 (FIX) UI/
Kg cada 48 hs. si fuera necesario para mantener niveles
>20 UI/mL (2C)
PFC 15-25 mL/Kg, repetir dosis de 10 mL/Kg cada 12
hs. si fuera necesario para mantener niveles >15-20
UI/mL Adicionalmente considerar transfusión de
plaquetas en sangrado severo o cirugía mayor (2C)
PFC15-25 mL/Kg y aporte suplementario de FVIII con
rFVIII 20-40 UI/Kg óDDAVP 0,3 μg/Kg para mantener
FV 15 UI/mL y FVIII >50 UI/mL (2C).
rFVIIa 15-30 μg/Kg, repetir si es necesario cada 4-6 hs.,
usualmente por 3 dosis (2B)
CCP 20-30 mL/Kg, repetir dosis de 10-20 mL/Kg cada
24 hs. si fuera necesario para mantener niveles >20
UI/mL (2C)
Alto riesgo de sangrado, CFXI 10-15 UI/Kg, sin uso de
AcTx adicional (2C).
Crio: 15-20 mL/Kg
PFC:15-30 mL/Kg
PFC:15-25 mL/Kg
CFVII: 30-40 mL/Kg
CCP: 20-30 UI/Kg
PFC: 15–25 mL/Kg.
Sin evidencia suficiente para
recomendar CFX(2C)
PFC: 15-25 mL/Kg más AcTx15-
20 mg/Kg ó 1 g c/6hs (2C).
1 g/L
10%
10%
40%
20%
40%
15-20%
0,5-1 g/L
20-30%
10-20%
10-15%
10-15%
10-20%
15-20%
Tabla 7. Alternativas de tratamiento en sangrado severo o cirugía mayor
Factor Primera elección
Recomendados
Otras alternativas
EN-RBD
Objetivo terapéutico (niveles en nadir)
5 | Desórdenes hemorrágicos raros del sistema de coagulación

99
• Profilaxis a largo plazo. La mayoría de los pacientes con déficit de factores reciben tratamiento “a demanda” (es decir, al momento de la hemorragia o previo a un procedimiento invasivo); sin embargo, en el déficit de FXIII se recomienda el tratamiento profiláctico (Grado 2B) y en otros déficits severos con historia familiar o personal de sangrado severo se debería considerar el tratamiento profiláctico a largo plazo (Grado 2C). Los regímenes sugeridos para cada déficit se muestran en la Tabla 8.
XIII Dependiendo del intervalo entre la última dosis de
profilaxis y la severidad del sangrado, considerar dosis
adicional de concentrado 10-40 UI/kg (2C).
Crio: 2-3 bolsas
PFC: 3 mL/Kg
rFXIII-A: 35 U/Kg
30%2-5%
(*) afibrinogenemia, hipofibrinogenemia o disfibrinogenemia (con sangrado). CFI: concentrado de fibrinógeno; Crio: crioprecipitados; PFC: plasma fresco congelado; CCP: concentrado de complejo protrombínico; rFVIIa: FVII recombinante activo; CFVII: concentrado de FVII; CFIX: concentrado de FIX; CFXI: concentrado de FXI; CFXIII: concentrado de FXIII; rFXIII-A: concentrado de FXIII recombinante; AcTx: ácido tranexámico.
Tabla 7. Cont.
Factor Primera elección
Recomendados
Otras alternativas
EN-RBD
Objetivo terapéutico (niveles en nadir)
CFXIII: Concentrado de FXIII; CFI: concentrado de fibrinógeno; CCP: concentrado de complejo protrombínico; PFC: plasma fresco congelado; rFVIIa: FVII activado recombinante.
FXIII
FI
FII
FV
FVII
FX
CFXIII 20–40 UI/Kg cada 28 días, ajustado para mantener niveles de FXIII en el valle de 10-30 UI/mL
(2B). Considerar el uso de CFXIII recombinante por sobre el FXIII derivado plasmático en los casos de
déficit de la subunidad A que no hayan sido expuestos a productos derivados del plasma (2C).
Dosis inicial de CFI 50-100 mg/Kg semanal y luego ajustar para mantener niveles >50 g/L (2C).
CCP 20-40 UI/Kg seminal ajustada para mantener niveles en valle >10 UI/mL (2C)
PFC 20 mL/Kg 2 veces por semana ajustados según respuesta clínica (2C)
rFVIIa 20-40 μg/Kg 3 veces por semana ajustados según respuesta clínica (2B)
CCP 2030 UI/Kg 2-3 veces por semana para mantener niveles en el valle 10-20UI/mL en niños (2C).
Tabla 8. Regímenes de profilaxis a largo plazo
Déficit Profilaxis
Desórdenes hemorrágicos raros del sistema de coagulación | 5
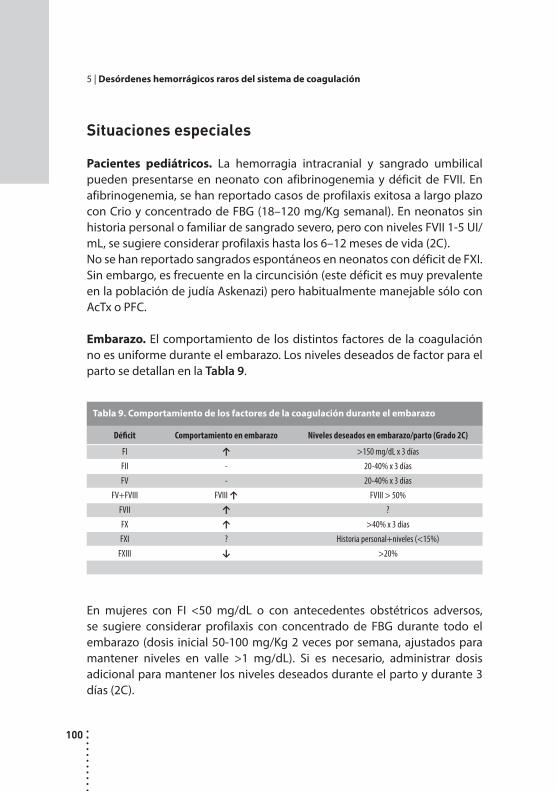
100
Situaciones especiales
Pacientes pediátricos. La hemorragia intracranial y sangrado umbilical pueden presentarse en neonato con afibrinogenemia y déficit de FVII. En afibrinogenemia, se han reportado casos de profilaxis exitosa a largo plazo con Crio y concentrado de FBG (18–120 mg/Kg semanal). En neonatos sin historia personal o familiar de sangrado severo, pero con niveles FVII 1-5 UI/mL, se sugiere considerar profilaxis hasta los 6–12 meses de vida (2C).No se han reportado sangrados espontáneos en neonatos con déficit de FXI. Sin embargo, es frecuente en la circuncisión (este déficit es muy prevalente en la población de judía Askenazi) pero habitualmente manejable sólo con AcTx o PFC.
Embarazo. El comportamiento de los distintos factores de la coagulación no es uniforme durante el embarazo. Los niveles deseados de factor para el parto se detallan en la Tabla 9.
En mujeres con FI <50 mg/dL o con antecedentes obstétricos adversos, se sugiere considerar profilaxis con concentrado de FBG durante todo el embarazo (dosis inicial 50-100 mg/Kg 2 veces por semana, ajustados para mantener niveles en valle >1 mg/dL). Si es necesario, administrar dosis adicional para mantener los niveles deseados durante el parto y durante 3 días (2C).
FI
FII
FV
FV+FVIII
FVII
FX
FXI
FXIII
↑-
-
FVIII ↑
↑ ↑?
↓
>150 mg/dL x 3 días
20-40% x 3 días
20-40% x 3 días
FVIII > 50%
?
>40% x 3 días
Historia personal+niveles (<15%)
>20%
Tabla 9. Comportamiento de los factores de la coagulación durante el embarazo
Déficit Comportamiento en embarazo Niveles deseados en embarazo/parto (Grado 2C)
5 | Desórdenes hemorrágicos raros del sistema de coagulación

101
Si el nivel de FII es <20 UI/mL en el tercer trimestre (3°T), considerar uso de CCP durante el parto o antes de la cesárea para lograr el nivel deseado. Puede ser necesaria una infusión adicional (10–20 UI/Kg) a las 48 hs. para mantener los niveles recomendados durante 3 días (2C).En mujeres con niveles de FV de 20-30 UI/mL, la infusión de PFC y rFVIIa administrados previo a la cesárea han sido efectivos. Cuando el déficit es <20 UI/mL, se sugiere PFC durante el parto o previo a la cesárea para lograr los niveles deseados. De ser necesario, repetir infusión de PFC (10 mL/Kg cada 12 hs.) para mantener niveles por 3 días (2C).Si el nivel de FVII en el 3°T es <20 UI/mL en mujeres con historia previa de sangrado o que requieren cesárea, considerar la administración de rFVIIa por 3 días. En los demás casos, sólo administrar rFVIIa en caso de sangrado anormal (2C).En pacientes con déficit de FX <30 UI/mL en el 3°T y con historia previa de sangrado o que requieren cesárea, considerar CCP para alcanzar los niveles de deseados y luego dosis menores (10–20 IU/Kg/día) para mantener los niveles deseados por al menos 3 días (2C).En pacientes con déficit de FXI los antecedentes personales de sangrado tienen especial relevancia. Si en el 3°T, la actividad de FXI es <15 UI/mL, considerar la infusión de concentrado (no disponible en nuestro país), PFC y AcTx en el parto y antes de la cesárea (2C). Si los niveles son >15 UI/mL en pacientes sin historia personal de sangrado o sin antecedentes de desafíos hemostáticos, considerar AcTx por 3 días (2C). En caso de que la mujer haya tenido desafíos hemostáticos sin sangrados, si los valores de FXI al 3°T son >15 UI/mL sólo considerar tratamiento en caso de sangrado anormal (2C).En la mujer con déficit de FXIII se recomienda continuar con la profilaxis, controlar los niveles de factor y realizar las infusiones con un intervalo menor (cada 14–21 días) con la finalidad de mantener niveles FXIII >20 UI/mL. En el momento del parto o previo a la cesárea, dependiendo del tiempo transcurrido desde la última aplicación, considerar una administración adicional de concentrado (10–40 UI/Kg) (2C).
Conflictos de interés
Cristina Duboscq es Asesora Científica de Werfen Medical-Argentina.Patricia Casais no presenta conflictos de interés.
Desórdenes hemorrágicos raros del sistema de coagulación | 5

102
Referencias
1. Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood 2015; 125: 2052-2061.
2. Mumford AD, Ackroyd S, Alikhan R, Bowles L, et al. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. Br J Haematol 2014; 167: 304-326.
3. Peyvandi F, Palla R, Menegatti M, et al., on behalf of the European Network of Rare Bleeding Disorders (EN-RBD) group. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost 2012; 10: 615–621.
4. Peyvandi F, Garagiola I, Menegatti M. Gynecological and obstetrical manifestations of inherited bleeding disorders in women. J Thromb Haemost 2011; 9 (Suppl 1): 236-245.
5. Fundamentos para el Manejo Práctico del Laboratorio de Hemostasia. Grupo CAHT. Editores en Jefe Kordich L. y Blanco A. Segunda Edición, 2013.
6. Kordich L. Calidad Analítica en el laboratorio de Hemostasia en Fundamentos para el Manejo Práctico del Laboratorio de Hemostasia. Grupo CAHT. Editores en Jefe Kordich L. y Blanco A. Segunda Edición, 2013.
7. CLSI Collection, transport and processing of blood Specimens for coagulation testing and general performance of coagulation assays. Aproved guideline-third Edition. NCCLS document H21-A5. 2008.
8. Mackie I, Cooper P, Lawrie A, Kitchen S, Gray E, Laffan M; British Committee for Standards in Haematology. Guidelines on the laboratory aspects of assays used in haemostasis and thrombosis. Int J Lab Hematol 2013; 35: 1-13.
9. NCCLS. H48-A Determination of Factor Coagulant Activities; Approved Guideline1997.
5 | Desórdenes hemorrágicos raros del sistema de coagulación

103
10. Mackie IJ, Kitchen S, Machin SJ, Lowe GD. Haemostasis and Thrombosis Task Force of the British Committee for Standards in Haematology. Guidelines on fibrinogen assays.Br J Haematol 2003; 121: 396-404.
11. CLSI H30-A2. Procedure for the Determination of Fibrinogen in Plasma; Approved Guideline - Second Edition, 2001.
12. Kohler HP, Ichinose A, Seitz R, AriensRas, Muszbek L, On behalf of the Factor XIII and Fibrinogen SSC Subcommittee of the ISTH. Diagnosis and classification of factor XIII deficiencies. J Thromb Haemost 2011; 9: 1404-1406.
Desórdenes hemorrágicos raros del sistema de coagulación | 5

104

105
Inhibidores adquiridos de factores de la coagulaciónPatricia Casais1 y Alicia N. Blanco2.1 Doctora en Medicina (UBA); Instituto de Investigaciones Epidemiológicas, Academia Nacional de Medicina; Centro de Hematología Pavlovsky.2 Doctora en Bioquímica (UBA); Jefe División Hemostasia, Instituto de Investigaciones Hematológicas “Mariano R. Castex”, Academia Nacional de Medicina
Introducción
Los inhibidores adquiridos de los factores de la coagulación son anticuerpos (ACs) neutralizantes que reconocen específicamente epitopes funcionales y bloquean su actividad. Pueden ser alo-Acs que aparecen en pacientes con déficit de factor en terapia de reemplazo o auto-Acs en individuos sin antecedentes de alteraciones de la coagulación que presentan súbitamente sangrados de diferente severidad y pueden o no estar asociados con patología subyacente o drogas. Existen además Acs (no-neutralizantes) que reconocen específicamente epitopes no funcionales; no inhiben la función, pero al formar complejos antígeno-Ac pueden modifcar la depuración y provocar déficit del factor. Los hallazgos de laboratorio son fundamentales en la confirmación de la sospecha diagnóstica.Son situaciones infrecuentes, pero potencialmente graves, por lo que requieren diagnóstico y tratamiento específico con urgencia.
6

106
6 | Inhibidores adquiridos de factores de la coagulación
Auto-Acs
La sospecha clínica se fundamenta en la aparición de sangrados, habitual-mente severos, en pacientes sin antecedentes previos. La frecuencia y características clínicas de los inhibidores adquiridos contra factores específicos no son uniformes y se detallan a continuación.
Inhibidor adquirido anti-FVIII (aFVIII) o hemofilia adquirida
Es el inhibidor adquirido más frecuente con una incidencia de 0,5-4/millón de habitantes/año. Se presentan habitualmente luego de la 5ta década de la vida y, en las mujeres, tiene un pico de incidencia entre los 20-30 años (post-parto). En niños es extremadamente rara.Como en toda patología rara, los registros aportan información sobre la forma de presentación clínica y la respuesta a las distintas opciones terapéuticas. El registro europeo EACH2 (European Acquired Haemophilia Registry) es el registro más completo y numeroso hasta la fecha (1).Etiología. Es idiopática en el 50% de los casos. Entre las enfermedades asociadas se describen los trastornos autoinmunes, embarazo, tumores sólidos y sindromes linfoproliferativos, enfermedades cutáneas, reacción a drogas (penicilina, sulfamida, cloranfenicol, difenilhidantoína, interferón, fludarabina, clopidogrel) y misceláneas como hepatitis, diabetes, enfermedades respi-ratorias, o transfusión múltiple.Clínica. Habitualmente se presenta con equimosis y hematomas espontáneos en tejidos blandos (80%) que puede conducir a un síndrome compartimental, epistaxis recurrente, hemorragia gastrointestinal o urogenital, hemorragia cerebral, sangrado post-exodoncia o procedimiento invasivo. A diferencia de la hemofilia congénita, las hemartrosis son infrecuentes. Alrededor del 9% se presentan con hemorragia severa o fatal (intracraneal o retroperitoneal).Diagnóstico diferencial. Desde el punto de vista clínico la sospecha de basa en la aparición de sangrados en pacientes previamente asintomáticos. A diferencia de la hemofilia congénita, en los pacientes con hemofilia adquirida, las hemartrosis son raras y predominan los sangrados mucocutáneos, de tejidos blandos, musculares o hematomas retroperitoneales. Esta presentación clínica hace recordar aquella de la CID crónica o la hiperfibrinolisis que acompaña a algunas neoplasias (próstata), como así también las equimosis extensas que

107
Inhibidores adquiridos de factores de la coagulación | 6
acompañan los excesos de anticoagulación oral. También deberá descartarse la púrpura amiloide (que también afecta a ancianos). En ocasiones se presenta con sangrado post-parto, sangrado excesivo asociado a traumatismos o cirugías. El laboratorio es fundamental para confirmar la sospecha clínica.Tratamiento. El manejo de estos pacientes se sostiene en dos pilares simultáneos: la erradicación del inhibidor y el manejo de la hemorragia.Las recomendaciones terapéuticas que se señalan a continuación están basadas en las guías de la United Kingdom Haemophilia Centre Doctors’ Organization (UKHCDO) y la Italian Association of Haemophilia Centres (AICE) (2, 3).Siempre que sea posible debe intentar resolver la condición subyacente que originó el desarrollo del inhibidor (tratamiento del cáncer o enfermedad reumática, suspensión de drogas, etc.) (Grado 2C, AICE).Manejo de la Hemorragia. No toda hemorragia requiere el uso de agentes hemostáticos, el 34% de los pacientes tiene hematomas que no requieren tratamiento específico. La severidad del sangrado, la elección del tratamiento y la respuesta al mismo son independientes del título de inhibidor.Agentes puente (by-pass): rFVIIa y FEIBA, ambos pueden ser indistintamente utilizados como primera línea de tratamiento (Grado 1B) (Tabla 1). La elección del agente inicial dependerá de la disponibilidad, conocimiento de la respuesta previa del paciente y costos. Si bien el rFVIIa tiene como ventaja ser recombinante (seguridad viral) y ser el más utilizado en el registro EACH2; la eficacia (90%) y la incidencia de eventos adversos es similar con ambos agentes (3).
1° línea
2° línea
rFVIIa 90 μg/Kg cada 2 h oFEIBA 50–100 UF/Kg cada 6-12 h
(dosis máxima: 200 UF/Kg/d)
Factor VIII y DDAVP
FVIII humano
FVIII humano (100 UI/Kg/d) + inmunoadsorción
DDAVP
Rotar al agente alternativo en caso de falta de
respuesta al agente inicial.
Menor chances de respuesta que a agentes by-passeantes.
Respuesta impredecible aún con bajo título de inhibidor. Deben administrarse dosis altas
para sobrepasar el efecto inhibitorio.
Respuesta aún con títulos altos de inhibidor. No disponible en todos los centros.
Podría ser útil si: inhibidor ≤2 UB/mL y FVIII >5%. Respuesta impredecible.
Opciones terapéuticas Comentarios
Tabla 1. Manejo hemostático del paciente con hemofilia adquirida

108
Las otras opciones terapéuticas se detallan en la Tabla 1. En los sangrados mucosos, excepto urinarios, puede usarse ácido tranexámico (AcTx) como terapia adyuvante (Grado 2C). Si bien se han reportado complicaciones con el uso concomitante de AcTx y FEIBA, son sumamente raras. Sin embargo, las recomendaciones italianas sugieren evitar la combinación en pacientes añosos y con factores de riesgo de trombosis adicionales (Grado 2B). No hay evidencias que sustenten el uso de inmunoglobulina endovenosa (Ig EV).Evaluación de la respuesta al tratamiento hemostático. Debe realizarse precozmente y, en caso de no ser adecuada, se debe rotar al otro agente (Grado 2C). La evaluación de la respuesta al tratamiento es principalmente clínica (cese del dolor, reducción del hematoma o de su tensión), pueden incluirse estudios de imágenes y la estabilidad de los niveles de hemoglobina para objetivar el cese/disminución del sangrado. Los niveles de FVIII u otras pruebas globales no necesariamente correlacionan con la respuesta hemostática.No hay criterios validados para decidir cuándo suspender el tratamiento hemostático; sin embargo, dependiendo del sitio y/o la gravedad del sangrado luego de 24-48 hs. de estabilidad clínica y de laboratorio podría evaluarse la suspensión. Al igual que en la hemofilia congénita, en los sangrados mayores (SNC, piso de boca, retroperitoneo), se sugiere mantener el tratamiento más tiempo.Complicaciones. Las complicaciones asociadas al tratamiento hemostático son las trombosis venosas y arteriales, especialmente de la terapia puentes. Dado que es una patología de pacientes añosos, debe evaluarse el riesgo trombótico individual y, una vez estabilizado el cuadro hemorrágico, considerar las medidas de tromboprofilaxis apropiadas (Grado 2C).Tratamiento inmunosupresor. El tratamiento debe iniciarse al momento del diagnóstico (Grado 1B), la atención al cuadro hemorrágico no debe demorar el inicio de la inmunosupresión. Ante la falta de evidencia concluyente sobre el mejor agente inmunosupresor, la elección del tratamiento debe considerar la edad y las comorbilidades del paciente (Tabla 2). La mortalidad asociada a la hemofilia adquirida se relaciona con la patología en sí misma (los pacientes añosos y con neoplasia tendrían peor pronóstico) y con las complicaciones del tratamiento inmunosupresor. La media del tiempo a la respuesta es de 3-5 semanas, por lo que se debe estar alerta sobre el riesgo de infecciones, a veces severas, y tener una actitud pro-activa para su búsqueda y tratamiento.
6 | Inhibidores adquiridos de factores de la coagulación

109
El objetivo del tratamiento es alcanzar la remisión completa, definida como títulos de inhibidor persistentemente indetectables (<0,6 Unidades Bethesda (UB)/mL) con niveles normales de FVIII (>70%) (Grado 2B, AICE).
El tratamiento de primera línea es corticoides con o sin ciclofosfamida (Grado 1B), con una tasa de remisión de 75% y una media de tiempo a la remisión de 32-40 días. En mujeres en edad fértil se recomienda en contra de la asociación con ciclofosfamida o cualquier otro agente alquilante (Grado 2C).El rituximab, solo o combinación con otros inmunosupresores, es el agente de segunda línea en caso de falta de respuesta luego de 3–5 semanas de tratamiento de primera línea (Grado 2B). Sólo en pacientes con contraindicación de terapia inmunosupresora convencional, podría utilizarse como primera línea (Grado 2B). El tiempo hasta la respuesta con rituximab es 65 días (29-144); según datos del registro EACH2, el 42% de los pacientes tratados sólo con rituximab alcanzaron remisión estable y 65% de quienes lo recibieron asociado con otros agentes.El uso de Ig EV, sola o en combinación con corticoides o agentes citotóxicos, no ha demostrado ningún beneficio y no debería utilizarse (Grado 1B). Tampoco sería costo-beneficioso el tratamiento de inducción de inmunotolerancia con FVIII según la guía británica; sin embargo, el consenso italiano considera que podría utilizarse en caso de fracaso al tratamiento de primera línea (Grado 2C).Seguimiento y recaídas. Luego de alcanzar la remisión, los pacientes deben continuar en seguimiento por al menos un año (Grado 1C) ya que el riesgo
1° línea
2° línea
Prednisolona 1 mg/Kg/d con (o sin) ciclofosfamida 1-2 mg/d vo (Grado 1B)
Rituximab 375 mg/m2 semanal por 4 semanas
Ciclosporina o tacrolimus asociado a corticoides
La combinación aumentaría la probabilidad de alcanzar RC pero sin diferencias en la sobrevida. Sería más eficaz en pacientes con enfermedades
autoinmunes.
No se asocia con RC más rápida.
Pequeñas series de casos reportan 90% eficacia en primera línea.
Opciones terapéuticas Comentarios
Tabla 2. Tratamiento inmunosupresor del paciente con hemofilia adquirida
RC: Remisión completa.
Inhibidores adquiridos de factores de la coagulación | 6

110
de recaídas es relativamente alto (10–20% a los 7,5 meses). Algunos autores sugieren control con tiempo de tromboplastina parcial activada (TTPA) y FVIII mensual durante 6 meses; luego cada 2-3 meses hasta cumplir el primer año y semestralmente durante el segundo año (4). La posibilidad de lograr una segunda remisión es alta (70%) pero los pacientes deberían continuar tratamiento inmunosupresor a largo plazo.
Situaciones especiales
EmbarazoEs una complicación muy infrecuente que se presenta habitualmente post-parto (3-12 meses), aunque se han comunicado inhibidores anti-FVIII durante el embarazo. Si bien se han observado remisiones espontáneas, debería iniciarse tratamiento inmunosupresor teniendo en cuenta la edad de la paciente (no asociar ciclofosfamida u otros alquilantes). Las guías italianas sugieren la posibilidad de una conducta expectante en caso de que el riesgo del tratamiento inmunosupresor se considere mayor que el riesgo de sangrado. Sin embargo, es importante señalar como criterio que la no instauración del tratamiento inmunosupresor en la embarazada/puérpera es una excepción que debe estar justificada por la situación clínica de cada paciente individual.El manejo hemostático sigue los mismos principios que en los demás casos. La recurrencia en subsiguientes embarazos es rara pero podría darse por lo que la paciente debe ser informada al respecto.Se han reportado casos de sangrado neonatal que sugerirían pasaje placentario del Ac (5). Aunque las guías internacionales no recomiendan el modo del nacimiento; parece prudente desde el punto de vista hematológico, recomendar la realización de una cesárea por la posibilidad de pasaje placentario del anticuerpo.
PediatríaEs muy infrecuente (0,045/millón/año). Las patologías asociadas son, entre otras, artritis reumatoidea juvenil, antibióticos, Sindrome de Goodpasture y hepatopatías. El patrón de sangrado es similar al de los adultos. Pueden pre-sentar remisiones espontáneas, especialmente si se asocian con infecciones.
6 | Inhibidores adquiridos de factores de la coagulación

111
Inhibidor adquirido contra otros factores de la coagulaciónSon extremadamente raros, muchas veces asintomáticos a pesar de las significativas alteraciones del laboratorio. Dada la escasa experiencia, cada paciente debería ser evaluado en forma individual para definir la conducta a seguir. Al igual que en el caso de hemofilia adquirida, estos pacientes deben ser evaluados por un especialista.En algunos casos, estos inhibidores son asintomáticos y transitorios, por lo que en el paciente asintomático un período de observación podría ser oportuno antes de comenzar el tratamiento inmunosupresor.La Tabla 3 resume las características clínicas más frecuentes en cada déficit adquirido.
aFI
aFII
aFV
aFVII
aFIXaFX
aFXI
aFXIII
Asociados con gammapatías. LES. Isoniacida. Sangrados habitualmente severos: GI, génito-urinarios,
retroperitoneal, SNC. También equimosis y hematomas.
Antecedentes: cirugía con exposición a trombina bovina (“fibrin glue”), enfermedades autoinmunes,
gammapatías. Asociados con inhibidor lúpico pueden dar hipoprotrombinemia, especialmente en niños
con cuadro viral previo a la aparición de equimosis o sangrados.
Clínica variable, incluso trombosis.
En pacientes añosos con neoplasia o tratamiento con antibióticos beta lactámicos o aminoglucósidos;
75% asociados a trombina bovina tópica. Paraproteínas.
Clínica variable, desde epistaxis y gingivorragia hasta sangrado retroperitoneal, GI, hematuria. Pueden coexistir con aFII.
Antecedentes: globulina anti-timocito (anemia aplástica), penicilina, ciclosporinas, sepsis, pancreatitis y neoplasia.
Clínica: habitualmente severa, sangrado SNC. Hemartrosis son raras.
Antecedentes y cuadro clínico similares a hemofilia adquirida (inhibidor aFVIII).
Extremadamente raros; asociados con infección respiratoria y neoplasia (incluso leucemia mieloide aguda). Lepra.
Clínica: GI, hematuria, equimosis y hemartrosis; 29% asintomáticos.
Asociados a colagenopatías, enfermedades oncohematológicas, enfermedades autoimunes intestinales y tratamiento
con fenotiazinas.
Clínica: impredecible; frecuentemente asintomáticos.
Patrón de sangrado más severo que en déficit congénito: hemartrosis, hematomas, menorragia, sangrado quirúrgico,
mala cicatrización de heridas y aborto recurrente.
Isoniacida.
Tabla 3. Características clínicas de los inhibidores contra factores de la coagulación
Características clínicas
LES: lupus eritematoso sistémico; GI: gastrointestinal; SNC: sistema nervioso central.
Inhibidores adquiridos de factores de la coagulación | 6

112
Las recomendaciones acerca de la erradicación del inhibidor son similares a las de la hemofilia adquirida (incluyen esteroides, ciclofosfamida o azatioprina así como el uso creciente de rituximab, aunque no hay evidencia de calidad que lo avale). El manejo de los sangrados es específico para cada factor y se detallan en la Tabla 4. Todas estas recomendaciones son Grado 2C.
Inhibidor anti-factor von Willebrand (aVWF)El síndrome de von Willebrand adquirido (AVWS) es una patología infrecuente que reconoce varios mecanismos fisiopatológicos. En pacientes con sindromes linfo y mieloproliferativos, neoplasias o enfermedades autoinmunes es causado por la presencia de Acs contra el factor von Willebrand (VWF) que generan inmunocomplejos que aceleran su clearance o inhiben su función. El patrón de sangrado es similar al de la enfermedad de von Willebrand (VWD) congénita.El tratamiento de la enfermedad de base es fundamental y paralelo al tratamiento de la hemorragia. Las alternativas de tratamiento hemostático incluyen: desmopresina (DDAVP), concentrados de FVIII/VWF. En estos casos la respuesta puede ser de breve duración por clearance acelerado. Altas dosis de Ig (0,4 g/Kg/día por 5 días o 1 g/Kg/día por 2 días, que serían más eficaces cuando el Ac no es de tipo IgM) y rFVIIa.
6 | Inhibidores adquiridos de factores de la coagulación
CFI: concentrado de Fibrinógeno; Crio: crioprecipitados; PFC: plasma fresco congelado; CCP: concentrado de complejo protrombínico; rFVIIa: factor FVII recombinante activo; CFIX: concentrado de factor IX; CFXI: concentrado de factor XI; CFXIII: concentrado de factor XIII.
aFIaFIIaFV
aFVIIaFIXaFXaFXI
aFXIII
CFI
FEIBA o CCP
Plaquetas±PFC
FEIBA
rFVIIa o FEIBA
FEIBA
rFVIIa o FEIBA
CFXIII
CFI±inmunoadsorción
FEIBA
PFC+inmunoadsorción
rFVIIa, FVII
Agente alternativo
CCP
Agente alternativo
CFXIII+inmunoadsorción
Crio±inmunoadsorción
FEIBA /CCP+inmunoadsorción
FEIBA
CCP±inmunoadsorción
CFIX±inmunoadsorción
FEIBA /CCP+inmunoadsorción
CFXI±inmunoadsorción
Crios±inmunoadsorción
Inhibidor 1° línea 2° línea Otras opciones
Tabla 4. Tratamiento hemostático en pacientes con Acs contra otros factores de la coagulación

113
Otras patologías
Existen otras patologías que pueden presentar Acs contra factores de la coagulación.Un caso particular es el Ac anti-protrombina, en el síndrome antifosfolípido (SAF). Es sabido que el SAF es un estado protrombótico, pero la presencia de Acs anti-protrombina no-neutralizantes, que provoquen déficit, puede ocasionar sangrados, inclusive severos. La respuesta al tratamiento hemostático suele ser favorable (ver Tabla 4).
Alo-Acs
El desarrollo de Acs contra el factor exógeno es la complicación más temida ya que la terapia de reemplazo se convierte en ineficaz lo que expone al paciente a sangrados severos con alta morbi-mortalidad.
Laboratorio: detección de inhibidores
Efecto sobre las pruebas de coagulaciónLos inhibidores adquiridos son Acs capaces de inhibir de un modo específico la función de los factores de la coagulación (2, 3, 6, 7). Según el factor inhibido, los Acs pueden afectar una o varias de las vías de la coagulación. Esto se traduce en alteración de las pruebas de laboratorio:Prolongación del tiempo de protrombina (TP). Cuando el factor inhibido pertenece a la vía extrínseca o común (VII, X, V, II, fibrinógeno).Prolongación del TTPA. En el caso de un inhibidor de un factor de la vía intrínseca o común (XII, XI, IX, VIII, X, V, II, fibrinógeno).Prolongación del tiempo de trombina (TT). En el caso de un inhibidor de la vía final (trombina, fibrinógeno).Prolongación del tiempo de reptilase (TR). En el caso de un inhibidor de fibrinógeno.Alteración de las pruebas de solubilidad (urea 5M o ácido monocloroacético al 1%). Con pruebas de coagulación normales (FXIII).Disminución de la actividad funcional. Del factor inhibido (métodos de
Inhibidores adquiridos de factores de la coagulación | 6
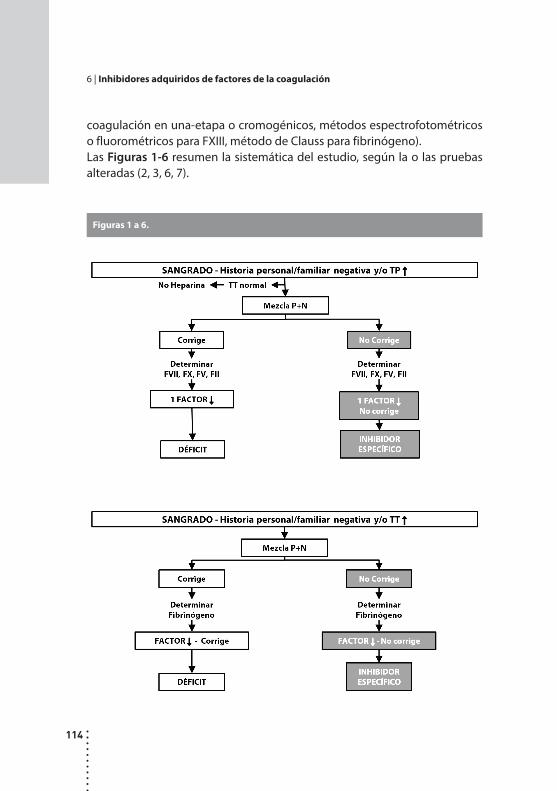
114
coagulación en una-etapa o cromogénicos, métodos espectrofotométricos o fluorométricos para FXIII, método de Clauss para fibrinógeno).Las Figuras 1-6 resumen la sistemática del estudio, según la o las pruebas alteradas (2, 3, 6, 7).
Figuras 1 a 6.
6 | Inhibidores adquiridos de factores de la coagulación

115
Inhibidores adquiridos de factores de la coagulación | 6

116
6 | Inhibidores adquiridos de factores de la coagulación

117
Corrección con plasma normalEl defecto producido por los inhibidores adquiridos no corrige por el agregado de plasma normal. Esto se observa tanto en la prueba global de la cual participa el factor inhibido, como en la determinación de la actividad coagulante del factor.El criterio para definir “no corrección” se basa en el cálculo del Índice de Rosner o ICA = [(P+N)-N/P]x100. Se considera que la prueba de mezcla (1:1) no corrige, cuando el valor del índice es mayor al valor de corte (6). Cada laboratorio debe establecer el valor de corte para los reactivos y el método de medida utilizados; se recomienda tomar el valor del percentil 99 de estudios de mezcla en al menos 40 plasmas de individuos sin inhibidor. Cuando se analiza la actividad funcional del factor, se considera que la mezcla (1:1) no corrige cuando la actividad observada es menor a la actividad teórica esperada (6). En el caso particular de los inhibidores aFVIII el efecto inhibitorio es tiempo-temperatura dependiente; a mayor tiempo de interacción del Ac con el FVIII a 37ºC, mayor inhibición de la actividad (2, 3, 6-8). La Figura 7 muestra el esquema de procedimiento (6) y su interpretación.
El plasma normal, utilizado en los ensayos de mezcla, puede ser de origen comercial (liofilizado) o un pool de voluntarios sanos, que puede ser conservado a -80ºC. Es importante el nivel de factores, en especial de aquellos lábiles como el FV y el FVIII, no sólo para las pruebas de corrección, sino
Inhibidores adquiridos de factores de la coagulación | 6
Figura 7.

118
también para la titulación, donde influye en la variabilidad inter-ensayo (7).Además de los inhibidores, hay Acs no-neutralizantes que reconocen de un modo específico a un factor de coagulación (formación de inmuno-complejos) pero no inhiben su función, sino que aceleran su depuración. Esto puede llevar a disminución del factor y prolongación de las pruebas de coagulación; defectos que corrigen con el agregado de plasma normal (2, 3, 6). Los Acs pueden ser evidenciados por técnicas de ELISA o immunoblotting (6).
Actividad coagulante - Curvas de diluciónLos inhibidores específicos son Acs de alta afinidad; la unión antígeno-Ac no se disocia por efecto de dilución de la muestra. Si se determina la actividad del factor en diluciones progresivas de la muestra con inhibidor, se obtiene una recta paralela a la del normal, semejante a lo observado en una deficiencia (Figura 8) (2, 6-8).
En teoría, en caso de un inhibidor específico de un factor, sólo debería verse disminuida la actividad del factor inhibido. Sin embargo, en presencia de
6 | Inhibidores adquiridos de factores de la coagulación
Figura 8.
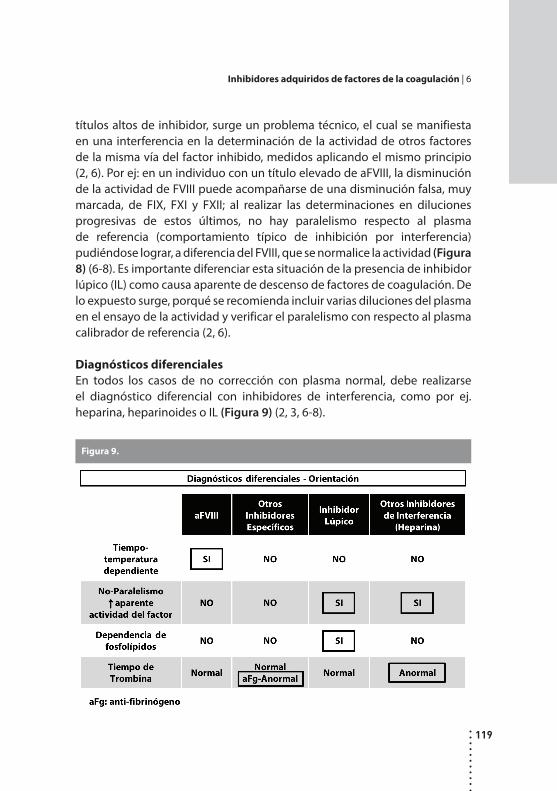
119
títulos altos de inhibidor, surge un problema técnico, el cual se manifiesta en una interferencia en la determinación de la actividad de otros factores de la misma vía del factor inhibido, medidos aplicando el mismo principio (2, 6). Por ej: en un individuo con un título elevado de aFVIII, la disminución de la actividad de FVIII puede acompañarse de una disminución falsa, muy marcada, de FIX, FXI y FXII; al realizar las determinaciones en diluciones progresivas de estos últimos, no hay paralelismo respecto al plasma de referencia (comportamiento típico de inhibición por interferencia) pudiéndose lograr, a diferencia del FVIII, que se normalice la actividad (Figura 8) (6-8). Es importante diferenciar esta situación de la presencia de inhibidor lúpico (IL) como causa aparente de descenso de factores de coagulación. De lo expuesto surge, porqué se recomienda incluir varias diluciones del plasma en el ensayo de la actividad y verificar el paralelismo con respecto al plasma calibrador de referencia (2, 6).
Diagnósticos diferencialesEn todos los casos de no corrección con plasma normal, debe realizarse el diagnóstico diferencial con inhibidores de interferencia, como por ej. heparina, heparinoides o IL (Figura 9) (2, 3, 6-8).
Inhibidores adquiridos de factores de la coagulación | 6
Figura 9.

120
La heparina se descarta si el TT es normal; esto no aplica frente a un inhibidor de fibrinógeno o de trombina, donde se requieren otras pruebas complementarias como el TR, entre otras (3, 6). Para excluir la presencia del IL, que puede afectar el TTPA y/o el tiempo de veneno de víbora Russell diluido (DRVVT), es preciso demostrar que el efecto no es dependiente de fosfolípidos (6).Hay situaciones complejas en las cuales puede presentarse más de un efecto inhibitorio (por ej: aFVIII+IL), efecto inhibitorio y déficit de factor (por ej: alo-Acs en pacientes deficientes) o concomitancia de Acs específicos no-neutralizantes e inhibidores de interferencia (por ej: hipoprotrombinemia+IL) (2, 6, 8). Casos en los cuales los resultados deben ser analizados de modo estricto, minucioso y requieren eventualmente pruebas o metodologías complementarias como la determinación de FVIII por sustratos cromogénicos para detectar y titular aFVIII en presencia de IL (6-8). Los defectos concomitantes complican la interpretación de los estudios de mezcla; siempre que se observe una corrección “parcial” debe sospecharse un inhibidor+déficit (6).
Titulación del inhibidorUna vez caracterizado el efecto inhibitorio y detectado el factor inhibido, puede realizarse la titulación del inhibidor, mediante la modificación de Nijmegen del método Bethesda que aumenta la sensibilidad (8). El método no es específico, puede ser afectado, por ej. por la presencia de IL (6-8).Fue descripto inicialmente para la titulación de inhibidores aFVIII, con efecto tiempo-temperatura dependiente; requiere la incubación del plasma problema con el plasma normal (aporta FVIII) durante dos horas a 37ºC (6-8). Se define como UB/mL a la cantidad de inhibidor que deja el 50% de FVIII residual, luego de 2 hs. de incubación a 37ºC (6). Títulos inferiores a 0,6 UB/mL son considerados negativos.La inter-variabilidad del ensayo es muy elevada (≤50%), al igual que los posibles falsos negativos o positivos (≤30%) (7, 8). El control estricto de variables pre-analíticas, analíticas y post-analíticas, con confirmación del ensayo en una nueva muestra, disminuye la variabilidad y mejora la exactitud (8).El método se adapta para titular otros inhibidores; la diferencia es que no requiere la incubación durante dos horas a 37ºC, dado que el efecto de otros inhibidores es inmediato (2, 3, 6).
6 | Inhibidores adquiridos de factores de la coagulación

121
Detección de inhibidores aFVIII y otrosAnte la sospecha clínica de un inhibidor, el laboratorio debe proceder a realizar, sin demora, los estudios de mezclas con plasma normal (1:1) (2, 3). La “no corrección” puede observase en la mezcla analizada en forma inmediata o, como en el caso del inhibidor aFVIII, requerir una incubación durante 1-2 horas a 37ºC detectar el máximo efecto inhibitorio (2, 6).aFVIII. No debe descartarse inhibidor aFVIII sin realizar los ensayos de mezclas con incubación prolongada a 37ºC, respetando estrictas condiciones de trabajo (control de pH, no-evaporación, etc.) (Figura 7) (6).aFV. Los inhibidores aFV, dependiendo del epitope reconocido por el anticuerpo, pueden alterar la función procoagulante (prolongación de TP, TTPA; disminución de FV-método en una-etapa) y/o la anticoagulante (resistencia a la acción de la proteína C activada) (2, 3, 9).aFXIII. Los inhibidores, al igual que los déficits de FXIII, no afectan las pruebas de coagulación (TP, TTPA, TT); pueden alterar las pruebas de solubilidad del coágulo (urea 5M o ácido monocloroacético al 1%) o inhibir la actividad del factor, medida por diferentes métodos (espectrofotométricos o fluorométricos) (2, 3, 6, 10). Hay además aFXIII no-neutralizantes que reconocen la molécula purificada o sus subunidades (2, 10).aVWF. Algunos casos de AVWS se producen por Acs. Los aVWF pueden inhibir al VWF, afectando una o varias de las diferentes pruebas (VWF:RCo, VWF:CB, VWF:Ag, TTPA/FVIII) según el epitope contra el cual estén dirigidos; las pruebas no corrigen por el agregado de plasma normal. Si los Acs no neutralizan la función, sino que incrementan la depuración, se observa un patrón semejante al del déficit congénito: disminución de la función que corrige con plasma normal; el tipo y duración de la respuesta al DDAVP o a los concentrados de FVIII/VWF puede evidenciarlos (2, 3, 6).
Algunas recomendaciones
• El diagnóstico de aFVIII (hemofilia adquirida) debe ser considerado en el caso de un individuo con sangrado inesperado, sin historia personal ni familiar de sangrado, que presente TTPA prolongado, que no corrige con plasma normal en mezcla 1:1 (Grado 1C) (2), luego de 2 horas a 37ºC (Grado 1B) (3).
Inhibidores adquiridos de factores de la coagulación | 6

122
• La presencia de inhibidores adquiridos contra epitopes funcionales de otros factores de coagulación debe ser considerada en el caso de un individuo con sangrado inesperado, sin historia personal ni familiar de sangrado, que presente TP y/o TTPA y/o TT prolongado, que no corrige por el agregado de plasma normal en mezcla 1:1 (Grado 1B) (3) (Grado 2C) (2). Si las pruebas de screening son normales, debería excluirse aFXIII mediante pruebas específicas (3).
• La detección de alo-Acs (aFVIII+hemofilia u otros inhibidores+déficit) debe ser considerada en el caso de un individuo deficiente, que no responda clínicamente a la terapéutica de reemplazo y/o no alcance los niveles de factor esperado post-tratamiento (7).
• El diagnóstico de laboratorio de pacientes con sospecha de inhibidores adquiridos contra un factor de coagulación debe ser confirmado por laboratorios especializados (Grado 1B), que puedan asegurar la rápida ejecución de las pruebas de mezclas y la determinación de la actividad de los factores, aún en un contexto de emergencia (Grado 1B) (3).
• Los laboratorios de coagulación deberían contar con algoritmos que aseguren evaluar en forma apropiada y rápida una prueba de screening anormal, en especial TTPA prolongado (Grado 2C). Así como informar con urgencia al clínico el significado potencial de los resultados anormales (Grado 2C) (2).
Conflictos de interés
Las autoras no presentan conflictos de interés.
6 | Inhibidores adquiridos de factores de la coagulación

123
Referencias
1. Knoebl P, Marco P, Baudo F, et al., on behalf of the EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10: 622–631.
2. Collins PW, Chalmers E, Hart D, et al. Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO. Br J Haematol 2013; 162: 758-773.
3. Franchini M, Castaman G, Coppola A, et al. Acquired inhibitors of clotting factors: AICE recommendations for diagnosis and management. Blood Transfus 2015; 13: 498-513.
4. Huth-Kühne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica 2009; 94: 566-575.
5. Tengborn L, Baudo F, Huth-Kühne A, et al. on behalf of the EACH2 registry contributors. Pregnancy-associated acquired haemophilia A: results from the European Acquired Haemophilia (EACH2) registry. BJOG 2012; 119: 1529-1537.
6. Blanco A. Inhibidores adquiridos de la coagulación y otros desórdenes inmunológicos. Fundamentos para el manejo práctico en el laboratorio de hemostasia. Segunda edición. Blanco A, Kordich L, Bermejo E, Forastiero R, Lauricella AM, Pieroni G, Quintana I, Scazziota A, 2013, 517-560. Federación Bioquímica de la Provincia de Buenos Aires, La Plata.
7. Kershaw G, Favaloro EJ. Laboratory identification of factor inhibitors: an update. Pathology 2012; 44: 293-302.
8. Favaloro EJ, Verbruggen B, Miller CH. Laboratory testing for factor inhibitors. Haemophilia 2014; 20: 94-98.
9. Matsumoto T, Nogami K, Shima M. Coagulation function and mechanisms in various clinical phenotypes of patients with acquired factor V inhibitors. J Thromb Haemost 2014; 12: 1503-1512.
Inhibidores adquiridos de factores de la coagulación | 6

124
10. Franchini M, Frattini F, Crestani S, Bonfanti C. Acquired FXIII inhibitors: a systtematic review. J Thromb Thrombolysis 2013; 36: 109-114.
6 | Inhibidores adquiridos de factores de la coagulación

125
Esta guía fue confeccionada en base a las sugerencias de la Guías Diagnósticas y Terapéuticas de la Sociedad Argentina de Hematología del año 2015, a cargo de María Cristina Rapetti, Hugo Donato, Daniel Fassi, Carlos Fondevila, Nora Goette, Marta Lavergne, Rosana Marta, Mónica Martinez, Miguel Raillon y Dardo Riveros.
Introducción
La Trombocitopenia Inmune, previamente llamada “Púrpura Trombo-citopénica Inmune” (PTI) se caracteriza por trombocitopenia aislada de origen auto-inmune. Debido al extenso uso de esta sigla en nuestro medio, en adelante continuaremos utilizándola para su denominación.
Definiciones
PTI primaria. Trombocitopenia menor a 100x109/L no asociada a patología reconocible. PTI secundaria. La que está relacionada a otra patología reconocible. Para
Trombocitopenia inmuneRosana Marta1, Mónica Martinez2 y Daniel Fassi3.1 Departamento Hematología Investigación. Instituto de Investigaciones Médicas Dr. Alfredo Lanari. Universidad de Buenos Aires. CONICET.2 Médica Hematóloga, Hospital de Niños Sor María Ludovica, La Plata.3 Jefe de Hematología. Hospital Universitario de la Universidad Abierta Interamericana (UAI).
7

126
definirla, debe incluirse el nombre de la misma al de PTI (por ej, “PTI asociada a lupus”). De acuerdo al tiempo transcurrido desde su diagnóstico, la PTI puede ser:• De reciente diagnóstico (menos de 3 meses desde el diagnóstico).• Persistente (3-12 meses).• Crónica (más de 12 meses).PTI severa. Es aquella en la que la magnitud clínica de la hemorragia exige tratamiento activo desde el comienzo de la enfermedad o requiere adicionar otros tratamientos al ya existente. PTI refractaria. Es la PTI severa que ocurre luego de la esplenectomía. Los pacientes que cumplen criterios de PTI severa pero que no han sido esplenectomizados no deben considerarse refractarios sino no respondedores a los fármacos utilizados hasta ese momento. Cortico-dependencia. Es el estado en el cual es necesaria la administración continua de corticoides para mantener el nivel de plaquetas >30x109/L y/o evitar la hemorragia. El tipo de respuesta debe ser valorado en 2 oportunidades separadas entre sí por 7 días. Las respuestas al tratamiento se definen como: • Respuesta completa (≥100x109/L y ausencia de hemorragia).• Respuesta (≥30x109/L, aumento >2 veces del inicial y no hemorragia).• No respuesta (<30x109/L, aumento <2 veces del inicial o hemorragia).
Diagnóstico
Anamnesis y examen físico• Historia familiar de trombocitopenia (descarta enfermedad congénita).• El patrón temporal de la trombocitopenia y de las hemorragias (agudos,
crónicos, recidivas).• Historia la enfermedad en relación a desórdenes autoinmunes,
infecciones ó neoplasias, viajes, transfusiones recientes, ingesta etílica, hábitos dietarios y drogas.
• La relevancia de la trombocitopenia en el paciente individual es variable y depende de la presentación clínica. El sangrado clínicamente significativo no suele ocurrir hasta que el recuento plaquetario cae a menos de 20x109/L. En el paciente ambulatorio, la trombocitopenia es frecuentemente aislada y asintomática hasta en un 25% de pacientes.
• Las manifestaciones clínicas en las formas primarias ó idiopáticas están
7 | Trombocitopenia Inmune

127
representadas por el sangrado cutáneo mucoso y visceral de grado variable:
1. Púrpura cutáneo-mucosa: petequias, equimosis, hematomas. 2. Sangrado mucoso: gingivorragias, epistaxis. 3. Sangrado visceral: melena, hematemesis, enterorragia, hematuria. 4. Raros: sangrados más graves (sistema nervioso central [SNC] y oculares) presentes generalmente con recuentos plaquetarios <10x109/L.• Las manifestaciones hemorrágicas pueden aparecer en cualquier
momento de la evolución de la PTI aunque el riesgo es mayor en los refractarios.
Estudios de laboratorio
En primer lugar se debe descartar una pseudo-trombocitopenia causada por la aglutinación de plaquetas debida a un efecto del anticoagulante usado, en general ácido etilendiaminotetraacético (EDTA), que está mediado por inmunoglobulinas (Ig) de tipo IgG y en menor medida IgM. Se debe observar al microscopio un extendido de sangre periférica obtenida por punción digital, para constatar si la trombocitopenia es verdadera o evidenciar la presencia de agregados plaquetarios. Ya que estas Igs pueden comportarse como aglutininas frías, de ser necesario se realizará una nueva extracción de sangre en tubo pre-calentado a 37ºC y manteniendo la muestra a esa misma temperatura hasta su análisis (1). La evaluación inicial del paciente con trombocitopenia debe incluir los estudios de laboratorio que se muestran en la Tabla 1.Los nuevos conceptos en relación a los mecanismos que conducen al descenso de plaquetas en la PTI han virado desde el clásico aumento de la destrucción periférica mediada por auto-anticuerpos (Acs), a mecanismos más complejos que incorporan tanto un defecto en la producción como el daño inducido por células T como responsables de la trombocitopenia. De todos modos, los auto-Acs mantienen una rol protagónico y a lo largo de los años se han desarrollado diferentes técnicas tendientes a su identificación. Hasta el momento no existe una técnica “gold standard” de laboratorio para establecer el diagnóstico de PTI, lo que se pone de manifiesto en el hecho de que las guías actuales no incluyen la determinación de Acs dirigidos contra plaquetas en la evaluación básica de esta enfermedad (2).
Trombocitopenia Inmune | 7

128
El estudio de los Acs en PTI puede estar dirigido a la detección de Igs asociadas a las plaquetas o a la identificación de la glicoproteína hacia la cual están dirigidos los auto-Acs.
Los ensayos que detectan la Igs asociada a la membrana plaquetaria se basan en el uso de Acs generados en especies distintas de la humana, que reconocen a dichas Igs. Estos Acs anti-Igs humana pueden estar unidos a peroxidasa, en el caso de la técnica de Leporrier, o a un fluorocromo, en el caso de la citometría de flujo. Esta última técnica tiene la ventaja de poder discriminar entre los isotipos de los auto-Acs (IgG o IgM). Estos ensayos pueden arrojar resultados positivos en trombocitopenias de origen no-inmune, así como detectar complejos inmunes unidos a la membrana de las plaquetas por el fragmento Fc, por lo cual su especificidad es baja (3) y se han calificado como de beneficio incierto para el diagnóstico de PTI. Los ensayos que identifican al blanco antigénico de los auto-Acs como los estudios de MAIPA (Monoclonal antibody-specific immobilization of platelet antigens), immunobead y PAICA (platelet-associated IgG characterization assay), se basan en el uso de Acs monoclonales dirigidos contra glicoproteínas plaquetarias que son usualmente blanco de los auto-Acs. Esto permite la captura del antígeno junto con el auto-Acs unido, que se pone de manifiesto usando un Acs dirigido contra IgG humana unido a una enzima que permite su detección. Estos ensayos pueden ser directos cuando se utilizan las
7 | Trombocitopenia Inmune
Hemograma con recuento de plaquetas.
Visualización del frotis de sangre periférica.
Pruebas de coagulación.
Prueba de Coombs directa (PCD) (en pacientes pediátricos y en adultos con anemia).
Hepatograma.
Proteinograma.
Dosaje de inmunoglobulinas. En niños.
Estudio de colagenopatías. En mayores de 10 años. Adecuarlo a cada paciente (sexo femenino, mayor edad al diagnóstico, trombocitopenia moderada al diagnóstico, etc.).
Serología para VIH, hepatitis B (VHB) y C (VHC), Helicobacter pylori, virus de Epstein Barr (VEB), Citomegalovirus (CMV). Según la edad del paciente.
Acs antifosfolípidos. Especialmente en mayores de 10 años.
Acs antitiroideos, especialmente en adultos.
Tabla 1. Estudios diagnósticos en PTI

129
plaquetas del paciente o indirectos cuando se usa una muestra de plasma o suero. Tienen mayor especificidad que los que detectan Ig asociada a plaquetas pero su sensibilidad es de alrededor del 60% y han sido calificados de potencial utilidad en el manejo de la PTI (4). Por lo tanto, si bien el estudio de Acs dirigidos hacia las plaquetas ha contribuido a profundizar el conocimiento de la fisiopatología de la PTI, su búsqueda no está incluida dentro de los estudios diagnósticos recomendados.Si la plaquetopenia se acompaña de otras citopenias deberá realizarse una punción aspirativa de médula ósea (PAMO) y/o biopsia de médula ósea (BMO) de inicio.
PTI y embarazo
En la evaluación de una trombocitopenia gestacional (Figura 1) es importante considerar: • el recuento plaquetario basal previo al embarazo,• el trimestre de aparición y• la severidad de la trombocitopenia.
Trombocitopenia Inmune | 7
Figura 1. Algoritmo de diagnóstico ante una plaquetopenia en el embarazo

130
Figura 2. Algoritmo para el tratamiento del paciente adulto con PTI
Tratamiento
Tratamiento inicial en adultos (ver Figura 2)Objetivos:Enfermedad reciente. Alcanzar un recuento plaquetario seguro lo antes posible, a fin de evitar el sangrado grave (SNC) o fatal. Enfermedad crónica. Mantener un recuento >30x109/L o >50x109/L en ancianos o pacientes con factores de riesgo adicionales para sangrado.
7 | Trombocitopenia Inmune
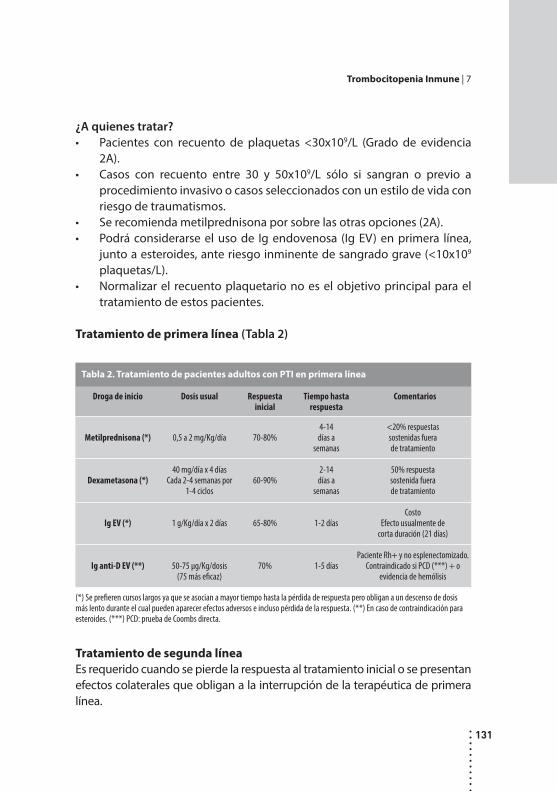
131
¿A quienes tratar?• Pacientes con recuento de plaquetas <30x109/L (Grado de evidencia
2A).• Casos con recuento entre 30 y 50x109/L sólo si sangran o previo a
procedimiento invasivo o casos seleccionados con un estilo de vida con riesgo de traumatismos.
• Se recomienda metilprednisona por sobre las otras opciones (2A).• Podrá considerarse el uso de Ig endovenosa (Ig EV) en primera línea,
junto a esteroides, ante riesgo inminente de sangrado grave (<10x109 plaquetas/L).
• Normalizar el recuento plaquetario no es el objetivo principal para el tratamiento de estos pacientes.
Tratamiento de primera línea (Tabla 2)
Tratamiento de segunda líneaEs requerido cuando se pierde la respuesta al tratamiento inicial o se presentan efectos colaterales que obligan a la interrupción de la terapéutica de primera línea.
Trombocitopenia Inmune | 7
Metilprednisona (*)
Dexametasona (*)
Ig EV (*)
Ig anti-D EV (**)
Droga de inicio
70-80%
60-90%
65-80%
70%
Respuesta inicial
0,5 a 2 mg/Kg/día
40 mg/día x 4 díasCada 2-4 semanas por
1-4 ciclos
1 g/Kg/día x 2 días
50-75 µg/Kg/dosis (75 más eficaz)
(*) Se prefieren cursos largos ya que se asocian a mayor tiempo hasta la pérdida de respuesta pero obligan a un descenso de dosis más lento durante el cual pueden aparecer efectos adversos e incluso pérdida de la respuesta. (**) En caso de contraindicación para esteroides. (***) PCD: prueba de Coombs directa.
Dosis usual
4-14 días a
semanas
2-14 días a
semanas
1-2 días
1-5 días
Tiempo hasta respuesta
<20% respuestas sostenidas fuera de tratamiento
50% respuesta sostenida fuera de tratamiento
CostoEfecto usualmente de
corta duración (21 días)
Paciente Rh+ y no esplenectomizado.Contraindicado si PCD (***) + o
evidencia de hemólisis
Comentarios
Tabla 2. Tratamiento de pacientes adultos con PTI en primera línea
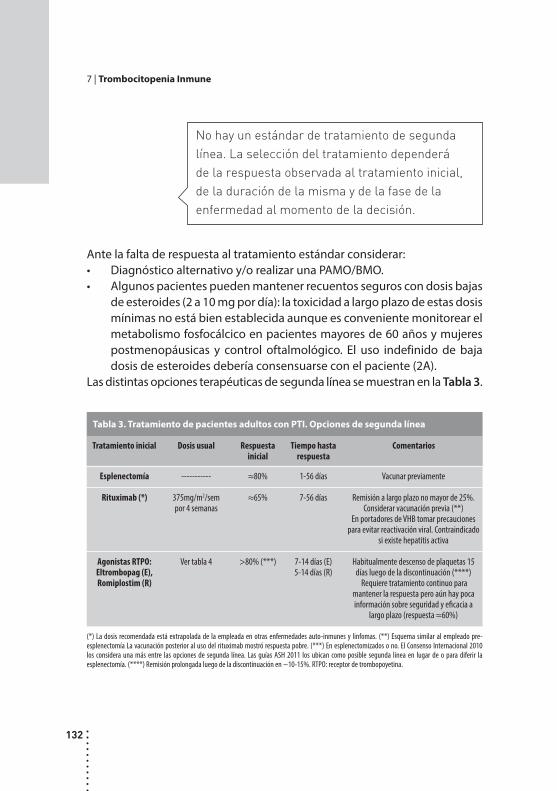
132
Ante la falta de respuesta al tratamiento estándar considerar:• Diagnóstico alternativo y/o realizar una PAMO/BMO.• Algunos pacientes pueden mantener recuentos seguros con dosis bajas
de esteroides (2 a 10 mg por día): la toxicidad a largo plazo de estas dosis mínimas no está bien establecida aunque es conveniente monitorear el metabolismo fosfocálcico en pacientes mayores de 60 años y mujeres postmenopáusicas y control oftalmológico. El uso indefinido de baja dosis de esteroides debería consensuarse con el paciente (2A).
Las distintas opciones terapéuticas de segunda línea se muestran en la Tabla 3.
No hay un estándar de tratamiento de segunda línea. La selección del tratamiento dependerá de la respuesta observada al tratamiento inicial, de la duración de la misma y de la fase de la enfermedad al momento de la decisión.
Esplenectomía
Rituximab (*)
Agonistas RTPO: Eltrombopag (E),Romiplostim (R)
Tratamiento inicial
≈80%
≈65%
>80% (***)
Respuesta inicial
-----------
375mg/m2/sempor 4 semanas
Ver tabla 4
(*) La dosis recomendada está extrapolada de la empleada en otras enfermedades auto-inmunes y linfomas. (**) Esquema similar al empleado pre-esplenectomía La vacunación posterior al uso del rituximab mostró respuesta pobre. (***) En esplenectomizados o no. El Consenso Internacional 2010 los considera una más entre las opciones de segunda línea. Las guías ASH 2011 los ubican como posible segunda línea en lugar de o para diferir la esplenectomía. (****) Remisión prolongada luego de la discontinuación en ~10-15%. RTPO: receptor de trombopoyetina.
Dosis usual
1-56 días
7-56 días
7-14 días (E)5-14 días (R)
Tiempo hasta respuesta
Vacunar previamente
Remisión a largo plazo no mayor de 25%. Considerar vacunación previa (**)
En portadores de VHB tomar precauciones para evitar reactivación viral. Contraindicado
si existe hepatitis activa
Habitualmente descenso de plaquetas 15 días luego de la discontinuación (****)
Requiere tratamiento continuo para mantener la respuesta pero aún hay poca información sobre seguridad y eficacia a
largo plazo (respuesta ≈60%)
Comentarios
Tabla 3. Tratamiento de pacientes adultos con PTI. Opciones de segunda línea
7 | Trombocitopenia Inmune

133
Se sugiere.• Agonistas RTPO sobre rituximab (2A). • Cualquiera de ambos por sobre realizar una esplenectomía antes del
año del diagnóstico (2A). • Esplenectomía por sobre tratamiento médico en PTI crónica (2B).El ajuste de la dosis para el tratamiento con agonistas de RTPO se muestra en la Tabla 4.
Esplenectomía en pacientes adultos con PTI
La esplenectomía es hoy el tratamiento más eficaz en el largo plazo (hasta 90% respuesta inicial; 70% respuesta sostenida a 5-10 años) siendo menor en pacientes añosos. El procedimiento convencional y el laparoscópico producen respuestas similares, pero la vía laparoscópica presenta menos complicaciones (9% vs. 12%) y menos mortalidad (0,2% vs. 1%). Se sugiere demorar la esplenectomía al menos 6 meses en adultos, salvo enfermedad severa sin respuesta a otras medidas o voluntad del paciente.• Si la captación de plaquetas marcadas con 111In es hepática o mixta la
respuesta a la esplenectomía es inferior, en cuyo caso debe evaluarse su beneficio respecto a otras opciones terapéuticas de segunda línea. En la Tabla 5 se describen consideraciones a tener en cuenta ante la esplenectomía.
Eltrombopag
(*) 25 mg/día en pacientes asiáticos.
Inicial: 50 mg/día, oral (*)
Ajuste cada 2 semanas• <50x109/L aumentar a 75 mg/día• 50-200x109/L seguir igual• 200-400x109/L reducir a 25 mg/día y luego a
25 mg/2 días• >400x109/L suspender y: - Aspirina - Reiniciar a dosis menor - Riesgo de rebote
•VigilarfunciónhepáticaconEltrombopag•Considerarfactoresderiesgotrombóticoycardiovasculares.
Inicial: 1 µg/Kg/semana, subcutáneo
Ajuste cada semana• <50x109/L aumentar de a 1 µg/Kg/semana
hasta 10 µg/Kg/semana como máximo• 50-200x109/L seguir igual• 200-400x109/L reducir de a 1 µg/Kg/semana• >400x109/L suspender y: - Aspirina - Reiniciar a dosis menor - Riesgo de rebote
Romiplostim
Tabla 4. Agonistas del RTPO y ajustes de dosis en adultos
Trombocitopenia Inmune | 7

134
El paciente adulto refractario
• Presentan un elevado riesgo de muerte hemorrágica. En casos sintomáticos o con un recuento plaquetario de riesgo (20-30x109/L), es recomendable discutir con el paciente las opciones de tratamiento que, en esta etapa, raramente serán curativas. Se recomienda asimismo, buscar diagnósticos alternativos, detectar y erradicar al H. Pylori y detectar la presencia de bazo accesorio. Algunos pacientes pueden presentar respuestas transitorias a esteroides o Ig EV.
• Los agonistas del receptor de trombopoyetina (RTPO) mostraron una respuesta plaquetaria sostenida en >60% de los casos seguidos por hasta 5 años con un perfil de seguridad aceptable.
Tabla 5. Consideraciones acerca de la esplenectomía
1. Vacunación:• H. influenzae, meningococo y neumococo, por lo menos 2 semanas previo a la esplenectomía.• Vacunación antigripal 1 vez por año.• Re-inmunización para neumococo cada 5 años.
2. Profilaxis antibiótica (ATB):• Beneficio incierto. El riesgo de infección tardía o sepsis fulminante en pacientes vacunados es bajo.• Profilaxis ATB: penicilina, amoxicilina (eritromicina o levofloxacina en caso de alergia). • Guías británicas recomiendan profilaxis ATB en: -Menores de 15 años y mayores de 50 años. -Respuesta inadecuada a la vacunación anti-neumocóccica o historia de infección grave por neumococo. -Durante al menos 2 años post-esplenectomía (período de mayor riesgo de infección sistémica fulminante, en especial en pediatría).
3. Otras complicaciones:• Complicaciones a corto plazo: mortalidad, sangrado, parálisis frénica, infección y tromboembolismo perioperatorio. A
largo plazo: eventración, adherencias, hipertensión pulmonar, enfermedad cardiovascular, infecciones a neumococo, sepsis fulminante.
• La PTI presenta tasas de infección, tromboembolismo venoso, hipertensión pulmonar y enfermedad cardiovascular mucho menores que cuando se la indica por otras causas.
Se recomienda. Consensuar la política de profi-laxis antibiótica con infectología. Los pacientes deben ser notificados sobre el riesgo de infección y la importancia de la consulta médica inmediata ante un cuadro febril (2A).
7 | Trombocitopenia Inmune
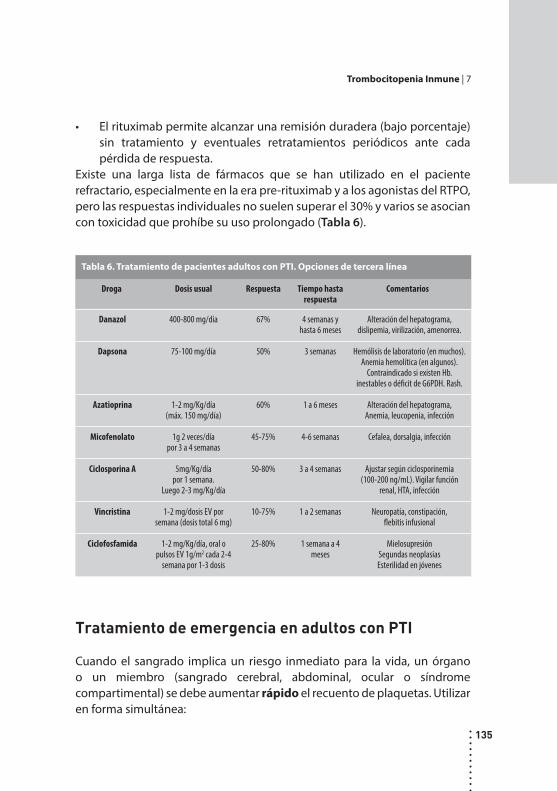
135
• El rituximab permite alcanzar una remisión duradera (bajo porcentaje) sin tratamiento y eventuales retratamientos periódicos ante cada pérdida de respuesta.
Existe una larga lista de fármacos que se han utilizado en el paciente refractario, especialmente en la era pre-rituximab y a los agonistas del RTPO, pero las respuestas individuales no suelen superar el 30% y varios se asocian con toxicidad que prohíbe su uso prolongado (Tabla 6).
Tratamiento de emergencia en adultos con PTI
Cuando el sangrado implica un riesgo inmediato para la vida, un órgano o un miembro (sangrado cerebral, abdominal, ocular o síndrome compartimental) se debe aumentar rápido el recuento de plaquetas. Utilizar en forma simultánea:
Danazol
Dapsona
Azatioprina
Micofenolato
Ciclosporina A
Vincristina
Ciclofosfamida
Droga
67%
50%
60%
45-75%
50-80%
10-75%
25-80%
Respuesta
400-800 mg/día
75-100 mg/día
1-2 mg/Kg/día (máx. 150 mg/día)
1g 2 veces/día por 3 a 4 semanas
5mg/Kg/día por 1 semana.
Luego 2-3 mg/Kg/día
1-2 mg/dosis EV por semana (dosis total 6 mg)
1-2 mg/Kg/día, oral o pulsos EV 1g/m2 cada 2-4
semana por 1-3 dosis
Dosis usual
4 semanas y hasta 6 meses
3 semanas
1 a 6 meses
4-6 semanas
3 a 4 semanas
1 a 2 semanas
1 semana a 4 meses
Tiempo hasta respuesta
Alteración del hepatograma, dislipemia, virilización, amenorrea.
Hemólisis de laboratorio (en muchos). Anemia hemolítica (en algunos).
Contraindicado si existen Hb. inestables o déficit de G6PDH. Rash.
Alteración del hepatograma, Anemia, leucopenia, infección
Cefalea, dorsalgia, infección
Ajustar según ciclosporinemia (100-200 ng/mL). Vigilar función
renal, HTA, infección
Neuropatía, constipación, flebitis infusional
MielosupresiónSegundas neoplasiasEsterilidad en jóvenes
Comentarios
Tabla 6. Tratamiento de pacientes adultos con PTI. Opciones de tercera línea
Trombocitopenia Inmune | 7

136
• Ig EV 1g/kg por 1-2 días consecutivos.• Bolos de metilprednisolona 1g EV por 2-3 días. • Como esta estrategia suele demorar entre 24 y 48 hs, también administrar
transfusión de concentrados plaquetarios 1-3 U/h o 10 U cada 4 hs y hasta 1 féresis cada 30 minutos. Control mecánico o quirúrgico en el sitio de sangrado, si es necesario y factible (abdomen, SNC).
• Esplenectomía. Será mandatoria en caso de sangrado que amerite laparotomía o neurocirugía. En otros, considerar caso por caso ante sangrado crítico persistente a pesar del tratamiento médico.
• Factor VII activo recombinante (rFVIIa). Su uso está limitado a falla del tratamiento médico y necesidad de cirugía, sangrado en SNC no quirúrgico, otro sangrado crítico con imposibilidad de cirugía. Dado que se requiere un número mínimo de plaquetas para que actúe, se debe administrar junto con concentrados plaquetarios.
Tratamiento de PTI gestacional
El tratamiento debe seguir pautas similares al resto de los adultos, enfatizándose el manejo conservador, especialmente en el primero y segundo trimestre. El uso de corticoides durante el embarazo presenta complicaciones especiales: diabetes gestacional, hipertensión, prematurez, abruptio placentae. Las referencias sobre el uso de Ig EV anti-D en el
Se recomienda. Triple terapia médica (Ig EV, bolos de metilprednisolona y concentrados de plaquetas). En el caso de ser necesaria la cirugía de urgencia, realizar esplenectomía previa (en caso de neurocirugía) o simultánea (en caso de laparotomía) (2A).Se sugiere. rFVIIa como último recurso en casos refractarios al tratamiento anterior (2A).El tratamiento de emergencia en niños sigue los mismos lineamientos que en adultos.
7 | Trombocitopenia Inmune

137
embarazo son escasas. La conducta terapéutica sugerida durante el embarazo se resume en la Tabla 7.
Tratamiento en pacientes pediátricos
La PTI en niños tiene una incidencia anual de 1/10.000. La forma aguda, es benigna, autolimitada, frecuentemente precedida por vacunación o infección viral. Los niños con diagnóstico de PTI tienen excelentes chances de recuperación espontánea, el recuento plaquetario se normaliza en los primeros tres meses en dos tercios de los pacientes. El 75-80% de los pacientes se recuperan en los primeros 6 meses. Aproximadamente en un 20% de los niños pueden evolucionar hacia las formas crónicas. Se consideran factores de riesgo: comienzo insidioso, sexo femenino y mayor de 10 años al momento del diagnóstico. Un tercio de los pacientes con PTI crónica pueden tener remisiones espontaneas años después del diagnóstico. Un 5% pueden
Condición
(*) La indicación de parto vaginal o cesárea deberá basarse en criterios obstétricos (2C).
PTI gestacional 1er/2do trimestre <30x109/L
PTI gestacional3er. trimestre y preparto (*)
PTI gestacional¡NO USAR!
PTI gestacionalMANEJO CONSERVADOR
PTI gestacionalPOST-PARTO o CESAREA
TEV: tromboembolismo venoso.ADMP: altas dosis de metilprednisolona.
1ra. Línea Esteroides, Ig EV2da. Línea ADMP + Ig EV o hidroxicloroquina (LES y AR)Si bien no hay evidencias fuertes de teratogenicidad, la azatioprina (categoría D de la FDA) sólo debería usarse en el embarazo luego de una rigurosa evaluación riesgo/beneficio
Monitoreo más frecuenteP. Vaginal >30x109/LCesárea >50x109/LPeridural >75x109/L
Danazol, rituximab, agonistas RTPO, ciclofosfamida, vincristina, inmunosupresores como metotrexato y leflunomida
NO esplenectomía (salvo de urgencia y, en lo posible, limitada a 2do. trimestre)
Considerar profilaxis del TEV mecánica o farmacológica (si plaquetas >50 x109/L)
Conducta terapéutica
Tabla 7. Tratamiento en el embarazo
Trombocitopenia Inmune | 7

138
presentar PTI recurrente caracterizada por periodos de trombocitopenia y períodos de remisión.No se considera necesaria la realización de la PAMO para confirmar el diagnóstico. Las indicaciones actuales para las que se reserva la realización del medulograma son: citopenia asociada (excepto anemia ferropénica en los niños), no respuesta a la Ig EV, no remisión a los 3-6 meses y previo al uso de corticoides, esta última se recomienda, pero es opcional.La evaluación de los niños con PTI crónica debe incluir: screening para inmunodeficiencias, Lupus eritematoso sistémico y otra enfermedades autoinmunes (ALPS, autoinmune lymphoproliferative syndrome), serología para VIH (virus de inmunodeficiencia humana). Excluir desórdenes familiares hereditarios (MYH9).
Tratamiento
PTI reciente diagnóstico. Se indica tratamiento con recuento plaquetario <20x109/L, sangrado activo y/o factores de riesgo (traumatismo craneano, politraumatismo, uso de antiagregantes 7 a 10 días previos, coagulopatía asociada) Ver Figura 3.PTI persistente. Recuentos plaquetarios sostenidos por debajo de 20x109/L y/o sangrado activo. Se pueden usar las diferentes opciones terapéuticas solas o combinadas por ej. corticoides EV + Ig EV.PTI crónica. Se aconseja el uso de tratamiento de segunda línea. • La esplenectomía es hasta hoy el tratamiento más eficaz con un
porcentaje de respuesta inicial de 90% y de respuesta sostenida de 70% a los 10 años de seguimiento. No se aconseja en menores de 6 años. Se recomienda vacunación previa y profilaxis antibiótica.
• El rituximab reporta una respuesta inicial 60-70% pero un descenso de recuentos esperable, siendo la respuesta a largo plazo del 25% aproximadamente.
• Los agonistas de RTPO en niños, según la experiencia publicada en la literatura, tiene una respuesta de alrededor del 60-65%, con pocos efectos adversos y no inmunosupresores. En la actualidad se los considera como opciones válidas previas a la esplenectomía o como terapia puente para demorarla. Las dosis y variaciones de dosis son iguales a las recomendadas en adultos.
7 | Trombocitopenia Inmune

139
Cabe destacar que recientemente la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT) ha aprobado el uso en Argentina de uno de estos agonistas (Eltrombopag) en niños mayores de 6 años con PTI crónica con respuesta insuficiente a los corticoides, Ig EV o esplenectomía. También se ha publicado la experiencia pediátrica con romiplostim, pero aún no tiene aprobación para uso pediátrico.En la Tabla 8 se detallan las opciones terapéuticas en PTI pediátrica.
Figura 3. Algoritmo para el tratamiento del paciente pediátrico con PTI
Trombocitopenia Inmune | 7

140
Conflictos de interés
Los autores no presentan conflictos de interés.
Prednisona
Metilprednisolona
Dexametasona
IgG
Ig anti-D(paciente Rh+)
Tratamiento
60-80%
60-90%
80%
90-95%
80%
Respuesta
1-2 mg/Kg/día x 14-21 días V. oral
4 mg/Kg/día x 4 días (dosis máxima 180 mg/día) V. oral
30 mg/Kg/día x 2-3 días EV (dosis máxima 1gr)
20-40 mg/m2/día (dosis máxima 40mg) V. oral
1 gr/Kg/día x 2 días EV0,8 gr/Kg/día x 5 días EV
50-75 µg/Kg/día EV o subcutánea
Dosis
Cushing, osteoporosis, cataratas, hipertensión,hiperglucemia, psicosis
Anafilaxia, cefalea, nauseas,febrícula,
meningitis aséptica
Anemia hemolítica inmune fatal con dosis de 75 µg/Kg/día
(Advertencia de FDA)
Efectos adversos
Tabla 8. Tratamiento en pediatría
7 | Trombocitopenia Inmune

141
Referencias
1. Fundamentos para el manejo práctico del laboratorio de hemostasia. Segunda edición 2012. Grupo CAHT.
2. Provan D, Stasi R, Newland AC et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115: 168-186.
3. McMillan R, Wang L, Tani P. Prospective evaluation of the immunobead assay for the diagnosis of adult chronic immune thrombocytopenic purpura (ITP). J Thromb Haemost 2003; 1: 485-491.
4. Brighton TA, Evans S, Castaldi PA, Chesterman CN, Chong BH. Prospective evaluation of the clinical usefulness of an antigen-specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood 1996; 88: 194-201.
5. Davies JM, Lewis MP, Wimperis J et al. Review of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen: prepared on behalf of the British standards in haematology by a working party of the haematoncology task force. Br J Haematol 2011; 155: 308-317.
6. Kühne T, Donato H, Espina B et al on behalf of the Intercontinental Cooperative ITP Study Group. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica 2011; 96: 1831-1837.
7. Andre Tichelli, Neunert C, Crowther M et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117: 4190-4207.
8. Rodeghiero F, Stasi R, Gernsheimer T, Provan D et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113: 2386-2393.
Trombocitopenia Inmune | 7

142
7 | Trombocitopenia Inmune
9. Trombocitopenia inmune. Guías terapéuticas SAH Edición 2015.

143
Manifestaciones clínicas
Las trombocitopatías congénitas y adquiridas suelen manifestarse con sangrado cutáneo mucoso, sin diferencias con los síntomas hemorrágicos que presentan los pacientes con diagnóstico de enfermedad de von Willebrand (VWD) y pueden presentarse en la niñez o en adultos (Tabla 1) (1).Si bien las escalas de sangrado son herramientas validadas para definir la severidad de los síntomas e identificar pacientes que requieren una evaluación más profunda, no existe una firme recomendación en las alteraciones plaquetarias.La historia familiar de sangrado es útil, pero puede estar ausente en las
Desórdenes plaquetariosSusana Meschengieser1, Emilse Bermejo2, Ana Glembotsky3 y María Esther Aris Cancela4.1 Médica Hematóloga. Ex-Jefa del Depto. de Hemostasia y Trombosis, Instituto de Investigaciones Hematólogas “Mariano R Castex”, Academia Nacional de Medicina. Médica del Centro de Hematología Pavlovsky.2 Div. Trombosis. Dpto. de Hemostasia y Trombosis, Instituto de Investigaciones Hematólogas “Mariano R Castex”, Academia Nacional de Medicina.3 Hematología Investigación. Instituto de Investigaciones Médicas “Alfredo Lanari”. Universidad de Buenos Aires. CONICET.4 Médica Hematóloga. Jefa Hematología del Instituto Cardiovascular de Buenos Aires. Staff de Servicio de Hematología Hospital Universitario Austral.
8

144
8 | Desórdenes plaquetarios
trombocitopatías severas de herencia recesiva con padres asintomáticos.Recabar el antecedente de consanguinidad es importante para orientar el diagnóstico.A veces la disfunción plaquetaria es un componente más de un desorden multisistémico, como el síndrome de Chediak Higashi o la anomalía de May Hegglin. La presencia de problemas esqueléticos, faciales, oculares, auditivos, neurológicos, renales o inmunológicos asociados a sangrado sugieren la existencia de tales patologías (Tabla 2).Lo mismo ocurre con las trombocitopenias hereditarias que pueden tener además defectos en la función de las plaquetas (2).El sangrado en sistema nervioso central, gastrointestinal o urinario es extremadamente raro, así como la hemartrosis o el sangrado con la caída del cordón.
Diagnóstico de trombocitopatías hereditarias (TPH)
La evaluación y el diagnóstico de los desórdenes plaquetarios sigue siendo una tarea compleja. Aún con la incorporación de nuevas tecnologías y
Equimosis fáciles, hematomas espontáneos, extensosEpistaxisMenorragia, hemorragia post-parto, folículo hemorrágicoSangrado con exodoncia o cirugía
Tabla 1. Trombocitopatías: manifestaciones clínicas
Signos clínicos
Sordera, catarata, insuficiencia renalEczema, infecciones recurrentesAlbinismo oculocutáneo, nistagmoHipoplasia de radio
Alteraciones relacionadas a MYH9WAS, trombocitopenia ligada al cromosoma XHermansky PudlakTrombocitopenia con ausencia de radio
Tabla 2. Signos asociados a patología de base que orientan al diagnóstico
Signos asociados a patología de base

145
Desórdenes plaquetarios | 8
con los intentos de estandarización (guía BJH, ISTH), un alto porcentaje de trombocitopatías quedan sin caracterizar y pasan a formar parte de un grupo denominado “trastornos plaquetarios” (Tabla 3). La causa de esta falencia se debe principalmente, a la baja frecuencia de ocurrencia (escaso número de pacientes), a la falta de un consenso que unifique criterios diagnósticos y a la imposibilidad de contar con tecnologías que permitan detectar fallas en los múltiples mecanismos plaquetarios (Figura 1).
Receptores
Gránulos a
Gránulos d
Metabolismo lipídico
Mecanismos de señalización
Proteínas activadoras de integrinas
Movilización de calcio
Generación de trombina(membrana plaquetaria)
Citoesqueleto
Anomalías en
Sindrome de Bernard Soulier (BSS)Enfermedad de VW tipo plaquetario (PT-VWD)
Tromboastenia de Glanzmann (TG)a2β1GPVIP2Y12P2X1TPa
Sindrome de plaquetas grises (SPG)Sindrome ARC
Sindrome Quebec
Sindrome de Hermansky–PudlakSindrome de Chediak–Higashi
Sindrome de Griscelli
Fosfolipasa A2 (cPLA2)Tromboxano Sintasa (TXA2)
Prostaglandin Endoperoxi Sintasa-1
Proteinas GProteina KinasaTirosina Kinasa
Kindlin-3CalDAG-GEFI
Sindrome de Stormorken
Sindrome de Scott
Desorden relacionado a MYH9 (DR-MYH9)Desorden relacionado a filamina (DR-FLNA)
Tubulina-β1WAS
Trombocitopatías hereditarias
GPIBA,GPIBB,GP9GPIBA
ITGA2B, ITGB3ITGA2 (SNPs?)
GP6P2RY12P2RX1
TBXA2R
NBEAL2, GFI1BVPS33B, VIPAS39
PLAU
HPS1, AP3B1, HPS3-9LYST
RAB27A,MLPH
PLA2G4ATBXAS1PTGS1
GNAQ, GNASPRKACG
FERMT3RASGRP2
STIM1/ORAI1
TMEM16F
MYH9FLNA
TUBB1WASP, WIPF1
Gen afectado
Tabla 3. Clasificación de las trombocitopatías hereditarias

146
8 | Desórdenes plaquetarios
Antes de comenzar con los pasos necesarios para el estudio de las alteraciones plaquetarias, es importante destacar la ayuda que una correcta anamnesis tiene para lograr un diagnóstico. Estudios familiares, identificación de consanguinidad, lugar y tipo de sangrado, conocimiento de otras patologías de base, ingesta de fármacos, son algunos de los datos que orientan para caracterizar las TPH.Proponemos un esquema para realizar los estudios plaquetarios que consta de tres etapas en donde los datos técnicos referidos a las variables pre-analíticas, técnicas y metodológicas se excluyen por estar referenciados en el manual de técnicas, capítulo V (3) y en las guías internacionales (4, 5, 6).
Primera etapaRecuento plaquetario (RP). Los contadores automáticos posibilitan obtener no solo el número de plaquetas sino también el volumen plaquetario medio (VPM), estos dos parámetros son el primer llamado de atención para el operador. Número de plaquetas inferiores a 150x109/L y/o VPM ≥12fL deben ser confirmados por recuento en cámara, además de la observación del frotis
Figura1.
THC: Trombocitopenia relacionada a ANKRD26; 22QDS: Sindrome de deleción 22q o Velocardiofacial.

147
Desórdenes plaquetarios | 8
de sangre periférica (Ver diagnóstico de Trombocitopenias Hereditarias).Tiempo de sangría. El tiempo de sangría (TS) evalúa in vivo el fun-cionamiento global de la hemostasia primaria, en donde participan las plaquetas, otras células sanguíneas y componentes de la pared vascular. En la actualidad la utilidad del TS es muy controvertida, en la mayoría de las guías internacionales no se recomienda su uso y a partir de 1990 se retiró la recomendación de su empleo para predecir hemorragias quirúrgicas. Sin embargo, el TS es una prueba que bien realizada orienta en la detección de patologías hemorrágicas (Tromboastenia de Glanzmann (TG), síndrome de Bernard-Soulier (BSS), enfermedad de pool de depósito (SPD), etc.) y que de acuerdo a nuestra experiencia debe formar parte de los estudios que conforman esta primera etapa (3).
Segunda etapaAgregación plaquetaria. El método óptico o por transmisión de luz descripto simultáneamente por O’Brian y Börn es el estudio gold standard para analizar la funcionalidad de las plaquetas en suspensión (plasma rico en plaquetas, plaquetas lavadas o filtradas). La observación del trazado de las curvas de agregación (cambio de forma, primera y segunda ola de agregación) y los hallazgos de la primera etapa será lo que permita la caracterización de un desorden plaquetario o lo que oriente al uso de otras técnicas confirmatorias. Es importante destacar que este análisis debe hacerse teniendo en cuenta el conjunto de curvas de agregación. Para llegar a una correcta caracterización, será necesario evaluar la información que cada agonista expresa en función del mecanismo que representa, de las concentraciones y de otros agonistas usados. En los desórdenes plaquetarios es necesario observar fallas en la agregación producidas por lo menos en dos de los agonistas, dado que el trazado anómalo de una sola curva no es suficiente para caracterizarlo.El cuadro que proponemos es solo una representación de lo que cada curva nos induce a pensar como una posible trombocitopatía y por lo ya explicado, debe ser interpretado no como un diagnóstico certero, sino como una orientación del camino a seguir para lograr la caracterización (Tabla 4).

148
8 | Desórdenes plaquetarios
Tercera etapaCada una de las técnicas que se mencionan a continuación complementan los hallazgos observados en la segunda etapa y en la mayoría de los casos confirman una trombocitopatía. Por ej, la ausencia de segunda ola de agregación con todos los agonistas puede sugerir un SPD, un déficit enzimático, la ingesta de un fármaco antiagregante o una falla en alguno de los mecanismos metabólicos y habrá que continuar el estudio para poder caracterizar la TPH (Figura 2).Brevemente se describen algunas de las posibilidades de diagnóstico que cada una de estas técnicas puede aportar:Reacción de liberación de ATP (lumi-agregómetro). Cuando se tiene ausencia de agregación secundaria, esta técnica permite evaluar el contenido de los gránulos densos. La disminución o ausencia de liberación deberá ser confirmada con otros estudios (citometría de flujo, biología molecular u otros como captación de serotonina).
ADP (2,5, 5 y 10 µM), Adrenalina (ADR 5, 10 y 50 µM), Colágeno (Col 1 y 8 µg/L), ácido araquidónico (AA 0,5 y1 mM), Ristocetina (Risto≤0,7 y 1,2 mg/L). TG: tromboastenia de Glanzmann, BSS: síndrome de Bernard-Soulier, SPD: síndrome de pool de depósito, DE: déficit enzimático, VWD2B: VWD tipo 2B, PT-VWD VWD tipo plaquetaria.
ADPADPADPADRADRADR
ColágenoColágenoColágeno
AAAAAA
Risto 1,2Risto 1,2
Risto ≤0,7Risto ≤0,7
NormalNormal
Normal o dism.
NormalNormal
Con latencia
NormalNormalAusenteNormalNormalAusente
NormalAusenteAusente
≥15%
NormalAusenteAusenteNormalAusenteAusenteNormalDism.
AusenteNormalDism.
AusenteNormalAusenteAusente
≥15%
NormalSPD?
TG? / P2Y12?Normal
SPD? / DE?TG?
NormalTG? / SPD?
TG? / SPD? / GP VI? / DE?Normal
TG? / SPD?SPD? / DE?
NormalBSS?
NormalVWD 2B? / PT-VWD?
Tabla 4. Patrones de agregación plaquetaria
Cambio de forma Agregac. primaria Agregac. secundaria Trombocitopatías probables

149
Desórdenes plaquetarios | 8
Agregación/liberación con otros agonistas. Muchas veces el uso de agonistas que complementen los descriptos en el cuadro, son necesarios para orientar la caracterización. Algunos propuestos son:• mimético sintético U46619 (1-4 µM) para evaluar receptores de
tromboxanos. • ionóforo de calcio A23187 (1-4 µM) para evaluar movilización de Ca2+.• PAR1-AP 10 µM para evaluar receptor PAR1 para trombina.• PAF (1-4 µM) para evaluar mecanismo de CalDAG-GEFI/Rap1.Citometría de flujo. La utilización de marcadores de superficie (CD42b-GPIb, CD41 y CD61-GP IIbIIIa) permiten confirmar BSS y TG, respectivamente.El contenido intraplaquetario de gránulos densos es evaluable usando Mepacrina como fluorocromo y en el caso de los gránulos alfa utilizando anti-fibrinógeno, anti-fibronectina y/o anti-VWF complejado con algún fluorocromo (por ej. FITC, PE), ambas marcaciones caracterizan SDP (d, a).Microscopía electrónica. Además de aportar datos sobre la morfología plaquetaria, los distintos tipos de marcación permiten evaluar el contenido granular, la presencia de receptores de membrana y de proteínas intraplaquetarias.Microscopía de inmunofluorescencia. Ver diagnóstico de TH.Enzimoinmunoanálisis. Permite evaluar el metabolismo del ácido araquidónico, generación de tromboxanos, actividad de ciclo-oxigenasa y de tromboxano sintetasa.Biología molecular. Para la mayoría de las trombocitopatías se cuenta
Figura 2.

150
8 | Desórdenes plaquetarios
con una base genómica que permite caracterizar las mutaciones correspondientes (Ver diagnóstico de TH).
Diagnóstico de trombocitopenias hereditarias (TH)
Las TH son desórdenes genéticos raros y sub-diagnosticados (Tabla 5). De presentación clínica heterogénea, pueden hallarse enfermedades graves con sangrado profuso que se presentan desde el nacimiento como también desórdenes donde los pacientes son asintomáticos y la trombocitopenia se detecta en exámenes de rutina de laboratorio durante la edad adulta.
Plaquetas de tamaño pequeño
Plaquetas de
tamaño normal
Macroplaquetas
Tamaño plaquetario
WASTrombocitopenia ligada al X
Trombocitopenia amegacariocítica congénita (CAMT)
Trombocitopenia Ausencia de radio (TAR)Trombocitopenia con sinostosis radio-ulnar
(CITRUS)Trombocitopenia relacionada a ANKRD26
Desorden plaquetario familiar con predisposición a leucemia
Trombocitopenia por mutación de ETV6
Sindrome de Bernard Soulier (BSS)Bialélico
MonoalélicoDi George/SindromeVelocardiofacial (VC)
PT-VWDDesorden relacionado a MYH9 (DR-MYH9)
Desorden relacionado a la filamina (DR-FLNA)Trombocitopenia relacionada a ITGA2B/ITGB3
Trombocitopenia asociada a sitosterolemia (STSL)
Enfermedad relacionada a GATA1 (DR-GATA1)Trombocitopenia Paris Trousseau (TCPT)
SPGBialélico
MonoalélicoSíndrome relacionado a TUBB1-RT
Trombocitopenias hereditarias
WASWAS
MPL
RBM8AHOXA11
ANKRD26RUNX1
ETV6
GPIBA,GPIBB, GP9GPIBAGPIBBGPIBAMYH9FLNA
ITGA2B/ITGB3ABCG5, ABCG8
GATA1FLI1
NBEAL2GFI1BTUBB1
Gen afectado
Tabla 5. Clasificación de las trombocitopenias hereditarias

151
Desórdenes plaquetarios | 8
Dado que estas entidades son poco reconocidas, es frecuente que sean diagnosticadas en forma errónea como Trombocitopenia Inmune (PTI). Hasta el momento se conocen 22 desórdenes diferentes, causados por 24 mutaciones distintas. El diagnóstico correcto de estas entidades evita el uso de tratamientos innecesarios y potencialmente perjudiciales, permite brindar un pronóstico y consejo genético, y guiar sobre las decisiones terapéuticas. Para ello se tiene en cuenta un algoritmo diagnóstico propuesto por el grupo italiano de estudio de las plaquetas (7), que consta de diferentes etapas. La mayoría de las TH son causadas por defectos en moléculas que participan de la biogénesis plaquetaria y solo 2 desórdenes por defectos en la vida media plaquetaria, Sindrome de Wiskott-Aldrich (WAS) y VWD2B / PT-VWD siendo el mecanismo mixto. Excepto en estos dos últimos desórdenes, el estudio de sobrevida plaquetaria es útil para distinguir una trombocitopenia de origen hereditario de una PTI.
Identificación de pacientes a evaluarEl primer paso para diferenciar si una trombocitopenia es hereditaria, es realizar una minuciosa historia clínica. La inclusión de la historia familiar de sangrados y trombocitopenia, indica claramente el origen genético de la trombocitopenia, aunque la ausencia de antecedentes familiares no la descarta, ya que se describen algunos casos donde las mutaciones resultan de novo o la herencia es autosómica recesiva, circunstancia en la cual pueden existir generaciones no afectadas. En las TH se incluyen desórdenes sindrómicos, donde los pacientes presentan trombocitopenia asociada a otras características clínicas, como infecciones recurrentes, eczemas, historia familiar de leucemia mieloide aguda o mielodisplasia. De esta manera, las infecciones o eczemas se asocian a WAS, signos de glomerulonefritis, pérdida de audición o cataratas, nos orientan a la sospecha de desorden relacionado al MYH9. Historia familiar de antecedentes de leucemia o síndrome mielodisplásico, al desorden plaquetario familiar con predisposición a leucemia mieloide aguda (DPF/LMA), desórdenes relacionados al gen ANKRD26 y trombocitopenia relacionada a ETV6, identificada recientemente. Por otro lado, existen entidades no sindrómicas, donde la única manifestación de la enfermedad es la trombocitopenia. Para la clasificación de estos desórdenes es útil la aplicación de un algoritmo diagnóstico (Figura 1).

152
8 | Desórdenes plaquetarios
Primera etapa del diagnósticoEl primer estudio que se realiza en el laboratorio y el más común, frente a la sospecha de pacientes con TH, es el RP y observación minuciosa del frotis de sangre periférica. La observación del frotis brinda información muy importante, la presencia de plaquetas de gran tamaño, grises, pálidas, nos orienta a pensar en la presencia de Síndrome de plaquetas grises (SPG), las inclusiones de Döhle en los neutrófilos en desorden relacionado al MYH9 y las anormalidades de la serie roja, a síndrome con mutación de GATA-1.La presencia de plaquetas de gran tamaño, macroplaquetas, puede subestimar el RP en los contadores hematológicos. En estas patologías, el RP utilizando cámara de Neubauer por microscopía óptica, es el método de referencia. La determinación del VPM en contadores hematológicos automatizados, facilita la orientación del diagnóstico mediante la clasificación de estas entidades en base al tamaño plaquetario. En laboratorios especializados, dado que es una determinación que requiere tiempo y un software específico de análisis de imagen, la medición del diámetro plaquetario medio (DPM) de las plaquetas contribuye a la confirmación del tamaño plaquetario en algunas patologías.
Agregación plaquetaria (3). La ausencia o disminución muy marcada de respuesta a la agregación plaquetaria frente a ristocetina, nos sugiere la presencia de BSS homocigota, que se confirmará mediante estudios de citometría de flujo y análisis mutacional.La presencia de agregación con bajas concentraciones de ristocetina, sugiere VWD2B o PT-VWD, que se confirma mediante estudio de mezcla de plasmas y análisis mutacional.El SPG puede presentar disminución de la respuesta a la agregación frente a trombina y/o colágeno que se confirmará luego por la ausencia o disminución marcada de los gránulos alfa plaquetarios por microscopía electrónica. La disminución de la agregación plaquetaria frente a la mayoría de los agonistas, plaquetas de tamaño normal y un sangrado mayor al esperado para el recuento plaquetario que presentan los pacientes, nos hace sospechar en DPF/LMA.Glicoproteínas (GP) de membrana plaquetaria por citometría de flujo.

153
Desórdenes plaquetarios | 8
Utilizando un panel de diferentes anticuerpos (Ac) dirigidos contra las GP de la superficie de la membrana de las plaquetas, se determina por citometría de flujo, la expresión de los diferentes complejos. La disminución marcada o ausencia de la GP Ib/IX sugiere la presencia de BSS clásico causado por mutación homocigota de la GP1BA mientras que una disminución del 50% indicaría BS monoalélico, en los pacientes portadores de la mutación heterocigota Bolzano (sustitución Ala156Val). En el desorden relacionado al MYH9 es frecuente hallar disminución de los niveles de la GP Ib/IX en relación a la GP IIIa, dicha disminución relativa probablemente sea consecuencia del gran tamaño de las plaquetas, característica de esta entidad.
Estudios confirmatoriosInmunofluorescencia para la detección de MYH9. Frente a la presencia de macroplaquetas, teniendo en cuenta que los desórdenes más frecuentes son el BSS y el desorden relacionado al MYH9, es importante la búsqueda de inclusiones tipo Döhle, inclusiones celestes o liliáceas, únicas o múltiples en los neutrófilos de los extendidos de sangre periférica de los pacientes. Estas inclusiones no siempre son evidentes y es difícil identificarlas con certeza. Son patognomónicas de los desórdenes relacionados al MYH9. Para su confirmación, se pueden detectar por inmunofluorescencia, utilizando Ac específico para miosina, donde se observan diferentes patrones de agregados de la proteína en el citoplasma de los neutrófilos de pacientes con este desorden, que se correlacionan con diferentes tipos de mutaciones, demostrando de esta manera, que esta determinación tiene un 100% de sensibilidad y un 95% de especificidad en el diagnóstico. Inmunofluorescencia para la detección de trombospondina (TSP). Para confirmar la disminución o ausencia de los gránulos alfa plaquetarios observada por microscopía electrónica, se evalúa por inmunofluorescencia la proteína adhesiva TSP-1 contenida en los gránulos alfa, en el caso que se tenga acceso a esta metodología. Este método es utilizado como herramienta para el diagnóstico en el SPG (8) y se establece como valor de corte la presencia de 5 o menos gránulos por plaqueta, como índice de deficiencia marcada de gránulos alfa.Microscopía electrónica. Ver diagnóstico de TPH.Biología molecular. Se utilizan técnicas de secuenciación convencionales de los genes implicados en los distintos desórdenes (Tabla 5). La utilización de nuevas técnicas genéticas como next generation sequencing (NGS),

154
8 | Desórdenes plaquetarios
empleando paneles de genes, promete la detección de nuevas formas de TH y la clasificación de un porcentaje de trombocitopenias que hasta el momento se encuentran sin diagnóstico etiológico.
Manejo terapéutico de desórdenes plaquetarios hereditarios
Medidas generales• Evitar aspirina y antiinflamatorios no esteroideos• Vacunación para hepatitis A y B• Control odontológico frecuente• Anticonceptivos orales o DIU hormonal para menorragiaAgentes terapéuticos• Desmopresina (DDAVP): induce liberación de VWF, activador tisular del
plasminógeno (tPA) y prostaciclina, mejora la adhesividad plaquetaria, no modifica la agregación. Solo es útil en trombocitopatías leves a moderadas (Tabla 6).
Su administración puede repetirse solo luego de 12 hs pero no antes por taquifilaxia y preferentemente a las 24 hs. Puede utilizarse en sangrado agudo o previo a procedimientos invasivos. No se puede predecir su eficacia con las pruebas de laboratorio.
• Agentes antifibrinolíticos: ácidos tranexámico (AcTx) o ε-aminocaproico, para sangrados menores, epistaxis, gingivorragia, menorragia. La dosis de AcTx es 10-15 mg/Kg cada 8 hs (1g en general cada 8 hs en adultos). La dosis del ácido ε- aminocaproico es de 100 mg/kg. Para procedimientos invasivos comenzar 12 a 24 hs antes y continuar 12 hs después. Además de la presentación oral se dispone de la presentación EV que puede
Administración
Efectos colaterales
Contraindicaciones
Endovenosa 0,3 µg/Kg en 20-50 mL de Sol. Fisiológica a pasar en 20 min
inmediatamente antes del procedimiento
Subcutánea 0,3 µg/Kg una hora antes
Retención hídrica, hipononatremia, enrojecimiento facial, cefalea ocasional
Antecedentes trombóticos, mayores de 70 años, convulsiones
Tabla 6. Desmopresina

155
Desórdenes plaquetarios | 8
utilizarse también localmente en sangrados bucales o post-exodoncias. La duración del tratamiento depende de la respuesta obtenida.
• Transfusión de plaquetas (Tabla 7).
• FVII recombinante activo (rFVIIa): induce una explosión de trombina. Útil cuando hay refractariedad a la transfusión de plaquetas o en ausencia de concentrados plaquetarios en la emergencia (Tabla 8). El uso de rFVIIa ha demostrado ser eficaz no solo en TG sino también en BSS y en PT-VWD. En casos de sangrados severos y recurrentes puede utilizarse como tratamiento profilactico (simil-hemofilia) con una administración semanal con buen resultado.
• ARTPO: si bien no se encuentran aprobados para el tratamiento de las TH, existen publicaciones acerca de su eficacia en pacientes con enfermedad relacionada el gen MYH9 y en WAS. La seguridad a largo plazo de estas drogas en estas entidades no está definida. Es importante considerar que algunos desórdenes plaquetarios congénitos cursan con predisposición a leucemia (DPF/LMA, ANKRD26, ETV6) o mielofibrosis (SPG), por lo cual el uso de estas drogas en estas entidades en particular debe ser evitado.
Manejo de exodoncias y cirugíasPara exodoncias considerar la severidad de la disfunción plaquetaria: Transfusión de plaquetas o rFVIIa en casos severos, AcTx y/o DDAVP en casos leves a moderados. De acuerdo a la cirugía evaluar la administración de concentrados plaquetarios a las 24 y/o 48 hs.
Recomendación
Riesgo
Donante único, leucodeplecionadas, HLA compatibles
Aloinmunización, desarrollo de Acs anti-GPIIbIIIa, anti-GPIb, reacción alérgica
Tabla 7. Transfusión de plaquetas
Indicación
Dosis
Tromboastenia de Glanzmann
90 µg/Kg EV cada 2-3 hs.
Tabla 8. FVII recombinante activo (rFVIIa)

156
EmbarazoEs necesario que la Institución prevista para el parto/cesárea cuente con la disponibilidad de concentrados plaquetarios. En general se requiere solo una transfusión previa a una cesárea y se repite a las 24 hs. Si hubiera hematoma en la herida sería necesario prolongar las transfusiones de plaquetas. Si no hubiera respuesta adecuada, utilizar rFVIIa y asociar AcTx.
Trombocitopatías adquiridas
Las trombocitopatías adquiridas pueden ser causadas por distintas etiologías (Tabla 9).
La inhibición plaquetaria inducida por drogas puede tener objetivo terapéutico o ser un efecto colateral (Tablas 10 y 11).Las drogas que inhiben el receptor P2Y12 son antitrombóticos potentes. La respuesta inhibitoria del clopidogrel es variable. La incidencia de eventos de sangrado es similar en pacientes que reciben clopidogrel y aspirina en forma aislada y aumenta con la combinación. Los nuevos antiagregantes, prasugrel y ticagrelor, son más efectivos en la reducción de eventos trombóticos que el clopidogrel, pero incrementan el riesgo de eventos de sangrado. El prasugrel está contraindicado en pacientes con antecedentes de ACV, mayores de 75 años y pacientes con peso menor a 60 Kg.
8 | Desórdenes plaquetarios
DrogasSuplementos dietarios
AlimentosSíndromes mieloproliferativos
ParaproteínasAcs antiplaquetarios
Circulación extracopóreaInsuficiencia renal
Tabla 9. Disfunciones plaquetarias adquiridas
Causas

157
Estudios randomizados sugieren que la incidencia de complicaciones de sangrado asociadas a inhibidores del receptor P2Y12 dependen más de la intensidad de la inhibición que del tipo de droga usada (9).Algunos suplementos dietarios, el alcohol y algunos alimentos pueden inhibir la función plaquetaria, aunque el significado clínico es poco claro; así en pacientes con alteraciones plaquetarias o defectos de coagulación conocidos se recomendaría evitarlos (10).
Insuficiencia renalEl deterioro de la función plaquetaria se evidencia por prolongación del TS. Los resultados de los estudios de agregación son inconsistentes. Si bien la diálisis (hemodiálisis o peritoneal) es la medida más apropiada para mitigar el sangrado inducido por la uremia, la corrección de la anemia, transfusional o con eritropoyetina, y la administración de DDAVP limitan el sangrado urémico. La eritropoyetina corrige la anemia reduciendo el sangrado, pero el efecto sobre las plaquetas puede ocurrir tempranamente por incremento del número de plaquetas reticuladas. El efecto de DDAVP es transitorio y estaría relacionado con el aumento de FVIII, VWF y adhesividad plaquetaria.
Desórdenes plaquetarios | 8
Inhibidores de la
ciclo-oxigenasa-1 (COX-1)
Inhibidores del P2Y12
Inhibidores de la GPIIbIIIa
Otros
Aspirina
Ticlopidina, clopidogrel, prasugrel
Abciximab
Colistazol, dipiridamol, antibióticos β-lactámicos, inhibidores de la recaptación de
serotonina, dextrán, alcohol
Ibuprofeno, naproxeno, indometacina,
diclofenac, ketorolac
Ticagrelor (ciclopentil-triazol-pirimidina)
Eptifibatide, tirofibán
Tabla 10. Antiplaquetarios con efecto sobre el sangrado clínico
Irreversibles Reversibles
Bloqueantes cálcicosAnestésicos regionales y generales
Beta-bloqueantesAntidepresivos tricíclicos
Nitratos
Ginkgo BIlobaGinseng
AjoJengibre
Tabla 11. Drogas sin efecto sobre el sangrado clínico
Drogas Hierbas

158
Los crioprecipitados (Crio) son una opción en pacientes urémicos con alto riesgo de sangrado o sangrado activo. Tienen efecto sobre el TS dentro de las primeras 4 a 12 hs. Las desventajas son los riesgos transfusionales. La respuesta es impredecible. Los estrógenos conjugados reducen la producción de óxido nítrico y promueven la reactividad plaquetaria. Se usan en dosis equivalente a la usada en terapia hormonal de reemplazo. Debe ser evaluado el riesgo trombótico en pacientes patología cardiovascular (11).
Enfermedad valvular aórticaLa estenosis aórtica puede tener sangrado excesivo secundario a VWD adquirida. El mecanismo es por alto shear y resuelve con la cirugía.
Disfunción plaquetaria adquirida transitoria en el contexto de cirugía cardiovascular (CCV)La circulación extracorpórea y las membranas de oxigenación del by-pass cardiopulmonar producen tanto trombocitopenia como disfunción plaquetaria. La disfunción plaquetaria en el post-operatorio de CCV con circulación extracorpórea es la causa más frecuente de sangrado excesivo no quirúrgico (aún con recuentos normales) descartadas entre otras etiologías como inadecuada neutralización de heparina, o rebote heparínico.El tratamiento es la transfusión de plaquetas o Crio. La DDAVP podría ser de utilidad, aunque hay reportes de complicaciones coronarias trombóticas en pacientes añosos con enfermedad ateromatosa.
Síndromes mieloproliferativosEl sangrado en los síndromes mieloproliferativos está asociado a múltiples mecanismos como activación plaquetaria y degranulación, defectos en los receptores de membrana, clivaje del VWF por proteasas, etc. Desde el punto de vista clínico recuentos superiores al millón de plaquetas se asocian a mayor riesgo de sangrado. La normalización del RP mejora la tendencia hemorrágica. La presencia de sangrado mayor es una indicación de tratamiento citoreductor.
Mielodisplasia El sangrado en general es por trombocitopenia, pero también puede ser
8 | Desórdenes plaquetarios

159
debido a disfunción plaquetaria por trombopoyesis anormal. Se describe una disminución de los gránulos alfa y anomalías del sistema tubular denso.
ParaproteínasTanto en el mieloma como en la macroglobulinemia de Wäldestrom, la disfunción plaquetaria se produce por la adhesión no específica de la inmunoglobulina a la superficie plaquetaria que dificulta la interacción agonista-receptor. En estas condiciones la transfusión de plaquetas es de eficacia limitada. La plasmaféresis es eficaz cuando se trata de una IgM.
PTIAcs dirigidos contra la GPIIbIIIa o el receptor GPIb-IX pueden producir disfunción plaquetaria. Si bien las pruebas de agregación plaquetaria con difíciles de realizar por la baja recuperación de plaquetas, muestran patrones similares a una TG o a un SPD. Estos pacientes presentan manifestaciones de sangrado que no condicen con el RP. En sangrado mucoso, los antifibrinolíticos pueden ser de utilidad.
Manejo de agentes antiplaquetarios en procedimientos invasivos
En caso de cirugía no cardíaca de pacientes tratados con terapia antiplaquetaria dual por implantes de stents coronarios es necesario considerar:• Riesgo de trombosis del stent (en caso de suspender el tratamiento
antiplaquetario dual)• consecuencias de retrasar el procedimiento quirúrgico • el riesgo de sangrado intra y post-procedimiento y las consecuencias
del mismo si se mantuviera el tratamiento antiplaquetario.Los stents de primera generación mostraban mayor riesgo de complicaciones trombóticas asociadas al stent entre la cuarta y sexta semana y continuaban elevadas hasta el primer año post-implante de un stent con drogas (DES), por lo que las cirugías cardíacas electivas se retrasaban hasta después del año. Los stents de nueva generación se asocian con menor riesgo de trombosis, por lo cual la recomendación clase I, de retrasar la cirugía a más de 1 año, se redujo a 6 meses en DES, y la de clase IIb que decía que podría
Desórdenes plaquetarios | 8

160
ser considerada después de 6 meses se acortó a 3 meses (12).El incremento del riesgo de sangrado en pacientes tratados con doble antiagregación que van a cirugía es incierto. Si es necesario suspender el inhibidor de P2Y12 se sugiere mantener la aspirina (basado en recomendación de expertos) y reiniciarlo lo más rápido posible en el post-operatorio. La rotación a agentes antiplaquetarios intravenosos (tirofibán y eptifibatide) en el pre-operatorio es una opción en pacientes de alto riesgo para la suspensión, si bien su eficacia y seguridad es controversial. El tratamiento antiplaquetario debe ser reiniciado a la brevedad. Los tiempos de suspensión se detallan en la Tabla 12.
La decisión de un procedimiento quirúrgico en un paciente con stent es compleja y debe ser consensuada con un equipo multidisciplinario y el paciente (Tabla 13).
8 | Desórdenes plaquetarios
Aspirina 100 mgTiclopidinaClopidogrel
PrasugrelTicagrelor
4 días5 a 7 días5 a 7 días5 a 7 días
Cirugía mayor 4 díasCirugía menor 2 días
Tabla 12. Tiempo de suspensión para procedimientos programados
Droga Tiempo de suspensión
Stent sin drogas
(BMS)
Stent con drogas
(DES)
<30 días
>30 días
<3 meses
3-6 meses
-Discontinuar tratamiento antiplaquetario dual
-Riesgo de retrasar cirugía >riesgo de trombosis del stent
> 6 meses
-Discontinuar tratamiento antiplaquetario dual
Retrasar cirugía
Proceder a cirugía
Retrasar cirugía
Rotar a inhibidores EV
Puede ser considerado proceder a la cirugía
Proceder a cirugía
Tabla 13. Cirugía no cardiaca electiva en pacientes con stent

161
Considerando el riesgo de sangrado de una cirugía bajo tratamiento antiplaquetario, un grupo de consenso identificó procedimientos quirúrgicos y/o invasivos de alto riesgo, para los que se requiere suspensión del mismo. La cirugía de catarata y odontológica son consideradas de bajo riesgo de sangrado por lo que no se recomienda la suspensión (13).Con respecto a la CCV, el uso de dosis menores a 325 mg no impacta en el sangrado a excepción de las cirugías de alto riesgo (Tabla 14).
Conflictos de interés
Las autoras no presentan conflictos de interés.
Desórdenes plaquetarios | 8
Cardíaca
General
Maxilofacial
Plástica
Torácica
Endoscópica digestiva
Ginecológica
Neurocirugía
Ortopédica
Urológica
Reoperación, endocarditis, disección aortica, post-angioplastia fallida
Resección hepática, duodeno pancreatectomía
Radical reconstructiva de cáncer de cabeza y cuello, sialoadenectomía submandibular
Reconstructiva, liposucción, quemaduras >15%
Esofagectomía, decorticación pulmonar, pleuronemonectomía, biopsia transbronquial,
broncoscopía con intervención
Dilatación por acalasia, mucosectomía, resección submucosa, aspiración con aguja fina
de quiste pancreático, ampulectomía
Histerectomía (útero grande), miomectomía, endometriosis, cirugías radicales de ovario
y cervix
Lesiones intradurales (intracerebrales, hemorragias intraparenquimatosas)
Cirugía protésica cadera, rodilla, fémur proximal, pelvis
Nefrectomía parcial, nefrostomía percutánea, penectomía, orquiectomía parcial,
prostatectomía radical y endoscópica
Tabla 14. Cirugías con alto riesgo de sangrado bajo tratamiento antiagregante

162
8 | Desórdenes plaquetarios
Referencias
1. Nurden AT, Freson K, Selighson U. Inherited platelet disorders. Haemophilia 2012; 18: 154-160.
2. Bolton-Maggs PHB, Chalmers EA, Collins PW et al. A review of inherited platelet disorders with guidelines for their management on behalf of UKHCDO. Br J Haematol 2006; 135: 603-633.
3. Fundamentos para el manejo práctico en el Laboratorio de Hemostasia. Grupo CAHT. Segunda Edición, 2014.
4. Harrison P, Mackie I, Mumford A, et al. Guidelines for the laboratory investigation of heritable disorders of platelet function. British Committee for Standards in Haematology Br J Haematol 2011; 155: 30-44.
5. Gresele P, for the Subcommittee on Platelet Physiology. Diagnosis of inherited platelet function disorders: guidance from the SSC of the ISTH. J Thromb Haemost 2015; 13: 314–322.
6. Cattaneo M, Cerletti C, Harrison P et al. Recommendations for the standardization of light transmission aggregometry: Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J Thromb Haemost 2013; 11: 1183–1189.
7. Balduini CL, Cattaneo M, Fabris F et al. Italian Gruppo di Studio delle Piastrine. Inherited thrombocytopenias: a proposed diagnostic algorithm from the Italian Gruppo di Studio delle Piastrine. Haematologica 2003; 88: 582–592.
8. Glembotsky AC, Marta RF, Pecci A et al. International collaboration as a tool for diagnosis of patients with inherited thrombocytopenia in the setting of a developing country. J Thromb Haemost 2012; 10: 1653–1661.
9. Cattaneo M. Bleeding manifestations of congenital and drug-induced defects of the platelet P2Y12 receptor for adenosine diphosphate. Thromb Haemost 2011; 105: S67–S74.

163
Desórdenes plaquetarios | 8
10. Konkle BA. Acquired disorders of platelet function. Hematology Am Soc Hematol Educ Program 2011; 1: 391-396.
11. Evidence-based treatment recommendations for uremic bleeding. Hedges SJ, Dehoney SB, Hooper JS, Amanzadeh J, Busti AJ. Nat Clin Pract Nephrol 2007; 3: 138-153.
12. Levine GN, Bates ER, Bittl JA k, et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Focused Update Writing Group. J Am Coll Cardiol 2016; pii: S0735-1097: 01699-5.
13. Capodanno D, Angiolillo DJ. Management of antiplatelet therapy in patients with coronary artery disease requiring cardiac and no cardiac surgery. Circulation 2013; 128: 2785-2798.

164

165
Trombocitopenia neonatalFlorencia Tisi Baña. Médica, Pediatra, Hematóloga, Hematóloga y Oncóloga Pediatra, UBA.
Introducción
La trombocitopenia (recuento plaquetario menor a 150x10⁹/L) es una alteración comúnmente hallada en neonatos (1/3 de los ingresos a la Unidad de Cuidados Intensivos Neonatal, UCIN) (1), especialmente en prematuros (2/3 de los prematuros de muy bajo peso, RN MBP) y en niños enfermos. Generalmente es leve o moderada y no requiere tratamiento, pero cuando es severa la mayoría de los neonatos recibe transfusiones de plaquetas profilácticas. La etiología es diversa, así como su evolución natural. Puede ser un marcador de enfermedad subyacente tanto como un factor de riesgo de sangrado.
Definición
Trombocitopenia: recuento plaquetario menor a 150x10⁹/L.Puede ser categorizada en:• Leve: 100-150x10⁹/L• Moderada: 50-100x10⁹/L• Severa: <50x10⁹/L
9

166
Consideraciones Generales. El recuento plaquetario normal es >150x10⁹/L desde la semana 15 de gestación. En cuanto a la función, las plaquetas de los recién nacidos de termino (RNT) sanos tienen menor respuesta a agonista plaquetarios, menor cantidad de receptores alfa-2 adrenérgicos en superficie, menor movilización de calcio y diferencias en la señalización del receptor de tromboxano. Sin embargo el tiempo de sangría, y el tiempo de oclusión en PFA100®, son más cortos, debido al hematocrito más alto, mayor volumen corpuscular medio, mayor concentración de Factor von Willebrand (VWF) y predominio de multímeros más largos. En recién nacidos pre-término (RNPT) la hiporeactividad es más marcada, el tiempo de sangría y el tiempo de oclusión de PFA100® son inversamente proporcionales a la edad gestacional.
Etiología
Los mecanismos que conducen a la plaquetopenia son la disminución de la producción y/o el aumento del consumo o destrucción.De acuerdo al momento de presentación, la trombocitopenia puede dividirse en dos grupos (Tabla 1), en los que se incluyen las distintas causas, algunas exclusivas de cada grupo, otras compartidas por ambos pero con distinta frecuencia (2, 3):• Temprana: desde el nacimiento hasta las 72 hs de vida.• Tardía: después de las 72 hs de vida.La patología materna y placentaria causa trombocitopenia temprana, mientras que hay causas fetales o neonatales en ambos grupos. De esta forma, la clasificación en uno de estos grupos permite orientarse a las posibles etiologías (maternas, placentarias o propias del paciente).
Diagnóstico
Laboratorio. Las extracciones de sangre pueden ser difíciles en neonatos enfermos y dar recuentos plaquetarios falsamente bajos por la formación de acúmulos de plaquetas (pseudotrombocitopenia), especialmente si la muestra es de punción de talón.El frotis de sangre periférica (tamaño y morfología plaquetarios,
9 | Trombocitopenia Neonatal

167
características de la serie mieloide y eritroide) puede orientar al diagnóstico.
Historia Clínica del paciente
Antecedentes familiares. Maternos (trombocitopenia inmune o lupus eritematoso sistémico –PTI o LES, respectivamente-), hermanos (antecedentes de trombocitopenia, sangrado, lesiones neurológicas sin diagnóstico de certeza).Características del embarazo. Preeclamsia, diabetes (DBT) gestacional, restricción de crecimiento intrauterino (RCIU).
Trombocitopenia Neonatal | 9
Un recuento plaquetario bajo debe siempre confirmarse. Se sugiere realizar frotis de sangre periférica en todos los neonatos con plaquetopenia.
En negrita se destacan las causas más frecuentes.
Insuficiencia Placentaria (Preeclampsia, RCIU, DBT)Asfixia perinatal
Infección perinatal (E. Coli, SGB, HI, LM)CID
Aloinmune (TFNA)Autoinmune (PTI, LES)
Infecciones congénitas (TORCH)Infecciones virales (HIV, enterovirus)
TrombosisPolicitemia
Reemplazo MO (Leucemia)Kasabach-Merritt
Congénitas/Hereditarias (TAR, TAMC, Fanconi)Aneuploidías (trisomía 18, 13, 21)
Enfermedad metabólica
Sepsis tardía.NEC.
Infecciones congénitas (TORCH)Infecciones virales (HSV, CMV)
TrombosisCID
AutoinmuneKasabach-Merritt
AneuploidíasEnfermedad metabólica
Asociada a drogas (antivirales, heparina)Congénitas/Hereditarias
Tabla 1. Clasificación de trombocitopenia neonatal por tiempo de presentación
Tempranas (<72hs) Tardías (>72 hs)

168
Características del parto y perinatológico. Parto dificultoso, apgar bajo u otros antecedentes de hipoxia perinatal, peso de nacimiento.Comorbilidades desde el nacimiento al momento de la consulta. Fiebre, cultivos, serologías virales, trombosis, enterocolitis necrotizante (NEC), cardiopatía, etc., sus tratamientos y evolución.
Examen Físico
Se evalúan:Signos de sangrado. Menor: tinción hemática de secreciones oral, nasofaríngea, endotraqueal, materia fecal, hematuria, petequias o babeo de sitios de punturas; Mayor: cerebral, hepático, pulmonar, gastrointestinal, renal.Condición general. Signos de infección, hepato o esplenomegalia, ictericia, signos de alteraciones cromosómicas u otras malformaciones congénitas, signos de trombosis.En la evaluación del paciente con trombocitopenia neonatal hay 3 puntos clave: • Tiempo de presentación (temprana o tardía).• Origen (causa materna, placentaria o fetal/neonatal).• Riesgo individual de sangrado.
Tiempo de Presentación:Trombocitopenia Temprana. Suele ser leve a moderada, autolimitada y no requerir intervención. Si es severa, las causas más comunes son infección (en niños enfermos) y trombocitopenia aloinmune (si no hay otra etiología probable que la explique y el bebé está por lo demás sano).Trombocitopenia Tardía. Suele anunciar una enfermedad grave, especialmente si el recuento plaquetario cae rápidamente. Sepsis (bacteriana o fúngica) o NEC, son las causas más comunes. La trombocitopenia puede anteceder los signos clínicos de sepsis 24 horas. Por lo general es prolongada, severa y se asocia a riesgo aumentado de sangrado, por lo que este grupo se transfunde plaquetas con mayor frecuencia.La Trombocitopenia Persistente (>14 días de duración), puede deberse a consumo crónico, inadecuada producción de trombopoyetina (TPO) por disfunción hepática (alimentación parenteral total prolongada, NEC con
9 | Trombocitopenia Neonatal

169
colestasis) y se asocia a aumento del riesgo de mortalidad. En niños por otro lado sanos, debe pensarse en trombocitopenias hereditarias.
Las Figuras 1 y 2, muestran los algoritmos diagnósticos sugeridos por esta guía para trombocitopenia temprana y tardía, respectivamente.
Según la evidencia disponible hasta el momento, se sugiere la utilización de la clasificación según el momento de aparición de la trombocitopenia ya que da orientación sobre la causa subyacente, la probable evolución y gravedad de la trombocitopenia y la necesidad de tratamiento transfusional.
Figura 1. Algoritmo diagnóstico en trombocitopenia temprana
Trombocitopenia Neonatal | 9

170
Riesgo de sangrado
Trombocitopenia y sangrado suelen estar asociados en neonatos, pero no necesariamente tienen relación causal. Muchos pacientes con trombocitopenia severa no tienen complicaciones hemorrágicas, y muchos pacientes con sangrado severo no tienen trombocitopenia. El sangrado en esta población parece ser multicausal. Entre los factores de riesgo conocidos más importantes se incluyen: la menor edad gestacional, la edad post-natal y el peso del nacimiento. Otros factores de riesgo son: apgar bajo, acidosis, requerimiento de asistencia respiratoria mecánica (ARM), NEC, coagulación intravascular diseminada (CID), alteraciones de la hemostasia.La trombocitopenia suele ser marcador de severidad de otra patología, que se asocia a aumento de la mortalidad neonatal tanto como a trombocitopenia, lo que explicaría la mayor mortalidad asociada a transfusiones de plaquetas.
Figura 2. Algoritmo diagnóstico en trombocitopenia tardía
9 | Trombocitopenia Neonatal

171
Manejo
La transfusión de plaquetas es la principal herramienta terapéutica en esta patología, profiláctica o como tratamiento de sangrado activo.El manejo de la trombocitopenia neonatal es heterogéneo, carece de evidencia científica y se basa en consensos de expertos (Recomendaciones Grado C Nivel IV), por lo que hay múltiples guías de transfusiones en neonatos, con diferentes valores de corte.El objetivo es evitar el sangrado mayor y disminuir la exposición a los hemoderivados.La transfusión de plaquetas profiláctica no ha mostrado reducir la mortalidad en neonatos. Los neonatos transfundidos tienen menor sobrevida (puede deberse a que los más enfermos son los que más se transfunden).
En base a las recomendaciones internacionales, se recomienda (Grado C, Nivel IV):• Transfundir plaquetas a los neonatos con sangrado activo mayor para
mantener 100x10⁹/L.• Otros valores de transfusión dependerán del riesgo de sangrado
percibido para cada paciente (Tabla 2).• Los neonatos con trombocitopenia aloinmune (TFNA) se consideran
en forma separada por su mayor riesgo de sangrado para el mismo recuento plaquetario, comparado con las otras etiologías.
• El uso de factores de crecimiento como los análogos de TPO no está recomendado por el momento para esta población.
Con la evidencia disponible actualmente, se recomienda que en la decisión clínica de la transfusión deberá evaluarse el riesgo individual de sangrado.
El efecto de la trombocitopenia en el riesgo de sangrado es desconocido.
Trombocitopenia Neonatal | 9

172
Producto a transfundir
Recomendaciones. Se sugiere transfundir concentrado de plaquetas:• ABO idéntico o compatible (Tabla 3). El uso de plaquetas de grupo O
para pacientes no-O debe tratar de ser evitado.• RhD idéntico o compatible.• Producidos con técnica estándar.• Irradiado, si el paciente tuviera una inmunodeficiencia, post-trasplante
de células progenitoras hematopoyéticas (TCPH), o si el donante fuera un familiar en 1° o 2° grado.
• Leucodeplecionado.• A 10-20 mL/Kg.
Recuento plaquetario (x 10⁹/L) Condición clínica
<20
<30
<50
<50
<100
Todos los RN
RNPT estables
RN peso <1.000 g
RN clínicamente inestables o con otro factor de riesgo:• antecedente de sangrado mayor• sangrado activo menor• NEC• peso <1.500 g• coagulopatía• previo a procedimientos invasivos (cirugía, exanguino transfusión)• 1as 72 hs de post-operatorio
Sangrado activo mayor.Previo a neurocirugía y en su post-operatorio.
Tabla 2. Valores recomendados para transfusión de plaquetas en neonatos
9 | Trombocitopenia Neonatal

173
Trombocitopenia feto/neonatal aloinmune (TFNA)
Resulta de la destrucción de las plaquetas fetales/neonatales por acción de anticuerpos maternos contra antígenos plaquetarios humanos (HPA) fetales heredados del padre. A diferencia de la incompatibilidad Rh, la alo-inmunización puede ocurrir en ese mismo embarazo. Los antígenos más comúnmente involucrados son HPA-1a y 5b (80% y 15% de los casos en caucásicos, respectivamente). Su incidencia está reportada entre 1:1000 y 1:1500 en RN. El 40-50% de los casos tiene afectación en el primer embarazo, y mayor severidad en el siguiente. La mitad se produce al final del embarazo, entre las semanas 30 y 35.De los pacientes sintomáticos, 10-20% presenta hemorragia intracraneal (HIC), con 10% de mortalidad y 20% de secuelas neurológicas. El 20% de los fetos de embarazos no tratados puede presentar HIC intraútero.Habitualmente resuelve en 1 a 3 semanas en forma espontánea, puede requerir hasta entonces transfusiones repetidas de plaquetas.
Grupo ABO del paciente Grupo ABO de las plaquetas
O1ª opción2ª opción
A1ª opción2ª opción3ª opción
B1ª opción2ª opción3ª opción
AB1ª opción2ª opción3ª opción
OA o B
AB*0*
BA*0*
ABA o B*
0*
Tabla 3. Grupo ABO para administración de plaquetas
Puede no haber concentrados disponibles de plaquetas B o AB.* Testeados negativos para alto título de anticuerpos anti-A y/o anti-B.
Trombocitopenia Neonatal | 9

174
Debe sospecharse en:• RN sin otra patología, con recuento plaquetario bajo (generalmente
<50x10⁹/L) en el 1er día de vida.• Trombocitopenia no explicada.• RN con lesiones purpúricas o con HIC y trombocitopenia, sin otra causa.• RNPT con HIC parenquimatosa y trombocitopenia.• Historia familiar de trombocitopenia neonatal transitoria.• Historia familiar de fetos con HIC o hidrocefalia de causa desconocida.La historia de un hermano con trombocitopenia neonatal hace el diagnóstico casi certero. Puede haber antecedentes de pérdidas fetales repetidas o nacidos muertos. El antecedente de hermano afectado es el único predictor de severidad aceptado.
Diagnóstico serológico
En las guías de diagnóstico y tratamiento de otros países, se recomienda enviar muestras de sangre anticoagulada con EDTA del padre (en su defecto del bebé) y de la madre, para genotipificación, sin que esto demore el inicio del tratamiento.
En Argentina no se hace genotipificación actualmente para manejo asistencial.El diagnóstico serológico consiste en: demostrar la presencia de un alo-anticuerpo plaquetario específico en suero materno y evidenciar la incompatibilidad materno/fetal o materno/paterna. En el Servicio de Hematología y Medicina Transfusional del Hospital Italiano Garibaldi, Rosario, Provincia de Santa Fé (STEM SRL), se realizan estudios serológicos para el diagnóstico de la incompatibilidad plaquetaria feto-materna que incluyen la detección de auto y alo-anticuerpos antiplaquetarios maternos, así como la prueba cruzada de la pareja por las técnicas de inmunofluorescencia directa
Recomendación internacional: para confirmación diagnóstica se debe realizar genotipificacion.
9 | Trombocitopenia Neonatal

175
e indirecta. (Se adjunta contacto al final del capítulo).Dado que esos estudios no están ampliamente disponibles en nuestro país, se recomienda ante la sospecha diagnóstica, la rápida implementación de medidas terapéuticas.
Manejo
De acuerdo con la evidencia publicada disponible, se recomienda (Grado C, Nivel IV):• Realizar ecografía cerebral.• Asegurarse de que se haya administrado vitamina K.• Transfundir plaquetas con: -Recuento <30x10⁹/L. -Recuento <50x10⁹/L con sangrado menor u otro factor de riesgo. -Recuento <100x10⁹/L ante sangrado mayor.• Producto a transfundir (4,5): -Si hay disponibilidad, las plaquetas HPA compatibles con la madre, son la primera opción. Si son de aféresis de la madre, deben ser lavadas y resuspendidas en solución fisiológica e irradiadas. -Si no hay plaquetas HPA compatibles, transfundir plaquetas de banco: ABO y Rh(D) compatibles con la madre. Evaluar la necesidad en ese caso de administrar Inmunoglobulina endovenosa (IgEV) 1-2 g/Kg. -En neonatos con recuentos mayores a 20-30x10⁹/L y ausencia de sangrado administrar Ig EV 1g/Kg/día por dos días. -Considerar otros diagnósticos si el recuento no mejora luego de transfusión de plaquetas compatibles. -Realizar recuento diario de plaquetas hasta que éste se mantenga estable por encima de 100x10⁹/L.Los siguientes embarazos comprometidos (50% de posibilidades si el padre es heterocigota para el antígeno comprometido, 100% si es homocigota), tendrán al menos la misma severidad. El equipo a cargo del seguimiento de la madre puede tener un manejo agresivo (transfusión de plaquetas intraútero), conservador (administración de Ig EV a la madre) o intermedio (Ig EV + corticoides). Pueden obtenerse plaquetas por aféresis de la madre
Trombocitopenia Neonatal | 9

176
antes del parto, en el caso de un parto o cesárea programados, si el equipo tratante lo considera apropiado.
Prevención
La prevalencia de personas HPA-1a negativas es 1-2% de la población general. El 10% de los embarazos en los que hay incompatibilidad HPA desarrolla Anticuerpos anti-HPA-1a. El 30% de los RN de esos embarazos tendrá trombocitopenia, 20% severa. En el 75% de las mujeres la sensibilización ocurre en el parto. Las embarazadas HPA-1a negativas, son las que más están en riesgo.Si bien la administración de Ig EV en embarazadas sensibilizadas a HPA en embarazos previos ha demostrado ser efectiva en reducir el grado de trombocitopenia y de sangrado fetal/neonatal, la Ig EV antenatal no ha logrado prevenir la sensibilización en primíparas con embarazos HPA incompatibles.No existe aún en el mundo ningún programa de screening dado que no hay disponible por el momento una prueba simple, segura, precisa y validada. El screening se justificaría si detectadas las embarazadas en riesgo, pudiera implementarse un tratamiento profiláctico que evitara la sensibilización. Hay varios estudios clínicos en marcha en busca de un tratamiento profiláctico efectivo. Por este motivo no se recomienda actualmente hacer screening de embarazadas.
Información de utilidad
Servicio de Hematología y Medicina Transfusional del Hospital Italiano Garibaldi, Rosario, Provincia de Santa Fé.Contacto: Daniel de la Vega, bioquímico, especialista en inmunohematología y transfusión. Tel/fax: (0341) 485-8825. e-mail: [email protected].
Conflictos de interés
La autora no presenta conflictos de interés.
9 | Trombocitopenia Neonatal

177
Referencias
1. Malte C, Hannes S, Pamela J, et al. Thrombocytopenia and platelet transfusion in the neonate. Semin Fetal Neonatal Med 2016; 21: 10-18.
2. Roberts I, Murray N. Neonatal thrombocytopenia: causes and management. Arch Dis Child Fetal Neonatal Ed 2003; 88: F359-364.
3. Gunnink S, Vlug R, Fijnvandraat K, et al. Neonatal thrombocytopenia: etiology, management and outcome. Expert Rev Hematol 2014; 7: 378-395.
4. British Committee for Standards in Hematology. Transfusion Guidelines for neonates and older children. Br J Haem 2004; 124: 433-453.
5. 2007 Amendments and corrections to the “Transfusion Guidelines for neonates and older children”.
6. Curtis B. Recent progress in understanding the pathogenesis of fetal and neonatal alloimmune thrombocytopenia. Br J Haematol 2015; 171: 671-682.
7. Brojer E, Husebekk A, Debska M, et al. Fetal/Neonatal alloimmune thrombocytopenia: pathogenesis, diagnostics and prevention. Arch Immunol Ther Exp 2015; DOI 10.1007/s00005-015-0371-9.
8. Salomon O, Rosenberg N. Predicting risk severity and response of fetal neonatal alloimmune thrombocytopenia. Br J Haematol 2013; 162: 304-312.
Trombocitopenia Neonatal | 9

178

179
Introducción
La coagulación intravascular diseminada (CID) es un estado disfuncional sistémico de la microvasculatura y del aparato hemostático, producido por un estímulo procoagulante intenso que sobrepasa al potencial anticoagulante y fibrinolítico normal. Constituye un mecanismo fisiopatológico intermedio, común a una gran cantidad de enfermedades o condiciones clínicas, y desencadenado por la acción conjunta del factor tisular (FT) y mediadores inflamatorios liberados o expresados en endotelio, células mononucleares sanguíneas u otras células extravasculares. Patológicamente se caracteriza por trombosis y hemorragias en diversos órganos, resultado de la activación sistémica de la coagulación que conduce al consumo de plaquetas y factores procoagulantes y al desequilibrio de la fibrinolisis. Los órganos más afectados son pulmón, riñón, cerebro, corazón e hígado (Figura 1).
10Coagulación intravascular diseminada Alicia Grinspon1 y Dardo Riveros2.1 Docente de la Universidad de Buenos Aires, Facultad de Farmacia y Bioquímica, Departamento de Bioquímica Clínica, Área Hemostasia.2 Consultor de Hematología. Profesor Asociado de Medicina Interna. Investigador Asociado. CEMIC (Centro de Educación Médica e Investigaciones Clínicas).

180
10 | Coagulación intravascular diseminada
Fisiopatología de la CID
En condiciones normales, la inmunidad innata y la hemostasia eliminan en forma inocua y coordinada los patógenos y las toxinas. En la CID hay una activación exagerada de la inmunidad innata, que lleva a activar también el mecanismo de la hemostasia. Sólo la intervención médica puede poner freno a este círculo de activación mutua. En las infecciones, los patógenos de cualquier origen exponen o liberan patrones moleculares asociados a patógenos (PAMPs), mientras que en diversas formas de trauma se liberan patrones moleculares asociados a daño (DAMPs). Tanto PAMPs como DAMPs se unen a los receptores tipo Toll de los macrófagos y de las células endoteliales (CE) e inician la activación de cascadas inflamatorias. El resultado es el aumento de interleuquinas (IL) inflamatorias: IL-1β y IL-6, factor de necrosis tumoral alfa (TNFα), intermediarios reactivos del oxígeno (ROIs) y la expresión de FT en macrófagos y CE. Los neutrófilos activados liberan elastasas, catepsina G, ROIs y dan origen a NETs, que son trampas extracelulares de neutrófilos (de allí su nomenclatura) formadas por DNA, histonas y proteasas.
Aspectos clínicos
Son numerosas las afecciones que pueden asociarse a la aparición de CID, pero según estudios clínicos retrospectivos, el 70% corresponden a infecciones, neoplasias y trauma (1, 2). Hay amplias variaciones individuales en la prevalencia de cada una de ellas según la fuente considerada, consecuencia del sesgo impuesto por el perfil asistencial de los diferentes
Figura 1.

181
Coagulación intravascular diseminada |10
centros que participaron en esos estudios. El 30% restante está asociado a entidades más heterogéneas y diversas, entre las que sobresale la patología obstétrica (Tabla 1).
Pueden distinguirse diferentes formas de presentación de la CID: una variante incipiente o compensada, también llamada CID “no-abierta”, otra variante completa, descompensada o CID “abierta” y CID crónica (1). La primera se caracteriza casi exclusivamente por alteraciones de laboratorio que son más evidentes con las pruebas de mayor complejidad, mientras que en la forma descompensada se expresan con distintas intensidades las anormalidades del laboratorio convencional o de rutina, asociadas a las manifestaciones clínicas características del síndrome (Tabla 2, Figura 2).
InfeccionesNeoplasias
Trauma
Reacciones tóxicas/inmunológicasMalformaciones vasculares
Destrucción/fallo de órganos
Complicaciones obstétricas
Cualquier microorganismoTumores sólidosLeucemiasEmbolia grasaQuemaduras Traumatismo cerebralMordeduras de víboraReacción transfusionalSíndrome de Kasabach-Merritt Aneurismas de aortaPancreatitisHepatopatíasDesprendimiento de placentaFeto muerto y retenidoEmbolia de líquido amniótico
Tabla 1. Condiciones clínicas que pueden complicarse con CID

182
10 | Coagulación intravascular diseminada
Forma compensada o “no abierta”Forma sobre-compensada en CID crónica, con “fenómeno de rebote”Forma descompensada o “abierta”• Hemorragias (64-87%)• Síndrome de desfibrinación (<10%)• Trombosis (8-47%)• Fallo orgánico -Renal (25-67%) -Respiratorio (16-78%) -Hepático (22-57%) -Cerebral (11-65%)
Tabla 2. Formas de presentación de la CID y frecuencia de las manifestaciones clínicas según diferentes series
Generación y diseminación de trombina. I: normal, activación localizada de la coagulación, por vía extrínseca e intrínseca, para generar trombina con un resultado balanceado entre lo procoagulante (por ej: conversión de fibrinógeno en fibrina) y lo anticoagulante (activación de la PC a PCa). El área superior derecha (grisada) denota las reacciones que son altamente dependientes de superficies fosfolipídicas. II: Incremento compensado de la activación de la coagulación con contribución de superficies celulares (células endoteliales, monocitos y plaquetas). III: generación descompensada y diseminada de trombina, con incremento de superficies con fosfolípidos aniónicos, provistas por micropartículas circulantes y lipoproteínas, liberadas en parte por la respuesta inflamatoria/estrés. (Adaptado de BMJ 2003; 327: 974-977).
Figura 2.

183
Coagulación intravascular diseminada |10
Hemorragias y trombosis (venosa, arterial o de la microcirculación) constituyen la expresión clínica más frecuente, y son también la causa del shock y de la falla funcional de distintos órganos (1). Predominan las hemorragias en casos de CID aguda o subaguda, en los que el estímulo procoagulante es particularmente potente y brusco (por ej. infecciones, trauma, complicaciones obstétricas), llevando a veces a situaciones máximas en el consumo del fibrinógeno (síndrome de desfibrinación). Por el contrario, puede haber una mayor tendencia a la trombosis en aquellos en los que la noxa es de menor intensidad, pero su efecto es más sostenido en el tiempo (CID crónica), como sucede en tumores sólidos.El impacto de la CID en los diferentes órganos produce fallo funcional y manifestaciones características: gangrena, bullas hemorrágicas y necrosis acral en piel; infarto y endocarditis trombótica no bacteriana en corazón; embolia y síndrome de distress respiratorio en pulmón; necrosis cortical y necrosis tubular aguda en riñón; infartos por embolia cardiogénica en cerebro, e insuficiencia por necrosis hemorrágica en adrenales. Algunos casos se distinguen por rasgos particulares y sobresalientes que dependen de la enfermedad asociada y tienen importancia en el pronóstico y el tratamiento (Tabla 3) (1).
Condicion asociada Características
Infecciones virales o bacterianas
Infección por S. pneumoniae
Leucemia promielocítica
Tumores sólidos
Politraumatismo
Malformaciones vasculares
Necrosis de piel (púrpura fulminans)Se asocia a trombofilia congénita (vía de la proteína C)
Síndrome de Waterhouse-Friderichsen en esplenectomizados
Coexisten CID, fibrinolisis primaria y proteólisis inespecíficaATRA corrige la coagulopatía
Trombosis venosa migratoria (síndrome de Trousseau)CID crónica sobrecompensada(“fenómeno de rebote”)
Efecto de citoquinas, fibrinolisis, acidosis e hipotensión (coagulopatía aguda del trauma)
CID crónica CID “localizada”
Tabla 3. CID: algunas situaciones especiales

184
10 | Coagulación intravascular diseminada
La mortalidad de la CID es elevada, entre 30 y 80%, contribuyendo a ello tanto la disfunción hemostática, como la enfermedad asociada. Existe una correlación directa entre mortalidad, severidad de la CID y edad. En infecciones complicadas con esta coagulopatía, la mortalidad a los 28 días es mayor.
Diagnóstico
No existe una única prueba diagnóstica. Se aconseja combinar la clínica más un algoritmo seriado, que es una combinación de pruebas a las que se le adjudica un puntaje (3). La CID es un proceso dinámico, mientras que los resultados son sólo una foto instantánea, por eso los exámenes deben ser seriados. Más que un dato único, lo importante es la tendencia. Características que deben tener las pruebas que integran el algoritmo: • Reflejar la situación clínica• Disponibles en todos los laboratorios• Económicas para permitir un seguimiento seriadoLas determinaciones que cumplen con estos requisitos son (4):• Recuento de plaquetas• Tiempo de protrombina (TP), expresado en segundos• Dímero D (DD) o productos de degradación de fibrinógeno/fibrina
(PDFs).• Dosaje de fibrinógeno
Otras pruebas. En CID por sepsis la modificación del tiempo de tromboplastina parcial activada (TTPA) puede adelantarse al aumento de los DD. Esta modificación puede ser detectada mediante coagulómetros automáticos, con sistema de detección óptica, que permiten graficar el proceso de coagulación. En casos de sepsis, se observa disminución de la transmitancia de la luz, antes de la formación del coágulo, dando un patrón de onda bifásica (BPW-biphasic waveform pattern), que se debe a la rápida formación de un precipitado, con cambio de turbidez, que coincide con la recalcificación (5). El precipitado contiene lipoproteínas de muy baja densidad (VLDL), proteína C reactiva (PCR) y Calcio, (VLDL y PCR están muy aumentados en sepsis).

185
Coagulación intravascular diseminada |10
Conclusión. La CID es una entidad elusiva, con un patrón de comportamiento cambiante y heterogéneo. No existe un estándar de oro para el diagnóstico, que se basa fundamentalmente en una combinación entre elementos clínicos y un conjunto de pruebas de laboratorio sencillas, rápidas y económicas que permitan efectuar determinaciones seriadas en centros de baja complejidad. Con la articulación de los datos clínicos y biológicos se han diseñado sistemas de puntuación o scores, que resultan orientadores y útiles al momento de tomar decisiones terapéuticas.Los tres scores para el diagnóstico de CID “abierta” (Tabla 4) han sido armonizados por consensos de expertos; en nuestro medio, el más utilizado es el de la ISTH (4). Lo mismo sucede para el diagnóstico de CID “no abierta” (Figura 3).
Figura 3.
cte: constante.
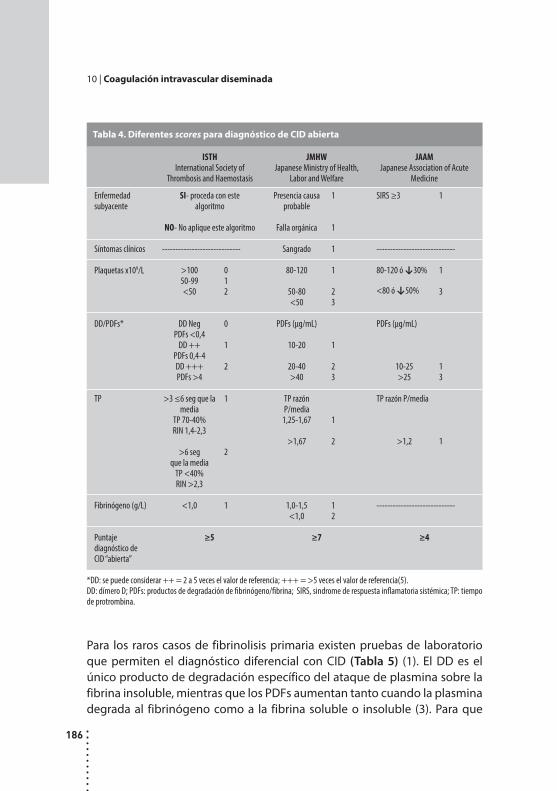
186
10 | Coagulación intravascular diseminada
Para los raros casos de fibrinolisis primaria existen pruebas de laboratorio que permiten el diagnóstico diferencial con CID (Tabla 5) (1). El DD es el único producto de degradación específico del ataque de plasmina sobre la fibrina insoluble, mientras que los PDFs aumentan tanto cuando la plasmina degrada al fibrinógeno como a la fibrina soluble o insoluble (3). Para que
ISTHInternational Society of
Thrombosis and Haemostasis
JMHWJapanese Ministry of Health,
Labor and Welfare
JAAMJapanese Association of Acute
Medicine
Enfermedad subyacente
Síntomas clínicos
Plaquetas x109/L
DD/PDFs*
TP
Fibrinógeno (g/L)
Puntaje diagnóstico deCID “abierta”
SI- proceda con estealgoritmo
NO- No aplique este algoritmo
Presencia causa probable
Falla orgánica
DD NegPDFs <0,4
DD ++ PDFs 0,4-4DD +++PDFs >4
>3 ≤6 seg que la media
TP 70-40%RIN 1,4-2,3
>6 segque la media
TP <40%RIN >2,3
<1,0
PDFs (μg/mL)
10-20
20-40>40
10-25>25
>1,2
80-120
50-80 <50
>10050-99<50
SIRS ≥3
PDFs (μg/mL)
TP razón P/media
-----------------------------
≥4≥7≥5
80-120 ó ↓30%
<80 ó ↓50%
1
1
0
1
2
1
2
1
TP razónP/media
1,25-1,67
>1,67
1,0-1,5<1,0
1
2
12
1
23
13
1
1
23
012
----------------------------- -----------------------------Sangrado 1
1
1
3
Tabla 4. Diferentes scores para diagnóstico de CID abierta
*DD: se puede considerar ++ = 2 a 5 veces el valor de referencia; +++ = >5 veces el valor de referencia(5).DD: dímero D; PDFs: productos de degradación de fibrinógeno/fibrina; SIRS, sindrome de respuesta inflamatoria sistémica; TP: tiempo de protrombina.

187
Coagulación intravascular diseminada |10
se produzca la fibrinolisis primaria tiene que haber formación excesiva de plasmina pero no de trombina. Lo cual puede ocurrir como consecuencia de diversas noxas, como cáncer de próstata, leucemia promielocítica, embolia de líquido amniótico, mordeduras de víboras o iatrogenia farmacológica.
Tratamiento
Ante la escasez de ensayos prospectivos y randomizados, las recomen-daciones terapéuticas se basan principalmente en los resultados de
Pruebas que no permiten diferenciar CID de fibrinolisis primaria
Prueba
Fibrinógeno
PDFs
Pruebas de coagulación
CID
Disminuido
Aumentado
Prolongadas
Fibrinolisis secundaria
Disminuido
Aumentado
Prolongadas
Pruebas de alta complejidad para diferenciar fibrinolisis primaria de secundaria
Prueba
α2 antiplasmina
Fragmento 1+2 y TAT
Fibrinopéptidos A y B
CID
Disminuida
Aumentados
Aumentados
Fibrinolisis primaria
Muy disminuida
Normal
Normal
Pruebas que permiten diferenciar fibrinolisis primaria de secundaria
Prueba
Recuento de plaquetas
DD
Esquistocitocitos en frotis
Antitrombina
Fibrinolisis primaria
Normal o disminuido, de acuerdo a la enfermedad asociada
Normal
Ausente
Normal
Fibrinolisis secundaria
Disminuido
Elevado
Presentes
Disminuida
Tabla 5. Diagnóstico diferencial entre fibrinolisis primaria y secundaria

188
10 | Coagulación intravascular diseminada
estudios retrospectivos o en opiniones de expertos (1, 2, 6, 7). La premisa fundamental ante la sospecha de CID es adoptar precozmente medidas de soporte general, tendientes a evitar la progresión a estadios más severos potencialmente irreversibles y proceder luego a realizar una aproximación diagnóstica, utilizando el sistema de puntuación de la Sociedad Internacional de Hemostasia y Trombosis (score ISTH) (Figura 4).
El soporte general debe centrarse en mantener la volemia y la perfusión con hidratación adecuada, controlar la acidosis y el equilibrio hidroelectrolítico, garantizar la oxigenación y vigilar estrechamente el funcionamiento de los órganos vitales. En esta etapa puede ser necesario el uso de inotrópicos, la asistencia respiratoria mecánica y la hemodiálisis. Garantizado este equilibrio general, se impone en forma simultánea, la necesidad de eliminar la causa de la CID; medida considerada como la piedra fundamental en el tratamiento. Aunque esto no siempre es posible, el intento debe realizarse en todos los casos sobre bases racionales y realistas. En este sentido, los ejemplos característicos son: la antibioticoterapia y la erradicación de focos sépticos en infecciones; la quimioterapia y la radioterapia en neoplasias; la evacuación uterina en caso de restos fetales retenidos; la corrección quirúrgica en malformaciones vasculares y la eliminación de tejidos necróticos en traumatismos.En aquellos casos que cumplen con los criterios de CID “abierta”, hay tres tipos de medidas terapéuticas adicionales: la reposición de componentes sanguíneos, la inactivación de la coagulación y la inhibición de la fibrinolisis (Figura 5).
Figura 4.

189
Coagulación intravascular diseminada |10
Reposición de componentes sanguíneos. Su beneficio es más notable en casos de CID aguda, en los que el estímulo de trombina es autolimitado y predominan clínicamente los efectos de la deficiencia hemostática. Se indica en casos de hemorragia activa o cuando se deben realizar procedimientos invasivos y si el nivel plaquetario es menor a 50x109/L, el tiempo de protrombina es mayor a 1,5 veces el valor superior del rango normal o el fibrinógeno es menor a 150 mg/dL. En general, no se aconseja transfundir sobre la base exclusiva de los parámetros de laboratorio sin tener en cuenta el escenario clínico, aunque puede haber excepciones a esta premisa en los casos raros, que se presentan asintomáticos y con severas alteraciones en las pruebas de hemostasia.Las dosis de los productos sanguíneos que se detallan a continuación pueden repetirse cada 6-8 hs dependiendo de la respuesta clínica.Plaquetas. 5-10 u de donantes múltiples o 1 concentrado obtenido por aféresis. Plasma fresco congelado: 10-15 mL/Kg de peso. Crioprecipitados (especialmente en déficits severos de fibrinógeno): 1-2 U/10 Kg de peso.El uso de concentrados comerciales de factores protrombínicos es tentador porque evita la posible sobrecarga hemodinámica que puede producir el plasma, pero son caros, carecen de FV, aumentan el riesgo trombótico y no están aprobados para CID. Consideraciones más o menos similares pueden hacerse con respecto a los concentrados comerciales de fibrinógeno.Inactivación de la coagulación. La heparina tiene actualmente un papel limitado en el tratamiento de la CID, ya que aumenta el riesgo de sangrado y no se ha comprobado fehacientemente que pueda revertir el proceso del consumo. Por estos motivos no se la recomienda o está contraindicada en la
Figura 5.

190
10 | Coagulación intravascular diseminada
mayor parte de los casos. Su uso se restringe exclusivamente a los cuadros de CID subaguda o crónica, en los que el consumo es menos severo debido a los mecanismos compensadores de la hemostasia, predominando las manifestaciones tromboembólicas sobre las hemorrágicas (tumores sólidos, infecciones con gangrena y necrosis acral, feto muerto retenido y embolia de líquido amniótico). También se la recomienda en anormalidades vasculares que van a ser corregidas y tienen evidencias de activación local de la coagulación y/o hipofibrinogenemia, lo que puede ser causa de hemorragia perioperatoria (síndrome de Kasabach-Merritt o aneurismas de aorta). En general se prefiere la heparina no fraccionada sobre las heparinas de bajo peso molecular, ya que se trata de enfermos lábiles con propensión a la hemorragia y es más seguro un fármaco que tenga una cinética predecible y un antídoto seguro. La dosis es de 18 U/Kg/hora por infusión intravenosa continua, aunque conviene comenzar con la mitad de esa dosis, para luego escalarla según la respuesta clínica, controlando el efecto biológico con heparinemia por anti-FXa (0,3-0,7 U/mL). Superado el episodio de CID, se aconseja continuar con heparina de bajo peso molecular como profilaxis del tromboembolismo venoso hasta la deambulación.Una manera más racional para inactivar el estímulo procoagulante, es restaurar los mecanismos anticoagulantes fisiológicos, con la utilización de concentrados de antitrombina (AT) o de proteína C activada (PCa). Sin embargo, los estudios y los meta-análisis que evaluaron la utilidad de estos agentes en CID secundaria a infecciones, no demostraron beneficios en la sobrevida global, aunque mejoraron algunos de los parámetros de laboratorio. Actualmente están en marcha estudios fase 3 para establecer si otras estrategias anticoagulantes tienen utilidad, como es el uso de trombomodulina (TM) o inhibidor de la vía del factor tisular (TFPI) recombinantes.Inhibición de la fibrinolisis. En general la fibrinolisis secundaria de la CID no debe ser inhibida, por el riesgo de fenómenos trombóticos. Pero en algunas situaciones en las que la respuesta fibrinolítica puede ser dominante, se recomienda la indicación de ácido tranexámico (AcTx) o ε-aminocaproico. Estos casos se pueden reconocer por el acortamiento notable de los tiempos de lisis de euglobulinas o del coágulo en sangre total; son más frecuentes en leucemia aguda promielocítica, cáncer avanzado de mama o próstata, hemangiomas gigantes y embolia de líquido amniótico.Finalmente, en los pacientes en los que no se cumplen los criterios de CID

191
Coagulación intravascular diseminada |10
“abierta” (Figura 6) deben realizarse controles clínicos y de laboratorio diariamente, para iniciar el tratamiento específico en cuanto se constate progresión del cuadro.
Aspectos de laboratorio
Cambios protrombóticos de las CE activadas en CID• Aumento de la expresión de FT• Expresión de selectinas, molécula de adhesión celular vascular (VCAM) y
molécula de adhesión intercelular (ICAM), que favorecen la interacción con neutrófilos y plaquetas
• Aumento de la síntesis de inhibidor del activador del plasminógeno tipo 1 (PAI-1)
• Disminución de la síntesis y expresión de TFPI• Disminución de síntesis y expresión de TM • Eliminación de TM por elastasas y liberación de TM soluble e inactiva• Disminución de la expresión del receptor endotelial de la proteína C
(EPCR)• Disminución de glicosaminoglicanos de membrana
Cambios en los inhibidores fisiológicos de la coagulación en CID• Disminución de PC, PS y AT por consumo y menor síntesis hepática• Disminución extra de AT y TFPI por las elastasas de los neutrófilos
Cambios en las plaquetas en CID• Activación, aumento de reactividad y consumo • Aumento de la exposición de fosfolípidos aniónicos
Figura 6.

192
10 | Coagulación intravascular diseminada
Cambios globales de la hemostasia• Disminución de la generación de PCa y aumento de la generación de
FVa y FVIIIa• Activación de la fibrinolisis en menor grado que la coagulación; en
CID infecciosa, suele haber desde el comienzo hipofibrinolisis debido al aumento de síntesis y liberación de PAI-1, esto empeora la isquemia tisular y se asocia con mayor tasa de mortalidad
• Aumento de micropartículas cargadas de FT provenientes de monocitos y CE y de micropartículas derivadas de plaquetas ricas en fosfolípidos aniónicos
• En sepsis aumentan las VLDL, matriz para la activación de la coagulación • La trombina se une a receptores PAR1, 2 y 4 activando plaquetas y CE• Aumento de la generación de trombina
Los inhibidores fisiológicos y la TM tienen una fuerte actividad anti-inflamatoria y anti-apoptótica, pero en la CID están muy descendidos, mientras que las cascadas de activación celular protrombó-ticas se imponen sobre los mecanismos de control, como se puede observar en las Figuras 2, 7 y 8 (8-10).
Los PAMPs y DAMPs provocan la generación de NETS por los neutrofilos y la expresión de FT por los monocitos, promoviendo la activación de la inmunidad innata y la trombosis. Las NETs pueden activar al FXII de la coagulación, inactivar al TFPI y proveer una matriz para que las plaquetas se unan y se agreguen. También se liberan micropartículas derivadas de monocitos que expresan FT. Todo esto promueve la activación de la coagulación. DAMPs: patrones moleculares asociados a daño; FT: factor tisular; NETs: trampas extracelulares de neutrófilos; PAMPs: patrones moleculares asociados a patógenos; TFPI: inhibidor de la vía del factor tisular. (Adaptado de J IntensiveCare 2014; 2:67).
Figura 7.

193
Coagulación intravascular diseminada |10
Pruebas del laboratorio de hemostasia que se alteran en CID
Debido a la naturaleza de la CID, se producen alteraciones de distinta magnitud en una gran variedad de parámetros de laboratorio (Tabla 6) (6).Recuento de plaquetas. Típicamente, en CID se observa trombocitopenia moderada a severa. Un recuento muy bajo de plaquetas resulta muy sensible para detectar CID, pero poco específico. Lo importante es evidenciar la tendencia, por eso es necesario el recuento seriado. El frotis de sangre periférica permite reflejar la microangiopatía trombótica por la presencia de esquistocitos, en general hasta un 10%.TP y TTPA. En el 50% de los pacientes con CID se observa prolongación del TP y del TTPA; no obstante, tiempos normales o ligeramente prolongados no excluyen la CID. El TP se debe expresar en segundos y en especial cuando se utiliza el algoritmo diagnóstico de la Sociedad Internacional de Hemostasia y Trombosis (ISTH) (4).
Las células endoteliales expresan varios anticoagulantes, incluyendo TM, TFPI y heparán sulfato. La trombina unida a TM pierde su capacidad de activar plaquetas, fibrinógeno y factores de la coagulación como FV, FVIII, FXI y FXIII. El complejo trombina-TM activa la PC. La PCa junto con la PS detiene la generación de trombina al inactivar a los FVa y FVIIIa. Las células endoteliales sintetizan y expresan en su superficie proteoglicanos (heparán sulfato) que se unen a AT y al TFPI; la primera inhibe a la trombina, el TFPI al complejo FVIIa-FT y al FXa. AT: antitrombina; PC: proteína C; PCa: proteína C activada; PS: proteína S; TFPI: inhibidor de la vía del factor tisular; TM: trombomodulina. (Adaptado de J IntensiveCare 2014; 2:67).
Figura 8.
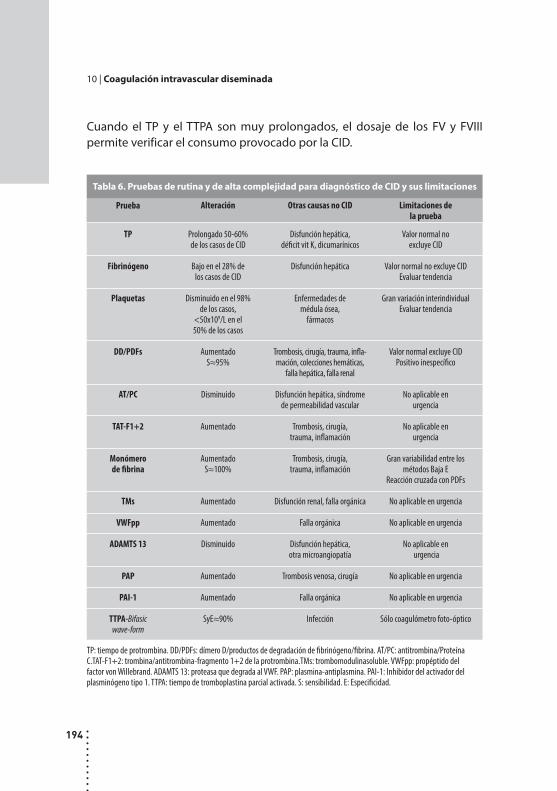
194
10 | Coagulación intravascular diseminada
Cuando el TP y el TTPA son muy prolongados, el dosaje de los FV y FVIII permite verificar el consumo provocado por la CID.
Prueba Alteración Otras causas no CID Limitaciones de la prueba
TP
Fibrinógeno
Plaquetas
DD/PDFs
AT/PC
TAT-F1+2
Monómero de fibrina
TMs
VWFpp
ADAMTS 13
PAP
PAI-1
TTPA-Bifasic wave-form
Prolongado 50-60%de los casos de CID
Bajo en el 28% delos casos de CID
Disminuido en el 98% de los casos,
<50x109/L en el50% de los casos
AumentadoS≈95%
Disminuido
Aumentado
AumentadoS≈100%
Aumentado
Aumentado
Disminuido
Aumentado
Aumentado
SyE≈90%
Disfunción hepática,déficit vit K, dicumarínicos
Disfunción hepática
Enfermedades de médula ósea,
fármacos
Trombosis, cirugía, trauma, infla-mación, colecciones hemáticas,
falla hepática, falla renal
Disfunción hepática, síndrome de permeabilidad vascular
Trombosis, cirugía,trauma, inflamación
Trombosis, cirugía,trauma, inflamación
Disfunción renal, falla orgánica
Falla orgánica
Disfunción hepática,otra microangiopatía
Trombosis venosa, cirugía
Falla orgánica
Infección
Valor normal no excluye CID
Valor normal no excluye CIDEvaluar tendencia
Gran variación interindividualEvaluar tendencia
Valor normal excluye CIDPositivo inespecífico
No aplicable en urgencia
No aplicable en urgencia
Gran variabilidad entre los métodos Baja E
Reacción cruzada con PDFs
No aplicable en urgencia
No aplicable en urgencia
No aplicable en urgencia
No aplicable en urgencia
No aplicable en urgencia
Sólo coagulómetro foto-óptico
Tabla 6. Pruebas de rutina y de alta complejidad para diagnóstico de CID y sus limitaciones
TP: tiempo de protrombina. DD/PDFs: dímero D/productos de degradación de fibrinógeno/fibrina. AT/PC: antitrombina/Proteína C.TAT-F1+2: trombina/antitrombina-fragmento 1+2 de la protrombina.TMs: trombomodulinasoluble. VWFpp: propéptido del factor von Willebrand. ADAMTS 13: proteasa que degrada al VWF. PAP: plasmina-antiplasmina. PAI-1: Inhibidor del activador del plasminógeno tipo 1. TTPA: tiempo de tromboplastina parcial activada. S: sensibilidad. E: Especificidad.

195
Coagulación intravascular diseminada |10
Dosaje de fibrinógeno. Teóricamente se esperaría que el fibrinógeno descendiera en la CID; por un lado, por acción de la trombina, que pasa el fibrinógeno a fibrina, y por otro lado, debido a la acción de la plasmina, que degrada al fibrinógeno en PDFs. Sin embargo, el dosaje del fibrinógeno es poco sensible en CID; su concentración depende del valor basal y además, el fibrinógeno es reactante de fase aguda, por ello en general su concentración al inicio de la CID es elevada y decrece a medida que progresa la CID. Un estudio demostró que en el 57% de los pacientes con CID, el fibrinógeno estaba en valores normales; en otro estudio, se mostró baja sensibilidad (28%) de los valores bajos de fibrinógeno para detectar CID. En cambio, un valor bajo tiene 90% de especificidad para consumo. Lo importante es el estudio seriado para determinar la tendencia.PDFs. El fibrinógeno pasa a fibrina por acción de la trombina y la fibrina se transforma en insoluble por el FXIIIa. La plasmina provoca la aparición de un producto soluble de degradación exclusivo de la fibrina, DD, lo que marca la acción previa de la trombina sobre el fibrinógeno y la posterior degradación de la fibrina insoluble por la plasmina. El dosaje de DD es la mejor prueba para el diagnóstico de CID. En cambio, los PDFs muestran la acción de la plasmina tanto sobre el fibrinógeno como sobre la fibrina soluble e insoluble, por lo que pueden estar aumentados en fibrinolisis primaria, y por eso son menos específicos que el DD. La determinación de monómeros de fibrina solubles (MFs) es la prueba más sensible, pero de baja especificidad y con mucha variabilidad entre distintos métodos, razón por la que no se ha generalizado su uso.Pruebas de mayor complejidad. Las pruebas de alta complejidad como son el dosaje de los inhibidores fisiológicos y de los marcadores moleculares de activación de la coagulación son muy sensibles, pero poco específicos y requieren laboratorios especializados. Las pruebas viscoelásticas como el tromboelastograma (TEG) y la tromboelastografía rotacional (ROTEM) utilizadas preferentemente para transplante hepático y cirugía cardiovascular (CCV), aportan valiosa información sobre la progresión de la CID. Se realizan simultáneamente distintas pruebas, desencadenándose la coagulación mediante reactivos ad hoc para el mecanismo extrínseco e intrínseco. Los equipos registran y grafican en forma continua, la activación de la coagulación, la formación del coágulo y su degradación por la fibrinolisis. Estos métodos permiten detectar: deficiencia de factores de ambas vías, déficit de plaquetas y

196
10 | Coagulación intravascular diseminada
fibrinógeno, hiperfibrinolisis y también coágulos friables por déficit de FXIII. Se definen 4 parámetros: • Tiempo de coagulación (TC) en segundos, da información sobre déficit
de factores o presencia de anticoagulantes• Tiempo de formación del coágulo (TFC), tiempo desde el inicio
del coágulo hasta una amplitud de 20 mm. Informa sobre factores, anticoagulantes, polimerización de la fibrina y estabilización del coágulo con plaquetas, fibrinógeno y FXIII.
• Máxima firmeza del coágulo (MCF), es la máxima amplitud de la curva, se mide en mm. Valora plaquetas, fibrinógeno y FXIII.
• Lisis máxima (ML), es la reducción de la amplitud del coágulo luego del MCF, se determina a los 30 y 60 min, y mide el grado de fibrinolisis.
Conflictos de interés
Los autores no presentan conflictos de interés.

197
Coagulación intravascular diseminada |10
Referencias
1. Levi M, Seligsohn U. Disseminated intavascular coagulation. In Kaushansky K, Lichtman MA, Prchal JT, Levi M, Press OW, Burns J, Caligiuri MA, eds. Williams Hematology, 9th ed. McGraw-Hill Education, 2016, 2199-2219.
2. Dempfle CE. Predicting disseminated intravascular coagulation in the intensive care unit. Hematology EHA Educ Program 2006; 2: 235-238.
3. Levi M, Meijers J. DIC: which laboratory tests are most useful. Blood Rev 2011; 25: 33-37.
4. Kim H, Hong K, Toh C. Application of the international normali-zed ratio in the scoring system for disseminated intravascular coagulation on behalf of the scientific and standardization committee on DIC of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2010; 8: 1116-1118.
5. Toh CH, Samis J, Downey C et al. Biphasic transmittance waveform in the APTT coagulation assay is due to the formation of a Ca-dependent complex of C-reactive protein with very-low–density lipoprotein and is a novel marker of impending disseminated intravascular coagulation. Blood 2002; 100: 2522-2529.
6. Wada H, Matsumoto T, Yamashita Y. Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J Intensive Care 2014; 15: 1-8.
7. Toh CH, Alhamdi Y. Current consideration and management of disseminated intravascular coagulation. Hematology Am Soc Hematol Educ Program 2013; 2013: 286-291.
8. Toh CH, Dennis M. Disseminated intravascular coagulation: old disease, new hope. BMJ 2003; 327: 974-977.
9. Ito T. PAMPs and DAMPs as triggers for DIC. J Intensive Care 2014; 2: 67.
10. Allen K, Sawheny E, Kinasewitz G. Anticoagulant modulation of inflammation in severe sepsis. World J Crit Care Med 2015; 4: 105-115.

198

199
Introducción
El hígado tiene un rol clave en la cascada hemostática, ya que es el órgano en cuyos hepatocitos se sintetizan la mayor parte de los factores (con el ciclo de óxido-reducción de la vitamina (Vit) K para la síntesis de los factores e inhibidores Vit K dependientes), inhibidores del sistema de coagulación, como así componentes del sistema fibrinolítico (Tabla 1).Adicionalmente, el hígado a través de su sistema reticuloendotelial participa activamente en la eliminación de factores activados, de complejos de activación y de productos de degradación de fibrinógeno/fibrina (PDFs).De esta manera, la falla hepática conduce a alteraciones concomitantes que involucran a los componentes pro y antitrombóticos (1).Desde los primeros trasplantes (Tx) hepáticos se describe una amplia variación en el manejo de la coagulopatía y las transfusiones sanguíneas.
11Coagulopatía en la enfermedad hepática y el trasplante hepáticoEstela S. Viñuales1 y Marta E. Martinuzzo2.1 Médica Hematóloga. Servicio de Clínica Médica del Hospital Italiano de Bs As.Docente del Instituto Universitario del Hospital Italiano de Bs As. Colaboradora docente del Curso Superior de Médicos Hematólogos de la SAH.2 Bioquímica. Dra en Ciencias Fisiológicas de la UBA. Grupo Bioquímico. Laboratorio Central del Hospital Italiano. Universidad Favaloro. Instituto Universitario del Hospital Italiano.

200
La experiencia en este sentido ha mostrado un aumento de la morbi-mortalidad con el uso masivo de hemoderivados, por lo cual se aplican principios para el manejo de los productos sanguíneos que pueden reducir el riesgo transfusional.Estos incluyen: el reconocimiento preoperatorio y el manejo de la anemia, la reducción de las pérdidas sanguíneas en el perioperatorio y el uso de medidas que controlen los disparadores de transfusión.Existen métodos: a) farmacológicos que controlan las pérdidas sanguíneas como los agentes antifibrinolíticos que reducen la fibrinolisis, concentrados de complejo protrombínico (CCP) que mejoran la generación de trombina y concentrados de fibrinógeno que optimizan la fuerza y estabilidad del coágulo, b) no farmacológicos que incluyen la técnica quirúrgica piggy back (hepatectomía total con preservación de la vena cava) manteniendo una presión venosa central baja.El rescate de células intraoperatorio (cell saver) y la hemodilución normovolémica son importantes medidas que reducen el aporte de sangre alogeneica (1).
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático
Lugar de síntesis
Factores del sistema de coagulación FibrinógenoFII, FV, FVII, FXFV, FXI, FXII y FXIIIBFVIII
Inhibidores del sistema de coagulación ATPC, PS, PZTM
Componentes del sistema fibrinolíticoPlasminógenoα2-antiplasminatPAPAI-1TAFI
MetaloproteinasasADAMTS 13
HepatocitoHepatocito (vit K dependiente)HepatocitoEndotelio, sinusoide hepático
HepatocitoHepatocito (vit K dependiente)Endotelio
HepatocitoHepatocitoHepatocitoEndotelio, hepatocito, adipocito, CML, fibroblastoHepatocito
Célula estrellada del hígado, CE, plaquetas, podocitos renales
Tabla 1. Síntesis hepática de proteínas importantes del sistema hemostático

201
Coagulopatía de la hepatopatía crónica
Las anormalidades de la coagulación que podrían resultar en sangrado son: trombocitopenia, disfunción plaquetaria, reducción de los factores de coagulación, disfibrinogenemia y niveles reducidos de α2-antiplasmina.Estos cambios tienen su contrapeso dado por las alteraciones con tendencia protrombótica: la síntesis disminuida de los anticoagulantes naturales proteína S (PS), antitrombina III (ATIII) y muy significativamente la proteína C (PC), niveles reducidos de plasminógeno, niveles muy incrementados de FVIII y factor von Willebrand (VWF), aumento de óxido nítrico y prostaciclina. El desbalance tan marcado de la relación FVIII/PC que puede llegar dar valores >5 en mediciones ex vivo y la resistencia consecuente a la disminución en la generación de trombina producida por el agregado de trombomodulina (TM) en ensayos in vitro han cambiado el paradigma de la noción de coagulopatía hemorrágica en el paciente cirrótico por la definición de un estado de hemostasia frágilmente equilibrado e incluso protrombótico (Figura 1) (2).
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11
Esquema del frágil balance hemostático y las variables involucradas en los pacientes con cirrosis hepática.
Figura 1.

202
La trombocitopatía y la trombocitopenia son comunes y su etiología es multifactorial:• hiperesplenismo secundario a hipertensión portal• disminución en la síntesis de trombopoyetina• complejos inmunes en relación al clearance y destrucción plaquetaria
por el sistema retículoendotelial (1, 3).La tendencia hemorrágica de la enfermedad hepática en estadío terminal podría ser explicada por mecanismos adicionales a los descriptos clásicamente de hipocoagulabilidad, como aquellas condiciones clínicas que gatillan el sangrado: las alteraciones hemodinámicas de la hipertensión portal, disfunción endotelial, infecciones bacterianas con posibilidad de liberación de sustancias heparina miméticas y falla renal.La principal causa de sangrado es la ruptura de las várices esofágicas, presentándose en un tercio de los pacientes.Un recuento plaquetario de 60x109/L en plasma rico en plaquetas de pacientes cirróticos, sin embargo, es suficiente para preservar la generación de trombina, siendo equivalente y menos sensible a la presencia de TM, que la hallada en personas sanas con plaquetas en el límite inferior normal (4).
Coagulopatía de la hepatopatía aguda
La injuria y la falla hepática aguda (FHA) es un síndrome progresivo y potencialmente fatal que se presenta en individuos sin signos previos de disfunción hepática reconocidos que lleva a una pérdida muy rápida de las funciones hepáticas. Puede ser producido por una variedad de noxas: por infección viral, tóxica por medicamentos como el acetominofeno, por solventes, o metabólica. Se caracteriza por necrosis masiva de los hepatocitos. La definición involucra distintos aspectos de la falla hepática, y es muy variada en la literatura, pero fundamentalmente incluye el desarrollo de encefalopatía y coagulopatía (como manifestación de laboratorio) en el paciente. Las alteraciones en la FHA son similares, pero no idénticas a las halladas en pacientes con cirrosis. En primer lugar, el grado de disminución en los factores de coagulación es mucho más marcado en la FHA lo que lleva a la presencia de tiempos de protrombina (TP) muy alterados que generan RINs (razón internacional normatizada) muy elevados. Si bien en la mayor parte de las definiciones de coagulopatía en FHA se considera un RIN>1,5, o
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático

203
TP <40%, ha sido reportado que el 14% de los pacientes con FHA se presenta con RIN entre 5 y 10 y el 5% con RIN>10(5). En la Tabla 2 se pueden ver los niveles de factores hallados en pacientes con FHA inducida por intoxicación con acetaminofeno en comparación con los hallados en pacientes con cirrosis (6). Una de las diferencias encontradas es que los niveles de FIX y FXII eran significativamente más elevados que los de FII, FV, FVII y FX en la injuria hepática aguda, pero no en cirrosis, y los niveles de FVIII eran 45 veces más altos a los de FVII en pacientes con FHA comparado conrelación FVIII/FVII <5 en cirróticos. Esto puede ser explicado por los niveles de citoquinas proinflamatorias, como interlequina (IL) 6 y factor de necrosis tumoral α (TNFα) muy elevados en FHA (probablemente por liberación a través de las células de Kupffer) en comparación con cirrosis que podrían inducir de manera importante la síntesis de FVIII por parte del endotelio activado.De manera interesante, a pesar de los valores de RIN francamente alterados, las complicaciones hemorrágicas espontáneas en estos pacientes son poco frecuentes.
Coagulopatía del Tx hepático
Históricamente, el Tx ortotópico de hígado, se asoció con sangrado mayor y el requerimiento masivo de productos sanguíneos; situación que ha mejorado en los últimos diez años dada por la implementación de mejores técnicas quirúrgicas y anestésicas y un mayor entendimiento de las anor-malidades de la hemostasia durante el proceso del Tx.Existen cuatro etapas bien definidas: 1) fase de disección, 2) fase anhepática,
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11
IIV
VIIX
VIIIIXXI
1,05 (1,02-1,33)0,93 (0,51-1,22)1,03 (0,70-1,39)1,03 (0,41-1,26)0,80 (0,55-1,29)0,94 (0,72-1,30)0,80 (0,59-1,04)
0,28 (0,05-0,82)0,16 (0,01-0,46)0,13 (0,01-0,76)0,19 (0,05-0,76)1,95 (0,40-4,90)0,51 (0,16-1,30)0,72 (0,26-1,15)
0,64 (0,48-0,94)0,69 (0,46-0,90)0,62 (0,16-1,06)0,66 (0,45-0,92)1,01 (0,40-1,70)0,60 (0,48-0,84)0,40 (0,29-0,52)
Tabla 2. Niveles de factores de coagulación en FHA y en cirrosis
Factor (UI/mL) Individuos sanos FHA Cirrosis

204
3) fase de reperfusión y 4) fase de post-perfusión.Fase de disección. El sangrado excesivo es debido al grado de dificultades que surgen durante la disección, a la hipertensión portal con grandes vasos colaterales dilatados y a la distensión esófago-gástrica secundaria a la compresión y al clampeo vascular.Fase anhepática. Presenta disminución marcada de la síntesis de los factores de coagulación y del clearance. Se incrementa la actividad fibrinolítica por falta de clearance del activador tisular del plasminógeno (tPA),mientras los niveles del inhibidor del activador del plasminógeno (PAI-1) no muestran mayores cambios, aumentando la probabilidad de desarrollo de la fibrinolisis.Fase de reperfusión. La coagulopatía es profunda y el resultado es un efecto similar a la presencia de heparina, conocido como heparin like effect. Hay atrapamiento de plaquetas en los sinusoides del hígado del donante, persiste la reducción global de los factoresde la coagulación y en esta etapa caen los niveles del PAI-1 y de otros factores antifibrinolíticos, mientras que simultáneamente se genera tPA. En esta etapa la fibrinolisis está discretamente balanceada por los factores pro y anti-fibrinolíticos.Fase post-reperfusión. Algunos pacientes tienen una liberación acelerada del tPA desde el endotelio del graft y causan hiperfibrinolisis que suele durar no más de una hora post-reperfusión. Si el graft es pobremente funcional puede prolongarse, requiriendo mantener las drogas antifibrinolíticas que mejoran esta situación.En este período no es inhabitual la trombocitopenia debido al consumo de plaquetas en el nuevo hígado, siendo contrarrestado por su activación y riesgo de hipercoagulabilidad (1, 7).
El laboratorio de hemostasia en las hepatopatías
Tiempo de protrombina (TP). Es la prueba más emblemática en la enfermedad hepática, debido a que es sensible a las deficiencias de FII, FV, FVII y FX, representa fielmente el deterioro de la síntesis proteica del hepatocito. Su expresión puede ser en tiempo (seg), % de actividad o RIN.El RIN se define como (TP del paciente (seg) / media geométrica de los normales)ISI. La expresión como RIN fue diseñada y el ISI (índice de sensibilidad de la tromboplastina) calibrado para los pacientes que reciben anticoagulación con anti-vitamina K permitiendo estandarizar los resultados
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático
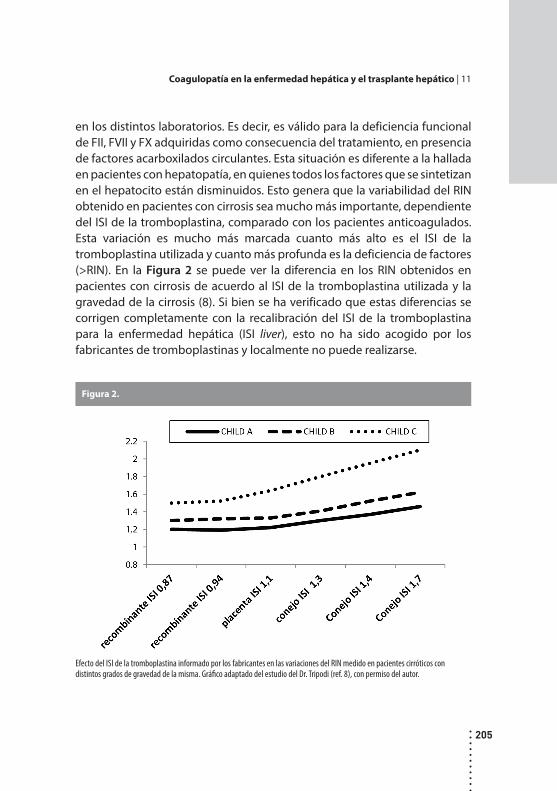
205
en los distintos laboratorios. Es decir, es válido para la deficiencia funcional de FII, FVII y FX adquiridas como consecuencia del tratamiento, en presencia de factores acarboxilados circulantes. Esta situación es diferente a la hallada en pacientes con hepatopatía, en quienes todos los factores que se sintetizan en el hepatocito están disminuidos. Esto genera que la variabilidad del RIN obtenido en pacientes con cirrosis sea mucho más importante, dependiente del ISI de la tromboplastina, comparado con los pacientes anticoagulados. Esta variación es mucho más marcada cuanto más alto es el ISI de la tromboplastina utilizada y cuanto más profunda es la deficiencia de factores (>RIN). En la Figura 2 se puede ver la diferencia en los RIN obtenidos en pacientes con cirrosis de acuerdo al ISI de la tromboplastina utilizada y la gravedad de la cirrosis (8). Si bien se ha verificado que estas diferencias se corrigen completamente con la recalibración del ISI de la tromboplastina para la enfermedad hepática (ISI liver), esto no ha sido acogido por los fabricantes de tromboplastinas y localmente no puede realizarse.
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11
Efecto del ISI de la tromboplastina informado por los fabricantes en las variaciones del RIN medido en pacientes cirróticos con distintos grados de gravedad de la misma. Gráfico adaptado del estudio del Dr. Tripodi (ref. 8), con permiso del autor.
Figura 2.

206
Recomendación (nivel de recomendación de expertos). Trabajar con tromboplastinas con ISI lo más cercano a 1, <1,2 e idealmente <1,1 para evitar variabilidades mayores entre los distintos laboratorios. Esto tiene relevancia, pues el RIN es uno de los factores pronósticos más importantes incluido en los criterios del King´s College en FHA e incorporado con un alto grado de ponderación en la fórmula del score de MELD (Figura 3) que es el mejor predictor de evolución y mortalidad en cirróticos por lo que se utiliza en la distribución de órganos para Tx. También estas diferencias deberían tratar de minimizarse por el hecho de que si bien el RIN no ha demostrado ser predictor de sangrado en cirrosis o en FHA, los algoritmos de transfusión en caso de sangrado claramente lo utilizan cuando el valor es mayor a 1,5 (9).
Variables analíticas y pre-analíticas a tener en cuenta en el TP y recomendaciones• la hiperbilirrubinemia extrema es uno de los problemas en instrumentos
foto-ópticos que posean filtros de lectura de densidad óptica <550 nm ya que interferirá en la detección del coágulo. En esos casos se sugiere realizar la determinación a través de un coagulómetro con detección electromagnética o por el método manual.
• la hipofibrinogenemia extrema (<50 mg/dL) hallada en pacientes terminales puede provocar coágulos no detectables por métodos ópticos o electromagnéticos, generando valores incoagulables. Se sugiere en esos casos llevarlos a cabo por método manual con movimientos muy sutiles del tubo. En caso de no detectar el coágulo ni siquiera por ese método, se sugiere dosar al menos dos factores de
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático
Descripción de la fórmula del score de MELD utilizado en pacientes con cirrosis.
Figura 3.

207
la vía extrínseca (por ej. FV y FVII) para poder dar una idea estimativa que ayudaría en las decisiones transfusionales de plasma además de sustituir fibrinógeno en caso de sangrado.
Medidas de funcionalidad plaquetaria. No se recomienda el tiempo de sangría (TS) dado que no ha demostrado ser predictor de sangrado.Tampoco se recomienda pruebas de funcionalidad plaquetaria debido a que por la trombocitopenia que presentan estos pacientes se hallarán tiempos de oclusión prolongados en el PFA-100® y plasma rico en plaquetas con insuficiente concentración de plaquetas para una correcta evaluación del estudio de agregación plaquetaria.Dosaje de FV. Es muy utilizado como indicador de síntesis hepática. Forma parte de los criterios de Clichy para FHA, y se utiliza en el seguimiento de la evolución en cirróticos. No tiene ningún valor predictivo de sangrado ni está incluido en algoritmos transfusionales.Dosaje de fibrinógeno. El método de Clauss es el de elección.Recomendación. En caso de obtener un valor disminuido se recomienda procesarlo con la técnica y la calibración para fibrinógeno bajo. Esto, en muchos coagulómetros automáticos se lleva a cabo automáticamente, es decir el equipo diluye menos la muestra, la interpola en la parte baja de la curva, y calcula el resultado considerando la menor dilución realizada. En otros equipos es indispensable establecer una técnica para fibrinógeno bajo (con menor dilución de la muestra) y calibrarla específicamente. En caso de realizar la técnica manual, se recomienda medirlo en una dilución más pequeña de la muestra o incluso directo y luego multiplicar el valor por el factor correspondiente de acuerdo a qué dilución se haya hecho.El fibrinógeno tampoco es predictor de sangrado, pero está mencionado en los algoritmos transfusionales en pacientes con sangrado activo, especialmente a valores <100 mg/dL.TTPA. No tiene ningún valor pronóstico de sangrado, en muchos casos se encuentra normal a pesar de niveles moderadamente disminuidos de factores de síntesis hepática, debido a los elevados niveles de FVIII que presentan en general los pacientes con enfermedad hepática aguda y crónica. No obstante, algunos algoritmos lo utilizan junto con el RIN en las decisiones terapéuticas de pacientes con sangrado activo, indicando transfusiones cuando la razón es >1,5.
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11

208
Medidas del sistema fibrinolítico
Lisis de euglobulinas. Laborioso, tarda más de 1 hora en dar resultados concretos, no es adaptable a la urgencia en la detección de hiperfibrinolisis.PDFs. No es extraño hallar valores aumentados de PDFs a valores moderados, en general <30 µg/mL, en caso de hallar valores superiores podría pensarse en una fibrinolisis exacerbada con fibrinogenolisis o CID.DD. En general presenta valores aumentados de niveles leves a moderados, en caso de hallar valores muy aumentados (>10.000 ng/mL) junto con otros parámetros podría indicar una CID asociada, situación que es muy poco frecuente.El mejor método para evaluar la hiperfibrinolisis en una paciente con hepatopatía que está sangrando o durante las distintas etapas del trasplante hepático es la tromboelastografía (TEG) o la tromboelastometría. TEG. Es una de las pruebas globales de la hemostasia que representa una técnica de medición en sangre entera en la que juegan un rol importante tanto los factores de coagulación como el nivel de fibrinógeno y el recuento plaquetario, ya que es una prueba de hemostasia viscoelástica. Mide la dinámica de formación y las características mecánicas del trombo y en su trazado se miden distintos parámetros descriptos en la Figura 4. Las alteraciones del TEG halladas en los cirróticos son menores que la alteración de las pruebas tradicionales de la hemostasia.
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático
Descripción de un trazado de tromboelastografía/tromboelastometría. Los distintos parámetros medibles con su significado se detallan a continuación:
Figura 4.

209
En la tromboelastometría rotacional ROTEM®, se utilizan distintas pruebas que se describen en la Tabla 3. En las distintas etapas del trasplante hepático se utilizan diferentes combinaciones de las mismas para evaluar el estado hemostático y seguir los algoritmos internaciones que guían los requerimientos transfusionales y terapéuticos de los pacientes en cada etapa. Las pruebas que se realizan a lo largo del trasplante hepático se muestran en la Tabla 4.
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11
EXTEM
INTEM
INTEM
APTEM
HEPTEM
FT + Ca2+ (posee inhibidor de heparina hasta
10 UI/mL)
Activador de carga negativa (ácido elágico) + Ca2+
FT + Ca2+ + Citocalasina D que inhibe el
mecanismo contráctil plaquetario (posee inhibidor
de heparina hasta 10 UI/mL)
FT + Ca2+ + aprotinina. Inhibición de la fibrinólisis
No mejora las alteraciones producidas por
deficiencia severa de FXIII. Se compara con EXTEM
Activador de carga negativa (ácido elágico) + Ca2+
+ heparinasa (degrada la heparina)
Sensible a deficiencias importantes de FII, V, VII y X
MCF disminuida con fibrinógeno y plaquetas disminuidas
Sensible a deficiencia severa de FXIII y a hiperfibrinolisis
Sensible a deficiencias moderadas y severas de FXII, XI, IX y VIII
MCF disminuida con fibrinógeno y plaquetas disminuidas
Sensible a deficiencia severa de FXIII y a hiperfibrinolisis
Sensible y muy buen estimador de la deficiencia de
fibrinógeno
Sensible para confirmar que las alteraciones son debido a un
estado de hiperfibrinolisis
Sensible para verificar que las alteraciones del INTEM son
debido a la presencia de heparina o sustancias “heparina like”
RAZON CT HEPTEM/INTEM <0,8 indica presencia de
heparina. Se compara al INTEM
Tabla 3. Descripción de las distintas pruebas utilizadas en el monitoreo de la hemostasia
Prueba Reactivo Sensibilidad

210
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático
En la Figura 5 se observa un ejemplo de algoritmo de decisiones terapéuticas de acuerdo a los distintos trazados hallados en EXTEM y FIBTEM.
Ejemplo de un algoritmo de decisiones terapéuticas en transplante hepático basado en los trazados de las pruebas tromboelastométricas de EXTEM y FIBTEM.
Figura 5.
Basal Fase anhepática ReperfusiónFinal de Qx
INTEM-EXTEM-FIBTEMEXTEM-FIBTEM-APTEMINTEM-EXTEM-FIBTEM-APTEMEXTEM-FIBTEM
Previo o al ingreso a quirófano15 min del inicio de fase anhepática15 min luego de reperfusión Previo al cierre de la Incisión quirúrgica
Etapa evaluada ROTEM-PRUEBA Tiempo de toma de muestra
Tabla 4. Descripción de las pruebas de tromboelastometría realizada en las distintas etapas del transplante hepático

211
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11
Factores predictores de sangrado en Tx hepático
Se han identificado numerosas variables como predictoras de transfusión.Factores de riesgo preoperatorio:• Características del receptor (R) - Severidad de la enfermedad hepática: Score de MELD ≥25 - Concentración de Hemoglobina (Hb) <10 mg/dL - Plaquetopenia - Concentración de fibrinógeno• Edad del R >50 años • Variables quirúrgicas.Factores de riesgo intraoperatorio: -Cirugía (Qx) abdominal previa (presencia de adherencias). Se ha identificado como factor de riesgo independiente transfusional. -Duración de la Qx en íntima relación con la experiencia y habilidad del grupo quirúrgico, especialmente en la fase de disección. La trombosis de la vena Porta incrementa el riesgo de sangrado, como así también la circulación hiperdinámica de la hipertensión portal (aumento de circulación colateral).La acidosis, hipotermia, hipocalcemia y toxicidad por citratos pueden contribuir a la coagulopatía intraoperatoria.Si bien ha sido dificultoso crear modelos predictivos de transfusión, es utilizado el índice de riesgo de McKlusky (siete variables):Edad >40 años, Hb <10 g/dL, RIN >2, recuento plaquetario <70x109/L, creatinina >1 mg/L para la mujer y >1,2 mg/L para el hombre y albúmina <2,4 mg/dL, además del re-Tx.
Tratamiento de la coagulopatia durante el Tx hepático
No existen trabajos randomizados controlados en este campo. Los datos provienen de reporte de casos y experiencia de centros expertos.El manejo del sangrado y de la coagulopatía varía entre los diferentes centros, sin embargo, existe evidencia que el número elevado de hemoderivados se asocia a una mayor tasa de morbi-mortalidad y existe consenso en minimizar el sangrado y la necesidad de transfusiones.

212
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático
Las pruebas clásicas, RIN y recuento plaquetario no son adecuados como guía transfusional (4) y pueden inducir terapias inapropiadas con escaso beneficio real.La terapia guiada por la información del ROTEM® ha logrado una reducción del 50% en las transfusiones de glóbulos rojos (GR), plasma fresco congelado (PFC) y concentrados de plaquetas.• No se recomienda el uso profiláctico de PFC. Los argumentos en contra
es que los Tx hepáticos en los últimos años se realizan sin productos sanguíneos; sin embargo, algunos centros realizan plasmaféresis previo al procedimiento cuando el TP es menor de 30%.
• Las drogas antifibrinolíticas se recomiendan en los momentos de hiperfibrinolisis evidenciada por sangrado de la microvasculatura o por medición de la máxima lisis del coágulo (TEG/ROTEM® >15%).
El uso profiláctico se recomienda sólo cuando se registra una reducción significativa de la MCF del coágulo <30 mm en el EXTEM.El ácido tranexámico (AcTx) y ε-aminocaproico reducen los requerimientos transfusionales; no hay evidencias que incrementen los eventos trombo-embólicos (8).Dosis: AxTx 15 mg/Kg en bolo, seguido por 10 mg/Kg x 4 hs, EV. Ácido ε-aminocaproico 4-5 g bolo en 1 hora. Infusión: 1-2 g/hora.• Concentrados de fibrinógeno. Han demostrado mejorar la hemostasia,
reducir el sangrado perioperatorio y las transfusiones. Indicado en hipofibrinogenemia con hiperfibrinolisis y sangrado.
Dosis inicial: 2-4 g/L cuando el nivel plasmático es <1 g/L. Las guías europeas aconsejan mantener niveles plasmáticos de 1,5-2 g/L. Ventajas: reduce la transmisión de enfermedades virales, la injuria pulmonar y la sobrecarga circulatoria.• CCP. Contienen hasta 25 veces las concentraciones de los FII, FVII, FIX,
FX y los inhibidores PC y PS. Logran la corrección de la coagulopatía usando volúmenes pequeños. Dosis: 25-30 UI/Kg. Actualmente hay en marcha un ensayo clínico multicéntrico rando- mizado (Protrom Trial).• Factor VII activo recombinante (rFVIIa). Mejora la hemostasia por
activación directa del FX, logrando la conversión de protrombina a trombina y formar un coágulo hemostático efectivo. Se une a la superficie de plaquetas activadas en los sitios de injuria vascular

213
Coagulopatía en la enfermedad hepática y el trasplante hepático | 11
incrementando la generación localizada de trombina.Los meta-análisis no han mostrado beneficio en la reducción del sangrado y del requerimiento transfusional. Incrementó hasta un 70% los eventos trombóticos arteriales. Las guías de la Sociedad Europea de Anestesiología (ESA) fuertemente no lo recomiendan en forma profiláctica.Se reserva su uso como terapia de rescate en sangrado masivo sin respuesta a las medidas previas. Dosis: 80 µg/Kg.
• Concentrados plaquetarios. La plaquetopenia caracteriza al can-didato al Tx hepático, pero la presencia de los multímeros de alto peso molecular del VWF incrementan la adhesividad de las mismas de manera importante y pueden generar trombosis, por lo que el uso preventivo para evitar sangrado está fuertemente no recomendado. Se asociaron a incremento en la mortalidad.
• Agonistas de los receptores de trombopoyetina. Eltrombopag fue usado en un ensayo clínico para optimizar el recuento plaquetario previo a procedimientos pero tuvo que ser suspendido prematuramente por asociarse a complicaciones trombóticas.
Conflictos de interés
Las autoras no presentan conflictos de interés.

214
Referencias
1. Clevenger B, Mallett S. Transfusion and coagulation Management in liver transplantation. World J Gastroenterol 2014; 20: 6148-6158.
2. Tripodi A, Manucci PM. The Coagulopathy of Chronic Liver Disease. N Engl J Med 2011; 365: 147-156.
3. Agarwal A, Sharma N, Vij V. Point of care coagulation monitoring during liver transplantation. Trends in Anaesthesia and Critical Care 2013; 3: 42-48.
4. Tripodi A, Primignani M, Chantarangkul V, et al. Thrombin generation in patients with cirrhosis: the role of platelets. Hepatology 2006; 44: 440-445.
5. MunozSJ, Stravitz RT, Gabriel DA. Coagulopathy of acute liver failure. Clin Liver Dis 2009; 13: 95-107.
6. Kerr R, Newsome P, Germain L, Thomson E, Dawson P, Stirling D, Ludlam CA. Effects of acute liver injury on blood coagulation. J Thromb Haemost 2003; 1: 754-759.
7. Krzanicki D, Sugavanam A,Mallett S. Intraoperative Hypercoagulability during liver transplantation as demons-trated by Thromboelastography. Liver Transplantation 2013; 852-861.
8. Tripodi A, Chantarangkul V, Primignani M et al. The International Normalized Ratio calibrated for cirrhosis (INR liver) normalizes prothrombin time results for Model for End-stage Liver disease Calculation. Hepatology 2007; 46: 520-527.
9. Tripathi D, Stanley AJ, Hayes PC, et al. UK guidelines on the management of variceal haemorrhage in cirrhotic patients. Gut 2015; 64: 1680–1704.
10. Massicotte L, Denault André,Beaulieu D. Transfusion rate for 500 consecutive liver Transplantations: Experience of one liver transplantation center. Transplantation 2012; 93: 1276-1281.
11 | Coagulopatía en la enfermedad hepática y el trasplante hepático

215
Introducción
Entre el 30 y el 60% de los pacientes con trauma sangran. El 50% será transfundido y de un 3 a 10% tendrá sangrado masivo. En las primeras 6 hs, se consumen el 80% de los hemocomponentes utilizados normalmente. La mortalidad en pacientes con sangrado trepa al 30-60%. La hemorragia es responsable del 40% de las muertes, en particular dentro de las primeras 24 hs del trauma (y 2 de cada 3 se producirán en las primeras 6 hs). Por otra parte, hasta el 80% de las muertes en las primeras horas intrahospitalarias se deberá a hemorragia. De este modo, la hemorragia es la segunda causa de muerte en la escena, la primera entre aquellos que viven lo suficiente como para llegar al hospital y la primera en quirófano. Hasta un tercio de los sangrados se deben a coagulopatía.
Sangrado en traumaCarlos Fondevila1 y Sebastián Marun2.1 Médico Hematólogo. Clínica Bazterrica. Facultad de Medicina. Universidad del Salvador.2 Bioquímico, Especialista en Hemostasia. Encargado del área hemostasia del Laboratorio de Bioquímica Clínica, Hospital Privado de Córdoba.
12

216
12 | Sangrado en trauma
Coagulopatía del paciente con trauma
La hemorragia traumática suele ocurrir, inicialmente, desde un sitio vascular o tisular, a partir de uno o varios vasos responsables. En estos casos, la hemostasia tiene un papel secundario: el cese del sangrado dependerá de soluciones quirúrgicas o mecánicas. La perpetuación del sangrado favorece la instalación progresiva de una coagulopatía, la cual ya no será solucionable quirúrgicamente. La hemorragia por falla hemostática se caracteriza por sangrado difuso en el lecho quirúrgico, en sitios donde los tejidos se encuentran razonablemente aproximados y más allá de la expectativa del cirujano, por babeo tisular continuado (una vez controlado el sangrado vascular) o por el sangrado de mucosas, serosas, sitios de heridas o de accesos vasculares y la aparición de equimosis sin trauma previo (Figura 1).
↑: aumentado; ↓: disminuido; ACoTS: acute coagulopathy of trauma shock; a2-AP: alfa2 anti-plasmina; CAT: coagulopatía aguda del trauma; CID-like: tipo-coagulación intravascular diseminada; crist/col: cristaloides/coloides; ISS: injury severity score; NET: neutrophil extracellular trap; PAI-1: inhibidor del activador del plasminógeno-1; PC: proteína C; PMNs: leucocitos polimorfonucleares; TAFI: inhibidor de la fibrinolisis activable por trombina; TIC: trauma induced coagulopathy; tPA: activador tisular del plasminógeno.
Figura 1.

217
Sangrado en trauma | 12
En ocasiones la coagulopatía es previa al trauma (coagulopatías congénitas, fármacos). Lo más frecuente es que la coagulopatía sea adquirida por diferentes mecanismos.
Coagulopatía de la resucitación
Es la causa más frecuente de coagulopatía y consecuencia directa del tratamiento del trauma. Las normas de resucitación ATLS (Advance Trauma Life Support) priorizan restaurar la volemia y el transporte de oxígeno. De allí la práctica usual de aportar inicialmente cristaloides y glóbulos rojos desplasmatizados (GRDs) postergando la infusión de plasma fresco congelado (PFC) o plaquetas luego de 4-6 / 8-12 unidades de GRDs, respectivamente. Esta forma secuencial de resucitar, resulta en hemodilución. Sin embargo, la coagulopatía por dilución per se, justifica el sangrado recién cuando se alcanza un nivel crítico de factores y plaquetas (usualmente luego del reemplazo de 1,5 a 2 volemias u 80 mL/Kg de resucitación).
Coagulación intravascular diseminada (CID)
En pacientes traumatizados, la CID puede presentar diferentes formas clínicas: • CID aguda clásica o trombohemorrágica. Se da en trauma tisular
extenso o descontrolado, trauma cerebral, embolismo graso o amniótico. Se debe a la acción de citoquinas que gatillan inflamación e hipofibrinolisis. Predomina la isquemia, con daño parenquimatoso y falla orgánica múltiple. El sangrado es por consumo.
• CID aguda hemorrágica. Se observa en trauma con sangrado
La coagulopatía de la resucitación, resulta de la presencia simultánea de la dilución junto a la hipotermia y la acidosis, consecuencias ambas de la hipoperfusión.

218
12 | Sangrado en trauma
sostenido y es una complicación de la resucitación prolongada, que ocasiona daño endotelial y falla de la microvasculatura. Se caracteriza por hiperfibrinolisis (HF) con sangrado incoercible. Con frecuencia coexisten hipotermia y/o acidosis. Hay poca respuesta al tratamiento con hemocomponentes.
• CID subaguda. Se instala días después del trauma. Suele ser isquémica, consecuencia de cirugía, inflamación sostenida o infección.
Hiperfibrinolisis
Hasta 10% de los pacientes con trauma grave, ISS (Injury Severity Score) >16, muestra hiperfibrinolisis al ingreso. La combinación de ISS >25, hipotensión, RIN (razón internacional normatizada) ≥1,3, temperatura (T) <36°C y pH <7,2 detectan la presencia de hiperfibrinolisis con una sensibilidad de 100%. La hiperfibrinolisis fulminante (<30 min desde el trauma) es parte constitutiva de la coagulopatía aguda del trauma (CAT) y se asocia con elevadísima mortalidad. En forma tardía (>60 min), se hace presente durante la etapa final de una resucitación prolongada y también se asocia con alta mortalidad. Los pacientes con trauma e hiperfibrinolisis presentan mayor mortalidad y mueren sangrando. La expansión con cristaloides la exacerba, mientras que la resucitación con PFC la atenúa.
Las diferentes formas clínicas de CID son poco evidentes en las pruebas de laboratorio de rutina. En cambio, pueden identificarse con pruebas como la generación de trombina o las pruebas viscoelásticas (TEG o ROTEM) que revelan tanto la presencia de hipo o hiperfibrinolisis, como la de coágulos friables.

219
Sangrado en trauma | 12
Coagulopatía aguda del trauma (CAT)
Hasta 25% de los pacientes con trauma grave presentan, desde el ingreso, descenso del tiempo de protrombina (TP) y de plaquetas; la prolongación del tiempo de tromboplastina parcial activada (TTPA) es menos frecuente. En presencia de CAT, el riesgo de muerte asociado a transfusión masiva (Tm) es del 30% (versus 5% en trauma con Tm y 0,6% en trauma sin Tm). La CAT es más prevalente en trauma grave (ISS >15) con daño tisular extenso, hipoperfusión y acidosis, con déficit de base (DB) menor a -6 mEq/L. Se debe a hipoperfusión tisular aguda y severa. Se asocia con un exceso de formación de trombina y de expresión de trombomodulina (TM), además de la presencia de TM soluble, como reflejo del daño endotelial. En presencia de acidosis, la trombina generada se “distrae” y, en lugar de formar fibrina, se une preferentemente a TM, resultando en un exceso de proteína C activada (PCa) que inactiva FV y FVIII, consume el inhibidor del activador del plasminógeno (PAI-1) y facilita la acción, sin oposición, del activador tisular del plasminógeno (tPA) liberado desde el endotelio dañado. El aporte de plasma u otros hemocomponentes es ineficaz, ya que no contribuye a la formación de fibrina y favorece la formación de más trombina y PCa.
La CAT es poco evidente en el laboratorio de rutina: TP <70%; RIN ≥1,4; plaquetas <100x109/L. Los marcadores de hiperfibrinolisis o de activación de la vía trombina-TM-PC (dímero D, fragmento 1+2 de la protrombina, aumento de TM soluble, descenso de PC) no se reflejan en las pruebas de rutina. Las pruebas viscoelásticas, realizadas aplicando el equipo ROTEM, han demostrado ser sensibles y específicas para evidenciar la hiperfibrinolisis.
La CAT se manifiesta dentro de los 30 min de producido el trauma, en pacientes con pocas medidas de resucitación y sin sangrado excesivo. No es por dilución; se debe a hipocoagulabilidad e hiperfibrinolisis. La expansión con solución salina agrava la acidosis (hiperclorémica) y empeora la situación.

220
12 | Sangrado en trauma
El factor tiempo (más tiempo de sangrado = más coagulopatía) y el punto de no-retorno
Todo sangrado prolongado, aún aquél inicialmente orgánico, evoluciona con el tiempo hacia una coagulopatía. Durante la resucitación de un paciente con sangrado sostenido hay un punto de no-retorno. Si el aporte de hemocomponentes comienza después de este punto crítico, ya no se podrá corregir la coagulopatía, ni detener el sangrado. El punto crítico se corresponde con la pérdida de, aproximadamente, una volemia en el caso del fibrinógeno y de dos volemias para plaquetas y factores como el FV. Aparece muy rápidamente si el sangrado es masivo, también con la infusión de grandes volúmenes de cristaloides/coloides/GRDs o cuando se mantiene una presión arterial elevada. Trabajos experimentales demostraron que admi-nistrando simultáneamente PFC y GRDs en relación >0,66 o plaquetas y GRDs en relación >0,8 se evita que los valores de TP, fibrinógeno y plaquetas alcancen la zona de no-retorno. Un trabajo clínico reciente confirmó estos datos.Por otra parte, cuando el sangrado y la resucitación se prolongan, la expansión se demora o el ritmo de la resucitación va desacompasado con el del sangrado, se produce hipoperfusión sostenida de la microvasculatura lo que causa hipoxia, acidosis e hipotermia, con la consecuencia de daño endotelial e hiperfibrinolisis. Alcanzado este punto, la transfusión de hemocomponentes ya no puede restaurar la hemostasia (coagulopatía del final).
Diagnóstico de laboratorio
En todo paciente gravemente traumatizado deberán realizarse las siguientes determinaciones de manera temprana y repetida debido a lo dinámico del proceso:• Estado ácido-base + ácido láctico + calcio iónico + electrolitos + glucosa • Hemoglobina + recuento plaquetario• TP/RIN + TTPA + fibrinógenoEn caso de ser posible, el panel de estudio debe incluir: • Pruebas viscoelásticas (ROTEM o TEG)Déficit de base y ácido láctico. Son utilizados como marcadores de shock, siendo a la vez factores pronóstico de transfusión masiva y muerte. Ayudan

221
Sangrado en trauma | 12
en el monitoreo de la reanimación. Los dos parámetros no correlacionan estrictamente en pacientes gravemente heridos; por lo tanto, se recomienda la medición independiente de ambos.Calcio. Es imprescindible para sostener una buena hemostasia. La hipocalcemia es frecuente en el contexto de transfusión masiva y puede perpetuar o agravar el sangrado.TP y TTPA. Estas pruebas globales clásicas son poco representativas del modelo celular de la coagulación; sólo reflejan el 4% inicial de toda la trombina generada para la formación del coágulo. No reflejan las alteraciones provocadas por hipotermia, acidosis o hipocalcemia en el paciente real. El TTPA suele verse afectado por la elevación del FVIII que es habitual en respuesta al trauma. En resumen, no son infrecuentes valores de TP/TTPA hemostáticos o normales, incluso si la hemostasia real del paciente está alterada. Sólo se alteran significativamente cuando la coagulopatía se encuentra avanzada: el valor predictivo positivo (VPP) de un RIN o un TTPA ≥1,5 es de 90% y 100%, respectivamente para deficiencia significativa (≈20%) de un factor sensible. También es baja la sensibilidad para detectar CAT o predecir Tm. Finalmente, el tiempo de procesamiento (usualmente 30-45 min) hace que estas pruebas resulten poco útiles para la toma de decisiones durante la etapa aguda, de inestabilidad hemodinámica y con el paciente sangrando activamente. Una vez controlado el sangrado y alcanzada la estabilidad hemodinámica, valores de TP/TTPA >1,5 permiten continuar con la resucitación.Fibrinógeno y recuento de plaquetas. Estas mediciones tienen especial importancia debido a que determinan la firmeza del coagulo. En particular, el fibrinógeno es el factor que primero desciende a causa de la reposición sanguínea masiva. El método usado para medir fibrinógeno debe ser el de Clauss, siempre referido a la curva de calibración local. Los coagulómetros ópticos pueden sobreestimar significantemente las concentraciones de fibrinógeno, en pacientes que han sido reanimados masivamente con coloides. Estudios recientes en cirugía cardiovascular (CCV) y en sangrado traumático u obstétrico mostraron que, valores por debajo de 200 mg/dL se acompañan de sangrado, en especial cuando coexisten con plaquetopenia. Durante el sangrado activo, se requieren al menos 100x109 plaquetas/L para sostener un coágulo firme.Pruebas viscoelásticas. Ambos equipos, TEG y ROTEM utilizan sangre entera y brindan resultados rápidamente (a partir de los 10 min), incluyendo información sobre la participación de plaquetas y la fibrinolisis en la forma-

222
12 | Sangrado en trauma
ción y firmeza del coágulo. Estas características justifican su uso creciente en centros de tratamiento de trauma. Las pruebas viscoelásticas no dan infor-mación sobre la participación del endotelio, del factor von Willebrand (VWF) ó de la presencia de antiagregantes plaquetarios (excepto los anti-GPIIbIIIa).TEG y ROTEM usan principios de medición similares pero no son equivalentes entre sí debido a diferencias en el equipamiento, los reactivos utilizados, los parámetros a medir y los algoritmos de interpretación. Estos algoritmos, al facilitar la identificación rápida de la falla hemostática, permiten una reposición dirigida, utilizando hemocomponentes o hemoderivados espe-cíficos (Figura 2).
Trazados de las pruebas viscoelásticas (ROTEM), realizadas con diferentes reactivos, según las condiciones clínicas del paciente. A10: amplitud 10 minutos (firmeza del coágulo); Crio: crioprecipitados; CC: concentrados; CCP: concentrados de complejo protrombínico; CT: tiempo de coagulación; EXTEM: prueba que analiza la vía extrínseca (coagulación, consolidación, fibrinolisis); FIBTEM: monitorización del fibrinógeno; MCF: máxima firmeza del coágulo; PFC: plasma fresco congelado.
Figura 2.

223
Sangrado en trauma | 12
Las pruebas viscoelásticas detectan más precozmente y más en detalle, las causas del sangrado en trauma. Resultados normales de estas pruebas, obligan a reconsiderar una causa orgánica.
Tratamiento
Valoración inicial del shock hemorrágicoEl shock es la reducción crítica de la perfusión tisular, que resulta en un desequilibrio entre aporte/demanda de oxígeno. En pacientes traumatizados, el shock hemorrágico es el más frecuente.Las prioridades en los minutos iniciales de su tratamiento serán:Reconocer la presencia de signos clínicos precoces de hipoperfusión tisular. Presión del pulso, trastornos del sensorio, frialdad acral, palidez, sudoración fría, taquicardia. Indican presencia de shock hipovolémico y pérdida de la volemia >30%. Una presión arterial sistólica <90 mmHg y una frecuencia cardíaca (FC) >120 latidos/min son indicadores tardíos, que aparecen cuando se ha perdido 40% de la volemia.Comenzar la resucitación con fluidos, evitar la acidosis y la hipotermia. Es importante observar la respuesta hemodinámica luego de la expansión inicial, a fin de inferir la gravedad de la lesión (Tabla 1). En caso de respuesta transitoria, mínima o nula, se deberá comenzar de inmediato el soporte hemostático y llamar a cirugía.
Respuesta Pérdida de volemia Condición clínica Conducta sugerida
Rápida
Transitoria
Mínima o nula
<20%
20-40%
>40%
Estable
Primero estable, luego vuelve a deteriorarse
Implica sangrado continuado
Shock sostenidoEmergencia médica y quirúrgicaImplica sangrado exsanguinante
No requiere transfusión
Comenzar transfusión de hemocomponentes
Avisar al cirujano para búsqueda/control del sitio de sangrado
Transfusión masiva de hemocomponentes (RCDH)
Cirugía precoz
Tabla 1. Respuesta hemodinámica luego de la expansión inicial
RCDH: resucitación con control del daño hemostático.

224
12 | Sangrado en trauma
Controlar el sitio de sangrado. En hemorragia controlable, hay que aplicar maniobras compresivas (incluyendo torniquetes). En sangrado no compresible, se procede a cirugía precoz (incluyendo cirugía de control de daño en casos con elevado riesgo de muerte); a fin de minimizar el tiempo/trauma operatorio: controlar el sangrado y/o la contaminación intestinal postergando la reparación definitiva. En hemorragia no controlable, y mientras se persigue una resolución quirúrgica, es preferible limitar la expansión (resucitación hipotensiva). Cuando el sitio de sangrado no sea evidente (fracturas no expuestas, pleura, abdomen, retroperitoneo) buscarlos mediante: Rx, FAST (Focus Assessment with Sonography in Trauma), TAC y/o proceder de inmediato a cirugía.Restaurar el transporte de oxígeno (transfusión de GRDs). Mantener una hemoglobina >7 g/dL. Reponer GRDs a partir de 1,5-2 L de resucitación.Valorar el grado de depleción circulatoria. Identificar dentro de los primeros minutos de arribo al shock-room al 10% de pacientes con posibilidad de necesitar Tm. Los términos Tm, sangrado masivo y exsangui-nante se han utilizado para caracterizar la magnitud del sangrado (Tabla 2).
Transfusión masiva
Sangrado masivo
Sangrado exsanguinante
• 1 volemia en 24 hs
• Sin definición uniforme - “Mucha sangre en poco tiempo” - >4u GRD en 4 hs y aun sangrando - >10u GRD en 4 hs - 50% de la volemia en 3 hs - >150 mL/min - >1,5mL/Kg/min >20 min
• Pérdida inicial >40% volemia• >50% volemia en 10 min• >250 mL/min• Criterios clínicos - No respuesta a la resucitación - Pulso débil o ausente (TAS <70) - Alteración del sensorio (TAS <60)
Tabla 2. Definiciones de sangrado
GRD: glóbulos rojos desplamatizados; TAS: tensión arterial sistólica
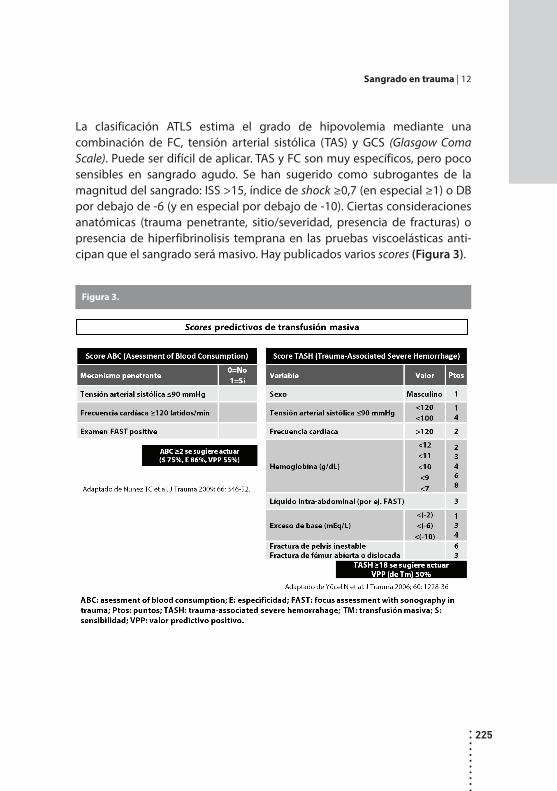
225
Sangrado en trauma | 12
La clasificación ATLS estima el grado de hipovolemia mediante una combinación de FC, tensión arterial sistólica (TAS) y GCS (Glasgow Coma Scale). Puede ser difícil de aplicar. TAS y FC son muy específicos, pero poco sensibles en sangrado agudo. Se han sugerido como subrogantes de la magnitud del sangrado: ISS >15, índice de shock ≥0,7 (en especial ≥1) o DB por debajo de -6 (y en especial por debajo de -10). Ciertas consideraciones anatómicas (trauma penetrante, sitio/severidad, presencia de fracturas) o presencia de hiperfibrinolisis temprana en las pruebas viscoelásticas anti-cipan que el sangrado será masivo. Hay publicados varios scores (Figura 3).
Figura 3.

226
12 | Sangrado en trauma
La importancia de identificar rápidamente un sangrado masivo, se debe a que la mortalidad conresucitación convencional es del 30-60%.La mortalidad se reduce significativamente tras implementar técnicas de resucitación proactivas. En sangrado masivo/exsanguinante, la experiencia e intuición clínica, así como la respuesta transitoria o nula a resucitación inicial, resultan más útiles y precoces que la cuantificación de sangrado (Tabla 3).
Al ingreso
Durante el examen clínico
Evidentes durante la resucitación
• Magnitud del sangrado
• Presencia de coagulopatía
• Presencia de hipoperfusión
• Pulso radial débil o ausente (sugiere TAS <70)• Status mental anormal (sugiere TAS <60)• Hipotermia o acidosis persistentes• Tipo de lesión - Trauma penetrante (con destrucción tisular masiva) - Amputaciones múltiples o proximales (muslo) - Sangrado de tronco + amputación proximal - Evisceración abdominal + hipotensión
• Respuesta transitoria o nula a la expasión inicial con fluidos y GRD (no sale del shock, no estabiliza la TA)
• Criterios de sangrado exsanguinante (pérdida >40% volemia, >50% volemia en 10 min, velocidad de sangrado >70-100 mL/min
• Más de 4 u GRD cada 4 hs o más de 6 u en 12 hs y aun sangrando (indicador tardío)
Shock/hipotensión (TAS <90)FC >100 latidos/minÍndice de shock >0,7Hb <11 g/dL (otros 9 g/dL)
TP <70%, RIN >1,4, plaquetas <100x109/L
Acidosis (DB <(-6) y en especial a (-10) o lactato >5 mmol/L)Temperatura <35-36°C
Tabla 3. Hallazgos sugestivos de la necesidad de transfusión masiva
DB: déficit de base; FC: frecuencia cardíaca; GRD: glóbulos rojos desplasmatizados; Hb: hemoglobina; RIN: razón internacional normatizada; TAS: tensión arterial sistólica; TP: tiempo de protrombina.

227
Sangrado en trauma | 12
Reconocer las diferentes formas de la coagulopatía del shock traumático. Como fue mencionado, el laboratorio tradicional tiene limitaciones para identificar tempranamente los cambios en la hemostasia que permiten identificar cada tipo de coagulopatía. Las pruebas viscoelásticas permiten caracterizar rápidamente el mecanismo de falla hemostática, en especial, la presencia de alteraciones funcionales atribuibles a fibrinógeno, plaquetas e hiperfibrinolisis. Los dispositivos point of care (POC) que utilizan sangre entera y permiten medir el TP, proveen información al costado de la cama y podrían ser de ayuda, una vez validados, para guiar la reposición.
Resucitación convencional (normas ATLS): normotensiva y secuencial
La Tabla 4 resume los pasos habituales que, como fue mencionado, comienzan con restaurar la volemia, luego detener el sangrado y mejorar el transporte de O2 para, finalmente, corregir la coagulopatía. El tipo de soporte hemostático y el momento de iniciarlo depende del perfil de laboratorio (ya que con este algoritmo, hay tiempo para procesar las muestras y disponer de los resultados). Como umbrales que gatillan la reposición se utilizan valores convencionales, extrapolados del nivel hemostático para casos de déficit único, medido a 37°C. En el trauma con sangrado activo y descenso simultáneo de varios factores, sumado a hipotermia y acidosis, se produce una deficiencia funcional; en esta situación, niveles bajos, aunque suficientes para el concepto hemostático convencional, de fibrinógeno, plaquetas y FXIII no son suficientes para formar un coágulo estable, en tiempo y forma. Estos coágulos friables sólo son evidentes mediante pruebas viscoelásticas. El fibrinógeno y en menor medida las plaquetas, son los factores limitantes; la hipotermia reduce la velocidad de síntesis del fibrinógeno y la acidosis promueve la fibrinolisis. Se ha demostrado que fibrinógeno <200 mg/dL y plaquetas <100x109/L predicen sangrado y mortalidad en trauma, trauma craneal, CCV, cirugía de aneurisma aorta abdominal roto y obstetricia. En consensos recientes se han modificado los umbrales para la reposición de fibrinógeno y de plaquetas; también se han formulado sugerencias para incorporar las pruebas viscoelásticas para la toma de decisiones.
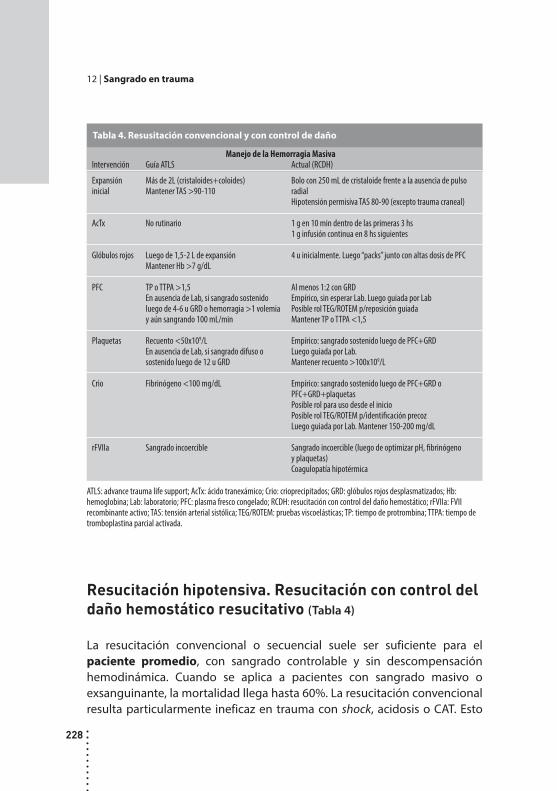
228
12 | Sangrado en trauma
Resucitación hipotensiva. Resucitación con control del daño hemostático resucitativo (Tabla 4)
La resucitación convencional o secuencial suele ser suficiente para el paciente promedio, con sangrado controlable y sin descompensación hemodinámica. Cuando se aplica a pacientes con sangrado masivo o exsanguinante, la mortalidad llega hasta 60%. La resucitación convencional resulta particularmente ineficaz en trauma con shock, acidosis o CAT. Esto
Intervención Guía ATLSManejo de la Hemorragia Masiva
Actual (RCDH)
Expansión inicial
AcTx
Glóbulos rojos
PFC
Plaquetas
Crio
rFVIIa
Más de 2L (cristaloides+coloides)Mantener TAS >90-110
No rutinario
Luego de 1,5-2 L de expansiónMantener Hb >7 g/dL
TP o TTPA >1,5En ausencia de Lab, si sangrado sostenido luego de 4-6 u GRD o hemorragia >1 volemia y aún sangrando 100 mL/min
Recuento <50x109/LEn ausencia de Lab, si sangrado difuso o sostenido luego de 12 u GRD
Fibrinógeno <100 mg/dL
Sangrado incoercible
Bolo con 250 mL de cristaloide frente a la ausencia de pulso radialHipotensión permisiva TAS 80-90 (excepto trauma craneal)
1 g en 10 min dentro de las primeras 3 hs1 g infusión continua en 8 hs siguientes
4 u inicialmente. Luego “packs” junto con altas dosis de PFC
Al menos 1:2 con GRDEmpírico, sin esperar Lab. Luego guiada por LabPosible rol TEG/ROTEM p/reposición guiadaMantener TP o TTPA <1,5
Empírico: sangrado sostenido luego de PFC+GRDLuego guiada por Lab. Mantener recuento >100x109/L
Empírico: sangrado sostenido luego de PFC+GRD o PFC+GRD+plaquetasPosible rol para uso desde el inicioPosible rol TEG/ROTEM p/identificación precozLuego guiada por Lab. Mantener 150-200 mg/dL
Sangrado incoercible (luego de optimizar pH, fibrinógeno y plaquetas)Coagulopatía hipotérmica
Tabla 4. Resusitación convencional y con control de daño
ATLS: advance trauma life support; AcTx: ácido tranexámico; Crio: crioprecipitados; GRD: glóbulos rojos desplasmatizados; Hb: hemoglobina; Lab: laboratorio; PFC: plasma fresco congelado; RCDH: resucitación con control del daño hemostático; rFVIIa: FVII recombinante activo; TAS: tensión arterial sistólica; TEG/ROTEM: pruebas viscoelásticas; TP: tiempo de protrombina; TTPA: tiempo de tromboplastina parcial activada.

229
Sangrado en trauma | 12
se ve en aproximadamente 30% de los casos de trauma de guerra y 5% de casos de trauma civil. La CCV (rotura AAA), el trasplante cardíaco o hepático y el sangrado obstétrico son posibles escenarios de sangrado masivo.
Este agravamiento es consecuencia de la expansión, en condiciones normotensas, con grandes volúmenes de solución salina que despega los coágulos incipientes y diluye marcadamente la concentración de factores y plaquetas, llegando tempranamente al punto de no-retorno. La solución salina agrava la acidosis, promueve la fibrinolisis y causa disfunción de la célula endotelial (inflamación, hiperpermeabilidad y apoptosis).Se han diseñado protocolos de resucitación con control del daño hemostático (RCDH) para aplicar en pacientes con trauma grave y riesgo de Tm. Consisten en el uso temprano de ácido tranexámico (AcTx, 1 g en bolo al inicio de la resucitación), así como la administración precoz de PFC±plaquetas y crioprecipitados (Crio), durante la resucitación inicial y en relación aproximadamente 1:1 con GRDs. El uso de PFC reduce la disfunción endotelial y la hiperfibrinolisis que acompañan al shock hemorrágico y al sangrado masivo. La reciente guía europea de manejo del sangrado en trauma, recomienda para el sangrado masivo el uso precoz de PFC a 15 mL/Kg. En cambio, es discutible el beneficio de administrar tempranamente plaquetas y Crio, considerando que las plaquetas caen a un nivel crítico tardíamente y que cada unidad de PFC aporta aproximadamente 400 mg de fibrinógeno. Muchos protocolos de resucitación hemostática comienzan sólo con PFC+GRDs postergando la infusión de plaquetas y Crio para el caso de sangrado sostenido luego de al menos 12 unidades de GRDs (Tabla 5).
En determinadas circunstancias, la resucitación convencional puede agravar la coagulopatía, en vez de mejorarla, produciendo un daño hemostático.
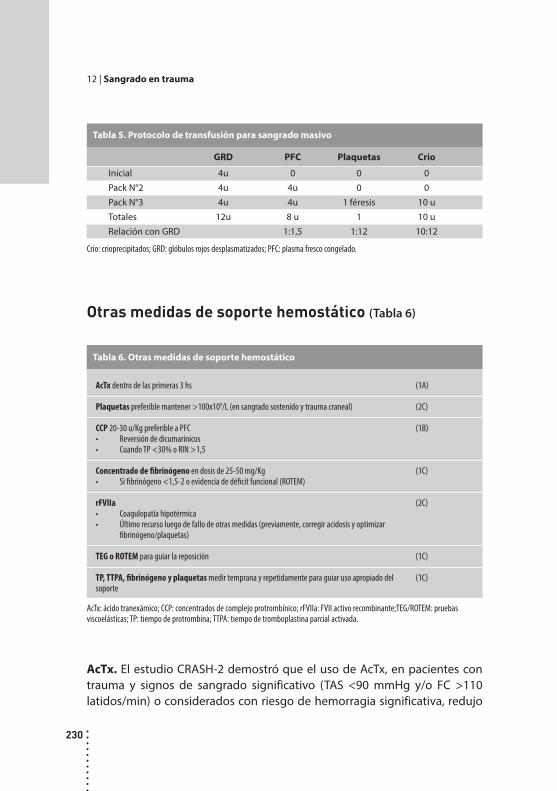
230
12 | Sangrado en trauma
Otras medidas de soporte hemostático (Tabla 6)
AcTx. El estudio CRASH-2 demostró que el uso de AcTx, en pacientes con trauma y signos de sangrado significativo (TAS <90 mmHg y/o FC >110 latidos/min) o considerados con riesgo de hemorragia significativa, redujo
AcTx dentro de las primeras 3 hs
Plaquetas preferible mantener >100x109/L (en sangrado sostenido y trauma craneal)
CCP 20-30 u/Kg preferible a PFC• Reversión de dicumarínicos• Cuando TP <30% o RIN >1,5
Concentrado de fibrinógeno en dosis de 25-50 mg/Kg• Si fibrinógeno <1,5-2 o evidencia de déficit funcional (ROTEM)
rFVIIa• Coagulopatía hipotérmica• Último recurso luego de fallo de otras medidas (previamente, corregir acidosis y optimizar
fibrinógeno/plaquetas)
TEG o ROTEM para guiar la reposición
TP, TTPA, fibrinógeno y plaquetas medir temprana y repetidamente para guiar uso apropiado del soporte
(1A)
(2C)
(1B)
(1C)
(2C)
(1C)
(1C)
Tabla 6. Otras medidas de soporte hemostático
AcTx: ácido tranexámico; CCP: concentrados de complejo protrombínico; rFVIIa: FVII activo recombinante;TEG/ROTEM: pruebas viscoelásticas; TP: tiempo de protrombina; TTPA: tiempo de tromboplastina parcial activada.
Crio: crioprecipitados; GRD: glóbulos rojos desplasmatizados; PFC: plasma fresco congelado.
InicialPack N°2Pack N°3TotalesRelación con GRD
4u4u4u
12u
00
1 féresis1
1:12
04u4u8 u
1:1,5
00
10 u10 u
10:12
Tabla 5. Protocolo de transfusión para sangrado masivo
GRD PlaquetasPFC Crio

231
Sangrado en trauma | 12
la mortalidad y la mortalidad por sangrado. El beneficio fue mayor en los tratados antes de 1 hora de producido el trauma y aún evidente hasta las 3 horas del mismo. En aquellos tratados luego de 3 horas, la mortalidad fue mayor. Todas las guías actuales recomiendan el uso precoz de AcTx (1 g en 10 minutos, seguido de 1 g por goteo en 8 hs) en trauma con sangrado mayor o riesgo de sangrado mayor. Carecemos de información respecto de otros fármacos con acción antifibrinolítica (ε-aminocaproico o aprotinina).Factor VII activado recombinante (rFVIIa). Los consensos recientes limitan el uso del rFVIIa. Se lo considera como último recurso ante coagulopatía hipotérmica o sangrado refractario al uso de otras medidas hemostáticas convencionales (cirugía, radiología intervencionista y transfusión de hemocomponentes). El uso de rFVIIa (dosis: 90-100 mg/Kg) mostró acortamiento del TP/TTPA, cese del sangrado y menor requerimiento de sangre y hemocomponentes; no redujo la mortalidad, ni la falla orgánica múltiple. Tampoco mostró eficacia en trauma penetrante. En pacientes añosos o con factores de riesgo cardiovascular o portadores de dispositivos endovasculares pueden producirse eventos trombóticos. Un Consenso Argentino sugirió el uso de rFVIIa en trauma (especialmente contuso) con sangrado masivo, sin respuesta al tratamiento convencional, sin vaso identificable o con sangrado coagulopático/laboratorio de CID.Concentrado de complejo protrombínico (CCP). Los CCP (20-30 U/Kg) se han utilizado en lugar del PFC en trauma con coagulopatía por dilución, durante la resucitación guiada por ROTEM. Resultan más eficaces y deberían preferirse al PFC, como tratamiento del sangrado asociado a exceso de dicumarínicos. Fuera de estas dos situaciones, su uso es controvertido. Por ej, en trauma experimental, dosis >50 U/Kg se asociaron a tromboembolismo y CID.Concentrado de fibrinógeno. Su principal ventaja sobre el PFC y los Crio es el poco volumen y la seguridad biológica. Se lo ha empleado en dosis de 25-50 mg/Kg en lugar del PFC en trauma con Tm o coagulopatía por dilución; en especial asociado al uso de hidroxietilamidón (HES) como expansor. También ha sido utilizado en protocolos de terapia dirigida por ROTEM/TEG, ante valores de fibrinógeno menores de 1,5-2 g/L o evidencia de defecto funcional. Carecemos de estudios que comparen eficacia y riesgo de tromboembolismo con otras fuentes de fibrinógeno como los Crio.Concentrado de FXIII. Siguiendo la expansión con coloides, se verifica una caída del FXIII cercana al 60%. En el paciente sangrando activamente,

232
12 | Sangrado en trauma
esta menor disponibilidad relativa se traduce en la formación de coágulos friables (baja MCF por ROTEM), a pesar de que posea un nivel de fibrinógeno suficiente. La evidencia actual para el uso de concentrados de FXIII (30 UI/Kg) es poca; se limita a casos de trauma con sangrado sostenido y actividad de FXIII <60% o cuando se verifica un coágulo friable a pesar de valores de fibrinógeno y plaquetas suficientes.
Conclusiones
• La coagulopatía es un evento frecuente en trauma grave. La muerte por sangrado es la segunda más frecuente y la primera entre aquellos que llegan al hospital.
• Las decisiones deben ser rápidas y el juicio clínico insustituible. • El laboratorio convencional es lento y refleja poco/tarde la situación
in vivo. • Una minoría de pacientes más gravemente traumatizados,
severamente hipoperfundidos y acidóticos, presenta CAT antes de la resucitación, consecuencia de hipocoagulabilidad e hiperfibrinolisis.
• En la mayoría de los sangrados, la coagulopatía será secundaria a la resucitación, resultando de la combinación de dilución, consumo, hipotermia y acidosis.
• La conducta convencional de posponer la transfusión de hemocomponentes hasta disponer de los resultados del laboratorio se ha mostrado ineficaz en trauma grave o con sangrado masivo. La terapia de reemplazo debe llegar en cantidad y tiempo adecuados: una vez pasada la ventana de oportunidad, es muy difícil detener el sangrado a pesar del uso agresivo de hemocomponentes.
• En sangrado grave, así como en CAT, el uso temprano de antifibrinolíticos y/o la resucitación proactiva mediante el uso temprano de PFC en relación >0,7 con GRDs ha logrado una reducción significativa de la mortalidad. La transfusión de plaquetas y de Crio deberán considerarse cuando el sangrado se mantiene a pesar de PFC+GRDs.
• Los beneficios de la resucitación proactiva se limitan a la población destinada a sangrar masivamente (más de 6-8 u de GRD). Deberá anticiparse la necesidad de Tm con el uso de distintas herramientas,

233
Sangrado en trauma | 12
entre las cuales el examen y la intuición clínica son principales, a fin de optimizar el uso de los recursos del banco de sangre.
• Evidencia reciente sugiere el tratamiento dirigido con concentrados específicos, utilizando la información brindada por pruebas viscoelásticas. El costo más elevado de estos concentrados exige disponer de estudios que comparen sus beneficios y riesgos antes de recomendar su uso por sobre PFC o Crio.
• Todo este esfuerzo será fútil si no se logra tempranamente el control del sitio de sangrado.
Conflictos de interés
Sebastián Marun es asesor técnico/científico del representante de ROTEM en Argentina (FELSAN SRL).Carlos Fondevila no presenta conflictos de interés.

234
12 | Sangrado en trauma
Referencias
Malone DL, Hess JR, Fingerhut A. Massive transfusion practices around the globe and a suggestion for a common massive transfusion protocol. J Trauma 2006; 60: S91-S96.
Gonzalez EA, Moore FA, Holcomb JB et al. Fresh frozen plasma should be given earlier to patients requiring massive transfusion. J Trauma 2007; 62: 112-119.
Hess JR, Dutton RB, Holcomb JB, Scalea TM. Giving plasma at a 1:1 ratio with red cells in resuscitation: who might benefit? Transfusion 2008; 48: 1763-1765.
Brohi K, Cohen MJ, Ganter MT et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma 2008; 64: 1211-1217.
Fries D, Innerhofer P, Schobersberger W. Time for changing coagulation management in trauma-related massive bleeding. Curr Opin Anaesthesiol 2009; 22: 267-274.
The CRASH-2 Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomized, placebo-controlled trial. The Lancet 2010; 376: 23-32.
Howard BM, Daley AT, Cohen MJ. Prohemostatic interventions in trauma: resuscitation-associated coagulopathy, acute traumatic coagulopathy, hemostatic resuscitation, and other hemostatic interventions. Semin Thromb Hemost 2012; 38: 250-58.
Kozek-Langenecker SA, Afshari A, Albaladejo P et al. Management of severe perioperative bleeding. Guidelines of the European Society of Anaesthesiology. Eur J Anaesthesiol 2013; 30: 270-382.
Spahn DR, Bouillon B, Cerny V et al. Management of bleeding and coagulopathy following major trauma: an updated European guideline. Critical Care 2013; 17: R76.
Hunt BJ, Allard S, Keeling D et al. A practical guideline for the haematological management of major haemorrhage. Br J Haematol 2015; 170: 788-803.
Cap A, Hunt BJ. The pathogenesis of traumatic coagulopathy. Anaesthesia 2015; 70: 96-101.

235
Aspectos clínicos en pacientes adultos
El sangrado post-operatorio (SPO) anómalo es una de las complicaciones inesperadas más frecuentes de las intervenciones quirúrgicas programadas.El correcto enfoque diagnóstico orientará a una terapia de soporte adecuada y a la acertada indicación de re-exploración cuando sea conveniente, por esto el hematólogo jugará un rol de fundamental importancia en la toma final de decisiones.
Definiciones
El término sangrado mayor o prolongado es amplio, y en un intento de armonización, se están empezando a usar las definiciones de sangrado mayor de estudios clínicos en pacientes que reciben antitrombóticos, definidas por la Sociedad Internacional de Hemostasia y Trombosis (ISTH), sin embargo, en la práctica el cirujano es quien va a definir el caso de sangrado anómalo.El SPO se puede considerar normal en ciertas cirugías: hematuria luego de resección transuretral de próstata o acumulación de 500 mL de sangre post-cadera, estas condiciones se tornan patológicas en el contexto de condiciones agravantes.En la Tabla 1 se describen situaciones que pueden afectar el SPO.
Sangrado quirúrgicoHugo H. Ferro1 y Cecilia C. Colorio2.1 Médico Hematólogo.2 Médica hematóloga. Servicio de Hematología de la Fundación Favaloro. Docente Adscripta UBA y Fundación Favaloro.
13
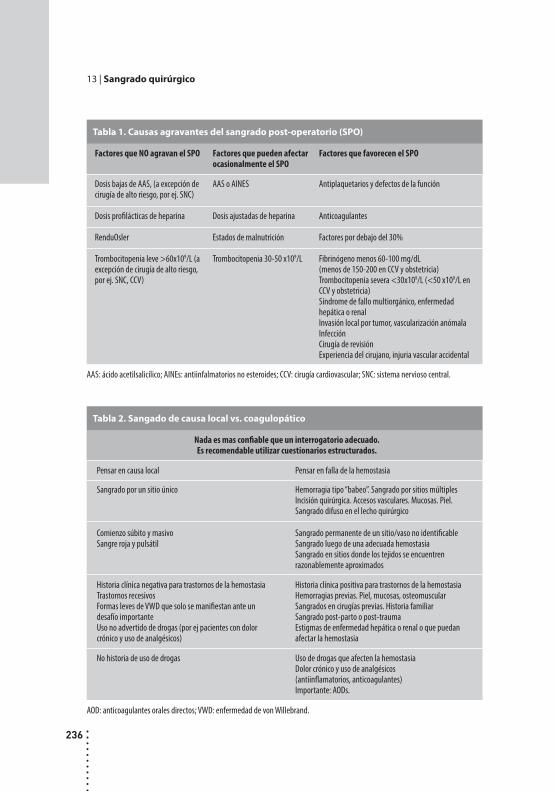
236
13 | Sangrado quirúrgico
Dosis bajas de AAS, (a excepción de cirugía de alto riesgo, por ej. SNC)
Dosis profilácticas de heparina
RenduOsler
Trombocitopenia leve >60x109/L (a excepción de cirugía de alto riesgo, por ej. SNC, CCV)
AAS o AINES
Dosis ajustadas de heparina
Estados de malnutrición
Trombocitopenia 30-50 x109/L
Antiplaquetarios y defectos de la función
Anticoagulantes
Factores por debajo del 30%
Fibrinógeno menos 60-100 mg/dL(menos de 150-200 en CCV y obstetricia)Trombocitopenia severa <30x109/L (<50 x109/L en CCV y obstetricia)Síndrome de fallo multiorgánico, enfermedad hepática o renal Invasión local por tumor, vascularización anómalaInfecciónCirugía de revisiónExperiencia del cirujano, injuria vascular accidental
Factores que NO agravan el SPO Factores que pueden afectar ocasionalmente el SPO
Factores que favorecen el SPO
AAS: ácido acetilsalicílico; AINEs: antiinfalmatorios no esteroides; CCV: cirugía cardiovascular; SNC: sistema nervioso central.
Tabla 1. Causas agravantes del sangrado post-operatorio (SPO)
Pensar en causa local
Sangrado por un sitio único
Comienzo súbito y masivoSangre roja y pulsátil
Historia clínica negativa para trastornos de la hemostasiaTrastornos recesivos Formas leves de VWD que solo se manifiestan ante un desafío importanteUso no advertido de drogas (por ej pacientes con dolor crónico y uso de analgésicos)
No historia de uso de drogas
Pensar en falla de la hemostasia
Hemorragia tipo “babeo”. Sangrado por sitios múltiplesIncisión quirúrgica. Accesos vasculares. Mucosas. Piel.Sangrado difuso en el lecho quirúrgico
Sangrado permanente de un sitio/vaso no identificableSangrado luego de una adecuada hemostasiaSangrado en sitios donde los tejidos se encuentren razonablemente aproximados
Historia clínica positiva para trastornos de la hemostasiaHemorragias previas. Piel, mucosas, osteomuscularSangrados en cirugías previas. Historia familiarSangrado post-parto o post-traumaEstigmas de enfermedad hepática o renal o que puedan afectar la hemostasia
Uso de drogas que afecten la hemostasiaDolor crónico y uso de analgésicos (antiinflamatorios, anticoagulantes)Importante: AODs.
Nada es mas confiable que un interrogatorio adecuado.Es recomendable utilizar cuestionarios estructurados.
AOD: anticoagulantes orales directos; VWD: enfermedad de von Willebrand.
Tabla 2. Sangado de causa local vs. coagulopático
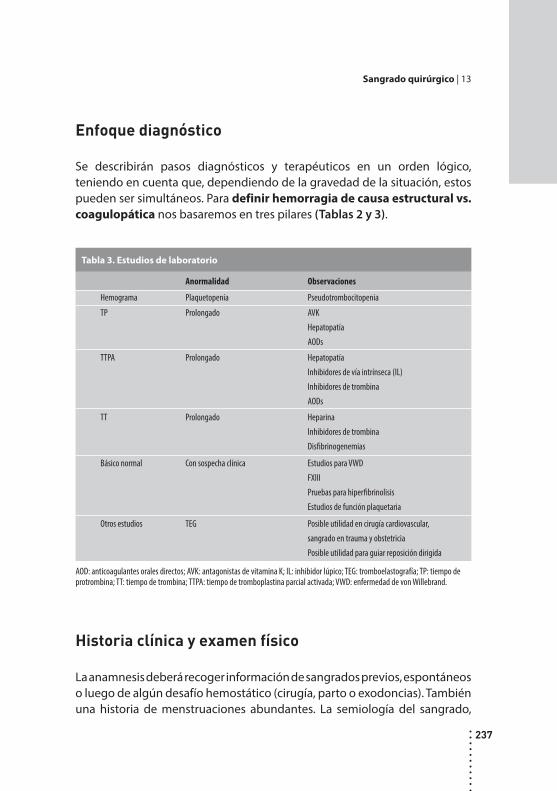
237
Sangrado quirúrgico | 13
Enfoque diagnóstico
Se describirán pasos diagnósticos y terapéuticos en un orden lógico, teniendo en cuenta que, dependiendo de la gravedad de la situación, estos pueden ser simultáneos. Para definir hemorragia de causa estructural vs. coagulopática nos basaremos en tres pilares (Tablas 2 y 3).
Historia clínica y examen físico
La anamnesis deberá recoger información de sangrados previos, espontáneos o luego de algún desafío hemostático (cirugía, parto o exodoncias). También una historia de menstruaciones abundantes. La semiología del sangrado,
AOD: anticoagulantes orales directos; AVK: antagonistas de vitamina K; IL: inhibidor lúpico; TEG: tromboelastografía; TP: tiempo de protrombina; TT: tiempo de trombina; TTPA: tiempo de tromboplastina parcial activada; VWD: enfermedad de von Willebrand.
Hemograma
TP
TTPA
TT
Básico normal
Otros estudios
Plaquetopenia
Prolongado
Prolongado
Prolongado
Con sospecha clínica
TEG
Pseudotrombocitopenia
AVK
Hepatopatía
AODs
Hepatopatía
Inhibidores de vía intrínseca (IL)
Inhibidores de trombina
AODs
Heparina
Inhibidores de trombina
Disfibrinogenemias
Estudios para VWD
FXIII
Pruebas para hiperfibrinolisis
Estudios de función plaquetaria
Posible utilidad en cirugía cardiovascular,
sangrado en trauma y obstetricia
Posible utilidad para guiar reposición dirigida
Tabla 3. Estudios de laboratorio
Anormalidad Observaciones

238
13 | Sangrado quirúrgico
si fue inmediato o retardado, si requirió internación o transfusión u otras medidas. Los problemas vinculados con la hemostasia primaria (plaquetas o enfermedad de von Willebrand -VWD-) se caracterizan por un patrón de sangrado inmediato y manifestaciones cutáneomucosas. En cambio, los relacionados con alteraciones en los factores de coagulación, se caracterizan por sangrado retardado, interno, profundo (articular o muscular) pero también se pueden ver equimosis, en este caso extensas. Un sangrado espontáneo de comienzo abrupto puede ser el signo inicial de la presencia de un inhibidor adquirido, en especial cuando se trata de alguien sin historia de sangrado a pesar de desafíos hemostáticos. Los sangrados más diferidos se pueden deber a una prematura caída de la escara de cicatrización (como por ej. en tonsilectomía). También se ve sangrado alejado varios días cuando hay defectos en la polimerización de fibrina (disfibrinogenemias, deficiencia de FXIII) y en casos de fibrinólisis excesiva.La presencia de enfermedad hepática o renal o de otra enfermedad concomitante podría explicar un trastorno adquirido. Es importante pesquisar el consumo de ciertos productos naturales (ajo, vitamina E, aceites de pescado, ginkgo), el consumo de fármacos que interfieran con la hemostasia y la función plaquetaria, así como los nuevos anticoagulantes que pudieron pasar inadvertidos en el laboratorio pre-quirúrgico. Finalmente, investigar signos y síntomas de sangrado en familiares de primer grado. Es importante recordar que la falta de antecedentes familiares no descarta un problema hereditario. Hay trastornos recesivos sin expresión clínica en los padres heterocigotas. Otros trastornos como la VWD, se caracterizan por una penetrancia clínica variable que explica la existencia de familiares a/oligosintomáticos. Además, ciertas coagulopatías leves sólo se manifiestan después del primer desafío importante de la hemostasia o trauma.Por lo antedicho, en ocasiones y en especial en las coagulopatías leves o en adolescentes que no han tenido aún desafíos hemostáticos, la historia de sangrado puede ser negativa.El examen físico estará centrado en las manifestaciones de sangrado así como en la presencia de estigmas sugestivos de enfermedades sistémicas que pudieran acompañarse de alteraciones de la coagulación. Por ej, la esplenomegalia y circulación colateral abdominal como indicadores de la presencia de hepatopatía. Una historia de falla renal, un hábito marfanoide o la presencia de albinismo parcial o total suelen asociarse con defectos de la coagulación y del número/función plaquetarios. Es clásica la secuela

239
Sangrado quirúrgico | 13
articular como característica de la hemofilia. Menos recordada, pero no menos significativa, es la presencia de hiperlaxitud y de cicatrices anormales características del síndrome de Ehlers-Danlos. También es posible observar signos de enfermedad hepática, como esplenomegalia y presencia de circulación colateral en pared abdominal. La presencia de anormalidades morfológicas (faciales u otras) y las anomalías cardiovasculares congénitas permiten sospechar la presencia de coagulopatía como acompañante de un síndrome genético (Noonan).La semiología del sangrado incluirá la identificación de sangrado cutáneo-mucoso como en petequias y equimosis, típicos de trastornos plaquetarios o vasculares. Los hematomas y las sufusiones hemorrágicas extensas orientan a defectos de la cascada. La inspección de la zona quirúrgica puede demostrar un sangrado pulsátil o en napa, orientando a origen arterial o venoso capilar del sangrado. El sangrado en zona quirúrgica acompañado de otros sitios orientará a trastornos sistémicos como una coagulación intravascular diseminada (CID).
Estudios de laboratorio (Tabla 3)
No se recomienda realizar estudios de screening pre-operatorios en pacientes sin antecedentes de sangrado que tuvieron desafíos hemostáticos.En aquellos pacientes que no tuvieron desafíos hemostáticos, o que han desarrollado patologías potencialmente asociadas con sangrado, así como pacientes con historia de sangrado, se comienza con un estudio basal que incluya hemograma, plaquetas, tiempo de protrombina (TP) y tiempo de tromboplastina parcial activada (TTPA), pudiéndose incluir también un tiempo de trombina (TT). En cuanto al dosaje de fibrinógeno, valores por debajo de 150-200 mg/dL anticipan el riesgo de sangrado en cirugía cardiovascular (CCV) y en obstetricia.
Manejo
Indicación de re-exploración y/o medidas de control local de la hemostasia y/o soporte con hemoderivados, ambas conductas se pueden

240
13 | Sangrado quirúrgico
tomar simultáneamente. Las características clínicas que favorecen pensar en sangrado de causa local comparado con trastornos de la hemostasia se describen en la Tabla 2.Causas de hemorragia. El segundo paso será investigar la causa del sangrado para tomar la conducta terapéutica definitiva, el análisis del tiempo evolutivo podrá ser clave, según se describe en la Tabla 4.
Enfoque terapéutico. El enfoque terapéutico inicial se tomará analizando las posibilidades etiológicas descriptas previamente, a modo de orientación y en la urgencia se sugieren las medidas descriptas en la Tabla 5. En el diagrama de flujo (Figura 1) se resumen las conductas diagnósticas y terapéuticas, remarcándose nuevamente que estos pasos secuenciales posiblemente se tengan que tomar en forma simultánea y en discusión con el equipo quirúrgico y anestesista.
Sangrado intraoperatorio
Problemas estructurales o defectos técnicos
CID
Sobredosis de heparina
Hiperfibrinolisis
Sangrado temprano día 0 a 2
Problemas estructurales o defectos técnicos
Trombocitopenia
Defectos de función plaquetaria
Trastornos hereditarios leves a moderados
Sangrado retrasado día 2 a 7
Trombocitopenia
AAS o AINES
Deficiencia de vitamina K
Síndrome falla multiorgánica
Anticuerpos anti-FV post-uso de trombina bovina
Sangrados más diferidos: caída de escara, defectos en la polimerización de fibrina, deficiencia FXIII
Causas frecuentes de hemorragia , diagnóstico clínico según su presentación
CID: coagulación intravascular diseminada; AAS: ácido acetilsalicílico; AINEs: antiinfalmatorios no esteroides.
Tabla 4. Causas de sangrado post-operatorio (SPO)

241
Sangrado quirúrgico | 13
Manejo del sangrado en cirugía cardiovascular (CCV)
El sangrado excesivo constituye una de las principales complicaciones de la cirugía cardiovascular (CCV) y eleva la morbimortalidad. Las causas de sangrado excesivo luego de CCV se resumen en la Tabla 6 y el porcentaje de re-operaciones por sangrado oscila entre el 2 y el 6%. Existen diferentes formas de cuantificar el sangrado (Tabla 7). Se acepta que un adulto pueda
AcTx: ácido tranexámico; CCV: cirugía cardiovascular; Crio: crioprecipitados; DDAVP: desmopresina; PFC: plasma fresco congelado; rFVIIa: factor VII activo recombinante; TEG: tromboelastografía; VWD: enfermedad de von Willebrand.
Reposición de volumen con adecuadas concentraciones de glóbulos rojos y plaquetas
Angiografía y eventual embolización
Re-exploración del sitio de sangrado
Uso juicioso de PFC, concentrados de factores, Crio y plaquetas, según determinaciones de laboratorio que confirmen el factor deficiente
Cc. plaquetarios para clopidogrel y nuevos antiplaquetarios
DDAVP (VWD, ciertas trombocitopatías congénitas o adquiridas, aspirina, uremia)
AcTx en CCV, cirugía ginecológica, sangrado obstétrico, sangrado en trauma y cirugía del trauma o evidencia de hiperfibrinolisis en TEG
rFVIIa en casos seleccionados, con sangrado a pesar de otras medidas hemostáticas (previamente, asegurar un mordiente adecuado)
Tabla 5. Opciones terapéuticas en sangrado post-operatorio (SPO)
AcTx: ácido tranexámico; Crio: crioprecipitados; PFC: plasma fresco congelado; POC: point of care; rFVIIa: factor VII activo recombinante; TP: tiempo de protrombina.
Figura 1.

242
13 | Sangrado quirúrgico
perder hasta 400 mL de sangre en las primeras 6 hs del post-operatorio.Hay diferentes escalas predictoras de sangrado, que toman en cuenta factores como hematocrito, recuento de plaquetas, género, edad, índice de masa corporal, ingesta de anticoagulantes orales, antiagregantes, etc. La mayoría de scores son similares ya que consideran variables semejantes.La CCV utiliza aproximadamente el 10% de los hemoderivados del banco de sangre. El número de transfusiones requeridas es variable y directamente proporcional a la experiencia del centro.Si bien no se dispone de consensos internacionales a seguir para manejo del sangrado en CCV, en los últimos 25 años se ha podido comprobar que los protocolos para transfusión basados en algoritmos resultan superiores a las decisiones individuales. Existen además una serie de productos de aplicación tópica que combinados con los que se utilizan por vía intravenosa son útiles para reducir el sangrado de la microvasculatura.
Causas quirúrgicas (50% de casos): identificadas durante la reoperación
Por coagulopatía
Coagulopatía pre-existente
Sangrado residual de la pared torácicaSangrado en líneas de suturaDehiscencia del graft, etc.
Tiempo de bomba prolongadoMayor grado o duración de hipotermiaEdad avanzadaDeterioro de la función renalReducido BMICirugías de emergenciaAntecedente de cirugía torácica previaEndocarditis infecciosaCirugía de aorta
No diagnosticada o no tratada
Tabla 6. Causas de sangrado excesivo en cirugía cardiovascular (CCV).
Según débito por drenajes
Según peso corporal
>400 mL en primeras 2 hs del POP >500 mL en la primera hora del POP>300 mL/h durante las 3 primeras hs del POP
>4 mL/Kg/h en 1 h>2 mL/Kg/h en 2 hs consecutivas>5 mL/Kg/h en las primeras 4 hs
Tabla 7. Cuantificación del sangrado excesivo en cirugía cardiovascular (CCV)

243
Sangrado quirúrgico | 13
Fisiopatología del sangrado por coagulopatía
Durante la CCV aparecen diferentes situaciones que favorecen el sangrado: el paciente es hemodiluído al ser conectado a la bomba de circulación extracorpórea (CEC); recibe una dosis importante de heparina (300 UI/Kg); la sangre entra en contacto con los tubos de plástico del circuito extracorpóreo causando activación de la fase contacto en grado variable, al igual que la circulación a través de la membrana semipermeable del oxigenador para el intercambio gaseoso, antes de retornar al sistema arterial del paciente. La CEC puede darse mediante bomba roller o centrífuga. Esta última causa mayor grado de activación plaquetaria y eventual formación de coágulos. A pesar de las altas dosis de heparina que se utilizan, las plaquetas resultan activadas, se consumen, y pierden sus gránulos alfa por haber entrado en contacto con material exógeno y por las condiciones del flujo a lo largo del circuito extracorpóreo. Luego de la inmediata reversión de la heparinización con protamina a la salida de bomba, a veces puede ocurrir un rebote de heparina en el paciente, a punto de partida de sitios de depósito de heparina (especialmente en obesos o cuando fueron utilizadas dosis de heparina excesivamente altas). El rebote se caracteriza por un aumento en la concentración de heparina libre, ocurrida 1 a 5 hs luego de la neutralización con protamina. Esto puede darse por la cinética diferente de ambas drogas: la heparina sufre una eliminación lenta mientras que la protamina es rápidamente degradada por proteasas. Así es que pueden resurgir pequeñas cantidades de heparina en la circulación evidenciables por prolongación del TTPA y TT. No obstante, en la literatura no existe evidencia convincente que relacione a estas pequeñas cantidades de heparina remanente con sangrado excesivo.La activación de la fase contacto desencadena además la activación de la vía alternativa del sistema complemento, la producción de interlequina (IL) 6 y la activación de leucocitos, generando una respuesta inflamatoria prolongada.Concomitantemente con la activación de la cascada de la coagulación se activa la fibrinólisis: la formación de trombina y los productos de degradación de la misma activan las células endoteliales que liberan activador tisular del plaminógeno (tPA) permitiendo mayor pasaje de plasminógeno a plasmina.

244
13 | Sangrado quirúrgico
Utilidad de la Historia Clínica (anamnesis)
El interrogatorio del paciente que será sometido a una CCV es esencial para recabar antecedentes personales y familiares que pudieran relacionarse con diátesis hemorrágica. Las pruebas de coagulación preoperatorias resultan insuficientes para identificar pacientes con alto riesgo de sangrado intraoperatorio. El principal factor predictor de sangrado en un paciente es haberlo desarrollado previamente. Por eso deberá investigarse si hubo sangrado con cirugías anteriores o extracciones dentarias, aparición de hematomas ante mínimos traumatismos, enfermedades asociadas (por ej. insuficiencia renal o hepática, anemia) o ingesta de medicamentos, ya que muchos de estos no afectan el coagulograma pre-quirúrgico. El interrogatorio pre-quirúrgico es fundamental ya que permite predecir qué pacientes se encontrarán en riesgo aumentado de sangrado durante y post-cirugía, tales como pacientes añosos, re-operaciones, pacientes con deterioro severo de la función ventricular, con endocarditis, urémicos, hepatópatas, etc. En estos casos lo recomendable es contactar al anestesiólogo hemoterapeuta y cirujano previo al acto quirúrgico, para adoptar medidas conjuntas tales como adecuado soporte transfusional, uso de recuperador celular, concen-trados de factores, desmopresina (DDAVP) y medidas hemostáticas locales.La decisión acerca de interrumpir el tratamiento antitrombótico que el paciente venía recibiendo, o la de administrar terapia puente para la CCV, requiere de una cuidadosa evaluación del riesgo/beneficio en cada caso particular. En caso de decidir suspender antes de la cirugía la terapia antiagregante que el paciente venía recibiendo, se deberá suspender la aspirina 7-8 días previos y 5 días antes los nuevos antiagregantes, pudiéndose reanudar la aspirina a las 6-8 hs del post-operatorio y a las 24-48 hs el resto de los antiagregantes, si no hubiere sangrado excesivo post-quirúrgico.
Rol de los estudios de laboratorio
Si bien las pruebas de laboratorio pueden tener excelente control de calidad, no están siempre disponibles en 15 a 20 min. Tampoco se dispone en la actualidad de métodos de laboratorio certeros y rápidos que detecten una hiperfibrinolisis.

245
Sangrado quirúrgico | 13
Por eso hay mayor tendencia actual hacia el uso de estudios basados en dispositivos point-of-care, tales como la tromboelastometría (ROTEM), y trom-boelastografía (TEG), que realizan pruebas de coagulación viscoelástica en sangre entera, si bien aún no se dispone de clara evidencia a favor de su uso.El National Institute of Health and Care Excellence (NICE) concluyó que estas pruebas mejoran el uso de hemocomponentes y concentrados de factores, reducen el número de transfusiones y son costo-efectivos durante el período intra y post-operatorio, pero no hay variaciones significativas en cuanto a mortalidad, número de re-esternotomías, o estadía en unidad de cuidados intensivos u hospitalaria, con su utilización.
Terapia hemostática de soporte
La integración de datos de laboratorio en algoritmos terapéuticos que combinen terapia medicamentosa y hemocomponentes es la clave para un adecuado manejo del sangrado excesivo en CCV (Figura 1). El soporte con hemocomponentes o fármacos básicamente dependerá de dos variables: del sangrado excesivo del paciente y de la anormalidad en las pruebas de laboratorio.Es esperable que el paciente que sale de bomba tenga una caída del orden del 50% en el recuento plaquetario y concentración de protrombina respecto de los valores pre-quirúrgicos, que se van normalizando dentro de las 6-12 hs posteriores a la salida de quirófano. Estos valores no ameritan tratamiento sustitutivo si el paciente está con parámetros hemodinámicos estables y sin evidencias de sangrado aumentado. La causa no quirúrgica más frecuente de sangrado en el paciente que sale de bomba de CEC es la hemodilución. Un paciente que sale de quirófano con débito aumentado por drenajes e inestabilidad hemodinámica obliga de inmediato a solicitar al banco de sangre plaquetas y plasma fresco congelado (PFC), si bien los datos de laboratorio o de los dispositivos point-of-care rápidamente orientarán hacia la coagulopatía (o no) que domina el cuadro.A pesar del uso difundido del PFC, hay limitada evidencia sobre su beneficio en estudios retrospectivos sobre transfusión masiva. Usualmente se utilizan en dosis de 15 mL/Kg. Se asume que 1 mL de plasma aporta 1mL de factores. En pacientes sometidos a CCV se deberá tener en cuenta la hemodilución y la potencial sobrecarga de volumen en aquéllos con mala función ventricular.

246
13 | Sangrado quirúrgico
Si bien los concentrados de complejo protrombínico (CCP) resultan útiles para aporte de estos factores cualquiera sea la causa del déficit, están universalmente aprobados para la reversión del efecto de dicumarínicos. Hay escasa evidencia que compare tratamiento con PFC vs. CCP para el tratamiento del sangrado post-operatorio en CCV. El principal beneficio de estos últimos consiste en su rápido efecto y el poco aporte de volumen. Las complicaciones tromboembólicas son del orden del 0,7 a 2% de los casos.No existe consenso en el valor límite de plaquetas por debajo del cual transfundir. El recuento puede ser inferior debido a la agregación de plaquetas si el paciente está hipotérmico o la muestra fue tomada durante o inmediatamente luego de pasaje por bomba de CEC. Además, se deberá considerar la disfunción de las mismas luego de su pasaje por la bomba de CEC, y las potenciales causas pre-quirúrgicas de disfunción tales como antiagregantes, uremia, etc.Los niveles bajos de fibrinógeno pueden ser corregidos utilizando crioprecipitados (Crio) o concentrados de fibrinógeno (obtenidos por pasteurización de plasma humano), Estos últimos contienen de 900 a 1300 mg de fibrinógeno por vial y su eficacia, en casos de hipofibrinogenemia, es superior a la de los Crio (hemocomponente obtenido por descongelamiento controlado del PFC para que precipiten proteínas de alto peso molecular, tales como fibrinógeno, FVIII, FXIII, factor von Willebrand (VWF) y fibronectina). Algunos estudios pequeños han sugerido que la utilización de los primeros reduce el número de transfusiones y de re-esternotomías. Las guías del NICE no hacen referencia a los Crio y directamente recomiendan el uso de concentrados de fibrinógeno, a pesar de no tener la licencia en el Reino Unido para el tratamiento de la coagulopatía adquirida con estos concentrados.El riesgo de transmisión de patógenos o de reacciones inmunológicas es muy bajo debido a los procesos de purificación e inactivación. Los riesgos de complicaciones tromboembólicas son infrecuentes (3,48 eventos cada 100.000 tratados). Se deberá descartar CID antes de su utilización.No hay consenso aún acerca del uso de pruebas para evaluar clínicamente la hiperfibrinolisis. La sensibilidad de los dispositivos point-of-care es baja. Los ácidos tranexámico (AcTx) y ε-aminocaproico son análogos de lisina que bloquean reversiblemente el sitio de unión del plasminógeno inhibiendo la lisis de fibrina polimerizada. Tienen vida media plasmática de alrededor de 2 hs y se excretan por orina. El AcTx es utilizado en dosis de 10 mg/Kg en bolo, seguido por infusión continua de 1 mg/Kg/h ó 2g en bolo seguido por

247
Sangrado quirúrgico | 13
0,5 g/h. El principal efecto adverso descripto es que puede, en ocasiones, favorecer la aparición de convulsiones. No han sido publicados eventos trombóticos con su uso. El ácido ε-aminocaproico se utiliza de rutina en muchos centros, en dosis de 100 mg/Kg en infusión de 10 min luego de heparinizar al paciente. Igual dosis se agrega al priming de la bomba de CEC, y se mantiene un goteo de 100 mg/Kg en infusión continua hasta el cierre de la esternotomía. Ambos agentes han demostrado reducir la pérdida de sangre cuando se los utiliza en forma profiláctica en CCV, de acuerdo con la Society of Thoracic Surgeons Guideline.La DDAVP es un análogo de la vasopresina que provoca liberación de VWF almacenado en los cuerpos de Weibel-Palade de las células endoteliales, así como también de FVIII, prostaciclina y tPA. Su efecto es agotable y raramente se utiliza la droga más de una vez en 24 hs. Su estructura ligeramente modificada respecto de la vasopresina reduce los efectos vasoactivos (disminución de la diuresis, retención hidrosalina). Si bien ha sido demostrada su utilidad en sangrado por disfunción plaquetaria en urémicos y cirróticos, en CCV sólo 2 trabajos de número limitado de pacientes demostraron reducción del sangrado con su uso. Algunos otros lo asociaron con AcTx para mejorar su eficacia, debido a la liberación concomitante de tPA.El uso de FVII activo recombinante (rFVIIa) es considerado una alternativa off label como terapia de rescate en el paciente que persiste con sangrado. La mayor experiencia se llevó a cabo en cirugía cardíaca no coronaria. Se ha publicado reducción del débito por drenajes y del sangrado del campo operatorio, del requerimiento transfusional y de la tasa de re-operación con su uso. Sin embargo, la ausencia de estudios con adecuado diseño (prospectivos, randomizados, controlados) no permite recomendarlo como tratamiento de rescate de rutina en caso de sangrado excesivo en CCV. Sólo debería ser utilizado como última alternativa debido al exceso de eventos protrombóticos y la carencia de evidencia en cuanto a reducción de mortalidad global con su utilización. Un metaanálisis que incluyó 4.468 pacientes mostró que el rFVIIa eleva el riesgo de trombosis arterial (5,5% vs 3,3%) especialmente en ancianos. El momento más adecuado para su utilización es luego de excluida la presencia de heparina circulante y el sangrado quirúrgico, y habiendo corregido (parcialmente) la hemostasia, la hipotermia, la hipocalcemia y la acidosis del paciente. La dosis recomendada es de 30 a 60 µg/Kg (se sugiere no exceder los 90 µg/Kg). En caso de ser necesario se podría administrar una segunda dosis de rFVIIa.

248
13 | Sangrado quirúrgico
En resumen, el manejo del sangrado excesivo en CCV es multidisciplinario y debería ajustarse a un algoritmo institucional que contemple métodos diagnósticos, soporte transfusional y drogas, teniendo en cuenta el posible efecto residual de la heparina de la bomba, la hemodilución y los antecedentes del paciente.
Conflictos de interés
Los autores no presentan conflictos de interés.

249
Sangrado quirúrgico | 13
Referencias
Kozek-Langenecker SA, ArashAfshari A, Albaladejo P et al. Management of severe perioperative bleeding. Guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol 2013; 30: 270-382.
Practice guidelines for perioperative blood management: an updated report by the American Society of Anesthesiologists Task Force on Perioperative Blood Management. American Society of Anesthesiologists Task Force on Perioperative Blood Management. Anesthesiology 2015; 122: 241-275.
Hunt BJ, Allard S, Keeling D, Norfolk D, Stanworth SJ, Pendry K and on behalf of the British Committee for Standards in Haematology. A practical guideline for the haematological management of major haemorrhage. Br J Haematol 2015; 170: 788-803.
Schulman S. Prolonged bleeding after surgery. En: The Coagulation Consult A Case-Based Guide. Ed: A. Lichtin, J Bartholomew. 2014: 151-161.
Kitchens C, Lawson J. Surgery and hemostasis. En: Consultative Hemostasis and Thrombosis. Ed: CS Kitchens, CM Kessler, BA Konkle. 2013: 651-672.
Besser M, Ortmann E, Klein A. Haemostatic management of cardiac surgical haemorrhage. Anesthesia 2015; 70: 87-95.
Shore-Lesserson L, Manspeizer H, De Perio M, Francis S, Vela-Cantos F, Ergin M. Thromboelastography-guided transfusion algorithm reduces transfusions in complex cardiac surgery. Anesth Analg 1999; 88: 312-319.
GorlingerK, Shore-Lesserson L, Dirkmann D, Hanke A, Rahe-Meyer N, Tanaka K. Management of hemorrhage in cardiothoracic surgery. J Cardiothorac Vasc Anesth 2013; 4: S20-S34.
National Institute for Healthcare and Excellence (NICE). Health Technologies Adoption Programme. NICE Diagnostic Support for Viscoelastometric Point-of-Care testing. 2014. http://guidance.nice.org.uk/htdg13.

250
Weber C, Klages M, Zacharowski K. Perioperative coagulation management during cardiac surgery. Curr Opin Anesthesiol 2013; 26: 60-64.
Pombo G. Sangrado crítico en la cirugía cardiovascular. Medicina Intensiva/Hematología 2008; 25: 74-78.
13 | Sangrado quirúrgico

251
Sangrado uterino excesivo (SUE) en la mujer
Introducción
El Sangrado Uterino Excesivo (SUE) es un síntoma. Es el término para describir la perdida excesiva de sangre durante el periodo menstrual que interfiere con la vida física, psíquica, social y/o emocional de la mujer. Aproximadamente un 5-10% de las mujeres en edad reproductiva buscan atención médica por este síntoma. La Organización Mundial de la Salud calcula que cerca de 18 millones de mujeres en todo el mundo son afectadas. Es una causa común de deficiencia de hierro, que no solo afecta la salud de la mujer, sino que también incrementa el riesgo de complicaciones durante el embarazo y nacimiento. Es importante tener en claro la definición y a su vez diferenciarlo de otros sangrados uterinos también anormales, pero no relacionados con la perdida menstrual.
MenorragiaBeatriz E. Grand1 y Diana Altuna2.1 Doctor en Medicina y Docente Adscripto, Facultad de Medicina, UBA. Dpto. Materno Infantil División Obstetricia. Hospital Juan A. Fernández.2 Hematóloga y Oncóloga Pediatra. Servicio de Hematología y Oncología Pediátrica. Trasplante de Médula Ósea. Hospital Italiano de Buenos Aires.
14

252
14 | Menorragia
Definición
La terminología para describir el SUE ha cambiado y los términos descriptivos tradicionalmente utilizados para caracterizar los distintos patrones de sangrado como: menorragia; sangrado uterino disfuncional; metrorragia ya no se utilizan más. Han sido reemplazado por el termino descriptivo “Sangrado Uterino Anormal” o SUA (1).El SUA, al ser un síntoma, ha sido difícil de valorar y clasificar. Para abordar el tema la Federación Internacional de Ginecología y Obstetricia (FIGO), diseñó el sistema de clasificación PALM-COEIN para las causas del SUA en los años reproductivos (1). Es una clasificación descriptiva, según se trate de una causa estructural (PALM) y/o funcional (COEIN), como se muestra en la Tabla 1.
La clasificación FIGO se introdujo en 2011 y fue aceptada por el Colegio Americano de Ginecología y Obstetricia (ACOG), primero clasifica a los SUA en: Agudo; Crónico; e Intermenstrual. Luego introduce el término “Sangrado Uterino Excesivo”(SUE) en vez de menorragia/hipermenorrea y
Sangrado Uterino Anormal
Sangrado Uterino Excesivo (SUA-SUE)
Sangrado Intermenstrual (SUA-SIM)
PALM (causas estructurales) Pólipo (SUA-P)
Adenomiosis (SUA-A)
Adenomiosis (SUA-A)
Leiomiomas (SUA-L)
-Submucosos (SUA-LSM)
-Otros (SUA-LO)
Malignidad e hiperplasiaCOEIN (causas no estructurales)
Coagulopatía (SUA-C)
Ovulatoria-disfunción (SUA-O)
Endometrial (SUA-E)
Iatrogénica (SUA-I)
No clasificada (SUA-N)
Tabla 1. Clasificación FIGO para el Sangrado Uterino Anormal (SUA)

253
Menorragia | 14
el de “Sangrado InterMenstrual” (SIM) en lugar de metrorragia. Se expresa SUA seguido de la letra que representa la etiología de la misma. El término disfuncional se deja fuera de esta clasificación.Dado que este tema será abordado desde lo hematológico exclusivamente y no se desarrollarán las patologías ginecológicas que pueden causar sangrado, es importante tener en cuenta la clasificación completa porque pueden coexistir varios factores al mismo tiempo. El sistema de clasificación se construyó precisamente reconociendo que cualquier paciente podría tener una o varias afecciones que pueden causar o contribuir a las molestias del SUA. Hay afecciones como la adenomiosis, los leiomiomas y los pólipos endocervicales o endometriales que pueden ser asintomáticos, pero que, asociados con algún problema hematológico puede manifestarse el sangrado. Es aquí donde probablemente el ginecólogo considere que no se justifica el sangrado por la lesión y el hematólogo evaluará cuál es el mejor mecanismo para manejar el SUE.
Causas
En cerca del 50% de las mujeres no se encuentra una causa. Los trastornos hemorrágicos más importantes relacionados con un SUE son la enfermedad de von Willebrand (VWD), los defectos plaquetarios y los déficits hereditarios de factores (2). Hay scores clínicos desarrollados que incluyen síntomas de sangrado que permiten al ginecólogo determinar en qué sangrados debe explorarse en forma más exhaustiva un problema de coagulación y de esta forma derivarlo al hematólogo para que complete la evaluación correspondiente. Este grupo entraría dentro del sangrado uterino no estructural por coagulopatía: SUE-C.
Coagulopatias (Corresponden a la letra C de la clasificación)
El SUE es el síntoma más común que experimentan las mujeres con trastornos hematológicos. La información de alta calidad demuestra que aproximadamente el 13% de las mujeres con SUE tienen trastornos de la hemostasia confirmados luego por estudios de laboratorio. El más frecuente

254
14 | Menorragia
es la VWD. Con frecuencia, cerca del 90% de estas pacientes ya vienen a la consulta con este antecedente que fue detectado por el pediatra debido a la presencia de sangrado abundante desde la menarca (ver menorragia en pediatría/adolescencia). En caso de no tener este antecedente, un interrogatorio dirigido nos permitirá sospechar la presencia de un trastorno de la hemostasia. Estos últimos incluyen: hemorragia post-parto, sangrado relacionado con procedimientos invasivos y extracciones dentarias, equimosis y epistaxis a repetición, gingivorragias y antecedentes familiares de enfermedades hemorrágicas. Es interesante comentar el punto considerado como Iatrogénica (SUA-I), dado que la presencia de un trastorno de coagulación puede agravarlo. Esta categoría incluye el Sistema Intra Uterino liberador de levonorgestrel (SIU-LNG) el cual puede presentar sangrado en los primeros meses, si el mismo se prolonga más hay que pensar en algún problema hemorrágico asociado (3).
SUE y anticoagulación
El sangrado que se asocia a los anticoagulantes entra dentro de la categoría SUE-C y es un punto importante a considerar. Las mujeres pueden estar recibiendo los anticoagulantes clásicos antagonistas de la vitamina K (AVK) en forma crónica o bien los nuevos anticoagulantes orales (NAO). En particular merece mencionarse la reciente publicación de Martinelli y col. (4) y el comentario editorial de Shulman (5). En su trabajo, Martinelli evalúa el riesgo adicional de continuar terapia hormonal en mujeres menores de 60 años bajo tratamiento con heparina, AVK o con rivaroxaban por el antecedente de tromboembolismo venoso (TEV) que participaron de un ensayo clínico fase 3. Su trabajo nos muestra una protección contra el TEV recurrente en mujeres con tratamiento hormonal que continúan con anticoagulantes, pero a su vez confirma mayor sangrado uterino con los NAO. De Crem y col.(6) también detectaron un mayor SUA en mujeres medicadas con rivaroxaban en relación a los AVK. Estas pacientes pueden manejarse con ácido tranexámico (AcTx) o tratamiento hormonal y en caso de no controlarse pasar a AVK (6).

255
Menorragia | 14
Cuantificación y valoración clínica de la pérdida
El gráfico sobre valoración de la perdida hemorrágica (Pictorial Blood loss Assessment Chart, PBAC) es un método propuesto para valorar el SUE (Figura 1). Los números de la parte superior son los días de la semana. Se solicita a las mujeres que marquen el recuadro adecuado cada vez que desechan una toalla o un tampón; 1 punto por cada toalla o tampón levemente saturado; 5 por cada toalla o tampón moderadamente saturado; 10 y 20 puntos por cada toalla o tampón respectivamente, totalmente saturado. Coágulos: 1 punto por coágulos pequeños; 5 puntos por coágulos grandes. Se ha demostrado que un puntaje mayor a 100 por ciclo constituye un pronóstico de una pérdida menstrual de más de 80 mL. Algunos autores cuestionan su validez.
Figura 1.

256
14 | Menorragia
Herramientas terapéuticas
Dado que el sangrado anormal puede ser signo de un problema ginecológico y no secundario a un trastorno de coagulación, es necesaria una valoración completa por el ginecólogo antes de iniciar un tratamiento.
Tratamiento hemostático
Las terapias hemostáticas son eficaces para el control de la hemorragia. Estos agentes constituyen la principal opción de tratamiento para las mujeres que desean tener hijos, ver luego algoritmo (7, 8).AcTx. Lethaby y col. han hecho en 2000 una exhaustiva revisión Cochrane del tema. El AcTx está aprobado por la FDA para esta indicación desde 2009. Por vía oral la dosis es de 1g 3 a 4 veces al día. En general la tolerancia es buena. Sus efectos secundarios incluyen nauseas, cefalea y diarrea (9). Nivel de evidencia: Grado A, Nivel 1b; aprobado por la FDA. Más de un estudio randomizado.Desmopresina (DDAVP). Este tema fue tratado en el capítulo 3 (VWD). Puede darse por vía endovenosa (EV), subcutánea o intranasal en forma de aerosol (no disponible en nuestro país). Los efectos secundarios están relacionados con el efecto vasomotor e incluyen taquicardia, rubefacción y cefalea. Es indispensable la restricción hídrica para evitar hiponatremia e intoxicación hídrica por su efecto antidiurético. Nivel de evidencia: Grado B, Nivel IIb. Los estudios son de baja calidad y no mostraron mayor beneficio; el mejor ensayo fue contra AcTx, si bien las dos drogas redujeron el sangrado, el AcTx fue mejor.
Terapia de reemplazo
En algunas mujeres con deficiencias graves de factores que no responden a otros tratamientos es necesaria la terapia de reemplazo del factor para el control del SUE. Concentrados de Factor von Willebrand (CVWF). Este tema fue tratado en capítulo 3 (VWD). Con respecto a SUE contamos con una investigación reciente sobre una evaluación de uso de CVWF en este tema. Una encuesta en 83 centros brindó datos para el análisis de 1.321 mujeres con VWD vistas

257
Menorragia | 14
durante 2011-2014, 816 (61,8%) tenían SUE. Los tratamientos recibidos incluyeron anticonceptivos combinados, AcTx y DDAVP como primera línea. El uso de CVWF se encontraba como tercera línea de tratamiento reportándose su uso en 13 mujeres (1,6%). Estos datos sumados a 88 mujeres en seis publicaciones previas nos permiten decir que el uso de CVWF en SUE es efectivo en reducirlo y seguro. Este dato fue obtenido en un total de 101 mujeres tratadas con dosis entre 33-100 UI/Kg, del día 1-6 del ciclo menstrual (10). Se planea efectuar un estudio prospectivo para confirmar estos datos. Nivel de evidencia: Grado B, Nivel III. No hay ensayos randomizados; solo estudios observacionales y descriptivos.
Tratamiento Hormonal
Sistema IntraUterino con Levonorgestrel (SIU-LNG); Anticonceptivos hormonales combinados; Progestágenos orales; Anticonceptivos de pro-gestina; Análogos de la hormona liberadora de la gonadotrofina (GnRH). Manejo a cargo del ginecólogo.
Tratamiento Quirúrgico
Incluye la histerectomía y ablación de endometrio. Manejo a cargo del ginecólogo.
Manejo sugerido
Seguir texto con el algoritmo (Figura 2).El tema ha sido ampliamente revisado en numerosas publicaciones (8, 9). Con fines prácticos se presenta un algoritmo que permite una aproximación al manejo de la mujer con SUE. Para aquella mujer con SUE que tiene un desorden hemorrágico (3) el enfoque va a depender de la edad y también del desorden hemorrágico. La edad tiene importancia en la valoración de cuál es su deseo de tener hijos, si ya tiene hijos, si desea tener hijos o qué planes tiene en cuanto a una maternidad próxima. Si la mujer desea

258
14 | Menorragia
preservar la fertilidad porque busca embarazo en corto plazo las opciones son: AcTx, DDAVP y terapia de reemplazo con concentrados de factores. Si la mujer decide postergar la maternidad hay opciones como el uso del SIU-LNG o píldoras con progesterona. Todas estas indicaciones con seguimiento a cargo del ginecólogo. En caso de haber completado su planificación familiar o de haber decidido no tener más embarazos y de ser el sangrado excesivo puede usarse la endoablación o histerectomía.
Manejo psicológico a largo plazo
Más allá del manejo clínico del SUE, no debemos olvidarnos que la mujer con estos problemas debe enfrentar por un lado el manejo de los síntomas y por otro la integración a su actividad diaria. Los profesionales de la salud tenemos que hacernos cargo de este aspecto. El apoyo incluye el trabajo con todo el equipo de profesionales especialistas en el tema en conjunto con el paciente y sus familiares.
Figura 2.
AcTx: ácido tranexámico; DDAVP: desmopresina; DIU: dispositivo intrauterino; SUE: sangrado uterino excesivo.

259
Menorragia en adolescentes
Comentario: todas las recomendaciones de menorragia en adolescencia se basan en opinión de expertos con evidencia Grado C, Nivel IV (Tabla 2).
Introducción
El sangrado menstrual abundante (SMA) es uno de los mayores problemas en la transición de la niñez a la adolescencia. Las irregularidades del ciclo menstrual relacionadas con ciclos anovulatorios en los primeros años luego de la menarca son habituales. Alrededor del 12% de estas pacientes requerirá intervención médica y/o hospitalización. El SMA crónico se asocia a alta morbilidad ya que tiene implicancias en la calidad de vida, pérdida de actividades habituales, complicaciones médicas (deficiencia de hierro y anemia, internaciones, transfusiones) y aumentos en los costos de salud. Hay un sub-reporte de su prevalencia y representa 30% de las consultas ginecológicas de este grupo etario.
Tabla 2. Clasificación FIGO para el Sangrado Uterino Anormal (SUA)
A
B
C
IaIb
IIaIIbIII
IV
Meta-análisis de estudios controlados y randomizadosAl menos un estudio controlado y randomizado
Al menos un estudio bien diseñado sin randomizaciónAl menos un estudio de otro tipo bien diseñado, cuasi-experimentalEstudios descriptivos no experimentales, como estudios comparativos, de correlación y de caso-control
Reportes de comités u opinión de expertos y/o experiencias clínicas de autoridades respetadas
Grado Nivel Evidencia obtenida de
Menorragia | 14

260
14 | Menorragia
Definiciones
SMA. Cualquier alteración de la menstruación normal (volumen, frecuencia, duración).En 2006 el ACOG y la American Academy of Pediatrics presentaron un Consenso en el que se definieron las características de una menstruación normal (Tabla 3).
Menorragia/SMA. Es el sangrado menstrual prolongado (más de 7 días) o excesivo (más de 80 mL por ciclo). La diferencia entre estos términos es que SMA describe el sangrado menstrual excesivo sin tener en cuenta el patrón del ciclo menstrual. Menorragia se refiere a ciclos regulares. En 2007 el National Institute of Clinical Excellence (NICE) definió SMA como el sangrado menstrual excesivo que interfiere con la calidad de vida física, social, emocional o material de la mujer, sin ningún énfasis en otras características de la menstruación.Identificar menorragia en una adolescente puede ser un desafío. La definición clásica es difícil de aplicar. Los hallazgos predictivos de pérdida >80 mL/ciclo se muestran en la Tabla 4. El gráfico para valorar la perdida menstrual (PBAC, Figura 1) es un método semi-cuantitativo propuesto para valorar el SMA. Si bien un score >100 de PBAC se asocia con pérdida menstrual >80 mL/ciclo, esto no ha sido validado en población adolescente. Se lo utiliza para la evaluación de la pérdida menstrual junto con la descripción detallada de la misma.
Tabla 3. Menstruación normal
Comienzo: 11-14 añosIntervalo: 20-45 días
Duración: ≤7 díasCantidad: hasta 80 mL por ciclo
No más de 3-6 toallas o tampones por día

261
Menorragia | 14
Etiología (Tabla 5)
La causa mayoritaria de un SMA está en la inmadurez del eje hipotálamo-hipófisis-ovario que resulta en ciclos anovulatorios (más del 85% en el primer año desde la menarca). La segunda causa son las alteraciones de la hemostasia (AH) y un amplio rango de prevalencia (7-48%) ha sido reportado en esta población. Estas causas no son mutuamente excluyentes por lo que las pacientes que presentan inmadurez del eje y AH sangran más severamente. Al contrario que en la mujer adulta, la patología anatómica es muy poco frecuente. La Tabla 6 resume el impacto de las AH en el SMA.
Tabla 4. Hallazgos predicitivos de pérdida >80 mL por ciclo
Coágulos >2,5 cmCambio frecuente (cada hora) de toalla o tampón
Manchar la ropa o la camaFerritina baja
Duración >7 díasACOG 2006 los cambios de paño cada 1-2 hs (especialmente en el contexto de menstruación que dura >7 días) debe ser considerada menstruación excesiva
Se sugiere chequear a través de las características de la menstruación obtenidas por interrogatorio dirigido y el PBAC, que la paciente tiene menorragia antes de solicitar estudios de laboratorio. (Grado C, Nivel IV).
El papel del hematólogo ante el SMA de la adolescente será:1. Diagnosticar la presencia de AH y planear su manejo.2. En adolescentes donde la AH ya sea conocida, deberá elaborar el plan de acción para la menar-ca, así como educar a la paciente y sus padres.

262
14 | Menorragia
Historia clínica y examen físico
La historia familiar y personal de sangrado ante desafíos hemostáticos (epistaxis, gingivorragia, equimosis fáciles, sangrado prolongado luego de procedimientos, anemia sintomática, historia de sangrado que requirió internación o transfusión) debe hacer sospechar la presencia de una alteración de la hemostasia. Una historia clínica negativa no descarta la presencia de AH. En ocasiones, en especial en las coagulopatías leves y en
Alteraciones de la hemostasia en pacientes con menorragia
Metrorragia en adolescentes con alteraciones de la hemostasia
VWD
Alteración de la función plaquetaria
Trombocitopenia
Déficit de factores
VWD
Alteración de la función plaquetaria
-Síndrome de Bernard Soulier
-Tromboastenia de Glanzmann
Déficit de factores (FI, FXI o FXIII)
5-36%
2-44%
13-20%
8%
32-100%
5-98%
51%
98%
35-70%
Tabla 6. Prevalencia de menorragia y alteraciones de la hemostasia en adolescentes
VWD: enfermedad de von Willebrand.
Endocrinas
Ciclos anovulatorios
Enfermedades tiroideas
Tumores y pólipos
Miomas
Cáncer
Traumatismos
Alteraciones de la hemostasia
VWD
Alteraciones plaquetarias (cuanti y cualitativas)
Déficit de factores de la coagulación
Sexualmente activas
Embarazo, aborto
ETS (clamidias)
Medicación
Anticoagulantes
Tabla 5. Causas de SMA en adolescentes
ETS: enfermedades de transmisión sexual; VWD: enfermedad de von Willebrand.

263
Menorragia | 14
adolescentes que no han tenido desafíos hemostáticos, la historia clínica puede ser negativa y el SMA puede ser la primera manifestación.
El examen físico buscará la presencia de manifestaciones de sangrado, así como los estigmas sugestivos de enfermedades sistémicas que pudieran acompañarse de alteraciones de la coagulación. Es importante rescatar cuándo sangra, cómo sangra (inmediato, alejado, tardío) y por dónde sangra (piel y mucosas vs. sangrado muscular o articular). La presencia de estigmas de hepatopatía o circulación colateral, de falla renal, laxitud articular, un hábito marfanoide, albinismo parcial o total, así como las anomalías cardiovasculares congénitas pueden asociarse con defectos de la coagulación y del número o función plaquetarias.
Laboratorio de hemostasia
Las diferentes sociedades de Ginecología y Obstetricia (ACOG 2001 y 2006, SOGC 2005) consideran que el screening de coagulación es apropiado como parte del estudio inicial en mujeres menores de 18 años con SMA, aún sin haber realizado el examen pelviano.
La presencia de menorragia desde la menarca es un signo altamente sugerente de coagulopatíaLa investigación de AH en adolescentes con SMA deberá incluir la historia personal y familiar de signos y síntomas de sangrado (Grado C, Nivel IV)
Según la evidencia publicada se recomienda el estudio de laboratorio de hemostasia en todas las adolescentes que se presenten con SMA. Las niñas con historia familiar de AH deben ser estudiadas ANTES de la menarca (Grado C, Nivel III).

264
14 | Menorragia
No ha sido claramente definido el panel ideal para el screening que permita descartar la presencia de alteraciones de la hemostasia en adolescentes con menorragia. Además de la hemostasia, sugerimos realizar los estudios necesarios para descartar embarazo y enfermedades de transmisión sexual, así como descartar las alteraciones hormonales como causa del sangrado. Se recomienda realizar estudios del estado del hierro en todas las adolescentes en estudio por menorragia a hematología, debido a la alta frecuencia de ferropenia y anemia por déficit de hierro asociadas a SMA crónico y a su impacto en la calidad de vida.
La información publicada hasta el momento recomienda que el laboratorio incluya un hemograma completo, TP, TTPA, panel para VWD, estudios de función plaquetaria y dosaje de factores individuales (Grado C, Nivel IV).
Se recomienda realizar los estudios en dos etapas: un primer nivel de screeningy una segunda etapa orientada por los resultados obtenidos en la primera etapa (Grado C, Nivel IV, Tabla 7).

265
Menorragia | 14
Algunas consideraciones en relación con el diagnóstico de laboratorio
Evaluación para VWD (ver capítulo 3): recordar el alto grado de variabilidad y la influencia de otros factores. Repetir las pruebas más de una vez antes de dar diagnóstico definitivo. Un estudio realizado en adolescentes con metrorragia antes de iniciar anticonceptivos hormonales mostró que el screening para VWD resultaba costo/efectivo (relación precio/calidad de vida). Esto nos sugiere que es razonable estudiar VWD en todas las pacientes adolescentes con SMA (11).Rol del ciclo menstrual. Los valores más bajos de VWF se presentan en los días 1-4 del ciclo. En especial en re-estudios, casos dudosos o con valores límite, se recomienda tomar la muestra durante estos días.El efecto de anticonceptivos orales combinados (COC) sobre valores de VWF es discutido. Una revisión sistemática de la literatura sugiere que pueden realizarse con el paciente recibiendo COC. Sería entonces razonable realizar el estudio bajo COC y no deberían ser suspendidos sólo para realizarlo, especialmente si la paciente continúa con sangrado.
Primer nivel: pruebas de screening
Segundo nivel: pruebas específicas
Hemograma y recuento de plaquetas
Frotis de sangre periférica
TP
TTPA
TT
Evaluación para VWD (VWF:Ag; VWF:RCo; FVIII)
Fibrinógeno
Agregación y liberación plaquetarias
Otros estudios de evaluación plaquetaria: ME, GP de membrana
Dosaje de otros factores
FXIII
Estudios de (hiper)fibrinólisis (PDFs, DD, PAI-1, α2-AP)
Tabla 7. Estudios de laboratorio en adolescentes con SMA
VWD: enfermedad de von Willebrand.

266
14 | Menorragia
Evaluación de la función plaquetaria. Las alteraciones de la función plaquetaria continúan siendo difíciles de diagnosticar. Debido a la alta prevalencia descripta entre adolescentes con SMA, algunos autores sugieren realizar estudios de agregación y liberación plaquetaria en el primer nivel de estudios de laboratorio. Los estudios de screening como PFA-100® y tiempo de sangría han mostrado baja sensibilidad y especificidad (12). Basados en estos datos se recomienda que, si los estudios de agregación y liberacion están disponibles, se realicen junto a los estudios de primer nivel. En caso contrario dejar esta evaluación para un segundo momento. Recordar que se requiere un número mínimo de plaquetas y que la muestra debe ser reciente. Los antiplaquetarios como aspirina, clopidogrel y otros, así como los antiinflamatorios no esteroides (AINEs) deben ser suspendidos 7-10 días antes del estudio (13). Los anticonceptivos hormonales pueden afectar los resultados de agregación por lo que se sugiere realizar el estudio en la semana de placebo. También deberá estudiarse la función plaquetaria tempranamente en aquellas adolescentes que presenten alteraciones del número o tamaño de las plaquetas así como ciertos estigmas de organicidad (albinismo, ectopía lentis, hábito marfanoide, insuficiencia aórtica, cicatrización anormal).
Manejo
El mejor tratamiento no está definido y carecemos de evidencia clínica que guíe el manejo óptimo de la menorragia en adolescentes con AH. Si bien las recomendaciones son extrapoladas de aquellas para adultos, los mecanismos de acción de las terapias que han mostrado ser útiles en mujeres adultas son presumiblemente similares en las adolescentes. Esto enfatiza la necesidad de no dejar de utilizar en la adolescente una terapia probada en mujeres adultas sólo por su edad. Un punto relevante en este grupo etario es la necesidad ineludible de preservar la fertilidad, por lo que los tratamientos quirúrgicos son excepcionales (riesgo de vida).Se recomienda que el manejo sea individualizado, basado en la etiología más probable y adaptado a la gravedad del sangrado.Se recomienda un manejo multidisciplinario que involucre a ginecólogos y hematólogos. Todos los tratamientos médicos disponibles podrán ser considerados (con la posible excepción de los AINEs, utilizados ocasionalmente en la mujer adulta).

267
Menorragia | 14
Manejo Hemostático
Antifibrinolíticos. Su uso en el tratamiento de la menorragia en adolescentes no ha sido evaluado sistemáticamente. Los estudios publicados muestran que el AcTx es más potente y presenta menores efectos adversos que el ácido ε-aminocaproico. Estudios en adolescentes que lo comparan con DDAVP intranasal muestran que disminuye el sangrado menstrual más efectivamente. Otros estudios comparativos con anticonceptivos hormonales muestran que el AcTx es igual de efectivo en disminuir el sangrado menstrual medido por PBAC y en mejorar la calidad de vida (aunque no disminuye la duración ni regula los ciclos) y presenta con una mayor compliance (100% vs. 43%).• La dosis de AcTx es de 10 mg/Kg EV cada 8 hs en menorragia aguda
y de 20-25 mg/Kg cada 8-12 hs VO. Máximo 5 días en tratamiento de mantenimiento. Efectos adversos gastrointestinales (causa de abandono del tratamiento).
El riesgo de trombosis es incierto. En estudios realizados en adolescentes no se ha presentado esta complicación, aunque el número de publicaciones es escaso. Se recomienda no usar si hay historia de trombosis (extrapolación de datos en adultos).DDAVP. Es utilizada para prevención y tratamiento de sangrado en algunos pacientes con alteraciones leves de la hemostasia, especialmente en VWD tipo 1. Se recomienda realizar una prueba de respuesta que demuestre su eficacia previamente a cualquier uso clínico. En SMA ha mostrado ser efectivo en reducir los scores PBAC y mejorar la calidad de vida, pero no es tan efectivo como el AcTx. También ha demostrado ser efectivo en mujeres con tiempo de sangría prolongado sin una alteración de la hemostasia conocida.• La dosis para uso intranasal es 150-300 µg (no disponible en nuestro
país a la concentración adecuada). Para uso EV o SC: 0,3 µg/Kg/dosis cada 12 hs. Presenta taquifilaxia por lo que su uso está limitado a 2-3 días del ciclo. Efectos adversos: cefalea, flushing en la región esclavina, taquicardia, hiponatremia.
Concentrado de factores o hemocomponentes. El tratamiento hormonal y los antifibrinolíticos son la base de la terapia del SMA en pacientes con AH y son generalmente efectivos. En pacientes con alteraciones severas de la hemostasia puede ser necesaria la profilaxis regular con infusión de

268
14 | Menorragia
factores específicos para controlar un SMA severo y refractario a otros tratamientos, pero dependerá de la disponibilidad de los mismos. Estos pueden ser administrados durante la menstruación o durante todo el ciclo para aquellas pacientes que presenten sangrado prolongado/irregular o sangrado recurrente en el momento de la ovulación (profilaxis regular).El tratamiento hemostático (AcTx solo o combinado con DDAVP) es una buena opción para el tratamiento del SMA por lo que se recomienda considerarlo:• En combinación con terapia hormonal, durante los períodos o en el
sangrado por suspensión.• En adolescentes con falla en el manejo hormonal (evaluación
ginecológica).• En adolescentes muy jóvenes y en aquellas que no aceptan el
tratamiento hormonal.
Manejo hormonal
Se encuentra a cargo del ginecólogo infanto-juvenil y debe ser indicado y controlado por el mismo. Se dan algunos datos de utilidad para el manejo conjunto y el abordaje multidisciplinario de las pacientes. El tratamiento hormonal no afecta el crecimiento ya que el 95% del mismo se ha completado para el momento de la menarca y no afecta la fertilidad futura. Estos son dos puntos de conflicto con los padres ya que generan temor.COC. Es el método de elección en las adolescentes porque regulan el ciclo, disminuyen los síntomas asociados, proveen anticoncepción y, en pacientes con alteraciones de la hemostasia, previenen el sangrado de ovulación que es causa de abdomen agudo recurrente y de hemoperitoneo. El régimen extendido de 63-84 días es el preferido ya que brinda control del momento de menstruación y menor interferencia con su vida social. El sangrado de suspensión se maneja con el uso concomitante de AcTx con o sin DDAVP.Progesterona (Progesterona oral, medroxiprogesterona). Utilizado en pacientes con contraindicación de COC. También en adolescentes con historia de trombosis y/o trombofilia por su menor impacto en los niveles de FVIII y VWF.

269
Menorragia | 14
DIU con levonorgestrel. Su eficacia en el manejo de menorragia en pacientes con AH ha sido comprobada. Por lo general no es considerado en las adolescentes por falta de datos sobre aceptabilidad y seguridad. Los temores son especialmente evidentes en nulíparas y mujeres sin actividad sexual. Se describen enfermedad inflamatoria pelviana, dificultad en la inserción (útero 5-6 cm, anestesia), riesgo de expulsión, dolor/perforación. El Committee on Adolescent Health Care del ACOG declara en 2007 que los datos disponibles en la literatura apoyan el uso del DIU para la mayoría de las mujeres, incluyendo las adolescentes. Se ha publicado su uso exitoso en adolescentes con menorragia, incluidas niñas con retraso mental y problemas neurológicos.Agonistas GnRH. Su uso se ha asociado a hipoestrogenemia y disminución de la densidad ósea. Siendo la adolescencia un período crítico, se deberá controlar la densidad ósea en esta población. Utilizado con terapia hormonal (estrógenos, progesterona) ofrece una alternativa a pacientes con alteraciones severas de la hemostasia que no respondan a otras terapias (tromboastenia de Glanzmann o síndrome de Bernard-Soulier). Si se planea su uso más allá de los 6 meses, será necesaria la terapia hormonal add-back para minimizar el riesgo de osteopenia.
Manejo del sangrado menstrual abundante agudo (ver
algoritmo Figura 3)
Los objetivos de la terapia son:• Detener o disminuir el sangrado• Obtener estabilidad hemodinámicaEl tratamiento médico se basa en altas dosis de hormonas (estrógenos de elección) y/o agentes hemostáticos. El tratamiento hormonal, así como la necesidad de intervenciones quirúrgicas serán manejados por el ginecó-logo. El tratamiento antifibrinolítico es una opción no hormonal útil para el tratamiento de la hemorragia aguda. Recomendamos AcTx 10 mg/Kg cada 8 hs EV o 1-1,5 g VO cada 6 a 8 hs hasta que cese el sangrado. Los antifibrinolíticos pueden ser utilizados en el SMA Agudo como tratamiento único o como adyuvante de la terapia hormonal.En aquellas adolescentes con AH especificas, deberá administrarse un trata-miento dirigido y específico, bajo control del hematólogo. El mismo incluirá:

270
14 | Menorragia
• DDAVP• Hemocomponentes (plasma fresco congelado, crioprecipitados,
concentrados plaquetarios)• Concentrados de factores • rFVIIa30 µg/Kg/dosis, por 3 dosis cada 1-2 hs en déficit de FVII o
tromboastenia de Glanzmann. En otras situaciones, su uso es off- label
Conflictos de interés
Las autoras no presentan conflictos de interés.
Figura 3.
AcTx: ácido tranexámico; COC: anticonceptivos orales combinados; DDAVP: desmopresina; DIU: dispositivo intrauterino; EV: endovenosa.

271
Menorragia | 14
Referencias
1. Munro MG, Critchley HO, Fraser IS. The FIGO systems for nomenclature and classification of causes of abnormal uterine bleeding in the reproductive years: who needs them? Am J Obstet Gynecol 2012; 207: 259-265.
2. Peyvandi F, Garagiola I, Menegatti M. Gynecological and obstetrical manifestations of inherited bleeding disorders. J Thromb Haemost 2011; 9: 236-245.
3. Lethaby A, Hussain M, Rishwoth Jr. Progesterone or progestogen-releasing intrauterine systems for heavy menstrual bleeding (Review). Cochrane Database Syst Rev 2015; 4: CD002126.
4. Martinelli I, Lensing AW, Middeldorpet al. Recurrent venous thromboembolim and abnormal uterine bleeding with anticoagulant and hormone therapy use. Blood 2016; 127: 1417-1425.
5. Shulman S.Less menorrhagia for women with VTE. Comment on Martinelli et al. Blood 2016; 127: 1378-1379.
6. De Crem N, Peerlinck K, Vanassche T et al. Abnormal uterine bleeding in VTE patients treated with rivaroxaban compared to vitamin K antagonists. Throm Res; 136: 749-753.
7. Ray S, Ray A. Non-surgical interventions for treating heavy menstrual bleeding (menorrhagia) in women with bleeding disorders (Review) Cochrane Library 2014; 11: CD010338.
8. Management of acute abnormal uterine bleeding in nonpregnant reproductive-aged women. Committee Opinion No 557. American College of Obstetricians and Gynecologists 2013.
9. Perriera L, Martin J, Ahuja S. Reducing heavy menstrual bleeding: safety and efficacy of tranexamic acid. Womens Health 2012; 5: 17-22.

272
14 | Menorragia
10. Ragni MV, Machin LM, Malec LM et al. Von Willebrand factor for menorrhagia: A survey and literature review. Haemophilia 2016; 22: 397-402.
11. Sidonio R, Smith K, Ragni M. Cost-utility analysis of von Willebrand disease screening in adolescents with menorrhagia. J Pediatr 2010; 157: 456–460.
12. Philipp C S. Antifibrinolytics in women with menorrhagia. Thrombosis Research 2011; 127: S113–S115.
13. Sokkary N, Dietrich J. Management of heavy menstrual bleeding in adolescents. Curr Opin Obstet Gynecol 2012; 24: 275-280.

273
Definición y clasificación
La definición de hemorragia involucra diferenciar el volumen de sangre que está perdiendo la parturienta con respecto a un máximo normal estimado en 500 mL para un parto vaginal. Sin embargo, la cantidad de sangrado es difícil de cuantificar por la lentitud en que cae el hematocrito y la posibilidad de sangrado pelviano o perineal. Algunas guías que correlacionan la visualización de sangre en compresas o derramada en la cama de la paciente son útiles para evitar una infra-estimación de la pérdida, común en esta situación (Tabla 1). En todo caso, la evaluación de severidad debe tener en cuenta la volemia a término (100 mL/Kg) y la hemoglobina (Hb) previa de la paciente. Se califica como hemorragia primaria post-parto (HPPP) la superior a 500 mL, siendo severa si es mayor a 1.000 mL o con hipotensión y masiva si es mayor a 2.000 mL. Son parámetros alternativos para estimar la severidad, la velocidad de caída del hematocrito (Hto), la necesidad de transfusión, la rapidez con que se produce metrorragia de consideración y la presencia de
Hemorragia post-partoJorge D. Korin.Sanatorio de los Arcos. Consultor en Hematología.
En memoria de mi amigo Pablo Porterie (QEPD), más rápido que un ROTEM y mucho más bueno, con quien hemos pasado noches despiertos, asistiendo pacientes en quirófano con HPP.
15

274
taquicardia, hipotensión y taquipnea. La condición de primaria se refiere a la que ocurre dentro de las 24 hs. del parto y la secundaria, a aquélla que se presenta entre 24 hs. y 12 semanas del alumbramiento. La hemoragia post-parto (HPP) secundaria se asocia habitualmente con endometritis con o sin retención de restos de la concepción y más raramente con defectos congénitos de hemostasia como enfermedad de von Willebrand (VWD), déficit de FXI, ser portadora hemofílica, padecer trombocitopatías severas o trombocitopenia inmune (PTI), o haber desarrollado un inhibidor de FVIII post-parto. El resto de esta guía se ocupará exclusivamente de la HPPP. Desde el punto de vista obstétrico, la HPPP puede clasificarse como del tercer estadío (antes de expulsar la placenta) o del cuarto estadío (post-alumbramiento).
Epidemiología
La prevalencia global en el mundo va del 6 al 11% y depende de la calidad de atención médica, condiciones socio-económicas y la capacidad de medición de la pérdida. En África ocurre en el 10,5% de los partos, en Australia en 10,4%, en Asia en el 2,6%, en Latinoamérica y Caribe en el 8,9% y en Europa y EE.UU. en el 6,3%. Si se considera la forma severa, las cifras globales van del 1,9 al 2,8%. Aún en países desarrollados las cifras no muestran tendencia a disminuir. La hemorragia sigue siendo la primera o segunda causa de muerte materna en el mundo (Tabla 2).
15 | Hemorragia post-parto
Compresa de 10x10 cm totalmente saturadaCompresa de 30x30 cm totalmente saturadaCompresa de 45x45 cm totalmente saturada 1 Kg de compresas saturadasCharco en el piso de 50 cm de diámetroCharco en el piso de 75 cm de diámetroCharco en el piso de 100 cm de diámetro Hemorragia limitada a la cama Hemorragia que se derrama de la cama al piso
60 mL140 mL350 mL1000 mL500 mL1000 mL1500 mL<1000 mL>1000 mL
Material a observar Pérdida
Tabla 1. Correlación entre material con sangre inspeccionado y magnitud de la HPPP. Guía para una estimación visual de la magnitud del sangrado

275
Etiología
Como regla mnemotécnica se mencionan las 4 T. HPPP puede estar asociada con anomalías de la contracción uterina (Tono). La atonía es responsable del 80% de los casos. La segunda etiología es la lesión del canal del parto, generalmente de origen instrumental (Trauma: frecuencia 12%). La tercera causa es la retención de productos placentarios vinculables con placentas invasivas (Tejido: frecuencia 7%) y la cuarta está relacionada con alteración de la coagulación (Trombina: 1%) (Tabla 3).
Hemorragia post-parto | 15
Fuente: Khan KS et al, Lancet 2006; 367, 1066-74.
HemorragiaHipertensión
InfecciónAborto
Tromboembolia
13,4%16,1%2,1%8,2%
14,9%
20,8%
25,7%
7,7%
12%
0,6%
33,9%9,1%9,7%3,9%2,0%
30,8%9,1%
11,6%5,7%0,4%
Etiología Países desarrollados América Latina y CaribeAfrica Asia
Tabla 2. Hemorragia obstétrica como causa de muerte según desarrollo socio-económico. Análisis de la WHO de las causas de muerte materna
*75% de las mujeres con HPPP no tienen ningún factor de riesgo
Tabla 3. Factores de riesgo para hemorragia post-parto*
Edad mayor a 30 añosMinoridad socio-económicaEstadío 3 de parto prolongadoAntecedente de hemorragia post-partoPre-eclampsia o HELLPPlacenta previa o retención de cotiledonesDesprendimiento de placenta normoinsertaMultiparidadMacrosomía fetalPolihidramniosParto inducidoCesárea de emergenciaParto instrumentado (forceps, vacuum, episiotomía)Enfermedad de von WillebrandAnemia (Hb <9 g/dL) prepartoFiebre intrapartoObesidad maternaCardiopatía materna
TonoTonoTonoTonoTrombinaTejidoTrombinaTonoTono/TraumaTono-TraumaTraumaTrombina-TrombinaTono-

276
Se suele agregar una quinta T para Teatro (o escenario), enfatizando la importancia del manejo obstétrico de esta patología en ambiente quirúrgico.Debe enfatizarse que si bien la inmensa mayoría de las causas están vinculadas con la producción de hipovolemia que puede llegar al shock por insuficiente reposición o falta de reconocimiento de la magnitud de la gravedad de la situación, la embarazada es mucho más proclive que la mujer no gestante en desarrollar activación secundaria del sistema de coagulación. Superados los sistemas inhibidores, esto implica el desarrollo de coagulación intravascular localizada (uterina, pelviana) o diseminada (CID, en la microcirculación sistémica). Tres condiciones en que el sistema de coagulación puede activarse inmediatamente en el parto y que son las responsables de la aparición precoz del factor T (Trombina) son la embolia de líquido amniótico, el desprendimiento de placenta normo-inserta y la sepsis (rotura prematura de membranas). En los otros casos, si ocurre, la activación del sistema de coagulación es tardía y las alteraciones de la hemostasia iniciales corresponden a hemodilución por inadecuada política transfusional (Tabla 4).
Causa de HPPP
% que requiere transfusión de PFC
Momento del sangrado
Dilución Consumo local
CID
Atonía uterina
Trauma genital
Desprendimiento placentario
Placenta ácreta
Ruptura uterina
Embolia de líquido amniótico
Pre-eclampsia/ HELLP
14
4
42
8
66
100
Mínimo
Tardío*
Tardío
Muy temprano
Temprano o tardío
Temprano
Temprano
Temprano (frecuentemente
preparto)
En casos severos
En casos severos
En casos severos
Frecuente
Mecanismo principal por la magnitud del
sangrado
Contribuye en sangrados severos
Contribuye en sangrados severos
En casos severos
En casos severos
Principal en casos leves y moderados
Ocasional
Ocasional
No
Ocasional
Muy rara
Muy rara
En casos severos
Rara excepto con infección concomitante
Muy rara
Causa principal
Ocasional
Tabla 4. Mecanismos y tiempo de desarrollo de la coagulopatía según etiología de la HPPP
*Sangrado tardío se entiende aquél que se produce luego de pérdida >2.000 mL.
15 | Hemorragia post-parto

277
Reconocimiento de la mujer en riesgo
Si bien algunos de los factores de riesgo para HPPP pueden reconocerse durante el embarazo (edad materma superior a 30 años, obesidad, pertenencia a minoridad socio-económica, antecedente de HPPP en parto previo, trastorno hipertensivo durante el embarazo, multiparidad, coagulopatía previamente detectada, etc), el concepto relevante es que cualquier parto puede complicarse con sangrado que amenace la vida. Algunos de las causas más prevalentes de riesgo de HPPP por las que podríamos ser consultados previo al parto son: • Empleo de fármacos anticoagulantes.• Coagulopatía hereditaria (VWF, tromboastenia, mujer portadora o con
inhibidor de FVIII, FIX, FXI).• PTI.• Placenta previa (RR x13).• Acretismo detectado durante el embarazo.• Antecedentes de sangrado post-parto, cirugía uterina o cesárea y
desprendimiento placentario.La Tabla 5 señala los pasos a seguir en las mujeres con mayor riesgo de HPPP.
Modificado de McLintock C. Thromb Res 1989; 123, S30–34.
Buscar y corregir anemia pre-parto
Toda cirugía sea electiva o de urgencia debe ser realizada por el equipo de mayor experiencia de la institución, con participación multidisciplinaria según complejidad y respuesta a las maniobras terapéuticas realizadas (obstetra, anestesista, intensivista, médico transfusional, hematólogo, radiólogo intervencionista, cirujano vascular)
La paciente debe tener al menos dos accesos IV de amplio calibre (14 o 16 G)
Hemoterapia debe proveer inmediatamente 4 u de sangre compatibles
El laboratorio debe proveer resultados de coagulación inmediatos para guiar la reposición de productos sanguíneos, o la institución debe proveer aparatos de point of care de coagulación
En lo posible debe existir un protocolo multidisciplinario de hemorragia masiva del cual participe Hemoterapia
Considerar con el equipo obstétrico colocación de catéter balón previo al sangrado
Tabla 5. Pasos esenciales a seguir en la mujer en la cual, anteparto, se identificó alto riesgo de HPPP
Hemorragia post-parto | 15

278
Implicancias de la HPPP
Las consecuencias inmediatas de la complicación hemorrágica son la magnitud del sangrado, el requerimiento transfusional, la necesidad de internación en cuidados intensivos y la duración de la misma, la posibilidad de necrosis tubular aguda o necrosis cortical renal, el eventual desarrollo de distress respiratorio, la posibilidad de infarto hipofisario o suprarrenal, la anemia residual y el mayor riesgo de complicación tromboembólica venosa en el puerperio. Consecuencias a mediano y largo plazo son la posibilidad de preservación uterina, la fertilidad futura, el impacto psicológico, los efectos secundarios de los tratamientos instituidos para el soporte hemodinámico y la corrección de la causa obstétrica y finalmente la mortalidad. Esta oscila entre el 1-2% en los cuadros masivos.
Tratamiento de la HPPP
Para ser efectivo, el manejo debe ser multidisciplinario y a cargo de los profesionales de mayor experiencia en las áreas de obstetricia, medicina transfusional, hematología, radiología intervencionista y cirugía vascular y ginecológica. Es esencial el rápido reconocimiento de la emergencia, la comunicación de la misma y el inicio simultáneo de las medidas de resuscitación, monitoreo, sostén hemodinámico y tratamiento dirigido a la causa obstétrica. La Tabla 6 muestra los pasos iniciales a tomar en pacientes en quien se presume requerirán protocolos de transfusión masiva.
15 | Hemorragia post-parto

279
La Figura 1 muestra el plan general de acción por los diferentes actores involucrados. Como hematólogos, nuestra tarea consiste en:• Verificar si existe alteración de coagulación, plaquetas o fibrinolisis que
justifique el sangrado que presenta la paciente (identificar si el sangrado es por coagulopatía).
• En caso negativo, asegurar al obstetra que las médidas quirúrgicas que deba tomar no se acompañarán de mayor producción de sangrado por defecto hemostático.
• Consensuar un plan transfusional acorde a la situación hemodinámica de esa paciente y a su grado de tolerancia a la pérdida sanguínea persistente (anemia previa, cardiopatías, etcétera).
• Monitorear en forma seriada si, como consecuencia del plan anterior o de las medidas de expansión, no se produce deterioro en la hemostasia por hemodilución o se ha activado en forma indeseable el sistema de coagulación o la fibrinolisis secundaria.
Si se prevee probable imposibilidad de Hemoterapia para cumplir necesidades transfusionales (placenta ácreta detectada pre-parto, miomatosis uterina extensa, dificultades de cross-match, paciente testigo de Jehová, etc.), emplear cell saver.
Monitoreo invasivo de presión venosa central y arterial media.
Repetir frecuentemente hemograma, estado ácido-base, función renal, electrolitos, calcio iónico y parámetros de coagulación clásicos o point of care.
Calentar soluciones a infundir y O2 8 a 12 L a respirar.
Bajar la cabecera de la cama o camilla de quirófano.
Emplear bombas de infusión.
Mantener una línea directa de comunicación con Hemoterapia y Laboratorio.
Solicitar rápidamente a Farmacia los vasopresores y útero-tónicos que se van a emplear.
Reponer cloruro de calcio durante la poli-transfusión.
Colocar sonda Foley vesical para medir flujo urinario y descartar retención vesical.
Pedir cama en Terapia Intensiva y compartir con el médico intensivista el manejo hemodinámico para evitar sobre-intervención con coloides.
Documentación del aporte de cristaloides, productos sanguíneos, diuresis y procedimientos en quirófano, horarios y secuencias de administración y la evolución de los parámetros vitales registrados cada 15 min y en relación con los pasos terapéuticos.
Una HPPP es traumática para la mujer y su familia. Se debe proveer adecuada información y soporte psicológico por el médico que comandó el operativo.
Se recomienda seguimiento para detectar coagulopatías pasado el período agudo y profilaxis de tromboembolismo venoso una vez lograda la hemostasia. Detección de panhipopituitarismo post-parto.
Tabla 6. Sugerencias de manejo en HPPP con con necesidad de transfusión masiva
Hemorragia post-parto | 15

280
• Indicar ácido tranexámico (AcTx) 1 g EV si la reposición inicial es inefectiva para controlar el sangrado.
• Corregir post-parto la anemia residual, mediante aporte de hierro (Fe)EV o transfusión de glóbulos rojos (GR) leucodepletados.
• Logrado el cese del proceso hemorrágico y con niveles plaquetarios hemostáticos, iniciar profilaxis de tromboembolismo venoso por medios mecánicos o farmacológicos.
Las alteraciones de las pruebas de coagulación en embarazadas a término deben analizarse ya sea por métodos estándar de laboratorio o por tromboelastografía (TEG) o tromboelastometría (ROTEM) en los sitios con experiencia en su uso. Los métodos son complementarios y no excluyentes
Figura 1. Algoritmo de tratamiento de hemorragia post-parto (HPP)
15 | Hemorragia post-parto

281
y ambos tienen valores normales establecidos para una embarazada a término. Ambos sirven de guía para el tratamiento sustitutivo específico (Tablas 7 y 8). En uno u otro caso debe tenerse en cuenta que los cambios hemostáticos en el embarazo son el resultado del aumento de factores procoagulantes FI, V, VIII, X y XII, la disminución de Proteína S, aumento de la resistencia a Proteína C activada y la disminución de fibrinolisis por aumento de PAI-1 y 2 (mayor generación de trombina y menor disolución de trombos). Cuando ocurre en forma excepcional CID clásica en la embarazada, ésta se acompaña de respuesta inflamatoria, hipofibrinolisis, daño en la microcirculación y signos cutáneos de isquemia acompañando al sangrado por múltiples sitios. Esta es la presentación en embolia de líquido amniótico, sepsis en embarazadas a término o pre-eclampsias complicadas con activación del sistema de coagulación.
Procedimiento/ Parámetro
RCOG AAGBI Autores
Monitoreo primario
PFC empírico
PFC dirigido por Lab de coagulación
Fibrinógeno
Plaquetas
Acido tranexámico
rFVIIa
Lab de coagulación
1L cada 6u de GR o si HPPP >4.500mL
15 mL/Kg si TP/TTPA >1,5 del valor normal
Criopreciptados si <1 g/L o pérdida >4.500 mL sin lab.
Disponible
Transfundir con<50x109/L
No
En sangrado que amenaza la vida si fibrinógeno >1g/L y
plaquetas >20x109/L
Lab de coagulación
1L si se anticipa transfusión masiva
15 mL/Kg para prevenir que TP/TTPA lleguen a >1,5 del valor normal
Mayor volumen si TP/TTPA >1,5 del valor normal
Crioprecipitados o concentrados de fibrinógeno para mantener
Clauss >1,5g/L
Transfundir con <75x109/L
Si
Si fibrinógeno normal.No hay acuerdo de protocolo
FIBTEM + Lab de coagulación
Excepcionalmente
15 mL/Kg si FIBTEM <12 mm o TP/TTPA anormales
Mayor volumen si TP/TTPA >1,5 el valor normal
Concentrado de fibrinógeno para mantener FIBTEM
>11 mm
Transfundir con <75x109/L
Si
Uso excepcional.Con fibrinógeno >2 g/L y
plaquetas >50 x109/L
Tabla 7. Diversas sugerencias para tratamiento sustitutivo de la coagulopatía de la HPPP
Fuentes: Royal College of Obstetricians and Gynaecologists; Association of Anaesthetists of Great Britain and Ireland; R. E. Collis y P. W. Collins.
Hemorragia post-parto | 15
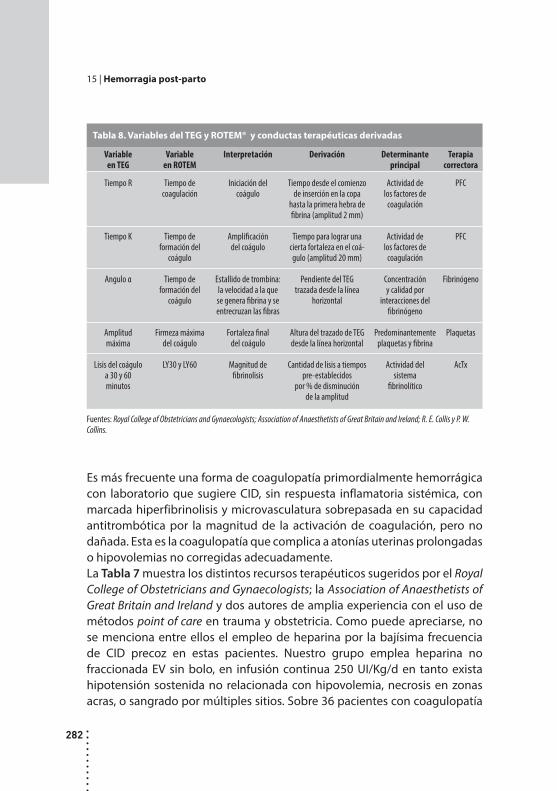
282
Es más frecuente una forma de coagulopatía primordialmente hemorrágica con laboratorio que sugiere CID, sin respuesta inflamatoria sistémica, con marcada hiperfibrinolisis y microvasculatura sobrepasada en su capacidad antitrombótica por la magnitud de la activación de coagulación, pero no dañada. Esta es la coagulopatía que complica a atonías uterinas prolongadas o hipovolemias no corregidas adecuadamente.La Tabla 7 muestra los distintos recursos terapéuticos sugeridos por el Royal College of Obstetricians and Gynaecologists; la Association of Anaesthetists of Great Britain and Ireland y dos autores de amplia experiencia con el uso de métodos point of care en trauma y obstetricia. Como puede apreciarse, no se menciona entre ellos el empleo de heparina por la bajísima frecuencia de CID precoz en estas pacientes. Nuestro grupo emplea heparina no fraccionada EV sin bolo, en infusión continua 250 UI/Kg/d en tanto exista hipotensión sostenida no relacionada con hipovolemia, necrosis en zonas acras, o sangrado por múltiples sitios. Sobre 36 pacientes con coagulopatía
Variable en TEG
Variable en ROTEM
Interpretación Derivación Determinante principal
Terapia correctora
Tiempo R
Tiempo K
Angulo α
Amplitud máxima
Lisis del coágulo a 30 y 60 minutos
Tiempo de coagulación
Tiempo de formación del
coágulo
Tiempo de formación del
coágulo
Firmeza máxima del coágulo
LY30 y LY60
Iniciación del coágulo
Amplificación del coágulo
Estallido de trombina: la velocidad a la que se genera fibrina y se entrecruzan las fibras
Fortaleza final del coágulo
Magnitud de fibrinolisis
Tiempo desde el comienzo de inserción en la copa
hasta la primera hebra de fibrina (amplitud 2 mm)
Tiempo para lograr una cierta fortaleza en el coá-gulo (amplitud 20 mm)
Pendiente del TEG trazada desde la línea
horizontal
Altura del trazado de TEG desde la línea horizontal
Cantidad de lisis a tiempos pre-establecidos
por % de disminución de la amplitud
Actividad de los factores de
coagulación
Actividad de los factores de
coagulación
Concentración y calidad por
interacciones del fibrinógeno
Predominantemente plaquetas y fibrina
Actividad del sistema
fibrinolítico
PFC
PFC
Fibrinógeno
Plaquetas
AcTx
Tabla 8. Variables del TEG y ROTEM® y conductas terapéuticas derivadas
Fuentes: Royal College of Obstetricians and Gynaecologists; Association of Anaesthetists of Great Britain and Ireland; R. E. Collis y P. W. Collins.
15 | Hemorragia post-parto

283
severa sólo empleamos heparina en 3, lo que demuestra que la mayoría de las pacientes no requieren más que reposición adecuada para corregir las alteraciones del laboratorio de coagulación y que rara vez hay signos de CID clásica.El otro punto a analizar es el empleo de AcTx en estas pacientes. Una revisión de Cochrane de 2010 concluye que el AcTx disminuye las pérdidas post-parto vaginal y cesárea sin HPPP. En esta entidad, se ha empleado una dosis de carga de 4g en 1 h, seguido por 1g/h durante 6 hs, demostrándose respecto al grupo control, menor duración de sangrado y menor requerimiento transfusional. El estudio WOMAN está en marcha, reclutando 20.000 mujeres con el objetivo de demostrar una reducción de 25% en mortalidad o necesidad de histerectomía mediante el empleo de 1g de AcTx EV, repetido a los 30 min si hay persistencia de hemorragia. Se esperan resultados para 2016/17.Como se menciona en la Tabla 7, se puede emplear rFVIIa intentando evitar histerectomía o en sangrado post-histerectomía que no responda a reposición, teniendo la precaución de asegurar niveles hemostáticos de fibrinógeno y plaquetas, como lo recomienda un Consenso Argentino sobre uso de rFVIIa y lo avala un trabajo randomizado recientemente publicado. En él se demuestra una reducción de necesidad de segundas líneas obstétricas luego de útero-tónicos en uno de cada 3 pacientes y se observa complicación trombótica en un 5%.El empleo de concentrados de fibrinógeno debe basarse en la presencia de sangrado y la demostración de un déficit del mismo. Su uso empírico no demostró reducción de requerimiento transfusional en un trabajo randomizado reciente en el que la media de concentración de fibrinógeno fue de 450 mg/dL.El correcto manejo de la causa obstétrica es fundamental para lograr el cese de la hemorragia. Con el fin de que el hematólogo esté familiarizado con los diversos procedimientos disponibles, se ha incluido la Tabla 9 con los resultados de los mismos.
Hemorragia post-parto | 15
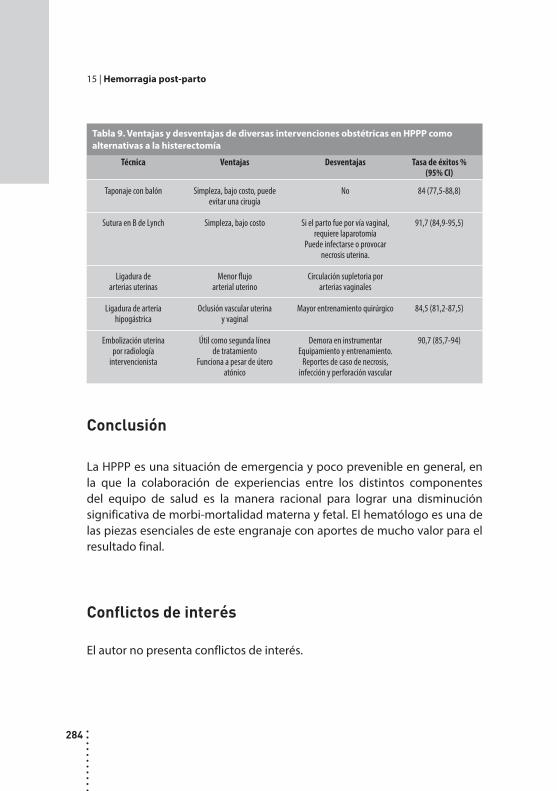
284
Conclusión
La HPPP es una situación de emergencia y poco prevenible en general, en la que la colaboración de experiencias entre los distintos componentes del equipo de salud es la manera racional para lograr una disminución significativa de morbi-mortalidad materna y fetal. El hematólogo es una de las piezas esenciales de este engranaje con aportes de mucho valor para el resultado final.
Conflictos de interés
El autor no presenta conflictos de interés.
Técnica Ventajas Desventajas Tasa de éxitos % (95% CI)
Taponaje con balón
Sutura en B de Lynch
Ligadura de arterias uterinas
Ligadura de arteria hipogástrica
Embolización uterina por radiología
intervencionista
Simpleza, bajo costo, puede evitar una cirugía
Simpleza, bajo costo
Menor flujo arterial uterino
Oclusión vascular uterina y vaginal
Útil como segunda línea de tratamiento
Funciona a pesar de útero atónico
No
Si el parto fue por vía vaginal, requiere laparotomía
Puede infectarse o provocar necrosis uterina.
Circulación supletoria por arterias vaginales
Mayor entrenamiento quirúrgico
Demora en instrumentarEquipamiento y entrenamiento.
Reportes de caso de necrosis, infección y perforación vascular
84 (77,5-88,8)
91,7 (84,9-95,5)
84,5 (81,2-87,5)
90,7 (85,7-94)
Tabla 9. Ventajas y desventajas de diversas intervenciones obstétricas en HPPP como alternativas a la histerectomía
15 | Hemorragia post-parto

285
Referencias
Collis RE, Collins PW. Haemostatic management of obstetric haemorrhage. Anaesthesia 2015; 70: 78-86.
Abdul-Kadir R, McLintock C, Ducloy AS et al. Evaluation and management of postpartum hemorrhage: consensus from an international expert panel. Transfusion 2014; 54: 1756-1768.
McLintock C, James AH. Obstetric Hemorrhage. J Thromb Haemost 2011; 9: 1441-1445.
Pavord S, Maybury H. How I treat post partum hemorrhage. Blood 2015; 125: 2759-2770.
WHO recommendations for the prevention and treatment of postpartum haemorrhage. 2012. Geneva: World Health Organization; 2012. ISBN-13: 978-92-4-154850-2 (NLM classification: WQ 330).
Allard S, Green L, Hunt BJ. How we manage the haematological aspects of major obstetric haemorrhage. Br J Haematol 2014; 164: 177-188.
Hill JS, Deveniet G, Powell M. Point-of-care testing of coagulation and fibrinolytic status during postpartum haemorrhage: deve-loping a thrombelastography®-guided transfusion algorithm. Anaesth Intensive Care 2012; 40: 1007-1015.
Bose P, Regan F, Paterson-Brown S. Improving the accuracy of estimated blood loss at obstetric haemorrhage using clinical reconstructions. BJOG 2006; 113: 919-924.
Hunt BJ. The current place of tranexamic acid in the management of bleeding. Anaesthesia 2015, 70: 50-53.
RCOG Green-top Guideline No. 52 May 2009; 1-24. © Royal College of Obstetricians and Gynaecologists.
Hemorragia post-parto | 15

286
Lavigne-Lissalde G, Aya AG, Mercier FJ et al. Recombinant human FVIIa for reducing the need for invasive second-lines therapies in severe refractory postpartum hemorrhage: a multicenter, randomized, open controlled trial. J Thromb Haemost 2015; 13: 520-529.
Wikkelso AJ, Edwards HM, Afshari A et al. Pre-emptive treatment with fibrinogen concentrate for postpartum haemorrhage: randomized controlled trial. Br J Anaesth 2015; 114: 623-633.
15 | Hemorragia post-parto

287
Manejo de las complicaciones hemorrágicas en pacientes con tratamiento antitrombótico
El aumento de la expectativa de vida, con el consiguiente crecimiento de la prevalencia de la fibrilación auricular y la introducción de nuevas drogas antitrombóticas, permite esperar un incremento continuo de pacientes que requieran terapia anticoagulante en los próximos años.Hoy sabemos que los pacientes anticoagulados con warfarina por cualquier motivo tienen un riesgo anual de sangrado mayor del 0,3 al 0,5% por año y de 0,2% por año de sangrado intracerebral. Hasta hace años la terapia anticoagulante oral estaba limitada a los dicumarínicos, conocidos también como antagonistas de la vitamina K (AVK). Hoy se han incorporado otras drogas, de inhibición directa a los factores de coagulación activados llamadas AODs, siendo DOACs la abreviatura aceptada de la nomenclatura en inglés (1).En nuestro país contamos con el inhibidor directo de trombina (FIIa),
16Complicaciones hemorrágicas del tratamiento antitrombóticoAlicia B. Vilaseca1 y Mirta Hepner2.1 Servicio de Hematología. Clínica San Camilo.2 Laboratorio de Hemostasia y Trombosis. Servicio de Hematología y Oncología. Hospital de Pediatría “Prof. Dr. Juan P. Garrahan”.

288
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
Dabigatran y con los inhibidores directos de FXa, Rivaroxaban y Apixaban. Estas drogas se caracterizan por un comienzo rápido de su acción, vida media corta, menor interacción con fármacos y alimentos, y una farmacocinética predecible, por lo cual no es necesario el monitoreo del efecto anticoagulante de rutina. Como desventaja, no existe un antídoto de uso clínico para su reversión en nuestro país hasta la fecha.
Recomendaciones para el manejo del sangrado en pacientes adultos bajo tratamiento antitrombótico
• Contar con protocolos o algoritmos de acción en todas las instituciones que manejen este tipo de pacientes (Grado 1A, Figura 1).
• Investigar alguna causa subyacente que podría determinar el sangrado (por ej. en un paciente con hematuria investigar la presencia de infección urinaria o pólipos vesicales, etc.) (recomendación de expertos).
• Hacer control mecánico del sangrado (Grado 1A). En sangrado localizado menor (por ej. taponaje, cauterización, etc.) no aplicar vasodilatadores como el agua oxigenada; recordar que el frío tiene efecto hemostático por vasoconstricción. Algunos sangrados como en nariz y cavidad bucal, donde la actividad fibrinolítica contribuye a una inadecuada hemostasia, el uso de ácido tranexámico (AcTx) 1g EV o por vía oral, continuando en caso necesario por 24 hs con 500 mg cada 8 hs EV u oral puede estar indicado. En muchos casos, estas medidas permiten no modificar el curso del tratamiento anticoagulante.
• Mantener las condiciones generales que garantizan una adecuada hemostasia: temperatura corporal del paciente cercana a 36°C (mantas térmicas, infusiones calientes, etc.), estabilidad hemodinámica, evitar la acidosis manteniendo el pH en 7,2, vigilancia estrecha del estado acido-base y la calcemia (mantener el nivel de calcio iónico alrededor de los 0,9 mmol/L) (Grado 1A) (2, 5). Administrar 1 g de AcTx dentro de las 3 hs de inicio del sangrado (Grado 1A) (2), luego continuar con las recomendaciones particulares que se detallan.

289
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
Figura 1.
AVK: antagonistas de vitamina K; HBPM: heparinas de bajo peso molecular; HNF: heparina no fraccionada; Inh: inhibidor; RIN: razón internacional normatizada; TTPA: tiempo de tromboplastina parcial activada.

290
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
Recomendaciones para situaciones de sangrado en adultos por AVK (Tabla 1)
Recomendaciones particulares para sangrado intracerebral en adultos por AVK
Cuando se sospecha sangrado intracerebral, es de buena práctica dejar suspendida la anticoagulación hasta la confirmación diagnóstica con tomo-grafía axial computarizada (TAC) cerebral (1).En sangrado intracraneal confirmado: revertir el efecto de los AVK teniendo en cuenta que la expansión del hematoma muchas veces puede ocurrir en las
RIN <5
RIN 5 a 9
RIN ≥9
RIN>4
Reducir dosis y monitoreo dentro de los 7 días. Si se identifica una causa de sangrado transitorio corregible podría no reducirse dosis (Grado 1C).
Discontinuar una o dos tomas, monitorear a las 48 hs y reiniciar con menor dosis cuando el RIN esté en valor terapéutico (Grado 1C).
Si el sangrado es de alto riesgo se sugiere interrumpir una dosis y administrar vitamina K oral controlando dentro de las 24 hs (1 a 2,5 mg)(Grado 2A).
Si se requiere reversión más rápida (por ej. cirugía) administrar vitamina K (≤5 mg EV) con la expectativa de reducción del RIN en 24 hs para warfarina y 4 a 8 hs para acenocumarol controlando en cada caso la respuesta. Si el RIN se mantiene todavía elevado se aconseja una dosis de vitamina K (1-2 mg) y nuevo
control con iguales intervalos (Grado 2C).
Suspender el anticoagulante transitoriamente hasta RIN terapéutico.Administrar una dosis más alta de vitamina K (2,5 a 5 mg EV) esperando reducción significativa del
RIN en 24 a 48 hs para warfarina y luego de 4 a 8 hs para acenocumarol (Grado 1B).Si el riesgo lo justifica puede considerarse una segunda dosis de vitamina K. Con el RIN adecuado reiniciar
el tratamiento con reducción de dosis y monitoreo dentro de los 5 a 7 días.En pacientes con RIN elevado sin sangrado mayor ni menor la administración de vitamina K
oral es buena alternativa.
Administrar una dosis elevada de vitamina K (5-10 mg EV) lenta, suplementada con CCP o PFC, en casos excepcionales si se agotaron las otras alternativas utilizar rFVIIa en dosisde 90 µg EV
(utilizar con precaución por su efecto protrombótico)(Grado 1C).Se recomienda la repetición de vitamina K cada 12 hs de continuar el RIN aumentado realizando monitoreos
de RIN. Las recomendaciones más recientes sugieren preferir el CCP al PFC en estos pacientes (1,2).
Sangrado menor por AVK
Sangrado mayor, no intracerebral y AVK
Tabla 1. Recomendaciones particulares para sangrado en adultos
AVK: antagonistas de la Vitamina K; CCP: concentrado de complejo protrombínico, EV: endovenosa; PFC: plasma fresco congelado; RIN: razón internacional normatizada.

291
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
primeras horas, pero el riesgo de sangrado se mantiene por lo menos hasta 72 hs después. Por este motivo debemos combinar la vitamina K 5 a 10 mg vía EV lenta combinado con concentrado de complejo protrombínico (CCP) si se cuenta con ellos, o en su defecto, con plasma fresco congelado (PFC). El factor VII activo recombinante (rFVIIa) es una segunda línea alternativa, pero con alto riesgo protrombótico (3). Se recomienda monitorear con RIN (razón internacional normatizada) cada 6 u 8 hs las primeras 24 y 48 hs hasta alcanzar un valor estable <1,4 y repetir vitamina K de ser necesario (Grado 1C).
Recomendaciones para el manejo de población pediátrica
Las recomendaciones particulares para el manejo de población pedíatrica con o sin sangrado en tratamiento con AVK (4) se detallan en la Tabla 2.
Sangrado por AODs recomendaciones particulares (4)
• Establecer la hora de la última dosis. Grado 1A.• Evaluar la función renal, en especial para dabigatran. Grado 1A y función
RIN 3-5sin sangrado
RIN 5-10sin sangrado
RIN ≥10sin sangrado
Sangrado mayor con cualquier RIN
Bajar u omitir una dosis, y controlar más frecuentemente; cuando el RIN se estabilice en valor terapéutico mantener con una dosis menor.
Omitir una o dos dosis, monitorear, cuando el RIN esté en valor terapéutico retomar el tratamiento a dosis menor.
Suspender la terapia con warfarina; administrar vitamina K 2,5 mg VO, esperando que el RIN tenga una reducción sustancial en 24 a 48 hs
con warfarina y en 6 a 8 hs con acenocumarol. Cuando el RIN vuelva a valores terapéuticos, reiniciar con dosis menor.
Suspender dicumarínicos; administrar vitamina K en infusión EV lenta (1-10 mg) y PFC o 4 CCP
Tabla 2. Recomendaciones para el manejo de población pediátrica con o sin sangrado en tratamiento con AVK
AVK: antagonistas de la Vitamina K; CCP: concentrado de complejo protrombínico; EV: endovenosa; PFC: plasma fresco congelado; RIN: razón internacional normatizada; VO: vía oral.
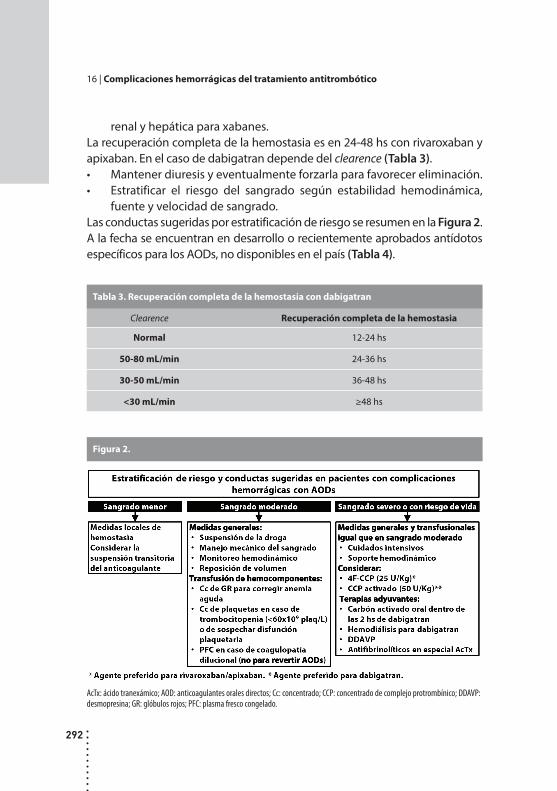
292
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
renal y hepática para xabanes.La recuperación completa de la hemostasia es en 24-48 hs con rivaroxaban y apixaban. En el caso de dabigatran depende del clearence (Tabla 3).• Mantener diuresis y eventualmente forzarla para favorecer eliminación.• Estratificar el riesgo del sangrado según estabilidad hemodinámica,
fuente y velocidad de sangrado.Las conductas sugeridas por estratificación de riesgo se resumen en la Figura 2.A la fecha se encuentran en desarrollo o recientemente aprobados antídotos específicos para los AODs, no disponibles en el país (Tabla 4).
Normal
50-80 mL/min
30-50 mL/min
<30 mL/min
12-24 hs
24-36 hs
36-48 hs
≥48 hs
Recuperación completa de la hemostasiaClearence
Tabla 3. Recuperación completa de la hemostasia con dabigatran
Figura 2.
AcTx: ácido tranexámico; AOD: anticoagulantes orales directos; Cc: concentrado; CCP: concentrado de complejo protrombínico; DDAVP: desmopresina; GR: glóbulos rojos; PFC: plasma fresco congelado.

293
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
Manejo del sangrado en pacientes adultos con heparina no fraccionada (HNF), heparina de bajo peso molecular (HBPM), pentasacáridos e Inhibidores directos de trombina parenteral (Bivalirudina)
En la Tabla 5 se detallan los mecanismos de acción y la farmacocinética de estos fármacos.
Aripazine (PER977)
Andexanet (PRT064445)
Idarucizumab o Praxbind
Molécula sintética pequeña
FXa recombinante modificado
Fragmento de anticuerpo humano
contra
Inhibidores orales directos de FXa
(apixaban, edoxaban, rivaroxaban)
Inhibidores directos de trombina (dabigatran)HNF y HBPM
Inhibidores orales de FXa HBPM y pentasacáridos
HNF y HBPM
No aprobado por FDA Probado en 80 voluntarios
sanos mostró reversión de efecto de xabanes y en
animales revirtió xabanes y dabigatran.
No aprobado por FDAEstudios en fase 2 con
voluntarios sanos mostraron corrección de la hemostasia y disminución del sangrado
por xabanes
Aprobado por FDA en octubre del 2015.
Revierte el dabigatran y normaliza la hemostasia en el 90% de pacientes
sometidos a procedimientos
Tipo de droga Objetivo EvidenciaNombre
Tabla 4. Antídotos en desarrollo para AODs
FDA: food and drugs administration; HBPM: heparinas de bajo peso molecular; HNF: heparina no fraccionada.

294
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
Recomendaciones generales
Las recomendaciones generales para el manejo del sangrado durante el tratamiento anticoagulante han sido detalladas al inicio del capítulo y se aplican a las drogas incluidas en la Tabla 5.
HNF
HBPM:Enoxaparina
HBPM:Nadroparine
Pentasacárido:Fondaparinux
Inhibidores directos de Trombina
parenteral:Bivalirudina
Se une a antitrombina potenciando el efecto inhibitorio a FXa y FIIa
Se une a antitrombina potenciando el efecto
inhibitorio a FXa (principalmente) y en
menor grado a FIIa
Se une a antitrombina potenciando el efecto
inhibitorio a FXa (principalmente) y en
menor grado a FIIa
Se une a antitrombina e inhibe el FXa
libre previniendo la formación del complejo
protrombinasa
Inhibición directa reversible de FIIa
incluyendo la adhesión y agregación plaquetaria mediada
por trombina
60-90 min
4-5 hs
3-5 hs
17-21 hsSe prolonga en ancianos y en pacientes con
insuficiencia renal
25 min con Cl normal34 min con Cl 30-5957 min con Cl 10-29
Renal
40% Renal
Renal
50-77% Renal
20% Renal
No
No
No
Si
Si
Mecanismo de acción Vida media Eliminación DializableDroga
Tabla 5. Mecanismos de acción y farmacocinética de las HNF y HBPM, pentasacáridos e inhibidores directos de trombina
Cl: Clearence; HBPM: heparinas de bajo peso molecular; HNF: heparina no fraccionada.

295
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
Recomendaciones particulares
• Para pacientes con sangrado menor o moderado deberá tratarse la causa del sangrado y valorar el riesgo/beneficio de discontinuar el tratamiento por unas horas.
• Para pacientes con sangrado mayor y/o intracerebral. Recomendación de buena práctica: suspender el tratamiento antitrombótico hasta superado el sangrado y riesgo de re-sangrado
• Las recomendaciones particulares para pacientes con sangrado mayor y/o intracerebral se detallan en la Tabla 6.
CCP: concentrado de complejo protrombínico; CCPa: concentrado de complejoprotrombínico activado; EV: endovenosa; HBPM: heparinas de bajo peso molecular; HNF: heparina no fraccionada; rFVIIa: factor VII activo recombinante.
HNF
HBPM:Enoxaparina
HBPM:Nadroparine
Pentasacárido:Fondaparinux
Inhibidores directos de Trombina parenteral:Bivalirudina
Administrar 100 mg de protamina por cada 100 U administradas en las 2-3 hs últimas (hasta 50 mg en una sola dosis) la administración es EV lenta por efecto hipotensor
Administrada en las 8 hs previas: 1 mg de protamina por cada mg de droga (hasta 50 mg de protamina como máximo)
Administrada entre las 8-12 hs previas: 0,5 mg de protamina por 1 mg de droga (hasta 50 mg de protamina como máximo)
Luego de las 12 hs la reversión no tendría utilidad.
Administrada entre 3-5 vidas medias antes utilizar 1 mg de protamina por cada 100 unidades anti-Xa (hasta 50 mg como máximo)
CCPa (FEIBA) 20 U/Kg EV o rFVIIa 90 µg/Kg EV
CCPa (FEIBA) 50 U/Kg o 4 CCP 50 U/kg EV
RecomendaciónDroga
Tabla 6. Recomendaciones para pacientes con sangrado mayor y/o intracerebral
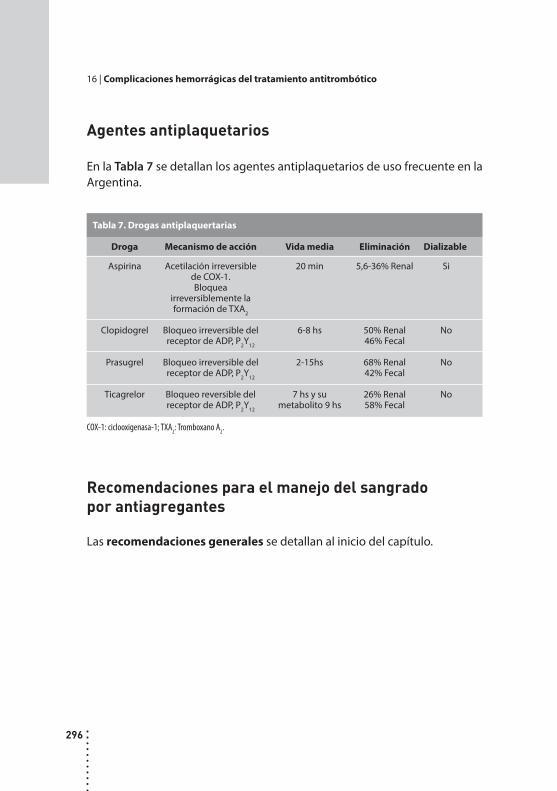
296
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
Agentes antiplaquetarios
En la Tabla 7 se detallan los agentes antiplaquetarios de uso frecuente en la Argentina.
Recomendaciones para el manejo del sangrado por antiagregantes
Las recomendaciones generales se detallan al inicio del capítulo.
Aspirina
Clopidogrel
Prasugrel
Ticagrelor
Acetilación irreversible de COX-1.Bloquea
irreversiblemente la formación de TXA2
Bloqueo irreversible del receptor de ADP, P2Y12
Bloqueo irreversible del receptor de ADP, P2Y12
Bloqueo reversible del receptor de ADP, P2Y12
20 min
6-8 hs
2-15hs
7 hs y su metabolito 9 hs
5,6-36% Renal
50% Renal46% Fecal
68% Renal42% Fecal
26% Renal58% Fecal
Si
No
No
No
Mecanismo de acción Vida media Eliminación DializableDroga
Tabla 7. Drogas antiplaquertarias
COX-1: ciclooxigenasa-1; TXA2: Tromboxano A2.

297
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
Recomendaciones particulares (Tabla 8)
Laboratorio de hemostasia en pacientes con tratamiento antitrombótico y complicaciones hemorrágicas
Antagonistas de la vitamina K (AVK). Los AVK requieren monitoreo para ajustar la dosis y optimizar la eficacia y seguridad. Para tal fin, la Organización Mundial de la Salud ha recomendado la utilización del RIN (Razón Internacional Normalizada). Los detalles técnicos se encuentran en el manual de procedimiento CAHT (7).Cuando un paciente bajo tratamiento con AVK presenta una complicación hemorrágica, los ensayos del laboratorio que deben estar incluidos además del RIN son: tiempo de protrombina (TP), tiempo de tromboplastina parcial activado (TTPA), fibrinógeno y hemograma con recuento de plaquetas. Este
Valorar el riesgo/beneficio de la eventual suspensión de la droga, comenzando por las medidas generales de control del sangrado.
• Discontinuar el agente plaquetario (conducta de buena práctica).• Si el paciente con sangrado mayor no requiere cirugía (incluyendo pacientes con sangrado intracerebral) y puede ser
estabilizado. No transfundir concentrado de plaquetas (1). Esta recomendación es independientemente del tipo de agente antiplaquetario utilizado, del ensayo de función plaquetaria, del volumen de sangrado intracerebral o del examen neurológico (recomendación condicional de baja calidad de evidencia).
• Se sugiere transfusión de plaquetas a aquellos que serán intervenidos quirúrgicamente de urgencia (recomendación condicional de baja calidad de evidencia). Si se transfunden plaquetas, el ideal es administrarlas 3-5 vidas medias después de la última dosis para evitar la inhibición farmacológica de las plaquetas transfundidas. Sugerimos sólo una unidad de plaquetas de aféresis (equivalente a 1 U de donante único cada 10 Kg del paciente) (5). Se sugiere realizar pruebas de función plaquetaria antes de transfundir plaquetas y no transfundir a pacientes con pruebas en límites normales o resistencia documentada.
• En candidatos a ser transfundidos, recomendamos fuertemente iniciar con una dosis de plaquetas de aféresis y sólo repetir si el sangrado continúa.
• Recomendamos considerar una dosis única de DDAVP en sangrado mayor y en especial hemorragia intracerebral (0,4 µg/Kg EV). El DDAVP puede utilizarse con el agregado de transfusión de plaquetas (recomendación condicional baja evidencia.
Sangrado menor y/o moderado
Sangrado mayor o intracerebral
Tabla 8. Recomendaciones particulares para el manejo del sangrado por antiagregantes
DDAVP: desmopresina; EV: endovenosa.

298
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
conjunto de ensayos representa una evaluación global del paciente. En estos casos, no se aconseja medir el RIN con un dispositivo portátil ya que además de no indicar otros parámetros hemostáticos, el rango de mejor correlación, comparado con el método tradicional, es entre 1,8 y 4,5.Si se sospecha que el paciente tiene la función hepática alterada, antes de la prescripción de la vitamina K, se aconseja medir el nivel de actividad de FV. Si estuviera en niveles anormales interpretar que la alteración del TP/RIN no es debida sólo a los AVK y por tal motivo, el tratamiento únicamente con vitamina K podría no ser efectivo.Utilizar reactivos de tromboplastina sin sensibilidad al inhibidor lúpico (IL) (3).Si bien muchos de los reactivos de tromboplastina contienen inhibidores de heparina (por ej. polibrene) se debe tener en cuenta que el RIN se modificará cuando la muestra tenga una concentración plasmática mayor de 1 UI/mL de HNF y mayor de 2 UI anti-Xa/mL de HBPM, no permitiendo una valoración adecuada de los factores vitamina K dependientes.Heparina no fraccionada (HNF). El TTPA y anti-Xa son los ensayos más ampliamente utilizados para monitorear la HNF.Cuando un paciente bajo tratamiento con HNF presenta una complicación hemorrágica, los ensayos de laboratorio que deben estar incluidos además de TTPA y/o anti-Xa son: TP, fibrinógeno y hemograma con recuento de plaquetas. Este conjunto de ensayos representa una evaluación global del paciente. Debido a que los diferentes reactivos comerciales de TTPA varían su respuesta a la HNF y que los resultados dependen de la combinación reactivo/instrumento, se sugiere que cada laboratorio defina la manera de establecer el rango terapéutico para sus pacientes.Se han descrito cuatro diferentes maneras de definir el rango terapéutico: (1) Segundos de TTPA, (2) Razón de TTPA (1,5-2,5 veces el valor basal), (3) TTPA calibrado frente al anti-Xa (TTPA del paciente correspondiente a 0,3-0,7 U/mL de anti-Xa y/o de una heparinemia de 0,2-0,4U/ml) y (4) Nivel de anti-Xa de 0,3-0,7 U/mL. Todos los métodos tienen sus limitaciones, aunque el primer método no se aconseja debido a que es el menos reproducible. Aun cuando el TTPA es de fácil realización, bajo costo, automatizable y se tiene mucha experiencia en los laboratorios, la mayor ventaja del ensayo de anti-Xa frente al TTPA es que está menos afectado por variables biológicas (por ej. niveles de los factores de coagulación o la presencia de IL). Por estos motivos, las asociaciones College of American Pathologists (CAP) y American College of Chest Physicians (ACCP) recomiendan definir el rango terapéutico

299
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
por el método del TTPA calibrado frente al anti-Xa, aunque el método óptimo para el seguimiento de los pacientes con HNF no está establecido hasta el momento (8). Los detalles técnicos se encuentran en el manual de procedimiento CAHT. (7)Se recomienda que las calibraciones se realicen con la misma heparina que recibe el paciente como tratamiento.El término heparinemia se refiere a medir la concentración de la heparina en U/mL por el método de neutralización con sulfato de protamina. Si bien es el patrón oro, no es utilizado en la práctica clínica diaria por considerarse un método muy laborioso y no automatizable.No se recomienda extraer muestras de sangre de catéter heparinizados. En caso que el catéter sea el único acceso vascular disponible, descartar de 5 a 10 mL de sangre antes de tomar la muestra.Heparina de bajo peso molecular (HBPM). Si bien la HBPM no requiere monitoreo en adultos, está indicado en pacientes: adultos mayores, en algunos casos en el embarazo, pacientes obesos, con falla renal o hepática, en pediatría y en pacientes con complicación hemorrágica.El método de anti-Xa cromogénico es el recomendado. La curva de calibración debe realizarse con la HBPM que está recibiendo el paciente.Cuando un paciente presenta una complicación hemorrágica, los ensayos de laboratorio que deben estar incluidos además del nivel anti-Xa son: TP, TTPA, fibrinógeno y un hemograma con recuento de plaquetas. Se debe determinar el tiempo que transcurrió desde la colocación de la última dosis hasta la extracción de la muestra de sangre. La interpretación del resultado dependerá de ese tiempo. Recuerde que la actividad anti-Xa máxima se alcanza entre las 3 y las 5 hs luego de la inyección subcutánea de la droga.No se recomienda extraer muestras de sangre de catéter heparinizados. En caso que el catéter sea el único acceso vascular disponible, descartar de 5 a10 mL de sangre antes de tomar la muestra.
Pruebas de función plaquetaria en pacientes tratados con drogas antiplaquetarias
Aspirina o ácido acetilsalicílico (AAS). El efecto inhibitorio de la aspirina, incluso a dosis bajas, es muy uniforme y el método que se debe utilizar

300
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
para medirlo es el de agregación por transmisión de luz (LTA) con ácido araquidónico como agonista. Existe muy baja evidencia de la capacidad que tienen las pruebas de función plaquetaria para predecir complicaciones de sangrado en pacientes con AAS.Se ha descrito resistencia a la aspirina, pero la verdadera resistencia química a la aspirina, sobre la base de la incapacidad de acetilar la ciclooxigenasa-1 (COX-1), es muy rara y no se refleja en las pruebas de función plaquetaria. Recientemente, Kovács y col. desarrollaron un nuevo método de referencia que detecta directamente la acetilación o la falta de acetilación de la COX-1 en las plaquetas por Western Blot. Aunque este método es demasiado laborioso para un laboratorio de rutina, es el ensayo específico disponible hasta el momento para detectar la resistencia a la aspirina real.Inhibidores P2Y12. En la actualidad, existen diferentes ensayos que se recomiendan para la evaluación del efecto antiagregante plaquetario de los inhibidores del receptor P2Y12: los dispositivos portátiles VerifyNow P2Y12, multiplaca ADP y también el VASP-fosforilado (citometría de flujo de la fosfoproteína activada por vasodilatadores).Numerosos estudios de datos primarios y secundarios con gran número de pacientes demostraron la existencia de la asociación entre la alta reactividad plaquetaria (ARP) medida con estos tres métodos y el riesgo de hemorragia, así como la baja reactividad plaquetaria (BRP) y el riesgo de complicaciones tromboticas. Se está acumulando evidencia de que existe un nivel de reactividad plaquetaria óptimo durante el tratamiento con inhibidores P2Y12, flanqueado por los puntos de corte de APR y BRP (ventana terapéutica) otorgando un beneficio clínico neto (9).El método de LTA con ADP como agonista, se recomienda cuando no se encuentran disponibles los dispositivos estandarizados o el ensayo pVASP.Recordar que el patrón de agregación plaquetaria inducida con ADP está influenciado por otros inhibidores como la aspirina.Anticoagulantes orales directos (AODs). Los pacientes con AODs no necesitan monitoreo de rutina. Una de las circunstancias donde es necesario medir la concentración de AODs es cuando el paciente presenta un sangrado mayor, espontáneo o traumático. Existen ensayos comerciales para medir la concentración en forma específica y sensible. En todos los casos deben ser utilizados calibradores y controles droga-específicos. Los resultados deben estar expresados en concentración de masa (ng/mL). Los detalles técnicos se encuentran en el manual de

301
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
procedimiento CAHT (7).Dabigatrán• Ensayo basado en trombina diluida• Ensayo basado en Ecarina (coagulométrico o cromogénico)• Ensayo anti-IIa cromogénico (en ausencia de heparina)Rivaroxabán• Ensayo anti-FXa cromogénico• Tiempo de coagulación inducido por protrombinasaApixabán• Ensayo anti-FXa cromogénicoLos ensayos de TTPA y TP tienen una limitada utilidad para determinar la presencia de AODs, tanto de anti-FIIa como de anti-FXa.Un tiempo de trombina (TT) normal excluye la presencia de dabigatrán.Si el TT es prolongado, se aconseja evaluar la presencia de heparina, disminución de los niveles de fibrinógeno o presencia de fibrinógeno anormal para realizar una correcta interpretación.El fibrinógeno debe ser determinado usando ensayos en los que dabigatrán tiene una mínima influencia.El uso de las pruebas de detección de drogas y el reporte de la concentración deben ser interpretados en relación a la historia clínica del paciente y al momento de la recolección de la muestra de sangre relativa a la última dosis de AOD que recibió el paciente (10).En los pacientes con AODs y complicaciones hemorrágicas se evalúan otras posibles causas de sangrado. Los AODs como inhibidores del FXa o FIIa influyen sobre los ensayos coagulométricos y cromogénicos dando resultados alterados que podrían impactar en la interpretación (Tabla 9) (11-12).Cada laboratorio debe conocer la sensibilidad de sus propios reactivos a los AODs y de esta manera asesorar al médico hematólogo sobre la interpretación (12).Actualmente, los ensayos de laboratorio son anticoagulante-específico. Un trabajo publicado recientemente muestra los resultados de un solo ensayo que podría ser útil para evaluar el efecto anticoagulante de warfarina, dabigatrán, rivaroxabán, apixabán y heparinas utilizando tromboplastina altamente diluida y el equipamiento disponible, aunque el valor clínico de este ensayo requiere más estudios (13).
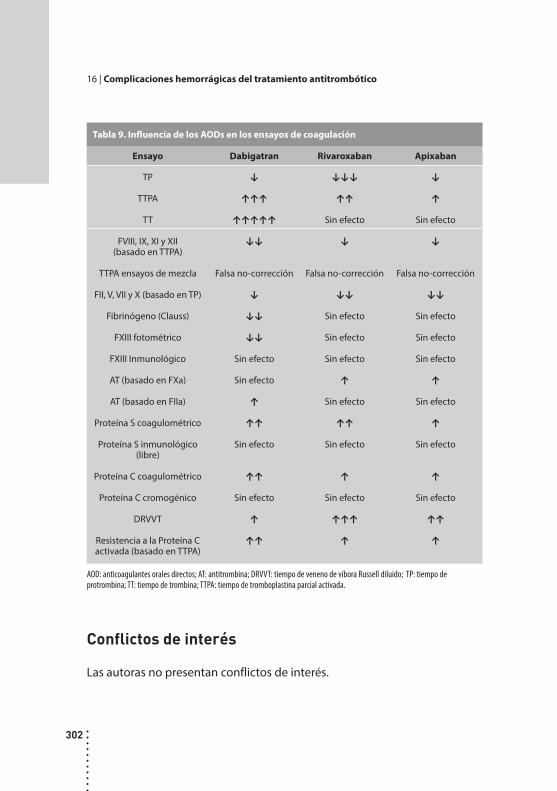
302
16 | Complicaciones hemorrágicas del tratamiento antitrombótico
Conflictos de interés
Las autoras no presentan conflictos de interés.
TP
TTPA
TT
FVIII, IX, XI y XII (basado en TTPA)
TTPA ensayos de mezcla
FII, V, VII y X (basado en TP)
Fibrinógeno (Clauss)
FXIII fotométrico
FXIII Inmunológico
AT (basado en FXa)
AT (basado en FIIa)
Proteína S coagulométrico
Proteína S inmunológico (libre)
Proteína C coagulométrico
Proteína C cromogénico
DRVVT
Resistencia a la Proteína C activada (basado en TTPA)
↓
↑↑↑
↑↑↑↑↑
↓↓
Falsa no-corrección
↓
↓↓
↓↓
Sin efecto
Sin efecto
↑
↑↑
Sin efecto
↑↑
Sin efecto
↑
↑↑
↓↓↓
↑↑
Sin efecto
↓
Falsa no-corrección
↓↓
Sin efecto
Sin efecto
Sin efecto
↑
Sin efecto
↑↑
Sin efecto
↑
Sin efecto
↑↑↑
↑
↓
↑
Sin efecto
↓
Falsa no-corrección
↓↓
Sin efecto
Sin efecto
Sin efecto
↑
Sin efecto
↑
Sin efecto
↑
Sin efecto
↑↑
↑
Dabigatran Rivaroxaban ApixabanEnsayo
Tabla 9. Influencia de los AODs en los ensayos de coagulación
AOD: anticoagulantes orales directos; AT: antitrombina; DRVVT: tiempo de veneno de víbora Russell diluido; TP: tiempo de protrombina; TT: tiempo de trombina; TTPA: tiempo de tromboplastina parcial activada.

303
Complicaciones hemorrágicas del tratamiento antitrombótico | 16
Referencias
1. Jennifer A. Frontera, John J, et al. Guideline for Reversal of Antithrombotics in Intracranial Hemorrhage. A Statement for Healthcare Professionals from the Neurocritical Care Society and Society of Critical Care Medicine. Neurocrit Care 2016; 24: 6-46.
2. Kozek-Langenecker SA, Afshari A, Albaladejo P et al. Management of severe perioperative bleeding: Guidelines from the European Society of Anaesthesiology European. Eur J Anaesthesiol 2013; 30: 270-382.
3. Holbrook A, Schulman S, Witt D et al. Evidence-Based Management of Anticoagulant Therapy. Chest 2012; 141: e152S-184S.
4. Law C, Raffini L. A Guide to the Use of Anticoagulant Drugs in Children. Paediatr Drugs 2015; 17: 105-114.
5. Siegal D, Garcia D, Crowther M. How I treat target-specific oral anticoagulant-associated bleeding. Blood 2014; 123: 1152-1158.
6. Li X, Sun Z, Zhao W et al. Effect of acetylsalicylic acid usage and platelet transfusion on postoperative hemorrhage and activities of daily living in patients with acute intracerebral hemorrhage. J Neurosurg 2013; 118: 94-103.
7. Fundamentos para el manejo práctico en el Laboratorio de Hemostasia. Grupo CAHT. Grupo Cooperativo Argentino de Hemostasia y Trombosis. Segunda Edición. Editoras en jefe: Blanco A, Kordich L. Editorial Fundación Bioquímica de la Provincia de Buenos Aires. Argentina. 2013.

304
8. Cuker A, Ptashkin B, Konkle BA et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7: 80-86.
9. Gross L, Aradi D, Sibbing D. Platelet Function Testing in Patients on Antiplatelet Medications. Semin Thromb Hemost 42: 306-320.
10. Gosselin R, Adcock DM. The laboratory’s 2015 perspective on direct oral anticoagulant testing. J Thromb Haemost 2016; 14: 886-893.
11. Kitchen S, Gray E, Mackie I, Baglin T, Makris M on behalf of the BCSH committee. Measurement of non-coumarin anticoagulants and their effects on tests of Haemostasis: Guidance from the British Committee for Standards in Haematology. Br J Haematol 2014; 166: 830-841.
12. Mani H. Interpretation of coagulation test results under direct oral anticoagulants. Int J Lab Hematol 2014; 36: 261-268.
13. Letertre LR, Gudmundsdottir BR, Francis CW et al. A single test to assay warfarin, dabigatran, rivaroxaban, apixaban, unfractionated heparin and enoxaparin in plasma. J Thromb Haemost. 2016; 14: 1043-1053.
16 | Complicaciones hemorrágicas del tratamiento antitrombótico


Este libro fue impreso en: “La Imprenta Digital SRL” www.laimprentadigital.com.ar
Calle Talcahuano 940 Florida, Provincia de Buenos Aires. En el mes de Septiembre del año 2016.

